







Rapport d'information n° 267 (2000-2001) de M. Claude HURIET , fait au nom de la commission des affaires sociales, déposé le 6 avril 2001
Disponible au format Acrobat (553 Koctets)
-
AVANT-PROPOS
-
I. LE RÔLE DES COMITÉS CONSULTATIFS DE
PROTECTION DES PERSONNES DANS LA RECHERCHE BIOMÉDICALE
-
II. LES OBJECTIFS ET LE DÉROULEMENT DE LA
MISSION
-
III. LES ENSEIGNEMENTS DE LA MISSION
-
A. LE CHAMP D'APPLICATION DE LA LOI : DES
DIFFICULTÉS RÉCURRENTES
-
1. L'examen des protocoles : le manque de
précision des statistiques disponibles
-
2. L'appréciation du bénéfice
direct ou indirect : un exercice délicat
-
3. L'agrément des lieux de
recherche : une procédure parfois redondante
-
4. La fourniture gratuite des moyens
nécessaires à la recherche : un frein persistant au
développement de la recherche biomédicale
-
1. L'examen des protocoles : le manque de
précision des statistiques disponibles
-
B. LE FONCTIONNEMENT DES COMITÉS : UN
PLURALISME ALÉATOIRE
-
C. L'INTERVENTION DES COMITÉS :
L'HÉTÉROGÉNÉITÉ DE L'ACTIVITÉ ET DES
MISSIONS
-
D. LES RELATIONS AVEC LES POUVOIRS PUBLICS :
UN DIALOGUE LIMITÉ
-
1. Des relations à réorganiser et
à approfondir avec les services de l'Etat
-
2. Une procédure d'affectation des moyens
budgétaires peu transparente
-
3. L'Agence française de
sécurité sanitaire des produits de santé (AFSSAPS)
souhaite approfondir ses relations avec les comités
-
4. La Conférence nationale des CCPPRB
s'affirme désormais comme un interlocuteur représentatif
-
1. Des relations à réorganiser et
à approfondir avec les services de l'Etat
-
A. LE CHAMP D'APPLICATION DE LA LOI : DES
DIFFICULTÉS RÉCURRENTES
-
IV. LES PROPOSITIONS DE LA MISSION
D'INFORMATION
-
A. L'ORGANISATION DE LA RECHERCHE
BIOMÉDICALE NÉCESSITE DES MODIFICATIONS
-
1. Clarifier la distinction entre les essais avec
et sans bénéfice individuel direct
-
2. Revenir sur l'obligation faite au promoteur de
fournir gratuitement les dispositifs médicaux
-
3. Associer davantage les personnes se
prêtant à la recherche
-
4. Assurer une meilleure régulation du
nombre des amendements
-
1. Clarifier la distinction entre les essais avec
et sans bénéfice individuel direct
-
B. LE FONCTIONNEMENT DES COMITÉS EST
SUSCEPTIBLE D'AMÉLIORATIONS
-
C. LES RELATIONS DES COMITÉS AVEC LES
AUTRES ACTEURS DE LA RECHERCHE BIOMÉDICALE DEMANDENT À ÊTRE
CLARIFIÉES
-
A. L'ORGANISATION DE LA RECHERCHE
BIOMÉDICALE NÉCESSITE DES MODIFICATIONS
-
I. LE RÔLE DES COMITÉS CONSULTATIFS DE
PROTECTION DES PERSONNES DANS LA RECHERCHE BIOMÉDICALE
-
TRAVAUX DE LA COMMISSION
-
ANNEXES
-
ANNEXE N° 1
-
QUESTIONS ÉCRITES DE M. CLAUDE HURIET
RELATIVES AUX CCPPRB ADRESSÉES
AU GOUVERNEMENT DEPUIS 1996
ET RESTÉES SANS RÉPONSE
-
ANNEXE N° 2
-
QUESTIONNAIRE ENVOYÉ AUX CCPPRB
-
ANNEXE N° 3
-
LISTE DES AUDITIONS
ET COMPTES RENDUS DES AUDITIONS
-
COMPTES RENDUS DES AUDITIONS
-
I. AUDITION DU MARDI 28 NOVEMBRE 2000
-
II. AUDITION DU MERCREDI 29 NOVEMBRE 2000
-
III. AUDITIONS DU LUNDI 4 DÉCEMBRE
2000
-
• M. JACQUES DUMONT, PRÉSIDENT, ET
MME CRESPON, DIRECTEUR DES AFFAIRES TECHNIQUES, AU SYNDICAT NATIONAL DE
L'INDUSTRIE DES TECHNOLOGIES MÉDICALES (SNITEM)
-
• M. ROBERT NAQUET, MME MARIE-CÉCILE
MASURE ET MME MARTINE LOIZEAU, REPRÉSENTANT LA CELLULE
ÉTHIQUE DU DÉPARTEMENT DES SCIENCES DE LA VIE DU CNRS, ET
PROFESSEUR JEAN-PAUL CAVERNI
-
• M. JACQUES DUMONT, PRÉSIDENT, ET
MME CRESPON, DIRECTEUR DES AFFAIRES TECHNIQUES, AU SYNDICAT NATIONAL DE
L'INDUSTRIE DES TECHNOLOGIES MÉDICALES (SNITEM)
-
IV. AUDITION DU JEUDI 7 DÉCEMBRE
2000
-
V. AUDITIONS DU MERCREDI 13 DÉCEMBRE
2000
-
• M. PASCAL PENAUD, CHEF DE SERVICE ADJOINT
AU DIRECTEUR GÉNÉRAL À LA DIRECTION GÉNÉRALE
DE LA SANTÉ ACCOMPAGNÉ DE MME CATHERINE GRILLOT-COURVALIN ET
MME MARIE-THÉRÈSE NUTINI
-
• PROFESSEUR G. HAZEBROUCQ, DIRECTEUR
PHARMACEUTIQUE DE LA PHARMACIE CENTRALE DES HÔPITAUX DE PARIS
-
• MM. JOËL MÉNARD,
DÉLÉGUÉ MÉDICAL, ET NICOLAS BEST, SECRÉTAIRE
GÉNÉRAL DE LA DIRECTION DE LA RECHERCHE CLINIQUE DE L'ASSISTANCE
PUBLIQUE DES HÔPITAUX DE PARIS (AP-HP)
-
• M. PASCAL PENAUD, CHEF DE SERVICE ADJOINT
AU DIRECTEUR GÉNÉRAL À LA DIRECTION GÉNÉRALE
DE LA SANTÉ ACCOMPAGNÉ DE MME CATHERINE GRILLOT-COURVALIN ET
MME MARIE-THÉRÈSE NUTINI
-
VI. AUDITION DU MERCREDI 20 DÉCEMBRE
2000
-
VII. AUDITION DU JEUDI 21 DÉCEMBRE
2000
-
VIII. AUDITION DU JEUDI 25 JANVIER 2001
-
I. AUDITION DU MARDI 28 NOVEMBRE 2000
-
ANNEXE N° 4
-
AGRÉMENTS ET RETRAITS D'AGRÉMENT DES CCPPRB PAR LA DIRECTION GÉNÉRALE DE LA SANTÉ (DGS)
-
ANNEXE N° 5
-
NOMBRES D'AVIS RENDUS PAR LES CCPPRB
EN 1998 ET 1999
-
ANNEXE N° 6
-
RÉPARTITION DU PRODUIT DES DROITS FIXES
EFFECTUÉE PAR LA DIRECTION GÉNÉRALE
DE LA SANTÉ EN 2000
-
ANNEXES N° 7
-
GRAPHIQUES REPRÉSENTANT LES TAUX DE PRÉSENCE PAR CATÉGORIE DES MEMBRES DES CCPPRB
-
ANNEXE N° 8
-
ORGANIGRAMME DE LA DIRECTION GÉNÉRALE
DE LA SANTÉ AVANT LA RÉFORME
-
ANNEXE N° 9
-
ORGANIGRAMME DE LA DIRECTION GÉNÉRALE
DE LA SANTÉ APRÈS LA RÉFORME
-
ANNEXE N° 10
-
TABLE DE CORRESPONDANCE
ENTRE L'ANCIENNE ET LA NOUVELLE NUMÉROTATION DU CODE DE LA SANTÉ PUBLIQUE
-
ANNEXE N° 11
-
DISPOSITIONS LÉGISLATIVES DU CODE DE LA SANTÉ PUBLIQUE APPLICABLES AUX CCPPRB
-
ANNEXE N° 12
-
TEXTE DE LA DIRECTIVE EUROPÉENNE
RELATIVE AUX ESSAIS CLINIQUES
N° 267
SÉNAT
SESSION ORDINAIRE DE 2000-2001
|
Rattaché pour ordre au procès-verbal de la séance du 5 avril 2001 Enregistré à la Présidence du Sénat le 6 avril 2001 |
RAPPORT D'INFORMATION
FAIT
au nom de la commission des Affaires sociales (1) sur le fonctionnement des comités consultatifs de protection des personnes dans la recherche biomédicale ,
Par M. Claude HURIET,
Sénateur.
(1) Cette commission est composée de : MM. Jean Delaneau, président ; Jacques Bimbenet, Louis Boyer, Mme Marie-Madeleine Dieulangard, MM. Guy Fischer, Jean-Louis Lorrain, Louis Souvet, vice-présidents ; Mme Annick Bocandé, MM. Charles Descours, Alain Gournac, Roland Huguet, secrétaires ; Henri d'Attilio, François Autain, Jean-Yves Autexier, Paul Blanc, Mme Claire-Lise Campion, MM. Jean-Pierre Cantegrit, Bernard Cazeau, Gilbert Chabroux, Jean Chérioux, Philippe Darniche, Claude Domeizel, Jacques Dominati, Michel Esneu, Alfred Foy, Serge Franchis, Francis Giraud, Alain Hethener, Claude Huriet, André Jourdain, Roger Lagorsse, Dominique Larifla, Henri Le Breton, Dominique Leclerc, Marcel Lesbros, Jacques Machet, Max Marest, Georges Mouly, Roland Muzeau, Lucien Neuwirth, Philippe Nogrix, Mme Nelly Olin, MM. Lylian Payet, André Pourny, Mme Gisèle Printz, MM. Henri de Raincourt, Bernard Seillier, Martial Taugourdeau, Alain Vasselle, Paul Vergès, André Vezinhet, Guy Vissac.
|
Santé publique. Recherche. |
AVANT-PROPOS
Mesdames, Messieurs,
Plus de dix ans après son adoption, les apports de la loi du 20 décembre 1988 relative à la protection des personnes qui se prêtent à des recherches biomédicales ne sont plus contestés. De l'avis général, cette loi a permis d'organiser une activité jusque-là mal encadrée et néanmoins indispensable aux progrès médicaux.
Ce texte était d'autant plus nécessaire qu'il existait auparavant une contradiction entre, d'une part, la réglementation de la procédure d'autorisation de mise sur le marché d'un médicament -les directives européennes imposant la mise en oeuvre d'essais thérapeutiques sur le volontaire sain- et, d'autre part, la loi qui interdit de porter atteinte à l'intégrité de la personne. Des médecins se livrant à de telles recherches pouvaient en effet voir engagée leur responsabilité civile et pénale sur la base des articles 309 (coups et blessures) ou 318 de l'ancien code pénal 1 ( * ) qui réprime la délivrance volontaire de substance qui " sans être de nature à donner la mort, sont nuisibles à la santé ", lorsqu'elles occasionnent à autrui " une maladie ou une incapacité de travail personnel " 2 ( * ) .
Dans ces conditions, l'absence de dispositions précises concernant l'organisation de la recherche biomédicale ne garantissait ni la sécurité des personnes, ni le niveau de responsabilité des promoteurs et investigateurs. Comme cela a pu être écrit : " jusqu'en 1988 la recherche clinique se pratiquait en France de manière " sauvage ", sous la seule responsabilité des médecins investigateurs, sans cadre normatif et le plus souvent sans l'accord explicite des personnes participant à ces recherches ". 3 ( * )
Instaurant un cadre clair et précis pour la recherche biomédicale, le législateur a placé le respect et la protection de la personne au coeur de ses préoccupations. En témoignent l'appréciation du bénéfice-risque, les prérequis, la nécessité de l'information et du consentement " libre, éclairé et exprès " ainsi que l'encadrement spécifique des recherches lorsque la personne qui s'y prête n'en tire aucun bénéfice direct.
Mais, ce faisant, la loi a également permis de renforcer la qualité scientifique des projets de recherche. Elle a d'ailleurs inspiré des textes législatifs et réglementaires au sein des autres pays de l'Union européenne et même au-delà. La directive du Parlement européen et du Conseil concernant le rapprochement des dispositions législatives, réglementaires et administratives des Etats-membres relatives à l'application de bonnes pratiques cliniques dans la conduite d'essais cliniques de médicaments à usage humain 4 ( * ) , adoptée le 26 février 2001, reprend également nombre de ses dispositions.
Les comités consultatifs de protection des personnes dans la recherche biomédicale (CCPPRB) sont chargés, de par la loi, d'émettre des avis sur les protocoles dont ils sont obligatoirement saisis. Ils sont " les piliers " sur lesquels repose une bonne application de la loi.
Pour autant, l'application de la loi du 20 décembre 1988 n'a pas été sans susciter des difficultés. Ainsi, le fonctionnement des 48 comités 5 ( * ) en activité début 2001 n'apparaît pas comme parfaitement homogène. La notion d'essais avec ou sans bénéfice direct fait l'objet d'interprétations différentes et le recueil du consentement apparaît parfois très difficile dans certaines circonstances pathologiques.
Il apparaît nécessaire aujourd'hui d'inventorier ces difficultés et de proposer des améliorations qui pourraient être débattues à l'occasion de la révision des lois dites de " bioéthique ".
C'est dans ce cadre que, lors de sa réunion du 9 mai 2000, la commission des Affaires sociales a confié à votre rapporteur la mission de réaliser une évaluation du fonctionnement des CCPPRB et de proposer éventuellement des améliorations 6 ( * ) .
Si les CCPPRB exercent leurs tâches convenablement malgré l'accroissement de leur charge de travail, il apparaît que leurs conditions de fonctionnement de même que l'exercice de la tutelle de l'administration doivent être revus dans le sens d'une meilleure efficacité.
I. LE RÔLE DES COMITÉS CONSULTATIFS DE PROTECTION DES PERSONNES DANS LA RECHERCHE BIOMÉDICALE
A. LA LOI : UN CADRE INDISPENSABLE POUR LA RECHERCHE BIOMÉDICALE
1. Un champ d'application très vaste
La loi n° 88-1188 du 20 décembre 1988 relative à la protection des personnes qui se prêtent à des recherches biomédicales couvre un champ d'application très large. Sont en effet concernés tous les essais ou expérimentations pratiqués sur l'être humain, en vue du développement des connaissances biologiques et médicales. Modifiée sensiblement à deux reprises 7 ( * ) , elle introduit dans le code de la santé publique un nouveau livre II bis relatif aux personnes qui se prêtent à des recherches biomédicales 8 ( * ) .
Les conditions dans lesquelles un protocole de recherche peut être engagé sont précisément définies ( article L. 209-2) . Aucune recherche biomédicale ne peut être effectuée sur l'être humain :
- si elle ne se fonde pas sur le dernier état des connaissances scientifiques et sur une expérimentation préclinique suffisante ;
- si le risque prévisible encouru par les personnes qui se prêtent à la recherche est hors de proportion avec le bénéfice escompté pour ces personnes ou l'intérêt de ces personnes ;
- si elle ne vise pas à étendre la connaissance scientifique de l'être humain et les moyens susceptibles d'améliorer sa condition.
Les conditions de la réalisation des protocoles sont en outre strictement réglementées (article L. 209-3) .
Les recherches biomédicales ne peuvent en effet être effectuées que :
- sous la direction et sous la surveillance d'un médecin justifiant d'une expérience appropriée ;
- dans des conditions matérielles et techniques adaptées à l'essai et compatibles avec les impératifs de rigueur scientifique et de sécurité des personnes qui se prêtent à ces recherches.
Des conditions plus restrictives ( cf. 2 ci-dessous ) sont posées pour les femmes enceintes ( art. L. 209-4 ), les personnes privées de liberté et les malades ne pouvant donner leur consentement ( art. L. 209-5 ) ainsi que pour les mineurs et les majeurs protégés ( art. L. 209-6 ).
Le promoteur, c'est-à-dire " la personne physique ou morale qui prend l'initiative d'une recherche biomédicale sur l'être humain " 9 ( * ) doit assurer l'indemnisation des conséquences dommageables de la recherche pour la personne qui s'y prête. Aussi, le promoteur doit-il souscrire une assurance garantissant sa responsabilité civile pouvant être engagée par ces recherches biomédicales.
Les règles à observer en matière de recueil du consentement sont précisées (art. L. 209-9) .
Préalablement à la réalisation d'une recherche biomédicale sur une personne, son consentement libre, éclairé et exprès doit être recueilli après que l'investigateur, c'est-à-dire " la ou les personnes physiques qui dirigent et surveillent la réalisation de la recherche " 1 ou un médecin qui le représente, fait connaître à la personne :
- l'objectif de la recherche ;
- les bénéfices attendus, les contraintes et les risques prévisibles, y compris en cas d'arrêt de la recherche avant son terme ;
- l'avis du comité consultatif de protection des personnes dans la recherche biomédicale (CCPPRB) ;
- et, le cas échéant, son inscription dans le fichier national prévu à l'article L. 209-17.
Quelques aménagements sont possibles pour les recherches en psychologie et dans les cas où, dans l'intérêt d'une personne malade, le diagnostic de sa maladie n'a pu lui être révélé.
Les informations communiquées doivent être résumées dans un document écrit remis à la personne dont le consentement est sollicité, ce consentement devant être recueilli par écrit.
Dans les situations d'urgence, le protocole présenté à l'avis du CCPPRB peut préciser que le consentement n'a pas été recherché et que seul a été sollicité celui des membres de sa famille, s'ils sont présents. L'intéressé est alors informé dès que possible et son consentement lui est demandé pour la poursuite éventuelle de la recherche.
|
Les différents acteurs de la recherche biomédicale Le promoteur Ce terme désigne la personne physique ou morale qui prend l'initiative d'une recherche biomédicale sur l'être humain. L' investigateur Ce terme désigne la ou les personnes physiques qui dirigent et surveillent la réalisation de la recherche. La personne Elle se prête à la recherche biomédicale. Le comité consultatif de protection des personnes dans la recherche biomédicale (CCPPRB) Il donne son avis sur les conditions de validité de la recherche au regard de la protection des personnes Le pharmacien de l'établissement où est effectuée la recherche Préalablement informé, il détient et dispense les produits, substances ou médicaments. Le directeur de l'établissement où est effectuée la recherche Préalablement informé, il signe une convention avec le promoteur. Le médecin inspecteur de la Santé Il veille au respect des dispositions de la loi. Le pharmacien inspecteur de la Santé Il veille au respect des dispositions de la loi. Il recherche et constate les infractions. Le ministre chargé de la Santé et les autorités administratives compétentes (ministère chargé de la Santé ou AFSSAPS) Le ministre agrée les CCPPRB. Les autorités administratives compétentes sont informées de la recherche et peuvent s'y opposer. |
2. La distinction entre les recherches avec ou sans bénéfice individuel direct
La loi du 20 décembre 1988 opère une nette distinction entre les recherches biomédicales selon qu'est attendu ou non un bénéfice direct pour la personne qui s'y prête.
Elle qualifie ainsi des " recherches biomédicales avec bénéfice individuel direct " les essais " dont on attend un bénéfice directe pour la personne qui s'y prête " 10 ( * ) .
A contrario , elle dénomme sans bénéfice individuel " toutes les autres recherches, qu'elles portent sur des personnes malades ou non ".
Cette distinction, claire et simple du moins dans son principe comporte des conséquences importantes.
Ces conséquences concernent tout d'abord l'appréciation du risque.
Si le principe général posé par l'article L. 209-2 du code de la santé proscrit toute recherche biomédicale si le risque prévisible encouru par les personnes qui s'y prêtent est " hors de proportion avec le bénéfice escompté pour ces personnes ou l'intérêt de cette recherche ", l'article L. 209-14 dispose que les recherches biomédicales sans bénéfice individuel direct " ne doivent comporter aucun risque prévisible sérieux pour la personne qui s'y prête ".
Elles concernent également les catégories de personnes pouvant se prêter à une recherche.
Si la recherche est sans bénéfice individuel direct, la loi interdit de solliciter les personnes privées de liberté par une décision judiciaire ou administrative, les malades en situation d'urgence et les personnes hospitalisées sans leur consentement. De même interdit-elle la recherche sur les personnes qui ne sont pas affiliées à un régime de sécurité sociale ou bénéficiaires d'un tel régime.
Elle pose des conditions supplémentaires pour deux autres catégories de personnes : les femmes enceintes, parturientes et mères qui allaitent d'une part, les mineurs, majeurs protégés et personnes admises dans un établissement sanitaire ou social 11 ( * ) d'autre part.
Outre la condition précitée de ne présenter " aucun risque prévisible sérieux ", la loi exige que la recherche sans bénéfice individuel direct :
- soit utile dans le premier cas " à la connaissance des phénomènes de la grossesse, de l'accouchement ou de l'allaitement " et dans le second cas " à des personnes présentant les mêmes caractéristiques d'âge, de maladie ou de handicap " ;
- ne puisse être réalisée autrement.
La distinction entre recherches avec ou sans bénéfice individuel direct entraîne en outre des régimes différents quant à la responsabilité du promoteur de la recherche 12 ( * ) .
Alors que pour les recherches biomédicales sans bénéfice individuel direct, le promoteur assume, même sans faute , l'indemnisation des conséquences dommageables de la recherche pour la personne qui s'y prête et celle de ses ayants droit. Pour les recherches biomédicales avec bénéfice individuel direct, le promoteur assume l'indemnisation des conséquences dommageables de la recherche pour la personne qui s'y prête et celle de ses ayants droit sauf preuve à sa charge que le dommage n'est pas imputable à sa faute ou à celle de tout intervenant .
Par ailleurs, si l'article L. 209-8 du code de la santé publique pose le principe général selon lequel " la recherche biomédicale ne donne lieu à aucune contrepartie financière directe ou indirecte pour la personne qui s'y prête " (hormis le remboursement des frais exposés), la loi (article L. 209-15 dudit code) tolère une exception pour les personnes se prêtant à une recherche sans bénéfice individuel direct.
Mais, dans le souci de prévenir toute dérive, elle prévoit un plafond aux indemnités que pourrait percevoir une même personne (plafond fixé réglementairement à 20.000 francs par an).
Elle interdit d'autre part qu'une personne puisse se prêter simultanément à plusieurs recherches.
Aussi prévoit-elle la tenue d'un fichier national des personnes se prêtant à des recherches sans bénéfice individuel direct.
Enfin, la distinction entre les recherches avec ou sans bénéfice individuel direct emporte une contrainte importante quant au lieu où se déroule la recherche. Les recherches sans bénéfice individuel direct ne peuvent en effet être réalisées que dans des lieux agréés selon les cas par l'Agence française de sécurité sanitaire des produits de santé ou par le ministre chargé de la santé, c'est-à-dire concrètement les directions régionales de l'action sanitaire et sociale. La loi (article L. 209-18 du code de la santé publique) précise que ces lieux doivent être " équipés des moyens matériels et techniques adaptés à la recherche et compatibles avec les impératifs de sécurité des personnes qui s'y prêtent ".
B. LES CCPPRB : LA PIERRE ANGULAIRE DU DISPOSITIF
Les comités consultatifs de protection des personnes dans la recherche biomédicale qui étaient, fin 2000, au nombre 13 ( * ) de 48, constituent la pierre angulaire du dispositif mis en place par la loi du 20 décembre 1988.
1. Un rôle déterminant
Les comités consultatifs de protection des personnes dans la recherche biomédicale (CCPPRB) jouent un rôle clef dans l'application de la loi.
L'article L. 209-18 du code de la santé publique définit précisément leurs attributions : ils sont chargés de donner leur avis sur les conditions de validité de la recherche au regard de la protection des personnes et notamment sur :
- la pertinence générale du projet ;
- l'adéquation entre les objectifs poursuivis et les moyens mis en oeuvre ;
- la qualification du ou des investigateurs ;
- la protection des participants ;
- les modalités de l'information des personnes se prêtant à la recherche, avant et pendant celle-ci ;
- les modalités de recueil du consentement ;
- les indemnités éventuellement versées par le promoteur en cas de recherche sans bénéfice individuel direct pour la personne.
Avant de réaliser une recherche sur l'être humain, tout investigateur (ou investigateur coordonnateur en cas d'étude multicentrique) est tenu d'en soumettre le projet à l'avis de l'un des CCPPRB compétents dans la région où il exerce son activité. Il ne peut solliciter qu'un seul avis par projet de recherche.
La liste des informations communiquées par l'investigateur au CCPPRB est détaillée par l'article R. 2029 du code de la santé publique.
|
- Identité du promoteur de la recherche et du fabricant du médicament ou matériel testé, - Titre et objectif de la recherche, - Informations utiles sur le produit, matériel ou méthode expérimentés, - Identité et expérience du ou des investigateurs, - Synthèse du dernier état des connaissances sur le sujet, - Protocole de la recherche, - Informations sur le ou les lieux de la recherche, - Nature des informations communiquées aux investigateurs. Sur les garanties prévues pour les personnes qui se prêtent à la recherche - Références des autorisations pour le médicament produit ou des certificats de conformité pour le matériel expérimenté, - Informations qui seront données aux personnes qui se prêtent à la recherche : . Objectif de la recherche, méthodologie, durée, . Bénéfices attendus, contraintes et risques prévisibles, . Droit de refus ou de retrait du consentement. - Modalités de recueil du consentement, y compris les documents qui seront remis aux personnes qui se prêtent à la recherche, - Copie de l'attestation d'assurance souscrite par le promoteur de la recherche. Pour les recherches sans bénéfice individuel direct - Autorisation pour chaque lieu de recherche, - Montant des indemnités éventuellement dues aux personnes qui se prêteront à la recherche, - Durée de la période d'exclusion. |
Le délai de réponse du CCPPPRB, fixé par la loi elle-même ( art. L. 209-12 ) est de cinq semaines au plus si le dossier est complet.
L'avis favorable ainsi qu'une lettre d'intention décrivant les données essentielles de la recherche doivent être transmis par le promoteur, et non par l'investigateur, à l'autorité administrative compétente, c'est-à-dire à l'Agence française de sécurité sanitaire des produits de santé (AFSSAPS) pour les produits mentionnés à l'article L. 793-1 (c'est-à-dire les produits sanitaires destinés à l'homme et certains produits à finalité cosmétique ou d'hygiène corporelle) ou au ministre chargé de la santé dans les autres cas.
Le CCPPRB peut également émettre un avis favorable à la réalisation d'une recherche sous réserve de la transmission d'informations complémentaires par l'investigateur, pendant le déroulement de cette recherche (art. L. 209-12-1, premier alinéa). A la suite de cette transmission, le comité peut maintenir ou modifier son avis. Son délai de réponse est de cinq semaines au plus. L'avis du comité doit être dans ce cas transmis par le promoteur à l'autorité administrative compétente, dans la semaine de son adoption.
Enfin, les projets ayant fait l'objet d'un avis défavorable sont directement communiqués par le CCPPRB à l'autorité administrative compétente (AFSSAPS pour les produits mentionnés à l'article L. 793-1 sinon ministère chargé de la Santé). Ils ne peuvent être mis en oeuvre avant un délai de deux mois à compter de leur réception par l'autorité administrative compétente qui seule peut en interdire la réalisation.
L'avis du CCPPRB représente donc une étape essentielle de la recherche incitant l'investigateur à présenter un dossier complet et clair qui réponde scrupuleusement aux conditions fixées à l'article R. 2029 du code de la santé publique. Il constitue également un outil précieux pour l'autorité administrative chargée d'apprécier les risques pour la santé publique et d'exercer la police sanitaire.
2. Une composition pluraliste
Selon l'article L. 209 du code de la santé publique, " les comités sont composés de manière à garantir leur indépendance et la diversité des compétences dans le domaine biomédical et à l'égard des questions éthiques, sociales, psychologiques et juridiques. Leurs membres sont nommés par le représentant de l'Etat dans la région où le comité siège. Ils sont choisis parmi les personnes figurant sur une liste établie sur proposition d'organismes ou d'autorités habilités à le faire, dans des conditions déterminées par décret ".
Les comités consultatifs de protection des personnes dans la recherche biomédicale comprennent ainsi douze membres titulaires et autant de membres suppléants ( art. R. 2001 du code de la santé publique ) répartis en huit catégories :
- quatre personnes, dont au moins trois médecins, ayant une qualification et une expérience approfondie en matière de recherche médicale,
- un médecin généraliste,
- deux pharmaciens dont l'un au moins exerce dans un établissement de soins,
- une infirmière ou un infirmier au sens des articles L. 473 à L. 477 du code de la santé publique,
- une personne qualifiée en raison de sa compétence à l'égard des questions d'éthique,
- une personne qualifiée en raison de son activité dans le domaine social,
- une personne autorisée à faire usage du titre de psychologue,
- une personne qualifiée en raison de sa compétence en matière juridique.
Deux principes sont ainsi énoncés : l'indépendance des comités et la diversité des compétences.
L'indépendance des comités est indispensable, elle constitue le fondement de leur crédibilité et de leur légitimité. Elle est garantie par l'attribution du pouvoir de nomination au préfet de région et par le pluralisme dans la composition qui permet d'assurer la confrontation des points de vue et d'éviter la constitution de " lobbies " au sein des comités.
La loi 14 ( * ) tire les conséquences de ce principe en prévoyant que " le ministre chargé de la santé peut retirer l'agrément d'un comité si les conditions d'indépendance, de composition ou de fonctionnement nécessaires pour assurer sa mission dans les meilleures conditions ne sont plus satisfaites ".
II. LES OBJECTIFS ET LE DÉROULEMENT DE LA MISSION
A. UNE ÉVALUATION NÉCESSAIRE DIX ANS APRÈS LA CRÉATION DES PREMIERS CCPPRB
1. Une première analyse utile : le rapport Mattei de 1994
La loi du 20 décembre 1988 constituait une condition nécessaire au développement de la recherche biomédicale. Elle a marqué en outre une indéniable avancée dans la protection des personnes.
Cependant, sa mise en oeuvre n'a pas été sans soulever des difficultés.
Six ans après le vote de la loi, M. Jean-François Mattei, député, a dressé un premier bilan de son application à l'invitation du Premier ministre de l'époque, M. Edouard Balladur 15 ( * ) . Ce bilan faisait apparaître deux sortes de difficultés : celles liées au champ d'application de la loi et celles résultant des procédures mises en oeuvre.
a) Le champ d'application de la loi
M. Jean-François Mattei évoquait tout d'abord les réticences des industriels face à l'inclusion dans le champ d'application de la loi des essais de médicaments dits de phase IV. Il remarquait que le recueil du consentement des personnes et le passage devant un CCPPRB constituaient une contrainte lourde. Il observait par ailleurs des difficultés concernant l'application de la loi dans le domaine du génie biomédical et de la cosmétique. Toutefois, il se prononçait pour le maintien de ces essais dans le champ d'application de la loi.
Il insistait également sur le problème posé par le coût des dispositifs médicaux soumis aux essais, rappelant que l'article R. 2038 du code de la santé publique posait le principe de la fourniture gratuite, par le promoteur, des matériels et produits pendant la période de l'essai. Il observait que cette disposition entraînait des coûts financiers très élevés pour les industriels en particulier dans le cas des prothèses implantables et des équipements lourds.
M. Jean-François Mattei observait en outre que les chercheurs en psychologie et en neurophysiologie avaient peu appliqué la loi du 20 décembre 1988 " bien que leurs travaux (entraient) sans conteste dans son champ ". Il expliquait ce phénomène par un déficit de formation des chercheurs et par le fait que la loi réservait la direction des recherches " aux seuls médecins, minoritaires dans ces disciplines " . 16 ( * )
Le professeur Mattei évoquait enfin les difficultés liées au consentement dans les domaines de " la pédiatrie, la gériatrie, la psychiatrie, la chirurgie et, à un moindre degré, toutes les recherches qui concernent des malades dont le pronostic est grave ou fatal " . Il observait que " le problème du consentement, qui porte là sur des personnes très fragilisées, se révèle particulièrement délicat " . Mais, il insistait sur la nécessité de continuer les recherches dans ces domaines et de les maintenir dans le champ d'application de la loi afin de ne pas priver de protection les personnes qui se prêtent aux expérimentations.
b) Les procédures mises en oeuvre
Concernant la mise en oeuvre des procédures, M. Jean-François Mattei soulignait la nécessité d'adapter le nombre de comités, afin que le nombre d'avis examinés par chacun soit compris entre 50 et 150 par an.
Il saluait l'action des membres des comités amenés à siéger " de manière bénévole, dans des conditions matérielles souvent déplorables, et d'après les observations des inspecteurs de l'IGAS, sans grand soutien des services déconcentrés des affaires sociales, lesquels n'avaient d'ailleurs reçu aucun moyen humain supplémentaire pour assurer le suivi de la loi " 17 ( * ) .
Mais il s'inquiétait de la dépendance matérielle des comités vis-à-vis des établissements de santé " hôtes " et du manque en moyens, de secrétariat et d'archivage qui en résultait.
Par ailleurs, il insistait sur les lacunes du dispositif de suivi par l'administration, observant " qu'il n'y a pas véritablement de suivi des effets graves non plus que d'organisation d'un lien avec la pharmacovigilance ". Il remarquait ainsi " qu'il n'est pas prudent que l'administration laisse sans suite la plupart des déclarations d'effets graves, non plus que de demander au promoteur des informations et de n'en rien faire " . Il estimait ainsi qu'il n'était " pas convenable, quelle que soit la qualité du laboratoire concerné, que le promoteur détermine seul ce qu'il considère ou non comme un effet grave, sans que l'administration lui fasse connaître ses réponses et prenne, le cas échéant, des mesures de protection " .
Il concluait que " la faiblesse du dispositif de suivi administratif (était) à l'origine d'une situation dangereuse, tant pour le public qui se croit à tort protégé, que pour l'Etat sur lequel ne manqueront pas de retomber les accusations en cas de complications mal mesurées d'une recherche qu'il avait la faculté d'interdire ".
|
Le suivi des " effets graves " La loi du 25 juillet 1994 modifiant le livre II bis du code de la santé publique relatif à la protection des personnes qui se prêtent à des recherches biomédicales a renforcé les obligations du promoteur concernant les suites à donner aux effets graves provoqués par la conduite de recherches biomédicales. La loi du 1er juillet 1998 relative au renforcement de la veille sanitaire et du contrôle sanitaire de produits destinés à l'homme a établi l'AFSSAPS comme autorité chargée d'assurer le suivi de ces effets dans la plupart des cas 18 ( * ) . Le sixième alinéa de l'article L. 209-12 fait obligation au promoteur d'informer l'autorité administrative compétente dès qu'il en a connaissance " de tout effet ayant pu contribuer à la survenue d'un décès, provoquer une hospitalisation ou entraîner des séquelles organiques ou fonctionnelles durables et susceptibles d'être dues à la recherche ". Il doit également transmettre " toute information relative à un fait nouveau concernant le déroulement de la recherche ou le développement du produit ou du dispositif faisant l'objet de la recherche lorsque ce fait nouveau est susceptible de porter atteinte à la sécurité des personnes qui se prêtent à la recherche ". Par ailleurs, le septième alinéa de l'article L. 209-12 autorise l'autorité administrative compétente à demander à tout moment des informations complémentaires sur la recherche. Cette autorité peut également suspendre ou interdire à tout moment une recherche biomédicale " en cas d'absence de réponse du promoteur, de risque pour la santé publique ou de non-respect des dispositions légales ". Ces modifications importantes apportées à la législation répondaient directement aux critiques formulées par le rapport de M. Jean-François Mattei concernant le suivi des " effets graves ". |
Le rapport de M. Jean-François Mattei a inspiré plusieurs dispositions de la loi du 25 juillet 1994 notamment en ce qui concerne la protection de certaines personnes, le recueil du consentement et l'organisation des comités.
Par ailleurs, la création de l'Agence française de sécurité sanitaire des produits de santé a permis d'améliorer le suivi des " effets graves ". Aussi, sept ans après la parution de ce rapport, était-il souhaitable de faire à nouveau le point sur le fonctionnement des CCPPRB.
2. La nécessité d'une évaluation complète à la veille de la révision des lois dites de bioéthique
Le bilan de l'application de la loi du 20 décembre 1988 réalisé en 1994 par M. Jean-François Mattei et la loi du 25 juillet 1994, que votre rapporteur a rapportée au nom de notre commission 19 ( * ) auraient dû permettre de corriger les quelques difficultés apparues dans l'application de ce texte novateur. Ce ne fut que partiellement le cas en matière de consentement et de protection des personnes.
De nombreux présidents et membres de comités ont ainsi attiré l'attention de votre rapporteur au fil des ans. L'opacité du financement, les carences en matière de suivi des nominations et les difficultés à déterminer le champ d'application de la loi étaient souvent citées parmi les dysfonctionnements les plus pénalisants touchant les comités.
Depuis 1996, à neuf reprises, au moyen de questions écrites, votre rapporteur a souhaité obtenir des éclaircissements de la part du Gouvernement sur l'ensemble de ces points. Fait particulièrement regrettable, aucune d'elles n'a reçu de réponse à ce jour.
La multiplication des interrogations de la part des comités et l'absence persistante de réponse du Gouvernement justifient que votre commission des Affaires sociales ait souhaité examiner le fonctionnement de ces structures dans le cadre de ses compétences relatives au suivi de la bonne application des lois.
Aussi, a-t-elle décidé, lors de sa réunion du 9 mai 2000, de procéder à une évaluation de ce texte et d'étudier les modifications nécessaires 20 ( * ) .
Dans ce cadre, trois questions principales ont retenu l'attention de votre rapporteur :
- dans sa définition, le champ d'application de la loi est-il suffisamment clair pour l'ensemble des recherches biomédicales ?
- le pluralisme dans la composition des comités est-il respecté et leur fonctionnement est-il satisfaisant ?
- selon quelles règles le financement des comités est-il assuré ?
B. LE DÉROULEMENT DE LA MISSION
1. Les sources d'information
a) L'envoi d'un questionnaire aux comités
Afin de recenser les difficultés d'application de la loi, votre rapporteur a adressé à chaque président des 48 comités existants 21 ( * ) un courrier en date du 1 er juin 2000 afin que lui soient communiquées toutes informations relatives au fonctionnement de ces structures ainsi que les suggestions permettant d'en améliorer le fonctionnement.
Un questionnaire 22 ( * ) était joint à ce courrier afin de préciser les deux centres d'intérêt de votre rapporteur. Il comprenait ainsi une dizaine de questions relatives à la composition des comités, à la périodicité de leurs réunions, au taux d'absentéisme, à la nature et au nombre des protocoles examinés, aux caractéristiques des promoteurs, aux modalités de financement ainsi qu'aux relations des comités avec les autres acteurs de la recherche biomédicale.
b) Les questions posées au Gouvernement
Souhaitant connaître les informations recueillies par l'administration, votre rapporteur a demandé, par courrier en date du 7 juin 2000, à Mme Dominique Gillot, secrétaire d'Etat à la santé et aux handicapés, de lui communiquer les données concernant l'activité, le fonctionnement et le financement de ces comités.
Ce courrier étant resté sans réponse, votre rapporteur a obtenu l'engagement de la Direction générale de la santé (DGS), lors de son audition le 13 décembre 2000, d'une prompte transmission de données quantitatives et qualitatives.
Cette promesse n'ayant pas eu de suite, votre rapporteur a réitéré sa demande à la ministre dans un second courrier en date du 16 janvier 2001.
A l'issue de ces multiples démarches, votre rapporteur a reçu de la Direction générale de la santé quelques éléments de nature essentiellement budgétaire.
c) Les auditions
Votre rapporteur a ensuite procédé à une série d'auditions 23 ( * ) afin de compléter son information.
Outre les services centraux de l'administration (DGS) votre rapporteur a souhaité rencontrer également certains services déconcentrés (DRASS) ainsi que les représentants de l'Agence française de sécurité sanitaire des produits de santé (AFSSAPS). Ces auditions des organismes de tutelle et de l'agence chargée d'assurer la sécurité sanitaire en matière de produits de santé devaient permettre de mieux comprendre les règles de fonctionnement et de suivi des comités.
Afin de mieux appréhender l'activité des comités, votre rapporteur a également souhaité rencontrer les " usagers " c'est-à-dire en particulier les promoteurs, qu'ils soient publics (AP-HP, INSERM, CNRS) ou privés (industrie pharmaceutique, dispositifs médicaux...).
Ce programme aurait été incomplet sans l'audition de personnalités reconnues en matière de recherche clinique. Enfin, votre rapporteur a auditionné le président de la Conférence nationale des CCPPRB.
2. Les réponses reçues par votre rapporteur
a) Un taux de réponse satisfaisant de la part des comités
Les deux tiers des comités ont répondu promptement et de manière détaillée au questionnaire envoyé par votre rapporteur.
Le tiers restant a été plus difficile à mobiliser pour des raisons tenant soit à des difficultés d'organisation (renouvellement en cours, secrétariat débordé) soit à une certaine mauvaise volonté.
Si les problèmes d'organisation ont pu donner lieu à un retard pouvant atteindre dans certains cas plusieurs mois, il convient d'observer que la mauvaise volonté manifestée par un petit nombre de comités a été plus dommageable puisqu'elle a donné lieu au renvoi de réponses hâtives voire à une absence totale de réponse de la part d'un comité (Necker).
Votre rapporteur souhaite remercier l'ensemble des membres des comités qui ont répondu à ce questionnaire afin d'éclairer le Parlement sur le fonctionnement d'une structure, les CCPPRB, à laquelle la loi du 20 décembre 1988 a confié un rôle pivot.
La plupart ont tenu à assortir leurs réponses de remarques et de propositions. Elles ont constitué une source d'information précieuse pour votre rapporteur qui a souhaité leur donner un large écho dans le présent rapport.
b) Une mobilisation laborieuse de la Direction générale de la santé (DGS)
Les difficultés rencontrées par votre rapporteur dans ses relations avec le ministère chargé de la Santé ont confirmé les craintes qu'avaient déjà soulevées l'absence de réponses du Gouvernement aux neuf questions écrites qu'il avait posées depuis 1996 sur le fonctionnement des CCPPRB 24 ( * ) .
Questions écrites de M. Claude Huriet
relatives aux CCPPRB
adressées au Gouvernement depuis 1996 et
restées sans réponse
|
Date parution JO |
N° de
|
Titre |
Suite |
|
18 avril 1996 |
14.993 |
Conditions de renouvellement des membres des CCPPRB |
sans réponse |
|
3 octobre 1996 |
17.785 |
Conditions de renouvellement des membres des CCPPRB |
sans réponse |
|
6 mars 1997 |
20.913 |
Conditions de renouvellement des membres des CCPPRB |
sans réponse |
|
13 mars 1997 |
21.043 |
Bilan financier du fonctionnement des CCPPRB |
sans réponse |
|
3 juillet 1997 |
00.669 |
Bilan d'activité des CCPPRB |
sans réponse |
|
10 juillet 1997 |
01.119 |
Bilan d'activité des CCPPRB |
sans réponse |
|
1 er octobre 1998 |
11.044 |
Représentation des médecins libéraux au sein des CCPPRB |
sans réponse |
|
21 octobre 1999 |
19.646 |
Dossiers examinés par les CCPPRB |
sans réponse |
|
27 octobre 2000 |
22.097 |
Fonctionnement des CCPPRB |
sans réponse |
Source : commission des Affaires sociales
Le contenu des réponses reçues amène votre rapporteur à considérer que le ministère n'a pas satisfait à son obligation d'assurer la pleine et entière information du Parlement.
En réalité, il apparaît que la pauvreté des réponses obtenues traduit moins une volonté de " rétention " que l'insuffisance des informations dont dispose la DGS et du suivi de l'application de la loi du 20 décembre 1988 qu'elle est censée assurer.
Ainsi le directeur général de la Santé, M. Lucien Abenhaïm, exprimait, dans un courrier en date du 7 février 2001, ses regrets " de ne pas pouvoir fournir les analyses plus détaillées (...) notamment concernant les différentes catégories de dossiers, pour des raisons de moyens affectés à ce type de travail ".
Il précisait toutefois que " pour l'avenir, la mise en place du suivi de ces dossiers au sein du bureau nouvellement créé 25 ( * ) " recherches et prospectives ", à la sous-direction des politiques de santé et stratégies de la Direction générale de la santé, devrait (...) permettre d'améliorer ce suivi et son exploitation ".
Votre rapporteur observe, au demeurant, que les difficultés rencontrées par la DGS ne se cantonnent pas à la simple tutelle des CCPPRB. En témoigne le dépôt le 4 janvier 2001 à l'Assemblée nationale d'une proposition de résolution tendant à créer une commission d'enquête sur l'aptitude matérielle et humaine de la Direction générale de la santé à assurer ses missions de santé publique et de sécurité sanitaire 26 ( * ) .
III. LES ENSEIGNEMENTS DE LA MISSION
Les informations recueillies auprès des comités, les observations que ces derniers ont formulées, les réflexions des personnalités auditionnées permettent à votre rapporteur de tirer de nombreux enseignements quant à l'application de la loi, quant au fonctionnement des comités, quant à l'évolution de leur activité et enfin quant à leur rapport avec l'Administration.
Ces enseignements fondés sur les préoccupations des différents acteurs de la recherche médicale constituent la base des propositions que formule le présent rapport.
A. LE CHAMP D'APPLICATION DE LA LOI : DES DIFFICULTÉS RÉCURRENTES
La première préoccupation a été d'apprécier les conditions dans lesquelles, dans le cadre de la loi du 20 décembre 1988, s'était développée la recherche biomédicale. Une première réflexion s'impose : dans un domaine pourtant aussi sensible que la protection des personnes, il est difficile de recueillir une information complète et cohérente sur l'activité des comités, les difficultés qu'ils rencontrent, les choix qu'ils sont amenés à opérer. L'Administration ne dispose guère d'une telle information ; les différentes données statistiques mais également qualitatives fournies par les comités ne sont pas aisées à agréger.
1. L'examen des protocoles : le manque de précision des statistiques disponibles
a) La part prépondérante de l'industrie pharmaceutique parmi les promoteurs
Le nombre total des avis rendus s'élevait en 1999 à 2.280 en baisse de 5,5 % par rapport à 1998 selon les données transmises par la DGS 27 ( * ) . Ces données distinguent deux catégories de promoteurs : d'une part, les personnes physiques ainsi que les établissements ou organismes de soins, de formation ou de recherche sans but lucratif ; d'autre part, les industriels.
Le choix de cette présentation sommaire s'explique probablement par l'appareil statistique relativement fruste de l'Administration qui se fonde sur la différence des droits applicables à chacune des deux catégories. La première acquitte en effet un droit réduit par rapport à la seconde 28 ( * ) .
Nombre d'avis rendus par les comités en 1998 et 1999
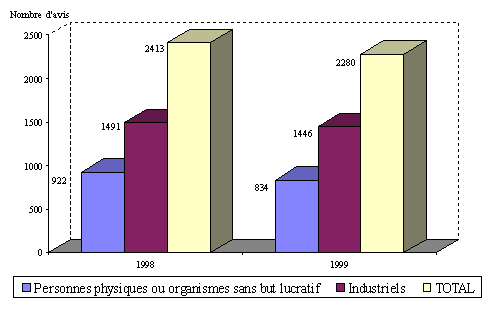
Source : DGS (février 2001)
Compte tenu de cette distinction, il apparaît que 63 % des protocoles examinés en 1999 avaient été déposés par des industriels et 37% par des personnes physiques ou des établissements ne poursuivant pas de but lucratif.
Se voulant plus précise, la DGS, dans une note du 18 janvier 2001, a estimé que " la nature des promoteurs dépend de la catégorie des recherches :
- pour les dossiers relevant de la compétence de l'AFSSAPS (produits relevant de l'article L. 5311-1 du code de la santé publique), les promoteurs sont essentiellement des industriels ;
- pour les dossiers relevant de la compétence de la DGS (physiologie, physiopathologie, épidémiologie, génétique), les promoteurs sont essentiellement des institutionnels (centres hospitaliers, INSERM, CNRS...) ".
Le questionnaire adressé à chacun des comités était plus ambitieux puisqu'il leur demandait de distinguer trois catégories de promoteurs : les firmes pharmaceutiques, les fabricants de dispositifs médicaux et les institutionnels (établissements hospitaliers, INSERM, CNRS...).
Les réponses portant sur trois années (1997-1999) sont cohérentes avec les données du ministère quant à la proportion des protocoles présentés par les industriels. Elles font apparaître également, parmi les promoteurs, la part prédominante des firmes pharmaceutiques (60 % des protocoles présentés).
Le nombre de projets déposés par des fabricants de dispositifs médicaux ne dépasserait pas en revanche 5 % du total. Nous reviendrons sur ce point. Quant à la troisième catégorie de promoteurs, hétérogène dans sa composition, car regroupant les institutions, associations et personnes physiques, elle représenterait plus du tiers des projets de recherche présentés 29 ( * ) .
Répartition des protocoles selon leurs
promoteurs*
(en unité et % du
total)
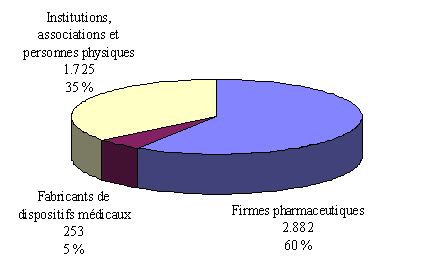
* 3 exercices cumulés (1997, 1998 et 1999) - 35 comités
b) La prédominance des essais sur les médicaments
Dans son questionnaire adressé aux comités, votre rapporteur avait également souhaité connaître la répartition des protocoles selon leur objet en distinguant quatre catégories : les médicaments, les dispositifs médicaux, les recherches cognitives-ergonomie et les recherches en psychologie.
Selon les réponses reçues 30 ( * ) , les protocoles de recherche que les comités sont amenés à examiner se répartissent comme suit : les médicaments représentent 78 % des projets de recherche, les dispositifs médicaux 10 %, les recherches cognitives 9 % et les recherches en psychologie 3 %.
Répartition des protocoles selon leur
objet*
(en unité et % du total)
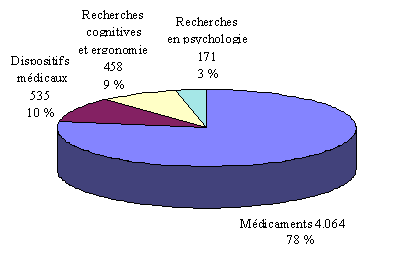
* 3 exercices cumulés (1997-1998-1999) - 42 comités
Le nombre des recherches en psychologie et comportements apparaît particulièrement faible (3 % du total). M. Jean-François Mattei expliquait dans son rapport 31 ( * ) au Premier ministre de 1994 cette situation par le fait que " de nombreux interlocuteurs de la mission semblaient ignorer que la loi Huriet ne s'appliquait pas seulement aux essais de médicaments, mais aussi aux recherches en psychologie ou aux expérimentations sur les réactions psychologiques dans des situations extrêmes ". 32 ( * )
Certes, la loi du 25 juillet 1994 a depuis introduit la possibilité pour un directeur de recherche d'" exercer conjointement " avec l'investigateur la direction de la recherche pour des recherches comportementales 33 ( * ) , mais les représentants de l'INSERM ont souligné, lors de leur audition, que cette évolution n'avait pas permis de " normaliser " les recherches en psychologie et " posait plus généralement la question des rôles respectifs du médecin et du responsable scientifique dans un certain nombre de recherches où le rôle du médecin pourrait être explicitement limité à sa compétence médicale stricte, le directeur de recherche exerçant la responsabilité scientifique pour la réalisation du projet de recherche ".
Aussi, les représentants du CNRS ont-ils pu proposer qu'un chercheur non médecin puisse présenter un projet de recherche en laissant au CCPPRB la possibilité de demander, le cas échéant, l'avis d'un médecin.
Enfin n'apparaissent pas dans la répartition des protocoles selon leur objet, les recherches portant sur les produits cosmétiques. Dans un précédent rapport 34 ( * ) , votre rapporteur avait pu estimer que " l'industrie des cosmétiques, dans sa grande majorité, n'appliquait pas la loi du 20 décembre 1988 lorsqu'elle entreprend des essais ou bien réalise ces essais hors du territoire national afin d'échapper à une législation qu'elle estime trop contraignante ".
Les réponses au questionnaire n'ont pas permis de clarifier la situation. Certains comités confondent médicaments et produits cosmétiques dans une même catégorie alors que la plupart ne les mentionne pas. Or, l'examen des listes de promoteurs permet toutefois d'observer que les grands laboratoires spécialisés en cosmétologie (L'Oréal, Shiseido, Rochas, Guerlain, Christian Dior...) se portent souvent promoteurs.
Au-delà de quelques aperçus utiles sur les caractéristiques des promoteurs et l'objet des protocoles, le premier enseignement de la mission concerne la faiblesse de l'appareil statistique permettant de bien connaître l'activité des comités.
L'appréhension de cette activité par l'administration semble se faire sur le seul fondement de la nature des droits acquittés par les promoteurs ; les rubriques retenues par les différents comités rendent très difficiles une agrégation un peu fine des données.
Cette constatation est particulièrement fondée s'agissant des recherches dans le domaine de la cosmétologie.
Il reste, en outre, un problème de fond qui est celui des recherches qui restent en marge du champ d'application de la loi.
Ainsi, M. François Chapuis, président de la Conférence nationale des CCPPRB a-t-il pu estimer, lors de son audition, que " 40 à 50 % des recherches biomédicales " n'entraient pas dans le cadre de la loi du 20 décembre 1988 ", en citant " le cas notamment des études épidémiologiques avec un prélèvement sanguin, ou des protocoles dont les modalités de mise en oeuvre de la recherche sont insuffisamment formalisées ".
2. L'appréciation du bénéfice direct ou indirect : un exercice délicat
Compte tenu de l'importance que la loi attache, à juste titre, à cette distinction, votre rapporteur a souhaité apprécier l'importance respective des protocoles de recherche selon que les personnes qui s'y prêtent peuvent en attendre ou non un bénéfice individuel direct. Mais parallèlement il a observé que les difficultés demeuraient quant à la qualification des protocoles.
a) La prédominance des essais avec bénéfice individuel direct
Sur les trois dernières années, 62 % des protocoles examinés concernaient des essais avec bénéfice individuel direct.
Répartition des essais
cliniques
" avec " ou " sans " bénéfice
individuel direct*

*3 exercices cumulés 1997, 1998 et 1999 - 36 comités
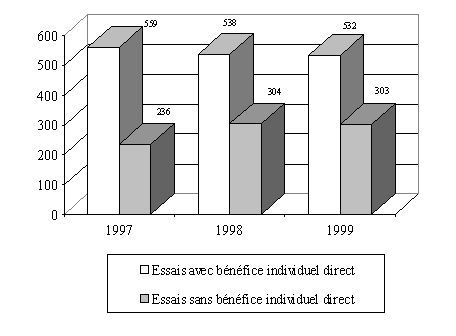
L'examen de l'évolution sur trois ans montre que la répartition entre les essais, selon qu'on peut en attendre ou non un bénéfice individuel direct, est assez stable. Il reste toutefois que seuls 13 comités ont détaillé cette information par exercice.
Evolution du nombre des essais
" avec " et
" sans " bénéfice individuel direct
(13
comités)
b) Des difficultés quant à la qualification des essais
La distinction entre essai " avec " ou " sans " bénéfice individuel direct est claire dans son principe et emporte des conséquences importantes 35 ( * ) mais elle est délicate dans son application comme en témoignent tant les réponses des comités que les observations des personnes auditionnées.
Le cas des volontaires sains est a priori simple : ils n'ont pas à attendre un bénéfice individuel direct de la recherche. Les autres situations le sont moins.
Il est ainsi significatif que l'Assistance Publique-Hopitaux de Paris ait estimé nécessaire d'éditer un guide pratique destiné aux praticiens de la recherche biomédicale 36 ( * ) qui propose une interprétation des dispositions législatives et réglementaires. Mais un tel guide, s'il peut éclairer les chercheurs, ne saurait s'imposer aux comités.
Un comité souligne les difficultés rencontrées concernant la catégorisation des essais " sans bénéfice individuel direct " lorsque l'essai bénéficie au patient mais que certains examens ne lui bénéficient pas (partie du protocole consacrée à une étude pharmacocinétique par exemple).
Un autre comité confirme les difficultés tenant aux limites du champ d'application de la loi et à la qualification du bénéfice direct sur les recherches non médicamenteuses. Il constate que " la loi a été conçue pour le médicament et des situations où il n'est pas difficile de savoir si l'on attend ou non un bénéfice direct " .
Ce même comité estime qu'il lui est souvent difficile de justifier la qualification d'essai " avec " bénéfice individuel direct dans le cas des recherches en matière cognitive, épidémiologique, psychologique et génétique.
Il déclare alors adopter l'une des trois attitudes suivantes :
" - classer les protocoles dans le champ d'application de la loi, sans bénéfice individuel direct ce qui a pour conséquence de rendre certaines études irréalisables compte tenu des conditions applicables dans ce cas notamment au regard du régime de la responsabilité ;
" - classer les protocoles dans le champ d'application de la loi en admettant un bénéfice individuel direct douteux, dans le but de rendre l'étude réalisable et considérant que le risque pour les personnes est nul ou cliniquement négligeable ;
" - ou, le plus souvent, les classer hors champ d'application de la loi, considérant qu'il n'y a pas de risque pour les personnes qui se prêtent à la recherche ".
Le comité, après avoir observé que tel n'était pas forcément son rôle, constate que les investigateurs potentiels lui demandent souvent de qualifier leur étude.
Ces réponses traduisent, à l'évidence, l'existence d'une grande incertitude quant à l'interprétation de la loi.
Aussi, le professeur G. Hazebroucq, directeur pharmaceutique à la Pharmacie centrale des hôpitaux de Paris, a-t-il estimé que les comités pourraient utilement se concerter avec l'AFSSAPS compte tenu de la complexité de cette notion en dehors des cas de volontaires sains.
De même le comité précité souhaite-t-il la définition de règles de conduite permettant de traiter des situations qui posent fréquemment problème, en mentionnant trois exemples :
" - est-ce qu'une simple prise de sang (non identifiante) suffit à mettre une étude dans le champ d'application de la loi ?
" - est-ce que le prélèvement d'un tube supplémentaire lors d'une prise de sang faite de toutes façons suffit à mettre une étude dans le champ d'application de la loi ?
" - où placer les enquêtes génétiques ? ".
Il convient néanmoins d'observer que tous les comités ne rencontrent pas les mêmes difficultés.
Ainsi, un comité remarque que " les études génétiques avec les problèmes nouveaux qu'elles posent (...) semblent pouvoir être analysées de manière satisfaisante dans le cadre de la loi actuelle dont le caractère adaptable apparaît remarquable ".
En revanche, il n'est pas douteux que la directive européenne adoptée formellement par le Parlement européen et le Conseil le 26 février 2001 " concernant le rapprochement des dispositions législatives, réglementaires et administratives des Etats membres relatives à l'application de bonnes pratiques cliniques dans la conduite d'essais cliniques de médicaments à usage humain 37 ( * ) " crée un nouveau contexte.
Lors de leur audition, les représentants de l'Institut national de la santé et de la recherche médicale (INSERM) ont ainsi rappelé que : " la qualification d'une recherche " avec bénéfice individuel direct ", telle que définie dans la loi, (était) limitée à une recherche dont on " attend un bénéfice direct pour la personne qui s'y prête " .
En ce sens, ils ont fait observer que " cette définition (était) plus restrictive que la notion inscrite dans le projet de directive européenne sur les essais cliniques de médicaments, en cours d'adoption, où la recherche avec bénéfice inclut la recherche dont on peut espérer un bénéfice sur une population de sujets souffrant d'une même affection que les sujets qui s'y prêtent ".
En tout état de cause, ces interrogations nombreuses de la part des comités font apparaître l'utilité d'établir un recueil des positions adoptées par les divers comités qui pourrait contribuer à établir une sorte de jurisprudence.
3. L'agrément des lieux de recherche : une procédure parfois redondante
Les difficultés rencontrées dans la qualification de la recherche " avec " ou " sans " bénéfice direct incluent également la procédure de l'agrément des lieux de recherche. Le principe de cet agrément est posé par l'article L. 209-18 du code de la santé publique pour les recherches biomédicales sans bénéfice individuel direct.
Cette disposition constitue une contrainte pour les chercheurs, notamment lorsqu'il s'agit de procéder à des actes simples (prise de sang, par exemple) mais aussi pour l'autorité administrative compétente.
Suivant les cas, le lieu de recherche doit bénéficier d'une autorisation émanant du ministère de la Santé ou du directeur général de l'AFSSAPS, voire des deux.
Les autorisations de lieu pour les recherches biomédicales portant sur la physiologie, la physiopathologie, l'épidémiologie, la génétique, les sciences du comportement, la nutrition (à l'exception des produits visés par l'article L. 793-1 alinéa 13 du code de la santé publique) relèvent ainsi de la compétence de la DGS.
En revanche, les autorisations de lieu de recherches biomédicales portant sur les produits et dispositifs définis à l'article L. 793-1 du code de la santé publique (à l'exception des produits des thérapies génique et cellulaire) relèvent de l'AFSSAPS.
Les procédures relatives à l'agrément des lieux de recherche sont lourdes, comme l'a confirmé l'audition des représentants de la DRASS d'Ile-de-France. Cette région comprend 550 lieux de recherche agréés répartis en 100 sites. L'essentiel de ces sites est formé d'établissements hospitaliers qui doivent respecter, en matière de sécurité, des exigences strictes pour recevoir des patients. Or, il s'avère que ces exigence sont identiques à celles requises pour autoriser un lieu de recherches sans bénéfice individuel direct sur des patients volontaires dans le domaine des activités habituelles du service.
Aussi l'agrément est toujours accordé quand l'établissement est autorisé à accueillir des patients. Il en est de même des recherches dans le domaine des sciences du comportement, réalisées dans les centres hospitaliers spécialisés, et des recherches génétiques, épidémiologiques et physiopathologiques nécessitant des structures autorisées à exécuter de tels prélèvements.
Pour de tels lieux de recherche, Mme Françoise Loeb, inspecteur régional de la pharmacie à la DRASS d'Ile-de-France, considère que " l'autorisation prévue à l'article L. 209-18 du code de la santé n'apporte pas dans les faits une sécurité supplémentaire, la protection des personnes restant assurée par l'obligation de fournir des informations concernant chaque recherche au CCPPRB et au ministre chargé de la santé ou au directeur de l'AFSSAPS " .
Compte tenu de la charge de travail que représente l'instruction des demandes d'autorisation de lieu de recherche 38 ( * ) , elle considère qu'il serait plus opportun :
" - d'accroître la vigilance lorsque les recherches se déroulent hors des établissements de santé et lorsqu'elles se déroulent en milieu hospitalier en dehors des activités habituelles de diagnostic, de surveillance et de traitement ;
" - de ne pas soumettre à autorisation, dans les établissements de santé, les services qui réalisent, dans le domaine de leurs activités habituelles, des recherches sans bénéfice individuel direct, sur leurs patients volontaires ;
" - d'envisager la possibilité de ne pas soumettre à autorisation les structures qui sont autorisées à exécuter des prélèvements sanguins lorsqu'un tel prélèvement est le seul acte subi par le volontaire. "
Les représentants de l'INSERM ont, quant à eux, estimé " urgent d'assouplir les conditions d'habilitation des lieux de recherches lorsque celles-ci se limitent à un prélèvement de sang ou un acte couramment pratiqué en médecine de ville ".
Ils ont considéré que cette mesure était " essentielle dans le cadre des recherches relevant en particulier des domaines de l'épidémiologie, de la pharmacoépidémiologie et de la génétique . Certaines recherches entrant dans ces catégories deviennent en effet dépassées avant d'être réalisées devant les délais extrêmement longs pour l'obtention de telles autorisations lorsque les recherches sont réalisées en dehors des environnements hospitaliers ".
4. La fourniture gratuite des moyens nécessaires à la recherche : un frein persistant au développement de la recherche biomédicale
L'article R. 2038 du code de la santé publique dispose que : " Les objets ou matériels ainsi que les médicaments ou produits mentionnés à l'article R. 5123 39 ( * ) sont fournis gratuitement, ou mis gratuitement à disposition pendant le temps de l'essai par le promoteur.
Il précise également que : " Le promoteur prend en charge les frais supplémentaires liés à d'éventuels fournitures ou examens spécifiquement requis par le protocole de l'essai.
Il stipule enfin que : " Si la mise en oeuvre du protocole est de nature à entraîner des frais supplémentaires de fonctionnement pour un établissement public ou privé, le promoteur prend ces frais en charge.
" Lorsque l'essai est réalisé dans un établissement public ou privé, la prise en charge des frais mentionnés aux deux précédents alinéas fait l'objet d'une convention conclue entre le promoteur et le représentant légal de cet établissement. "
Déjà en 1994, votre rapporteur appelait de ses voeux " une modification de cette réglementation, particulièrement pénalisante lorsque les personnes se prêtant à un essai en retirant un bénéfice individuel direct auraient dû en toute hypothèse utiliser ou se voir implanter le matériel faisant l'objet de l'essai ". 40 ( * )
Dans une réponse 41 ( * ) à une question écrite 42 ( * ) de votre rapporteur, le ministre avait annoncé que : " des modifications pourraient être envisagées " au vu des conclusions d'un rapport de l'Inspection générale des affaires sociales.
De fait, le rapport de l'IGAS 43 ( * ) constatait que la gratuité de la fourniture du matériel pendant la durée de l'expérimentation pouvait " représenter un surcoût net impossible à supporter " lorsque les promoteurs étaient des personnes physiques ou un établissement de recherche ou un organisme sans but lucratif.
L'IGAS considérait que cette réglementation était " d'autant plus insupportable, ne pouvant qu'inciter les industriels à ne pas effectuer les essais en France, que ces matériels (étaient) inévitablement implantés sur des sujets qui en (avaient) besoin, pour lesquels l'assurance maladie aurait, dans tous les cas, à supporter une charge financière due aux soins ".
Votre rapporteur a pu constater, au cours des auditions, que cette exigence constituait toujours un obstacle au développement de la recherche biomédicale en France 44 ( * ) .
Ainsi, selon le Syndicat national de l'industrie des technologies médicales (SNITEM), cette disposition a amené de nombreuses entreprises internationales à effectuer leurs recherches dans d'autres pays. Il considère par ailleurs qu'il existe aujourd'hui un certain consensus pour revenir sur le principe de gratuité. Selon ce syndicat professionnel, une solution pourrait consister à indemniser le promoteur sur la base du tarif de responsabilité de la sécurité sociale applicable aux dispositifs ou produits auxquels se substituent les dispositifs ou produits faisant l'objet de la recherche.
Le professeur Patrice Jaillon, président de l'Association pour le développement de la pharmacologie clinique, a confirmé que le principe de gratuité posé par l'article R. 2038 constituait un handicap pour le développement de l'innovation dans le domaine de la recherche biomédicale. Il a suggéré, quant à lui, une prise en charge financière par le Programme hospitalier de recherche clinique (PHRC).
Les représentants de l'INSERM ont estimé que l'obligation faite au promoteur de fournir gratuitement des médicaments ou produits utilisés dans l'essai " devrait pouvoir être levée, pour tous les médicaments ou produits mentionnés explicitement dans le protocole, dès lors qu'ils concernent un traitement ou un produit déjà autorisé, quand celui-ci est administré dans les indications correspondant aux autorisations de mise sur le marché ".
Selon eux : " cette exemption devrait pouvoir être étendue aux médicaments ou produits utilisés couramment dans une indication non mentionnée dans l'autorisation de mise sur le marché, dès lors que la recherche vise justement à évaluer leur rapport bénéfice/risque dans cette situation et que le promoteur est une institution publique ou reconnue d'utilité publique ", ajoutant qu'" actuellement certains coûts de recherches étaient incompatibles avec les contraintes de la recherche publique, ce qui freinait la réalisation d'études pourtant nécessaires, notamment dans le domaine de l'évaluation de stratégies en thérapeutique et en santé publique ".
B. LE FONCTIONNEMENT DES COMITÉS : UN PLURALISME ALÉATOIRE
La qualité du fonctionnement des comités est fonction de l'effectivité du pluralisme de sa composition. Il dépend donc de la présence des représentants de chaque catégorie qui le compose.
Votre rapporteur a voulu s'en assurer.
1. Le constat d'un important absentéisme
La fréquence des réunions des comités n'est pas précisée par les textes légaux et réglementaires, elle relève donc des comités qui fixent le rythme des réunions dans leur règlement intérieur. Le rythme mensuel est retenu par trente-six des comités (87 %) sur les quarante et un ayant répondu. Seuls cinq comités (13 %), ont adopté un rythme de deux réunions par mois.
a) Des difficultés méthodologiques pour évaluer l'absentéisme
Concernant l'absentéisme aux réunions, quinze comités ont répondu en faisant état d'un taux de présence moyen de l'ensemble des membres se situant entre 50 % et 80 %.
Cette donnée est peu significative car le bon fonctionnement des comités tient à une bonne représentation des huit catégories de membres qui les composent et à la satisfaction du quorum prévu par le code de la santé publique qui exige en outre un l'équilibre entre les différentes catégories de membres présents.
|
L'exigence d'un quorum L'article R. 2015 du code de la santé publique prévoit que " les délibérations ne sont valables que si six membres au moins sont présents, dont au moins quatre appartiennent aux catégories 1 à 4 mentionnées à l'article R. 2001 et au moins un appartenant aux autres catégories ". L'article R. 2001 du code de la santé publique répartit les douze membres titulaires des comités en huit catégories : I. quatre personnes, dont au moins trois médecins, ayant une qualification et une expérience approfondie en matière de recherche biomédicale ; II. un médecin généraliste ; III. deux pharmaciens dont l'un au moins exerce dans un établissement de soins ; IV. une infirmière ou un infirmier au sens des articles L. 473 à L. 477 du code de la santé publique ; V. une personne qualifiée en raison de sa compétence à l'égard des questions d'éthique ; VI. une personne qualifiée en raison de son activité dans le domaine social ; VII. une personne autorisée à faire usage du titre de psychologue ; VIII. une personne qualifiée en raison de sa compétence en matière juridique. |
Un seul comité a fait état de réunions qui ont dû être annulées, faute de quorum.
Vingt-quatre comités ont fourni des données suffisamment précises pour permettre d'évaluer le taux de présence des membres de chacune des huit catégories 45 ( * ) .
L'interprétation de ces résultats partiels se doit cependant d'être prudente. Il n'est pas possible, en effet, d'extrapoler des résultats concernant la moitié des comités à l'ensemble des comités.
Certains comités, qui ont seulement fourni une moyenne globale des présences, sans distinguer selon les catégories, ont pu estimer que les écarts entre les différentes catégories n'étaient pas significatifs.
Mais votre rapporteur s'est heurté dans sa démarche à une difficulté méthodologique : la confusion dans les réponses reçues entre l'absentéisme et la composition lacunaire de certains comités.
Certains comités, constatant en effet que des catégories n'étaient pas pourvues en représentants (catégories V et VI relatives respectivement aux personnes qualifiées en matière d'éthique et dans le domaine social), ont sans doute considéré qu'il ne leur était pas possible de fournir des données autres qu'une moyenne sans grande signification.
Cette dernière analyse est corroborée par les remarques formulées par plusieurs comités qui font état d'un taux de présence satisfaisant à l'exception d'une ou plusieurs catégories non pourvues ou dont les membres démissionnaires n'ont pas été remplacés. Ces catégories concernent en particulier les médecins généralistes, les juristes et les personnes qualifiées en matière d'éthique.
Les comités n'établissent pas de distinction entre des absences occasionnelles et les postes non pourvus. L'examen de la liste des membres des comités montre aussi qu'un nombre de postes de suppléants ne sont pas pourvus ; il en est de même pour certains postes de titulaires, ce qui est plus grave.
Bien que les éléments quantitatifs ne soient pas disponibles pour en évaluer l'importance respective, il est essentiel d'observer que deux phénomènes distincts expliquent l'absence de certaines catégories de membres dans les réunions des comités : d'une part, les déficiences dans la procédure et le suivi des nominations et des renouvellements et d'autre part le manque d'assiduité de certains membres pourtant dûment nommés.
b) Un phénomène qui concerne particulièrement les membres exerçant des professions libérales
Même si elles ne constituent que des données partielles, les réponses détaillées transmises par vingt-quatre comités concernant le taux de présence des membres par catégorie peuvent être analysées.
L'annexe n° 7 comporte une représentation graphique des données rassemblées.
Compte tenu des informations recueillies, il apparaît que le taux de présence des personnalités ayant une qualification et une expérience approfondie en matière de recherche médicale (catégorie I) est satisfaisant puisqu'il dépasse 90 % dans 60 % des réunions organisées par les comités ayant répondu de manière détaillée. Ces résultats s'expliquent notamment par le fait que les représentants de cette catégorie sont particulièrement motivés par l'activité des CCPPRB.
Le taux de présence des médecins généralistes (catégorie II) est moins satisfaisant. Ils sont quasiment absents dans près de 9 % des réunions organisées. Ils ne sont présents qu'à une réunion sur deux dans 30 % des cas.
Le taux de présence des pharmaciens (catégorie III) dépasse 60 % dans 76 % des réunions organisées par les CCPPRB.
Bien que plus faible que celui des pharmaciens, le taux de présence des infirmiers (catégorie IV) reste assez élevé puisqu'il dépasse 60 % dans 71 % des cas.
On observe, en revanche, des taux de présence souvent très faibles pour les représentants des catégories " non médicales ".
Ainsi, pour les personnes qualifiées en matière d'éthique , ce taux ne dépasse les 60 % que dans 48 % des réunions organisées par les comités. Elles sont pratiquement absentes dans 26 % des réunions des comités. Il existe même des comités où cette catégorie n'a jamais été pourvue ou pour laquelle les intéressés ne se sont jamais présentés.
Cette faible représentation des catégories non médicales est très regrettable puisqu'elle est contraire à la volonté de pluralisme du législateur et qu'elle risque de compromettre la validité des avis des comités. Les représentants de la DRASS d'Ile-de-France ont ainsi estimé que " le principe de la loi n'était plus respecté ".
L'important absentéisme constaté pour les personnes qualifiées en matière d'éthique (catégorie V) se retrouve en effet pour les personnes qualifiées dans le domaine social (catégorie VI) et pour les psychologues (catégorie VII).
Ainsi, les personnes qualifiées dans le domaine social sont peu ou pas représentées dans un quart des réunions des comités tandis que les psychologues sont peu ou pas représentés dans 16 % des réunions.
La présence des personnes compétentes en matière juridique (catégorie VIII) est également très irrégulière.
2. Les causes d'un tel phénomène
Des causes communes sont souvent à l'origine tant de l'absentéisme que des démissions qui affectent certaines catégories de membres. Il reste que l'absence d'un véritable suivi par l'Administration de " l'état civil " des comités accentue fortement ce phénomène.
a) Les limites du bénévolat
Certaines démissions semblent pouvoir s'expliquer par un déficit d'information initiale concernant tant le rôle des comités que la charge de travail qui incombe aux membres.
Un comité insiste sur la nécessité de " pré-information " et de formation des nouveaux membres lors des renouvellements. Il précise que plusieurs de ses membres ont été des " volontaires désignés d'office " alors qu'ils n'étaient pas informés de leur candidature, voire de l'existence même de la loi et de l'existence des CCPPRB !
L'absence d'indemnisation des membres des comités pour compenser les contraintes que leur imposent leurs fonctions et les pertes financières éventuelles constitue également un obstacle à la participation des professions libérales.
Comme le note, avec quelque ironie, un des comités dans sa réponse : " Le bénévolat et le volontariat ont de grandes vertus, mais peuvent générer aussi des dysfonctionnements au sein des Comités ".
Un autre comité observe que la participation des professions libérales est contrariée par les horaires des réunions lorsqu'elles ont lieu l'après-midi, ce que confirment les réponses au questionnaire concernant l'absentéisme.
Ce comité remarque en outre que " le seul médecin généraliste qui y participe régulièrement (une fois sur deux) met un remplaçant à son cabinet ce jour-là ". En 1997, il avait été décidé de lui rembourser les frais occasionnés par ce remplacement avant que la DGS ne fasse savoir que cette procédure n'était pas légale, ce qui est d'ailleurs contestable.
En effet, le principe de gratuité de l'exercice des fonctions de membre d'un comité n'a pas été déterminé par la loi mais par voie réglementaire 46 ( * ) . L'utilisation dans le décret de l'adverbe " notamment " amène à s'interroger sur le fait de savoir si la prise en compte des frais occasionnés par un remplacement n'est pas autorisée par l'article R. 2013 qui fait référence aux frais supportés par un membre " à l'occasion de sa participation aux travaux du comité ", sans limiter ces frais au seul déplacement. La Direction générale de la santé n'a pas apporté de réponse satisfaisante à cette question.
L'absentéisme ne tient pas seulement au manque d'information et à l'absence de rémunération ou d'indemnisation ; il s'explique aussi par un manque de motivation.
Un comité fait ainsi remarquer que " les modifications apportées en 1994 au mode de désignation des membres ont été bénéfiques, en évitant de tirer au sort des candidats désignés, en fait peu motivés par l'activité ". Il poursuit en observant cependant que ces modifications " n'ont pas suffi à supprimer totalement les candidatures de personnes qui trouvent peut-être la fonction valorisante sur une carte de visite mais ne sont pas prêtes à en assumer la charge ".
La motivation des membres suppléants semble être un souci particulier pour plusieurs comités.
Un comité s'interroge ainsi sur l'utilité de nommer des suppléants. Il recommande de ramener le nombre total des membres à 16 en supprimant la distinction entre titulaires et suppléants. Cette évolution permettrait, selon lui, de réduire l'absentéisme et l'absence de motivation des membres, notamment des suppléants.
Plusieurs comités soulignent qu'ils s'attachent à convoquer les suppléants à toutes les réunions comme le code de la santé publique 47 ( * ) en donne la possibilité.
b) Les carences en matière de nomination et de renouvellement des membres
La Direction générale de la santé estime, dans les réponses écrites qu'elle a adressées à votre rapporteur, que " le pluralisme paraît assuré ". Elle reconnaît toutefois que " les services déconcentrés (DRASS), chargés de constituer les comités rencontrent des difficultés pour obtenir des candidatures pour certaines catégories : par exemple, la catégorie VII (personnes qualifiées en matière d'éthique), du fait de la non-réponse des rectorats d'académie, ou pour les personnes exerçant à titre libéral, par exemple dans la catégorie II (les médecins généralistes), du fait de la gratuité des fonctions de membre d'un comité ".
Le mode de nomination des membres des comités est particulièrement lourd. La loi ( art. L. 209-11 du code de la santé publique ) prévoit qu'ils sont choisis " parmi les personnes figurant sur une liste établie sur proposition d'organismes ou d'autorités habilités à le faire ".
Il revient aux DRASS, agissant au nom du préfet de Région, d'opérer ce choix, la liste devant comporter un nombre de noms au moins deux fois supérieur au nombre de postes à pourvoir.
Concrètement, le nombre des " organismes ou autorités habilités " à établir les propositions est impressionnant et l'on comprend les difficultés à gérer la procédure pour chaque renouvellement, du moins lorsque la DRASS n'est pas elle-même l'autorité chargée de formuler les propositions (cas des infirmiers, voire des personnes compétentes dans le domaine social).
|
Organismes ou autorités habilités
à établir les listes de propositions pour la nomination des
membres des comités
Catégorie I ( personnes qualifiées en matière de recherche médicale ) : les directeurs d'unités de formation et de recherche médicale de la région, le directeur général de l'INSERM ou son représentant dans la région, les directeurs des principaux établissements de soins et des autres établissements ou organismes compétents en matière de formation ou de recherche biomédicale dans la région ; Catégorie II ( médecins généralistes ) : les présidents des conseils départementaux de l'Ordre des médecins ; Catégorie III ( pharmaciens ) : les directeurs des principaux établissements de soins, de formation ou de recherche biomédicale dans la région, le président du conseil régional de l'Ordre des pharmaciens, et le président du conseil central de la section D de l'Ordre national des pharmaciens ; Catégorie IV ( infirmiers ) : le directeur régional des affaires sanitaires et sociales (DRASS), les directeurs d'établissements de soins de la région ; Catégorie V ( personnes qualifiées en matière d'éthique ) : le recteur de l'académie ; Catégorie VI ( personnes qualifiées dans le domaine social ) : l'union régionale des organisations de consommateurs, l'union régionale des associations familiales, le président du comité régional des retraités et des personnes âgées (ou, à défaut, le directeur régional des affaires sanitaires et sociales (DRASS)) ; Catégorie VII ( psychologues ) : les directeurs des principaux établissements de soins de la région, les organisations professionnelles les plus représentatives au niveau de la région ; Catégorie VIII ( juristes ) : le premier président de la Cour d'appel, le président du tribunal de grande instance, le bâtonnier du barreau près le tribunal de grande instance dans le ressort duquel siège le comité, les présidents des universités de la région. |
De fait, les DRASS rencontrent des difficultés pour exercer leur mission, particulièrement en matière de renouvellement. La DRASS de Lorraine considère à cet égard ne pas disposer d'instructions claires concernant le suivi des nominations. Dans son esprit, il ne lui appartient pas de s'assurer de la présence effective des membres pour chaque catégorie, ce que semble contredire l'article R. 2013 du code de la santé publique 48 ( * ) .
Un comité remarque que les rectorats, auxquels il incombe notamment de présenter des candidats " qualifiés en matière d'éthique ", ne s'acquittent pas de leur tâche. Ce dernier point est confirmé par la DGS.
De façon plus générale, la DRASS d'Ile-de-France souligne l'existence de difficultés à pourvoir les catégories autres que la catégorie I (personnes ayant une qualification et une expérience approfondie en matière de recherche biomédicale) du fait notamment du manque de diligence des rectorats et des conseils de l'ordre à présenter des candidats.
Un comité suggère dans ces conditions d'" ouvrir la possibilité d'une cooptation, même partielle afin de susciter des candidatures réellement motivées qui offriraient plus de souplesse pour faire face aux démissions en cours de mandat ou pour appliquer réellement la mesure d'exclusion vis-à-vis des absents impénitents ".
Cependant, toutes les démissions ne sont pas le fait de personnes qui ne prennent pas leur mission au sérieux. Ainsi, un comité fait état de la démission d'une infirmière consécutive aux réticences de son employeur -un CHU- qui tolérait mal ses absences.
Les comités s'étonnent du non-renouvellement des démissionnaires et de la non-démission d'office des absents par l'autorité administrative. Ils estiment que ces non-renouvellements sont d'autant plus gênants que " le mandat des membres des comités est de six ans " ( art. R. 2005 du code de la santé publique ).
Pourtant, les textes sont précis. L'article R. 2207 dispose qu'" en cas de vacances survenant en cours de mandat, le siège d'un membre titulaire est pourvu par son suppléant ". Le même article prévoit également que le siège d'un membre suppléant devenu vacant au cours des cinq premières années de mandat doit être renouvelé. Enfin, selon le dernier alinéa de l'article R. 2013, " au-delà de trois absences consécutives non justifiées d'un membre titulaire aux séances du comité, le préfet de région peut mettre fin au mandat de ce membre ".
3. Des réformes attendues
Chacun est conscient qu'un pluralisme effectif est indispensable pour que les comités puissent remplir pleinement leur rôle, avec une diversité des points de vue qui ne doit pas se limiter à ceux exprimés par les spécialistes en recherche biomédicale.
La multiplication des suggestions formulées par les comités eux-mêmes et les personnes auditionnées mais également les solutions pratiques parfois retenues témoignent à l'évidence des difficultés rencontrées.
Concernant la rémunération des membres déjà évoqués précédemment 49 ( * ) , un comité fait observer qu'il ne lui semblerait pas inconcevable " que les membres des Comités soient réglementairement rémunérés pour effectuer un travail de qualité qui pourrait alors être mieux précisé et évalué. Un règlement intérieur type incluant notamment ce qui touche à la rémunération des différents acteurs participant au fonctionnement des Comités devrait être proposé par la DGS ".
Les comités ont par ailleurs conscience des problèmes que pose l'absentéisme de certaines catégories de membres. Ils observent que ce phénomène s'explique pour partie par les modalités de nomination.
Dans ces conditions, un comité propose de désigner dans les milieux universitaires les membres de certaines catégories (personnes qualifiées en raison de leurs compétences en matière juridique, par exemple).
Un autre comité a pris l'habitude de désigner trois rapporteurs (un médecin, un pharmacien et un non-médecin non-pharmacien) afin de permettre " une analyse exhaustive des dossiers présentés avec un regard multidisciplinaire garant d'une véritable éthique ".
Un comité remarque enfin que " le recrutement des membres de CCPPRB, tel qu'il est aujourd'hui, ne semble pas satisfaisant (...). La proposition de candidatures par une autorité extérieure, suivie de la désignation par le préfet, est une procédure lourde, lente et qui ne tient pas compte de la motivation de celui qui est proposé ".
Ce comité précise qu'il a lui-même procédé au recrutement de personnalités auxquelles il a expliqué ce que l'on attendait d'elles. Il explique que " les candidatures ont ensuite été soumises à la procédure administrative habituelle, ce qui a permis de remplir toutes les fonctions souhaitées par le législateur avec des personnalités qualifiées ".
Enfin, allant au-delà des voies et moyens pour assurer le pluralisme des comités dans ses exigences actuelles, certains interlocuteurs souhaitent diversifier davantage la composition des comités.
Ainsi, le SNIP a-t-il proposé, lors de son audition, que les associations de patients puissent être représentées dans les comités, tandis que Mme Wintreberg de la DRASS d'Ile-de-France a constaté que, depuis 1994, les représentants des cultes ne pouvaient plus appartenir à un comité. Elle a fait observer que cette évolution était peut-être regrettable compte tenu de la forte motivation de ces personnes.
C. L'INTERVENTION DES COMITÉS : L'HÉTÉROGÉNÉITÉ DE L'ACTIVITÉ ET DES MISSIONS
Les comités ne se bornent pas à donner un avis sur les protocoles de recherche. " En amont ", certains comités ont accepté un rôle de conseil qui contribue à améliorer la qualité des projets. " En aval ", les comités sont de plus en plus souvent saisis d'amendements aux protocoles. En outre, certains s'interrogent sur le contenu de l'avis qu'ils doivent rendre, et notamment sur la légitimité et l'opportunité qu'ils auraient à se prononcer sur le fondement scientifique des projets de recherche.
1. Le développement d'un rôle de conseil
Le rôle de conseil ne figure pas parmi les attributions confiées aux CCPPRB par le code de la santé publique et c'est un apport important de la présente mission que d'avoir mis en évidence l'émergence de cette activité qui correspond à une demande des " usagers ". Si le rôle de conseil a priori apparaît " naturel " à bon nombre de comités, leurs réserves portent davantage sur le rôle de conseil à " caractère scientifique ".
a) Le conseil a priori est devenu habituel
Lors de son audition, le docteur François Chapuis, président de la Conférence nationale des CCPPRB a ainsi confirmé que des comités étaient consultés notamment quant à la qualification " avec " ou " sans " bénéfice individuel direct des protocoles dont ils allaient être saisis.
Dans sa réponse au questionnaire, un comité fait état de " demandes de plus en plus fréquentes émanant d'investigateurs désireux de savoir si leur recherche entre ou non dans le champ d'application de la loi ". Il déclare que " ces dossiers (sont) présentés en séance plénière afin que l'avis soit celui du comité et non de son seul président ".
Après avoir observé qu'il ne pouvait " se dessaisir d'un dossier sans donner de réponse ", ce même comité s'interroge : " cette position est-elle en conformité avec le rôle des CCPPRB ou un tel avis doit-il être donné selon le cas par la DGS ou l'AFSSAPS ? ".
Un autre comité remarque qu'outre " les fonctions définies par la loi et pour lesquelles ils ont été créés, et sans prétendre se substituer à un conseil scientifique ou à un comité d'éthique, les CCPPRB sont également sollicités à titre de conseil par les investigateurs. Cette demande est devenue très fréquente, peut-être parce que la grande majorité des dossiers que nous examinons émanent d'investigateurs locaux et sont promus par leurs établissements de santé. Il est de plus en plus fréquent que les investigateurs consultent le comité dès les premières étapes de la rédaction de leurs projets ".
Le rôle de conseil des comités est sans doute utile puisqu'il permet d'améliorer la qualité des projets. Néanmoins, il n'est pas sans susciter des interrogations. En effet, l'implication d'un comité dans la préparation ou la présentation d'un dossier sur lequel il devra émettre un avis n'est pas dépourvue d'un risque de confusion des rôles.
Si cette activité d'assistance ou de conseil est exercée par certains des membres du comité, ces derniers deviendront naturellement les " avocats " du dossier dès lors que leurs conseils auront été suivis.
Si, dans un souci de rigueur, les conseils sont formulés collégialement par le comité, l'avis de ce dernier risque d'apparaître le moment venu, comme une simple formalité et le recours à ses conseils comme un préalable indispensable.
Dès lors, formaliser le rôle de conseil des comités peut apparaître comme une garantie de transparence mais peut poser également une question de principe.
Les auditions auxquelles a procédé votre rapporteur ne permettent pas toutefois de considérer qu'un tel risque, théoriquement possible, se soit réalisé jusqu'à présent.
b) Le conseil sur la méthodologie suscite des interrogations
Certains comités sont tentés de se prononcer sur les aspects méthodologiques, voire scientifiques du projet de recherche. Ainsi, un comité souligne qu'" exceptionnellement, il s'autorise des remarques sur la méthodologie. Celles-ci sont présentées comme des suggestions sans valeur contraignante, sauf cas particulier tenant à la protection des personnes ".
Les comités doivent-ils s'intéresser à la méthodologie de la recherche ? La question est posée dans la mesure où leur avis porte prioritairement sur la sécurité des personnes se prêtant à la recherche et que c'est au regard de cette seule préoccupation qu'ils doivent apprécier la finalité et l'intérêt de la recherche. L'article L. 209-12 qui détermine les compétences des CCPPRB en fixant la nature des avis qu'ils rendent ne fait pas référence par exemple à un rôle de conseil a priori , il ne prévoit pas non plus de compétence relative à l'examen de la méthodologie retenue. La seule référence à la méthodologie de la recherche se trouve dans l'article L. 209-9 du code de la santé au nombre des éléments qui doivent être portés à la connaissance de la personne participant à la recherche pour obtenir son consentement.
Si l'article L. 209-12 reconnaît aux comités la mission d'examiner pour avis " les modalités de recueil de leur consentement ", il n'est pas prévu que cet examen doive porter sur la méthodologie elle-même.
Lors de son audition, le professeur Patrice Jaillon, président de l'Association pour le développement de la pharmacologie clinique (ADPC) a estimé que certains comités avaient pu donner un avis favorable à des protocoles qui n'étaient pas fondés scientifiquement. Il a observé par ailleurs qu'il n'y avait pas de contrôle sur la méthode, postérieurement à l'avis rendu par le comité.
Les comités ne disposent pas d'ailleurs nécessairement en leur sein des compétences requises pour apprécier des questions relatives à la méthodologie, et le législateur n'avait pas souhaité que les comités exercent une fonction de conseil scientifique.
Le professeur G. Hazebroucq, directeur pharmaceutique de la Pharmacie centrale des hôpitaux de Paris, a ainsi estimé que " pour des essais de phase I voire IIa, une attestation d'expert toxicologique (voire pharmaceutique pour des produits de biotechnologie) serait la bienvenue car les compétences nécessaires ne figurent pas dans tous les comités ". Le professeur Patrice Jaillon a abondé dans ce sens en proposant de prévoir la présence de pharmacologues dans les comités.
|
Les quatre phases du développement d'un médicament 50 ( * ) La phase I a pour but essentiel d'apprécier la tolérance chez l'homme du médicament, en fonction de la dose et de la voie d'administration. Une première évaluation des propriétés pharmacocinétiques de la molécule est habituellement conduite simultanément. La phase II vise à confirmer chez l'homme les propriétés pharmacologiques potentiellement thérapeutiques observées chez l'animal (II.a). Ces essais portent habituellement sur un petit nombre d'individus, le plus souvent volontaires sains. Ils visent à déterminer la posologie efficace (II.b). Dès lors que la posologie efficace est connue, les essais de la phase III ont pour objet la confirmation des propriétés thérapeutiques sur des effectifs de patients plus importants et avec des durées d'administration du médicament plus prolongées. Ils permettent d'établir le ratio bénéfices/risques du médicament, notamment par une meilleure appréciation de son profil de tolérance à moyen et long termes (analyse des effets indésirables). A la fin de la phase III, le médicament est évalué par la Commission d'autorisation de mise sur le marché (AMM) et devient alors une spécialité pharmaceutique qui peut être commercialisée. Les essais de phase IV sont réalisés après la mise sur le marché du médicament (on parle d'essais post-AMM). Ces essais doivent être effectués dans les conditions habituelles de prescription. Ces derniers essais, qui se font souvent en collaboration avec les centres régionaux de pharmacovigilance, nécessitent des effectifs de patients très importants. |
2. La multiplication des avis favorables " sous réserve "
La multiplication des réserves formulées par les comités peut traduire soit une augmentation de leurs exigences qui illustre leur " savoir faire ", soit une hétérogénéité des critères d'examen des protocoles préjudiciable à l'activité des promoteurs.
Le premier alinéa de l'article L. 209-12-1 du code de la santé publique dispose que le comité peut émettre un avis favorable à la réalisation d'une recherche " sous réserve de la transmission d'informations complémentaires par l'investigateur pendant le déroulement de celle-ci ".
Les comités n'hésitent pas à faire usage de cette prérogative.
En effet, les 2/5 des avis favorables sont donnés " sous réserve ". Cette proportion peut même être bien plus importante dans certains cas. Ainsi, un comité précise que " plus de 60 % des dossiers (protocoles et amendements confondus) font l'objet d'une demande de compléments ".
Avis donnés par les
comités*
(en unité et % du total)
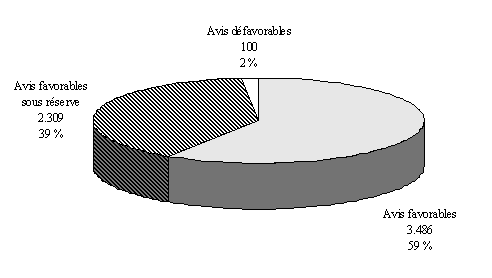
*3 exercices cumulés (1997-1998-1999) - 38 comités
Les réserves portent le plus souvent sur les points suivants :
- documents destinés aux personnes participant à la recherche incomplets (consentement, information),
- demande d'informations complémentaires sur les contraintes ou risques encourus par les participants,
- demande d'une argumentation plus précise concernant le bénéfice individuel direct,
- demande de précision sur le protocole (nombre de cas, durée de l'étude).
Certains promoteurs ont attiré l'attention de votre rapporteur sur le fait que les réserves émises par les CCPPRB étaient quelquefois consécutives à l'hétérogénéité des critères d'examen des protocoles propres aux différents comités. Ainsi l'INSERM a estimé que les réserves dont faisaient l'objet ses protocoles avaient " pour origine l'hétérogénéité des modes de fonctionnement et de présentation des avis des différents comités ".
3. La forte hausse du nombre d'amendements
L'article R. 2630 du code de la santé publique établit que " toute modification du projet de recherche affectant de manière substantielle les informations communiquées au comité doit faire l'objet d'une demande d'avis complémentaire accompagnée des justifications appropriées ".
Le nombre des amendements a fortement augmenté au cours des dernières années. On observe ainsi une hausse de plus 50 % du nombre des amendements en trois ans pour les treize comités qui ont communiqué des données par année.
Evolution du nombre d'amendements
examinés*
* 13 comités.
Pour les 36 comités qui ont répondu de façon suffisamment détaillée, le nombre des amendements est désormais en moyenne équivalent à celui des protocoles.
Nature de l'activité des comités*
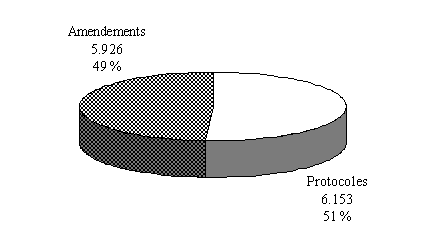
* 3 exercices cumulés (1997-1998-1999) - 39 comités.
Cette moyenne recouvre toutefois des situations contrastées comme le montre le graphique ci-dessous dans lequel chaque point représente un comité.
Représentation graphique, pour chaque
comité, du nombre de protocoles
et d'amendements
examinés*
3 exercices cumulés (1997-1998-1999) - 36 comités
Près d'un quart des comités présente une situation atypique par rapport à la moyenne, caractérisée, comme il a été dit, par un nombre équivalent de protocoles et d'amendements.
Ainsi tel comité examine-t-il trois amendements pour un protocole (près de 250 amendements pour 80 protocoles environ), tandis qu'un autre examine 0,6 amendement pour un protocole (pas moins de 150 amendements pour plus de 250 protocoles).
De façon générale, la hausse du nombre des amendements modifie l'activité de ces comités.
Par ailleurs, les amendements ne faisant pas l'objet de redevance, il n'y a aucune " incitation " pour les promoteurs à en limiter le nombre. Enfin, ces amendements n'étant pas rémunérateurs et la DGS ne tenant pas compte de cette activité dans le calcul de dotations qu'elle attribue aux différents comités, ils peuvent être une source de difficultés pour certains comités en termes de moyens de fonctionnement.
Comme le remarque un comité, " le nombre des amendements s'est considérablement accru au cours de ces dernières années. Certains de ces amendements demandent un travail de recherche considérable et cette activité n'est pas vraiment prise en compte (dans le calcul de la dotation budgétaire) ".
Outre les incidences budgétaires, la hausse importante du nombre des amendements complique la " lecture " des protocoles.
En effet, selon ce même comité, " la succession d'amendements les uns derrière les autres pour un même protocole peut amener, par un phénomène de glissement, à donner un avis sur un document qui n'a plus rien à voir avec le projet initial. Cette dérive n'est pas sensible d'un amendement à l'autre et ne peut être perceptible que si l'on considère l'ensemble des amendements ".
Certains comités en viennent ainsi à déplorer l'existence d'un biais qui amène les promoteurs à présenter des protocoles inachevés qui seront complétés ultérieurement par voie d'amendements.
Toutefois, certains interlocuteurs, se plaçant il est vrai du côté des " usagers ", tempèrent la difficulté que représente le développement des amendements.
Ainsi, lors de son audition, le professeur Patrice Jaillon, président de l'Association pour le développement de la pharmacologie clinique (ADPC), a rappelé que le promoteur " ne pouvait pas penser à tout lors de la rédaction du protocole ". Les modifications présentées dans les amendements seraient en outre généralement peu importantes (modifications concernant des limites d'âges ou des posologies) et certains comités considèrent que le recours accru aux amendements leur permet de mieux suivre l'évolution des projets car ils déplorent de ne pas être toujours informés du résultat des recherches et des conditions de leur déroulement, voire des difficultés rencontrées.
Ainsi, un comité regrette de " n'avoir aucun retour d'information sur les suites données aux avis, d'éventuelles reclassifications d'essais (avec ou sans bénéfice individuel direct), ou sur les jugements émis par les promoteurs ou investigateurs à notre égard. Il en résulte un sentiment d'isolement et une ignorance de la qualité de notre travail ".
4. La cohabitation avec les comités d'experts des grands organismes de recherche
Le rôle des comités devient d'autant plus variable que l'hétérogénéité des promoteurs et des protocoles s'accentue. Tel comité est confronté à un protocole très technique pour lequel il s'interroge sur sa capacité d'expertise alors que tel autre est saisi en amont et peut participer à la conception du projet ou tout du moins donner des conseils avisés de présentation.
Cette diversité de situations s'explique en particulier par le fait que certains promoteurs se sont organisés pour développer " en interne " des structures d'évaluation et de conseil qui agissent en amont comme un filtre à la fois quantitatif et qualitatif.
a) L'Institut national de la santé et de la recherche médicale (INSERM)
L'INSERM peut se porter promoteur de recherches biomédicales dans le cadre de ses missions et de sa politique de soutien au développement de la recherche clinique. C'est pourquoi il a mis en place des procédures internes particulières permettant d'évaluer la qualité des projets qui lui sont soumis.
Afin d'aider les investigateurs à monter leurs projets, l'INSERM a créé en 1991 un bureau " recherches biomédicales " qui s'appuie sur un Comité d'experts en recherches biomédicales. Ce comité de neuf experts examine la qualité méthodologique et scientifique des protocoles.
Par ailleurs, la conformité aux dispositions législatives et réglementaires (qualification des investigateurs, information des personnes...) est vérifiée par l'INSERM.
Le comité sert aussi d'instance de réflexion et de proposition pour les questions concernant le champ et les modalités d'application de la loi du 20 décembre 1988.
La nature des renseignements demandés par le comité d'experts (qualité méthodologique et scientifique, qualification des investigateurs, informations délivrées par les investigateurs...) en font, comme l'a reconnu l'INSERM lors de son audition, un " quasi-CCPPRB " .
Par ailleurs, le bureau " recherches biomédicales " accompagne les projets du début jusqu'à la diffusion des données et assure un suivi renforcé par des visites sur sites du service en charge de la qualité à l'INSERM.
A bien des égards, les procédures mises en oeuvre au sein de l'INSERM sont donc plus précises et plus exigeantes que les procédures de droit commun.
b) L'Assistance publique - Hôpitaux de Paris (AP-HP)
L'AP-HP peut également prendre l'initiative de la promotion d'un projet de recherche ou reprendre à son compte un projet élaboré par un tiers 51 ( * ) .
Lorsque ce tiers est un médecin d'un établissement de l'AP-HP, la décision de se porter promoteur est prise par le Directeur de la Politique médicale (DPM) sur le fondement de la demande transmise par le directeur d'établissement.
L'AP-HP n'accepte d'être promoteur que de projets n'entraînant pas de frais supplémentaires ou bénéficiant d'un financement clairement identifié et couvrant les surcoûts. Ce financement peut être obtenu auprès d'organismes publics (INSERM, CNAM...) ou privés (industrie pharmaceutique) ou au sein de l'établissement dans le cadre de procédures d'appels d'offres.
Sur le plan technique, les dossiers sont instruits par la délégation à la recherche clinique (DRC) qui :
- examine le contenu du dossier par rapport aux exigences légales et réglementaires. Elle demande, le cas échéant, de fournir les pièces ou les informations manquantes ;
- fait évaluer le projet par deux experts anonymes (qui examinent la qualité scientifique, l'intérêt clinique, l'originalité, la méthodologie, les aspects éthiques du projet et la pertinence pour l'AP-HP d'être le promoteur) ;
- analyse la cohérence et la pertinence globale du protocole et des documents annexes ;
- évalue les surcoûts induits par la recherche et vérifie l'existence d'un financement spécifique.
La délégation à la recherche clinique adresse les avis des experts (en maintenant leur anonymat) à l'initiateur du projet. Si des remarques ou des objections ont été soulevées par l'un ou l'autre des experts, l'investigateur doit fournir des réponses argumentées et si nécessaire une nouvelle version du protocole.
En fonction des réponses fournies par l'investigateur, la délégation à la recherche clinique notifie ou non un avis favorable pour la promotion par l'AP-HP. En cas d'accord, la délégation à la recherche clinique fournit à l'investigateur par courrier un avis favorable identifiant l'AP-HP comme promoteur de la recherche biomédicale, une attestation d'assurance, et une attestation de versement du droit fixe pour la saisine du CCPPRB.
Cette organisation rigoureuse doit néanmoins veiller à maintenir chacun dans son rôle. On peut rappeler que l'article L. 209-1 du code de la santé publique désigne par le terme " promoteur " la personne physique ou morale qui prend l'initiative d'une recherche biomédicale sur l'être humain et par " investigateur " la ou les personnes physiques qui dirigent et surveillent la réalisation de la recherche.
Lors de leur audition, les représentants de l'AP-HP ont insisté sur l'importance du filtre scientifique que constitue l'intervention de la DRC. Ils ont également insisté sur les moyens mis à disposition de la recherche clinique, à travers notamment le Programme hospitalier de recherche clinique (PHRC) qui représente 100 millions de francs par an pour l'AP-HP.
5. Le risque d'un " Gault-Millau " des CCPPRB
Une critique, parfois formulée à l'encontre du fonctionnement des CCPPRB, ne concerne pas leurs compétences mais plutôt le fait que les comités n'auraient pas tous les mêmes exigences. Cette remarque prend une dimension particulière dans le contexte évoqué précédemment d'une hétérogénéité croissante des promoteurs et des protocoles qui influe sur le rôle de chaque comité.
a) Une critique qui se répand
L'article L. 209.12 dispose dans son premier alinéa qu' " avant de réaliser une recherche biomédicale sur l'être humain, tout investigateur est tenu d'en soumettre le projet à l'avis de l'un des comités consultatifs de protection des personnes dans la recherche biomédicale compétents pour la région où l'investigateur exerce son activité ". 52 ( * )
Par ailleurs, le deuxième alinéa de l'article L. 209-11 énonce que " le ministre fixe par arrêté le nombre de comités dans chaque région " et que " le champ de compétence territorial d'un comité peut être étendu à plusieurs régions ".
Les textes législatifs reconnaissent ainsi une certaine liberté à l'investigateur dans le choix du comité devant lequel il souhaite déposer son projet de protocole.
Selon certains, il serait plus " facile " d'obtenir un avis favorable à un projet de recherche en s'adressant à tel ou tel comité, ce qui amène à évoquer l'existence d'un " Gault-Millau " des CCPPRB.
On peut en effet s'interroger devant le volume très important d'avis rendus par certains comités concernant tant les protocoles que les amendements, qui peut atteindre 150 par an.
Ces avis peuvent-ils être rendus avec tout le soin nécessaire ? Que dire par ailleurs d'un phénomène de concentration des promoteurs sur certains comités ? Ne peut-on pas penser qu'un tel phénomène peut refléter une moindre rigueur dans l'examen des dossiers ?
Lors de son audition, M. Joël Ménard, délégué médical de l'AP-HP, évoquant ce risque, a estimé qu'il existait parfois une trop forte proximité entre les comités et certains investigateurs.
Comme le note par ailleurs un comité : " la diminution progressive, mais nette, du nombre de protocoles présentés à notre comité l'amène à s'interroger. Est-ce que le nombre de protocoles de recherche diminue dans notre région ? Est-ce que notre comité est " trop " rigoureux ? Est-ce que les investigateurs et les promoteurs, ayant l'aval d'institutions ou de sociétés savantes, pour mener leurs recherches, jugent inutile de demander l'avis du comité ? ".
b) Des indices concordants
Les données statistiques transmises par la DGS à votre rapporteur, concernant le nombre d'avis rendus en 1998 et 1999, ne permettent pas de vérifier cette hypothèse compte tenu d'un trop faible recul.
La situation de la région Ile-de-France, selon les données transmises par la DRASS, appelle en revanche un commentaire particulier quant à l'évaluation globale du nombre d'avis rendus mais également au regard des contrastes qui apparaissent dans l'activité des différents comités.
Ces données font apparaître une forte baisse du nombre d'avis rendus ces dernières années dans cette région (- 43 % entre 1993 et 2000). Cette baisse s'est accélérée particulièrement en 2000 (- 27 % pour cette seule année). Elle concerne les protocoles déposés par des promoteurs tant industriels qu'institutionnels.
La DRASS d'Ile-de-France considère qu'il est trop tôt pour avancer sans risque d'erreur un ou plusieurs facteurs explicatifs.
Evolution du nombre d'avis rendus par les 14 CCPPRB d'Ile-de-France
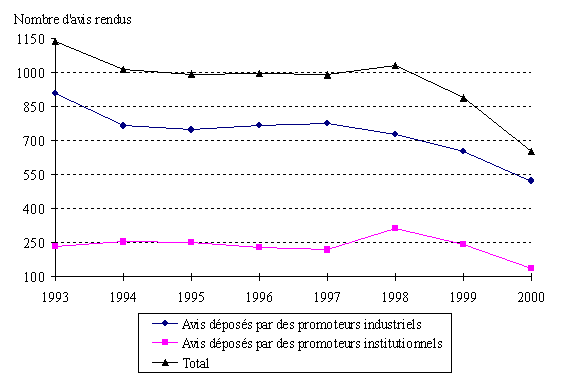
Toutefois, cette baisse générale en Ile-de-France ne concerne pas tous les comités de la région. Deux comités sur quatorze ont rendu plus d'avis en 2000 qu'en 1993 ; six ont subi une érosion proche de la moyenne régionale et six ont connu une baisse du nombre des avis supérieure à 50 % (pour trois comités, la baisse dépasse les 70 %).
L'effondrement de l'activité de certains comités retient l'attention dans la mesure où elle coïncide avec une consolidation, voire une progression, de l'activité d'autres comités. Ce phénomène de concentration que l'on observe sur une durée assez longue peut ne pas être sans lien avec l'hypothèse évoquée ci-dessus d'une hétérogénéité des exigences des comités quant aux conditions d'examen des protocoles.
Cette tendance a été confirmée par la DGS lors de son audition.
A cet égard, votre rapporteur observe que les comités s'identifient, ne serait-ce que dans leur appellation, à la structure hospitalière qui les héberge. On parle ainsi du CCPPRB " Cochin " ou " Lyon II ".
L'identification des comités à l'hôpital est désapprouvée par la DGS qui estime que cette confusion nuit à leur indépendance et favorise la polarisation du dépôt des protocoles au profit des comités implantés dans des hôpitaux réputés.
Un comité propose dans ces conditions que la loi oblige les investigateurs à présenter leurs protocoles dans un CCPPRB non rattaché à l'établissement dans lequel ils exercent. Il s'agit ce faisant d'éviter une trop grande proximité entre l'investigateur et le comité qui pourrait porter atteinte à l'indépendance de ce dernier.
Cette mesure ne saurait pourtant suffire à elle seule à prévenir le syndrome du " Gault-Millau " des CCPPRB si il est confirmé.
De fait, la loi ne prévoit pas une " sectorisation " stricte du champ de compétences des comités. Elle prévoit seulement que " tout investigateur est tenu (de soumettre son projet) à l'avis de l'un des comités consultatifs de protection des personnes dans la recherche biomédicale compétents pour la région où l'investigateur exerce son activité " 53 ( * ) et dispose que " dans le cas d'une recherche confiée à plusieurs investigateurs, cet avis est demandé par l'investigateur coordinateur (...). " 54 ( * )
En outre, comme évoqué précédemment, une base de données retraçant la " jurisprudence " des différents comités et permettant d'harmoniser leurs positions fait à l'évidence défaut.
D. LES RELATIONS AVEC LES POUVOIRS PUBLICS : UN DIALOGUE LIMITÉ
Les comités se situent au centre d'un réseau qui met en relation les différents acteurs de la recherche biomédicale.
Ils entretiennent des relations tant avec les autorités de tutelle (ministère chargé de la Santé, à travers la DGS et les DRASS) qu'avec l'autorité de sécurité sanitaire (AFSSAPS).
Afin de mieux se faire entendre de l'Administration, les comités se sont organisés en Conférence nationale, ce qui leur permet également de partager leurs expériences et d'améliorer leur fonctionnement.
1. Des relations à réorganiser et à approfondir avec les services de l'Etat
a) La Direction générale de la santé réorganise ses relations avec les comités
Comme votre rapporteur a déjà eu l'occasion de le souligner 55 ( * ) , la Direction générale de la santé (DGS) n'avait pas jusqu'à présent mis en place les moyens permettant le suivi effectif de la loi du 20 décembre 1988.
Cette mission était assurée, au sein de la DGS, par un seul conseiller technique disposant d'une (trop ?) grande autonomie par rapport à la direction " Santé des populations " 56 ( * ) .
Afin de rendre plus effective la tutelle, une nouvelle organisation du suivi des CCPPRB a été décidée à l'occasion de la réforme de la DGS.
Cette mission est désormais dévolue à un bureau spécifique, le bureau 1E " Recherche et prospective " rattaché à la première sous-direction " Politiques de santé et stratégies " de la première direction " Politique de santé et qualité du service de santé " de la DGS.
Le bureau 1E est en voie de constitution, ce qui peut expliquer, sans les excuser, les difficultés rencontrées pour informer le Parlement sur l'activité des CCPPRB. Il devrait recevoir à terme le renfort de plusieurs fonctionnaires spécialisés dans des domaines de compétence qui jusqu'alors faisaient apparemment défaut (médical, juridique, budgétaire).
Lors de son audition, la DGS a estimé que cette réorganisation était destinée à permettre au ministère de la Santé de poursuivre trois objectifs :
" - faire connaître et prendre en compte les orientations et les besoins des politiques de santé publique par le monde de la recherche ;
- mieux connaître et prendre en compte les apports de la recherche dans la définition des politiques de santé ;
- exercer dans des conditions administratives satisfaisantes les tâches qui lui incombent : mise en oeuvre et suivi de la loi Huriet, participation au conseil d'administration de l'INSERM... ".
Cette évolution était nécessaire ; elle témoigne d'une volonté d'intégrer le suivi de l'application de la loi du 20 décembre 1988 dans le " droit commun " de l'organisation administrative.
Votre rapporteur souhaite que cette nouvelle organisation puisse répondre aux attentes des acteurs de la recherche biomédicale, notamment en matière d'information (textes et procédures applicables) et d'organisation (fonctionnement et financement des CCPPRB).
Les difficultés rencontrées dans la mise en oeuvre de cette nouvelle organisation 57 ( * ) démontrent que le ministère chargé de la Santé doit maintenant fournir des efforts importants pour atteindre ces objectifs.
b) La mobilisation insuffisante des services déconcentrés du ministère de la Santé (DRASS)
Les relations entre les DRASS et les CCPPRB restent limitées même si le président de la Conférence nationale des CCPPRB, le docteur François Chapuis, les qualifie de relations " quasi tutélaires " en évoquant leur rôle dans l'attribution des budgets et dans la nomination des membres des comités.
Le terme de tutelle n'est pas exagéré dans la mesure où l'on fait référence à une tutelle administrative. Le régime juridique des CCPPRB est en effet d'ordre légal et réglementaire. Le deuxième alinéa de l'article L. 209-11 du code de la santé publique dispose en particulier que : " les comités exercent leur mission en toute indépendance. Ils sont dotés de la personnalité juridique ".
Cette disposition a été introduite par la loi n° 94-630 du 25 juillet 1994 contre l'avis de votre commission des Affaires sociales. Votre commission estimait en effet qu'il n'était " pas opportun de doter une structure de la personnalité morale lorsque rien ne le (justifiait) et qu'il (convenait) d'éviter que les comités ne puissent avoir à répondre des conséquences de leurs avis devant les juridictions pénales, le nouveau code pénal ayant institué le principe de la responsabilité pénale des personnes morales " 58 ( * ) .
Au demeurant, l'expression de " personnalité juridique " reste imprécise et ne suffit pas à déterminer le statut des comités.
En droit, la tutelle administrative des DRASS apparaît étroite. L'article R. 2002 du code de la santé publique précise ainsi que " chaque comité a son siège au sein de la direction régionale des affaires sanitaires et sociales ou d'une direction départementale des affaires sanitaires et sociales ".
Il établit également que c'est la DRASS qui " peut passer convention avec un établissement hospitalier public aux fins de donner aux comités les moyens en locaux, matériels, et éventuellement en secrétariat, nécessaires pour assurer leur mission moyennant une rémunération forfaitaire versée par le comité intéressé. "
Les DRASS exercent également, au nom du préfet de région, le pouvoir de nomination.
Le second alinéa de l'article L. 1123-2 du code de la santé publique (anc. L. 209-11) dispose en effet que les membres du comité sont nommés par le représentant de l'Etat de la région où le comité a son siège. Le texte précise qu'" ils sont choisis parmi les personnes figurant sur une liste établie sur proposition d'organismes ou d'autorités habilités à le faire, dans des conditions déterminées par décret ".
De même, comme mentionné plus haut, le préfet de région doit pourvoir aux vacances qui surviennent parmi les suppléants ( art. R. 2007 ). Il lui appartient en outre " au-delà de trois absences consécutives, non justifiées, d'un membre titulaire aux séances du comité, (de) mettre fin au mandat de ce membre " ( art. R. 2013 ).
Dans les faits, votre rapporteur a constaté 59 ( * ) une faible mobilisation des DRASS et une vigilance insuffisante quant au bon fonctionnement des comités.
En témoigne notamment le suivi pour le moins lacunaire de " l'état civil " des comités qui relève pourtant de la responsabilité et de l'initiative des services déconcentrés.
Il souhaite à cet égard que la réforme en cours de la DGS puisse se traduire sans tarder par une meilleure organisation des DRASS dans leurs relations avec les comités.
2. Une procédure d'affectation des moyens budgétaires peu transparente
L'examen des conditions de financement des comités constituait pour votre rapporteur un des objectifs prioritaires de la mission compte tenu des difficultés apparues, depuis plusieurs années, dans les relations entre les comités et la Direction générale de la santé (DGS) 60 ( * ) .
Les difficultés rencontrées pour réunir, dans le cadre de la présente mission, des informations précises sur le financement des comités expliquent rétrospectivement l'absence de réponses aux questions écrites posées précédemment par votre rapporteur.
Car ces difficultés révèlent non pas une rétention d'informations mais la nécessité dans laquelle s'est trouvée l'Administration de reconstituer, ou plutôt de constituer, ces informations suite aux demandes pressantes de votre rapporteur.
Une telle situation traduit l'absence d'un interlocuteur capable de suivre l'activité des comités et permet de comprendre le climat d'incompréhension, voire de méfiance, qu'une telle carence a provoqué au sein des comités.
Ce n'est en définitive que la veille de l'examen du présent rapport par la commission que votre rapporteur a pu obtenir des informations à peu près cohérentes. Encore convient-il d'observer que ces informations démentent celles fournies quelques mois plus tôt.
a) Le régime juridique du financement des comités
L'organisation du financement des CCPPRB repose sur plusieurs principes. Elle met à contribution les promoteurs qui doivent s'acquitter d'une redevance. Elle distingue selon la nature des promoteurs en prévoyant une redevance réduite pour les personnes physiques et les organismes sans but lucratif. Telle qu'elle résulte des pratiques de la DGS depuis 1991, elle se traduit implicitement par une forme de mutualisation entre les moyens des comités afin de permettre la couverture du territoire et la diffusion de la recherche biomédicale.
Les frais de fonctionnement des comités sont financés par le produit d'un droit fixe versé par les promoteurs pour chacun des protocoles faisant l'objet d'une demande d'avis.
Ce produit est rattaché par voie de fonds de concours 61 ( * ) au budget du ministère chargé de la Santé (chapitre 34-98, art. 93) qui impute sur ce chapitre les subventions versées aux différents comités 62 ( * ) .
Le montant de ce droit fixe, arrêté par le ministre chargé de la Santé, n'a pas varié depuis 1990. Il est de 9.500 francs par protocole. Ce droit est réduit à 900 francs pour les protocoles dont le promoteur est une personne physique ou un établissement ou organisme de soins, de formation ou de recherche sans but lucratif.
Tant le tarif du droit que son assiette soulèvent les réserves des comités.
Ainsi, l'existence de deux tarifs différents selon la nature du promoteur fixée -faut-il le rappeler ?- par voie réglementaire s'explique probablement par la nécessité de ne pas pénaliser la recherche institutionnelle.
Un comité constate toutefois qu'" il y a un très grand déséquilibre entre la tarification selon la nature du promoteur, entre un promoteur industriel et un promoteur institutionnel. Le travail du comité est beaucoup plus important pour un promoteur institutionnel, car il s'agit souvent d'un médecin seul ou d'une équipe peu structurée au niveau logistique et qui n'est souvent pas rompue au travail de rédaction des protocoles. Les dossiers sont souvent renvoyés plusieurs fois avant de pouvoir aboutir ".
S'agissant de l'assiette du droit, la principale question est celle des amendements qui en sont exonérés.
L'article R. 2030 du code de la santé publique dispose que " toute modification du projet de recherche affectant de manière substantielle les informations communiquées au comité doit faire l'objet d'une demande d'avis complémentaire accompagnée des justifications appropriées ". L'article 2 de l'arrêté du 27 décembre 1990 relatif au montant du droit fixe versé par les promoteurs de recherches biomédicales précise que " les demandes d'avis complémentaires prévues à l'article R. 2030 du code de la santé publique ne donnent pas lieu au versement du droit ".
Afin d'évaluer cette activité, la DGS demande que le nombre d'amendements soit mentionné dans les rapports d'activité. Elle ne tient pas compte pour autant de ce critère pour calculer les dotations budgétaires.
Selon les informations rassemblées par votre rapporteur, le financement des comités est établi sur la base de documents qu'ils fournissent chaque année à la DGS.
L'article R. 2020 du code de la santé publique dispose en effet qu'avant le 31 mars de chaque année, les comités doivent adresser au ministre de la santé et au préfet de région un rapport d'activité et une copie du rapport financier relatif à l'année civile précédente.
Les régies de recettes des DRASS sont chargées d'informer la DGS du montant de droits fixes perçus.
b) La réforme du financement de 1998
L'audition de la DGS a permis de clarifier l'évolution intervenue depuis 1998 dans les modalités de financement des comités.
Jusqu'en 1998, la subvention accordée à chaque comité était calculée en fonction du nombre de droits fixes perçus.
Ce mode de calcul prenait donc en compte l'activité de chaque comité (nombre d'avis rendus) et non son " chiffre d'affaires " car il neutralisait les caractéristiques de cette activité selon que les protocoles étaient présentés par des industriels (droits de 9.500 francs) ou des personnes physiques ou organismes sans but lucratif (droits de 900 francs).
Pratiquement, la procédure de répartition faisait intervenir le niveau régional mais de façon transparente : selon les critères définis, les dotations étaient attribuées à chaque région puis réparties par elle entre les comités.
Ayant observé que certains comités disposaient d'importantes réserves financières, la DGS a considéré que les versements ne tenaient pas suffisamment compte des charges réelles des comités.
A partir de 1998, le système a donc été modifié sans concertation avec les comités, semble-t-il.
La DGS retient désormais deux critères 63 ( * ) :
" - les charges salariales et de fonctionnement ;
- la situation des comités au vu des comptes de résultats et des bilans financiers. "
De fait, pour la répartition des dotations entre les comités, la DGS est passée d'un mode mécanique mais clair à des modalités " souples " mais opaques !
Ce changement dans la politique d'attribution des dotations budgétaires a pris de court certains comités qui ont vu " fondre " leurs moyens de fonctionnement. Plusieurs comités ont vu leur dotation supprimée (Marseille I) ou fortement réduite (Clermont-Ferrand) au motif qu'ils disposaient de réserves. L'origine de ces réserves peut être diverse, un comité remarque par exemple qu'il a pu constituer des réserves suite à sa décision de ne pas rémunérer les rapporteurs.
Lors de son audition, la Direction générale de la santé a reconnu que le financement des comités avait toujours constitué un exercice difficile. Elle a souligné l'inégalité de gestion des différents comités. Elle a observé que les DRASS n'avaient pas toujours les moyens d'appréhender cette question, ce qui pouvait donner lieu à des décisions contestables dans la répartition des moyens financiers.
Mais l'introduction des nouveaux critères à partir de 1998 n'a pas eu pour seul effet de répartir différemment entre les comités l'enveloppe des droits fixes. Elle a eu pour première conséquence de faire apparaître un écart croissant entre le montant de cette enveloppe et les sommes globalement distribuées aux comités.
c) La non-restitution intégrale des droits perçus
Les réponses fournies à votre rapporteur dans les quelques jours qui ont précédé l'examen du présent rapport par la commission et, pour les dernières d'entre elles, la veille de cet examen, permettent de formuler un certain nombre de constatations.
(1) Les droits fixes font l'objet d'une double ponction
Le produit des droits fixes versés par les promoteurs qui, depuis 1992, est resté stable aux environs de 15,5 millions de francs fait l'objet d'une double ponction.
Une première ponction par le ministère de l'Economie et des Finances : les crédits ouverts en faveur des comités ne sont pas égaux au montant des droits fixes dont ils sont pourtant la contrepartie. Ils subissent en effet un prélèvement de 10 % au titre, semble-t-il, des frais de gestion.
Une seconde ponction est opérée par le ministère des Affaires sociales. Selon la Direction du budget 64 ( * ) " les modalités de rattachement du produit du droit versé par les promoteurs ont été fixées (...) par l'arrêté du 28 septembre 1994. Ce dernier arrêté prévoit qu'après rattachement au chapitre 31-96 " Autres rémunérations " dans la limite des crédits inscrits au budget voté, les recettes excédant cette limite sont imputées sur le chapitre 34-98 " Moyens de fonctionnement des services ".
Il faut comprendre de cette explication que le produit des droits fixes, après le prélèvement susmentionné de 10 %, abonde deux chapitres du budget de l'Emploi et de la Solidarité : l'un couvrant à nouveau des frais de gestion (chapitre 31-96), l'autre comportant les dotations budgétaires des comités (chapitre 34-98).
Ces frais de gestion qui s'élèvent à 300.000 francs depuis 1994 (soit environ 2 % du produit brut des droits fixes) correspondent, selon la Direction générale de la santé 65 ( * ) , aux frais de personnel engagés par le ministère pour traiter -comprend-on- la répartition entre les comités des crédits du chapitre 34-98, soit :
" - un instructeur des dossiers à temps partiel (agent contractuel hors échelle B3), soit environ 195.000 francs annuels ;
" - un agent contractuel à temps incomplet pour assurer le secrétariat (IM 254 base 149 heures), soit 105.000 francs annuels ".
A la lecture de cette réponse, votre rapporteur comprend mieux les difficultés rencontrées pour obtenir une information complète sur le financement des comités. Mais il s'étonne également du poids des prélèvements (de l'ordre au total de 12 %) opérés par les administrations sur le produit du droit fixe qui devrait bénéficier intégralement aux comités.
(2) Les crédits des comités ne sont pas suffisamment identifiables
Le chapitre 34-98 précité " Moyens des services ", au sein duquel figure un article 93 consacré spécifiquement aux comités, comporte bien d'autres dépenses réparties en 23 autres articles. Les moyens des comités (14 millions de francs) sont ainsi dilués dans une masse de crédits supérieurs à 1 milliard de francs.
Ainsi que l'indique la Direction du budget 66 ( * ) :
" D'un point de vue réglementaire, les fonds de concours ne font pas l'objet d'un suivi de consommation distinct du suivi de l'exécution des crédits budgétaires, avec lesquels ils sont fongibles au sein du chapitre budgétaire de rattachement. Les crédits de fonds de concours représentent des suppléments de crédits qui s'ajoutent aux crédits budgétaires prévus par la loi de finances, l'ensemble constituant la dotation du chapitre prise en compte pour l'année considérée, dans le cadre des procédures d'ordre dont la Direction du budget à la charge.
" Les mouvements de crédits (transferts et virements) qui, sur le fondement de l'article 14 de l'ordonnance du 2 janvier 1959 modifiée relative aux lois de finances, modifient la répartition des crédits entre chapitres sont examinés au vu de la dotation du chapitre après adjonction des fonds de concours et sans qu'il soit possible dès lors de déterminer la part des fonds de concours mouvementée par la modification réglementaire opérée.
" Il en va de même pour les procédures de report des crédits disponibles au titre du budget suivant, effectués en application de l'article 17 de l'ordonnance du 2 janvier 1959 modifiée précitée.
Et de conclure :
" Cela étant, chaque ordonnateur peut mettre en place, à son niveau, des modalités de suivi spécifiques des divers crédits de fonds de concours bénéficiant au budget dont il est responsable, s'il juge utile de disposer de données d'analyse sur l'emploi réservé à tel ou tel versement à titre de fonds de concours. Le ministère de l'Emploi et de la Solidarité est donc peut-être en mesure de fournir les indications recherchées.
On conçoit au terme de cette explication qu'il soit fort difficile de suivre un article au sein d'un chapitre budgétaire surtout lorsque cet article ne représente que 1,4 % des crédits du chapitre et ce, en dépit du fait que cet article est alimenté exclusivement par une redevance perçue sur l'usager à l'exclusion de tout abondement par le budget général.
(3) Les crédits des comités ne leur sont pas intégralement reversés
L'apparition de crédits conservés par le budget de l'Etat et donc non distribués aux comités est un phénomène antérieur à la réforme du financement de ces derniers.
Toutefois, l'écart entre les crédits disponibles et les dotations budgétaires versées se creuse très nettement au moment de la mise en oeuvre de la réforme.
Le tableau qui suit fait apparaître au 31 décembre 2000 un montant de 12,9 millions de francs de réserves sur le chapitre 31-98. Ces crédits non versés aux comités ne s'élevaient qu'à 4,7 millions de francs fin 1997. En trois ans, cette réserve a quasiment triplé.
Chapitre 34-98 art. 93
|
(2)
|
(3)
|
(4)
|
Dotations pour fonctionnement versées au CCPPRB en n |
|
|
1993 |
14.123.250 |
nd |
nd |
nd |
|
1994 |
14.763.700 |
nd |
nd |
nd |
|
1995 |
13.450.299 |
nd |
17.187.610 |
12.184.005 |
|
1996 |
14.402.447 |
5.185.913 |
19.588.360 |
13.264.193 |
|
1997 |
13.980.254 |
6.324.167 |
20.304.421 |
15.616.640 |
|
1998 |
13.807.641 |
4.838.936 |
18.646.577 |
11.376.170 |
|
1999 |
14.298.029 |
7.327.032 |
21.625.061 |
8.645.000 |
|
2000 |
13.570.039 |
13.230.994 |
26.801.033 |
13.880.000 |
Source : Direction du Budget et Direction générale de la Santé (avril 2001)
Le premier enseignement tiré de ce tableau est, d'une certaine façon, rassurante : les crédits non versés aux comités sont reportés sur les exercices suivants. Ils ne sont pas détournés de leur objet ou utilisés à d'autres fins.
La rigueur des informations fournies n'est toutefois pas totale. Ainsi, la différence entre les crédits disponibles de l'exercice n et les dotations versées aux comités pour le même exercice ne correspond pas parfaitement au montant des crédits reportés. La différence reste toutefois minime (de l'ordre de 50.000 francs à 250.000 francs selon les exercices) et va de surcroît dans le bon sens : les crédits reportés sont légèrement supérieurs aux dotations non versées 67 ( * ) .
En revanche, les chiffres fournis par la Direction du budget font apparaître un écart avec ceux de la Direction générale de la santé, s'agissant des crédits ouverts pour 2000. Les premiers sont inférieurs de 500.000 francs aux seconds. Cet écart révèle en réalité une opération de plus grande ampleur ainsi décrite par la Direction générale de la santé 68 ( * ) .
" Pour l'année 2000, il a été procédé, dans le cadre d'une décision modificative et pour des raisons liées à l'urgence sanitaire en fin de gestion, à une avance à hauteur de 8 millions de francs dont 7,5 millions de francs ont fait l'objet d'un décret de virement dès le 10 novembre 2000 et dont le solde, soit 0,5 million de francs, est en cours de remboursement. Au 31 décembre 2000, les crédits disponibles représentent 26,801 millions de francs, sachant que les 0,5 millions de francs susvisés ne sont pas encore intégrés dans ce montant " .
Ainsi, selon cette explication un peu embarrassée, courant 2000, 8 millions de francs ont été " empruntés " sur les crédits destinés aux comités dont 7,5 millions de francs ont été " remboursés " en cours d'année. De sorte que la DGS fait apparaître dans le montant tant des crédits ouverts que des crédits disponibles en 2000 une " créance " de 500.000 francs que détiennent les comités sur le budget....
Cette opération " d'emprunt ", rapidement et presque complètement régularisée, est néanmoins significative des tentations que peuvent susciter les réserves constituées sur les dotations des comités, alors même que rien ne vient justifier une telle politique.
Dans ces conditions, il n'est guère étonnant que des craintes persistent car des crédits inutilisés sont rapidement considérés par les autorités budgétaires comme des crédits inutiles et donc mobilisables à d'autres fins.
La constitution de réserves sur le chapitre budgétaire consacré au fonctionnement des comités n'a fait en effet l'objet d'aucune concertation avec ces derniers, ni même d'une information ou encore d'une explication quant à la raison d'être de ces réserves ou à leurs possibles utilisations.
En outre, l'apparition concomitante d'un nouveau mode de répartition des dotations et d'une ponction sur les crédits disponibles conduit à s'interroger sur la justification des nouveaux critères de financement définis par la DGS.
En effet, l'article R. 2012 du code de la santé publique prévoit que le ministère chargé de la Santé répartit le produit du droit fixe " en fonction notamment de leurs charges et de leur activité ". L'activité doit donc être prise en compte, ce qui n'est plus le cas, puisque la DGS substitue à cette notion d'activité celle de charges et de " situation ".
Votre rapporteur observe à cet égard que l'ensemble de la réforme du financement des comités ne repose sur aucun texte 69 ( * ) . La circulaire n° 2000/537 du 25 octobre 2000 relative au financement des CCPPRB détermine le montant total de la dotation 70 ( * ) pour cette année (11,86 millions de francs) mais n'explicite ni la clé de répartition, ni les modalités de calcul de ce total.
d) Les comités se heurtent au manque de transparence dans leur financement
Les réponses aux questionnaires, comme les auditions auxquelles a procédé votre rapporteur, ont confirmé l'existence de difficultés concernant les modalités du financement des comités.
Le docteur François Chapuis, président de la Conférence nationale des CCPPRB remarque que les comités ne reçoivent aucune information sur la manière dont les dotations sont calculées. Selon lui, l'Administration ne tient pas compte du budget prévisionnel présenté par les comités, ni de l'importance de leur activité. Il regrette enfin la complète opacité qui entoure la procédure de financement et remarque que l'insuffisance de ressources explique l'absence de suivi par les comités de l'évolution des protocoles et notamment du nombre de protocoles abandonnés comme de ceux qui font l'objet d'une publication.
Plusieurs comités ont confirmé les observations formulées par le président de la Conférence nationale des CCPPRB.
Le témoignage suivant apparaît à cet égard comme particulièrement significatif :
" Depuis la création du comité, le montant des subventions accordées a suivi une évolution inquiétante : excédentaire dans les toutes premières années, calqué sur le montant du budget prévisionnel les années suivantes, en retrait par rapport à celui-ci depuis deux ans. En 1998, la subvention accordée ne permettait même pas de prendre en charge la rémunération de l'unique secrétaire mi-temps du comité.
" Les subventions sont, de tout temps, attribuées très tardivement, au mieux au mois de décembre de l'année, au pire l'année suivante. Le comité ne peut survivre que grâce à la ligne de crédit que lui accorde le CHU, ce qui n'est pas sain.
" En l'absence d'information sur les critères de répartition des subventions, leur montant annuel selon les comités, l'activité des autres comités, nous éprouvons des difficultés certaines lors de la rédaction du budget prévisionnel et des doutes, fondés ou non, sur la qualité de la péréquation faite à l'échelle nationale ou régionale. Dans le cas (de ce) comité, malgré un budget prévisionnel calculé au plus juste, tenant compte des facilités qui nous sont accordées par l'établissement d'accueil et réduisant de façon drastique certaines lignes (pas de recours aux experts extérieurs, un seul déplacement d'une personne à une manifestation extérieure en 1999, limitation au minimum des abonnements documentaires, réduction du montant des indemnités accordées aux rapporteurs), le budget accuse un déficit pour la deuxième année consécutive ".
Ce témoignage illustre l'incompréhension des comités vis-à-vis de l'organisation de leur financement qui, pour être désormais " souple ", semble désormais arbitraire.
A l'évidence, la nouvelle politique d'attribution des dotations ne satisfait pas les comités.
En outre, l'existence d'une réserve non distribuée contraste avec le constat qu'établissent les comités selon lequel leurs moyens sont insuffisants.
Un comité remarque qu'" une dotation non consommée entièrement sera réduite par le ministère l'année suivante ". Il observe qu'" il serait plus logique que les comités puissent disposer des financements excédentaires pour soutenir des projets de recherche régionaux en collaboration avec les délégations à la recherche clinique ".
Par ailleurs, les comités ne sont pas tous sur un pied d'égalité. Certains bénéficient de moyens mis à disposition par les hôpitaux qui les accueillent (locaux, personnels...). La plupart doivent leur verser une compensation financière qui peut être à l'origine de dettes impayées du fait d'une dotation budgétaire insuffisante. Le seul cas de contentieux impliquant un comité concerne d'ailleurs un problème de rétribution des moyens mis à disposition du comité par la structure accueillante.
Arbitraire des modalités de calcul des dotations, moyens insuffisants et inégalité des comités devant les charges de fonctionnement constituent trois caractéristiques du financement des CCPPRB auxquelles il convient d'ajouter l'existence d'une " cagnotte " ministérielle dont l'emploi reste à déterminer.
Ce constat appelle impérativement une " remise à plat " des modalités de financement des CCPPRB.
Si les règles définies en 1991 qui prévoyaient une répartition intégrale des droits perçus en fonction de l'activité ont pu provoquer des effets pervers (incitation à examiner le plus d'avis possible, constitution de réserves...), la définition des dotations pour chaque comité depuis Paris a démontré l'existence d'un risque d'arbitraire préjudiciable aux comités.
Outre les règles de répartition des droits fixes, c'est leur montant qu'il conviendra de réexaminer en s'interrogeant sur le tarif et l'assiette du droit, compte tenu des excédents constatés depuis trois ans, mais également des financements nécessaires à un fonctionnement satisfaisant des comités.
Lors de son audition, M. Pascal Penaud, adjoint au directeur général de la DGS, a considéré qu'" une réflexion était nécessaire pour éviter que ne se créent des excédents inutilisés. Une des pistes en est l'adaptation des redevances. Mais il est également nécessaire de mieux adapter les financements aux besoins réels de fonctionnement des CCPPRB. Ainsi, une modulation des indemnités versées aux rapporteurs en fonction de la complexité des dossiers pourrait être envisagée ".
3. L'Agence française de sécurité sanitaire des produits de santé (AFSSAPS) souhaite approfondir ses relations avec les comités
a) Des relations juridiquement limitées
L'AFSSAPS est un établissement public de l'Etat créé par la loi n° 98-535 du 1 er juillet 1998 relative au renforcement de la veille sanitaire et du contrôle de la sécurité sanitaire des produits destinés à l'homme . Cette agence s'est inscrite dans la continuité de l'Agence du médicament, avec des missions étendues à l'ensemble des produits de santé et des moyens renforcés.
Les relations directes entre l'AFSSAPS et les CCPPRB sont assez limitées.
L'article L. 209-12 du code de la santé publique dispose que les CCPPRB doivent communiquer à l'AFSSAPS tout avis défavorable donné à un projet de recherche concernant les produits mentionnés à l'article L. 793-1 71 ( * ) .
L'AFSSAPS et les comités ont également des relations indirectes. L'article L. 209.12 fait obligation au promoteur de transmettre à l'Agence, pour les produits mentionnés à l'article L. 793-1, " avant la mise en oeuvre d'une recherche biomédicale sur l'être humain ", une lettre d'intention décrivant les données essentielles de la recherche, accompagnée de l'avis du comité consulté, tandis qu'en vertu de l'article L. 209-12-1 le promoteur doit transmettre à l'autorité administrative compétente 72 ( * ) l'avis du CCPPRB sur les amendements.
b) Des relations à développer
Lors de son audition, l'AFSSAPS a confirmé que ses relations avec les comités " étaient ponctuelles, voire rares " . Elle a remarqué qu'aucune disposition du code de la santé publique ne prévoyait l'existence de dialogue entre elle et les comités.
L'AFSSAPS considère qu'il existe des incertitudes sur les compétences respectives. Elle souligne en particulier qu'il lui arrive de se prononcer sur le rapport bénéfice/risque d'un produit même si cela ne relève pas formellement de ses compétences.
A l'occasion de cette audition, l'AFSSAPS a fait part de son souhait de renforcer ses relations avec les comités.
Elle a souhaité pouvoir disposer des mêmes informations que les comités (contenu du protocole, brochure pour l'investigateur, note d'information pour le patient, dates réelles de début et de fin de l'essai) et a suggéré, à cet effet, la réalisation d'un document unique dont elle serait également destinataire.
Elle a remarqué qu'elle n'était pas en mesure de savoir si un essai était en cours ou non et a préconisé que les comités lui transmettent leurs avis parallèlement à leur transmission aux promoteurs.
4. La Conférence nationale des CCPPRB s'affirme désormais comme un interlocuteur représentatif
La Conférence nationale des CCPPRB est une association " loi 1901 " qui regroupe 42 des 48 CCPPRB. Elle poursuit trois objectifs : assurer la formation des membres des comités, favoriser la diffusion des bonnes pratiques et permettre la représentation des CCPPRB notamment auprès des pouvoirs publics.
La Conférence nationale organise des réunions annuelles qui permettent aux membres des CCPPRB d'échanger leurs expériences.
Le VIII e colloque, organisé en mai 1999, avait pour objet la présentation d'un bilan des dix années d'application de la loi du 20 décembre 1988. Il comprenait sept ateliers de travail thématiques concernant : le champ d'application de la loi, le consentement des personnes vulnérables, la qualification de la recherche " avec " ou " sans " bénéfice individuel direct, l'analyse du fondement des avis, l'application de la loi du 20 décembre 1988 aux dispositifs médicaux, les modalités de fonctionnement des comités et le devenir d'un essai thérapeutique après un avis favorable. Par ailleurs, ce colloque a été l'occasion d'élaborer un règlement intérieur " type " pour les comités.
La Conférence nationale joue donc un rôle important dans la diffusion des " bonnes pratiques ". Cette fonction d'accompagnement et de conseil ne lui est cependant pas officiellement reconnue.
IV. LES PROPOSITIONS DE LA MISSION D'INFORMATION
Si la mission a confirmé les effets bénéfiques de la législation mise en place depuis 1988 dans le développement de la recherche biomédicale en France, elle n'en a pas moins mis en évidence l'existence d'insuffisances et de contraintes qui pourraient s'avérer préjudiciables à l'avenir.
Ces obstacles, qui concernent tant l'organisation de la recherche biomédicale que celle des CCPPRB, ont inspiré à votre rapporteur plusieurs propositions d'ajustements et de réformes qui ouvriraient une nouvelle étape dans la mise en place d'une législation adaptée à la recherche biomédicale et à la protection des personnes qui y participent.
A. L'ORGANISATION DE LA RECHERCHE BIOMÉDICALE NÉCESSITE DES MODIFICATIONS
Quatre aspects relatifs à l'organisation de la recherche biomédicale appellent des modifications. Il s'agit tout d'abord de la distinction parfois difficile entre les essais avec ou sans bénéfice individuel direct. Vient ensuite la question de la gratuité des dispositifs médicaux fournis par le promoteur. Une autre difficulté concerne les relations de partenariat à développer entre les chercheurs et les personnes participant à la recherche. Enfin, une meilleure régulation du nombre des amendements pourrait être recherchée.
1. Clarifier la distinction entre les essais avec et sans bénéfice individuel direct
La distinction entre " les recherches biomédicales dont on attend un bénéfice direct pour la personne qui s'y prête " et les autres recherches, posée par le deuxième alinéa de l'article L. 209-1 du code de la santé publique, constitue le fondement du cadre juridique mis en place par le législateur pour organiser la recherche biomédicale.
Elle emporte des conséquences pratiques, en ce qu'elle implique des procédures distinctes dans la conduite des recherches, et juridiques, puisque la mise en cause de la responsabilité du promoteur diffère dans l'un ou l'autre cas.
C'est pourquoi il importe que les différents acteurs de la recherche biomédicale (CCPPRB, AFSSAPS, DGS, promoteurs, investigateurs) puissent disposer d'éléments d'information suffisamment précis pour pouvoir, chacun en ce qui les concerne, appliquer la loi.
Ce n'est pas toujours le cas aujourd'hui. Votre rapporteur a, en effet, pu constater l'existence d'incertitudes concernant le champ d'application de la loi. Les interrogations semblent particulièrement nombreuses concernant les recherches cognitives, épidémiologiques, psychologiques et génétiques.
La modification de la législation dans le sens d'une plus grande précision des définitions des essais avec ou sans bénéfice individuel direct devrait pouvoir intervenir lors de la révision des lois dites bioéthiques..
En outre, l'adoption par le Parlement européen et le Conseil, le 26 février 2001, d'une directive " concernant le rapprochement des dispositions législatives, réglementaires et administratives des Etats membres relatives à l'application de bonnes pratiques cliniques dans la conduite d'essais cliniques de médicaments à usage humain " 73 ( * ) pourra nécessiter des adaptations de notre législation.
Il reste que les incertitudes des comités et des promoteurs trouvant bien souvent leur origine dans un manque d'information, il est souhaitable d'organiser une meilleure diffusion des bonnes pratiques et le recensement des différents essais sur des bases de données afin de permettre une homogénéisation des critères de qualification.
Naturellement ces bases de données ne se limiteraient pas à la seule question de la frontière entre recherches avec ou sans bénéfice direct mais comporteraient, de manière générale, les informations nécessaires à une meilleure connaissance de la teneur des avis des comités.
Qui serait chargé de gérer ces bases de données ? Parmi les différentes possibilités, il convient de mentionner l'AFSSAPS et les CCPPRB à travers, par exemple, la Conférence nationale ou l'établissement public qu'il est proposé de créer (voir ci-dessous).
L'AFSSAPS a montré, à travers la réalisation d'un guide relatif aux études précliniques, qu'elle pouvait étendre ses compétences à une activité de cette nature. Une telle évolution présenterait néanmoins l'inconvénient de placer les CCPPRB dans une position de dépendance vis-à-vis de l'AFSSAPS, ce que nul ne souhaite.
Ainsi, la Conférence nationale des CCPPRB pourrait-elle prendre une initiative dans ce domaine en lien avec l'AFSSAPS.
2. Revenir sur l'obligation faite au promoteur de fournir gratuitement les dispositifs médicaux
Le principe posé par l'article R. 2038 du code de la santé publique constitue un obstacle au développement de la recherche biomédicale en France, comme en a convenu l'IGAS 74 ( * ) il y a déjà plusieurs années et comme l'ont rappelé les personnes auditionnées.
L'internationalisation des entreprises travaillant dans le domaine médical comme l'expatriation de certains chercheurs renforcent les effets négatifs de cette disposition réglementaire de telle sorte que sa modification apparaît aujourd'hui encore plus indispensable.
Un aménagement de ce principe pourrait être obtenu en favorisant une prise en charge partielle par la sécurité sociale du coût des dispositifs médicaux faisant l'objet d'une recherche, sur le fondement du tarif de responsabilité des dispositifs existants ou proches de l'objet de la recherche. Cette suggestion nécessiterait une disposition législative afin d'autoriser la prise en charge, même partielle, de ces dispositifs par la sécurité sociale.
3. Associer davantage les personnes se prêtant à la recherche
Le développement de la recherche biomédicale est étroitement lié à la participation des personnes qui s'y prêtent. Cette participation repose sur la confiance, la transparence et la conviction que les recherches menées contribueront aux progrès de la médecine.
Certaines personnes participant à des protocoles manifestent le souhait d'être tenues informées du déroulement et des résultats des recherches, et cette information n'étant pas aujourd'hui de droit, il convient d'en reconnaître le principe dans les textes.
La personne participant à la recherche pourrait faire valoir, à l'occasion du recueil du consentement, sa volonté de recevoir, ou non, des informations relatives aux résultats de la recherche.
Les articles L. 209-9 et R. 2029 du code de la santé publique seraient ainsi modifiés afin que soit incluse, au nombre des pièces ou éléments d'information transmis par l'investigateur au comité, la possibilité reconnue à la personne se prêtant à la recherche d'être informée sur ses résultats.
4. Assurer une meilleure régulation du nombre des amendements
L'augmentation du nombre des amendements constitue une des caractéristiques de l'évolution de l'activité des comités. Le nombre des amendements examinés par les comités est aujourd'hui du même ordre que celui des protocoles.
Ce phénomène n'est pas, en lui-même, préjudiciable à la conduite des projets de recherche, pour autant qu'il ne traduit pas l'existence d'un " biais " qui inciterait les investigateurs à multiplier les amendements du fait de l'absence de droit fixe.
Or, une telle évolution n'a pas été exclue par nombre des interlocuteurs auditionnés. Cette situation pourrait porter atteinte à la qualité du travail des comités si elle les amenait à consacrer moins de temps à l'examen des protocoles.
La création d'un droit fixe spécifique aux amendements pourrait être envisagée. Elle inciterait en effet les intéressés à présenter des protocoles les plus complets possible en première intention.
La tarification des amendements n'ayant pas pour objet d'augmenter les ressources des comités mais de réguler le nombre des amendements, elle pourrait s'accompagner d'une baisse du montant des droits fixes associé au dépôt des protocoles afin de maintenir stable le montant total des redevances.
La création d'un droit fixe pour les amendements nécessiterait la modification de l'article L. 209-11 du code de la santé publique.
B. LE FONCTIONNEMENT DES COMITÉS EST SUSCEPTIBLE D'AMÉLIORATIONS
La plupart des acteurs de la recherche biomédicale sont satisfaits de la manière dont les CCPPRB exercent leur mission. Des améliorations restent néanmoins à la fois possibles et nécessaires, tant en ce qui concerne leur régime juridique que leurs modes d'organisation et de fonctionnement.
1. Doter les CCPPRB d'un statut adapté
Votre commission des Affaires sociales n'était pas favorable en 1994 à ce que les comités soient dotés de la " personnalité juridique " 75 ( * ) . Ce pas ayant néanmoins été franchi à l'occasion de l'adoption de la loi du 25 juillet 1994, il convient aujourd'hui d'en tirer les conséquences et de réfléchir au statut dont pourraient être dotés les CCPPRB.
La définition d'un statut pour les CCPPRB permettrait de mettre un terme aux difficultés rencontrées par certains d'entre eux dans leurs relations avec les services déconcentrés du ministère de l'Economie et des Finances qui ont eu tendance quelquefois à évoquer l'absence de statut pour retarder le versement de la subvention publique qui leur revient.
Plusieurs statuts sont envisageables parmi lesquels on peut citer celui des associations dites " loi 1901 ", celui des établissements publics ainsi que celui des groupements d'intérêt public.
Les comités fonctionnent de fait aujourd'hui comme de quasi-associations. Le statut des associations dites " loi 1901 " est certes réputé pour sa souplesse, mais il reste peu adapté aux organismes bénéficiant de fonds publics et placés sous la tutelle de l'Etat, compte tenu en particulier des difficultés rencontrées pour contrôler l'utilisation des fonds et des exigences particulières qui incombent à des personnes morales participant à des missions de service public.
Le statut de groupement d'intérêt public ne semble pas davantage approprié puisqu'il est destiné à servir de cadre pendant une période de temps limitée à la coopération entre personnes publiques et privées.
Le statut d'établissement public permettrait de concilier l'autonomie des CCPPRB et le maintien de la tutelle administrative de l'Etat. Il est cependant difficilement envisageable de créer 48 établissements publics en lieu et place des comités existant aujourd'hui.
Aussi conviendrait-il d'examiner à tout le moins la possibilité de créer un établissement public au niveau régional, ou interrégional pour les régions ne comptant qu'un seul comité, auquel seraient rattachés les CCPPRB dans leur forme actuelle, voire au niveau national.
En effet, la création d'un établissement public national ne serait toutefois pas sans présenter un réel avantage :
- il pèserait d'un poids accru face à l'Administration, mais aussi vis-à-vis des grands organismes de recherche ;
- il pourrait être l'affectataire du droit fixe qui échapperait ainsi au mécanisme budgétaire complexe des fonds de concours ; il en garantirait de manière transparente sa restitution aux comités et sa répartition selon des règles du jeu qui associeraient directement ces derniers ;
- il serait le bon niveau pour l'organisation d'actions d'informations ou éventuellement de conseil, à l'égard des chercheurs, et de formation pour les membres des comités ou encore pour la mise en place de bases de données sur l'activité des comités et la teneur de leurs avis ;
- il se substituerait plus facilement à l'Administration lorsque la carence de cette dernière est manifeste, notamment dans la gestion de l'état civil des comités : mobilisation des autorités chargées de proposer les candidatures, suivi des démissions, etc.
Naturellement, une telle démarche, si elle était comprise comme une volonté de réduire l'autonomie des comités, ne serait pas acceptable : l'établissement public national devrait être conçu non comme une tutelle centralisatrice qui pèserait sur les comités mais comme une sorte d'organe central à leur service . Il se rapprocherait, à ce titre, de l'actuelle Conférence des CCPPRB.
Il serait le point d'impact adéquat de la tutelle de l'Etat aujourd'hui exercée dans un certain désordre à l'égard de chaque comité individuellement.
Mais la composition équilibrée de son conseil d'administration, et, le cas échéant, de son conseil de surveillance, en ferait également un lieu d'échanges et de propositions entre les représentants des comités, qui devraient y occuper une place importante, les représentants de l'Etat, des agences sanitaires, des organismes de recherche mais également des associations exprimant le point de vue des personnes se prêtant aux recherches biomédicales.
2. Réformer l'organisation du financement des CCPPRB
La mission a pu confirmer l'existence de graves difficultés dans l'organisation du financement des CCPPRB.
La répartition du produit des redevances au prorata du nombre d'avis rendus qui existait jusqu'en 1998 était soupçonnée d'inciter les comités à " faire du chiffre ".
Toutefois, la réforme décidée par la DGS en 1998, qui prévoit une répartition des crédits en fonction des budgets prévisionnels établis par les comités, si elle a eu le mérite de rechercher une gestion plus rigoureuse de certains comités, n'a pas permis d'établir des règles suffisamment transparentes pour qu'elles ne soient pas contestées.
Outre le fait que certains comités ont connu une chute brutale de leur subvention, voire une suppression pure et simple sur plusieurs exercices, votre rapporteur a constaté qu'une partie des crédits budgétaires ouverts en contrepartie des droits fixes perçus n'était pas attribuée. Ces crédits sont reportés d'année en année sur le fonds de concours correspondant.
Votre rapporteur considère, dans ces conditions, qu'une remise à plat du financement des comités est nécessaire, de paire avec la réflexion sur un statut des CCPPRB, afin de mieux prendre en compte les besoins des comités et d'établir des règles de financement plus transparentes.
Observant qu'il n'est pas acceptable qu'une partie des crédits reste en attente d'affectation sur un fonds de concours, votre rapporteur estime par ailleurs que la réforme du financement devrait permettre de mieux ajuster le montant des redevances aux besoins des comités, afin soit d'augmenter le montant des subventions versées aux comités, soit de réduire le montant des redevances.
Le minimum indispensable serait d'adapter la nomenclature budgétaire de telle sorte qu'un chapitre soit exclusivement consacré aux CCPPRB. Actuellement, en effet, ces derniers ne font l'objet que d'un paragraphe au sein d'un chapitre qui comporte bien d'autres dépenses. De fait, il est difficile, ou même impossible, de suivre aujourd'hui de façon précise au sein de ce chapitre la consommation des crédits destinés aux comités.
Une étape supplémentaire gagnerait à être franchie en faisant échapper le droit fixe à la lourde machine du budget général pour l'affecter à une personne morale différente de l'Etat, dont l'une des missions importantes serait de gérer cette ressource et de la répartir entre les comités, selon des règles claires à la définition desquelles ces derniers seraient étroitement associés.
La réforme du financement nécessiterait des modifications d'ordre réglementaire afin de clarifier son organisation et les règles de répartition des crédits. L'article L. 209-11 76 ( * ) du code de la santé publique ne semble pas quant à lui devoir être nécessairement modifié 77 ( * ) dans le cadre de la " première étape " de la réforme du financement.
3. Reconnaître le rôle de conseil des comités
C'est un apport important de cette mission que d'avoir mis en évidence le rôle de conseil des CCPPRB.
Comme l'ont souligné plusieurs comités dans leurs réponses ainsi que les personnes auditionnées 78 ( * ) , les comités sont de plus en plus souvent sollicités préalablement au dépôt du protocole pour avis pour qualifier la recherche (avec ou sans bénéfice individuel direct). Les investigateurs leur demandent également quelquefois de se prononcer sur la méthodologie retenue. Cette pratique est aujourd'hui largement répandue.
La crainte selon laquelle les " conseils " donnés en " amont " de l'avis pourraient lier le comité lors de l'examen de l'avis n'apparaît pas fondée au regard des auditions réalisées par votre rapporteur.
Reconnaître le rôle de conseil des comités permettrait, dans ces conditions, de dissiper les interrogations de certains comités quant à la licéité de cette activité.
Dans la pratique, le rôle de conseil pourrait être reconnu soit au comité dans son entier, soit à son seul président. Cependant, le souci du respect du pluralisme dans la composition des comités ne devrait pas permettre de retenir cette dernière solution. En revanche, il semble possible d'imaginer des solutions intermédiaires comme la création au sein de chaque comité d'une formation réduite associant trois ou quatre membres des différentes catégories qui les composent qui serait chargée d'assurer cette fonction de conseil.
Une telle évolution, qui nécessiterait une intervention du législateur, devrait toutefois se garder de toute confusion des genres entre le conseil préalable et l'avis officiel. Elle ne permettra pas, de toutes les façons, de faire l'économie d'une meilleure information des chercheurs grâce à une documentation ad hoc les guidant dans la présentation des protocoles et à la constitution de bases de données leur permettant de mieux connaître la " jurisprudence " des comités. Bon nombre de demandes de conseils adressées aux comités résultent en effet autant d'un déficit général d'information que de la nécessité de faire trancher en amont des points très spécifiques.
4. Garantir l'effectivité du pluralisme des comités
Les résultats de la mission ont fait apparaître des craintes quant au respect du principe de pluralisme posé par le législateur. Celles-ci trouvent leurs origines dans les modalités de nomination et de formation de leurs membres ainsi que dans l'absence de mécanisme d'indemnisation de certaines catégories de membres.
a) Modifier la composition des CCPPRB
Votre rapporteur ne considère pas qu'il soit nécessaire de modifier l'article L. 209-11 du code de la santé publique 79 ( * ) qui pose le principe du pluralisme dans la composition des comités.
Il s'interroge néanmoins sur la mise en oeuvre de ce principe telle qu'elle résulte de l'article R. 2001 qui prévoit que chaque comité comprend douze membres titulaires et douze membres suppléants répartis en huit catégories. Il fait siennes les remarques d'un comité qui proposait de supprimer la distinction entre membres titulaires et suppléants et de porter le nombre des membres des comités à seize. Cette mesure permettrait sans doute de favoriser le présentéisme.
b) Assurer l'effectivité des nominations et des remplacements des membres des comités
L'absentéisme de certaines catégories de membres constitue le " talon d'Achille " des CCPPRB. Il est dû pour partie au défaut de suivi de la part des DRASS qui exercent cette mission au nom du préfet de région.
Les résultats de la mission ont révélé l'hétérogénéité des procédures de suivi des différentes DRASS comme la très grande inégalité des moyens consacrés à la bonne application de la loi du 20 décembre 1988. Ce constat amène votre rapporteur à demander que les moyens des DRASS soient renforcés et que des dispositions soient adoptées pour assurer le suivi des nominations qui incombe, de par la loi, à l'administration déconcentrée des Affaires sociales à travers le préfet de région. A défaut, votre rapporteur proposera une nouvelle répartition de compétences en matière de nomination 80 ( * ) .
c) Prévoir la formation des nouveaux membres
Si les absences des membres des comités s'expliquent dans certains cas par les carences de l'administration à assurer les nominations et les renouvellements, ces difficultés trouvent également leur origine dans l'insuffisance des candidatures et la proportion élevée des démissions en cours de mandat.
Ces démissions s'expliquent notamment par l'information insuffisante sur le rôle des comités et la charge de travail inhérente à la fonction de membre. Une information préalable devrait être prévue afin de permettre le recrutement de personnes motivées et au fait de leurs obligations futures.
Par ailleurs, la plupart des membres de comités ne sont pas formés pour exercer leurs fonctions. Ils apprennent " sur le tas ". Compte tenu des nouveaux enjeux que représente, par exemple, le développement de la génétique et afin d'assurer l'effectivité du pluralisme, il apparaît nécessaire d'organiser des formations régulières à l'attention des membres des comités.
Ces formations pourraient être coordonnées ou organisées par la Conférence nationale des CCPPRB qui exerce déjà ce rôle de manière ponctuelle ou par le ou les établissements publics dont votre rapporteur propose la création.
d) Mettre en place un mécanisme d'indemnisation de certains membres des comités
L'absentéisme des membres des comités exerçant une profession libérale constitue un obstacle important à la bonne application de la loi.
Afin de résoudre cette difficulté, votre rapporteur propose la création d'une indemnité spécifique dont pourraient bénéficier, par exemple, les médecins généralistes (catégorie II), les pharmaciens (catégorie III) et les juristes (catégorie VIII) exerçant une profession libérale.
Cette indemnité pourrait être financée au moyen d'une majoration des dotations des comités.
Le montant actuel du produit des droits fixes devrait facilement permettre une telle augmentation des subventions étant donné la différence structurelle observée entre le montant total des droits perçus et celui des dotations accordées aux comités par la DGS 81 ( * ) .
5. Informer les comités du suivi des protocoles
Les comités ont acquis un véritable " savoir faire " qui en fait des partenaires à part entière de la recherche biomédicale. Pourtant, rien n'est prévu aujourd'hui pour leur permettre de connaître les résultats des recherches qu'ils ont été amenés à examiner.
Cette situation est regrettable dans la mesure où elle prive les comités d'une source d'information susceptible d'améliorer la qualité des avis qu'ils rendent.
De la même manière que la création de bases de données répertoriant les essais et les avis serait susceptible de favoriser la diffusion des " bonnes pratiques ", une meilleure information des comités des résultats des recherches devrait permettre d'améliorer la protection des personnes participant à ces recherches.
C. LES RELATIONS DES COMITÉS AVEC LES AUTRES ACTEURS DE LA RECHERCHE BIOMÉDICALE DEMANDENT À ÊTRE CLARIFIÉES
L'ensemble des propositions présentées précédemment, qu'elles concernent l'organisation de la recherche biomédicale ou celle des CCPPRB, illustre bien la nécessité de redéfinir les relations qui existent entre les comités et l'administration. La Conférence nationale des CCPPRB apparaît à cet égard aujourd'hui comme un partenaire incontournable dans l'organisation de ces relations.
1. Préciser les compétences respectives de l'AFSSAPS, de la DGS et des CCPPRB
Concernant les relations entre l'AFSSAPS et les comités, votre rapporteur a pu constater l'existence d'une volonté réciproque d'accroître les échanges à travers, par exemple, la création de bases de données et de guides répertoriant les différents essais. Cette évolution semble nécessaire pour autant qu'elle ne porte pas atteinte à l'indépendance des comités.
La nature des rapports entre la DGS et les comités pose des problèmes d'une autre ampleur compte tenu des carences de la tutelle mises en évidence par la mission.
Les modifications à apporter concernent autant l'organisation du suivi des comités par la DGS que le contenu même de la relation de tutelle. Votre rapporteur a proposé précédemment de renforcer l'autonomie des comités en confiant l'organisation de leur financement à un ou plusieurs établissements publics.
La tutelle du ministère chargé de la Santé, telle qu'elle est exercée par les DRASS au niveau déconcentré, ayant elle aussi révélé des carences, notamment au regard des nominations, une clarification des compétences respectives des DRASS et du ou des établissements publics à créer pour accompagner le fonctionnement des comités devrait être recherchée. Si le préfet de région doit continuer à nommer les membres des CCPPRB, la gestion, du régime particulièrement complexe des propositions de candidatures, pourrait être utilement de la responsabilité du ou des établissements publics qu'il est proposé de mettre en place.
2. Valoriser la Conférence nationale des CCPPRB
La représentativité de la Conférence nationale est aujourd'hui établie puisqu'elle rassemble 42 des 48 CCPPRB existants.
Cette association s'est révélée utile pour favoriser le dialogue entre les différents CCPPRB. Elle devrait être reconnue comme un interlocuteur privilégié de l'administration.
La Conférence nationale apparaît comme la structure idoine pour organiser la formation des membres des CCPPRB. Elle pourrait également être associée à l'organisation des bases de données rassemblant les protocoles et les avis dont votre rapporteur propose la création.
Enfin, la Conférence nationale pourrait jouer un rôle consultatif dans l'organisation du financement des CCPPRB. Dans le cadre actuel, il serait en effet utile qu'elle puisse donner son avis sur le projet de répartition des crédits établi chaque année par la DGS.
A terme, la création d'un véritable statut pour les CCPPRB, qui pourrait s'inspirer par exemple du régime des établissements publics, pourrait amener la Conférence nationale à évoluer pour intégrer pleinement cette nouvelle organisation en s'inspirant par exemple de la " Conférence des doyens ".
*
* *
Telles sont les principales propositions que formule votre rapporteur. Certaines sont d'ordre pratique, d'autres appellent une évolution des textes réglementaires, mais certaines exigent des modifications législatives. La révision des lois dites de " bioéthique ", qui devrait être déjà intervenue depuis juillet 1999, pourrait être l'occasion d'en débattre.
TRAVAUX DE LA COMMISSION
I. RÉUNION DU MARDI 9 MAI 2000
Au cours de la réunion de commission du mardi 9 mai 2000, sous la présidence de M. Jean Delaneau, président, au titre des questions diverses, M. Jean Delaneau, président, a fait part à la commission d'une demande de M. Claude Huriet concernant l'application de la loi du 20 décembre 1998 relative à la protection des personnes qui se prêtent à des recherches biomédicales.
M. Claude Huriet , en tant que rapporteur de cette loi pour la commission des affaires sociales, a fait état des nombreuses questions, restées sans réponse, qu'il avait adressées au Gouvernement quant au fonctionnement des comités consultatifs de protection des personnes qui se prêtent à des recherches biomédicales (CCPPRB).
Il a estimé qu'il relevait particulièrement des missions de la commission de réaliser un bilan de ce fonctionnement et d'avancer des propositions d'amélioration ; il a souligné en effet l'importance d'un suivi attentif de l'application des lois mais il a également souhaité inscrire ce bilan et ces propositions dans la perspective d'une révision des lois dites " bioéthiques " dont le Gouvernement tarde à prendre l'initiative en dépit des dispositions votées par le Parlement.
M. Jean Delaneau, président , a proposé à la commission, qui l'a approuvé, de confier à M. Claude Huriet la mission d'établir un bilan du fonctionnement des CCPPRB et des propositions d'amélioration ; en plein accord avec M. Claude Huriet, il a demandé aux membres de la commission intéressés par cette mission de lui faire savoir dans les prochains jours afin qu'ils puissent être informés du programme de travail du rapporteur et participer à tout ou partie des auditions conduites par ce dernier.
M. Francis Giraud a évoqué son expérience de président d'un CCPPRB et a fait part, d'ores et déjà, de son vif intérêt pour le travail de Claude Huriet, dont il a pleinement approuvé l'initiative.
II. RÉUNION DU MERCREDI 4 AVRIL 2001
Réunie le mercredi 4 avril 2001, sous la présidence de M. Jean Delaneau, président, la commission a tout d'abord entendu une communication de M. Claude Huriet sur le fonctionnement des comités consultatifs de protection des personnes dans la recherche biomédicale (CCPPRB).
M. Claude Huriet, rapporteur, a rappelé que la mission qui lui avait été confiée en mai dernier par la commission consistant à établir le bilan du fonctionnement des comités consultatifs de protection des personnes dans la recherche biomédicale (CCPPRB) répondait à un double souci : rassembler les informations disponibles concernant l'activité de ces structures créées par la loi du 20 décembre 1988 et proposer des réformes susceptibles d'être discutées à l'occasion de la révision des lois dites de " bioéthique ".
Evoquant les neuf questions écrites relatives au fonctionnement des CCPPRB qu'il avait adressées au ministre chargé de la santé depuis 1996, dont aucune n'avait reçu à ce jour de réponse, il a précisé que cette mission constituait également une réponse au défaut d'information du Parlement par le Gouvernement.
M. Claude Huriet a indiqué qu'afin de recueillir une information complète, il avait envoyé un questionnaire à l'ensemble des comités et avait conduit un important programme d'auditions associant des personnalités reconnues de la recherche biomédicale.
M. Claude Huriet a ensuite rappelé les grands principes de la loi du 20 décembre 1988 relative à la protection des personnes qui se prêtent à des recherches biomédicales et le rôle des CCPPRB.
Il a précisé tout d'abord que cette loi avait permis d'organiser une activité jusque-là mal encadrée et néanmoins indispensable aux progrès de la médecine.
Il a rappelé que le code de la santé publique prévoyait que les recherches devaient se fonder sur le dernier état des connaissances scientifiques ainsi que sur une expérimentation préclinique suffisante et devaient également avoir pour objet d'étendre la connaissance scientifique de l'être humain et les moyens susceptibles d'améliorer sa condition. Il a ajouté que le risque prévisible encouru par les personnes qui se prêtent à la recherche ne devait pas être hors de proportion avec le bénéfice escompté.
M. Claude Huriet, rapporteur, a souligné également que les conditions de la réalisation des protocoles étaient également strictement réglementées, ces recherches ne pouvant être effectuées que sous la direction et sous la surveillance d'un médecin justifiant d'une expérience appropriée et dans des conditions matérielles et techniques adaptées à l'essai et compatibles avec les impératifs de rigueur scientifique et de sécurité des personnes qui se prêtent à ces recherches.
Il a remarqué que des conditions plus restrictives étaient, par ailleurs, posées pour les femmes enceintes, les personnes privées de liberté et les malades ne pouvant donner leur consentement ainsi que pour les mineurs et les majeurs protégés.
Il a insisté sur le fait que, dans tous les cas, le promoteur, c'est-à-dire " la personne physique ou morale qui prend l'initiative d'une recherche biomédicale sur l'être humain ", devait assurer l'indemnisation des conséquences dommageables de la recherche pour la personne qui s'y prête, et devait donc souscrire une assurance.
Par ailleurs, M. Claude Huriet a rappelé que des règles strictes avaient été définies pour recueillir le consentement " libre, éclairé et exprès " de la personne qui se prête à la recherche, l'investigateur, c'est-à-dire " la ou les personnes physiques qui dirigent et surveillent la réalisation de la recherche " ou un médecin qui le représente, devant faire connaître à la personne l'objectif de la recherche, les bénéfices attendus, les contraintes et les risques prévisibles, l'avis du CCPPRB et, le cas échéant, son inscription dans un fichier national.
M. Claude Huriet, rapporteur, a rappelé qu'un des principaux apports de la loi du 20 décembre 1988 résidait dans la distinction qu'elle opérait entre les " recherches biomédicales avec bénéfice individuel direct " qui désignent les essais " dont on attend un bénéfice direct pour la personne qui s'y prête " et a contrario " toutes les autres recherches, qu'elles portent sur des personnes malades ou non " que la loi qualifie de " sans bénéfice individuel ".
Il a expliqué que cette distinction, claire et simple dans son principe, emportait des conséquences importantes en particulier au regard du régime applicable en matière de responsabilité.
M. Claude Huriet, rapporteur, a estimé que les CCPPRB constituaient les " piliers " sur lesquels reposait la bonne application de la loi.
Il a rappelé qu'ils étaient chargés de donner leur avis sur les conditions de validité de la recherche au regard de la protection des personnes et notamment sur la pertinence générale du projet, l'adéquation entre les objectifs poursuivis et les moyens mis en oeuvre, la qualification du ou des investigateurs, la protection des participants, les modalités de l'information des personnes se prêtant à la recherche, les modalités de recueil du consentement et les indemnités éventuellement versées par le promoteur en cas de recherche sans bénéfice individuel direct pour la personne.
Il a souligné qu'avant de réaliser une recherche sur l'être humain, tout investigateur était tenu d'en soumettre le projet à l'avis de l'un des CCPPRB compétents dans la région où il exerçait son activité.
Evoquant les enseignements de la mission, M. Claude Huriet a constaté tout d'abord le manque de précision des statistiques disponibles.
La direction générale de la santé (DGS) connaît le nombre total d'avis rendus par les comités (2.280 en 1999, en baisse de 5,5 % par rapport à 1998). Selon les réponses au questionnaire adressé aux comités, 63 % des avis ont été demandés par les industriels contre 37 % par les personnes physiques ou les organismes sans but lucratif.
S'agissant de la nature des essais, M. Claude Huriet, rapporteur, a observé que 78 % des protocoles concernaient des médicaments, 10 % des dispositifs médicaux, 9 % des recherches cognitives et 3 % des recherches en psychologie. En revanche, il a indiqué que les données disponibles n'apparaissaient pas suffisamment précises puisqu'elles ne permettaient pas d'apprécier, par exemple, le nombre de recherches en cosmétique. Il a estimé que des doutes continuaient à subsister quant à l'application de la loi dans le cas de certaines recherches comme les études épidémiologiques nécessitant un prélèvement sanguin.
M. Claude Huriet, rapporteur, a observé que les comités comme les promoteurs déclaraient rencontrer des difficultés pour qualifier les essais " avec " ou " sans " bénéfice individuel direct.
Il a souligné qu'un des enseignements importants de la mission résidait dans la nécessité d'établir un recueil des positions adoptées par les divers comités afin d'établir une sorte de " jurisprudence ".
Il a remarqué qu'un autre sujet de préoccupation avait trait à l'obligation faite aux fabricants de dispositifs médicaux de fournir gratuitement les moyens nécessaires à la recherche. Il a rappelé que l'ensemble des interlocuteurs questionnés à ce sujet avaient confirmé que cette situation pénalisait le développement de la recherche biomédicale en France.
M. Claude Huriet, rapporteur, a évoqué ensuite les défaillances propres à l'organisation des comités.
Il a rappelé que la qualité du fonctionnement des CCPPRB était fonction de l'effectivité du pluralisme de leur composition et dépendait donc de la présence des représentants de chacune des huit catégories de membres qui les composaient.
Il a observé que la présence de certaines catégories de membres était loin d'être assurée dans nombre de comités, ces absences concernant en particulier les médecins généralistes, les personnes qualifiées en matière d'éthique, les personnes qualifiées dans le domaine social, les psychologues, ainsi que les personnes compétentes en matière juridique.
Il a constaté que les causes de ce fort absentéisme semblaient tenir au nombre élevé des démissions, à l'absence de formation des membres ainsi qu'à l'absence de rémunération de ceux d'entre eux qui exercent une profession libérale. Il a estimé que cet absentéisme devait également beaucoup aux carences de l'administration en matière de nomination et de renouvellement des membres comme au manque de diligence de certaines autorités habilitées à proposer des candidats.
M. Claude Huriet, rapporteur, a souligné que l'hétérogénéité de l'activité et des missions des comités constituait le deuxième enseignement de la mission concernant les CCPPRB.
Il a, en particulier, constaté que les comités ne se bornaient pas à donner un avis sur les protocoles de recherche mais qu'" en amont ", certains comités avaient accepté un rôle de conseil qui contribuait à améliorer la qualité des projets.
Il a observé qu'" en aval ", les comités étaient de plus en plus fréquemment saisis d'amendements aux protocoles, le nombre d'amendements étant aujourd'hui équivalent à celui des protocoles, une telle situation n'étant pas sans inconvénient si elle devait amener les comités à consacrer moins de temps à l'examen des protocoles.
Il a remarqué que l'hétérogénéité de l'activité des comités était par ailleurs accentuée par le fait que certains grands organismes de recherche avaient mis en place des comités d'experts qui exerçaient une fonction de " filtre scientifique ".
M. Claude Huriet, rapporteur, a rappelé que la forte variation du nombre des avis rendus par certains comités, comme la polarisation de l'activité sur quelques-uns, avaient pu laisser craindre que certains avis ne soient pas rendus avec tout le soin nécessaire, d'où la référence à l'existence d'un " Gault-Millau " des CCPPRB. Il a estimé que les données disponibles ne permettaient pas de confirmer ou d'infirmer une telle hypothèse qui n'avait cependant pas été contredite par la DGS lors de son audition.
Il a estimé ensuite que les relations des comités avec les pouvoirs publics constituaient l'un des points les plus préoccupants.
Il a remarqué que le suivi des comités par la DGS, malgré la réforme de ses services intervenue récemment, n'était pas satisfaisant, de même que la mobilisation des DRASS, qui exercent la tutelle de l'Etat au nom du préfet de région.
Il a souligné en outre que la procédure d'affectation des moyens budgétaires aux comités semblait particulièrement opaque.
Il a rappelé que le financement des comités reposait sur une redevance dont les promoteurs devaient s'acquitter pour chaque protocole et que les crédits ouverts en contrepartie, selon la procédure des fonds de concours, étaient redistribués aux comités en fonction de leur activité respective, cette activité étant appréciée au regard du nombre d'avis rendus.
Il a insisté sur le fait que, depuis 1998, les modalités de répartition des crédits avaient été modifiées de manière unilatérale, la DGS retenant désormais deux critères : les charges salariales et de fonctionnement et la situation des comités au vu des comptes de résultats et des bilans financiers.
M. Claude Huriet, rapporteur, a considéré que, de fait, la direction générale de la santé était passée, pour la répartition des dotations entre les comités, d'un mode mécanique mais transparent à des modalités " souples " mais opaques.
Ayant rappelé que la DGS entendait, ce faisant, introduire davantage de rigueur dans la gestion de certains comités, il a souligné qu'elle avait elle-même reconnu que ces modifications avaient pu porter préjudice à des comités dont la gestion n'était pas discutée.
Il a indiqué que cette " réforme " du financement s'était également traduite par un écart croissant entre le montant total des redevances et les sommes globalement distribuées aux comités en précisant que contrairement à une interrogation répandue parmi les comités, les crédits non distribués n'étaient pas reversés au budget général, mais reportés d'année en année sur le fonds de concours correspondant.
Il a considéré que cette situation appelait impérativement une " remise à plat " des modalités de financement des CCPPRB.
M. Claude Huriet, rapporteur, a ensuite évoqué ses propositions en commençant par celles relatives à l'organisation même de la recherche biomédicale.
Il a insisté sur la nécessité de clarifier la distinction entre les essais " avec " ou " sans " bénéfice individuel direct.
Il a ensuite proposé de revenir sur l'obligation faite au promoteur de fournir gratuitement les dispositifs médicaux et d'associer davantage les personnes se prêtant à la recherche.
Il a aussi évoqué les moyens d'assurer une meilleure régulation du nombre des amendements aux protocoles.
M. Claude Huriet, rapporteur, a ensuite souligné la nécessité de réformer le fonctionnement même des comités.
A cet égard, il a détaillé ses propositions permettant de doter les CCPPRB d'un statut adapté et de réformer l'organisation de leur financement.
Il a estimé nécessaire de reconnaître le rôle de conseil des comités et de garantir le pluralisme de leur composition.
Compte tenu des causes multiples qui concourent à l'absentéisme, il a évoqué plusieurs pistes pour y mettre un terme comme la modification de la composition des CCPPRB, l'augmentation des moyens des directions régionales des affaires sanitaires et sociales (DRASS), une meilleure information des candidats, la formation des membres, ainsi qu'un mécanisme d'indemnisation de certaines catégories de membres.
M. Claude Huriet a enfin suggéré une dernière série de propositions concernant les relations des comités avec les autres acteurs de la recherche biomédicale.
Il a souligné la nécessité de préciser les compétences respectives de l'association française de sécurité sanitaire et des produits de santé (AFSSAPS), de la direction générale de la santé (DGS) et des CCPPRB et de valoriser la conférence nationale des CCPPRB.
M. Claude Huriet, rapporteur, a considéré, en conclusion, que certaines de ses propositions étaient d'ordre pratique, que d'autres appelaient une évolution des textes réglementaires, que d'autres enfin nécessitaient des modifications législatives. Il a estimé que la révision des lois dites de " bioéthique ", qui aurait dû intervenir dès juillet 1999, pourrait être l'occasion de débattre de ces dernières.
M. Francis Giraud, faisant état de son expérience d'ancien président de CCPPRB a souligné alors le rôle indispensable de ces structures créées à l'initiative de MM. Claude Huriet et Franck Sérusclat, en insistant sur l'apport majeur que représentait leur composition pluraliste associant des médecins et des non-médecins. Il s'est inquiété des constatations du rapporteur faisant apparaître que des menaces pesaient sur ce pluralisme.
Il a évoqué les difficultés qui pouvaient être soulevées par le fait que la loi avait prévu des procédures uniformes quelle que soit la nature des médicaments faisant l'objet d'une recherche. Il a confirmé que la question du financement des CCPPRB avait toujours posé des problèmes. Il a, enfin, insisté sur la nécessité de conserver et de conforter ces structures indispensables au développement de la recherche biomédicale.
La commission a décidé d'autoriser la publication des conclusions du rapporteur sous la forme d'un rapport d'information .
ANNEXES
Annexe n° 1 : Questions écrites de M. Claude Huriet relatives aux CCPPRB adressées au Gouvernement depuis 1996 et restées sans réponse
Annexe n° 2 : Questionnaire envoyé aux CCPPRB
Annexe n° 3 : Liste des auditions et comptes rendus des auditions
Annexe n° 4 : Agréments et retraits d'agrément des CCPPRB par la Direction générale de la Santé (DGS)
Annexe n° 5 : Nombres d'avis rendus par les CCPPRB en 1998 et 1999
Annexe n° 6 : Répartition du produit des droits fixes effectuée par la Direction générale de la Santé en 2000
Annexe n° 7 : Graphiques représentant les taux de présence par catégorie des membres des CCPPRB
Annexe n° 8 : Organigramme de la Direction générale de la Santé avant la réforme
Annexe n° 9 : Organigramme de la Direction générale de la Santé après la réforme
Annexe n° 10 : Table de correspondance entre l'ancienne et la nouvelle numérotation du code de la santé publique
Annexe n° 11 : Dispositions législatives du code de la santé publique applicables aux CCPPRB
Annexe n° 12 : Texte de la directive européenne relative aux essais cliniques
ANNEXE
N° 1
-
QUESTIONS ÉCRITES DE M. CLAUDE
HURIET
RELATIVES AUX CCPPRB ADRESSÉES
AU GOUVERNEMENT DEPUIS
1996
ET RESTÉES SANS RÉPONSE
Question écrite n° 14993 du 18/04/1996 : Conditions de renouvellement des membres des CCPPRB
M. Claude Huriet attire l'attention de M. le secrétaire d'Etat à la santé et à la sécurité sociale sur les modalités d'application des dispositions administratives contenues dans l'article L. 209-11 du code de la santé publique, modifié par la loi n° 94-630 du 25 juillet 1994 quant aux conditions de renouvellement des membres des Comités consultatifs de protection des personnes dans la recherche biomédicale (CCPPRB).
Selon cet article, les membres des CCPPRB doivent être " choisis parmi les personnes figurant sur une liste établie sur proposition d'organismes ou d'autorités habilités à le faire, dans des conditions déterminées par décret ". En outre, l'article R. 2006 stipule que " ces comités sont renouvelés par moitié tous les trois ans ". A l'heure actuelle, dans l'attente de la parution du décret prévu à l'article L. 209-11, les préfets de région ont prolongé le mandat, venu à expiration de la première moitié des membres des CCPPRB, sans toutefois pouvoir procéder à leur renouvellement. Si cette situation devait perdurer quelques mois encore, l'ensemble des membres de certains comités devrait être renouvelés en même temps, empêchant ainsi le partage des compétences et d'expertise entre anciens et nouveaux membres que le renouvellement par moitié devait permettre. C'est pourquoi il lui demande de bien vouloir lui indiquer dans quel délai ce décret pourra être publié.
Question écrite n° 17785 du 03/10/1996 : Conditions de renouvellement des membres des CCPPRB
M. Claude Huriet rappelle à M. le secrétaire d'Etat à la santé et à la sécurité sociale sa question n° 14993, parue au Journal officiel du 18 avril 1996, question qui avait trait aux modalités d'application des dispositions administratives contenues dans l'article L. 209-11 du code de la santé publique, modifié par la loi n° 94-630 du 25 juillet 1994 quant aux conditions de renouvellement des membres des comités consultatifs de protection des personnes dans la recherche biomédicale (CCPPRB). Selon cet article, les membres des CCPPRB doivent être " choisis parmi les personnes figurant sur une liste établie sur proposition d'organismes ou d'autorités habilités à le faire, dans des conditions déterminées par décret ". D'autre part, l'article R. 2006 stipule que " ces comités sont renouvelés par moitié tous les trois ans ". A l'heure actuelle, dans l'attente de la parution du décret prévu à l'article L. 209-11, les préfets de région ont prolongé le mandat, venu à expiration de la première moitié des membres des CCPPRB, sans toutefois pouvoir procéder à leur renouvellement. Si cette situation devait perdurer quelques mois encore, l'ensemble des membres de certains comités devrait être renouvelé en même temps, empêchant ainsi le partage des compétences et d'expertise entre anciens et nouveaux membres, que le renouvellement par moitié devait permettre. En outre, le risque existe de voir, par là même, certaines décisions des comités contestées pour illégalité. C'est pourquoi il lui demande, une nouvelle fois, de bien vouloir lui indiquer dans quel délai ce décret pourra être publié.
Question écrite n° 20913 du 06/03/1997 : Conditions de renouvellement des membres des CCPPRB
M. Claude Huriet rappelle à M. le secrétaire d'Etat à la santé et à la sécurité sociale sa question n° 17785 parue au Journal officiel du 3 octobre 1996, question qui avait trait aux modalités d'application des dispositions administratives contenues dans l'article L 209-11 du code de la santé publique, modifié par la loi du 25 juillet 1994 quant aux conditions de renouvellement des membres des comités consultatifs de protection des personnes dans la recherche biomédicale (CCPPRB). Selon cet article, les membres des CCPPRB doivent être " choisis parmi les personnes figurant sur une liste établie sur proposition d'organismes ou d'autorités habilités à le faire, dans des conditions déterminées par décret ". D'autre part, l'article R. 2006 stipule que " ces comités sont renouvelés par moitié tous les trois ans ". A l'heure actuelle, dans l'attente de la parution du décret prévu à l'article L. 209-11, les préfets de région ont prolongé le mandat venu à expiration, de la p remière moitié des membres des CCPPRB, sans toutefois pouvoir procéder à leur renouvellement. Si cette situation devait perdurer quelques mois encore, l'ensemble des membres de certains comités devrait être renouvelé en même temps, empêchant ainsi le partage des compétences et d'expertise entre anciens et nouveaux membres, que le renouvellement par moitié devait permettre. En outre, le risque existe de voir, par là-même, certaines décisions des comités contestées pour illégalité. C'est pourquoi il lui demande, pour la troisième fois, de bien vouloir lui indiquer dans quel délai ce décret pourra être publié.
Question écrite n° 21043 du 13/03/1997 : Bilan financier du fonctionnement des CCPPRB
M. Claude Huriet appelle l'attention de M. le secrétaire d'Etat à la santé et à la sécurité sociale sur le bilan financier du fonctionnement des comités consultatifs de protection des personnes dans la recherche biomédicale (CCPPRB). Dans ces organismes, les dépenses sont essentiellement les salaires et les charges salariales du secrétariat ainsi que les indemnisations des membres du comité. Quant aux recettes, elles proviennent des versements de droits par les promoteurs des protocoles. Ces versements sont effectués auprès des directions départementales des affaires sanitaires et sociales (DDASS) qui réallouent ensuite annuellement aux comités une somme en fonction du nombre de protocoles examinés. Dans le cas du CCPPRB de Lyon B, il a été informé que, pour l'exercice 1996, il existe une grande différence entre la somme allouée par la DDASS et le total des sommes que celle-ci a encaissé au nom du comité. La DDASS a ainsi encaissé en 1996 484.500 francs (pour un total de 62 protocoles) et la somme reversée par cette dernière au titre de 1996 s'est élevée à 198.000 francs seulement. En outre, cette somme est en général versée au comité plus d'un an après les premiers versements des droits, obligeant le comité à recourir au crédit auprès de ses membres et des hospices civils de Lyon. Cet exemple n'est pas un cas isolé et de nombreux dysfonctionnements de ce type lui ont déjà été signalés. L'opacité actuelle du système de financement des CCPPRB n'est pas admissible. C'est pourquoi il lui demande de bien vouloir lui faire connaître les raisons des distorsions qui ont pu être constatées et quelles dispositions il entend prendre pour que le financement de ces comités puissent s'effectuer dans la plus grande transparence.
Question écrite n° 00669 du 03/07/1997 : Bilan d'activité des CCPPRB
M. Claude Huriet demande à M. le secrétaire d'Etat à la santé de lui indiquer, à l'occasion du renouvellement des membres des comités de protection des personnes dans la recherche biomédicale (CCPPRB), quelle est l'importance de la représentation des médecins libéraux au sein de ces instances.
Question écrite n° 01119 du 10/07/1997 : Bilan d'activité des CCPPRB
M. Claude Huriet attire l'attention de M. le secrétaire d'Etat à la santé sur le bilan d'activité des comités consultatifs de protection des personnes pour la recherche biomédicale (CCPPRB), comités issus de la loi du 20 décembre 1988. Selon une enquête effectuée pour l'année 1995, les CCPPRB ont examiné 2.501 nouveaux dossiers. Sur ce total, 2.303 avis favorables ont été accordés. En outre, 1.537 dossiers ont été déposés visant à amender un protocole déjà autorisé. Il lui demande de bien vouloir lui faire connaître la ventilation pour chaque CCPPRB du nombre de nouveaux dossiers examinés, d'avis favorables accordés et de demandes d'amendements à un protocole.
Question écrite n° 11044 du 01/10/1998 : Représentation des médecins libéraux au sein des CCPPRB
M. Claude Huriet rappelle à M. le secrétaire d'Etat à la santé le texte de sa question n° 669 parue au Journal officiel du 3 juillet 1997 et qui concernait la représentation des médecins libéraux au sein des comités consultatifs de protection des personnes dans la recherche biomédicale, question à laquelle il n'a pas encore reçu de réponse à ce jour.
Question écrite n° 19646 du 21/10/1999 : Dossiers examinés par les CCPPRB
M. Claude Huriet attire l'attention de Mme le secrétaire d'Etat à la santé et à l'action sociale sur le bilan d'activité des comités consultatifs de protection des personnes pour la recherche biomédicale, comités issus de la loi du 20 décembre 1988. Selon une enquête effectuée pour l'année 1995, les CCPPRB ont examiné 2.501 nouveaux dossiers. Sur ce total, 2.303 avis favorables ont été accordés. En outre, 1.537 dossiers ont été déposés visant à amender un protocole déjà autorisé. Il lui demande de bien vouloir lui faire connaître la ventilation pour chaque CCPPRB du nombre de nouveaux dossiers examinés, d'avis favorables accordés et de demandes d'amendements à un protocole.
Question écrite n° 22097 du 27/01/2000 : Fonctionnement des CCPPRB
M. Claude Huriet appelle l'attention de Mme le secrétaire d'Etat à la santé et à l'action sociale sur le bilan financier du fonctionnement des CCPPRB. Les comités consultatifs de protection des personnes dans le cadre de la recherche biomédicale perçoivent des recettes qui proviennent des versements de droits dus par les promoteurs des protocoles. Ces versements sont effectués auprès des directions départementales de l'action sanitaire et sociale (DDASS) qui réallouent ensuite aux comités annuellement une somme en fonction du nombre de protocoles examinés. Il souhaite savoir quel a été, pour 1997, 1998 et 1999, le montant des sommes versées par les promoteurs auprès de la CCPPRB et quel budget a été attribué à chaque CCPPRB. Il lui demande également s'il est exact que certaines DDASS, au-delà du pourcentage de droits qu'elles prélèvent pour couvrir leurs frais de gestion, aient conservé par devers elles la totalité des sommes collectées.
ANNEXE N° 2
-
QUESTIONNAIRE ENVOYÉ AUX
CCPPRB
1. Localisation du comité
2. Composition du comité
3. Périodicité des réunions
4. Taux moyen de présence des membres du comité (si possible par catégorie : professions de santé , juristes, sociologues etc.)
5. Nombre de protocoles examinés :
- au cours des trois dernières années :
- médicament :
- dispositifs médicaux :
- recherches cognitives-ergonomie :
- recherches en psychologie :
- avec bénéfice individuel direct :
- sans bénéfice individuel direct :
- avis favorables :
- avis favorables " sous réserve " (motifs les plus fréquents) :
- avis défavorables (motifs les plus fréquents) :
- nombre d'amendements examinés au cours des trois dernières années
6. Les promoteurs (Nombre de protocoles sur trois ans) :
- firmes pharmaceutiques : lesquelles ?
- fabricants de dispositifs médicaux :
- institutionnels : établissements hospitaliers, INSERM, CNRS ... :
7. Recours à des experts extérieurs :
8. Modalités de suivi des protocoles :
9. Contentieux :
10. Montant des frais de fonctionnement (par an, sur 3 ans) et observations sur le financement des comités :
11. Nombre de personnes employées par le comité :
12. " Relations extérieures " du comité :
- ministère(s) : DGS, DRASS, Fichier national des essais :
- AFSSAPS :
- Comité consultatif national d'éthique et comités " locaux " d'éthique :
- assureurs :
13. Autres observations ou réflexions sur le fonctionnement des CCPPRB :
ANNEXE
N° 3
-
LISTE DES AUDITIONS
ET COMPTES RENDUS DES
AUDITIONS
• Secrétariat d'Etat à la santé, Direction générale de la santé :
M. Pascal Penaud, chef de service adjoint au directeur général à la Direction générale de la santé
Mme Catherine Grillot-Courvalin, chef de bureau " Recherche et prospective (1E), Mme Marie-Thérèse Nutini, conseiller technique
• Agence française de sécurité sanitaire des produits de santé (AFSSAPS) :
Mme Chantal Belorgey et M. Philippe Vella
• Conférence nationale des CCPPRB
Docteur François Chapuis, président
• Assistance publique - Hôpitaux de Paris (AP-HP) :
M. Joël Ménard, délégué médical, et M. Nicolas Best, secrétaire général de la Direction de la recherche clinique
• INSERM
Mmes Anne Bisagni, responsable de la mission d'animation de la recherche clinique et thérapeutique et Mme Estelle Mottez, responsable du bureau " recherches biomédicales "
• CNRS
M. Robert Naquet, Mme Marie-Cécile Masure et Mme Martine Loizeau, représentant la cellule " éthique " du département des sciences de la vie et Professeur Jean-Paul Caverni
• Professeur Patrice Jaillon, président de l'Association pour le développement de la pharmacologie clinique et chef du service de pharmacologie du CHU Saint-Antoine à Paris
• Professeur G. Hazebroucq, directeur pharmaceutique de la Pharmacie centrale des hôpitaux de Paris
• SNIP
Docteur Olivier Amédée-Manesme, directeur des affaires scientifiques, pharmaceutiques et médicales
• SNITEM
M. Jacques Dumont, président, Mme Crespeau, directeur des Affaires techniques
• DRASS Ile-de-France
Mmes Brigitte Wintrebert, secrétaire administratif, et Françoise Loeb, pharmacien inspecteur en chef
COMPTES RENDUS DES AUDITIONS
I. AUDITION DU MARDI 28 NOVEMBRE 2000
• PROFESSEUR PATRICE JAILLON, PRÉSIDENT DE L'ASSOCIATION POUR LE DÉVELOPPEMENT DE LA PHARMACOLOGIE CLINIQUE (ADPC) ET CHEF DU SERVICE DE PHARMACOLOGIE DU CHU SAINT-ANTOINE À PARIS
Selon le professeur Patrice Jaillon, les investigateurs considèrent que les CCPPRB fonctionnent bien comme en témoigne le respect des délais (cinq semaines). Les comités sont disponibles, ce qui permet à un investigateur d'être entendu par un comité. Ils jouent leur rôle de protection des personnes en étant attentifs à ce que les informations données soient complètes et loyales. Néanmoins, alors qu'ils sont sensés évaluer la pertinence des projets qui leur sont présentés, il arrive qu'ils donnent un avis favorable à des projets de recherche qui ne sont pas scientifiquement fondés. Cela pose un problème compte tenu du fait qu'il n'y a pas de contrôle postérieur à l'avis favorable du comité. Peut-être les comités ne se considèrent-ils pas suffisamment compétents pour s'occuper de méthodologie ? Dans ce cas, il pourrait être utile de leur adjoindre la compétence de pharmacologues. De fait, les comités ont recours à des experts plusieurs fois par an.
Le professeur Patrice Jaillon reconnaît que les investigateurs ont peut-être tendance à privilégier certains comités par habitude. Pour autant, cela ne veut pas dire que des projets pourraient échapper à un examen rigoureux, l'AFSSAPS conserve en effet un droit de regard sur la qualification des projets de recherche. Concernant les essais sans bénéfice individuel direct, les comités demandent toujours la composition de l'équipe chargée de conduire la recherche ainsi que des précisions sur les conditions de la rémunération des personnes se prêtant à la recherche.
Peu de comités s'intéressent au suivi et aux résultats des recherches. Ils devraient être informés des effets indésirables graves, par exemple par les services de pharmacovigilance des promoteurs des essais cliniques (ce qui pose un problème pour les promoteurs publics -INSERM ou hôpitaux- qui n'ont pas en propre de système de pharmacovigilance des essais dont ils sont les promoteurs).
De même l'information des personnes participant à la recherche varie selon les équipes, mais elle demeure peu répandue, en règle générale, ce qui donne lieu à des plaintes de la part des personnes. Le principe d'une information de la personne des résultats de la recherche pourrait être inscrit dans le formulaire qui recueille les consentements.
Le recours à un investigateur coordinateur ne pose pas de problème particulier. Il faut seulement communiquer aux autres investigateurs la photocopie de l'avis favorable. Dans ce cas, lorsqu'un comité a donné son avis, les autres l'acceptent.
Les amendements concernent généralement des points peu importants. Le comité est informé et rend un avis si l'amendement a pour conséquence de changer le sens du projet de recherche. Il n'y a pas de redevance pour les amendements. Leur développement s'explique par le fait qu'un investigateur ne peut pas penser à tout lorsqu'il rédige un protocole.
Le contrôle est réalisé à la fois par l'AFSSAPS et, de fait, par le directeur de l'établissement dans lequel est réalisée la recherche. Le professeur Patrice Jaillon ne croit pas qu'il y ait de tricherie car la loi a prévu des sanctions et les promoteurs n'y ont pas intérêt. Le système est en fait autocontrôlé. Il est entré dans les moeurs et constitue même un modèle au niveau européen.
La gratuité des dispositifs médicaux fournis par le promoteur pose un problème lorsque l'innovation coûte très cher. La CNAM prend pour base le prix de journée alors que dans d'autres pays il existe une certaine prise en charge par le système de sécurité sociale. Le Programme hospitalier de recherche clinique (PHRC) pourrait participer au financement de certaines de ces recherches. Le problème se pose tout particulièrement lors des essais cliniques à promotion publique (INSERM ou hôpitaux) lorsque des investigateurs veulent comparer plusieurs médicaments ou veulent étudier les effets de dispositifs médicaux et que ni ces médicaments, ni ces dispositifs ne peuvent être fournis gratuitement par les firmes.
II. AUDITION DU MERCREDI 29 NOVEMBRE 2000
• DOCTEUR FRANÇOIS CHAPUIS, PRÉSIDENT DE LA CONFÉRENCE DES CCPPRB
La Conférence nationale représente 42 comités sur 47 ce qui est bien compte tenu du renouvellement en cours des différents comités. Après avoir renouvelé son conseil d'administration en mai 1998, la Conférence nationale des CCPPRB a trouvé ses marques et fonctionne aujourd'hui correctement comme une plate-forme sui generis de coordination, de formation et d'échanges entre les Comités.
1. Le fonctionnement interne des comités
Le docteur Chapuis considère que le fonctionnement des comités est satisfaisant et conforme aux dispositions du code de la santé publique. Concernant leur composition, bien que les membres ne soient pas des experts, la réflexion collective donne lieu à un travail de qualité qui surpasse souvent la prestation d'un expert individuel. Le pluralisme dans la composition constitue une richesse qui dépend de la personnalité et de l'expérience de chacun des membres. Les membres bénévoles acquièrent progressivement une certaine compétence, mais on peut regretter que rien n'ait été prévu pour assurer une formation initiale ni pour les faire progresser. Cette absence de suivi, combinée à l'absence de reconnaissance de cette activité entièrement bénévole et au niveau important de la charge de travail, peut expliquer pour une partie le nombre parfois élevé de démissions. Les membres exerçant une profession libérale ont comparativement plus de difficultés à conjuguer leur activité professionnelle et la charge de leur mandat. Certains membres sont désignés, conformément aux textes, par les instances habilitées mais sans toujours avoir été suffisamment informés de la charge de travail correspondant à cette fonction.
Certains comités indemnisent les membres en fonction du nombre de protocoles examinés mais ce n'est pas un cas général ; le montant de l'indemnité versée varie globalement de 0 à 500 francs pour chaque protocole examiné, parfois plus pour un rapporteur extérieur au Comité. Cette indemnité est alors imputée sur les frais de fonctionnement. Cette différence entre les comités s'explique historiquement par le fait que certains comités ont dû adapter leur budget à leurs charges de fonctionnement, afin de tenir compte de la dotation budgétaire annuelle qui leur était allouée, sur une base qui n'a jamais été rendue explicite.
Les comités n'ont pas de statut juridique identifié, bien qu'ils possèdent a priori la personnalité morale, ce qui conduit l'administration à considérer qu'ils réalisent pour elle un simple travail préparatoire.
Le docteur Chapuis fait deux constats d'ordre quantitatif. Il estime d'une part que le nombre de protocoles étudiés augmente chaque année et d'autre part que la proportion des protocoles médicamenteux a tendance à diminuer compte tenu du développement de nouvelles formes de recherche biomédicale (chirurgie, biomatériaux, recherches cliniques non médicamenteuses).
Les promoteurs sont essentiellement de trois types : entreprises du secteur pharmaceutique, centres hospitaliers, établissements publics de recherche. Il y a peu de personnes physiques parmi les promoteurs, compte tenu notamment de la durée des recherches qui nécessite des moyens administratifs et financiers pour assurer le contrôle et le suivi des recherches promues.
Les comités exercent de façon plutôt satisfaisante l'ensemble des missions qui leur ont été confiées par la loi mais il existe un problème concernant le suivi des protocoles. Aujourd'hui personne ne connaît vraiment l'évolution des protocoles et notamment le nombre de protocoles abandonnés, ceux qui arrivent à terme et ceux qui font l'objet d'une publication. Les comités ne sont pas en mesure d'assurer ce suivi compte tenu en particulier des faibles ressources mises à leur disposition.
2. Les relations extérieures des comités
Les DRASS exercent une relation " quasi tutélaire " sur les comités pourtant indépendants ; elles attribuent des dotations budgétaires et assurent la nomination des membres. Les comités n'ont cependant pas de retour sur l'exploitation des budgets prévisionnels qu'ils soumettent ni sur leurs rapports d'activité annuels. Les DRASS ne donnent pas d'indication sur les modalités de calcul des dotations. Ces modalités sont déterminées par la DGS qui entend semble-t-il assurer une péréquation entre les différents comités. Une certitude pourtant : les dotations ne sont pas liées à l'activité et peuvent varier d'une année à l'autre. Il n'y a donc pas de transparence sur le financement de l'activité des Comités dont les ressources ont pourtant été spécifiquement prévues par la loi.
Le docteur Chapuis considère que l'AFSSAPS joue un rôle utile pour aider à l'interprétation des textes applicables à la recherche biomédicale. Cependant, il ne lui revient pas, compte tenu de son statut d'administration, d'assurer la centralisation des données relatives à l'activité des comités. Cette fonction est partiellement assurée par la Conférence nationale des CCPPRB.
Le système mis en place par la loi de 1988 est purement déclaratif. A l'heure actuelle aucun contrôle n'est exercé pour empêcher une recherche qui ne serait pas déclarée.
Des problèmes ont pu apparaître pour distinguer les essais avec ou sans bénéfice individuel direct, mais aujourd'hui des formations sont organisées sur ce point et ces questions se posent avec moins d'acuité. Les comités assurent plus largement des consultations méthodologiques à l'attention des investigateurs. La Conférence nationale organise à cet égard des séances de formation initiale à destination des membres des comités nommés à l'issue de chaque renouvellement.
Selon le docteur Chapuis, il faudrait davantage valoriser l'appartenance à un comité. Une réflexion pourrait être menée sur le profil type des candidats, les conditions de nomination ainsi que sur les règles de fonctionnement interne. La création d'une " plate-forme " afin de faciliter les échanges d'informations, le conseil et la formation pourrait être utilement envisagée.
Enfin, le docteur Chapuis s'est interrogé plus largement sur la protection des personnes participant à des recherches biomédicales qui n'entrent apparemment pas dans le cadre de la loi du 20 décembre 1988. Ce peut être le cas des études épidémiologiques avec un prélèvement sanguin, ou des protocoles dont les modalités de mise en oeuvre de la recherche sont insuffisamment formalisées. Il estime que 40 à 50 % des recherches biomédicales sont dans ce cas.
III. AUDITIONS DU LUNDI 4 DÉCEMBRE 2000
• M. JACQUES DUMONT, PRÉSIDENT, ET MME CRESPON, DIRECTEUR DES AFFAIRES TECHNIQUES, AU SYNDICAT NATIONAL DE L'INDUSTRIE DES TECHNOLOGIES MÉDICALES (SNITEM)
Les industriels du secteur des technologies médicales sont satisfaits du fonctionnement des comités et de l'organisation de la recherche biomédicale à l'exception du principe de gratuité des dispositifs médicaux ou produits mis à disposition pendant le temps de l'essai par le promoteur. Cette disposition réglementaire a amené de nombreuses entreprises nationales et internationales à se détourner de la France pour mettre en oeuvre leurs projets de recherche. Cela représente un risque pour la recherche française, d'autant plus élevé que l'AFSSAPS accepte, dorénavant, les études internationales.
Les modalités de prise en charge sont très différentes dans les autres pays européens. Un compromis pourrait consister à un remboursement sur la base du tarif de la sécurité sociale correspondant à un dispositif équivalent.
Concernant les relations extérieures, l'AFSSAPS est l'interlocuteur unique. Elle fait preuve d'une véritable volonté de dialogue et d'une bonne connaissance du secteur des dispositifs médicaux. Les contacts avec la direction des hôpitaux restent limités, ce qui est regrettable.
• M. ROBERT NAQUET, MME MARIE-CÉCILE MASURE ET MME MARTINE LOIZEAU, REPRÉSENTANT LA CELLULE ÉTHIQUE DU DÉPARTEMENT DES SCIENCES DE LA VIE DU CNRS, ET PROFESSEUR JEAN-PAUL CAVERNI
Les représentants de la cellule éthique font observer :
- qu'il n'y a plus de réunions communes organisées avec les comités depuis 1996. De même, des membres de la cellule éthique ont pu participer dans le passé aux travaux de la Conférence nationale des CCPPRB, mais ce n'est plus le cas aujourd'hui.
- que le rôle du CNRS consiste à se porter promoteur de protocoles qui lui sont proposés par les chercheurs. Dans ce cadre, la cellule éthique a pour charge de vérifier les dossiers.
- que, lors de consultations officieuses, il a pu arriver que des membres de deux comités différents donnent des avis contradictoires, ce qui pose un problème de cohérence.
M. Jean-Paul Caverni fait en outre observer que la législation pose un problème particulier dans le cas des recherches comportementales puisqu'il faut être médecin pour présenter un protocole, ce qui est rarement le cas. Le CNRS propose qu'un scientifique " non-médecin " puisse saisir un comité et qu'il incombe à ce dernier de demander, le cas échéant, la saisine d'un médecin.
Concernant la composition des comités, il serait souhaitable que le recrutement des membres des comités ayant une qualité scientifique soit davantage diversifié et comprenne, notamment des chercheurs en sciences du comportement.
IV. AUDITION DU JEUDI 7 DÉCEMBRE 2000
• MME CHANTAL BELORGEY ET M. PHILIPPE VELLA DE L'AGENCE FRANÇAISE DE SÉCURITÉ SANITAIRE ET DES PRODUITS DE SANTÉ (AFSSAPS)
L'AFSSAPS n'est pas chargée de suivre le fonctionnement des comités, c'est le rôle de la Direction générale de la santé (DGS). Les relations sont donc ponctuelles et rares, ce qui constitue un problème. Il n'est écrit nulle part que les comités et l'Agence doivent entretenir des relations particulières. Or, les compétences de chacun ne sont pas clairement définies. L'Agence revendique en particulier des compétences qui ne lui sont pas reconnues explicitement par les textes au motif, en particulier, que les comités ne sont pas en mesure de les exercer pleinement, n'ayant pas notamment toute l'information, en particulier celle portant sur la tolérance des produits de santé soumis à l'essai.
L'Agence reçoit communication des effets graves de la part des promoteurs (35.000 par an) et des faits nouveaux pouvant mettre en danger la sécurité des patients. Les lettres d'intention ne font pas l'objet d'une instruction systématique, sauf si l'Agence a connaissance d'un risque particulier avec un produit donné.
Elle est amenée à examiner le rapport bénéfice/risque d'un produit alors que cela ne relève pas formellement de sa compétence. Cela lui semble pourtant nécessaire, même si cela peut provoquer des difficultés avec certains comités. Il n'est pas de leur domaine, cependant, de chercher à détecter les risques alors que c'est précisément une des missions dévolue à l'Agence.
Afin de permettre une certaine homogénéisation des avis, l'Agence a diffusé un guide relatif aux études précliniques devant être réalisées avant tout essai clinique. Elle travaille à la réalisation d'autres guides dans le but d'améliorer la recherche biomédicale. Elle souhaite par ailleurs développer ses relations avec les comités et reconnaît la nécessité de distinguer ses compétences de celles de ces derniers. Alors que les comités doivent s'attacher à la protection des personnes et à l'évaluation ou à la pertinence des projets, l'AFSSAPS pourrait se voir reconnaître une compétence relative à l'évaluation de la validité scientifique des protocoles et des produits.
Afin de ne pas retarder les essais cliniques, l'Agence propose une évaluation simultanée des protocoles par elle-même et par les CCPPRB. Elle souhaite pouvoir disposer des mêmes informations que les comités, c'est-à-dire, notamment outre le contenu du protocole, la brochure pour l'investigateur, la note d'information pour le patient et les dates réelles de début et de fin d'essai. Par ailleurs, le comité pourrait lui transmettre directement son avis. Ce nouveau mode de fonctionnement s'inspire de la directive " concernant le rapprochement des dispositions législatives, réglementaires et administratives des Etats membres relatives à l'application de bonnes pratiques cliniques dans la conduite d'essais cliniques de médicaments à usage humain " en voie d'adoption, qui prévoit notamment un dispositif d'autorisations d'essais cliniques délivrées par les autorités de tutelle.
Il arrive que des comités interrogent à juste titre l'Agence alors que celle-ci ne dispose pas du protocole concerné et que le droit ne lui reconnaît pas la compétence d'autoriser les essais.
L'AFSSAPS souhaiterait un aménagement de la législation qui lui reconnaîtrait le pouvoir d'exiger des modifications de tout document relatif à la recherche. Elle ne peut aujourd'hui que suspendre ou interdire les essais qui présentent un risque pour la santé publique. Cette évolution lui semble naturelle compte tenu du fait qu'elle centralise déjà l'information concernant les événements indésirables qui surviennent lors de la réalisation d'essais de médicaments en France comme à l'étranger et les faits nouveaux pouvant mettre en danger la sécurité des personnes participant à la recherche.
Afin d'améliorer l'information des différents acteurs de la recherche biomédicale, l'Agence propose de diffuser des registres des essais cliniques, notamment des essais relatifs à certaines pathologies graves ou rares. Cela permettrait par exemple une meilleure information sur les traitements des maladies rares. Or, il peut exister de nombreuses réticences de la part des promoteurs. Aux Etats-Unis, il existe une loi qui oblige les autorités de tutelle à diffuser les informations relatives aux essais (titre de la recherche, produit testé, maladie concernée, coordonnées du promoteur et de l'investigateur...). Une telle pratique permettrait d'éviter des essais redondants. L'AFSSAPS est la seule structure à même d'établir et de gérer une telle base de données.
V. AUDITIONS DU MERCREDI 13 DÉCEMBRE 2000
• M. PASCAL PENAUD, CHEF DE SERVICE ADJOINT AU DIRECTEUR GÉNÉRAL À LA DIRECTION GÉNÉRALE DE LA SANTÉ ACCOMPAGNÉ DE MME CATHERINE GRILLOT-COURVALIN ET MME MARIE-THÉRÈSE NUTINI
Dans le cadre de la réforme de la Direction générale de la santé (DGS), le suivi des comités consultatifs de protection des personnes dans la recherche biomédicale (CCPPRB) a été attribué à un nouveau bureau intitulé " recherche et prospective " qui dépend de la première sous-direction " politique de santé et stratégies ". Cette réorganisation, selon M. Pascal Penaud, est destinée à permettre à la DGS, en étant dotée des moyens supplémentaires nécessaires, de :
- faire connaître et prendre en compte les orientations et les besoins des politiques de santé publique par le monde de la recherche,
- mieux connaître et prendre en compte les apports de la recherche dans la définition des politiques de santé ;
- exercer dans des conditions administratives satisfaisantes les tâches qui lui incombent : mise en oeuvre et suivi de la loi Huriet, participation au conseil d'administration de l'INSERM...
1. L'organisation des comités
Mme Marie-Thérèse Nutini constate l'existence de problèmes pour nommer les membres des comités. Certaines des catégories sont sous-représentées (professions libérales, personnes qualifiées en matière d'éthique). Les différentes réformes relatives aux modalités de nomination n'ont pas permis de résoudre cette difficulté. Un décret du 1 er octobre 1997 a prévu qu'au delà de trois absences consécutives non justifiées d'un membre titulaire aux séances du comité le préfet de région pouvait mettre fin au mandat de ce membre, et cette possibilité a permis, dans certains cas, de limiter les problèmes d'absentéisme.
Alors que le mode de nomination devrait constituer une garantie d'indépendance, elle remarque que, dans la réalité, les DRASS sont sollicitées par les membres des professions médicales à chaque renouvellement. Par ailleurs, certains membres restent en fonction trop longtemps.
Afin de tenir compte de la charge de travail que représente la fonction de membre de comité, la DGS ne serait pas hostile à la création d'une compensation financière sous la forme d'une vacation hospitalière pour les professions libérales.
2. Le financement des comités
La loi prévoit le paiement d'une redevance pour chaque protocole faisant l'objet d'un avis de la part d'un CCPPRB. Le pouvoir réglementaire a fixé à 9.500 francs le montant de la redevance lorsque l'avis est déposé par un industriel et à 900 francs dans les autres cas. Ces droits servent à financer les frais de fonctionnement des comités. Ils transitent par un fonds de concours géré par le ministère de l'Economie, des Finances et de l'Industrie. Il existe un décalage d'un an entre le moment où les sommes sont recouvrées et celui où elles sont attribuées.
Mme Marie-Thérèse Nutini observe que l'aspect financier a toujours constitué une difficulté dans le fonctionnement des CCPPRB. Un premier mode de financement privilégiant le nombre de dossiers examinés par comité n'a pas donné satisfaction puisqu'il incitait à multiplier les avis. Certains comités avaient pu constituer d'importantes réserves financières. Par ailleurs, les frais de gestion n'étaient pas toujours en rapport avec la réalité de l'activité.
Depuis 1998, la dotation est calculée en fonction des budgets prévisionnels préparés par les comités. Les sommes qui ne sont pas attribuées sous forme de dotation sont conservées sur le fonds de concours.
La dotation est faite sur la base des dépenses de l'année antérieure et donc au second semestre de l'année. Toutefois chaque comité dispose d'une année de fonctionnement de " trésorerie " de sorte que cette date de mise à disposition des fonds ne doit pas poser de difficulté aux comités.
M. Pascal Penaud considère qu'une réflexion est nécessaire pour éviter que ne se créent des excédents inutilisés. Une des pistes en est l'adaptation des redevances. Mais il est également nécessaire de mieux adapter les financements aux besoins réels de fonctionnement des CCPPRB. Ainsi, une modulation des indemnités versées aux rapporteurs en fonction de la complexité des dossiers pourrait être envisagée.
Mme Marie-Thérèse Nutini estime que les DRASS n'ont pas les moyens humains suffisants pour contrôler les besoins réels des comités, ce qui a pu donner lieu à des incompréhensions concernant le calcul des dotations. C'est pourquoi la DGS ajuste dans certains cas le montant des dotations en cours d'exercice pour tenir compte des difficultés rencontrées par les comités.
3. Le fichier national des recherches et les relations avec l'AFSSAPS
Le fichier national des personnes qui se prêtent à des recherches biomédicales sans bénéfice individuel direct comprend deux mille lieux de recherche autorisés. Il est géré par la DGS qui estime entre 300 et 400.000 le nombre de personnes qui participent à des recherches sans bénéfice individuel direct.
Les relations entre la DGS et l'AFSSAPS sont quotidiennes. Elles permettent une amélioration de la qualité des dossiers à travers une meilleure circulation de l'information et l'échange des expériences réciproques.
• PROFESSEUR G. HAZEBROUCQ, DIRECTEUR PHARMACEUTIQUE DE LA PHARMACIE CENTRALE DES HÔPITAUX DE PARIS
Les modalités de recueil du consentement dans les cas d'urgence ne sont pas suffisamment précises. Le rôle des comités devrait être clarifié concernant les malades dans le coma notamment lorsqu'il n'est pas possible de joindre la famille. Les comités pourraient accepter protocole par protocole le principe de l'intégration en urgence de patients dans une recherche si celle-ci est susceptible de leur être bénéfique.
Le Professeur G. Hazebroucq évoque également un assouplissement de la règle de l'accord des deux parents lorsque le danger est faible au regard du bénéfice attendu de la recherche en pédiatrie.
Evoquant les prérequis, le Professeur Hazebroucq a souhaité qu'un dossier pharmaceutique soit joint en complément de la brochure réalisée par l'investigateur.
Afin d'améliorer la qualité de l'examen par les comités des dossiers concernant des essais de phase I et II, il faudrait prévoir un rapport systématique d'expert toxicologue. Ceci compenserait l'absence assez fréquente de pharmacotoxicologue dans des comités. Pour les mêmes phases précoces, un rapport d'expert pharmaceutique devrait être également joint pour les produits de biotechnologie ou de haute technologie. Ces rapports d'experts les engageraient sur le fait que le données disponibles ne s'opposent pas à l'entreprise de la recherche clinique envisagée.
La formation des membres et des présidents de comité devrait être obligatoire.
Les présidents de comité pourraient se voir reconnaître un rôle de conseil en recherche biomédicale (aspects juridiques, éthiques, méthodologiques) et être formés dans cette perspective.
Afin de permettre une meilleure représentation des professions libérales, il conviendrait d'instaurer un système d'indemnisation qui pourrait prendre la forme d'une vacation hospitalière.
Le Professeur Hazebroucq a insisté sur les problèmes soulevés par la multiplication des amendements. Le rôle des comités reste ambigu à leur égard. Leur examen demande beaucoup de temps et il n'est pas sûr qu'il soit réalisé avec toute l'attention nécessaire. Les procédures d'examen des amendements n'ont pas fait l'objet d'une harmonisation entre les différents comités.
Le principe posé par l'article R. 2038 du code de la santé publique de la gratuité des médicaments ou produits mis à disposition par le promoteur pendant le temps d'essai constitue un obstacle au développement de la recherche clinique, sur les dispositifs médicaux en France.
Par ailleurs, le Professeur Hazebroucq estime que la distinction entre les essais avec ou sans bénéfice individuel direct n'est pas suffisamment claire notamment en ce qui concerne les médicaments. Il considère qu'une plus grande concertation entre les comités et l'AFSSAPS serait utile.
Le Professeur Hazebroucq pense enfin que les déclarations d'intérêt des membres et des experts devraient être systématiques.
• MM. JOËL MÉNARD, DÉLÉGUÉ MÉDICAL, ET NICOLAS BEST, SECRÉTAIRE GÉNÉRAL DE LA DIRECTION DE LA RECHERCHE CLINIQUE DE L'ASSISTANCE PUBLIQUE DES HÔPITAUX DE PARIS (AP-HP)
Lorsque certains CHU se portent promoteurs, la gestion administrative et financière des dossiers est assurée par la délégation à la recherche clinique. Le financement de ces recherches repose pour partie sur les crédits prévus par le programme hospitalier de recherches cliniques (PHRC) qui s'élèvent à 100 millions de francs par an.
Les études multicentriques font l'objet de convention entre les différents hôpitaux qui participent à la recherche.
Lorsque les recherches présentent des risques médicaux importants, l'AH-HP met en place un " monitoring " qui assure à l'investigateur une assistance à la recherche clinique.
Afin d'examiner la pertinence scientifique de la recherche, l'AP-HP a créé une commission d'experts qui examine les projets qui lui sont proposés. Ce système multiplie les contrôles. Ainsi quand un promoteur institutionnel dépose un projet devant un CCPPRB, celui-ci a déjà été vu par au moins trois personnes (2 experts et le rapporteur).
Les hôpitaux ont créé un filtre scientifique avant l'examen de l'avis par le CCPPRB, celui-ci a pour fonction d'examiner les modalités du consentement et la nature de l'essai (avec ou sans bénéfice individuel direct). Les industriels n'ont pas mis en place de tels dispositifs.
M. Joël Ménard estime que les sources de financement de la recherche clinique ont été augmentées ces dernières années.
Le principe posé par l'article R. 2038 du code de la santé publique selon lequel le promoteur doit fournir gratuitement les médicaments ou les produits nécessaires à la recherche concerne essentiellement les promoteurs industriels, et ne devrait pas être applicable aux promoteurs institutionnels pour les essais de dispositifs médicaux.
Evoquant le risque d'un Gault et Millau des CCPPRB, M. Joël Ménard a estimé qu'il existait dans certains cas une trop forte proximité entre les comités et certains investigateurs. Par ailleurs, il a considéré que le nombre d'avis devait atteindre un certain équilibre afin de permettre aux comités d'acquérir une certaine expérience et de pouvoir néanmoins consacrer suffisamment de temps à chaque avis.
VI. AUDITION DU MERCREDI 20 DÉCEMBRE 2000
• DOCTEUR OLIVIER AMÉDÉE-MANESME, DIRECTEUR DES AFFAIRES SCIENTIFIQUES, PHARMACEUTIQUES ET MÉDICALES DU SYNDICAT NATIONAL DE L'INDUSTRIE PHARMACEUTIQUE (SNIP)
1. Le fonctionnement des comités
Le docteur Olivier Amédée-Manesme souligne que le fonctionnement des CCPPRB a eu tendance à s'améliorer ces deux dernières années. Ce fonctionnement n'est toutefois pas encore homogène et le délai d'examen des protocoles dépasse dans certains cas les cinq semaines prévues par les textes.
Il propose d'harmoniser les règles de fonctionnement des comités et notamment les procédures écrites internes, la formation de leurs membres, les conditions d'appel des décisions et les modalités de réponse en cas de transmission de données complémentaires en cours de recherche (amendements).
Lorsqu'un comité ne s'estime pas suffisamment compétent pour examiner un projet de recherche, il devrait être possible de présenter le projet à un autre comité qui pourrait donc être différent de celui dont l'investigateur principal relève.
La formulation des avis rendus par les comités devrait être plus complète et préciser notamment la liste des documents qui ont été revus et approuvés ainsi que la liste des membres votants.
Une base de données pourrait être créée sous la forme d'un site internet afin de mettre à disposition des comités des éléments de réponse aux questions qu'ils peuvent se poser lors de l'examen des avis.
Concernant la composition des comités, celle-ci pourrait comprendre des spécialistes de maladies rares ainsi que des représentants de patients.
Le docteur Olivier Amédée-Maresme a considéré que les règles de recueil du consentement devraient être aménagées notamment pour les malades en situation d'urgence et les patients mineurs ou majeurs protégés par la loi. Il a considéré que certaines pathologies comme les accidents vasculaires aigus devraient être considérées comme des situations d'urgence. Il a fait état de difficultés à obtenir le consentement des deux parents dans le cadre des études pédiatriques et pour obtenir la signature du représentant légal d'un majeur protégé par la loi notamment lors d'une recherche en psychiatrie.
2. Les relations extérieures des comités avec l'AFSSAPS et la DGS.
Concernant la qualification des études avec ou sans bénéfice individuel direct, il existe des désaccords entre les comités et l'AFSSAPS. Par ailleurs, en matière d'évaluation des faits nouveaux, il arrive que l'AFSSAPS demande l'arrêt d'une étude alors que celle-ci a fait l'objet d'un amendement qui a reçu un avis favorable de la part du CCPPRB. Ces divergences posent des problèmes de crédibilité pour soutenir le développement des essais cliniques par l'industrie pharmaceutique sur le sol français.
Il existe également des différences d'appréciation entre les comités et la DGS. Alors que les comités hésitent à qualifier avec ou sans bénéfice individuel direct les études pharmacogénétiques, la DGS leur donne systématiquement la qualité d'études sans bénéfice individuel direct. La requalification de certains essais pose également un problème aux industriels lorsqu'elle intervient après le commencement de l'étude.
VII. AUDITION DU JEUDI 21 DÉCEMBRE 2000
• MMES ANNE BISAGNI, RESPONSABLE DE LA MISSION D'ANIMATION DE LA RECHERCHE CLINIQUE ET THÉRAPEUTIQUE ET ESTELLE MOTTEZ, RESPONSABLE DU BUREAU " RECHERCHES BIOMÉDICALES " DE L'INSTITUT NATIONAL DE LA SANTÉ ET DE LA RECHERCHE MÉDICALE (INSERM)
1. Les procédures particulières mises en place à l'INSERM
L'INSERM peut se porter promoteur de recherches biomédicales dans le cadre de ses missions et de sa politique de soutien au développement de la recherche clinique. C'est pourquoi il a mis en place des procédures internes particulières permettant d'évaluer la qualité des projets qui lui sont soumis.
La majorité de ses projets de recherche concerne le domaine de la physiologie-physiopathologie, des neurosciences cognitives, de l'épidémiologie et de la génétique.
L'Institut s'est déclaré promoteur de 48 nouveaux projets en 1999 82 ( * ) . Les investigateurs sont le plus souvent des hospitalo-universitaires ou des chercheurs émargeant au profil d'une unité ou équipe INSERM (77 %). Les autres projets sont conduits par un médecin coordinateur ou un médecin délégué d'un centre d'investigation clinique (12 %) ou un médecin, non inscrit au profil d'une formation INSERM ou d'un centre d'investigation clinique (CIC), qui bénéficie d'un contrat sur appel d'offres (11 %).
Ces projets, qui sont menés principalement en Ile-de-France (64 %), sont financés sur les crédits propres des formations INSERM (54 %) ou par appel d'offres (46 %).
Afin d'aider les investigateurs à monter leurs projets, l'INSERM a créé en 1991 un bureau " recherches biomédicales " qui s'appuie sur un Comité d'experts en recherches biomédicales. Ce comité de neuf experts examine la qualité méthodologique et scientifique des projets. Par ailleurs, les dispositions législatives et réglementaires (qualification des investigateurs, information des personnes, ...) sont vérifiées par l'INSERM.
Le comité sert aussi d'instance de réflexion et de proposition pour les questions concernant le champ et les modalités d'application de la loi du 20 décembre 1988.
La nature des renseignements demandés par le comité d'experts (qualité méthodologique et scientifique, qualification des investigateurs, informations délivrées par les investigateurs...) en font un quasi CCPPRB. Par ailleurs, le bureau " recherches biomédicales " accompagne les projets du début jusqu'à la diffusion des données et assure un suivi, renforcé par des visites sur sites effectuées par le service en charge de la qualité à l'INSERM.
A bien des égards, les procédures mises en oeuvre au sein de l'INSERM sont donc plus précises et plus exigeantes que les procédures de droit commun.
2. Les relations avec les CCPPRB
L'INSERM souhaiterait être destinataire de l'ensemble des informations contenues dans les avis rendus par les CCPPRB. Il constate que les investigateurs n'ont pas le réflexe d'informer les promoteurs.
La majorité des protocoles présentés par l'INSERM fait l'objet de réserves de la part des comités. Celles-ci ont pour origine l'hétérogénéité des modes de fonctionnement et de présentation des avis des différents comités.
La multiplication des amendements pose un problème pour la conduite des essais compte tenu du délai important nécessaire à leur examen (3 à 14 mois).
La forte atomisation des lieux de recherche constitue un obstacle pour l'INSERM de même que les exigences de l'administration pour leur agrément. Un traitement particulier devrait être réservé en particulier pour les recherches menées en population.
La Conférence nationale des CCPPRB pourrait être dotée d'un secrétariat permanent qui jouerait un rôle d'interlocuteur vis à vis des promoteurs et des investigateurs.
VIII. AUDITION DU JEUDI 25 JANVIER 2001
• MMES BRIGITTE WINTREBERT, SECRÉTAIRE ADMINISTRATIF, ET FRANÇOISE LOEB, PHARMACIEN INSPECTEUR EN CHEF À LA DIRECTION RÉGIONALE DES AFFAIRES SANITAIRES ET SOCIALES (DRASS) D'ILE-DE-FRANCE
1. Le fonctionnement des comités
Dans la région Ile-de-France, 14 comités consultatifs pour la protection des personnes dans la recherche biomédicale ont été installés depuis 1991. Leur siège est à la direction régionale des affaires sanitaires et sociales, mais pour des raisons évidentes de locaux, certains hôpitaux de la région (AP-HP et hôpitaux généraux) ont bien voulu les accueillir. Loyer, charges, éventuellement mise à disposition de personnel font alors l'objet de conventions signées entre les comités et la direction de ces établissements.
Le financement de ces comités est assuré par une dotation annuelle (fonds de concours alimenté par les droits fixes versés par les promoteurs). Depuis 1999, le mode d'attribution des subventions a changé (accord DGS/DRASS) et repose uniquement sur le compte de bilan et le budget prévisionnel. Jusque-là, la dotation des différents comités était calculée au prorata des avis rendus, avec le risque d'inciter les comités à augmenter leur activité au détriment peut-être de la qualité des avis rendus.
L'année 2000 s'est caractérisée par une baisse sensible de l'activité des comités sans que l'on puisse faire un rapprochement avec le changement de mode d'attribution des subventions.
Mme Brigitte Wintrebert observe que l'absence de statut juridique des CCPPRB pose des difficultés lors de l'attribution des dotations. Le contrôleur financier conteste en effet la légalité du versement d'une subvention à de telles structures.
Concernant la composition des comités, il existe une insuffisance des candidatures pour certaines catégories de membres (médecin généraliste, pharmacien d'officine, personne qualifiée en matière d'éthique, juriste). Une indemnité pourrait être créée sous la forme d'une vacation afin de dédommager les membres pour leur présence. Par ailleurs, il apparaît nécessaire d'harmoniser les indemnisations versées aux rapporteurs qui varient de 500 à 800 francs et aux experts qui, lorsqu'ils ne sont pas bénévoles, peuvent percevoir jusqu'à 3.000 francs.
Mme Brigitte Wintrebert considère en outre qu'il existe un problème de frontières dans la qualification des essais avec ou sans bénéfice individuel direct.
2. L'agrément des lieux de recherche
Les procédures relatives à l'agrément des lieux de recherche sont particulièrement lourdes. L'Ile-de-France comprend 550 lieux de recherche répartis en 100 sites. L'essentiel de ces sites est formé d'établissements à caractère hospitalier qui sont déjà tenus à certaines exigences pour recevoir des patients. Or, il s'avère que ces exigences sont identiques à celles requises pour autoriser un lieu de recherches sans bénéfice individuel direct sur des patients volontaires dans le domaine des activités habituelles du service.
Il apparaît que l'agrément est toujours accordé quand l'établissement est autorisé à accueillir des patients. Il en est de même des recherches dans le domaine des sciences du comportement qui sont réalisées dans les centres hospitaliers spécialisés et des recherches génétiques, épidémiologiques et physiopathologiques nécessitant des prélèvements sanguins dans des structures qui sont autorisées à exécuter de tels prélèvements.
Pour ces types de lieux, Mme Françoise Loeb, pharmacien inspecteur en chef à la DRASS d'Ile-de-France, considère que l'autorisation prévue à l'article L. 209-18 du code de la santé n'apporte pas dans les faits une sécurité supplémentaire, la protection des personnes restant assurée par l'obligation de fournir des informations concernant chaque recherche au CCPPRB et au ministre chargé de la santé ou au directeur de l'AFSSAPS.
Compte tenu de la charge de travail que représente l'instruction des demandes d'autorisation de lieu de recherche 83 ( * ) , elle considère qu'il serait plus opportun :
- d'accroître la vigilance lorsque les recherches se déroulent hors des établissements de santé et lorsqu'elles se déroulent en milieu hospitalier en dehors des activités habituelles de diagnostic, de surveillance et de traitement ;
- de ne pas soumettre à autorisation, dans les établissements de santé, les services qui réalisent, dans le domaine de leurs activités habituelles, des recherches sans bénéfice individuel direct, sur leurs patients volontaires ;
- d'envisager la possibilité de ne pas soumettre à autorisation les structures qui sont autorisées à exécuter des prélèvements sanguins lorsqu'un tel prélèvement est le seul acte subi par le volontaire.
ANNEXE
N° 4
-
AGRÉMENTS ET RETRAITS D'AGRÉMENT DES
CCPPRB PAR LA DIRECTION GÉNÉRALE DE LA SANTÉ (DGS)
|
RÉGIONS |
AGRÉMENTS |
RETRAITS D'AGRÉMENT |
|
|
ALSACE |
Alsace n° 1 Strasbourg |
21 octobre 1991 |
|
|
Alsace n° II, Colmar |
16 septembre 1991 |
5 octobre 1995 |
|
|
AQUITAINE |
Bordeaux A |
29 mars 1991 |
|
|
Bordeaux B |
29 mars 1991 |
||
|
Pays de l'Adour |
29 mars 1991 |
5 janvier 1995 |
|
|
AUVERGNE |
Auvergne |
23 juillet 1991 |
|
|
BOURGOGNE |
Dijon |
29 mars 1991 |
|
|
BRETAGNE |
Brest |
3 mai 1991 |
|
|
Rennes |
3 juillet 1991 |
||
|
CENTRE |
Tours |
24 avril 1991 |
|
|
CHAMPAGNE-ARDENNES |
Champagne-Ardennes |
19 juillet 1991 |
|
|
FRANCHE-COMTÉ |
Franche-Comté |
8 avril 1991 |
|
|
ILE-DE-FRANCE |
Saint-Germain-en-Laye |
19 juillet 1991 |
|
|
Paris Cochin |
19 juillet 1991 |
||
|
Créteil Henri-Mondor |
23 juillet 1991 |
||
|
Paris Bichat-Claude Bernard |
23 juillet 1991 |
||
|
Paris-Pitié-Salpétrière |
25 juillet 1991 |
||
|
Paris Saint-Antoine |
25 juillet 1991 |
||
|
Paris Hôtel-Dieu |
25 juillet 1991 |
||
|
Paris Saint-Louis |
21 juillet 1991 |
||
|
Aulnay-sous-Bois |
21 juillet 1991 |
||
|
Paris Bicêtre |
21 juillet 1991 |
||
|
Paris Necker Enfants-Malades |
22 juillet 1991 |
||
|
Boulogne Ambroise Paré |
8 novembre 1991 |
||
|
Versailles |
10 octobre 1991 |
||
|
Paris Boucicaut |
12 mai 1992 |
||
|
LANGUEDOC- |
Montpellier Saint-Eloi |
10 décembre 1991 |
|
|
ROUSSILLON |
Nîmes |
19 novembre 1991 |
|
|
Montpellier-Lapeyronie |
10 décembre 1991 |
2 février 1995 |
|
|
LIMOUSIN |
Limousin |
24 avril 1991 |
|
|
LORRAINE |
Nancy |
22 mars 1991 |
|
|
Metz |
22 mars 1991 |
23 mai 1996 |
|
|
MIDI-PYRÉNÉES |
Toulouse 1 |
22 avril 1991 |
|
|
Toulouse 2 |
22 avril 1991 |
||
|
NORD-PAS-DE-CALAIS |
Lille |
30 août 1991 |
|
|
Lens |
30 août 1991 |
30 juin 1994 |
|
|
Valenciennes |
30 août 1991 |
30 juin 1994 |
|
|
BASSE-NORMANDIE |
Caen |
3 mai 1991 |
|
|
HAUTE-NORMANDIE |
Haute-Normandie |
10 juin 1991 |
|
|
PAYS-DE-LA-LOIRE |
Pays-de-la-Loire 1 (Angers) |
8 avril 1991 |
|
|
Pays-de-la-Loire 2 (Nantes) |
26 février 1991 |
||
|
Orléans |
6 mai 1991 |
17 avril 1997 |
|
|
PICARDIE |
Picardie |
30 août 1991 |
|
|
POITOU-CHARENTES |
Poitou-Charentes |
11 mars 1991 |
|
|
Poitier |
11 mars 1991 |
||
|
PROVENCE-ALPES- |
Marseille 1 |
3 mai 1991 |
|
|
CÔTE D'AZUR |
Marseille 2 |
24 avril 1991 |
|
|
Nice |
15 mai 1991 |
||
|
Aix en Provence |
3 mai 1991 |
21 juillet 2000 |
|
|
Toulon |
3 mai 1991 |
26 avril 1994 |
|
|
RHÔNE-ALPES |
Lyon CRLCC |
3 mai 1991 |
|
|
Lyon A |
23 avril 1991 |
||
|
Lyon B |
23 avril 1991 |
||
|
Rhône-Alpes-Loire |
24 avril 1991 |
||
|
Grenoble 1 |
3 juillet 1991 |
29 mars 1994 |
|
|
Grenoble 2 |
3 juillet 1991 |
||
|
Lyon C |
15 mai 1991 |
29 mars 1994 |
|
|
GUADELOUPE |
Guadeloupe |
18 mars 1994 |
|
|
MARTINIQUE |
Martinique |
28 juin 1993 |
Source : Direction générale de la santé (février 2001).
ANNEXE
N° 5
-
NOMBRES D'AVIS RENDUS PAR LES CCPPRB
EN 1998 ET
1999
|
(1) |
(2) |
(3) |
(4) |
(5) |
(6) |
(7) |
(8) |
(9) |
|||||||||||||
|
1998 |
1999 |
1998 |
1999 |
1999/
|
1999/
|
1999/
|
|||||||||||||||
|
Redevances à 9.500 F |
Redevances à 900 F |
Redevances à 9.500 F |
Redevances à 900 F |
Total redevances |
Total redevances |
Redevances à 9.500 F |
Redevances à 900 F |
Total redevances |
|||||||||||||
|
ALSACE |
Alsace n° 1 Strasbourg |
49 |
45 |
55 |
38 |
94 |
93 |
12 % |
- 16 % |
- 1 % |
|||||||||||
|
AQUITAINE |
Total |
63 |
43 |
55 |
37 |
106 |
92 |
- 13 % |
- 14 % |
- 13 % |
|||||||||||
|
Bordeaux A |
48 |
37 |
39 |
33 |
85 |
72 |
- 19 % |
- 11 % |
- 15 % |
||||||||||||
|
Bordeaux B |
15 |
6 |
16 |
4 |
21 |
20 |
7 % |
- 33 % |
- 5 % |
||||||||||||
|
AUVERGNE |
Auvergne |
25 |
31 |
29 |
14 |
56 |
43 |
16 % |
- 55 % |
- 23 % |
|||||||||||
|
BOURGOGNE |
Dijon |
14 |
7 |
6 |
19 |
21 |
25 |
- 57 % |
171 % |
19 % |
|||||||||||
|
BRETAGNE |
Total |
35 |
32 |
45 |
31 |
67 |
76 |
29 % |
- 3 % |
13 % |
|||||||||||
|
Brest |
22 |
9 |
22 |
10 |
31 |
32 |
0 % |
11 % |
3 % |
||||||||||||
|
Rennes |
13 |
23 |
23 |
21 |
36 |
44 |
77 % |
- 9 % |
22 % |
||||||||||||
|
CENTRE |
Tours |
16 |
19 |
10 |
16 |
35 |
26 |
- 38 % |
- 16 % |
- 26 % |
|||||||||||
|
CHAMPAGNE-ARDENNE |
Champagne Ardenne |
8 |
8 |
7 |
4 |
16 |
11 |
- 13 % |
- 50 % |
- 31 % |
|||||||||||
|
FRANCHE-COMTÉ |
Franche Comté |
17 |
10 |
13 |
17 |
27 |
30 |
- 24 % |
70 % |
11 % |
|||||||||||
|
ILE-DE-FRANCE |
Total |
680 |
311 |
637 |
210 |
991 |
847 |
- 6 % |
- 32 % |
- 15 % |
|||||||||||
|
Saint-Germain-en-Laye |
61 |
12 |
32 |
23 |
73 |
55 |
- 48 % |
92 % |
- 25 % |
||||||||||||
|
Paris Cochin |
96 |
56 |
113 |
39 |
152 |
152 |
18 % |
- 30 % |
0 % |
||||||||||||
|
Créteil Henri-Mondor |
19 |
23 |
24 |
11 |
42 |
35 |
26 % |
- 52 % |
- 17 % |
||||||||||||
|
Paris Bichat Claude Bernard |
14 |
16 |
10 |
7 |
30 |
17 |
- 29 % |
- 56 % |
- 43 % |
||||||||||||
|
Paris Pitié Salpétrière |
133 |
50 |
134 |
31 |
183 |
165 |
1 % |
- 38 % |
- 10 % |
||||||||||||
|
Paris Saint Antoine |
21 |
17 |
25 |
16 |
38 |
41 |
19 % |
- 6 % |
8 % |
||||||||||||
|
Paris Hôtel Dieu |
24 |
7 |
28 |
6 |
31 |
34 |
17 % |
- 14 % |
10 % |
||||||||||||
|
Paris Saint-Louis |
77 |
33 |
71 |
21 |
110 |
92 |
- 8 % |
- 36 % |
- 16 % |
||||||||||||
|
Aulnay-sous-Bois |
40 |
3 |
24 |
12 |
43 |
36 |
- 40 % |
300 % |
- 16 % |
||||||||||||
|
Paris Bicêtre |
27 |
50 |
46 |
17 |
77 |
63 |
70 % |
- 66 % |
- 18 % |
||||||||||||
|
Paris Necker Enfants malades |
29 |
26 |
22 |
18 |
55 |
40 |
- 24 % |
- 31 % |
- 27 % |
||||||||||||
|
Boulogne Ambroise Paré |
41 |
14 |
31 |
9 |
55 |
40 |
-24 % |
- 31 % |
- 27 % |
||||||||||||
|
Versailles |
49 |
3 |
17 |
0 |
52 |
17 |
- 65 % |
- 100 % |
- 67 % |
||||||||||||
|
Paris Boucicault |
49 |
1 |
60 |
0 |
50 |
60 |
22 % |
- 100 % |
20 % |
||||||||||||
|
LANGUEDOC- |
Total |
59 |
38 |
42 |
24 |
97 |
66 |
- 29 % |
- 37 % |
- 32 % |
|||||||||||
|
ROUSSILLON |
Montpellier Saint-Eloi |
37 |
31 |
nc |
nc |
68 |
|||||||||||||||
|
Nîmes |
22 |
7 |
nc |
nc |
29 |
||||||||||||||||
|
LIMOUSIN |
Limousin |
10 |
13 |
5 |
13 |
23 |
18 |
- 50 % |
0 % |
- 22 % |
|||||||||||
|
LORRAINE |
Nancy |
27 |
24 |
60 |
35 |
51 |
95 |
122 % |
46 % |
86 % |
|||||||||||
|
MIDI |
Total |
59 |
26 |
49 |
39 |
85 |
88 |
- 17 % |
50 % |
4 % |
|||||||||||
|
PYRÉNÉES |
Toulouse 1 |
35 |
14 |
27 |
31 |
49 |
58 |
- 23 % |
121 % |
18 % |
|||||||||||
|
Toulouse 2 |
24 |
12 |
22 |
8 |
36 |
30 |
- 8 % |
- 33 % |
- 17 % |
||||||||||||
|
NORD PAS DE CALAIS |
Lille |
41 |
7 |
60 |
6 |
48 |
66 |
46 % |
- 14 % |
38 % |
|||||||||||
|
BASSE NORMANDIE |
Caen |
29 |
17 |
23 |
26 |
46 |
49 |
- 21 % |
53 % |
7 % |
|||||||||||
|
HAUTE NORMANDIE |
Haute Normandie |
26 |
32 |
22 |
36 |
58 |
58 |
- 15 % |
13 % |
0 % |
|||||||||||
|
PAYS DE LOIRE |
Total |
36 |
45 |
42 |
44 |
81 |
86 |
17 % |
- 2 % |
6 % |
|||||||||||
|
Pays de Loire 1
|
8 |
16 |
9 |
12 |
24 |
21 |
13 % |
- 25 % |
- 13 % |
||||||||||||
|
Pays de Loire 2
|
28 |
29 |
33 |
32 |
57 |
65 |
18 % |
10 % |
14 % |
||||||||||||
|
PICARDIE |
Picardie |
7 |
1 |
12 |
6 |
8 |
18 |
71 % |
500 % |
125 % |
|||||||||||
|
POITOU-CHARENTES |
Poitou-Charentes |
44 |
14 |
35 |
17 |
58 |
52 |
- 20 % |
21 % |
- 10 % |
|||||||||||
|
PROVENCE ALPES |
Total |
84 |
48 |
80 |
103 |
132 |
183 |
- 5 % |
115 % |
39 % |
|||||||||||
|
CÔTE D'AZUR |
Marseille 1 |
26 |
9 |
23 |
42 |
35 |
65 |
- 12 % |
367 % |
86 % |
|||||||||||
|
Marseille 2 |
36 |
11 |
40 |
45 |
47 |
85 |
11 % |
309 % |
81 % |
||||||||||||
|
Nice |
19 |
22 |
16 |
13 |
41 |
29 |
- 16 % |
- 41 % |
- 29 % |
||||||||||||
|
Aix en Provence |
3 |
6 |
1 |
3 |
9 |
4 |
- 67 % |
- 50 % |
-56 % |
||||||||||||
|
RHÔNE-ALPES |
Total |
161 |
146 |
159 |
92 |
307 |
251 |
- 1 % |
- 37 % |
- 18 % |
|||||||||||
|
Lyon CRLCC |
35 |
31 |
37 |
24 |
66 |
61 |
6 % |
- 23 % |
- 8 % |
||||||||||||
|
Lyon A |
58 |
13 |
31 |
12 |
71 |
43 |
- 47 % |
- 8 % |
- 39 % |
||||||||||||
|
Lyon B |
41 |
16 |
70 |
12 |
57 |
82 |
71 % |
- 25 % |
44 % |
||||||||||||
|
Rhône-Alpes-Loire |
3 |
15 |
5 |
12 |
18 |
17 |
67 % |
- 20 % |
- 6 % |
||||||||||||
|
Grenoble 2 |
24 |
71 |
16 |
32 |
95 |
48 |
- 33 % |
- 55 % |
- 49 % |
||||||||||||
|
GUADELOUPE |
Guadeloupe |
0 |
0 |
0 |
4 |
0 |
4 |
||||||||||||||
|
MARTINIQUE |
Martinique |
1 |
5 |
0 |
3 |
6 |
3 |
- 100 % |
- 40 % |
- 50 % |
|||||||||||
|
TOTAL GÉNÉRAL |
1.491 |
922 |
1.446 |
834 |
2.413 |
2.280 |
- 3 % |
- 10 % |
- 6 % |
||||||||||||
Source : DGS (février 2001)
ANNEXE N°
6
-
RÉPARTITION DU PRODUIT DES DROITS FIXES
EFFECTUÉE
PAR LA DIRECTION GÉNÉRALE
DE LA SANTÉ EN 2000
|
RÉGIONS |
Dotation pour chaque comité (en francs) |
Dotation par région (en francs) |
|
|
ALSACE |
Strasbourg |
620.000 |
620.000 |
|
AQUITAINE |
Bordeaux A |
400.000 |
|
|
Bordeaux B |
100.000 |
500.000 |
|
|
AUVERGNE |
Auvergne |
50.000 |
50.000 |
|
BOURGOGNE |
Dijon |
100.000 |
100.000 |
|
BRETAGNE |
Brest |
270.000 |
|
|
Rennes |
100.000 |
370.000 |
|
|
CENTRE |
Tours |
200.000 |
200.000 |
|
CHAMPAGNE-ARDENNES |
Champagne-Ardenne |
70.000 |
70.000 |
|
FRANCHE-COMTÉ |
Franche-Comté |
0 |
0 |
|
ILE-DE-FRANCE |
Saint-Germain-en-Laye |
450.000 |
|
|
Paris Cochin |
800.000 |
||
|
Créteil Henri-Mondor |
320.000 |
||
|
Paris Bichat-Claude Bernard |
350.000 |
||
|
Paris-Pitié-Salpétrière |
850.000 |
||
|
Paris Saint-Antoine |
400.000 |
||
|
Paris Hôtel-Dieu |
320.000 |
||
|
Paris Saint-Louis |
150.000 |
||
|
Aulnay-sous-Bois |
350.000 |
||
|
Paris Bicêtre |
450.000 |
||
|
Paris Necker Enfants-Malades |
400.000 |
||
|
Boulogne Ambroise Paré |
360.000 |
||
|
Versailles |
200.000 |
||
|
Paris Boucicaut |
0 |
5.400.000 |
|
|
LANGUEDOC- |
Montpellier Saint-Eloi |
300.000 |
|
|
ROUSSILLON |
Nîmes |
250.000 |
550.000 |
|
LIMOUSIN |
Limousin |
130.000 |
130.000 |
|
LORRAINE |
Nancy |
0 |
0 |
|
MIDI-PYRÉNÉES |
Toulouse 1 |
300.000 |
|
|
Toulouse 2 |
200.000 |
500.000 |
|
|
NORD-PAS-DE-CALAIS |
Lille |
200.000 |
200.000 |
|
BASSE-NORMANDIE |
Caen |
150.000 |
150.000 |
|
HAUTE-NORMANDIE |
Haute-Normandie |
350.000 |
350.000 |
|
PAYS-DE-LA-LOIRE |
Pays-de-la-Loire 1 (Angers) |
100.000 |
|
|
Pays-de-la-Loire 2 (Nantes) |
450.000 |
550.000 |
|
|
PICARDIE |
Picardie |
150.000 |
150.000 |
|
POITOU-CHARENTES |
Poitou-Charentes |
100.000 |
100.000 |
|
PROVENCE-ALPES- |
Marseille 1 |
0 |
|
|
CÔTE D'AZUR |
Marseille 2 |
0 |
|
|
Nice |
300.000 |
300.000 |
|
|
RHÔNE-ALPES |
Lyon CRLCC |
350.000 |
|
|
Lyon A |
300.000 |
||
|
Lyon B |
300.000 |
||
|
Rhône-Alpes-Loire |
200.000 |
||
|
Grenoble 2 |
300.000 |
1.450.000 |
|
|
MARTINIQUE |
Martinique |
50.000 |
50.000 |
|
GUADELOUPE |
Guadeloupe |
70.000 |
70.000 |
|
TOTAL |
11.860.000 |
Source : DGS (janvier 2001)
ANNEXES
N° 7
-
GRAPHIQUES REPRÉSENTANT LES TAUX DE
PRÉSENCE PAR CATÉGORIE DES MEMBRES DES CCPPRB
|
Les graphiques suivants représentent le nombre de réunions des comités correspondant à des taux de présence des membres par vingtile. Dans le premier graphique, par exemple, il apparaît que le taux de présence des membres de la catégorie I, c'est-à-dire les personnalités ayant une compétence en matière de recherche médicale dépasse les 80 % dans la moitié des réunions organisées par les comités. Ce taux de présence est encore supérieur à 60 % dans les trois-quarts des réunions. |
Présentéisme des personnalités
ayant une compétence
en matière de recherche
médicale
(catégorie I)
Proportion des comités
pour chaque vingtile (%)
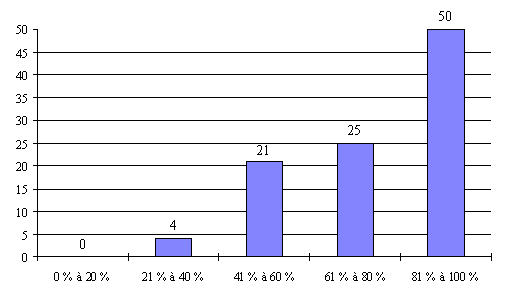
Taux de présence
aux réunions par vingtile
Présence des médecins
généralistes
(catégorie II)
Proportion des comités
pour chaque vingtile (%)
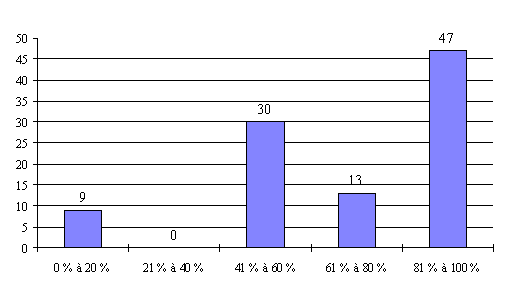
Taux de présence
aux réunions par vingtile
Présence des pharmaciens
(catégorie
III)
Proportion des comités
pour chaque vingtile (%)
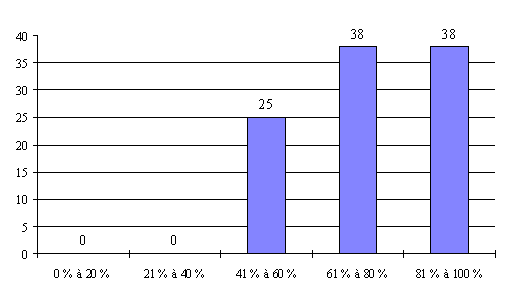
Taux de présence
aux réunions par vingtile
Présence des infirmiers
(catégorie
IV)
Proportion des comités
pour chaque vingtile (%)
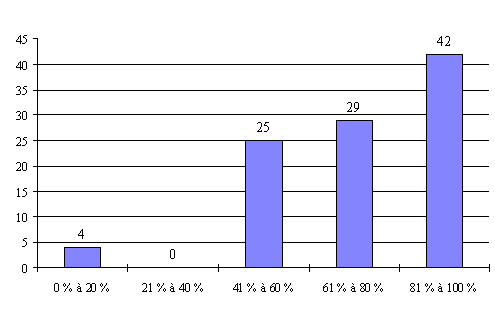
Taux de présence
aux réunions par vingtile
Présence des personnes qualifiées en
matière d'éthique
(catégorie V)
Proportion des comités
pour chaque vingtile (%)
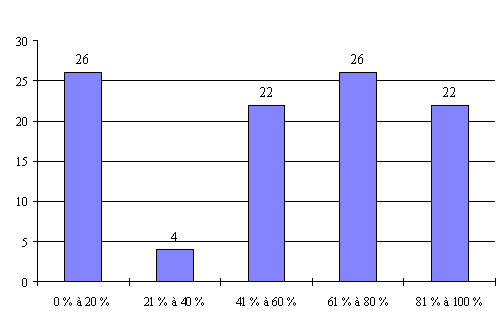
Taux de présence
aux réunions par vingtile
Présence des personnes qualifiées dans le
domaine social
(catégorie VI)
Proportion des comités
pour chaque vingtile (%)
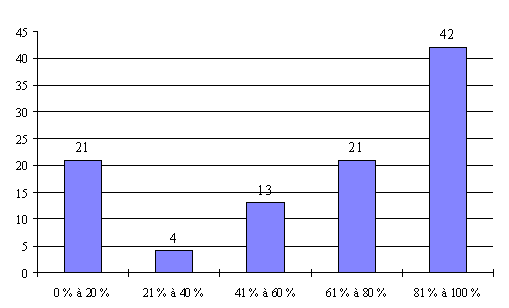
Taux de présence
aux réunions par vingtile
Présence des psychologues
(catégorie
VII)
Proportion des comités
pour chaque vingtile (%)
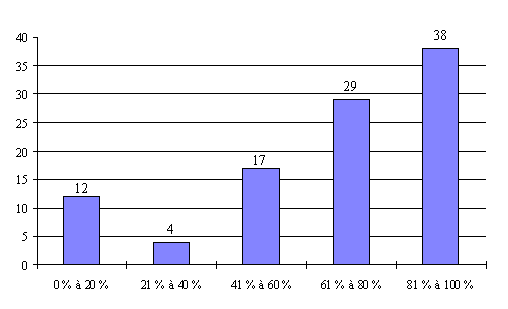
Taux de présence
aux réunions par vingtile
Présence des personnes qualifiées en
matière juridique
(catégorie VIII)
Proportion des comités
pour chaque vingtile (%)
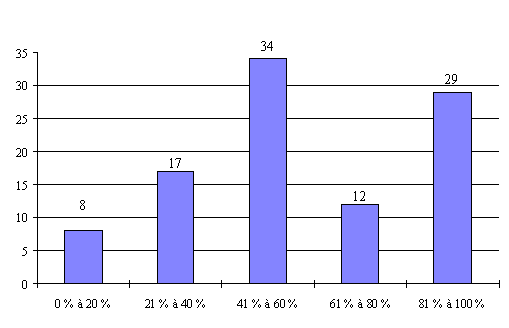
Taux de présence
aux réunions par vingtile
ANNEXE
N° 8
-
ORGANIGRAMME DE LA DIRECTION
GÉNÉRALE
DE LA SANTÉ AVANT LA RÉFORME
|
Lucien ABENHAIM Communication Christiane Dollander Affaires générales et financières Thierry Breton
Relations internationales J. Antourville-Harari Affaires européennes André Ernst
ADJOINTE AU DIRECTEUR Emmanuèle MENGUAL
Rose-Marie Lubet
Christine d'Autume SP1 Promotion et prospective en santé Danièle Mischlich SP2 Ages de la vie et population Robert Simon SP3 Santé mentale, toxicomanies, dépendances Katherine Cornier |
Conseillers techniques :
A. Lordier-Brault
( périnatalité )
M. T.Nutini
( loi Huriet )
P. Gardeur
( personnes âgées)
|
SQ. SYSTÈME DE SANTÉ ET QUALITÉ DES SOINS Anne Bourjade Adjointe : Dominique Varagne SQ 2 Organisation des soins et urgences Christine Courault
Techniques médicales SQ 4 Transfusion sanguine et produits biologiques Félix Faucon |
Conseillers techniques :
A. Pinteaux
( RSS )
S. Deschaumes
( économie de la santé)
|
PH. PHARMACIE Alice Slonimski Adjointe : Hélène Sainte-Marie PH. 2 Affaires professionnelles Jacques Cerda PH. 3 Affaires juridiques Patrick Guyot PH. 4 Médicaments et cosmétiques Françoise Cabane |
Conseillers techniques :
M. P. Baron
( pharmacovigilance )
|
Directeur de Cabinet : N. Relations avec les services déconcentrés Bernard Basset Dominique Tricard Coordination avec les agences Catherine Toussaint Normalisation réseau santé social Michel Rousseau Bioéthique et droits des malades Hélène Khodoss
Yves coquin Adjointe : Françoise Houel
Observation de la santé
Maladies transmissibles Martine Lequellec-Nathan
Eaux Charles Saout
Radioprotection Jean-Luc Godet
François Vareille Adjoint : P. Botreau-Roussel
Ethique et droit Chantal Brault
Professions médicales Isabelle Venencie |
Conseiller technique
N. Quivron
( soins infirmiers )
|
Suzanne Guglielmi Adjoint : N
Stratégies Jean-Jacques Nansot
Actions de proximité Nicole Labrosse-Solier
Risques des milieux, alimentation Daniel Marchand
Professions paramédicales Nathalie Cuvillier |
ANNEXE
N° 9
-
ORGANIGRAMME DE LA DIRECTION
GÉNÉRALE
DE LA SANTÉ APRÈS LA RÉFORME
|
Directeur général Lucien ABENHAIM C Cellule des affaires A européennes et internationales E André ERNST I Jeannine ANTOURVILLE-HARARI D G Service politique de santé et qualité S du système de santé 1 Pascal PENAUD 1 ère sous-direction Sous direction politiques de santé et stratégies Pascal ABRAHAM Adjointe : Anne-Carole BENSADON 2 ème sous-direction Sous-direction de la qualité du système de santé Anne BOURJADE Adjointe : Dominique VARAGNE 3 ème sous-direction Sous-direction de la politique des produits de santé Hélène SAINTE-MARIE (f.f) Adjoint : N... 4 ème sous-direction Sous-direction coordination des services et affaires juridiques Pierre-Marie DETOUR (f.f) Adjoint : N.... 1 Analyse des besoins A et objectifs de santé N... 1 Démocratie sanitaire B Jean-Jacques NANSOT 1 Evaluation des C programmes Gérard PELE 1 Systèmes d'information D Freddy BITAN 1 Recherche et prospective E Catherine GRILLOT- COURVALIN 2 Systèmes de santé A Stéphanie DESCHAUME 2 Qualité des pratiques B Mireille FONTAINE 2 Formations des professions C de santé Jean-Marc BRAICHET 3 Médicament A Caroline GARDETTE-HUMEZ 3 Dispositifs médicaux et B autres produits de santé Patrick GUYOT
3 Produits de santé
Félix FAUCON 4 Éthique et droit A Chantal BRAULT 4 Services déconcentrés B et agences Françoise LACOMBE 4 Ressources humaines et C Affaires générales N... C A Cellule d'appui scientifique S William DAB C C Cellule communication O Christiane DOLLANDER M 6 ème sous-direction Sous-direction santé et société Bernard BASSET (f.f) Adjoint : N... 6 ème sous-direction Sous-direction santé et société Bernard BASSET (f.f) Adjoint : N... 7 ème sous-direction Sous-direction gestion des risques des milieux Thierry MICHELON (f.f) Adjointe : Geneviève BESSE 6 Lutte contre le VIH A Suzanne GUGLIELMI 6 Pratiques addictives B Katherine CORNIER 6 Santé mentale C Nathalie CUVILLIER 6 Santé des populations, D précarité et exclusion Robert SIMON 7 Eaux et aliements A Charles SAOUT 7 Air, sols et déchets B Hugues MALECKI 7 Bâtiments, bruit et C milieu de travail Soraya KOMPANY 7 Rayonnements D Jean-Luc GODET 6 ème sous-direction Sous-direction santé et société Bernard BASSET (f.f) Adjoint : N... D G Service prévention, programmes de S santé et gestion des risques 2 Christine DE MASSON D'AUTUME 5 ème sous-direction Sous-direction pathologies et santé Martine LEQUELLEC-NATHAN (f.f) Adjointe : Juliette BLOCH Adjointe : Laurence LEFEVRE 5 Développement de A programmes de santé Sixte BLANCHY 5 Alerte et problèmes B émergents Vincent PIERRE 5 Maladies infectieuses et C politique vaccinale Thanh LE LUONG 5 Maladies chroniques D enfants et vieillissement Martine VACARIE 6 ème sous-direction Sous-direction santé et société Bernard BASSET (f.f) Adjoint : N... 7 ème sous-direction Sous-direction gestion des risques des milieux Thierry MICHELON (f.f) Adjointe : Geneviève BESSE 6 Lutte contre le VIH A Suzanne GUGLIELMI 6 Lutte contre le VIH A Suzanne GUGLIELMI 6 Lutte contre le VIH A Suzanne GUGLIELMI 6 Pratiques addictives B Katherine CORNIER 6 Santé mentale C Nathalie CUVILLIER
6 Santé des populations,
Robert SIMON 7 Eaux et aliments A Charles SAOUT 7 Air, sols et déchets B Hugues MALECKI 7 Bâtiments, bruit et C milieu de travail Soraya KOMPANY 7 Rayonnements D Jean-Luc GODET |
ANNEXE N° 10
-
TABLE DE
CORRESPONDANCE
ENTRE L'ANCIENNE ET LA NOUVELLE NUMÉROTATION DU CODE
DE LA SANTÉ PUBLIQUE
|
Livre II bis
|
Titre II
|
|
Art. L. 209-1 |
Art. L. 1121-1 |
|
Titre premier
|
|
|
Art. L. 209-2 |
Art. L. 1121-2 |
|
Art. L. 209-3 |
Art. L. 1121-3 |
|
Art. L. 209-4 |
Art. L. 1121-4 |
|
Art. L. 209-5 |
Art. L. 1121-5 |
|
Art. L. 209-6 |
Art. L. 1121-6 |
|
Art. L. 209-7 |
Art. L. 1121-7 |
|
Art. L. 209-8 |
Art. L. 1121-8 |
|
Titre IV
|
|
|
Art. L. 209-13 |
Art. L. 1121-9 |
|
Loi n° 88-1138 du 20.12.88
|
Art. L. 1121-10 |
|
Titre II
|
Chapitre II
|
|
Art. L. 209-9 |
Art. L. 1122-1 |
|
Art. L. 209-10 |
Art. L. 1122-2 |
|
Titre III
|
Chapitre III
|
|
Art. L. 209-11
|
Art. L. 1123-1 |
|
Art. L. 209-11
|
Art. L. 1123-2 |
|
Art. L. 209-11
|
Art. L. 1123-3 |
|
Art. L. 209-11
|
Art. L. 1123-4 |
|
Art. L. 209-11
|
Art. L. 1123-5 |
|
Art. L. 209-12
|
Art. L. 1123-6 |
|
Art. L. 209-12
|
Art. L. 1123-7 |
|
Art. L. 209-12
|
Art. L. 1123-8 |
|
Art. L. 209-12-1 |
Art. L. 1123-9 |
|
Art. L. 209-12
|
Art. L. 1123-10 |
|
Art. L. 209-11
|
Art. L. 1123-11 |
|
Art. L. 209-13-1 |
|
|
Loi n° 88-1138
|
|
|
Titre IV
|
Chapitre IV
|
|
Art. L. 209-14 |
Art. L. 1124-1 |
|
Art. L. 209-15 |
Art. L. 1124-2 |
|
Art. L. 209-16 |
Art. L. 1124-3 |
|
Art. L. 209-17 |
Art. L. 1124-4 |
|
Art. L. 209-18-1 |
Art. L. 1124-5 |
|
Art. L. 209-18 |
Art. L. 1124-6 |
|
Loi n° 88-1138 du 20.12.88 |
Art. L. 1124-7 |
|
art. 6, 2° et 3° |
|
|
Titre IV bis
|
Chapitre V
|
|
Art. L. 209-18-2 |
Art. L. 1125-1 |
|
Art. L. 209-18-3 |
Art. L. 1125-2 |
|
Art. L. 209-18-4 |
Art. L. 1125-3 |
|
Art. L. 209-18-5 |
Art. L. 1125-4 |
|
Art. L 209-18-2
|
Art. L. 1125-5 |
|
Art. L. 209-18-5
|
|
|
Titre V
|
Chapitre VI
|
|
Art. L. 209-19
|
Art. L. 1126-1 |
|
Art. L. 209-19
|
Art. L. 1126-2 |
|
Art. L. 209-19-1
|
Art. L. 1126-3 |
|
Art. L. 209-19-1
|
Art. L. 1126-4 |
|
Art. L. 209-20 |
Art. L. 1126-5 |
|
Art. L. 209-21 |
Art. L. 1126-6 |
|
Titre VI
|
|
|
Art. L. 209-22 |
Art. L. 1126-7 |
|
Art. L. 209-23 |
Art. L. 1512-1 |
ANNEXE
N° 11
-
DISPOSITIONS LÉGISLATIVES DU CODE DE LA
SANTÉ PUBLIQUE APPLICABLES AUX CCPPRB
TITRE II
RECHERCHES BIOMÉDICALES
Chapitre premier
Principes généraux
Article L. 1121-1
Les essais ou expérimentations organisés et pratiqués sur l'être humain en vue du développement des connaissances biologiques ou médicales sont autorisés dans les conditions prévues au présent livre et sont désignés ci-après par les termes : "recherche biomédicale".
Les recherches biomédicales dont on attend un bénéfice direct pour la personne qui s'y prête sont dénommées recherches biomédicales avec bénéfice individuel direct. Toutes les autres recherches, qu'elles portent sur des personnes malades ou non, sont dénommées sans bénéfice individuel direct.
La personne physique ou morale qui prend l'initiative d'une recherche biomédicale sur l'être humain est dénommée le promoteur. La ou les personnes physiques qui dirigent et surveillent la réalisation de la recherche sont dénommées les investigateurs.
Lorsque plusieurs personnes prennent l'initiative d'une même recherche, elles peuvent désigner une personne physique ou morale qui aura la qualité de promoteur et assume les obligations correspondantes en application du présent livre.
Lorsque le promoteur d'une recherche confie sa réalisation à plusieurs investigateurs, il désigne parmi eux un investigateur coordonnateur.
Article L. 1121-2
Aucune recherche biomédicale ne peut être effectuée sur l'être humain :
- si elle ne se fonde pas sur le dernier état des connaissances scientifiques et sur une expérimentation préclinique suffisante ;
- si le risque prévisible encouru par les personnes qui se prêtent à la recherche est hors de proportion avec le bénéfice escompté pour ces personnes ou l'intérêt de cette recherche ;
- si elle ne vise pas à étendre la connaissance scientifique de l'être humain et les moyens susceptibles d'améliorer sa condition.
Article L. 1121-3
Les recherches biomédicales ne peuvent être effectuées que :
- sous la direction et sous la surveillance d'un médecin justifiant d'une expérience appropriée ;
- dans des conditions matérielles et techniques adaptées à l'essai et compatibles avec les impératifs de rigueur scientifique et de sécurité des personnes qui se prêtent à ces recherches.
Dans les sciences du comportement humain, une personne qualifiée, conjointement avec l'investigateur, peut exercer la direction de la recherche.
Les recherches biomédicales concernant le domaine de l'odontologie ne peuvent être effectuées que sous la direction et la surveillance d'un chirurgien-dentiste et d'un médecin justifiant d'une expérience appropriée.
Article L. 1121-4
Les recherches sans bénéfice individuel direct sur les femmes enceintes, les parturientes et les mères qui allaitent ne sont admises que si elles ne présentent aucun risque sérieux prévisible pour leur santé ou celle de leur enfant, si elles sont utiles à la connaissance des phénomènes de la grossesse, de l'accouchement ou de l'allaitement et si elles ne peuvent être réalisées autrement.
Article L. 1121-5
Les personnes privées de liberté par une décision judiciaire ou administrative, les malades en situation d'urgence et les personnes hospitalisées sans consentement en vertu des articles L. 3212-1 et L. 3213-1 qui ne sont pas protégées par la loi ne peuvent être sollicités pour se prêter à des recherches biomédicales que s'il en est attendu un bénéfice direct et majeur pour leur santé.
Article L. 1121-6
Les mineurs, les majeurs protégés par la loi et les personnes admises dans un établissement sanitaire ou social à d'autres fins que celle de la recherche ne peuvent être sollicités pour une recherche biomédicale que si l'on peut en attendre un bénéfice direct pour leur santé.
Toutefois, les recherches sans bénéfice individuel direct sont admises si les trois conditions suivantes sont remplies :
- ne présenter aucun risque sérieux prévisible pour leur santé ;
- être utiles à des personnes présentant les mêmes caractéristiques d'âge, de maladie ou de handicap ;
- ne pouvoir être réalisées autrement.
Article L. 1121-7
Pour les recherches biomédicales sans bénéfice individuel direct, le promoteur assume, même sans faute, l'indemnisation des conséquences dommageables de la recherche pour la personne qui s'y prête et celle de ses ayants droit, sans que puisse être opposé le fait d'un tiers ou le retrait volontaire de la personne qui avait initialement consenti à se prêter à la recherche.
Pour les recherches biomédicales avec bénéfice individuel direct, le promoteur assume l'indemnisation des conséquences dommageables de la recherche pour la personne qui s'y prête et celle de ses ayants droit, sauf preuve à sa charge que le dommage n'est pas imputable à sa faute, ou à celle de tout intervenant sans que puisse être opposé le fait d'un tiers ou le retrait volontaire de la personne qui avait initialement consenti à se prêter à la recherche.
La recherche biomédicale exige la souscription préalable, par son promoteur, d'une assurance garantissant sa responsabilité civile telle qu'elle résulte du présent article et celle de tout intervenant, indépendamment de la nature des liens existant entre les intervenants et le promoteur. Les dispositions du présent article sont d'ordre public.
Article L. 1121-8
La recherche biomédicale ne donne lieu à aucune contrepartie financière directe ou indirecte pour les personnes qui s'y prêtent, hormis le remboursement des frais exposés et sous réserve de dispositions particulières prévues par l'article L. 1124-2 relatif aux recherches sans bénéfice individuel direct.
Article L. 1121-9
Les médecins inspecteurs de santé publique et, dans la limite de leurs attributions, les inspecteurs de l'Agence française de sécurité sanitaire des produits de santé ont qualité pour veiller au respect des dispositions du présent titre et des textes réglementaires pris pour son application.
Article L. 1121-10
Les modalités d'application des dispositions du présent chapitre sont déterminées par décret en Conseil d'Etat et notamment les minima de garanties pour l'assurance prévue au troisième alinéa de l'article L. 1121-7.
Chapitre II
Consentement de la personne
Article L. 1122-1
Préalablement à la réalisation d'une recherche biomédicale sur une personne, le consentement libre, éclairé et exprès de celle-ci doit être recueilli après que l'investigateur, ou un médecin qui le représente, lui a fait connaître :
- l'objectif, la méthodologie et la durée de la recherche ;
- les bénéfices attendus, les contraintes et les risques prévisibles, y compris en cas d'arrêt de la recherche avant son terme ;
- l'avis du comité mentionné à l'article L. 1123-6 ;
- le cas échéant, son inscription dans le fichier national prévu à l'article L. 1124-4.
Il informe la personne dont le consentement est sollicité de son droit de refuser de participer à une recherche ou de retirer son consentement à tout moment sans encourir aucune responsabilité.
L'objectif d'une recherche en psychologie, ainsi que sa méthodologie et sa durée, peuvent ne faire l'objet que d'une information préalable succincte dès lors que la recherche ne porte que sur des volontaires sains et ne présente aucun risque sérieux prévisible. Une information complète sur cette recherche est fournie à l'issue de celle-ci aux personnes s'y étant prêtées. Le projet mentionné au premier alinéa de l'article L. 1123-6 mentionne la nature des informations préalables transmises aux personnes se prêtant à la recherche.
A titre exceptionnel, lorsque dans l'intérêt d'une personne malade le diagnostic de sa maladie n'a pu lui être révélé, l'investigateur peut, dans le respect de sa confiance, réserver certaines informations liées à ce diagnostic. Dans ce cas, le protocole de la recherche doit mentionner cette éventualité.
Les informations communiquées sont résumées dans un document écrit remis à la personne dont le consentement est sollicité.
Le consentement est donné par écrit ou, en cas d'impossibilité, attesté par un tiers. Ce dernier doit être totalement indépendant de l'investigateur et du promoteur.
Toutefois, en cas de recherches biomédicales à mettre en oeuvre dans des situations d'urgence qui ne permettent pas de recueillir le consentement préalable de la personne qui y sera soumise, le protocole présenté à l'avis du comité instauré par l'article L. 1123-1 peut prévoir que le consentement de cette personne ne sera pas recherché et que seul sera sollicité celui des membres de sa famille s'ils sont présents, dans les conditions prévues ci-dessus. L'intéressé est informé dès que possible et son consentement lui est demandé pour la poursuite éventuelle de cette recherche.
Article L. 1122-2
Lorsqu'une recherche biomédicale est effectuée sur des mineurs ou des majeurs protégés par la loi :
- le consentement doit être donné, selon les règles prévues à l'article L. 1122-1, par les titulaires de l'exercice de l'autorité parentale pour les mineurs non émancipés. Pour les mineurs ou les majeurs protégés par la loi, le consentement est donné par le représentant légal pour les recherches avec bénéfice individuel direct ne présentant pas un risque prévisible sérieux et, dans les autres cas, par le représentant légal autorisé par le conseil de famille ou le juge des tutelles ;
- le consentement du mineur ou du majeur protégé par la loi doit également être recherché lorsqu'il est apte à exprimer sa volonté. Il ne peut être passé outre à son refus ou à la révocation de son consentement.
Chapitre III
Comités consultatifs de protection des personnes
Article L. 1123-1
Dans chaque région, le ministre chargé de la santé agrée un ou, selon les besoins, plusieurs comités consultatifs de protection des personnes dans la recherche biomédicale.
Le ministre fixe par arrêté le nombre de comités dans chaque région. Le champ de compétence territoriale d'un comité peut être étendu à plusieurs régions.
Les comités exercent leur mission en toute indépendance. Ils sont dotés de la personnalité juridique.
Les comités sont compétents au sein de la région où ils ont leur siège.
Article L. 1123-2
Les comités sont composés de manière à garantir leur indépendance et la diversité des compétences dans le domaine biomédical et à l'égard des questions éthiques, sociales, psychologiques et juridiques.
Leurs membres sont nommés par le représentant de l'Etat dans la région où le comité a son siège. Ils sont choisis parmi les personnes figurant sur une liste établie sur proposition d'organismes ou d'autorités habilités à le faire, dans des conditions déterminées par décret.
Article L. 1123-3
Les membres des comités, les personnes appelées à collaborer à leurs travaux, et les agents relevant du statut général des fonctionnaires qui en sont dépositaires sont tenus, dans les conditions et sous les peines prévues aux articles 226-13 et 226-14 du code pénal, de garder secrètes les informations dont ils peuvent avoir connaissance à raison de leurs fonctions et qui sont relatives à la nature des recherches, aux personnes qui les organisent ou qui s'y prêtent ou aux produits, objets ou méthodes expérimentés.
Ne peuvent valablement participer à une délibération les personnes qui ne sont pas indépendantes du promoteur et de l'investigateur de la recherche examinée.
Article L. 1123-4
Les frais de fonctionnement des comités sont financés par le produit d'un droit fixe versé par les promoteurs pour chacun des projets de recherches biomédicales faisant l'objet d'une demande d'avis. Le montant de ce droit est arrêté par le ministre chargé de la santé.
Article L. 1123-5
Le ministre chargé de la santé peut retirer l'agrément d'un comité si les conditions d'indépendance, de composition ou de fonctionnement nécessaires pour assurer sa mission dans les meilleures conditions ne sont plus satisfaites.
Article L. 1123-6
Avant de réaliser une recherche biomédicale sur l'être humain, tout investigateur est tenu d'en soumettre le projet à l'avis de l'un des comités consultatifs de protection des personnes dans la recherche biomédicale compétents pour la région où l'investigateur exerce son activité. Il ne peut solliciter qu'un seul avis par projet de recherche.
Dans le cas d'une recherche confiée à plusieurs investigateurs, cet avis est demandé par l'investigateur coordonnateur, qui soumet le projet dans les conditions définies au premier alinéa du présent article.
Article L. 1123-7
Le comité rend son avis sur les conditions de validité de la recherche au regard de la protection des personnes, notamment la protection des participants, leur information avant et pendant la durée de la recherche et les modalités de recueil de leur consentement, les indemnités éventuellement dues, la pertinence générale du projet et l'adéquation entre les objectifs poursuivis et les moyens mis en oeuvre ainsi que la qualification du ou des investigateurs. Dans un délai de cinq semaines, il fait connaître par écrit son avis à l'investigateur. Il communique à l'Agence française de sécurité sanitaire des produits de santé pour les produits mentionnés à l'article L. 5311-1 ou au ministre chargé de la santé dans les autres cas tout avis défavorable donné à un projet de recherche.
Article L. 1123-8
Avant la mise en oeuvre d'une recherche biomédicale sur l'être humain, le promoteur transmet à l'Agence française de sécurité sanitaire des produits de santé pour les produits mentionnés à l'article L. 5311-1 ou au ministre chargé de la santé dans les autres cas une lettre d'intention décrivant les données essentielles de la recherche, accompagnée de l'avis du comité consulté. Cet avis ne le dégage pas de sa responsabilité. Les projets ayant fait l'objet d'un avis défavorable ne peuvent être mis en oeuvre avant un délai de deux mois à compter de leur réception par l'Agence française de sécurité sanitaire des produits de santé pour les produits mentionnés à l'article L. 5311-1 ou par le ministre chargé de la santé dans les autres cas.
Le promoteur informe, dès qu'il en a connaissance, l'autorité administrative compétente de tout effet ayant pu contribuer à la survenue d'un décès, provoquer une hospitalisation ou entraîner des séquelles organiques ou fonctionnelles durables et susceptibles d'être dues à la recherche. Le promoteur transmet également à l'Agence française de sécurité sanitaire des produits de santé pour les produits mentionnés à l'article L. 5311-1 ou au ministre chargé de la santé dans les autres cas toute information relative à un fait nouveau concernant le déroulement de la recherche ou le développement du produit ou du dispositif faisant l'objet de la recherche lorsque ce fait nouveau est susceptible de porter atteinte à la sécurité des personnes qui se prêtent à la recherche. Il informe, selon le cas, l'Agence française de sécurité sanitaire des produits de santé ou le ministre chargé de la santé enfin de tout arrêt prématuré de la recherche en indiquant le motif de cet arrêt.
L'Agence française de sécurité sanitaire des produits de santé pour les produits mentionnés à l'article L. 5311-1 ou le ministre chargé de la santé dans les autres cas peut, à tout moment, demander au promoteur des informations complémentaires sur la recherche. En cas d'absence de réponse du promoteur, de risque pour la santé publique ou de non-respect des dispositions du présent livre, l'Agence française de sécurité sanitaire des produits de santé pour les produits mentionnés à l'article L. 5311-1 ou le ministre chargé de la santé dans les autres cas peut également à tout moment suspendre ou interdire une recherche biomédicale.
Article L. 1123-9
Le comité consultatif de protection des personnes peut émettre, dans les conditions prévues aux articles L. 1123-6, L. 1123-7 et L. 1123-8, un avis favorable à la réalisation d'une recherche sous réserve de la transmission d'informations complémentaires par l'investigateur pendant le déroulement de celle-ci.
A la suite de cette transmission, le comité peut maintenir ou modifier son avis. Cette décision est transmise par écrit à l'investigateur dans un délai de cinq semaines ; elle est adressée par le promoteur à l'Agence française de sécurité sanitaire des produits de santé pour les produits mentionnés à l'article L. 5311-1 ou au ministre chargé de la santé dans les autres cas dans un délai d'une semaine après sa réception.
Article L. 1123-10
Lorsque la recherche doit se dérouler dans un ou plusieurs établissements publics ou privés, le promoteur en informe le ou les directeurs de ces établissements avant que cette recherche ne soit mise en oeuvre.
Article L. 1123-11
Les modalités d'application des dispositions du présent chapitre sont déterminées par décret en Conseil d'Etat et notamment :
1° La composition et les conditions d'agrément, de financement, de fonctionnement et de nomination des membres des comités consultatifs de protection des personnes dans la recherche biomédicale ainsi que la nature des informations qui doivent leur être communiquées par l'investigateur et sur lesquelles ils sont appelés à émettre leur avis ;
2° Les conditions minimales d'activité en deçà desquelles le champ de compétence territoriale d'un comité peut être étendu à plusieurs régions ;
3° La nature des informations qui doivent être communiquées par le promoteur à l'autorité administrative compétente, dans la lettre d'intention mentionnée à l'article L. 1123-8 ;
4° Les modalités de consultation des comités consultatifs de protection des personnes dans la recherche biomédicale en ce qui concerne les recherches à caractère militaire.
Chapitre IV
Recherches sans bénéfice individuel direct
Article L. 1124-1
Les recherches biomédicales sans bénéfice individuel direct ne doivent comporter aucun risque prévisible sérieux pour la santé des personnes qui s'y prêtent.
Elles doivent être précédées d'un examen médical des personnes concernées. Les résultats de cet examen leur sont communiqués préalablement à l'expression de leur consentement par l'intermédiaire du médecin de leur choix.
Article L. 1124-2
Dans le cas d'une recherche sans bénéfice individuel direct, le promoteur peut verser aux personnes qui s'y prêtent une indemnité en compensation des contraintes subies. Le montant total des indemnités qu'une personne peut percevoir au cours d'une même année est limité à un maximum fixé par le ministre chargé de la santé.
Les recherches effectuées sur des mineurs, des majeurs protégés par la loi ou des personnes admises dans un établissement sanitaire ou social à d'autres fins que celle de la recherche ne peuvent en aucun cas donner lieu au versement de l'indemnité prévue au premier alinéa du présent article.
Article L. 1124-3
Toute recherche biomédicale sans bénéfice individuel direct sur une personne qui n'est pas affiliée à un régime de sécurité sociale ou bénéficiaire d'un tel régime est interdite.
L'organisme de sécurité sociale dispose contre le promoteur d'une action en paiement des prestations versées ou fournies.
Article L. 1124-4
Nul ne peut se prêter simultanément à plusieurs recherches biomédicales sans bénéfice individuel direct.
Pour chaque recherche sans bénéfice individuel direct, le protocole soumis à l'avis consultatif du comité consultatif de protection des personnes dans la recherche biomédicale détermine une période d'exclusion au cours de laquelle la personne qui s'y prête ne peut participer à une autre recherche sans bénéfice individuel direct. La durée de cette période varie en fonction de la nature de la recherche.
En vue de l'application des dispositions du présent article, le ministre chargé de la santé établit et gère un fichier national.
Article L. 1124-5
Aucune recherche biomédicale ne peut être effectuée sur une personne en état de mort cérébrale sans son consentement exprimé directement ou par le témoignage de sa famille.
Les dispositions de l'article 225-17 du code pénal ne sont pas applicables à ces recherches.
Article L. 1124-6
Les recherches biomédicales sans bénéfice individuel direct ne peuvent être réalisées que dans un lieu équipé des moyens matériels et techniques adaptés à la recherche et compatibles avec les impératifs de sécurité des personnes qui s'y prêtent et autorisé, à ce titre, par l'Agence française de sécurité sanitaire des produits de santé pour les produits mentionnés à l'article L. 5311-1 ou par le ministre chargé de la santé dans les autres cas.
Article L. 1124-7
Les modalités d'application des dispositions du présent chapitre sont déterminées par décret en Conseil d'Etat et notamment :
1° Les conditions de la constitution, de la gestion et de la consultation du fichier national prévu à l'article L. 1124-4 ;
2° Les conditions de l'autorisation prévue à l'article L. 1124-6.
Chapitre V
Dispositions particulières à certaines recherches
Article L. 1125-1
Les protocoles d'essais cliniques concernant les produits mentionnés à l'article L. 1261-1 ne peuvent être réalisés que dans des établissements de santé ou de transfusion sanguine ayant reçu l'autorisation mentionnée à l'article L. 1262-1. Cette autorisation vaut pour l'application de l'article L. 1124-6.
Les dispositions de la troisième phrase du premier alinéa de l'article L. 1123-8 ne s'appliquent pas aux protocoles mentionnés au présent article. Ces protocoles ne peuvent être mis en oeuvre qu'après avoir été autorisés par l'Agence française de sécurité sanitaire des produits de santé en fonction du respect des dispositions relatives aux essais de médicaments et, le cas échéant, de la loi n° 92-654 du 13 juillet 1992 relative au contrôle de l'utilisation et de la dissémination des organismes génétiquement modifiés et modifiant la loi du 19 juillet 1976 relative aux installations classées pour la protection de l'environnement.
L'autorisation ou le refus d'autorisation est prononcé dans un délai de quatre-vingt-dix jours à compter de la réception de la demande. L'autorisation vaut agrément au sens de l'article 6 et autorisation au sens de l'article 11 de la loi du 13 juillet 1992 précitée.
La méconnaissance des dispositions précitées fonde, à tout moment, les mesures de suspension ou d'interdiction mentionnées au dernier alinéa de l'article L. 1123-8 L'autorisation est alors suspendue ou retirée.
Article L. 1125-2
L'utilisation à des fins thérapeutiques d'organes, de tissus ou de cellules d'origine animale qui ne sont ni des dispositifs médicaux, ni destinés à des thérapies génique ou cellulaire, ni à des médicaments n'est possible que dans le cadre de recherches biomédicales soumises aux dispositions du présent titre. Par dérogation aux dispositions de l'article L. 1123-8, les recherches cliniques portant sur l'utilisation thérapeutique de tels organes, tissus ou cellules chez l'être humain ne peuvent être mises en oeuvre qu'après autorisation du ministre chargé de la santé, après avis de l'Agence française de sécurité sanitaire des produits de santé et de l'Etablissement français des greffes. L'autorisation peut être assortie de conditions particulières, portant notamment sur la surveillance à long terme des patients.
Des règles de bonne pratique relatives au prélèvement, à la conservation, à la transformation, au transport et à l'utilisation des organes, tissus et cellules animaux sont préparées par l'Agence française de sécurité sanitaire des produits de santé après avis de l'Etablissement français des greffes et homologuées par le ministre chargé de la santé.
Des arrêtés du ministre chargé de la santé, pris sur proposition de l'Agence française de sécurité sanitaire des produits de santé, après avis de l'Etablissement français des greffes et de l'Agence française de sécurité sanitaire des aliments, fixent :
- les règles de bonne pratique relatives à la sélection, à la production et à l'élevage des animaux ;
- les conditions sanitaires auxquelles doivent répondre les animaux dont proviennent les organes, tissus et cellules utilisés ;
- les règles d'identification de ces animaux, organes, tissus et cellules permettant d'assurer la traçabilité des produits obtenus.
Article L. 1125-3
Par dérogation aux dispositions du premier alinéa de l'article L. 1123-8, les investigations cliniques portant sur des dispositifs médicaux cités à l'article L. 5211-4 ne peuvent être mises en oeuvre avant un délai de deux mois à compter de la réception de la lettre d'intention par l'Agence française de sécurité sanitaire des produits de santé.
Article L. 1125-4
Sans préjudice des dispositions de l'article L. 1125-1, les dispositions de la troisième phrase du premier alinéa de l'article L. 1123-8 ne s'appliquent pas aux protocoles des essais cliniques concernant les cellules issues du corps humain. Ces protocoles ne peuvent être réalisés que dans des établissements de santé ayant reçu l'autorisation prévue au deuxième alinéa de l'article L. 1243-4. Cette autorisation vaut pour l'application de l'article L. 1124-6.
Ces protocoles ne peuvent être mis en oeuvre qu'après avoir été autorisés par l'Agence française de sécurité sanitaire des produits de santé.
L'autorisation ou le refus d'autorisation est prononcé dans un délai de quatre-vingt-dix jours à compter de la réception de la demande.
La méconnaissance des dispositions précitées fonde, à tout moment, les mesures de suspension ou d'interdiction mentionnées au dernier alinéa de l'article L. 1123-8.
L'autorisation est alors suspendue ou retirée.
Article L. 1125-5
Les modalités d'application des dispositions du présent chapitre sont déterminées par décret en Conseil d'Etat et notamment les conditions dans lesquelles l'Agence française de sécurité sanitaire des produits de santé autorise :
1° Les protocoles concernant les produits mentionnés à l'article L. 1261-1 et prévus à l'article L. 1125-1 ;
2° Les protocoles des essais cliniques concernant les cellules issues du corps humain prévus à l'article L. 1125-4.
Chapitre VI
Dispositions pénales
Article L. 1126-1
Comme il est dit à l'article 223-8 du code pénal ci-après reproduit :
Art. 223-8 - " Le fait de pratiquer ou de faire pratiquer sur une personne une recherche biomédicale sans avoir recueilli le consentement libre, éclairé et exprès de l'intéressé, des titulaires de l'autorité parentale ou du tuteur dans les cas prévus par les dispositions du code de la santé publique est puni de trois ans d'emprisonnement et de 300.000 F d'amende.
" Les mêmes peines sont applicables lorsque la recherche biomédicale est pratiquée alors que le consentement a été retiré. "
Article L. 1126-2
Comme il est dit à l'article 223-9 du code pénal ci-après reproduit :
" Les personnes morales peuvent être déclarées responsables pénalement, dans les conditions prévues par l'article 121-2, de l'infraction définie à l'article 223-8.
" Les peines encourues par les personnes morales sont :
" 1° L'amende, suivant les modalités prévues par l'article 131-38 ;
" 2° Les peines mentionnées à l'article 131-39.
" L'interdiction mentionnée au 2° de l'article 131-39 porte sur l'activité dans l'exercice de laquelle ou à l'occasion de laquelle l'infraction a été commise. "
Article L. 1126-3
Le fait de pratiquer ou de faire pratiquer une recherche biomédicale en infraction aux dispositions des articles L. 1121-4 à L. 1121-6 et du dernier alinéa de l'article L. 1122-1 est puni de trois ans d'emprisonnement et de 300 000 F d'amende.
Les personnes physiques coupables de l'infraction prévue à l'alinéa précédent encourent également les peines suivantes :
1° L'interdiction des droits civiques, civils et de famille, suivant les modalités prévues par l'article 131-26 du code pénal ;
2° L'interdiction, pour une durée de cinq ans au plus, d'exercer l'activité professionnelle ou sociale à l'occasion de laquelle ou dans l'exercice de laquelle l'infraction a été commise ;
3° La confiscation définie à l'article 131-21 du code pénal ;
4° L'exclusion des marchés publics à titre définitif ou pour une durée de cinq ans au plus.
Article L. 1126-4
Les personnes morales peuvent être déclarées responsables pénalement, dans les conditions prévues par l'article 121-2 du code pénal, de l'infraction définie à l'article L. 1126-3.
Les peines encourues par les personnes morales sont :
1° L'amende, suivant les modalités prévues par l'article 131-38 du code pénal ;
2° Les peines mentionnées à l'article 131-39 du code pénal.
L'interdiction mentionnée au 2° de l'article 131-39 du code pénal porte sur l'activité dans l'exercice de laquelle ou à l'occasion de laquelle l'infraction a été commise.
Article L. 1126-5
Est puni d'un an d'emprisonnement et de 100 000 F d'amende le fait de pratiquer ou de faire pratiquer une recherche biomédicale :
1° Sans avoir obtenu l'avis préalable prévu par l'article L. 1123-6 ;
2° Dans des conditions contraires aux dispositions des deux premiers alinéas de l'article L. .1124-4 ;
3° Dont la réalisation a été interdite ou suspendue par le ministre chargé de la santé ou par l'Agence française de sécurité sanitaire des produits de santé pour les produits mentionnés à l'article L. 5311-1.
L'investigateur qui réalise une telle recherche en infraction aux dispositions de l'article L. 1124-6 est puni des mêmes peines.
Article L. 1126-6
Le promoteur dont la responsabilité civile n'est pas garantie par l'assurance prévue à l'article L. 1121-7 est puni d'un an d'emprisonnement et de 100 000 F d'amende.
Le promoteur qui réalise ou fait réaliser une recherche biomédicale sans avoir transmis au ministre chargé de la santé ou à l'Agence française de sécurité sanitaire des produits de santé pour les produits mentionnés à l'article L. 5311-1 la lettre d'intention prévue à l'article L. 1123-8 est puni des mêmes peines.
Article L. 1126-7
Par dérogation à l'article 13 de la loi des 16 et 24 août 1790 sur l'organisation judiciaire, le tribunal de grande instance est seul compétent pour statuer sur toute action en indemnisation des dommages résultant d'une recherche biomédicale ; cette action se prescrit dans les conditions prévues à l'article 2270-1 du code civil.
ANNEXE N°
12
-
TEXTE DE LA DIRECTIVE EUROPÉENNE
RELATIVE AUX ESSAIS
CLINIQUES84
(
*
)
DIRECTIVE DU PARLEMENT EUROPÉEN ET DU CONSEIL CONCERNANT LE RAPPROCHEMENT DES DISPOSITIONS LÉGISLATIVES, RÉGLEMENTAIRES ET ADMINISTRATIVES DES ETATS MEMBRES RELATIVES À L'APPLICATION DE BONNES PRATIQUES CLINIQUES DANS LA CONDUITE D'ESSAIS CLINIQUES DE MÉDICAMENTS À USAGE HUMAIN
CHAPITRE I
Champ d'application et définitions
Article premier
Champ d'application
1. La présente directive fixe des dispositions spécifiques concernant la conduite des essais cliniques, y compris des essais multicentriques, effectués sur des êtres humains et portant sur les médicaments définis à l'article 1er de la directive 65/65/CEE, en particulier en ce qui concerne l'application de bonnes pratiques cliniques. La présente directive ne s'applique pas aux essais non-interventionnels.
2. Les bonnes pratiques cliniques constituent un ensemble d'exigences de qualité dans les domaines éthique et scientifique, reconnues au plan international, qui doivent être respectées lors de la planification, la mise en oeuvre, l'enregistrement et la notification des essais cliniques auxquels des êtres humains participent. Le respect de ces bonnes pratiques garantit la protection des droits, de la sécurité et du bien-être des participants à des essais cliniques, ainsi que la crédibilité des résultats des essais cliniques.
3. Les principes des bonnes pratiques cliniques et les lignes directrices détaillées conformes à ces principes sont adoptés et, le cas échéant, révisés pour tenir compte des progrès scientifiques et techniques, conformément à la procédure visée à l'article 21, paragraphe 2. La Commission publie ces lignes directrices détaillées.
4. Tous les essais cliniques, y compris les études de biodisponibilité et de bioéquivalence, sont conçus, mis en oeuvre et notifiés conformément aux principes des bonnes pratiques cliniques.
Article 2
Définitions
Aux fins de la présente directive, on entend par :
a) " essai clinique ": toute investigation menée chez l'homme, afin de déterminer ou de confirmer les effets cliniques, pharmacologiques et/ou les autres effets pharmacodynamiques d'un ou de plusieurs médicaments expérimentaux, et/ou de mettre en évidence tout effet indésirable d'un ou de plusieurs médicaments expérimentaux, et/ou d'étudier l'absorption, la distribution, le métabolisme et l'élimination d'un ou de plusieurs médicaments expérimentaux, dans le but de s'assurer de leur innocuité et/ou efficacité ;
Sont compris les essais cliniques réalisés sur un site unique ou sur des sites multiples, dans un ou plusieurs Etats membres ;
b) " essai clinique multicentrique " : essai clinique réalisé selon un même protocole, mais sur des sites différents et donc par plusieurs investigateurs, les sites de l'essai pouvant se trouver dans un seul Etat membre, dans plusieurs Etats membres et/ou dans des Etats membres et des pays tiers;
c) " essai noninterventionnel ": étude dans le cadre duquel le ou les médicaments sont prescrits de la manière habituelle conformément aux conditions fixées dans l'autorisation de mise sur le marché. L'affectation du patient à une stratégie thérapeutique donnée n'est pas fixée à l'avance par un protocole d'essai, elle relève de la pratique courante et la décision de prescrire le médicament est clairement dissociée de celle d'inclure le patient dans l'étude. Aucune procédure supplémentaire de diagnostic ou de surveillance ne doit être appliquée aux patients et des méthodes épidémiologiques sont utilisées pour analyser les données recueillies ;
d) " médicament expérimental " : principe actif sous forme pharmaceutique ou placebo expérimenté ou utilisé comme référence dans un essai clinique, y compris les produits bénéficiant déjà d'une autorisation de mise sur le marché, mais utilisés ou formulés (présentation ou conditionnement) différemment de la forme autorisée, ou utilisés pour une indication non autorisée ou en vue d'obtenir de plus amples informations sur la forme autorisée ;
e) " promoteur " : personne, entreprise, institut ou organisme responsable du lancement, de la gestion et/ou du financement d'un essai clinique ;
f) " investigateur " : un médecin ou une personne exerçant une profession agréée dans l'Etat membre aux fins de travaux d'investigation en raison des connaissances scientifiques et de l'expérience dans le domaine des soins aux patients qu'elle requiert. L'investigateur est responsable de la conduite de l'essai clinique sur un site. Si, sur un site, l'essai est réalisé par une équipe, l'investigateur est le responsable de l'équipe et peut être appelé investigateur principal ;
g) " brochure pour l'investigateur ": ensemble des données cliniques ou non concernant le ou les médicaments expérimentaux, et qui sont pertinentes pour l'étude de ce(s) produit(s) chez l'homme ;
h) " protocole " : document décrivant le ou les objectifs, la conception, la méthode, les aspects statistiques et l'organisation d'un essai. Le terme protocole recouvre le protocole, ses versions successives et ses modifications ;
i) " participant " : personne qui participe à un essai clinique, qu'il reçoive le médicament expérimental ou serve de témoin ;
j) " consentement éclairé ": décision, qui doit être écrite, datée et signée, de participer à un essai clinique, prise de plein gré après avoir été dûment informé de la nature, de la portée, des conséquences et des risques et avoir reçu une documentation appropriée, par une personne capable de donner son consentement ou, s'il s'agit d'une personne qui n'est pas en mesure de le faire, par son représentant légal; si la personne concernée n'est pas en mesure d'écrire, elle peut donner, dans des cas exceptionnels prévus par la législation nationale, son consentement oral en présence d'au moins un témoin ;
k) " comité d'éthique " : organe indépendant, dans un Etat membre, composé de professionnels de la santé et de membres non médecins, chargé de préserver les droits, la sécurité et le bien-être des participants à un essai et de rassurer le public à ce sujet, notamment en formulant un avis sur le protocole d'essai, l'aptitude des investigateurs et l'adéquation des installations, ainsi que sur les méthodes et les documents à utiliser pour informer les participants aux essais en vue d'obtenir leur consentement éclairé ;
l) " inspection " : activité menée par une autorité compétente et consistant à procéder à l'examen officiel des documents, installations, enregistrements, systèmes d'assurance qualité et de tout autre élément qui, de l'avis de l'autorité compétente, ont trait à l'essai clinique et qui peuvent se trouver sur le site de l'essai, dans les locaux du promoteur et/ou de l'organisme de recherche sous-traitant ou dans tout autre établissement que l'autorité compétente juge nécessaire d'inspecter ;
m) " événement indésirable " : toute manifestation nocive chez un patient ou un participant à un essai clinique traité par un médicament, et qui n'est pas nécessairement liée à ce traitement;
n) " effet indésirable " : toute réaction nocive et non désirée à un médicament expérimental, quelle que soit la dose administrée ;
o) " événement indésirable grave ou effet indésirable grave " : événement indésirable ou effet indésirable qui, quelle que soit la dose, entraîne la mort, met en danger la vie du participant, nécessite une hospitalisation ou la prolongation de l'hospitalisation, provoque un handicap ou une incapacité importants ou durables, ou bien, se traduit par une anomalie ou une malformation congénitales ;
p) " effet indésirable inattendu " : effet indésirable dont la nature ou la gravité ne concorde pas avec les informations relatives au produit (par exemple, la brochure pour l'investigateur pour un produit expérimental non autorisé ou, dans le cas d'un produit autorisé, la notice jointe au résumé des caractéristiques du produit).
CHAPITRE II
Article 3
Protection des participants aux essais cliniques
1. La présente directive s'applique sans préjudice des dispositions nationales relatives à la protection des participants à des essais cliniques dès lors que ces dispositions ont une portée plus large que celles de la présente directive et pour autant qu'elles soient conformes aux procédures et délais prévus par cette dernière. Les Etats membres adoptent, dans la mesure où ils ne l'ont pas encore fait, des règles détaillées en vue de protéger contre des abus les personnes qui sont incapables de donner leur consentement éclairé.
2. Un essai clinique ne peut être entrepris que si, notamment:
a) les risques et inconvénients prévisibles ont été pesés au regard du bénéfice attendu pour le sujet participant à l'essai et pour d'autres patients actuels et futurs. Un essai clinique ne peut commencer que si le comité d'éthique et/ou l'autorité compétente conclut que les bénéfices attendus sur le plan thérapeutique et en matière de santé publique justifient les risques et ne peut se poursuivre que si le respect de cette exigence est constamment surveillé ;
b) le sujet participant à l'essai ou, lorsque cette personne n'est pas en mesure de donner son consentement éclairé, son représentant légal a eu la possibilité, par le biais d'un entretien préalable avec l'investigateur ou un membre de l'équipe d'investigation, de comprendre les objectifs de l'essai, ses risques et ses inconvénients, ainsi que les conditions dans lesquelles il sera réalisé, et a, en outre, été informé de son droit de se retirer des essais à tout moment ;
c) sont garantis le droit du participant au respect de son intégrité physique et mentale tout comme le droit du participant à la vie privée, ainsi qu'à la protection des données le concernant selon les modalités prévues par la directive 95/46/CE ;
d) le sujet participant à l'essai ou, lorsque cette personne n'est pas en mesure de donner son consentement éclairé, son représentant légal a donné son consentement écrit après avoir été informé de la nature, de la portée, des conséquences et des risques de l'essai clinique; si la personne concernée n'est pas en mesure d'écrire, elle peut, dans des cas exceptionnels prévus par la législation nationale, donner son consentement oral en présence d'au moins un témoin ;
e) le participant peut, à tout moment et sans qu'il n'encoure aucun préjudice de ce fait, se retirer de l'essai clinique du fait de la révocation de son consentement éclairé.
f) il existe des dispositions relatives à l'assurance ou à l'indemnité couvrant la responsabilité de l'investigateur et du promoteur.
3. Les soins médicaux dispensés aux participants et les décisions médicales prises à leur égard sont de la responsabilité d'un médecin dûment qualifié ou, le cas échéant, d'un dentiste qualifié.
4. Le participant dispose d'un point de contact, auprès duquel il peut obtenir de plus amples informations.
Article 4
Essais cliniques sur les mineurs
Outre toute autre restriction pertinente, un essai clinique sur des mineurs ne peut être entrepris que si:
a) le consentement éclairé des parents ou du représentant légal a été obtenu; ce consentement doit exprimer la volonté présumée du mineur et peut être annulé à tout moment sans que ce dernier en pâtisse;
b) le mineur a reçu des informations, en fonction de sa capacité de compréhension, de la part d'un personnel pédagogiquement qualifié, au sujet de l'essai, des risques et des bénéfices ;
c) le souhait explicite d'un mineur, capable de se former une opinion et d'évaluer ces informations, de refuser de participer à l'essai clinique ou d'en être retiré à tout moment est examiné par l'investigateur ou, le cas échéant, l'investigateur principal ;
d) aucun encouragement ni avantage financier n'est accordé hormis des compensations ;
e) certains avantages directs résultant de l'essai clinique sont obtenus pour le groupe de patients, et seulement dans le cas où cette recherche est essentielle pour valider des données obtenues dans des essais cliniques sur des personnes capables de donner leur consentement éclairé ou par d'autres méthodes de recherche; en outre, cette recherche doit soit se rapporter directement à une condition clinique dont le mineur concerné souffre, soit être telle qu'elle ne puisse être conduite que sur des mineurs ;
f) les orientations scientifiques correspondantes de l'Agence ont été suivies ;
g) les essais cliniques ont été conçus pour minimiser la douleur, les désagréments, la peur et tout autre risque prévisible lié à la maladie et au niveau de développement; le seuil de risque et le degré d'atteinte doivent être expressément définis et constamment réexaminés ;
h) le protocole a été adopté par un comité d'éthique doté de compétences en pédiatrie, ou après consultation sur des problèmes cliniques, éthiques et psychosociaux liés à la pédiatrie ;
i) les intérêts du patient priment toujours ceux de la science et de la société.
Article 5
Essais cliniques sur les incapables majeurs non en mesure
de donner
leur consentement éclairé légal
Toutes les exigences pertinentes énumérées pour les personnes capables de donner leur consentement éclairé légal s'appliquent à d'autres personnes qui ne sont pas en mesure de donner un tel consentement. Outre ces exigences, la participation à un essai clinique des incapables majeurs qui n'ont pas donné ou pas refusé de donner leur consentement éclairé avant le début de leur incapacité n'est possible que si :
a) le consentement éclairé du représentant légal a été obtenu; ce consentement doit exprimer la volonté présumée du patient et peut être annulé à tout moment sans que ce dernier en pâtisse ;
b) la personne qui n'est pas en mesure de donner un consentement éclairé légal a reçu des informations, en fonction de sa capacité de compréhension, au sujet de l'essai, des risques et des bénéfices ;
c) le souhait explicite d'un sujet, capable de se former une opinion et d'évaluer ces informations, de refuser de participer à l'essai clinique ou d'en être retiré à tout moment est examiné par l'investigateur ou, le cas échéant, l'investigateur principal ;
d) aucun encouragement ni avantage financier n'est accordé hormis des compensations ;
e) cette recherche est essentielle pour valider des données obtenues dans des essais cliniques sur des personnes capables de donner leur consentement éclairé ou par d'autres méthodes de recherche et elle se rapporte directement à une condition clinique mettant la vie en danger, ou débilitante dont souffre l'incapable majeur concerné ;
f) les essais cliniques ont été conçus pour minimiser la douleur, les désagréments, la peur et tout autre risque prévisible lié à la maladie et au niveau de développement; le seuil de risque et le degré d'atteinte sont expressément définis et constamment réexaminés ;
g) le protocole a été adopté par un comité d'éthique doté de compétences quant à la maladie et à la population concernées, ou après consultation sur des problèmes cliniques, éthiques et psychosociaux liés à la maladie et à la population concernées ;
h) les intérêts du patient priment toujours ceux de la science et de la société ;
i) il existe un espoir justifié que l'administration du médicament à tester offre un bénéfice plus grand que le risque pour le patient concerné ou ne présente aucun risque.
Article 6
Comité d'éthique
1. En vue de la mise en oeuvre des essais cliniques, les Etats membres prennent les mesures nécessaires à la mise en place et au fonctionnement de comités d'éthique.
2. Le comité d'éthique est tenu d'émettre son avis avant le commencement de tout essai clinique au sujet duquel il a été sollicité.
3. Le comité d'éthique formule son avis en prenant en compte, notamment, les éléments suivants :
a) la pertinence de l'essai clinique et de sa conception ;
b) le caractère satisfaisant de l'évaluation des bénéfices et des risques attendus, telle que prévue à l'article 3, paragraphe 2, point a), et le bien-fondé des conclusions ;
c) le protocole ;
d) l'aptitude de l'investigateur et de ses collaborateurs ;
e) la brochure pour l'investigateur ;
f) la qualité des installations ;
g) l'adéquation et l'exhaustivité des informations écrites à fournir ainsi que la procédure à suivre pour obtenir le consentement éclairé, et la justification de la recherche sur des personnes incapables de donner leur consentement éclairé en ce qui concerne les restrictions spécifiques visées à l'article 3 ;
h) les dispositions prévues en vue de la réparation ou de l'indemnisation en cas de dommages ou de décès imputables à l'essai clinique ;
i) toutes assurances ou indemnités couvrant la responsabilité de l'investigateur et du promoteur ;
j) les montants et les modalités de rétribution ou d'indemnisation éventuelles des investigateurs et des participants à l'essai clinique et les éléments pertinents de tout contrat prévu entre le promoteur et le site ;
k) les modalités de recrutement des participants.
4. Nonobstant les dispositions du présent article, un Etat membre peut décider de charger l'autorité compétente qu'il a désignée aux fins de l'article 9 d'examiner les éléments visés au paragraphe 3, points h), i) et j), du présent article et d'émettre un avis à ce sujet.
Lorsqu'un Etat membre se prévaut de la présente disposition, il en informe la Commission, les autres Etats membres et l'agence.
5. Le comité d'éthique dispose d'un délai maximum de 60 jours à compter de la date de la réception de la demande en bonne et due forme, pour communiquer son avis motivé au demandeur ainsi qu'à l'autorité compétente de l'Etat membre concerné.
6. Pendant la période d'examen de la demande d'avis, le comité d'éthique ne peut formuler qu'une seule demande de renseignements en complément des informations déjà fournies par le demandeur. Le délai prévu au paragraphe 5 est suspendu jusqu'à la réception des renseignements complémentaires.
7. Aucune prolongation du délai de 60 jours visé au paragraphe 5 ne peut être accordée sauf s'il s'agit d'essais impliquant les médicaments de thérapie génique et de thérapie cellulaire somatique et tous les médicaments contenant des organismes génétiquement modifiés. Dans ce cas une prolongation maximale de 30 jours peut être accordée. Pour ces produits, cette période de 90 jours peut être prolongée de 90 jours supplémentaires en cas de consultation d'un groupe ou d'un comité conformément aux réglementations et procédures de l'Etat membre concerné. Il n'existe pas de limitation de la durée du délai d'autorisation pour la thérapie cellulaire xénogénique.
Article 7
Avis unique
Pour les essais cliniques multicentriques limités au territoire d'un seul Etat membre, les Etats membres définissent une procédure prévoyant, nonobstant le nombre de comités d'éthique, la formulation d'un avis unique pour cet Etat membre.
Dans le cas d'essais cliniques multicentriques effectués dans plusieurs Etats membres à la fois, il y a autant d'avis uniques que d'Etats membres concernés par cet essai clinique.
Article 8
Indications détaillées
La Commission, en consultation avec les Etats membres et les parties concernées, formule et publie des indications détaillées concernant la présentation de la demande et les documents à fournir pour solliciter l'avis du comité d'éthique, en particulier en ce qui concerne les informations communiquées aux participants, ainsi que les garanties appropriées pour assurer la protection des données personnelles.
Article 9
Commencement d'un essai clinique
1. Les Etats membres prennent les mesures nécessaires pour que le commencement d'un essai clinique s'effectue suivant la procédure prévue au présent article.
Le promoteur ne peut commencer un essai clinique qu'après délivrance d'un avis favorable de la part du comité d'éthique et pour autant que l'autorité compétente de l'Etat membre concerné n'ait pas signifié au promoteur d'objections motivées. Les procédures visant à la prise de ces décisions peuvent ou non se dérouler en parallèle selon ce que souhaite le promoteur.
2. Avant le commencement de tout essai clinique, le promoteur est tenu de présenter à l'autorité compétente de l'Etat membre dans lequel il envisage de conduire un essai clinique une demande d'autorisation en bonne et due forme.
3. Si l'autorité compétente de l'Etat membre signifie au promoteur qu'elle a des objections motivées, le promoteur peut, une fois et une seule, modifier le contenu de la demande visée au paragraphe 2 afin de prendre en compte les objections qui lui ont été signifiées. Si le promoteur ne modifie pas en conséquence ladite demande, cette dernière est alors considérée comme rejetée et l'essai clinique ne peut pas commencer.
4. L'examen d'une demande d'autorisation en bonne et due forme par l'autorité compétente visée au paragraphe 2 est achevé le plus rapidement possible et ne dépasse pas 60 jours. Les Etats membres peuvent, dans leur domaine de compétence, fixer un délai inférieur à 60 jours, si cela est conforme à la pratique habituelle. L'autorité compétente peut néanmoins notifier au promoteur, avant la fin de cette période, qu'elle n'a pas de motif de ne pas accepter.
Aucune nouvelle prolongation du délai visé au premier alinéa ne peut être accordée sauf s'il s'agit d'essais impliquant les médicaments énumérés au paragraphe 6, pour lesquels une prolongation maximale de 30 jours est accordée. Pour ces produits, cette période de 90 jours peut être prolongée de 90 jours supplémentaires en cas de consultation d'un groupe ou d'un comité conformément aux réglementations et procédures de l'Etat membre concerné. Il n'existe pas de limitation de la durée du délai d'autorisation pour la thérapie cellulaire xénogénique.
5. Sans préjudice du paragraphe 6, peuvent toutefois être soumis à une autorisation écrite préalable à leur commencement, les essais cliniques des médicaments qui n'ont pas d'autorisation de mise sur le marché au sens de la directive 65/65/CEE et qui sont visés à la partie A de l'annexe du règlement (CEE) n° 2309/93 ainsi que des autres médicaments répondant à des caractéristiques particulières, tels que les médicaments dont l'(les) ingrédient(s) actif(s) est(sont) un(des) produit(s) biologique(s) d'origine humaine ou animale ou contient (contiennent) des composants biologiques d'origine humaine ou animale, ou dont la fabrication nécessite de tels composants.
6. Sont soumis à une autorisation écrite préalable à leur commencement, les essais cliniques impliquant les médicaments de thérapie génique, de thérapie cellulaire somatique, y compris de thérapie cellulaire xénogénique, ainsi que tous les médicaments contenant des organismes génétiquement modifiés. Aucun essai thérapeutique génique aboutissant à des modifications de l'identité génétique du participant ne peut être conduit.
7. Cette autorisation est délivrée sans préjudice de l'application éventuelle des directives 90/219/CEE du Conseil du 23 avril 1990 relative à l'utilisation confinée de micro-organismes génétiquement modifiés 1 et 90/220/CEE du Conseil du 23 avril 1990 relative à la dissémination volontaire d'organismes génétiquement modifiés dans l'environnement.
8. En consultation avec les Etats membres, la Commission formule et publie des indications détaillées concernant :
a) la présentation et le contenu de la demande visée au paragraphe 2, ainsi que les documents à fournir à l'appui de cette demande, portant sur la qualité et la fabrication du médicament expérimental, les essais toxicologiques et pharmacologiques, le protocole et les informations cliniques relatives au médicament expérimental, notamment la brochure pour l'investigateur ;
b) la présentation et le contenu de la proposition de modification visée à l'article 10, point a), relative aux modifications substantielles apportées au protocole ;
c) la déclaration de fin de l'essai clinique.
Article 10
Conduite d'un essai clinique
La conduite d'un essai clinique peut être modifiée selon les modalités suivantes :
a) après le commencement de l'essai clinique, le promoteur peut apporter des modifications au protocole. Lorsque ces modifications sont substantielles et de nature à avoir des incidences sur la sécurité des participants ou à changer l'interprétation des pièces scientifiques qui viennent appuyer le déroulement de l'essai, ou si elles sont significatives de quelque autre point de vue que ce soit, le promoteur notifie les raisons et le contenu de ces modifications aux autorités compétentes du ou des Etats membres concernés et en informe le ou les comités d'éthique concernés conformément aux articles 6 et 9.
Sur la base des éléments visés à l'article 6, paragraphe 3, et conformément à l'article 7, le comité d'éthique rend un avis dans un délai maximum de 35 jours à compter de la date de la réception de la proposition de modification en bonne et due forme. Si cet avis n'est pas favorable, le promoteur ne peut pas mettre en oeuvre la modification du protocole.
Si l'avis du comité d'éthique est favorable, et si les autorités compétentes des Etats membres n'ont pas émis d'objections motivées à l'encontre de ces modifications substantielles, le promoteur poursuit la conduite de l'essai clinique en suivant le protocole modifié. Dans le cas contraire, soit le promoteur tient compte de ces objections et adapte, en conséquence, la modification envisagée du protocole, soit il retire sa proposition de modification,
b) sans préjudice du point a), et selon les circonstances, notamment la survenue de tout fait nouveau concernant le déroulement de l'essai ou le développement du médicament expérimental lorsque ce fait nouveau est susceptible de porter atteinte à la sécurité des participants de l'essai, le promoteur ainsi que l'investigateur prennent les mesures urgentes de sécurité appropriées afin de protéger les participants contre un danger immédiat. Le promoteur informe sans délai les autorités compétentes de ces faits nouveaux et des mesures prises et s'assure que le comité d'éthique est informé simultanément,
c) dans un délai de 90 jours suivant la fin d'un essai clinique, le promoteur avise les autorités compétentes du ou des Etats membres concernés ainsi que le comité d'éthique, que l'essai clinique est terminé. Lorsque l'arrêt de l'essai clinique doit être anticipé, ce délai est ramené à 15 jours et les raisons qui le motivent clairement exposées.
Article 11
Echange d'informations
1. Les Etats membres, sur le territoire desquels l'essai clinique a lieu, introduisent dans une base européenne de données accessible uniquement aux autorités compétentes des Etats membres, à l'agence et à la Commission :
a) des données extraites de la demande d'autorisation visée à l'article 9, paragraphe 2 ;
b) d'éventuelles modifications apportées à cette demande, conformément à l'article 9, paragraphe 3 ;
c) d'éventuelles modifications apportées au protocole, conformément à l'article 10, point a) ;
d) l'avis favorable du comité d'éthique ;
e) la déclaration de fin de l'essai clinique ;
f) la mention des inspections réalisées sur la conformité aux bonnes pratiques cliniques.
2. À la demande justifiée d'un Etat membre, de l'agence ou de la Commission, l'autorité compétente à laquelle a été adressée la demande d'autorisation fournit tous les renseignements complémentaires autres que ceux déjà introduits dans la base européenne de données concernant l'essai clinique en question.
3. En consultation avec les Etats membres, la Commission formule et publie des indications détaillées concernant les données à introduire dans cette base européenne de données dont elle assure le fonctionnement avec le concours de l'agence, ainsi que les méthodes à utiliser pour l'échange, par voie électronique, de ces données. Ces indications détaillées sont élaborées dans le strict respect de la confidentialité des données.
Article 12
Suspension de l'essai ou infractions
1. Si un Etat membre a des raisons objectives de considérer que les conditions de la demande d'autorisation visée à l'article 9, paragraphe 2, ne sont plus réunies ou s'il détient des informations qui suscitent des doutes quant à la sécurité ou au bien-fondé scientifique de l'essai clinique, cet Etat membre peut procéder à la suspension ou à l'interdiction de l'essai clinique en question qu'il signifie au promoteur.
Avant de prendre une décision, l'Etat membre, sauf en cas de risque imminent, demande l'avis du promoteur et/ou de l'investigateur; cet avis doit lui être notifié dans un délai d'une semaine.
Dans ce cas, l'autorité compétente concernée informe immédiatement les autres autorités compétentes, le comité d'éthique concerné, l'agence ainsi que la Commission de sa décision de suspension ou d'interdiction et des raisons qui l'ont motivée.
2. Si une autorité compétente a des raisons objectives de considérer que le promoteur ou l'investigateur ou tout autre intervenant dans l'essai ne répond plus aux obligations qui lui incombent, elle l'en informe immédiatement et lui expose le plan d'action qu'il doit mettre en oeuvre pour remédier à cet état de fait. L'autorité compétente concernée informe immédiatement le comité d'éthique, les autres autorités compétentes et la Commission de ce plan.
Article 13
Fabrication et importation des médicaments expérimentaux
1. Les Etats membres prennent toutes les mesures appropriées pour que la fabrication et l'importation de médicaments expérimentaux soient soumises à la possession d'une autorisation. En vue d'obtenir cette autorisation, le demandeur, de même qu'ultérieurement le titulaire, devront satisfaire à des exigences au moins équivalentes à celles qui seront définies conformément à la procédure visée à l'article 21, paragraphe 2.
2. Les Etats membres prennent toutes les dispositions utiles pour que le titulaire de l'autorisation visée au paragraphe 1 dispose d'une façon permanente et continue d'au moins une personne qualifiée, responsable notamment de l'exécution des obligations spécifiées au paragraphe 3 du présent article, répondant aux conditions prévues à l'article 23 de la deuxième directive 75/319/CEE du Conseil du 20 mai 1975 concernant le rapprochement des dispositions législatives, réglementaires et administratives relatives aux spécialités pharmaceutiques.
3. Les Etats membres prennent toutes les dispositions utiles pour que la personne qualifiée visée à l'article 21 de la directive 75/319/CEE, sans préjudice de ses relations avec le fabricant ou l'importateur, ait la responsabilité, dans le cadre des procédures visées à l'article 25 de ladite directive, de veiller :
a) dans le cas de médicaments expérimentaux fabriqués dans l'Etat membre concerné, que chaque lot de médicament a été fabriqué et contrôlé conformément aux exigences de la directive 91/356/CEE de la Commission du 13 juin 1991 établissant les principes et lignes directrices de bonnes pratiques de fabrication pour les médicaments à usage humain, au dossier de spécification du produit et à l'information notifiée conformément à l'article 9, paragraphe 2, de la présente directive ;
b) dans le cas de médicaments expérimentaux fabriqués dans un pays tiers, que chaque lot de fabrication a été fabriqué et contrôlé selon des normes de bonnes pratiques de fabrication au moins équivalentes à celles prévues par la directive 91/356/CEE de la Commission, conformément au dossier de spécification du produit et que chaque lot de fabrication a été contrôlé conformément à l'information notifiée conformément à l'article 9, paragraphe 2, de la présente directive ;
c) dans le cas d'un médicament expérimental qui est un médicament de comparaison en provenance de pays tiers et ayant une autorisation de mise sur le marché, lorsque la documentation attestant que chaque lot de fabrication a été fabriqué selon des normes de bonnes pratiques de fabrication au moins équivalentes à celles précitées ne peut être obtenue, que chaque lot de fabrication a fait l'objet de toutes les analyses, essais ou vérifications pertinents et nécessaires pour confirmer sa qualité conformément à l'information notifiée conformément à l'article 9, paragraphe 2, de la présente directive.
Les indications détaillées concernant les éléments à prendre en compte lors de l'évaluation des produits en vue de la libération des lots dans la Communauté sont élaborées selon les lignes directrices de bonnes pratiques de fabrication, et notamment de leur annexe 13. Ces indications seront adoptées conformément à la procédure visée à l'article 21, paragraphe 2, de la présente directive et publiées conformément à l'article19 bis de la directive 75/319/CEE.
Si les points a), b) ou c) sont respectés, les médicaments expérimentaux sont dispensés des contrôles ultérieurs lorsqu'ils sont importés dans un autre Etat membre accompagnés des certificats de libération des lots signés par la personne qualifiée.
4. Dans tous les cas, la personne qualifiée doit attester dans un registre ou un document équivalent que chaque lot de fabrication répond aux dispositions du présent article. Ledit registre ou document équivalent doit être tenu à jour au fur et à mesure des opérations effectuées et mis à la disposition des agents de l'autorité compétente pendant une période spécifiée par les dispositions des Etats membres concernés. Cette période ne sera, en tout état de cause, pas inférieure à 5 ans.
5. Tout personne qui, à la date de mise en application de la présente directive, exerce dans l'Etat membre où elle se trouve, les activités de la personne qualifiée visée à l'article 21 de la directive 75/319/CEE, en ce qui concerne les médicaments expérimentaux, mais sans toutefois remplir les conditions prévues à ses articles 23 et 24, est autorisée à poursuivre ces activités dans l'Etat membre concerné.
Article 14
Etiquetage
Les renseignements devant figurer, au moins dans la ou les langues officielles de l'Etat membre, sur l'emballage extérieur des médicaments expérimentaux ou, à défaut d'emballage extérieur, sur le conditionnement primaire, sont publiés par la Commission dans le guide des bonnes pratiques de fabrication des médicaments expérimentaux, adopté conformément à l'article 19 bis de la directive 75/319/CEE.
En outre, le guide arrête des dispositions appropriées sur l'étiquetage des médicaments expérimentaux destinés à des essais cliniques ayant les caractéristiques suivantes :
- la conception de l'essai ne requiert pas de fabrication ou de conditionnement particuliers ;
- l'essai est conduit avec des médicaments bénéficiant, dans les Etats membres concernés par l'étude, d'une autorisation de mise sur le marché au sens de la directive 65/65/CEE, et fabriqués ou importés conformément aux dispositions de la directive 75/319/CEE ;
- les patients participant à l'essai présentent les mêmes caractéristiques que ceux qui sont couverts par l'indication mentionnée dans l'autorisation précitée.
Article 15
Vérification de la conformité avec les bonnes
pratiques cliniques et
de fabrication des médicaments
expérimentaux
1. En vue de vérifier le respect des dispositions relatives aux bonnes pratiques cliniques et aux bonnes pratiques de fabrication, les Etats membres désignent à cet effet des inspecteurs chargés de procéder à l'inspection des lieux concernés par la conduite d'un essai clinique, en particulier: le ou les sites où se déroule l'essai clinique, le site de fabrication du médicament expérimental, tout laboratoire d'analyses utilisé pour l'essai clinique et/ou les locaux du promoteur.
Les inspections sont diligentées par l'autorité compétente de l'Etat membre concerné qui en informe l'agence; elles sont effectuées au nom de la Communauté et leurs résultats sont reconnus par tous les autres Etats membres. La coordination de ces inspections est assurée par l'agence, dans le cadre de ses compétences prévues au règlement (CEE) n° 2309/93. Un Etat membre peut à ce sujet demander assistance à un autre Etat membre.
2. À la suite de l'inspection, un rapport d'inspection est établi. Ce rapport doit être tenu à la disposition du promoteur tout en sauvegardant les aspects confidentiels. Il peut être mis à la disposition des autres Etats membres, du comité d'éthique ainsi que de l'agence sur demande motivée.
3. La Commission peut, sur demande de l'agence, dans le cadre de ses compétences prévues au règlement (CEE) n° 2309/93, ou d'un Etat membre concerné, et après consultation des Etats membres concernés, demander une nouvelle inspection si la vérification de la conformité avec la présente directive fait apparaître des différences d'un Etat membre à l'autre.
4. Sous réserve des accords qui ont pu être passés entre la Communauté et des pays tiers, la Commission, sur demande motivée d'un Etat membre ou de sa propre initiative, ou un Etat membre, peuvent proposer une inspection sur le site d'essai et/ou dans les locaux du promoteur et/ou chez le fabricant établis dans un pays tiers. Cette inspection est effectuée par des inspecteurs dûment qualifiés de la Communauté.
5. Les lignes directrices détaillées concernant la documentation se rapportant à l'essai clinique, qui constitue le dossier permanent de l'essai, les méthodes d'archivage, la qualification des inspecteurs et les procédures d'inspections destinées à vérifier la conformité de l'essai clinique en question avec la présente directive sont adoptées et révisées conformément à la procédure visée à l'article 21, paragraphe 2.
Article 16
Notification des événements indésirables
1. L'investigateur notifie immédiatement au promoteur tous les événements indésirables graves, à l'exception de ceux qui sont recensés dans le protocole ou dans la brochure de l'investigateur comme ne nécessitant pas une notification immédiate. La notification immédiate est suivie de rapports écrits détaillés. Dans cette notification comme dans les rapports ultérieurs, les participants sont identifiés par un numéro de code.
2. Les événements indésirables et/ou les résultats d'analyse anormaux définis dans le protocole comme déterminants pour les évaluations de la sécurité sont notifiés au promoteur, conformément aux exigences de notification et dans les délais spécifiés dans le protocole.
3. En cas de décès notifié d'un participant, l'investigateur communique au promoteur et au comité d'éthique tous les renseignements complémentaires demandés.
4. Le promoteur tient des registres détaillés de tous les événements indésirables qui lui sont notifiés par le ou les investigateurs. Ces registres sont remis aux Etats membres sur le territoire desquels l'essai clinique est conduit, à leur demande.
Article 17
Notification des effets indésirables graves
1. a) Le promoteur s'assure que toutes les informations importantes concernant les suspicions d'effets indésirables graves inattendus ayant entraîné ou pouvant entraîner la mort sont enregistrées et notifiées le plus rapidement possible aux autorités compétentes de tous les Etats membres concernés, ainsi qu'au comité d'éthique, en tout état de cause, dans un délai maximum de 7 jours à compter du moment où le promoteur a eu connaissance de ce cas, et que des informations pertinentes concernant les suites soient ensuite communiquées dans un nouveau délai de 8 jours.
b) Toutes les suspicions d'autres effets indésirables graves inattendus sont notifiées aux autorités compétentes concernées, ainsi qu'au comité d'éthique concerné le plus rapidement possible, mais au plus tard dans un délai maximum de 15 jours à compter du jour où le promoteur en a eu connaissance pour la première fois.
c) Chaque Etat membre s'assure que toutes les suspicions d'effets indésirables graves inattendus d'un médicament expérimental qui ont été portées à sa connaissance sont enregistrées.
d) Le promoteur informe également les autres investigateurs.
2. Une fois par an pendant toute la durée de l'essai clinique, le promoteur fournit aux Etats membres sur le territoire desquels l'essai clinique est conduit et au comité d'éthique une liste de toutes les suspicions d'effets indésirables graves survenus au cours de cette durée, ainsi qu'un rapport concernant la sécurité des participants.
3. a) Chaque Etat membre veille à ce que toutes les suspicions d'effets indésirables graves inattendus d'un médicament expérimental qui ont été portées à sa connaissance soient immédiatement introduites dans une banque européenne de données accessible uniquement, conformément à l'article 11, paragraphe 1, aux autorités compétentes des Etats membres, à l'Agence et à la Commission.
b) L'information notifiée par le promoteur est mise à la disposition des autorités compétentes des Etats membres par l'agence.
Article 18
Indications concernant les rapports
La Commission, en consultation avec l'agence, les Etats membres et les parties concernées, formule et publie des indications détaillées concernant l'établissement, la vérification et la présentation des rapports sur les événements/effets indésirables, ainsi que les modalités de décodage concernant les effets indésirables graves inattendus.
Article 19
Dispositions générales
La présente directive ne préjuge pas de la responsabilité civile et pénale du promoteur ou de l'investigateur. À cette fin, le promoteur ou un représentant légal du promoteur doit être établi dans la Communauté.
Les médicaments expérimentaux et, le cas échéant, les dispositifs utilisés pour les administrer sont fournis gratuitement par le promoteur, à moins que les Etats membres n'aient fixé des conditions précises applicables dans des cas exceptionnels.
Les Etats membres informent la Commission des conditions qu'ils auraient fixées.
Article 20
Adaptation au progrès scientifique et technique
La présente directive est adaptée au progrès scientifique et technique conformément à la procédure visée à l'article 21, paragraphe 2.
Article 21
Comité
1. La Commission est assistée par le comité permanent des médicaments à usage humain, ci-après dénommé " comité ", institué à l'article 2 ter de la directive 75/318/CEE.
2. Dans les cas où il est fait référence au présent paragraphe, les articles 5 et 7 de la décision 1999/468/CE s'appliquent, dans le respect des dispositions de l'article 8 de celle-ci.
La période prévue à l'article 5, paragraphe 6, de la décision 1999/468/CE est fixée à trois mois.
3. Le comité adopte son règlement intérieur.
Article 22
Mise en application
1. Les Etats membres adoptent et publient avant le ... * les dispositions législatives, réglementaires et administratives nécessaires pour se conformer à la présente directive. Ils en informent immédiatement la Commission.
Ils appliquent ces dispositions au plus tard à partir du ... **
Lorsque les Etats membres arrêtent ces dispositions, celles-ci contiennent une référence à la présente directive ou sont accompagnées d'une telle référence lors de leur publication officielle. Les modalités de cette référence sont arrêtées par les Etats membres.
2. Les Etats membres communiquent à la Commission le texte des dispositions de droit interne qu'ils adoptent dans le domaine régi par la présente directive.
Article 23
Entrée en vigueur
La présente directive entre en vigueur le jour de sa publication au Journal officiel des Communautés européennes.
Article 24
Destinataires
Les Etats membres sont destinataires de la présente directive.
|
LA PROTECTION DES PERSONNES SE
PRÊTANT
Le rôle des comités : un bilan et des propositions Les comités consultatifs de protection des personnes dans la recherche biomédicale, " pierre angulaire " de la loi du 20 décembre 1988, sont chargés d'émettre un avis sur les protocoles de recherche biomédicale sur l'être humain. A la veille de la révision bioéthique, la commission des Affaires sociales a souhaité disposer d'un bilan précis de l'activité de ces comités et des difficultés qu'ils peuvent rencontrer dans l'exercice de leur mission. Pour mener à bien la mission qui lui a été confiée en mai 2000, M. Claude Huriet a interrogé l'ensemble des comités consultatifs et a procédé à un grand nombre d'auditions. Les enseignements qu'il tire de ce travail le conduisent à formuler une dizaine de propositions pour renforcer le rôle des comités et clarifier leur mode de fonctionnement. |
* 1 Les dispositions de l'article 309 de l'ancien code pénal correspondent aux articles 222-11 à 222-13 du nouveau code pénal, tandis que celles de l'article 318 correspondent à l'article 222-15 du nouveau code pénal.
* 2 Voir à cet égard le rapport n° 307 du Sénat (1993-1994) fait au nom de la commission des Affaires sociales sur la proposition de loi de MM. Claude Huriet et Franck Sérusclat tendant à réformer la loi n° 88-1138 du 20 décembre 1988 modifiée (par la loi n° 90-86 du 23 janvier 1990 et la loi n° 91-73 du 18 janvier 1991), relative à la protection des personnes qui se prêtent à des recherches biomédicales, M. Claude Huriet, rapporteur, p. 11 et suivantes.
* 3 Cf. M. Funck-Brentano " Difficultés d'application des lois encadrant la recherche clinique en France ", La lettre du pharmacologue - volume 12 - n° 2 - Février 1998.
* 4 Voir annexe n° 12
* 5 Voir liste en annexe n° 4.
* 6 Voir travaux de la commission avant les annexes.
* 7 Une première fois par la loi n° 90-86 du 23 janvier 1990 portant diverses dispositions relatives à la sécurité sociale et à la santé. Une seconde fois par la loi n° 94-630 du 25 juillet 1994 modifiant le livre II bis du code de la santé publique relatif à la protection des personnes qui se prêtent à des recherches biomédicales.
* 8 Les références du présent rapport renvoient au code de la santé publique en vigueur au 15 juin 2000. Figure en annexe une table de concordance avec les articles du nouveau code de la santé publique publié par ordonnance n° 2000-548 en date du 15 juin 2000. Cette ordonnance a fait l'objet d'un projet de loi de ratification n° 461 (1999-2000) déposé sur le bureau du Sénat le 13 juillet 2000 qui n'a pas encore été examiné par le Parlement.
* 9 Selon les termes de l'article L. 209-1 du code de la santé publique.
* 10 Selon les termes de l'article L. 209-1 du code de la santé publique.
* 11 Hormis le cas où cette admission a pour objet même l'engagement d'une recherche.
* 12 Selon les termes de l'article L. 209-7 du code de la santé publique.
* 13 Voir annexe n° 4.
* 14 Selon l'article L. 209-11 du code de la santé publique.
* 15 " La vie en questions : pour une éthique biomédicale " par M. Jean-François Mattei, député en mission, rapport au Premier ministre, La documentation française, collection des rapports officiels, 1994.
* 16 Idem, p. 64.
* 17 Idem, p. 67.
* 18 C'est-à-dire pour les produits mentionnés à l'article L. 793-1 du code de la santé publique. Dans tous les autres cas, l'autorité compétente demeure le ministère chargé de la Santé.
* 19 Voir rapport n° 307 du Sénat (1993-1994) fait au nom de la commission des Affaires sociales sur la proposition de loi de MM. Claude Huriet et Franck Sérusclat tendant à réformer la loi n° 88-1138 du 20 décembre 1988 modifiée (par la loi n° 90-86 du 23 janvier 1990 et la loi n° 91-73 du 18 janvier 1991), relative à la protection des personnes qui se prêtent à des recherches biomédicales, M. Claude Huriet, rapporteur.
* 20 Voir les travaux de la commission avant les annexes.
* 21 Le nombre des comités a été ramené de 59 en 1992 à 48 en 2000, voir annexe n° 4.
* 22 Voir annexe n° 2.
* 23 Voir la liste et le compte rendu des auditions en annexe n° 3.
* 24 Voir le texte de ces questions écrites restées sans réponse en annexe n° 1.
* 25 Voir le III-D ci-après ainsi que les annexes n° 8 et n° 9.
* 26 Proposition AN n° 2841 présentée par M. Bernard Accoyer, député.
* 27 Voir le détail du nombre d'avis rendus pour chaque comité en annexe n° 5.
* 28 Voir ci-après D-2. une procédure peu transparente d'affectation des moyens budgétaires.
* 29 Les réponses reçues ne sont pas suffisamment précises pour distinguer entre les institutions, les associations et les personnes physiques.
* 30 Ces informations reposent sur les réponses selon les cas de 35, 36 ou 42 CCPPRB. Celles provenant des comités restants ne sont pas suffisamment précises pour être exploitées.
* 31 " La vie en questions : pour une éthique biomédicale " par M. Jean-François Mattei, député en mission, rapport au Premier ministre, La documentation française, collection des rapports officiels, 1994, p. 58 et p. 131.
* 32 Idem, p. 58
* 33 Le dernier alinéa de l'article L. 209-3 du code de la santé publique prévoit ainsi que : " dans les sciences du comportement humain, une personne qualifiée, conjointement avec l'investigateur, peut exercer la direction de la recherche ".
* 34 Voir rapport n° 307 du Sénat (1993-1994) fait au nom de la commission des Affaires sociales sur la proposition de loi de MM. Claude Huriet et Franck Sérusclat tendant à réformer la loi n° 88-1138 du 20 décembre 1988 modifiée (par la loi n° 90-86 du 23 janvier 1990 et la loi n° 91-73 du 18 janvier 1991), relative à la protection des personnes qui se prêtent à des recherches biomédicales, M. Claude Huriet, rapporteur, p. 26.
* 35 Cf. ci-après.
* 36 Les guides de l'AP-HP, " Loi du 20 décembre 1988 modifiée sur la protection des personnes qui se prêtent aux recherches biomédicales ", 1997.
* 37 Voir à cet égard l'annexe n° 12 qui présente le texte de la directive européenne relative aux essais cliniques adoptée mais non encore publiée au Journal officiel des communautés européennes.
* 38 85 enquêtes sur place en Ile-de-France en 2000 ayant nécessité 115 jours de travail (équivalent mi-temps d'un inspecteur de santé publique).
* 39 C'est-à-dire " les médicaments ou produits soumis à l'essai et les éventuels médicaments de référence ou produits de référence ou placebos ".
* 40 Voir rapport n° 307 du Sénat (1993-1994) fait au nom de la commission des Affaires sociales sur la proposition de loi de MM. Claude Huriet et Franck Sérusclat tendant à réformer la loi n° 88-1138 du 20 décembre 1988 modifiée (par la loi n° 90-86 du 23 janvier 1990 et la loi n° 91-73 du 18 janvier 1991), relative à la protection des personnes qui se prêtent à des recherches biomédicales, M. Claude Huriet, rapporteur, p. 25.
* 41 JO Questions Sénat du 1 er juillet 1993, p. 1062 .
* 42 JO Questions Sénat du 29 avril 1993, p. 719 .
* 43 Rapport de l'IGAS n° 93-121, " L'expérimentation sur l'Homme : la loi du 20 décembre 1988 modifiée dite loi Huriet ", tome I, p. 272.
* 44 Voir à cet égard le II-A-1-a) du présent rapport .
* 45 Voir annexe n° 7 relative à une représentation graphique des taux de présence par catégorie de membres.
* 46 Le premier alinéa de l'article R. 2013 du code de la santé publique prévoit que " les fonctions de membre d'un comité consultatif de protection des personnes dans la recherche biomédicale sont gratuites. Les frais, notamment de déplacement, supportés par un membre à l'occasion de sa participation aux travaux du comité lui sont remboursés sur justification ".
* 47 L'article R. 2014 prévoit que " les membres suppléants peuvent assister aux séances du comité sans prendre part aux délibérations lorsque le membre titulaire qu'ils suppléent est présent ".
* 48 Le dernier alinéa de l'article R. 2013 dispose qu'" au-delà de trois absences consécutives, non justifiées, d'un membre titulaire aux séances du comité, le préfet de région peut mettre fin au mandat de ce membre ".
* 49 Voir le 2 a) ci-dessus.
* 50 Guide pratique de l'Assistance Publique - Hôpitaux de Paris, " loi du 20 décembre 1988 modifiée sur la protection des personnes qui se prêtent aux recherches biomédicales ", 1997, p. 17.
* 51 Les procédures internes à l'AP-HP pour l'examen des protocoles telles qu'elles sont présentées ici sont reprises du guide pratique sur la loi du 20 décembre 1988 publié par l'AP-HP en 1997 (p. 51 et suivantes).
* 52 Voir en annexe n° 4 la liste des agréments et des retraits d'agrément des CCPPRB par la DGS.
* 53 Selon les termes du premier alinéa de l'article L. 209-12 du code de la santé publique.
* 54 Selon les termes du deuxième alinéa de l'article L. 209-12 du code de la santé publique .
* 55 Voir le II-B-2-b) du présent rapport.
* 56 Voir en annexes 8 et 9 l'ancien et le nouvel organigrammes de la DGS.
* 57 Le chef de bureau entré en fonction en janvier a démissionné début mars après avoir estimé qu'il ne disposait pas des moyens nécessaires à l'accomplissement de sa mission.
* 58 Rapport n° 307 du Sénat (1993-1994) fait au nom de la commission des Affaires sociales sur la proposition de loi de MM. Claude Huriet et Franck Sérusclat tendant à réformer la loi n° 88-1138 du 20 décembre 1988 modifiée (par la loi n° 90-86 du 23 janvier 1990 et la loi n° 91-73 du 18 janvier 1991), relative à la protection des personnes qui se prêtent à des recherches biomédicales, M. Claude Huriet, rapporteur, p. 48.
* 59 Cf. III-B-2b) de ce rapport.
* 60 Voir notamment sur ce sujet l'annexe n° 1 relative aux questions écrites adressées au Gouvernement depuis 1996 et restées sans réponse.
* 61 Le deuxième alinéa de l'article 19 de l'ordonnance n° 59-2 du 2 janvier 1959 portant loi organique relative aux lois de finances dispose que " les fonds versés par des personnes morales ou physiques pour concourir avec ceux de l'Etat à des dépenses d'intérêt public, ainsi que les produits de legs et donations attribués à l'Etat ou à diverses administrations publiques, sont directement portés en recettes au budget. Un crédit supplémentaire de même montant est ouvert par arrêté du ministre des Finances au ministre intéressé. L'emploi des fonds doit être conforme à l'intention de la partie versante ou du donateur. Des décrets pris sur le rapport du ministre des Finances peuvent assimiler le produit de certaines recettes de caractère non fiscal à des fonds de concours pour dépenses d'intérêt public. "
* 62 Voir l'annexe n° 6 qui présente la répartition du produit des droits fixes effectuée par la DGS en 2000.
* 63 Réponse écrite de la DGS du 18 janvier 2001 au questionnaire adressé par votre rapporteur.
* 64 Réponse à votre rapporteur en date du 30 mars 2001.
* 65 Réponse à votre rapporteur en date du 3 avril 2001.
* 66 Réponse précitée du 30 mars 2001.
* 67 Ainsi, les crédits disponibles pour 1999 s'élevaient (colonne 3 du tableau) à 21.625.061 francs. Les dotations versées (colonne 4) se sont élevées à 8.645.000 francs. Les crédits reportés (colonne 3) en 2000 apparaissent pour 13.230.994 francs alors qu'ils devraient s'élever à 12.980.081 francs (21.625.061 francs - 8.645.000 francs), soit une différence de 250.913 francs.
* 68 Réponse précitée en date du 3 avril 2001.
* 69 Votre rapporteur n'a en tout cas reçu aucune indication de la DGS lui permettant de penser qu'il existait une base juridique déterminant les modalités détaillées de calcul des dotations.
* 70 L'annexe n° 6 détaille la répartition de cette enveloppe par région et par comité.
* 71 Dans les autres cas, l'autorité administrative compétente est le ministère chargé de la Santé.
* 72 AFSSAPS ou ministère chargé de la Santé, selon qu'il s'agit ou non des produits mentionnés à l'article L. 793-1 du code de la santé publique.
* 73 Voir à cet égard le III-A-2-b) du présent rapport et l'annexe n° 12 qui présente le texte de cette directive.
* 74 Rapport de l'IGAS n° 93-121, " L'expérimentation sur l'Homme : la loi du 20 décembre 1988, modifiée dite loi Huriet ", tome I, p. 272.
* 75 Voir à cet égard le III-1-b) du présent rapport.
* 76 L'article L. 209-11 dispose que " les frais de fonctionnement des comités sont financés par le produit d'un droit fixe versé par les promoteurs pour chacun des projets de recherche biomédicales faisant l'objet d'une demande d'avis. Le montant de ce droit est arrêté par le ministre chargé de la santé ".
* 77 On peut toutefois rappeler que la proposition faite au IV. A. 4 de mieux réguler le nombre d'amendements grâce à la création d'un droit fixe nécessiterait, quant à elle, la modification de ce même article L. 209-11.
* 78 Voir à cet égard le III C 1 a) du présent rapport.
* 79 Cet article prévoit dans son cinquième alinéa que " les comités sont composés de manière à garantir leur indépendance et la diversité des compétences dans le domaine biomédical et à l'égard des questions éthiques, sociales, psychologiques et juridiques ".
* 80 Voir ci-après le C-1).
* 81 Voir à cet égard le III-D-2) du présent rapport.
* 82 L'INSERM reçoit en moyenne 50 à 60 nouveaux projets par an, ce qui représente un stock de 120 dossiers en cours auxquels il faut ajouter 40 à 50 amendements.
* 83 85 enquêtes sur place en Ile-de-France en 2000 ayant nécessité 115 jours de travail (équivalent mi-temps d'un inspecteur de santé publique).
* 84 La présente directive adoptée par le Parlement européen et le Conseil le 26 février 2001 n'ayant pas été encore publiée au Journal officiel des communautés européennes, le texte reproduit ci-après correspond aux dispositions adoptées par le Conseil à l'issue de l'examen de la proposition de directive par le Parlement européen.