







Rapport n° 44 (2011-2012) de M. Bernard CAZEAU , fait au nom de la commission des affaires sociales, déposé le 19 octobre 2011
Disponible au format PDF (1 Moctet)
Tableau comparatif au format PDF (611 Koctets)
-
AVANT-PROPOS
-
EXAMEN DES ARTICLES
-
Article 1er (art. L. 1451-1,
L. 1451-1-1, L. 1451-2 et L. 1451-3 (nouveaux) du code
de la santé publique) Règles déontologiques et
expertise sanitaire
-
Article 1er bis (nouveau) (art.
L. 1415-4 du code de la santé publique) Conditions de
nomination des personnels dirigeants de l'institut national du
cancer
-
Article 1er (art. L. 1451-1,
L. 1451-1-1, L. 1451-2 et L. 1451-3 (nouveaux) du code
de la santé publique) Règles déontologiques et
expertise sanitaire
-
CHAPITRE II - Avantages
-
CHAPITRE III - Sanctions
pénales
-
TITRE II - GOUVERNANCE DES PRODUITS DE
SANTÉ
-
Article 4 (art. L. 5311-1,
L. 5311-2, L. 5312-4, L. 5311-4-1 (nouveau) et
L. 5421-8 à L. 5421-11 (nouveaux) du code de la santé
publique) Création et prérogatives de l'agence nationale de
sécurité du médicament et des produits de santé
-
Article 4 bis A (nouveau) Observatoire
national des prescriptions et consommations des médicaments
-
Article 4 bis (art. L. 5122-15,
L. 5122-16 et L. 5323-4 du code de la santé
publique) Coordinations
-
Article 5 (art. L. 5322-1,
L. 5324-1 (nouveau) et L. 1413-8 du code de la santé
publique) Composition du conseil d'administration et publicité des
travaux de l'agence nationale du médicament et des produits de
santé
-
Article 5 bis (art. L. 161-40 du
code de la sécurité sociale) Base de données mise en
oeuvre par la Haute Autorité de santé
-
Article 4 (art. L. 5311-1,
L. 5311-2, L. 5312-4, L. 5311-4-1 (nouveau) et
L. 5421-8 à L. 5421-11 (nouveaux) du code de la santé
publique) Création et prérogatives de l'agence nationale de
sécurité du médicament et des produits de santé
-
TITRE III - LE MÉDICAMENT
À USAGE HUMAIN
-
Article 6 (art. L. 5121-8 du code
de la santé publique) Réalisation d'études
après l'autorisation de mise sur le marché
-
Article 6 bis (art. L. 5121-8-2
(nouveau) du code de la santé publique) Information de l'agence en
charge de la sécurité du médicament relative aux
essais cliniques pré-AMM
-
Article 7 (art. L. 5121-9 du code de la
santé publique) Conditions de suspension, de retrait ou de
modification de l'autorisation de mise sur le marché
-
Article 8 (art. L. 5121-9-2 et
5121-9-3 (nouveaux) du code de la santé publique) Obligations
du titulaire de l'autorisation de mise sur le marché
-
Article 9 (art. L. 5124-11 du code
de la santé publique) Conditions d'interdiction de l'exportation de
médicaments ayant fait l'objet d'un retrait d'autorisation de mise
sur le marché
-
Article 9 bis (art. L. 162-17
du code de la sécurité sociale) Conditions de fixation du
service médical rendu des médicaments
-
Article 6 (art. L. 5121-8 du code
de la santé publique) Réalisation d'études
après l'autorisation de mise sur le marché
-
CHAPITRE II - La
prescription
-
Article 10 (art. L. 5121-1 du code
de la santé publique) Encadrement de la préparation
magistrale
-
Article 10 bis (art. L. 5125-1-1 et
L. 5125-1-1-1 (nouveau) du code de la santé publique) Régime
d'autorisation des préparations en pharmacie
-
Article 11 (art. L. 5121-12-1
(nouveau) du code de la santé publique et L. 162-4 du code de
la sécurité sociale) Encadrement des prescriptions en dehors
des indications de l'autorisation de mise sur le marché
-
Article 12 (art. L. 5121-1-2
(nouveau) du code de la santé publique) Prescription en
dénomination commune
-
Article 13 (art. L. 162-17-4-1
(nouveau) du code de la sécurité sociale) Contrôle et
sanction des prescriptions hors autorisation de mise sur le marché
par le comité économique des produits de santé
-
Article 10 (art. L. 5121-1 du code
de la santé publique) Encadrement de la préparation
magistrale
-
CHAPITRE III - La délivrance des
médicaments
-
CHAPITRE IV - L'autorisation
temporaire d'utilisation
-
CHAPITRE V - La prise en charge
hors autorisation de mise sur le marché
-
CHAPITRE VI - La
pharmacovigilance
-
CHAPITRE VII - Information et
publicité sur le médicament à usage humain
-
Article 18 (art. L. 5122-2,
L. 5122-3, L. 5122-5, L. 5122-6, L. 5122-9,
L. 5122-9-1 (nouveau), L. 5122-16, L. 5422-3,
L. 5422-4, L. 5422-6 et L. 5422-11 du code de la santé
publique et art. L. 613-5 du code de la propriété
intellectuelle) Réglementation de la publicité pour les
médicaments à usage humain
-
Article 19 (art. L. 162-17-8 du code de la
sécurité sociale) Encadrement de la visite
médicale
-
Article 20 (art. L. 5121-14-3
(nouveau) du code de la santé publique) Garantie par l'industrie
pharmaceutique du bon usage des médicaments
-
Article 20 bis (nouveau)
(art. L. 245-2 du code de la sécurité sociale)
Garantie par l'industrie pharmaceutique du bon usage des médicaments
-
Article 18 (art. L. 5122-2,
L. 5122-3, L. 5122-5, L. 5122-6, L. 5122-9,
L. 5122-9-1 (nouveau), L. 5122-16, L. 5422-3,
L. 5422-4, L. 5422-6 et L. 5422-11 du code de la santé
publique et art. L. 613-5 du code de la propriété
intellectuelle) Réglementation de la publicité pour les
médicaments à usage humain
-
CHAPITRE VIII - Les logiciels d'aide
à la prescription et à la dispensation
-
CHAPITRE IX - Les études en
santé publique
-
TITRE IV - DISPOSITIFS
MÉDICAUX
-
Article 23 (art. L. 5213-1 à
L. 5213-7, L. 5461-6, L. 5461-7, L. 5461-8
(nouveaux), L. 5212-1 et L. 5222-2 du code de la santé
publique et art. L. 165-8 du code de la sécurité
sociale) Publicité pour les dispositifs médicaux
-
Article 24 (art. L. 165-1-2 (nouveau)
du code de la sécurité sociale) Conformité des
dispositifs médicaux aux spécifications requises pour
pouvoir être remboursés
-
Article 25 (art. L. 114-10,
L. 162-1-14 et L. 162-1-20 (nouveau) du code de la
sécurité sociale) Contrôle par les agents
assermentés de l'assurance maladie de la conformité des
dispositifs médicaux aux règles de facturation et de
tarification
-
Article 26 (art. L. 165-11,
L. 165-12 et L. 165-13 (nouveaux) du code de la
sécurité sociale) Evaluation de certains dispositifs
médicaux
-
Article 23 (art. L. 5213-1 à
L. 5213-7, L. 5461-6, L. 5461-7, L. 5461-8
(nouveaux), L. 5212-1 et L. 5222-2 du code de la santé
publique et art. L. 165-8 du code de la sécurité
sociale) Publicité pour les dispositifs médicaux
-
TITRE V - DISPOSITIONS DIVERSES
-
Article 27 Habilitation du Gouvernement
à prendre par ordonnance les mesures nécessaires à la
transposition d'une directive « médicaments »
-
Article 28 Habilitation du Gouvernement
à prendre par ordonnance les mesures nécessaires pour harmoniser
les sanctions pénales et administratives avec les dispositions de
la présente loi
-
Article 29 Habilitation du Gouvernement
à prendre par ordonnance des dispositions relatives à la
mise en cohérence du droit métropolitain avec le droit de
l'Outre-mer
-
Article 30 Dispositions transitoires
-
Article 30 bis (nouveau) Nom et pouvoirs de
la commission de la transparence de la Haute Autorité de
santé
-
Article 30 ter (nouveau) Rapport sur la
profession de visiteur médical
-
Article 31 (art. L. 713-7 du code de la
propriété intellectuelle) Apparence et texture des
médicaments génériques
-
Article 32 (art. L. 5312-4-2 (nouveau)
du code de la santé publique) Protection des lanceurs
d'alerte
-
Article 33 (art. L. 5134-1 du code de la
santé publique) Prescription de contraceptifs par les sages-femmes
et délivrance de médicaments contraceptifs dans les services
de médecine de prévention des universités
-
Article 34 (art. L. 713-7 du code de la
propriété intellectuelle) Contrôle des exportations
parallèles de médicaments
-
Article 27 Habilitation du Gouvernement
à prendre par ordonnance les mesures nécessaires à la
transposition d'une directive « médicaments »
-
TRAVAUX DE LA COMMISSION
-
LISTE DES PERSONNES AUDITIONNÉES
-
TABLEAU COMPARATIF
N° 44
SÉNAT
SESSION ORDINAIRE DE 2011-2012
|
Enregistré à la Présidence du Sénat le 19 octobre 2011 |
RAPPORT
FAIT
au nom de la commission des affaires sociales (1) sur le projet de loi , ADOPTÉ PAR L'ASSEMBLÉE NATIONALE APRÈS ENGAGEMENT DE LA PROCÉDURE ACCÉLÉRÉE, relatif au renforcement de la sécurité sanitaire du médicament et des produits de santé ,
Par M. Bernard CAZEAU,
Sénateur
|
(1) Cette commission est composée de : Mme Annie David , présidente ; M. Jacky Le Menn, Mme Catherine Génisson, MM. Jean-Pierre Godefroy, Claude Jeannerot, Alain Milon, Mme Isabelle Debré, MM. Jean-Louis Lorrain, Jean-Marie Vanlerenberghe, Gilbert Barbier , vice-présidents ; Mmes Claire-Lise Campion, Aline Archimbaud, M. Alain Gournac, Mme Catherine Deroche, M. Marc Laménie , secrétaires ; Mmes Jacqueline Alquier, Natacha Bouchart, Marie-Thérèse Bruguière, MM. Jean-Noël Cardoux, Luc Carvounas, Mme Caroline Cayeux, M. Bernard Cazeau, Mmes Karine Claireaux, Laurence Cohen, M. Yves Daudigny, Mme Christiane Demontès, MM. Gérard Dériot, Jean Desessard, Mme Muguette Dini, M. Jean-Léonce Dupont, Mmes Odette Duriez, Anne-Marie Escoffier, MM. Guy Fischer, Michel Fontaine, Mme Samia Ghali, M. Bruno Gilles, Mmes Colette Giudicelli, Christiane Hummel, M. Jean-François Husson, Mmes Chantal Jouanno, Christiane Kammermann, MM. Ronan Kerdraon, Georges Labazée, Jean-Claude Leroy, Hervé Marseille, Mmes Michelle Meunier, Isabelle Pasquet, M. Louis Pinton, Mmes Gisèle Printz, Catherine Procaccia, MM. Gérard Roche, René-Paul Savary, Mme Patricia Schillinger, MM. René Teulade, Michel Vergoz, André Villiers, Dominique Watrin. |
Voir le(s) numéro(s) :
|
Assemblée nationale ( 13 ème législ.) : |
3714 , 3725 et T.A. 741 |
|
|
Sénat : |
5 et 45 (2011-2012) |
|
AVANT-PROPOS
Mesdames, Messieurs,
L'affaire du Mediator marque la dernière étape en date dans la remise en cause de notre système de police sanitaire des produits de santé. L'insouciance liée à cinquante ans de progrès scientifiques passés en relative sécurité a été perdue à partir des années quatre-vingt-dix. Elle a progressivement laissé la place à des interrogations croissantes sur les conséquences des produits sanitaires destinés à l'homme. Les malades ont désormais pris conscience du dilemme devant lequel ils sont placés : accéder facilement à tous les médicaments produits et prendre le risque d'effets indésirables parfois graves, ou accepter de limiter les options thérapeutiques pour garantir leur sécurité.
Les doutes des patients ne peuvent qu'être accentués par l'affaiblissement du lien qui les unit à leur médecin. La multiplication des alternatives prétendument thérapeutiques à la médecine et la plus grande difficulté d'accès aux soins sur certaines parties du territoire ont distendu la confiance qui constitue pourtant la base de la relation médecin-patient.
Dès 2006, le Sénat avait appelé à restaurer la confiance dans le médicament. En 2011, à la suite de l'affaire du Mediator, un nouveau rapport d'information sénatorial 1 ( * ) a dressé un bilan complet des problèmes de notre système de sécurité sanitaire et fait de nombreuses propositions. Votre rapporteur, qui était vice-président de la mission d'information, ne peut que regretter que, de même qu'en 2006, les propositions sénatoriales aient été peu reprises par le Gouvernement.
Après avoir présenté, à l'instar de la mission d'information sénatoriale sur le Mediator, un bref panorama du cadre actuel dans lequel s'exerce la police sanitaire, il demeure nécessaire de faire valoir à nouveau ce que les Français sont en droit d'attendre de la réforme de la sécurité sanitaire et de mesurer à cette aune les propositions du Gouvernement, complétées par l'Assemblée nationale.
I. PRÉSERVER LES ACQUIS DU SYSTÈME ACTUEL DES AGENCES SANITAIRES
Les critiques adressées à l'actuel système de sécurité sanitaire sont déjà anciennes. Elles ont été formulées notamment par le rapport sénatorial d'information de 2006 intitulé, déjà, Médicament, restaurer la confiance 2 ( * ) . Il convient cependant de garder à l'esprit que le pouvoir des autorités sanitaires est loin d'être discrétionnaire et que plusieurs acquis importants en matière de sécurité des produits de santé doivent être préservés.
A. DES DÉCISIONS SANITAIRES NATIONALES SOUS CONTRAINTE
Les autorités de police sanitaire doivent, dans leur action de protection de la santé publique, respecter deux niveaux de contrainte sous peine de voir leurs décisions annulées par le juge et de faire condamner l'Etat à verser de lourdes indemnités aux laboratoires.
Ces contraintes sont issues du droit communautaire pour ce qui concerne l'autorisation de mise sur le marché (AMM) et du respect dû au droit de propriété ainsi qu'aux prérogatives des médecins pour ce qui concerne le retrait ou la suspension de l'AMM.
1. L'encadrement européen de la mise sur le marché
a) Une compétence communautaire en développement
Bien qu'elle relève à l'origine, comme l'ensemble de la santé publique, de la compétence souveraine des Etats membres, la police sanitaire a progressivement été encadrée par le droit européen, au travers de l'édiction de normes communes de plus en plus contraignantes sur les conditions de mise sur le marché des médicaments. C'est en s'appuyant sur la double nature du médicament, bien de santé publique mais aussi bien commercial, que la Commission européenne a justifié sa première intervention en la matière - la directive du Conseil du 26 janvier 1965 3 ( * ) - concernant le rapprochement des dispositions législatives, réglementaires et administratives, relatives aux spécialités pharmaceutiques. Celle-ci dispose que l'objectif de santé publique, relevant des Etats membres, « doit être atteint par des moyens qui ne puissent pas freiner le développement de l'industrie pharmaceutique et les échanges de produits pharmaceutiques au sein de la Communauté » ; de plus, les disparités des dispositions nationales « ont pour effet d'entraver les échanges des spécialités pharmaceutiques au sein de la Communauté et [...] ont de ce fait une incidence directe sur l'établissement et le fonctionnement du marché commun » .
Il serait inexact de voir dans le développement des normes communautaires depuis cette date une simple volonté d'accroissement des compétences communautaires. C'est d'abord la volonté des Etats, confrontés aux premiers scandales sanitaires mondiaux, dont celui du thalidomide, de trouver ensemble un équilibre entre sécurité du médicament et accès des patients aux traitements, qui a été le moteur d'une harmonisation puis d'une intégration des conditions d'autorisation de mise sur le marché (AMM) au niveau communautaire.
Malgré la création, dès 1975 4 ( * ) , d'une procédure européenne d'AMM et d'un Comité des spécialités pharmaceutiques (CSP) composé de représentants des Etats membres, à la fois structure d'arbitrage et lieu d'échanges, la part de l'Europe dans la mise sur le marché des médicaments est restée faible jusqu'à la relance du marché intérieur dans les années 1980.
C'est il y a vingt ans seulement, en 1993 5 ( * ) , qu'une Agence européenne d'évaluation des médicaments a été mise en place afin de gérer les deux nouvelles procédures d'AMM communautaires : la procédure centralisée, qui permet à un laboratoire d'obtenir pour l'ensemble des Etats membres de l'Union européenne une AMM en déposant une demande unique auprès de l'agence, et la procédure de reconnaissance mutuelle, obligatoire pour toute demande d'AMM dans plus d'un Etat membre pour une entreprise qui ne veut ou ne peut utiliser la procédure centralisée.
Une nouvelle étape a été franchie en 2001 par la directive 2001/83/CE instituant un code communautaire relatif aux médicaments à usage humain, puis en 2004, par le règlement (CE) n° 726/2004 établissant des procédures communautaires de mise sur le marché et de surveillance des médicaments et relatif à l'Agence européenne des médicaments (EMA). Ces textes ont en effet organisé un système européen de pharmacovigilance, fondé sur la nécessité d'une appréciation commune des bénéfices et des risques pour éviter toute entrave aux échanges. La pharmacovigilance constitue aujourd'hui l'axe privilégié de développement de la politique européenne du médicament, consacré par l'adoption, le 15 décembre 2010, d'une nouvelle directive (2010/84/UE), dont le projet de loi soumis à l'examen du Sénat prévoit la transposition, et d'un nouveau règlement (n° 1235/2010).
b) Les procédures de mise sur le marché existantes
Il existe deux types de procédures d'AMM : les procédures européennes et la procédure nationale. Le nombre de procédures nationales tend à décroître, les laboratoires pharmaceutiques ayant intérêt à recourir à une procédure européenne pour pouvoir diffuser simultanément leurs médicaments dans plusieurs pays. En 2010, les procédures nationales ne représentent ainsi plus qu'un tiers des nouvelles demandes d'AMM déposées.
|
Bilan des AMM délivrées en procédure nationale en France et en procédures européennes |
|||||
|
2006 |
2007 |
2008 |
2009 |
2010 |
|
|
Nouvelles demandes d'AMM déposées |
1 365 |
1 345 |
1 808 |
1 855 |
1 620 |
|
dont procédure nationale |
719 |
662 |
754 |
859 |
556 |
Les procédures européennes sont utilisées lorsque le médicament est destiné à plusieurs Etats membres. L'accès d'un médicament au marché européen peut emprunter soit la voie de la procédure centralisée, soit celle de la procédure de reconnaissance mutuelle ou de la procédure décentralisée.
La procédure centralisée est obligatoire pour les médicaments issus de la biotechnologie, les médicaments de thérapie innovante et les médicaments orphelins. Il en est de même pour tous les nouveaux médicaments destinés au traitement du VIH, du cancer, du diabète, des maladies neurodégénératives ou des maladies rares. En dehors de ces obligations, la procédure est facultative pour les autres médicaments contenant une nouvelle substance active et ceux constituant une innovation thérapeutique, scientifique ou technique majeure ou « présentant un intérêt au niveau communautaire ».
Pour l'instruction de ces dossiers, le comité compétent de l'Agence européenne désigne parmi ses membres un Etat rapporteur et un Etat co-rapporteur, dont les agences ou autorités compétentes vont conduire un premier travail d'expertise. Les Etats ont la possibilité de faire acte de candidature. Le rapport d'évaluation des pays rapporteurs est ensuite soumis à la délibération des autres membres. Des objections, des demandes de précision peuvent être formulées ; une période d'échanges entre le pays rapporteur qui centralise les demandes d'informations de ses partenaires et le laboratoire peut alors s'instaurer. Ces évaluations doivent se dérouler en 210 jours (soit sept mois). Le comité se prononce par un vote à la majorité simple. Son avis est transmis à la Commission européenne, qui octroie ou non l'AMM. Celle-ci est valable pour l'ensemble des pays membres de l'Union européenne. Le pays rapporteur a par la suite la responsabilité du suivi du médicament.
La procédure de reconnaissance mutuelle et la procédure décentralisée sont applicables à tous les médicaments non soumis à la procédure centralisée. Dans le cadre de ces deux procédures, l'Agence européenne du médicament n'interviendra qu'en cas d'arbitrage, si un ou plusieurs Etats s'opposent à la commercialisation sur leur territoire d'un médicament autorisé par d'autres.
La procédure de reconnaissance mutuelle peut être utilisée lorsque l'autorité sanitaire compétente d'un Etat membre a déjà procédé à l'évaluation d'un médicament et lui a délivré une AMM nationale. Le laboratoire peut alors demander que l'autorisation soit étendue aux autres Etats membres. Ces derniers ont la possibilité de refuser la mise sur le marché en motivant leur décision.
La procédure décentralisée est utilisée lorsqu'aucune AMM initiale n'a été délivrée par un Etat membre. Elle consiste alors, pour le laboratoire, à déposer un dossier simultanément dans tous les Etats membres souhaités. Un Etat est nommé comme référent pour conduire l'évaluation, qui ensuite peut être reconnue par les autres Etats membres sollicités selon les règles de la reconnaissance mutuelle.
2. L'obligation de respecter les droits acquis
L'action de police sanitaire dans le domaine des produits de santé et de la cosmétique est consacrée au plus haut niveau normatif puisqu'elle découle d'un des « principes particulièrement nécessaires à notre temps » proclamés par le Préambule de la Constitution de 1946 : la Nation « garantit à tous, notamment à l'enfant, à la mère et aux vieux travailleurs, la protection de la santé » 6 ( * ) .
Ce principe doit être concilié avec un autre, doublement consacré par la Déclaration des droits de l'homme et du citoyen : le droit de propriété 7 ( * ) . En l'occurrence, ce droit est celui des firmes pharmaceutiques titulaires d'un brevet leur garantissant l'exclusivité d'exploitation d'une molécule pendant une période de vingt à vingt-cinq ans.
Les brevets relatifs aux médicaments sont une création relativement récente liée au développement de l'industrie du médicament dans l'après-guerre et ils ne sont entrés dans le droit commun de la propriété intellectuelle que récemment, en 1968 8 ( * ) . En l'espèce, les brevets découlent d'une double nécessité publique : favoriser l'investissement dans la recherche, par la garantie d'un retour sous forme de profits commerciaux, et encourager l'innovation en assortissant la protection des droits de propriété de l'obligation de divulguer l'invention.
Le droit de propriété des industriels sur les médicaments qu'ils élaborent et produisent est limité par la nécessité d'obtenir l'AMM, soumise à l'évaluation du rapport bénéfices-risques lié au produit. Le Leem (les entreprises du médicament), représentant des industries du médicament, estime à dix ans en moyenne le temps d'étude puis d'élaboration du dossier d'AMM, ce qui réduirait la durée de l'exploitation commerciale exclusive à dix ans, un brevet d'exploitation commerciale étant accordé pour vingt ans. En raison de cette spécificité liée à la sécurité du médicament qui impose une phase pré-commerciale particulièrement longue, les industriels peuvent obtenir un certificat complémentaire de protection d'une durée de cinq ans qui prolonge leur droit d'exploitation. La nécessité d'obtenir une AMM a donc des conséquences importantes sur l'exploitation d'un médicament, conséquences prises en compte par la possibilité de prolongement des droits exclusifs de propriété intellectuelle.
La suspension ou le retrait de l'AMM a également un impact commercial notable. Elles sont donc étroitement contrôlées par le juge communautaire et par le Conseil d'Etat. La décision de la Commission européenne de retirer un médicament du marché se prend sur recommandation de l'Agence européenne du médicament et est soumise à la nécessité pour l'administration communautaire d'apporter la preuve que des connaissances scientifiques ou des données nouvelles, issues notamment de la pharmacovigilance, ont modifié les bases sur lesquelles l'AMM avait été accordée 9 ( * ) .
En France, l'article L. 5121-8 du code de la santé publique dispose que l'AMM est suspendue ou retirée dans des conditions déterminées par voie réglementaire et, en particulier, « lorsqu'il apparaît que l'évaluation des effets thérapeutiques positifs du médicament ou produit au regard des risques tels que définis au premier alinéa n'est pas considérée comme favorable dans les conditions normales d'emploi, que l'effet thérapeutique annoncé fait défaut ou que la spécialité n'a pas la composition qualitative et quantitative déclarée ». La nécessité d'établir que l'une ou plusieurs de ces conditions sont réunies incombe à l'agence en charge de la police sanitaire, l'agence française de sécurité sanitaire des produits de santé (Afssaps).
Cette nécessité est renforcée pour la suspension d'AMM qui, conformément au régime général des dispositions administratives pour lesquelles la procédure est allégée, est soumise au fait de prouver l'urgence de la décision. Il incombe dès lors à l'Afssaps d'apporter les « éléments établissant l'existence d'indices sérieux et concluants d'un risque grave pour la santé des patients, pour que la mesure de suspension [soit] justifiée par une situation d'urgence », sous peine d'être regardée comme entachée d'une erreur manifeste d'appréciation 10 ( * ) .
Le contrôle du juge administratif est ainsi plus étroit pour les conditions de suspension que pour celles de retrait, à l'égard desquelles il se fonde sur l'appréciation des commissions de l'Afssaps et les résultats des études scientifiques qui ont servi de base à la décision. La contestation des signaux de pharmacovigilance, l'absence d'unanimité des experts ou d'étude scientifique prouvant l'existence d'un risque nouveau ou aggravé lié à l'usage du médicament fragilisent donc les fondements juridiques des décisions de suspension et de retrait d'un médicament de l'Afssaps. Il en résulte qu'en l'état du droit, de simples doutes ne peuvent pas permettre de retirer un médicament mis sur le marché, et ce quel que soit son niveau d'efficacité, dès lors que celle-ci n'est pas nulle. Ceci tend, de fait, à subordonner la sécurité sanitaire à la protection du droit de propriété.
A cette première limite légale à l'action de l'Afssaps, qui doit donc respecter le droit de propriété des industriels en apportant des preuves scientifiques suffisantes à l'appui de ses décisions de suspension ou de retrait d'un médicament, s'en ajoute une autre concernant l'usage du médicament .
Les médecins, en effet, ne sont pas tenus de suivre dans leurs prescriptions les usages déterminés par l'AMM. La liberté de prescription, fixée par l'article 8 du code de déontologie médicale (article R. 4127-8 du code de la santé publique) est également protégée par l'article L. 162-2 du code de la sécurité sociale. La prescription dite « hors AMM » apparaît comme une nécessité pour soigner certaines pathologies ou certains malades, suivant les données de la science. Un exemple classique d'utilisation hors AMM est celui de l'aspirine utilisée pour soigner des maladies cardiovasculaires alors que sa mise sur le marché ne comportait que l'utilisation antalgique. L'Afssaps ne dispose donc d'aucun moyen d'interdire ces prescriptions et peut simplement agir par voie d'information des professionnels de santé et du public sur les effets indésirables, ainsi qu'elle l'a fait le 6 juin 2011 à propos de l'utilisation hors AMM du baclofène dans le traitement de l'alcoolo-dépendance. Ainsi, même si le directeur général de l'Afssaps décide, comme le prévoit l'article R. 5121-47 du code de la santé publique, de modifier d'office une AMM afin d'en restreindre le champ, l'agence ne peut obliger les praticiens à restreindre en conséquence leurs prescriptions. C'est alors la responsabilité du médecin prescripteur qui est en cause puisqu'il lui est interdit, en application de la loi du 4 mars 2002 11 ( * ) , de soumettre son patient à un risque inconsidéré et qu'il doit l'informer des risques qu'il encourt en acceptant le traitement.
B. LA MISE EN oeUVRE D'UNE EXPERTISE PUBLIQUE
Le système des agences sanitaires a été critiqué pour sa complexité. Celle-ci est cependant plus apparente que réelle. D'une part, le système français maintient un équilibre entre l'exclusion totale du pouvoir politique des décisions concernant les produits de santé, tel qu'il se pratique dans les systèmes anglo-saxon et scandinave d'agences indépendantes, et la gestion pleinement ministérielle et administrative qui est encore pratiquée dans de nombreux Etats européens. D'autre part, la distinction entre les fonctions des différents acteurs, alliée à la multiplicité des regards portés sur les produits de santé, apparaît comme la meilleure garantie d'une expertise publique de qualité.
1. Les acteurs de la politique du médicament
Concrètement, trois acteurs publics agissent pour déterminer l'usage des produits de santé : l'agence française de sécurité sanitaire des produits de santé (Afssaps), la Haute Autorité de santé (HAS) et le Comité économique des produits de santé (Ceps).
L'Afssaps a été créée par la loi d'origine sénatoriale du 1 er juillet 1998 relative au renforcement de la veille sanitaire et du contrôle de la sécurité sanitaire des produits destinés à l'homme 12 ( * ) afin de rassembler, au sein d'une même agence, le contrôle de tous les produits de santé ou à finalité sanitaire ainsi que des produits cosmétiques. Elle s'était alors donc substituée à l'Agence du médicament créée en 1993 13 ( * ) . Etablissement public de l'Etat placé sous la tutelle du ministre chargé de la santé, l'Afssaps est un organisme administratif dépositaire d'un pouvoir autonome pour prendre des décisions individuelles en matière de sécurité sanitaire des produits de santé et des cosmétiques. Elle n'est donc pas qu'une instance d'expertise. C'est une instance décisionnaire dont le directeur général a pour mission de prendre les mesures nécessaires à la sécurité sanitaire. Le directeur général est libre de se déterminer par rapport aux avis des experts mais son pouvoir s'exerce dans le cadre légal exposé précédemment.
S'agissant tant de la mise sur le marché du médicament que de son retrait, les décisions du directeur général s'appuient sur les avis émis par les experts réunis au sein d'une commission dite commission d'AMM. Celle-ci se prononce en fonction du rapport bénéfices-risques du médicament et se doit d'autoriser la mise sur le marché si le bilan est favorable. Cette autorisation est pour le moment fondée sur la notion de non infériorité au sens où une firme, pour obtenir l'AMM d'un nouveau médicament, doit apporter la preuve qu'il est plus efficace qu'un placebo et, pour certains médicaments, au moins aussi efficace et aussi bien toléré que les médicaments déjà disponibles, grâce à des essais cliniques dits de non-infériorité.
Les signaux liés aux effets indésirables des médicaments sont pour leur part étudiés par une autre des commissions de l'Afssaps, la commission nationale de pharmacovigilance (CNPV). Comme le notait la mission d'information sénatoriale sur le Mediator, la CNPV « se prononce sur le risque lié à un médicament, mais ne peut formuler d'avis relatif à l'AMM. Dans les faits, elle fonctionne donc actuellement comme une commission d'information de la commission d'AMM ».
Le directeur général de l'Afssaps a fait part à votre rapporteur de sa volonté de réunion au sein d'une « assemblée plénière » des experts en charge de l'AMM et de la pharmacovigilance, afin de déterminer à l'avenir les conditions d'octroi, de retrait et de suspension des AMM. Si les modalités concrètes de cette réorganisation sont encore à déterminer, l'évolution projetée semble pouvoir limiter le risque que les experts en charge de l'AMM ne négligent les signaux de pharmacovigilance et que les deux commissions ne s'engagent dans une opposition stérile. Votre rapporteur insiste cependant sur le fait que l'essentiel est que le directeur général de l'Afssaps assume pleinement les compétences qui sont les siennes et qu'il prenne les décisions qu'il juge nécessaires à la santé publique sans être lié par les contradictions entre experts ou les aléas d'un vote majoritaire.
Parallèlement au pouvoir de police dont dispose l'Afssaps, la Haute Autorité de santé , créée en 2004 14 ( * ) , porte un deuxième regard sur l'intérêt thérapeutique des médicaments au travers d'une de ses commissions spécialisée, la commission de la transparence . Celle-ci détermine le service médical rendu (SMR) d'un médicament et l'éventuelle amélioration du service médical rendu (ASMR) qu'il offre pour déterminer son efficacité thérapeutique absolue et, surtout, comparative, afin de juger si le médicament doit être pris en charge par la collectivité, c'est-à-dire remboursé par la Sécurité sociale. Pour établir l'intérêt relatif d'un médicament, la commission est cependant le plus souvent contrainte de recourir à des comparaisons indirectes à partir des études publiées sur le médicament soumis et de celles publiées sur les médicaments de référence. En effet, le dossier qui lui est présenté ne comporte que rarement des études nouvelles par rapport au dossier d'AMM. Il s'agit donc pour la commission d'examiner à nouveau le dossier en complétant son information par les autres publications scientifiques utiles à son évaluation. Le caractère nécessairement imparfait des comparaisons ainsi dressées est regretté tant par la commission que par l'industrie pharmaceutique, avec des objectifs souvent inverses. Votre rapporteur relève pour sa part le caractère rigoureux du travail de la commission de la transparence, qui utilise au mieux les moyens dont elle dispose. La diffusion d'un médicament étant essentiellement liée à son remboursement, les avis de cette commission ont une importance déterminante sur l'exposition de la population.
Comme l'avait indiqué la mission d'information sénatoriale sur le Mediator, votre rapporteur estime que le double regard, porté par la commission d'AMM puis par la commission de la transparence, sur l'évaluation du progrès thérapeutique d'un médicament pourrait devenir une garantie importante de sécurité si, à l'avenir, une place institutionnelle est faite à l'avis de la commission de la transparence sur ce sujet. L'absence de procédure lui permettant de faire valoir son analyse sur la nocivité d'un médicament limite, à l'heure actuelle, la possibilité de remettre en cause une AMM. Elle devrait pouvoir saisir le ministre de la santé de ses doutes concernant le rapport bénéfices-risques d'un médicament, à charge pour celui-ci de demander à l'Afssaps de répondre aux arguments présentés. Ce système aurait l'intérêt de préserver l'indépendance de la commission de la transparence, de permettre l'information du ministre sur les débats relatifs à l'efficacité d'un médicament et de l'amener à demander à l'Afssaps un approfondissement de la question.
Le troisième acteur à intervenir pour réglementer la diffusion des médicaments est le comité économique des produits de santé (Ceps) créé en 1996 15 ( * ) . Organisme interministériel placé sous l'autorité conjointe des ministres chargés de la santé, de la sécurité sociale et de l'économie, il a pour mission légale de fixer les prix des médicaments et les tarifs des dispositifs médicaux à usage individuel pris en charge par l'assurance maladie obligatoire, ainsi que de suivre les dépenses et la régulation financière du marché. Pour mener cette action, le comité a conclu avec les entreprises du médicament un accord cadre portant sur le prix des médicaments et son évolution, sur les remises, sur les engagements des entreprises concernant le bon usage des médicaments et les volumes de vente, sur les modalités de participation des entreprises à la mise en oeuvre des orientations ministérielles. C'est, dans le cadre du droit actuel, au Ceps qu'il convient d'appliquer les pénalités financières prononcées contre les laboratoires qui ne respectent pas leurs engagements au titre de la police sanitaire. Critiqué pour son approche « industrielle » des questions de santé, le Ceps semble, sous l'impulsion de son nouveau président, Gilles Johanet, déterminé à nouer des liens étroits avec la HAS et à mettre en place des procédures plus transparentes qui permettront notamment une meilleure compréhension du lien entre les avis émis par la commission de la transparence sur le service médical rendu et le prix fixé par le Ceps.
Le triple regard ainsi porté sur les médicaments remboursables, et l'autonomie des différents acteurs chargés d'examiner les dossiers, sont des garanties pour permettre une politique de sécurité sanitaire efficace, à une double condition : l'information réciproque et le fait pour les acteurs, tant administratifs que politiques, d'assumer leurs responsabilités. En effet, c'est au Gouvernement et en son sein plus précisément aux ministres en charge de la santé et de la sécurité sociale qu'il appartient de s'assurer de la cohérence du système de sécurité sanitaire et d'éviter les blocages porteurs de risques pour la santé publique.
2. L'expertise
L'expertise publique repose aujourd'hui sur un système ancien et sans doute obsolète qui fait appel à la bonne volonté et au bénévolat des scientifiques les plus qualifiés, abnégation censée être compensée par le prestige national et international associé à la reconnaissance publique du statut d'expert. Or, la remise en cause régulière de l'expertise et le soupçon qui semble s'attacher trop souvent à son exercice réduisent à peu de choses la gratification symbolique que représente le fait d'être choisi pour conseiller les décideurs publics.
Fixer le cadre déontologique de l'expertise ne peut donc suffire. Il est nécessaire, pour maintenir l'expertise publique française au plus haut niveau de qualité, d'offrir aux experts un statut attractif tant en terme de rémunération que de déroulement de carrière. Il convient également, afin d'enrichir les débats au sein des instances d'expertise, de diversifier les profils des experts publics : ils ne doivent pas être exclusivement recrutés parmi les médecins hospitaliers et il convient que siègent également dans les instances d'expertise les médecins de terrain, les autres professions médicales et des représentants des sciences humaines.
En ne proposant pas de statut de l'expertise, le projet de loi soumis au Sénat reste en retrait et ne permettra pas de résoudre l'ensemble des difficultés pointées pourtant de manière récurrente par les rapports successifs sur l'expertise, les crises et la sécurité sanitaire.
II. RENFORCER LA SÉCURITÉ DES PRODUITS DE SANTÉ
Il convient de reconnaître que le projet de loi cherche à maintenir l'équilibre existant. Il reste cependant trop souvent au niveau des symboles.
A. TROUVER L'ÉQUILIBRE ENTRE ACCÈS AUX THÉRAPIES ET SÉCURITÉ
Ainsi que le soulignent à juste titre les associations de patients, la mise en oeuvre de la sécurité sanitaire ne doit pas aboutir à une perte de chance pour les patients. Un médicament nocif en population générale peut avoir un effet positif pour certains malades dans le cadre d'un usage contrôlé. C'est le cas du thalidomide, dont les effets secondaires dramatiques pour les femmes enceintes et leurs enfants ont, on l'a été rappelé, provoqué le premier scandale sanitaire européen et qui a cependant fait l'objet de plusieurs autorisations temporaires d'autorisations depuis 1997 16 ( * ) . Depuis 2009, il fait en effet l'objet d'une indication pour le traitement de première ligne des patients âgés de plus de soixante-cinq ans présentant un myélome multiple non traité ou présentant une contre-indication à la chimiothérapie à haute dose.
Personne ne saurait contester la nécessité de préserver l'accès aux traitements les plus adaptés et les plus innovants. Il convient cependant que les autorités sanitaires se mettent en capacité d'anticiper les risques futurs liés aux produits de santé autres que le médicament.
1. Ne pas faire perdre de chances aux malades
A côté de la procédure de mise sur le marché, qui répond aux sollicitations des laboratoires dans le cadre de leur logique commerciale, des dérogations ont toujours existé pour permettre la diffusion de médicaments non ou non encore autorisés aux patients pour lesquels ils présentent un intérêt thérapeutique majeur. Ces demandes émanant des médecins souvent spécialistes de maladie rares ou de pathologies particulièrement sévères visent à faire profiter leurs patients au plus vite des avancées de la science. Plusieurs mécanismes permettent de leur apporter une réponse.
L'article L. 5121-12 du code de la santé publique dispose que l'Afssaps peut délivrer, à titre exceptionnel, des autorisations temporaires d'utilisation ATU pour des médicaments ne disposant pas d'AMM centralisée ou nationale et destinés à traiter des maladies graves ou rares lorsqu'il n'existe pas de traitement approprié.
Deux types d'ATU sont à distinguer.
L'ATU de cohorte doit être demandée par le titulaire des droits d'exploitation pour un groupe de patients lorsque l'efficacité et la sécurité du médicament sont fortement présumées au regard des résultats des essais thérapeutiques auxquels il a été procédé en vue d'une demande d'AMM. Le titulaire des droits d'exploitation doit avoir déposé une demande d'AMM ou s'engager à le faire dans un délai déterminé. Il conclut également avec l'Afssaps un protocole d'utilisation thérapeutique et de recueil d'informations.
L'ATU nominative peut être accordée pour un patient déterminé, à la demande et sous la responsabilité du médecin, lorsque le médicament est susceptible de présenter un bénéfice pour le patient et que soit son efficacité et sa sécurité sont présumées en l'état des connaissances scientifiques, soit une issue fatale pour le patient est, en l'état des thérapeutiques disponibles, inéluctable. L'ATU nominative suppose une information adaptée du patient ou de son représentant légal et peut être subordonnée par l'Afssaps à la conclusion d'un protocole d'utilisation thérapeutique et de recueil d'informations.
Si l'article L. 5121-12 dispose en son quatrième alinéa que les ATU sont attribuées pour une durée limitée, la partie réglementaire du code de la santé publique précise que les ATU de cohorte sont délivrées pour une année et les ATU nominatives pour la durée du traitement, dans la limite maximale d'un an. Les deux types d'autorisation peuvent être renouvelés à l'issue de ces délais 17 ( * ) . L'Afssaps a le pouvoir de suspendre ou retirer l'ATU si les conditions prévues à l'article L. 5121-12 ne sont plus remplies ou pour des motifs de santé publique.
Le coût des médicaments sous ATU est libre et entièrement pris en charge par l'assurance maladie. L'article L. 162-16-5-1 du code de la sécurité sociale oblige le laboratoire titulaire des droits d'exploitation à déclarer au Comité économique des produits de santé (Ceps) le montant de l'indemnité demandée auprès des établissements de santé pour le médicament sous ATU. Si, dans le cadre d'une AMM ultérieure, le Ceps fixe un prix ou un tarif de remboursement inférieur au prix déclaré, le laboratoire doit reverser tout ou partie de la différence aux organismes de recouvrement. L'indemnité est ensuite affectée aux régimes d'assurance maladie.
Les ATU sont le plus souvent délivrées pour :
- des médicaments anciens bénéficiant généralement d'une AMM à l'étranger mais pour lesquels aucune demande n'a été déposée en France en raison d'un marché jugé insuffisamment rentable par le titulaire de l'autorisation ;
- des médicaments en cours de développement, avec un projet de dépôt d'AMM ;
- des médicaments en cours d'évaluation d'AMM, le plus souvent en procédure centralisée ;
- des médicaments pour lesquels l'ATU permet de pallier une suspension, un retrait ou un refus d'AMM, un arrêt de commercialisation ou l'impossibilité d'inclure les patients dans les essais cliniques.
Les données chiffrées présentées ci-dessous sont révélatrices de la diversité des situations que recouvrent les deux types d'ATU. Alors que les ATU de cohorte sont, de façon générale et de plus en plus, attribuées pour une durée limitée, en vue d'une AMM future, les ATU nominatives sont souvent accordées pour des durées très longues. En 2009, seuls 9 % des médicaments disponibles en ATU nominatives ont obtenu une AMM.
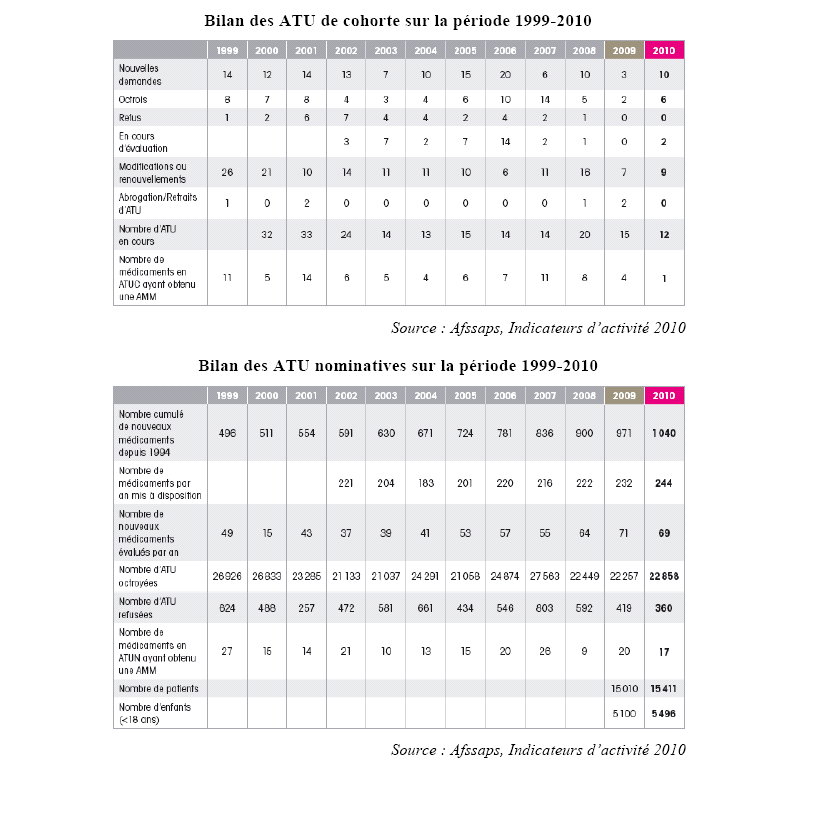
Il résulte de ces éléments, comme l'a souligné le directeur général de l'Afssaps à votre rapporteur, qu'une clarification des conditions dans lesquelles sont accordées les ATU nominatives est nécessaire. En effet, l'intérêt collectif étant que tous les patients puissent bénéficier le plus rapidement possible des innovations thérapeutiques, la transformation des ATU nominatives en ATU de cohorte et de celles-ci en AMM doit être encouragée. Il n'est en tous cas pas souhaitable que perdure la tendance actuelle de pérennisation de fait du régime des ATU.
2. Une pharmacovigilance proactive
Le projet de loi soumis à l'examen du Sénat entend fonder une pharmacovigilance active, en confiant à l'agence en charge du médicament la mission de réévaluer régulièrement les médicaments sur le marché. Cette évolution, dont la nécessité a été soulignée par l'ensemble des rapports relatifs à l'affaire du Mediator, ne doit pas faire oublier le risque lié aux autres produits de santé . L'une des plus grandes catastrophes sanitaires que notre pays ait connues était liée à des produits de santé, le sang et le plasma, qui n'étaient pas des médicaments. Votre rapporteur ne peut qu'être inquiet de l'analyse faite par plusieurs des acteurs du système de santé lors de ses auditions, selon laquelle « le prochain Mediator sera un dispositif médical » . Cette inquiétude est renforcée par le fait que la proposition de loi sénatoriale dont l'adoption, en 1998, avait entraîné le remplacement de l'agence du médicament par l'Afssaps était motivée par la nécessité de réunir, au sein d'une même agence, l'ensemble des compétences sur les produits de santé et d'améliorer les « règles de droit applicables à certains produits, tels que les dispositifs médicaux, aujourd'hui insuffisamment réglementés » 18 ( * ) .
Aux termes de l'article L. 5211-1 du code de la santé publique, constitue un dispositif médical « tout instrument, appareil, équipement, matière, produit, à l'exception des produits d'origine humaine, ou autre article utilisé seul ou en association, y compris les accessoires et logiciels nécessaires au bon fonctionnement de celui-ci, destiné par le fabricant à être utilisé chez l'homme à des fins médicales et dont l'action principale voulue n'est pas obtenue par des moyens pharmacologiques ou immunologiques ni par métabolisme, mais dont la fonction peut être assistée par de tels moyens. Constitue également un dispositif médical le logiciel destiné par le fabricant à être utilisé spécifiquement à des fins diagnostiques ou thérapeutiques » .
Cette définition recouvre une grande variété de produits, allant du stéthoscope au fauteuil roulant en passant par la pompe à insuline ou le stimulateur cardiaque 19 ( * ) . Les accessoires des dispositifs médicaux constituent également des dispositifs médicaux (les solutions pour lentilles par exemple).
Le marché mondial des dispositifs médicaux connaît un développement récent lié à l'essor de technologies de plus en plus performantes. Il est aujourd'hui estimé à près de 200 milliards d'euros, soit un tiers de celui du médicament 20 ( * ) . Les dispositifs médicaux représentent en France 12 % de la consommation totale de biens et soins médicaux. En vertu du principe de libre circulation et contrairement aux médicaments, les dispositifs médicaux ne font l'objet d'aucune autorisation avant leur commercialisation . Les règles qui entourent leur mise sur le marché ainsi que leur surveillance a posteriori ont pour l'essentiel été fixées au niveau communautaire avant d'être transposées en droit national.
La commercialisation des dispositifs médicaux s'inscrit dans le cadre de la « nouvelle approche » engagée en 1985 par le Conseil de l'Union européenne, destinée à assurer la libre circulation d'un ensemble de produits industriels et fondée sur le principe de reconnaissance mutuelle. Plusieurs directives sont venues adapter cette « nouvelle approche » aux dispositifs médicaux, notamment pour fixer les exigences essentielles que doivent respecter ces produits en termes de sécurité et de rapport bénéfices/risques 21 ( * ) . Ces textes ont été révisés par la directive 2007/47/CE du 5 septembre 2007 dans le sens d'un renforcement de l'évaluation clinique des dispositifs médicaux. C'est sur le fabricant ou son mandataire que repose la responsabilité d'apposer un marquage CE attestant du respect des exigences essentielles prévues par les directives. Ces dernières varient selon la classification des dispositifs médicaux au sein des quatre catégories prévues par les directives :
|
Classes |
Niveau de risque |
Exemples |
|
Classe 1 |
Faible degré de risque |
Lits médicaux, fauteuils roulants, seringues... |
|
Classe II a |
Degré moyen de risque |
Prothèses auditives, échographe, scalpels à usage unique... |
|
Classe II b |
Potentiel élevé de risque |
Lentilles intra-oculaires, scanner, pompes à insuline externes... |
|
Classe III |
Potentiel très sérieux de risque |
Valves cardiaques, cathéters héparinés, stents coronaires... |
|
Source : Igas, Evolution et maîtrise de la dépense des dispositifs médicaux novembre 2010, Tome II, p. 20 |
||
Pour les dispositifs de classe I non stériles, le fabricant procède à une auto-certification. Dans les autres cas, la certification doit être effectuée par un organisme notifié, parmi ceux figurant dans une liste établie par la Commission européenne.
En France, la procédure de mise sur le marché et de matériovigilance peut être schématisée de la façon suivante :
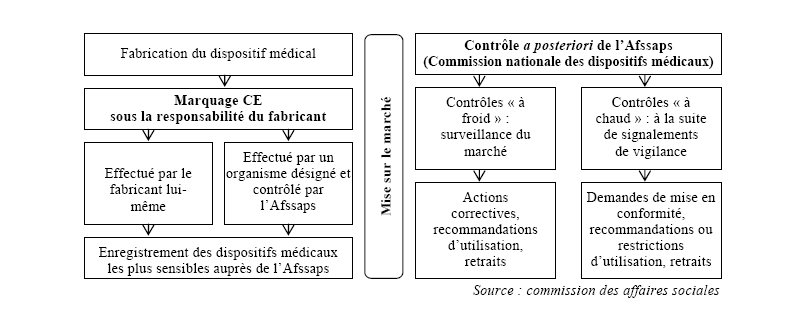
En 2008, l'Afssaps a enregistré 7 799 signalements de matériovigilance.
Une réforme du système particulièrement complexe relatif aux dispositifs médicaux, avec la mise en place d'une évaluation comparative pour garantir leur sécurité, doit être une priorité de l'action des autorités sanitaires.
B. NE PAS SE LIMITER AUX SYMBOLES
Votre rapporteur regrette que, bien qu'il comporte quelques mesures utiles et que certains points aient été précisés par l'Assemblée nationale, le projet de loi soumis à l'examen du Sénat ne soit pas à la hauteur des ambitions qu'il affiche ni des annonces faites par le ministre de la santé à l'occasion de l'affaire du Mediator.
1. Le projet de loi ne propose que des progrès limités
Tout en donnant une base légale à l'action de pharmacovigilance proactive déjà entamée par l'Afssaps à la suite de l'affaire du Mediator, le texte ne propose que des apports assez limités au droit existant et écarte toutes les réformes de fond, préconisées notamment par le rapport de la mission d'information sénatoriale sur le Mediator, qui sont susceptibles d'entrainer des coûts, comme la création déjà évoquée d'un véritable statut de l'expert. Cette attitude est d'autant plus dommageable que le Parlement, soumis aux dispositions de l'article 40 de la Constitution, se voit interdit de remédier aux insuffisances du texte d'origine si l'initiative n'en est pas prise par le Gouvernement.
Il apparaît d'abord que l'unification des dispositions concernant les déclarations d'intérêts des experts n'ajoute pas grand-chose au droit en vigueur, des dispositions analogues étant déjà prévues dans les textes législatifs spécifiques à ces instances sauf, semble-t-il, dans les cas de l'Institut national du cancer (INCa) et des agences régionales de santé (ARS). Au contraire même, elle a pour corollaire la suppression de ces dispositions spécifiques, souvent plus complètes et plus détaillées. Elle aboutit, en outre, compte tenu de la diversité des organismes concernés, à un texte inévitablement confus et imprécis. De plus, cet article ne prévoit ni définition des liens et des conflits d'intérêts, ni système de contrôle centralisé des déclarations, de sorte qu'il est permis de douter de l'efficacité du dispositif proposé.
L'attachement du Gouvernement au changement de nom de l'Afssaps paraît lui aussi largement symbolique, de plus au prix d'approximations et pour un coût non nul. Afin de pouvoir affirmer qu'il crée une « nouvelle » agence, ce qui est juridiquement inexact, le Gouvernement a décidé de revenir sur ce qui avait été l'un des points les plus importants de loi de 1998 sur le renforcement de la sécurité sanitaire : l'unification du contrôle sur l'ensemble des produits de santé , fondé sur le fait que « les nouvelles générations de produits de santé font éclater les distinctions traditionnelles entre produits telles que celles qui séparent, par exemple, le médicament, les produits sanguins, les greffes et les dispositifs médicaux » 22 ( * ) . Affirmer que la crise du Mediator impose un changement de nom de l'agence revient non seulement à masquer l'ensemble du travail qu'elle a accompli depuis 1998 mais également s'exposer, en l'état actuel de la proposition gouvernementale, au risque de devoir changer le nom de l'agence à chaque crise. La réforme de l'Afssaps, si elle est réelle, se vérifiera dans les faits, pas dans les intitulés.
Concernant les procédures d'ATU, le texte soumis à l'examen du Sénat apporte des modifications dont il est difficile d'apprécier la portée. Il crée en particulier une procédure dérogatoire pour les ATU nominatives qui pourrait potentiellement s'appliquer à un grand nombre de situations. Si toute action sur le prix de ces autorisations comporte inévitablement des effets pervers, il n'est pas certain que les évolutions proposées par le texte permettent réellement d'éviter le recours aux ATU comme moyen de contourner la procédure d'AMM et la fixation du prix des médicaments par le Comité économique des produits de santé.
Plus fondamentalement, ce texte est peu lisible, sa rédaction particulièrement imprécise et il renvoie de manière quasi systématique à des textes règlementaires, ce qui crée une incertitude quant à la traduction concrète des dispositions qu'il contient. Il laisse d'autant plus de prise aux procédures contentieuses que ne manqueront pas d'engager les industries du médicament et risque de rendre rapidement l'agence en charge de la sécurité sanitaire des produits de santé, quel que soit son nom, aussi frileuse que l'a été l'Afssaps à une période trouble de son histoire.
2. Les apports de l'Assemblée nationale
L'Assemblée nationale, principalement à l'initiative du groupe socialiste, radical, citoyen et divers gauche, a apporté plusieurs compléments importants au texte du Gouvernement. Le premier est la création d'une base publique informatique des maladies et de leurs traitements confiée à la HAS. Ce portail public répondra à une aspiration profonde de la population à une information sanitaire indépendante, de qualité et facilement accessible.
L'apport essentiel est cependant constitué par l'article 9 bis , qui conditionne l'inscription d'un médicament sur la liste des médicaments remboursables à la conduite d'essais comparatifs . Cette disposition permet, en toute conformité avec le droit communautaire, de demander enfin le plus haut niveau de preuve scientifique pour qu'un médicament soit pris en charge par la collectivité. Ce dispositif, qui devrait être étendu aux dispositifs médicaux une fois leur régime légal mieux défini, doit encore être conforté et précisé. Il constitue, aux yeux de votre rapporteur, le point le plus important du projet de loi soumis et ce qui, sous réserve d'autres amendements présentés, pourra fonder son adoption par le Sénat.
EXAMEN DES ARTICLES
TITRE
IER - TRANSPARENCE DES LIENS D'INTÉRÊTS
CHAPITRE
IER - Liens d'intérêts
Article 1er (art. L. 1451-1, L. 1451-1-1, L. 1451-2 et L. 1451-3 (nouveaux) du code de la santé publique) Règles déontologiques et expertise sanitaire
Objet : Cet article organise les obligations pesant sur les experts et les membres des autorités sanitaires et prévoit les modalités de publicité des débats en leur sein.
I - Le dispositif proposé
Cet article se compose de trois parties.
La première partie comporte huit points.
Le 1° modifie, au sein du livre IV de la première partie du code de la santé publique, qui traite de l'administration générale de la santé, l'intitulé du titre V, relatif aux conseils et commissions.
Le titre V prend l'intitulé de son premier chapitre actuel « Règles déontologiques » complété par une référence à l'expertise sanitaire.
Le 2° modifie en conséquence l'intitulé du chapitre 1 er dont le contenu est précisé. Il devient « Liens d'intérêts et transparence ».
Le 3° réécrit l'article L. 1451-1 en y regroupant, au côté de celle figurant déjà pour les membres des commissions et conseils siégeant auprès des ministres chargés de la santé et de la sécurité sociale, l'obligation pour l'ensemble des dirigeants, personnels de direction et d'encadrement et les membres des instances collégiales, commissions et conseils des autorités sanitaires d'établir des déclarations d'intérêts et de les actualiser si nécessaire.
La déclaration fait mention des intérêts directs ou indirects avec les entreprises, établissements ou organismes dont les activités, les techniques ou les produits entrent dans le champ de compétences de l'instance au sein de laquelle siège le déclarant. La formulation retenue est plus large que celle figurant dans l'article actuel du code, qui vise les entreprises susceptibles de soumettre des dossiers.
La possibilité de participer aux délibérations et aux votes est désormais soumise au dépôt et à l'actualisation d'une déclaration d'intérêts. L'interdiction de participer aux délibérations et aux votes en cas de conflit d'intérêts sous peine de sanction pénale demeure, de même que l'obligation de discrétion professionnelle.
L'obligation d'établir une déclaration pèsera également sur les agents des autorités dont les missions ou fonctions auront été considérées comme le justifiant et portées sur une liste établie par décret en Conseil d'Etat.
Le 4° insère un article L. 1451-1-1 pour encadrer par décret en Conseil d'Etat la possibilité de rendre publics, d'enregistrer et de diffuser les comptes rendus et les enregistrements des séances et débats des autorités sanitaires.
Le 5° modifie l'article L. 1451-2 pour procéder à des coordinations.
Le 6° crée un article L. 1451-3 pour renvoyer les conditions d'application du chapitre et plus particulièrement celles relatives à la forme et au contenu des déclarations d'intérêts à un décret en Conseil d'Etat.
Le 7° procède à des mises en cohérence au sein du code afin de permettre l'insertion des dispositions relatives à l'expertise sanitaire.
Le 8° insère un nouveau chapitre relatif à l'expertise sanitaire, composé de deux articles L. 1452-1 et L. 1452-2.
L'article L. 1452-1 prévoit l'élaboration d'une charte de l'expertise, approuvée par décret en Conseil d'Etat, pour déterminer les modalités de choix des experts appelés à donner leur avis aux ministres ou aux autorités sanitaires et la gestion des conflits d'intérêts.
L'article L. 1452-2 dispose que les experts désignés déposent une déclaration d'intérêts selon les modalités prévues à l'article L. 1451-3.
La deuxième partie de l'article se compose de quatre points. Elle procède à des coordinations en supprimant au sein du code de la santé publique, du code de la sécurité sociale, de la loi créant une agence française de sécurité sanitaire 23 ( * ) et de la loi relative à la sécurité sanitaire 24 ( * ) , les mentions éparses relatives aux déclarations d'intérêts des membres des différentes autorités sanitaires.
La troisième partie de l'article procède à une renumérotation au sein du code de la santé publique.
II - Les dispositions adoptées par l'Assemblée nationale
L'Assemblée nationale a adopté plusieurs amendements pour :
- préciser que les liens d'intérêts indirects devant figurer dans les déclarations prévues par cet article sont ceux qui concernent les liens indirects personnels mais aussi ceux des conjoints, ascendants ou descendants, établis au cours des cinq dernières années ;
- limiter l'obligation de publicité des délibérations à celles qui fondent une décision administrative ;
- prévoir, selon des modalités déterminées par décret en Conseil d'Etat, qu'une commission d'éthique constituée au sein de chaque agence contrôle la véracité des informations contenues dans les déclarations d'intérêts ;
- supprimer une coordination inutile.
III - Le texte adopté par la commission
Votre commission est réservée sur la portée de cet article qui n'ajoute rien au droit existant et fait au contraire disparaître plusieurs dispositions plus contraignantes qui figuraient parmi celles relatives à chacune des agences.
Elle a donc adopté, à l'initiative de son rapporteur, un amendement rédactionnel ainsi que huit amendements afin de :
- clarifier la définition de la déclaration publique d'intérêts en précisant que celle-ci est aussi nécessaire pour les dirigeants des autorités sanitaires ;
- interdire au président de la HAS et au directeur général de l'Afssaps, dans les trois ans précédant leur mandat et au cours de celui-ci, tout lien susceptible d'affecter ou de paraître affecter l'exercice indépendant, impartial et objectif de leurs fonctions ;
- préciser les conditions de la publicité des débats au sein des autorités sanitaires en supprimant la référence au secret industriel et commercial et en prévoyant, parallèlement à la captation vidéo, l'obligation de diffuser gratuitement en ligne un procès verbal intégral comprenant le détail et les explications des votes, y compris les opinions minoritaires ;
- supprimer la disposition, juridiquement inutile et peu efficace prévoyant la création au sein de chaque autorité sanitaire d'une instance de déontologie chargée de contrôler les déclarations d'intérêt public de ses membres ;
- supprimer le recours à une charte de l'expertise, dont le caractère normatif paraissait contestable, pour la définition de choix des experts externes et de la gestion des liens et des conflits d'intérêts ;
- prévoir la présence de médecins généralistes et d'experts en sciences humaines au sein des instances collégiales des autorités sanitaires ;
- étendre la compétence de la commission de déontologie créée par la loi « Sapin » du 29 janvier 1993 25 ( * ) , à l'égard des personnels des conseils et agences sanitaires soumis à déclaration publique d'intérêts. Cette commission, que le projet de loi relatif à la déontologie et à la prévention des conflits d'intérêts dans la vie publique propose de transformer en une « Autorité de la déontologie de la vie publique » aux compétences élargies, sera chargée de la centralisation des déclarations publiques d'intérêts ;
- confier le contrôle du respect des secrets protégés par la loi à la commission d'accès aux documents administratifs (Cada) afin d'éviter toute invocation abusive tendant à limiter la publicité des débats au sein des agences.
A l'initiative d'Isabelle Pasquet, de Laurence Cohen, d'Annie David, de Dominique Watrin et de Guy Fischer, la commission a étendu l'interdiction de liens d'intérêts directs au directeur général de l'institut national de la santé et de la recherche médicale (Inserm) et au président de l'institut national du cancer (INCa) dans les mêmes conditions que pour le président de la HAS et le directeur général de l'Afssaps.
La commission a adopté cet article ainsi modifié.
Article 1er bis (nouveau) (art. L. 1415-4 du code de la santé publique) Conditions de nomination des personnels dirigeants de l'institut national du cancer
Objet : Cet article additionnel détermine les modalités et les conditions de choix du président du conseil d'administration de l'INCa et du président de son conseil scientifique.
Cet article additionnel, inséré sur amendement présenté par Isabelle Pasquet, Laurence Cohen, Annie David, Dominique Watrin et Guy Fischer, tend à soumettre le choix, par le ministre, du président du conseil d'administration et du président du conseil scientifique de l'INCa à un appel à candidature préalable et à l'absence de tout lien d'intérêts entre les personnalités choisies et les entreprises dont l'activité entre dans le champ de compétences de l'institut.
La commission a adopté cet article additionnel ainsi rédigé.
CHAPITRE II - Avantages
Article 2 (art. L. 1453-1 (nouveau) et L. 4113-6 du code de la santé publique) Obligation de publication des avantages consentis par les entreprises au profit des acteurs du champ des produits de santé
Objet : Cet article fixe les conditions de publicité des relations entre les acteurs du système de santé et les entreprises.
I - Le dispositif proposé
Cet article se compose de deux parties.
La première partie crée un nouveau chapitre au sein du code de la santé publique consacré aux « avantages consentis par les entreprises ». Il se compose d'un nouvel article L. 1453-1 divisé en trois parties :
- le paragraphe I impose aux entreprises dont l'activité relève de la compétence de l'agence française de sécurité sanitaire des produits de santé de rendre publique l'existence des conventions conclues avec les professionnels de santé visés par le code de la santé publique, leurs associations, les établissements de santé, les fondations, sociétés savantes et organismes de conseil, les étudiants en médecine et en odontologie, les associations de patients et la presse spécialisée ;
- le paragraphe II rend obligatoire la publication des avantages en nature ou en espèces accordés par les entreprises au-delà d'un seuil fixé par décret ;
- le paragraphe III renvoie les modalités d'application de l'article à un décret en Conseil d'Etat.
La deuxième partie procède, pour les étudiants en médecine et en odontologie, à des coordinations avec l'article L. 4113-6 du code de la santé publique qui prévoit le principe de l'interdiction d'avantages en espèces ou en nature sauf exceptions définies.
II - Les dispositions adoptées par l'Assemblée nationale
L'Assemblée nationale a adopté plusieurs amendements pour étendre l'obligation de publication aux conventions conclues par les entreprises avec :
- les associations et groupements représentant les étudiants se destinant aux professions de santé régies par le code de la santé publique ;
- les services de radio ou de télévision et les éditeurs de communication au public en ligne ;
- les éditeurs de logiciels d'aide à la prescription et à la délivrance.
III - Le texte adopté par la commission
Votre commission estime que le texte de cet article n'offre pas les mêmes garanties concrètes en matière de transparence que le « Physician Payments Sunshine Act » américain dont il s'inspire. Pour renforcer ce dispositif et de garantir l'indépendance des étudiants face aux entreprises, votre commission a adopté, à l'initiative de son rapporteur, un amendement rédactionnel et cinq amendements afin de :
- garantir la publicité annuelle des conventions passées avec les entreprises et non pas seulement la mention de leur existence ;
- assurer la publicité des liens entre les entreprises et les établissements qui assurent la formation initiale des futurs professionnels de santé ou qui concourent à cette formation ;
- imposer la publication des rémunérations versées ;
- centraliser les informations recueillies sur un site internet unique et gratuit et garantir aux patients et aux responsables d'établissements de santé un accès aux informations précises qu'ils cherchent concernant les personnes et les entreprises ;
- interdire aux entreprises de passer des conventions d'hospitalité avec les étudiants ou de leur octroyer des avantages.
À l'initiative d'Isabelle Pasquet, Laurence Cohen, Annie David, Dominique Watrin et Guy Fischer, la commission a adopté deux amendements : l'un tendant à supprimer tout seuil en deçà duquel la déclaration des avantages reçus ne serait pas obligatoire ; l'autre tendant à rendre publics les avantages versés par les parlementaires nationaux et européens, les membres des cabinets du ministère en charge de la santé et de la sécurité sociale ainsi que les membres du gouvernement.
A l'initiative de Gilbert Barbier, la commission a adopté un amendement tendant à ce que les entreprises informent les organismes ordinaux compétents de la mise en oeuvre des conventions qu'elles ont déclarées.
La commission a adopté cet article ainsi modifié.
Article 2 bis Rapport sur le financement des associations de patients
Objet : Cet article, introduit par l'Assemblée nationale, entend permettre une plus grande transparence sur le financement des associations de patients.
I - Le dispositif proposé par l'Assemblée nationale
Cet article demande la transmission au Parlement d'un rapport sur le financement des associations d'usagers du système de santé avant le 30 juin 2012.
II - Le texte adopté par la commission
Votre commission est sensible à la question du financement des associations de patients, qui ne peut trouver de solution pérenne qu'au travers de la mise en place de subventions publiques. Bien que sans illusion sur la portée d'un nouveau rapport sur ce sujet, elle estime que celui-ci peut être l'occasion de dresser un état des lieux et de permettre à toutes les associations de donner leur point de vue.
La commission a adopté cet article sans modification.
CHAPITRE III - Sanctions pénales
Article 3 (art. L. 1454-2 à L. 1454-4 (nouveaux) du code de la santé publique) Dispositions pénales
Objet : Cet article prévoit les sanctions pénales applicables en cas de non-respect des obligations de déclaration pesant sur les personnes ou les entreprises.
I - Le dispositif proposé
Cet article se compose de trois parties.
La première propose d'insérer quatre articles après l'article L. 1454-1 du code de la santé publique.
L'article L. 1454-2 prévoit une amende de 30 000 euros pour les personnes qui omettraient sciemment d'établir ou d'actualiser leur déclaration d'intérêts ou y feraient figurer une information mensongère qui porte atteinte à sa sincérité.
L'article L. 1454-3 punit de 45 000 euros d'amende l'absence de publication par les entreprises concernées des conventions et avantages qu'elles accordent.
L'article L. 1454-4 ouvre la possibilité de condamnation des personnes physiques à des peines complémentaires prévues par le code pénal : publicité de la décision, interdiction des droits civiques, interdiction d'exercer une fonction publique. Une peine complémentaire d'interdiction de fabriquer, conditionner, importer ou mettre sur le marché, pour une durée maximale de cinq ans, des médicaments ou dispositifs médicaux est également créée.
L'article L. 1454-5 prévoit la possibilité de peines complémentaires pour les personnes morales condamnées. Ces peines reprennent pour partie celles prévues par l'article 131-39 du code pénal, soit :
- l'interdiction, à titre définitif ou pour une durée de cinq ans au plus, d'exercer directement ou indirectement une ou plusieurs activités professionnelles ou sociales ;
- le placement, pour une durée de cinq ans au plus, sous surveillance judiciaire ;
- la fermeture définitive ou pour une durée de cinq ans au plus des établissements ou de l'un ou de plusieurs des établissements de l'entreprise ayant servi à commettre les faits incriminés ;
- l'exclusion des marchés publics à titre définitif ou pour une durée de cinq ans au plus ;
- l'interdiction, à titre définitif ou pour une durée de cinq ans au plus, de procéder à une offre au public de titres financiers ou de faire admettre ses titres financiers aux négociations sur un marché réglementé ;
- l'interdiction, pour une durée de cinq ans au plus, d'émettre des chèques autres que certifiés ou qui permettent le retrait de fonds par le tireur auprès du tiré ou d'utiliser des cartes de paiement ;
- la peine de confiscation des biens utilisés pour commettre l'infraction ou obtenus grâce à elle, dans les conditions et selon les modalités prévues à l'article 131-21 du code pénal ;
- l'affichage de la décision prononcée ou la diffusion de celle-ci soit par la presse écrite, soit par tout moyen de communication au public par voie électronique.
La deuxième partie étend la compétence des agents habilités par l'article L. 4163-1 à constater les infractions prévues par le code de la santé publique en matière de perception d'avantages ou de conventions de partage d'honoraires au contrôle des relations entre pharmaciens et entreprises.
La troisième partie procède à des coordinations au sein de l'article L. 4163-2 relatif aux sanctions applicables à la perception d'avantages en espèces ou nature par les professionnels de santé.
II - Les dispositions adoptées par l'Assemblée nationale
L'Assemblée nationale a adopté un amendement de coordination à cet article.
III - Le texte adopté par la commission
Votre commission estime nécessaire que des sanctions puissent désormais être prononcées par le juge pour non-respect des obligations relatives à la transparence ou aux avantages consentis par les entreprises.
A l'initiative de son rapporteur, elle a adopté deux amendements de cohérence à cet article.
A l'initiative d'Isabelle Pasquet, Laurence Cohen, Annie David, Dominique Watrin et Guy Fischer, elle a également adopté deux amendements , l'un afin de sanctionner tout oubli dans les déclarations rendues obligatoires pour les entreprises et l'autre de cohérence.
La commission a adopté cet article ainsi modifié.
TITRE II - GOUVERNANCE DES PRODUITS DE SANTÉ
Article 4 (art. L. 5311-1, L. 5311-2, L. 5312-4, L. 5311-4-1 (nouveau) et L. 5421-8 à L. 5421-11 (nouveaux) du code de la santé publique) Création et prérogatives de l'agence nationale de sécurité du médicament et des produits de santé
Objet : Cet article modifie le nom de l'agence française de sécurité sanitaire des produits de santé et lui accorde le pouvoir de prononcer des amendes administratives.
I - Le dispositif proposé
Cet article se compose de huit parties.
La première propose de modifier l'article L. 5311-1 pour changer le nom de l'agence française de sécurité sanitaire des produits de santé (Afssaps) afin de mieux faire apparaître sa compétence en matière de médicament et lui accorde de nouvelles compétences en matière d'étude sur l'efficacité et sur la tolérance des médicaments ainsi qu'en matière de recueil d'informations. Elle se compose de quatre points :
- le 1° transforme l'Afssaps en « agence nationale de sécurité du médicament » ; son statut reste celui d'établissement public de l'Etat. La rédaction reprend celle de l'alinéa 23 de l'article L. 5311-1 actuel et attribue comme première mission de l'agence, l'évaluation et la réévaluation des bénéfices et des risques liés à l'usage des produits à finalité sanitaire destinés à l'homme et des produits à finalité cosmétique. La surveillance du risque lié à ces produits est explicitement prévue parmi les missions de l'agence ;
- le 2° procède à un regroupement d'alinéas ;
- le 3° opère à une coordination avec le texte proposé par l'article 5 du projet de loi pour l'article L. 5324-1 nouveau ;
- le 4° coordonne les mesures de publicité des délibérations de l'agence.
La deuxième partie propose de modifier l'article L. 5311-2 relatif aux activités de l'agence. Il se compose de deux points :
- le 1° ajoute à l'activité de conseil scientifique et technique de l'agence celle nécessaire à l'élaboration et à la mise en oeuvre des plans de santé publique ;
- le 2° complète la liste des activités énumérées, par l'encouragement de la recherche et l'éventuelle mise en place, par voie de convention, d'études de suivi de patients, d'efficacité et de tolérance.
Il propose en outre d'accorder une nouvelle prérogative à l'agence : celle d'accéder à toute information nécessaire à l'exercice de ses missions, sans que puisse lui être opposée aucune forme de secret, à charge pour elle de préserver la confidentialité de ces données à l'égard des tiers.
La troisième partie se compose de trois points :
- le 1° modifie l'article L. 5312-4 pour prévoir que l'agence informe l'opinion publique dans tous les cas où la santé publique l'exige ;
- le 2° prévoit que les professionnels de santé sont informés par l'agence parallèlement à l'opinion publique ;
- le 3° impute le coût des mesures d'information à la personne physique ou morale responsable de la mise sur le marché, de la mise en service ou de l'utilisation du ou des produits concernés.
La quatrième partie procède aux coordinations rendues nécessaires par le changement de nom de l'Afssaps.
La cinquième partie propose d'insérer dans le code un article L. 5312-4-1 octroyant à l'agence le pouvoir de prononcer des amendes administratives, éventuellement assorties d'astreintes journalières. Le prononcé de l'amende est soumis à la possibilité, pour la personne physique ou morale concernée, de présenter ses observations et de régulariser la situation, éventuellement sous astreinte. Lorsque l'infraction concerne une publicité, celle-ci pourrait être interdite.
Le nouvel article précise également le régime et le mode de recouvrement des amendes.
La sixième partie complète le nom du livre IV de la cinquième partie du code de la santé publique pour préciser que les sanctions applicables en matière d'infraction aux règles relatives aux produits de santé sont non seulement pénales mais aussi financières.
La septième partie propose d'ajouter quatre articles au sein des dispositions générales relatives au médicament à usage humain, après l'article L. 5421-7.
L'article L. 5421-8 dresse la liste de onze manquements susceptibles d'être sanctionnés par une amende prononcée par l'agence. Les six premiers manquements (1° à 6°) concernent le non-respect d'obligations liées à la pharmacovigilance. Les manquements définis aux points 7° à 10° sont relatifs aux autorisations temporaires d'utilisation ou à l'usage compassionnel des médicaments. Enfin le 11° sanctionne le non-respect des dispositions relatives à la publicité pour les dispositifs médicaux.
L'article L. 5421-9 rappelle le pouvoir de sanction financière octroyé à l'agence et fixe les montants maximaux exigibles.
L'article L. 5421-10 précise les sanctions complémentaires susceptibles d'être prononcées à l'encontre des personnes physiques : affichage, interdiction d'exercice, confiscation.
L'article L. 5421-11 fixe les peines complémentaires encourues par les personnes morales.
La huitième partie procède à des coordinations au sein de l'article L. 162-17-4 du code de la sécurité sociale relatives aux pénalités prononcées par le comité économique des produits de santé (Ceps) à la suite d'une sanction en matière de publicité.
II - Les dispositions adoptées par l'Assemblée nationale
L'Assemblée nationale a adopté, outre trois amendements de coordination, plusieurs amendements de fond.
Le premier précise que l'agence en charge de la sécurité du médicament est placée sous la tutelle du ministre de la santé.
Le deuxième ouvre la faculté à l'agence de demander que les essais cliniques des médicaments soient effectués sous forme d'essais contre comparateurs. L'entreprise s'opposant à cette comparaison devra justifier ce choix.
Le troisième prévoit que la mise en demeure de la personne physique ou morale que l'agence envisage de soumettre à une amende administrative comporte l'indication de la possibilité de se faire assister d'un conseil.
III - Le texte adopté par la commission
Votre commission est favorable à ce que la loi confie une mission renforcée de pharmacovigilance à l'agence en charge de la sécurité du médicament. Elle estime cependant que substituer à l'Afssaps une « agence nationale de sécurité du médicament et des produits de santé » ne contribuera guère à remédier aux causes qui ont été à l'origine des dysfonctionnements mis en évidence dans l'affaire du Mediator. L'idée qu'un changement de nom entraîne la création d'une nouvelle agence est par ailleurs juridiquement faux.
En outre, la nouvelle dénomination proposée, si longue qu'est prévu un sigle « abrégé » (ANSM), présente deux inconvénients sérieux :
- qualifier l'agence de nationale ne facilitera guère son identification au sein de l'Union européenne, par rapport à l'Agence européenne du médicament et aux autres agences « nationales » ;
- il n'est pas approprié de distinguer le médicament et les produits de santé car les médicaments sont des produits de santé. Il serait question de répondre à l'inquiétude vis-à-vis des médicaments suscitée par l'affaire du Mediator. Mais si demain apparaissait un problème grave de sécurité d'un dispositif médical, faudra-t-il une nouvelle fois modifier la loi pour souligner que l'agence est en charge de la sécurité du médicament, des dispositifs médicaux et des produits de santé ?
Si l'on peut comprendre l'intérêt psychologique tant pour une partie, limitée, de l'opinion publique que pour les personnels de l'agence, que présente le fait de lui donner un nouveau nom, il faut retenir une dénomination qui ne présente pas les mêmes inconvénients - et soit aussi plus facile à mémoriser.
A l'initiative de son rapporteur, la commission a donc adopté un amendement tendant à renommer l'agence « agence française de sécurité des produits de santé », ou Afseps .
Elle a également adopté, toujours à l'initiative de son rapporteur, un amendement tendant à prévoir que les demandes d'essais comparatifs faits par l'agence se font contre des comparateurs « actifs », excluant ainsi les placebos, ainsi qu' un amendement de coordination .
A l'initiative d'Isabelle Pasquet, Laurence Cohen, Annie David, Dominique Watrin et Guy Fischer, la commission a également adopté un amendement tendant à renforcer le niveau des sanctions administratives prononcées par l'agence.
Elle a adopté cet article ainsi modifié.
Article 4 bis A (nouveau) Observatoire national des prescriptions et consommations des médicaments
Objet : Cet article additionnel tend à réactiver l'observatoire national des prescriptions et consommations des médicaments.
Cet article additionnel, inséré sur amendement d'Isabelle Pasquet, Laurence Cohen, Annie David, Dominique Watrin et Guy Fischer, tend à réactiver et à conférer un statut législatif à l'observatoire national des prescriptions et consommations des médicaments, créé par un arrêté du 19 septembre 1996 et supprimé par un arrêté du 10 mars 2006.
La mission confiée à l'observatoire est celle de rassembler les informations relatives aux prescriptions et d'analyser les déterminants médicaux, sociaux, culturels et promotionnels de la prescription et de remettre un rapport annuel au ministre en charge de la santé et de la sécurité sociale.
La commission a adopté cet article additionnel ainsi rédigé.
Article 4 bis (art. L. 5122-15, L. 5122-16 et L. 5323-4 du code de la santé publique) Coordinations
Objet : Cet article, inséré par l'Assemblée nationale, procède à des coordinations.
I - Le dispositif proposé par l'Assemblée nationale
Cet article opère des coordinations relatives à la publicité des dispositifs médicaux.
II - Le texte adopté par la commission
La commission a adopté cet article sans modification.
Article 5 (art. L. 5322-1, L. 5324-1 (nouveau) et L. 1413-8 du code de la santé publique) Composition du conseil d'administration et publicité des travaux de l'agence nationale du médicament et des produits de santé
Objet : Cet article modifie la gouvernance de l'agence en charge de la sécurité du médicament ainsi que les règles de publicité des débats au sein de ses commissions.
I - Le dispositif proposé
Cet article se compose de trois parties.
La première modifie l'article L. 5322-1 du code de la santé publique pour prévoir une nouvelle composition du conseil d'administration de l'agence en charge de la sécurité du médicament où ne siègent aujourd'hui, outre son président et à parité, que des représentants de l'Etat et des personnalités qualifiées. Aux côtés de représentants de l'Etat détenant la moitié des droits de vote, le nouveau conseil d'administration comprend : un député et un sénateur, des représentants des caisses nationales d'assurance maladie, des représentants des entreprises fournissant les produits de santé, des représentants des professionnels de santé autorisés à prescrire et à dispenser, des représentants d'associations agréées, des personnalités qualifiées et des représentants des personnels de l'agence. Le président de l'agence conserve une voix prépondérante en cas de partage égal des voix.
La deuxième introduit un nouveau chapitre, « commissions », dans le titre du code de la santé publique relatif à l'organisation de l'agence en charge de la sécurité du médicament. Composé d'un article unique, L. 5324-1, ce chapitre prévoit la publicité de l'ordre du jour, des comptes rendus, des votes et de leurs explications, sous réserve du respect de la confidentialité industrielle et commerciale et du secret médical. Un décret en Conseil d'Etat fixe les modalités de cette publicité.
La troisième prévoit des dispositions spécifiques pour la composition du conseil d'administration de l'institut de veille sanitaire (InVS), qui est actuellement composé selon les règles qui s'appliquent à celui de l'Afssaps. Il est proposé de modifier l'article L. 1413-8 du code de la santé publique pour prévoir que le conseil d'administration de l'InVS sera composé pour moitié de représentants de l'Etat et pour l'autre moitié de personnalités qualifiées et de représentants du personnel.
II - Les dispositions adoptées par l'Assemblée nationale
L'Assemblée nationale a adopté trois amendements :
- le premier tend à faire siéger trois députés et trois sénateurs au sein du conseil d'administration de l'agence en charge de la sécurité du médicament ;
- le deuxième supprime la participation de représentants de l'industrie pharmaceutique au conseil d'administration ;
- le dernier procède à une coordination.
III - Le texte adopté par la commission
Votre commission est favorable à l'élargissement du conseil d'administration de l'agence en charge de la sécurité du médicament à condition que soient garanties les compétences du conseil sur la politique de l'agence et son programme de travail. Elle a donc adopté, à l'initiative de son rapporteur, un amendement à cette fin ainsi qu' un amendement tendant à garantir que ne soient représentées au sein du conseil d'administration que les associations d'usagers du système de santé sans lien avec l'industrie, puis un amendement de cohérence .
A l'initiative d'Isabelle Pasquet, Laurence Cohen, Annie David, Dominique Watrin et Guy Fischer, la commission a également adopté un amendement tendant à ce que les associations représentant exclusivement les victimes d'accidents médicamenteux figurent au sein du conseil d'administration de l'agence.
Elle a adopté cet article ainsi modifié.
Article 5 bis (art. L. 161-40 du code de la sécurité sociale) Base de données mise en oeuvre par la Haute Autorité de santé
Objet : Cet article, inséré par l'Assemblée nationale, confie à la HAS la mission de gérer une base de données de référence sur les maladies et leurs traitements.
I - Le dispositif proposé par l'Assemblée nationale
Cet article confie à la HAS, en liaison avec l'agence en charge de la sécurité du médicament et les caisses d'assurance maladie, la mission de créer une base de données de référence sur les maladies et leurs traitements. Cette base sera accessible aux professionnels de santé et au grand public.
II - Le texte adopté par la commission
Votre commission est très favorable à la création d'une base publique accessible sur internet et susceptible d'offrir une information scientifique de qualité contrebalançant la multiplicité des opinions diffusées en ligne sur les questions de santé. A l'initiative de son rapporteur, elle a complété par voie d'amendement les informations contenues sur cette base pour y inclure celles relatives au bon usage des produits de santé.
La commission a également adopté, à l'initiative d'Isabelle Pasquet, Laurence Cohen, Annie David, Dominique Watrin et Guy Fischer, un amendement tendant à préciser que la base créée par cet article sera consultable gratuitement.
Elle a adopté cet article ainsi modifié.
TITRE
III - LE MÉDICAMENT À USAGE HUMAIN
CHAPITRE
IER - L'autorisation de mise sur le marché
Article 6 (art. L. 5121-8 du code de la santé publique) Réalisation d'études après l'autorisation de mise sur le marché
Objet : Cet article tend à permettre à l'agence en charge de la sécurité du médicament de demander des études de sécurité et d'efficacité après l'autorisation de mise sur le marché.
I - Le dispositif proposé
Cet article se compose de deux parties :
la première introduit dans l'article L. 5121-8, relatif aux compétences de l'agence, la mention de l'obligation qui lui est faite de demander des études de sécurité et d'efficacité post-autorisation au laboratoire responsable de la mise sur le marché d'un médicament ;
la deuxième partie introduit un nouvel article L. 5121-8-1 et précise les conditions dans lesquelles l'agence peut demander au titulaire de l'autorisation de mise sur le marché des études complémentaires. Les études de sécurité peuvent être demandées s'il existe des craintes concernant les risques liés au médicament. Les études d'efficacité sont liées à un progrès significatif des connaissances ou des méthodes de prise en charge clinique de la maladie. Un décret en Conseil d'Etat fixe les conditions supplémentaires.
II - Les dispositions adoptées par l'Assemblée nationale
L'Assemblée nationale a adopté plusieurs amendements à cet article.
Le premier transforme la faculté accordée à l'agence de demander des études complémentaires dans certains cas précis en obligation, assortie de la fixation de délais de réalisation de celles-ci.
Le deuxième précise que les études de sécurité sont demandées dès que des signalements d'effets indésirables ont été enregistrés par la pharmacovigilance.
Le troisième précise que les études sont effectuées en comparaison avec les traitements de référence disponibles.
Le quatrième procède à des modifications relatives au renouvellement de l'autorisation temporaire de mise sur le marché, qui précède l'octroi d'une AMM sans limitation de durée, en supprimant la nécessité d'une raison de pharmacovigilance pour renouveler le caractère temporaire de la mise sur le marché et en limitant à cinq ans la nouvelle autorisation temporaire.
III - Le texte adopté par la commission
Votre commission approuve le renforcement des pouvoirs de l'agence en matière de contrôle des médicaments post-AMM. Elle rappelle cependant qu'une réforme de l'AMM est nécessaire en droit européen et qu'il est important que la France continue à oeuvrer en ce sens.
A l'initiative de son rapporteur, la commission a adopté un amendement rédactionnel à cet article.
Elle a adopté cet article ainsi modifié.
Article 6 bis (art. L. 5121-8-2 (nouveau) du code de la santé publique) Information de l'agence en charge de la sécurité du médicament relative aux essais cliniques pré-AMM
Objet : Cet article, inséré par l'Assemblée nationale, tend à rendre obligatoire l'inscription des essais cliniques pré-AMM sur la base de données nationales des recherches biomédicales.
I - Le dispositif proposé par l'Assemblée nationale
Cet article crée un article L. 5121-8-2 dans le code de la santé publique pour prévoir l'obligation d'inscrire les essais pré-AMM sur la liste nationale des essais cliniques prévue à l'article L. 1121-15.
II - Le texte adopté par la commission
Votre commission soutient cette mesure qui devrait permettre plus de transparence dans l'information sur la conduite des essais cliniques.
Elle a adopté cet article sans modification.
Article 7 (art. L. 5121-9 du code de la santé publique) Conditions de suspension, de retrait ou de modification de l'autorisation de mise sur le marché
Objet : Cet article élargit et précise les cas de suspension ou de retrait de l'autorisation de mise sur le marché pour en assurer la conformité avec la directive 2010/84/UE du 15 décembre 2010 relative à la pharmacovigilance.
I - Le dispositif proposé
Cet article réécrit le quatrième alinéa de l'article L. 5121-9 relatif aux causes de suspension ou de retrait de l'autorisation de mise sur le marché (AMM). Il renvoie les conditions de ces mesures à un décret en Conseil d'Etat. Il complète et modifie l'énumération partielle des motifs de suspension et retrait afin de les mettre en conformité avec la directive 2010/84/UE du 15 décembre 2010 relative à la pharmacovigilance. Il est ainsi précisé que l'autorisation est suspendue ou retirée si le médicament est nocif ou si le titulaire de l'AMM ne se conforme pas aux demandes d'informations et d'études émises par l'agence en charge de la sécurité du médicament.
S'agissant des sanctions liées à un rapport bénéfices-risques devenu défavorable ou à l'absence d'effet thérapeutique du médicament, deux éléments nouveaux sont apportés :
- la nécessité d'évaluer le rapport entre bénéfices et risques dans le cadre de l'usage normal du médicament est supprimée ;
- l'absence d'effet thérapeutique est définie comme le fait de considérer « que le médicament ne permet pas d'obtenir de résultats thérapeutiques » .
II - Les dispositions adoptées par l'Assemblée nationale
L'Assemblée nationale a adopté un amendement tendant à permettre à une association agréée d'usagers du système de santé de saisir l'agence en charge de la sécurité du médicament d'une demande d'application de l'article L. 5121-9 aux fins de modification, de suspension ou de retrait d'un médicament ou produit de santé, dans des conditions fixées par décret en Conseil d'Etat. Le refus éventuel et ses motifs sont rendus publics.
III - Le texte adopté par la commission
Votre commission juge nécessaires les précisions apportées par cet article et important l'amendement adopté par l'Assemblée nationale.
A l'initiative de son rapporteur, elle a adopté un amendement de rectification d'une erreur matérielle, puis cet article ainsi modifié.
Article 8 (art. L. 5121-9-2 et 5121-9-3 (nouveaux) du code de la santé publique) Obligations du titulaire de l'autorisation de mise sur le marché
Objet : Cet article fixe les obligations du titulaire de l'autorisation de mise sur le marché (AMM) en matière de sécurité.
I - Le dispositif proposé
Cet article propose d'insérer deux articles après l'article L. 5121-9-1 du code de la santé publique.
L'article L. 5121-9-2 fait peser sur le titulaire de l'AMM l'obligation d'informer immédiatement l'agence en charge de la sécurité du médicament de toute interdiction ou restriction prononcée par l'autorité compétente d'un pays où le médicament est distribué ainsi que de toute information susceptible d'influencer l'évaluation du rapport bénéfices-risques.
L'article L. 5121-9-3 permet à l'agence en charge de la sécurité du médicament de demander à tout moment au titulaire de l'autorisation de transmettre des données démontrant que le rapport bénéfices-risques reste favorable.
II - Les dispositions adoptées par l'Assemblée nationale
L'Assemblée nationale a adopté un amendement tendant à préciser à nouveau que tout arrêt de commercialisation à l'étranger d'un médicament commercialisé en France doit être notifié et motivé à l'agence en charge de la sécurité du médicament.
III - Le texte adopté par la commission
Votre commission juge nécessaires les mesures contenues dans cet article.
A l'initiative de son rapporteur, elle a adopté un amendement tendant à supprimer des dispositions redondantes ou déclaratoires susceptibles de nuire à l'efficacité juridique du texte.
La commission a adopté cet article ainsi modifié.
Article 9 (art. L. 5124-11 du code de la santé publique) Conditions d'interdiction de l'exportation de médicaments ayant fait l'objet d'un retrait d'autorisation de mise sur le marché
Objet : Cet article complète les cas où l'agence en charge de la sécurité du médicament peut s'opposer à leur exportation.
I - Le dispositif proposé
Cet article complète l'article L. 5124-11 relatif aux conditions d'exportation des médicaments pour préciser que l'agence en charge du médicament peut interdire l'exportation de ceux dont l'AMM n'a pas été renouvelée pour des raisons de santé publique.
L'Assemblée nationale a adopté cet article sans modification.
II - Le texte adopté par la commission
Votre commission estime que cet article apporte un complément utile aux dispositions existantes.
Elle a adopté cet article sans modification.
Article 9 bis (art. L. 162-17 du code de la sécurité sociale) Conditions de fixation du service médical rendu des médicaments
Objet : Cet article, inséré par l'Assemblée nationale, soumet l'inscription d'un médicament sur la liste des médicaments remboursables à la réalisation d'essais cliniques comparatifs.
I - Le dispositif proposé par l'Assemblée nationale
Cet article tend à soumettre l'inscription d'une spécialité pharmaceutique sur la liste des médicaments remboursables à la conduite d'essais cliniques versus stratégie thérapeutique pour la même pathologie, afin de déterminer plus exactement l'amélioration du service médical rendu.
II - Le texte adopté par la commission
Votre commission juge fondamental cet article. En effet, si les exigences opposables aux demandeurs d'une AMM relèvent de la compétence communautaire, les autorités nationales sont seules compétentes pour définir les éléments d'information que doivent comporter les dossiers de demande de prise en charge ou de remboursement par l'assurance maladie.
L'article 9 bis , adopté par l'Assemblée nationale, représente donc un progrès important. Au-delà du fait qu'il permettra une amélioration de la sécurité sanitaire et des conditions d'évaluation de l'amélioration du service médical rendu, il pourra contribuer à convaincre l'Union européenne de la nécessité d'imposer des essais comparatifs.
A l'initiative de son rapporteur, la commission a adopté une nouvelle rédaction de cet article, avec le souci de la rendre plus claire, plus concise et plus cohérente avec la terminologie utilisée.
Au vu des échanges que votre rapporteur a pu avoir avec les experts en la matière, l'application de la mesure ainsi rédigée ne posera pas de réelles difficultés.
En effet, les deux objections qui avaient été avancées contre cette mesure paraissent désormais infondées :
- le cas des médicaments pour lesquels il n'existe pas de traitement de référence est réglé par le texte même de l'article ;
- le cas de ceux pour lesquels il existerait un « comparateur » actif, mais trop récent pour que l'essai clinique comparatif soit possible est également pris en compte : on peut penser que l'instance chargée d'instruire la demande - c'est-à-dire la commission de la transparence - sera alors parfaitement capable d'apprécier cette situation et de considérer que les comparateurs actifs doivent non seulement exister mais exister depuis suffisamment longtemps pour qu'un essai clinique comparatif soit réalisable.
Au niveau de la loi, il paraît donc suffisant de poser un principe clair, dont les parties en présence seront capables de définir la portée et qu'elles pourront interpréter en se fondant sur le simple bon sens : à l'impossible, nul ne peut être tenu.
La commission a adopté cet article ainsi modifié.
CHAPITRE II - La prescription
Article 10 (art. L. 5121-1 du code de la santé publique) Encadrement de la préparation magistrale
Objet : Cet article précise la définition des préparations magistrales et préparations hospitalières.
I - Le dispositif proposé
Cet article modifie les deux premiers alinéas de l'article L. 5121-1 qui définit les différents types de préparations et de médicaments. Il se compose de deux points.
Le 1° précise qu'une préparation magistrale n'est élaborée qu'en l'absence de spécialité pharmaceutique disponible disposant d'une AMM ou d'une autorisation temporaire de mise sur le marché, prévue à l'article L. 5121-12.
Le 2° précise qu'une préparation hospitalière n'est élaborée qu'en absence de spécialité pharmaceutique ne disposant pas d'une autorisation temporaire d'utilisation.
L'Assemblée nationale a adopté cet article sans modification.
II - Le texte adopté par la commission
La commission a adopté cet article sans modification.
Article 10 bis (art. L. 5125-1-1 et L. 5125-1-1-1 (nouveau) du code de la santé publique) Régime d'autorisation des préparations en pharmacie
Objet : Cet article, inséré par l'Assemblée nationale, précise le régime d'autorisation des officines pour les préparations pharmaceutiques.
I - Le dispositif proposé par l'Assemblée nationale
L'Assemblée nationale a adopté cet article tendant à simplifier le régime d'autorisation des officines pour les préparations pharmaceutiques. Ce régime, prévu par l'article L. 5125-1-1 du code de la santé publique, est issu de l'ordonnance du 23 février 2010. Il soumet la réalisation de préparations stériles ou dangereuses à une autorisation du directeur général de l'agence régionale de santé (ARS).
Or, ce régime complexe n'a pu jusqu'à présent être mis en oeuvre faute d'une définition précise de ce que sont les substances dangereuses visées. La première partie de cet article propose donc de simplifier le régime d'autorisation en le limitant aux préparations pouvant présenter un risque pour la santé et figurant sur une liste fixée par un arrêté du ministre en charge de la santé.
La deuxième partie de cet article introduit dans le même code un article L. 5125-1-1-1 pour définir le régime d'interdiction de préparation des substances ne présentant pas de risque et celui du retrait de l'autorisation pour les substances présentant un risque pour la santé. Dans les deux cas, la décision du directeur général de l'ARS se fondera sur le non-respect des bonnes pratiques ou la réalisation des préparations dans des conditions dangereuses pour la santé. La possibilité pour le pharmacien d'officine concerné de faire valoir ses observations avant la mise en oeuvre de la décision est également prévue, sauf cas d'urgence.
II - Le texte adopté par la commission
La commission des affaires sociales et le Sénat dans son ensemble ont déjà adopté cette disposition, insérée à l'initiative du Gouvernement dans la proposition de loi modifiant certaines dispositions de la loi HPST 26 ( * ) mais ensuite censurée par le Conseil constitutionnel en tant que cavalier 27 ( * ) .
Elle est favorable à ce dispositif plus simple et mieux à même de protéger la santé des malades mais aussi des praticiens.
Elle a donc adopté cet article sans modification.
Article 11 (art. L. 5121-12-1 (nouveau) du code de la santé publique et L. 162-4 du code de la sécurité sociale) Encadrement des prescriptions en dehors des indications de l'autorisation de mise sur le marché
Objet : Cet article fixe les conditions dans lesquelles une prescription hors AMM peut être effectuée.
I - Le dispositif proposé
Cet article se compose de deux parties.
La première insère un article additionnel après l'article L. 5121-12 du code de la santé publique.
Cet article L. 5121-12-1 pose une condition alternative à la prescription hors AMM ou autorisation temporaire d'autorisation (ATU). Soit :
- soit l'utilisation de la spécialité a fait l'objet d'une recommandation temporaire d'utilisation établie par l'agence en charge de la sécurité du médicament ;
- soit le prescripteur juge indispensable, au regard des données acquises de la science, le recours à cette spécialité pour stabiliser ou améliorer l'état du patient.
L'article précise également que le patient est informé de la prescription hors AMM ou ATU et que la décision du prescripteur est motivée dans le dossier médical. La mention « prescription hors AMM » doit figurer sur la prescription.
Les recommandations temporaires d'utilisation émises par l'agence en charge de la sécurité du médicament sont soumises à la conclusion préalable d'une convention avec le titulaire de l'autorisation sur les conditions d'utilisation du médicament et au recueil des données permettant d'établir le rapport bénéfices-risques. Ces conventions peuvent prévoir l'engagement pour le titulaire de l'AMM d'en demander la modification dans un délai déterminé.
La deuxième partie complète l'article L. 162-4 du code de la sécurité sociale, relatif au remboursement des médicaments, pour préciser que la mention « prescription hors AMM » vaut non-remboursement.
II - Les dispositions adoptées par l'Assemblée nationale
L'Assemblée nationale a adopté plusieurs amendements pour prévoir :
- que les recommandations temporaires d'utilisation ne peuvent excéder trois ans ;
- que ces recommandations sont mises à disposition des prescripteurs ;
- que le prescripteur d'un médicament hors AMM doit faire la preuve qu'il a informé le patient, son représentant légal ou la personne de confiance, de l'absence d'alternative médicamenteuse ainsi que des risques, contraintes et bénéfices susceptibles d'être apportés par le médicament prescrit.
III - Le texte adopté par la commission
Votre commission est pleinement convaincue de la nécessité d'encadrer mieux la prescription hors AMM et estime intéressantes les dispositions contenues dans cet article, dont il faudra toutefois surveiller l'application sur le terrain. A l'initiative de son rapporteur, elle a adopté deux amendements , l'un supprimant une redondance et l'autre prévoyant l'information du patient sur le caractère remboursable ou non du médicament qui lui est prescrit hors AMM.
A l'initiative de Gilbert Barbier, la commission a également adopté un amendement limitant l'obligation d'information qui pèse sur le praticien au premier prescripteur.
Elle a adopté cet article ainsi modifié.
Article 12 (art. L. 5121-1-2 (nouveau) du code de la santé publique) Prescription en dénomination commune
Objet : Cet article prévoit l'obligation, limitée, pour le prescripteur de faire figurer la dénomination scientifique des substances prescrites.
I - Le dispositif proposé
Cet article introduit un nouvel article L. 5121-1-2 après l'article L. 5121-1-1 du code de la santé publique pour rendre obligatoire le recours à la dénomination commune internationale ou à défaut la dénomination en pharmacopée européenne ou française ou, enfin, à la dénomination commune, pour la prescription de toute spécialité pharmaceutique comprenant jusqu'à trois spécialités pharmaceutiques. Le nom commercial de la spécialité peut être précisé en sus.
Pour les spécialités comprenant plus de trois principes actifs, seul le nom commercial est requis.
II - Les dispositions adoptées par l'Assemblée nationale
L'Assemblée nationale a adopté plusieurs amendements pour :
- prévoir l'obligation de prescription en dénomination commune internationale ou, à défaut, en pharmacopée européenne ou française ;
- la simple faculté de mentionner la dénomination de fantaisie de la spécialité ;
- l'obligation pour le titulaire de l'autorisation de mettre à disposition du public sur son site internet, la désignation des principes actifs de la spécialité selon la dénomination commune internationale ou, à défaut, en pharmacopée européenne ou française.
III - Le texte adopté par la commission
A l'initiative d'Isabelle Pasquet, Laurence Cohen, Annie David, Dominique Watrin et Guy Fischer, la commission a adopté un amendement renforçant l'obligation d'établir les prescriptions en dénomination commune internationale.
Elle a adopté cet article ainsi modifié.
Article 13 (art. L. 162-17-4-1 (nouveau) du code de la sécurité sociale) Contrôle et sanction des prescriptions hors autorisation de mise sur le marché par le comité économique des produits de santé
Objet : Cet article prévoit les conditions de respect des obligations fixées dans les conventions relatives à l'usage hors autorisation d'un médicament.
I - Le dispositif proposé
Cet article insère dans le code de la sécurité sociale un article L. 162-17-4-1 qui autorise les conventions encadrant l'usage hors autorisation d'un médicament à prévoir l'obligation, pour le titulaire de l'autorisation, de mener des campagnes d'information tendant à limiter l'usage hors recommandation et de contrôler cet usage.
Le non-respect de ces obligations est sanctionné par une pénalité prononcée par le Ceps et pouvant aller jusqu'à 10 % du chiffre d'affaires réalisé en France au titre du médicament faisant l'objet de la convention. Les conditions de mise en oeuvre de ces sanctions sont fixées par décret en Conseil d'Etat.
L'Assemblée nationale a adopté cet article sans modification.
II - Le texte adopté par la commission
A l'initiative d'Isabelle Pasquet, Laurence Cohen, Annie David, Dominique Watrin et Guy Fischer, la commission a adopté un amendement permettant au Ceps de prononcer des sanctions sous la forme de baisses de prix et non d'amendes.
Elle a adopté cet article ainsi modifié.
CHAPITRE III - La délivrance des médicaments
Article 14 (art. L. 5121-14-2 (nouveau) du code de la santé publique) Interdiction de délivrance des médicaments
Objet : Cet article définit les motifs d'interdiction de délivrance ou de retrait du marché d'une spécialité pharmaceutique.
I - Le dispositif proposé
Cet article insère un article L. 5121-14-2 dans le code de la santé publique. Il se compose de deux parties.
La première affirme la capacité de l'agence en charge de la sécurité du médicament pour interdire la délivrance d'une spécialité pharmaceutique ou la retirer du marché. Un décret en Conseil d'Etat doit en déterminer les modalités. Quelques-uns des motifs qui peuvent justifier cette décision reprennent les termes de la directive 2010/84/UE du 15 décembre 2010 relative à la pharmacovigilance. L'absence de contrôle sur la fabrication ou l'absence d'autorisation est également citée comme motif de retrait.
La seconde permet à l'agence de moduler l'interdiction qu'elle prononce. Celle-ci peut ainsi être limitée à certains lots de médicaments. De plus, dans des conditions définies par décret en Conseil d'Etat, le prolongement de traitements suivis par certains patients peut être autorisé avec un médicament interdit.
II - Les dispositions adoptées par l'Assemblée nationale
L'Assemblée nationale a adopté un amendement rédactionnel ainsi qu'un amendement tendant à prévoir que la décision d'interdiction de délivrance ou la décision de retrait sont rendues publiques aux frais du titulaire de l'AMM.
III - Le texte adopté par la commission
La commission a adopté cet article sans modification.
Article 14 bis Accès du Conseil national de l'ordre des pharmaciens aux données contenues dans le dossier pharmaceutique
Objet : Cet article, inséré par l'Assemblée nationale, autorise le Conseil national de l'ordre des pharmaciens à accéder, pour raison de santé publique, aux données anonymes relatives aux médicaments contenues dans le dossier pharmaceutique.
I - Le dispositif proposé par l'Assemblée nationale
Cet article permet, sur demande expresse des autorités chargées de la veille sanitaire et pour raison de santé publique, au Conseil national de l'ordre des pharmaciens d'accéder aux données anonymes relatives aux médicaments contenues dans le dossier pharmaceutique.
II - Le texte adopté par la commission
La commission mesure l'intérêt que peut représenter le dossier pharmaceutique pour la sécurité sanitaire. A l'initiative de son rapporteur, elle a cependant adopté un amendement tendant à définir de façon limitative les autorités qui pourront avoir accès aux données qu'il contient.
Elle a adopté cet article ainsi modifié.
CHAPITRE IV - L'autorisation temporaire d'utilisation
Article 15 (art. L. 5121-12, L. 1121-16-1 et L. 1123-14 du code de la santé publique) Modification des procédures d'octroi des autorisations temporaires d'utilisation nominative
Objet : Cet article renforce l'encadrement des procédures d'autorisations temporaires d'utilisation (ATU).
I - Le dispositif proposé
L'ATU constitue une procédure dérogatoire à l'AMM . Aux termes de l'article L. 5121-12 du code de la santé publique, les ATU sont délivrées à titre exceptionnel par l'actuelle Afssaps pour des médicaments ne disposant pas d'AMM centralisée ou nationale, destinés à traiter des maladies graves ou rares pour lesquelles il n'existe pas de traitement approprié.
L'article L. 5121-12 définit deux types d'ATU :
- l'ATU de cohorte peut être délivrée pour un groupe de patients lorsque les essais thérapeutiques menés en vue de la demande d'AMM permettent de présumer fortement de l'efficacité et de la sécurité du médicament. Si aucune demande d'AMM n'a été déposée, le demandeur doit s'engager à le faire dans un délai déterminé ;
- l'ATU nominative est délivrée pour un patient nommément désigné et ne pouvant participer à une recherche biomédicale. Deux conditions cumulatives doivent être remplies pour que l'ATU soit accordée :
- le médicament doit être susceptible de présenter un bénéfice pour le patient ;
- soit son efficacité et sa sécurité sont présumées en l'état des connaissances scientifiques, soit le pronostic vital du patient est en jeu à court terme en l'état des thérapeutiques disponibles (ATU dite « compassionnelle »).
L'ATU nominative engage la responsabilité du médecin qui doit justifier que le patient, son représentant légal ou la personne de confiance qu'il a désignée a reçu une information adaptée. La procédure suivie est en outre inscrite dans le dossier médical.
C'est le titulaire des droits d'exploitation du médicament (pour l'ATU de cohorte) ou le médecin prescripteur (pour l'ATU nominative) qui dépose auprès de l'Afssaps la demande d'ATU.
L'ATU de cohorte doit être sollicitée dans le cadre d'un protocole d'utilisation thérapeutique et de recueil d'informations établi avec le titulaire des droits d'exploitation et concernant notamment les conditions réelles d'utilisation et les caractéristiques de la population bénéficiant du médicament ainsi autorisé. Le demandeur de l'ATU de cohorte adresse systématiquement à l'agence, après octroi de l'autorisation, toute information concernant notamment les conditions réelles d'utilisation et les caractéristiques de la population bénéficiant du médicament.
Le suivi des patients est plus souple concernant l'ATU nominative, l'Afssaps ayant le pouvoir de demander la mise en place d'un protocole d'utilisation thérapeutique et de recueil d'informations, sans que cette démarche revête un caractère systématique.
Attribuées pour une durée limitée, les ATU peuvent être suspendues ou retirées par l'Afssaps si les conditions prévues à l'article L. 5121-12 ne sont plus remplies, ou pour des motifs de santé publique.
L'ATU constitue une procédure nécessaire en ce qu'elle permet aux patients de bénéficier de traitements médicaux les plus adaptés possible à leur situation en l'absence d'alternative disposant de l'AMM, notamment grâce à un accès précoce à l'innovation thérapeutique. En 2010, dix nouvelles demandes d'ATU de cohorte ont été présentées à l'Afssaps et six ont été octroyées. Neuf ATU de cohorte ont fait l'objet d'une modification ou d'un renouvellement. 22 858 ATU nominatives ont été accordées en 2010, couvrant 15 411 patients dont plus du tiers étaient des enfants 28 ( * ) .
Dans les faits, les ATU de cohorte sont généralement attribuées pour des durées courtes et débouchent le plus souvent sur une AMM. Tel n'est cependant pas le cas de certaines ATU nominatives qui tendent à se pérenniser. Or ces dernières font l'objet d'un suivi limité, comme cela a été souligné plus haut.
En outre, les rapports d'information de l'Assemblée nationale 29 ( * ) et du Sénat 30 ( * ) , tout comme le groupe du travail des Assises du médicament consacré à ces questions, ont insisté sur le fait que l'ATU ne doit pas constituer un moyen pour un laboratoire de contourner la procédure d'AMM.
De surcroît, le mode de fixation des prix des médicaments sous ATU étant libre et intégralement pris en charge par l'assurance maladie, il existe un risque que la procédure d'ATU soit utilisée dans le but de contourner la fixation des prix par le Ceps. Pour cette raison, la mission commune d'information du Sénat avait recommandé la suppression de la procédure actuelle de déclaration au Ceps par le laboratoire titulaire des droits d'exploitation du montant de l'indemnité demandée aux établissements de santé pour le médicament sous ATU 31 ( * ) afin de confier au Ceps la fixation du prix des médicaments sous ATU (proposition n° 21).
Le présent article tente de parvenir à un équilibre entre accès des patients à des soins adaptés et nécessité de mieux encadrer les procédures d'ATU , en particulier d'ATU nominative.
A l'alinéa premier de l'article L. 5121-12, la condition de l'absence de traitement approprié pour le dépôt d'une demande d'ATU est complétée par celle de l'impossibilité de différer la mise en oeuvre du traitement.
L'octroi d'une ATU nominative sera subordonné au dépôt d'une demande d'ATU de cohorte ou d'AMM dans l'indication thérapeutique sollicitée. A défaut, des essais cliniques devront être conduits en France sur le médicament pour l'indication thérapeutique sollicitée. En cas de rejet d'une de ces demandes, l'ATU nominative sera retirée pour les indications sollicitées dans la demande.
La conclusion d'un protocole d'utilisation thérapeutique et de recueil d'informations entre l'agence et le titulaire des droits d'exploitation du médicament devient désormais une condition nécessaire de l'octroi de l'autorisation. Jusqu'à présent, ce protocole était obligatoire pour l'ATU de cohorte mais facultatif pour l'ATU nominative.
L'ATU compassionnelle est cependant préservée au paragraphe IV de l'article L. 5121-12. Les règles prévues pour l'ATU nominative ne seront pas applicables « lorsqu'une issue fatale à court terme pour le patient est, en l'état des thérapeutiques disponibles, inéluctable » . La même dérogation s'appliquera lorsque la spécialité pharmaceutique aura fait l'objet d'un arrêt de commercialisation mais que la demande d'ATU porte sur une indication autre que celle à l'origine de l'arrêt et qu'il existe de fortes présomptions d'efficacité et de sécurité du médicament dans l'indication sollicitée.
Ces ATU ne seront pas soumises à la conclusion d'un protocole d'utilisation thérapeutique et de recueil d'informations. Cependant, à l'expiration de l'autorisation et, le cas échéant, à chaque renouvellement, des données de suivi des patients traités seront transmises par les médecins prescripteurs à l'agence. Cette dernière devra préciser la nature de ces données.
L'article 30 du présent projet de loi prévoit des dispositions transitoires. Les ATU accordées sur le fondement de la rédaction actuelle de l'article L. 5121-12 du code de la santé publique demeureront régies par ces dispositions, y compris pour leur renouvellement, pendant une période de trois ans à compter de la publication de la loi. Ce dispositif transitoire s'appliquera également pour les demandes d'ATU nominatives lorsque des autorisations auront déjà été accordées dans la même indication pour le médicament concerné.
II - Les dispositions adoptées par l'Assemblée nationale
A l'initiative du Gouvernement, l'Assemblée nationale a apporté plusieurs modifications à cet article :
- elle a précisé, par un vote unanime, que les ATU peuvent être renouvelées par l'agence en charge de la sécurité du médicament. La députée socialiste Catherine Lemorton a souligné que cet amendement permettait de contrecarrer l'idée selon laquelle, du fait de l'encadrement accru des ATU, « les Français ne pourraient plus accéder à l'innovation, notamment dans des cas dramatiques ou des maladies très rares ou très douloureuses » ;
- elle a assoupli les contraintes pesant sur l'ATU nominative. Dans le texte initial, celle-ci ne pouvait être délivrée que si une demande d'ATU de cohorte ou une demande d'AMM avaient été déposées ou si des essais cliniques étaient conduits en France sur le médicament dans l'indication thérapeutique sollicitée. Désormais, l'ATU nominative pourra également être accordée lorsque le titulaire des droits d'exploitation s'engage à déposer dans un délai déterminé une demande d'AMM ou d'ATU de cohorte ou qu'une demande d'autorisation d'essai clinique a été déposée en France ;
- elle a autorisé l'octroi d'une ATU nominative de façon dérogatoire pour un médicament qui, dans l'indication thérapeutique sollicitée, se serait vu refuser une demande d'ATU de cohorte ou pour lequel une demande d'autorisation d'essai clinique aurait été refusée. L'ATU ne pourra être délivrée qu'après information du patient et du praticien sur les motifs du refus de la demande et sous réserve d'un bénéfice individuel pour le patient ;
- elle a précisé les conditions d'octroi de l'ATU compassionnelle, l'expression « lorsqu'une issue fatale à court terme pour le patient est, en l'état des thérapeutiques disponibles, inéluctable » ayant été remplacée par « lorsque le pronostic vital du patient est engagé, en l'état des thérapeutiques disponibles » .
III - Le texte adopté par la commission
Votre commission a estimé nécessaire, d'une part, d'affirmer le caractère temporaire des ATU, d'autre part de mieux encadrer les procédures d'ATU nominatives.
Elle a donc adopté un amendement, présenté par Isabelle Pasquet, Laurence Cohen, Annie David, Dominique Watrin et Guy Fischer, qui limite la durée des ATU à une année renouvelable deux fois.
A l'initiative de son rapporteur, elle a également adopté un amendement qui revient sur certains assouplissements de la procédure d'ATU nominative adoptés à l'Assemblée nationale. Une telle demande ne pourra désormais être acceptée que si une demande d'ATU de cohorte ou d'autorisation de mise sur le marché a été déposée par le titulaire des droits d'exploitation ou si celui-ci procède en France à des essais cliniques.
Enfin, elle a rétabli, sur amendement de son rapporteur, la possibilité pour l'agence de subordonner l'octroi d'une ATU nominative dérogatoire à la conclusion d'un protocole avec le titulaire des droits d'exploitation, ce qui permettra d'assurer un meilleur suivi des patients.
Votre commission a adopté cet article ainsi modifié.
CHAPITRE V - La prise en charge hors autorisation de mise sur le marché
Article 16 (art. L. 162-17-2-1 du code de la santé publique) Prise en charge des produits prescrits hors autorisation de mise sur le marché par l'assurance maladie
Objet : Cet article complète les conditions de prise en charge ou de remboursement des médicaments prescrits hors AMM.
I - Le dispositif proposé
Cet article modifie l'article L. 162-17-2-1 du code de la sécurité sociale relatif à la prise en charge ou au remboursement des médicaments prescrits hors AMM pour intégrer les spécialités pharmaceutiques faisant l'objet de recommandations temporaires d'utilisation faites par l'agence en charge de la sécurité du médicament.
L'Assemblée nationale a adopté cet article sans modification.
II - Le texte adopté par la commission
La commission estime nécessaire que les médicaments faisant l'objet d'une recommandation temporaire d'utilisation puissent bénéficier d'un remboursement.
Elle a donc adopté cet article sans modification.
CHAPITRE VI - La pharmacovigilance
Article 17 (art. L. 5121-22 à L. 5121-26 (nouveaux) et L. 5421-6-1 du code de la santé publique) Dispositions relatives à la pharmacovigilance
Objet : Cet article apporte une définition de la pharmacovigilance et fixe la compétence de l'agence en charge de la sécurité du médicament.
I - Le dispositif proposé
Cet article se compose de trois parties.
La première introduit un chapitre I er bis « pharmacovigilance » dans le titre II du livre I er de la cinquième partie du code de la santé publique, relative aux produits de santé. Ce nouveau chapitre reprend et complète au niveau législatif les dispositions générales relatives à la pharmacovigilance figurant actuellement dans la partie réglementaire du même code 32 ( * ) .
Il se compose de cinq articles.
L'article L. 5121-22 reprend et élargit la définition de la pharmacovigilance contenue à l'article R. 5121-150. La pharmacovigilance n'est plus limitée à la surveillance des effets indésirables liés aux médicaments mais s'étend également à l'évaluation, la prévention et la gestion du risque de ces effets.
L'article L. 5121-23 trouve sa source dans le premier alinéa de l'article R. 5121-155 concernant les compétences de l'agence en charge de la sécurité du médicament. Les obligations de l'agence sont cependant renforcées, en cohérence avec la définition étendue de la pharmacovigilance. Outre l'évaluation, l'agence doit, au travers du système de pharmacovigilance, examiner les options permettant de prévenir les risques ou de les réduire et, si nécessaire, prendre les mesures appropriées. La participation de l'agence aux travaux de la pharmacovigilance européenne est explicitement inscrite dans l'article.
L'article L. 5121-24 reprend les principes contenus dans l'article R. 5121-171. Il oblige les entreprises ou organismes exploitant un médicament à se soumettre aux règles encadrant la pharmacovigilance, notamment en ce qui concerne la remise des études post-AMM.
L'article L. 5121-25 est relatif au signalement des effets indésirables. Il reprend, dans son premier alinéa, le contenu de l'article R. 5121-170, qui fixe l'obligation de signalement des médecins, chirurgiens-dentistes, sages-femmes et pharmaciens.
Le second alinéa de l'article ouvre la faculté de signalement à tous les professionnels de santé ainsi qu'aux patients et aux associations agréées, conformément à l'article 83 de la loi HPST 33 ( * ) et au décret du 10 juin 2011 34 ( * ) .
L'article L. 5121-26 renvoie la définition des règles applicables à la pharmacovigilance à un décret en Conseil d'Etat, conformément aux dispositions du 13° de l'article L. 5121-20 actuel.
La deuxième partie supprime par coordination le 13° de l'article L. 5121-20.
La troisième partie renforce les sanctions pénales associées au non-respect des dispositions de pharmacovigilance en réécrivant l'article L. 5421-6-1.
II - Les dispositions adoptées par l'Assemblée nationale
L'Assemblée nationale a adopté un amendement pour insérer un article L. 5121-27 nouveau destiné à préserver de toute discrimination professionnelle les personnes ayant informé de bonne foi leur employeur ou les autorités sanitaires de la possibilité d'effets indésirables dont elles auraient pu avoir connaissance à l'occasion de leur fonction.
III - Le texte adopté par la commission
A l'initiative de son rapporteur, la commission a adopté un amendement tendant à supprimer des dispositions redondantes avec l'article 32 relatif à la protection des « lanceurs d'alerte ».
Elle a adopté cet article ainsi modifié.
CHAPITRE VII - Information et publicité sur le médicament à usage humain
Article 18 (art. L. 5122-2, L. 5122-3, L. 5122-5, L. 5122-6, L. 5122-9, L. 5122-9-1 (nouveau), L. 5122-16, L. 5422-3, L. 5422-4, L. 5422-6 et L. 5422-11 du code de la santé publique et art. L. 613-5 du code de la propriété intellectuelle) Réglementation de la publicité pour les médicaments à usage humain
Objet : Cet article encadre de façon accrue la publicité portant sur les médicaments dans leur ensemble, et les vaccins en particulier, et aligne le régime de la publicité destinée aux professionnels de santé sur celui de la publicité pour le grand public.
I - Le dispositif proposé
Conformément au code communautaire relatif aux médicaments à usage humain, le principe fixé à l'article L. 5122-6 du code de la santé publique est celui de l'interdiction des campagnes publicitaires pour les médicaments faisant l'objet d'un remboursement par l'assurance maladie. Pour les autres médicaments ainsi que pour les vaccins et les médicaments anti-tabac, le chapitre II du titre II du livre I er de la cinquième partie du code de la santé publique prévoit un encadrement strict que le présent article vise à compléter et renforcer.
La publicité pour l'ensemble des médicaments
Le paragraphe I complète le premier alinéa de l'article L. 5122-2 du même code par une disposition prévoyant que la présentation du médicament effectuée dans le cadre d'une publicité doit respecter les stratégies thérapeutiques recommandées par la HAS.
Le paragraphe II complète les règles fixées à l'article L. 5122-3. Celui-ci dispose jusqu'à présent que seuls les médicaments faisant l'objet d'une AMM ou d'une autorisation équivalente peuvent faire l'objet d'une publicité. Il est désormais ajouté que lorsque, à la suite d'un signalement de pharmacovigilance, le médicament fait l'objet d'une réévaluation du rapport bénéfices-risques, sa publicité est interdite.
Le paragraphe III opère des changements de référence à l'article L. 5122-5 liés aux modifications introduites au paragraphe V concernant la publicité à destination des professionnels de santé.
La publicité portant sur les vaccins et les médicaments anti-tabac
A l'exception des campagnes vaccinales institutionnelles, le troisième alinéa de l'article L. 5122-6 du code de la santé publique autorise les campagnes publicitaires auprès du public pour des vaccins, à la condition qu'elles soient assorties, « de façon clairement identifiée, des mentions minimales obligatoires in extenso facilement audibles et lisibles, selon le support du message publicitaire concerné et sans renvoi, que le Haut Conseil de la santé publique détermine sur la base de ses avis » .
En pratique, l'encadrement de la publicité portant sur les vaccins apparaît insuffisant. L'étude d'impact du projet de loi mentionne des exemples de campagnes publicitaires menées par des laboratoires pour des indications autres que celles pour lesquelles le Haut Conseil de la santé publique (HCSP) avait émis une recommandation, ce qui nuit à la qualité du message apporté au public. Afin de rétablir la confiance dans les informations diffusées, le présent article vise à limiter les campagnes de vaccination aux seules indications d'autorisation de mise sur le marché pour lesquelles le HCSP a émis une recommandation de vaccination.
Le paragraphe IV prévoit par conséquent que les vaccins faisant l'objet de campagnes publicitaires non institutionnelles doivent figurer sur une liste établie pour des motifs de santé publique par arrêté du ministre chargé de la santé pris après avis du HCSP.
Le contenu des campagnes doit être conforme à l'avis du HCSP et assorti, de façon clairement identifiée, des mentions minimales obligatoires déterminées par cette instance. Ces mentions sont reproduites in extenso , facilement audibles et lisibles, selon le support du message publicitaire concerné. Elles sont sans renvoi et conformes à des caractéristiques définies par arrêté du ministre chargé de la santé.
Les quatrième, cinquième et sixième alinéas de l'article L. 5122-6, relatifs aux médicaments qui ne sont plus remboursés par l'assurance maladie et peuvent faire, sous certaines conditions, l'objet d'une publicité, sont supprimés.
L'alignement de la publicité destinée aux professionnels de santé sur la publicité non dédiée
Le paragraphe V instaure un contrôle a priori de la publicité à destination des professionnels de santé. Cet alignement a fait l'objet de nombreuses recommandations, notamment à l'occasion des Assises du médicament. Le groupe de travail n° 4, chargé de réfléchir au développement de l'information sur les produits de santé à destination des professionnels et du grand public a, en particulier, souligné dans son rapport l'impact qu'avaient pu avoir les pratiques promotionnelles des laboratoires Servier dans l'envolée des prescriptions hors AMM du Mediator. Il a également estimé qu'un contrôle a priori des documents utilisés par les laboratoires Servier aurait sans doute permis d'interdire les publicités qui ont été diffusées 35 ( * ) .
Jusqu'à présent, l'article L. 5122-9 du code de la santé publique dispose que la publicité destinée aux professionnels de santé doit faire l'objet, dans un délai de huit jours après sa diffusion, d'un dépôt auprès de l'Afssaps. Cette dernière peut ordonner sa suspension, exiger sa modification ou l'interdire en exigeant éventuellement la diffusion d'un rectificatif lorsque la publicité ne respecte pas les règles fixées aux articles L. 5122-2 et L. 5122-3. La publicité pour des vaccins doit être assortie, de façon clairement identifiée et sans renvoi, des recommandations in extenso de l'avis du HCSP.
Le paragraphe V remplace ce dispositif : il prévoit de soumettre la publicité effectuée auprès des professionnels de santé à un visa de publicité, c'est-à-dire à une autorisation préalable délivrée par l'agence en charge de la sécurité du médicament. Le visa ne peut excéder la durée de l'AMM. En cas de méconnaissance des articles L. 5122-2 36 ( * ) et L. 5122-3 37 ( * ) , il peut être suspendu ou retiré. La publicité portant sur les vaccins continue de devoir être assortie des recommandations in extenso de l'avis du HCSP.
Le paragraphe VI insère un nouvel article L. 5122-9-1 qui prévoit que les demandes de visa sont effectuées selon un calendrier et durant une période déterminés par décision du directeur général de l'agence.
Le paragraphe VII abroge un alinéa et deux articles du code de la santé publique :
- le 5° de l'article L. 5122-16, dans sa rédaction issue de l'article 21 de la loi n° 2007-248 du 26 février 2007 portant diverses dispositions d'adaptation au droit communautaire dans le domaine du médicament autorisait la publicité de rappel. Or, le décret en Conseil d'Etat devant définir les modalités d'application de cette disposition n'a jamais été pris. Dans un contexte de renforcement des règles applicables en matière de publicité, le Gouvernement propose l'abrogation de cette disposition, l'étude d'impact annexée au projet de loi la jugeant conforme au droit communautaire, qui laisse les Etats membres libres d'autoriser, ou non, la publicité de rappel ;
- la suppression des articles L. 5422-3 et L. 5422-4, qui définissent les dispositions pénales applicables à la publicité auprès des professionnels de santé, est cohérente avec les dispositions prévues aux paragraphes V et VIII.
Le paragraphe VIII remplace les dispositions des deux articles précités par un alignement des nouvelles sanctions prévues concernant la publicité destinée aux professionnels de santé sur celles relatives à la publicité auprès du public, soit une amende de 37 500 euros pour les publicités qui portent mention d'indications thérapeutiques interdites selon les modalités de l'article L. 5122-9.
Les dispositions prévues pour la publicité à l'égard des professionnels de santé font l'objet de dispositions transitoires définies au paragraphe VII de l'article 30. Pendant une année, l'obligation d'une demande de visa préalable ne s'applique pas aux publicités qui ont fait l'objet d'un dépôt dans les conditions antérieures à la publication de la loi.
II - Les dispositions adoptées par l'Assemblée nationale
L'Assemblée nationale a modifié ainsi cet article :
- elle a adopté une rédaction fixant clairement une interdiction de principe pour les médicaments faisant l'objet d'une réévaluation de leur rapport bénéfices-risques ;
- elle a ajouté un paragraphe IX pour compléter les dispositions pénales applicables en matière de publicité à destination des professionnels de santé figurant à l'article L. 5422-11 du code de la santé publique : une publicité n'ayant pas obtenu son visa ou effectuée malgré la décision de suspension ou de retrait de celui-ci encourt une amende de 37 500 euros ;
- à l'initiative du Gouvernement, elle a inséré un paragraphe X tendant à compléter l'article L. 613-5 du code de la propriété intellectuelle afin de prévoir que les droits conférés par un brevet ne peuvent s'opposer aux démarches engagées pour l'obtention du visa pour les publicités destinées aux professionnels de santé.
III - Le texte adopté par la commission
A l'initiative de son rapporteur, votre commission a adopté un amendement posant une interdiction de principe des campagnes de publicité portant sur des vaccins, effectuées par les industriels. Il est en effet nécessaire d'affirmer que la prévention en matière de vaccinations doit être assurée par la puissance publique. Elle a ensuite adopté un amendement de coordination avec la suppression de la publicité pour les vaccins.
La commission a adopté cet article ainsi modifié.
Article 19 (art. L. 162-17-8 du code de la sécurité sociale) Encadrement de la visite médicale
Objet : Cet article met en place, à titre expérimental et pour une durée maximum de deux ans, la visite médicale collective dans les établissements de santé et renforce la portée de la charte de la visite médicale.
I - Le dispositif proposé
La visite médicale constitue une source d'information non négligeable pour les médecins et l'outil de promotion privilégié des laboratoires pharmaceutiques. Ainsi, selon les données du Leem (Les entreprises du médicament) citées dans l'étude d'impact du projet de loi, la visite médicale représente environ 75 % des dépenses consacrées par l'industrie pharmaceutique à la promotion du médicament, soit 12 % de son chiffre d'affaires réalisé en France pour l'année 2010.
Or, les conditions dans lesquelles s'effectue la visite médicale ainsi que ses conséquences sur les prescriptions des médecins font l'objet de critiques répétées . L'observatoire mis en place par la revue Prescrire permet de dresser un bilan contrasté de la qualité des informations transmises à l'occasion des visites médicales. Les données collectées pour la période 1999-2007 montrent en effet que 70 % des visites médicales n'ont pas donné lieu à une présentation spontanée des contre-indications, interactions médicamenteuses, précautions d'emploi et effets indésirables. En outre, dans 20 % des cas, le résumé des caractéristiques du produit n'a pas été spontanément remis au médecin, ce qui constitue pourtant une obligation prévue à l'article R. 5122-11 du code de la santé publique. Ainsi que le souligne le rapport de la mission commune d'information du Sénat, « rien n'assure que les objectifs de vente des médicaments convergent avec les impératifs de leur bon usage » 38 ( * ) . Une telle situation est d'autant plus regrettable dans un pays où la surconsommation médicamenteuse et les risques de maladies iatrogéniques qui en découlent sont avérés.
Jugeant impossible la conciliation entre le caractère informatif et promotionnel de la visite médicale, la mission commune d'information du Sénat préconisait la suppression de la visite médicale au profit d'une information objective qui serait délivrée par des personnes placées sous la responsabilité de la Haute Autorité de santé (proposition n° 51). Les visiteurs médicaux dont l'emploi aurait été supprimé auraient été confiés à la HAS. Le rapport de l'Igas publié en juin 2011 allait également dans cette direction 39 ( * ) .
Les Assises du médicament et le rapport de l'Assemblée nationale ont préféré recommander une évolution des pratiques et un meilleur encadrement des visiteurs médicaux, mettant notamment en avant les conséquences sociales qu'aurait la suppression d'un métier exercé par plus de 18 000 personnes.
Pour l'ensemble de ces motifs, le présent article prévoit, à titre expérimental et pour une durée maximum de deux ans, que la visite médicale effectuée dans les établissements de santé ne sera plus individuelle mais collective . Les conditions de la visite doivent être définies par une convention conclue entre chaque établissement de santé et les entreprises concernées.
Avant toute pérennisation et adaptation éventuelle à la médecine de ville, un rapport dressant le bilan de l'expérimentation devra être présenté d'ici le 1 er janvier 2013 au Parlement par le Gouvernement.
Introduit par la loi n° 2004-810 du 13 août 2004 relative à l'assurance maladie, l'article L. 162-17-8 du code de la sécurité sociale prévoit la signature d'une charte de qualité relative à la visite médicale entre le Ceps et un ou plusieurs syndicats représentatifs des entreprises du médicament. Cette charte a été signée le 22 décembre 2004.
Le deuxième alinéa du même article L. 162-17-8 dispose que la charte vise notamment à mieux encadrer les pratiques commerciales et promotionnelles qui pourraient nuire à la qualité des soins. Le présent article y ajoute une disposition prévoyant que le Ceps peut, à cet effet, fixer des objectifs annuels chiffrés d'évolution des pratiques, le cas échéant pour certaines classes pharmaceutiques.
Enfin, le texte complète cet article par trois alinéas selon lesquels le Ceps sera en mesure, à l'issue d'une procédure contradictoire, d'infliger une pénalité financière à l'entreprise ne respectant pas les objectifs fixés par lui. Cette pénalité ne pourra être supérieure à 10 % du chiffre d'affaires hors taxes réalisé en France par l'entreprise au titre du dernier exercice clos sur le ou les produits considérés. Les règles, délais de procédure et modes de calcul de la pénalité financière devront être fixés par décret en Conseil d'Etat.
II - Les dispositions adoptées par l'Assemblée nationale
Cinq modifications ont été apportées à cet article :
- une clarification rédactionnelle concernant l'affectation du produit de la pénalité prévue au présent article sera affecté aux régimes d'assurance maladie ;
- la suppression d'une disposition redondante de renvoi à un décret en Conseil d'Etat ;
- la fixation du modèle de convention pour les visites médicales collectives selon des modalités définies par arrêté du ministre chargé de la santé, pris après avis de la HAS ;
- la précision selon laquelle le champ de l'expérimentation englobe aussi bien les visites médicales effectuées pour les médicaments que pour les dispositifs médicaux ;
- l'exclusion de l'expérimentation de trois des cinq types de médicaments entrant dans la catégorie des médicaments soumis à prescription restreinte définie à l'article R. 5121-77 : les médicaments de réserve hospitalière, de prescription hospitalière et de prescription initiale hospitalière.
III - Le texte adopté par la commission
S'il est permis de douter de l'opportunité et de la faisabilité de la visite médicale collective en établissements de santé, votre commission a estimé qu'exclure certains médicaments de l'expérimentation diminuait considérablement la portée de celle-ci. Elle a donc adopté, à l'initiative de son rapporteur, un amendement de suppression des dispositions introduites à l'Assemblée nationale limitant le champ des médicaments soumis à la visite médicale collective, puis un amendement accordant un rôle pilote à la HAS dans l'évaluation de cette expérimentation.
Concernant les objectifs qui pourront être fixés par le Ceps en termes d'évolution des pratiques de la visite médicale et toujours sur proposition de son rapporteur, elle a adopté un amendement qui les étend à certains produits et non plus seulement à certaines classes pharmaceutiques.
Pour les sanctions qui pourront être décidées par le Ceps, elle a adopté un amendement déposé par Isabelle Pasquet, Laurence Cohen, Annie David, Dominique Watrin et Guy Fischer, qui en modifie la nature : plutôt que de fixer des sanctions financières portant sur un pourcentage du chiffre d'affaires de l'entreprise sur le ou les médicaments en cause, le comité pourra imposer une baisse du prix de ces médicaments. Ce changement a également été apporté aux articles 24 et 26 qui donnent au Ceps le même pouvoir de sanction pour les dispositifs médicaux.
Votre commission a adopté cet article ainsi modifié.
Article 20 (art. L. 5121-14-3 (nouveau) du code de la santé publique) Garantie par l'industrie pharmaceutique du bon usage des médicaments
Objet : Cet article fait obligation aux entreprises exploitant une spécialité pharmaceutique de contribuer à son bon usage.
I - Le dispositif proposé
Cet article insère un article L. 5121-14-3 dans le code de la santé publique dans un double objectif : d'une part, faire obligation aux entreprises titulaires d'une autorisation d'exploiter une spécialité pharmaceutique de veiller à la conformité de sa prescription à l'autorisation obtenue ; d'autre part, préciser que l'entreprise doit aviser sans délai l'agence en charge de la sécurité du médicament lorsqu'elle constate des prescriptions non conformes au bon usage. Elle doit également prendre des mesures d'information des professionnels de santé.
L'Assemblée nationale a adopté cet article sans modification.
II - Le texte adopté par la commission
La commission a adopté cet article sans modification.
Article 20 bis
(nouveau)
(art. L. 245-2 du code de la sécurité
sociale)
Garantie par l'industrie pharmaceutique du bon usage des
médicaments
Objet : Cet article additionnel tend à soumettre l'achat d'espaces publicitaires dans la presse médicale à la taxe sur la promotion.
Cet article additionnel, inséré sur amendement présenté par Isabelle Pasquet, Laurence Cohen, Annie David, Dominique Watrin et Guy Fischer, tend à soumettre tout achat d'espaces publicitaires par les laboratoires pharmaceutiques à la contribution due au titre de leurs dépenses de promotion et d'information à l'intention des prescripteurs. Cette contribution, crée en 1983, est prévue par l'article L. 245-2 du code de la sécurité sociale. La loi de financement de la sécurité sociale pour 2003 40 ( * ) a élargi son assiette mais exclut de celle-ci les frais de publication et d'achats d'espaces publicitaires dans la presse médicale. Cette exclusion était fondée sur le caractère d'information que pouvait avoir la publicité à destination des professionnels.
Le rapport de la mission commune d'information du Sénat sur le Mediator a néanmoins souligné les effets de dépendance que pouvait créer la publicité sur certains organes de la presse médicale. De plus, les professionnels ne doivent pas être tributaires de la publicité pour leur information concernant les médicaments. Il a donc paru à la commission nécessaire de faire entrer l'ensemble des dépenses liées aux frais de publication et d'achat d'espace publicitaires dans l'assiette de la contribution.
La commission a adopté cet article additionnel ainsi rédigé.
CHAPITRE VIII - Les logiciels d'aide à la prescription et à la dispensation
Article 21 (art. L. 161-38 du code de la sécurité sociale) Certification des logiciels d'aide à la prescription et à la dispensation
Objet : Cet article précise les objectifs de la certification des logiciels d'aide à la prescription et met en place une procédure du même type concernant les logiciels d'aide à la dispensation.
I - Le dispositif proposé
Actuellement, l'article L. 161-38 du code de la sécurité sociale charge la HAS d'établir une procédure de certification des logiciels d'aide à la prescription. Ces logiciels doivent contenir un certain nombre de bonnes pratiques au nombre desquelles figurent l'intégration des recommandations et avis médico-économiques identifiés par la HAS, la possibilité de prescrire directement en dénomination commune internationale, l'affichage du prix des produits au moment de la prescription et du montant total de cette dernière, l'indication de l'appartenance au répertoire des génériques et une information relative à leur concepteur et à la nature de leur financement.
Depuis le 1 er janvier 2006, la procédure de certification des logiciels d'aide à la prescription doit être mise en oeuvre et délivrée par un organisme accrédité attestant du respect des règles de bonne pratique édictées par la HAS.
Le présent article complète l'article L. 161-38 par trois alinéas :
- le premier précise les objectifs de la procédure de certification des logiciels d'aide à la prescription. Ces derniers doivent participer à l'amélioration des pratiques de prescription médicamenteuse et garantir la conformité du logiciel à des exigences minimales en termes de sécurité, de conformité et d'efficience de la prescription ;
- le deuxième alinéa prévoit une procédure de certification pour les logiciels d'aide à la dispensation et en définit les objectifs. De la même façon que pour les logiciels d'aide à la prescription, la certification des logiciels d'aide à la dispensation, cette fois, doit contribuer à l'amélioration des pratiques de dispensation officinale et garantir le respect, par le logiciel, d'exigences minimales en termes de sécurité et de conformité de la dispensation ;
- le troisième alinéa prévoit une période transitoire, la certification des deux types de logiciels devant être effectuée d'ici au 1 er janvier 2015. Les règles qui entourent cette obligation seront fixées par décret en Conseil d'Etat.
Enfin, le présent article apporte des précisions quant à la nature des organismes certificateurs des logiciels d'aide à la prescription. Le second alinéa de l'article L. 161-38 prévoit jusqu'à présent que l'organisme certificateur doit être accrédité et attester du respect des règles de bonne pratique définies par la HAS. Il devra désormais s'agir d'organismes « accrédités par le comité français d'accréditation ou par l'organisme compétent d'un autre Etat membre de l'Union européenne » .
II - Les dispositions adoptées par l'Assemblée nationale
L'Assemblée nationale a apporté une amélioration rédactionnelle à cet article.
III - Le texte adopté par la commission
Les multiples modifications introduites depuis plusieurs années à cet article relatif à la certification l'ont rendu peu lisible. Votre commission a donc adopté, sur proposition de son rapporteur, un amendement de forme qui vise à en améliorer la rédaction.
Cette nouvelle rédaction a en outre le mérite de fixer clairement des règles de certification équivalentes pour les sites informatiques dédiés à la santé et les logiciels d'aide à la prescription et à la dispensation.
La commission a adopté cet article ainsi modifié.
CHAPITRE IX - Les études en santé publique
Article 22 (art. L. 5121-28 (nouveau) du code de la santé publique) Groupement d'intérêt public compétent en matière d'études de santé publique
Objet : Cet article crée un groupement d'intérêt public chargé d'autoriser l'accès au système national d'informations inter-régimes de l'assurance maladie lorsque des études le nécessitent et pouvant lui-même en mener ou en superviser la réalisation.
I - Le dispositif proposé
Le présent article crée un chapitre I er ter au sein du titre II du livre I er de la cinquième partie du code de la santé publique intitulé « Etudes en santé publique ». Ce nouveau chapitre est composé d'un article L. 5121-28 qui prévoit qu'un GIP, régi par les dispositions de la loi n° 2011-525 du 17 mai 2011 de simplification et d'amélioration de la qualité du droit, pourra autoriser l'accès au système national d'informations inter-régimes de l'assurance maladie (Sniiram) ou une extraction de ses données lorsque la réalisation d'études de pharmacovigilance ou de pharmaco-épidémiologie le nécessite.
Créé par l'article 21 de la loi n° 98-1194 du 23 décembre 1998 de financement de la sécurité sociale codifié à l'article L. 161-28-1 du code de la sécurité sociale, le Sniiram est mis en place par les régimes d'assurance maladie qui lui transmettent les données nécessaires. Ces données doivent préserver l'anonymat des personnes ayant bénéficié de prestations de soins. Le Sniiram contribue :
- à la connaissance des dépenses de l'ensemble des régimes d'assurance maladie par circonscription géographique, par nature de dépenses, par catégorie de professionnels responsables de ces dépenses et par professionnel ou établissement ;
- à la transmission en retour aux prestataires de soins d'informations pertinentes relatives à leur activité et leurs recettes et, s'il y a lieu, à leurs prescriptions ;
- à la définition, à la mise en oeuvre et à l'évaluation des politiques de santé publique.
Le présent article précise les considérations qui doivent guider le GIP au moment de délivrer ou non une autorisation : la contribution que les études menées sont susceptibles d'apporter à la santé publique ou à l'efficience des dépenses d'assurance maladie doit être mise en regard du respect de la protection des données personnelles et notamment du secret médical.
Enfin, il renvoie au pouvoir réglementaire le soin de fixer les conditions de son application. Un décret en Conseil d'Etat devra être pris après avis de la commission nationale de l'informatique et des libertés.
II - Les dispositions adoptées par l'Assemblée nationale
Outre le vote d'un amendement rédactionnel, l'Assemblée nationale a élargi les prérogatives du GIP : celui-ci pourra lui-même mener des études ou lancer des appels d'offres pour la réalisation d'études, lesquelles ne pourront être financées par des entreprises commercialisant des produits autorisés par l'Afssaps.
Les études ainsi menées feront l'objet d'un rapport d'activité remis chaque année au Parlement.
III - Le texte adopté par la commission
A l'initiative de son rapporteur, la commission a adopté trois amendements :
- deux d'entre eux délimitant le champ des produits qui entreront dans le champ de compétences du GIP : il s'agit de l'ensemble des produits mentionnés à l'article L. 5311-1 du code de la santé publique relatif aux compétences de l'actuelle Afssaps ;
- le troisième précisant la composition du GIP de façon à clairement affirmer sa vocation à constituer un pôle d'expertise sanitaire public. Les membres du GIP seront l'Etat, la HAS, l'agence en charge de la sécurité du médicament, l'InVS et la Cnam.
Votre commission a adopté cet article ainsi modifié.
TITRE IV - DISPOSITIFS MÉDICAUX
Article 23 (art. L. 5213-1 à L. 5213-7, L. 5461-6, L. 5461-7, L. 5461-8 (nouveaux), L. 5212-1 et L. 5222-2 du code de la santé publique et art. L. 165-8 du code de la sécurité sociale) Publicité pour les dispositifs médicaux
Objet : Cet article tend à aligner le régime juridique applicable à la publicité portant sur les dispositifs médicaux sur les règles fixées pour les médicaments.
I - Le dispositif proposé
Les dispositifs médicaux occupent aujourd'hui une place importante dans les soins des patients. La prise en charge croissante de certaines affections à domicile, l'augmentation ou le meilleur repérage de certaines pathologies ainsi que le vieillissement de la population contribuent au développement du marché des dispositifs médicaux et à une imbrication renforcée entre ceux-ci et les médicaments. Cette évolution est accélérée par les améliorations technologiques importantes que connaissent ces produits depuis plusieurs années.
Il apparaît par conséquent légitime d'aligner, dans la mesure du possible, le régime juridique applicable aux dispositifs médicaux sur celui des médicaments. Tel est l'objectif du titre IV du projet de loi.
Le présent article porte plus particulièrement sur la publicité. En effet, contrairement aux médicaments, les dispositifs médicaux ne sont pas soumis à des règles spécifiques en matière de publicité, la directive 93/42/CEE du Conseil du 14 juin 1993 relative aux dispositifs médicaux ne prévoyant aucune disposition particulière sur ce point.
Le paragraphe I complète le titre I er du livre II de la cinquième partie du code de la santé publique par un chapitre III intitulé « Publicité » et composé de sept articles.
L'article L. 5213-1 définit la notion de publicité pour les dispositifs médicaux. Celle-ci est quasi identique à celle énoncée à l'article L. 5122-1 du même code pour les médicaments.
Doit être considérée comme une publicité « toute forme d'information, y compris le démarchage, de prospection ou d'incitation qui vise à promouvoir la prescription, la délivrance, la vente ou l'utilisation de ces dispositifs, à l'exception de l'information dispensée, dans le cadre de leurs fonctions, par les pharmaciens gérant une pharmacie à usage intérieur » .
Ne relèvent pas de la publicité :
- l'étiquetage et la notice d'instruction des dispositifs médicaux ;
- la correspondance, accompagnée le cas échéant de tout document non publicitaire nécessaire pour répondre à une question précise sur un dispositif médical en particulier ;
- les informations relatives aux mises en garde, aux précautions d'emploi et aux effets indésirables relevés dans le cadre de la matériovigilance, ainsi que les catalogues de ventes et listes de prix s'il n'y figure aucune information sur le dispositif médical ;
- les informations relatives à la santé humaine ou à des maladies humaines, pour autant qu'il n'y ait pas de référence même indirecte à un dispositif médical.
L'article L. 5213-2 dispose que seuls les dispositifs médicaux pour lesquels a été établi un certificat de conformité par le fabricant ou par des organismes désignés par l'agence pourront faire l'objet d'une publicité. Celle-ci doit présenter le produit de façon objective, respecter les dispositions du certificat de conformité et favoriser le bon usage du dispositif médical. Elle ne peut ni être trompeuse, ni présenter un risque pour la santé publique.
L'article L. 5213-3 prévoit que, tout comme les médicaments, les dispositifs médicaux pris en charge ou financés, même partiellement, par les régimes obligatoires d'assurance maladie ne peuvent faire l'objet d'une publicité auprès du public.
Cet article met fin à une ambigüité née de la rédaction de l'article L. 165-8 du code de la sécurité sociale relatif au remboursement par l'assurance maladie des dispositifs médicaux. Son premier alinéa disposant que « la publicité auprès du public pour les produits ou prestations inscrits sur la liste prévue à l'article L. 165-1 ne peut mentionner que ces produits ou ces prestations peuvent être remboursés par l'assurance maladie ou par un régime complémentaire » , on pouvait en conclure l'existence d'une autorisation tacite de la publicité sur les dispositifs médicaux pris en charge par l'assurance maladie tant qu'il n'était pas fait mention de cette prise en charge. Le nouvel article L. 5213-3 pose clairement le principe de l'interdiction.
Le paragraphe III modifie en conséquence le premier alinéa de l'article L. 165-8 du code de la sécurité sociale.
L'article L. 5213-4 indique que, pour certains dispositifs présentant un risque important, la publicité devra faire l'objet d'une autorisation préalable délivrée par l'agence en charge de la sécurité du médicament pour une durée de cinq ans renouvelable. L'autorisation pourra être suspendue ou retirée par décision motivée de l'agence. La liste de ces dispositifs sera fixée par arrêté du ministre chargé de la santé
L'article L. 5213-5 prévoit la possibilité pour l'agence de mettre en place, en matière de publicité, la procédure de sanctions administratives créée à l'article 4 du projet de loi par l'insertion d'un article L. 5312-4-1 au code de la santé publique. Après mise en demeure de l'entreprise concernée, l'agence pourra prononcer des sanctions allant jusqu'à l'interdiction de la publicité.
L'article L. 5213-6 précise que les dispositions du nouveau chapitre ne s'appliquent pas aux contraceptifs, l'article L. 5134-2 du même code prévoyant un alignement de la publicité les concernant sur les règles prévues pour les médicaments.
Il est enfin mentionné à l'article L. 5213-7 que les modalités d'application du chapitre seront fixées par décret en Conseil d'Etat.
Le paragraphe II met en place un système de sanctions pénales en cas de publicité de caractère trompeur ou de nature à porter atteinte à la santé publique. Pour les dispositifs médicaux soumis à une autorisation préalable de l'agence, ces sanctions seront également applicables lorsque l'agence n'aura pas délivré, refusé de délivrer, suspendu ou retiré l'autorisation en question. La création d'un délit en cas de non-respect de l'obligation d'autorisation préalable constituait une proposition des Assises du médicament.
Sont également prévues, pour les personnes physiques et morales reconnues coupables des infractions précitées, des peines complémentaires permettant de rendre publique la sanction pénale et de prévoir la fermeture du ou des établissements de l'entreprise ayant servi à commettre les faits incriminés et l'interdiction de fabriquer, conditionner, importer ou mettre sur le marché des dispositifs médicaux. Ces deux dernières peines pourront avoir une durée maximale de cinq ans.
II - Les dispositions adoptées par l'Assemblée nationale
Outre des modifications rédactionnelles et la correction d'une erreur de référence, l'Assemblée nationale a :
- complété les articles L. 5212-1 et L. 5222-2 qui imposent aux personnes souhaitant revendre un dispositif médical ou un dispositif médical de diagnostic in vitro de faire établir une attestation technique justifiant du maintien des performances du produit concerné. Il est désormais précisé que cette obligation s'applique aussi aux cessions à titre gratuit ;
- prévu une dérogation au principe de l'interdiction de la publicité sur les dispositifs médicaux pris en charge par l'assurance maladie. Un arrêté des ministres chargés de la santé et de la sécurité sociale devra fixer une liste de dispositifs médicaux présentant un faible risque pour la santé humaine et pouvant faire l'objet de publicité.
III - Le texte adopté par la commission
La commission a adopté cet article sans modification.
Article 24 (art. L. 165-1-2 (nouveau) du code de la sécurité sociale) Conformité des dispositifs médicaux aux spécifications requises pour pouvoir être remboursés
Objet : Cet article renforce le contrôle de la conformité des dispositifs médicaux inscrits sur la liste des produits et prestations remboursables à des spécifications techniques minimales.
I - Le dispositif proposé
L'article L. 165-1 du code de la sécurité sociale dispose que le remboursement par l'assurance maladie des dispositifs médicaux s'effectue après leur inscription sur la liste des produits et prestations remboursables. Deux modalités d'inscription sur cette liste existent :
- la procédure de droit commun consiste à inscrire le dispositif médical par description générique. Elle s'applique aux dispositifs médicaux présentant des spécifications techniques identiques à celles de produits ou prestations déjà inclus dans la liste des produits et prestations remboursables. Les produits inscrits sur une même ligne générique sont alors remboursés par l'assurance maladie sur la base d'un tarif unique ;
- la seconde procédure permet l'inscription du dispositif médical sous forme de marque ou de nom commercial. Elle est utilisée quand le fabricant considère que le caractère innovant de son produit justifie un prix supérieur à celui de la ligne générique ou quand l'impact sur les dépenses d'assurance maladie, les impératifs de santé publique ou le contrôle des spécifications techniques minimales nécessitent un suivi particulier du produit.
Si une simple déclaration à l'actuelle Afssaps est suffisante pour l'inscription d'un produit sur une ligne générique, l'inscription sous forme de marque ou de nom commercial doit faire l'objet d'un avis de la part de la commission nationale des dispositifs médicaux et des technologies de santé de la HAS.
L'objectif du présent article est de renforcer le contrôle du respect de spécifications techniques minimales par les dispositifs médicaux inscrits sur la liste des produits et prestations remboursables.
A cet effet, il insère un article L. 165-1-2 qui prévoit la possibilité pour l'agence de contrôler, ou faire contrôler par des organismes compétents, le respect des spécifications techniques auxquelles l'inscription sur la liste est subordonnée. Dans le cas où l'agence saisirait certains produits, le fabricant, le mandataire ou le distributeur doit compenser la perte financière qui en résulte pour ceux qui ont acheté les produits.
Ce nouvel article L. 165-1-2 prévoit la possibilité pour l'agence d'engager une procédure pouvant conduire à des sanctions. L'agence pourra en premier lieu adresser au fabricant, à son mandataire ou au distributeur un courrier notifiant les manquements retenus à son encontre. Une copie de ce courrier sera adressée aux ministres chargés de la santé et de la sécurité sociale ainsi qu'au directeur général de l'union nationale des caisses d'assurance maladie (Uncam) et au Ceps. Ce dernier aura le pouvoir, à l'issue d'une procédure contradictoire, d'infliger une pénalité financière qui ne peut être supérieure à 10 % du chiffre d'affaires hors taxes réalisé en France sur le ou les produits concernés.
La procédure de recouvrement de l'indu prévue à l'article L. 133-4 du même code sera engagée quand le non respect par le produit des spécifications techniques auxquelles il est soumis aura conduit à un remboursement indu par l'assurance maladie.
Lorsque ce manquement aura rendu nécessaire la dispensation d'actes de soins, de prestations ou de produits de santé à un assuré, le professionnel ou l'établissement de santé qui en aura connaissance et aura accompli les actes en cause en informera, dans le respect du secret médical, l'organisme local d'assurance maladie auquel l'assuré est affilié.
Un décret en Conseil d'Etat précisera les conditions d'application de l'article.
II - Les dispositions adoptées par l'Assemblée nationale
L'Assemblée nationale a adopté des modifications rédactionnelles ainsi qu'une correction sur l'affectation aux régimes d'assurance maladie des pénalités prévues par l'article.
Elle a aussi précisé qu'au moment où l'agence en charge de la sécurité du médicament notifie au contrevenant les manquements retenus à son encontre, elle doit lui signaler les risques qu'il encourt.
III - Le texte adopté par la commission
La commission a adopté un amendement présenté par Isabelle Pasquet, Laurence Cohen, Annie David, Dominique Watrin et Guy Fischer, relatif au pouvoir de sanction du Ceps, par analogie avec sa position sur l'article 19, puis un amendement de son rapporteur réparant une omission de référence.
Elle a adopté cet article ainsi modifié.
Article 25 (art. L. 114-10, L. 162-1-14 et L. 162-1-20 (nouveau) du code de la sécurité sociale) Contrôle par les agents assermentés de l'assurance maladie de la conformité des dispositifs médicaux aux règles de facturation et de tarification
Objet : Cet article instaure un contrôle par des agents assermentés de l'assurance maladie de la conformité des dispositifs médicaux aux règles de facturation et de tarification.
I - Le dispositif proposé
Le présent article permet de renforcer le contrôle de la conformité aux règles de facturation et de tarification des dispositifs médicaux et des médicaments et produits soumis à autorisation.
Ces contrôles doivent être effectués par les agents dont il est fait mention à l'article L. 114-10 du code de la sécurité sociale, qui autorise des agents désignés par les directeurs des organismes de sécurité sociale à procéder à toutes vérifications ou enquêtes administratives concernant l'attribution des prestations et la tarification des accidents du travail et des maladies professionnelles. Le même article mentionne également que des praticiens-conseil peuvent mener les enquêtes. Le présent article y adjoint les auditeurs comptables.
Ces agents pourront réaliser leurs enquêtes sur pièces et sur place. Les documents et informations auxquels ils pourront avoir accès sont ceux mentionnés à l'article L. 114-19 du même code qui dispose que doit être communiquée, sans que puisse s'y opposer le secret professionnel, toute pièce permettant aux enquêteurs de contrôler la sincérité et l'exactitude des déclarations souscrites ou l'authenticité des pièces produites en vue de l'attribution et du paiement des prestations servies par les organismes de sécurité sociale.
Le droit de visite s'imposera aux établissements de santé ou à toute autre personne physique ou morale autorisée à réaliser une prestation de service ou à délivrer les produits faisant l'objet de l'enquête, à condition que ces derniers aient été préalablement informés de la venue du ou des agents dans un délai et dans des formes qui doivent être définis pas décret en Conseil d'Etat. Ils devront en particulier avoir été informés de leur droit à se faire assister pendant l'enquête du conseil de leur choix.
Le présent article précise que lorsque l'enquête a trait à la fraude, l'agent est dispensé de fournir une information préalable.
Enfin, il opère un renvoi à l'article L. 162-1-20 dans l'article L. 162-1-14 pour permettre au directeur de l'organisme local d'assurance maladie de prononcer une pénalité en cas de refus d'accès à une information, d'absence de réponse ou de réponse fausse, incomplète ou abusivement tardive à toute demande de pièce justificative, d'information, d'accès à une information ou à une convocation dans le cadre de l'enquête prévue.
II - Les dispositions adoptées par l'Assemblée nationale
L'Assemblée nationale a adopté des modifications rédactionnelles.
III - Le texte adopté par la commission
Votre commission a adopté cet article sans modification.
Article 26 (art. L. 165-11, L. 165-12 et L. 165-13 (nouveaux) du code de la sécurité sociale) Evaluation de certains dispositifs médicaux
Objet : Cet article instaure un dispositif d'évaluation des dispositifs médicaux intégrés dans les forfaits des groupes homogènes de séjour.
I - Le dispositif proposé
Depuis la mise en place de la tarification à l'activité (T2A), la procédure de droit commun pour le remboursement des dispositifs médicaux en secteur hospitalier consiste en leur intégration dans les forfaits de groupes homogènes de séjour (GHS). Certains dispositifs médicaux demeurent cependant financés en sus de la tarification à l'activité et sont pour cela inscrits sur la liste des produits et prestations remboursables. Il s'agit d'une dérogation prévue par la loi n° 2003-1199 du 18 décembre 2003 qui permet le financement en sus des médicaments et dispositifs médicaux coûteux et innovants. Les dispositifs médicaux inscrits sur une liste en sus fixée par arrêté du ministre après avis du conseil de l'hospitalisation peuvent ainsi bénéficier d'un remboursement intégral de la part prise en charge par les régimes d'assurance maladie obligatoire. Ils sont inscrits à la liste des produits et prestations remboursables sur la base de tarifs fixés par le Ceps. Subsistent donc, dans les établissements de santé, deux modalités de remboursement et de fixation des prix pour les dispositifs médicaux.
Cette dualité a des conséquences sur la connaissance des caractéristiques techniques et de sécurité des dispositifs médicaux : les produits inclus dans les forfaits de GHS ne sont soumis à aucune étude particulière tandis que l'intégration de dispositifs médicaux sur la liste en sus doit faire l'objet d'un avis de la commission nationale des dispositifs médicaux et des technologies de santé de la HAS.
L'étude d'impact du présent projet de loi met en lumière plusieurs des effets pervers qui peuvent résulter, pour les établissements de santé, de cette situation :
- le manque d'information sur les dispositifs médicaux inclus dans les GHS ne permet pas aux acheteurs de faire clairement la différence entre deux produits ; les décisions d'achat se fondent alors essentiellement sur les prix plutôt que sur une comparaison des bénéfices et risques de chacun ;
- le maintien sur la liste en sus peut être justifié non par le seul caractère innovant ou coûteux du dispositif médical, mais par la nécessité de disposer d'informations exhaustives sur celui-ci.
L'objectif du présent article est de rendre obligatoire l'évaluation par la HAS des dispositifs médicaux relevant d'un financement dans les GHS. Cette évaluation aurait pour but de valider leur intérêt thérapeutique et leur efficience par rapport aux alternatives mais également d'identifier un besoin éventuel d'études complémentaires.
A cet effet, il insère, à la suite de l'article L. 165-10 du code de la sécurité sociale, trois articles nouveaux.
L'article L. 165 - 11 dispose que l'achat, la fourniture, la prise en charge et l'utilisation par les établissements de santé de dispositifs médicaux inclus dans les groupes homogènes de séjour est limité aux produits inscrits sur une liste établie par arrêté des ministres chargés de la santé et de la sécurité sociale après avis de la commission nationale d'évaluation des dispositifs médicaux et technologies de santé de la HAS.
Les fabricants de dispositifs médicaux, leurs mandataires ou leurs distributeurs devront donc déposer auprès de la commission nationale des dispositifs médicaux et des technologies de santé une demande d'inscription sur la liste précitée, l'inscription déterminant l'intégration ou non dans les GHS.
La commission nationale des dispositifs médicaux et des technologies de santé rendra ses avis en fonction de trois critères :
- la validation de leur efficacité clinique ;
- la définition de spécifications techniques particulières ;
- l'appréciation de leur efficience au regard des alternatives thérapeutiques disponibles.
L'inscription sur la liste sera limitée dans le temps et renouvelable. L'inscription ou le renouvellement pourront conduire à ce que soient émises des conditions de prescription et d'utilisation et seront subordonnées à la réalisation, par les fabricants, leurs mandataires ou leurs distributeurs, d'études complémentaires demandées sur les produits.
L'article L. 165 - 12 prévoit des sanctions financières pour les établissements de santé qui achèteraient des dispositifs médicaux appartenant aux GHS sans être inscrits sur la liste qui doit être fixée par arrêté.
La sanction sera prononcée par le directeur général de l'ARS à l'issue d'une procédure contradictoire et de contrôles effectués sur pièces et sur place par les inspecteurs de santé publique ou les praticiens-conseils des organismes d'assurance maladie. Le montant de la sanction ne pourra excéder le coût total d'achat par l'établissement des produits considérés durant l'année précédant la constatation du manquement.
L'article L. 165-13 prévoit quant à lui des sanctions financières à l'encontre des fabricants, mandataires ou distributeurs qui ne feraient pas procéder aux études prévues par l'article L. 165-11 nouveau ou ne le feraient pas dans les délais.
La sanction financière sera prononcée par les ministres chargés de la santé et de la sécurité sociale à l'issue d'une procédure contradictoire. Le montant de la pénalité ne pourra excéder 10 % du chiffre d'affaires hors taxes réalisé en France sur le ou les produits considérés durant les douze mois précédant la constatation du manquement.
Le présent article prévoit deux décrets en Conseil d'Etat, pour préciser respectivement les articles L. 165-11 et L. 165-13. Il renvoie également, comme cela a été écrit précédemment, à un arrêté pris par les ministres chargés de la santé et de la sécurité sociale.
L'étude d'impact table sur une période transitoire estimée entre dix-huit et vingt-quatre mois qui doit permettre d'inclure progressivement les dispositifs médicaux dans le champ de la mesure en tenant compte de leur degré de risque tel qu'il est établi par la classification communautaire.
II - Les dispositions adoptées par l'Assemblée nationale
L'Assemblée nationale a adopté des amendements rédactionnels.
III - Le texte adopté par la commission
Sur proposition d'Isabelle Pasquet, Laurence Cohen, Annie David, Dominique Watrin et Guy Fischer, la commission a adopté un amendement relatif au pouvoir de sanction des ministres chargés de la santé et de la sécurité sociale, par coordination avec sa position à l'article 19 puis cet article ainsi modifié.
TITRE V - DISPOSITIONS DIVERSES
Article 27 Habilitation du Gouvernement à prendre par ordonnance les mesures nécessaires à la transposition d'une directive « médicaments »
Objet : Cet article tend à habiliter le Gouvernement à prendre par ordonnance certaines mesures de transposition de la directive 2011/62/UE du 8 juin 2011.
I - Le dispositif proposé
Cet article se compose de trois parties.
La première prévoit l'habilitation du Gouvernement à prendre par ordonnance les dispositions de transposition de la directive du 8 juin 2011 relative au médicament, qui touchent à l'introduction, dans la chaîne d'approvisionnement légal, de médicaments falsifiés et celles relatives à l'information et au commerce électronique relatives aux autres produits de santé.
La deuxième prévoit que l'ordonnance comporte les mesures d'extension nécessaire aux îles Wallis et Futuna ainsi qu'à la Nouvelle-Calédonie et à la Polynésie française.
La troisième prévoit le dépôt d'un projet de loi de ratification devant le Parlement dans les quatre mois qui suivent la publication de l'ordonnance.
L'Assemblée nationale a adopté cet article sans modification.
II - Le texte adopté par la commission
La commission est opposée au recours aux ordonnances quand celles-ci n'ont pour but que de pallier une mauvaise organisation du travail gouvernemental.
A l'initiative conjointe de son rapporteur et d'Isabelle Pasquet, Laurence Cohen, Annie David, Dominique Watrin et Guy Fischer, la commission a donc supprimé cet article .
Article 28 Habilitation du Gouvernement à prendre par ordonnance les mesures nécessaires pour harmoniser les sanctions pénales et administratives avec les dispositions de la présente loi
Objet : Cet article tend à habiliter le Gouvernement à procéder à certaines mesures d'harmonisation au sein du code de la santé publique consécutives à l'adoption du projet de loi.
I - Le dispositif proposé
Cet article propose d'habiliter le Gouvernement à prendre par ordonnance les mesures d'harmonisation et de mise en cohérence des sanctions administratives et financières relatives aux produits de santé ainsi que les mesures d'adaptation des prérogatives des agents chargés de constater les manquements punis par ces sanctions et de mettre celles-ci en oeuvre.
Un projet de loi de ratification doit être déposé devant le Parlement dans les quatre mois après la publication de l'ordonnance.
L'Assemblée nationale a adopté cet article sans modification.
II - Le texte adopté par la commission
Là encore, la commission est opposée au recours aux ordonnances quand celles-ci n'ont pour but que de pallier une mauvaise organisation du travail gouvernemental.
A l'initiative conjointe de son rapporteur et d'Isabelle Pasquet, Laurence Cohen, Annie David, Dominique Watrin et Guy Fischer, la commission a donc supprimé cet article .
Article 29 Habilitation du Gouvernement à prendre par ordonnance des dispositions relatives à la mise en cohérence du droit métropolitain avec le droit de l'Outre-mer
Objet : Cet article vise à habiliter le Gouvernement à prendre par ordonnance les mesures d'application de la présente loi aux îles Wallis et Futuna et, en tant qu'elles relèvent des compétences de l'Etat, à la Nouvelle-Calédonie et à la Polynésie française.
I - Le dispositif proposé
En application de l'article 38 de la Constitution, le présent article habilite le Gouvernement à prendre par ordonnance les mesures d'extension et d'adaptation de la loi à Wallis-et-Futuna et, en tant qu'elles relèvent des compétences de l'Etat, à la Nouvelle-Calédonie et à la Polynésie française. Ces mesures doivent intervenir dans les douze mois suivant la publication de la loi.
Le projet de loi de ratification devra être déposé dans un délai de quatre mois à compter de la publication de l'ordonnance.
L'Assemblée nationale a adopté l'article sans modification.
II - Le texte adopté par la commission
Si le recours aux ordonnances de l'article 38 pour étendre et adapter les dispositions d'une loi aux collectivités d'outre-mer régies par le principe de spécialité et à la Nouvelle-Calédonie tend à devenir une pratique courante, votre commission estime qu'un tel dessaisissement du Parlement n'est pas satisfaisant.
A l'initiative de son rapporteur, elle a par conséquent adopté un amendement de façon à prévoir l'application des dispositions du projet de loi à Wallis-et-Futuna et, en tant qu'elles relèvent des compétences de l'Etat, à la Nouvelle-Calédonie et à la Polynésie française, sans renvoyer à une ordonnance. Cela devrait inciter le Gouvernement à expliciter, devant le Parlement, les mesures d'adaptation qu'il entend mettre en oeuvre dans ces territoires.
La commission a adopté cet article ainsi modifié.
Article 30 Dispositions transitoires
Objet : Cet article propose un ensemble de dispositions transitoires applicables à plusieurs des articles du projet de loi.
I - Le dispositif proposé
Aux termes des paragraphes I, II et III du présent article, entrent en vigueur à compter de la publication des décrets prévus par la loi pour leur application ou, au plus tard, le 31 juillet 2012 :
- les dispositions relatives aux agents des autorités et organismes de sécurité sanitaire dont les missions ou la nature des fonctions justifient une déclaration d'intérêts (paragraphe II de l'article 1 er ) ;
- les dispositions punissant d'une amende les entreprises qui ne rendent pas publiques les conventions conclues avec les personnes énumérées aux 1° à 8° du I de l'article L. 1453-1 nouveau et les avantages qui leur sont consentis (article 3) ; les conventions conclues avant l'entrée en application des dispositions mais qui continuent de produire leurs effets après cette date verront s'appliquer les règles nouvelles ;
- les dispositions relatives à la nouvelle agence prévues aux articles 4 (à l'exception des V, VI et VII) et 5.
Le paragraphe IV dispose que les pouvoirs de sanction administrative dont disposera l'agence, prévus à l'article 4, entrent en vigueur le 21 juillet 2012.
Le paragraphe V aligne la mise en application de l'article 12 relatif à la prescription en dénomination commune internationale sur celle de l'article 21 qui instaure une obligation de certification des logiciels d'aide à la dispensation.
Le paragraphe VI fixe un régime transitoire pour les demandes d'ATU. Les autorisations déjà accordées avant le changement de régime juridique restent régies par les anciennes règles, y compris pour leur renouvellement, pendant un délai de trois ans. Ce même délai s'applique aux demandes d'ATU nominatives de même nature que des ATU déjà accordées pour le médicament concerné avant la publication de la loi.
Le paragraphe VII est relatif aux publicités destinées aux professionnels de santé. L'obligation d'une demande de visa préalable ne s'applique pas pendant un an aux publicités qui ont fait l'objet d'un dépôt dans les conditions antérieures à la publication de la loi.
II - Les dispositions adoptées par l'Assemblée nationale
L'Assemblée nationale a reporté la date limite prévue aux paragraphes I, II et III du 31 juillet au 1 er août 2012. Elle a également précisé au paragraphe III que jusqu'à la mise en place de la nouvelle agence, les compétences et pouvoirs qui lui sont attribués seront exercés par l'Afssaps.
Outre des modifications rédactionnelles, elle a enfin demandé au Gouvernement de remettre au Parlement, avant le 1 er janvier 2013, un rapport formulant des propositions sur la réparation des dommages dus à la réalisation d'un risque lié à un médicament.
III - Le texte adopté par la commission
Sur proposition de son rapporteur, la commission a adopté trois amendements :
- le premier permet de tenir compte des modifications introduites à l'article 2 concernant les avantages consentis par les entreprises au profit de certains acteurs du champ des produits de santé ;
- le deuxième prévoit que l'article 9 bis , qui impose la réalisation d'essais contre comparateurs actifs aux entreprises demandant la prise en charge d'un médicament par l'assurance-maladie, entrera en vigueur le 1 er janvier 2013 ;
- le troisième requiert de la HAS la remise au Parlement, avant le 30 juin 2012, d'un rapport dressant le bilan des règles applicables à la sécurité des dispositifs médicaux et présentant les mesures susceptibles de l'améliorer.
La commission a adopté cet article ainsi modifié.
Article 30 bis (nouveau) Nom et pouvoirs de la commission de la transparence de la Haute Autorité de santé
Objet : Cet article additionnel modifie le nom de la commission de la transparence de la Haute Autorité de santé et renforce la portée de ses avis.
Cet article additionnel est issu d'un amendement présenté par Isabelle Pasquet, Laurence Cohen, Annie David, Dominique Watrin et Guy Fischer.
Il vise en premier lieu à remplacer le nom de la commission de la transparence de la HAS par celui de « Commission du progrès thérapeutique ».
En second lieu, il rend opposables au ministre chargé des affaires sociales les avis de celle-ci sur l'inscription de médicaments sur la liste des produits et prestations remboursables, sauf opposition de sa part dans un délai de quinze jours.
Votre commission a adopté cet article additionnel ainsi rédigé.
Article 30 ter (nouveau) Rapport sur la profession de visiteur médical
Objet : Cet article additionnel demande au Gouvernement la remise d'un rapport au Parlement sur l'évolution de la profession de visiteur médical.
Cet article additionnel est issu d'un amendement présenté par Isabelle Pasquet, Laurence Cohen, Annie David, Dominique Watrin et Guy Fischer.
Il prévoit que le Gouvernement remettra au Parlement, avant le 1 er janvier 2013, un rapport relatif à l'éventuelle extinction de la profession de visiteur médical et à son remplacement par un corps public rattaché auprès de la HAS.
Votre commission a adopté cet article additionnel ainsi rédigé.
Article 31 (art. L. 713-7 du code de la propriété intellectuelle) Apparence et texture des médicaments génériques
Objet : Cet article, inséré par l'Assemblée nationale, tend à permettre aux spécialités génériques, pour éviter les erreurs de prise, de se présenter sous des formes pharmaceutiques d'apparence similaire à celle du médicament princeps.
I - Le dispositif proposé par l'Assemblée nationale
Cet article reprend une disposition adoptée une première fois dans le projet de loi de financement de la sécurité sociale pour 2010 puis une seconde lors de la discussion de la proposition de loi modifiant certaines dispositions de la loi HPST du 21 juillet 2009 41 ( * ) mais invalidée à deux reprises par le Conseil constitutionnel en tant que cavalier 42 ( * ) .
Il prévoit une exception au droit des marques, au profit du producteur de génériques, afin de lui permettre de donner au médicament qu'il fabrique la même apparence que celle du princeps.
II - Le texte adopté par la commission
La rédaction de cet article reprend le texte proposé par le Gouvernement lors de la discussion de la proposition de loi précitée. Tout en partageant pleinement l'objectif poursuivi, le Sénat demeure réservé sur l'effectivité de cette rédaction.
En effet, la présentation des médicaments est généralement protégée à titre de dessin ou modèle et une dérogation au seul droit de la protection des marques, telle que prévue par cet article, serait donc inefficace. De plus, si, comme l'avait souligné le Gouvernement lors des précédentes discussions de cette disposition, la législation communautaire ne prévoit pas d'exception à la protection des dessins et modèles, le droit français autorise la limitation du droit de propriété lorsqu'elle est justifiée par l'intérêt général.
Dès lors, une exception au droit de propriété intellectuelle protégeant les dessins ou modèles permettant la reproduction de la forme ou de la couleur d'un cachet ou d'une gélule ne paraît pas disproportionnée au regard des enjeux en matière de santé publique et de préventions d'accidents iatrogéniques graves. D'autant plus que la protection des droits de propriété intellectuelle sur les dessins et modèles est sans doute moins vitale pour l'industrie pharmaceutique que pour d'autres secteurs d'activité économique.
Enfin, l'inscription de la disposition proposée dans le code de la santé publique paraît beaucoup plus justifiée, en termes d'accessibilité du droit, que son inscription dans le code de la propriété intellectuelle. Qui plus est, il ne faudrait pas, si elle devait être inscrite dans le code de la propriété intellectuelle, qu'elle ne le soit que dans les dispositions de ce code relatives au droit des marques.
A l'initiative de son rapporteur, la commission a donc adopté un amendement tendant à rétablir la rédaction précédemment adoptée par le Sénat pour cette disposition.
Elle a adopté cet article ainsi modifié.
Article 32 (art. L. 5312-4-2 (nouveau) du code de la santé publique) Protection des lanceurs d'alerte
Objet : Cet article, introduit par l'Assemblée nationale, vise à insérer dans le code de la santé publique des dispositions relatives à la protection des lanceurs d'alerte.
I - Le texte adopté par l'Assemblée nationale
Introduit en séance à l'Assemblée nationale à l'initiative du Gouvernement, le présent article ajoute un titre VI intitulé « Protection des personnes » au sein du livre IV de la première partie du code de la santé publique.
Il s'agit de créer des dispositions protectrices pour les lanceurs d'alerte, c'est-à-dire pour les personnes qui relatent ou témoignent de bonne foi, à leur employeur ou aux autorités judiciaires ou administratives, de faits qui pourraient porter atteinte à la sécurité sanitaire des produits de santé dont ils auraient eu connaissance dans l'exercice de leurs fonctions.
L'article s'applique pour l'ensemble des produits mentionnés entrant dans le champ de compétence de l'agence.
Il est précisé que le lanceur d'alerte :
- ne peut faire l'objet d'une mesure discriminatoire, être écarté d'une procédure de recrutement ou de l'accès à un stage ou à une période de formation professionnelle ;
- il ne peut pas non plus être sanctionné ou faire l'objet d'une mesure discriminatoire, directe ou indirecte, notamment en matière de rémunération, de traitement, de formation, de reclassement, d'affectation, de qualification, de classification, de promotion professionnelle, de mutation ou de renouvellement de contrat
En cas de litige entre le lanceur d'alerte et la partie défenderesse, la charge de la preuve reposera sur cette dernière.
Des dispositions semblables sont prévues notamment dans le code du travail en matière de corruption 43 ( * ) ou dans le code de l'action sociale et des familles concernant le signalement des cas de mauvais traitements ou privations constatés dans les établissements médico-sociaux 44 ( * ) .
II - Le texte adopté par la commission
Sur amendement proposé par son rapporteur, la commission a déplacé ces dispositions dans la partie du code de la santé publique relative aux prérogatives de l'actuelle Afssaps. Outre une plus grande cohérence du code, ce changement a le mérite d'éviter la création d'un nouveau titre dont l'intitulé apparaissait peu adapté.
Elle a en outre, toujours à l'initiative de son rapporteur, apporté une amélioration rédactionnelle en substituant au terme « attentatoire » les mots « susceptibles de porter atteinte ».
Elle a adopté cet article ainsi modifié.
Article 33 (art. L. 5134-1 du code de la santé publique) Prescription de contraceptifs par les sages-femmes et délivrance de médicaments contraceptifs dans les services de médecine de prévention des universités
Objet : Cet article, introduit par l'Assemblée nationale, modifie le régime juridique applicable à la prescription de contraceptifs par les sages-femmes et à la délivrance et à l'administration de médicaments ayant pour but la contraception dans les services de médecine de prévention des universités.
I - Le dispositif proposé par l'Assemblée nationale
Le présent article supprime toute forme de tutelle du médecin traitant sur les sages-femmes lorsqu'elles délivrent des contraceptifs locaux et hormonaux. Est en conséquence abrogée la deuxième phrase du premier alinéa du paragraphe III de l'article L. 5134-1 du code de la santé publique qui dispose que la surveillance et le suivi biologique sont assurés par le médecin traitant.
Par ailleurs, il modifie les règles applicables dans les services de médecine de prévention des universités. Le même article L. 5134-1 prévoit actuellement que peuvent être délivrés des médicaments ayant pour but la contraception et notamment la contraception d'urgence. Le champ est désormais limité aux seuls cas d'urgence. En contrepartie, il est proposé que les infirmiers qui exercent dans ces services puissent procéder à la délivrance et à l'administration de ces médicaments. L'intervention des infirmiers est déjà prévue au I de cet article qui leur permet, dans les établissements d'enseignement du second degré, lorsqu'un médecin, une sage-femme ou un centre de planification ou d'éducation familiale n'est pas immédiatement accessible, à titre exceptionnel et en application d'un protocole national déterminé par décret, dans les cas d'urgence et de détresse caractérisés, d'administrer aux élèves une contraception d'urgence.
II - Le texte adopté par la commission
La commission rappelle que ces dispositions, déjà adoptées par le Parlement lors de l'examen de la proposition de loi devenue la loi n° 2011-940 du 10 août 2011 modifiant certaines dispositions de la loi HPST, ont été censurées par le Conseil constitutionnel en tant que cavalier législatif, en dépit du lien manifeste qu'elles entretenaient avec l'objet de ce texte.
Sans sous-estimer le risque de voir le même sort réservé à cet article, mais compte tenu de l'utilité des mesures ici introduites, la commission a adopté cet article sans modification.
Article 34 (art. L. 713-7 du code de la propriété intellectuelle) Contrôle des exportations parallèles de médicaments
Objet : Cet article, inséré par l'Assemblée nationale, tend à encadrer les exportations parallèles de médicaments.
I - Le dispositif proposé par l'Assemblée nationale
Cet article reprend des dispositions précédemment insérées par l'Assemblée nationale dans le projet de loi de financement de la sécurité sociale pour 2010 mais ensuite invalidées par le Conseil constitutionnel en tant que cavalier 45 ( * ) . Il vise à encadrer les exportations parallèles de médicaments.
Ces exportations sont le fait de distributeurs qui tirent parti d'un prix fabricant inférieur en France à celui pratiqué dans certains pays étrangers. Afin de les limiter, les laboratoires imposent aux distributeurs des quotas, qui peuvent avoir pour effet d'entraîner des difficultés d'approvisionnement des officines.
Pour décourager cette pratique, cet article fait obligation aux distributeurs qui recourent à des exportations parallèles de déclarer leur activité, ce qui aura pour effet de soustraire les quantités ainsi vendues ou revendues à l'étranger de l'assiette de la taxe annuelle perçue au profit de l'Afssaps (article L. 5127-17 du code de la santé publique) et de celle de la contribution sur le chiffre d'affaires des entreprises pharmaceutiques (article L. 245-6 du code de la sécurité sociale), qui sont assises sur les ventes en France. Parallèlement, une modification apportée à l'article L. 5123-1 du code de la santé publique permet aux entreprises pharmaceutiques de fixer librement leurs prix à l'exportation.
II - Le texte adopté par la commission
Votre commission est sceptique quant à l'efficacité du dispositif proposé par cet article pour limiter les exportations parallèles de médicaments. Le Gouvernement ayant indiqué s'être engagé dans une démarche de négociations qui devrait aboutir à de meilleurs résultats, la commission a , à l'initiative de son rapporteur, supprimé cet article .
*
* *
Réunie le mercredi 19 octobre 2011, la commission a adopté le texte du projet de loi dans la rédaction résultant de ses travaux.
TRAVAUX DE LA COMMISSION
Réunie le mercredi 19 octobre 2011 , sous la présidence d' Annie David, présidente, la commission examine le rapport de Bernard Cazeau sur le projet de loi n° 5 (2011-2012) relatif au renforcement de la « Sécurité sanitaire du médicament et des produits de santé ».
Bernard Cazeau , rapporteur . - Notre mission commune d'information a intitulé son rapport « La réforme du médicament, enfin » pour regretter que les préconisations, formulées par le Sénat dès 2006 en matière de sécurité sanitaire, n'aient pas été prises au sérieux avant l'affaire du Mediator. La majorité de l'époque n'avait pas jugé bon d'ajouter un point d'interrogation. Pourtant, à considérer le projet de loi qui nous est soumis, nos doutes quant à la volonté du Gouvernement de mener une réforme ambitieuse étaient pleinement justifiés. D'abord, la rédaction imprécise, le renvoi fréquent à des textes réglementaires et la transposition d'une directive communautaire ne facilitent pas la compréhension de ce texte. Ensuite, il ne concerne qu'une partie du cadre législatif du médicament, sans apporter de véritables innovations.
Le titre premier du projet de loi, « Transparence des liens d'intérêt », recoupe, en partie, le projet de loi « Sauvé ». Ce dernier texte, qui porte le nom du président de la commission chargée d'élaborer un rapport sur la prévention des conflits d'intérêts, a été déposé quatre jours avant le nôtre. Cette dichotomie, qui se justifierait par la spécificité du secteur sanitaire, est gênante. De fait, le projet de loi « Sauvé » vise les directeurs d'hôpitaux ou les praticiens hospitaliers. Surtout, il prévoit la création d'une autorité de la déontologie de la vie publique qui couvrira aussi le secteur sanitaire.
L'article 1 er impose une déclaration publique d'intérêts aux membres des organes consultatifs placés auprès des ministres, aux dirigeants et aux membres des organes collégiaux de tous les organismes compétents en matière de sécurité sanitaire ou de produits de santé. Cette disposition n'ajoute presque rien au droit en vigueur puisque la plupart des autorités sanitaires pratiquaient déjà un régime de déclaration d'intérêts. Qui plus est, ce regroupement, dont on attend plus de lisibilité, peut aussi bien produire un effet inverse à celui recherché étant donné, entre autres, la diversité des organismes visés, des comités de protection des personnes, à l'institut de radioprotection et de sécurité nucléaire en passant par l'agence française de sécurité sanitaire des produits de santé (Afssaps).
S'il est difficile de remédier à tous ces inconvénients dans les délais impartis, il faut, au moins, revoir la définition des liens d'intérêts, prévoir la déclaration des liens noués au cours des cinq années précédentes et interdire tout lien d'intérêts pour le président de la Haute Autorité de santé (HAS) et le directeur général de l'Afssaps. Je propose également d'étendre les compétences de la commission de déontologie créée par la loi Sapin aux personnels des conseils et agences sanitaires sans attendre qu'elle devienne l'autorité de déontologie de la vie publique prévue le projet de loi Sauvé. A mon sens, celle-ci doit, dès à présent, exercer un rôle de conseil. J'aurais voulu faire davantage, mais nous sommes tenus par l'article 40 de la Constitution...
S'agissant de la publicité des débats des instances d'expertise, mieux vaut prévoir des procès-verbaux précisant le détail des votes et des opinions dissidentes diffusés en ligne, en plus des enregistrements audiovisuels qui s'avèrent difficiles à visionner dès que la réunion dure un peu. Afin d'éviter une invocation abusive des secrets protégés par la loi, confions à la commission d'accès aux documents administratifs (Cada) le soin de contrôler l'application de ces dispositions.
Pourquoi renvoyer à une charte approuvée par décret l'encadrement de l'expertise sanitaire ? Outre que la définition des notions de lien d'intérêts et de conflit d'intérêts doit relever du législateur, la mesure est dépourvue de portée normative. Supprimons-la.
Enfin, pour prévenir tout biais intellectuel et le risque avéré de pensée de groupe, au-delà même des conflits d'intérêts, je souhaite ouvrir les instances d'expertise à des médecins généralistes et à des experts en sciences humaines.
L'article 2 traite du Sunshine Act à la française. En fait de sunshine, il s'agit plutôt de sunset ! Des modifications substantielles sont nécessaires. L'innovation consistant à autoriser les étudiants se destinant aux professions de santé à passer des conventions avec les laboratoires ou à les faire bénéficier de prestations d'hospitalité n'est pas souhaitable. Je proposerai que toute relation avec les entreprises passe par les organismes de formation.
Enfin, l'article 3 prévoit des dispositions pénales pour non-respect des obligations énoncées aux articles 1 er et 2.
Nous en arrivons au titre II, consacré à l'Afssaps. Aux termes de l'article 4, celle-ci s'appellera désormais l'agence nationale de sécurité du médicament et des produits de santé (ANSM), un changement cosmétique qui pose difficulté. Elle pourra désormais demander des essais contre comparateurs actifs et contre placebo, prononcer des sanctions financières et disposera d'un droit d'accès aux informations qu'elle juge « nécessaires à sa mission ».
L'article 5 prévoit les règles de publicité du travail de l'agence et élargit son conseil d'administration aux acteurs du système de santé et aux parlementaires - l'Assemblée nationale a voulu trois sénateurs et trois députés. Cette réforme est intéressante sous réserve que le conseil d'administration puisse réellement suivre l'activité de police sanitaire et que l'on garantisse l'indépendance des associations représentées en son sein.
L'article 5 bis, introduit par l'Assemblée nationale, confie à la HAS le soin de créer un portail public sur les maladies et leur traitement. Le but est d'apporter une information gratuite et de qualité face aux forums qui se multiplient sur internet. Pour prévenir son détournement, je propose que nous ajoutions une information sur le bon usage des produits de santé.
Le titre III procède à la transposition de la directive européenne de décembre 2010 relative à la pharmacovigilance. Les articles 6 à 9 entendent renforcer la mission de pharmacovigilance de la nouvelle agence, mais seulement après l'autorisation de mise sur le marché. Impossible de réformer la mise sur le marché quand celle-ci dépend à 80 % du droit communautaire : toute mesure contraire aux normes européennes serait automatiquement écartée par le juge. Le ministre de la santé s'est engagé à faire évoluer la législation communautaire ; nous en rediscuterons en séance.
En revanche, un dispositif très intéressant a été inséré à l'initiative des députés socialistes : l'article 9 bis, qui impose les essais comparatifs, non pour la mise sur le marché puisque cela est impossible, mais pour le remboursement des médicaments. Sachant qu'un médicament non remboursé intéresse moins les laboratoires, cet article est fondamental ; reste à renforcer sa rigueur juridique.
Les articles 10 à 13 encadrent les conditions de prescription. J'insisterai sur l'article 11 qui autorise l'agence à proposer des recommandations temporaires d'utilisation (RTU) lorsque cela est utile pour la santé publique. Les prescriptions hors du cadre de l'autorisation de mise sur le marché sont nécessaires et répandues. Prenons un des cas les plus connus : l'aspirine, qui est indiqué comme antalgique, est prescrit dans le traitement des maladies cardio-vasculaires. En revanche, le patient doit savoir si le médicament lui sera ou non remboursé ; cette information doit lui être délivrée.
Les articles 14 à 16 concernent l'accès au médicament et, surtout, les autorisations temporaires d'utilisation (ATU). Contrairement aux RTU, celles-ci consistent à autoriser un patient à prendre un traitement avant son autorisation formelle : ce fut le cas, en son temps, de la trithérapie. Mais cette procédure dérogatoire ne doit pas être l'occasion pour les laboratoires de contourner les procédures. Les évolutions contenues dans le texte de loi sont perfectibles ; nous devrons notamment débattre de la question du prix des médicaments sous le régime d'ATU.
L'article 19 invente la visite médicale collective dans les établissements de santé. Cette innovation ne me semble pas très réaliste : comment réunir des médecins en amphithéâtre pour une présentation groupée des produits ? Ce système ne fonctionnera jamais dans les petits hôpitaux. De plus, les députés ont restreint la portée du texte en excluant certains produits du champ de la visite collective. Revenons sur ces dispositions et confions à la HAS l'évaluation de ce dispositif expérimental. En d'autres termes, nous n'avons pas résolu le problème de l'encadrement de la visite médicale...
Enfin, l'article 21 rend obligatoire la certification des logiciels d'aide à la prescription et à la délivrance de médicaments. Cela va dans le bon sens : ces logiciels qu'utilisent les médecins sont privés, et parfois créés par des laboratoires qui en profitent pour se mettre en avant. C'est la loi du commerce !
Le titre IV aligne, dans la mesure du possible, le régime juridique des dispositifs médicaux sur celui des médicaments. Par dispositif médical, on entend les prothèses de hanche, les pacemakers, les lunettes, les appareils auditifs ou encore les fauteuils roulants. Je garde en mémoire cet avertissement qu'on m'a lancé en audition : « le prochain Mediator sera un dispositif médical ». Je vous soumettrai donc un amendement demandant à la HAS de dresser le bilan sur leur encadrement.
Enfin les articles 27 à 34 sont des dispositions hétérogènes : habilitation du Gouvernement à prendre des ordonnances, dates d'entrée en vigueur, protection des lanceurs d'alerte, insertion de dispositions précédemment censurées par le Conseil constitutionnel. Je proposerai d'y voir plus clair dans cet ensemble hétéroclite.
Pour conclure, la réforme du système de sécurité sanitaire n'a que trop tardé. Ce projet de loi en propose une version a minima, qui nous interdit de créer un véritable statut de l'expertise publique en raison de l'article 40. Néanmoins, nous pouvons faire preuve d'une attitude plutôt positive à l'égard de ce texte que nous chercherons à améliorer, après les députés. Considérons-le comme une première étape vers un système de sécurité sanitaire qui réponde, enfin, à l'attente des Français !
Jacky Le Menn . - Je félicite Bernard Cazeau pour ce premier rapport. Ce texte, s'il comporte des avancées, est effectivement en retrait par rapport aux conclusions de notre mission sur le Mediator. Il n'apporte pas de remèdes aux dysfonctionnements du système du médicament dont nous avons eu vent tout au long des auditions. Certes, nous sommes tenus par le droit communautaire. Mais que cela ne nous empêche pas d'être ambitieux ! Le Gouvernement s'est montré trop timide concernant la mise sur le marché alors qu'il est possible, comme le montre l'article 9 bis, de jongler avec les règles européennes. Avec les améliorations apportées à l'Assemblée nationale et les amendements du rapporteur, nous pouvons écrire un bon texte. Quel dommage que le Gouvernement n'ait pas tenu compte du rapport de notre mission ! Il y avait matière dans ce texte, adopté à l'unanimité, à nourrir substantiellement ce projet de loi.
Isabelle Pasquet . - A mon tour de féliciter Bernard Cazeau pour ce premier rapport sur un sujet assez technique. Après le scandale du bisphénol A, du Vioxx et du Mediator, les parlementaires investissent de plus en plus le champ de la sécurité sanitaire jusqu'alors réservé aux experts. Ce projet de loi peut-il prévenir un nouveau scandale ? Malgré les améliorations apportées par les députés, le texte reste insuffisant. L'importance de la question des conflits d'intérêts, après les affaires du Mediator et celle du vaccin de la grippe A, n'est plus à démontrer. Or, la déclaration publique d'intérêts ne suffira pas à la résoudre. Il faut interdire à toute personne entretenant un lien d'intérêts, direct ou indirect, avec l'industrie pharmaceutique de participer aux décisions. Mettons fin aux cadeaux et prestations d'hospitalité. S'agissant des essais comparatifs, le texte les rend obligatoires pour l'obtention du remboursement. Je m'en réjouis : les cotisations sociales n'ont pas à subventionner l'industrie pharmaceutique mais à financer les seuls produits dans l'intérêt réel du patient. Enfin, le débat sur la visite médicale n'est pas clos.
Le groupe CRC, vous l'aurez compris, est très réservé sur ce texte. Pour autant, nous présenterons des amendements dans le but d'apporter plus de transparence et de renforcer la sécurité sanitaire.
Bruno Gilles . - Pour avoir été visiteur médical en hôpital et en ville, puis responsable régional durant une vingtaine d'années, je connais bien le secteur. L'article 19, au-delà des problèmes qu'il pose pour l'emploi - la France compte 18 000 visiteurs, dont 4 000 visiteurs hospitaliers -, est techniquement inapplicable : imagine-t-on que l'on va réunir des médecins de différentes spécialités dans un même amphithéâtre pour une présentation groupée des produits ? Trouvons une porte de sortie : oui à l'encadrement, car il ne faut pas laisser faire tout et n'importe quoi, mais ne supprimons pas la profession. A mon sens, il faut au moins conserver le texte de l'Assemblée nationale et prévoir de maintenir la visite individuelle pour les médicaments de réserve hospitalière, les prescriptions hospitalières et les prescriptions d'initiative hospitalière.
Jean-Louis Lorrain . - Monsieur le rapporteur, je m'attendais au pire ! Malgré quelques adjectifs qualificatifs tels que le mot « hétéroclite » qui marque votre appartenance à l'opposition, votre rapport est intéressant. Nous y apporterons une réponse argumentée en séance.
En attendant, je m'interroge. Toujours plus de transparence ? A force, on finit par ne plus voir la vitre et s'y heurter... Le Sunshine Act est une dynamique tout à fait respectable : les Etats-Unis, mais aussi les Anglais et les Espagnols l'ont adoptée.
La nouvelle agence relèverait seulement de la cosmétique ? Son directeur général et les techniciens ont l'enthousiasme, la volonté d'agir, ils veulent bien faire, ils ont envie de se servir de la loi pour modifier les comportements !
Avec la transposition de la directive de décembre 2010, nous avons l'occasion de revoir notre rapport à l'Europe. Notre système de pharmacovigilance était certes tardif, mais très performant. En revanche, notre système de pharmaco-épidémiologie était très pauvre. D'une manière générale, il faut davantage de coordination, de dialogue entre les différents acteurs du système du médicament. C'est un élément essentiel pour renforcer la sécurité sanitaire.
Enfin, la visite médicale collective n'est effectivement peut-être pas adaptée. S'agissant de la publicité des travaux de l'agence, que faire lorsqu'elle croule sous les dossiers ? Les dispositifs médicaux sont une question importante : évitons des dérives telles qu'un joueur de tennis vantant les mérites d'une prothèse de genou...
Pour conclure, ce texte, parce qu'il est technique, nécessite des renvois au décret et au règlement. Je note que les députés socialistes ne l'ont pas vu d'un si mauvais oeil. Comme vous, j'ai en revanche des doutes sur les ajouts de dernière minute à l'Assemblée nationale.
Jean-Marie Vanlerenberghe . - Monsieur le rapporteur, j'approuve vos réserves. On regrette souvent le manque de réactivité du Gouvernement ; en l'occurrence, nous aurions préféré qu'il soit moins rapide et tienne compte des soixante-cinq préconisations de notre mission sur le Mediator. Quid de la place et de l'information du ministre ? Quid de l'interdiction totale des liens d'intérêts pour les présidents de commission et l'encadrement des instances d'expertise ? Concernant la mise sur le marché, soyons clairs : il faut contourner la directive européenne. Autre réforme : la formation du prix du médicament si peu lisible, si complexe actuellement. Le poids des laboratoires dans la formation continue des médecins n'est pas abordé dans le rapport ; peut-être faudrait-il y remédier. Si la visite collective paraît singulière, la supprimer reviendrait à renoncer à tout encadrement. Trouvons la bonne formule. Enfin, mettons l'assurance maladie, qui dispose aujourd'hui de capacités de détection, au coeur du système de pharmacovigilance. Nous sommes plutôt favorables à ce texte. A nous de réparer ses insuffisances.
Gilbert Barbier . - Merci de ce rapport. A ma grande surprise, vous êtes finalement plutôt favorable à ce texte qui ne tient pas assez compte, j'en conviens, des conclusions de notre mission.
Je souligne la multiplicité des intervenants, entre l'Afssaps, la commission de la transparence et le comité économique des produits de santé (Ceps). L'autonomie de ce dernier nous ayant étonnés, je souhaite qu'il soit réformé.
Il me semble bon que cette agence soit financée non via l'AMM, mais grâce à une taxe pesant sur les laboratoires, même s'ils n'en sont pas d'accord...
A propos des experts, quel crédit pourrait-on accorder à une personne n'ayant jamais expérimenté de médicament ? Il faut surtout garantir la sincérité des déclarations.
Sur un autre plan, je ne vois pas l'intérêt de faire siéger des parlementaires au conseil d'administration.
En définitive, la transparence accrue est une bonne chose mais je regrette le caractère précipité du débat parlementaire.
Ronan Kerdraon . - Notre rapporteur mérite l'hommage qui lui a été rendu ce matin car sa tâche n'était pas facile.
Pour rétablir la confiance ébranlée des patients potentiels, il fallait certes tirer les leçons du Mediator et du Vioxx, mais la réactivité quelque peu excessive face au Mediator explique les insuffisances du texte, qui a cependant le mérite d'exister.
Il me semble nécessaire de conduire une réflexion approfondie sur le rôle des 18 000 à 20 000 visiteurs médicaux, qui ont pour mission de commercialiser un produit et vanter ses vertus thérapeutiques. Nous devons également nous pencher sur les progrès thérapeutiques procurés par les nouveaux médicaments. Enfin, notre réflexion devra inclure la pharmacovigilance.
Annie David , présidente . - J'ajoute mes félicitations au rapporteur pour la rapidité de son travail. Le texte dont nous discutons est attendu par de nombreuses personnes que l'affaire du Mediator a ébranlées. Il est nécessaire qu'une loi éclaire le monde obscur du médicament.
La transparence est nécessaire, tout comme l'élimination des conflits d'intérêts. Il faudra débattre de la question des visiteurs médicaux. En ce domaine, le dispositif proposé semble complexe. Faut-il conserver cette profession ? Pourquoi pas. Mais comment éviter alors un lobbying en faveur de médicaments au service rendu insuffisant ?
Alain Milon . - Qu'est-ce qu'un service médical rendu suffisant ?
Bernard Cazeau , rapporteur . - Le défi à relever tient en peu de mots : les délais sont brefs, les auditions sont donc limitées, mais nous avons beaucoup travaillé. Nombre de mes amendements répondent à vos interrogations.
Après celui de l'Igas, le rapport sénatorial a proposé de mettre fin aux visiteurs médicaux. L'effectif de cette profession m'avait conduit à formuler des réserves, mais il reste à trouver une solution. Le ministre n'en a pas ; celles auxquelles je pense ne sont pas encore parvenues à maturité.
Monsieur Lorrain, je ne suis là ni pour défendre le Gouvernement, ni pour établir un rapport dénué de fondement, mais je ne peux que constater que les mesures finales du texte sont hétéroclites, d'autant que ce projet de loi en recouvre partiellement un autre qui viendra ultérieurement. Changer le nom de l'Afssaps est une mesure cosmétique, à moins qu'elle ne soit purement psychologique, mais nos concitoyens attendent surtout des changements d'attitude et de comportements.
Monsieur Vanlerenberghe, il me semble que lorsqu'il a élaboré son projet, le ministre a négligé les préconisations de notre rapport sénatorial. J'ai donc repris certaines de nos suggestions dans mes amendements.
Monsieur Barbier, l'ancien directeur du Ceps cédait aux laboratoires dans certains domaines pour obtenir des contreparties dans d'autres, mais son successeur n'a pas la même disposition d'esprit. Je note enfin moi aussi l'enthousiasme du nouveau directeur de l'Afssaps et sa volonté de changement. Avec sa verve marseillaise, il ferait au demeurant un excellent visiteur médical.
Alain Milon . - C'est déjà un excellent médecin !
EXAMEN DES AMENDEMENTS
Article additionnel avant l'article 1 er
Bernard Cazeau , rapporteur. - L'amendement n° 1 concerne les nouvelles associations agréées de patients. Si l'on menait la logique à son terme, l'amendement tomberait sous le coup de l'article 40.
Isabelle Debré . - Il n'y a donc pas lieu de le discuter.
Annie David , présidente . - L'article 40 de me semble pas applicable puisque l'amendement ne sollicite pas explicitement les deniers publics.
Bernard Cazeau , rapporteur . - Ne nous voilons pas la face : si le financement n'est pas privé, il est public.
Jean-Louis Lorrain . - Nous nous abstiendrons sur tous les amendements du rapporteur car leur analyse et leur discussion supposent un travail de fond à conduire à tête reposée.
Alain Milon . - L'amendement n° 1 ne laisse en effet qu'une alternative : tout financement privé conduit au conflit d'intérêts ; l'appel au financement public déclenche l'article 40.
Bernard Cazeau , rapporteur . - Les laboratoires peuvent aussi financer une association en sous-main.
Laurence Cohen . - Je ne comprends pas qu'on évoque l'éventuelle application de l'article 40 contre un amendement qui reprend une conclusion figurant dans le rapport de l'Igas.
Annie David , présidente . - Dès lors qu'une suggestion de l'Igas est susceptible d'accroître la dépense publique, c'est au Gouvernement qu'il revient de la reprendre, pas au Parlement. Ceci étant, en l'espèce, je ne suis pas certaine que l'article 40 s'applique.
Isabelle Pasquet . - Nous allons retirer l'amendement aujourd'hui, quitte à le redéposer en séance.
Bernard Cazeau , rapporteur . - Amendement n° 3 : accroître les missions du service central de prévention de la corruption (SCPC) augmenterait ses charges. Mieux vaut s'en remettre à la commission de déontologie. Seriez-vous disposés à le retirer ?
Isabelle Pasquet . - Nous nous interrogeons sur l'indépendance d'une commission n'ayant rendu qu'un seul avis négatif. Par ailleurs, cette instance dispose-t-elle aujourd'hui des moyens qui font défaut au SCPC ?
L'amendement n° 3 est retiré.
Bernard Cazeau , rapporteur. - Amendement n° 5 : la meilleure solution consiste à créer un corps d'experts indépendants mais comment appliquer cet amendement du jour au lendemain?
Isabelle Pasquet . - Il faut une discussion en séance publique sur ce point.
Aline Archimbaud . - La qualité de notre travail est entravée par la brièveté des délais qui nous sont accordés. J'invite les auteurs des amendements que la commission ne peut intégrer dans son texte à les représenter en séance publique.
Isabelle Debré . - Il sera manifestement impossible d'examiner ce matin les 134 amendements déposés. Est-il prévu de tenir une autre réunion de commission ?
Annie David , présidente . - Nous prolongerons notre réunion jusqu'à 13 heures. Le cas échéant, la commission se réunira cet après-midi, voire mardi.
Isabelle Debré . - Il n'est pas possible de nous réunir mardi pour établir le texte de la commission qui sera discuté mercredi en séance publique !
Bernard Cazeau , rapporteur . - Mon amendement n° 108 clarifie la déclaration publique d'intérêts.
L'amendement n° 108 est adopté.
Bernard Cazeau , rapporteur . - L'amendement n° 8 sera satisfait par l'amendement n° 115, que je présenterai, sur la centralisation des déclarations publiques d'intérêts.
Bernard Cazeau , rapporteur . - Mon amendement n° 109 s'inspire des conclusions de la mission d'information sénatoriale sur le Mediator.
L'amendement n° 109 est adopté.
Alain Milon . - Nous sommes hélas contraints de partir. Au demeurant, nous vous avons indiqué que les membres de mon groupe s'abstenaient sur l'ensemble des amendements.
L'amendement rédactionnel n° 110 est adopté.
Bernard Cazeau , rapporteur. - Amendement n° 15 : je suis très partagé. Les groupes de travail sont caractérisés par des discussions très libres. La publicité des débats pourrait porter atteint à cette spontanéité. Mieux vaut retirer cet amendement.
Isabelle Pasquet . - Soit, mais nous le redéposerons en séance car il aborde un vrai sujet.
Dominique Watrin . - L'amendement est justifié par le fait que les groupes décisionnels se réfèrent aux groupes de travail.
Christiane Demontès . - Je partage la crainte formulée par le rapporteur car les groupes de travail débroussaillent les dossiers.
Annie David , présidente. - Il faudrait peut-être en modifier la rédaction, pour viser les instances décisionnelles.
L'amendement n° 15 est retiré.
Bernard Cazeau , rapporteur. - Amendement n° 12 : ce dispositif est large au point de rendre publiques même des décisions purement administratives. Ce ne serait guère conciliable avec le fonctionnement normal de l'institution.
L'amendement n° 12 est retiré.
Bernard Cazeau , rapporteur. - L'amendement n° 10 est satisfait par le n° 110. En outre, la notion de confidentialité commerciale est trop floue. Enfin, il est inutile de mentionner le respect des secrets protégés, qui s'impose déjà.
L'amendement n° 10 est retiré.
L'amendement rédactionnel n° 111 est adopté.
L'amendement n° 18 est retiré.
Bernard Cazeau , rapporteur. - L'alinéa 17, que l'amendement n° 112 tend à supprimer, insère dans le code de la santé publique un article L. 1451-4 renvoyant à un décret en Conseil d'Etat la fixation des conditions dans lesquelles une commission éthique mise en place dans chaque agence contrôlerait la véracité des informations délivrées dans la déclaration d'intérêt.
Outre qu'il est très mal rédigé, cet alinéa souffre d'une faible portée juridique. Il n'est pas de nature législative. Enfin, la commission éthique instituée n'a pas les caractéristiques de la « cellule de veille déontologique » dont la mission sénatoriale avait préconisé la création dans les instances d'expertise.
Par ailleurs, les organismes du secteur sanitaire ne pouvant pas tous assurer de suivi déontologique, ni contrôler les déclarations publiques d'intérêts, je proposerai par amendement qu'ils puissent s'appuyer sur la future autorité de la déontologie de la vie publique.
L'amendement n° 112 est adopté.
L'amendement n° 21 devient sans objet.
Bernard Cazeau , rapporteur . - En supprimant l'alinéa 22, l'amendement n° 113 écarte la création d'une charte de l'expertise sanitaire, disposition qui s'analyse comme un catalogue d'intentions destiné à masquer l'absence d'expertise indépendante.
L'amendement n° 113 est adopté.
En conséquence, l'amendement n° 25 devient sans objet.
Bernard Cazeau , rapporteur. - L'amendement n° 28 me paraît irréaliste.
Isabelle Pasquet . - Nous le redéposerons en séance.
L'amendement n° 28 est retiré.
Bernard Cazeau , rapporteur . - Amendement n° 114 : la commission d'enquête sénatoriale souhaitait ouvrir l'expertise sanitaire aux médecins généralistes et à des représentants des sciences humaines.
Christiane Demontès . - Il est bon que ce ne soient pas toujours les mêmes qui décident en vase clos.
L'amendement n° 114 est adopté.
Bernard Cazeau , rapporteur . - L'amendement n° 115 précise les compétences dévolues à la nouvelle autorité de la déontologie de vie publique, qui doit succéder à la commission de déontologie créée par la loi Sapin du 29 juin 1993.
Je propose en outre que les déclarations publiques d'intérêts soient communiquées à cette nouvelle autorité, dont le rôle pédagogique pourrait être joué dès à présent par l'actuelle commission de déontologie.
L'amendement n° 115 est adopté.
Bernard Cazeau , rapporteur . - Amendement n° 116 : l'intervention de la commission d'accès aux documents administratifs (Cada) permettrait de concilier la transparence des débats et le respect des secrets protégés par la loi.
L'amendement n° 116 est adopté.
Bernard Cazeau , rapporteur . - Je propose de rectifier l'amendement n° 109 en y intégrant le président de l'institut national du cancer et le directeur général de l'Inserm visés par l'amendement n° 31.
L'amendement n° 31 rectifié est adopté.
L'amendement n° 109 rectifié est adopté.
La commission adopte l'article 1 er dans la rédaction issue de ses travaux.
Article additionnel après l'article 1 er
Bernard Cazeau , rapporteur . - Amendement n° 34 : je vois bien l'intérêt d'un appel à candidatures mais le dispositif proposé est complexe. Au demeurant, l'amendement est déjà satisfait pour ce qui est du président de la HAS.
Christiane Demontès . - Notre groupe est plutôt favorable à la transparence. Je propose de rectifier l'amendement pour supprimer la première partie concernant la HAS et conserver la seconde.
Bernard Cazeau , président . - Le président de la HAS préside aussi ses instances internes, qu'il s'agisse du conseil d'administration ou du conseil scientifique. Ces larges compétences ont pour but de rendre le poste attractif pour des médecins.
Annie David , présidente - Je propose d'adopter l'amendement, quitte à le modifier, voire à le retirer, en séance.
Bernard Cazeau , rapporteur . - Le débat sera identique.
Christiane Demontès . - Non car le ministre répondra.
Bernard Cazeau , rapporteur . - Pourquoi ne pas le présenter comme amendement extérieur ?
Annie David , présidente . - Autant l'inclure dans le texte dès lors que la commission y est favorable.
L'amendement n° 34 rectifié est adopté et devient article additionnel.
Bernard Cazeau , rapporteur. - A l'instar du « sunshine act », l'amendement n° 117 fixe à un an la périodicité des informations sur les liens d'intérêts, en précisant que les conventions devront être publiées.
L'amendement n° 117 est adopté.
Bernard Cazeau , rapporteur . - Amendement n° 118 : pour éviter toute ambiguïté, il faut que le financement des activités de recherche dans la préparation d'un diplôme passent par les organismes de formation.
L'amendement n° 118 est adopté.
L'amendement rédactionnel n° 119 est adopté.
L'amendement n° 37, auquel le rapporteur a donné un avis favorable, est adopté.
Bernard Cazeau , rapporteur . - Amendement n° 120 : pour ne pas neutraliser le « sunshine act » à la française, il importe de connaître les rémunérations versées.
L'amendement n° 120 est adopté.
L'amendement n° 39, auquel le rapporteur a donné un avis favorable, est adopté.
Bernard Cazeau , rapporteur . - Amendement n° 121 : les liens d'intérêts doivent être consultables sur internet.
L'amendement n° 121 est adopté.
L'amendement n° 40 est retiré.
Bernard Cazeau , rapporteur . - Amendement n° 43 : nous retrouvons la multiplication des tâches confiées au SCPC. Avis défavorable
L'amendement n° 43 est retiré.
Bernard Cazeau , rapporteur . - Amendement n° 72 : je ne suis pas sûr qu'il faille multiplier les contrôles. Avis défavorable.
L'amendement n° 72 est rejeté.
Bernard Cazeau , rapporteur . - Amendement n° 122 : il s'agit d'interdire aux laboratoires de distribuer directement des subventions aux étudiants en médecine.
L'amendement n° 122 est adopté.
L'amendement n° 45 est retiré.
Bernard Cazeau , rapporteur . - Amendement n° 73 : je suis réservé sur l'intervention des instances ordinales.
L'amendement n° 73 est rejeté.
Bernard Cazeau , rapporteur . - Je suis favorable à l'amendement n° 74 qui tend à combler une lacune du dispositif anti-cadeaux.
L'amendement n° 74 est adopté.
La commission adopte l'article 2 dans la rédaction issue de ses travaux.
L'article 2 bis est adopté sans modification.
Article 3
L'amendement de coordination n° 123 est adopté.
L'amendement n° 48, auquel le rapporteur a donné un avis favorable, est adopté.
L'amendement n° 124 est adopté.
L'amendement n° 51, auquel le rapporteur a donné un avis favorable, est adopté.
La commission adopte l'article 3 dans la rédaction issue de ses travaux.
Bernard Cazeau , rapporteur . - Amendement n° 125 : quitte à changer la dénomination de l'Afssaps, choisissons un intitulé simple et qui indique que le médicament est un produit de santé.
L'amendement n° 125 est adopté.
Bernard Cazeau , rapporteur . - Amendement n° 4 : il me semble que le financement de l'agence doit être traité dans la loi de financement de la sécurité sociale.
Aline Archimbaud . - Mais nous pouvons prendre dès aujourd'hui une position de principe sur ses sources de financement.
Bernard Cazeau , rapporteur . - L'utilisation de l'adverbe « intégralement » interdit les dons et legs. Je crains que cette formulation ne soit pas la plus opérationnelle.
Aline Archimbaud . - Elle traite néanmoins d'une question de fond.
Annie David , présidente . - Je vous propose de retirer cet amendement et d'en revoir la rédaction d'ici la séance publique.
Bernard Cazeau , rapporteur . - Le dispositif de l'amendement n° 6 est flou. Il serait préférable de le retirer et d'aborder à nouveau la question en séance.
Isabelle Pasquet . - Soit.
Bernard Cazeau , rapporteur. - Amendement n° 126 : il faut garantir que la future agence pourra demander la réalisation d'essais cliniques avec des comparateurs actifs et pas uniquement avec des placebos.
L'amendement n° 126 est adopté.
Bernard Cazeau , rapporteur . - Amendement n° 54 : on ne peut accepter cette modification des AMM, dont 80 % relèvent du niveau européen. En revanche, nous pourrons demander au ministre d'intervenir pour renforcer le cadre communautaire.
L'amendement n° 54 est retiré.
Bernard Cazeau , rapporteur . - L'amendement n° 58 est satisfait.
L'amendement n° 58 devient sans objet.
Bernard Cazeau , rapporteur . - L'amendement n° 60 pose des difficultés rédactionnelles.
L'amendement n° 60 est retiré.
Bernard Cazeau , rapporteur . - Amendement n° 62 : mieux vaut supprimer le I de l'article 13 que l'alinéa 41, qui sanctionne le non-respect des conventions.
L'amendement n° 62 est retiré.
L'amendement de coordination n° 92 est adopté.
Bernard Cazeau , rapporteur . - Amendement n° 64 : je comprends la préoccupation de ses auteurs mais la réforme du code pénal intervenue en 1992 exclut les seules peines minimales.
Dominique Watrin . - Il existe pourtant bien des peines plancher ! Ici, la pénalité financière serait au minimum fixée à 10 % du chiffre d'affaires.
Bernard Cazeau , rapporteur . - La règle veut que le juge fixe une amende dans la limite d'un maximum légal, soit 500 000 euros dans le cas présent.
Aline Archimbaud . - Je ne vois pas où se situe l'impossibilité juridique d'établir une peine plancher égale à 10 % du chiffre d'affaires.
Annie David , présidente . - On peut en effet concevoir que le juge fixera l'amende entre 10 % du chiffre d'affaires au minimum et 500 000 euros au maximum.
Christiane Demontès . - Concrètement, si le chiffre d'affaires du laboratoire atteint 6 millions d'euros, l'amende sera de 500 000 euros.
Bernard Cazeau , rapporteur . - Mais l'amendement n° 64 se borne à fixer une peine minimale. L'absence de maximum serait source d'inconstitutionnalité.
Aline Archimbaud . - D'où vient la limite de 500 000 euros ?
Annie David , présidente . - Elle figure dans la version initiale du texte.
Bernard Cazeau , rapporteur . - Peut-être peut-on imaginer de conserver la borne de 500 000 euros, tout en introduisant un minimum égal à 10 % du chiffre d'affaires.
Annie David , présidente . - On peut aussi augmenter le plafond si on le juge insuffisant.
Christiane Demontès . - Pourquoi ne pas le porter à un million d'euros ?
Bernard Cazeau , rapporteur . - Parce que cette somme serait fixée au hasard.
Christiane Demontès . - La limite de 500 000 euros ne l'est-elle pas tout autant ?
Annie David , présidente . - Si le Gouvernement veut rétablir le plafond de 500 000 euros, il devra le justifier. En attendant, je vous propose de rectifier l'amendement pour conserver le plancher et porter le plafond à un million d'euros.
Christiane Demontès . - Et en plus, nous fournissons des recettes supplémentaires au Gouvernement !
L'amendement n° 64 rectifié est adopté.
La commission adopte l'article 4 dans la rédaction issue de ses travaux.
Article additionnel après l'article 4
L'amendement n° 9 est adopté et devient article additionnel.
L'article 4 bis est adopté sans modification.
Bernard Cazeau , rapporteur . - Amendement n° 13 : le conseil d'administration de la nouvelle agence comprendra un représentant de l'assurance maladie parce que c'est la Cnam qui détient l'ensemble des données permettant les études de pharmaco-épidémiologie. Cet argument justifierait le retrait de l'amendement.
L'amendement n° 13 est retiré.
Bernard Cazeau , rapporteur . - Même raisonnement pour l'amendement n° 16.
L'amendement n° 16 est retiré.
Bernard Cazeau , rapporteur . - Amendement n° 82 : pour garantir la neutralité du futur conseil d'administration de l'agence, seuls y siégeront les représentants d'associations d'usagers ne recevant aucun avantage de l'industrie pharmaceutique.
L'amendement n° 82 est adopté.
L'amendement n° 19, auquel le rapporteur a donné un avis favorable, est adopté.
Bernard Cazeau , rapporteur . - Amendement n° 22 : favorable sur le fond, mais je ne peux approuver sa rédaction.
L'amendement n° 22 est retiré.
Bernard Cazeau , rapporteur . - Grâce à l'amendement n° 83, le conseil d'administration aura un droit de regard sur le programme de travail de l'agence.
L'amendement n° 83 est adopté.
L'amendement rédactionnel n° 127 est adopté.
Bernard Cazeau , rapporteur . - L'amendement n° 24 est satisfait par l'amendement n° 110 à l'article 1 er .
L'amendement n° 24 devient sans objet.
Bernard Cazeau , rapporteur . - L'amendement n° 33 est satisfait par l'amendement n° 127.
L'amendement n° 33 devient sans objet.
L'amendement n° 30 est retiré.
Bernard Cazeau , rapporteur . - L'amendement n° 27 est peu précis.
Annie David , présidente . - Pourtant, il précise le dispositif !
Bernard Cazeau , rapporteur . - La référence à l'article L. 5311-1, relatif aux missions de l'agence, est trop vague.
L'amendement n° 27 est retiré.
La commission adopte l'article 5 dans la rédaction issue de ses travaux.
Bernard Cazeau , rapporteur . - Amendement n° 84 : la base publique de référence constituée par la HAS permettra de favoriser le bon usage des produits de santé.
L'amendement n° 84 est adopté.
L'amendement n° 35, auquel le rapporteur a donné un avis favorable, est adopté.
La commission adopte l'article 5 bis dans la rédaction issue de ses travaux.
Bernard Cazeau , rapporteur . - Amendement n° 49 : il ne me semble pas bienvenu de remplacer la formulation de l'AMM, « autorisation de mise sur le marché », qui est connue de tous, par une « autorisation d'utilisation ».
Annie David , présidente . - Selon nous, l'intérêt du patient prime : nous voulions l'affirmer plutôt que de mettre en avant l'aspect commercial et financier qu'évoque la notion de mise sur le marché.
Bernard Cazeau , rapporteur . - C'est pourtant bien la réalité des choses que de parler de la mise du produit sur le marché.
L'amendement n° 49 est retiré.
Bernard Cazeau , rapporteur . - Amendement n° 47 : nous avons déjà débattu des conditions de l'AMM, dont 80 % sont délivrées au niveau européen.
L'amendement n° 47 est retiré.
Bernard Cazeau , rapporteur . - Amendement n° 53 : les dispositions du projet de loi permettent une réévaluation de l'AMM à tout moment. L'amendement comporte un alourdissement des procédures risquant d'entraver le travail de l'agence : il est inutile de renouveler tous les cinq ans l'AMM de l'aspirine. En revanche, les produits dangereux ne doivent pas échapper à tout contrôle pendant cinq années.
Dominique Watrin . - Cette proposition figurait dans notre rapport.
Bernard Cazeau , rapporteur . - Elle est quelque peu maximaliste...
Annie David , présidente . - Elle avait été adoptée à l'unanimité !
Isabelle Pasquet . - Nous représenterons cet amendement en séance.
L'amendement n° 53 est retiré.
Bernard Cazeau , rapporteur . - Même raisonnement pour l'amendement n° 56.
L'amendement n° 56 est retiré.
Bernard Cazeau , rapporteur . - Amendement n° 59 : je ne suis pas certain que l'harmonisation proposée favorise un examen plus strict du rapport bénéfices-risques.
L'amendement n° 59 est retiré.
L'amendement rédactionnel n° 128 est adopté.
Bernard Cazeau , rapporteur . - L'amendement n° 63 est satisfait.
L'amendement n° 63 devient sans objet.
Bernard Cazeau , rapporteur . - Je m'interroge sur la portée pratique de l'amendement n° 66.
L'amendement n° 66 est retiré.
Bernard Cazeau , rapporteur . - Intéressant sur le fond, l'amendement n° 68 pose des difficultés rédactionnelles.
L'amendement n° 68 est retiré.
La commission adopte l'article 6 dans la rédaction issue de ses travaux.
L'article 6 bis est adopté sans modification.
Bernard Cazeau , rapporteur . - Outre qu'il est sans doute incompatible avec le droit communautaire, l'amendement n° 69 limiterait considérablement le nombre de médicaments disponibles.
Le Ceps estime que les nouveaux médicaments n'améliorant pas le service rendu sont habituellement moins chers que les spécialités déjà disponibles, dont les prix tendent alors à diminuer.
Annie David , présidente . - Encore faut-il avoir conduit des essais comparatifs complets...
Bernard Cazeau , rapporteur . - Si on raisonne à service rendu comparable et à risque identique. Il faut revoir la rédaction de cet amendement.
Annie David , présidente . - L'amendement peut en tout cas permettre d'ouvrir le débat en séance.
L'amendement n° 69 est retiré.
L'amendement rédactionnel n° 129 est adopté.
Bernard Cazeau , rapporteur . - Amendement n° 70 : il faudrait préciser les cas d'application.
L'amendement n° 70 est retiré.
La commission adopte l'article 7 dans la rédaction issue de ses travaux.
Bernard Cazeau , rapporteur . - L'amendement n° 85 supprime des dispositions déclaratoires et redondantes.
L'amendement n° 85 est adopté.
L'amendement n° 2 est retiré.
La commission adopte l'article 8 dans la rédaction issue de ses travaux.
L'article 9 est adopté sans modification.
Article 9 bis
L'amendement rédactionnel n° 130 est adopté.
Bernard Cazeau , rapporteur . - Je suggère le retrait de l'amendement n° 7 au profit de l'amendement n° 130.
L'amendement n° 7 est retiré.
L'amendement n° 130 est adopté.
L'amendement n° 11 est retiré.
La commission adopte l'article 9 bis dans la rédaction issue de ses travaux.
Les articles 10 et 10 bis sont adoptés sans modification.
Article 11
L'amendement n° 86 est adopté.
Bernard Cazeau , rapporteur . - L'amendement n° 75 apporte une précision utile.
L'amendement n° 75 est adopté.
Bernard Cazeau , rapporteur . - Amendement n° 87 : il importe que les patients ne découvrent pas après coup que le médicament acheté n'est pas remboursé.
L'amendement n° 87 est adopté.
Bernard Cazeau , rapporteur . - Je crains que l'amendement n° 14 ne facilite la vie des laboratoires en reportant sur l'agence la constitution - qui plus est gratuite ! - du dossier tendant à modifier l'AMM.
L'amendement n° 14 est retiré.
La commission adopte l'article 11 dans la rédaction issue de ses travaux.
Article 12
L'amendement n° 17, auquel le rapporteur a donné un avis favorable, est adopté.
La commission adopte l'article 12 dans la rédaction issue de ses travaux.
Article additionnel après l'article 12
Bernard Cazeau , rapporteur . - L'amendement n° 29 pose une vraie question mais qui relève du domaine réglementaire. Nous interrogerons donc le ministre en séance.
L'amendement n° 29 est retiré.
Article 13
L'amendement n° 32, auquel le rapporteur a donné un avis favorable, est adopté.
La commission adopte l'article 13 dans la rédaction issue de ses travaux.
L'article 14 est adopté sans modification.
Article 14 bis
L'amendement n° 131 de précision est adopté.
La commission adopte l'article 14 bis dans la rédaction issue de ses travaux.
Article additionnel après l'article 14 bis
Bernard Cazeau , rapporteur . - L'amendement n° 71 est un cavalier : avis défavorable.
L'amendement n° 71 est rejeté.
Article 15
L'amendement n° 20, auquel le rapporteur a donné un avis favorable, est adopté.
Bernard Cazeau , rapporteur . - L'amendement n° 93 précise qu'une ATU nominative, c'est-à-dire destinée à un seul patient souffrant d'une maladie rare, sera recevable seulement si son titulaire a déposé une demande d'ATU pour une cohorte de malades ou une demande d'AMM. Par cohorte, on entend un groupe de malades affectés par la même maladie.
L'amendement n° 93 est adopté.
Bernard Cazeau , rapporteur . - L'amendement n° 23 est satisfait par le n° 93.
L'amendement n° 23 devient sans objet.
Bernard Cazeau , rapporteur . - Demande de retrait de l'amendement n° 26 : il ne faut pas confondre les RTU, visées à l'article 11, et les ATU qui donnent lieu à une AMM.
L'amendement n° 26 est retiré.
Bernard Cazeau , rapporteur . - L'amendement n° 94 maintient la possibilité de subordonner l'octroi d'une ATU à la conclusion d'un protocole.
L'amendement n° 94 est adopté.
La commission adopte l'article 15 dans la rédaction issue de ses travaux.
Article additionnel après l'article 15
Bernard Cazeau , rapporteur . - L'amendement n° 36 vise à rendre gratuits les médicaments bénéficiant d'une ATU de cohorte, comme en Allemagne. J'y vois un inconvénient majeur : le laboratoire, refusant la fourniture de médicaments à titre non onéreux, pourrait priver les malades de soins.
Annie David , présidente . - Au contraire, la gratuité poussera les laboratoires à demander une AMM.
Bernard Cazeau , rapporteur . - Nous en débattrons en séance.
L'amendement n° 36 est retiré.
Bernard Cazeau , rapporteur . - La crainte du groupe CRC est infondée : le texte n'étend pas l'ATU à la médecine de ville. Retrait de l'amendement de suppression n° 38 ?
L'amendement n° 38 est retiré.
L'article 16 est adopté sans modification.
Article 17
L'amendement n° 76 est rejeté.
Bernard Cazeau , rapporteur . - L'amendement n° 95 supprime des dispositions redondantes avec l'article 32.
L'amendement n° 95 est adopté.
La commission adopte l'article 17 dans la rédaction issue de ses travaux.
Bernard Cazeau , rapporteur . - Les vaccins ne doivent pas faire l'objet de campagnes publicitaires. D'où l'amendement n° 96.
L'amendement n° 96 est adopté.
Bernard Cazeau , rapporteur . - L'amendement n° 41 est satisfait par le n° 96.
L'amendement n° 41 devient sans objet.
Bernard Cazeau , rapporteur . - L'avis est défavorable aux amendements n os 79 et 80 de Gilbert Barbier.
Les amendements n os 79 et 80 sont rejetés.
L'amendement n° 97 de coordination est adopté.
La commission adopte l'article 18 dans la rédaction issue de ses travaux.
Bernard Cazeau , rapporteur . - Par l'amendement n° 98, nous revenons sur l'exclusion de certains médicaments du champ de la visite médicale votée par les députés, ce qui réduit la portée de l'expérimentation.
L'amendement n° 98 est adopté.
Bernard Cazeau , rapporteur . - C'est à la HAS qu'il revient d'évaluer le principe expérimental de la visite collective. D'où l'amendement n° 99.
L'amendement n° 99 est adopté.
Bernard Cazeau , rapporteur . - L'amendement n° 100 autorise le Ceps à fixer des objectifs d'évolution des pratiques produit par produit.
L'amendement n° 100 est adopté.
L'amendement n° 42 de coordination, auquel le rapporteur a donné un avis favorable, est adopté.
La commission adopte l'article 19 dans la rédaction issue de ses travaux.
Article additionnel après l'article 20
Bernard Cazeau , rapporteur . - L'amendement n° 44 trouverait mieux sa place dans le projet de loi de financement de la sécurité sociale. Pour autant, j'y suis favorable sur le fond.
L'amendement n° 44 est adopté et devient un article additionnel.
Bernard Cazeau , rapporteur . - L'amendement n° 101 est de forme.
L'amendement n° 101 est adopté.
La commission adopte l'article 21 dans la rédaction issue de ses travaux.
Bernard Cazeau , rapporteur . - L'amendement n° 102 précise le champ des études qui entreront dans la compétence du GIP.
L'amendement n° 102 est adopté.
Bernard Cazeau , rapporteur . - L'amendement n° 103 précise la composition du GIP.
L'amendement n° 103 est adopté.
L'amendement n° 104 de coordination est adopté.
La commission adopte l'article 22 dans la rédaction issue de ses travaux.
L'article 23 est adopté sans modification.
Article 24
L'amendement n° 46 de coordination, auquel le rapporteur a donné un avis favorable, est adopté.
L'amendement n° 105 de cohérence est adopté.
La commission adopte l'article 24 dans la rédaction issue de ses travaux.
L'article 25 est adopté sans modification.
Article 26
L'amendement n° 50 de coordination, auquel le rapporteur a donné un avis favorable, est adopté.
La commission adopte l'article 26 dans la rédaction issue de ses travaux.
Bernard Cazeau , rapporteur . - L'amendement n° 88 propose de supprimer l'article 27 : par principe, je refuse les demandes d'habilitation à légiférer par ordonnance. Le Parlement n'a pas à se dessaisir.
Les amendements de suppression n os 88 et 52 sont adoptés et l'article 27 est supprimé.
Bernard Cazeau , rapporteur . - Même logique qu'à l'article précédent.
Les amendements de suppression n os 89 et 55 sont adoptés et l'article 28 est supprimé.
Bernard Cazeau , rapporteur . - L'objet de l'amendement n° 135 est d'éviter le recours à une ordonnance de l'article 38 en précisant les conditions d'application de ce texte à Wallis-et-Futuna, à la Nouvelle-Calédonie et à la Polynésie française.
L'amendement n° 135 est adopté.
Bernard Cazeau , rapporteur . - L'amendement n° 57 est satisfait.
L'amendement n° 57 devient sans objet.
La commission adopte l'article 29 dans la rédaction issue de ses travaux.
Article 30
L'amendement n° 132 de coordination est adopté.
L'amendement n° 77 est rejeté.
Bernard Cazeau , rapporteur . - L'amendement n° 133 ménage un délai avant l'application de l'article 9 bis.
L'amendement n° 133 est adopté.
L'amendement n° 78 est rejeté.
Bernard Cazeau , rapporteur . - Par l'amendement n° 134, nous demandons un rapport à la HAS sur la sécurité des dispositifs médicaux.
L'amendement n° 134 est adopté.
La commission adopte l'article 30 dans la rédaction issue de ses travaux.
Articles additionnels après l'article 30
L'amendement n° 61, auquel le rapporteur a donné un avis favorable, est adopté et devient article additionnel.
Bernard Cazeau , rapporteur . - Je suis plutôt favorable à l'amendement n° 65 mais il présente un problème de rédaction. Retrait ?
L'amendement n° 65 est retiré.
Bernard Cazeau , rapporteur . - Un rapport sur les visiteurs médicaux pour le 30 juin 2012 ? Avec les élections présidentielle et législatives, la date est mal choisie. Mieux vaut la décaler au 1 er janvier 2013. Sous réserve de cette modification, l'avis est favorable à l'amendement n° 67.
Isabelle Pasquet . - Nous acceptons la rectification.
L'amendement n° 67 rectifié est adopté et devient article additionnel.
Bernard Cazeau , rapporteur . - L'amendement n° 90 reprend une disposition de la proposition de loi Fourcade censurée par le Conseil constitutionnel.
L'amendement n° 90 est adopté.
La commission adopte l'article 31 dans la rédaction issue de ses travaux.
Bernard Cazeau , rapporteur . - L'amendement n° 106 vise à rassembler les dispositions relatives aux lanceurs d'alerte au sein de cet article.
L'amendement n° 106 est adopté.
L'amendement n° 107 rédactionnel est adopté.
La commission adopte l'article 32 dans la rédaction issue de ses travaux.
Article 34
L'amendement de suppression n° 91 est adopté et l'article 34 est supprimé.
La commission adopte l'ensemble du projet de loi dans la rédaction issue de ses travaux.
|
Article additionnel avant l'article 1 er |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Pasquet, Cohen, David, Watrin et Fischer |
1 |
Agrément des seules associations d'usagers n'ayant pas de lien avec l'industrie |
Retiré |
|
Pasquet, Cohen, David, Watrin et Fischer |
3 |
Centralisation des informations relatives aux conflits d'intérêts par le service central de prévention de la corruption (SCPC) |
Retiré |
|
Article 1 er Règles déontologiques et expertise sanitaire |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Pasquet, Cohen, David, Watrin et Fischer |
2 |
Interdiction de tous liens d'intérêts depuis trois ans pour les membres et dirigeants des autorités sanitaires |
Retiré |
|
Rapporteur |
108 |
Clarification de la définition de la déclaration publique d'intérêt |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
8 |
Centralisation des déclarations publiques d'intérêts par le SCPC |
Retiré |
|
Rapporteur |
109 |
Interdiction de liens d'intérêts pendant trois ans avant leur mandat et pendant celui-ci pour le président de la HAS et le directeur général de l'Afssaps |
Adopté |
|
Rapporteur |
110 |
Précision des modalités de publicité des débats |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
15 |
Publicité des groupes de travail |
Retiré |
|
Pasquet, Cohen, David, Watrin et Fischer |
12 |
Publicité de tous les débats pendant une décision administrative |
Retiré |
|
Pasquet, Cohen, David, Watrin et Fischer |
10 |
Suppression de la restriction de la publicité du fait du secret industriel et commercial |
Retiré |
|
Rapporteur |
111 |
Publicité des avantages accordés aux organismes de formation |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
18 |
Compétence du SCPC |
Retiré |
|
Rapporteur |
112 |
Suppression du contrôle des déclarations par des commissions éthiques |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
21 |
Compétence du SCPC |
Satisfait |
|
Rapporteur |
113 |
Suppression du contrôle des déclarations par des commissions éthiques |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
25 |
Détermination des moyens de résoudre des conflits d'intérêts par la charte |
Satisfait |
|
Pasquet, Cohen, David, Watrin et Fischer |
28 |
Interdiction de tout lien d'intérêts aux experts externes |
Retiré |
|
Rapporteur |
114 |
Présence de médecins généralistes et de représentants des sciences sociales au sein des collèges d'experts |
Adopté |
|
Rapporteur |
115 |
Compétence de la commission de déontologie créée par la loi Sapin pour centraliser les déclarations publiques d'intérêts |
Adopté |
|
Rapporteur |
116 |
Contrôle de la Cada en cas de contestation de la publicité des propos tenus lors des débats |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
31 |
Interdiction au dirigeant des autorités sanitaires d'avoir pendant trois ans après la fin de leurs fonctions des activités sources de conflits d'intérêts |
Adopté |
|
Article additionnel après l'article 1 er |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Pasquet, Cohen, David, Watrin et Fischer |
34 |
Appel à candidature pour les postes de direction de la HAS et de l'INCa |
Adopté |
|
Article 2 Obligation de publication des avantages consentis par les entreprises au profit des acteurs du champ des produits de santé |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Rapporteur |
117 |
Publication annuelle du contenu des conventions avec l'industrie |
Adopté |
|
Rapporteur |
118 |
Publication des avantages perçus par les organismes de formation initiale des professionnels de santé |
Adopté |
|
Rapporteur |
119 |
Précision rédactionnelle |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
37 |
Suppression d'un seuil plancher pour les déclarations |
Adopté |
|
Rapporteur |
120 |
Obligation de publication des rémunérations |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
39 |
Publicité des avantages accordés au personnel politique |
Adopté |
|
Rapporteur |
121 |
Diffusion gratuite sur un site unique et facilement consultable des informations relatives aux avantages |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
40 |
Publication du contenu des conventions passées avec l'industrie |
Satisfait |
|
Pasquet, Cohen, David, Watrin et Fischer |
43 |
Compétence du SCPC pour contrôler les déclarations |
Retiré |
|
Barbier |
72 |
Participation des ordres professionnels |
Rejeté |
|
Rapporteur |
122 |
Interdiction des liens entre étudiants et industrie |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
45 |
Publication des données par le SCPC |
Retiré |
|
Barbier |
73 |
Soumission des conventions aux conseils des ordres compétents |
Rejeté |
|
Barbier |
74 |
Obligation pour l'entreprise d'informer l'Ordre de la mise en oeuvre des conventions |
Adopté |
|
Article 3 Dispositions pénales |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Rapporteur |
123 |
Amendement de cohérence |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
48 |
Sanction de toute omission de déclaration par les entreprises |
Adopté |
|
Rapporteur |
124 |
Amendement de conséquence |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
51 |
Amendement de cohérence |
Adopté |
|
Article 4 Création et prérogatives de l'Agence nationale de sécurité des produits de santé |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Rapporteur |
125 |
Dénomination de l'agence |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
4 |
Financement intégralement public de l'agence |
Retiré |
|
Pasquet, Cohen, David, Watrin et Fischer |
6 |
Contrôle des autorités nationales sur les fonds destinés à la pharmacovigilance |
Retiré |
|
Rapporteur |
126 |
Obligation de conduire les essais comparatifs contre des comparateurs actifs |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
54 |
Essais comparatifs pré-AMM |
Retiré |
|
Pasquet, Cohen, David, Watrin et Fischer |
58 |
Sanction des informations erronées sur les risques |
Retiré |
|
Pasquet, Cohen, David, Watrin et Fischer |
60 |
Sanction de l'absence de signalisation d'un usage hors AMM |
Retiré |
|
Pasquet, Cohen, David, Watrin et Fischer |
62 |
Suppression de la sanction relative au non-respect des conventions de régulation de l'usage hors AMM |
Retiré |
|
Rapporteur |
92 |
Coordination |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
64 |
Montant des amendes prononcées par l'agence |
Adopté |
|
Article additionnel après l'article 4 |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Pasquet, Cohen, David, Watrin et Fischer |
9 |
Missions de l'observatoire national des prescriptions |
Adopté |
|
Article 5 Coordinations |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Pasquet, Cohen, David, Watrin et Fischer |
13 |
Suppression de la participation de la Cnam au conseil d'administration de l'agence |
Retiré |
|
Pasquet, Cohen, David, Watrin et Fischer |
16 |
Participation des mutuelles au conseil d'administration |
Retiré |
|
Rapporteur |
82 |
Représentation des associations d'usagers ne recevant pas de subventions de l'industrie |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
19 |
Représentation des associations de victimes des accidents médicamenteux |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
22 |
Appel à candidature et limitation de la durée du mandat pour le poste de directeur général |
Retiré |
|
Rapporteur |
83 |
Compétences du conseil d'administration |
Adopté |
|
Rapporteur |
127 |
Simplification rédactionnelle |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
24 |
Publication d'un relevé nominatif des votes |
Satisfait |
|
Pasquet, Cohen, David, Watrin et Fischer |
33 |
Suppression de la mention du secret industriel et commercial |
Satisfait |
|
Pasquet, Cohen, David, Watrin et Fischer |
30 |
Coordination |
Retiré |
|
Pasquet, Cohen, David, Watrin et Fischer |
27 |
Coordination |
Retiré |
|
Article 5 bis Base de données mise en oeuvre par la Haute Autorité de santé |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Rapporteur |
84 |
Information sur le bon usage des produits de santé |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
35 |
Gratuité d'accès à la base d'information publique |
Adopté |
|
Article 6 Réalisation d'études après l'autorisation de mise sur le marché |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Pasquet, Cohen, David, Watrin et Fischer |
49 |
Changement de nom de l'AMM |
Retiré |
|
Pasquet, Cohen, David, Watrin et Fischer |
47 |
Essais comparatifs pré-AMM |
Retiré |
|
Pasquet, Cohen, David, Watrin et Fischer |
53 |
Renouvellement quinquennal de l'AMM |
Retiré |
|
Pasquet, Cohen, David, Watrin et Fischer |
56 |
Renouvellement quinquennal de l'AMM |
Retiré |
|
Pasquet, Cohen, David, Watrin et Fischer |
59 |
Harmonisation rédactionnelle |
Retiré |
|
Rapporteur |
128 |
Simplification |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
63 |
Amendement rédactionnel |
Retiré |
|
Pasquet, Cohen, David, Watrin et Fischer |
66 |
Suivi spécifique des médicaments causant un effet indésirable grave |
Retiré |
|
Pasquet, Cohen, David, Watrin et Fischer |
68 |
Mise en place d'études de sécurité dans les cas d'usage hors AMM |
Retiré |
|
Article 7 Conditions de suspension, de retrait ou de modification de l'autorisation de mise sur le marché |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Pasquet, Cohen, David, Watrin et Fischer |
69 |
Mise sur le marché des seuls médicaments apportant un progrès thérapeutique |
Retiré |
|
Rapporteur |
129 |
Rectification d'une erreur matérielle |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
70 |
Etude spécifique en cas de retrait d'un médicament |
Retiré |
|
Article 8 Obligations du titulaire de l'autorisation de mise sur le marché |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Rapporteur |
85 |
Suppression de dispositions redondantes |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
2 |
Sanction pour non-information d'un retrait |
Retiré |
|
Article 9 bis Conditions de fixation du service médical rendu des médicaments |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Rapporteur |
130 |
Nouvelle rédaction |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
7 |
Nouvelle rédaction |
Retiré |
|
Pasquet, Cohen, David, Watrin et Fischer |
11 |
Nouvelle rédaction |
Retiré |
|
Article 11 Encadrement des prescriptions en dehors des indications de l'autorisation de mise sur le marché |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Rapporteur |
86 |
Suppression d'une redondance |
Adopté |
|
Barbier |
75 |
Information du patient par le premier prescripteur |
Adopté |
|
Rapporteur |
87 |
Information du patient sur les conditions de remboursement |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
14 |
Possibilité de modification d'office de l'AMM |
Retiré |
|
Article 12 Prescription en dénomination commune |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Pasquet, Cohen, David, Watrin et Fischer |
17 |
Obligation de prescription en dénomination commune internationale |
Adopté |
|
Article additionnel après l'article 12 |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Pasquet, Cohen, David, Watrin et Fischer |
29 |
Mention manuscrite du caractère non substituable d'un médicament |
Retiré |
|
Article 13 Contrôle et sanction des prescriptions hors autorisation de mise sur le marché par le comité économique des produits de santé |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Pasquet, Cohen, David, Watrin et Fischer |
32 |
Possibilité pour le comité économique des produits de santé (Ceps) de prononcer des baisses de prix |
Adopté |
|
Article 14 bis Accès du Conseil national de l'ordre des pharmaciens aux données contenues dans le dossier pharmaceutique |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Rapporteur |
131 |
Amendement rédactionnel |
Adopté |
|
Article additionnel après l'article 14 bis |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Couderc |
71 |
Accès des pharmaciens aux dossiers pharmaceutiques |
Rejeté |
|
Article 15 Modification des procédures d'octroi des autorisations temporaires d'utilisation nominative |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Pasquet, Cohen, David, Watrin et Fischer |
20 |
Limitation de la durée des ATU |
Adopté |
|
Rapporteur |
93 |
Encadrement de la procédure de demande d'ATU nominative |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
23 |
Encadrement de la procédure de demande d'ATU nominative |
Satisfait |
|
Pasquet, Cohen, David, Watrin et Fischer |
26 |
Restrictions à la procédure dérogatoire de demande d'ATU nominative |
Retiré |
|
Rapporteur |
94 |
Possibilité de conclure un protocole d'utilisation thérapeutique pour les ATU nominatives obtenues par la procédure dérogatoire |
Adopté |
|
Article additionnel après l'article 15 |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Pasquet, Cohen, David, Watrin et Fischer |
36 |
Gratuité des ATU de cohorte |
Retiré |
|
Article 16 Prise en charge des produits prescrits hors autorisation de mise sur le marché par l'assurance maladie |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Pasquet, Cohen, David, Watrin et Fischer |
38 |
Amendement de suppression |
Retiré |
|
Article 17 Dispositions relatives à la pharmacovigilance |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Barbier |
76 |
Règles de signalement par un professionnel de santé des effets indésirables d'un produit de santé |
Rejeté |
|
Rapporteur |
95 |
Suppression de dispositions redondantes avec l'article 32 |
Adopté |
|
Article 18 Réglementation de la publicité pour les médicaments à usage humain |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Rapporteur |
96 |
Interdiction des campagnes publicitaires menées auprès du public pour des vaccins |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
41 |
Interdiction des campagnes publicitaires menées auprès du public pour des vaccins |
Satisfait |
|
Barbier |
79 |
Conditions de délivrance des visas pour les publicités destinées aux professionnels de santé |
Rejeté |
|
Barbier |
80 |
Délais de délivrance des visas pour les publicités destinées aux professionnels de santé |
Rejeté |
|
Rapporteur |
97 |
Coordination |
Adopté |
|
Article 19 Encadrement de la visite médicale |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Rapporteur |
98 |
Champ de l'expérimentation de la visite médicale collective |
Adopté |
|
Rapporteur |
99 |
Évaluation de l'expérimentation par la HAS |
Adopté |
|
Rapporteur |
100 |
Étendue des pouvoirs du Ceps |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
42 |
Pouvoir de sanction du Ceps |
Adopté |
|
Article additionnel après l'article 20 |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Pasquet, Cohen, David, Watrin et Fischer |
44 |
Taxe sur les dépenses promotionnelles des laboratoires pharmaceutiques |
Adopté |
|
Article 21 Certification des logiciels d'aide à la prescription et à la dispensation |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Rapporteur |
101 |
Amendement rédactionnel |
Adopté |
|
Article 22 Groupement d'intérêt public compétent en matière d'études de santé publique |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Rapporteur |
102 |
Champ de compétence du GIP |
Adopté |
|
Rapporteur |
103 |
Composition du GIP |
Adopté |
|
Rapporteur |
104 |
Coordination |
Adopté |
|
Article 24 Conformité des dispositifs médicaux aux spécifications requises pour pouvoir être remboursés |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Pasquet, Cohen, David, Watrin et Fischer |
46 |
Pouvoir de sanction du Ceps |
Adopté |
|
Rapporteur |
105 |
Amendement rédactionnel |
Adopté |
|
Article 26 Evaluation de certains dispositifs médicaux |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Pasquet, Cohen, David, Watrin et Fischer |
50 |
Pouvoir de sanction des ministres chargés de la santé et de la sécurité sociale |
Adopté |
|
Article 27 Habilitation du Gouvernement à prendre par ordonnance les mesures nécessaires à la transposition d'une directive « médicaments » |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Rapporteur |
88 |
Amendement de suppression |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
52 |
Amendement de suppression |
Adopté |
|
Article 28 Habilitation du Gouvernement à prendre par ordonnance les mesures nécessaires pour harmoniser les sanctions pénales et administratives avec les dispositions de la présente loi |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Rapporteur |
89 |
Amendement de suppression |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
55 |
Amendement de suppression |
Adopté |
|
Article 29 Habilitation du Gouvernement à prendre par ordonnance des dispositions relatives à la mise en cohérence du droit métropolitain avec le droit de l'Outre-mer |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Rapporteur |
135 |
Application directe de la loi outre-mer |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
57 |
Amendement de suppression |
Satisfait |
|
Article 30 Dispositions transitoires |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Rapporteur |
132 |
Date d'entrée en vigueur des dispositions de l'article 2 |
Adopté |
|
Barbier |
77 |
Date d'entrée en vigueur des dispositions de l'article 2 |
Rejeté |
|
Rapporteur |
133 |
Date d'entrée en vigueur de l'article 9 bis |
Adopté |
|
Barbier |
78 |
Date d'entrée en vigueur de l'article 9 bis |
Rejeté |
|
Rapporteur |
134 |
Rapport sur les dispositifs médicaux |
Adopté |
|
Article additionnel après l'article 30 |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Pasquet, Cohen, David, Watrin et Fischer |
61 |
Décisions d'inscription au remboursement des médicaments par la HAS |
Adopté |
|
Pasquet, Cohen, David, Watrin et Fischer |
65 |
Application du principe de précaution aux médicaments |
Retiré |
|
Pasquet, Cohen, David, Watrin et Fischer |
67 |
Rapport sur les visiteurs médicaux |
Adopté |
|
Article 31 Apparence et texture des médicaments génériques |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Rapporteur |
90 |
Apparence des médicaments génériques |
Adopté |
|
Article 32 Protection des lanceurs d'alerte |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Rapporteur |
106 |
Amendement rédactionnel |
Adopté |
|
Rapporteur |
107 |
Amendement rédactionnel |
Adopté |
|
Article 34 Contrôle des exportations parallèles de médicaments |
|||
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Rapporteur |
91 |
Amendement de suppression |
Adopté |
LISTE DES PERSONNES AUDITIONNÉES
Mardi 11 octobre 2011
• Jean-Luc Harousseau , président de la Haute Autorité de santé (HAS) et Dominique Maigne, directeur
Mercredi 12 octobre 2011
• Gilles Johanet , président du comité économique des produits de santé (Ceps)
• Christophe Duguet , Association française contre les myopathies, Franck Barbier , Associations AIDES, le Ciss
• Pierre Chirac , vice-président, Bruno Toussaint , directeur, Revue Prescrire
• Dominique Maraninchi , directeur général de l'Agence française de sécurité sanitaire et des produits de santé (Afssaps), François Hébert , directeur général adjoint
Jeudi 13 octobre 2011
• Christian Lajoux, président, et Philippe Lamoureux, secrétaire général du Leem
• Cabinet du ministre et services du ministère de la santé
TABLEAU COMPARATIF
* 1 La réforme du système du médicament, enfin : rapport d'information de Marie-Thérèse Hermange, fait au nom de la mission commune d'information sur le Mediator n° 675 (2010-2011).
* 2 Rapport d'information d'Anne-Marie Payet et Marie-Thérèse Hermange n° 382 (2005-2006), fait au nom de la commission des affaires sociales.
* 3 Directive 65/65/CEE.
* 4 Directive 75/319/CEE.
* 5 Règlement (CEE) n° 2309/93 du Conseil du 22 juillet 1993 établissant des procédures communautaires pour l'autorisation et la surveillance des médicaments à usage humain et à usage vétérinaire et instituant une Agence européenne pour l'évaluation des médicaments.
* 6 Alinéa 11 du Préambule de la Constitution de 1946.
* 7 L'article 2 de la Déclaration des Droits de l'Homme et du Citoyen place la propriété parmi les droits naturels et imprescriptibles de l'homme, l'article 17 déclare qu'il s'agit d'un droit inviolable et sacré et soumet l'expropriation à un constat légal de sa nécessité publique et à la condition d'une juste et préalable indemnité.
* 8 Loi n o 68-1 du 2 janvier 1968 sur les brevets d'invention.
* 9 Ce dispositif résulte de l'article 11 de la directive 65/65/CEE du Conseil du 26 janvier 1965 concernant le rapprochement des dispositions législatives, réglementaires et administratives, relatives aux spécialités pharmaceutiques selon lequel la Commission européenne, en l'absence de données scientifiques ou d'informations nouvelles, ne peut revenir sur l'appréciation positive qu'elle a émise de l'efficacité des substances en cause.
* 10 Voir en dernier lieu la décision n° 335101 du Conseil d'Etat, sous-sections réunies, en date du 7 juillet 2010, annulant la suspension par l'Afssaps du Ketum.
* 11 Loi n° 2002-303 du 4 mars 2002 relative aux droits des malades et à la qualité du système de santé.
* 12 Loi n° 98-535 du 1 er juillet 1998.
* 13 Loi n° 93-5 du 4 janvier 1993.
* 14 Loi n° 2004-810 du 13 août 2004 relative à l'assurance maladie.
* 15 Loi n° 96-452 du 28 mai 1996 portant diverses mesures d'ordre sanitaire, social et statutaire.
* 16 Bilan de l'expérience du thalidomide dans le cadre des ATU. Février 1997/Février 2009, www.afssaps.fr.
* 17 Articles R. 5121-72 à R. 5121-74 du code de la santé publique.
* 18 Rapport Sénat n° 413 (1996-1997) de Claude Huriet, au nom de la commission des affaires sociales, sur la proposition de loi tendant au renforcement de la veille sanitaire et du contrôle de la sécurité sanitaire des produits destinés à l'homme.
* 19 Une définition plus précise des dispositifs médicaux en fonction de leurs objectifs est donnée aux articles R. 5211-1 à 3 du code de la santé publique.
* 20 Igas, Evolution et maîtrise de la dépense des dispositifs médicaux, novembre 2010, Tome II, p. 15-18.
* 21 La directive 90/385/CEE pour les dispositifs médicaux implantables actifs ; la directive 93/42/CEE pour les dispositifs médicaux autres que ceux mentionnés précédemment ; la directive 98/79/CE pour les dispositifs de diagnostic in vitro.
* 22 Claude Huriet, rapport précité.
* 23 Loi n° 2001-398 du 9 mai 2001.
* 24 Loi n° 2006-686 du 13 juin 2006.
* 25 Loi n° 93-122 du 29 janvier 1993 relative à la prévention de la corruption et à la transparence de la vie économique et des procédures publiques.
* 26 Devenue la loi n° 2011-940 du 10 août 2011.
* 27 Décision n° 2011-640 DC du 4 août 2011.
* 28 Source : Afssaps, Indicateurs d'activité 2010.
* 29 Gérard Bapt, Jean-Pierre Door, Rapport d'information AN n° 3552, Mediator : comprendre pour réagir, juin 2011, p. 51-54.
* 30 François Autain, Marie-Thérèse Hermange, Rapport d'information Sénat n° 675, La réforme du système du médicament, enfin, juin 2011, p. 124-125.
* 31 L'article L. 162-16-5-1 du code de la sécurité sociale oblige le laboratoire titulaire des droits d'exploitation à déclarer au Ceps le montant de l'indemnité demandée auprès des établissements de santé pour le médicament sous ATU. Si, dans le cadre d'une AMM ultérieure, le Ceps fixe un prix ou un tarif de remboursement inférieur au prix déclaré, le laboratoire doit reverser tout ou partie de la différence aux organismes de recouvrement. L'indemnité est ensuite affectée aux régimes d'assurance maladie.
* 32 Section 13 du chapitre I er du titre II du livre I er de la cinquième partie du code de la santé publique relative à la pharmacovigilance des médicaments à usage humain (articles R. 5121-150 à R. 5121-201).
* 33 Loi n° 2009-879 du 21 juillet 2009 portant réforme de l'hôpital et relative aux patients, à la santé et aux territoires.
* 34 Décret n° 2011-655 du 10 juin 2011 relatif aux modalités de signalement par les patients ou les associations agréées de patients d'effets indésirables susceptibles d'être liés aux médicaments et produits mentionnés à l'article L. 5121-1 du code de la santé publique.
* 35 Travaux disponibles sur : http://www.sante.gouv.fr/IMG/pdf/VOLUME_I_Rapport_des_six_groupes_de_travail-3.pdf.
* 36 La publicité ne doit pas être trompeuse ni porter atteinte à la santé publique. Elle doit présenter le médicament ou produit de façon objective et favoriser son bon usage. Elle doit respecter les dispositions de l'autorisation de mise sur le marché.
* 37 Le médicament a obtenu l'autorisation de commercialisation à laquelle il est soumis.
* 38 François Autain, Marie-Thérèse Hermange, Rapport d'information Sénat n° 675, La réforme du système du médicament, enfin, juin 2011, p. 173.
* 39 Inspection générale des affaires sociales, Rapport sur la pharmacolovigilance et la gouvernance de la chaîne du médicament, juin 2011.
* 40 Loi n° 2002-1487 du 20 décembre 2002 de financement de la sécurité sociale pour 2003.
* 41 Devenue loi n° 2011-940 du 10 août 2011.
* 42 Décisions n os 2009-596 DC du 22 décembre 2009 et 2011-640 DC du 4 août 2011.
* 43 Article L. 1161-1 du code du travail.
* 44 Article L. 313-24 du code de l'action sociale et des familles.
* 45 Décision n° 2009-596 DC du 22 décembre 2009.