







Rapport d'information n° 490 (2011-2012) de MM. Claude BIRRAUX, député et Jean-Louis TOURAINE, député, fait au nom de l'Office parlementaire d'évaluation des choix scientifiques et technologiques, déposé le 28 mars 2012
Disponible au format PDF (1,1 Moctet)
N° 4484 N° 490
____ ___
ASSEMBLÉE NATIONALE SÉNAT
CONSTITUTION DU 4 OCTOBRE 1958
TREIZIÈME LÉGISLATURE SESSION ORDINAIRE DE 2011 - 2012
____________________________________ ___________________________
Enregistré à la présidence de l'Assemblée nationale Enregistré à la présidence du Sénat
le 28 mars 2012 le 28 mars 2012
________________________
OFFICE PARLEMENTAIRE D'ÉVALUATION
DES CHOIX SCIENTIFIQUES ET TECHNOLOGIQUES
________________________
RAPPORT
sur
LES MALADIES MONOGÉNIQUES : ÉTAT DES LIEUX
Compte rendu de l'audition publique du 7 juin 2011
et de la présentation des conclusions, le 25 janvier 2012
Par MM. Claude Birraux et Jean-Louis Touraine,
Députés.
__________ __________
Déposé sur le Bureau de l'Assemblée nationale Déposé sur le Bureau du Sénat
par M. Claude BIRRAUX, par M. Bruno SIDO,
Premier Vice-Président de l'Office Président de l'Office
_________________________________________________________________________
INTRODUCTION
M. le président Claude Birraux. Mon collègue Jean-Louis Touraine et moi-même sommes heureux de vous accueillir, au nom de l'Office parlementaire d'évaluation des choix scientifiques et technologiques, pour évoquer les enjeux des maladies monogéniques.
L'OPECST, créé en 1983 et seul organe commun à l'Assemblée nationale et au Sénat, a pour mission de permettre au Parlement d'évaluer, en toute indépendance, les enjeux stratégiques et sociaux des avancées scientifiques et technologiques. Il réunit dix-huit députés et dix-huit sénateurs, désignés à la proportionnelle par les groupes politiques des deux assemblées. Ces trente-six parlementaires, aidés par un conseil scientifique, s'appuient sur une méthode rigoureuse qui garantit la qualité de leurs travaux. Pour réaliser une étude, l'Office doit être saisi par un organe interne au Parlement : commission permanente, groupe politique ou Bureau de l'une des deux assemblées.
S'il est un domaine où les progrès permis par la science sont assez évidents, c'est bien celui de la santé. Néanmoins des événements tragiques et inacceptables, comme l'affaire du Mediator, nous rappellent qu'un dialogue permanent entre science et société est indispensable. L'Office y participe. Ce dialogue a d'abord lieu à travers son conseil scientifique, lequel réunit vingt-quatre scientifiques reconnus dans leurs disciplines respectives. Par ailleurs, les membres de l'Office vont régulièrement à la rencontre des chercheurs dans les laboratoires et instituts de recherche. Ainsi, depuis plusieurs années, un partenariat avec l'Académie des sciences permet, en associant jeunes chercheurs et parlementaires, d'améliorer la compréhension entre scientifiques et politiques, et de mieux prendre en compte les préoccupations des uns et des autres. Enfin, chacun des travaux de l'Office est l'occasion d'échanges, à travers notamment les auditions et les visites organisées par les rapporteurs. Avant l'été, vont ainsi se conclure des travaux sur la coopération scientifique dans les terres australes ou bien encore la biodiversité en Méditerranée. Sont par ailleurs en cours des travaux sur la biologie de synthèse, les neurosciences, l'innovation à l'épreuve des peurs et des risques, la sûreté nucléaire et l'avenir de la filière nucléaire.
J'en viens au thème de l'audition d'aujourd'hui. La commission des affaires sociales de l'Assemblée nationale, par l'intermédiaire de son président Pierre Méhaignerie, nous a chargés en octobre 2010 d'une étude sur la drépanocytose, saisine que l'Office a élargie à l'ensemble des maladies monogéniques.
Ce faisant, l'Office prolonge les débats qu'il a lancés depuis longtemps sur les sciences du vivant et la bioéthique. Depuis les premières lois de bioéthique, il est chargé d'en réaliser l'évaluation et de formuler des recommandations. L'audition d'aujourd'hui prend place dans le cycle de nos travaux sur les biotechnologies et la bioéthique.
Contrairement à la plupart des maladies monogéniques, la drépanocytose n'est pas une maladie rare puisque l'on estime à plusieurs dizaines de millions le nombre de malades de par le monde, et qu'on dénombre cinq cents nouveaux cas par an en France, ce qui en fait dans notre pays la première maladie monogénique par le nombre de malades atteints. Cette forte prévalence s'explique par le fait que le gène muté responsable de la maladie protège des formes sévères de malaria, notamment de ses atteintes neurologiques.
Les maladies monogéniques représentent un enjeu important pour nos sociétés. Elles sont très invalidantes, parfois mortelles. Elles touchent tous les champs de la vie des malades et altèrent souvent profondément leur qualité de vie. Leur prise en charge est difficile, vu que leur incidence est au final faible par rapport à d'autres maladies. Nous avons souhaité dans cette audition publique en aborder tous les aspects : aussi avons-nous invité des spécialistes de domaines très divers.
Nous traiterons d'abord de la recherche et de la prise en charge des patients, puis de la place des associations de malades et du financement de la recherche, avant d'évoquer des questions juridiques, notamment celle de la protection et de l'utilisation des données de santé.
Accessible sur le portail Internet de l'Office, où pourra dès ce soir être consultée sa vidéo et où sera disponible dans quelque temps son compte rendu, cette audition doit éclairer le Parlement. Elle donnera lieu à la publication d'un rapport qui reprendra intégralement le contenu de nos débats, et sera complété des conclusions que Jean Louis Touraine et moi-même présenterons devant nos collègues de l'OPECST, pour fixer les grandes lignes d'analyse et ouvrir des perspectives sur les leçons à tirer et les mesures à prendre.
M. Jean-Louis Touraine. Notre réunion d'aujourd'hui a ceci d'original qu'elle donne l'occasion à des spécialistes de domaines très divers de se rencontrer, d'échanger et de confronter leur expérience et leurs compétences dans le domaine des maladies monogéniques. Une telle rencontre n'est pas si fréquente car les aspects scientifiques et médicaux sont souvent traités indépendamment des aspects sociétaux ou juridiques. Une approche pluridisciplinaire présente pourtant un intérêt tout particulier pour la prise en charge globale des malades.
Cette audition publique arrive à point nommé, alors que le projet de loi relatif à la bioéthique vient tout juste d'être examiné en deuxième lecture par l'Assemblée nationale. Je me félicite que ce texte permette une meilleure prise en compte de toutes les dimensions de la génétique, garantisse une meilleure protection dans l'examen des caractéristiques génétiques de la personne, qui sera désormais plus strictement encadré. La définition du consentement des personnes a été précisée, de même que les conditions de l'information de la parentèle en cas de découverte d'une anomalie génétique grave. Je regrette en revanche la méfiance infondée qui a prévalu, conduisant l'Assemblée nationale à refuser, pour la recherche sur les cellules souches embryonnaires, le régime d'autorisation encadrée en faveur duquel s'étaient pourtant prononcés le Conseil d'État et le Comité consultatif national d'éthique, et qu'avait voté le Sénat. Restera donc en vigueur le régime actuel d'interdiction, assorti de dérogations pour les projets ayant reçu l'aval de l'Agence de la biomédecine.
Sur le sujet qui nous occupe aujourd'hui, nous pouvons être relativement optimistes. Le séquençage génétique à haut débit est très prometteur et conduira tout naturellement à l'identification de nouveaux gènes responsables de maladies encore mal connues. Nos connaissances théoriques progresseront et de nouvelles thérapeutiques, reposant sur une médecine personnalisée, pourront voir le jour. La prise en charge des patients pourra être adaptée en fonction de leur « carte génétique ». Celle-ci permettra par exemple de savoir si tel traitement a ou non une chance d'être efficace, d'en prévoir les éventuels effets secondaires, et donc de mieux les anticiper. Aura alors vécu l'approche empirique où ce n'était qu'essai thérapeutique après essai thérapeutique que pouvait être mis au point un traitement adapté à chaque malade. L'utilisation, certes courante depuis déjà un certain temps, des puces à ADN permet d'explorer les différents gènes et d'identifier leur responsabilité dans telle ou telle maladie. Demain, une chirurgie du gène permettra d'envisager une véritable médecine « à la carte ».
Plus il devient facile de séquencer le génome complet d'une personne et moins cela coûte cher, plus il importe de protéger ces données. Une connaissance plus fine des prédispositions génétiques a nécessairement des conséquences éthiques, juridiques et sociétales, dont il importe de prendre la mesure. Nous avons le devoir d'éviter que de telles données soient utilisées à des fins autres que médicales et scientifiques. Cela est d'autant plus délicat que la mondialisation de la biologie rend quasiment impossible de s'assurer que ce qui est strictement réglementé, voire interdit, dans notre pays, ne sera pas pratiqué sous l'empire d'une réglementation beaucoup plus lâche, voire autorisé sans restrictions, dans d'autres pays - pour un coût souvent modique. Que faire avec les pays qui autorisent plus librement que nous l'accès aux données génétiques ?
Les progrès médicaux ont permis à des personnes porteuses de certains gènes qui les auraient auparavant condamnées dès l'enfance, de vivre plus longtemps, et donc d'avoir des enfants, ce qui avait fait dire à Sir Peter Medawar, futur prix Nobel de médecine, que l'efficacité à laquelle était parvenue la médecine dans la seconde moitié du 20 ème siècle aboutirait mécaniquement à augmenter la fréquence des gènes responsables de certaines maladies. Loin d'être pessimiste, dès les années soixante, il soulignait que ce même progrès permettrait un jour de les corriger. Nous y sommes. Les espoirs sont grands. Des craintes aussi se font jour. Il nous appartient de rassurer nos concitoyens en encadrant les pratiques médicales et de recherche.
Dans le cas des maladies monogéniques, la seule approche médico-scientifique ne suffit pas. En effet, celles-ci ne concernent pas seulement la personne qu'elles affectent. Elles ont des conséquences pour tout l'entourage, en particulier la famille avec de multiples implications en matière de conseil génétique mais aussi de prise en charge et d'accompagnement, beaucoup d'entre elles se développant dès l'enfance. Notre société doit aider à cette prise en charge. Les associations de malades ont là tout leur rôle à jouer.
Si chacune de ces maladies est par elle-même rare, leur très grand nombre fait qu'au total, leur occurrence est importante : les maladies rares, toutes formes comprises, sont aussi fréquentes que le cancer. Une réflexion pluridisciplinaire n'en est que plus indispensable, de sorte que ces maladies monogéniques, d'une extrême diversité, ne soient pas chacune l'affaire d'un seul spécialiste mais que toutes les compétences puissent s'additionner afin de faire progresser la science, en encadrer les avancées, et surtout apporter une dimension humaine à la prise en charge des malades.
Je souhaite enfin m'élever en faux contre certains propos que je qualifierais d'obscurantistes. Sans nul doute par ignorance, certains considèrent comme de l'eugénisme le diagnostic pré-implantatoire (DPI) et le diagnostic prénatal (DPN) qui peuvent être pratiqués dans le cas de maladies génétiques d'une extrême gravité, la découverte de la présence du gène défectueux autorisant une interruption médicale de grossesse. Si la recherche de tels gènes devait être tenue pour de l'eugénisme, il devrait logiquement en être de même de la règle qui veut qu'on dissuade les mariages consanguins, qui n'a elle aussi d'autre finalité que d'éviter l'apparition de maladies ! S'opposer au DPN, c'est aussi oublier qu'il peut déboucher sur des traitements in utero . Dès 1988, nous procédions aux premières greffes de cellules souches sur des foetus atteints de déficit immunitaire, et l'année suivante sur des malades atteints de bêta-thalassémie. D'autres thérapies existent également qui peuvent être utilisées chez le foetus dès les tout premiers stades de son développement si le diagnostic a été assez précoce. La recherche des gènes causes de maladies graves n'a aucune visée eugéniste.
En dépit des bénéfices incontestables qu'apportent tous ces progrès, on se méfie encore trop souvent de la recherche. Il nous faut à la fois prévoir un encadrement propre à rassurer nos concitoyens et faire oeuvre de pédagogie pour que l'ensemble de la société française retrouve confiance en le progrès.
PREMIÈRE TABLE RONDE : LES MALADIES MONOGÉNIQUES ET LEUR PRISE EN CHARGE
LES AVANCÉES SCIENTIFIQUES ET MÉDICALES
M. Jean-Louis Touraine. Je donne immédiatement la parole au Professeur Alain Fischer, qui a développé les premières thérapies géniques dans notre pays.
M. Alain Fischer, professeur d'immunologie pédiatrique à l'université Paris Descartes, membre de l'Académie des sciences, directeur de l'Institut hospitalo-universitaire Imagine. Merci de nous donner l'occasion de traiter ainsi, d'une manière plus large que strictement médicale et scientifique, des maladies monogéniques dites rares.
Il ne serait pas illogique de s'interroger sur l'intérêt pour la nation d'investir dans la recherche sur des maladies rares. En réalité, vous l'avez dit, bien que chacune d'entre elles soit rare, ces maladies constituent un réel problème de santé publique, puisqu'elles touchent au total des dizaines de milliers de personnes.
Leur médecine est difficile puisqu'elle embrasse toutes les spécialités, parfois plusieurs en même temps, et tous les âges de la vie. Beaucoup de maladies génétiques se révèlent dès l'enfance mais certaines n'apparaissent qu'à l'âge adulte, et d'autres, qui se sont déclenchées tôt dans la vie, deviennent chroniques et exigent une prise en charge particulière. Elles engagent souvent le pronostic vital ou sont source de handicaps lourds qui retentissent gravement sur la vie de l'entourage des malades.
La rareté de chacune de ces maladies fait que les connaissances sur chacune sont limitées - très variables selon les cas. On ne peut pas exiger de chaque praticien qu'il les connaisse toutes, alors même que pour certaines d'entre elles, il existe des traitements qui, à défaut de guérir, peuvent au moins stabiliser ou améliorer l'état des malades. La phase « d'errance diagnostique », qui peut durer des années, est douloureuse pour les malades et leurs familles. C'est un véritable problème qui soulève celui de l'information médicale, de l'organisation des filières de soins...
Je le dis à dessein de manière quelque peu provocatrice : les maladies rares sont les plus faciles à étudier. En effet, elles ont en gros un facteur déclenchant unique en la mutation d'un gène. « Un gène, une maladie » - même si la réalité est un peu plus complexe - voilà qui est plus simple que les mécanismes de déclenchement et de développement de maladies multifactorielles comme le cancer, l'asthme ou les maladies neuro-dégénératives, bien difficiles à élucider.
Les malades atteints d'une maladie monogénique sont d'une certaine façon des exemples naturels de ce à quoi on aboutit en laboratoire quand on active ou inactive l'expression d'un gène chez un animal pour étudier le phénotype qui en résulte. Ici, à l'inverse, on identifie un gène à partir d'un phénotype. Les maladies rares ont ainsi largement contribué au développement des connaissances fondamentales.
On commence à penser que certaines maladies fréquentes résultent peut-être de la conjonction de myriades de maladies rares. Ainsi la maladie de Crohn, dont la fréquence augmente, est-elle certainement liée à des facteurs environnementaux mais on ignore si l'élément déterminant n'est pas d'origine génétique, multigénique ou non. On se pose la même question pour certains retards mentaux chez l'enfant, dont on connaît peu de chose et pour lesquels il n'existe pas de traitement. Le séquençage à haut débit apportera sans doute une réponse. L'étude de maladies rares peut servir à celle de maladies beaucoup moins rares.
Ces maladies, parce qu'elles constituent en quelque sorte des « idéals-type », peuvent aussi servir de modèle pour le développement de nouvelles thérapeutiques, y compris de maladies fréquentes. C'est l'une des premières fois où on peut mettre au point une thérapeutique à partir de la physiopathologie et non par une approche empirique : on définit des cibles, des molécules et des voies de substitution ; on peut aussi chercher à modifier l'expression des gènes, stabiliser les ARN messagers, des protéines... Un très grand nombre de pistes thérapeutiques, ouvertes par la recherche sur les maladies rares, sont aujourd'hui en train d'être explorées. C'est aussi le premier champ où se sont développées la thérapie cellulaire et la thérapie génique.
Les maladies rares sont également le domaine par excellence où utiliser les cellules souches embryonnaires. A ce sujet, je regrette moi aussi que la révision des lois de bioéthique n'ait pas été l'occasion de modifier le statut de la recherche sur ces cellules. L'argument parfois avancé selon lequel les cellules pluripotentes induites (iPS) permettraient de se passer des cellules souches embryonnaires est fallacieux. On peut entendre des objections éthiques ou religieuses, mais pas cet argument pseudo-scientifique, totalement inexact. Nous avons impérativement besoin de la recherche sur les cellules souches embryonnaires et j'espère qu'un jour, en France elle sera autorisée de manière encadrée, comme elle l'est dans de très nombreux pays développés.
La recherche sur les maladies rares n'est pas vouée à demeurer marginale sur le plan économique. Il existe plusieurs exemples de réussites industrielles autour de médicaments liés à ces maladies. Je pense à Genzyme, société de biotechnologies, qui, en développant avec succès des molécules substitutives pour quelques maladies rares, a connu un succès économique qui lui a valu d'être rachetée par Sanofi. D'autres grands groupes pharmaceutiques comme Novartis ou Roche ont eux aussi compris l'intérêt d'investir dans les maladies rares, qu'il s'agisse de trouver un traitement ou parce que l'étude des mécanismes d'une maladie rare peut déboucher sur le traitement de maladies plus fréquentes.
Je n'en prendrai qu'un exemple dans ma spécialité, celui d'une pathologie auto-inflammatoire gravissime de l'enfant, extrêmement rare, décrite dans les années 70 et dont le gène a été identifié au début des années 2000. Dès que l'on a compris qu'elle était due à la production excessive d'interleukine-1, a très vite été disponible sur le marché un inhibiteur de cette cytokine. L'administration de cette protéine a, du jour au lendemain, transformé la vie de malades qui n'étaient guère plus d'une cinquantaine dans le monde. Mais de là, les laboratoires se sont demandé si l'interleukine-1 ne jouait pas un rôle dans toute une série de maladies inflammatoires immunologiques comme le psoriasis, certaines formes de rhumatismes, la maladie de Crohn, l'uvéite... Et aujourd'hui Novartis et d'autres développent, sous forme d'anticorps monoclonaux ou de protéines recombinantes, des traitements du psoriasis ou de l'uvéite, qui ne sont pas des maladies rares. Cet exemple montre l'intérêt de la recherche sur ces maladies pour les malades eux-mêmes et pour l'industrie. Des sociétés de biotechnologies développent des produits au départ ciblés sur des maladies rares en espérant que leur utilisation puisse un jour s'élargir.
Avant d'identifier le gène responsable d'une maladie, a fortiori de comprendre les mécanismes physiopathologiques de celle-ci, il faut disposer de cohortes. Il faut donc des lieux disposant de l'expertise médicale, du plateau technique et de l'environnement intellectuel permettant d'évaluer dans les meilleures conditions les patients atteints de maladies rares. Même si beaucoup reste à faire, la création des centres de référence maladies rares a facilité la tâche. Dès lors que l'on a dans chaque centre des médecins ayant une connaissance approfondie de la maladie en question et que les malades ont réellement accès à ces centres, sans discrimination financière - ce qui n'est pas toujours le cas : j'ai encore en mémoire le cas d'un malade dont la Sécurité sociale refusait de rembourser le déplacement jusqu'à un centre -, il est possible de regrouper une expertise autour d'un phénotype donné et de constituer des cohortes de patients pour lesquelles seront collectées des informations cliniques, biologiques, d'imagerie et maintenant génétiques, de plus en plus sophistiquées. Cette annotation de cohortes est un long travail de fourmi qui exige d'importants moyens humains et financiers. Il faut aussi que le personnel ait été correctement formé à ce recueil de données. L'importance de ce travail, indispensable tant pour soigner le mieux possible les malades en l'état des connaissances qu'identifier de nouveaux gènes, est souvent sous-estimée. Les moyens actuels de la génomique permettent d'aller beaucoup plus loin et beaucoup plus vite. La France n'est, hélas, pas leader en ce domaine, largement distancée par les Etats-Unis, où tout a débuté, et par la Chine qui développe elle aussi maintenant des outils très performants. Nous ne faisons que suivre. Sans doute n'était-ce pas la meilleure solution que de se lancer dans la génomique en France à travers la constitution de gros centres, certes utiles mais pas bien adaptés à ce type de recherches qui n'ont pas besoin d'être faites partout mais plutôt dans certains centres pointus. Inutile de dire que derrière doit suivre aussi la biologie des protéines, etc.
Croiser leurs connaissances est extrêmement fructueux pour les chercheurs. Ainsi une pathologie des structures ciliées des cellules explique certaines maladies du poumon, du rein, du cerveau..., n'ayant a priori rien à voir. De telles découvertes peuvent déboucher sur la mise au point d'outils diagnostiques et thérapeutiques d'intérêt commun. Cela justifie que les centres de référence maladies rares atteignent une certaine taille critique : nous avons cette chance à Necker. Toute une série de pistes peuvent être explorées dans le domaine académique mais aussi industriel. Il faut collaborer avec les industriels.
Le besoin d'organisation est plus important encore que pour la recherche fondamentale : centres de références, cohortes et outils d'exploitation afférents, notamment informatiques, étroite coopération entre médecins, chercheurs et médecins-chercheurs, réseaux de collaboration nationaux et internationaux... Sur ce dernier point, je regrette que l'Union européenne ne joue pas un rôle suffisant : on pourrait réfléchir à une organisation de la recherche sur les maladies rares au niveau de l'Union. Il existe certes quelques projets communs, mais ils demeurent limités et leurs circuits sont trop complexes. Il y a une dizaine d'années, j'ai essayé, en vain, de plaider cette cause auprès de la direction générale « Recherche » à Bruxelles. Rien ou presque n'a avancé depuis. Vu la rareté, l'extrême diversité, la répartition géographique particulière des maladies rares, une structure de recherche au niveau européen serait pourtant indispensable.
M. Jean-Louis Touraine. Je donne maintenant la parole au professeur Nicolas Lévy, dont l'équipe a identifié le gène de la progeria, cette redoutable maladie qui provoque un vieillissement accéléré.
M. Nicolas Lévy, professeur de génétique à la faculté de médecine de Marseille, directeur du groupement d'intérêt scientifique « Maladies rares ». Après avoir dit d'un mot que je partage totalement les vues d'Alain Fischer, je voudrais insister sur la nécessité absolue d'une recherche translationnelle dans le domaine des maladies rares, dont la plupart sont d'origine génétique, avec pour cause un gène facilement identifiable. Le point de départ reste la constitution de cohortes de patients faisant l'objet d'explorations clinico-biologiques, qui permettent un suivi longitudinal et prospectif selon des critères identiques dans tous les centres. Cela suppose le stockage de prélèvements dans des biobanques, des possibilités d'évaluer l'histoire naturelle de ces maladies, d'identifier leurs bases moléculaires grâce à la génomique, l'épigénomique, la transcriptomique et la métabolomique, de disposer de modèles cellulaires ou animaux afin d'en étudier les mécanismes physiopathologiques les plus fins. Ces modèles sont également indispensables aux études précliniques, une fois identifiée une cible thérapeutique potentielle, afin de pouvoir passer aux essais cliniques. Beaucoup de traitements dans ce type de maladies ne peuvent encore prétendre guérir les malades mais améliorent leur qualité de vie et allongent leur espérance de vie, ce qui est toujours intéressant dans l'attente de la mise au point éventuelle de thérapies plus efficaces.
On ne peut aujourd'hui faire l'impasse sur les développements industriels. Chacun sait que les financements publics seront insuffisants pour couvrir l'ensemble des besoins pour tous les malades. D'où la nécessité de partenariats avec l'industrie privée pour pouvoir conduire ces recherches translationnelles dans de bonnes conditions.
La recherche dans le domaine des maladies rares exige une approche pluridisciplinaire. Il faut faire avec le nombre très important de ces affections, leur extrême hétérogénéité - la complexité de la tâche est encore accrue par le fait qu'une même pathologie peut être liée à un déficit d'une multitude de gènes différents : on en connaît aujourd'hui plus de 150 impliqués dans le retard mental par exemple -, la difficulté du recueil des informations phénotypiques et de matériel biologique, la dispersion des informations et l'inégalité des moyens dédiés à chacune de ces maladies.
Un autre problème a longtemps été la faiblesse de l'investissement de l'industrie pharmaceutique dans ce domaine. Ce n'est plus le cas. Les industriels sont beaucoup moins réticents aujourd'hui à financer certaines recherches sur les maladies rares, dans la mesure où il est devenu évident qu'elles permettent de mieux connaître des mécanismes physiopathologiques à l'oeuvre dans des maladies beaucoup plus fréquentes. Tout cela rend indispensable un effort spécifique à long terme et une coordination des actions.
Une Fondation Maladies Rares doit être créée en 2011, dont nous espérons qu'elle permettra de combler certains des manques actuels dans le domaine de la clinique comme de la recherche. Il faut se féliciter qu'un deuxième plan national maladies rares ait été annoncé le 28 février dernier, après qu'un premier plan avait couvert la période 2004-2008.
Ce plan comporte trois axes principaux. Le premier est d'améliorer la qualité de la prise en charge des patients. Cela suppose tout d'abord de faciliter l'accès au diagnostic. Les centres de référence, mis en place dans le cadre du premier plan national, ont été une excellente initiative. La plupart des patients ont ainsi pu trouver un lieu d'expertise sur leur maladie dans un rayon maximal de 100 à 150 km de leur domicile. Les médecins et les chercheurs ont, pour leur part, pu constituer les indispensables cohortes qu'évoquait Alain Fischer. L'objectif est maintenant d'organiser de véritables filières de soins pour mettre au point des protocoles nationaux de diagnostic et de soins, et de parvenir à ce que les différents centres travaillant sur des pathologies semblables coopèrent. Il faut ensuite donner toute leur place à la biologie et aux nouvelles techniques de séquençage du génome. Lorsqu'un patient s'adresse à nous, dont nous ignorons quel gène peut être responsable de sa maladie, nous procédons à des tests gène par gène et il arrive que, même après plusieurs années, nous ne soyons toujours pas parvenus à identifier le gène en cause. Le séquençage à haut débit qui permet de tester simultanément l'ensemble des gènes devrait permettre d'identifier beaucoup plus vite les mutations en cause. Par manque de moyens, la France a pris un retard considérable dans ces techniques, y compris par rapport à l'Allemagne, la Belgique, l'Espagne ou les Pays-Bas, sans même parler des Etats-Unis et de la Chine. En dépit de tous les efforts aujourd'hui consentis avec nos maigres moyens, ce retard ne pourra pas être comblé.
Il faudrait aussi que toutes les structures dédiées aux maladies rares disposent du même système d'information ou du moins que leurs systèmes soient compatibles et donc exploitables conjointement. Il existe certes une base de données, Cemara, regroupant des données issues des centres de référence, mais il existe une multitude d'autres bases de données autres que cliniques, qui, hélas, ne peuvent communiquer avec Cemara. Le projet est donc de créer une banque nationale de données maladies rares, qui regrouperait l'ensemble des données cliniques issues des centres de référence mais aussi l'ensemble des données biologiques et thérapeutiques issues d'autres bases, et d'assurer l'interopérabilité de tous ces outils, de façon que partout il soit possible d'en extraire les données que l'on souhaite. Ce peut être par exemple d'identifier très vite une population de patients immédiatement éligibles à un essai thérapeutique en cours de démarrage ou d'établir une corrélation entre certaines anomalies cliniques et certaines anomalies génétiques. Cette démarche, novatrice, exige des moyens et beaucoup d'énergie.
Un mot du projet Radico, fédération des cohortes de patients atteints de maladies rares. Des moyens du grand emprunt y ont été affectés. Il sera piloté par la future fondation de coopération scientifique. La création de cette fondation est l'un des axes principaux du plan national maladies rares. Il s'agit de créer une structure nationale d'impulsion de la recherche, en interface avec les acteurs publics et privés. Le statut juridique du GIS maladies rares ne permet pas de conclure de partenariats avec le monde industriel. Ce GIS a donc vocation à évoluer en une structure autorisant de tels partenariats et ayant des missions plus larges.
Les objectifs principaux de cette fondation, qui sera probablement hébergée par l'INSERM, seront de structurer et d'harmoniser les recherches sur les maladies rares ; de coordonner et d'articuler les missions du GIS avec celles de la base nationale de données maladies rares et d'Orphanet ; de développer et fédérer les expertises ainsi que les structures existantes ; de financer des projets, notamment ceux qui requièrent l'accès à des plates-formes technologiques de séquençage, de mise au point de modèles, cellulaires ou animaux, de criblage thérapeutique de molécules, de fabrication de cellules pluripotentes induites, d'autant plus nécessaires que la recherche sur les cellules souches embryonnaires demeure en France interdite sauf dérogation.
Cette fondation aura également des objectifs plus ciblés, parmi lesquels le développement de partenariats avec l'industrie. Des industriels ont fait part de leur intérêt pour certains projets. D'autres se sont d'ores et déjà engagés à nos côtés : les conventions sont en cours d'élaboration. Aux financements publics et en provenance du milieu associatif s'ajouteront donc des financements privés.
Cette fondation aura aussi pour mission d'identifier les besoins de recherche dans le domaine des maladies rares, y compris en sciences humaines et sociales, de proposer des mesures incitatives pour que des équipes spécialisées mènent certaines recherches indispensables, par exemple en matière d'épidémiologie, où nous manquons cruellement de données chiffrées. Aucune évaluation du coût sociétal des maladies rares n'a jamais été réalisée. De telles études nous aideront, nous l'espérons, éléments chiffrés à l'appui, à faire comprendre aux pouvoirs publics que les maladies rares représentent, au même titre que le cancer ou la maladie d'Alzheimer, un véritable enjeu de santé publique, contrairement à ce que pourrait laisser penser leur qualification de « rares ». Cette fondation sera également chargée de développer l'information sur ces maladies et les recherches qui y ont trait, ainsi que d'assurer la pérennité des bases de données et des registres. Sur ce dernier point, j'insiste sur le danger qu'il y aurait à financer la constitution des bases de données avec des crédits exceptionnels. Il faut des financements sécurisés à long terme, de façon que les personnels affectés à ces tâches puissent être embauchés en CDI ou appartiennent à des institutions établies, publiques si possible.
La fondation bénéficiera du soutien des organismes de recherche publics déjà engagés dans la recherche sur les maladies rares - INSERM, CNRS, universités et hôpitaux. Elle associera des partenaires privés et associatifs.
Elle doit être un outil pour les associations, lesquelles doivent se reconnaître dans ses missions, pour les chercheurs, pour les médecins et pour les industriels.
M. Jean-Louis Touraine. Le recensement des maladies rares reste très imprécis. Leur nombre oscille dans une fourchette de 5 000 à 8 000. On ne connaît pas non plus pour chacune d'elles le nombre exact de malades atteints. On ne dispose d'estimations que pour les plus fréquentes : on sait ainsi qu'en France quelque 15 000 personnes souffrent de drépanocytose, 5 000 à 6 000 de mucoviscidose et 5 000 de myopathie de Duchenne. Un meilleur recensement serait en effet un préalable indispensable pour progresser.
Nous allons maintenant entendre le professeur Paul-Henri Roméo.
M. Paul-Henri Roméo, directeur de l'Institut de radiobiologie cellulaire et moléculaire du CEA et de l'ITMO IHP (Institut thématique multi-organismes Immunologie, Hématologie et Pneumologie). Je traiterai de la drépanocytose qui, bien que n'étant pas une maladie rare, est délaissée des chercheurs et des pouvoirs publics. Le coût de ce délaissement est considérable pour les patients et pour la société.
Cette maladie touche les globules rouges, cellules parmi les plus simples du corps humain. En effet, ceux-ci n'ont pas de noyau, ni donc d'ADN, et ne contiennent que de l'hémoglobine, molécule qui véhicule l'oxygène depuis les poumons jusqu'à l'ensemble des tissus. C'est une maladie génétique monogénique à transmission autosomique récessive. Bien que la cause en soit connue depuis plus de cinquante ans, quasiment aucun progrès thérapeutique n'a eu lieu depuis lors. Il n'existe aujourd'hui qu'un seul traitement curatif, la greffe de moelle. Les autres traitements disponibles ne sont que palliatifs. En dépit de sa forte prévalence, très peu de recherches sont consacrées à la drépanocytose. Celles de Philippe Leboulch, que nous entendrons tout à l'heure, font exception.
Chez les malades drépanocytaires, une mutation dans le sixième codon du gène codant pour la chaîne bêta de l'hémoglobine fait que celle-ci, en hypoxie, se polymérise de façon irréversible, entraînant une déformation caractéristique des globules rouges - c'est pourquoi elle est dite aussi anémie falciforme - qui les rend rigides et très fragiles. En résulte chez les patients homozygotes une anémie hémolytique, une atteinte vasculaire systémique grave et irréversible pouvant toucher tous les tissus - rein, foie, cerveau... -, des risques infectieux majorés durant les premières années de la vie, diverses complications consécutives à des infarctus tissulaires comme des ostéonécroses, des ulcères de jambe... La maladie réduit l'espérance de vie de trente à quarante ans.
Le gène de la drépanocytose confère aux individus hétérozygotes, porteurs du gène de la maladie sans être eux-mêmes atteints, une résistance à la malaria. La carte des zones du monde où sévit de manière endémique la malaria et celle où se rencontrent majoritairement les personnes atteintes ou vectrices de la drépanocytose, se superposent. En Afrique subsaharienne, 2% des nouveau-nés sont homozygotes pour la mutation responsable de la maladie et 10 à 40% de la population, selon les pays, est hétérozygote. Du fait des migrations, on retrouve des patients drépanocytaires aux Etats-Unis, au Brésil et dans la Caraïbe, en France, en Espagne, en Italie, ainsi qu'au Moyen-Orient, en Arabie saoudite et en Turquie.
La drépanocytose n'est pas une maladie rare. L'OMS en a d'ailleurs fait une priorité de santé publique depuis 2006. C'est la première maladie génétique en France par sa fréquence. Les cas se concentrent principalement en Île-de-France, où on dénombre plus de 5 000 cas, et outre-mer, en particulier en Martinique et en Guadeloupe où l'on en compte plus de 2 500. A l'horizon 2020, quelque 20 000 patients devraient être atteints dans notre pays.
La drépanocytose fait l'objet d'un dépistage néonatal et peut même faire l'objet d'un dépistage prénatal (DPN). Le premier DPN a d'ailleurs été réalisé pour cette maladie. Un programme de dépistage néonatal existe en France depuis 1995, ciblé sur les populations à risque. En métropole, il n'est effectué que lorsque les origines des parents peuvent faire craindre l'apparition de la pathologie. Il est en revanche systématique dans les DOM-TOM. L'incidence de la maladie est d'environ un cas pour 2 000 naissances dans l'Hexagone, mais d'un cas sur 200 à 300 en Guadeloupe, Martinique et Guyane. Les familles acceptent très bien le dépistage. Dans les populations à risque, un conseil génétique est proposé.
Hormis la greffe de moelle et dans l'attente de la mise au point d'une thérapie génique, il n'existe aujourd'hui aucun traitement curatif. Un seul médicament, l'hydroxyurée, permet d'atténuer les symptômes. Il diminue la fréquence et la sévérité des crises vaso-occlusives chez l'adulte et chez l'enfant, ainsi que celles des syndromes thoraciques aigus. L'administration d'hydroxyurée permet de réduire le nombre des hospitalisations et des transfusions. Une étude parue dans le New England Journal of Medicine fait état d'une diminution de 40% de la mortalité dans un groupe de patients traités par cette molécule.
Pour terminer, je voudrais dire un mot du coût de la prise en charge. D'après les données disponibles d'un fichier Medicaid de Floride, il a pu être établi qu'en 2009, la prise en charge d'un patient drépanocytaire revenait aux Etats-Unis à 2 000 dollars par mois environ, tous âges confondus, avec un minimum de 900 dollars entre zéro et neuf ans et un maximum de près de 3 000 dollars entre 30 et 40 ans. Avec un coût annuel moyen de plus de 10 000 dollars pour les enfants, montant jusqu'à près de 35 000 dollars pour les sujets plus âgés, le coût total s'élève, pour une espérance de vie moyenne de 45 ans, à plus de 950 000 dollars par patient. Avec dans notre pays quelque 20 000 patients atteints à l'horizon 2020, comme il est prévu, on voit le coût qui en résultera. Il est donc paradoxal qu'on continue à ne pas investir dans la recherche sur cette maladie.
Au-delà de son coût financier pour notre système de santé, cette maladie est lourde pour la société. La qualité de vie des malades est très fortement dégradée. Beaucoup de soins pourtant indispensables ne sont pas pris en charge par l'assurance maladie. La « productivité sociale » des patients est très affaiblie, de même que celle de leur entourage, notamment des mères qui doivent prodiguer des soins continus à leurs enfants lorsqu'ils sont atteints.
Quatre-vingts pour cent des coûts totaux sont consacrés à l'hospitalisation, 3,2% aux passages aux urgences, 0,9% à la consultation de généralistes, 3,6% aux médicaments, 11,7% à d'autres soins, infirmiers ou médicaux spécialisés. Le traitement palliatif par l'hydroxyurée qui diminue par deux à trois le nombre des hospitalisations, surtout chez les sujets les plus âgés, permet de substantielles économies.
Débat
M. le Président Claude Birraux. Monsieur Lévy, vous avez insisté sur l'importance des données épidémiologiques et la nécessité de conserver sur le long terme diverses données cliniques, biologiques et génétiques à long terme. Y a-t-il des difficultés avec la CNIL ? Si oui, comment les résoudre ? Il ne faudrait pas en prendre prétexte pour ne pas constituer ces fichiers.
Chacun sait que l'épidémiologie a toujours été le parent pauvre de la recherche médicale dans notre pays. La rareté des crédits fait qu'on préfère les affecter à des recherches plus visibles et en apparence plus porteuses. « A quoi bon faire des études épidémiologiques puisqu'au final, on sait bien qu'on ne découvrira pas grand-chose ? », a-t-on souvent entendu. A quoi j'ai toujours répondu que « pas grand-chose » était déjà quelque chose et toujours mieux que l'ignorance totale !
M. Nicolas Lévy. L'équipe de la future fondation de coopération scientifique sur les maladies rares comporte des spécialistes des bases de données. C'est le professeur Paul Landais, actuel responsable de Cemara à l'hôpital Necker, qui sera chargé de la banque nationale de données sur les maladies rares. Il a déjà sécurisé l'ensemble des données de Cemara, qui regroupe à peu près la moitié de celles provenant des centres de référence. Les bases et les fichiers ont été déclarés à la CNIL. D'autres sources de données en-dehors de Cemara ont vocation à alimenter la banque nationale qu'il faut voir comme une sorte d'entrepôt de données remontant de diverses sources, chacune de ces sources pouvant être consultée pour en extraire des informations-clés pour la connaissance des maladies et la constitution de cohortes de patients susceptibles d'être incluses dans des essais cliniques. Tout cela est déclaré à la CNIL et il n'y a quasiment plus de problème dans la mesure où les données sont anonymisées et la confidentialité totalement garantie. En tout cas, ce n'est pas l'obstacle principal.
M. Jean-Louis Touraine. Vous avez insisté, monsieur Fischer, sur l'intérêt que présenterait une mutualisation des moyens à l'échelon européen. Il est très frustrant que les coopérations qui seraient si utiles ne voient pas le jour. Où en est-on exactement aujourd'hui ? Que pourrait-on faire pour renforcer celles qui existent et en encourager de nouvelles ?
M. Alain Fischer. Les situations sont très variées selon les pathologies. Il est des domaines où les équipes, à partir du terrain, se sont fédérées, souvent autour de sociétés savantes ou de groupes de recherche académique. Il est arrivé qu'elles constituent des bases de données communes, mais ce n'est pas le cas le plus fréquent. On travaille encore souvent au seul échelon national. Or, il serait parfaitement imaginable qu'un dispositif analogue à celui que l'on s'apprête à mettre en place en France, et que vient de décrire Nicolas Lévy, soit développé au niveau européen, rattaché à la direction générale « Recherche », avec une organisation et un budget dédiés. Ce serait un formidable pas en avant : le gain serait beaucoup plus que proportionnel au changement d'échelle. Mais les scientifiques peuvent avoir du mal à renoncer à certaines prérogatives qui étaient les leurs à l'échelle régionale ou nationale. Et il est sans doute encore plus difficile pour les autorités européennes de modifier leur façon de travailler...
M. le président Claude Birraux. Monsieur Roméo, comment expliquez-vous que les recherches sur la drépanocytose n'aient que si peu avancé alors que cette maladie n'est pas rare ?
M. Paul-Henri Roméo. Il y a à cela plusieurs raisons. La première est qu'elle affecte une cellule très particulière, difficile à appréhender. Le globule rouge en effet ne vit pas plus de 120 jours dans le sang, si bien que pour corriger de manière pérenne l'anomalie génétique en cause, il faudrait intervenir en amont, sur les cellules souches hématopoïétiques de la moelle osseuse. Une autre raison tient à ce qu'elle a longtemps touché, en France des populations qui ne vivaient pas en métropole. La prise de conscience de sa prévalence et de sa gravité est récente. Seules cinq unités de l'INSERM travaillent actuellement sur la drépanocytose. Ce n'est pas une maladie « à la mode », il est plus porteur aujourd'hui de travailler sur le cancer par exemple. Enfin, beaucoup d'essais se sont soldés par des échecs, si bien que les chercheurs ont délaissé le sujet. Le DPN a marqué une avancée majeure mais des personnes continueront d'être atteintes. Il faut inciter les chercheurs à travailler sur la connaissance et le traitement de cette maladie.
M. le président Claude Birraux. Le plan national maladies rares n'a pas suscité de vocations ?
M. Paul-Henri Roméo. Aucune.
M. le président Claude Birraux. Vu la prévalence de cette maladie, un projet européen, voire international, ne se justifierait-il pas ?
M. Paul-Henri Roméo. Une prise de conscience a eu lieu aux Etats-Unis, où des moyens importants ont été dégagés. Je ne pense pas en revanche qu'en Europe, la drépanocytose intéresse beaucoup d'autres pays que la France. Ce n'y est clairement pas une priorité.
M. Alain Fischer. Il y a tout de même eu des progrès en matière de recherche clinique, notamment après la mise en place des centres de référence. Cela reste insuffisant, mais ce premier pas est encourageant.
M. Paul-Henri Roméo. Tout à fait.
TRAITER ET ACCOMPAGNER LES MALADES : LA DIVERSITÉ DES APPROCHES
M. Jean-Louis Touraine. Je donne maintenant la parole au professeur Philippe Leboulch.
M. Philippe Leboulch, professeur des universités à la faculté de Paris-XI, praticien hospitalier, chef de l'Institut des maladies émergentes et des thérapies innovantes (IMETI) de la direction des sciences du vivant du CEA et d'une unité mixte de recherche INSERM-CEA . Je vais essayer de faire devant vous le point sur les thérapies géniques de ces deux maladies-soeurs que sont la â-thalassémie et la drépanocytose. Ces deux hémoglobinopathies, relativement fréquentes de par le monde, présentent l'intérêt de pouvoir constituer d'intéressants modèles pour la thérapie génique.
Ces maladies sont faussement simples. Bien qu'il s'agisse des premières dans l'histoire de la médecine dont le mécanisme a été identifié au niveau moléculaire, on ne sait pas pour autant les traiter de manière efficace et durable. Beaucoup de temps a été nécessaire pour accomplir les premiers progrès. Ceux-ci s'accélèrent désormais.
Dans notre sang circulent à chaque instant plus de vingt mille milliards de globules rouges. Il n'est pas simple de les traiter quand ils sont, comme dans le cas des hémoglobinopathies, déformés. Si on parvenait à traiter ces maladies par thérapie génique, il y aurait des retombées non seulement pour d'autres maladies rares mais pour l'ensemble de la médecine et de la biologie. L'essai clinique que nous avons mené a permis de mieux comprendre comment les cellules souches hématopoïétiques donnaient naissance à l'ensemble des éléments figurés du sang et conduit à remettre en question les modèles simples qui nous étaient enseignés en faculté de médecine.
La drépanocytose et la â-thalassémie sont surtout répandues en Afrique sub-saharienne et en Asie, sur toute la bande équatoriale et sub-tropicale. Cela tient au fait que le parasite du paludisme, transmis par un moustique, a besoin d'une hémoglobine normale pour pouvoir se reproduire. Les individus hétérozygotes, porteurs sains du gène, se trouvent être résistants aux formes sévères du paludisme, ce qui explique que cette particularité génétique se soit amplifiée dans certaines zones d'Afrique jusqu'à être présente chez 80%, voire 90%, de la population. Les mouvements migratoires expliquent l'essaimage de ces maladies aux Antilles, en Amérique du Nord et en Europe.
On dénombrerait environ 500 000 nouveau-nés par an dans le monde atteints de formes sévères d'hémoglobinopathies. Il n'y en a quasiment pas en France atteints de â-thalassémie grâce à l'efficacité du diagnostic prénatal, mais en Thaïlande, pays de population comparable à la nôtre, on compte environ 3 000 nouveaux cas par an. Les patients aujourd'hui atteints en métropole sont originaires d'Afrique du Nord, du Bassin méditerranéen et d'Asie. On compte en revanche dans notre pays, DOM-TOM inclus, 15 000 patients atteints de drépanocytose, essentiellement antillais ou africains, et ce nombre devrait, avec environ 400 nouvelles naissances d'enfants atteints par an, passer à 20 000 vers 2020. En l'absence de traitement, ces maladies sont mortelles. Il n'existe, hélas, presque que des traitements palliatifs, le seul traitement curatif résidant dans une greffe de moelle osseuse avec donneur compatible.
Ces maladies sont d'origine génétique. Chez les individus malades, chacun des deux chromosomes codant pour la â-globine présente une ou plusieurs mutations - diverses dans le cas de la â-thalassémie, toujours la même dans le cas de la drépanocytose - empêchant le fonctionnement normal des globules rouges. Lorsque chacun des parents possède sur l'un des deux chromosomes concernés le gène muté, si l'enfant n'hérite d'aucun des chromosomes porteurs de la mutation, il est sain et ne risque pas de transmettre la maladie. S'il hérite d'un seul chromosome atteint, celui de son père ou celui de sa mère, il est sain également mais, porteur de la mutation, il peut transmettre la maladie - le seul avantage pour lui est que cela le protège des formes sévères de paludisme. Si par malchance, il hérite de chacun de ses parents du chromosome porteur du gène muté, il est atteint.
La â-globine est l'un des principaux constituants protéiques du tétramère d'hémoglobine, lequel comporte deux molécules de â-globine, une molécule d'á-globine et une molécule de fer. Les globules rouges ne contiennent quasiment rien d'autre que de l'hémoglobine, qui assure tous les échanges gazeux depuis les poumons jusque dans les tissus.
Chez les malades atteints de â-thalassémie, la production de â-globine est insuffisante, que le gène codant pour cette protéine présente une mutation qui l'empêche de s'exprimer ou ait disparu par délétion. En l'absence de traitement, les individus homozygotes, qui ne peuvent fabriquer de globules rouges, meurent dans la petite enfance, alors même qu'ils sont nés normaux. En effet, le foetus est protégé pendant la vie intra-utérine grâce au gène foetal de la globine, dit ã, lequel cesse de s'exprimer dans les deux ou trois premières années de la vie.
Chez les malades atteints de drépanocytose, la production de â-globine est suffisante mais la protéine fabriquée est anormale. Elle polymérise dans les globules rouges qu'elle déforme - ils prennent la forme d'une faucille - et rigidifie, si bien qu'ils adhèrent à la paroi des vaisseaux qui s'obstruent, provoquant d'atroces douleurs, puis des micro-infarctus dans les différents tissus de l'organisme.
Dans la â-thalassémie majeure, on observe un retard de croissance, des problèmes endocriniens, une hypertension pulmonaire, des thromboses veineuses, des atteintes cardiaques, une cirrhose, un diabète, une arthropathie, des déformations osseuses, une hépatosplénomégalie... Bref, tous les organes sont touchés. Non traitée, la maladie entraîne le décès avant l'âge de deux ans. Mal traitée, ce qui est le cas dans beaucoup de pays moins favorisés que le nôtre, elle provoque tous ces symptômes. Et même lorsqu'elle est traitée de la meilleure manière, cela n'empêche pas un décès prématuré, en général entre 30 et 40 ans.
Dans la drépanocytose, les crises vaso-occlusives et hémolytiques détruisent peu à peu les tissus, ce qui, dans les formes sévères, peut raccourcir de beaucoup l'espérance de vie.
Le seul traitement curatif actuellement disponible consiste en une greffe de moelle prélevée chez un donneur compatible, membre de la fratrie possédant les mêmes caractéristiques HLA d'histo-compatibilité. Le taux de succès peut atteindre 80% à long terme si le patient greffé ne développe pas une maladie du greffon contre l'hôte (GVHD), toujours possible même avec un donneur dit compatible. C'est d'ailleurs pourquoi on ne sollicite quasiment jamais de donneurs extra-familiaux, même compatibles. Outre que 75% des patients n'ont pas de donneur compatible, il faut compter avec le risque de réaction du greffon contre l'hôte. Ce traitement est donc loin d'être la panacée.
D'où l'intérêt de la thérapie génique. On prélève des cellules des patients, on y corrige le gène déficient, puis on les leur réinjecte une fois la correction effectuée. Dans la mesure où il s'agit d'une greffe autologue, il n'y a pas de risque de rejet. On réserve pour l'instant cette thérapie, encore balbutiante, aux cas les plus graves et où le patient n'a pas de donneur compatible. Si les succès se confirment, elle pourra être étendue.
On pourrait imaginer de corriger in situ le gène codant de façon erronée pour la â-globine dans les cellules souches hématopoïétiques de la moelle osseuse, capables d'auto-renouvellement. Mais ces cellules sont très difficiles à isoler, à manipuler et à conserver sans qu'elles perdent leurs propriétés spécifiques de cellules souches. Il faudrait en outre s'assurer que le nouveau gène ne s'exprimera bien ultérieurement que dans les globules rouges. On arrive aujourd'hui à corriger in vitro le gène déficient, pas encore in vivo.
L'autre méthode, celle utilisée aujourd'hui, consiste à introduire, grâce à un vecteur, un gène dans les cellules souches hématopoïétiques destiné à s'exprimer dans les globules rouges. Des essais ont déjà eu lieu chez l'animal. J'en profite pour dire que ce serait une folie que de prétendre se passer des expérimentations sur l'animal : le modèle animal demeure indispensable. Les vecteurs qui marchent chez l'animal pour les cellules souches hématopoïétiques sont les lentivirus. Tout l'art de la vectorologie est de ne conserver du rétrovirus que les parties mobilisant le gène jusqu'au chromosome.
Il nous faut mieux comprendre le fonctionnement des cellules souches hématopoïétiques comme des globules rouges pour pouvoir les modifier, savoir comment y insérer un gène, comprendre comment la moelle « corrigée » pourra se substituer à la moelle d'origine, savoir comment fabriquer les gènes-médicaments avec toute la sécurité nécessaire et en juste quantité. Une souris est environ trois mille fois plus petite qu'un homme. Ce qu'on réussit chez elle, il faut être capable de le reproduire en trois mille fois plus grand chez l'homme ! C'est ainsi que pour un seul patient on produit de vingt à quarante litres de vecteur, ensuite concentrés. Les premières souris traitées avec succès par thérapie génique pour des hémoglobinopathies l'ont été en 2000-2001 et le premier patient en 2007. En réalité, contrairement à ce que l'on entend parfois dire, c'est allé très vite pour faire tous les essais nécessaires, prendre toutes les garanties en matière de toxicité, obtenir l'aval des agences sanitaires et fabriquer en grande quantité les vecteurs dans des conditions GMP ( Good manufacturing process) .
Comment cela se passe-t-il en pratique ? On prélève des cellules de moelle osseuse, soit par voie intra-veineuse soit par ponction de l'os sous anesthésie locale. On les mélange pendant quelques heures avec le vecteur, auparavant préparé, congelé puis décongelé. On congèle les cellules puis on les analyse pour s'assurer que tout est normal. Le patient revient plus tard à l'hôpital où lui est administrée une chimiothérapie, beaucoup moins lourde que celle normalement pratiquée lors d'une greffe de moelle, dans la mesure où on n'a pas besoin de déprimer son immunité. Et on lui réinjecte ses propres cellules qui ont été mêlées auparavant au vecteur.
M. le président Claude Birraux. Il faut préparer trente à quarante litres de vecteur pour un seul patient. Mais combien lui en injecte-t-on ?
M. Philippe Leboulch. Quelques dizaines de millilitres seulement. On concentre la préparation jusqu'à n'obtenir plus que cette quantité qu'on mélange avec les cellules.
C'est en juin 2007 que nous avons traité de la sorte un premier patient, après avoir obtenu l'aval de l'Agence française de sécurité sanitaire des produits de santé (AFSSAPS). Il était atteint d'une thalassémie majeure de type â E /â 0 , devait être transfusé tous les mois depuis l'âge de trois ans et prendre des chélateurs du fer pour éliminer la surcharge en fer, inéluctable lorsqu'on est obligé d'être transfusé à un rythme aussi rapproché - jusqu'à l'apparition de formes orales beaucoup mieux tolérées de chélateurs du fer ; ce patient, depuis l'enfance, avait une perfusion avec une pompe branchée dans son abdomen cinq nuits par semaine. Il était arrivé que son taux d'hémoglobine tombe avant transfusion à 4,5 g/dl. Il souffrait d'une hépatomégalie et d'une splénomégalie qui avait nécessité l'ablation de la rate à l'âge de six ans. Les tentatives de traitement à l'hydroxyurée avaient échoué et il n'avait pas de donneur intra-familial compatible qui aurait permis une greffe de moelle. Quatre ans après cette thérapie génique, il n'est pas guéri mais son état s'est très nettement amélioré. Depuis trois ans, il n'a pas eu besoin d'une seule transfusion et son taux d'hémoglobine oscille entre 9 et 10 g/dl. Il travaille à plein temps. Et pour éliminer le fer qui s'est accumulé dans son organisme au cours des années antérieures, il suffit de lui faire régulièrement des saignées, ce qui est beaucoup moins lourd que d'administrer des chélateurs. Ce patient reste suivi de près mais le bénéfice de la thérapie génique a été pour lui considérable.
En conclusion, je voudrais insister sur la nécessité que les politiques et les associations de patients continuent de nous aider.
Même si tout est loin d'être parfait en France et que du retard a été pris dans le domaine de la génomique, notamment par rapport aux Etats-Unis et à la Chine qui ont réalisé des investissements colossaux, notre pays s'est donné les moyens d'être leader en matière de thérapie génique et le demeure aujourd'hui - je pense aux travaux d'Alain Fischer à l'hôpital Necker ou bien encore à ceux de José Sahel sur les pathologies rétiniennes à l'hôpital des Quinze-Vingts. Ces recherches ont besoin de financements pérennes.
On ne peut pas non plus se passer des modèles animaux, y compris avec les grands singes, les seuls vraiment pertinents avant de passer à l'essai clinique chez l'homme.
Enfin, l'AFSSAPS, sans se départir de son indispensable regard critique, doit comme elle l'a fait pour les premiers travaux de thérapie cellulaire et de thérapie génique, accompagner la phase ultérieure du développement de médicaments géniques pour les patients auprès de l'EMA ( European Medicine Agency) .
Il faut aussi nouer des partenariats industriels car il n'est pas envisageable de développer autrement ce type de thérapie jusqu'au stade du médicament. Les petites sociétés de biotechnologies sont le lieu idéal de développement des initiatives aux tous débuts. Les idées, les projets y sont foisonnants mais il faut aider ces sociétés à conclure des partenariats avec les grands laboratoires pharmaceutiques. On a évoqué le rachat de Genzyme par Sanofi. Il faut favoriser ce type d'alliances.
Il pourrait, à première vue, sembler que les traitements classiques de la drépanocytose coûtent moins cher que des thérapies plus innovantes. Il n'en est rien car les malades doivent être soignés durant toute leur vie. Ces thérapies ne sont donc pas réservées aux pays riches. Un malade atteint de la maladie de Gaucher, qui est une mucopolysaccharidose, coûte aux Etats-Unis - le coût est voisin en France - de 250 000 à 800 000 dollars par an pour la seule protéine recombinante qui devra lui être administrée sa vie durant. Si cette maladie pouvait être traitée par thérapie génique, sans effets secondaires majeurs, cela reviendrait au final bien meilleur marché car, même coûteux, le traitement n'aurait à être administré qu'une fois. Si le développement des thérapies géniques a un coût élevé, leur utilisation permettrait ensuite des économies considérables par rapport aux médicaments classiques. En Thaïlande, on dénombre chaque année 3 000 nouveaux cas de â-thalassémie. Depuis plusieurs années, le gouvernement thaïlandais a décidé de prendre en charge le coût des greffes de moelle allogéniques chez les enfants atteints, pour la raison simple que cette technique est au final financièrement plus avantageuse que des transfusions et l'administration de chélateurs du fer à vie.
M. Jean-Louis Touraine. Je donne maintenant la parole à Mme Cécile Martinat qui dirige à I-STEM l'équipe qui travaille sur les maladies du motoneurone.
Mme Cécile Martinat, chargée de recherche à l'INSERM et à l'Institut des cellules souches pour le traitement et l'étude des maladies monogéniques (I-STEM). Je me réjouis de pouvoir vous présenter aujourd'hui une partie de nos travaux sur un thème débattu récemment à l'occasion de la révision des lois de bioéthique, à savoir le potentiel des cellules souches embryonnaires humaines et des cellules pluripotentes induites (iPS) dans le cadre de maladies rares. Je profite de l'occasion pour dire combien je regrette que la législation française n'ait pas évolué vers une autorisation encadrée des recherches sur les cellules souches embryonnaires. Je m'inquiète de ce que pourra être la position de la France sur le plan international dans ce domaine de recherche.
Je vais vous présenter les avancées qui ont pu être obtenues à la suite de travaux menés à I-Stem sur les cellules souches embryonnaires humaines pour la modélisation pathologique de maladies monogéniques et comment ces nouveaux modèles ont pu être utilisés pour identifier de nouvelles thérapeutiques.
Les lignées de cellules souches embryonnaires sont dérivées d'une cellule extraite de la masse interne du blastocyte, aux tous premiers stades du développement embryonnaire -les embryons utilisés sont les embryons surnuméraires des fécondations in vitro . Les cellules pluripotentes induites (iPS), elles, s'obtiennent à partir d'une cellule somatique adulte dédifférenciée puis reprogrammée pour retrouver des propriétés proches de celles d'une cellule souche embryonnaire.
Ces deux types de cellules ont la capacité de s'autorenouveler : elles constituent donc en théorie une source illimitée de matériel de recherche. Elles ont également la capacité de se différencier en tous les types cellulaires de l'organisme - neurones, cellules du muscle, de la peau... Leur premier champ d'application est de remplacer les cellules détruites ou endommagées dans le cas de maladies dégénératives. Une autre application, qui est celle dont je traiterai aujourd'hui, découle de la possibilité d'obtenir des lignées de cellules souches embryonnaires ou de cellules iPS portant la mutation causale d'une maladie monogénique. Il y a deux moyens d'y parvenir. Le diagnostic pré-implantatoire permet d'identifier les embryons porteurs du gène et d'en dériver des lignées de cellules souches embryonnaires porteuses de l'anomalie. Avec les iPS, il est possible de reprogrammer des cellules somatiques adultes de façon qu'elles contiennent le gène en question.
Pourquoi créer de nouveaux modèles ? Tout d'abord, parce que pour mettre au point des traitements adaptés, il faut mieux connaître les mécanismes de certaines pathologies. Dans le cas des maladies monogéniques, il faut comprendre comment la mutation aboutit au développement de la maladie. Ensuite, les modèles animaux utilisés jusqu'à présent, qu'il s'agisse de la souris, de la drosophile ou du ver, présentent l'inconvénient d'une restriction liée à l'espèce. Quant aux modèles cellulaires, qu'il est possible d'élaborer à partir de biopsies humaines, ils présentent eux aussi des difficultés. Il est parfois difficile d'effectuer les prélèvements nécessaires, notamment dans le cas de maladies neurologiques. Il l'est également de maintenir en culture les cellules prélevées. Et je ne m'étendrai pas sur la difficulté intrinsèque d'utiliser des cellules immortalisées par génie génétique, les propriétés de ces cellules se trouvant modifiées du fait de cette immortalité même. Tout cela explique qu'il soit nécessaire de disposer de nouveaux modèles plus pertinents.
Quel est le principe de la modélisation pathologique à partir de cellules souches embryonnaires humaines prélevées au cours d'un DPI ? Notre stratégie a été de faire se différencier ces cellules en progénies d'intérêt pour les pathologies auxquelles nous nous intéressions, de démontrer la relevance pathologique en recherchant la présence de stigma caractéristiques de ces maladies et de voir si, forts de ces résultats, nous pouvions utiliser ces modèles à la fois pour mieux comprendre les mécanismes pathologiques jusqu'au niveau moléculaire et identifier de nouveaux composés potentiellement thérapeutiques. Nous avons travaillé en laboratoire sur des cellules souches embryonnaires humaines portant la mutation causale de la myotonie dystrophique de type I (MD1), maladie neuro-musculaire d'origine génétique.
Nous avons d'abord différencié ces cellules en précurseurs mésenchymateux et recherché la présence des signes caractéristiques de la MD1 : agrégation de transcrits mutés au niveau du noyau, défaut d'expression de l'une des isoformes du gène codant pour le récepteur à l'insuline... Nous nous sommes ensuite demandés si nous pouvions utiliser ces modèles pour mieux comprendre les mécanismes de la maladie et faire du criblage thérapeutique.
Nous avons recherché tous les gènes qui s'exprimaient de manière différente dans les cellules malades et les cellules témoins. Grâce à la transcriptomique, nous avons pu en identifier toute une liste. Deux nous ont paru particulièrement intéressants, qui appartiennent à la famille des SLITRK, connue pour coder des protéines impliquées dans la communication entre neurones et myocytes. La mauvaise communication entre ces deux types de cellules est à l'origine même du syndrome myotonique associée à la DM1.
Exploitant au maximum le potentiel de pluripotence des cellules souches embryonnaires, nous les avons différenciées en neurones de la moelle épinière et avons étudié la capacité des cellules malades à interagir avec des cellules musculaires. Nous avons ainsi pu montrer que le défaut d'expression des gènes SLITRK résultait d'un défaut de communication entre les neurones et leurs cibles musculaires. Ce travail, qui a fait l'objet d'une publication récente dans une revue internationale, a apporté la preuve que des cellules souches embryonnaires humaines porteuses de la mutation causale d'une maladie monogénique pouvaient servir à identifier de nouveaux mécanismes pathologiques.
Nous avons également utilisé les cellules souches embryonnaires humaines pour identifier de nouvelles molécules thérapeutiques. Nous avons développé une plate-forme de criblage thérapeutique pour identifier des composés capables de faire disparaître les agrégats nucléaires et améliorer l'expression du récepteur à l'insuline. Une molécule est actuellement en phase d'étude pré-clinique.
Alors que depuis 1998, une trentaine de pathologies ont pu être « capturées » grâce à des cellules souches embryonnaires humaines obtenues à partir de DPI - les limites tiennent d'une part à la pratique du DPI elle-même, peu de maladies encore en faisant l'objet, d'autre part à la difficulté de dériver des cellules souches embryonnaires à partir des embryons recueillis -, depuis 2007 et la découverte des iPS par le professeur Yamanaka au Japon, plus d'une vingtaine l'ont été avec ces iPS.
Mais des problèmes sont apparus, liés au processus même de la reprogrammation. On force des cellules somatiques adultes à retrouver les propriétés de cellules embryonnaires en leur faisant surexprimer des gènes normalement impliqués dans le contrôle de l'état embryonnaire. On utilise pour cela différents vecteurs, notamment lentiviraux. Plusieurs études ont montré que cette reprogrammation induisait dans les iPS un grand nombre de défauts, notamment dans le contrôle de l'ADN - certaines régions du génome sont, à l'état embryonnaire, éteintes, mais deviennent actives lorsqu'on pousse la cellule à se différencier. Une cellule iPS n'a plus de zones éteintes et garde la mémoire de la cellule somatique adulte dont elle est issue. Une autre conséquence de la reprogrammation est l'apparition de nombreuses mutations dans les chromosomes. Environ 126 ont été identifiées, sur lesquelles 50 affectent des gènes potentiellement impliqués dans la survenue de cancers.
Après la parution d'articles sur le sujet en mars 2011, les mises en garde de la communauté scientifique internationale se sont multipliées sur les dangers de l'utilisation des iPS, invitant en tout cas à la précaution.
Pour terminer, je dresserai un tableau comparatif des avantages et inconvénients respectifs des cellules souches embryonnaires humaines et des cellules iPS.
Ces deux types de cellules présentent l'intérêt de permettre de dériver des lignées à grande échelle, de disposer de phénotypes très divers, et de pouvoir être cultivées de façon standardisée.
Tous deux présentent l'inconvénient d'une certaine instabilité génomique en culture. Se posent également la question de la pertinence du modèle - est-il pertinent d'utiliser des cellules souches embryonnaires pour modéliser par exemple des pathologies dégénératives survenant à l'âge adulte ? - et la difficulté de modéliser des maladies multifactorielles.
Les iPS offrent l'avantage que la reprogrammation permet de choisir les génotypes afin de répliquer exactement les maladies génétiques, dans leur extrême diversité. Elles présentent en revanche l'inconvénient que la reprogrammation entraîne des altérations de l'ADN. Il semble aussi maintenant qu'elles n'aient pas tout à fait la même pluripotence, et donc les mêmes capacités de différenciation que les cellules souches embryonnaires.
Un dernier mot sur I-Stem, premier institut français à avoir obtenu les autorisations nécessaires pour travailler sur des cellules souches embryonnaires humaines. I-Stem a été créé par Marc Peschanski en 2005, sous l'impulsion de l'INSERM, de l'université d'Evry et de l'AFM, l'Association française contre les myopathies. Alors qu'il ne comptait que quinze personnes à son lancement, I-STEM, qui a été labellisé en 2007 et reconnu à la fois par l'INSERM et par l'AFM comme bras armé, compte aujourd'hui 90 chercheurs répartis en onze équipes, dont le seul objectif est d'explorer le potentiel offert par les cellules souches embryonnaires humaines, notamment dans l'espoir de trouver des traitements pour les maladies monogéniques.
M. Jean-Louis Touraine. Je donne enfin la parole au professeur Soumeya Bekri, qui a notamment travaillé sur le traitement enzymatique de la maladie de Fabry.
Mme Soumeya Bekri, professeur des universités, praticien hospitalier, responsable du dépistage et du suivi des maladies métaboliques au CHU de Rouen. J'ai le plaisir de vous présenter aujourd'hui de nouvelles thérapies de maladies monogéniques, notamment celles dues à un déficit enzymatique. Je me focaliserai sur les maladies lysosomales de surcharge. C'est un groupe hétérogène qui comprend plus de cinquante maladies, dont la prévalence combinée est de un sur cinq mille naissances. Elles se caractérisent toutes par un défaut de catabolisme ou de transport d'un substrat.
Leur caractéristique commune est l'accumulation progressive de différentes macromolécules dans les lysosomes. Ce sont des pathologies graves, entraînant des atteintes multiviscérales - hépatosplénomégalie, problèmes osseux et souvent une atteinte neurologique... Ces manifestations cliniques ne sont pas immédiates, n'apparaissant qu'après accumulation suffisante du substrat non dégradé, mais elles sont alors inexorables. La morbi-mortalité est très élevée. Il est important de pouvoir porter un diagnostic devant des signes cliniques évocateurs, non nécessairement spécifiques, car il existe des traitements pour certaines de ces maladies.
Les maladies lysosomales de surcharge les plus connues et les plus fréquentes sont la maladie de Gaucher et la maladie de Fabry.
On dénombre un cas de maladie de Gaucher sur 50 000 naissances. Cette maladie, de transmission autosomique récessive, entraîne des atteintes osseuses et hématologiques, ainsi qu'une hépatosplénomégalie. Dans les types II et III, on trouve également des atteintes neurologiques. Le type I est connu pour être la forme « non neurologique ».
La maladie de Fabry est une pathologie héréditaire du métabolisme des glycosphingolipides, de transmission récessive liée au chromosome X, et due à un déficit en alpha-galactosidase A, enzyme lysosomale. Ce défaut enzymatique conduit à l'accumulation du substrat non dégradé dans les tissus et le plasma. On dénombre un cas sur 80 000 naissances. Dans sa forme classique, la maladie touche plus sévèrement les hommes hémizygotes. Les femmes hétérozygotes, conductrices de la maladie, présentent souvent des symptômes, mais plus variables et généralement moindres que les hommes. La maladie entraîne des atteintes neurovasculaires, avec notamment des accidents vasculaires cérébraux chez le sujet jeune, des atteintes cardiaques avec une hypertrophie ventriculaire gauche, des atteintes du rein aboutissant à une insuffisance rénale, parfois aussi des atteintes ophtalmologiques et dermatologiques.
Les maladies lysosomales de surcharge ont constitué un modèle pour le développement de nouvelles thérapies ces dernières années. Il est possible de remplacer l'enzyme manquante ou déficiente par une enzyme exogène - c'est le principe de l'enzymothérapie substitutive - ; de limiter la production du substrat non dégradé ; de stabiliser l'enzyme mutante et d'en augmenter l'activité résiduelle grâce à des molécules chaperons.
Le lysosome est un compartiment cellulaire au pH acide, dont la principale fonction est de dégrader des macromolécules intra et extracellulaires. C'est une composante du système endosome/lysosome, impliqué dans l'homéostasie générale de la cellule, et qui permet la communication entre milieu intracellulaire et milieu extracellulaire.
Pour comprendre le principe de l'enzymothérapie substitutive, il faut revenir brièvement sur la synthèse et l'adressage des enzymes lysosomales. La plupart d'entre elles sont synthétisées au niveau du réticulum endoplasmique puis migrent vers l'appareil de Golgi, où elles subissent des modifications dites post-traductionnelles qui leur permettent ensuite de fixer le résidu que constitue le mannose-6-phosphate. L'ensemble constitué du mannose-6-phosphate, de son récepteur spécifique et de l'enzyme est adressé au compartiment cellulaire de l'endosome tardif, lequel, en s'acidifiant, devient un lysosome. L'enzyme ainsi adressée est active. Les récepteurs mannose-6-phosphate, eux, sont recyclés soit vers l'appareil de Golgi pour recruter de nouvelles enzymes, soit vers la membrane plasmique.
Dans le cas d'une maladie lysosomale, l'enzyme soit n'est pas synthétisée soit est mal adressée, en tout cas elle n'est pas assez active dans le lysosome. Dans l'enzymothérapie substitutive, on apporte une enzyme exogène, synthétisée in vitro , qui va être reconnue par le récepteur au mannose-6 phosphate, internalisée par endocytose dans une vésicule qui va rejoindre l'endosome précoce puis l'endosome tardif, appelé à devenir lysosome. Une fois parvenue dans le lysosome, cette enzyme exogène pourra y remplacer l'enzyme déficiente.
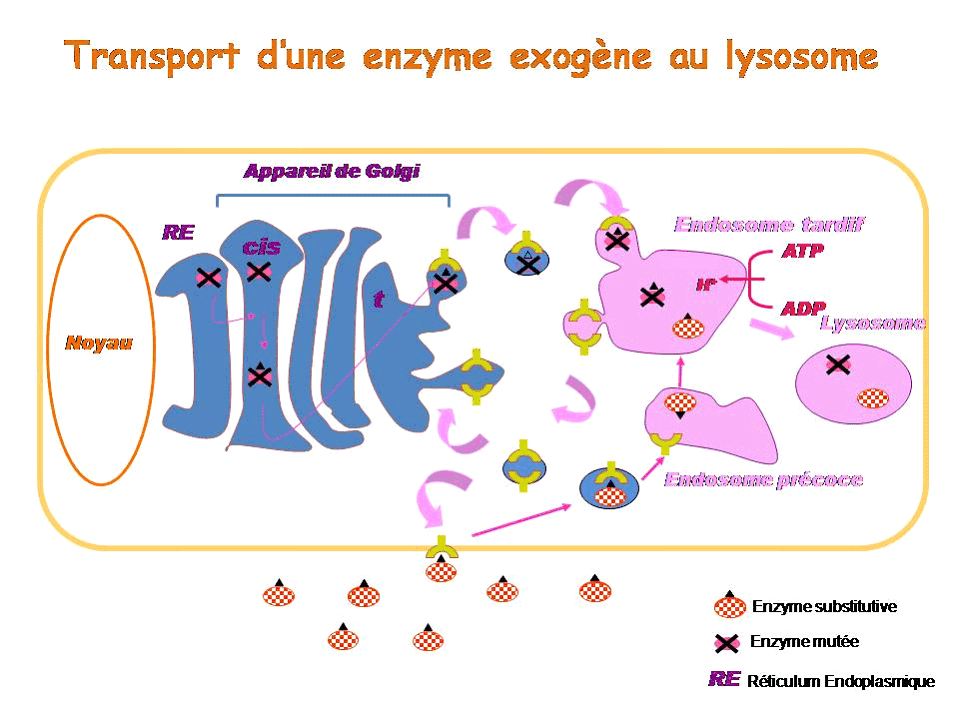
Il existe actuellement une enzymothérapie substitutive pour la maladie de Gaucher, la maladie de Fabry, les mucopolysaccharidose de type I, II et VI, et la maladie de Pompe. Son efficacité est variable selon la pathologie. Les résultats sont très bons pour la maladie de Gaucher, plus discutés pour la maladie de Fabry, et le recul est encore insuffisant pour les mucopolysaccharidoses. L'efficacité varie également selon le stade de la maladie et l'organe atteint. L'enzymothérapie est très efficace sur la viscéralomégalie, beaucoup moins sur les atteintes osseuses. Les enzymes en question ne peuvent pas être administrées pour l'instant par voie orale car elles sont trop volumineuses pour passer la barrière intestinale. Elles le sont donc par voie intra-veineuse et leur administration doit se poursuivre à vie. Une enzyme de substitution, mise au point en Israël sur des cellules de carottes et qui vient d'être approuvée par la Food and drug administration (FDA) américaine, semblerait toutefois pouvoir être administrée par voie orale : une étude clinique est en cours.
Un autre inconvénient de l'enzymothérapie substitutive est que, comme il s'agit de protéines, elle ne passe pas la barrière hémato-encéphalique : les atteintes neurologiques ne sont donc pas accessibles au traitement.
L'administration de ces enzymes peut en outre entraîner une réaction immunologique avec le développement d'anticorps neutralisants, susceptibles de réduire l'efficacité du traitement.
Enfin, le coût de cette thérapie est très élevé, d'au moins 200 000 euros par an et par patient, et ce à vie. Le traitement d'une mucopolysaccharidose de type VI coûte 500 000 euros par an pour un enfant.
En sus de l'enzymothérapie substitutive, il existe d'autres thérapies en cours d'évaluation ou venant d'être approuvées. Elles visent à réduire la quantité de substrat ou font appel à des molécules chaperons. Leur avantage est qu'elles peuvent être administrées par voie orale et qu'elles ont une meilleure distribution tissulaire. Il semble aussi qu'elles passent la barrière hémato-encéphalique et qu'elles n'entraînent pas de réaction immunologique. Leur coût de production serait également moins élevé. Cela étant, on manque encore beaucoup de recul.
Dans un métabolisme normal, un substrat B, synthétisé à partir d'une molécule A, est dégradé en une molécule C. Dans le cas d'une maladie enzymatique, l'enzyme qui permet la dégradation de B en C, est déficiente, si bien que le substrat B s'accumule, alors même que sa synthèse se poursuit normalement. Le principe de la réduction de substrat est de limiter la synthèse.
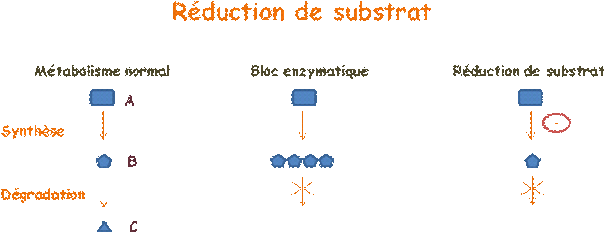
Un médicament, le Miglustat, est aujourd'hui disponible pour la maladie de Gaucher de type I et la maladie de Niemann-Pick de type C. Un autre, l'Eliglustat, commercialisé par Genzyme pour la maladie de Gaucher de type I, est en phase d'essai clinique. La Genistein, destinée à traiter la mucopolysaccharidose de type III, elle aussi en cours d'essai clinique, présente un intérêt tout particulier car les manifestations de cette maladie étant surtout neurologiques, on ne peut faire appel à l'enzymothérapie substitutive.
Un mot des traitements par molécules chaperons. Ces molécules permettent la stabilisation tridimensionnelle des enzymes qui sont synthétisées au niveau du réticulum endoplasmique mais ne se replient pas pour prendre leur conformation normale. Ce sont pour la plupart des analogues de substrat ayant une très forte affinité pour le site catalytique. Elles aident au repliement de l'enzyme et l'adressent au lysosome. En milieu acide, les inhibiteurs se détachent de l'enzyme, laquelle peut alors dégrader le substrat. Ces traitements, porteurs d'espoir, ne peuvent toutefois servir qu'à traiter les maladies résultant d'une mutation induisant un défaut de repliement. Aujourd'hui, on teste in vitro l'efficacité de la molécule chaperon avant de traiter le patient. Deux molécules sont en cours d'essai clinique : le Migalastat dans la maladie de Fabry et l'Isofogamine pour la maladie de Gaucher de type I et de type III.
En conclusion, je voudrais insister sur le caractère trompeur de la dénomination « maladies rares » pour les maladies lysosomales de surcharge car leur incidence globale est loin d'être négligeable : nous rencontrons un grand nombre de cas au quotidien. Ces maladies, connues pour être pédiatriques, concernent un nombre croissant d'adultes. Certaines d'entre elles se manifestent dès la vie in utero. Ces maladies rares font néanmoins l'objet de recherches actives, dont les retombées sont beaucoup plus larges. Grâce à ces recherches, plusieurs approches thérapeutiques innovantes ont pu être mises au point.
Débat
M. Jean-Louis Touraine. Ces nouvelles thérapeutiques, qui se sont développées en peu de temps, sont fascinantes. Dans les années 70, où rien de tout cela ne pouvait même être imaginé, nous pratiquions des greffes de cellules de foie foetal chez des patients atteints de maladie de Fabry ou de Gaucher. Cela permettait d'enrayer la progression de la maladie, sans toutefois permettre d'en faire régresser les symptômes. Mais des malades ainsi traités ont pu « attendre » jusqu'à la mise au point de l'enzymothérapie substitutive, qui a pris le relais. Tous les traitements, même ceux qui n'ont pas encore fait la preuve d'une efficacité totale sur tous les symptômes de la maladie, sont intéressants, ne serait-ce que parce qu'ils peuvent permettre à certains malades de survivre jusqu'à ce qu'un traitement plus efficace soit disponible. Toutes ces thérapies innovantes sont porteuses de beaucoup d'espoirs.
Mme Soumeya Bekri. Être en mesure de proposer une solution partielle ou d'attente change complètement la donne. C'est un espoir considérable pour les malades. J'ai en tête le cas d'une fillette atteinte de mucopolysaccharidose de type VI, dont l'intelligence n'avait pas été atteinte par la maladie, et qui a pu bénéficier de l'une de ces thérapies. Sa vie et son pronostic en ont été révolutionnés.
M. Jean-Louis Touraine. Il est très encourageant que de nouveaux traitements semblent franchir la barrière hémato-encéphalique. En effet, jusqu'à présent, en cas d'atteinte neurologique lourde, on ne disposait d'aucun outil, ne serait-ce que pour stopper, sans parler même de réversibilité, les atteintes cérébrales. Il était très frustrant de ne pas pouvoir protéger le système nerveux.
M. le président Claude Birraux. Je trouve moi aussi tous ces progrès très encourageants. Des résultats ont d'ores et déjà été obtenus, il faut le faire savoir plus largement... y compris à M. Bergé, qui s'en était pris au Téléthon.
Le coût élevé de ces traitements tient, entre autres, à celui des recherches nécessaires à leur mise au point. Quid de la propriété intellectuelle des découvertes ?
Mme Soumeya Bekri. Nous avons acquis une culture du brevet, que nous n'avions pas auparavant. Les universités comme l'INSERM nous incitent de plus à plus à protéger nos découvertes avant de les publier.
M. Philippe Leboulch. Qu'on le déplore ou qu'on s'en réjouisse, force est de constater que s'il n'y avait pas d'application industrielle en aval des découvertes fondamentales, on ne pourrait pas aller très loin. Il faut en effet des moyens matériels et financiers considérables pour en arriver jusqu'aux essais cliniques puis à la mise à disposition de nouveaux médicaments pour les patients. Les étapes-clés des découvertes doivent donc être protégées par des brevets, de façon que les industriels puissent non seulement rentrer dans leurs frais mais faire des bénéfices leur permettant de continuer à investir.
Le prix des médicaments dont on a parlé aujourd'hui est très élevé, peut-être trop élevé. Mais leur coût même de production fait qu'il ne pourra pas s'abaisser au-delà d'un certain seuil. La thérapie génique, elle, marque une rupture car le traitement, dans la plupart des cas, est destiné à n'être administré qu'une seule fois. Une transplantation de moelle avec transfert de gène coûtera nécessairement davantage qu'une transplantation de moelle simple. Mais le surcoût sera au final quasiment négligeable par rapport aux thérapies innovantes non géniques qui nous ont été présentées ou aux thérapies palliatives qui doivent durer toute la vie du patient.
M. Jean-Louis Touraine. Les retombées potentielles de la thérapie génique sont considérables. Elle pourrait être utilisée dans le traitement de maladies comme le cancer, le diabète, le sida. Le coût d'une thérapie génique unique, aussi onéreuse soit-elle, serait bien moindre au final chez les malades du sida, dont l'espérance de vie avoisine désormais la normale, que l'administration à vie des trithérapies actuelles, dont l'observance sur le long terme est par ailleurs quotidienne et imparfaite chez de nombreux malades. Il faut donc encourager la recherche sur les thérapies géniques, quel que soit le montant des investissements nécessaires, car en résulteront des progrès médicaux considérables pour les malades mais aussi, à terme, des économies pour nos systèmes de santé.
M. Philippe Leboulch. Des équipes travaillant sur le sida ont découvert un corécepteur dont l'absence ou la mutation expliquent que certaines personnes soient comme protégées de l'infection par le VIH. En Allemagne, un patient atteint à la fois d'une leucémie et du sida a reçu une greffe de moelle à partir d'un donneur compatible qui se trouvait posséder la mutation de ce corécepteur. Ce patient a non seulement été guéri de sa leucémie mais semble l'avoir été aussi du sida, les cellules qu'on lui a greffées n'étant plus infectables. Un essai clinique est en cours aux Etats-Unis, utilisant le même vecteur que celui utilisé pour les hémoglobinopathies pour exprimer dans les cellules un gène empêchant ce corécepteur d'être présent et ainsi immuniser « de l'intérieur » les cellules du patient contre le VIH. Voilà l'exemple-type d'une retombée d'une recherche conduite initialement pour des maladies génétiques rares dans le traitement d'une maladie fréquente, d'ailleurs infectieuse et non génétique.
M. Nicolas Lévy. Je voudrais abonder dans le sens de mes collègues. Certaines des molécules qui ont fait la preuve d'une certaine efficacité dans le traitement des maladies lysosomales de surcharge - je pense en particulier au Miglustat et à toutes les molécules capables d'induire le repliement de certaines protéines - peuvent trouver des applications dans bien d'autres champs. Elles pourront servir à traiter toutes les maladies liées à une anomalie du « contrôle de qualité » ou de repliement des protéines du fait d'une mutation génétique. Il importe donc de développer la recherche dans ces domaines.
Certains opposent thérapies pharmacologiques classiques et thérapies innovantes, dont la thérapie génique et la thérapie cellulaire. Cette opposition n'a pas de sens. Tout d'abord, les secondes n'excluent pas de recourir aux premières, y compris les plus classiques. Mais, pour certaines maladies du muscle, de la rétine, du sang ..., les thérapies pharmacologiques n'ont pas apporté de solution et sont vraisemblablement vouées à demeurer impuissantes. Il faut donc absolument soutenir les approches innovantes, parmi lesquelles la thérapie génique, par transfert de gène, ou, plus « à la carte », par saut d'hexon ou modification génomique grâce à des molécules anti-sens. Les deux approches s'opposent d'autant moins que certaines formes de thérapie génique sont appelées à être administrées comme un médicament classique. C'est le cas des petites molécules d'ADN anti-sens d'ores et déjà utilisées dans certains essais cliniques.
M. le président Claude Birraux. Si je puis me permettre en conclusion un trait d'humour sur un sujet aussi sérieux, au moins dans ce domaine n'y a-t-il pas de faucheurs volontaires pour ruiner les recherches !
DEUXIÈME TABLE RONDE : LES LIENS ENTRE RECHERCHE, PRATICIENS ET ASSOCIATIONS : QUESTIONS JURIDIQUES ET ÉTHIQUES
LA PLACE DES DIFFÉRENTS ACTEURS
M. Jean-Louis Touraine. La deuxième table ronde va porter sur les liens entre la recherche, les praticiens et les associations. Nous abordons, en premier lieu, la place des différents acteurs.
Je donne la parole à Mme le docteur Anne Cambon Thomsen, directrice de recherche au Centre national de la recherche scientifique (CNRS), spécialisée en immunogénétique humaine. Elle dirige l'équipe « génomique, santé et société » au sein de l'unité « épidémiologie et analyses en santé publique » - unité mixte INSERM (Institut national de la santé et de la recherche médicale)-Université Paul Sabatier, à Toulouse.
Mme Anne Cambon-Thomsen, directrice de recherche, membre du Public and professional policy committee (Société européenne de génétique humaine), ancien membre du Groupe européen d'éthique. J'interviendrai aujourd'hui en tant que membre du conseil d'administration de la Société européenne de génétique humaine ( European society of human genetics, ESHG), de membre de son Comité sur les politiques publiques et professionnelles ( Public and professional policy committee ) et d'ancien membre du Groupe européen d'éthique (GEE).
Créée en 1967, l'ESHG est une société scientifique, mais aussi professionnelle. Elle organise un maillage de comités sur les divers aspects de la génétique, en se préoccupant aussi bien de l'organisation de la profession que de la recherche, de l'éducation des professionnels, ou encore des contacts avec les associations de patients et les patients.
La génétique humaine, qu'elle soit de recherche ou clinique, est par nécessité une activité internationale, et notamment européenne. La France est très active au sein de l'ESHG : deux Français l'ont présidée, le prochain président sera également un Français et des membres français font partie des différents comités. L'ESHG est un fédérateur et un catalyseur d'acteurs divers, au-delà des acteurs professionnels. Elle a en particulier porté le projet de la reconnaissance officielle, au niveau européen, de la spécialité de génétique médicale - reconnaissance qui est effective depuis l'adoption par la Commission, le 3 mars 2011, d'un règlement sur le sujet.
L'ESGH a également une activité de veille et de réponse aux consultations publiques, par exemple au sujet de la recommandation du Conseil européen sur une action dans le domaine des maladies rares, mais aussi au sujet des propositions de révision de la directive sur les diagnostics in vitro .
Le Comité pour les politiques publiques et professionnelles est ouvert à d'autres spécialistes que des généticiens, notamment à des philosophes, et assure l'interface avec des associations de patients. Il produit des documents de base ( background documents ), souvent accompagnés de recommandations, qui sont publiés sur le site web de l'ESHG et dans le Journal européen de génétique humaine (EJHG) et qui deviennent souvent des points de référence. Ces documents et recommandations ont porté, notamment, sur les tests génétiques chez les mineurs (enfants asymptomatiques) ; sur les aspects éthiques, légaux et sociaux en matière de brevets et de licences dans le domaine des tests génétiques ; sur le conseil génétique ; sur l'organisation des services génétiques en Europe ; sur les programmes screening de dépistages génétiques ; sur les cohortes et les biobanques d'échantillons et de cellules... S'agissant des biobanques d'ADN et d'autres éléments du corps humain, dont on a vu l'importance pour les maladies rares, l'infrastructure qui se met en place associe les biobanques qui rassemblent des échantillons provenant de patients atteints de maladies rares. La mise en place de cette organisation témoigne de l'importance accordée aux maladies rares, et facilitera les échanges au niveau européen.
J'en viens aux projets européens.
Parmi les derniers appels d'offres, beaucoup sont dirigés vers les maladies rares. Dans le domaine du séquençage, qui prend actuellement une grande importance dans les recherches en génétique, quatre projets européens sont pilotés par l'Espagne, la Hollande, l'Angleterre et l'Allemagne. Mon équipe de l'INSERM à Toulouse ne participe qu'au titre des aspects sociétaux et éthiques. Autrement dit, en France la réflexion sur l'utilisation de ces techniques est très poussée, au regard de la réalité de la recherche et de la pratique clinique - mais après tout, il n'est pas forcément inutile de réfléchir avant d'agir !
Parmi les projets européens, ceux lancés par la DG SANCO (Direction générale santé et consommateurs) portent avant tout sur l'établissement de guidelines (guides de bonnes pratiques). L'un d'eux a pour objectif d'étudier la possibilité d'intégrer davantage la génétique dans la santé publique.
Ces projets sont des actions structurantes avec des acteurs variés, pouvant inclure des associations de patients. La Commission encourage en effet beaucoup la participation de ces dernières. Le deuxième projet Eurogentest, par exemple, qui a démarré l'année dernière, est un réseau d'excellence dont les branches sont dirigées vers l'industrie, la pratique de laboratoire, le conseil génétique, l'information des patients, mais aussi la participation d'associations.
Petit à petit, la structuration progresse - même si on n'en est pas, comme je l'avais proposé, à une infrastructure européenne de recherche sur les maladies rares... La reconnaissance officielle de la profession de génétique médicale au niveau européen, dont je parlais à l'instant, devrait permettre d'avancer.
Paradoxalement, le Groupe européen d'éthique - qui existe depuis 1991 et dont j'ai eu le bonheur de faire partie de 2005 à 2010 - s'est peu préoccupé de génétique. Il a rendu un avis en 2004 sur les tests génétiques dans le monde du travail, de multiples avis sur les cellules souches (notamment embryonnaires) ainsi que sur les inventions biotechnologiques ; il a rédigé une lettre sur les tests génétiques proposés directement au consommateur ; mais en dehors de cela, il n'a pas rendu d'avis sur les tests génétiques.
Néanmoins, des initiatives ont été prises au niveau de la Commission et du Conseil de l'Europe.
En 2004, un groupe d'experts, dont j'ai été le rapporteur, a rédigé un rapport sur les aspects éthiques, légaux et sociaux des tests génétiques, incluant 25 recommandations, dont un grand nombre sont particulièrement pertinentes au regard des maladies rares.
En 2008, un protocole additionnel à la Convention d'Oviedo sur les Droits de l'homme et la biomédecine, relatif aux tests génétiques dans le domaine de la santé, a été adopté par le Conseil de l'Europe. Même si peu de pays l'ont signé jusqu'à présent, il commence à faire référence en matière de bonnes pratiques dans différents domaines.
En conclusion, il existe au niveau européen un ensemble de professionnels actifs. La Société européenne de génétique humaine est un carrefour d'acteurs. Les maladies monogéniques et les maladies rares tiennent une grande place au sein de la génétique médicale. Mais la génétique étant extrêmement diverse, des régulations adaptées s'imposent ; il faut distinguer la génétique médicale pour les maladies monogéniques, la génétique dans le cadre de la pharmacogénétique et la génétique associée aux maladies multifactorielles.
M. Jean-Louis Touraine. Merci. Je donne maintenant la parole à Mme Martine Bungener, présidente du Groupe de réflexion avec les associations de malades (GRAM), directrice de recherche au CNRS, directrice adjointe du Centre de recherche médecine, sciences, santé, santé mentale et société (CERMES).
Mme Martine Bungener, directrice adjointe du Centre de recherche médecine, sciences, santé, santé mentale et société (CERMES), (CNRS, INSERM, EHESS), et présidente du Groupe de réflexion avec les associations de malades (GRAM). Le Groupe de réflexion avec les associations de malades (GRAM) a été mis en place en 2003 par l'Institut national de la santé et de la recherche médicale (INSERM), dont il est l'instance de réflexion et de proposition à la direction générale. Il est constitué de représentants de dix associations et de dix personnes représentant d'une part les chercheurs dans les différents volets de la recherche, d'autre part l'administration de l'INSERM.
La Mission INSERM Associations, créée dans la foulée, en est l'instance opérationnelle : elle met en place les opérations en direction des associations. À l'écoute de ces dernières, elle fait également remonter au GRAM puis à la Direction générale et maintenant à la présidence de l'INSERM leurs souhaits et les accords qui ont pu être négociés avec elles.
Le lien est établi avec 469 organisations - associations de patients, fédérations ou fondations, dont le nombre total en France est sans doute compris entre 2000 et 3000. 137 concernent des maladies rares, 111 des maladies génétiques et 76 des maladies d'enfants. Ces maladies ont donc un poids très important dans la demande adressée à la recherche - car leur nombre ne résulte pas d'une sélection par l'INSERM lors de son appel à candidatures.
Au moment où ces associations se sont rapprochées de l'INSERM, la moitié d'entre elles avaient déjà un conseil scientifique, les deux tiers étaient déjà impliquées dans la recherche, pour certaines avec un financement direct, pour d'autres à travers un soutien logistique, pour d'autres encore en participant à des projets de recherche et à des essais thérapeutiques. Dans le cadre des liens que nous développons avec elles depuis 2003, les formes d'engagement de ces associations sont très diversifiées. Il s'agit tout aussi bien de financer la recherche, de la stimuler par du lobbying, de l'orienter vers des domaines délaissés, d'apporter des connaissances propres, de participer à la recherche, d'en diffuser les résultats, d'avoir une influence sur les formes d'application de ces résultats, ou encore d'informer le monde « profane » - y compris le monde médical, tous les médecins généralistes ne connaissant pas les maladies rares -, en particulier sur de nouvelles thérapies.
L'engagement de ces associations est accompagné par différentes actions, mises en place en concertation avec elles par le GRAM et la Mission INSERM Associations : formations ciblées, débats réunissant chercheurs et associations. L'enjeu est en effet de renforcer les capacités de dialogue entre chercheurs et associations. Les sessions de formation sur les tests génétiques sont celles qui ont été le plus souvent demandées. Sur 30 sessions depuis 2004, 12 ont porté sur les tests génétiques, mobilisant 85 associations et, entre 2008 et 2010, 190 participants.
Une autre action du GRAM est de promouvoir la participation de membres associatifs aux instances. Au conseil d'administration de l'INSERM, il y a dorénavant une place pour un membre associatif. Le GRAM, je l'ai dit, comprend dix membres représentants d'associations. Le Comité d'orientation stratégique et de suivi des essais cliniques (COSSEC) compte six membres associatifs, ce qui permet une discussion en son sein sur les formes d'essais et leur orientation ; il a d'ailleurs favorisé l'essor du département de recherche clinique de l'INSERM ces quatre dernières années. Le Comité de qualification institutionnel (CQI), qui donne un avis éthique sur les recherches, comprend quatre membres associatifs. Le Collège de relecteurs, rattaché au département de recherche clinique, mobilise de façon tournante 69 associations. Il vérifie tous les documents transmis aux patients qui vont participer aux essais cliniques, évalue les enjeux éthiques, vérifie les niveaux de compréhension, et conseille éventuellement les promoteurs de ces essais cliniques que sont les chercheurs.
Après la participation de membres d'associations aux conseils d'orientation des programmes de recherche INSERM, une mise en place progressive d'associations se fait dans les conseils de réflexion des instituts thématiques multi-organismes (ITMO), nés de la création de l'Alliance nationale pour les sciences de la vie et de la santé (AVIESAN). Enfin, les associations ont leur place dans les expertises collectives, dont celle sur la drépanocytose.
Les associations ont donc un rôle de plus en plus important pour relier la recherche, le monde professionnel et la société civile. Elles diffusent des informations sur la recherche, notamment sur le lancement de projets, ainsi que des informations biomédicales ; elles valorisent leurs propres compétences et leurs savoirs profanes ; et elles font valoir leurs attentes pour que les projets de recherche soient mieux construits.
M. Jean-Louis Touraine. Merci. Nous allons maintenant entendre Mme Jenny Hippocrate Fixy, présidente de l'Association pour l'information et la prévention de la drépanocytose (APIPD). Elle a été assistante sociale et a fait des études de psychologie.
Mme Jenny Hippocrate-Fixy, présidente de « Ensemble contre la drépanocytose », et de l'Association pour l'information et la prévention de la drépanocytose (APIPD), directrice française de l'Organisation européenne pour les anémies rares (EORA). Je me présente devant vous au nom du Collectif ECD, Ensemble contre la drépanocytose, qui regroupe une centaine d'associations, en tant que directrice française de l'European organisation for rare aneamias (EORA), basée en Grèce, et en tant que présidente de l'Association pour l'information et la prévention de la drépanocytose (APIPD), qui existe depuis 23 ans. Dans ces associations, je suis bénévole mais je travaille à plein temps. Et j'ai un deuxième travail à plein temps, celui de maman d'un adolescent atteint de drépanocytose. Pour pouvoir m'occuper de mon enfant et de toutes ces structures, j'ai dû démissionner de mes fonctions de psychologue dans le secteur carcéral. Je vis grâce aux bénéfices de la vente de mes ouvrages - mais je les reverse le plus souvent aux associations, notamment à l'APIPD - et au salaire de mon mari, éducateur à l'hôpital.
L'APIPD est une association dont les objectifs, depuis sa création en 1988, sont : faire reconnaître la lutte contre la drépanocytose comme une priorité dans les actions à mener par les gouvernements ; faire avancer la recherche ; accroître l'information sur cette maladie et y sensibiliser le public, le personnel de santé, les enseignants et les politiques ; conforter et associer les différents acteurs qui mènent souvent, à petite échelle et de manière confidentielle, des actions intéressantes qui méritent d'être développées dans un esprit de solidarité et d'humanisme ; assurer la diffusion de l'information sur la drépanocytose ; servir de trait d'union entre les malades drépanocytaires et les aider, eux et leur famille, à résoudre les diverses difficultés matérielles et morales engendrées par cette maladie ; contribuer à la diffusion des informations concernant le dépistage et les méthodes modernes de traitement de la maladie, ainsi que favoriser et organiser leur application ; faciliter la scolarité et la formation professionnelle de ces malades ; faciliter la recherche scientifique sur cette maladie ; être une association ouverte, se refusant à tout prosélytisme politique, religieux ou racial ; sensibiliser l'opinion publique sur cette maladie afin que les moyens soient alloués aux médecins pour la recherche ; établir une liaison avec les associations locales, nationales ou internationales analogues.
L'APIPD est gérée par un conseil d'administration de 46 membres. Elle dispose d'un conseil scientifique composé de 30 membres parmi lesquels figurent d'éminents professeurs spécialistes de la drépanocytose, en particulier Frédéric Galacteros et Robert Girot. Il y a aussi une APIPD Guadeloupe, une APIPD Martinique, une APIPD Mayotte, une APIPD Bordeaux Sud-Ouest et quarante-deux antennes à travers le monde.
L'APIPD est membre de : Eurordis ; l'Alliance maladies rares ; la Fédération des associations de lutte contre la drépanocytose (FALD), que j'ai également l'honneur de présider ; le Collectif « Ensemble contre la drépanocytose » ; l'Association des bénévoles AP-HP de Paris ; l 'European organisation for rare aneamias (EORA) ; la Commission des relations avec les usagers et de la qualité de la prise en charge (CRUQPC), dont je suis une des titulaires ; le Comité de lutte contre la douleur (CLUD) ; le Collectif interassociatif sur la santé (CISS).
L'APIPD a l'agrément nécessaire pour intervenir en tant qu'association de malades, au sein des instances hospitalières. Elle travaille en collaboration avec : La Mutuelle nationale des hospitaliers et des personnels de santé (MNH) ; l'Assistance publique-Hôpitaux de Paris (AP-HP) ; le ministère de l'outre-mer ; le ministère de la santé ; la Mairie de Paris ; les associations analogues nationales et internationales ; une centaine d'associations culturelles et communautaires ; les Laboratoires Novartis ; les Laboratoires Addmedica ; l'Éducation nationale - en tant que formatrice, j'interviens auprès des élèves de terminale ES ; je suis directrice de mémoire et participe comme membre de jury à la soutenance de mémoires de pharmacie, de psychologie, etc.
L'Association enregistre environ 7 000 adhérents et sympathisants, et plus de 20 000 passages chaque année. Notre site Internet est très visité.
Nous accompagnons les malades, sans distinction. Nous sommes d'ailleurs très fréquemment sollicités par les assistantes sociales.
Nous avons pris l'initiative de sensibiliser le monde du travail aux contraintes liées à la drépanocytose, en travaillant avec UNIRH et Hanploi.com. Il faut en particulier éviter aux drépanocytaires les courants d'air, le chaud et le froid.
Les malades ou les familles s'adressent prioritairement à nous pour avoir des informations sur la drépanocytose. Nous assurons très souvent l'information et rédigeons des documents ad hoc pour informer et sensibiliser non seulement les malades, mais aussi le grand public, notamment en matière de dépistage.
En France, le dépistage est systématique, mais ciblé - donc au faciès ou au nom. Je pense donc qu'il y a - excusez-moi de froisser quelques susceptibilités - une question de racisme dans le fait de ne pas beaucoup s'occuper de la drépanocytose dans notre pays. À l'hôpital où j'ai emmené mon fils, j'ai entendu dire : « le petit drogué à la morphine », « le petit négro qui est dans la chambre d'à côté », « il en veut encore parce qu'il est drogué, il n'a pas si mal que ça »... Nous nous battons pour ne plus entendre ce genre de réflexion. Mon fils a frôlé la mort huit fois en étant victime de sept syndromes thoraciques aigus et d'un infarctus. La drépanocytose est une maladie qui accable les familles et qui met les malades au ban de la société ! Ce n'est pas normal !
La drépanocytose en chiffres, c'est 50 millions de dépenses par an, 17 % des lits d'hôpital de l'Ile-de-France, 20 jours d'hospitalisation minimum par malade. Et on ne fait rien. Mais nous ne devrions pas parler d'argent car nous avons tous droit à la santé.
L'APIPD joue un rôle capital au sein des hôpitaux. Nous sommes sollicités par les médecins, les assistantes sociales, les psychologues ; nous siégeons dans les commissions ; nous recevons les malades et les familles pour les aider à faire valoir leurs droits.
Il est faux de dire que la drépanocytose est une maladie de la population noire. L'EORA recense 2 000 malades en Grèce. La drépanocytose est certes plus fréquente dans les pays chauds ; des malades d'origine africaine sont donc présents en France. Les parents parlent encore de malédiction, de mauvais sort ; et quand nous arrivons à leur faire comprendre que c'est une maladie génétique, ils disent qu'ils ont cassé la chaîne de bonne santé de toute une famille. Ma ligne téléphonique est accessible 24 heures sur 24, on m'appelle à 2 heures du matin. Je connais un malade qui vit dans un squat et qui n'arrête pas de hurler : il faut l'emmener à l'hôpital, mais il a peur car il n'a pas de papiers, et il est dans le déni. Notre travail est fatigant, mais nous le faisons pour tous ces malades qui souffrent. En ce qui me concerne, je suis lasse, mais pas lassée : je vais continuer à me battre car on ne peut pas admettre une telle situation au pays des droits de l'homme.
Nos associations se substituent souvent - trop souvent - à l'État dans son rôle d'information et de formation sur la drépanocytose. À ce titre, elles sont officiellement reconnues, puisque sollicitées par l'État (ministères de l'éducation nationale, de la santé), mais insuffisamment, semble-t-il, pour prétendre à des subventions. Dans un souci de commodité pour les malades et leur famille, chaque association a un site Internet qu'elle met à jour et alimente à ses frais. L'APIPD écoute, soutient, informe, éduque. Elle sensibilise le grand public à travers des colloques organisés chaque année à ses frais. Elle soutient la recherche, notamment en exprimant les attentes et les besoins des malades auprès des instances de santé et des partenaires pharmaceutiques. On ne guérit pas encore la drépanocytose. Cependant, l'APIPD doit lutter chaque jour pour l'éradication des préjugés et des tabous liés à cette maladie discriminante et discriminatoire. Elle est dans l'action, immédiate et à long terme. Indéniablement, elle est un acteur et un partenaire de santé à part entière. Merci de nous aider !
M. Jean-Louis Touraine. Votre témoignage nous touche beaucoup, Madame. Nous avons bien conscience des discriminations qui peuvent exister et de la possibilité d'une répartition inéquitable de l'effort de recherche entre les maladies. Il est par exemple probable que si le paludisme avait été une maladie fréquente en Amérique du Nord et en Europe occidentale, la recherche aurait été plus active. Cette réunion a justement pour but d'identifier les priorités en matière de maladies monogéniques.
M. le président Claude Birraux. Vous pourrez faire savoir, Madame, que l'OPECST s'est saisi de cette question. Initialement, nous pensions étudier le seul cas de la drépanocytose, puis nous avons élargi nos travaux à l'ensemble des maladies rares. Le professeur Leboulch a tracé quelques pistes de recherche. Je retiens des diverses interventions qu'une avancée scientifique et médicale sur une maladie permet souvent de traiter d'autres maladies : c'est une leçon d'espoir.
Merci beaucoup pour votre témoignage, Madame.
M. Jean-Louis Touraine. Nous en venons à Mme Vololona Rabeharisoa, professeur au Centre de sociologie de l'innovation (CSI) de Mines Paris Tech, docteur en socio-économie de l'innovation, et qui s'intéresse en particulier à l'hybridation des savoirs entre génétique et psychiatrie à propos de l'autisme.
Mme Vololona Rabeharisoa, professeur de sociologie, Centre de sociologie de l'innovation (CSI) de Mines-ParisTech. La mobilisation de la recherche par les associations de malades est un sujet auquel je m'intéresse depuis une quinzaine d'années.
Mais tout d'abord, qu'est-ce qu'une association de malades ?
Les associations sont très diverses, tant par les maladies autour desquelles elles se mobilisent que par leur taille, leurs ressources humaines et financières, leur mode d'organisation ou leur gouvernance. Les chercheurs en sciences sociales, français et étrangers, qui s'intéressent aux associations de malades, ont convenu de retenir une définition relativement restrictive des associations de malades, afin de ne pas les confondre avec les associations professionnelles ou avec les associations caritatives du tiers secteur. Nous considérons donc qu'une association de malades est une organisation sans but lucratif, qui regroupe des personnes directement concernées par une maladie, un handicap ou un problème de santé, et au sein de laquelle le pouvoir de décision est entre les mains des personnes concernées.
La mobilisation de la recherche par les associations de malades, dans le monde occidental, est un phénomène récent. C'est à partir des années 1980 et 1990, avec les associations de lutte contre le sida, que les associations ont commencé à s'intéresser à la recherche. Mais les associations ont quantité d'autres missions. En France, celles constituées autour des personnes handicapées ont été longtemps, par délégation de service public, gestionnaires d'établissements. Historiquement, le rôle des associations est d'organiser l'entraide et de défendre les droits des malades et de leurs familles. L'engagement dans la recherche est donc le fait d'associations qui, à un moment de leur histoire, ont jugé nécessaire de contribuer à la lutte contre « leurs » maladies.
La mobilisation de la recherche par les associations doit être entendue au sens large.
Tout d'abord, elle ne concerne pas seulement la recherche génétique ou biologique. Elle porte aussi sur la recherche clinique, translationnelle, thérapeutique, technologique, en sciences humaines et sociales, médico-économique, éthique...
Ensuite, la mobilisation prend des formes très différentes. La forme la plus connue est le soutien financier à des équipes de recherche, mais il faut aussi mentionner la contribution à la collecte du matériau biologique - les gènes des malades par exemple, pour constituer des collections -, le recrutement de patients pour les essais cliniques, ou encore la collecte et la mise en forme d'informations scientifiques et médicales à destination des familles et du grand public.
L'engagement des associations dans la recherche et la lutte contre les maladies repose sur des bases légitimes.
D'une part, les membres des associations ont une expérience singulière de la maladie puisqu'ils la vivent au jour le jour. Les associations constituent donc des « experts d'expérience » et estiment qu'elles peuvent informer les professionnels de certaines manifestations de la maladie que les médecins, parfois, ignorent. Les associations portent là une revendication épistémologique.
Il s'y ajoute une revendication politique. Les associations étant les premiers groupes concernés par la recherche sur « leur maladie », elles réclament légitimement de participer aux actions de recherche menées en leur nom.
Les raisons historiques pour lesquelles les associations concernées par des maladies monogéniques s'intéressent à la recherche ne sont que des déclinaisons des deux raisons précédentes. Un grand nombre de maladies monogéniques sont rares, même si toutes les maladies rares ne sont pas d'origine génétique. Le fait qu'elles affectent des enfants, qu'elles soient mal connues, qu'il n'y ait pas de traitement, ni même de prise en charge adéquate, a conduit les associations à s'investir dans la recherche - parce que peu de chercheurs et de cliniciens s'y intéressaient. Ce n'est pas un hasard si en France, les associations constituées autour des maladies monogéniques sont très impliquées dans la recherche. Dans d'autres pays, comme le Royaume-Uni, le Portugal ou l'Italie, ces maladies sont prises en charge d'une façon différente - et au Royaume-Uni, par exemple, il y a une longue tradition de charities qui apportent un soutien significatif à la recherche médicale.
Les mécanismes au travers desquels les associations mobilisent la recherche sont au nombre de trois.
Le premier est la délégation. Les associations apportent leur soutien à la recherche mais confient aux scientifiques le choix de définir les recherches à soutenir : elles n'interviennent pas dans la définition des projets susceptibles d'aider les malades et les familles. C'est un mécanisme courant. Aux États-Unis, il prend la forme de ce qu'on appelle advocacy research : les lobbys professionnels aident les associations à obtenir que des lignes du budget fédéral soient affectées à un certain nombre de maladies, à charge pour les institutions scientifiques de définir les programmes de recherche sur telle ou telle pathologie.
Le deuxième mécanisme est l'expertise profane. La délégation ne permettant pas de contrôler l'utilisation de l'argent, de plus en plus d'associations se forment à la recherche, afin de pouvoir discuter avec les chercheurs, leur faire des propositions ou, au moins, avoir une lecture critique de ce qui leur est proposé. C'est maintenant un mécanisme général, voire institutionnalisé. Comme l'a dit Mme Bungener, la Mission INSERM-Associations y contribue en proposant des sessions de formation à la recherche aux associations.
Le troisième mécanisme, un peu plus avancé et qui nécessite l'acquisition de l'expertise profane, est le partenariat. Les associations, reconnues comme étant porteuses d'une connaissance particulière, collaborent avec des spécialistes dans des projets de recherche, mènent des enquêtes sur la qualité de vie des patients, participent à la lecture des clinical guidelines, etc. Elles apportent ainsi au monde scientifique à la fois leur force de mobilisation, mais aussi leur propre expertise basée sur leur expérience des maladies.
En conclusion, je tiens tout d'abord à souligner que les associations de malades n'ont pas pour objectif de soutenir la recherche pour la recherche : elles se mobilisent pour défendre un intérêt collectif qu'elles estiment négligé par les acteurs institutionnels. Leur soutien à la recherche ne correspond donc ni au modèle des politiques publiques, ni au modèle du marché, ni au modèle caritatif.
La mobilisation de la recherche par les associations est une action stratégique qui demande des moyens et qui doit s'articuler avec leurs autres missions. Ce choix stratégique peut d'ailleurs être temporaire : elles peuvent décider de s'engager dans le soutien à la recherche pendant un an ou deux avec un objectif précis, puis s'atteler ensuite à d'autres missions.
Enfin, certaines associations de malades ne souhaitent pas s'engager dans la recherche, et certains malades ne souhaitent pas se constituer en association ou adhérer à une association. Ceci est parfaitement légitime, et il est intéressant de comprendre pourquoi. Des études menées par des collègues américains ont montré qu'il y a quelques années aux États-Unis, des malades souffrant de drépanocytose, en majorité Africains-Américains, ne voulaient pas devenir membres d'associations, et encore moins participer à la recherche sur cette maladie. Ils craignaient en effet que la découverte du « gène de la drépanocytose » et son éventuelle qualification comme « gène Africain-Américain » n'accroisse la stigmatisation de cette population. D'une façon générale, une association peut s'abstenir de s'engager dans la recherche biomédicale afin de ne pas mettre en péril l'identité et l'existence sociale des malades.
M. Jean-Louis Touraine. Je donne à présent la parole à M. David Bardey, professeur d'économie à l'Université de Bogota, titulaire d'un doctorat de l'Université de Besançon sur les assurances santé et la concurrence.
M. David Bardey, professeur d'économie à l'Université de Bogota, et « visiting fellow » à l'Ecole d'économie de l'Université de Toulouse. Merci beaucoup de me donner l'opportunité de présenter nos travaux sur les problèmes que peut poser l'existence de tests génétiques sur les marchés d'assurance maladie, que ce soit des marchés au « premier euro » ou des marchés d'assurance complémentaire.
En matière de génétique, deux avancées sont susceptibles d'avoir des conséquences relativement importantes sur la gestion du risque santé. La première est la baisse du coût : le génome pourra bientôt être accessible de façon massive. La deuxième est la personnalisation des traitements, qu'ils soient curatifs ou préventifs. Mais les généticiens semblent travailler beaucoup plus rapidement que les économistes qui s'intéressent à ce domaine : nous sommes un peu « à la traîne » pour comprendre les conséquences de ces avancées sur la gestion du risque santé.
Examinons donc quelles pourraient être les conséquences, dans un avenir qui n'est peut-être pas si éloigné, d'une utilisation massive des tests génétiques par les assurés.
Considérons, dans un premier temps, que les individus ne peuvent pas modifier leur risque. Ils ont la possibilité de faire des tests génétiques qui vont les classer dans les « bons risques » ou les « mauvais risques ». La théorie du risque nous enseigne que la valeur de l'information est négative ; les tests génétiques peuvent donc réduire le bien-être économique des assurés.
En effet, avant de réaliser un test génétique, les individus sont caractérisés par un risque moyen. Or, s'ils s'assurent, c'est précisément parce qu'ils ont de l'« aversion à l'égard du risque ». Par conséquent, leur bien-être économique est supérieur lorsqu'ils ne prennent pas le risque de devenir un « mauvais risque » aux yeux des assureurs, ce qui se traduirait par des contrats beaucoup plus onéreux. C'est ce que les économistes ont coutume d'appeler « le bonheur derrière le voile d'ignorance ».
L'information fournie par les tests génétiques peut conduire à une variation des conditions dans lesquelles la personne va être assurée. Du côté des assureurs, on craint des phénomènes d'anti-sélection - les individus qui ont obtenu par des tests une information sur leur risque santé choisissant leur couverture en conséquence -, susceptibles de déstabiliser les marchés d'assurance, comme l'ont montré des chercheurs aussi confirmés que Rothschild et Stiglitz.
Finalement, un arbitrage se fait entre une efficacité ex ante - on préfère économiquement sa situation avant le test - et une efficacité ex post. le fait que les assurés peuvent entreprendre des actions qui leur permettent de réduire leur risque.
La meilleure des situations peut aujourd'hui paraître de faire le test, et de garder le résultat pour soi s'il révèle un risque supérieur à la moyenne. Mais si dans cinq ou dix ans, les tests génétiques deviennent une pratique courante, alors les personnes qui ne les produiront pas devant leur assureur santé seront suspectées d'avoir un risque plus élevé que la moyenne.
Les tests génétiques peuvent également donner une information utile pour entreprendre des actions de prévention primaire - permettant de réduire la probabilité de certaines maladies - et des actions de prévention secondaire - dépistage, ou encore pour personnaliser les traitements.
Bien entendu, l'efficacité de la prévention dépend du niveau de risque de la personne, révélé par le test. La valeur économique de l'information peut alors redevenir positive si la réduction du risque liée à la prévention est suffisante. Mais les efforts de prévention ne sont pas observables par les assureurs. Ceux-ci doivent inciter les assurés à effectuer des actes de prévention - par des franchises ou des co-paiements. Par ailleurs, les contrats d'assurance santé ne couvrent pas l'intégralité du risque. Cette couverture partielle peut encore réduire la valeur de l'information des tests génétiques.
En résumé, d'un point de vue économique, la valeur de l'information fournie par les tests génétiques est négative en l'absence de prévention ; elle peut redevenir positive si la prévention est suffisamment efficace ; mais elle peut à nouveau diminuer en raison de problèmes d'asymétrie d'information liés au risque moral sur les marchés d'assurance santé.
Les politiques publiques qui réduisent les coûts de la prévention peuvent avoir des effets ambigus. En effet la décision de faire un test n'est pas sans lien avec le coût de la prévention. Si ce coût est très faible, les individus ne seront pas incités à faire le test puisqu'ils pratiqueront de toute façon cette prévention ; s'il est très élevé, ils ne feront pas non plus le test car ils renonceront de toute façon à la prévention ; mais s'il se situe à un niveau intermédiaire, c'est le test génétique qui déterminera la réalisation des actes de prévention.
Quelle législation envisager concernant l'usage des tests génétiques ?
La législation ne peut pas être la même si l'usage des tests génétiques devient commun ou s'il concerne un très faible pourcentage de la population. Dans le premier cas, les individus ne pourront plus affirmer qu'ils n'ont pas fait le test car leur assureur les soupçonnera de cacher des résultats négatifs (négatif au sens économique du terme).
Le premier objectif que doit poursuivre la législation me paraît être de protéger la société contre la discrimination génétique. Personne n'étant responsable de ses gènes, cet objectif fait consensus.
Le deuxième objectif devrait être d'encourager l'usage de la génétique à des fins thérapeutiques et de prévention. Ce n'est pas parce que la valeur de l'information des tests génétiques peut être négative d'un point de vue économique qu'il faut oublier les autres dimensions de ces tests.
En troisième lieu, il faudrait faire en sorte d'éviter les problèmes d'anti-sélection sur les marchés d'assurance : il ne faut pas que les contrats d'assurance santé se trouvent totalement déséquilibrés par l'information privée obtenue par les assurés grâce aux tests.
Les différentes régulations en vigueur ne permettent pas de concrétiser ces différents objectifs. Elles s'attachent principalement à lier les mains des assureurs, c'est-à-dire à éviter qu'ils ne demandent systématiquement les tests génétiques à leurs assurés. Elles ne prennent pas en compte le comportement « proactif » des assurés ; si dans cinq ou dix ans, la plupart des individus présentent le résultat de leurs tests, la situation sera totalement différente, et il faudra impérativement repenser les législations actuelles.
Il faut éviter le piège de l'opposition classique entre la protection des assureurs et la protection les assurés. Les problèmes d'anti-sélection dont peuvent pâtir les assureurs finissent par se retourner contre les assurés. Il est donc nécessaire, à la fois, de protéger les assurés contre la discrimination génétique et de permettre aux marchés d'assurance de continuer à fonctionner. Quand le voile d'ignorance que se partagent les assurés et les assureurs tend à se déchirer de par la possibilité d'effectuer des tests génétiques, il faut élaborer de nouvelles législations.
M. Jean-Louis Touraine. Votre souci de protéger le fonctionnement des assurances est tout à fait légitime. Pour autant, la préoccupation première du législateur est de protéger l'individu. Nous préférerions maintenir une formule de solidarité par mutualisation. Sinon, nous nous acheminons vers un monde où les différents risques seront si bien identifiés que toute une frange de la population sera très malheureuse. Mais vous avez raison : il faudra adapter les textes aux évolutions à venir ; il est évidemment très différent de faire des tests pour chercher un petit nombre de choses au sein d'une catégorie limitée de personnes, ou d'en faire dans toute la population pour identifier la totalité des facteurs de risque. Cette deuxième voie serait quelque peu effrayante, mais nécessiterait de continuer à protéger l'individu, afin qu'il puisse, malgré telle ou telle faiblesse, trouver un emploi, contracter un emprunt pour se loger ou souscrire une assurance.
M. le président Claude Birraux. Je suis tout à fait d'accord : le rôle des politiques est d'assurer l'égalité des citoyens, en organisant la solidarité en faveur des faibles et des moins bien portants. Nous connaissons ce principe de répartition aussi bien dans les assurances que dans les systèmes de retraite.
La généralisation des tests génétiques pourrait certes avoir de lourdes conséquences. L'OPECST avait déjà mis en garde contre les tests sur Internet, dont la fiabilité n'est pas garantie. Par ailleurs, nous nous devons de protéger l'individu contre les inégalités, notamment devant l'emploi : comment faire pour qu'un employeur engage une personne s'il peut déduire de son profil génétique qu'elle sera absente une semaine par mois à partir de 40 ans ? C'est le rôle du politique de réfléchir à cela.
Dans le cadre d'une étude sur l'innovation face aux peurs et aux risques, Jean-Yves Le Déaut et moi-même avons rencontré dans nos circonscriptions de jeunes lycéens. Je leur ai expliqué le risque que représentera, à l'avenir, la possibilité pour des sociétés spécialisées de dégager leur profil psychologique à partir de celui qu'ils auront eux-mêmes mis sur les réseaux sociaux, d'en déduire leur aptitude à exercer tel ou tel travail et de décider de ne pas les embaucher.
Comment assurer une égale protection à tous nos concitoyens ? Cette question, peut-être la plus importante qui est posée aux politiques, ne concerne pas que les assurances. Une banque a été condamnée par la Cour de cassation pour avoir refusé d'accorder un petit prêt à un retraité sous prétexte qu'il avait 75 ans - prêt qu'il avait les moyens de rembourser et dont il avait besoin pour réaliser des travaux dans sa maison. On pourrait transposer cette jurisprudence dans le domaine de l'assurance en matière de profils génétiques.
M. David Bardey. Je partage totalement votre préoccupation. L'objectif est d'éviter toute discrimination génétique.
La législation est adaptée à la situation actuelle. Mais le sera-t-elle encore dans le futur, avec des assurés de plus en plus proactifs, qui multiplient les tests génétiques ? Pour l'instant, on lie les mains aux assureurs, et les choses se passent bien. Mais ne faudra-t-il pas également une régulation du côté des assurés, pour éviter un déséquilibre ? Il me semble nécessaire d'anticiper les problèmes futurs car, pour le bien-être économique des assurés, il faut que les marchés fonctionnent.
M. le président Claude Birraux. Madame Cambon-Thomsen, pourriez-vous nous dire pourquoi la France n'a pas encore ratifié le protocole additionnel à la Convention d'Oviedo ?
Vous avez dit que les équipes françaises ne pilotent pas les quatre projets européens, à l'exception des aspects médico-sociaux. Mais ces équipes ne sont-elles pas très spécialisées, alors que les appels européens sont formulés en termes très généraux ?
Mme Anne Cambon-Thomsen. La France ne peut pas signer et ratifier le protocole additionnel car elle n'a pas encore signé et ratifié la Convention d'Oviedo elle-même. Une des raisons pour lesquelles elle n'a pas signé la Convention est l'interprétation à donner à certains articles, susceptibles d'empêcher certains types d'engagement ou de recherche. Le problème a été discuté par différents pays mais paraît désormais levé. D'après mes informations, il est donc prévu que la France signe la Convention ; il serait assez logique qu'elle signe également les protocoles additionnels.
Les projets européens dont j'ai parlé portent sur le séquençage, sujet sur lequel d'autres pays que la France sont sur le front. Mais il existe bien d'autres projets européens qui sont pilotés par des Français.
Permettez-moi de poser une question à notre collègue économiste.
Actuellement, la plupart des tests disponibles sur Internet portent sur des maladies qui ne sont pas monogéniques : ce sont des tests de susceptibilité, dont on ne peut pas faire grand-chose au niveau individuel et dont l'interprétation peut changer d'un jour à l'autre en fonction des publications qui paraissent. Il existe même une société qui réinterprète les mêmes tests au fur et à mesure de l'apparition de nouvelles publications, si bien qu'une personne peut être classée à risque un jour pour telle maladie, et considérée protégée six mois après. Mais tel que vous le présentez, monsieur Bardey, le risque est toujours négatif ; or dans notre génome, nous avons autant de gènes qui nous protègent que de gènes qui nous fragilisent - et l'interprétation est particulièrement difficile...
M. David Bardey. En parlant des comportements de prévention, primaire ou secondaire, qui peuvent respectivement réduire soit la probabilité d'occurrence d'une maladie, soit les conséquences de la maladie, je voulais nuancer ma vision pessimiste d'économiste sur le caractère plutôt négatif de la valeur d'information des tests, économiquement parlant. Cette valeur peut devenir positive s'il y a changement de comportement. Une personne qui présente un terrain favorable aux maladies cardiovasculaires adoptera plus facilement un comportement de prévention - comme s'abstenir de fumer - si elle a fait le test. Mais si l'on ne prend pas en compte les comportements de prévention, du point de vue des économistes il faut éviter les tests.
D'un point de vue sanitaire, bien sûr, ces tests génétiques apportent des informations précieuses. Certes, sur Internet, beaucoup laissent à désirer, mais je pense qu'une régulation sera mise en place ou que le marché opérera une sélection entre eux , ce qui nous permettra de disposer de tests assez fiables dans un futur assez proche.
M. Jean-Louis Touraine. Il faudra reprendre cette discussion dans quelques années...
Madame Cambon-Thomsen, la réglementation en matière d'accès aux tests génétiques est très différente d'un pays à l'autre, y compris à l'intérieur de l'Europe. On peut très bien aller faire ailleurs un test qui est interdit chez soi. Comment régler ce problème ?
Mme Anne Cambon-Thomsen. Ce sujet a fait l'objet de discussions dans divers cercles.
La mobilisation de la législation sur la consommation et l'information des personnes constituent une forme de contrôle. Dire que des tests qui ne servent à rien sont utiles est une fausse information qui peut être combattue ! Et si l'on dit d'emblée qu'ils ne servent à rien, on peut faire confiance aux consommateurs pour en tirer les conclusions... On voit mal comment une loi pourrait réguler la vente des tests sur Internet ; je pense donc que l'essentiel est d'apporter aux consommateurs une information exacte, indépendante des sociétés qui vendent les tests.
M. Jean-Louis Touraine. Qu'en est-il des tests de paternité ?
Mme Anne Cambon-Thomsen. Même s'il lui est interdit d'utiliser les résultats, je ne vois pas comment on peut empêcher une personne qui veut savoir si elle va aller ou non au procès de faire un test de paternité dans un autre pays.
Mme Martine Bungener. Ce test-là a une valeur pour la personne ; ce n'est pas la même chose qu'un test de prédisposition.
M. Jean-Louis Touraine. Il reste qu'on doit tenir compte de l'environnement international.
LES INTERROGATIONS JURIDIQUES ET ÉTHIQUES
M. Jean-Louis Touraine. Pour aborder notre dernière table ronde, je donne la parole à M. Bertrand Mathieu, agrégé de droit public, professeur à l'École de droit de la Sorbonne - Université de Paris I.
M. Bertrand Mathieu, professeur à l'École de droit de la Sorbonne (Université Paris I), membre du Conseil supérieur de la magistrature, président de l'Association française de droit constitutionnel. En France, les dispositions réglementaires précisant les hypothèses dans lesquelles on peut recourir à l'examen des caractéristiques génétiques ont pour pendant l'article du code civil selon lequel « nul ne peut faire l'objet de discriminations en raison de ses caractéristiques génétiques », prescription reprise dans le code pénal, le code du travail, le code des assurances et le code de la sécurité sociale. Cette énumération est elle-même significative : si l'on affirme un principe, c'est parce que l'on ressent qu'il est menacé.
Plusieurs principes, notamment constitutionnels, sont en jeu. Les principes du droit au travail, de non-discrimination, de respect de la vie privée doivent être conciliés avec ceux de sécurité des salariés et de liberté contractuelle.
On retrouve la même construction dans les textes internationaux. La Déclaration sur le génome humain de l'UNESCO interdit les discriminations fondées sur ses caractéristiques génétiques. La Convention bioéthique du Conseil de l'Europe prohibe toute forme de discrimination à l'encontre d'une personne en raison de son patrimoine génétique ; il est précisé qu'il ne peut être dérogé à ce principe alors même qu'il s'agirait de « mesures nécessaires, dans une société démocratique, à la sûreté publique, à la prévention des infractions pénales, à la protection de la santé publique ou à la protection des droits et libertés d'autrui ». Cependant, le rapport explicatif fait état de l'ambiguïté de la rédaction de la version anglaise du texte, qui ne vise que les « discriminations injustifiées » : or si l'on sait ce qu'est une discrimination, on sait moins ce qu'est une discrimination injustifiée ! De manière plus générale, le recours à la notion anglo-saxonne de discrimination, comme se référant seulement aux discriminations injustifiées, est susceptible d'affaiblir la portée de l'interdiction.
Le protocole additionnel à cette Convention, relatif aux tests génétiques à des fins médicales, prohibe toute forme de discrimination à l'encontre d'une personne en raison de son patrimoine génétique.
Par ailleurs, une résolution de 1989 du Parlement européen préconise l'interdiction de l'usage de tests génétiques en matière d'emploi et d'assurance.
Ainsi, sur le plan moral, le principe de non-discrimination semble relativement clair. Cependant, sur le plan juridique, les choses sont un peu plus compliquées.
En effet, le droit applicable à l'utilisation des tests génétiques est plus flou qu'une lecture rapide des textes pertinents pourrait le laisser supposer. Par ailleurs, l'utilisation de tests génétiques concernant une personne par un tiers doit être envisagée à plusieurs niveaux : Soit un employeur ou un assureur peut exiger d'être informé des résultats d'un test génétique ; soit l'employeur ou l'assureur peut demander à la personne de subir un test génétique ; soit encore la personne est autorisée à divulguer spontanément ses propres caractéristiques génétiques - ce qui peut conduire aux mêmes conséquences que la possibilité d'exiger des informations génétiques car, dès lors que l'accès aux tests est aisé, le fait qu'une personne ne divulgue pas les résultats sera nécessairement considéré comme une information défavorable.
En matière d'emploi, l'interdiction de recourir à des tests génétiques est fondée sur l'interdiction des discriminations opérées à partir des caractéristiques génétiques. Cependant le recours aux résultats de tests génétiques est susceptible d'être légitimé dans deux hypothèses : d'une part, lorsqu'un test génétique démontrera l'existence d'une prédisposition particulière à une maladie liée à l'environnement de travail ; d'autre part, lorsqu'une prédisposition génétique est susceptible de révéler un risque de danger pour autrui - par exemple chez les pilotes de ligne ou les chauffeurs routiers. Dans ces hypothèses, la connaissance de la prédisposition génétique a pour objet non pas d'opérer une discrimination entre des individus, mais de protéger les droits de l'individu lui-même ou la vie d'autres individus.
Le Comité européen d'éthique, dans un avis de 2003, a analysé de manière prospective les conditions d'une réglementation et d'un encadrement au niveau européen du recours aux tests génétiques en matière d'emploi. Autrement dit, si un jour les tests sont fiables, on les utilisera ; ce qui conduit à affirmer que, premièrement, il est interdit de discriminer, mais que, deuxièmement, on recherche les moyens de discriminer le moins possible !
Si l'on fait abstraction de la question de la protection des droits d'autrui, il reste à s'interroger sur le point de savoir jusqu'à quel point on peut protéger une personne contre elle-même et opérer à sa place un choix entre le risque de développer certaines pathologies et la perte d'un emploi ou l'abandon d'une profession.
En matière d'assurance, la question de la discrimination génétique se pose dans des termes voisins. Cependant, les intérêts liés à la protection d'autrui ou de l'intéressé ne sont pas ici des justificatifs pertinents à l'utilisation des tests. Sont en jeu, dans ce cas, l'intérêt des assureurs et, si l'assureur est la sécurité sociale, celui de la collectivité.
En l'état, il est interdit aux assureurs d'utiliser les résultats de tels tests, quelle que soit la manière dont ils sont susceptibles de se les procurer. Mais pourquoi faut-il réserver un sort particulier à ces tests ? Plusieurs réponses sont possibles.
On peut considérer les informations génétiques comme échappant par nature au domaine des informations auxquelles l'assureur peut avoir accès. Cette solution très protectrice se heurte cependant à certaines objections. D'une part, elle conduit à opérer une distinction incertaine entre les maladies génétiques et les autres maladies : pourquoi pourrait-on être obligé de fournir des informations relatives, par exemple, aux maladies des parents, et ne pas pouvoir donner des informations génétiques ? On n'est pas plus responsable des unes que des autres... D'autre part, cette solution se heurte à un environnement juridique favorable à la communication à l'assureur d'informations relatives à la santé : en France, le code pénal exclut expressément du cadre des infractions pénales « les discriminations fondées sur l'état de santé, lorsqu'elles consistent en des opérations ayant pour objet la prévention et la couverture du risque décès, des risques portant atteinte à l'intégrité physique de la personne ou des risques d'incapacité de travail ou d'invalidité ». Dans toutes ces hypothèses, la discrimination génétique est permise. Par ailleurs, la Convention bioéthique du Conseil de l'Europe ne s'oppose pas à ce qu'un candidat à l'assurance fasse spontanément état de tests génétiques favorables.
Une autre logique peut conduire à distinguer les différents types d'assurance. La possibilité offerte à l'assureur de connaître l'ensemble des données disponibles relatives à l'état de santé relève de la logique du contrat de droit privé et du mécanisme même de l'assurance, qui implique notamment la bonne foi et l'égalité de traitement entre les assurés. L'interdiction faite à l'assureur de connaître les résultats de tests génétiques obéit à une logique de protection des droits de l'individu, notamment de son droit à ne pas faire l'objet de discrimination. Or le principe de non-discrimination ne joue pas de manière générale dans le droit des assurances, même en matière de santé. En revanche, l'accès aux soins représente un droit fondamental pour lequel il ne peut être opéré de distinction. Il est assez logique qu'une personne souscrivant une assurance-vie soit conduite à faire état des données relatives à leur santé ; et de ce point de vue, les données génétiques ne sont que des données parmi d'autres. Les assurances liées à l'accès au logement doivent obéir à la même logique, dès lors que l'accès au logement est un objectif constitutionnel. Il appartient à la société de définir ce qui relève de la solidarité et ce qui relève des lois du marché.
En tout cas, le développement des tests génétiques s'inscrit dans une logique de renforcement du contrôle social sur l'individu, sujet qu'il conviendra d'approfondir.
M. Jean-Louis Touraine. Merci beaucoup. Je donne maintenant la parole à Mme Emmanuelle Prada-Bordenave, directrice générale de l'Agence de la biomédecine.
Mme Emmanuelle Prada-Bordenave, directrice générale de l'Agence de la biomédecine. Les missions de l'Agence de la biomédecine sont définies aux articles L.1418-1 et suivants du code de santé publique, au sein duquel est codifiée la loi de bioéthique, actuellement réexaminée par le législateur.
En matière de génétique, seule la génétique médicale fait partie des missions de l'Agence de la biomédecine. Nous ne sommes pas compétents sur les aspects judiciaires ou d'identification des personnes. Nous ne le sommes pas non plus pour contrôler ou censurer les dérapages de l'activité de génétique.
Nous envisageons la génétique médicale comme une activité médicale au service des patients. Comme dans les autres domaines dont elle a la charge, l'Agence a une mission d'encadrement, par la définition de règles ; d'accompagnement des professionnels dans leur travail ; d'évaluation de l'activité et des résultats de ces professionnels ; enfin, d'information du Parlement et du Gouvernement, mais aussi de la société à travers les médias et la presse. L'information est en effet une ardente obligation en matière de génétique -car celle-ci quitte à peine le laboratoire pour devenir une science médicale.
En ce qui concerne les maladies monogéniques, les activités de l'Agence sont au nombre de trois : le diagnostic préimplantatoire, qui a concerné 71 naissances en 2008 ; le diagnostic prénatal, avec 3150 tests de maladies aujourd'hui disponibles et proposés aux couples ; le diagnostic postnatal, tout au long de la vie, réalisé dans près de 862 laboratoires et qui concerne en France 1 060 maladies, ayant donné lieu à environ 270 000 analyses de génétique moléculaire en 2 009 - je ne parle pas ici de la cytogénétique, qui ne concerne pas les maladies monogéniques.
Jusqu'à présent, l'Agence a eu une activité d'agrément des praticiens des laboratoires de génétique postnatale. Elle va continuer à l'exercer. Néanmoins, en la matière, le médecin prescripteur - qui informe, conseille, rend le résultat et apporte des informations sur la maladie au vu du résultat rendu - a un rôle essentiel.
Même si nous l'envisageons à la lumière des activités que nous encadrons depuis longtemps - comme la greffe ou l'assistance médicale à la procréation -, la génétique médicale est une discipline tout à fait nouvelle. Elle fait rencontrer des patients symptomatiques, c'est-à-dire malades, mais aussi des patients asymptomatiques - et c'est cela qui est nouveau -, c'est-à-dire des personnes qui ne sont pas, qui ne se sentent pas malades.
La prise en charge de ces dernières doit donner lieu à une véritable réflexion. Comment accueillir une personne asymptomatique susceptible d'être porteuse de la maladie et, s'il s'agit d'une maladie récessive, de la transmettre ? Comment lui annoncer qu'elle est porteuse d'une maladie et la prendre en charge ? Comment la conseiller sur la suite à donner pour elle-même, mais aussi pour ses proches ? Des consultations pluridisciplinaires se mettent en place, ce qui est souvent une nouveauté pour les établissements hospitaliers. Les équipes pluridisciplinaires doivent se déclarer à l'Agence, mais n'ont commencé à le faire que timidement.
Je voudrais insister sur le lien entre la recherche et la médecine en matière de génétique. S'agissant de la génétique moléculaire portant sur les maladies rares, le passage n'est pas encore terminé entre le laboratoire de recherche et l'équipe médicale. Cela pose des problèmes, notamment d'accès aux soins. L'Agence doit donc s'interroger sur la meilleure façon de rendre disponibles des tests tout récents, qui viennent d'être découverts par des équipes de recherche. Devons-nous forcer la main à ces équipes pour que ces tests soient très rapidement diffusés dans un grand nombre de laboratoires ? Ou devons-nous, s'agissant de maladies rares, nous contenter d'adresser les patients à ces laboratoires, qui ne sont pas encore de vraies équipes médicales, mais qui savent bien faire les tests, pour permettre à ces patients de disposer le plus rapidement possible d'un test fiable ?
Il convient également de s'interroger sur le rôle du réseau. Devons-nous développer des laboratoires nombreux, avec le risque qu'ils soient moins compétents ? Devons-nous, au contraire, nous orienter vers une politique de réseau ? C'est la voie que notre pays a privilégiée jusqu'à présent. Maintenant, le relais doit être assuré notamment par les schémas régionaux d'organisation sanitaire en matière de génétique, en cours d'élaboration.
Deux évolutions techniques suscitent de la part de l'Agence quelques inquiétudes.
La première est le développement très rapide de l'analyse du génome entier - même si cela nous fait un peu sortir de la problématique des maladies monogéniques. Que devra-t-on faire si, en voulant soigner un patient atteint d'une maladie, l'analyse de son génome amène à trouver autre chose ? Nous devons réfléchir avec les professionnels à la conduite à tenir face à ces découvertes fortuites.
La deuxième est la génétique « récréative », c'est-à-dire les tests sur Internet. Pour qu'ils permettent de gagner de l'argent, on ne les cible pas sur des maladies très rares... Ils portent donc sur diverses prédispositions, mais ils nous posent de vrais problèmes, à nous agence de régulation, en termes d'accompagnement des patients. Qui interprétera le résultat ? Quel médecin prendra en charge le patient si quelque chose est découvert ? Comment éviter l'errance diagnostique - autrement dit que la personne avec son test ne sache pas à qui s'adresser ? Enfin, quelle est l'utilité clinique d'un test qui révèle une prédisposition dont on ne sait pas quoi faire ?
Face à cela, le rôle de l'Agence reste constant : rédiger des règles de bonnes pratiques en génétique pour les professionnels, diffuser ces règles et l'information qui les accompagne à la population tout entière, afin que celle-ci soit bien informée sur les conditions de passage de la recherche au diagnostic, sur les conditions de l'accès aux soins en génétique, enfin sur les modalités d'information du patient et de recueil de son consentement, un rôle majeur revenant au médecin. La génétique est en effet de la médecine et doit le rester ; le médecin doit accompagner le patient, qu'il ne doit pas laisser seul face à une maladie, une suspicion de maladie ou une prédisposition à une maladie. L'exclusion de la génétique médicale du champ de la médecine aboutirait, en termes de santé publique, à des résultats très mauvais et très coûteux. Il s'agit donc là d'une responsabilité lourde pour l'Agence.
M. Jean-Louis Touraine. Merci beaucoup, Madame.
Nous allons maintenant entendre Mme Dominique Stoppa-Lyonnet, membre du Comité consultatif national d'éthique (CCNE), professeur de génétique, chef du service de génétique oncologique de l'Institut Curie.
Mme Dominique Stoppa-Lyonnet, membre du Comité consultatif national d'éthique (CCNE), professeur de génétique, chef du service de génétique oncologique de l'Institut Curie. Entre 1985 et 2009, le Comité national d'éthique a rendu 12 avis touchant aux tests génétiques, qu'il s'agisse de diagnostic préimplantatoire, de diagnostic prénatal ou de test postnatal sur les prédispositions aux maladies. L'ensemble de ces travaux fait ressortir l'importance de l'information concernant les enjeux et les limites des tests génétiques. C'est une information un peu particulière puisqu'elle porte sur des éléments constitutifs de l'individu - une information sur l'être plus que sur l'avoir, qui relie l'individu à sa famille et qui dit quelque chose de son destin. On voit d'ailleurs aujourd'hui le succès sur Internet du test sur la longueur des télomères : le fait qu'elle pourrait être proportionnelle à l'espérance de vie a été aussitôt traduit par les médias en possibilité de connaître l'heure de la mort. Je ne reviens pas sur les risques de discrimination, qui ont été évoqués. Information, autonomie, consentement sont les notions les plus présentes dans les différents avis.
Le CCNE s'est par ailleurs autosaisi du problème des tests génétiques de susceptibilité - multigéniques. Ce travail va démarrer.
Quels sont les problèmes éthiques soulevés par les tests génétiques ?
Tout d'abord, il faut impérativement s'interroger à la fois sur la validité scientifique du test et sur son utilité clinique. Dans le cas des tests de susceptibilité, la validité scientifique est très grande, mais l'utilité clinique est quasi-nulle. Pour les maladies monogéniques, des gènes sont identifiés, mais on ne sait pas encore précisément quelle va être leur pénétrance - c'est-à-dire le risque de maladie associée ; pourtant, on pressent bien que ce sont des gènes importants pour la santé. J'insiste donc sur l'importance de l'épidémiologie génétique : il est crucial d'examiner les manifestations associées à la mutation d'un gène. Cela suppose la participation des patients : c'est grâce à eux, il faut en être bien conscient, que les gènes ont pu être identifiés et que la recherche peut progresser.
En deuxième lieu, il apparaît, notamment avec le séquençage, que des gènes de maladie présentent des variations génétiques, dont on ne connaît pas toujours la signification biologique. La compréhension de ces « variants » doit devenir un sujet très important de recherche clinique et de recherche fondamentale ; on parle maintenant de génomique ou génétique fonctionnelle.
S'agissant des résultats d'un test, se pose tout d'abord le problème de la diffusion de l'information à la parentèle. Les dispositions proposées dans le cadre du projet de loi bioéthique devraient constituer un progrès, mais tout n'est pas réglé - notamment le cas où une personne ne souhaite pas la diffusion d'une information qui pourrait être importante pour ses apparentés.
Autre problème : les tests génétiques pour les maladies monogéniques sont en train de sortir de l'histoire familiale. Jusqu'à présent, on répondait à une demande ; c'est ainsi qu'ont été mis en place les tests sur la maladie de Huntington, dans des conditions très encadrées. Nous sommes maintenant dans une situation nouvelle : des facteurs génétiques vont être identifiés chez des personnes qui n'avaient pas demandé grand-chose. Ainsi en va-t-il pour le diagnostic postnatal de mucoviscidose, avec les tests génétiques à rebours chez les parents puis les apparentés des sujets porteurs. C'est toute la problématique des tests pratiqués chez les porteurs sains ; l'avis n° 97 du CCNE, relatif aux questions éthiques posées par la délivrance de l'information génétique néonatale à l'occasion du dépistage de maladies génétiques , témoigne d'une très grande prudence quant à la communication aux parents des résultats des tests génétiques pratiqués sur l'enfant. Bien entendu, il est difficile de s'assurer que les parents seront en mesure de décider s'ils veulent savoir ou ne pas savoir. On peut aussi citer l'exemple des études « génome entier » ou « exome » : on identifie des gènes altérés qui, dans le cas d'une maladie dominante, concernent la personne elle-même, mais qui, dans celui d'une maladie récessive, pourront concerner sa descendance ; on pourrait ainsi être amené, si ces personnes ont un projet parental, à proposer un test au conjoint pour savoir s'il est également porteur de l'altération.
Ces tests sont aujourd'hui réalisés dans un cadre de recherche. L'anticipation est sans doute le maître mot : il s'agit d'anticiper les données génétiques incidentes non recherchées. Par ailleurs, on va faire des progrès dans l'analyse du génome ; il va falloir anticiper le fait que l'interprétation du génome est appelée à évoluer.
En ce qui concerne les maladies récessives, une question m'est très souvent posée dans ma pratique. Un enfant est atteint d'une maladie récessive ; on identifie la mutation à l'état hétérozygote chez un frère ou une soeur, à l'âge adulte, qui a un projet parental ; que doit-on dire à son conjoint ? Faut-il qu'il fasse un test, même si le risque qu'il soit porteur est faible ? Pour répondre, il faut réfléchir au coût, et par ailleurs être conscient que l'on peut trouver des variants, de signification biologique inconnue, dont on ne va pas savoir quoi faire.
M. le président Claude Birraux. M. Mathieu a évoqué les problèmes de traduction de la Convention du Conseil de l'Europe, mais il existe une différence plus fondamentale encore entre les Britanniques et les Français : en Grande-Bretagne, il n'y a pas de règlements ni de circulaires d'interprétation ; c'est la jurisprudence qui vaut règlement. La vision britannique du droit est toujours en perspective.
M. Bertrand Mathieu. La Cour européenne des droits de l'homme raisonne de plus en plus comme vous dites, c'est-à-dire en considérant que la Convention européenne des droits de l'homme n'est qu'un texte attributif de compétences, qu'elle interprète totalement librement. Il suffit donc pour elle d'estimer que l'équilibre entre plusieurs droits fondamentaux n'est pas assuré comme il convient en France pour qu'elle lève certaines interdictions : nous ne sommes pas du tout à l'abri de cette évolution.
Mme Emmanuelle Prada-Bordenave. Permettez-moi une dernière remarque : nous n'avons pas en France beaucoup de médecins généticiens compétents ; il ne faut pas gaspiller cette ressource rare en l'utilisant à des fins de génétique récréative. Le colloque qui s'est tenu il y a deux ans sur le diagnostic prénatal a bien montré qu'en France, comme le veut la loi, ce diagnostic est réservé aux maladies graves et incurables, alors qu'aux Etats-Unis on n'hésite pas à dépenser beaucoup d'argent pour des designer babies . Nous devons veiller à ce que les ressources dédiées à la génétique dans notre pays ne soient pas, par un dérapage insidieux, détournées des personnes qui en ont vraiment besoin.
M. le président Claude Birraux. Nous en sommes certainement tous pleinement d'accord.
Merci à tous pour votre participation à cette audition, dont nous ne manquerons pas de tirer les conclusions.
CONCLUSIONS ET RECOMMANDATIONS (EXTRAIT DE LA RÉUNION DE L'OPECST DU 25 JANVIER 2012)
PRÉSENTATION DES CONCLUSIONS DE L'AUDITION PUBLIQUE SUR LES MALADIES MONOGÉNIQUES
M. Claude Birraux. Saisi par la commission des affaires sociales de l'Assemblée nationale d'une étude sur la drépanocytose, l'OPECST a souhaité élargir cette saisine à l'ensemble des maladies monogéniques. Une audition publique ouverte à la presse intitulée « Les maladies monogéniques : état des lieux », a été organisée le 7 juin 2011 par Claude Birraux, député, Président de l'OPECST et Jean-Louis Touraine, député. Elle a réuni des chercheurs de domaines variés et des représentants d'associations de malades pour aborder aussi bien les aspects scientifiques et médicaux de ces maladies que leurs enjeux sociétaux et juridiques.
Cette approche pluridisciplinaire était essentielle : en effet, les maladies monogéniques se développent dès l'enfance et ne concernent pas seulement la personne affectée, ou celle qui risque de le devenir un jour, mais aussi son entourage, notamment sa famille, qui se trouve confrontée à de multiples difficultés d'accompagnement et de soins du malade ainsi qu'à des risques de stigmatisation liés à l'origine génétique de la maladie.
L'audition publique a montré qu'il s'agissait d'un phénomène d'une ampleur non négligeable, nécessitant des efforts de recherche dans de multiples directions et soulevant des problèmes éthiques et juridiques.
M. Jean-Louis Touraine. Si certaines de ces maladies sont par elles-mêmes rares, leur très grand nombre fait qu'au total, les maladies monogéniques, toutes formes comprises, sont aussi fréquentes que le cancer et concernent des dizaines de milliers de personnes, elles représentent un enjeu de santé publique important. En fait, le qualificatif de « rare » est impropre, nuit à leur prise en charge et les pénalise. Ces maladies sont en réalité fort nombreuses. Auparavant, elles étaient même souvent mortelles avant l'âge de la procréation et leur fréquence a augmenté grâce aux progrès de la médecine qui ont contribué à limiter la sélection naturelle.
La drépanocytose, objet de la saisine, est la maladie génétique la plus répandue en France : on compte aujourd'hui 15 000 malades et leur nombre devrait atteindre 20 000 à l'horizon 2020. Au-delà de son coût financier pour notre système de santé, la qualité de vie des malades est très fortement dégradée. Bien des soins pourtant indispensables ne sont pas pris en charge par l'assurance maladie. L'insertion sociale des patients atteints de cette maladie en termes d'éducation et d'accès à l'emploi est difficile, de même que celle de leur entourage, notamment des mères qui doivent prodiguer des soins continus à leurs enfants lorsqu'ils sont atteints. Les malades de la drépanocytose s'estiment victimes de discriminations.
Les données disponibles d'un fichier Medicaid de Floride établissent qu'en 2009, la prise en charge d'un patient drépanocytaire revenait aux États-Unis à 2 000 $ par mois environ, tous âges confondus, avec un coût annuel moyen de plus de 10 000$ pour les enfants, atteignant près de 35 000 $ pour les sujets plus âgés ; le coût total s'élève, pour une espérance de vie moyenne de 45 ans, à plus de 950 000 $ par patient. Ces données s'appliquent aussi en Europe. En France, 80% des coûts totaux sont consacrés à l'hospitalisation, 3,2% aux passages aux urgences, 0,9% à la consultation de généralistes, 3,6% aux médicaments, 11,7% à d'autres soins infirmiers ou médicaux spécialisés.
Les maladies monogéniques, parce qu'elles constituent en quelque sorte des « expériences de la nature », qui permettent de comprendre la signification d'un gène, peuvent aussi servir de modèle pour le développement de nouvelles thérapeutiques, y compris pour des maladies fréquentes, comme le cancer, le diabète ou le sida. Les traitements, qu'ils relèvent de la pharmacologie classique, de la thérapie génique, ou de la thérapie cellulaire, et ou de la thérapie enzymatique sont complémentaires et peuvent être successifs. Même si certaines de ces thérapies innovantes n'ont pas encore fait la preuve d'une efficacité totale, elles sont utiles dès maintenant, elles améliorent la qualité de vie des patients, permettant à certains d'entre eux de survivre jusqu'à ce qu'un traitement plus efficace soit disponible. L'opposition avancée par certains entre thérapies pharmacologiques classiques et thérapies innovantes n'a pas de sens : les secondes n'excluent pas de recourir aux premières, y compris les plus classiques. Les deux approches s'opposent d'autant moins que certaines formes de thérapie génique sont appelées à être administrées comme des médicaments classiques.
Concernant la recherche, de nombreuses difficultés et insuffisances ont été relevées, telles les difficultés de recourir au diagnostic préimplantatoire. Le maintien du régime d'interdiction avec dérogation des recherches sur l'embryon et les cellules souches embryonnaires humaines par la loi relative à la bioéthique de juillet 2011 a été unanimement dénoncé par la communauté scientifique. L'argument parfois avancé selon lequel les cellules pluripotentes induites (iPS) permettraient de se passer des cellules souches embryonnaires reste fallacieux. L'OPECST 1 ( * ) a montré combien ce régime pénalisait les recherches en France. Il n'a pas été entendu et continuera de suivre les modalités d'application comme les conséquences de ce regrettable principe d'interdiction.
L'importance des modèles animaux, y compris des grands singes -les seuls vraiment pertinents avant de passer à l'essai clinique chez l'homme-, a été soulignée. Les tentatives de l'Union Européenne d'en limiter trop strictement l'utilisation inquiètent.
Le manque de biobanques reste un frein alors qu'elles sont nécessaires à la recherche translationnelle, l'essentiel de ces maladies étant d'origine génétique, avec en cause un gène facilement identifiable. Pour progresser, il est en effet nécessaire de constituer des cohortes de patients faisant l'objet d'explorations clinico-biologiques ce qui suppose le stockage de prélèvements dans des biobanques. Ceci permet d'évaluer l'histoire naturelle de ces maladies, d'identifier leurs bases moléculaires grâce à la génomique, l'épigénomique, la transcriptomique et la métabolomique. On disposerait ainsi de modèles cellulaires ou animaux afin d'en étudier les mécanismes physiopathologiques les plus fins .
Le retard de la France face à l'importance prise par les nouvelles techniques de séquençage du génome handicape. Le séquençage à haut débit qui permet de tester simultanément l'ensemble des gènes devrait permettre d'identifier beaucoup plus vite les mutations en cause. Par manque de moyens, la France a pris un certain retard dans les techniques de thérapie génique par rapport à l'Allemagne, la Belgique, l'Espagne ou les Pays-Bas, sans parler des États-Unis ou de la Chine.
L'absence de financements sécurisés à long terme avec des partenariats industriels, corollaires indispensables des financements publics, n'est pas assez prise en considération. On a d'ailleurs toujours des difficultés à comprendre pourquoi il est plus difficile de faire cela en France qu'à l'étranger. Or, il n'est pas envisageable de développer autrement ce type de thérapie jusqu'au stade du médicament, et les investissements de l'industrie pharmaceutique sont jusqu'à présent restés trop timides. Pourtant la recherche sur les maladies rares n'est pas vouée à demeurer marginale sur le plan économique.
M. Claude Birraux. Par ailleurs, les maladies monogéniques soulèvent des problématiques éthiques liées à leur origine génétique. Le coût de plus en plus réduit du séquençage du génome à haut débit et le développement de tests génétiques diagnostics mais aussi prédictifs, parfois en libre accès sur Internet, accroissent la pertinence de ces interrogations.
L'information génétique a un caractère particulier car elle porte sur des éléments constitutifs de l'individu, qui le relient à sa famille, et est en relation avec son destin. Aussi cette connaissance plus fine de prédispositions génétiques a-t-elle nécessairement des conséquences éthiques, juridiques et sociétales, dont il importe de prendre la mesure. Quand elle est fournie par les tests génétiques, elle peut conduire à une variation des conditions dans lesquelles la personne sera assurée, embauchée etc...
Quelle législation envisager concernant l'usage des tests génétiques ? La législation ne peut pas être la même si l'usage des tests génétiques devient commun ou s'il concerne un très faible pourcentage de la population. Le Comité national consultatif d'éthique, comme l'Agence de la biomédecine, s'inquiètent du développement très rapide de l'analyse du génome humain qualifié de « génétique récréative » et de la multiplication de tests génétiques en libre accès sur Internet, dont on ne peut pas garantir la fiabilité.
Actuellement, la plupart des tests disponibles sur Internet portent sur des maladies qui ne sont pas monogéniques : tests de susceptibilité, sans utilité au niveau individuel et dont l'interprétation peut évoluer d'un jour à l'autre en fonction des publications, car dans le génome, on trouve autant de gènes qui protègent que de gènes qui fragilisent, ce qui rend son interprétation particulièrement difficile. Or les tests génétiques portent sur diverses prédispositions, et posent des problèmes en termes d'accompagnement des patients. Qui interprétera le résultat ? Quel médecin prendra en charge le patient si un élément pathologique est découvert ? Quelle est l'utilité clinique d'un test qui révèle une prédisposition dont on ne sait que faire ?
S'agissant des tests génétiques pour les maladies monogéniques, jusqu'à présent, on répondait à une demande ; c'est ainsi qu'ont été mis en place les tests sur la maladie de Huntington, par exemple, dans des conditions très encadrées. Or on se trouve désormais face à une situation nouvelle : des facteurs génétiques seront identifiés chez des personnes qui n'avaient rien demandé. Il en va ainsi pour le diagnostic postnatal de la mucoviscidose, avec les tests génétiques à rebours chez les parents, puis les apparentés des sujets porteurs, posant la problématique des tests pratiqués chez les porteurs sains. Il sera difficile de s'assurer que ceux-ci seront en mesure de décider s'ils veulent savoir ou ne pas savoir.
Le développement très rapide de l'analyse du génome entier, ajoute des interrogations. Que devra-t-on faire si, en voulant soigner un patient atteint d'une maladie, l'analyse de son génome conduit à en trouver une autre ? En génétique, on rencontre des malades et des personnes qui ne sont pas ou qui ne se sentent pas malades. Par ailleurs, on progressera dans l'analyse du génome ; il faudra donc anticiper l'évolution de l'interprétation du génome.
Le développement des tests génétiques, comme le séquençage du génome à haut débit, s'inscrivent dans une logique de renforcement du contrôle social sur l'individu, sujet qu'il conviendra d'approfondir. Plus il devient facile de séquencer le génome complet d'une personne et moins cela coûte cher, plus il importe de protéger ces données pour éviter que celles-ci ne soient utilisées à des fins autres que médicales et scientifiques. Cela est d'autant plus délicat que la mondialisation de la biologie rend quasiment impossible de s'assurer que ce qui est strictement réglementé, voire interdit, dans un pays, ne sera pas pratiqué sous l'empire d'une réglementation plus laxiste ou inexistante dans d'autres pays pour un coût souvent modique. À cet égard, l'Agence de la biomédecine joue son rôle, elle veille à une information et un suivi de l'usage des tests génétiques en libre accès sur Internet. Cependant est-ce suffisamment dissuasif ?
M. Jean-Louis Touraine. Ces questions ont été évoquées au cours de missions, notamment aux États-Unis et au Royaume-Uni, effectuées par des membres de l'OPECST sur d'autres thématiques. Elles inquiètent associations de malades, généticiens et juristes de nombreux pays. Les instruments internationaux de protection des personnes, comme la Convention sur les droits de l'Homme et la biomédecine (Convention d'Oviedo), ne sont pas ratifiés par tous les États signataires. La France a déposé les instruments de ratification de la Convention d'Oviedo le 13 décembre 2011, l'article 1er de loi relative à la bioéthique de juillet 2011 ayant décidé de sa ratification. Cette Convention prévoit une protection générale encore insuffisante. Aussi, un protocole additionnel spécifique relatif aux tests génétiques à des fins médicales, ouvert à la signature en novembre 2008, a-t-il été élaboré. Il n'est pas encore entré en vigueur, la France qui a participé à son élaboration l'a également signé en décembre. Un instrument, en cours d'élaboration au Conseil de l'Europe, concerne la prédictibilité des tests génétiques et les assurances ; la France participe à son élaboration à un haut niveau car elle dirige le Comité directeur pour la bioéthique du Conseil de l'Europe.
En France, le code civil dispose que « nul ne peut faire l'objet de discriminations en raison de ses caractéristiques génétiques » et cette prescription est reprise dans le code pénal, le code du travail, le code des assurances et le code de la sécurité sociale. S'agissant des résultats d'un test, le problème de la diffusion de l'information à la parentèle, notamment dans le cas où une personne ne souhaite pas révéler à ses apparentés des éléments qui pourraient être importants pour eux, est délicat et a été réglé de manière équilibrée par l'article 2 de loi relative à la bioéthique de juillet 2011. Cependant, un suivi de l'application de ces dispositions s'impose.
Cette audition publique a démontré la nécessité de continuer à étudier l'impact du séquençage du génome à haut débit ; elle constitue donc une étape dans la réflexion à laquelle l'OPECST devra contribuer.
Débat sur les conclusions de l'audition
Mme Catherine Procaccia, sénatrice. Je m'étonne de la remise en cause de certains choix faits dans des lois très récentes déjà discutées et adoptées par notre Parlement, notamment les lois sur la bioéthique et la recherche. Certes, à l'OPECST, on met toujours l'accent sur l'aspect scientifique, mais je suis gênée d'entendre qu'il faut revenir sur des textes votés.
Par ailleurs, je ne comprends pas pourquoi il faudrait craindre les assureurs et les suspecter de ne pas appliquer les lois. Il ne me paraît pas justifié de leur faire des procès d'intention, d'autant que la démarche auprès d'un assureur est volontaire.
M. Claude Birraux. Les positions exposées sur les tests génétiques et le diagnostic pré-implantatoire ressortent des débats qui ont eu lieu au sein de notre Office. Nous avons présenté ces propositions dans le rapport de l'OPECST sur l'évaluation de la loi de bioéthique rédigé par nos collègues Alain Claeys et Jean-Sébastien Vialatte ; elles n'ont pas été suivies in fine par le Parlement, mais s'interroger sur les lois votées ne signifie pas les remettre en cause. De plus, la nouvelle loi de bioéthique prévoit un suivi de ses dispositions par notre Office et par l'Agence de la biomédecine. Notre rôle est bien de relayer les questions et les inquiétudes exprimées par les chercheurs lors d'une audition publique.
Par ailleurs, la multiplication des tests via Internet exige qu'on se montre très méfiant, car on n'a aucune garantie sur le résultat ni sur la confidentialité. On fait une recherche génétique sur un enfant, on remonte ensuite aux parents et si les résultats circulent et que les parents ont besoin d'une assurance-vie pour acheter une maison, l'assureur peut être amené à demander et utiliser les informations médicales contenues dans les tests.
M. Jean-Pierre Brard, député. On ne peut pas écarter ce débat. Les conditions politiques peuvent changer et évoluer pour donner satisfaction au monde des chercheurs. Nous avons créé les conditions d'une évolution de la loi sans drame. Nous sommes certes des modérateurs de la science, mais en débattre est important.
M. Jean-Louis Touraine. Il est normal de laisser des portes ouvertes pour le futur, d'autant que le Sénat, s'alignant souvent sur les positions du rapport de l'OPECST, avait été réticent à adopter certaines dispositions de la nouvelle loi de bioéthique. Tous ces sujets touchant à l'humain sont à juste titre soumis à révisions régulières. Sur les assurances, je me souviens encore du débat féroce mené à l'époque où le sida était systématiquement mortel et où les assureurs refusaient d'assurer les personnes qui avaient subi des transfusions sanguines.
M. Bruno Sido, sénateur, Président de l'OPECST. Je constate que le débat est clos et mets aux voix les conclusions de l'audition publique.
Les conclusions de l'audition publique ont été adoptées à l'unanimité des membres présents.
RECOMMANDATIONS
On peut identifier quelques orientations pour assurer une prise en charge adéquate de ces maladies. Ainsi, il appartient au Gouvernement, et aux instances compétentes :
- de réunir des données chiffrées précises (aucune évaluation du coût sociétal des maladies rares n'a jamais été réalisée).
- d'identifier les priorités en s'assurant que les ressources financières soit réparties équitablement.
- de ne négliger aucune approche thérapeutique, et d'éviter qu'elles ne soient gaspillées ou détournées de leur objet médical et scientifique.
- de soutenir la mise en oeuvre de la Fondation Maladies Rares 2 ( * ) , car ses objectifs répondent à une demande forte des patients et des équipes soignantes.
- d'appliquer rapidement le deuxième plan national maladies rares présenté le 28 février 2011 (après le premier plan 2004-2008) car il a pour mission d'organiser de véritables structures nationales de diagnostic et de soins.
- d'encourager la création d'une banque nationale de données sur les maladies rares regroupant les données cliniques issues des centres de référence et les données biologiques et thérapeutiques issues d'autres bases pour permettre d'identifier rapidement une population de patients éligibles à un essai thérapeutique en cours de démarrage ou d'établir une corrélation entre certaines anomalies cliniques et certaines anomalies génétiques.
- de mutualiser les moyens à l'échelon européen en créant une structure de recherche européenne, rattachée à la direction générale « Recherche » de la Commission européenne, qui regrouperait toutes les bases de données existantes et permettrait de créer une véritable banque européenne de données maladies rares.
- de participer plus activement à l'harmonisation des règles et des pratiques au niveau international en ratifiant rapidement les instruments internationaux additionnels à la Convention d'Oviedo qui concernent la génétique.
ANNEXES
ANNEXE 1 : EXTRAITS DE LA LOI N° 2011-814 DU 7 JUILLET 2011 RELATIVE À LA BIOÉTHIQUE
Article 1
Est autorisée la ratification de la convention du Conseil de l'Europe pour la protection des droits de l'homme et de la dignité de l'être humain à l'égard des applications de la biologie et de la médecine : convention sur les droits de l'homme et la biomédecine, signée à Oviedo le 4 avril 1997.
Titre Ier : Examen des caractéristiques génétiques à des fins médicales
Article 2 ...... sont insérés des articles L. 1131-1-2 et L. 1131-1-3 ainsi rédigés :
« Art. L. 1131-1-2.-Préalablement à la réalisation d'un examen des caractéristiques génétiques d'une personne, le médecin prescripteur informe celle-ci des risques qu'un silence ferait courir aux membres de sa famille potentiellement concernés si une anomalie génétique grave dont les conséquences sont susceptibles de mesures de prévention, y compris de conseil génétique, ou de soins était diagnostiquée. Il prévoit avec elle, dans un document écrit qui peut, le cas échéant, être complété après le diagnostic, les modalités de l'information destinée aux membres de la famille potentiellement concernés afin d'en préparer l'éventuelle transmission. Si la personne a exprimé par écrit sa volonté d'être tenue dans l'ignorance du diagnostic, elle peut autoriser le médecin prescripteur à procéder à l'information des intéressés dans les conditions prévues au quatrième alinéa.
« En cas de diagnostic d'une anomalie génétique grave, sauf si la personne a exprimé par écrit sa volonté d'être tenue dans l'ignorance du diagnostic, l'information médicale communiquée est résumée dans un document rédigé de manière loyale, claire et appropriée, signé et remis par le médecin. La personne atteste de cette remise. Lors de l'annonce de ce diagnostic, le médecin informe la personne de l'existence d'une ou plusieurs associations de malades susceptibles d'apporter des renseignements complémentaires sur l'anomalie génétique diagnostiquée. Si la personne le demande, il lui remet la liste des associations agréées en application de l'article L. 1114-1.
« La personne est tenue d'informer les membres de sa famille potentiellement concernés dont elle ou, le cas échéant, son représentant légal possède ou peut obtenir les coordonnées, dès lors que des mesures de prévention ou de soins peuvent leur être proposées.
« Si la personne ne souhaite pas informer elle-même les membres de sa famille potentiellement concernés, elle peut demander par un document écrit au médecin prescripteur, qui atteste de cette demande, de procéder à cette information. Elle lui communique à cette fin les coordonnées des intéressés dont elle dispose. Le médecin porte alors à leur connaissance l'existence d'une information médicale à caractère familial susceptible de les concerner et les invite à se rendre à une consultation de génétique, sans dévoiler ni le nom de la personne ayant fait l'objet de l'examen, ni l'anomalie génétique, ni les risques qui lui sont associés. Le médecin consulté par la personne apparentée est informé par le médecin prescripteur de l'anomalie génétique en cause.
« Lorsque est diagnostiquée une anomalie génétique grave dont les conséquences sont susceptibles de mesures de prévention, y compris de conseil génétique, ou de soins chez une personne qui a fait un don de gamètes ayant abouti à la conception d'un ou plusieurs enfants ou chez l'un des membres d'un couple ayant effectué un don d'embryon, cette personne peut autoriser le médecin prescripteur à saisir le responsable du centre d'assistance médicale à la procréation afin qu'il procède à l'information des enfants issus du don dans les conditions prévues au quatrième alinéa.
« Art. L. 1131-1-3.-Par dérogation au deuxième alinéa de l'article L. 1111-2 et à l'article L. 1111-7, seul le médecin prescripteur de l'examen des caractéristiques génétiques est habilité à communiquer les résultats de cet examen à la personne concernée ou, le cas échéant, aux personnes mentionnées au second alinéa de l'article L. 1131-1. »
Article 3
L'article L. 1131-2 du même code est ainsi rédigé :
« Art. L. 1131-2.-Un arrêté du ministre chargé de la santé, pris sur proposition de l'Agence de la biomédecine et de la Haute Autorité de santé, définit les règles de bonnes pratiques applicables à la prescription et la réalisation de l'examen des caractéristiques génétiques d'une personne et de son identification par empreintes génétiques à des fins médicales. Cet arrêté définit également les règles de bonnes pratiques applicables, le cas échéant, au suivi médical de la personne. »
Article 4
I.- Il est inséré un article L. 1131-2-1 ainsi rédigé :
« Art. L. 1131-2-1.-L'examen des caractéristiques génétiques d'une personne ou son identification par empreintes génétiques à des fins médicales ne peuvent être pratiqués que dans des laboratoires de biologie médicale autorisés à cet effet dans les conditions prévues au chapitre II du titre II du livre Ier de la sixième partie et accrédités dans les conditions prévues au chapitre Ier du titre II du livre II de la même partie.
« Lorsque le laboratoire dépend d'un établissement de santé, l'autorisation est délivrée à cet établissement.
« Un laboratoire de biologie médicale établi dans un autre Etat membre de l'Union européenne ou partie à l'accord sur l'Espace économique européen peut réaliser la phase analytique de l'examen des caractéristiques génétiques ou de l'identification par empreintes génétiques s'il est autorisé dans cet Etat à pratiquer cette activité, sous réserve qu'il ait adressé une déclaration si les conditions d'autorisation dans cet Etat ont été préalablement reconnues comme équivalentes à celles qui résultent du premier alinéa ou, à défaut, qu'il ait obtenu une autorisation après vérification que ses normes de fonctionnement sont équivalentes à celles qui résultent du premier alinéa.
« Les autorisations et accréditations prévues aux trois premiers alinéas peuvent être retirées ou suspendues, respectivement dans les conditions prévues aux articles L. 6122-13 et L. 6221-2 ou en cas de manquement aux prescriptions législatives et réglementaires applicables à l'examen des caractéristiques génétiques d'une personne ou à son identification par empreintes génétiques. » ..
Article 6
I.- Après l'article 226-28 du code pénal, il est inséré un article 226-28-1 ainsi rédigé :
« Art. 226-28-1.-Le fait, pour une personne, de solliciter l'examen de ses caractéristiques génétiques ou de celles d'un tiers ou l'identification d'une personne par ses empreintes génétiques en dehors des conditions prévues par la loi est puni de 3 750 € d'amende. »
II.- Après l'article L. 1133-4 du code de la santé publique, il est inséré un article L. 1133-4-1 ainsi rédigé :
« Art. L. 1133-4-1.-Le fait, pour une personne, de solliciter l'examen de ses caractéristiques génétiques ou de celles d'un tiers ou l'identification d'une personne par ses empreintes génétiques en dehors des conditions prévues par la loi est puni de la peine prévue à l'article 226-28-1 du code pénal. »
ANNEXE 2 : PLAN NATIONAL MALADIES RARES 2
SYNTHÈSE
Le Plan Maladies Rares 2 (2011-2014) a été rendu public et présenté le 28 février 2011 par Madame Nora Berra, secrétaire d'Etat chargée de la santé et Madame Valérie Pécresse, ministre de l'enseignement supérieur et de la recherche.
CONTENU DU PLAN
Quatre points majeurs ont été identifiés ("focus")
- Création de la Fondation "Maladies Rares" : abritée par une Fondation de Coopération Scientifique "Santé", la fondation aura pour objectif de structurer et coordonner les missions du GIS-Institut des Maladies Rares, de la Base nationale de données Maladies Rares et d'Orphanet. Dans le cadre de ses missions, elle devra être le point de contact national pour tous les acteurs impliqués ; elle sera en charge du développement des partenariats avec les industriels.
- Création d'une banque nationale de données maladies rares : partie prenante de la fondation, elle comprendra les données cliniques (puis biologiques et thérapeutiques) recueillies dans les centres de référence et de compétence, et à partir des registres de maladies rares. Le recueil sera réalisé en utilisant la nomenclature Orphanet. Une interface avec les données de suivi de cohortes spécifiques, notamment RADICO, est prévu.
- Prise en compte des besoins spécifiques des patients d'Outre-mer , avec une attention particulière pour les patients souffrant de drépanocytose.
- Soutien à l'action des associations maladies rares .
Articulation autour de trois axes
Axe A : Améliorer la qualité de la prise en charge du patient
Ø Améliorer l'accès au diagnostic et la prise en charg e
En structurant les filières, en identifiant des plateformes de laboratoires de diagnostic dédiés et en développant des approches à haut débit pour identification des bases moléculaires des pathologies, en déployant un système d'information unique, en favorisant le développement de la télémédecine, en améliorant le dépistage pré-implantatoire, prénatal ou néonatal.
Ø Optimiser l'évaluation et le financement des centres de références
En simplifiant le dispositif d'évaluation et de labellisation des centres de référence, en améliorant l'identification des crédits dédiés, en prenant en compte le caractère complexe et pluridisciplinaire des consultations pour le financement.
Ø Intensifier la rédaction des PNDS
En priorisant le programme, en simplifiant la méthodologie d'élaboration.
Ø Garantir la qualité de la prise en charge médicamenteuse adaptée à chaque patient
En facilitant l'accès et le remboursement de produits non remboursés ou hors AMM, en surveillant les projets d'arrêts de commercialisation, en élargissant l'accès aux médicaments expérimentaux, en recensant et en documentant les utilisations hors AMM, en sécurisant les prescriptions hospitalières.
Ø Développer les liens entre les acteurs de la prise en charge et de l'accompagnement
Ø Améliorer les pratiques des professionnels de santé
En renforçant les connaissances et en développant de nouvelles compétences, par la formation initiale et la formation continue, par l'élaboration de recommandations pour les situations d'urgence.
Ø Rendre accessible l'information et la diffuser
Ø Développer Orphanet
En intégrant la nomenclature dans les bases existantes (PMSI, CepiDC, CNAM...), en poursuivant son enrichissement en termes de contenu et d'ergonomie.
Axe B : Développer la recherche sur les maladies rares
Ø Créer une fondation "Maladies Rares" pour améliorer les connaissances et accélérer le développement des thérapies
Ø Assurer à la recherche sur les maladies rares un financement minimum et développer les outils nécessaires à l'utilisation des échantillons biologiques, du séquençage à haut débit
Ø Promouvoir le développement des essais thérapeutiques
Ø Favoriser la recherche clinique et translationnelle
Axe C : Amplifier les coopérations européennes et internationales
Ø Partager l'expertise via les réseaux européens de référence
Ø Améliorer la capacité de mise en oeuvre d'essais cliniques multinationaux
En évaluant le dispositif ECRIN pour tendre vers une optimisation, en mutualisant et en standardisant les tests diagnostics.
Ø Structurer les coopérations européennes et internationales
En sécurisant la collecte et le maintien des collections d'échantillons biologiques, en accompagnant les centres de référence dans leurs recherches et en contribuant au financement et à la pérennisation du projet E-rare.
SUIVI DU PLAN
Le suivi du plan reposera sur 3 types de structures
Un comité de suivi et de prospective
Ø Créé pour s'assurer de la mise en oeuvre des mesures et proposer des évolutions si nécessaire.
Ø Composition
o présidence : directrice générale de l'offre de soins (Annie Podeur)
o vice-présidence : volet "santé" Sabine Sarnacki (AP-HP) ; volet "recherche" Hélène Dollfus (CHU de Strasbourg)
Un secrétaire général
Ø Suivi des aspects opérationnels
Ø Nomination : Alain Garcia
Les ARS
Identification d'un interlocuteur dédié et lien avec le "terrain"
FINANCEMENT DU PLAN
Un budget de 86,4 millions d'euros a été alloué spécifiquement pour le financement de ce second plan.
Ce budget se répartit de la façon suivante :
Améliorer la qualité de la prise en charge du patient : 30,4 millions d'euros
Développer la recherche sur les maladies rares : 51 millions d'euros
Amplifier les coopérations européennes et internationales : 5 millions d'euros
ANNEXE 3 : CHARTE DU RÉSEAU CEMARA RÉSEAU DES CENTRES MALADIES RARES




ANNEXE 4 : SAISINE DE L'OPECST

* 1 rapport de l'OPECST sur l'évaluation de l'application de la loi n° 2004-800 du 6 août 2004 relative à la bioéthique n° 1325 déposé le 17 décembre 2008 - http://www.assemblee-nationale.fr/13/rap-off/i1325.asp
* 2 Elle a été créée le 7 mars 2012