







b) L'accroissement considérable des connaissances
Si plusieurs de mes interlocuteurs ont qualifié la biologie des systèmes d'instrument indispensable de la BS, c'est précisément parce que la biologie des systèmes tente de parvenir à une vision plus globale et prédictive du système cellulaire (comparé à la biologie moléculaire classique), qui peut revêtir un intérêt pour la BS.
L'objectif d'une vision globale est ancien puisque, par exemple, après la seconde guerre mondiale, François Jacob et Jacques Monod avaient déjà introduit la notion de système en biochimie, comme un processus susceptible d'expliquer la différenciation cellulaire, c'est-à-dire les modalités selon lesquelles des cellules au génome identique peuvent exprimer des formes et des propriétés aussi différentes que celles d'un globule rouge et de cellules cardiaques. Ainsi, leurs recherches et celles d'autres chercheurs sur les mécanismes de régulation moléculaires ont-elles permis, fût-ce à une petite échelle, de découvrir les composants moléculaires et la logique sous-tendant les processus cellulaires, souvent en parallèle avec les caractérisations des macromolécules individuelles.
Le séquençage du génome, le développement des technologies à haut débit et la modélisation mathématique et informatique ont conduit les biologistes des systèmes à s'intéresser à l'identification et à la modélisation des réseaux, grâce auxquels gènes et protéines interagissent pour remplir les fonctions cellulaires.
Or, de tels champs d'étude sont d'autant plus importants que des mécanismes aussi différents que la synthèse de l'ADN, la production d'ATP (Adénosine Triphosphate) 90 ( * ) et la maturation de l'ARN sont tous accomplis par des « machines moléculaires » impliquant des interactions entre de nombreuses protéines, parfois plusieurs dizaines, produisant soit des relations stables, soit des liaisons transitoires.
C'est pourquoi les biologistes de synthèse estiment que l'intérêt de la biologie des systèmes réside dans la possibilité de comprendre et de fabriquer des réseaux biologiques, qu'ils concernent les mécanismes de régulation intracellulaires ou les rapports entre les cellules et leur environnement physique et chimique 91 ( * ) . Estimant que l'ingénierie biologique implique de concevoir des systèmes entiers et des circuits, outre le fait de standardiser et de mélanger des modules protéiques destinés à l'accomplissement de tâches spécifiques, Petra Schwille, professeure à l'Université de Dresde, souligne que « pour réussir, la BS, quelle que soit son approche, doit unir ses forces à la biologie des systèmes » 92 ( * ) .
Cette affirmation est d'autant plus fondée qu'un second aspect de la biologie des systèmes - l'objectif d'élaborer des modèles prédictifs - ne peut manquer d'intéresser les biologistes de synthèse.
Une telle méthode illustre bien l'application de la démarche de l'ingénieur à la biologie, notamment par le recours à la bio-informatique, caractéristique essentielle de la biologie de synthèse.
Ce souci de prédiction répond également à celui que les biologistes de synthèse assignent aux circuits génétiques et même, au-delà, à la BS 93 ( * ) . Des banques de données - généralistes ou spécialisées - concourent à la poursuite de cet objectif de prédiction. Elles fournissent des informations sur la structure et la fonction de la protéine codée, en vue de procéder à ce que l'on appelle l'annotation des gènes.
Cette annotation se déroule en deux étapes : la première, l'annotation structurelle, permet d'identifier les zones de la séquence génomique qui déterminent les séquences protéiques. Une seconde étape cherche à associer une information aux zones identifiées et, en particulier, la ou les fonctions de la protéine dans l'organisme. Cette deuxième étape, l'annotation fonctionnelle, se réalise en précisant les réactions biochimiques auxquelles la protéine participe ou ses rôles dans les processus biologiques. La plupart des informations concernant l'annotation d'une protéine sont générées par des prédictions basées sur le calcul ou par des comparaisons avec des protéines similaires.
Concrètement, pour associer une annotation à une séquence protéique, l'annotateur exploite différents critères et met en oeuvre plusieurs étapes. Lorsque l'on suppose l'annotation structurale résolue, annoter fonctionnellement une protéine consiste à identifier :
- ses caractéristiques intrinsèques directement calculables à partir de la séquence protéique ;
- les caractéristiques issues des prédictions apportées par l'analyse des résultats des logiciels bio-informatiques ;
- une protéine déjà annotée et dont la séquence en acides aminés présente des similitudes.
Les paramètres intrinsèques directement calculables concernent, par exemple, la taille en acides aminés, la séquence, le début et la fin de la traduction.
Les paramètres prédits nécessitent le lancement d'un logiciel bio-informatique et l'analyse des résultats obtenus. Pour procéder à celle-ci, l'annotateur s'appuie sur les valeurs scores proposées par les logiciels, si elles sont suffisamment discriminantes. On observe les résultats plus en détail dans le cas contraire avant de conclure. Les prédictions concernent, par exemple, la localisation sub-cellulaire de la protéine dans les différents compartiments cellulaires. Les prédictions touchent aussi aux informations issues du contexte génique, telles que les annotations fonctionnelles associées aux gènes présents en amont ou en aval du gène codant pour la protéine en cours d'annotation.
La prédiction de la structure tridimensionnelle de toutes les protéines, ce à quoi tente de parvenir la protéomique structurale, fournit une autre illustration de cette démarche prédictive de la biologie des systèmes.
La connaissance de la structure 3D des protéines apporte une information particulièrement pertinente pour permettre de comprendre son mode d'action : activité enzymatique, interaction avec d'autres protéines. La détermination de la structure d'une seule protéine demande un travail de laboratoire qui peut durer plusieurs années pour chaque structure. Par conséquent, dans le cadre de la génomique structurale, il est essentiel d'automatiser chacune des étapes décrites ci-après.
|
|
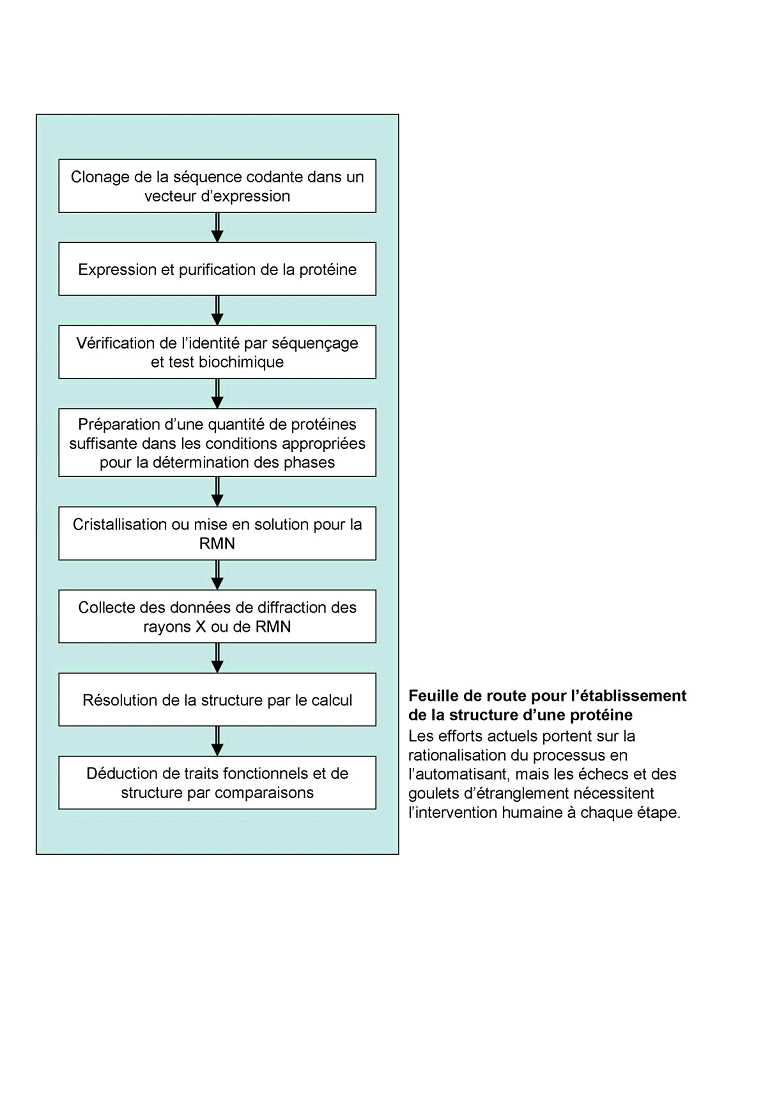

Source : Greg Gibson et Spencer V. Muse, Précis de génomique, 2005, p. 207
Les deux méthodes expérimentales pour résoudre les structures des protéines sont la radiocristallographie ou diffraction des rayons X par des cristaux et la spectroscopie par résonance magnétique nucléaire (RMN).
Trois méthodes sont utilisées pour prédire la structure tridimensionnelle des protéines :
La prédiction ab initio , dans laquelle la structure est directement déduite de la séquence, en acides aminés, c'est-à-dire en prédisant la probabilité qu'une sous-séquence se replie en une hélice alpha 94 ( * ) ou en un feuillet bêta 95 ( * ) à l'aide notamment de paramètres physico-chimiques. Une contrainte majeure de ce type de modélisation théorique est le nombre considérable de calculs à effectuer pour déterminer les fonctions d'énergie de tous les contacts possibles. Pour répondre à cet objectif, IBM a construit en 2005 un superordinateur appelé « Blue Gene » , qui effectue 280 600 milliards d'opérations de calcul par seconde.
L'identification d'un repliement : le repliement désigne le processus par lequel les chaînes d'acides aminés qui constituent les protéines se replient en une spirale tridimensionnelle plus complexe. En comprenant comment les protéines se replient et quelles structures finales elles sont susceptibles d'adopter, les chercheurs espèrent en prédire la fonction. L'identification d'un repliement désigne une méthode de prédiction de la structure tertiaire 96 ( * ) d'une protéine. On combine les données de prédiction de structure secondaire avec des données de similitude de séquence pour rechercher le domaine dont le repliement est déjà connu et qui s'apparente au mieux avec la structure de la protéine inconnue.
Les méthodes de calcul de la modélisation du repliement des protéines existent depuis une vingtaine d'années. Mais le professeur Jérôme Waldispühl, chercheur au centre de bio-informatique de l'Université MacGill (Montréal) a réussi à développer, avec des collaborateurs du MIT, des algorithmes adaptables à un portable pour étudier les propriétés chimiques fondamentales d'une protéine, puis évaluer les différentes formes qu'elle peut prendre avant de prédire la structure finale qu'elle est susceptible d'adopter.
L'ajustement sur un modèle ( threading ) : cette approche de la prédiction de la structure protéique se base sur la conjonction de similarités de la structure secondaire et la vérification des énergies probables de liaison des repliements potentiels.
Une fois publiées, les structures des protéines sont déposées dans une banque de données, le Protein Data Bank (PDB). PDB est une collection mondiale de données sur la structure 3D de macromolécules biologiques : protéines, principalement, et acides nucléiques. Ces structures sont principalement déterminées par cristallographie aux rayons X ou par spectroscopie RMN. Ces données expérimentales sont déposées par des chercheurs du monde entier et appartiennent au domaine public. Leur consultation est gratuite et peut être effectuée depuis le site internet de la banque. La PDB est la principale source de données de biologie structurale et permet surtout d'accéder à des structures 3D de protéines d'intérêt pharmaceutique. La PDB contenait, au 31 janvier 2012, 78952 structures.
Pour ce qui est du rôle des modèles prédictifs élaborés en biologie des systèmes, une récente étude 97 ( * ) fait état des avancées suivantes :
L'étude de la régulation du cycle cellulaire chez Caulobacter crescentus 98 ( * ) , du chimiotactisme bactérien 99 ( * ) , de l'organisation subcellulaire des protéines et de l'ADN dans les cellules bactériennes. A ce jour, selon les auteurs, les réseaux de régulation de la transcription sont un exemple des systèmes les mieux caractérisés par des modèles prédictifs à l'échelle du génome. Même le procaryote le plus étudié, E. coli , a bénéficié et bénéfice encore de l'analyse systémique de ces réseaux.
Des avancées substantielles sont également intervenues dans la reconstruction du métabolisme de divers procaryotes. Le modèle métabolique d' E. coli , par exemple, contient maintenant 48 % de l'ensemble des gènes ayant des fonctions déterminées expérimentalement. Ces modèles ont été utilisés pour fabriquer des souches, en vue d'une production de métabolite, et ce afin d'identifier des gènes putatifs 100 ( * ) pour des réactions orphelines. Il est important de relever que des approches adoptées pour la reconstruction de réseaux métaboliques peuvent être étendues aux systèmes complexes eucaryotes, comme dans le cas de la levure, Aspergillus nodulans 101 ( * ) , ou de Caenorhabditis elegans. 102 ( * )
* 90 L'ATP est une substance chimique qui fournit l'énergie à de nombreux processus cellulaires et qui est l'un des précurseurs de l'ARN.
* 91 Eric Young et Al Halper, « Synthetic Biology: Tools to Design, Build and Optimize cellular process», Journal of Biomedicine and Biotechnology, janvier 2010.
* 92 Petra Schwille, «Bottom up Synthetic Biology: engineering in a Tinkerer's world», Science, 2 septembre 2011.
* 93 « La capacité à concevoir un système biologique qui se comporte de façon prédictible et fonctionne mieux que son équivalent naturel est le rêve des biologistes de synthèse. » , Jian Liang et al. , article précité.
* 94 Hélice alpha : une des structures secondaires possibles des polypeptides, dans laquelle la chaîne d'acides aminés prend une conformation spiralée (hélicoïdale) .
* 95 Feuillet bêta : une des structures secondaires d'un polypeptide, dans laquelle plusieurs plages bêta sont parallèles les unes aux autres, donnant ainsi la disposition en feuille.
* 96 La structure tertiaire d'une protéine correspond au repliement de la chaîne polypeptidique dans l'espace. On parle plus couramment de structure tridimensionnelle ou structure 3D.
* 97 Tic Kode et al. , « The role of predictive modelling in rationally reengineering biological systems », PubMed Central, avril 2009.
* 98 Caulobacter crescentus est une bactérie dont le pédicule a une grande capacité adhésive due à des polysaccharides.
* 99 Le chimiotactisme est le phénomène par lequel les cellules somatiques, les bactéries et autres organismes cellulaires ou pluricellulaires dirigent leurs mouvements en fonction de certains produits chimiques dans leur environnement. Pour les bactéries, il est important de trouver de la nourriture (par exemple le glucose) en nageant vers la plus forte concentration de molécules alimentaires ou pour fuir des poisons (par exemple, le phénol).
* 100 Gènes putatifs : encore appelés gènes hypothétiques, les gènes putatifs sont des fragments d'ADN considérés comme étant des gènes, en se fondant sur leur séquence. Mais ni leur produit, ni leur fonction ne sont connus.
* 101 Il s'agit de l'une des nombreuses espèces de champignons filamenteux du genre aspergillus. Il a beaucoup été utilisé comme matériel de recherche pour des études sur les eucaryotes.
* 102 Caenorhabditis elegans est un petit vers transparent d'un millimètre de longueur, hermaphrodite ou mâle, se reproduisant environ tous les trois jours et dont la durée de vie est d'environ trois semaines. Il a été introduit dans les laboratoires de génétique dans les années 70 pour répondre au besoin d'un modèle génétique destiné à comprendre l'élaboration d'un organisme pluricellulaire.