







Rapport n° 306 (2013-2014) de MM. Alain CLAEYS et Jean-Sébastien VIALATTE, fait au nom de l'Office parlementaire d'évaluation des choix scientifiques et technologiques, déposé le 22 janvier 2014
Disponible au format PDF (4,1 Moctets)
-
OUVERTURE
PAR MM. ALAIN CLAEYS ET JEAN-SÉBASTIEN VIALATTE,
DÉPUTÉS, RAPPORTEURS
-
PROPOS INTRODUCTIFS
-
PREMIÈRE TABLE RONDE :
POUR QUELLES PATHOLOGIES ?
-
DEUXIÈME TABLE RONDE :
AVEC QUELS OUTILS ?
-
APRÈS-MIDI
-
PROPOS INTRODUCTIFS
-
TROISIÈME TABLE RONDE :
AVEC QUELLE VALORISATION DE LA RECHERCHE ?
-
QUATRIÈME TABLE RONDE :
AVEC QUELS MODELES DE RECHERCHE-DEVELOPPEMENT ?
-
CONCLUSION
-
OUVERTURE
PAR MM. ALAIN CLAEYS ET JEAN-SÉBASTIEN VIALATTE,
DÉPUTÉS, RAPPORTEURS
-
PROPOS INTRODUCTIFS
-
PREMIÈRE TABLE
RONDE :
MÉDECINE DE PRÉVENTION OU DE PRÉDICTION : QUELLE RELATION MÉDECIN-MALADE ?
-
DEUXIÈME TABLE RONDE :
QUELLE PROTECTION DES PERSONNES, DE LEUR VIE PRIVÉE
ET DES DONNÉES PERSONNELLES ?
-
APRÈS-MIDI
-
PROPOS INTRODUCTIFS
-
TROISIÈME TABLE RONDE :
QUELLE ORGANISATION DU SYSTÈME DE SANTÉ ?
-
QUATRIÈME TABLE RONDE :
QUELS REGARDS DES PATIENTS ET DES ASSOCIATIONS DE MALADES ?
|
N° 1724 |
N° 306 |
|
|
ASSEMBLÉE NATIONALE |
SÉNAT |
|
|
CONSTITUTION DU 4 OCTOBRE 1958 QUATORZIÈME LÉGISLATURE |
SESSION ORDINAIRE 2013 - 2014 |
|
|
Enregistré à la présidence de l'Assemblée nationale |
Enregistré à la présidence du Sénat |
|
|
le 22 janvier 2014 |
le 22 janvier 2014 |
RAPPORT
au nom de
L'OFFICE PARLEMENTAIRE D'ÉVALUATION
DES CHOIX SCIENTIFIQUES ET TECHNOLOGIQUES
sur
LES PROGRÈS DE LA GÉNÉTIQUE : VERS UNE MÉDECINE DE PRÉCISION ?
LES ENJEUX SCIENTIFIQUES, TECHNOLOGIQUES, SOCIAUX ET ÉTHIQUES DE LA MÉDECINE PERSONNALISÉE
PAR
MM. Alain CLAEYS et Jean-Sébastien VIALATTE,
députés
|
Déposé sur le Bureau de l'Assemblée nationale par M. Jean-Yves LE DÉAUT, Premier Vice-président de l'Office |
Déposé sur le Bureau du Sénat par M. Bruno SIDO, Président de l'Office |
Composition de l'Office parlementaire d'évaluation des choix scientifiques et technologiques
Président
M. Bruno SIDO, sénateur
Premier Vice-président
M. Jean-Yves LE DÉAUT, député
Vice-présidents
M. Christian BATAILLE, député M. Roland COURTEAU, sénateur
Mme Anne-Yvonne LE DAIN, députée M. Marcel DENEUX, sénateur
M. Jean-Sébastien VIALATTE, député Mme Virginie KLÈS, sénatrice
|
DÉPUTÉS |
SÉNATEURS |
|
M. Gérard BAPT M. Christian BATAILLE M. Denis BAUPIN M. Alain CLAEYS M. Claude de GANAY Mme Anne GROMMERCH Mme Françoise GUEGOT M. Patrick HETZEL M. Laurent KALINOWSKI Mme Anne-Yvonne LE DAIN M. Jean-Yves LE DÉAUT M. Alain MARTY M. Philippe NAUCHE Mme Maud OLIVIER Mme Dominique ORLIAC M. Bertrand PANCHER M. Jean-Louis TOURAINE M. Jean-Sébastien VIALATTE |
M. Gilbert BARBIER Mme Delphine BATAILLE M. Michel BERSON Mme Corinne BOUCHOUX M. Marcel-Pierre CLÉACH M. Roland COURTEAU Mme Michèle DEMESSINE M. Marcel DENEUX Mme Dominique GILLOT Mme Chantal JOUANNO Mme Fabienne KELLER Mme Virginie KLÈS M. Jean-Pierre LELEUX M. Jean-Claude LENOIR M. Christian NAMY M. Jean-Marc PASTOR Mme Catherine PROCACCIA M. Bruno SIDO |
SAISINE

INTRODUCTION
La Commission des affaires sociales de l'Assemblée nationale a saisi l'OPECST d'une demande d'étude sur les enjeux scientifiques, technologiques, éthiques et juridiques de la médecine personnalisée. Cette saisine s'inscrit dans la vaste lignée des travaux de l'Office sur les sciences du vivant, la santé et plus généralement la bioéthique.
Dès 1999, le sénateur Frank Sérusclat présentait devant l'OPECST un rapport intitulé « Génomique et informatique : l'impact sur les thérapies et sur l'industrie pharmaceutique » (1 ( * )) , qui décrivait les principales avancées et leurs impacts futurs. Une grande partie des concepts, des analyses et recommandations est encore d'actualité car les évolutions à l'oeuvre depuis une quinzaine d'années montrent leur pertinence. Il en va de même des travaux menés, dès 2001, au sein de l'OPECST par l'un de vos rapporteurs sur la brevetabilité du vivant.
Les évaluations successives des lois relatives à la bioéthique de 1994 puis de 2004 ont souligné l'impact juridique et éthique des avancées scientifiques et techniques. Lors de l'évaluation de la loi d'août 2004, vos rapporteurs s'interrogeaient sur les recherches en cours sur les cellules souches et les possibilités de traitements personnalisés offerts par la thérapie cellulaire. Ils mettaient également en exergue le rôle des tests génétiques à l'oeuvre dans ce que l'on nomme la médecine personnalisée et se demandaient, par exemple, pourquoi des personnes tentaient de connaître leur patrimoine génétique en recourant à l'achat de tests en libre accès sur Internet.
L'audition publique de l'OPECST sur les maladies monogéniques du 7 juin 2011 insistait sur les difficultés inhérentes au traitement des maladies rares, démontrant la nécessité de recourir à une médecine personnalisée avec des traitements curatifs très ciblés mais coûteux. Cependant, elle révélait aussi les risques de discrimination, voire de stigmatisation, bien réels pour les personnes atteintes par de telles pathologies, suscitant des interrogations sur le degré de protection offert par la législation au regard des assurances, de l'emploi, etc .
Lors de leur étude de 2012 sur les « Enjeux des nouvelles technologies d'exploration du cerveau », vos rapporteurs soulignaient l'importance de la fiabilité des machines et des modèles d'interprétation des images et des données numérisées. Ils plaçaient en outre au coeur de leur réflexion les difficultés suscitées par la protection de la masse de données personnelles identifiantes ainsi générées. Or, le développement de nouvelles pratiques médicales en produisent, et en produiront bien plus.
Il reste que la définition de la notion de médecine personnalisée n'est pas aisée. Le caractère flou et polysémique du concept se prête à de nombreuses interprétations. Toute bonne pratique médicale est par essence « personnalisée », le praticien cherchant toujours à s'adapter au profil de son patient. Ce colloque singulier patient-médecin, caractéristique de la médecine, est évidemment une démarche personnalisée. En quoi cette « personnalisation » aurait-elle donc changé de nature ?
L'expression médecine personnalisée apparaît ainsi quelque peu singulière et donne légitimement à penser au malade qu'il s'agit d'une médecine sur mesure, s'adressant spécifiquement à lui, d'une prise en charge plus individuelle, plus proche, d'une plus grande empathie à son égard de la part du médecin. En réalité, dans l'acception médicale du concept, il s'agit plutôt de définir des sous-groupes étroits de patients grâce à des biomarqueurs (2 ( * )) permettant de trouver les molécules soignantes les plus appropriées et donc d'aboutir à une plus grande efficacité médicale avec un taux d'échec limité. Cependant, ces procédés relèvent plutôt d'une médecine stratifiée.
La médecine personnalisée passe par l'utilisation de nouvelles méthodes d'analyse moléculaire en vue d'assurer une meilleure compréhension de la maladie dont souffre un patient, ou encore de sa prédisposition à cette maladie. En fonction du profil génétique et environnemental du malade, elle permet aux médecins et patients de choisir, parmi les diverses options thérapeutiques, celles qui sont susceptibles de donner les meilleurs résultats.
Le Pr Anne Fagot-Largeault (3 ( * )) , explique ainsi le succès populaire du concept de médecine personnalisée et les confusions qu'il engendre : « a publicité faite pour la médecine dite personnalisée tombe dans un contexte où l'on constate une aspiration à un contact plus personnel, voire à une empathie du médecin, qu'on ne trouve pas, qu'on ne rencontre plus. On s'abrite derrière une formule attirante, rassurante, qui donne une image attractive, mais fausse et c'est très préoccupant ». La médecine personnalisée est née de la publicité qu'ont fait certains laboratoires pharmaceutiques pour leur traitement innovant.
Le concept de médecine personnalisée développé voici deux décennies par le laboratoire pharmaceutique suisse Roche, est fondé sur le fait qu'un même médicament peut provoquer des réactions différentes selon les malades, et que pour un patient donné, certains médicaments sont efficaces et d'autres pas. Avec l'introduction dans les années 1990 de l'Herceptine pour traiter le cancer du sein, Roche a démontré qu'il était possible d'anticiper le résultat, grâce à un simple test génétique sur les patients, et de montrer ceux pour qui le traitement serait le plus bénéfique, et ceux pour qui il ne le serait probablement pas. L'Herceptine, qui traite le cancer du sein et agit seulement sur les cancers des femmes qui surexpriment le gène HER-2, a été le premier médicament autorisé issu de la médecine stratifiée. On disposait ainsi du traitement et du test compagnon correspondant et certains ont alors considéré que le modèle de médecine personnalisée permettait de trouver et de délivrer le médicament le mieux adapté. Il devenait désormais possible de personnaliser les traitements en n'administrant un médicament qu'aux patients réagissant positivement. L'impact de cette nouvelle approche est énorme : efficacité accrue, effets secondaires réduits, et absence de temps perdu et de ressources gaspillées dans un traitement inopérant.
Cette médecine, fondée sur une analyse des caractéristiques biologiques et génétiques de la personne et de son environnement spécifique, nécessite un recours massif à des techniques de pointe et des outils nouveaux d'analyses biologiques. La médecine personnalisée est issue du transfert progressif vers la clinique de ces progrès scientifiques et technologiques considérables en termes de performance des techniques de séquençage et d'imagerie, initiée dans les années quatre-vingts à une période de crise du modèle de développement de nouveaux médicaments. C'est la recherche académique, plus que celle des grandes sociétés pharmaceutiques, pour qui le coût de fabrication de médicaments destinés à un nombre limité de malades est apparu souvent trop lourd, qui a le plus contribué à la diffusion de ce modèle. En effet, pour développer un médicament, plus le marché est large, plus le coût est faible. Or, les nouvelles thérapies s'adressent à de petites populations, ce qui conduira à une transformation complète de notre modèle socio-économique traditionnel.
Dans un rapport récent, l 'European Science Foundation (4 ( * )) , organisation européenne qui a mandaté un groupe de chercheurs reconnus, formule une définition très large : la médecine personnalisée serait une pratique médicale dont l'individu est le centre, à la fois précise et adaptée à ses caractéristiques biologiques qui englobent ses données génétiques, ses taux de protéines, ses biomarqueurs et prend en compte son environnement, son mode de vie, ses habitudes alimentaires. Les chercheurs de l 'European Science Foundation se réfèrent à une médecine dite des « 4P » : « Personnalisée », au sens du traitement d'une maladie ; « Préventive », la dimension de prévention étant importante ; « Prédictive », car le patient peut recourir à des tests ; « Participative », au sens où l'individu deviendrait acteur de sa santé. Cette définition large va de la prévention et du diagnostic au traitement et au suivi post-traitement. Ce rapport opère une distinction entre la médecine personnalisée et la médecine génomique basée uniquement sur la personnalisation des traitements à partir de l'information génétique. Il distingue donc médecine personnalisée, médecine génomique et médecine stratifiée, cette dernière catégorisant les individus selon certaines de leurs caractéristiques génétiques pour élaborer des traitements ciblés selon ces éléments.
La polysémie du concept prête à confusion car, dans le cadre de la médecine stratifiée, la médecine personnalisée n'est pas la médecine personnelle d'un seul individu mais celle d'un groupe ciblé en fonction du profil médical des individus qui le composent. On prévoit les risques associés à une pathologie ou un médicament en ciblant les patients ayant une probabilité de subir des effets secondaires. La médecine personnalisée, lorsqu'elle implique la mise au point de traitements adaptés à un groupe de personnes ou de patients présentant des caractéristiques médicales génétiques communes, est une médecine stratifiée souvent appliquée au cancer. Lorsque l'on cible, parmi les patients atteints de cancers, le groupe de ceux chez qui on peut identifier l'efficacité thérapeutique maximale, on entre dans le cadre de la médecine stratifiée.
Les nombreux experts entendus par vos rapporteurs ont chacun donné leur définition personnelle du concept de médecine personnalisée et de ses nombreux avatars.
Selon le Pr Axel Kahn (5 ( * )) , « la médecine prédictive est celle qui permet de prévoir une susceptibilité particulière à certaines maladies ou à l'action de certains agents pathogènes, à partir de déterminants individuels de santé, génétiques ou autres. Mieux vaut parler de médecine de prévision ». Pour le Pr Bertrand Jordan (6 ( * )) , la médecine personnalisé, c'est « le bon traitement pour le bon patient au bon moment grâce aux avancées dans la connaissance du génome, de la génétique et des corrélations entre le génome et la physiologie ». M. Didier Tabuteau (7 ( * )) explique : « le concept de médecine personnalisée me semble englober d'une part, la pharmacogénétique et le ciblage des traitements, et d'autre part, le suivi et l'analyse de tests génétiques, de marqueurs biologiques sur l'état de santé de la personne, ou pour identifier des probabilités de développer des pathologies, voire pour détecter de futures maladies très en amont des signes cliniques... C'est une médecine qui se basera sur une analyse des caractéristiques biologiques et génétiques de la personne ».
Le Pr Agnès Buzyn (8 ( * )) précise « dans le cas du cancer, on dénomme médecine personnalisée le fait de génotyper la tumeur et de cibler les traitements sur ce génotypage ». Elle estime toutefois que cette approche est restrictive : « la médecine personnalisée doit comprendre la personnalisation des approches de prévention, de dépistage et pas uniquement des traitements de chimiothérapie. Aujourd'hui, on est au stade d'une médecine stratifiée, qui concerne un groupe de patients ; ce n'est pas encore de la médecine personnalisée, adaptée à un individu ».
Pour Mme Simone Bateman (9 ( * )) , le concept est flou, car il n'y a de consensus ni sur une définition, ni sur le champ que le terme recouvre, ni sur le point d'entrée qu'il convient de privilégier pour comprendre son développement. Toutefois elle relève que quelques idées reviennent avec insistance : « il s'agit d'une médecine faite sur mesure, en anglais, customised , ou tailored , par analogie avec les tailleurs, l'habillement ; des technologies rendent possibles cette nouvelle vision d'une médecine sur mesure ».
Quant au rapport de la Commission « Innovation 2030 », qui intègre la médecine personnalisée dans l'ambition n° 5, il évoque le concept de médecine individualisée partant du constat que la manière de se soigner en 2025 sera très différente de celle que nous connaissons actuellement. « Ainsi, il est d'ores et déjà acquis que la médecine saura personnaliser son diagnostic en fonction des caractéristiques propres de chaque individu et notamment de son génome . L'individu et ses caractéristiques propres seront, plus que jamais, au coeur de la médecine de demain avec une forte diminution des risques associés aux soins ».
On pourrait multiplier les exemples de tentatives de définition, lesquelles se recoupent en grande partie. Le concept « médecine personnalisée » devient une référence assez commune dans la publicité des grandes entreprises pharmaceutiques, voire dans la presse quotidienne et/ou hebdomadaire récente.
En réalité, la plupart des scientifiques et praticiens rencontrés par vos rapporteurs se sont accordés sur l'ambiguïté de la terminologie, sur la confusion qu'elle engendre mais aussi sur le fait que la «vraie» médecine personnalisée permettant d'identifier qu'un patient est différent parmi les milliards d'autres sur la planète est encore du domaine d'un futur proche pour certains, plus lointain pour d'autres. Pour nombre de nos concitoyens, la médecine personnalisée est porteuse d'une promesse, celle d'améliorer l'innocuité et l'efficacité du traitement médical que reçoit chaque patient en présence de maladies graves.
Cependant, une des dimensions, reconnue par tous et qui soulève de nombreuses questions éthiques, est le caractère prédictif de cette médecine. Fondant son approche sur le patrimoine génétique et l'environnement spécifique de chaque individu et, donc, sa susceptibilité particulière à certaines maladies ou à l'action de certains médicaments, la médecine personnalisée vise aussi à estimer le risque de survenue de telle ou telle maladie en fonction de ses déterminants génétiques croisés avec ses modes de vie. Quelles seront dès lors les attentes des patients surinformés de leurs éventuelles prédispositions ?
La saisine de l'OPECST arrive donc à un moment particulier où la médecine se trouve en transition. Elle doit recourir à des moyens technologiques de plus en plus sophistiqués dont les performances sont en augmentation constante. Ces techniques exigent de vastes connaissances dans des domaines variés. L'interdisciplinarité des équipes médicales devient progressivement une nécessité pour traiter des pathologies graves, ce qui provoque déjà et provoquera des changements de paradigme dans tous les secteurs de la santé. Aussi, vos rapporteurs se félicitent-ils d'emblée que l'impact de l'innovation soit reconnu et encouragé dans les grands axes de la stratégie nationale de santé lancée en février 2013 et actuellement débattue (10 ( * )) . En effet, il est de notre responsabilité de comprendre, d'informer et d'accompagner au mieux ces évolutions.
La médecine personnalisée, quelle que soit la définition retenue, implique un changement de paradigme dans l'approche du traitement et de la maladie qui entraîne des bouleversements socio-économiques importants dans le secteur de la santé et exige de garantir l'égalité d'accès au soin et la protection des citoyens.
I. UN CHANGEMENT DE PARADIGME DANS L'APPROCHE DU TRAITEMENT ET DE LA MALADIE
Les évolutions scientifiques et technologiques ont eu un retentissement considérable sur la médecine et la prise en compte rapide de ce processus est essentielle. On a découvert la complexité intrinsèque du vivant. « Plus on comprend le fonctionnement normal et pathologique des gènes, des cellules, des tissus, des organes, plus on constate la très grande diversité de leurs altérations, et donc de leur protection ou de leur réparation. En d'autres termes, la médecine devient individualisée parce que le vivant est individualisé » observe le Pr André Syrota (11 ( * )) . La découverte de nouvelles cibles, de nouvelles voies de signalisation, de leur modification au cours de telle ou telle pathologie, ouvre des perspectives nouvelles pour le traitement des patients. Progressivement et selon les pathologies traitées, on s'oriente vers un changement d'approche dans la conception des traitements, voire même des pathologies.
Ainsi est-on passé d'une pratique médicale fondée sur des données publiées pour parvenir à un jugement basé sur des statistiques irréprochables ( evidence-based medicine ) à la médecine de précision ( precision based medicine ) . La médecine de précision se réfère à la multiplication des paramètres d'analyse du patient, et surtout à l'utilisation des « omiques » en clinique, elle vise à l'analyse globale du patient et induit un changement de paradigme dans l'approche du traitement et de la maladie.
A. DES PROGRÈS SCIENTIFIQUES ET TECHNOLOGIQUES CONSIDÉRABLES
Les progrès des méthodes, des outils et des connaissances, couvrent un vaste champ, les avancées très rapides des technologies ayant accru les possibilités de mieux comprendre les processus pathologiques. L'analyse biologique dans le domaine expérimental et dans ses applications médicales est passée récemment d'une approche ponctuelle limitée à la détermination d'un ou d'un petit nombre de paramètres, à une approche globale c'est-à-dire à la prise en compte simultanée de l'ensemble du génome ou de ses différents niveaux d'expression.
Ces avancées techniques ont permis l'apparition d'« omiques ». Comme l'observe le rapport du Pr Jean-Yves Le Gall (12 ( * )) , « à la génomique, éventuellement limitée à ?l'exomique? , c'est à dire à la seule étude des séquences codantes et des séquences immédiatement environnantes, sont venues s'ajouter la ?transcriptomique? (analyse des ARN messagers ou transcriptome), la ?protéomique? (analyse des protéines), la ?métabolomique? (analyse des métabolites du métabolisme intermédiaire) ».
Cette évolution résulte d'une augmentation considérable et rapide des performances technologiques qui, relayée par la puissance de calcul de l'informatique permet aujourd'hui des analyses globales très rapides, qualifiées de « haut débit ». Le séquençage de l'ADN, les puces à ADN, la spectrométrie de masse, la résonance magnétique nucléaire, sont autant de nouveaux outils qui conjugués à la rapidité des avancées en numérisation, en bio-informatique, en nanotechnologies permettent de réaliser des analyses biologiques de plus en plus détaillées sur des surfaces de plus en plus réduites, d'enregistrer, de conserver, de transférer, de croiser, de grandes quantités de données biomédicales et de cibler, avec une extrême précision les thérapies.
1. L'évolution des techniques de séquençage du génome
L'ADN (13 ( * )) est une molécule, présente dans toutes les cellules vivantes, elle contient l'ensemble des informations nécessaires au développement et au fonctionnement d'un organisme. Transmis lors de la reproduction, il est le support de l'information génétique et de l'hérédité. La réplication de l'ADN, notamment lorsqu'une cellule se divise, implique un brin d'ADN à copier et une protéine spécifique, l'ADN polymérase. Cette protéine « lit » le brin modèle et met un A en face d'un T, etc . Le séquençage de l'ADN, consiste à déterminer l'ordre de la succession linéaire des bases A, C, G et T d'un fragment d'ADN donné.
Le séquençage d'ADN humain représente 3 milliards d'éléments groupés en 46 chromosomes, et sur ces 3 milliards d'éléments, on comptabilisera environ 10 % de différences entre les génomes de deux individus. Ces différences sont à l'origine de notre apparence, notre phénotype extérieur, mais aussi de nos maladies, de nos réponses aux médicaments et de nombreux autres phénomènes. Il existe deux outils majeurs pour observer l'ADN : le génotypage et le séquençage. Le génotypage vise à déterminer la nature d'une variation génétique à une position spécifique dans le génome, pour un individu. Il consiste à observer des endroits précis du génome et permet de connaître les variations génétiques d'une partie ou d'un génome entier. Le séquençage détermine l'ordre d'enchaînement des nucléotides pour un fragment d'ADN donné.
a. L'ampleur des évolutions
D'après Patrick Winkler, chef du laboratoire de séquençage du Génoscope (Institut de génomique du CEA) (14 ( * )) , la première version du génome entier fut rendue publique en 2001. En 2006, une nouvelle technique de séquençage plus rapide a permis une utilisation pour la recherche et offert des possibilités d'application. À partir de 2006 jusqu'à nos jours des techniques sont apparues pour séquencer de plus en plus vite, et on assiste actuellement à une diversification des méthodes de séquençage qui influe sur les pratiques.
i. Des méthodes de séquençage de plus en plus rapides
C'est Frederick Sanger, deux fois lauréat du prix Nobel de chimie (1958 et 1980) qui a mis au point la première méthode de séquençage de l'ADN : la méthode Sanger, encore très employée dans le monde. Cette méthode (15 ( * )) , basée sur l'interruption de la synthèse enzymatique d'un brin d'ADN complémentaire, comporte plusieurs limites. La plus importante est son faible rendement, car les étapes de préparation de l'échantillon d'ADN et d'amplification sont lentes et coûteuses, et la taille des séquences lisibles (inférieur à 1 000 nucléotides) est limitée.
Quelque temps après la publication du premier génome humain, des machines de nouvelle génération de séquençage à haut-débit (ou sous l'acronyme « Next Génération Sequencing » NGS ), bien plus rapides, ont été proposées par deux sociétés Solexa (depuis rachetée par Illumina) et 454 Life Science appartenant à Roche, marquant la première révolution du domaine (16 ( * )) . Elles utilisent un séquençage massif parallèle permettant de réduire les coûts et d'augmenter la rapidité des analyses.
La compagnie 454 Life Science , a mis au point la plateforme 454, qui s'appuie sur la technique de pyroséquençage, technique de séquençage par synthèse qui repose sur la détection de la libération de pyrophosphate lors de l'incorporation de nucléotides (17 ( * )) . La société Solexa (Illumina) a opté pour une stratégie plus proche de la méthode de Sanger. La molécule d'ADN à séquencer est mise en présence des quatre types de nucléotides, chacun associés à un fluorophore différent. Le nucléotide nécessaire à l'élongation du brin complémentaire est alors inséré, mais une modification de sa structure empêche l'élongation de se poursuivre. Un signal lumineux spécifique du nucléotide présent est alors émis.
Une évolution bien plus impressionnante est à l'oeuvre, la société Oxford Nanopore Technology (ONT) a présenté en 2012 un nouveau dispositif de séquençage : le Minion. La technologie de ce dispositif s'appuie sur des nano-pores (un pore d'un diamètre compris entre 1 et 100 nm), ce qui comporte deux avantages : la préparation des échantillons est très simple et rapide et il sera possible de lire des dizaines de milliers de bases en une seule fois. Cependant, le taux d'erreur (4 %) reste pour l'instant trop élevé pour la plupart des applications envisagées, dont le diagnostic médical.
Le Minion est une grosse clé USB jetable capable de lire directement l'ADN (jusqu'à 1Gb de données) à partir d'un échantillon de sang ; il a été testé sur des génomes simples (virus, bactéries) et a démontré son efficacité en décodant leur ADN en quelques secondes. Ce procédé, utilisé par la firme britannique, fait appel à une technique de séquençage dans laquelle les chaînes ADN traversent des micropores (2 000 dans le Minion) au niveau desquels chaque nucléotide est identifié.
ii. Des effets à court terme sur les connaissances et à plus long terme sur les thérapeutiques
Selon l'expression du Dr Laurent Alexandre (18 ( * )) , « on assiste actuellement à un véritable tsunami technologique dans ce domaine. Cela ne veut pas dire que l'on va découvrir un univers génomique déterministe, cela signifie simplement que personne n'avait prévu ce qui nous arrive. En 1990, on avait un consensus mondial dans la communauté des généticiens sur le fait qu'on ne saurait jamais séquencer intégralement les 3 milliards de paires de bases » .
En outre, l'on a aussi découvert que l'idée répandue depuis les années soixante-dix que 98,5 % de l'ADN ne servait à rien était erronée. Cette partie contient en réalité de nombreuses zones régulatrices très importantes qui influencent entre autres le câblage neuronal, la neuro-embryogénèse et l'immunité.
Pour le Pr Dominique Stoppa-Lyonnet (19 ( * )) , les progrès dus au séquençage du génome humain au cours de ces dix dernières années, et à la technologie de séquençage à très haut débit ou NGS ( Next Generation Sequencing ) ont multiplié les capacités d'analyse par un facteur de 50 000. « On est passé presque brutalement d'un séquençage besogneux de quelques gènes pour un patient dans le cadre d'une maladie donnée, à la capacité de séquencer un très grand nombre de gènes pour un grand nombre de personnes. Cela permet d'effectuer des tests et des lectures ciblés, voire même la lecture du génome entier, ou au moins les séquences qui codent pour ces gènes ».
Ainsi peut-on obtenir la séquence complète du génome, les modifications de l'expression des gènes, leur produit (protéome) dans les différents milieux biologiques, ainsi que le métabolome, (ensemble des métabolites ou des auto-anticorps, et la séquence des microbes peuplant notre tube digestif qui forment le microbiote intestinal. On est en mesure d'analyser ces données dans leur globalité.
Pour autant, pour la plupart des experts, si les méthodes d'analyse bénéficient très vite de ces sauts technologiques, pour la médecine, les progrès restent incrémentaux, car selon le Pr Florent Soubrier (20 ( * )) , « il faut que toute la chaîne soit impliquée, depuis l'imagerie, les paramètres biologiques, jusqu'aux traitements ». Néanmoins, on observe un renforcement du lien entre développement du séquençage à haut débit et extension de la médecine personnalisée.
b. La baisse drastique des coûts
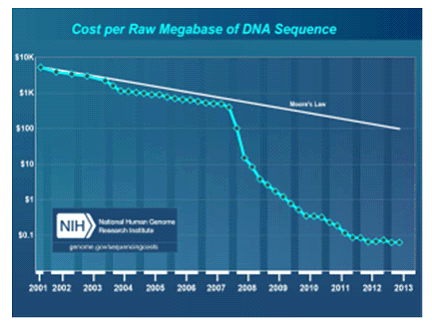
Le 26 juin 2000, M. Bill Clinton et M. Tony Blair annoncèrent que le consortium international public de séquençage Human Genome Project (HGP) et son concurrent privé lancé par M. Craig Venter , Celera Genomics Corp, avaient tous deux décrypté une première version de génomes humains ; cependant il faudra attendre avril 2003, avant que le premier génome humain soit déclaré entièrement séquencé. Au total, HGP aura coûté, sur quinze ans, environ 2,7 milliards de dollars (2 milliards d'euros). Dix ans plus tard, le séquençage des 3 milliards de paires de bases qui constituent le patrimoine héréditaire humain tend à coûter 3 millions de fois moins cher, soit entre 1 000 et 3 000 dollars et s'effectue en quelques heures seulement. La courbe qui décrit l'évolution du prix du génome décroit bien plus vite que celle suivie par la loi de Moore, laquelle prédit le doublement tous les dix-huit mois de la puissance de calcul informatique.

D'après M. Jean-François Deleuze (21 ( * )) , selon les moyens dont on dispose et l'évolution des coûts, on peut analyser la séquence totale, pour 3 000 euros, mais l'on ne dispose pas actuellement d'outils d'analyse des données aussi performants et rapides que les séquenceurs. Cependant, une évolution se profile dans les mois et années à venir. Actuellement, le Centre national de génotypage peut analyser de façon localisée des marqueurs, ce qui revient beaucoup moins cher, examiner des gènes ou un ensemble de gènes. Des efforts majeurs devront être fournis dans le domaine informatique en matière d'analyse, de capacité de calcul et de stockage. La baisse des coûts de séquençage, conjuguée à l'augmentation de la rapidité de réalisation, retentissent sur la personnalisation du traitement, dans les pathologies graves notamment.
Selon le Pr Bertrand Jordan (22 ( * )) , s'agissant du cancer, on peut se contenter d'analyser les 50 gènes les plus probables, ce qui est peu coûteux ; on peut également cibler jusqu'à 100 gènes et les séquencer en quelques jours pour quelques centaines d'euros en opérant sur 50 ou 100 patients à la fois, car une analyse individuelle serait trop chère. En revanche, si l'on veut séquencer le génome complet (25 0000 gènes) avec les séquences codantes, c'est bien plus long et bien plus cher : deux ou trois semaines et de 5 000 euros à 10 000 euros pour l'instant, sachant que l'interprétation n'est pas encore totalement automatisée et exigera des bases de données de qualité et une bonne infrastructure informatique. Il apparait d'ores et déjà que l'analyse des données issues du séquençage est plus coûteuse que le séquençage lui-même.
i. L'état des lieux en France
En France on constate une accélération ces dernières années dans l'évolution des équipements des hôpitaux, grâce au financement des projets « équipements d'excellence » (EQUIPEX) dans le cadre du grand emprunt ; de nombreuses équipes qui travaillaient sur la génétique, en bénéficient pour développer des plateformes d'analyses génétiques. S'y ajoutent 28 plateformes de l'INCa, (voir infra ) dédiées aux traitements du cancer. Les infrastructures françaises en génomique sont donc variées et réparties sur tout le territoire ; mais la question de la création d'un centre de référence d'intérêt national se pose.
• Créer un centre national de référence ?
Pour certains, comme le Pr Philippe Froguel (23 ( * )) , les infrastructures existantes sont suffisantes pour effectuer des diagnostics et point n'est besoin de créer un équipement national de plus qui exigera un certain temps pour le créer et des moyens supplémentaires. Ce point de vue semblait partagé par le Dr Catherine Bourgain (24 ( * )) . D'autres experts, ils sont les plus nombreux, tels le Pr. Jean-Paul Moatti (25 ( * )) , Pr Thierry Frébourg (26 ( * )) , le Pr Hélène Dolfus (27 ( * )) , le Pr Soumeya Bekri (28 ( * )) , militent fortement pour la création d'une plateforme d'intérêt national de qualité diagnostique, très performante, couvrant les besoins de tout le territoire, et représentant un niveau supérieur de diagnostic.
Cette plateforme doit être capable d'effectuer une analyse génétique globale et pour cela être pourvue de séquenceurs à très haut débit de troisième génération. Disposant de moyens d'analyse et de stockage, elle serait un pôle unique de référence afin de doter la France d'une infrastructure nationale évitant l'absence de diagnostic, voire l'errance diagnostique, notamment pour des patients atteints de maladies rares, car certaines maladies métaboliques doivent être traitées en urgence.
Il s'agit également là d'un problème de souveraineté nationale, on évitera de sous-traiter l'analyse de génomes entiers à l'étranger et l'on sauvegardera en France les données y afférentes. Il s'agit d'éviter de décrocher par rapport au Royaume-Uni, à la Chine, et aux États-Unis. Ces experts ont explicitement fait référence à une extension possible à l'horizon 2015 des capacités de l'Institut de génomique du CEA que vos rapporteurs ont visité dans le cadre de leur déplacement au Génopole d'Évry (29 ( * )) .
• Le Génopole d'Évry
Le Centre national de génotypage (CNG) avec le Centre national de séquençage (CNS), fait partie de l'Institut de génomique du CEA et compte 17 machines qui coûtent chacune un demi-million d'euros, selon son directeur, M. Jean-François Deleuze. En séquençage, le CNG est probablement le 2 ème centre européen, après le Sanger Institute du Royaume-Uni, en termes de capacité pour le nombre de machines localisées dans un contexte de plateforme. Cependant, en tant que séquenceur, il intervient dans le domaine de la recherche et n'a pas à l'heure actuelle de capacité en termes de diagnostic. Il est capable de séquencer quasiment trois génomes humains par jour. Le prix d'un séquençage de génome humain varie et continue sans cesse à baisser. Il avoisinerait 3 000 euros au CNG. Le CNG constitue donc un organe solide d'un grand apport pour la communauté scientifique française, conformément à sa mission.
Par ailleurs, le CNG est leader européen en matière de génotypage, c'est-à-dire pour l'examen d'altérations spécifiques. Le génotypage est la technologie qui permet de découvrir les prédispositions génétiques aux maladies. Il fonctionne depuis les années quatre-vingts, mais cette technologie en elle-même fera place au séquençage total du génome humain qui fournit une information globale. Le saut technologique apparu ces dernières années, permet maintenant le séquençage total du génome.
M. Jean-François Deleuze (30 ( * )) précise : « il coûte beaucoup plus cher et renseigne sur la totalité de l'information présente dans les gènes, non seulement les éléments fonctionnels, mais les éléments régulateurs de l'expression des gènes. Ce qui freine aujourd'hui le passage du génotypage au séquençage total, ce sont le coût et les outils d'analyse. En génotypage, on analyse 5 millions de marqueurs, alors qu'avec le séquençage, c'est 3 milliards de marqueurs » . Le CNG est capable de produire le séquençage, mais manque d'outils d'analyse suffisants pour traiter entièrement les informations recueillies.
Le CNG dispose de données sur des milliers d'exomes, qui occupent des quantités impressionnantes de stockage informatique. Les données produites proviennent d'échantillons totalement anonymes et aucune n'est exposée dans le domaine public ; pourtant certains estiment que regrouper les données permettrait d'acquérir une certaine puissance statistique, mais le risque demeure si des correspondants n'ont pas la même éthique. Le CNG s'est spécialisé dans la compréhension du séquençage ; il est partenaire de plusieurs projets internationaux notamment avec l'Allemagne, le Royaume-Uni, l'Espagne, la Norvège, l'Italie... car il existe plusieurs consortiums à travers les frameworks européens.
Le centre est doté d'un très grand centre de calcul disposant de deux salles de machines de 1 300 m 2 , avec des supers calculateurs 120 000 coeurs, 2-3 Pflop/s. Il est au service de la recherche et de l'industrie et dispose d'une extension dédiée à la communauté France-Génomique de 3.000 coeurs et 5 Po (gain x 10 sur les applications exomes). Les mutualisations sont possibles sur l'ensemble des calculateurs (voir photographie ci-dessous).
Au niveau financier, le centre a pour mission de favoriser la recherche nationale, mission de service public pour laquelle il dispose d'une dotation importante par rapport à celles des autres institutions du CEA. Cela lui permet de développer des projets qui lui sont propres et qui le rendent compétitif.
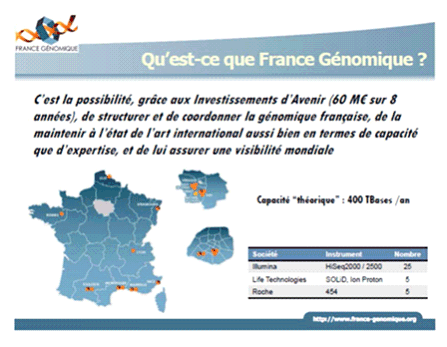
Le projet France Génomique coordonné par le CEA et comprenant l'INRA, le CNRS, l'INSERM, a pour mission d'intégrer et d'animer, à partir des possibilités nationales en génomique, un réseau d'expertise et de service destiné à la communauté scientifique pour lui fournir des capacités technologiques de pointe dans l'analyse du génome et le traitement informatique des données à haut débit. France Génomique a été dotée d'un financement de 60 millions d'euros sur dix ans dans le cadre des investissements d'avenir. C'est une initiative de structuration regroupant toutes les thématiques d'études y compris les questions logistiques qui sont très importantes.

Vos rapporteurs ont visité le Génopole d'Évry (31 ( * )) et ont été reçus par son directeur général, M. Pierre Tambourin, le directeur du CNG, M. Jean-François Deleuze et celui du CNS-Génoscope, le Pr Jean Weissenbach, qui ont observé qu'on assistait à une véritable révolution ayant des conséquences considérables au plan scientifique, comme au niveau de la société dans son ensemble.
Ces responsables ont reconnu que la France n'avait pas suffisamment anticipé le tournant du séquençage à très haut débit. Ils ont regretté le manque de masse critique en bio-informatique pour progresser et souligné la difficulté d'attirer, sur le long terme, des bio-informaticiens en raison de la durée trop brève des contrats que les centres sont tenus de leur proposer. Ils ont insisté sur le besoin de transdisciplinarité et de formation. Pour autant selon eux, le CNG et le CNS disposent d'atouts pour répondre à la nécessité, exprimée par plusieurs chercheurs, de créer un centre d'intérêt national. Il conviendrait de réfléchir à la mise en oeuvre rapide d'un tel centre auquel vos rapporteurs sont favorables.
La place de la France
Sur la place de la France dans ce domaine et les perspectives d'avenir, les opinions divergent quelque peu. Comme certains experts l'ont souligné, tels les Dr Laurent Alexandre et le Dr Christophe Le Tourneau (32 ( * )) , l'évolution actuelle de la bio-informatique n'est pas satisfaisante. Chaque centre refait à sa façon son logiciel d'analyse de variants, choisit son matériel s'adapte aux nouveaux systèmes etc . Selon le Dr Laurent Alexandre, « nous avons besoin de moins de bio-informaticiens qu'il n'y en a. En revanche, nous avons besoin de plus de concepteurs de logiciels bio-informatiques ». Cependant, définir une stratégie politique et médicale sur ces sujets, est délicat car les évolutions technologiques sont difficiles à anticiper et on ignore comment on se servira des nouvelles technologies, car l'on ne maîtrise quasiment pas en Europe la fabrication des appareils de séquençage qui viennent tous des États-Unis. De ce fait une grande partie des logiciels d'interprétation y sont développés, ce qui complique encore l'analyse des données.
ii. L'état des lieux à l'étranger
• En Europe
Le principal centre de séquençage en Europe est le Wellcome Trust Sanger Institute installé en Grande-Bretagne à Cambridge. Environ 500 chercheurs et techniciens y travaillent. Le centre dispose d'une quarantaine de machines de seconde génération et de machines de troisième génération. Les bio-informaticiens constituent le tiers des collaborateurs du centre. En outre, l'Institut de bio-informatique européen (EBI), situé sur le même campus que le Sanger Institute, dispose de 400 collaborateurs dont les thématiques s'articulent avec celles du centre. C'est un institut de référence qui représente le niveau supérieur de diagnostic.
Le Wellcome Trust Sanger analyse et rassemble les données de séquençage réalisées. Il s'agit de permettre l'exploitation, centralisée par le National Health Service (NHS ), des opportunités offertes par les technologies de génomique dans le but d'améliorer le diagnostic et les traitements, de faciliter et d'accélérer les résultats de recherche et leur translation en termes de bénéfices pour la santé et de retombées économiques, en soutenant le développement des entreprises britanniques de génomique et de bio-informatique. La taille importante des échantillons permet des études statistiques pouvant conduire à la découverte de nouveaux traitements et/ou moyens de prévention.
L'Espagne a développé un centre important spécialisé dans le domaine de la génomique du cancer : le CNAG ( Centro Nacional de Análisis Genómica ) à Barcelone. Son directeur, le Pr Ivo Gut, en a fait la présentation lors du 2 ème colloque de santé publique de l'ITMO (33 ( * )) . C'est un centre de 1 200 m 2 , intégré au Parco Cientifico de Barcelona (PCB). Il dispose d'une équipe pluridisciplinaire de 55 personnes, dont plus de la moitié des membres sont des informaticiens. Le CNAG possède 11 machines de séquençage de dernière génération. Il est le deuxième site en Europe ( ex-aequo avec le CNG d'Évry) en termes de concentration de séquenceurs. Le CNAG peut séquencer 4 à 5 génomes par jour. Ce centre a été imaginé à l'occasion de la création, en 2008, du consortium international de génomique du cancer ( International Cancer Genome Consortium - ICGC ), dont l'Espagne est l'un des neuf fondateurs.
• Dans le monde
Le premier centre mondial est le Beijing Genomics Institute ( BGI ), qui dispose à lui seul de 128 séquenceurs Solexa. Il constitue la plus grande plateforme mondiale de séquençage avec 3 000 bio-informaticiens et, à ce titre, il est un contributeur scientifique de premier ordre. Ses plateformes de haute-technologie lui permettent de générer des volumes de séquences et de typages à de très bas coûts. Avec des débits de 8,5 Tbases/jours, soit plus de 1 000 équivalents génomes humains par jour, le BGI a eu plusieurs premières à son actif, telles le séquençage du premier homme préhistorique. Il a contribué à hauteur de 1 % au projet génome humain de référence, et a démontré la faisabilité du séquençage du micro-biome du tube digestif humain. Première institution chinoise à séquencer le virus du SRAS, il a été un acteur clé dans l'analyse de l'épidémie d'Escherichia coli entéro-hémorragique qui a frappé l'Allemagne en 2011. Actuellement, le BGI développe un laboratoire de génétique cognitive avec pour ambition de répondre à des questions sur le fonctionnement du cerveau humain, sur la manière dont les gènes affectent la capacité cognitive ; l'éthique de la démarche scientifique ainsi engagée est cependant problématique.
Aux États-Unis (34 ( * )) , il n'y a pas véritablement de centre d'intérêt national de référence à l'échelon fédéral, les centres importants sont adossés aux grandes universités. Ils sont le fruit de la vaste collaboration entre les différents instituts de recherche du pays induite par le projet de séquençage du génome humain. C'est dans ce contexte que plusieurs instituts dotés de capacité en génomique ont vu le jour. Il en est ainsi, par exemple, du Broad Institute, cofondé en mai 2004 par l'Université de Harvard et le Massachusetts Institute of Technology (MIT). Il compte plus de 150 enseignants chercheurs de ces deux universités et plus de 1 500 scientifiques ont à ce jour participé aux projets collaboratifs du Broad Institute . L'un des principaux objectifs de cet Institut est l'exploitation des résultats du séquençage du génome humain. Centré sur la recherche fondamentale, il laisse aux industriels le soin de développer les molécules et les outils de diagnostic, et assure à toutes les communautés de recherche le libre accès à ses données scientifiques. Le Broad Institute projette l'ouverture d'un nouveau centre consacré à la recherche sur l'ensemble des circuits biochimiques intervenant à l'intérieur des cellules humaines. Il vise ainsi à dépasser le séquençage du génome en dénombrant et cartographiant l'ensemble des voies biomoléculaires qui régissent l'activité cellulaire et qui permettent d'expliquer comment le génome contrôle les cellules humaines.
Le projet du Broad Institute semble faire des émules car plusieurs autres grandes institutions de recherche, comme la Mount Sinai School of Medicine à New York, le Salk Institute à La Jolla, en Californie, ou l'Université de Californie à San Diego, préparent des plans similaires. Il est envisagé de créer un centre dont le but serait de coordonner les différents laboratoires de recherche et d'organiser le partage des données recueillies en s'inspirant de la Human Genome Organisation (HGO) constituée pour le projet de séquençage du génome humain.
Créé en août 2010, le New York Genomic Center (NYGC) (35 ( * )) est devenu un acteur important en génomique. Organisme indépendant à but non lucratif, il tend à fédérer l'expertise et les ressources d'universités, de centres médicaux, de compagnies pharmaceutiques, et de philanthropes privés pour mutualiser les efforts en recherche médicale et soins cliniques. Parmi les partenaires du NYGC ou membres fondateurs, on compte de grandes unités de recherche ou institutions de la région de New York: le Cold Spring Harbor Laboratory , l'Université de Columbia, la Cornell University avec le Weill Cornell Medical College , le Jackson Laboratory , le Memorial Sloan-Kettering Cancer Center , le Mount Sinai Medical Center , le NewYork Presbyterian Hospital , l'Université de New York avec la New York School of Medicine , l'Université Rockefeller ou l'Université Stony Brook.
Le NYGC cherche à développer des collaborations au sein des institutions impliquées dans de la recherche biomédicale à New York, avec une mission de recherche dans les domaines de la génomique et de la bio-informatique. L'organisme ne cache pas ses ambitions. Il entend bien devenir l'un des plus grands centres de génomique en Amérique du Nord et dans le monde en établissant un modèle de collaboration en médecine génomique à une échelle sans précédent.
Ce centre vise à promouvoir la médecine personnalisée au sein des membres fondateurs du centre qui sont amenés à traiter des millions de patients chaque année. Il souhaite favoriser le développement de produits thérapeutiques et de diagnostic en faisant le lien entre l'industrie pharmaceutique et la recherche au sein des hôpitaux afin d'accélérer le processus de recherche translationnelle en permettant à toutes les parties prenantes de mener des projets communs. En 2012, le NYGC a lancé un nouveau laboratoire pilote hébergé au Memorial Sloan-Kettering Cancer Center permettant à ses membres fondateurs d'accéder aux nouvelles technologies de séquençage. Avec des équipements de pointe dans le domaine du séquençage, le NYGC veut créer une synergie entre les différents établissements impliqués pour favoriser la recherche de nouvelles technologies de séquençage et de nouvelles applications Son centre d'innovation équipé de quatre séquenceurs Proton Ion de la société Life Technologies devrait pouvoir permettre, en quelques heures et à coût très limité (moins de 1.000 dollars), de séquencer un génome humain. L'accès aux technologies les plus récentes est une véritable opportunité pour les chercheurs des institutions new-yorkaises.
À ce contexte de leadership américain sur les technologies de séquençage s'ajoute la création récente de Calico , en septembre 2013. Sur ce point, le Dr Laurent Alexandre (36 ( * )) a expliqué que Google, qui en a annoncé le 18 septembre 2013 la création, nourrit de grandes ambitions dans le domaine de la génomique et du traitement de certaines pathologies. Calico sera une entreprise de biotechnologies dont le but est la lutte contre les effets du vieillissement ; elle sera indépendante de Google et dirigée par Arthur Levinson, spécialiste de biotechnologies, membre du conseil d'administration de Genentech et d'Apple. Cette création dans le domaine de la biologie, complète le domaine de la filiale de Google 23andMe spécialisée dans le séquençage ADN.
La naissance de Calico apparaît lourde de conséquences pour le monde de la santé, selon le Dr Laurent Alexandre. Liée à l'accroissement des potentialités techniques à l'oeuvre dans le domaine du séquençage à très haut débit, l'apparition d'une telle entreprise au sein d'une société qui gère des masses de données identifiantes peut inquiéter dans cette période charnière où le rôle des tests génétiques et des biomarqueurs dans les traitements s'accroît et où les biobanques s'arrogent un rôle et une place importants.
2. L'extension de l'usage des puces à ADN et la nano-médecine
Les nouvelles technologies permettent des études sur le génome entier qu'il s'agisse des puces à ADN ou des méthodes de séquençage haut débit, elles ciblent en plus des gènes, des régions non codantes qui sont pour certaines, impliquées dans la pathogenèse et représentent un apport non négligeable pour comprendre les maladies pour lesquelles la cause génétique n'est pas identifiée.
Le recours à des matériaux nouveaux et aux possibilités offertes par les nanotechnologies accroissent les capacités de ciblage et de suivi des patients.
a. L'usage des puces à ADN
Pour identifier les facteurs génétiques responsables de prédispositions (ou de protection) vis-à-vis des maladies communes, on utilise des puces constituées de fragments d'ADN (oligonucléotides) correspondant à des séquences données, déposées sur un support solide selon une disposition ordonnée ( array ). Le fonctionnement de ces puces repose sur une hybridation par des séquences complémentaires marquées par un fluorochrome. Les modèles sont basés sur des études effectuées à grande échelle (plusieurs milliers de malades et de témoins) par des études GWAS ( Genome Wide Associations Studies ) qui recherchent des déséquilibres de liaison entre le trait pathologique et des SNPs ( Single Nucleotide Polymorphism ) (37 ( * )) localisés dans une même région du génome.
L'évolution technique récente des puces ADN a été considérable. La miniaturisation par la technique des puces à ADN permet de mesurer et de visualiser très rapidement les différences d'expression entre les gènes et ce à l'échelle d'un génome complet. L'utilisation des puces à ADN comme outil de diagnostic présente l'avantage de faire appel à de nombreux marqueurs : plusieurs milliers de gènes peuvent être ciblés simultanément pour fournir une signature du type cellulaire étudié. Les puces à ADN permettent donc de comparer l'expression des gènes de deux types cellulaires différents, d'étudier les gènes exprimés sur un grand nombre de patients pour observer l'effet d'un médicament sur l'expression des gènes, et de comparer tissus sains contre tissus malades, traités contre non-traités, etc .
L'utilisation des biopuces permet en une seule expérience, qui dure environ deux jours, d'obtenir une estimation sur l'expression de plus de 30 000 gènes. Les puces à ADN de dernière génération ont la capacité de contenir et d'analyser environ 50 000 polymorphismes. La gestion de la masse d'informations obtenues repose sur des outils informatiques puissants permettant de collecter, traiter et archiver les signaux. Ces techniques sont en évolution constante vers l'amélioration de la sensibilité, l'augmentation de la capacité et de la rapidité, et la diminution des coûts.
D'après le Pr Bertrand Jordan (38 ( * )) , on peut désormais mesurer la sensibilité individuelle aux médicaments par une analyse sur le patient soit par séquençage, soit grâce à des puces à ADN, qui coûtent quelques centaines d'euros et permettent de savoir si le patient concerné réagira comme la majorité ou présentera des réactions particulières. Certaines puces à ADN sont conçues pour analyser des mutations dans certains gènes qui interviennent le plus fréquemment dans la résistance ou l'hypersensibilité à tels ou tels médicaments.
Ces puces sont développées en production industrielle et le passage du laboratoire à l'industrie s'est accompagné d'une amélioration de la qualité. Aux États-Unis, certaines sont accréditées uniquement pour la recherche, mais parfois utilisées par des laboratoires cliniques comme outils cliniques. La Food and Drug Administration ( FDA) s'y est intéressée et l'accréditation est nécessaire.
b. L'impact du développement de la nano-médecine sur la détection, le traitement et le suivi des patients
Les nanomatériaux sont à l'échelle nanométrique, 1 000 fois plus petits qu'une cellule ; ils ont des propriétés physiques, chimiques et biologiques particulières. Les nano-dispositifs, les nano assemblages moléculaires, se trouvent donc à la même échelle de taille que les biomolécules, c'est-à-dire toutes les molécules intervenant dans des processus pathologiques. Ces dispositifs sont plus sophistiqués que les puces ADN. Ils permettent d'effectuer du séquençage à haut débit.
Les nanotechnologies innovent au niveau du diagnostic personnalisé grâce à des microsystèmes pouvant être multi-paramétrés, plus rapides, capables de traiter un plus grand nombre d'échantillons ou de travailler sur des échantillons de bien plus petite taille. Ceux-ci sont accessibles dans les laboratoires centraux, comme au lit du patient, au cabinet du médecin traitant ou dans d'autres configurations.
L'utilisation de nano-vecteurs dans les thérapies ciblées permet le transport de petites molécules médicamenteuses, après une administration veineuse, nasale ou orale, et conduisent ainsi le traitement thérapeutique au bon endroit dans l'individu. D'après le Pr Patrick Boisseau (39 ( * )) , « l'encapsulation dans des nanoparticules permet de transporter des drogues extrêmement toxiques jusqu'au point souhaité, et pas à côté » et de limiter des effets secondaires délétères. Le troisième domaine d'utilisation est celui du suivi thérapeutique, où des capteurs peuvent être portés sur la personne, voire implantés dans la personne, sachant qu'à l'échelle nanométrique ou micrométrique et nanométrique, on arrive à les réduire pour mesurer des biomarqueurs ou la variation de paramètres physiques ou physico-chimiques. Les nano assemblages ont la même taille que les particules biologiques.
c. Un projet de carte d'identité métabolique pour adapter la posologie des médicaments
Selon M. Jean-Francois Deleuze (40 ( * )) , la posologie d'une drogue devrait être adaptée à notre contribution génétique par rapport an polymorphisme de nos enzymes. « C'est assez déterministe. C'est vrai pour une grande partie des enzymes du métabolisme des médicaments. Ainsi le cytochrome 2D6, enzyme hépatique prenant en charge de nombreux médicaments pour les détoxifier, est selon lui, très polymorphe et prend des formes très variables selon les individus. Certaines personnes n'en ont aucune copie et accumulent les drogues sans les métaboliser ; d'autres en ont de nombreuses, et vont donc métaboliser très rapidement . » Il importe de le savoir. On pourrait selon lui systématiser ces analyses pour adapter la posologie des médicaments au métabolisme de chaque patient pour un coût réduit.
Pour M. Jean-Marc Grognet (41 ( * )) , « plus que la seule détection des patients non-répondeurs à un traitement, le vrai Graal de la médecine personnalisée est d'administrer la bonne dose du traitement le plus efficace pour un patient donné. C'est l'ambition du projet de Carte d'identité métabolique (CIME) ». L'inefficacité d'un médicament peut s'expliquer de deux façons : soit l'interaction avec sa cible dans l'organisme est mauvaise, soit la concentration de molécules actives n'est pas optimale car elle est métabolisée trop vite par l'organisme ou elle n'est pas arrivée à passer les différentes barrières biologiques.
Si l'interaction avec la cible est liée au design du médicament, sa cinétique est variable d'un individu à l'autre. Elle dépend de l'action de protéines appelées « cytochromes P450 » pour le métabolisme et « transporteurs » pour le passage des barrières. « Il existe des centaines de cytochromes P450. Nous analysons la fonctionnalité d'une dizaine d'entre eux, essentiels dans le métabolisme des médicaments » explique Henri Bénech (iBiTec-S), responsable du projet CIME. « On administre à un sujet un cocktail d'une dizaine de médicaments commercialisés et dont le métabolisme est spécifique d'un cytochrome P450 ou d'un transporteur, puis en analysant les urines et le plasma, on détermine la quantité de médicament qui a réagi » (42 ( * )) . Ces données sont destinées à être intégrées, en plus des indications sur le patient et sa pathologie, dans un logiciel d'aide à la décision pour le médecin. Parallèlement à ces développements, d'autres instruments interviennent et leur perfectionnement permet des avancées notables.
3. Le perfectionnement des autres instruments
a. La spectrométrie de masse
Le principe de la spectrométrie de masse repose sur l'éventuelle fragmentation de la ou des grosses molécules (protéines) à analyser en ions qui sont ensuite séparés en fonction de leurs masses et de leurs charges. La technique ne pouvant analyser que des molécules ionisées et en phase gazeuse, son champ d'application ne concernait à l'origine que de petites molécules (sucres, acides aminés, stéroïdes, ...). Cependant, il a été ensuite considérablement élargi par la mise au point de techniques permettant de vaporiser et de ioniser les protéines ; encore plus récemment des développements technologiques ont étendu la méthode à des échantillons solides ou à des coupes de tissus.
La puissance des logiciels informatiques couplés à ces spectromètres permet d'établir la composition en acides aminés et même leur séquence. Les principales applications de la spectrométrie de masse portent sur la « protéomique», la « métabolomique », et l'analyse des structures complexes. C'est est un moyen très utile d'analyse fine du protéome (43 ( * )) .
b. La résonance magnétique nucléaire (RMN)
La résonance magnétique nucléaire (RMN) étudie des propriétés magnétiques des noyaux atomiques ; constitués de protons, neutrons entourés d'électrons, ils possèdent un moment dipolaire magnétique et un moment cinétique, responsables du phénomène de « spin ». Le moment magnétique de l'atome dépend du nombre de protons et de neutrons ; si ces deux nombres sont pairs ce moment est nul, ce qui explique que les atomes ou leurs isotopes étudiés par RMN correspondent à des chiffres impairs dans la classification de Mendeléiev.
D'après le rapport de l'Académie nationale de médecine sur les applications médicales de ces techniques (44 ( * )) , la RMN se prête en biologie à différents types d'applications, telles que l'identification et la quantification de composés, l'analyse du mécanisme de réactions chimiques ou enzymatiques ou celle de liquides biologiques, ainsi que les mesures de distances interatomiques et la reconstitution de structures 3D ou le criblage de molécules pharmacologiques. La sensibilité de la méthode peut cependant être améliorée par l'utilisation d'une sonde cryogénique, par l'étude de deux atomes différents ou par la transformation chimique préalable des molécules à analyser, ce qui est potentiellement utilisable soit pour le diagnostic de maladies héréditaires du métabolisme, soit dans une approche globale dite « métabolomique ».
4. La diversité des thérapeutiques personnalisées
La découverte d'un gène et de sa physiopathologie amène à envisager une thérapeutique personnalisée, qui peut relever de biothérapies : thérapie génique, thérapie cellulaire.
a. La thérapie génique
C'est une des voies privilégiées pour traiter les maladies génétiques et certains cancers. Elle consiste à insérer, dans les cellules du malade, une version normale d'un gène qui ne fonctionne pas et cause la maladie. Le gène fonctionnel permet alors au patient de produire à nouveau la protéine dont la déficience était la source de sa maladie. Pour réussir, l'opération est soumise à trois conditions : connaître la fonction du gène responsable de la maladie pour pouvoir « réparer » la cellule, permettre au gène d'entrer dans la cellule à l'aide d'un « vecteur », fréquemment un virus rendu inoffensif pour le malade, et associer le gène à une petite séquence d'ADN pour permettre son fonctionnement une fois dans la cellule. Dans ce champ nouveau qu'est la thérapie génique, les problèmes de sécurité de l'utilisation des rétrovirus demeurent.
À cet égard, on distingue deux périodes en thérapie génique. De 1997 à 2006, l'utilisation de rétrovirus a apporté quelques bénéfices indiscutables, des inefficacités et un certain degré de toxicité. De 2007 à 2013, on en a démontré l'efficacité en l'absence de toute toxicité liée aux vecteurs viraux modifiés utilisés. Ainsi, on compte pour la seule association AFM-Telethon, près de 36 essais cliniques en cours ou en développement pour différents organes. En ophtalmologie, ils portent sur le traitement de l'amaurose de Leber, une rétinite pigmentaire congénitale qui conduit à une quasi-cécité très précocement chez l'enfant, et sur la neuropathie optique héréditaire de Leber, maladie mitochondriale rare qui entraîne une perte de la vision brutale et asymétrique chez les adolescents et les jeunes adultes. Grâce à une approche de thérapie génique, le Pr José-Alain Sahel, directeur scientifique de l'Institut de la vision, espère empêcher la détérioration sensorielle, voire permettre une récupération après l'apparition des premiers symptômes.
Au niveau des déficits immunitaires innés, le Pr Alain Fisher, chef du service d'immunologie et hématologie de l'Hôpital Necker à Paris, participe à des recherches cliniques sur deux pathologies héréditaires : le syndrome de Wiskott-Aldrich (SWA), qui se traduit par des hémorragies, des infections récurrentes et des maladies auto-immunes, et la granulomatose septique chronique (GSC), qui inhibe la réponse aux infections microbiennes. Contre le SWA, les équipes françaises de Généthon travaillent en coopération avec l'Italie, la Grande-Bretagne et les États-Unis et ont développé un vecteur lentiviral afin de transférer des cellules souches hématopoïétiques. Contre la GSC, les chercheurs ont élaboré un vecteur lentiviral afin de restaurer la fonction de l'enzyme défaillante ; cet essai de phase I/II, mené en collaboration avec des médecins britanniques, allemands et suisses, devrait débuter courant 2014.
D'après le Pr Marina Cavazzana (45 ( * )) , la thérapie génique reste une source d'espoir grandissante pour toutes les maladies génétiques, monogéniques et autosomiques récessives. La thérapie génique représente non seulement un exemple extraordinaire de médecine personnalisée, mais également une approche thérapeutique importante pour les maladies acquises et les tumeurs. En effet, ces dernières années, l'apport des connaissances provenant des maladies héréditaires a également permis des progrès pour les maladies acquises.
b. La thérapie cellulaire
Elle consiste à greffer des cellules pour réparer ou régénérer un organe ou un tissu endommagé. Elle vise à restaurer les fonctions d'un tissu ou d'un organe lorsqu'elles sont altérées par un accident, une pathologie ou le vieillissement. La thérapie cellulaire utilise des cellules issues du corps humain, le plus souvent après une transformation réalisée dans un laboratoire pour traiter des maladies spécifiques ; le produit de thérapie cellulaire est en général préparé pour un patient donné. La thérapie cellulaire se distingue donc de l'ingénierie cellulaire et tissulaire qui conçoit au laboratoire un produit d'origine cellulaire et/ou tissulaire destiné à un usage thérapeutique pour un certain nombre de malades.
La thérapie cellulaire par greffe cellulaire est pratiquée couramment depuis plusieurs dizaines d'années, les greffes de moelle osseuse sont ainsi destinées au traitement des maladies du sang comme par exemple les leucémies et les anémies. Les greffes autologues, réalisées à partir de cellules du malade lui-même, ne posent en effet pas de problème d'immunotolérance. Les cellules souches adultes de la moelle osseuse donnent ainsi naissance à tous les types de cellules sanguines, les hématies (globules rouges), les leucocytes (globules blancs) et les plaquettes sanguines. De même, la greffe d'épiderme fait appel aux cellules du receveur lui-même, pour éliminer les risques de rejet. En vue de la greffe, on met en culture un petit fragment de peau afin d'en obtenir une plus grande surface.
Ces thérapies ont bénéficié des avancées scientifiques récentes sur les cellules souches et nourrissent chez des millions de patients l'espoir d'une médecine régénérative.
c. L'état des lieux des recherches : les potentialités de la thérapie cellulaire
La thérapie cellulaire repose sur les trois types de cellules souches : les cellules souches « adultes », les cellules souches embryonnaires humaine (CSEh) et les cellules souches pluripotentes induites (cellules iPS). Les CSEh ont une double propriété, de prolifération indéfinie (auto-renouvellement), et de différenciation dans tous les types de tissus (pluri-potence), elles offrent un nombre illimité de cellules capables de multiples destins cellulaires. Plusieurs centaines de lignées de CSEh ont été dérivées et caractérisées dans le monde ; elles sont utilisées par de très nombreux laboratoires. Les cellules souches « adultes » sont capables de se régénérer avec un moindre potentiel de renouvellement et de différenciation que les cellules souches embryonnaires. Cependant les cellules souches adultes participent au renouvellement des tissus, et peuvent être prélevées sur le patient, être cultivées puis réinjectées, sans risque de rejet. Quant aux cellules souches pluripotentes induites iPS connues sous leur acronyme anglais ( Induce Pluripotent Stem Cell) , elles permettent depuis 2007 à partir de cellules souches adultes de produire des cellules souches ayant les caractéristiques des cellules souches embryonnaires.
Depuis leur mise au point en 2006-2007 par le Pr Shinya Yamanaka, prix Nobel de médecine en 2012, les cellules souches iPS soulèvent de grands espoirs dans le domaine de la médecine. La stratégie du Pr Yamanaka consiste, grâce à un cocktail de quatre gènes, à faire retourner des cellules matures, à un stade proche de celui des cellules souches embryonnaires (dédifférenciation). Dès lors ces cellules induites, dites pluripotentes, peuvent donner naissance à tous les types de cellules présentes dans un embryon, et donc à tous les organes et tissus d'un organisme, sans pouvoir régénérer un embryon entier, avec ses annexes (placenta, sang de cordon), comme le font les cellules souches totipotentes, présentes au stade de blastocyste - moins de huit cellules - du développement.
i. Les recherches en cours
L'étude des CSEh, seules cellules souches pluripotentes physiologiques, a permis des avancées considérables dans la connaissance de l'embryologie, des gènes et réseaux moléculaires qui définissent et contrôlent l'état pluripotent, et qui conduisent une cellule souche vers un programme de cellule différenciée.
Quinze ans après la publication de la première lignée de CSEh aux États-Unis (1998), deux essais cliniques de phase I chez l'homme sont en cours. Ils visent à tester la sécurité, la faisabilité, et non l'efficacité, de l'approche. Ils sont menés par la société de biotechnologie ACT ( Advanced Cell Technology ) dans deux pathologies rétiniennes : la dystrophie maculaire de Stagardt, liée à une mutation génétique et la dégénérescence maculaire liée à l'âge (DMLA). En septembre 2011, la compagnie ACT a reçu l'autorisation d'étendre son protocole à des patients européens au Royaume- Uni. Des premiers éléments d'analyse ont été publiés en 2012 pour deux patients atteints de Stagardt, deux ans après l'administration des cellules ; ils confirment l'innocuité de l'injection des cellules et constatent une amélioration de la vision pour l'une des deux patientes, mais ce résultat doit être considéré avec grande prudence.
Un autre essai, dont Géron Corporation aux États-Unis était le promoteur a débuté en octobre 2011. L'indication était celle de traumatismes de la moelle épinière. Quatre patients ont été inclus, avant que l'essai ne soit arrêté par cette société pour des raisons financières. Selon l'Agence de la biomédecine, il reste cependant « toujours très difficile, même après quinze ans d'efforts de recherche, d'obtenir à partir de CSEh des cellules différenciées de type ?adulte? répondant aux stimuli qu'un organisme adulte émet. Un second défi, qui devra être résolu à l'échelle internationale, est celui de la nécessaire mise au point de tests pour définir et analyser les critères requis avant l'utilisation clinique de ces produits cellulaires. Or, ces cellules créent une situation nouvelle : aucun test standardisé n'est actuellement validé pour évaluer la sécurité, la distribution dans l'organisme et l'efficacité des produits cellulaires issus de ces CSEh » (46 ( * )) .
Quant aux cellules souches pluripotentes induites, (iPS) le fait que leur potentiel soit modifié par l'expérimentateur n'est pas dénué d'inconvénient : le risque d'instabilité et de cancérogenèse créé par ce procédé doit être évalué, et l'induction de programmes de différenciation efficaces à partir des iPS se heurte des difficultés, telle que la possible « mémoire » que pourraient avoir les iPS de leur tissu d'origine. La question cruciale de l'identité, ou de la divergence, entre CSEh et iPS est l'objet d'un important traitement scientifique. « Il reste à démontrer avec certitude si ces divergences - faibles - retentissent ou non sur le potentiel de ces iPS et sur leur possible exploitation thérapeutique » (47 ( * )) .
En théorie, les iPS pourraient en effet permettre des greffes autologues puisqu'elles peuvent être dérivées à partir d'un petit échantillon de peau ou d'un prélèvement de sang, ce qui évite l'administration d'immunosuppresseurs que requiert l'utilisation thérapeutique de cellules dérivées de CSEh allogéniques. Cependant on méconnait encore les risques réels que présentent les iPS dont on ne connaît ni la stabilité génétique in vivo , ni l'efficacité en l'absence totale de standardisation des préparations d'iPS en conditions de bonnes pratiques de fabrication (Good Manufacturing Approach GMP ). En revanche, les iPS restent un outil incomparable de modélisation de maladies humaines (plus de 150 lignées issues de maladies différentes) et de criblage en pharmacologie ou toxicologie, avec des résultats tangibles (48 ( * )) .
Quant aux cellules souches adultes, il apparaît qu'hormis les cellules souches hématopoïétiques et les cellules souches épidermiques, reconnues comme produits thérapeutiques en routine, peu d'autres types de cellules souches adultes sont en cours d'essais cliniques pour une thérapie de remplacement cellulaire. En revanche, les progrès des connaissances concernant leur fonctionnement in vivo ont ouvert des perspectives qui pourraient être très bénéfiques.
ii. L'état des lieux en France
La loi du 6 aout 2013 autorisant sous certaines conditions la recherche sur l'embryon et les cellules souches embryonnaires (49 ( * )) met fin à l'hypocrisie des lois relatives à la bioéthique du 6 août 2004 et de 2011 qui posaient un principe d'interdiction de la recherche sur les cellules souches embryonnaires mais avec la possibilité d'y déroger, « à titre exceptionnel » , avec l'accord du couple concerné, lorsque les recherches envisagées « sont susceptibles de permettre des progrès thérapeutiques majeurs et qu'aucune méthode alternative ne peut être envisagée, en l'état des connaissances scientifiques ». Ce régime dérogatoire illisible avait été dénoncé par toutes les instances interrogées, les experts et à diverses reprises par vos rapporteurs (50 ( * )) .
La loi de 2013 inverse totalement le principe et comme vos rapporteurs l'avaient préconisé, passe du régime d'interdiction à l'autorisation étroitement encadrée. On n'autorise pas n'importe quelle recherche, et le législateur maintient la condition imposée en 2004, selon laquelle seules seront autorisées les recherches qui ne peuvent pas être développées par d'autres moyens. De même, l'accord du couple à l'origine de la création de ces embryons est toujours exigé par la loi. Ces conditions restent contrôlées par l'Agence de la biomédecine qui délivre l'autorisation de recherche, en appréciant évidemment la pertinence scientifique de la recherche et les compétences de ceux qui l'entreprennent. Cette réforme devrait sécuriser les équipes et susciter de nouvelles vocations en évitant d'opposer les recherches sur les divers types de cellules CSEh, iPS et cellules souches adultes, les unes aux autres.
En France, un laboratoire public de 2 000 m 2 , CELLforCURE, consacré à la fabrication des médicaments issus de thérapies cellulaires pour traiter des pathologies graves sans alternative thérapeutique été inauguré en septembre 2013. Cette plateforme permettra dans un premier temps de valider cinq médicaments de thérapie cellulaire en cours de développement dans le cadre du projet C4C qui vise à produire des traitements de thérapie cellulaire en France et d'y développer une filière industrielle. C4C est porté par CELLforCURE et soutenu par Oséo.
Il s'agit de réparer des tissus abîmés grâce à des cellules souches pouvant devenir des cellules de peau, de muscle ou encore de coeur. Selon le président de cette plateforme, le Pr Pierre-Noël Lirsac, l'industrialisation des thérapies cellulaires, aujourd'hui réservées à des laboratoires d'excellence devrait démocratiser ce type de traitement. Les thérapies cellulaires, encore en phase d'essai clinique, concernent pour l'instant le cancer du sang, le diabète, l'incontinence anale, le mélanome et insuffisance cardiaque.
La plateforme de CELLforCURE est une filiale du Laboratoire français du Fractionnement et des Biotechnologies (LFB) et offre un programme de 80 millions d'euros, dont plus de 30 apportés par la Banque publique d'investissement au titre des Investissements d'avenir ; il s'agit de créer une passerelle entre recherche amont et production industrielle dans le domaine des thérapies innovantes.
À cet égard, c'est en oncologie que le concept de médecine personnalisée, voire de médecine génomique personnalisée selon les termes employés par le Pr Dominique Stoppa Lyonnet (51 ( * )) , a pris son essor car le lien entre l'analyse génétique et l'oncologie a été systématiquement effectué par tous les experts rencontrés par les rapporteurs.
B. L'ONCOLOGIE, DOMAINE ESSENTIEL DE LA MÉDECINE PERSONNALISÉE
Le terme de médecine personnalisée est né en cancérologie avec l'émergence des thérapies ciblées à la fin des années quatre-vingt-dix. Le Dr Christophe Le Tourneau (52 ( * )) , a d'ailleurs rappelé que l'Institut national du cancer américain définit la médecine personnalisée en cancérologie par l'utilisation des données biologiques des patients, afin de prévenir, diagnostiquer ou traiter.
Selon le Pr Axel Kahn (53 ( * )) , « La médecine personnalisée des cancers consiste à savoir en cas d'anomalies de gènes quelles modifications engendreront des signaux anormaux et quels médicaments inhibiteurs donner. C'est déjà largement une réalité dans le traitement des cancers et depuis longtemps pour déterminer les bases génétiques des effets des drogues ».
Pour le Pr Agnès Buzyn (54 ( * )) , dans le cas du cancer, on qualifie de médecine personnalisée le fait de classifier les tumeurs à partir d'une analyse génétique approfondie pour détecter les anomalies génétiques qui seront la cible de traitements ciblés. Elle ajoute cependant que cette approche est très restrictive et estime que cette médecine doit comprendre non seulement les traitements de chimiothérapie, mais encore la personnalisation des approches de prévention et de dépistage. Elle explique : « actuellement en cancérologie pour un grand nombre de patients on se situe dans le domaine de la médecine stratifiée ; ce n'est pas encore une médecine personnalisée adaptée à un individu ».
Si l'on en croit le Pr Agnès Buzyn, le terme médecine personnalisée en oncologie sera véritablement approprié dans le futur quand il sera possible de personnaliser les traitements au vu non seulement du génotypage de la tumeur, mais aussi du génotypage constitutionnel des patients pour déterminer ceux qui seront plus sensibles à la chimiothérapie que d'autres. Il ne suffira plus de génotyper la tumeur pour guider le traitement comme on le fait actuellement, on pourra aussi génotyper l'ensemble des cellules pour adapter les traitements de chimiothérapie, les doses en fonction du métabolisme des drogues... Ainsi, même pour un message de prévention, on peut imaginer selon elle qu'il sera possible d'être guidé par un génotypage. L'aide au sevrage pourra être adaptée à une addiction au tabac plus ou moins forte, selon le génotypage.
La plupart des oncologues interrogés s'accordent sur le fait qu'on entre dans l'ère de la médecine personnalisée, mais que l'on pratique plutôt actuellement une médecine stratifiée. L'objectif de l'Institut national du cancer INCa est d'assurer le passage d'une médecine stratifiée à une médecine personnalisée.
1. Le rôle moteur de l'INCa dans le développement des thérapies ciblées
Créé par la loi de santé publique du 9 août 2004, dans le cadre du premier Plan cancer 2003-2007, pour coordonner les actions de lutte contre le cancer, l'INCa a joué un double rôle d'une part, le développement d'expertises dans le domaine des cancers et la programmation scientifique, et d'autre part, l'évaluation et le financement de projets. Placé sous la tutelle des ministères chargés de la santé et de la recherche, l'Institut rassemble l'ensemble des acteurs de la lutte contre le cancer en France pour contribuer à diminuer la mortalité par cancer et améliorer la qualité de vie des personnes atteintes d'un cancer.
Son programme de travail actuel s'inscrit dans les axes du deuxième Plan cancer 2009-2013 dont la médecine personnalisée a été l'un des axes majeurs car les rédacteurs estimaient que la génétique somatique des cancers guiderait et personnaliserait les traitements ; il fallait donc s'organiser pour rendre accessible à tous les patients du territoire cette médecine personnalisée.
a. Le deuxième Plan cancer : le développement des plateformes et des thérapies ciblées.
L'objectif affiché de ce plan est de favoriser l'accès aux thérapeutiques ciblées, développées dès les années 2000 avec l'arrivée de l'Imatinib, d'aider à une meilleure compréhension de l'oncogenèse des tumeurs, et de développer des marqueurs pronostiques ou prédictifs de certaines tumeurs. Les thérapies ciblées sont des médicaments synthétisés de novo par les industriels pour inhiber une cible thérapeutique bien précise dans la cellule tumorale ou dans son environnement.
Selon la présidente de l'INCa, le Pr Agnès Buzyn (55 ( * )) , ce plan a abouti à la création de plateformes dédiées, financées pour partie par la Direction générale de l'offre de soins (DGOS) du ministère de la santé, par l'INCa, et parfois par une participation de l'industrie pharmaceutique. L'INCa en 2007 a identifié 28 centres ou laboratoires de génétique somatique, ceux qui réalisaient des analyses de génétique constitutionnelle. Avec des financements spécifiques, l'INCa a organisé un maillage territorial pour tous les patients dont la tumeur pouvait bénéficier d'un traitement ciblé, permettant l'envoi de leur prélèvement tumoral sur une plateforme de génétique moléculaire par le laboratoire d'anatomopathologie ayant effectué le prélèvement. Que ces patients soient traités dans le secteur privé ou public, l'INCa finance les tests compagnons et l'envoi des prélèvements par les anatomopathologistes. Il les rétribue pour l'organisation que représente le transport des fragments de biopsie.
Les premiers types de plateformes ont été consacrés à l'oncogénétique, avec des consultations d'oncogénétique associées, ciblant les facteurs de risque du cancer du sein et du cancer colorectal, afin de mieux identifier les familles à risque et donc les individus à haut risque de cancer. Le deuxième type de plateformes a été dédié à la génétique somatique.
Actuellement on compte 28 plateformes de génétique moléculaire labellisées INCa réparties sur l'ensemble du territoire (56 ( * )) , permettant de fournir la signature génétique de la tumeur de chaque patient. Il s'agit de rationaliser la prescription de médicaments coûteux sur la base de cette signature génétique, et d'améliorer l'accès aux thérapies ciblées. Aujourd'hui, l'INCa dispose de 17 thérapies ciblées dans le cancer, et les tests compagnons nécessaires à ces thérapies ciblées sont effectués sur ces 28 plateformes. D'après le Pr Agnès Buzyn, « le principe repose sur une égalité d'accès à ces tests sur tout le territoire. Tous les patients atteints de tumeur en France, qu'ils soient pris en charge dans le privé ou dans le public, ont accès à ces tests. Ils sont fiables, reposent sur un contrôle qualité, et ces plateformes ont un objectif de réactivité, c'est-à-dire que les tests compagnons doivent être prêts dès qu'une nouvelle thérapeutique est sur le point d'obtenir une autorisation de mise sur le marché (AMM) dans une indication donnée ».
Ces plateformes de génétique moléculaire ont permis de structurer l'accès aux thérapeutiques ciblées, elles fonctionnent à un niveau accéléré car on dispose de 17 thérapeutiques ciblées ayant une autorisation de mise sur le marché pour certains cancers exprimant certaines anomalies moléculaires qui nécessitent un test compagnon. On constate une montée en charge continue sur ces plateformes, avec plus de 60 000 patients ayant eu un test de génétique moléculaire de leur tumeur, et jusqu'à 155 000 patients testés chaque année si l'on considère le diagnostic et le suivi moléculaire de certaines hémopathies.
Le Pr Agnès Buzyn (57 ( * )) , a rappelé que « la structuration française est unique au monde, puisque tous les patients du territoire, où qu'ils soient traités, ont leur tumeur testée pour la présence d'une éventuelle anomalie génétique. Cela concerne les cancers colorectaux, du poumon, les leucémies, les cancers du sein. On prévoit, à l'évidence, une augmentation de ces tests dans les années à venir ».
Pour l'instant, cette organisation permet de réaliser un, deux, trois tests par tumeur, des tests unitaires, alors que les tumeurs exigeront la recherche de dix ou vingt anomalies à terme, pour accéder aux futurs médicaments. L'INCa estime qu'il faudra être en capacité de séquencer le génome des tumeurs des patients qui en auront besoin. Pour cela il faudra un changement d'échelle stratégique. Le deuxième Plan cancer a anticipé le fait que les traitements allaient cibler des anomalies moléculaires spécifiques des tumeurs pour pratiquer en quelque sorte une médecine de précision, stratifiée, car des patients ayant des tumeurs présentant les mêmes types d'anomalies pouvaient être accessibles aux mêmes traitements. Pour qu'ils puissent bénéficier de ces traitements ciblés, la mise en place des tests compagnons accessibles sur des plateformes de qualité était nécessaire et a pu être réalisée grâce à ce plan.
b. Vers le 3 ème Plan cancer
Selon le Pr Agnès Buzyn, l'INCa accompagnera l'évolution des plateformes de génétique moléculaire en capacité de séquençage, afin d'absorber l'identification de nouvelles cibles d'anomalies moléculaires pour de nouveaux tests, et d'anticiper des traitements en développement sur le point d'obtenir une autorisation de mise sur le marché. On estime qu'entre 90 ou 100 nouveaux médicaments ciblant des anomalies spécifiques seront disponibles dans les années à venir. « Jusqu'à présent les plateformes étaient adaptées à une ou deux nouvelles molécules de chimiothérapie qui arrivaient sur le marché, et obligeaient à faire des essais de phase 3 randomisés sur des milliers de patients pour juger de l'efficacité d'un traitement. Si nous avons à absorber l'arrivée de 90 nouvelles molécules en cancérologie dans les cinq ans qui viennent ciblées sur de petites populations de patients, il va falloir repenser notre modèle d'essais cliniques et d'évaluation du médicament » estime le Pr Agnès Buzyn (58 ( * )) .
« Faut-il s'intéresser uniquement à la tumeur, au diagnostic ou suivre l'évolution des métastases qui peuvent présenter des évolutions clonales avec d'autres anomalies génétiques apparaissant au fur et à mesure de la vie de la tumeur ? » s'interroge-t-elle (59 ( * )) .
D'autres questions se posent à l'INCa : celle du rythme de la mise en oeuvre du séquençage à haut débit dans les plateformes de génétique moléculaire et celle des possibilités techniques et en ressources humaines de bio-informatique de ces plateformes.
Selon l'INCa, le 3 ème Plan cancer, devra tenir compte du tournant du séquençage à haut débit, voire à très haut débit, et adapter la façon de faire de la recherche clinique. La prise en charge individualisée du patient devra prendre en considération des facteurs de génétique constitutionnelle aussi bien pour la prévention, (addiction à certains toxiques comme le tabac ou l'alcool) que pour les traitements.
Dans son rapport sur le 3 ème Plan cancer, le Pr Jean-Pierre Vernant (60 ( * )) rejoint les préoccupations exprimées par l'INCa. Il propose une simplification et une amélioration de la lisibilité de l'organisation territoriale de la lutte contre le cancer en conservant à l'INCa toutes ses responsabilités nationales, en donnant aux cancéropôles de larges responsabilités interrégionales. Dans le domaine de la prévention, du dépistage comme du soin, il propose l'usage d'indicateurs autorisant la mesure et le contrôle de ces inégalités pour mener des actions correctrices. Selon les auteurs du rapport, « le cancer peut être le terrain privilégié de la mise en place d'une nouvelle conception du soin qui se réfère autant au ?prendre soin? qu'à l'action de ?soigner? ... Les attentes du patient comme de son entourage doivent être prises en compte dans l'organisation du parcours de soin. Le rôle des associations peut contribuer à la mise en place de cette nouvelle conception du soin. Ainsi peut-on espérer faire reculer l'exclusion sociale et les regards stigmatisants ».
Observant que dans le domaine de la cancérologie, une adaptation rapide à des innovations continues est impérative, le rapport suggère que soit mis en place un comité indépendant chargé de proposer des ajustements et de nouvelles recommandations au fur et à mesure que des problèmes surgissent à condition qu'une évaluation en temps réel de l'impact des actions inscrites dans le 3 ème plan cancer soit effectuée. « Un plan cancer élaboré et corrigé de façon ininterrompue permettrait que soient exploitées rapidement et au bénéfice de chacun toutes les innovations » (61 ( * )) .
Vos rapporteurs se félicitent des efforts de l'INCa, ils demandent que le 3 ème Plan cancer prenne notamment en compte les inégalités d'accès sociales et territoriales et renforce les possibilités d'accès rapide aux innovations.
En effet, les avancées dans la connaissance du génome des tumeurs commencent à produire d'importantes évolutions dans la compréhension, l'analyse, la classification et le traitement des cancers.
2. Les tentatives de modélisation
a. Comprendre l'action de traitements efficaces sur les mécanismes moléculaires de la leucémie aiguë promyélocytaire
Cette leucémie est une maladie rare (100 cas par an en France) et grave du fait de l'existence de troubles de la coagulation qui mettent en péril la vie du malade dès les premières heures du diagnostic. Un taux de guérison de 25 % avait été obtenu par des chimiothérapies conventionnelles proposées par le Pr Jean Bernard en 1967. L'association de deux médicaments, une hormone, l'acide rétinoïque (1988, Wang) et un toxique l'arsenic (1995, Chen) conduit à la guérison de tous les malades.
Selon le Pr Hugues de Thé (62 ( * )) , l'acide rétinoïque in vivo provoque un changement de la morphologie des cellules malignes et la disparition progressive de celles-ci. Or la mise en évidence de la sensibilité de cette leucémie à cette hormone a permis la caractérisation de la translocation chromosomique à l'origine de cette maladie. Cette leucémie est provoquée par la fusion de deux protéines créant une protéine de fusion, marqueur moléculaire de la maladie qui induit son déclenchement. Or cette protéine comprend l'un des récepteurs de l'acide rétinoïque qui se trouve en fait être un traitement ciblé.
Le Pr Hugues de Thé a rappelé que les travaux de plusieurs équipes ont montré que l'acide rétinoïque et l'arsenic produisent la dégradation de la protéine en ciblant chacune de ses extrémités constitutives. « On se trouve donc face à ce que les théoriciens chinois appelaient combattre l'ennemi dans ses plans », en ciblant la protéine responsable du déclenchement de cette maladie, et ce de manière intelligente. Il existe des biomarqueurs permettant d'effectuer un diagnostic quasi immédiat de cette pathologie.
Ce modèle de thérapie ciblée assez extraordinaire au niveau des résultats cliniques offre un haut degré de compréhension des mécanismes moléculaires de ce cancer rare. Les mécanismes moléculaires à l'oeuvre dans cette pathologie pourraient aider à la compréhension d'autres types de leucémies, selon le Pr Hugues de Thé et Mme Valérie Lallemand (63 ( * )) .
On observe que des modèles souris très efficaces ont été obtenus et reproduisent parfaitement la physiopathologie, l'évolution, la réponse thérapeutique de cette maladie. Ils ont montré la synergie formidable de l'association des deux médicaments, permettant de détruire complètement la maladie et de guérir toutes les souris dans un temps record, alors que d'autres tests, non fondés sur des souris, avaient conclu à l'antagonisme fonctionnel de ces médicaments, conduisant à ne pas les associer chez les patients.
On commence d'ailleurs à explorer ce type de modélisation, en construisant sur la souris, des répliques biologiques de patients malades, pour pouvoir tester différents traitements sur ces cobayes.
b. Introduire dans des rongeurs les clones du cancer du patient
En janvier 2011, un article publié dans Molecular Cancer Therapeutics a relaté le cas d'un homme de 61 ans, atteint d'un cancer du pancréas avec des métastases résistantes à la chimiothérapie couramment utilisée dans ce type de cancer. L'équipe médicale a décidé d'implanter des morceaux de sa tumeur dans plusieurs cohortes de souris, créant ainsi des clones du malade et testant sur eux plusieurs traitements. La mitomycine C fonctionnant bien fut administrée au patient avec d'autres médicaments. Alors que la plupart des personnes atteintes de ce genre de tumeur ne survivent en moyenne que quelques mois, l'homme, au moment de la publication de l'article, avait fêté, sans aucun symptôme, ses trois ans de rémission.
L'utilisation d'un avatar personnalisé de cancer offre la possibilité d'essayer plusieurs combinaisons et même de faire des erreurs, avant de commencer un traitement. C'est la direction que prennent plusieurs équipes de recherche. Cette technique du double biologique ne se limite pas au cancer, elle est exploitée pour analyser in vivo le cas de personnes souffrant ou risquant de souffrir de diabète de type 1 selon le Pr Philippe Froguel (64 ( * )) .
La même opération est également tentée en créant, à partir des cellules souches du patient, des iPS afin de simuler la réplique à petite échelle d'un organe artificiel atteint d'une tumeur (65 ( * )) . Cette forme de modélisation a été décrite aux rapporteurs lors de leur visite à Gustave Roussy, elle pourrait se révéler utile pour tester la réaction aux traitements avant de les administrer aux patients.
Ces avancées de la connaissance couplées aux capacités d'analyses sans cesse accrues en termes de rapidité et d'efficacité conduisent à de nouvelles approches des cancers.
3. Des évolutions considérables en cours en oncologie : vers une nouvelle nosologie
L'hétérogénéité des tumeurs est apparue bien plus grande à mesure que des avancées scientifiques et technologiques ont montré que des pathologies telles que les cancers du sein ou du poumon, « ne sont pas une maladie unique, mais un mélange de 20 ou 30 maladies différentes (voire plus), liées à des mécanismes génétiques et moléculaires différents, et qu'il faudra donc traiter de façon différente », comme l'a souligné le Pr Thomas Tursz (66 ( * )) . Pour celui-ci et la plupart des experts, « en cancérologie, le point-clé est le bouleversement de la classification des tumeurs, qui jusqu'ici reposait avant tout sur l'examen anatomo-pathologique des échantillons tumoraux au microscope, et donc sur des aspects purement morphologiques » .
La classification histologique des tumeurs a permis d'établir des protocoles thérapeutiques permettant de guérir aujourd'hui environ 50 % des cancers, mais elle connait des limites. Elle ne permet pas de prédire qui rechutera après un traitement, qui développera des métastases, et qui répondra à tel ou tel médicament ou à une radiothérapie. C'est pourquoi, en recevant le même traitement, certains malades guériront et d'autres rechuteront.
Aussi, si le premier niveau de décision reste encore la localisation de la tumeur dans le corps pour décider du traitement, actuellement la communauté scientifique s'interroge. Faut-il toujours donner les traitements, en particulier les thérapies ciblées, en fonction de la localisation de la tumeur des patients ? Ou bien faut-il faire abstraction de la localisation tumorale et uniquement prendre en compte le profil moléculaire de la tumeur ? Cela implique de rechercher l'ensemble des anomalies moléculaires connues, prédictives de réponses à des thérapies ciblées, chez tous les patients, quel que soit leur cancer, afin de déterminer s'il existe une thérapie ciblée à proposer.
Cette approche, envisageable aujourd'hui, ne l'était pas il y a deux ans, car la technologie n'était pas prête. Il n'était pas possible en un coup d'oeil d'observer toutes les anomalies moléculaires d'une tumeur d'un patient parce que c'était trop coûteux et surtout trop long, donc incompatible avec la prise en charge des patients à l'hôpital.
Désormais certains oncologues se demandent si donner le traitement ciblé correspondant à la détermination du profil moléculaire de la tumeur chez chacun des patients atteints d'un cancer, est une stratégie plus efficace que la détermination des traitements à partir de la localisation tumorale comme cela se fait actuellement à l'hôpital. Cette question encore ouverte, est soulevée dans un essai intitulé SHIVA initié à l'Institut Curie et dans six autres centres (67 ( * )) .
a. L'essai SHIVA à l'Institut Curie
Lors de leur visite à l'Institut Curie, les rapporteurs ont pu s'entretenir avec le Pr Dominique Stoppa-Lyonnet et le Dr Christophe Le Tourneau (68 ( * )) . Ils ont visité les installations de ce centre de référence pour les cancers du sein, les tumeurs de l'oeil et les cancers pédiatriques auxquels deux unités hospitalières sont rattachées et qui traite en outre des patients atteints d'autres types de cancers.
Berceau de la radiothérapie, l'Institut Curie continue à innover dans les techniques de radiothérapie de haute précision, de protonthérapie, de curiethérapie, d'oncoplastie, d'oncogénétique. Son centre de recherche composé de plus de 80 équipes, au sein de 15 unités associées au CNRS, à l'INSERM et à des universités, rassemble biologistes, chimistes, physiciens, bio-informaticiens et médecins. Leurs recherches sont pluridisciplinaires et les équipes s'appuient sur des plateformes de pointe en imagerie cellulaire, bio-informatique, génomique et protéomique que la mission a visitées. L'Institut tend à assurer le passage de la découverte scientifique à l'innovation médicale, il valide les concepts permettant de mettre au point des techniques diagnostiques et des approches thérapeutiques nouvelles, plus efficaces et mieux tolérées.
C'est pour valider un nouveau concept qu'est organisé l'essai SHIVA. Il s'agit d'un essai comparatif dont le Dr Christophe Le Tourneau est à l'origine. On extrait l'ADN des cellules cancéreuses pour l'envoyer à des plateformes de séquençage qui réalisent la carte génétique de la tumeur, produisent des données qui sont ensuite transférées à des bio-informaticiens qui les analysent pour transmettre un compte-rendu compréhensible par les médecins et interprété par les biologistes. Ainsi les médecins, disposent d'un compte-rendu de la carte génétique permettant de savoir si la tumeur présente une anomalie moléculaire pertinente pour laquelle on peut donner un traitement.
Si on découvre une anomalie moléculaire, le traitement ciblé qui lui correspond est proposé au patient. En effet, si une thérapie ciblée est efficace contre un type de cancer, pourquoi ne le serait-elle pas pour les autres porteurs de la même anomalie moléculaire ? L'essai SHIVA lancé en septembre 2012 vise à répondre à cette question. Il sort du dogme du traitement de l'organe pour traiter la maladie en fonction de son profil moléculaire. On pose l'hypothèse que chaque individu est unique et a besoin d'un traitement individualisé, adapté à son propre organisme, à son profil génétique, en fonction de son âge et de son environnement. Lorsqu'une anomalie est identifiée, les patients bénéficient d'une thérapie dite ciblée.
Contrairement aux chimiothérapies classiques qui agissent sur toutes les cellules en division induisant des effets secondaires comme la perte de cheveux ou les nausées, les thérapies ciblées reconnaissent spécifiquement l'anomalie présente sur les cellules tumorales. Ces thérapies ne peuvent être proposées qu'aux patients porteurs de l'anomalie en question. Jusqu'à présent les anomalies recherchées dépendaient de la localisation tumorale initiale. « Nous changeons totalement d'approche en recherchant systématiquement chez les patients toutes les anomalies biologiques pour lesquelles il existe des thérapies ciblées, quelle que soit la localisation de leur maladie » a expliqué le Dr Christophe Le Tourneau, coordinateur de cet essai de nouvelle génération.
Au total, la recherche d'une quarantaine d'anomalies moléculaires sera réalisée dans les prélèvements de 1 000 patients. « Environ 20 % des patients devraient être porteurs d'une anomalie pour laquelle on possède une thérapie ciblée » précise le Dr Le Tourneau. Cette analyse permettra ensuite de proposer l'une des thérapies ciblées actuellement disponibles (11 dont 3 hormonothérapies).
Dans cet essai de phase II, il s'agit d'évaluer l'efficacité d'une médecine reposant entièrement sur le profil moléculaire de la tumeur. Les patients seront donc répartis dans deux groupes par tirage au sort, l'un recevant le traitement conventionnel, la chimiothérapie validée à ce jour en fonction de la localisation et de l'évolution tumorale, l'autre bénéficiant d'une des onze thérapies ciblées selon leur profil biologique.
Le recrutement des 1 000 patients s'effectuera dans six centres de lutte contre le cancer (Centre Léon Bérard, Lyon, Institut de Cancérologie de l'Ouest-René Gauducheau, Nantes, Institut Paoli Calmettes, Marseille, Centre Georges-François Leclerc, Dijon, Institut Claudius Regaud, Toulouse, Centre Alexis Vautrin, Nancy). Les analyses seront centralisées à l'Institut Curie.
Cet essai implique une dizaine de personnes : des chirurgiens, des radiologues, des anatomo-pathologistes, des d'oncologues médicaux, des radiologues, des généticiens et des bio-informaticiens. Il repose sur la capacité à établir le profil moléculaire de la tumeur et à en tirer des conclusions dans un temps compatible avec la prise en charge médicale.
Une partie des prélèvements sera également séquencée complètement grâce aux technologies de séquençage à haut débit acquises dans le cadre du projet ICGex « Équipement de biologie intégrative du cancer pour une médecine personnalisée » pour des analyses complémentaires.
« Avec l'essai SHIVA, nous allons explorer de nouveaux territoires de la médecine personnalisée », explique Dr Christophe Le Tourneau, « pour la première fois le choix thérapeutique sera fonction du profil moléculaire de la tumeur sans tenir compte de l'organe dans lequel elle s'est développée ».
Cette approche est comparée aux traitements
donnés au patient qui ne sont pas inclus dans cette étude. Depuis
le mois d'octobre 2012, un peu plus de 130 patients ont été
inclus dans ce protocole, et les inclusions continuent. L'essai SHIVA
génère un flux de données qui partent des données
cliniques des patients et vont jusqu'à celles de la carte
génétique de la tumeur, puis jusqu'à la décision.
«
Ces données doivent être envoyées à
certains moments de façon automatisée. C'est un réel
défi, car à la fin, il faut que le compte-rendu de la carte
génétique du patient, non seulement corresponde bien au bon
patient, mais encore que celui-ci soit validé et soit de qualité.
Tout cela nécessite des compétences que nous n'utilisons pas
d'habitude en pratique à l'hôpital
» a
expliqué le Dr Christophe
Le Tourneau
(69
(
*
))
.
L'essai SHIVA, ou d'autres essais qui démarreront en France ou à l'étranger ouvrent la voie à une stratégie qui consistera effectivement à adapter les traitements en fonction de la carte génétique des cancers des patients, et non plus en fonction de la localisation tumorale primitive, comme l'a souligné le Pr Véronique Trillet-Lenoir (70 ( * )) qui, elle aussi, constate un démantèlement de la nosologie des tumeurs : « à l'heure actuelle, rien ne peut ressembler autant à un cancer du sein qu'un cancer du poumon ou de la prostate, pour peu qu'ils partagent les mêmes anomalies moléculaires. On n'assiste donc pas tant à l'émergence de tumeurs rares et de niches, mais plutôt à une juxtaposition de tumeurs rares. Le cancer du poumon est actuellement la juxtaposition de 150 formes rares de cancers du poumon ». Fort de ces constats plusieurs autres essais cliniques d'ampleur sont en cours, la mission s'est intéressée à un essai clinique mené dans le cadre de Gustave Roussy Cancer Campus.
b. Les essais cliniques MOSCATO à Gustave Roussy
i. Le rôle de Gustave Roussy Cancer Campus
La mission a visité Gustave Roussy (71 ( * )) , lieu vaste et impressionnant, premier centre européen de prise en charge du cancer : il compte 2 600 professionnels, 46 000 patients suivis, dont 12 000 nouveaux chaque année, réunis sur le même site. Le Pr Alexander Eggermont, directeur général, a présenté le centre. Gustave Roussy prend en charge les adultes atteints de cancers rares ou fréquents, à tous les stades de la maladie, au sein d'un établissement où soins, recherche et enseignement sont étroitement liés. Il est spécialisé dans les cancers du sein, seule pathologie pour laquelle les patientes consultent directement à Gustave Roussy ; pour les cancers rares et les cancers de la tête et du cou, ceux relevant de l'ORL, les tumeurs solides, les patients sont adressés par d'autres centres. Il dispose d'une pharmacie robotisée, ce qui lui permet une fabrication en interne des chimiothérapies.
C'est un centre de référence en France, mais aussi à l'échelle internationale, avec une base de données concernant des milliers de patients. Ses missions vont du soin aux personnes atteintes de cancer à la mise au point des thérapies nouvelles et à la diffusion des connaissances dans les communautés médicales et scientifiques françaises et internationales.
Gustave Roussy fonde sa spécificité sur l'innovation thérapeutique, le développement de la médecine personnalisée, la qualité et la sécurité des soins, en plaçant toujours le patient au centre de ses missions (72 ( * )) . Il s'est doté d'un nouveau bâtiment de médecine moléculaire opérationnel depuis septembre dernier. Gustave Roussy et Sanofi ont conclu un partenariat de recherche clinique dédié à la médecine personnalisée en oncologie afin de faciliter l'accès de patients aux nouvelles opportunités thérapeutiques les plus adaptées aux caractéristiques moléculaires de leur tumeur dans les cancers avancés. Conclu pour une durée de trois ans avec une extension possible, ce partenariat prévoit que Sanofi confiera la réalisation d'essais cliniques de phase I ou I/II au service des innovations thérapeutiques précoces (SITEP), centre créé en 2008 par Gustave Roussy pour la prise en charge des patients participant à des études cliniques de phase précoce.
Selon le Pr Éric Solary, directeur de la recherche, les grands axes de la recherche portent sur les mécanismes de réparation de l'ADN, la réponse immunitaire tumorale, les tumeurs du sein et du poumon. Les gains en termes de survie sont possibles dès lors que les traitements sont administrés dans un délai raisonnable. On modélise et on utilise des algorithmes adaptés au profil du patient.
ii. L'essai MOSCATO
C'est dans ce cadre que Gustave Roussy a lancé l'essai MOSCATO qui vise à valider, dans la pratique clinique, la mise en oeuvre des techniques d'analyses à haut débit utilisées pour dresser le portrait moléculaire de la tumeur de chaque patient. L'essai MOSCATO cherche à sélectionner les patients porteurs d'une anomalie moléculaire contre laquelle il est possible d'agir. On tend à une validation clinique du concept de médecine personnalisée.
L'étude préliminaire porte sur 129 patients atteints d'un cancer du poumon dont 112 ont bénéficié d'un traitement. On a retrouvé dans près d'un cas sur deux, soit chez 53 malades des mutations ponctuelles et des aberrations chromosomiques impliquées dans les mécanismes de la formation des tumeurs. Pour 33 d'entre eux, ces anomalies se sont avérées correspondre à une cible thérapeutique, ce qui leur a permis de recevoir rapidement un traitement ciblé attaquant spécifiquement les cellules tumorales, à la différence des chimiothérapies classiques agissant aussi sur les cellules saines. Près de 50 % des patients ont accepté de subir une biopsie et 33 % ont pu recevoir un traitement approprié avec un taux de réponse de 20 % supérieur aux valeurs habituelles, L'objectif de recrutement total porte sur 900 patients et cette étude pourrait durer trois ans.
c. Les progrès espérés grâce au séquençage des tumeurs
On constate que même les tumeurs primaires sont hétérogènes et il n'est pas rare de découvrir des mutations différentes au sein de la même tumeur. Dans ce cas on risque de ne traiter qu'une partie des cellules tumorales et la tumeur reprend après un traitement efficace qui l'a réduite, car interviennent quelques clones non sensibles au premier agent de traitement. Selon le Pr Thomas Tursz (73 ( * )) , la découverte des oncogènes et de leurs mutations permet de comprendre les mécanismes de la transformation d'une cellule normale en cellule cancéreuse. Grâce aux techniques nouvelles de séquençage du génome on analyse rapidement les anomalies génétiques des cellules cancéreuses d'un malade précis et il devient possible de prédire, dans une certaine mesure, quelles devraient être la structure et les propriétés d'un médicament potentiellement efficace pour contrecarrer l'effet de ces anomalies génétiques.
Les biologistes du cancer demandent aux chimistes de préparer « à façon » un médicament dont les cibles moléculaires précises et les propriétés devront être prédéfinies. On ne fabriquera plus « à l'aveugle » des milliers de molécules, mais on synthétisera à la demande la molécule qui ira se fixer exactement à tel endroit, dans telle poche, dans tel repli de telle cellule.
Pour le Pr Bertrand Jordan (74 ( * )) , le traitement standard pour chaque cancer s'éloigne la distinction des cancers selon les organes du corps. Les mutations trouvées dans ces différents cancers par organe ne sont pas spécifiques. « Il existe une cinquantaine ou une centaine de gènes souvent mutés dans les cancers et on retrouve les mêmes mutations dans différents cancers de différents organes. C'est le résultat de milliers d'analyses moléculaires faites ces deux ou trois dernières années. On ne peut plus se permettre aujourd'hui de spécialiser l'oncologie strictement par organe ».
Ainsi la nouvelle nosologie du cancer est une révolution qu'il faudra assimiler car elle aura des conséquences lourdes sur les concepts, les technologies, l'organisation des soins, les traitements dont les premiers médicaments ciblés, apparus depuis quelques années, ne sont que les précurseurs. De plus, selon le Pr Thomas Tursz (75 ( * )) , il sera assez fréquent de devoir traiter la tumeur d'un patient avec l'association de plusieurs molécules différentes parfois synthétisées par des laboratoires pharmaceutiques différents ce qui exige des collaborations efficaces qui n'ont toujours pas cours actuellement.
Pour le Pr Laurent Degos (76 ( * )) , il ne s'agira plus « de prescrire des traitements du cancer du sein, de l'intestin ou d'un autre organe mais de traiter en fonction des anomalies de la cellule maligne quel que soit l'organe. L'on a déjà connu un tel changement de paradigme médical lors de la découverte des antibiotiques : le traitement est spécifique du germe et non de l'organe ».
C. LA DIVERSITÉ DES PATHOLOGIES CONCERNÉES PAR LA MÉDECINE PERSONNALISÉE
Les changements de paradigme induits par la personnalisation des traitements concernent aussi des pathologies diverses dans leurs causes et leur impact, telle les maladies infectieuses, certaines maladies chroniques, et un grand nombre de maladies rares qui ont longtemps servi de modèle de Recherche & Développement de traitement.
1. Les maladies infectieuses
Les maladies infectieuses, au sens très large du terme, demeurent la deuxième grande cause de mortalité dans le monde.
a. Les marqueurs d'évolutivité et de pronostic des maladies infectieuses
On constate une inégalité devant les maladies infectieuses. Une petite fraction de la population est atteinte d'une forme très grave de l'infection, alors que la majorité n'aura qu'une forme intermédiaire, quand d'autres n'auront rien du tout. Il s'agit de savoir qui sera atteint de formes graves. La France est très en pointe dans la détermination de ceux qui risquent d'être atteints d'une forme sévère de tuberculose ou de choc septique grave. Des équipes françaises ont mis en évidence des marqueurs, en particulier des récepteurs de cytokine.
On progresse sur les facteurs liés à l'hôte, on enregistre déjà un certain nombre de succès notamment sur la réponse dans le traitement des hépatites, en particulier des hépatites C car il existe un marqueur de réponse à l'Interféron, avec une expression génique, et ensuite protéique. Associées à ce traitement à l'Interféron, arrivent depuis deux ans, d'autres molécules, plus ciblées, et qui surpassent l'efficacité de l'Interféron. Ainsi, selon la puissance des agents infectieux dont on dispose au départ, le marqueur de sensibilité aura un rôle plus ou moins important.
b. Les réactions aux vaccins
On ignore pourquoi, environ 3 % des personnes ne répondent pas à un vaccin ; on a beau les vacciner, les revacciner. Ils ne répondent pas à tel vaccin, mais répondent à tel autre. On constate qu'il existe une relation entre la génétique de l'hôte et les peptides des bactéries ou virus contre lesquels on vaccine. Selon le Pr Jean François Delfraissy (77 ( * )) , on a mis en place, dans le cadre de l'Institut thématique multi-organisme ITMO « Microbiologie et maladies infectieuses », une banque de données sur les non-répondeurs pour essayer d'expliquer pourquoi certaines personnes ne répondent pas aux vaccins, car la capacité de comprendre ce type de phénomènes permettra éventuellement de développer des vaccins du futur. De même certains vaccins ont des effets secondaires sur des personnes, il importe de comprendre pourquoi (78 ( * )) .
c. L'identification des pathogènes et le suivi des traitements
Selon le Pr Bertand Jordan (79 ( * )) , on séquence la variété d'un virus ou d'une bactérie qui affecte un malade à un moment donné pour connaître ses résistances aux antibiotiques, aux différents traitements et suivre l'évolution du pathogène au cours de l'infection. Cela permet notamment dans le cas d'une infection hospitalière de pister ce qui se passe, d'identifier précisément la souche. On identifie un staphylocoque de façon très précise par l'ensemble de son génome, on peut voir qu'il est présent chez des porteurs asymptomatiques. À titre d'exemple, on peut comprendre comment une unité hospitalière qu'on a nettoyée, se réinfecte car un membre du personnel est porteur asymptomatique.
Pour le Sida, on génotype le virus, on dispose d'une quinzaine de molécules et on choisit pour chaque malade les trois molécules contre lesquelles il n'a pas de résistance. Mais cela varie parfois dans le temps. Si le patient n'observe pas toujours bien son traitement, le virus se remultiplie en opposition au produit donné et acquiert une mutation de résistance. Les traitements se restreignent, d'autant plus que la personne peut ensuite transmettre à autrui ce virus déjà mutant, déjà résistant. On assiste donc à une course à la recherche de nouvelles molécules. Chaque mutation du virus signifie une résistance à une molécule. On se trouve en fait face à une personnalisation plus tellement sur le malade, mais sur le virus. C'est bien sûr indissociable de l'identification d'un marqueur.
Sur les effets secondaires des médicaments utilisés dans le traitement du VIH, on constate que l'Abacavir entraîne des intolérances graves, voire quasi mortelles chez certains patients qui ont un HLA ( Human Leukocyte Antigene , antigène de leucocyte humain) particulier. Un marqueur permettant de ne pas recourir à l'Abacavir, est utilisé en pratique courante, pour éviter les effets secondaires.
En outre, la « Precision Medicine » s'adapte parfaitement bien au cas de patients déjà traités pour des maladies virales chroniques, comme l'hépatite B ou le VIH. Ils se portent bien, mais les effets secondaires de ces traitements sont lourds. Une certaine forme de sous-classification de ces patients, est nécessaire. Des biomarqueurs permettraient d'obtenir des éléments plus ciblés autour de tel ou tel type de médicament.
d. Le développement de petites unités de microbiologie au lit du patient
La possibilité d'effectuer de la microbiologie sans culture, au lit du patient, et d'obtenir en moins d'une heure des conditions de diagnostic marque un tournant dans la personnalisation des traitements anti-infectieux. Cela n'est pas encore très courant, ni en France, ni même aux États-Unis. En France des biomarqueurs de sensibilité à différentes molécules, d'identification et de résistance, devraient être disponibles d'ici deux à trois ans, et dans les hôpitaux d'ici cinq ans. Grâce à la technique dite « GeneXpert» il est possible d'effectuer le diagnostic de la tuberculose et de la sensibilité de la tuberculose en moins d'une demi-heure. « Auparavant, il fallait six semaines de culture. Cela change complètement la donne, que ce soit au nord ou au sud. Au niveau du séquençage de bactéries, on peut aujourd'hui le faire dans un cadre hospitalier, rapidement, pour un coût tout à fait bon marché et cela change les dimensions du possible » a expliqué le Pr Bertrand Jordan (80 ( * )) .
À Marseille le regroupement des laboratoires hospitaliers de biologie médicale dans une seule unité s'est accompagné de la création de petites unités de microbiologie équipées pour effectuer rapidement et près du lit du malade les analyses urgentes (81 ( * )) .
Selon le Pr François Delfraissy (82 ( * )) , pour aborder ces problèmes, il ne sera peut-être plus nécessaire d'effectuer de grands essais randomisés longs et coûteux pendant une très longue période. Si l'on sélectionne les patients au départ, on peut progresser bien plus vite avec des essais de plus petite taille. Les critères d'enregistrement des médicaments peuvent en être profondément bouleversés.
2. Les maladies chroniques
Le diabète le plus fréquent est celui de type 2 ; caractérisé par un excès de glucose dans le sang et une résistance croissante à l'insuline, il touche 300 millions de personnes dans le monde, dont 3 millions en France. Ce chiffre risque de doubler dans les prochaines années en raison de l'épidémie d'obésité et de la modification des modes de vie. Cette augmentation est provoquée par les effets conjugués d'une alimentation riche en graisses et glucides, d'un manque d'activité physique, de certains facteurs génétiques qui peuvent favoriser l'obésité.
Le Pr Philippe Froguel (83 ( * )) et ses équipes ont identifié de nombreux gènes responsables du diabète et de l'obésité. Il a expliqué : « j 'ai travaillé sur l'obésité parce que 80 % des obèses sont diabétiques et je pensais que la génétique de l'obésité ferait avancer celle du diabète mais finalement la génétique a décrit d'autres processus. Ce qu'on sait de la génétique aujourd'hui nous permet de mieux comprendre les maladies et d'envisager une médecine personnalisée ». Mais il ajoute « pour le bien portant, la génétique ne prédit rien ». Il considère qu'il faut adapter le traitement en fonction des caractéristiques génétiques du patient.
Pour preuve, il cite le cas de patients qui n'arrivaient pas à réguler leur diabète avec l'insuline, avaient une qualité de vie médiocre et des difficultés d'observance du traitement. Son équipe a identifié une mutation du gène impliqué dans le transport du potassium, mécanisme sur lequel agissent, les sulfamides qui sont des molécules anciennes. « La prescription de sulfamide à ces patients a transformé leur vie » a expliqué le Pr Philippe Froguel (84 ( * )) .
Le diabète chronique qui selon la Haute autorité de santé (HAS), touche aujourd'hui au moins 15 millions de Français constitue un champ immense. On peut citer le diabète de type 1 et encore plus celui de type 2. Une approche de médecine stratifiée est apparue opportune, elle permet d'adapter le traitement au patient en fonction de ses caractéristiques biologiques, mais aussi de son comportement, de ses habitudes. Cela a abouti à définir des profils dont les particularités permettent de mieux savoir quelles seront les réponses au traitement et les préconisations. On est donc conduit à des soins plus personnalisés du diabète que ce qui était traditionnel.
C'est à cela que s'emploie l'équipe du Pr Froguel qui anime, depuis 2000, un laboratoire européen franco-britannique, situé conjointement à Lille et à Londres. Par ailleurs, il est l'un des cofondateurs de l'Institut Européen de Génomique du Diabète, (EGID ou « European Genomic Institute for Diabetes », qui est le premier institut de recherche dédié spécifiquement au diabète en France. Il développe une recherche de renommée internationale pluridisciplinaire, associant des compétences scientifiques et médicales, de la recherche fondamentale et de la recherche thérapeutique appliquée au malade.
L'EGID a pour mission d'identifier les facteurs de risque des diabètes, de comprendre les mécanismes d'apparition des complications, de prévenir la survenue de cette maladie invalidante, de mettre en place des thérapies individualisées. Au sein d'EGID, les chercheurs du CNRS, de l'INSERM et de l'université Lille 2 mutualisent leurs compétences avec trois équipes fondatrices : l'Unité « Génomique et physiologie moléculaire des maladies métaboliques » dirigée par le Pr Philippe Froguel (Université Lille 2 - CNRS - Institut Pasteur de Lille), l'Unité « Thérapie cellulaire du diabète » dirigée par le Pr François Pattou (Université Lille 2 CHRU de Lille - INSERM), l'Unité « Récepteurs nucléaires, maladies cardiovasculaires et diabète » dirigée par le Pr Bart Staels (Université Lille 2 - Institut Pasteur de Lille - INSERM).
Ce pôle français de recherche sur le diabète, l'EGID, a obtenu les financements et les labels « laboratoire d'excellence » (Labex) et « équipements d'excellence » (Equipex) dans le cadre des investissements d'avenir.
Vos rapporteurs saluent cette initiative très centrée sur une pathologie fréquente et qui développe des thérapies innovantes et ciblées ; l'EGID est un modèle de R&D qui met les avancées scientifiques et technologiques directement au service du patient en s'inspirant du modèle maladie rare .
3. Des évolutions à l'oeuvre dans les maladies rares
Les maladies rares sont définies par leur prévalence. La définition retenue en France est celle d'une prévalence inférieure à 1/2000. Ce taux est différent dans d'autres pays (1/1000 aux États-Unis). Ainsi, en France, moins de 30 000 personnes sont concernées pour une maladie donnée mais elles affecteraient 3 millions de personnes en France ; au niveau européen, cela concernerait 30 millions de patients ; cependant ces estimations sont incertaines. Les avancées technologiques ont suscité des progrès dans la connaissance de ces maladies. Il y a 25 ans, on connaissait environ 20 myopathies, actuellement on sait qu'il existe 250 formes différentes, autant de gènes en cause.
Selon Mme Laurence Thiennot-Herment (85 ( * )) , les maladies rares sont au nombre de 6 à 8 000. Elles sont à plus de 80 % d'origine génétique et ont donné lieu à des découvertes nombreuses. En 1986 on connaissait 6 gènes à l'origine de maladies rares, et aujourd'hui plus de 3 800. Des bases de données cliniques/génétiques, extrêmement importantes pour permettre des thérapies ciblées, commencent à se développer grâce au séquençage à grande échelle et à haut débit.
Certaines de ces maladies sont connues : drépanocytose, sclérose latérale amyotrophique, mucoviscidose ; d'autres, bien plus rares, comme la progéria, sont caractérisée par un vieillissement très précoce et des troubles cardiovasculaires sévères. Le Pr Nicolas Levy (86 ( * )) , à Marseille, a isolé le gène responsable de la progéria, le Pr Hélène Dolfus (87 ( * )) a identifié un nouveau gène responsable du syndrome Bardet-Bied, caractérisé par une obésité précoce, une rétinopathie pigmentaire, une polydactylie, des troubles cognitifs. D'autres maladies rares, qui atteignent le métabolisme, sont étudiées et traitées notamment par le Pr Soumeya Bekri (88 ( * )) à Caen. S'y ajoutent les cancers rares dont le Pr Thierry Frébourg (89 ( * )) est l'un des spécialistes à Caen.
a. D'importants enjeux de recherche
Les maladies rares soulèvent de multiples enjeux, en termes de recherche, de connaissance de diagnostic, et de santé publique, comme l'a relevé le Pr Odile Kremp (90 ( * )) .
Comme l'ont montré l'audition publique sur les maladies monogéniques organisée par l'OPECST (91 ( * )) , les auditions des rapporteurs mentionnées ci-dessus, en termes de recherche, ces maladies dites rares sont de plus en plus prises en compte. Un consortium international de recherche sur les maladies rares a été mis en place récemment, l'IRDiRC ( International Rare Diseases Research Consortium ), dont l'Agence nationale de la recherche (ANR) fait partie.
Ces pathologies peuvent servir de modèle pour des maladies plus fréquentes, que ce soit en termes de physiopathologie ou de développement de médicaments. Elles ont aussi une dimension européenne très importante pour l'organisation des soins et de la recherche. Elles représentent un enjeu scientifique important : 6 000 à 7 000 maladies rares sont identifiées à ce jour. On décrit de nouveaux syndromes chaque semaine dans la littérature internationale.
Environ 80 % sont d'origine génétique, et l'explosion actuelle des techniques diagnostiques permet une meilleure approche de leur origine et parfois de leur traitement. D'après les données de la base Orphanet, une grande partie des maladies rares est liée à des anomalies du développement (16,40 %), dont des anomalies morphologiques et des troubles intellectuels ; il y a des maladies oncologiques (10,87 %), neurologiques (10,78 %) et tous les grands champs pathologiques sont concernés.
En Europe les recherches s'intensifient avec plus de 4 500 projets de recherche, près de 1800 essais cliniques ; les tests diagnostiques ainsi que les laboratoires experts dans différents pays sont répertoriés dans Orphanet. Dans certains pays européens, plus de 500 gènes peuvent être testés pour permettre de diagnostiquer de plus en plus de maladies (fin 2011 : 1 129 en France, 1 449 en Allemagne). Le consortium IRDiRC vise à ce que la plupart des maladies rares puissent être diagnostiquées et à ce que 200 nouveaux médicaments soient disponibles d'ici 2020 à l'échelle internationale.
b. Un enjeu de santé publique reconnu par les plans nationaux maladies rares
Les maladies rares sont graves, puisque le pronostic vital est en jeu dans la moitié des cas, et elles sont à l'origine d'un tiers de la mortalité infantile (35 % des décès avant l'âge d'1 an), 10 % de la mortalité entre 1 et 5 ans, 12 % entre 5 et 15 ans. Nombre d'entre elles sont des maladies chroniques, avec des déficits sensoriels, intellectuels et moteurs associés. C'est pourquoi, elles exigent une prise en charge globale qui s'est organisée à partir de la prise en compte du médicament orphelin.
i. Un enjeu de santé publique
D'après le Pr Odile Kremp et les généticiens auditionnés, on constate une rareté de l'expertise des professionnels, un déficit de connaissance et d'information des professionnels de santé et des patients, à l'origine d'une errance diagnostique importante génératrice d'inégalités et de retards dommageables dans la prise en charge, le remboursement, et l'accès aux produits de santé. Ces malades sont mal repérés dans le système de soins. Or, pour pouvoir participer à la recherche et être traité, il faut que le malade soit diagnostiqué.
Orphanet a développé à l'INSERM, avec le soutien de la Direction générale de la santé (DGS), une plateforme d'information destinée aux professionnels, aux malades et aux institutions. La France a été leader dans la prise en charge des maladies rares, et elle le reste en Europe. C'est à partir d'une réflexion sur la recherche dans ce domaine commencée en 1995 autour des médicaments orphelins au ministère de la santé menée par Annie Wolf et grâce aux deux Plans nationaux maladies rares (2005-2008 et 2011-2014), que s'est construite la prise en charge des patients atteints de maladies rares.
ii. Les Plans nationaux maladies rares
Le premier Plan national maladies rares a mis en place un réseau de centres de référence et de compétences qui a permis aux patients d'avoir accès au diagnostic.
Le Plan national maladies rares 2011-2014 a mis aussi l'accent sur la recherche, avec la création de la Fondation maladies rares début 2012, présidée par le Pr Nicolas Lévy (92 ( * )) , qui a indiqué que les recherches sur les maladies rares pouvaient ouvrir des perspectives cliniques étendues. Ses recherches sur la progéria (maladie très rare provoquant un vieillissement prématuré), dont il a découvert le gène, suscitent un fort intérêt dans le cadre de l'étude des processus de vieillissement. Plusieurs appels à projet ont été lancés en recherche fondamentale pour des modèles thérapeutiques sur les souris et aussi en recherche en sciences sociales, car les données sur les maladies rares sont insuffisantes. L'amélioration de l'accès aux soins et l'accélération de la recherche sont l'un des grands axes du plan maladies rares. Le soutien au développement des plateformes de génétique moléculaire est affirmé ; les modalités de ce développement doivent être définies.
Cet effort doit être accompli pour combattre l'errance diagnostique qui est une souffrance pour les patients et leur proches a insisté le Pr Helène Dolfus (93 ( * )) . Il faut pouvoir intervenir très en amont dans le cas notamment des maladies métaboliques. Comme l'a souligné le Pr Soumeya Bekri (94 ( * )) , en diagnostiquant très rapidement les causes d'un coma inaugural, on inverse un pronostic des plus graves par un traitement, parfois un régime alimentaire ; ceci suppose un suivi des familles et un centre de référence.
En termes de traitement, il reste de nombreuses lacunes. Au niveau de la Haute autorité de santé (HAS), seuls 50 protocoles nationaux de diagnostic et de soins ont été mis en place pour 6 000 maladies rares, on est loin du compte.
c. Des pathologies difficiles à traiter exigeant une mobilisation internationale et générant un nouveau modèle de R&D
Même si la connaissance génétique des maladies rares se développe, ce qui permet de mieux les caractériser, de comprendre leur physiopathologie et de chercher de nouvelles thérapeutiques, le défi reste celui du faible nombre de traitements disponibles. Pour de nombreuses maladies rares, on ne dispose d'aucun traitement. Le marché européen en 2012 comptait 68 médicaments orphelins, dont 9 avec une autorisation de mise sur le marché (AMM) cette année-là, et 75 médicaments ayant une indication pour une maladie rare ou un groupe de maladies rares.
i. La prise en compte européenne
Elle est extrêmement importante ; elle se manifeste par des recommandations pour disposer de stratégies sur les maladies rares dans différents pays par la constitution de réseaux européens d'experts de référence répertoriés pour que les malades pris en charge participent à l'ensemble des essais. Des tentatives pour répartir les recherches sur différentes pathologies sont en cours.
ii. Le rôle des associations de patients
Les associations de patients jouent un rôle majeur dans les maladies rares, notamment l'Association française contre les myopathies (AFM), mais aussi l'Alliance maladies rares, qui fédère plus de 200 associations de malades en France, et Eurordis, qui rassemble plus de 560 associations membres, dont plusieurs Alliances nationales maladies rares dans 51 pays. Elles participent aux essais thérapeutiques et aux réflexions en cours sur les maladies rares.
iii. L'importance du modèle maladies rares
Selon Mme Christine Pezel (95 ( * )) , « les maladies rares donnent naissance, avec ces premières thérapies ciblées, à de vraies innovations de rupture, à partir de thérapies géniques, de l'ARN, cellulaires. Á chaque fois, on le constate, ces innovations de rupture, bien sûr sont efficaces pour des petites populations dans des maladies rares, mais peuvent également s'appliquer à différentes maladies rares, et aussi être des innovations pour des maladies fréquentes ». Ce constat est partagé par la plupart des experts.
Dans le cas de la mucoviscidose, maladie génétique provoquée par un dysfonctionnement de la protéine CFTR, le génotype du malade permet de prévoir ce que sera le dysfonctionnement de la cellule. Un registre national de la mucoviscidose existe depuis vingt ans et recense 95 % des patients atteints de cette maladie, c'est un atout majeur pour une thérapie ciblée. Certaines formes de mucoviscidose nécessitent une surveillance cardiaque particulière, et il faut connaître le gène très exactement en cause pour la mettre en oeuvre. Le diagnostic est donc crucial.
Pour bénéficier des thérapies ciblées, il faut non seulement connaître le type de maladie en cause, mais aussi le type de mutation ou de délétion dans cette maladie ; or même dans une maladie rare il existe des sous-groupes de maladies. Ceci exige une organisation de soins spécifiques ; aussi des évolutions des structures de soins seront-elles nécessaires.
Dans ce contexte, la médecine personnalisée modifiera la différence entre les maladies rares et les maladies dites fréquentes en faisant de ces dernières une myriade de maladies rares pour les traitements. Aussi faudra-t-il repenser la manière dont on recherchera les traitements adaptés.
I. II. UN BOULEVERSEMENT SOCIO-ÉCONOMIQUE DU SECTEUR DE LA SANTÉ
L'impact considérable des technologies et l'augmentation des savoirs à l'oeuvre dans les nouvelles conceptions de la médecine obligent à une rénovation de la formation des personnels de santé, à une diversification des parcours et des métiers. Les enseignements doivent s'adapter à ces nouvelles approches qui exigent une grande spécialisation et une forte transdisciplinarité.
A. UNE ÉVOLUTION DE LA RELATION MÉDECIN-MALADE IMPLIQUANT UNE RÉFORME DE LA FORMATION DES PERSONNELS
Les nouveaux outils technologiques utilisés par la médecine personnalisée génèrent une quantité d'informations considérable, désormais largement en possession du patient lui-même. « De fait, tout un corps numérique est en train d'apparaître à côté du corps réel. Allons-nous céder à la croyance que nous pourrions tout voir/mesurer, donc tout prévoir, tout dépister, tout prévenir, tout traiter ou tout remplacer ? » se demandait le Pr Hervé Chneiweiss (96 ( * )) .
Comme le remarquait le Pr Jean-Claude Ameisen (97 ( * )) dans son intervention lors de l'audition publique de l'OPECST du 25 juin 2013 : « c ela interroge sur le rôle que la société veut voir jouer au médecin. C'est un problème de formation. Comment interpréter et pouvoir transmettre cette masse énorme de données, comment faire ré-émerger la personne derrière cette approche médicale qui paradoxalement en fait une identité absolument abstraite, un point dans un nuage de points ? Faut-il repenser le rôle de la médecine en faisant du médecin un médiateur, un interlocuteur parmi tous les spécialistes qui recueillent et interprètent les données ? Doit-il être un homme-orchestre qui sait à la fois interpréter les résultats du séquençage du génome et de toutes les données, et en plus parler à la personne ? Cela amène à s'interroger sur un risque paradoxal, pas si étonnant que cela, de dépersonnalisation lié à l'approche de la médecine personnalisée. Au fond, la véritable singularité de la personne, son histoire, ses espoirs, ses craintes, apparaissent comme une gêne, une interférence avec un processus qui justement a essayé d'établir un profil-type par rapport aux groupes où on l'a rangé ».
1. Une évolution considérable de la relation médecin-malade
La relation médecin-malade se trouve perturbée soit parce que le médecin en sait trop par rapport à un patient qui souhaite ne pas savoir, soit parce que le patient vient le consulter avec son génome dans son téléphone portable. Auparavant, le patient était conscient que le médecin ne pouvait pas tout savoir, mais désormais, quand celui-ci sait que le malade va contracter telle ou telle maladie, cela modifie leurs rapports. S'il est utile pour le praticien de disposer de nouveaux examens qui enrichissent sa connaissance de la maladie dont souffre son patient, il se retrouve enfermé dans un carcan, bien plus dépendant que par le passé des modèles et données que lui fournissent des technologies de plus en plus performantes. Le praticien se voit également contraint de respecter un modus operandi et des protocoles plus ou moins rigides de soins, ce qui ne lui laisse qu'une marge réduite d'appréciation. Or, il est essentiel qu'il puisse rester le référent du malade pour l'orienter et le guider, ce qui implique qu'il soit suffisamment formé aux nouvelles technologies et qu'il suive une formation continue.
a. Vers un bouleversement de la consultation traditionnelle ?
Selon le Pr Philippe Amouyel (98 ( * )) , lors de la consultation du spécialiste, on obtiendra une meilleure description de chaque patient qui répond à une demande, les nouveaux examens complexes ajoutent à la précision du diagnostic, à l'amélioration de la prise en charge. On se situe alors dans une médecine de précision. Selon lui, « on est vraiment toujours dans la même relation, avec un apport d'information supplémentaire ». En revanche, « p our ce qui est de la consultation du généraliste, on change complètement de modèle », car pour un individu donné il y aura un énorme potentiel d'informations, qui fournira des probabilités plus ou moins importantes de survenue d'évènements.
i. L'impact du degré d'information du patient
La relation médecin-malade sera un colloque singulier avec une augmentation colossale du volume d'informations, essentiellement sous forme de valeurs probabilistes. « On passe vraiment de la notion d'art ?médical? comme la voyait Auguste Comte à celle de science ». Il faudra commencer à utiliser des outils scientifiques, ce qui pose la question de l'accès aux données du patient par le médecin, du dossier numérique et des coûts générés par le stockage. Il faudra aussi que le médecin généraliste gère le patient surinformé voire parfois, mal informé qui disposera de résultats de tests génétiques obtenus par Internet dans son téléphone portable. Parallèlement comment gérera-t-il le patient qui exerce son droit de ne pas savoir ?
ii. Des risques de dépersonnalisation de la relation médecin malade
Comment le lien entre la pratique d'une médecine personnalisée dans de grands centres souvent des canceropôles et la médecine de ville individuelle s'établira-t-il ? se demande Mme Anne-Yvonne Le Dain (99 ( * )) ; elle précise « Ce lien me paraît compliqué et malaisé à construire. En tant que parlementaires, notre ambition est de construire un projet de société autour de cette question de la médecine personnalisée. Comment va-t-on le construire ? » Cette question importante est centrale du point de vue du patient et nous préoccupe car on se trouve face à des attentes et des perceptions contradictoires parfaitement relevées par le Pr Anne Fagot-Largeault (100 ( * )) : « je me place maintenant dans la position du malade, du patient, et je fais la remarque générale suivante : il y a souvent à l'heure actuelle une expression des patients qui se plaignent d'une frustration à l'égard de la médecine, qui souffrent d'être pris pour des machines et d'être traités comme tels . Vous téléphonez à l'hôpital, c'est une machine qui répond, vous devez discuter avec une machine. Vos données personnelles, vos paramètres individuels sont numérisés. Ils sont une colonne de chiffres. La pratique des médecins est standardisée ». Elle observe que nombre de patients demandent une médecine qui les comprendrait mieux, personnellement, avec un abord des médecins ou des services médicaux qui prendraient toutes leurs dimensions en compte.
Derrière ce concept de médecine personnalisée assez engageant qui implique une individualisation souhaitable des traitements, se profile une forme de dépersonnalisation de la médecine que le Dr Catherine Bourgain (101 ( * )) a soulignée. La montée en puissance de toutes les technologies qui permettent ces progrès conduisent à l'introduction de tiers dans la relation médecin- malade.
b. L'intervention d'un tiers dans la relation médecin malade
L'intervention fréquente d'un tiers scientifique dans le colloque singulier médecin-malade devient déterminante au travers des analyses génétiques et biologiques, qui vont bien au-delà des analyses de sang classiques. En effet, la médecine personnalisée multiplie les données sur le patient et l'usage de technologies complexes qui exigent l'implication d'experts capables de les interpréter. Pour leur mise au point comme pour leur utilisation, un travail en équipe interdisciplinaire est nécessaire, souvent jusqu'au lit du malade.
Doivent s'allier les compétences de mathématiciens, de physiciens, d'ingénieurs en microélectronique, d'informaticiens spécialisés dans la mise au point de logiciels, le transfert et la gestion de données, afin de rendre les résultats multiples sous forme visible, lisible et interprétable pour les bio-informaticiens et les médecins. Les utilisateurs eux-mêmes doivent en fait être formés à diverses disciplines.
« Si l'on intègre dans la médecine personnalisée la pharmaco-génomique, la thérapie cellulaire, ainsi que les traitements partant du patient avec réinjection de ses propres cellules, qui sera maître de ces technologies ? Si le laboratoire arrive au chevet du malade, les liens entre techniciens et patients deviendront très étroits à travers les équipes médicales hospitalières qui disposent de plateaux techniques médicaux » observe M. Didier Tabuteau (102 ( * )) . Le patient se trouvera face à de multiples interlocuteurs. Cette évolution, déjà largement initiée dans les cancéropoles, parait inéluctable comme l'a confirmé le Pr Marie-Christine Favrot (103 ( * )) , adjointe au directeur général de la santé, chef des politiques de santé.
Quand les traitements deviennent complexes même pour des pathologies de masse, est-ce alors le généraliste isolé qui sera le point d'entrée ? Ne va-t-on pas s'adresser d'abord à d'autres structures, comme les centres de santé ou les organismes travaillant en équipes ? Qui décidera des traitements à appliquer au malade ? Qui sera maître des technologies ? Qui en sera responsable ? Qu'exigera-t-on du patient ?
c. Vers une responsabilisation croissante du patient ?
« La notion de médecine personnalisée donne une image attractive mais fausse, d'une vérité à la fois attirante et préoccupante : nous avons été habitués à croire que notre médecine, notre système de santé, sont les mêmes pour tous. Or, l'époque de la même médecine pour tous est révolue. Elle devient personnalisée, c'est-à-dire différente pour chacun, ?customised?, comme on dit, et on le craint, proportionnelle aux ressources de l'individu qui sera capable ou non de se l'offrir » prévient le Pr Anne Fagault-Largeault (104 ( * )) .
L'intervention de la médecine se fera plus tôt dans la vie des personnes : même en étant bien portant, sans signe clinique apparent, le patient aura noué une relation avec le médecin, car les possibilités médicales permettront d'anticiper une pathologie susceptible d'apparaître dans cinq ou dix ans. Ce n'est plus la maladie qui crée le lien avec le médecin comme aujourd'hui. La limite entre le normal et le pathologique deviendra de plus en plus délicate. Ce changement profond de la place du médecin et des centres de santé est la conséquence directe de la disparition de la frontière entre prévention et soin. Les prévisions du Dr Knock seront mises en oeuvre, « l'avenir sera sombre pour les hypocondriaques ! » s'exclamait le Pr Axel Kahn (105 ( * )) .
Le médecin généraliste disposera d'informations génétiques délicates à communiquer au patient, qui le concerneront aussi bien lui-même que ses apparentés et que celui-ci n'aura pas nécessairement envie de connaître. Le praticien devra-t-il les communiquer ou non à son patient ? Devra-t-il faire appel à un généticien, plus formé que lui à l'exposé des conséquences induites par le résultat d'un test génétique ?
Comme le remarquait encore M. Didier Tabuteau (106 ( * )) : « sur un plan juridique ou administratif, la médecine personnalisée remet en cause des frontières traditionnelles, les distinctions prévention-soins, actes-produits, médicaments-dispositif médical. Ces distinctions vont voler en éclat vu les perspectives scientifiques et médicales ouvertes par les thérapeutiques et les pratiques qu'elles permettront. Or, ce sont ces frontières qui structurent notre organisation administrative et juridique. Les codes de la santé publique et de la sécurité sociale s'appuient sur ces concepts traditionnels pour les définitions, les processus d'autorisation, de contrôle, d'évaluation. Il faudra dépasser ces clivages, ces frontières traditionnelles. Ce ne sera pas simple car nous y sommes habitués ».
La multiplicité des informations dont disposera le patient induit aussi une plus grande participation, une responsabilité accrue de sa part dans la prise en charge de sa propre santé. On lui explique qu'il peut encore être mieux soigné dès lors qu'on séquence son génome, qu'on cible ses modes de vie plutôt que de lui donner un médicament standard qui ne lui conviendra peut-être pas, et qu'ainsi on trouvera le dosage parfait, voire son propre médicament.
Pour M. Christian Byk (107 ( * )) , « La contrepartie est qu'il devra être encore plus coopératif, il devra donner des informations sur son mode de vie et, en plus, une fois qu'on aura analysé le traitement approprié, il ne pourra pas se permettre de ne pas le suivre . On évoque parfois le caractère pervers, sinon à risque potentiel de la médecine personnalisée » . On peut dissimuler des informations, mettre en avant la mise en place de médicaments personnalisés et, en pratique aboutir à une médecine pour une catégorie de la population, une médecine stratifiée.
Faut-il prendre en compte une demande, susceptible d'accroître les devoirs des patients envers eux-mêmes ? Certains seront demandeurs et arriveront bientôt chez leur médecin avec le séquençage de leur génome sur leur téléphone portable. D'autres ne seront pas prêts à se plier à des techniques d'analyses médicales au sens large, voire de vérifications du degré d'observance de traitements ciblés prescrits. On risque le contrôle intrusif des patients.
Des populations à risques connus plus faibles que d'autres, pourraient demander une tarification médicale en fonction du risque. On l'observe déjà à l'étranger avec les assurances privées qui personnalisent la tarification du risque et évoluent en fonction du risque et de l'observance du traitement par le malade.
Plus on multiplie les informations sur la connaissance du risque et les moyens d'y pallier, plus la tentation de lier l'assurance-maladie au comportement du patient sera forte : le risque de remise en cause de la médecine de solidarité entre les biens portants et les malades augmentera.
La médecine personnalisée déchire le voile d'ignorance de l'avenir pour le patient, sa famille, voire pour les tiers, ce qui n'est pas sans conséquences éthiques et juridiques. L'information génétique et identifiante, bien que personnelle par nature, touche l'ensemble des membres d'une famille, voire d'une communauté ethnique avec des particularismes locaux.
Certains traitements tout en étant utiles peuvent aussi être sources de discriminations, voire comprises ou vécues comme telles. « La personnalisation de la médecine peut en ce sens entraîner des distinctions au sein de la population générale si des traitements et diagnostics individuels se dégage un constat collectif. Cependant, si toute distinction entre groupes d'individus au sein d'une population générale n'implique pas nécessairement une stigmatisation des individus, il existe des risques de rupture d'égalité entre les patients, voire des risques de discrimination dans certains cas » soulignait Mme Audrey Aboukrat (108 ( * )) .
À l'appui de sa démonstration elle citait d'une part le cas du BiDil, un médicament destiné au traitement de l'insuffisance cardiaque au sein de la population africaine américaine exclusivement, et d'autre part, le test génétique commercialisé par l'entreprise Myriad Genetics pour le dépistage d'une prédisposition aux cancers du sein et des ovaires chez les femmes juives ashkénazes exclusivement. Elle rappelait aussi que ces ciblages avaient été en plus figés dans les revendications d'un brevet intensifiant à la fois le risque de discrimination et la responsabilisation des patients concernés aux États-Unis.
Ces évolutions scientifiques et technologiques provoquent et provoqueront des bouleversements considérables dans tout le secteur de santé. Ils exigeront rapidement des réformes afin de l'adapter à ces nouvelles réalités. C'est d'ailleurs le sens des recommandations du rapport de Commission « Innovation 2030 et de la « Stratégie nationale de santé » .
2. Une réforme de la formation des personnels de santé à mettre en oeuvre
Les avancées décrites nécessitent de repenser la formation des personnels de santé et notamment des médecins en France ; il s'agit de pallier la pénurie de généralistes dont le rôle sera de plus en plus important pour orienter les patients, d'éviter les errances diagnostiques dommageables. Partant du principe que la médecine était en transition, tous les experts rencontrés ont insisté sur la nécessité d'un décloisonnement des disciplines scientifiques entre elles, sur l'importance d'un enseignement en informatique adapté et sur la nécessité d'introduire des sciences humaines et sociales dans ce contexte évolutif.
a. L'exigence d'une réforme des études de santé
La question de la formation des futurs professionnels de santé apparait cruciale. C'est d'ailleurs l'un des cinq piliers de la stratégie nationale de santé, lancée par le Premier ministre en 2013 (109 ( * )) . Elle en définit les priorités dans le cadre de son axe 2 « Mieux organiser les soins pour les patients, garantir l'égalité d'accès, en privilégiant une logique territoriale ». La formation initiale doit mieux préparer les futurs professionnels à un exercice en équipe, en favorisant les passerelles. Si toute la formation doit s'adapter à cette nouvelle donne, leur mode de recrutement aussi. Les études de santé ne répondent pas à ces attentes, car l'un des écueils de la formation médicale aujourd'hui réside dans l'explosion énorme du corpus de connaissances. Selon le Doyen Patrick Berche (110 ( * )) , « p ersonne n'est plus capable d'appréhender l'ensemble des connaissances médicales ». Or, on forme actuellement ceux qui exerceront dans moins d'une décennie.
i. Les fondements dépassés du dispositif actuel
Mis en place dans les années soixante-dix, le numerus clausus visait à contenir la démographie médicale pour limiter les dépenses de santé. Le contrecoup de cette mesure une quarantaine d'années plus tard provoque de véritables pénuries dans certaines spécialités médicales, étend les déserts médicaux et n'a pas eu l'effet escompté, comme l'ont rappelé la plupart des experts auditionnés, les députés et les sénateurs lors des débats sur la loi n° 2013-660 relative à l'enseignement supérieur et à la recherche du 22 juillet 2013. Les axes de la stratégie nationale de santé lancée par le Premier ministre procèdent eux aussi de ce constat.
Malgré le développement de déserts médicaux, le recours à des médecins formés à l'étranger, l'exil d'étudiants français en Belgique, voire en Roumanie où la première année d'étude s'effectue en français, on a maintenu un système de numérus clausus en première année en augmentant certes quelque peu le nombre de places.
Selon le Pr Georges Leonetti (111 ( * )) , le maintien actuel de ce dispositif peu satisfaisant serait également dû, d'une part à la baisse du nombre des lits dans les CHU chargés d'accueillir les étudiants dès leur deuxième année d'études et d'autre part, à la désaffection des étudiants pour les études scientifiques, qu'un appel d'air pour les seules études de médecine pourrait accroître.
ii. Un cursus des études de médecine à réexaminer face à l'échec de la première année commune aux études de santé (PACES)
La réforme de la première année commune d'études de santé (PACES) de 2009 n'a pas produit les résultats escomptés ; d'ailleurs l'élaboration de la loi n°2013-660 relative à l'enseignement supérieur et à la recherche de juillet 2013 a été l'occasion d'en débattre.
La loi du 7 juillet 2009 qui a instauré la première année commune aux études de santé (PACES) en France, est née d'un constat unanime : l'organisation de ces études engendre pour les étudiants trop d'échecs, de frustration, de stress et de temps perdu. Près de 77 % des étudiants n'ont pas pu intégrer une deuxième année, toutes filières confondues, à l'issue de l'année universitaire 2010-2011, et 76 % étaient dans le même cas en 2011-2012.
Sur l'ensemble des étudiants inscrits pour la première fois en PACES en 2011-2012, 61 % échouent à l'issue de deux présentations aux concours, 14,1 % sont admis à poursuivre des études en vue d'un diplôme d'État de sage-femme, de docteur en pharmacie, de docteur en chirurgie dentaire ou de docteur en médecine, 52,3 % redoublent, 12,2 % sont inscrits dans une première année de licence et 0,7 % en deuxième année ; 2,5 % sont inscrits dans une première année de cursus hors système licence-master-doctorat ; enfin, 18,2 % sortent de l'université. La PACES demeure l'un des cursus les plus sélectifs en France. Le taux d'échec est beaucoup trop lourd, il entraine une perte de temps considérable, voire un véritable « décrochage » pour certains étudiants.
La mise en place de la PACES visait à offrir aux étudiants un nombre élargi de débouchés et une orientation adaptée afin de réduire le taux d'échec en première année. Il s'agissait de décloisonner les études de santé, de forger une culture scientifique commune aux professions médicales et pharmaceutiques, et de diversifier le recrutement des futurs professionnels de santé. Or les premiers bilans disponibles ont montré que la mise en place de la PACES n'a permis ni de rendre cette année d'études réellement formatrice, ni de remédier au taux d'échec massif aux épreuves de sélection organisées à son issue. Les taux de réussite n'ont pas progressé, les systèmes de réorientation fonctionnent mal. Le principal objectif de la PACES, qui est de remédier au gâchis humain, n'est pas atteint.
Comme le note le rapport final des Assises de l'enseignement supérieur et de la recherche remis par M. Vincent Berger en décembre 2012, « si cette première année marque un progrès par rapport à l'organisation précédente, il reste encore beaucoup à faire pour endiguer le «gâchis humain». Avec un chantier prioritaire : les réorientations en cours d'année ».
b. Les propositions des réformes
i. Le rapport de M. Jean-Yves Le Déaut « Refonder l'université, dynamiser la recherche, mieux coopérer pour réussir »
Dans son rapport au Premier ministre, M. Jean-Yves Le Déaut, Premier vice-président de l'OPECST, parlementaire en mission, part du constat que dans le système français rien n'est rattrapable, rien n'est fait pour ceux qui échouent, et que les études de santé sont celles qui obéissent le plus à cette logique. Selon ce rapport, le cursus actuel des études médicales en France cumule plusieurs défauts. Le concours de fin de première année, très sélectif entraîne encore pour la majorité des étudiants une perte de temps, particulièrement problématique pour ceux qui ne sont pas classés en rang utile à l'issue des épreuves terminales et qui obtiennent une moyenne inférieure à 10/20. L'organisation de la PACES engendre une sélection par l'échec d'étudiants souvent brillants et motivés.
Le rapport de M. Jean-Yves Le Déaut recommande une spécialisation plus progressive au cours des trois années de licence, en préservant au début une pluridisplinarité compatible avec le besoin d'une éventuelle réorientation et en repoussant les concours d'accès aux formations master et doctorat d'exercice en santé de la fin de première année à la fin de la troisième année. Il demande de créer des passerelles d'entrée dans les premières années d'études en santé à partir des autres filières universitaires, comme elles existent déjà à partir des écoles d'ingénieurs, d'augmenter le nombre des missions d'enseignement contractuelles des doctorants en vue de renforcer l'encadrement des étudiants de premier cycle, de multiplier d'une façon générale les équivalences et les passerelles, offrant des possibilités de rebond dans le parcours universitaire et professionnel afin de préserver le capital humain en dépit des erreurs d'aiguillage. Il souhaite redonner à l'État la responsabilité de toutes les formations sanitaires et sociales inscrites dans le cadre du système Licence Master Doctorat (LMD).
ii. Les propositions du Pr Patrick Berche, Doyen de l'Université de Paris- Descartes (112 ( * ))
Rendant hommage à la qualité de la formation clinique (trois ans de formation à mi-temps pour les externes, et quatre ou cinq ans de formation à plein temps à l'hôpital) et au recours aux nouvelles techniques de communication à l'université : Internet, simulations sur mannequin, jeux électroniques, E-learning, cours en ligne, le Doyen Berche a énuméré les faiblesses des formations actuelles qui vont de l'obsolescence des programmes actuels, avec une approche mono-disciplinaire, à une formation scientifique confinée à la première année commune aux études de santé (PACES) souffrant de l'absence des sciences humaines et sociales (SHS). Certains domaines de la médecine sont selon lui négligés tels la médecine préventive, la douleur, les soins palliatifs, les addictions, les maladies chroniques, le handicap et le vieillissement. Ces études encadrées par deux concours archaïques, celui de la PACES suivi du concours de l'examen commun national (ECN), ne permettent pas de sélectionner de futurs médecins capables de travailler en équipe et de faire preuve d'empathie.
S'inspirant partiellement du système américain, il propose la suppression du concours de la PACES et un nouveau cursus : trois ans de licence, avec une approche multidisciplinaire, notamment avec l'addition de SHS, mais aussi de sciences médicales et sciences fondamentales (renforçant la formation scientifique des étudiants français). À ce stade, les étudiants seraient classés par les notes cumulées pendant les trois ans de cursus ; les meilleurs d'entre eux seraient soumis à des interviews, à l'admission par des jurys d'enseignants, d'étudiants et de psychologues, la France étant d'ailleurs le seul pays en Occident où il n'y a pas d'interviews des étudiants en médecine.
Après ces trois années, ils suivraient un externat de quatre ans, et un internat de quatre ans, comme c'est le cas actuellement. S'y ajouteraient des enseignements modulaires complémentaires, puisqu'en étalant l'enseignement clinique sur quatre ans, les étudiants auront plus de temps à lui consacrer. Ces enseignements modulaires complémentaires optionnels (diplômes universitaires, master, thèse d'université...) seraient validés par l'université ou le ministère et comptés sous forme de bonus.
On construirait ainsi un cursus personnalisé comme à l'École de médecine de Harvard. C'est un enseignement parallèle précoce, orientant vers les filières. La validation de modules d'enseignement selon l'orientation souhaitée aurait lieu pendant le temps libre, les modules pouvant être acquis au sein des facultés (de médecine, des sciences, de SHS, psychologie, droit, sciences po, etc .) ou d'Instituts de recherche (psychologie, sociologie, santé publique, économie, recherche...). On pourrait créer des pôles pédagogiques (virtuels) qui dépasseraient les institutions sur des problématiques particulières. Ce melting-pot améliorerait les études de médecine et donc la qualité de l'enseignement. Cet enseignement pourrait être multi-universités dans les TICE ou sous forme d'un enseignement en présentiel pendant de courtes périodes.
Ainsi pourrait-on, selon le Doyen Berche, supprimer le concours de l'internat et le remplacer par un examen régional, associé à un numerus clausus pour la région. Cet examen régional anonyme serait segmenté par discipline (médecine, chirurgie, psychiatrie, santé publique, biologie...), chaque discipline ayant son numerus clausus défini par le ministère de la santé en fonction des besoins régionaux.
Le dispositif souple adopté par la loi sur la recherche et l'enseignement supérieur ne s'inspire que très partiellement des propositions ainsi formulées sans pour autant les rejeter totalement.
iii. Le dispositif mis en oeuvre à titre expérimental par la loi sur la recherche et l'enseignement supérieur
Lors des débats sur la loi recherche et enseignement supérieur, certains parlementaires ont critiqué la PACES. Ils ont mis en cause son efficacité, le mode de sélection quasi fondé sur les questionnaires à choix multiples (QCM). Cette organisation favorise le bachotage et le développement d'officines privées de préparation parallèle souvent coûteuses. La PACES est vécue comme une épreuve d'autant plus redoutable que l'étudiant n'a droit qu'à un seul échec.
Ils ont cité certains universitaires qui préconisent la sélection à l'entrée des étudiants, car dans d'autres pays les étudiants sont sélectionnés soit directement sur dossier à l'issue du baccalauréat (Allemagne), soit après un premier cycle universitaire (États-Unis). Le système instauré en 2009 pour la PACES n'a pas remis en cause la hiérarchie entre les différentes disciplines, voire même, a pu nuire à l'attractivité de certaines spécialités, comme les études pharmaceutiques. Enfin, les passerelles «entrantes» et «sortantes» sont aujourd'hui insuffisamment développées.
Tenant en partie compte de ces critiques, la loi relative à la recherche et à l'enseignement supérieur maintient le principe d'un numerus clausus qu'elle aménage quelque peu. En effet, la loi rappelle que « l e nombre des étudiants admis en deuxième année après la première année commune et le nombre des étudiants admis directement en deuxième ou en troisième année sont fixés, pour chaque université concernée et pour chacune des filières, par arrêté conjoint des ministres chargés de l'enseignement supérieur et de la santé ».
Dans ses articles 39 et suivants, elle prévoit la mise en oeuvre de dispositions expérimentales pendant six ans dans des formations paramédicales, dans des études médicales, odontologiques, pharmaceutiques et de maïeutique.
Ces dispositions fixées par décret, ouvrent une possibilité de réorientation, soit dès le début de la PACES (huit semaines), soit à l'issue de la PACES pour un pourcentage d'étudiants mal classés. Il est prévu que : « l'université assure dans tous les cas la réorientation de ces étudiants en leur proposant une inscription dans une formation qui les accueille dès l'année universitaire en cours ». Cette réorientation précoce doit permettre de diminuer le flux d'étudiants, d'augmenter le taux d'encadrement des étudiants et d'améliorer la pédagogie. Elle facilitera aussi la réorientation, dès le début de l'année universitaire, dans des conditions plus efficaces, des étudiants dont les résultats aux premières épreuves montrent qu'ils n'ont pratiquement aucune chance d'intégrer des études de santé. Cependant, le droit au redoublement demeure.
La nouvelle loi prévoit un système d'admission en deuxième ou en troisième année des études médicales, odontologiques, pharmaceutiques ou de maïeutique après une à trois années d'un premier cycle universitaire adapté conduisant à un diplôme national de licence. La loi vise à diversifier le recrutement des professions médicales et pharmaceutiques en développant des sortes de passerelles entrantes. Parmi les diplômes requis, les arrêtés du 26 juillet 2010 évoquent notamment les masters scientifiques ou littéraires, les diplômes des écoles de commerce qui confèrent le grade de master et les diplômes des instituts d'études politiques. Ces nouvelles voies s'ajoutent à la procédure d'admission directe en troisième année prévue par l'arrêté du 26 mars 1993. Ces étudiants sont recrutés sur dossier et sur entretien, dans le cadre d'un numerus clausus spécifique fixé par arrêté.
La loi ne fige pas complètement le mode de sélection et incite à des réformes puisqu'elle stipule que « d ans un délai de six mois suivant la promulgation de la présente loi, le Gouvernement remet au Parlement un rapport formulant des propositions en vue d'améliorer le mode de sélection et de formation des futurs médecins et d'élargir les origines sociales et géographiques des étudiants. Ce rapport analyse la faisabilité de l'organisation d'épreuves classantes interrégionales pour les études de médecine » (113 ( * )) .
Le Gouvernement n'a pour l'instant pas transmis ce rapport au Parlement mais des propositions de réformes en profondeur ont été faites. De nouveaux métiers exigent un décloisonnement des disciplines scientifiques. L'usage massif de technologies toujours plus performantes produisant des masses de données difficiles à interpréter nécessitent des connaissances en mathématiques, en informatique, en biochimie, etc . On voit apparaître de plus en plus de métiers liés aux systèmes d'informations : ingénieur en informatique décisionnelle de la santé, ingénieur clinique, ingénieur en systèmes embarqués et télésanté, manager data santé. Cependant le cloisonnement des disciplines, le manque de bio-informaticiens notamment, constitue un handicap. Ce constat n'est pas nouveau.
3. Assurer des formations adéquates
Déjà, en 2010, le rapport de M. Emmanuel Tunon De Lara (114 ( * )) préconisait de décloisonner les cursus et de professionnaliser les enseignements par le développement de l'apprentissage notamment, mais aussi de rassembler industriels et universités autour de plateformes partagées de recherche et de formations en sciences du vivant et de la santé. Le modèle de ce qui a été mis en place à Grenoble ou Bordeaux fut mis en exergue.
À Grenoble, l'école de biotechnologies, au sein de l'Université Joseph Fourrier, a signé des accords de partenariats avec les PME locales permettant à ses étudiants de trouver facilement des stages correspondant à leurs attentes, et aux industriels de recruter de nouveaux talents répondant à leurs besoins. À Bordeaux, un cursus alterné en bio-santé industrielle existe.
Selon M. Jean-Marc Grognet (115 ( * )) , il faudra former à de nouveaux métiers, de vrais ingénieurs biomédicaux, à un niveau plus avancé qu'actuellement, car il faut gérer de l'apport d'énergie, du traitement de données, du stockage. Il convient d'être attentif à plusieurs secteurs. La formation est essentielle. Certaines écoles des Mines proposent déjà des doubles formations (pharmacien-ingénieur par exemple), mais c'est rare.
D'après le Dr Jean-Marc Egly (116 ( * )) , de nombreuses écoles de médecine aux États-Unis intègrent à présent, dans le cursus médical, des formations aux nouvelles technologies dans le domaine de la génomique. Le programme TRIG ( Training Résidents In Genomics ), lancé il y a quatre ans au sein du Département de pathologie du Beth Israël Deaconess Medical Center de Boston, comprend une formation obligatoire en génomique/génétique et médecine personnalisée pour les futurs pathologistes.
À cet égard, une nouvelle infrastructure nationale de service en bio-informatique, devrait voir le jour. Il s'agit de « l'Institut français de bio-informatique » (IFB). Issu d'une réponse du réseau national des plateformes de bio-informatique (ReNaBi) à l'appel à propositions « Infrastructures en Biologie et Santé » du programme « Investissements d'avenir » lancé par le ministère de l'enseignement supérieur et de la recherche, ce projet coordonné par le CNRS a débuté en novembre 2013.
i. Favoriser le décloisonnement entre les disciplines scientifiques
La plupart des experts et des études attachent une importance considérable au décloisonnement entre les disciplines scientifiques.
« Le biologiste, observateur qui décrivait le monde du vivant, utilise désormais les données biologiques pour construire des modèles prédictifs » (117 ( * )) . Ce changement conduit à l'émergence de nouveaux métiers à l'interface entre la science et l'ingénierie et entre l'ingénierie et l'innovation. Il induit un rapprochement des technologies des sciences de la vie (biotechnologies) et de la santé (médecine, pharmacie...) avec les sciences de l'ingénieur. « De nouvelles disciplines apparaissent, à moyen terme, qui transcendent deux ou plusieurs disciplines existantes ou qui réalisent la fusion entre un domaine scientifique et un ensemble de technologies ».
Outre la mise en place de passerelles lors des années de formation des étudiants, il est essentiel aussi bien d'après les experts du monde académique que ceux du monde industriel d'opérer de manière interdisciplinaire.
À cet égard, les entretiens avec M. Pierre Tambourin (118 ( * )) et son équipe, lors de notre visite Génopole d'Evry, et particulièrement au Centre national de génotypage et au Genoscope, furent instructifs. Dans ce biocluster qui compte 71 entreprises innovantes, 21 laboratoires académiques de recherche et 21 plateformes technologiques connus pour leurs travaux de recherches fondamentales et appliquées, ce point focalisait l'attention. Étaient soulignées les difficultés d'attirer des mathématiciens alors que la France dispose d'une école de mathématiques de renommée mondiale, de retenir pour une durée excédant un an des bio-informaticiens rapidement happés par des laboratoires privés. Malgré le contexte pluridisciplinaire et tourné vers les sciences du vivant de ce grand centre cette problématique du cloisonnement restait prégnante.
ii. Adapter le système de formation continue
D'après M. Didier Tabuteau (119 ( * )) , « au-delà de la formation médicale initiale, qui doit certes évoluer à certaines étapes de la carrière médicale, il est nécessaire de prévoir des cycles de formation et d'adaptation pour des professions qui sont amenées à prendre des décisions aussi essentielles. C'est sans doute plus difficile à mettre en oeuvre sur des professionnels libéraux isolés, que sur des équipes hospitalières ou des professionnels organisés en groupe ».
Le constat est général, la rapidité des évolutions à l'oeuvre exige un effort de formation continue des personnels de santé, indépendant des laboratoires pharmaceutiques et à la hauteur des enjeux.
L'article L.4133-1 du code de la santé publique introduit par la loi n° 2009-879 du 21 juillet 2009 portant réforme de l'hôpital et relative aux patients, à la santé et aux territoires prévoit : « l e développement professionnel continu (DPC) a pour objectifs l'évaluation des pratiques professionnelles, le perfectionnement des connaissances, l'amélioration de la qualité et de la sécurité des soins ainsi que la prise en compte des priorités de santé publique et de la maîtrise médicalisée des dépenses de santé. Il constitue une obligation pour les médecins » . Le décret n° 2011-2116 du 30 décembre 2011, relatif au développement professionnel continu des médecins a mis en place un cadre.
Des mesures ont été prises au sein de la Haute autorité de santé (HAS) car depuis janvier 2013, les professionnels de santé doivent satisfaire, tous les ans, à une obligation de développement professionnel continu (DPC), et doivent s'inscrire dans un programme annuel ou pluriannuel de DPC. La HAS a validé la liste des méthodes et des modalités de DPC pour améliorer la qualité et la sécurité des soins en associant la formation continue et l'évaluation des pratiques professionnelles. Selon la HAS, pour satisfaire à l'obligation de DPC, les professionnels de santé devront s'inscrire dans un programme annuel ou pluriannuel de DPC qui comporte deux activités : l'analyse des pratiques, l'acquisition ou l'approfondissement de connaissances ou compétences.
Cependant, ce système n'a pas encore pris sa pleine mesure alors qu'une formation continue adaptée aux besoins nouveaux est une urgence pour la médecine libérale, si l'on veut garantir un accès équitable de tous à la même qualité de soins comme le préconise la feuille de route sur la Stratégie nationale de santé (rapport de M. Alain Cordier, remis le 23 septembre 2013) (120 ( * )) .
Vos rapporteurs soutiennent les propositions formulées dans ce rapport sur les formations qui « seront renforcées en qualité et surtout décloisonnées afin de s'adapter aux évolutions des pratiques et des modes d'exercice, et répondre aux problèmes de la démographie médicale. Différentes modalités d'accès aux études médicales et paramédicales seront mises en place à la rentrée 2014 comme le prévoit la loi sur l'enseignement supérieur et la recherche du 22 juillet 2013.
Une réflexion sur la modernisation et l'évolution de l'examen classant national (ECN) débutera avant la fin de l'année afin d'assurer une meilleure adéquation de la formation aux besoins des territoires ».
Les évolutions, voire les révolutions à l'oeuvre, selon les termes mêmes de M. Pierre Tambourin (121 ( * )) , conduisent les industries concernées à s'interroger sur leur modèle de recherche et développement (R&D).
B. UN NOUVEAU MODÈLE DE RECHERCHE ET DEVELOPPEMENT (R&D) POUR L'INDUSTRIE PHARMACEUTIQUE
Le Pr Axel Kahn (122 ( * )) considère l'émergence d'un nouveau modèle de R&D comme très probable : « La médecine personnalisée amènera dans le champ économique, l'industrie pharmaceutique à se reposer la question de ses modèles, puisque, à terme, la subdivision de tout champ pathologique en un très grand nombre de sous-domaines, chacun justifiable d'un traitement particulier, marque la fin presque généralisée du modèle de blockbuster sur lequel l'industrie pharmaceutique reposait jusqu'à présent. D'un côté, cela permet de réhabiliter des médicaments qui n'auraient pas démontré leur efficacité dans le champ pathologique qui est global, de l'autre, cela diminue le champ d'utilisation d'une drogue donnée, et par conséquent, pose des problèmes de rentabilité généralisée ».
La médecine personnalisée va à l'encontre de la logique du grand nombre, car elle procède par subdivision de la population générale de patients en groupes, en sous-groupes, de plus en plus réduits, afin de choisir les traitements, les plus adaptés à chaque profil. L'évolution vers une médecine personnalisée passe par une phase de stratification des populations. Cette médecine stratifiée multiplie les sous-groupes de pathologies abolissant dans certains cas la frontière entre maladie courante et maladie rare.
Or cette évolution se produit alors que parallèlement on observe une baisse de l'innovation et des bénéfices de l'industrie pharmaceutique. En effet, la plupart des médicaments les plus vendus tombent ou tomberont dans le domaine public à court ou moyen terme. Cette industrie se trouve confrontée à un nombre insuffisant de nouveaux médicaments de type blockbuster pour compenser cette perte, d'autant que la régulation des essais cliniques et la durée des brevets évoluent lentement.
La médecine personnalisée bouleverse les conditions du transfert de technologies, les modèles de R&D de l'industrie pharmaceutique ainsi que le droit des brevets et de la propriété industrielle.
1. L'évolution des conditions du transfert de R&D
« Le premier grand mouvement auquel on assiste aujourd'hui, et qui rend le transfert de technologie particulièrement crucial dans le domaine de la santé, est une ouverture des industriels petits, moyens, et grands, vers plus petit que soi quand il s'agit de partenariat entre une grande industrie pharmaceutique et une petite société de biotechnologie, ou d'un partenariat entre tous les acteurs privés et la recherche académique » soulignait d'emblée Mme Cécile Tharaud (123 ( * )) .
La complexité croissante des technologies, est un grand problème de R&D dans le domaine de la santé en termes d'efficacité plutôt que de rentabilité. Ceci induit un mouvement de recherche public-privé, privé-privé, en réseau, à l'échelle nationale, internationale, européenne. Ce mouvement, qui représente pour la recherche académique et pour les offices de transfert de technologie une opportunité et un défi, nécessite que les professionnels du transfert de technologie comprennent la demande industrielle, la demande sociétale, et les contraintes réglementaires et de propriété intellectuelle.
a. L'accès au patient
« L'industrie, de la même façon que la recherche translationnelle, redéfinit ses efforts de R&D autour du patient. L'innovation est au coeur des questions que se pose l'industrie, depuis la compréhension de la maladie, vers la compréhension de la question posée en matière de diagnostic, de prévention, d'autres thérapeutiques ou de vaccin. Cette question, est directement celle que soulève la médecine personnalisée, implique que le développement d'un produit se fasse avec accès au patient, et donc à des cohortes plus ou moins importantes » précisait Mme Cécile Tharaud (124 ( * )) . Selon elle, tout au long du développement du médicament, y compris en post-inscription d'autorisation de mise sur le marché (AMM) et, donc, en période de commercialisation, il y a un retour incessant aux populations de patients.
En écho à ce constat, le Pr Philippe Monteyne (125 ( * )) ajoute : « nous travaillons de plus en plus avec ce qu'on appelle la médecine translationnelle, c'est-à-dire en ayant accès à du matériel humain, en rapprochant le lit du malade de la paillasse, et inversement ». Il considère que depuis les années quatre-vingt-dix, on a attaché beaucoup trop d'importance aux modèles animaux ; aussi, parfois, dans des essais en phase 3 on ne connaissait pas assez le mode d'action du médicament, parce qu'une trop grande partie de la connaissance était basée sur des modèles animaux. Selon lui, « on perçoit bien qu'il faudra aller vers une médecine fortement stratifiée, avec des outils de médecine translationnelle, avec un accès à des matériels humains et avec des biomarqueurs ».
Les industriels du médicament estiment nécessaire de comprendre plus tôt la maladie chez l'homme et de conduire des recherches en partant du traitement symptomatique vers le mode d'action des maladies, et finalement vers la génétique quand il y a une base génétique. En agissant de la sorte, on doit découper les maladies. Pour diminuer le taux d'attrition en phase 3, les industriels doivent se rapprocher bien plus du monde académique et du monde hospitalo-universitaire pour accéder au patient et à l'innovation. Aussi considèrent-t-ils qu'il faut effectuer des études cliniques dès la phase zéro, avant même d'avoir un médicament en développement, pour mieux comprendre le malade, pour avoir accès à des échantillons. Et il faut d'ailleurs que le patient donne son autorisation pour qu'on utilise le matériel de façon assez large.
b. La preuve de concept, un élément-clé du transfert
La preuve de concept, mécanisme général sur tous les segments thérapeutiques, est encore plus essentielle en médecine personnalisée, puisque tous les biomarqueurs ne font pas la preuve de leur utilité. Avant d'avancer dans un développement, il faut réunir nombre de preuves de concept pour valider les marqueurs. Or, en France on manque de financement de plateformes de prototypage. Ce manque est encore plus important que sur les cohortes, puisque les investissements d'avenir ont financé cette dernière demande, qui venait à la fois du secteur académique et du secteur privé. Le financement de plateformes de prototypage, sans doute moins aidé, est pourtant très nécessaire aujourd'hui. Pour Mme Cécile Tharaud (126 ( * )) , « c ontrairement à l'Allemagne, la France n'est pas un pays bien équipé pour prototyper des tests de diagnostic. Pourtant cela permet d'avancer plus rapidement dans le développement et le transfert industriel de certains tests, quand le marqueur, ou la série de marqueurs, est validée et en vaut la peine. Il faut donc encore financer les recherches amont du secteur industriel ». Elle regrette d'ailleurs que, lorsque des biomarqueurs ont montré la preuve de leur utilité, ce soient les industriels étrangers, dont les industries de service du diagnostic sont très développées, qui se présentent les premiers.
2. Vers une réorientation de la R&D sur des médicaments de niche ?
Les progrès de la médecine expérimentale moderne ont été fondés sur la découverte de médicaments à très large spectre, les blockbusters. Ainsi, les vaccins, les antibiotiques ont été en quelque sorte les premiers blockbusters de l'industrie pharmaceutique. Ces blockbusters assuraient un certain équilibre dans le portefeuille de brevets détenus par un industriel, mais leurs prix sont en baisse en raison des génériques et de l'arrivée à leur terme des brevets. Aussi les médicaments de niche issus de la pharmaco-génomique tendent-ils à être privilégiés par les laboratoires.
Cependant, comme le rappelait le Pr Hervé Chneiweiss (127 ( * )) , « les différents acteurs, au lieu de s'engager sur la voie royale de cette médecine de demain, ont tendance à ne pas se précipiter . L'industrie pharmaceutique est réticente à s'adapter car la médecine personnalisée réduit la taille du marché et les bénéfices liés à ses médicaments vedettes ». Il précise : « Le système prudentiel résiste aux changements car le retour sur investissement des diagnostics et des thérapies adaptées à chaque individu n'est tout simplement pas là ».
Selon M. Jean-Louis Touraine (128 ( * )) , « l'exploitation des connaissances liées à la recherche sur le génome humain et les biotechnologies permettent d'espérer que des médicaments soient administrés de façon ciblée, et que des thérapies cellulaires individualisées soient développées. C'est le tournant des biotechnologies par rapport à la pharmacie chimique ».
« Quel sera dès lors le modèle de retour sur investissement, notamment pour les sociétés pharmaceutiques et biotechnologiques, qui travaillent sur des traitements de niche ? Les thérapies les plus avancées coûtent des dizaines, des centaines de milliers d'euros par an, comment financera-t-on leur accès ? » s'interrogeait le Pr André Syrota (129 ( * )) .
Le développement de nouveaux traitements coûte de plus en plus cher, avec un délai de mise sur le marché de plus en plus long, et une probabilité de succès de plus en plus difficile à estimer à l'avance. Pour autant, cette mutation était à l'oeuvre depuis une dizaine d'années.
a. Une mutation liée à la crise de l'innovation dans l'industrie pharmaceutique
i. Les déboires de l'industrie pharmaceutique
Le Pr Laurent Degos (130 ( * )) a rappelé que la mutation du monde pharmaceutique a commencé en 2004-2005, indépendamment de la médecine personnalisée : à la suite du scandale du médicament Vioxx, Merck, l'un des premiers laboratoires pharmaceutiques, a été mis en péril par les class actions et les procès intentés contre lui.
Selon lui, cette affaire a conduit les différents laboratoires à mener des réflexions séparément et presque tous ont abouti à la même conclusion : arrêter voire limiter la recherche de traitements innovants pour de grandes populations, et s'en tenir à des traitements pour des populations de 1 000 à 2 000 personnes dans chaque pays, car si un accident survient une fois sur 20 000 patients, il faudra dix ans pour qu'il survienne et vingt ans pour la survenue de deux cas. Avec ce raisonnement, le risque d'être poursuivi pour un accident causé par un médicament devient quasi nul. Dès lors l'industrie pharmaceutique a considéré que pour le moment, les vieilles molécules, souvent des génériques, étaient suffisantes pour traiter de grandes populations et des maladies fréquentes.
ii. La nécessité de développer de nouvelles molécules
Les quinze groupes mondiaux du secteur pharmaceutique affrontent depuis quelques années une période sans précédent d'expiration de brevets. Dans le même temps, le secteur pharmaceutique est confronté à une politique de maîtrise des coûts plus rigoureuse de la part des gouvernements et à une pression sur la demande. Cependant, peu de médicaments génèrent des gains suffisants pour couvrir l'ensemble des coûts de recherche et de développement engagés, d'où l'importance capitale du respect de la propriété intellectuelle que confère le brevet.
Les prochains médicaments dont les brevets vont tomber dans le domaine public sont des médicaments issus des biotechnologies, dont les génériques, et sont beaucoup plus coûteux à produire ; l'impact sur les ventes de médicaments princeps sera donc moins important que celui des génériques.
Pour surmonter la crise de l'innovation médicamenteuse, les industries pharmaceutiques prennent le virage de la médecine personnalisée pour essayer de faire évoluer le cadre réglementaire en préconisant le modèle du médicament orphelin pour les essais cliniques. « Le travail bibliographique mené par le LEEM montre qu'il existe plus de 400 projets pour lesquels on a un couple molécule et test prospectif pour la phase clinique 2 et à peu près 170 molécules pour des tests de phase 3. De ces innovations en développement, des avancées seront effectivement proposées dans un avenir proche » indiquait M. Oliver Perche (131 ( * )) .
b. L'émergence du modèle des médicaments de niche
Désormais, l'industrie du médicament cherche à développer des médicaments ultra ciblés, agissant sur une mutation ou une anomalie génétique particulière. Selon une étude du LEEM (132 ( * )) , ce type de traitement présente l'avantage d'induire moins d'effets secondaires tout en ayant un meilleur taux de réponse au traitement . « Associés aux médicaments, les biomarqueurs améliorent la qualité et l'efficacité des solutions thérapeutiques. Ils permettent non seulement d'offrir des traitements ciblés et à la carte, mais sont également utilisés pour le dépistage et la collecte d'informations prédictives visant à toujours mieux soigner les patients ».
C'est une approche différente de la R&D, explique Mme Corinne Blachier-Poisson (133 ( * )) , car la cible des médicaments se restreint au fur et à mesure que la recherche avance. Au final, la population concernée ne dépasse parfois pas quelques milliers d'individus. Mais pour les industriels, le potentiel d'innovation est important. Selon elle, c'est un changement pour l'industrie pharmaceutique, la médecine personnalisée oblige à mieux cibler les populations, ce qui permet de raccourcir les délais de développement. Parallèlement, cela exige une recherche multifactorielle, une recherche ciblée sur un récepteur et un essai pour identifier le bio-marqueur qui joue un rôle important. C'est une autre façon de faire de la recherche.
Pour autant, à force de restreindre et d'individualiser les traitements, on individualise les malades, mais on individualise aussi les maladies orphelines. On risque de créer très rapidement des catégories de maladies orphelines et ultra-orphelines, observe le Dr Pierre Attali (134 ( * )) ; il indique que la prévalence est de 5/10 000 sujets en Europe pour les maladies orphelines, avec une contribution majeure à la santé publique. Cependant, pour les maladies ultra-orphelines, le nombre de patients est extrêmement restreint et il n'y a pas de définition : ce serait moins de 20 000 patients dans le monde et une prévalence en Europe inférieure à 1/50 000 sujets. En Grande-Bretagne, le National Institute for Health and Care Excellence (NICE ) les définit à moins de 1 000 patients. Si on reporte ce chiffre à la population européenne, on obtient une prévalence inférieure à 1/50 000 sujets.
i. Le recours « aux anges déchus »
La médecine personnalisée stratifiée, constitue une opportunité pour l'industrie pharmaceutique de ressortir certains médicaments qui ne semblaient guère efficaces statistiquement, « les anges déchus ». Grâce à des biomarqueurs, on identifie les patients qui seraient les plus sensibles à ces molécules et les maladies, souvent des maladies rares, pour lesquels ils fonctionneraient. Le modèle médicament orphelin intéresse les industriels tant pour recycler d'anciennes molécules que pour en développer de nouvelles.
ii. Le modèle du médicament orphelin
Comme le rappelle le Pr Laurent Degos (135 ( * )) , il existe une conjonction « entre le milieu académique qui trouve des mécanismes assez restreints, et des cibles et des médicaments pour de petites populations, et une réflexion industrielle qui ne souhaite pas prendre trop de risques pour ne pas mettre toute la maison en danger. De plus, puisque la découverte est faite au niveau académique, il n'y a plus besoin de recherche fondamentale pour ces médicaments orphelins ».
La mise sur le marché est plus simple, le remboursement est en général accepté, et il n'y a pas besoin de publicité, car il s'agit de traiter une maladie orpheline ; cela permet d'alléger le budget de R&D. En passant par la médecine personnalisée, les industriels espèrent faire évoluer les instances de régulation pour qu'elles acceptent des critères d'essais moins exigeants que les essais classiques randomisés avec des standards assez hauts, et ainsi bénéficier des législations et dérogations spécifiques accordées aux médicaments orphelins.
iii. Vers une législation sur les essais cliniques plus incitative ?
La législation sur les médicaments orphelins a pour but d'inciter les entreprises pharmaceutiques à mettre au point et à commercialiser des médicaments ciblés, et donc plus efficaces, pour traiter des maladies rares.
En 1983, les États-Unis avaient créé avec l'Orphan Drug Act , la première loi sur les médicaments orphelins. Dans les années quatre-vingt-dix, le Japon, puis l'Australie ont adopté une législation similaire. En Europe, le Parlement européen a adopté le Règlement (CE) n° 141/2000 du 16 décembre 1999 sur les médicaments orphelins, texte entré en vigueur en janvier 2000.
Lorsqu'une autorisation de mise sur le marché (AMM) est accordée pour un médicament orphelin, celui-ci bénéficie, avec certaines dérogations et limitations, d'un droit d'exclusivité commerciale pendant dix ans. « Les médicaments orphelins peuvent bénéficier de mesures d'incitation prises par la Commission et les États-membres pour promouvoir leur recherche, leur développement et leur mise sur le marché. Il s'agit de mesures d'aide de nature fiscale à la recherche en faveur des petites et moyennes entreprises », observe Mme Hélène Gaumont-Prat (136 ( * )) . Une réflexion devra être menée sur le modèle de recherche si les médicaments de niche deviennent la voie normale de mise au point de traitements.
Les laboratoires pharmaceutiques, les chercheurs et agences de régulation explorent des pistes de réforme, pour les études de phase 3, de plus en plus longues et coûteuses pour les laboratoires. On envisagerait une autorisation de mise sur le marché (AMM) plus rapide des médicaments mais avec un suivi plus strict grâce à un recours massif à des bases de données transmettant les résultats du traitement. L'évaluation ne se ferait pas seulement au moment de l'enregistrement, mais tout au long de la vie du médicament. On tendrait à recourir massivement à des AMM conditionnelles, assorties de l'obligation de fournir des éléments complémentaires, comme cela existe déjà, pour certaines molécules.
Certains, comme le Pr Bruno Falissard (137 ( * )) , biostatisticien, plaide pour un système assez original : une « pré-AMM » assez précoce, après les essais de phase 2, puis une prescription limitée à des centres experts (type CHU), qui permettrait de continuer l'évaluation avec un financement public. Ce n'est que dans un troisième temps que l'usage serait étendu en médecine de ville. On éviterait selon lui que ce soit l'industrie pharmaceutique qui évalue les médicaments qu'elle développe.
Les nouvelles technologies, et les bases de données, constituent un autre moyen d'optimiser les essais cliniques, grâce à des gélules à puce RFID ou des patchs surveillant la prise des médicaments ; les données recueillies en ligne peuvent être envoyées à un smartphone , ou à une interface Internet ; encore faut-il s'assurer de la protection de ces données.
En outre, des essais virtuels utilisent des principes de modélisation. On modélise un patient : un modèle mathématique permet de déterminer les facteurs influençant les résultats positifs ou négatifs d'un essai et de mieux cibler les critères d'inclusion. On peut ainsi obtenir des informations avec 1 000 patients au lieu de 10 000, avec des délais de réalisation plus courts et des coûts considérablement abaissés.
Actuellement, on attend le règlement du Parlement européen et du Conseil européen concernant les essais cliniques de médicaments à usage humain, dont le vote est prévu au printemps 2014. Il impliquera une simplification administrative en matière de délais d'autorisation, en particulier en garantissant des délais d'autorisation des essais compétitifs au niveau international tout en préservant la sécurité des patients. Il assurera une plus grande transparence concernant les résultats de ces essais, qui devront être rendus publics dans un délai d'un an. La mise en place d'un portail européen, véritable guichet unique, conduira à une réduction significative des délais de mise en place des essais dans les différents pays européens, lesquels ne pourront plus dépasser 60 jours.
Pour l'instant, force est de constater que les dispositions relatives aux « recherches impliquant la personne humaine » édictées en 1988 par la loi Huriet sont destinées à évoluer avec la loi Jardé de mars 2012, qui distingue trois types de recherche selon le niveau de risque pour le patient. Cependant, les décrets d'application ne sont toujours pas sortis et devront s'accorder avec le futur règlement européen.
D'un côté, les industriels souhaitent un guichet unique d'accès aux bases de données, de l'autre, les patients veulent être rassurés quant à l'usage de leurs données qui sont essentielles au développement des thérapies ciblées.
Cependant, la régulation protégée pour les maladies orphelines génétiques risque d'être remise en question. D'après M. Didier Tabuteau (138 ( * )) , la médecine personnalisée remplace progressivement le terme de médicament orphelin, élargissant le spectre certes, mais occultant la philosophie du médicament orphelin. La multiplication des médicaments de niche, des tests, des marqueurs pose inévitablement la question d'un nouvel équilibre. Le financement de la recherche évoluera, cela remettra aussi en cause les conditions initiales d'évaluation des médicaments et une transition, certes difficile, devra se faire.
c. Une R& D dépendante des tests diagnostiques et des tests compagnons
Au niveau de la R&D, l'industrie s'intéresse non plus au seul médicament, mais au couple médicament-test diagnostique (le test compagnon). Ceci l'oblige à avoir une vision intégrant médicament, test diagnostique, méthodes de stockage de l'information. Cette évolution exige une diversification de compétences allant du diagnostic in vitro au dispositif médical. Pour un médicament donné, il s'agit par ces tests, d'identifier les sujets répondeurs et non répondeurs ainsi que ceux qui pourraient provoquer un effet indésirable afin d'adapter la dose à chaque individu. Cependant, d'après Mme Christine Guillen (139 ( * )) , les taux d'attrition concernant les biomarqueurs sont élevés ; « la recherche et la validation de biomarqueurs ayant une vraie valeur médicale ajoutée sont extrêmement longues et difficiles ».
i. Identifier les groupes cibles par des tests compagnons
Ces biomarqueurs compagnons permettent de mesurer l'efficacité d'un nouveau médicament, de sélectionner les patients auxquels la thérapie sera la plus adaptée et de surveiller le développement de la maladie et les effets du traitement. Ils constituent des outils d'orientation thérapeutique.
Les entreprises du médicaments (le LEEM) (140 ( * )) considèrent que la découverte et la validation de biomarqueurs visant à identifier et sélectionner des groupes de patients homogènes, éligibles à des médicaments adaptés très précisément à leur maladie, constitue un nouvel enjeu pour les industriels engagés dans la mise au point de traitements de plus en plus personnalisés. L'association médicament/biomarqueur s'inscrit dans une dimension stratégique et économique.
Le biomarqueur compagnon a la capacité de transformer le développement et la mise à disposition d'un nouveau médicament en un processus plus rapide et moins coûteux. À l'appui de leur démonstration, les industriels citent le Glivec qui a permis le traitement de la leucémie myéloïde chronique avec 88 % des patients traités encore en vie six ans après le diagnostic contre 20 % avant l'arrivée de cette molécule.
ii. Le biomarqueur compagnon : un marché d'avenir ?
Les industriels qui développent un médicament cherchent le test compagnon, car ce qu'ils perdront en vente du médicament, ils espèrent le regagner en vente du test compagnon qui sera fait, a priori, sur une population beaucoup plus large.
Selon eux, la médecine personnalisée implique le développement simultané d'un test compagnon, un deuxième produit qui permet de déterminer si le médicament sera efficace ou non sur tel ou tel patient. Le marché du diagnostic compagnon pourrait largement s'accroitre dans les cinq prochaines années. Cependant, si pour le médicament, à la Haute autorité de santé (HAS), le processus est bien banalisé, et répond aux questions posées pour mettre des molécules sur le marché, pour le test diagnostique in vitro , des paramètres restent bloquants dans le processus d'instruction des dossiers.
Un industriel ne peut pas demander la mise sur le marché d'un test diagnostique en même temps que celle du médicament correspondant et des synchronisations s'en suivent. L'évaluation technique et médicale du médicament est séparée et désynchronisée du test. Pour le test, ces critères ne sont pas publics, ni connus des industriels eux-mêmes et il en est de même pour les délais de mise sur le marché. Il n'y a pas à ce jour d'évaluation économique du couple médicament-test diagnostique, et les acteurs sont différents.
Selon Mme Corinne Blachier-Poisson (141 ( * )) , pour les laboratoires qui comme Amgen, ne développent que des médicaments avec pour les diagnostics des partenariats avec des sociétés extérieures, ou des centres universitaires, il faut s'assurer que les médicaments soient développés au même rythme que les bio-marqueurs et tests. Or ceux-ci sont souvent développés plus rapidement que les médicaments, dont l'enregistrement exige des procédures plus lourdes d'autorisation de mise sur le marché (AMM) que celles d'un test. Ce ne sont pas les mêmes structures qui décident des deux.
M. Olivier Perche (142 ( * )) a suggéré une harmonisation du processus d'assemblage test et molécule en demandant que l'évaluation technique et économique du test et de la molécule associée, soit réalisée conjointement, ce qui représenterait une nouveauté en particulier pour les industriels du diagnostic. Il souhaite en outre « un encadrement des délais, l'obligation d'utiliser des produits marqués CE, une autorisation temporaire d'utilisation avec un financement associé en attente du remboursement. L'objectif étant de construire un nouvel acte comprenant une partie test et une partie activité médical e ».
La Haute autorité de santé (HAS) est consciente du problème. Selon le Pr Gilles Bouvenot (143 ( * )) , deux évaluations conjointes devront être effectuées : celle du médicament par la Commission de transparence et celle du test compagnon par la Commission nationale d'évaluation des dispositifs médicaux et des technologies de santé (CNEDiMTS), ce qui, selon lui, ne pose pas de problème particulier, « la HAS étant déjà coutumière de ce type d'évaluation transversale . La CT va devoir réfléchir à l'évaluation spécifique de ces thérapies ciblées qui ne pourra plus être la simple déclinaison de l'évaluation standard des produits ?traditionnels? ».
S'agissant de l'évaluation concernant le cancer, il précise : « nos évaluations et les critères pris en compte pour la réaliser dépendront du type de cancer et de sa rareté, du type de cible et, même dans certains cas, la survie en fonction de l'évolution spontanée de la maladie ou sous traitement habituel pourra être retenue. Pour autant, nos critères de jugement devront toujours répondre aux deux exigences actuelles retenues par la Commission européenne : être objectifs et vérifiables ».
Il s'agira de mieux discriminer les types d'essais cliniques considérés comme pertinents pour démontrer l'efficacité du type de médicament et accepter des essais d'effectifs limités. Pour la HAS, il faut, dans l'intérêt des malades, arriver à un compromis entre le désir d'une mise à disposition rapide des nouveaux produits supposant parfois des dossiers insuffisamment étayés et le désir d'une mise à disposition la plus sécuritaire possible, sans transiger sur la sécurité des patients face à des autorisations de mise sur le marché (AMM) un peu trop précipitées.
En revanche, selon le Pr Gilles Bouvenot, la HAS s'efforcera de raccourcir les délais d'évaluation, au-dessous des 90 jours imposés par la directive européenne. Encore faut-il que la durée de la négociation pour la fixation du prix par le comité économique des produits de santé (CEPS) n'allonge pas trop, du fait des revendications des industriels, cette mise à disposition.
Pour le Pr Thomas Tursz (144 ( * )) , les combinaisons de drogues dans le cancer, offrent des possibilités de traitement en raison du nombre d'anomalies, de résistances, car actuellement on donne à près de 90 % de la population des médicaments qui ne marchent pas, coûtent chers, et sont toxiques pour tout le monde. Il observe que « le schéma actuel de mise sur le marché, comme le schéma que proposent les industriels, vise à mettre un médicament sur le marché tout seul, avant de permettre de faire des associations, en particulier avec des médicaments issus de firmes concurrentes. Ceci est une catastrophe scientifique et économique et amène à de gros problèmes industriels : sur dix substances qui entrent en phase 1, une seule deviendra réellement un médicament. Les industriels jettent donc à la poubelle neuf médicaments sur dix, sur lesquels ils ont déjà énormément investi sur les plans recherche et essais précliniques ».
Il préconise un système permettant très précocement de tester sur un financement européen des combinaisons de médicaments en fonction des anomalies du génome.
La régulation à appliquer pour le développement de traitements ciblés reste un enjeu, car ces médicaments feront sans doute l'objet de brevets, destinés à assurer le retour sur investissement de la R&D et poseront les questionnements liés à la brevetabilité du vivant. La question de la propriété intellectuelle du test diagnostique développé par un industriel pour son médicament se pose avec une acuité de plus en plus forte.
3. L'impact sur la brevetabilité
« La médecine personnalisée est essentiellement comprise comme une méthode de traitement. Donc il faudra à nouveau que la jurisprudence évolue, de façon à ce que la médecine personnalisée prenne une forme concrète que les spécialistes du droit de la propriété intellectuelle arrivent à décrire selon leur arsenal juridique » rappelait Mme Cécile Tharaud (145 ( * )) .
L'Office européen des brevets (OEB) contrairement à celui des États-Unis, ne permet pas aujourd'hui de breveter des méthodes de traitement. Or, en médecine personnalisée , a priori le recours au test compagnon relève de la décision de prescrire un médicament basé sur un test de diagnostic. Au sens de la propriété intellectuelle européenne, on est encore face à une interrogation.
Les questions de brevetabilité, complexes mais cruciales, se posent avec une acuité particulière dès lors que l'on souhaite développer des traitements ciblés impliquant des dispositifs médicaux, notamment des tests qui sont le pendant de la médecine personnalisée. L'incitation à la recherche et au développement de tests génétiques prédictifs ou diagnostiques assortis à une thérapie ciblée dépend en grande partie des solutions qui seront trouvées pour résoudre les questions de brevetabilité et de propriété industrielle.
Cette problématique provoque une tension que M. Maurice Cassier, décrit ainsi (146 ( * )) : « je rappellerai que l'application du droit des brevets dans le champ de la médecine, dans le champ médical, a toujours été problématique et difficile ». Il observe que depuis vingt ans, on se trouve face à une continuité de ces interrogations et contestations. « La particularité est que des discussions juridiques ont lieu à la fois à l'intérieur de l'Office européen des brevets et aujourd'hui aux États-Unis. Ce qui est intéressant, c'est aussi l'engagement, que ce soit en Europe ou aux États-Unis, des associations médicales, des médecins, dans la discussion de ces brevets et de leurs effets. Toutes les sociétés de médecine moléculaire, alliées aux associations de patients, puisqu'il s'agit de brevets sur les gènes de prédisposition au cancer du sein, se sont portées plaignantes contre ces brevets pour demander leur annulation » .
a. Les procédures judiciaires aux États-Unis
Deux affaires emblématiques : le cas Mayo Collaborative Services contre Prometheus Laboratories , et le cas Association for Molecular Pathology et autres contre Myriad Genetics instruites par la Cour Suprême des États-Unis portent sur la brevetabilité du vivant et notamment sur celle des gènes.
i. L'affaire Mayo Collaborative Services contre Prometheus Laboratories
L'affaire Mayo Collaborative Services contre Prometheus Laboratories a initié les débats. Le laboratoire Prometheus a mis au point une méthode de dosage à la suite de la prise du médicament pour le traitement de la maladie de Crohn, ce qui a permis le développement d'un test déterminant la dose à administrer au patient pour qu'une réponse positive soit observée sans effets secondaires. Mayo Collaborative Services qui achetait et utilisait les kits de tests de la société Prometheus Laboratories, a décidé de vendre et de commercialiser son propre test. Le laboratoire Prometheus , en juin 2004, a alors poursuivi Mayo pour violation de brevet devant un tribunal de district qui a conclu que les tests de Mayo portaient effectivement atteinte aux brevets de Prometheus . Après une procédure en appel, l'affaire a été portée devant la Cour Suprême qui a rendu son verdict le 19 mars 2012, concluant à la non-brevetabilité de ce test comme reposant sur les lois de la nature.
L'affaire suivante est encore plus emblématique. Elle met aux prises, d'un côté, des représentants de la communauté scientifique et des associations de citoyens, notamment l'Association of Molecular Pathology , l'American Civil Liberties Union et, de l'autre, Myriad Genetics.
ii. L'affaire Myriad Genetics contre l' Association of Molecular Pathology et l' American Civil Liberties Union
Elle a commencé en 2009 par le dépôt d'une plainte auprès de la Cour fédérale du district sud de New York contre l'United States Patent and Trademark , ( USPTO ) l'Office américain des brevets, à qui les plaignants reprochaient d'avoir accordé à Myriad Genetics une quinzaine de brevets qui auraient dû être invalidés.
Les différents brevets délivrés à cette société sur les gènes BRCA1 et BRCA2 à des fins d'applications diagnostiques pour les cancers du sein et de l'ovaire s'étendaient à toute méthode utilisant une comparaison de la séquence d'une personne à risque avec la séquence de référence décrite par le brevet. Ainsi, tout laboratoire contrevenant aux termes de ce brevet s'exposait à un procès pour contrefaçon, quelle que soit la technique de détection utilisée. Les intervenants contre Myriad Genetics considéraient que ces brevets contribuaient à étouffer la recherche menée dans ce domaine et limitaient les options offertes aux patients quant à leurs soins médicaux.
En première instance en 2010, les brevets de Myriad Genetics ont été invalidés, au motif que la séquence d'ADN isolée couverte par les brevets est très proche de l'ADN tel qu'il est présent dans le corps humain et n'est donc pas brevetable. Cette décision a été infirmée une première fois en appel. La Cour suprême, saisie de l'affaire, a décidé de renvoyer cette affaire à la Cour d'appel en se référant explicitement à la jurisprudence de l'affaire Prometheus Laboratories contre Mayo Collaborative Services . Cependant, la Cour d'appel fédérale a tranché en faveur de l'acceptation de la brevetabilité du test dans une décision du 16 août 2012 (147 ( * )) .
De nouveau saisie de ce litige, la Cour suprême a rendu sa décision le 13 juin 2013. Le résumé communiqué par le rapporteur de la décision a été commenté par Mme Elisabeth Thouret-Lemaitre (148 ( * )) . Il s'agissait, pour la Cour suprême, de répondre à deux interrogations concernant la brevetabilité de séquence d'ADN. Un segment d'ADN existant dans la nature, dès lors que ce segment est isolé du reste du génome humain, est-il brevetable ? L'ADN synthétique ou complémentaire contenant la même information codante pour la protéine que celle du segment d'ADN naturel, mais sans contenir la portion d'ADN ne codant pas pour les protéines, peut-il être lui aussi brevetable ? « Nous considérons que le segment d'ADN existant dans la nature est un produit de la nature et n'est pas brevetable simplement parce qu'il a été isolé. En revanche l'ADN complémentaire est brevetable car il n'existe pas dans la nature » répond la Cour suprême. La décision de la Cour d'appel est donc en partie confirmée et en partie annulée.
Selon Mme Elisabeth Thouret Lemaitre (149 ( * )) , la décision n'a examiné ni les revendications concernant la méthode, ni les brevets relatifs aux nouvelles applications de la connaissance des gènes BRCA 1 et 2. Elle n'a pas non plus évalué la brevetabilité d'un ADN dans lequel l'ordre des nucléotides naturels aurait été modifié. D'ailleurs elle a mentionné ce qui n'avait pas été traité. Il s'agit d'une décision limitée aux gènes existant dans la nature.
Ces deux affaires soulèvent la question de l'avenir de la médecine personnalisée et témoignent de la difficulté inhérente à la définition de la brevetabilité du vivant. Dans le cadre de la médecine personnalisée, dont le principe même est de développer un médicament adapté à un type de patient, des tests similaires à ceux de Myriad Genetics sont fréquemment développés. En rappelant au travers de l'affaire Mayo Collaborative Services contre Prometheus Laboratories la non-brevetabilité des lois de la nature, la Cour Suprême entend protéger le développement de la recherche vis-à-vis de brevets qui offriraient une protection trop étendue à leurs bénéficiaires en leur conférant une situation quasi monopolistique.
Ces décisions ont entraîné une réaction de l'USPTO qui a diffusé un mémorandum à l'attention des examinateurs américains visant à guider les parties prenantes du secteur lors de leurs demandes de brevet : ce mémorandum annonce que la décision « Myriad genetics » change désormais la politique d'examen de l'USPTO relève Mme Hélène Gaumont-Prat (150 ( * )) .
Le monopole d'exploitation dont pouvait se prévaloir Myriad Genetics entrainait une perte d'expertise et d'information pour les médecins et les chercheurs européens qui n'étaient plus en mesure d'améliorer les techniques et les méthodes diagnostiques, et donc de poursuivre leurs recherches dans des conditions acceptables.
En outre, grâce à l'envoi obligatoire chez Myriad Genetics d'échantillons d'ADN provenant de personnes à haut risque, cette firme américaine constituait à son profit l'unique banque de données génétiques au monde. Elle disposait alors d'une maîtrise totale des principaux matériaux de recherche sur les gènes de prédisposition aux cancers du sein et de l'ovaire, et avait une ainsi une avance conséquente pour réaliser d'autres découvertes et déposer éventuellement d'autres brevets sur leurs applications. L'arrêté de la Cour suprême aura des effets en Europe et en France.
Le Pr Dominique Stoppa-Lyonnet (151 ( * )) considère que la décision est assez claire concernant l'identification de gènes car la séquence de gènes, qu'ils soient mutés ou pas, ne peut plus entrer dans le domaine privé et être brevetée. Elle s'interroge : « ne peut-on pas considérer que cela ouvre l'ensemble du marché des tests génétiques, que ce soit en génétique constitutionnelle ou somatique ? D'ailleurs, il ne faut pas se leurrer, cette décision arrive à point nommé en même temps que le séquençage très haut débit. Il y a eu, j'imagine, une pression extrêmement forte des sociétés de biotechnologies qui développent le séquençage très haut débit, devenu impossible si plus de 20 % des gènes sont brevetés ».
Ces affaires révèlent une évolution aux États-Unis : s'il y a vingt ou trente ans, la croissance de l'industrie biotechnologique s'est fondée sur les brevets, le Département d'État et la justice américaine aujourd'hui commencent à penser qu'il faudrait que cette régulation évolue. Cette réflexion porte, sur les brevets et la connaissance. Elle se base sur des articles de la Constitution, sur les progrès de la science, sur la liberté de la recherche, elle est soutenue par des associations de patients et une partie de la communauté scientifique aux États-Unis et dans le monde.
La situation de monopole de Myriad Genetics a fait émerger aux États-Unis un marché de la génétique qui tend à dissocier la réalisation proprement dite des tests, de la consultation génétique et du suivi des personnes à risque. Cette logique se heurte à la conception de la santé publique, en France et plus généralement en Europe. Les pratiques des cliniciens européens se font, en général, sur un modèle intégrant recherche biologique, exploration clinique et prise en charge des personnes. En outre, on tient compte des aspects médicaux et psychologiques du diagnostic ainsi que des trajectoires cliniques des personnes à risque et de leur famille.
b. Les pratiques européennes et l'Office européen des brevets (OEB)
La bataille autour de la brevetabilité du vivant s'est étendue à l'Europe. Après avoir longtemps résisté à l'extension des principes de brevetabilité aux gènes, la directive 98/44/CE du Parlement européen et du Conseil du 6 juillet 1998, relative à la protection juridique des inventions biotechnologiques a transposé la conception issue de l'Office américain des brevets dans le droit européen. Cette directive dispose que « t out élément isolé du corps humain ou produit par un procédé technique, y compris la séquence totale ou partielle d'un gène, peut constituer une invention brevetable ». La décision de la Cour Suprême américaine a donc une incidence.
En Europe, les brevets sont délivrés pour des inventions nouvelles, impliquant une activité inventive, et susceptibles d'applications industrielles. La protection conférée par un brevet à une séquence de gènes est limitée à la fonction technique de cette séquence, telle que décrite dans la demande de brevet.
Dans la nouvelle convention européenne sur les brevets, un article important concerne les exceptions à la brevetabilité. Il est bien précisé que les brevets ne sont pas délivrés pour les méthodes de diagnostic appliquées au corps humain ou animal. En revanche, les produits pour la mise en oeuvre des méthodes peuvent être brevetés à condition bien sûr, d'être nouveaux et d'avoir une activité inventive.
Selon Mme Elisabeth Touret-Lemaitre (152 ( * )) , la question principale pour les déposants et les offices de brevets est la nouveauté de l'invention ; elle se pose pour le traitement du groupe de patients possédant un biomarqueur spécifique, une nouvelle revendication et représente une limitation par rapport au traitement déjà connu antérieurement pour tous les patients, ceux qui possèdent ledit biomarqueur et ceux qui ne le possèdent pas. L'OEB a décidé que, dans un test en deux étapes, s'il y a recouvrement en partie des groupes de patients, et que le nouveau groupe est arbitraire, la brevetabilité n'est pas possible, car la revendication n'est pas nouvelle. Il faut démontrer que le groupe de patients nouvellement déterminé par ce biomarqueur n'est pas compris dans ce groupe ou ne recoupe pas le groupe précédent.
Une décision de 1986 T19/86 appelée « Cochons 2 » portait sur un traitement prophylactique des animaux ; la brevetabilité a été reconnue car la population traitée était différente sur le plan immunologique. La décision essentielle est la décision T 233/96. Elle a conclu que le groupe de patients n'était pas distinctif, et donc de ce fait, la revendication n'était pas acceptable. Le groupe de patients ne peut pas être arbitraire.
La brevetabilité est acceptée par l'OEB si les groupes de patients ne se recouvrent pas et s'il existe une relation fonctionnelle entre le biomarqueur et le traitement thérapeutique. Trois décisions de chambres de recours techniques de 2001, 2004 et 2006 : T83/01, T1399/04 et T1642/06 concluent à la brevetabilité de ces traitements pour certains groupes de patients, parce qu'il y a une relation fonctionnelle entre le biomarqueur et le traitement thérapeutique.
Des incertitudes subsistent pour Mme Thouret-Lemaitre qui fait un parallèle avec les décisions relatives au régime de dosage. « Il a été accepté au niveau de l'OEB, dans une décision de la Grande chambre de recours, qu'une posologie particulière, avec une administration particulière, était brevetable. Malheureusement, les tribunaux nationaux en France, en Allemagne et en Grande-Bretagne qui ont eu à traiter de brevets relatifs à telles posologies, n'ont pas tous pris les mêmes décisions. La France n'a malheureusement pas accepté ce brevet ».
Pour ce qui est de la médecine personnalisée, on a peu de décisions. Selon Mme Hélène Gaumont-Prat (153 ( * )) , ces affaires judiciaires américaines récentes reflètent une réalité : celle de la législation qui peine à suivre le rythme imposé par les avancées de la biotechnologie.
En France, la question de la brevetabilité se pose essentiellement pour les tests in vitro . Le droit américain requiert des exigences similaires de nouveauté et d'activité inventive pour l'obtention d'un brevet. En revanche, si le droit français exige que l'application de l'invention soit industrielle (article L.611-15 du Code de la propriété intellectuelle), le droit américain reconnaît, en principe, la brevetabilité des inventions dès lors qu'elles sont utiles à la société. Les exclusions, déterminées par les Cours de justice américaines, concernent les lois naturelles, les phénomènes physiques et les idées abstraites.
Il reste que pour l'instant l'OEB délivre toujours des brevets de gènes ; aucun renversement n'a eu lieu, malgré plusieurs oppositions ponctuelles ; aussi la pratique de la prise de brevets n'a-t-elle pas changé.
Cependant, comme l'a souligné M. Maurice Cassier (154 ( * )) , la question de la brevetabilité est donc à la fois une question juridique et de politique de santé. Dès le début des années 2000, elle a été évoquée à plusieurs reprises à l'Assemblée nationale, en 2002, en 2003, dans des questions écrites. Lors de la révision de la loi de bioéthique, elle a même fait l'objet d'un amendement particulier. L'OPECST s'en est saisi et l'un de vos rapporteurs a présenté un rapport à ce sujet dès 2004 sur « Les conséquences des modes d'appropriation du vivant sur les plans économique, juridique et éthique » (155 ( * )) . C'est la conjonction de la mobilisation des médecins en France et en Europe à cette époque, de plusieurs États qui se sont portés opposants contre ces brevets, et de la discussion sur la transposition qui a conduit à cet amendement.
Le développement des traitements ciblés, base de la médecine personnalisée exigera sans doute le recours à des systèmes de régulation des brevets qui favorisent la R&D et l'accès aux soins.
c. Les solutions envisageables
i. Les procédés limitant les effets nocifs des brevets
• Les procédures d'opposition
En Europe, les procédures d'opposition ont été fréquemment utilisées dans le domaine de la biologie. Des médecins, parfois des ministères de la santé inquiets des conséquences à la fois sur le prix et sur la disponibilité de ces technologies, se portent opposants pour demander l'annulation de brevets. Ces procédures d'opposition ont été utilisées par les généticiens, par les médecins, mais ces actions sont limitées car on formule une opposition contre un brevet particulier, sur une technologie particulière. Il faudrait multiplier les actes d'opposition pour arriver à des régulations plus stables et plus durables.
Il arrive aussi qu'une fondation médicale prenne un brevet sur un des gènes et distribue gratuitement des licences à tous les laboratoires médicaux susceptibles de pratiquer des tests génétiques. Dans ce cas, les institutions médicales utilisent le brevet, en quelque sorte pour contrarier et empêcher ses effets nocifs.
• Les licences d'office pour raison de santé publique
Pour surmonter les blocages dus à l'existence de brevets trop larges ou qui sont exploités au détriment de la santé publique, les procédures de licences obligatoires permettent d'utiliser l'invention sans l'autorisation de son propriétaire dès lors que le prix, la qualité s'avèrent contraire à l'intérêt de la santé publique. Cette procédure existe en France depuis 1953 dans le domaine du médicament mais cet outil est rarement utilisé car il constitue un frein à l'innovation. Ce sont des mesures ponctuelles visant un brevet spécifique.
• La construction de communs
Le malaise suscité par la possession de brevets de gènes très large par des laboratoires en situation de monopole aux États-Unis permet l'émergence de nouveaux systèmes comme le système des communs décrit par M. Maurice Cassier (156 ( * )) . Les communs sont des entités juridiques qui proposent de nouveaux usages des ressources qu'offre le droit de la propriété intellectuelle, en fractionnant les différents attributs de cette propriété pour les partager entre les différentes parties prenantes, d'après M. Benjamin Cariat (157 ( * )) .
Il s'agit en pratique de licences très larges, voire de licences libres qui partent du principe que si la connaissance est générée par un très grand nombre de personnes, la redistribution de royalties sur tous ces consortiums devient pratiquement impossible. Cela permet de gérer les brevets existants et de choisir de mettre en oeuvre d'autres politiques : soit on publie, soit on organise la circulation avec des licences ouvertes comme l'ont fait plusieurs consortiums.
ii. Les possibilités offertes par les regroupements de brevets dans des patents pools
Des regroupements volontaires de brevets dans des patents pools ont l'avantage d'offrir un guichet unique pour négocier les licences de brevet ; l'accessibilité des gènes et des nouvelles technologies en serait facilité (158 ( * )) . Cela répondrait aux problèmes des tests multi-marqueurs qui pourraient bouleverser les systèmes de brevetabilité : « Comment fait-on sur un test multi-marqueurs pour lever les droits de propriété ? » s'est demandé M. Maurice Cassier (159 ( * )) . Si divers propriétaires disposent de plusieurs marqueurs, il apparaitra nécessaire de négocier un regroupement de brevets. En matière de biotechnologies, on a souvent recours à des pools de brevets. Des entreprises s'associent pour mettre en commun leurs brevets essentiels et en délivrer des licences d'exploitation, afin de faciliter l'adoption de la technologie et de permettre aux utilisateurs de signer une licence globale.
Selon Mme Hélène Gaumont-Prat (160 ( * )) , ces pools de brevets présentent un réel intérêt économique car ils induisent une baisse du coût des transactions et limitent des litiges potentiels malgré les risques d'effets anticoncurrentiels qu'ils soulèvent et les difficultés que pose leur mise en place. Il convient de s'assurer que les pools n'incluent pas plus de brevets que ceux qui sont nécessaires à l'exploitation du standard, et que ces brevets sont bien complémentaires.
Pour une pathologie, une autorité médicale définit un test standard, comprenant la liste des marqueurs ou des gènes nécessaires pour le réaliser. Cela constitue la base d'un pool de brevets à partir duquel on peut attribuer des licences. En septembre 2012, les National Institutes of Health (NIH), l'Université de Pennsylvanie, l'Université de Californie ont créé un pool de brevets commun (400 brevets), pour la médecine personnalisée. Les NIH ont confié la gestion de ce pool à l'institution déjà en charge de pools de brevets dans d'autres domaines comme l'électronique. Mais le regroupement de brevets peut être attaqué et il faut donc prendre en compte les lois anti-trust aux États-Unis et les textes sur la concurrence en Europe.
En effet, on assiste aux États-Unis à une prise de conscience assez large des risques de blocage par les brevets sur les gènes humains, de la recherche en médecine personnalisée même si les avis les plus divers se côtoient à propos de cette question. « Certains considèrent qu'il existe un risque réel de blocage de la recherche par les brevets... Si des brevets couvrent en effet l'ensemble des fonctions d'un gène humain, l'analyse et l'exploitation des variations du gène risquent de constituer un acte de contrefaçon » relève Mme Audrey Aboukrat (161 ( * )) .
Les alliances de recherche comme l'AVIESAN pourraient s'intéresser à ce système qui permet de trouver des structures souples de gestion, car de nombreux brevets ne sont pas utilisés et coûtent plus chers qu'ils ne rapportent. On pourrait alors constituer des paniers de brevets si ceux-ci contiennent des bases de connaissances qu'il est possible de rassembler. On effectuerait ensuite un tri des brevets réellement intéressants et novateurs.
iii. La durée de protection conférée par le brevet
Certains industriels s'interrogent sur la durée de protection des brevets eu égard à la lenteur du processus de développement. Limitée à 20 ans à compter du jour de dépôt de la demande, dans la plupart des secteurs industriels, l'invention brevetée sera disponible sur le marché deux ou trois ans après le dépôt de brevet, parfois moins. Le détenteur du brevet dispose donc de 17 ou 18 ans d'exclusivité commerciale.
Cependant, une nouvelle molécule, dont le brevet vient d'être déposé, fera encore l'objet de recherches, de mises au point et d'essais pendant une dizaine d'années avant que les autorités sanitaires n'autorisent sa mise à disposition pour les malades. Le médicament ne serait donc en réalité protégé par le brevet qu'une dizaine d'années. Afin de compenser la durée exceptionnellement longue de sa recherche, le médicament bénéficie d'un « certificat complémentaire de protection » (CCP) qui prolonge la durée du brevet, au maximum pour cinq ans complémentaires.
En pratique, le médicament est en moyenne protégé commercialement pendant environ une quinzaine d'année (durée de validité du brevet au moment de la mise sur le marché prolongée du CCP). Lorsque les droits de propriété intellectuelle ont expiré, l'invention tombe dans le domaine public. Dans le cas du médicament le prix diminue en conséquence.
On assistera donc à des évolutions que commande la tension entre d'une part le coût et la protection de la R&D et d'autre part la nécessité d'innover et de permettre un accès équitable à l'innovation. Des pratiques nouvelles permettant de sauvegarder la recherche et le développement de traitements innovants sont mises en oeuvre, et il convient de les encourager.
4. Les possibilités offertes par l'Union européenne
L'Union européenne est impliquée dans le développement de la médecine personnalisé grâce au programme européen Horizon 2020 et à l'Initiative médicament innovants (IMI) (162 ( * )) .
a. Le programme européen Horizon 2020
Le programme de recherche et d'innovation de l'Union européenne est doté de 79 milliards d'euros pour la période de 2014-2020. Il est entré en vigueur le 1 er janvier 2014 et recentre les financements sur trois priorités : l'excellence scientifique, la primauté industrielle et les défis sociétaux. Il regroupe l'actuel programme-cadre de recherche et développement technologique (PCRDT), Euratom, le programme-cadre pour la compétitivité et l'innovation (CIP), ainsi que l'Institut européen d'innovation et de technologie (IET).
Il vise notamment à doter l'Europe d'infrastructures de recherche d'envergure mondiale accessibles à tous les chercheurs d'Europe et d'ailleurs et à soutenir la mobilité des chercheurs européens et originaires des pays tiers vers et hors d'Europe. C'est un programme unique regroupant les financements en matière de recherche et d'innovation. Il met en oeuvre un accès simplifié aux financements européens par un système d'attribution accélérée, et un modèle de coûts simplifié. L'initiative médicaments innovants (IMI) s'inscrit dans cette perspective.
b. L'initiative médicaments innovants (IMI)
L'IMI est un partenariat public-privé entre l'Union européenne et l'Association de l'industrie pharmaceutique (EFPIA) qui vise à accélérer le développement de médicaments plus sûrs et de meilleure qualité pour les patients. Il soutient des projets de recherche et anime des réseaux d'experts industriels et universitaires afin de stimuler l'innovation pharmaceutique en Europe.
L'IMI dispose d'un budget de 2 milliards d'euros pour soutenir des projets de recherche. Agissant en tant que tierce partie dans la création de partenariats novateurs, il encourage les collaborations pour la recherche et développement (R&D) pharmaceutique. L'IMI a pour but de renforcer la compétitivité de l'Europe dans le monde et de rendre l'Europe plus attractive pour la R&D pharmaceutique.
Les consortiums de recherche participant aux projets IMI se composent de grandes sociétés biopharmaceutiques et d'autres partenaires tels que de petites PME, les associations de patients, les universités et les autres organismes de recherche, les hôpitaux, etc .
C. UN BOULEVERSEMENT DU SYSTÈME SOLIDAIRE DE SANTÉ PUBLIQUE
On estime que la demande mondiale pour les produits et services de santé serait à l'heure actuelle de 5 500 milliards de dollars, et qu'elle devrait atteindre 12 000 milliards de dollars à l'horizon 2030. Comment assurer la durabilité des assurances-maladies solidaires face aux coûts représentés par les technologies nouvelles, les dépenses accrues dues à des médicaments destinés à de petites populations ?
L'assurance-maladie devrait-elle se cantonner à prendre en charge les risques importants qui impliquent des traitements très coûteux pour certains malades, laissant le patient payer pour les petits risques liés aux maladies fréquentes et chroniques traitées par des médicaments génériques peu coûteux ? L'assurance-maladie solidaire ne prend déjà en charge qu'environ 50 % des dépenses de ville. Que serait une assurance pour une minorité délaissant la majorité, d'autant que les tests génétiques permettront dans certains cas de définir cette minorité dès la naissance ? Les « autres » paieront-ils pour un risque qui ne les concerne pas ? Chaque sous-population mutualisera-t-elle alors son risque ?
Au sein de l'Europe les pays ayant peu de moyens n'auront accès à ces médicaments très coûteux que grâce à la participation des pays plus riches au nom de l'égalité de l'accès aux technologies. Cela alourdit d'autant la charge des assurances solvables des pays riches de l'Europe, alors que la Commission européenne s'intéresse à la médecine personnalisée (voir supra ).
1. L'impact sur les coûts
Le passage dans le domaine public des médicaments à large spectre qui faisaient la fortune des laboratoires et le développement de nouvelles molécules ciblées pèseront sur les systèmes de santé qui devront s'adapter dans un contexte caractérisé par une croissance forte des dépenses de santé. Comme le relève Mme Valérie Seror (163 ( * )) , le cas du cancer par exemple est exemplaire, puisqu'entre 2005 et 2009, l'histogramme montre une augmentation des dépenses en médicaments de 220 % environ.
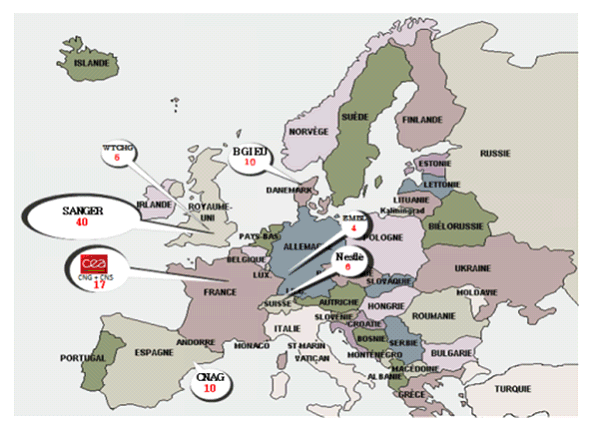
a. Des traitements ciblés coûteux
Le retour sur investissement des coûts de développement de la molécule et du test compagnon pour un nombre réduit de patients risque d'être limité, une cible étroite, signifiant moins de ventes. Pour rentrer dans ses frais, le laboratoire réclame donc des prix très élevés .
i. Des prix très élevés
D'après le Pr Laurent Degos (164 ( * )) , les prix de ces médicaments de petite population montrent une constante. « Si l'on traite 300 personnes, cela revient à 350 000 euros par an par personne en France pour le Soliris. Si l'on traite 3 000 personnes, comme pour le Glivec, c'est 30 000 euros par an par personne. Si l'on traite 30 000 personnes, ce sera 3 000 euros, etc . Quand on multiplie le prix et le volume, on obtient le même nombre ». En Allemagne et au Royaume-Uni, les autorités ont refusé le remboursement de certains de ces médicaments. Aux États-Unis, les assurances privées sont moins regardantes, mais pour les patients non couverts, ces médicaments sont tout simplement inabordables.
Les laboratoires, eux, affirment au contraire que la
médecine personnalisée peut faire économiser de l'argent.
Les traitements habituels sont inefficaces sur
38 à 75 % des
patients, fait valoir la
Personalized Medicine Coalition
, une
association américaine qui rassemble chercheurs académiques,
entreprises, assurances et associations de patients. Grâce aux
pré-diagnostics et à un meilleur ciblage, il serait possible
d'éviter les prescriptions inutiles et les hospitalisations dues aux
effets néfastes des médicaments mal administrés.
«
Les autorités sanitaires sont de plus en plus sensibles
au rapport coût-efficacité
» assure
Mme Corinne Blachier-Poisson. Dans certains cas, elles ont mis en place un
paiement « à la performance », où seuls les
traitements des patients ayant répondu positivement sont
remboursés au laboratoire.
ii. Le surcoût des tests est-il compensé par un meilleur ratio coût/efficacité ?
D'après le Pr Jean-Paul Moatti (165 ( * )) , on émet l'hypothèse que le surcoût induit par les tests sera compensé par des économies sur le traitement. Or, selon lui, ces progrès thérapeutiques, qu'il faut saluer, obéissent généralement à la loi des rendements décroissants car on tend, et c'est éthiquement nécessaire, à les étendre à des cas de plus en plus difficiles et complexes.
S'agissant par exemple des thérapies ciblées, on reste encore frustre dans la détermination précise des patients à traiter. Des patients bons, moins bons, voire mauvais répondeurs, les reçoivent ; il est clair que si certains tests ciblent mieux les bons répondeurs, leur surcoût sera compensé mais ce n'est pas toujours aussi évident même si dans certains cas le surcoût fonctionne de manière vertueuse. Il a pris comme exemple le cas du cancer du poumon (non à petites cellules) : le traitement coûte 1 million d'euros par année de vie gagnée rapporté au PNB par tête d'habitants en France qui est de 27 000 euros par an par habitant. Prenant lui aussi l'exemple du cancer avancé du poumon, le Pr Thomas Tursz (166 ( * )) , a indiqué qu'en quarante ans, on a augmenté la survie moyenne des malades de huit mois. Pendant le même temps, le coût moyen du traitement a augmenté entre 100 et 1 000 fois. Sans cynisme, on peut se poser la question de savoir si ces huit mois de vie supplémentaires valent vraiment de tels surcoûts.
Il explique que la façon dont se font les essais thérapeutiques est une addition ; on montre qu'A + B est mieux qu'A, qu'A+B+C est mieux qu'A+B, etc . L'on cherche à ajouter des médicaments, qui sont en général coûteux, à des thérapeutiques qui existent déjà. Cela ne fait qu'alourdir le traitement, le rendre plus toxique, plus coûteux, et pas beaucoup plus efficace.
Il serait beaucoup plus rationnel et avantageux de réserver le médicament A aux malades susceptibles de répondre à A, le médicament B aux seuls malades potentiellement répondeurs à B, etc . Ceci pose la question du modèle de régulation des dépenses dans le système de santé, car le progrès thérapeutique en grande partie obéit à la loi des rendements décroissants, comme le montre le schéma ci-dessous transmis par le Pr Jean-Paul Moatti.
Le schéma ci- dessous, transmis par le Pr Jean- Paul Moatti (167 ( * )) , indique le niveau de prix pour lequel le test génomique est coût-efficace. Il distingue selon que le médicament est générique ou pas.
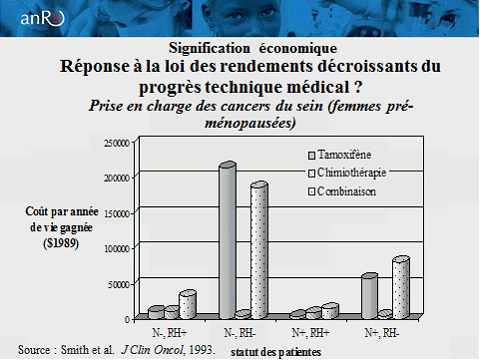
Selon Mme Valérie Seror (168 ( * )) , « il y a une interdépendance entre la cotation des tests et le prix des médicaments, une complémentarité entre tests et thérapies ciblées, puisque l'utilisation conjointe du test et du médicament conduit à un bénéfice clinique supérieur à leur utilisation séparée. Il en ressort que les enjeux de cotation dépassent la question de l'équilibre des comptes de l'assurance maladie. Celle-ci est soumise à des arbitrages entre faciliter l'adoption d'une amélioration thérapeutique, et avoir les moyens de l'offrir à ses patients ».
Les décisions de cotation ont un impact direct sur le marché des médicaments et des tests. Une cotation d'un test trop basse peut être comprise comme une désincitation au développement d'autres tests, alors que trop élevée, elle pourrait conduire à des rentes de situation payées par les adhérents de l'assurance maladie. Par ailleurs, la complémentarité entre les tests et les thérapies met en lumière des barrières organisationnelles, qui ne sont pas propres à la France.
Plusieurs travaux publiés en Europe et aux États Unis montrent qu'il faut davantage de coopération dans l'évaluation de la valeur ajoutée des tests et dans les décisions de prix et de cotation, dans la mesure où la complémentarité clinique de ces deux éléments, devrait se traduire dans une évaluation conjointe de leur valeur ajoutée, ou du prix. Cependant ces travaux militent dans le sens d'analyses coût-efficacité systématiquement intégrés aux essais cliniques, et non pas effectués après.
iii. Des interrogations sur l'évaluation des thérapies ciblées : quelle rationalisation ?
En France il existe, un pacte de solidarité nationale qui fonctionne et permet un accès des patients à l'innovation assez facile, sinon systématique, même en cas de médicaments particulièrement onéreux. Comme l'a souligné le Pr Gilles Bouvenot (169 ( * )) , la Commission de transparence (CT) de la HAS est chargée de se prononcer sur le service médical rendu des médicaments, c'est-à-dire sur le bien-fondé de leur prise en charge ou non par la solidarité nationale. Ses avis, consultatifs, constituent une aide à la décision pour le ministre.
« La CT se prononce sur l'amélioration du service médical rendu (ASMR) par les nouveaux médicaments, c'est-à-dire sur le degré de progrès que leur utilisation est susceptible d'induire. En fonction de l'appréciation, plusieurs niveaux d'ASMR ont été définis : ASMR I, majeure, ASMR II, importante, ASMR III, modérée, ASMR IV, mineure, ASMR V, inexistante, signifie ?absence de progrès thérapeutique? ». Le niveau d'ASMR intervient dans la fixation du prix d'un médicament remboursable de 1 à 5, la commission de transparence de la HAS n'effectuant qu'une l'évaluation médicotechnique.
Le Pr Gilles Bouvenot (170 ( * )) précise : « D'une certaine manière, il faut se réjouir de l'indépendance dans notre pays, entre l'évaluation médicotechnique pure, que je représente aujourd'hui, et l'évaluation médico-économique. Ce n'est pas le cas dans d'autres pays . Il ajoute : « Le NICE (National Institute for Health and Clinical Excellence), pour ce qui concerne l'Angleterre et le Pays de Galles, a par exemple refusé le Xalkori au motif qu'il n'est pas coût-efficace. Ce n'est pas encore le cas dans notre pays où il n'y a pas de rationnement, mais seulement des efforts, pas toujours efficaces, de rationalisation ».
Mme Hélène Gaumont-Prat (171 ( * )) observe que, d'ores et déjà, en France, « on ne rembourse plus que les médicaments qui apportent un réel avantage par rapport aux traitements déjà en place ; leur prix est encadré. Pour les spécialités remboursables, le critère actuel est le service médical rendu (SMR), ce qui suppose pour un dossier à monter, une procédure à respecter, l'avis du comité économique des produits de santé ».
Selon l'article R.163-3 du Code de la sécurité sociale, les médicaments sont inscrits sur la liste prévue au premier alinéa de l'article L.162-17 « au vu de l'appréciation du service médical rendu qu'ils apportent indication par indication ». Cette appréciation prend en compte l'efficacité et les effets indésirables du médicament, sa place dans la stratégie thérapeutique notamment au regard des autres thérapies disponibles, la gravité de l'affection à laquelle il est destiné, le caractère préventif, curatif ou symptomatique du traitement médicamenteux et son intérêt pour 1a santé publique. Les médicaments dont le service médical rendu est insuffisant au regard des autres médicaments ou thérapies disponibles ne sont pas inscrits sur la liste.
Chaque pays cherche à endiguer la montée en charge des nouveaux médicaments En France, on joue le jeu du volume, avec les plateformes de l'INCa notamment, on cherche l'indication précise en ne donnant le médicament qu'à ceux qui en ont besoin. Au Royaume-Uni, on cherche l'utilité et les médicaments anticancéreux sont très peu mis sur le marché, car cela apporte assez peu de survie, ou de vie augmentée, pour le patient. Les Allemands travaillent sur le prix lui-même et mettent un plafond de prix.
b. Des économies possibles sur les traitements inutiles
Le développement des tests et des thérapies ciblées pourrait permettre de prescrire certains traitements aux seuls patients susceptibles d'en bénéficier, d'éviter des traitements inutiles, coûteux, toxiques, notamment en oncologie dans un contexte d'augmentation très rapide des dépenses de santé et de médicament. Cela pourrait freiner ou atténuer l'escalade thérapeutique et les coûts associés.
Le Pr Gilles Bouvenot (172 ( * )) indique : « à la Haute autorité de santé comme à l'Académie de médecine, nous partageons l'idée que les traitements ciblés ont beaucoup plus de chance d'être efficaces que les traitements standards et sont même susceptibles d'être source d'économie pour l'assurance maladie dès lors qu'on ne les prescrit qu'à des patients qui, a priori, ont toutes les chances d'y être répondeurs ».
Néanmoins des questions doivent être posées : est-ce le traitement fonctionne ? Et pour combien de temps marche-t-il ? Car si les traitements ciblés peuvent être source d'économies dès lors que leur emploi rationnel évite tout gaspillage, encore faut-il disposer à leur endroit de règles d'arrêt, afin de ne pas prolonger inutilement leur utilisation une fois que le patient leur est devenu résistant.
Il s'agira, selon la HAS, d'estimer les performances d'un nouveau produit dans le cadre d'un parcours séquentiel ou en association à d'autres produits et pour une durée de temps limitée, au vu des résistances qui ne manqueront pas de survenir et de l'arrivée de produits encore plus nouveaux à lui associer ou à lui substituer. La connaissance des règles d'arrêt de ces traitements est indispensable.
D'après le Pr Thomas Tursz (173 ( * )) , on sur-traite certainement un bon nombre de malades, dont le cancer n'évoluerait pas, avec trop de radios et de chimiothérapie, mais on ignore lesquels à l'avance. Or l'une des principales conséquences attendues de la nouvelle médecine sera d'éviter les traitements inutiles ou inefficaces. « Même si nous sommes encore loin des traitements réellement individualisés pour chaque malade, de la haute couture ou du sur-mesure, il nous faut quitter l'ère du prêt-à-porter et de la taille unique pour tous et toutes ».
2. L'impact sur l'organisation de l'assurance-maladie
L'évolution de l'assurance-maladie inquiète certains experts comme M. Didier Tabuteau (174 ( * )) : « p hilosophiquement, le modèle actuel repose sur un voile d'ignorance : tout le monde cotise au cas où. On ignore ce qui va nous arriver, on entre dans la plupart des pays d'Europe dans une assurance maladie obligatoire et solidaire et l'on cotise à proportion de ses revenus. La médecine personnalisée déchire ce voile d'ignorance et change considérablement la donne. La multiplication des informations réduit la zone d'ignorance et peut induire des effets terribles sur l'organisation de l'assurance-maladie. Des populations à risques connus plus faibles que d'autres, pourraient demander une tarification en fonction du risque ».
L'assurance maladie est concernée par ces médicaments de niche si coûteux. Ils sont pour l'instant peu nombreux et concernent très peu de patients, mais la montée en charge est grande. L'assurance-maladie doit-elle continuer à tarifer systématiquement des produits, ou doit-elle entrer dans une réflexion sur des tarifications de protocoles, ou même de résultats thérapeutiques ? Certes, selon le Pr Laurent Degos (175 ( * )) , « il faut promouvoir cette médecine personnalisée, mais aussi s'interroger sur le prix qu'elle représente, et essayer de conforter les laboratoires pharmaceutiques à ne pas abandonner les traitements des grandes populations ».
Pour M. Didier Tabuteau (176 ( * )) , « Si ces thérapeutiques restent à la marge, très spécialisées, localisées, elles ne modifieront pas le fonctionnement du système quand elles s'y intègreront. En revanche, si cette approche de la médecine concerne progressivement de 5, 10, 30 % du système de santé, elle produira des bouleversements considérables ».
La médecine préventive incluse dans la médecine personnalisée apparaît comme une solution à long terme contre l'explosion des dépenses de santé car il est beaucoup moins cher de dépister et de prévenir une maladie que de la traiter lorsqu'elle s'est déclarée. « Cela aurait aussi un impact sur le remboursement des médicaments, car il est probable qu'à l'avenir, les systèmes de la santé ne paieront plus pour des médicaments, mais pour des résultats » relève Mme Hélène Gaumont-Prat (177 ( * )) .
a. La restructuration de l'offre de soins
La médecine personnalisée se traduit inévitablement par une hyperspécialisation de l'offre de soins et incite à la création de grands centres nationaux, ou de centres très spécialisés. Elle pose la question de l'évolution des plateaux techniques et du système hospitalier dans son ensemble. Si elle devient une part significative de l'activité médicale, elle peut conduire à une restructuration considérable du système hospitalier et même du système de soins.
Le parcours de soins sera plus complexe dans une médecine hyperspécialisée du fait de la personnalisation des thérapeutiques ou des processus de prise en charge. On assistera sans doute à l'émergence de pôles de compétences, sur le modèle des cancéropôles avec des centres d'analyses de génétique et d'autres systèmes. « Pour les thérapeutiques les plus complexes et les plus liées à la personne, on peut s'interroger sur la façon dont la séquence prise en charge-soins-élaboration des produits- administration au patient, s'organisera. Respectera-t-on toujours une séparation franche entre les entreprises pharmaceutiques, celles de dispositifs médicaux, et les prestataires de soins ? » se demande M. Didier Tabuteau (178 ( * )) .
Ces évolutions se traduiront par l'émergence d'un besoin accru d'orientation médicale et d'accompagnement performant, difficile à organiser dans un système peu régulé comme le système français. Il faudra se prémunir contre la tentation d'acteurs plus ou moins bien intentionnés de tenir ce rôle d'accompagnement par le coaching ou le conseil à des fins purement économiques. Qui sera à l'origine de l'orientation du patient ? Le médecin généraliste et/ou le médecin référent ? Sera-t-il chargé du suivi du patient qui consultera dans de grands centres spécialisés ?
La médecine personnalisée remet en cause des frontières traditionnelles, les distinctions prévention-soins, actes-produits, médicaments-dispositif médical. Ces frontières, ces concepts structurent l'organisation administrative et juridique du système de santé en France et fonde les définitions, les processus d'autorisation, de contrôle, d'évaluation figurant dans le code de la santé publique ou de la sécurité sociale. « Il faudra donner un statut juridique ou administratif à des pratiques beaucoup plus complexes que celles auxquelles nous étions habitués, et c'est toujours difficile » souligne M. Didier Tabuteau.
b. Résister à la tentation d'une assurance-maladie conditionnelle.
Plus on multiplie les informations sur la connaissance du risque et les moyens d'y pallier, plus la tentation de lier l'assurance-maladie au comportement du patient sera forte : le risque de remise en cause de la médecine de solidarité entre les biens portants et les malades pourra augmenter, dans une période de contraintes fortes sur l'assurance-maladie. Plus le comportement individuel, le suivi personnel en temps réel de la prévention sera possible, étayé scientifiquement, diffusé publiquement, plus pourra exister la tentation de lier la prise en charge aux risques connus et aux comportements des patients.
Cette assurance maladie conditionnelle était jusqu'à présent extrêmement restreinte en France. Le droit des affections de longue durée (ALD) permet depuis longtemps de subordonner la prise en charge à 100 % à des conditions d'observance et de comportement et de la supprimer si celles-ci ne sont pas respectées. Mais en pratique ce n'est pas utilisé.
Cependant on observe déjà à l'étranger des assurances privées qui personnalisent la tarification du risque et évoluent en fonction du risque et de l'observance du traitement par le malade. Certains systèmes introduisent un lien entre le comportement et la prise en charge, ce qui est porteur de très profondes inégalités sociales et socioculturelles. Si elles ne sont pas prohibées, ces pratiques se développeront. Cela est d'autant plus inquiétant que ces processus conduisent à la banalisation et la dissémination des centres utilisateurs et détenteurs de données sensibles. Les transferts d'informations multiplieront les risques d'atteinte au secret des données médicales.
Il faudra donc mesurer ce qui est supportable par l'assurance-maladie, s'orientant vers une approche bénéfice/risque raisonnable, ce qui est le plus utile en termes de santé publique, explique M. Didier Tabuteau (179 ( * )) .
I. III. GARANTIR L'ÉQUITÉ ET L'INFORMATION DES CITOYENS FACE AUX ENJEUX ETHIQUES DE LA MEDECINE PERSONNALISEE
Le potentiel de la médecine personnalisée et notamment de la pharmaco génomique ne pourra se réaliser que si les chercheurs peuvent avoir accès à l'information génétique de nombreux individus dans des essais cliniques de grande envergure, ciblés sur tel ou tel type de maladie.
Quand on disposera de tests prédictifs ou de marqueurs biologiques suffisamment convaincants au niveau scientifique, nombre de questions devront être soulevées souligne M. Didier Tabuteau (180 ( * )) . Quand effectuer les tests ? À quel âge ? Avec quelle périodicité ? Pour quelle partie de la population et avec quelle prise en charge ? Quel sera l'accès à la prévention pour quel coût, sachant que la prévention précoce pourrait être efficace, mais qu'il ne faudrait pas pour autant inquiéter les patients inutilement ? Or, d'ores et déjà une partie de ces questions se pose, car le rôle des tests génétiques à valeur prédictive s'accroît. La médecine prédictive passe d'abord par un calcul de surplus de risque d'avoir une maladie par rapport à la population générale.
Le déclenchement d'une pathologie n'est pas systématique : il est conditionné par l'exposition à des facteurs environnementaux et/ou comportementaux spécifiques.
A. L'IMPACT DU RECOURS AUX TESTS GENETIQUES
Toute médecine diagnostique comporte une dimension prédictive car elle s'intéresse à l'évolution d'une maladie. Le concept de médecine prédictive qualifie la médecine ayant pour but de détecter une prédisposition biologique à une maladie, afin de retarder, d'atténuer, voire d'empêcher son apparition. Cette médecine préventive personnalisée repose le plus souvent sur l'identification de variations génétiques, qui signalent une susceptibilité individuelle à déclencher une maladie dans un environnement donné. En 1997, le film « Bienvenue à Gattaca » décrivait une société où les choix de vie sont orientés, voire conditionnés par les facteurs de risques génétiques déterminés à la naissance. Quinze ans plus tard, même si la réalité n'a pas rejoint la fiction, les progrès considérables accomplis en matière de connaissance du génome ont rendu plus crédibles les projets d'une médecine dite prédictive.
Cette médecine s'applique aux maladies polygéniques (dues à plusieurs gènes) et poly-factorielles (certains facteurs du milieu ambiant influencent leur développement). Il s'agit d'affections relativement communes comme le diabète, l'hypertension, les maladies cardio-vasculaires ou encore la maladie d'Alzheimer. La médecine personnalisée inclut largement la médecine prédictive ou de prévision dans son champ. Largement fondée sur l'analyse génétique, elle conduit à un recours large aux tests génétiques, ce qui induit des interrogations d'ordre éthique et sociétal que vos rapporteurs ont abordées en 2008 dans le cadre de l'OPECST lors de l'évaluation de la loi relative à la bioéthique (181 ( * )) et au cours de l'audition publique organisée en 2011 par MM. Claude Birraux et Jean-Louis Touraine dans le cadre de l'OPECST sur les maladies monogéniques (182 ( * )) . Les interrogations, observations et recommandations faites alors restent encore d'actualité alors même que l'usage des tests s'étend.
1. Les tests génétiques, outils d'une médecine prédictive
À mesure que l'accès au séquençage à haut débit, voire à très haut débit se développe, le recours aux tests génétiques tendra à s'accroitre. Qu'on vise à prédire l'apparition d'une pathologie, à personnaliser la prescription d'un traitement, ou encore à favoriser la prévention de la pathologie anticipée, le développement de ces analyses génétiques conduit à une individualisation croissante des soins et de la prévention des maladies ; mais ces analyses suscitent un questionnement éthique et exigent une vigilance quant à leurs impacts sur l'individu et la société.
En effet, comme le rappelle le Pr Axel Kahn (183 ( * )) : « L'idée selon laquelle un gène code un destin est une idée fausse, qui procède d'une méconnaissance du gène. Un gène code des propriétés d'une cellule, ou, si l'on connaît parfaitement le jeu des gènes travaillant ensemble, les propriétés d'un organisme. Aussi le destin est-il la résultante d'un être vivant qui dispose de propriétés données, confrontées à un environnement qui lui, n'est pas génétiquement déterminé » . Pour autant, on constate un recours accru aux tests génétiques dans le cadre de la prévision.
a. Le recours accru aux tests génétiques
Mme Emmanuelle Prada-Bordenave (184 ( * )) a insisté sur l'augmentation du nombre de tests génétiques pratiqués en France : « On compte en 2012 en France 82 756 tests de cytogénétique et 416 767 tests de génétique moléculaire, qui couvrent aujourd'hui plus de 1 500 maladies. Il est intéressant de se reporter en 2009, car ces tests couvraient simplement à peu près 1000 maladies. Il s'en est rajouté un peu plus de 500, et surtout les tests de génétique moléculaire étaient seulement 271 330 ». Elle constate une augmentation vertigineuse du nombre de tests entre 2009 et 2012. 228 laboratoires réalisent ces tests, et adressent régulièrement leurs rapports d'activité à l'Agence de la biomédecine. Ces chiffres éloquents montrent clairement un engouement nouveau du public pour la connaissance des déterminants de santé, qu'ils soient génétiques ou autres.
Dans de nombreux cas, l'apport de ces examens est selon Mme Prada-Bordenave, considérable et l'avantage individuel précieux, car il permet de proposer des traitements préventifs et une hygiène de vie limitant le risque. Ainsi dès qu'un test est inscrit à la nomenclature, cela se traduit par une augmentation au décuple du nombre de prescriptions. Cependant, l'interprétation d'une mutation pathogène par rapport à une simple variation du génome reste complexe car cette notion évolue au fur et à mesure de l'avancée des connaissances.
i. L'accès à des tests génétique des personnes asymptomatiques
Pour des personnes malades, une analyse génétique conduit à un diagnostic. Cependant, certaines personnes considérées comme patients, ne sont pas vraiment malades ou n'ont aucun signe pathologique, tels les membres de la famille de la personne qui se présente spontanément pour une maladie.
Certaines personnes sont asymptomatiques, mais une analyse génétique révélera un risque de transmission à la descendance pouvant conduire à un conseil génétique, si un projet parental existe. Parmi les personnes asymptomatiques, certaines sont pré-symptomatiques car elles ont dans leur génome la trace d'une maladie déjà présente qui se révèlera plus tardivement, mais avec certitude dans le futur. Les tests pré-symptomatiques sont réalisés au sein d'une équipe pluridisciplinaire, dans le cadre d'un protocole. En France, l'équipe est déclarée, et le médecin délivre une attestation certifiant qu'il a délivré des informations à la personne concernée et a recueilli son consentement.
Des tests de prédisposition peuvent être prescrits à une personne totalement asymptomatique pour déterminer un facteur de risque de développer une maladie avec une forte probabilité. On identifie un risque élevé mais inférieur à 100 %, de développer la maladie dans le futur car l'anomalie génétique identifiée est nécessaire mais pas suffisante pour avoir cette pathologie. Pour être légitimes, les tests de prédisposition doivent révéler un risque supérieur aux influences environnementales, et face auquel une action préventive ou curative est possible. C'est le cas par exemple des gènes de prédisposition aux cancers du sein familiaux (BRCA1 et BRCA2).
Des tests de susceptibilité sont proposés à des personnes asymptomatiques qui se portent bien pour déterminer leur facteur de risque d'être malades. Pour elles, le risque est faible, parfois très faible. L'anomalie génétique n'est ni nécessaire, ni suffisante pour avoir la maladie. L'utilité médicale des tests n'est alors pas établie.
Selon le Pr Florent Soubrier (185 ( * )) , « on peut réaliser des tests de confirmation de la maladie, chez une personne qui a déjà des symptômes, et chez qui on va découvrir une maladie héréditaire et l'identifier, on peut réaliser des tests prédictifs chez un apparenté, ou des tests prénataux, pour une maladie génétique de l'enfant avec des symptômes particulièrement sévères et des dépistages néonataux ».
La prescription de test dans un cadre médical est faite pour une personne qui veut savoir si elle est porteuse d'une anomalie génétique et, en France, il existe dans un cadre juridique bien précis.
ii. L'encadrement des tests en France : le rôle de l'Agence de la biomédecine (ABM) et de la Haute autorité de santé (HAS)
La loi règlemente le recours aux examens génétiques, leur prescription, le rendu des résultats ; on notera que le terme « test » n'est pas utilisé dans la loi. Ces dispositions introduites pour la plupart d'entre elles par la loi relative à la bioéthique de 2004 ont été peu modifiées par la loi de 2011, sauf en ce qui concerne l'information de la parentèle. Elles figurent aux articles 16-10 à 16-13 du code civil et L. 1131-1 et suivants du code de la santé publique. Des sanctions pénales sont édictées dans les cas de recours à ces techniques, hors du cadre légal (articles 225-25 et suivants du code pénal). L'article 16-13 du code civil pose le principe que « nul ne peut faire l'objet de discriminations en raison de ses caractéristiques génétiques ». L'article 16-10 du code civil précise les finalités auxquelles doivent répondre de tels examens et les modalités de consentement exigées.
Les articles L.1131-1 et suivants du code de la santé publique, après avoir renvoyé aux dispositions du code civil, pour rappeler les modalités de recueil du consentement (préalable, exprès et écrit), et de l'information de la personne concernée par l'examen, consacre la compétence de l'Agence de la biomédecine pour délivrer l'agrément aux praticiens habilités à procéder aux examens des caractéristiques génétiques.
L'article L.2131-1 du même code précise les conditions du recours au diagnostic prénatal (DPN) dont le but est de détecter, in utero chez l'embryon ou le foetus une « affection d'une particulière gravité ». Il s'effectue dans des centres pluridisciplinaires de diagnostic prénatal (CPDPN).
Dès 2008 (186 ( * )) nous constations que la législation ne pouvait pas être la même si l'usage des tests génétiques devient commun ou s'il ne concerne qu'un très faible pourcentage de la population. Nous observions que le Comité consultatif national d'éthique, CCNE comme l'Agence de la biomédecine, s'inquiétaient du développement très rapide de l'analyse du génome humain et de la multiplication de tests génétiques en libre accès sur Internet qualifiée de « génétique récréative », et dont on ne peut pas garantir la fiabilité.
La loi relative à la bioéthique a confié à l'Agence de la biomédecine un une mission générale en matière de génétique (article L.1418-1 du code de la santé publique), et un rôle particulier sur la réflexion en amont et l'élaboration d'un arrêté de « bonnes pratiques applicables à la prescription et la réalisation de l'examen des caractéristiques génétiques d'une personne et de son identification par empreintes génétiques à des fins médicales » (article L.1131-2 du même code). Cependant, d'autres autorités publiques interviennent : la Haute autorité de santé (HAS), l'Institut national du cancer (INCa) pour le cancer, l'Agence nationale de sécurité du médicament et des produits de santé (ANSM) pour le contrôle de la qualité des dispositifs médicaux car les tests génétiques sont des dispositifs médicaux de diagnostic in-vitro au sens des textes.
La sphère de compétence de l'Agence de la biomédecine se concentre sur la mise en oeuvre de bonnes pratiques en génétique constitutionnelle, ce qui couvre tout un champ de l'histoire d'un individu, parfois avant même son commencement, puisque le diagnostic préimplantatoire est pratiqué avant la grossesse, et le diagnostic prénatal en cours de grossesse. L'article R. 1131-4 du code de la santé publique prévoit que : « préalablement à l'expression écrite de son consentement, la personne est informée des caractéristiques de la maladie recherchée, des moyens de la détecter, du degré de fiabilité des analyses ainsi que des possibilités de prévention et de traitement. En outre, elle est informée des modalités de transmission génétique de la maladie recherchée et de leurs possibles conséquences chez d'autres membres de sa famille ». Ce code fixe les conditions de prescription et de réalisation des examens des caractéristiques génétiques d'une personne à des fins médicales.
L'Agence de la biomédecine a élaboré, en liaison avec la Haute autorité de santé, un arrêté en mai 2013 (187 ( * )) définissant les règles de bonnes pratiques applicables à l'examen des caractéristiques génétiques d'une personne à des fins médicales. Ce texte rappelle que l'individu doit rester au centre des préoccupations des acteurs, qu'une importance particulière doit être accordée à l'information, au consentement, aux modalités de rendu d'un résultat notamment pour toutes les découvertes incidentes, et tous les variants qui n'ont pas de portée, ou dont on ignore l'impact. L'arrêté insiste sur le fait que les caractéristiques de génétique constitutionnelle sont définitives, et que les résultats ont des conséquences non seulement pour le patient mais aussi pour sa famille.
Ainsi les examens génétiques ne doivent être prescrits que lorsqu'ils ont une utilité clinique et sont souhaités par le patient. Le seul fait qu'un examen soit disponible et réalisable ne justifie ni sa prescription, ni sa réalisation. Cependant, la prescription n'est pas soumise à des restrictions, elle doit être effectuée par quelqu'un qui connaît la maladie et ses aspects génétiques, éventuellement un médecin spécialiste, car l'apport d'un test de susceptibilité est variable, souvent mineur, et contribue assez peu, à la prise en charge de la personne. On a attiré l'attention des prescripteurs sur la retenue dont ils doivent faire preuve.
iii. La transmission des résultats et le conseil génétique
La loi française encadre particulièrement la prescription, la réalisation et le rendu des résultats de tests génétiques. Cet encadrement vise à protéger les patients et à leur assurer l'information nécessaire, notamment par la mise en place de consultations pluridisciplinaires pour les patients asymptomatiques.
La transmission du résultat est très importante, elle doit être faite par le prescripteur et non pas être envoyée par courrier et l'on doit toujours prévoir un accompagnement psychologique, à la différence de ce qui se passe pour les tests en libre accès par Internet pour lesquels le résultat n'est ni rendu par un médecin, ni accompagné d'une explication, ou d'un suivi psychologique, alors que cela peut être souhaité et s'avérer nécessaire.
En effet, le point 4 de l'arrêté précité définissant les règles de bonnes pratiques applicables à l'examen des caractéristiques génétiques d'une personne à des fins médicales intitulé « communication du résultat » définit avec précision les modalités de communication du résultat au patient : « le résultat d'un examen génétique ne doit pas être directement communiqué au patient par le laboratoire de biologie médicale mais par le prescripteur. Il s'agit d'une dérogation à l'article L.6211-2 du code de la santé publique (issu de l'ordonnance Ballereau) . Les modalités de communication de ce résultat doivent être préalablement définies, notamment au cours de la consultation qui a donné lieu à la prescription. La personne peut exprimer, par écrit, sa volonté d'être tenue dans l'ignorance d'un diagnostic conformément à l'article L.1131-1-2 du code de la santé publique ».
« La communication du résultat par le prescripteur est résumée dans un document rédigé de manière loyale, claire et appropriée. Elle doit s'accompagner d'information sur les conséquences pour l'individu ; les conséquences familiales du résultat de l'examen (complète les informations données en amont) ; les modalités d'information de la parentèle (complète les informations données en amont) ; le cas échéant, la transmission du résultat au responsable du centre d'assistance médicale à la procréation si la personne a fait don de ses gamètes ou appartient à un couple dont les embryons ont été destinés à être accueillis ; une copie du résultat du laboratoire de biologie médicale doit être donnée au patient ».
Le point 7.2 intitulé « le compte rendu » précise que « le laboratoire qui réalise l'examen de génétique doit transmettre le résultat et le compte rendu au prescripteur . Dans la situation où plusieurs laboratoires interviennent pour la réalisation des examens génétiques (travail en réseau), le laboratoire qui rédige le compte rendu et rend le résultat au prescripteur en transmet une copie aux autres laboratoires impliqués (laboratoire ayant reçu initialement le prélèvement ainsi que les laboratoires ayant participé au diagnostic). C'est le LBM (laboratoire de biologie médicale) qui a réalisé le prélèvement qui demeure responsable de l'examen et doit communiquer le compte rendu au prescripteur ».
Le conseil génétique, implique une information du patient précise et comprise par lui-même et toute la famille. Il faut également recueillir correctement les consentements des membres concernés de la famille, mettre en place un suivi, voire une prise en charge, éventuellement proposer des diagnostics préimplantatoires (DPI), et des diagnostics prénataux (DPN), et enfin protéger les données de santé.
Ainsi l'évolution rapide des technologies doit s'accompagner de la volonté d'assurer aux patients une information fiable sur les bénéfices attendus des examens génétiques pour leur permettre de faire la différence entre des examens génétiques prescrits dans un contexte médical et cette sorte de génétique récréative que suscite les tests en libre accès sur Internet.
b. Des tests génétiques en libre accès sur Internet : une tendance à la biologisation ?
« Le phénomène de la mise à disposition sur Internet d'une génomique dite personnalisée fait écho semble-t-il à une autre pratique déjà bien installée celle de la promotion de produits pharmaceutiques directement auprès du consommateur plutôt qu'auprès des professionnels de santé » observent P. Ducournau, A. Cambon-Thomsen et E. Rial-Sebag (188 ( * )) . Dans un article intitulé « Tests génétiques en accès libre sur Internet - Stratégies commerciales et enjeux éthiques et sociétaux », ils recensent, en 2011, 42 sites proposant des analyses génétiques avec des sociétés comme 23andMe, DecodeMe, Navigenics très présentes en Europe sur le marché des tests génétiques individuels sur Internet.
Ces entreprises étudient le génome pour en déduire les risques de développer une vingtaine de maladies courantes pour lesquelles des facteurs génétiques ont été identifiés. 23andMe et DecodeMe proposent également à leur client de retracer leur généalogie, de comparer son information génétique à celle de proches, d'amis, de vedettes, pour une mise en réseau sur Internet par le biais de réseaux sociaux encore inédits. Rappelons que 23andMe est une filiale de Google qui s'est lancé en septembre 2013 avec la création de Calico dans la lutte contre le vieillissement à l'aide de l'usage des biotechnologies et du séquençage à très haut débit. Il est ainsi à la source de données personnelles de santé et peut-être en mesure de les domestiquer, certes pour lutter de façon personnalisée contre la maladie, mais aussi pour créer des banques de données.
i. Le mode de fonctionnement
Les trois sociétés fonctionnent de la même
façon. Le Dr Catherine Bourgrain
(189
(
*
))
a expliqué que pour
trois ou quatre génomes par 24 heures, si on cible une certaine
catégorie de mutations pour un panel de 90 maladies par exemple, il
existe des tests disponibles sur Internet. Ces tests proposent pour
100 ou
200 dollars de rechercher les mutations les plus fréquentes, ce qui est
finalement assez rapide, les machines existant à l'étranger pour
traiter l'information en quantité.
En pratique, le client reçoit un kit pour racler quelques cellules de sa paroi buccale. Les sites ne sont pas en mesure de vérifier si le prélèvement appartient à celui qui l'envoie ou s'il l'a effectué pour un tiers avec ou non son accord. Le client effectue lui-même la démarche sans intermédiaire médical. « Pour devenir de véritables acteurs de notre propre santé » dit la publicité. Il s'agit de responsabiliser la personne dans la prise en charge de sa santé. On part du postulat que des informations personnalisées ne peuvent qu'influencer positivement le comportement sanitaire de ces personnes et les inciter à utiliser au mieux les « potentialités de leur génome ». On ignore si ces sites offrent des garanties de confidentialité sur les données sensibles qu'ils recueillent sur les caractéristiques génétiques de leur client.
ii. Des résultats transmis sans encadrement
Une fois le l'examen réalisé, l'information est engrangée dans une base de données sécurisée par la société. Au bout d'un mois, le client accède par Internet à ses informations personnalisées, qui seront mises à jour au gré des nouvelles découvertes scientifiques. Ces tests qui sont généralement des tests de susceptibilité peuvent être dangereux, car en dehors de tout cadre, on tend à faire croire aux personnes qui les utilisent qu'elles ne risquent pas de développer telle ou telle maladie ou, à l'inverse, on les angoisse à tort sur d'autres pathologies dont elles ne seront pas atteintes.
Certains sites rendent en plus des résultats de prédisposition ou de diagnostic (cancer du sein, mucoviscidose) sans l'encadrement médical et psychologique indispensable pour ce type d'annonce. Le plus souvent, les résultats sont fournis sous forme brute, sans aucun moyen d'en décoder le véritable sens pour chaque individu. Or, la majorité des pathologies évoquées mettent en jeu plusieurs gènes et de nombreux facteurs, notamment environnementaux. La valeur prédictive de ces tests n'est donc que relative. La révélation brutale d'un pronostic défavorable peut modifier la perception que l'individu se fait de son existence, indépendamment de l'imprécision et du caractère aléatoire du pronostic ; elle pourrait aussi modifier la perception que la société ou les autres individus se feront de lui si cela est connu. Les généticiens français de la société de génétique humaine ont dénoncé vigoureusement ces démarches.
Selon eux, ces pratiques sont mensongères car, d'une part, les calculs de risques que l'on peut faire en utilisant uniquement l'information génétique sont totalement faussés à cause du manque de prise en compte de tous les facteurs environnementaux, et d'autre part, les résultats bruts communiqués en pourcentages de prédispositions à une collection de maladies ne sauraient avoir de sens sans être accompagnés du commentaire éclairé d'un médecin généticien.
Ainsi que le constate M. Didier Tabuteau (190 ( * )) , « ces sociétés ne peuvent qu'alimenter les perspectives fantasmées ou réelles de la génétique et générer une espèce d'hypocondrie collective dangereuse pour le bien-être de l'individu, mais également pour le fonctionnement de la société, le fonctionnement du système d'assurance-maladie et du système de santé ». Ces dérives anxiogènes justifient pleinement la mise en place du site de références sur la génétique suggéré par le Pr Axel Kahn (191 ( * )) sur le modèle de celui annoncé par l'Agence de la biomédecine, auprès duquel le public pourrait vérifier la pertinence des informations obtenues avec des pratiques mercantiles et être mis en garde contre les risques d'atteinte à la vie privée.
L'article précité paru dans Médecine/Sciences en janvier 2011 essaie de cerner les motivations de ceux qui y recourent. Les auteurs constatent une tendance de nos sociétés à placer la santé et la longévité au panthéon des valeurs ; ces démarches répondent à une anxiété et un désir de maitrise et manifestent une volonté d'être acteurs et auteurs de décisions de santé.
La figure ci-dessous donne un exemple de résultat de test en libre accès. En orange, les pathologies pour lesquelles l'individu (« you ») a un surplus de risque par rapport à la population générale (« avg ») (192 ( * )) .
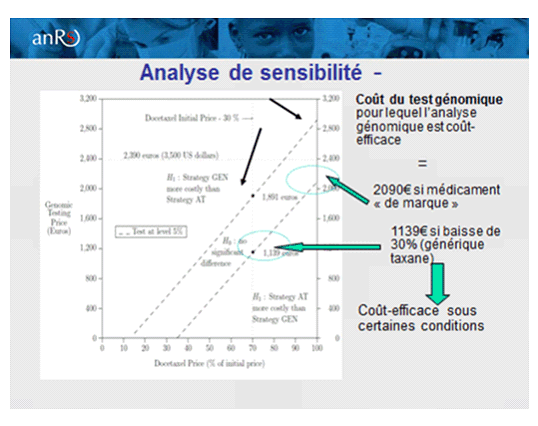
Mme Simone Bateman (193 ( * )) s'interroge également sur l'impact des tests « direct to consumer » dont le bien-fondé est remis en cause « Doit-on restreindre l'accès aux tests au seul contexte médical ? Quelles raisons avons-nous de le restreindre ou non ? Quelle est la qualité du conseil qui est donné aux personnes qui vont faire ces tests ? ». La soif d'information biologique sur soi est sans limite. L'enjeu est de savoir sur quoi l'on fonde la limitation et à qui laisse-t-on le soin de donner l'information accompagnant des tests éventuellement disponibles.
Les tests génétiques produisent de l'information génétique : ils renforcent en quelque sorte des tendances lourdes de nos sociétés à revendiquer un droit individuel à l'information que favorisent Internet et le développement des réseaux sociaux. À partir du moment où la possibilité de connaître l'information existe, l'accès à cette dernière fait l'objet d'une revendication, malgré la nécessité d'accompagnement de l'information génétique. Mais les choses ne se passent pas toujours ainsi et l'on voit poindre une « automédication génétique », les patients disposant d'un droit à l'information sur leur santé et pouvant aussi exiger que des tests soient disponibles. Nous sommes dans la société de l'Internet ; ce qui est interdit ou indisponible en France n'est guère difficile à obtenir dans les pays voisins où des possibilités d'accès supplémentaires existent.
iii. Une régulation difficile à mettre en oeuvre : la réforme de la directive européenne sur les dispositifs médicaux
Il suffit d'utiliser Internet, voire de lire la presse quotidienne pour apprendre l'existence de laboratoires étrangers qui proposent des tests : tests de paternité, analyses génétiques destinées à satisfaire la curiosité de chacun sur ses origines familiales, les diverses migrations de sa famille afin de construire sa lointaine généalogie, tests de susceptibilité à telle ou telle maladie (cancers, maladies neurodégénératives, etc .). On constate une attirance à l'égard de cette information génétique, toujours considérée comme solide, parce que basée sur des structures scientifiques.
Corollaire de cette évolution, une attention croissante au niveau national et européen s'est portée sur l'encadrement de l'ensemble des « activités » de génétique. Les préoccupations essentielles se situent à trois niveaux : la qualité des tests génétiques, les conditions d'utilisation des tests génétiques, l'utilisation des résultats de ces tests.
• En Europe
Or la situation est très hétérogène dans les pays d'Europe. Peu de pays disposent d'une législation spécifique, comme l'Autriche, ou plus récemment la Suisse et le Portugal. On trouve parfois des dispositions concernant les tests génétiques dans des lois couvrant le champ général de la bioéthique, comme en France, ou relatives aux droits des patients, comme au Danemark, ou encore à la protection des données, comme en Allemagne. Toutefois, ces dispositions ne définissent pas toujours un cadre juridique complet pour les tests génétiques.
Cependant, dans la plupart des pays, des lignes directrices ont été développées par différents secteurs, notamment les sociétés de génétique humaine ou de cliniciens généticiens. Quelques pays ont, par ailleurs, mis en place des instances consultatives chargées de conseiller le gouvernement sur les questions ayant trait à la génétique tels le Royaume Uni, l'Autriche, et l'Espagne. Aucun des instruments juridiques en vigueur dans les différents pays d'Europe ne s'applique en dehors des frontières nationales, et notamment ne couvre les offres commerciales accessibles sur Internet qui sont proposées par des laboratoires situés en dehors de ces pays, majoritairement aux États-Unis.
Les tests génétiques sont considérés couverts par la Directive 98/79/CE, relative aux dispositifs médicaux de diagnostic in vitro. La Directive porte essentiellement sur les aspects de sécurité et de qualité et ne traite pas des conditions d'utilisation des tests génétiques, ni de celles de leurs résultats. Les exigences définies par la Directive varient selon le niveau de risque présenté par les dispositifs concernés. Or, les tests génétiques y sont généralement considérés comme présentant un niveau de risque peu élevé, pour lesquels les procédures d'évaluation de la conformité peuvent, en règle générale, rester sous la responsabilité du fabricant.
Toutefois, le 26 septembre 2012, la Commission européenne a publié deux textes de révision des directives européennes sur les dispositifs médicaux qu'elle a soumis au Parlement européen et au Conseil de l'Union européenne. Ces deux propositions de règlements européens viendront remplacer les directives actuellement en vigueur (90/385/CEE - 93/42/CEE - 98/79/CE). Elles pourraient être adoptées en 2014 et mises en application entre 2015 et 2019. La Commission souhaite accroître le niveau de la protection de la santé humaine et de la sécurité et renforcer les exigences relatives à la preuve clinique en fonction du risque. Elle prévoit un chapitre sur les exigences des évaluations cliniques, et le renforcement de la surveillance post-commercialisation ainsi qu'un chapitre sur la traçabilité de tous les dispositifs.
D'autres initiatives ont été prises au niveau européen, comme celle d' Eurogentest qui est un réseau européen d'excellence dans le domaine des tests génétiques, dans le but d'harmoniser et d'améliorer la qualité générale des services génétiques en Europe.
• Aux États-Unis,
Selon Mme Mathilde Reynaudi (194 ( * )) , ces questions sont débattues : en témoignent les débats induits par les propositions de certaines universités américaines comme Berkeley et Stanford, à leurs étudiants de bénéficier d'un test génétique pour moins de 50 dollars au cours l'année scolaire 2011-2012. À cette offre était associée un module de cours « Genomics and personalized medicine » dont l'objectif était de débattre des enjeux éthiques, de l'encadrement juridique, des limites et des bénéfices potentiels des tests. Ce cours était destiné aux étudiants médecins, aux doctorants chercheurs en génétique humaine et plus généralement à toute personne intéressée par la médecine prédictive.
Des professeurs et plusieurs groupes de recherche et de réflexion ont alerté les universités des potentielles retombées négatives d'un tel programme, qui semblait cautionner l'achat de tests en vente directe. Cependant dans certaines universités américaines, il est proposé aux étudiants en médecine et en génétique de séquencer eux-mêmes leur propre génome. En fait, la loi varie d'un État à l'autre, la loi californienne sur les tests biologiques interdit que ceux-ci soient réalisés en dehors d'une prescription médicale.
• Des normes internationales
Sur les aspects éthiques relatifs aux tests génétiques, le seul texte juridique international contraignant est la Convention sur les Droits de l'Homme et la biomédecine du Conseil de l'Europe, dite Convention d'Oviedo (STE n°164, 4 avril 1997) qui établit un certain nombre de principes fondamentaux s'appliquant aux conditions d'utilisation des tests génétiques et de leurs résultats. Elle limite notamment l'utilisation des tests génétiques prédictifs de maladie aux seules fins médicales ou de recherches médicales et interdit toute forme de discrimination à l'encontre d'une personne sur la base de son patrimoine génétique.
Sur la base de ces principes, le comité directeur pour la bioéthique (CDBI) du Conseil de l'Europe a élaboré un nouvel instrument juridique, complétant les dispositions de la Convention dans le domaine spécifique des tests génétiques à des fins médicales : le Protocole additionnel à la Convention sur les Droits de l'Homme et la biomédecine, relatif aux tests génétiques à des fins médicales Strasbourg, du 27 novembre 2008 (STCE n° 203) (195 ( * )) .
Ce protocole porte notamment sur l'information et le consentement préalable, le conseil génétique ; il établit des règles générales pour la conduite des tests génétiques, des règles de protection de la vie privée, et assure le droit à l'information recueillie au moyen de tests génétiques. La France qui fut très active lors de son élaboration, l'a signé le 13 décembre 2011 quand elle a enfin ratifié la convention d'Oviedo, mais elle ne l'a pas encore ratifié. Ce Protocole n'est pas encore en vigueur car, sur sept États qui l'ont signé, trois seulement l'ont ratifié et son entrée en vigueur est soumise à cinq ratifications. La France s'honorerait à le ratifier, ce serait un signal fort.
Le recours à des tests de susceptibilité sur Internet est difficile à réguler, les personnes qui les effectuent n'ont aucune garantie de qualité et de confidentialité ; elles obtiennent des résultats difficiles à interpréter parfois inutilement anxiogènes. Le développement de ces examens engage d'importants intérêts financiers, alors que les conséquences individuelles de l'annonce d'une susceptibilité génétique soulèvent de nombreuses questions.
Selon la HAS et l'ABM, il faut déployer un effort de formation et d'information pour porter à la connaissance des professionnels et du public les conséquences délétères du recours à des tests sans encadrement, hors de toute utilité clinique avérée et de contrôle de qualité bien documenté. C'est aussi l'avis de vos rapporteurs.
c. La problématique de la prédiction
La valeur prédictive des tests et son impact est débattue de manière récurrente.
i. La précision et la fiabilité des technologies
Pour le Dr Catherine Bourgain (196 ( * )) , le développement des tests pose des questions sur la précision technologique et la fiabilité des analyses. Dans quelle mesure les outils disponibles permettent-ils de détecter avec une bonne fiabilité la présence de mutations ? Est-on sûr de ce que l'on détecte ? Ne risque-t-on pas des erreurs de technique ? On sait effectivement que celles-ci sont assez nombreuses. Les valeurs de pénétrance employées jusqu'ici, c'est-à-dire la probabilité de tomber malade pour les personnes porteuses de ces mutations, sont- elles fiables ? Elle constate : « en analysant un nombre croissant de personnes qui ne sont pas malades, on se rend compte que, dans certains cas, des individus porteurs de mutations, parfois en double dose, auraient dû être considérés comme malades alors qu'ils n'ont pas développé la maladie ». L'essor de ces technologies interroge aussi notre capacité à bien mesurer les risques, même dans le cas de maladies censées avoir un très fort déterminisme du point de vue génétique.
Quant aux maladies multifactorielles qui résultent d'une combinaison de facteurs génétiques et de facteurs d'environnement, au sens le plus large de ce terme, c'est-à-dire couvrant à la fois la vie foetale et tous les facteurs non génétiques, des mesures de composante génétique sont souvent mises en avant dans les débats pour justifier les mesures de risque basées sur la seule génétique. Quand on mesure l'héritabilité, on estime que, pour telle maladie, cela vaut la peine de rechercher des facteurs de risque génétique, car l'héritabilité de cette maladie atteint 30 % ou 40 %.
ii. Les débats autour de l'héritabilité
La notion d'héritabilité est extrêmement discutée dans la communauté scientifique, parce qu'elle présuppose l'absence d'interaction entre les effets génétiques et les effets environnementaux ; or on sait pertinemment que c'est faux. Le génome d'un individu, selon l'environnement dans lequel celui-ci sera plongé tout au long de sa vie, présentera un risque très différent d'induire telle ou telle pathologie.
Face à un génome entier en séquence, ou bien à un exome, on dispose de capacités très variées d'interprétation des différents points de ce génome ou exome. On sait générer toute une série d'informations sans savoir les interpréter.
À propos de cette prédictivité, le Dr Anne Cambon-Thomsen (197 ( * )) citait une phrase du Pr Didier Sicard, ancien président du Comité consultatif national d'éthique (CCNE) : « la médecine s'engouffre dans cette prédiction en pensant que plus on prédira un risque, moins il aura de chances de survenir. Il existe une confusion permanente entre l'usage que l'on fera de cette prédiction et la prédiction elle-même ».
Elle s'interroge : « peut-on considérer, réguler, expliquer tout type de test génétique, de biomarqueur, de la même façon ? La réponse est négative ». Selon elle, l'information génétique peut être classée dans une catégorie un jour donné et puis se retrouver le lendemain dans une autre catégorie car on disposera d'un peu plus d'informations sur l'environnement, sur les interactions, ou bien parce que l'on aura progressé dans la capacité à interpréter certains domaines du génome. Aussi l'information génétique est-elle à prendre en considération avec d'autres biomarqueurs, en tenant compte notamment de la dimension familiale de cette information, ainsi que de la dimension de catégorie ou de groupe.
Le Pr Jean-Claude Ameisen (198 ( * )) présente ainsi la prédiction : « Elle n'est jamais qu'une extrapolation du passé. Si les conditions changent, les mêmes relations de causalité n'induiront pas les mêmes conséquences. Encore une fois, la prédiction consiste à inclure le patient dans un groupe, sans prendre spécifiquement en considération son avenir. On écrase l'avenir, dans la mesure où on donne un poids démesuré à une forme de prévision, par rapport à l'ensemble des autres évènements imprévisibles ». Il s'interroge : « dès lors, libère-t-on, augmente-t-on le degré de liberté de la personne, ou risquons-nous de l'enfermer ? Que fait-on d'une information quand la prédiction ne peut pas se traduire en prévention, et qu'elle est simplement un dire de l'avenir ? ».
2. Un recours croissant à des tests prénataux et néonataux : l'impact sur la procréation.
Aujourd'hui, les diagnostics prénataux (DPN) et préimplantatoires DPI) sont strictement encadrés en France. L'article L.2131-4 du code précité détermine les conditions du diagnostic biologique effectué à partir de cellules prélevées sur l'embryon in vitro , diagnostic préimplantatoire (DPI) autorisé à titre exceptionnel quand le couple a une forte probabilité de donner naissance à un enfant atteint « d'une maladie génétique d'une particulière gravité reconnue comme incurable au moment du diagnostic ». Les deux membres du couple expriment par écrit leur consentement à la réalisation du diagnostic. Le DPI, qui consiste à rechercher sur un embryon in vitro une maladie monogénique, n'est pratiqué que si le couple est reconnu comme risquant de transmettre une maladie grave. Cette technique est alors restreinte à la recherche d'anomalies génétiques d'une particulière gravité, c'est-à-dire reconnues comme menant à une maladie incurable au moment du diagnostic. Or le séquençage à très haut débit et les possibilités nouvelles de prélever l'ADN du foetus dans le sang maternel changent la donne.
a. L'état des lieux
i. Les tests pré-conceptionnels
Il s'agit de proposer aux membres d'un couple, avant la procréation, de se soumettre à ce type d'analyses pour voir si, par hasard, ils seraient tous les deux porteurs d'une mutation impliquée dans une même maladie. Le cas échéant, des analyses de diagnostic prénatal, voire préimplantatoire, pourraient leur être proposées. Du fait du développement des analyses génomiques, le nombre de personnes potentiellement concernées par des tests pré-conceptionnels peut être considérablement élargi, puisque le public auquel on s'adresserait serait, non plus uniquement des enfants malades ou des couples ayant déjà eu un enfant atteint, mais potentiellement tous les couples désireux de procréer. L'appariement, certes minimal, existe déjà dans les banques du sperme en France, afin que l'enfant ait quelques caractéristiques physiques du père pour faciliter son intégration dans la famille.
Selon une étude du Commissariat général à la stratégie et à la prospective (199 ( * )) , à l'étranger, l'usage de tests visant à établir si les couples désireux de fonder une famille sont « biologiquement compatibles » ou s'ils risquent de transmettre une maladie à leur descendance se répand, soit sous l'impulsion des ministères de la santé, soit par décision individuelle.
Ces sortes de tests génétiques prénuptiaux visent principalement à lutter contre la transmission de maladies génétiques à forte prévalence dans certaines populations, comme la thalassémie à Chypre ou la maladie de Tay-Sachs dans la population juive ashkénaze. En 2004, l'Arabie saoudite a par exemple rendu obligatoire le test pour la drépanocytose et la thalassémie.
Progressivement, ces tests se diversifient et couvrent de plus en plus de pathologies génétiques. Ils se diffusent notamment aux États-Unis, en Israël en Asie et dans les pays du Golfe.
ii. Les tests prénataux
La notion de risque d'apparition d'une maladie commence à être prise en compte dans le cadre du diagnostic préimplantatoire (DPI), bien que ne soient pour l'instant concernées que les maladies monogéniques à pénétrance incomplète. Ainsi, des cas de cancers familiaux précoces ont déjà justifié la sélection d'embryons lors d'une fécondation in vitro en France. Cependant, il n'est pas à exclure qu'un tel usage puisse se développer quand le DPI sera moins risqué en terme de survie de l'embryon, car la recherche de facteurs de risques génétiques sur le foetus commence à être facilitée grâce au développement des analyses effectuées à partir du sang maternel.
En effet, la présence de fragments d'ADN du foetus dans le sang de la femme enceinte est connue depuis plusieurs années, or il est désormais possible d'isoler l'ADN du foetus dans le sang de la mère pour l'analyser. Pour disposer de fragments d'ADN foetal bien identifié, il est possible de rechercher des cellules foetales dans le sang des femmes enceintes. Plusieurs obstacles avaient reporté leur utilisation : leur caractérisation, leur qualité, le fait qu'elles peuvent persister d'une grossesse à l'autre, la durée d'obtention d'un résultat incompatible avec le diagnostic prénatal, etc .
Or « les évolutions techniques récentes, reposant sur la généralisation des méthodes de séquençage à haut débit (NGS, New Generation Sequencing), élargissent considérablement les options offertes aux parents désireux de s'assurer une descendance de la meilleure ?qualité? possible. Ces avancées, dont certaines sont déjà largement utilisées aux États-Unis tandis que d'autres n'en sont qu'à la démonstration de faisabilité, vont sans nul doute poser des questions éthiques inédites, ou tout au moins rendre bien plus aigus des problèmes jusque-là plutôt théoriques » précise le Pr Bertrand Jordan dans une chronique de génomique intitulée « en route vers l'enfant parfait (200 ( * )) ».
Actuellement, selon l'Agence de la biomédecine (201 ( * )) , deux diagnostics sont proposés « en routine » en France : la détermination du sexe foetal dans le cadre du diagnostic prénatal de maladies associées au chromosome X et la détermination du rhésus foetal lorsque la femme enceinte est rhésus négatif (dans l'objectif de prévenir les allo-immunisations foeto-maternelles ou de surveiller la grossesse de femmes déjà allo-immunisées). La recherche du chromosome supplémentaire foetal intervient vers la 10 ème semaine, avec une réponse dans la semaine, soit dans le délai légal autorisant une interruption volontaire de grossesse.
Cette nouvelle technique permet d'éviter l'amniocentèse qui reste un examen diagnostique invasif, à l'origine de pertes foetales et d'anxiétés importantes. Son extension au dépistage de la trisomie 21 a fait l'objet de travaux sur des populations à risque identifiées sur la base du dépistage du premier trimestre.
iii. Les tests néonataux
Le séquençage du génome de tous les nouveau-nés pourrait devenir un test de routine aux États-Unis. Un projet de recherche est lancé pour en évaluer les enjeux médicaux et éthiques. Quatre projets de recherche ont été lancés sur cinq ans par le National Institute of Health pour analyser les bénéfices et les risques du séquençage du génome de tous les nouveau-nés du point de vue technique, médical et éthique, et savoir s'il serait pertinent de séquencer le génome de tous les bébés à leur naissance. Le dépistage précoce de certaines anomalies génétiques qui prédisposent à certaines maladies peut-il changer le destin des enfants ? Telle est l'une des questions majeures qui se pose. Faudra-t-il réactualiser ces données ?
Début 2014, les nouveau-nés et leurs familles qui feront partie de ce programme de recherche, seront recrutés, leur génome sera séquencé, pour quelques centaines de dollars. Mais, comment ces données seront-elles utilisées et interprétées car à l'heure actuelle, il y a certaines mutations génétiques dont on ne connaît pas les conséquences ? Par ailleurs, dépister une maladie à la naissance ne signifie pas qu'on l'on est capable de la soigner, et encore moins de la guérir. Dans ces conditions, faut-il annoncer aux parents que leur enfant développera une maladie ? Et les médecins doivent-ils faire le tri dans les informations recueillies ? Ces tests posent la question d'une possible dérive eugéniste ou du désir d'enfant parfait que nous évoquions dans notre rapport précité concernant l'évaluation de la loi relative à la bioéthique de 2004 (202 ( * )) .
b. Le questionnement éthique
« On peut facilement imaginer que, dans un futur proche, à l'échelle de quelques années, il soit techniquement possible de procéder au ?contrôle de qualité? d'embryons obtenus par fécondation in vitro. Contrôle de qualité qui serait fondé sur l'examen de leur séquence intégrale, et permettrait non seulement d'éviter certaines anomalies mais aussi de choisir le ?meilleur? embryon en fonction des desiderata des parents : sexe bien sûr, mais aussi taille à l'âge adulte, autres caractéristiques physiques et, peut-être, tendances comportementales. Certes, comme je ne cesse de le souligner dans ces chroniques, notre capacité à définir le phénotype à partir de l'ADN est encore assez imparfaite, bien plus que ne l'imagine le public en général » observe le Pr Bertrand Jordan (203 ( * )) dans la chronique précitée.
i. Les avis successifs du Comité consultatif national d'éthique (CCNE)
Depuis une dizaine d'années le CCNE a rendu plusieurs avis sur les diagnostics anténataux : avis n° 72 : réflexion sur l'extension du diagnostic préimplantatoire (juillet 2002), avis n° 83 sur le dépistage prénatal généralisé de la mucoviscidose (mars 2004), avis n° 97 : questions éthiques posées par la délivrance de l'information génétique néonatale à l'occasion du dépistage de maladies génétiques (janvier 2007), avis n° 107 sur les problèmes éthiques liés au diagnostic prénatal et au diagnostic préimplantatoire (novembre 2009).
Dans l'avis n° 107, le CCNE considérait qu'en l'état actuel du droit et des pratiques, le système utilisé, limité à des maladies d'une particulière gravité et incurables au moment du diagnostic, était globalement satisfaisant car il reposait sur le choix personnel libre et informé des personnes, c'est-à-dire la décision de la femme enceinte ou du couple dans le cadre d'un accompagnement médical. Le Comité s'est interrogé sur l'acceptabilité de tests génétiques réalisés en l'absence de signes d'appel, notamment la naissance d'un premier enfant atteint d'une maladie grave et incurable, et sur les problèmes éthiques liés aux diagnostics ultra-précoces effectués sur sang maternel. Le CCNE avait d'ailleurs rappelé que l'interrogation sur la valeur prédictive des mutations en termes de gravité et d'incurabilité doit rester centrale et qu'elle limite la généralisation rapide des tests.
Le CCNE s'inquiétait également de « l'ouverture des tests anténataux de diagnostic génétique sur prélèvement de sang maternel à une exploitation commerciale, notamment sur Internet, qui serait alors difficilement contrôlable et pourrait conduire à la mise en place d'un véritable tourisme prédictif laissant les couples seuls et désemparés face à des tests non validés » ; mais il constatait qu'une harmonisation des législations à l'échelle internationale était un objectif aléatoire, du fait des spécificités culturelles . « En effet, les préférences individuelles sont liées aux normes sociétales, à l'image construite d'une personne saine et à l'acceptation du handicap » .
ii. L'avis n° 120 du CCNE : questions éthiques associées au développement des tests génétiques foetaux sur sang maternel
Dans cet avis d'avril 2013, le CCNE constate qu'une technique non invasive existe, qu'elle sera probablement utilisée dans un contexte de baisse des coûts. Il souligne le caractère probabiliste du savoir que les tests délivrent. « Ce que nous pouvons déduire de la séquence de l'ADN est, notamment, qu'existent, dans la séquence d'un gène, certaines mutations qui, héritées de chacun des deux parents, conduiront, avec une forte probabilité, à une maladie, la mucoviscidose par exemple. Mais nous ne pouvons pas déduire, la gravité clinique de l'affection à laquelle conduira l'anomalie génétique constatée ».
Il insiste sur le caractère complexe de ces données qui nécessitent une information rigoureuse et scientifiquement pertinente. Le conseil génétique a donc un rôle crucial « dans les choix et décisions libres des femmes et des couples en attente d'enfant ». Cette problématique de l'information tient une place centrale dans la réflexion du CCNE, et la nécessité de sa mise en oeuvre figure au rang de recommandations premières.
Le dépistage de la trisomie 21 à partir du séquençage de l'ADN foetal sur sang maternel constitue un progrès car il correspond à une amélioration technique du dépistage qui limite largement les effets secondaires, et diminue le nombre de prélèvements invasifs et potentiellement dangereux. Le CCNE estime que « cette simple amélioration devrait être accompagnée d'une prise en charge du test par la solidarité nationale, à supposer que le coût en soit devenu acceptable » . Néanmoins si les problèmes techniques, organisationnels et de coût sont résolus, l'extension de ce test nécessiterait « qu'un ensemble de conditions en assure la pertinence, la sécurité, l'égalité d'accès sans conditions de ressources, ainsi que la qualité de l'information et de l'accompagnement ».
Selon le CCNE, l'extension des prescriptions de tests génétiques foetaux sur sang maternel sera à terme inéluctable ; il faudra accompagner cette extension. « Les sciences et les techniques nous placent dans une situation où un test donné, correspondant à un handicap ou une maladie d'origine génétique particulière, ne peut plus être considéré indépendamment de tout un ensemble d'autres tests, voire du déchiffrage de l'ensemble de notre patrimoine génétique ». La dimension emblématique et exceptionnelle de la trisomie 21 s'estompera devant un nombre croissant d'altérations chromosomiques et de mutations associées aux maladies et handicaps génétiques, parfois gravissimes, qui seront identifiés. Le CCNE préconise de « pouvoir effectuer soit une lecture sélective des séquences d'ADN alors que les évolutions techniques nous entraînent à en effectuer une lecture globale, soit une lecture totale mais une communication sélective et adaptée ».
La lecture de l'ADN foetal entier, lorsqu'elle pourra être réalisée dans des conditions économiques et de qualité clinique reconnue, devrait être restituée de manière sélective sur des critères pertinents et rigoureux telle la particulière gravité et l'incurabilité de la maladie au moment du diagnostic.
Cependant devant un risque de handicap ou de maladie « d'une particulière gravité et incurable au moment du diagnostic », mais de probabilité de survenue faible, comment faire la part entre gravité et probabilité de survenue ? Quand considérer qu'elle serait trop faible pour devoir être prise en considération dans le cadre d'une demande d'interruption médicale de grossesse (IMG), et donc comment fixer un seuil ?
Le CCNE s'interroge sur le devenir de la séquence dont « l'interprétation s'affinera au cours du temps, laissant ouverte, après la naissance, la question de l'actualisation de l'information auprès des futurs parents, puis de l'enfant lui-même et de l'adulte qu'il pourra devenir. Conviendra-t-il de conserver les données brutes ? Sous quelle forme ? Sous la responsabilité de qui ? Et quand, comment, et à qui les communiquer le cas échéant ? ». Cette problématique de la conservation et de l'utilisation ex post des données nous interroge.
Face à ces développements, le CCNE souhaite que « soit valorisé ce que la génomique apporte(ra) à la thérapeutique » et insiste sur la dimension humaine de la prise en charge des personnes porteuses d'un handicap ou atteintes d'une maladie, notamment chronique et/ou évolutive,
Il constate que les futurs parents souhaitent « non pas l'enfant parfait, mais un enfant en bonne santé, et, pour beaucoup de parents, un enfant qui ne soit pas obligatoirement condamné, dès la naissance, à un handicap ou une maladie incurable et d'une particulière gravité ». Il rappelle que la convention de l'ONU de décembre 2006 sur les droits des personnes handicapées, ratifiée par la France, considère que le handicap ne résulte pas uniquement des problèmes physiologiques que présente une personne, mais aussi des obstacles que la société met à l'exercice de ses droits, de ses capacités et de son autonomie.
iii. Comment informer sur les découvertes fortuites ?
Mme Simone Bateman (204 ( * )) observe que si on applique ces nouvelles techniques génomiques aux DPI et DPN, à la naissance, on aura accès à bien plus de données que ce que l'on veut savoir dans l'immédiat. Or même si on cible l'individu, la famille sera immédiatement au courant en cas de maladie génétiquement transmissible, les secrets de famille apparaitront au grand jour. Des découvertes fortuites pourront indiquer des risques d'être atteint d'autres maladies et entraîner d'autres types d'examens.
Elle précise : « De nombreuses études portent sur le cancer héréditaire du sein et des ovaires car c'est un sujet très conversé. Des études sur des personnes ayant effectué un test montrent que si on peut faire ces tests pour pouvoir éventuellement mieux traiter la personne potentiellement malade ou en vue d'une prévention, cela entraine nécessairement un souci de prévention pour la descendance ».
Le Pr Jean-Claude Ameisen (205 ( * )) , commentant l'avis n° 120 du CCNE, observe : « ce serait peut-être la première fois dans la médecine que des médecins s'interdiraient d'acquérir une information par peur de ne pas savoir quoi en faire. Cette masse d'informations et la difficulté de l'interpréter engendre des problèmes nouveaux particulièrement en matière d'organisation et de réflexion sur le conseil génétique. On se trouve face à un décalage de plus en plus grand entre ce à quoi la technique donne accès, les règles mises en place sur le choix libre et informé, et ce que la personne peut faire de l'information ».
3. L'impact sur les assurances et l'embauche
La médecine prédictive pourrait être détournée de sa fonction sanitaire et être exploitée à des fins non médicales, dans le domaine des assurances et du travail.
a. L'impact sur les assurances
L'individualisation des risques, pourrait avoir un impact important sur les mécanismes d'assurance, car la prédiction lèverait le « voile d'ignorance » sur lequel repose le principe de la mutualisation du risque dans le domaine de la santé (206 ( * )) . « Le développement des capacités prédictives de la médecine pourraient inciter les assureurs à faire une sélection génétique des assurés. Ces informations nouvelles seraient susceptibles de diminuer l'assurabilité des uns et de réduire les primes des autres ».
Certes actuellement, l'état de santé est déjà pris en compte dans les contrats d'assurance. Contrevenir au principe de bonne foi en cachant une pathologie, des antécédents médicaux et familiaux à un assureur peut provoquer une annulation du contrat. Ce point, mal connu des usagers, entraîne des contentieux très lourds au détriment des assurés. En France, afin de permettre au plus grand nombre de s'assurer, différents mécanismes ont été mis en place pour que la santé n'obère pas les possibilités d'assurance, telle la convention Aeras qui vise à permettre aux personnes à risque aggravé de santé de pouvoir s'assurer et emprunter.
Face aux calculs de risques encore peu fiables découlant des tests prédictifs, de nombreux États en ont limité, voire interdit, l'utilisation par les assureurs. Pour l'instant, la majorité des assureurs mondiaux ne s'est pas opposée à l'interdiction de l'utilisation de la génétique dans leur secteur. En effet, les questionnaires sur l'état de santé suffisent pour l'instant à établir un profil de risque proche, en général, de la réalité.
Comme en Espagne et en Belgique, en France les assureurs français ne peuvent faire payer une surprime en fonction de résultats génétiques. De la même manière, nul ne peut se prévaloir d'un profil peu risqué pour bénéficier de primes moins élevées.
Certains pays ont quant à eux fixé un seuil de couverture au-delà duquel les assurances peuvent avoir accès aux résultats d'un test génétique. C'est le cas de la Suisse ou encore au Royaume-Uni. Ce type de législation risque toutefois d'être contre-productif en matière de santé publique car il peut décourager les personnes de se faire tester.
Au Royaume-Uni, l'utilisation des tests génétiques prédictifs dans le cadre de l'assurance est très limitée : en vertu d'un moratoire, signé en 2001 et reconduit depuis jusqu'en 2017, entre le ministère de la santé et l'Association des assureurs britanniques (ABI), les résultats de tests génétiques prédictifs et pré-symptomatiques ne peuvent affecter la capacité d'un individu à s'assurer, en dehors des assurances-vie excédant les 500 000 Livres (3 % des contrats). En outre, au-delà de ce seuil, seuls les tests approuvés par le ministère peuvent être utilisés, ce qui a été le cas en 2012 pour le test de la maladie de Huntington.
Même si les compagnies d'assurances ne peuvent pour l'instant pas exiger de leurs clients qu'ils subissent un test génétique, l'assureur cherchera à obtenir l'information au même titre que les autres antécédents médicaux de son client si celui-ci a déjà subi un tel test.
C'est la raison pour laquelle le Conseil de l'Europe s'est penché sur le problème. Son Comité de bioéthique (DH-BIO) a effectué une consultation publique sur le document de consultation sur la prédictivité, les tests génétiques et l'assurance élaboré par son Comité directeur pour la bioéthique (CDBI) (207 ( * )) . L'objectif de cette consultation était de solliciter les réponses et commentaires, notamment des associations et organismes représentatifs des différents secteurs concernés (patients, consommateurs, assureurs et réassureurs, médecins, généticiens et médiateurs), au niveau national et international, sur des questions et propositions concernant l'utilisation des données prédictives liées à la santé, en particulier, les données génétiques, dans le domaine des assurances.
Ce travail s'inscrit dans une activité plus large entreprise par le Conseil de l'Europe en vue d'élaboration un cadre juridique pour la protection des droits fondamentaux à l'égard de l'utilisation des tests génétiques, dans le domaine médical comme en dehors de celui-ci, dont la première étape fut l'adoption du Protocole additionnel précité à la Convention sur les Droits de l'Homme et la Biomédecine, relative aux tests génétiques à des fins médicales.
b. L'impact sur l'embauche
La détermination de risques de santé pourrait aussi intéresser les employeurs, soit pour introduire une sélection à l'embauche, soit pour gérer leurs ressources humaines. Cela pourrait conduire à une sélection à l'embauche des personnes à « bon profil génétique », afin, par exemple, de limiter les jours d'absence des salariés ou de privilégier des capacités physiques ou intellectuelles utiles à l'entreprise.
De nombreux pays ont donc adopté des réglementations afin de se prémunir contre ce type de discriminations. En France, cette protection relève du Code pénal qui sanctionne les discriminations, quelles qu'elles soient. L'Allemagne et les États-Unis ont, quant à eux, adopté une réglementation plus spécifique.
Entrée en vigueur en 2009 aux États-Unis, la loi Gina ( Genetic Information Non-Discrimination Act ) protège les usagers contre la discrimination génétique dans le domaine de l'assurance santé et de l'emploi. Certains États fédérés ont par ailleurs étendu cette protection. La Californie, par exemple, a voté la loi CalGINA, qui, à partir de 2012, empêchera toute utilisation de tests génétiques préalables à l'accès aux soins, à l'assurance dépendance, aux prêts immobiliers, au logement ou à tout type de services et commerces.
B. LA PROTECTION ET LA GESTION DES DONNÉES PERSONNELLES
Les données génétiques sont personnelles, identifiantes, et pour partie invariantes tout au long de la vie. Elles constituent un dossier qui portera sur les possibilités d'évolution de l'état de santé d'une personne pendant toute sa vie. En outre, même si la médecine personnalisée cible l'individu, la famille sera informée en cas de maladie génétiquement transmissible. Le secret médical prend donc une dimension encore plus importante que par le passé, mais ses contours sont moins nets dès lors que l'information de la parentèle est obligatoire dans certaines circonstances.
Les risques d'obtention indue de données s'accroissent d'autant plus qu'il ne s'agit plus des fiches cartonnées du généraliste, mais d'un système informatisé, donc fragile, consultable à distance, intégrant durablement des données personnelles. Cela est préoccupant, car l'anonymisation de ces données doit pouvoir être garantie pour assurer une protection rigoureuse du secret médical, éviter des discriminations préjudiciables aux personnes ou aux familles. En avons-nous les capacités techniques à l'heure actuelle, surtout à un moment où l'on découvre que la surveillance numérique s'effectue à notre insu dans de très nombreuses circonstances et que l'anonymisation n'est peut-être pas absolue ? Nos concitoyens sont-ils informés de ces risques ? Se posent-ils suffisamment la question de la sécurité de leur intimité numérique ? Un dossier médical personnel sur clé USB serait-il plus sûr ?
1. Un cadre juridique protecteur des données génétiques
Le droit français a cherché à offrir une protection particulière garantissant la confidentialité des données de santé, lesquelles touchent à l'intimité de la vie privée. Pour autant la circulation des données de santé ne se limite pas au seul territoire de la France. Des tentatives de protections à l'échelon international sont en cours d'élaboration.
a. Un cadre français protecteur des données personnelles
La loi du 6 janvier 1978 modifiée, relative à l'informatique, aux fichiers et aux libertés reconnaît aux données personnelles, un caractère de données sensibles. À ce titre elles sont soumises à un principe d'interdiction de traitement, assorti cependant d'un certain nombre d'exceptions prévues par la loi, moyennant des garanties que la Commission nationale de l'informatique et des libertés (CNIL) est chargée de faire respecter.
L'article 8 de la loi stipule : « Il est interdit de collecter ou de traiter des données à caractère personnel qui font apparaître, directement ou indirectement, les origines raciales ou ethniques, les opinions politiques, philosophiques ou religieuses ou l'appartenance syndicale des personnes, ou qui sont relatives à la santé ou à la vie sexuelle de celles-ci ».
Les exceptions prévues concernent le consentement de la personne elle-même, les traitements à des fins de suivi médical individuel, à des fins d'intérêt public, de recherche médicale et d'évaluation de pratiques de soin, ou encore l'anonymisation des données prévue à bref délai. Á l'intérieur de ces exceptions, les caractéristiques particulières des données génétiques, leur caractère prédictif notamment, justifient que le droit français leur confère un statut très protecteur.
Les « collections d'échantillons biologiques
humains » et la constitution d'ensembles de collections, dites
biothèques, sont encadrées par les articles
L.1131-4, L.
1243-3 et 4 du code de la santé publique. Elles sont soumises, soit
à un système de déclaration à la CNIL dans le cadre
de soins, soit à un régime lourd de double autorisation par un
comité scientifique et par la CNIL pour les programmes de recherches. La
CNIL vérifie notamment la pertinence des données
collectées et leur proportionnalité au regard du but de la
recherche, la durée limitée de conservation des données
compte tenu de la finalité de la recherche. Elle effectue
régulièrement des contrôles sur dossiers et sur place.
Les droits des personnes doivent être respectés (article L.1122-1-1 du code de la santé publique, article 56 modifié de la loi Informatique et libertés). Une note d'information doit leur expliquer clairement à qui et à quoi sont destinées les recherches, comment elles vont être mises en oeuvre, quels sont leurs droits, de rectification notamment, et comment elles peuvent les exercer. C'est ce que l'on appelle le consentement libre et éclairé, mais la question de ses limites dans le temps se pose, car la technologie évolue très vite et nécessitera de plus en plus des consentements larges et homogènes.
b. Des mesures de protection au niveau européen
Au niveau européen, un projet de règlement sur la protection des données, actuellement devant le Parlement européen prévoit de mentionner, pour la première fois au nombre des données sensibles, les données génétiques. La Commission des libertés civiles, de la justice et des affaires intérieures (LIBE) du Parlement européen a adopté le 21 octobre 2013 à une forte majorité la proposition de règlement de l'Union européenne sur la protection des données personnelles, ainsi que la proposition de directive présentée en parallèle en matière de police et de justice.
Ce vote de la Commission LIBE renforce les principes de la protection des données et les droits des citoyens, ainsi que les obligations des responsables de traitement et des sous-traitants, avec des sanctions plus dissuasives. Le texte introduit un contrôle des autorités de protection sur les demandes d'autorités administratives et judiciaires de pays tiers d'accéder aux données relatives à des citoyens européens.
Mais, le plus important à vérifier reste la sécurité des données génétiques : il faut veiller à ce que les données soient conservées et traitées par le responsable du traitement dans des conditions de nature à garantir leur confidentialité, mais aussi leur intégrité.
c. Le changement d'échelle du respect de la confidentialité
La CNIL contrôle scrupuleusement le respect des règles de confidentialité qui sont très précises et portent sur le codage, la dissociation de données, les habilitations spéciales d'accès, l'anonymisation. Cependant, cet ensemble de contraintes est aujourd'hui mis à mal par un changement considérable d'échelle dans le temps et l'espace avec des projets internationaux de recherche médiatisés et avec le comportement des personnes elles-mêmes qui souhaitent disposer de leurs données personnelles et les faire figurer sur des supports, voire les diffuser sur Internet.
Selon M. Ashween Peerbaye (208 ( * )) : « on se trouve face à un ?déluge des données? dans les sciences du vivant. Ce déluge de données peut être lu comme la conséquence d'une technologie de rupture. Il s'agit en effet d'une rupture dans les pratiques et dans les significations accordées aux données produites. Les chercheurs qui se sont intéressés à ces questions semblent faire état d'une certaine ?paralysie? des scientifiques, parfois, face à l'incapacité de traiter des données, et à un sentiment de gaspillage ».
.
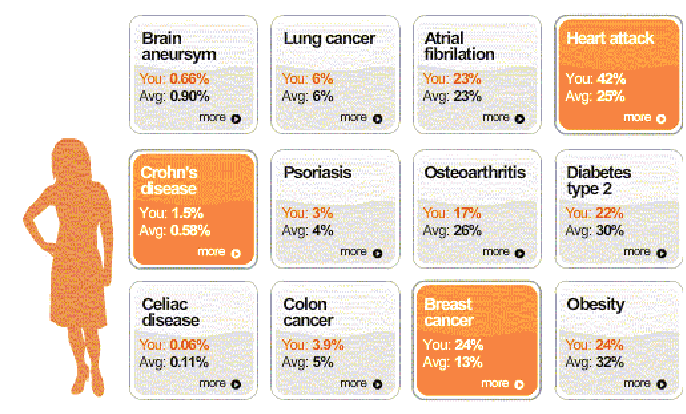
Le graphique ci-dessus illustre les besoins de stockage nécessaires compte tenu des volumes de données biologiques produits et souligne l'importance des moyens à mettre en place pour faire face à l'augmentation massive de ces données.
i. La capacité de protection des grandes bases de données
La constitution de vastes bases de données génétiques de populations, à la dimension d'un pays, afin de mutualiser les moyens et de les mettre à disposition des chercheurs, des autorités sanitaires pour effectuer une surveillance sanitaire de population, est utile mais conduit à s'interroger sur la capacité à les protéger. Comment en effet sécuriser la masse extraordinaire de données ainsi recueillies, d'autant que des travaux récents indiquent que l'idée de garantir l'anonymat des données génétiques est sans doute illusoire ?
Vis-à-vis de ces vastes bases de données qui se constituent aujourd'hui grâce aux outils technologiques puissants dont on dispose, de nombreuses règles de notre législation informatique et libertés sont remises en cause, telle la durée de conservation des données limitée à la finalité de la recherche ; il faudra mettre les données à disposition de tous les chercheurs si la recherche devient une recherche fondamentale dont on ne peut appréhender toutes les finalités. Tous les principes sont mis à l'épreuve. Comment assurer le respect du droit des personnes, quand la recherche est appelée à durer longtemps et à évoluer ? On ne pourra pas redemander un consentement à chaque étape et les personnes disparaîtront.
ii. Les limites de l'anonymisation des données
Des exemples de ré-identification, à partir de données minimales anonymisées contenues dans des bases à accès ouvert ont été récemment décrits. Un article, qui commence à être célèbre, paru dans le magazine Science (209 ( * )) en janvier 2013, expose qu'il a été possible, en croisant plusieurs éléments, d'identifier une personne à partir de son patrimoine génétique et de données généalogiques et de géolocalisation. L'article de Science a été fait à partir de séquences répétées du chromosome Y, c'est peu. On n'a donc pas besoin de tout le génome pour rompre la protection de l'identité.
Un autre exemple de difficulté concerne l'anonymisation des données transférées qui ne devra pas être faite de façon non irréversible, car d'une part celles relatives à l'évolution clinique du patient sont essentielles à la recherche elle-même, et d'autre part, les résultats de la recherche tendent à être restitués au patient, si celui-ci le souhaite. On devra alors informer les personnes de cette possibilité de réutilisation de leurs données, mais aussi de l'éventualité de ré-identification à partir de données de séquences croisées avec d'autres types de données accessibles publiquement.
Il faudra certainement compléter les règles actuelles, faire peser sur les titulaires de vastes bases de données d'autres types d'obligations de sécurité. Il existe heureusement en matière de santé des hébergeurs de données agréés présentant un haut niveau de sécurité. Pourquoi ne pas avoir recours aussi, dans les liens entre les laboratoires et les personnes, à des tiers de confiance indépendants qui détiendraient les données d'identification permettant de faire le lien entre l'identité et la donnée médicale ? suggère Mme Frédérique Lesaulnier (210 ( * )) .
Au surplus, il est impératif que ces données ne tombent pas entre des mains non autorisées, sur Internet par exemple. Or, ces processus vont conduire à la banalisation et à la dissémination des centres utilisateurs et détenteurs de données sensibles. Les transferts d'informations multiplieront forcément les risques d'atteinte au secret des données médicales.
On devra donc trouver un système qui garantisse à la fois une qualité de la recherche, une certaine réactivité de la recherche, et en même temps la confidentialité des données du patient. Par ailleurs, on ne fera pas l'économie d'une nouvelle réflexion sur l'anonymat des dons de gamètes qui sera difficile à assurer et à justifier .
iii Le dossier médical personnel : une solution possible
• En France
Toutes les données personnelles du dossier médical doivent être sauvegardées, protégées de la même façon. Le « serpent de mer » du dossier médical personnel (DMP) (211 ( * )) , en cours d'informatisation, devra en plus inclure les données génétiques. Il semble d'ailleurs qu'un dossier médical personnalisé n° 2 soit en cours d'élaboration. Cependant le fait que ces informations médicales durables n'apparaissent pas forcément en routine et que l'informatisation des dossiers médicaux ne soit pas encore généralisée constitue un risque considérable et un frein à la prévention comme à la médecine personnalisée.
Interrogée à ce sujet, Mme Marie-Christine Favrot (212 ( * )) a convenu que la mise en oeuvre du DMP n'avait guère progressé. La mise en place du DMP a, dès le départ, été mal engagée au plan informatique car on n'a pas su sensibiliser correctement les généralistes ; or bien sécurisé et sous la responsabilité du patient, le DMP pourrait contenir les données de santé indispensables aux traitements, permettre de soigner mieux en renforçant la prévention, en favorisant l'adaptation des traitements au métabolisme du patient, en évitant des examens inutiles.
• Aux États-Unis
D'après le Dr Jean-Marc Egly (213 ( * )) , la mise en place du Dossier médical électronique, ou DME, (en anglais EMR, Electronic Medical Record ou EHR, Electronic Health Record ) au sein des services hospitaliers se fait progressivement depuis une vingtaine d'années à la suite des recommandations de l'Institute of Medicine . Les instances fédérales ont débloqué jusqu'à 27 milliards de dollars pour développer la mise en place des DME et démontrer leur efficacité au niveau clinique et hospitalier. Les incitations financières sont conditionnées par la qualité de la mise en place, l'utilisation des données concernant le patient ainsi que les prestations cliniques subies. On peut imaginer y trouver le quotidien clinique de chaque patient, vu par l'entourage médical dans son ensemble. En 2014, les hôpitaux ayant mis en place les DME devront y enregistrer les données de chaque patient, et en 2015, en cas de retard dans l'établissement et l'utilisation de ce système électronique, ils s'exposeront à des pénalités financières.
En dépit de ces incitations financières, la mise en place de ce système reste encore très coûteuse pour les établissements. Deux milliards de dollars ont également été débloqués, le pourcentage des hôpitaux qui utilisent le dossier médical électronique étant passé de moins de 10 % en 2006-2007 à près de 40 % en 2011-2012 aux États-Unis. Cela requiert des accords inter-hospitaliers préalables.
La mise en place des DME a pour but de permettre de rassembler toutes les informations d'un patient, des résultats du dernier examen médical de routine aux analyses les plus poussées, effectuées à la suite d'une hospitalisation. Leur accès tant pour les patients que pour le personnel médical pose la question du contrôle des informations, de leur harmonisation et classification.
Sous les pressions exercées par les instances fédérales pour le développement du DME, l'amélioration de la qualité et de l'efficacité des soins délivrés au patient et la réduction des coûts associés aux soins de santé, les hôpitaux américains se regroupent au sein de réseaux le plus souvent régionaux mais parfois nationaux. C'est le cas du réseau eMERGE, pour Electronic Medical Records and Genomics, consortium national lancé par le National Human Genomic Research Institute (NHGRI) du NIH (National Institutes of Health) sur l'ensemble des États-Unis. Il met en réseau des institutions qui disposent de banques d'ADN et possèdent des dossiers médicaux électroniques (DME).
Cette opération permet le développement de recherches génétiques à grande échelle et à haut débit à partir de prélèvements opérés sur des patients (après signature d'un consentement éclairé) dont les bénéficiaires seront, outre le patient lui-même, les établissements de soins qui collecteront les données génomiques et analytiques. Ainsi, d'après le Dr Jean-Marc Egly (214 ( * )) , aux États-Unis les programmes de médecine personnalisée se développent, dans un contexte de généralisation de l'utilisation des dossiers médicaux électroniques par les hôpitaux.
2. Les défis du stockage et du partage d'énormes banques de données
Le Dr Jean-Marc Egly (215 ( * )) a insisté d'emblée sur le rôle des banques de données : « la biobanque ne peut être utile que dans la mesure où elle est constituée dans l'intérêt du patient et non pour la recherche du scientifique. Cela signifie qu'il faut impliquer les cliniciens dans les biobanques. Une biobanque, n'est pas uniquement une collection d'échantillons. Elle est constituée de trois éléments : le dossier médical du patient, les analyses du patient, toutes les actions du clinicien pour insérer dans la biobanque le suivi du malade ».
Le Dr Anne Cambon-Thomsen (216 ( * )) a expliqué qu'« étant donné la rapidité de ces nouvelles découvertes, il est important que des bio-banques et des bases de données soient créées, avec des informations à jour sur les génotypes et de l'information phénotypique sur les variants et les patients. Il ne faut pas oublier cette notion : on constitue des bases en même temps que l'on traite des patients . Les ressources nécessaires pour ces bases doivent évidemment être trouvées quelque part, d'autant que ces bio-banques devront être alimentées par toutes les découvertes récentes en recherche. Comment parvenir à ce résultat ?
a. Des outils de stockage d'une dimension nouvelle, difficiles à contrôler
Le traitement numérique des données du séquençage à haut débit du génome est une opération complexe qui repose en grande partie sur la comparaison avec des données similaires récoltées auprès d'autres patients ou d'individus non malades. Son coût risque de devenir supérieur à celui du séquençage proprement dit.
Ainsi, loin d'être un aboutissement, la démocratisation du séquençage apparaît de plus en plus comme un point de départ. Désormais, pouvoir obtenir, pour un coût relativement modique, le génome de vastes populations impliquera des analyses à grande échelle qui, seules, permettront de comprendre le comportement et le rôle des gènes, à condition toutefois de disposer des ressources informatiques adéquates. Dès lors, la gestion, le stockage, le contrôle, la protection des bases de données, deviennent un enjeu crucial et des efforts importants doivent être fournis en matière de stockage et de capacité de calcul.
Comment collecter, rassembler et archiver les informations ? Qui aura obligation de stocker les données, alors que celles susceptibles de circuler exigent des espaces considérables ? Pour être utiles, interprétables, ces données transitent, ce qui suppose une fiabilité extrême des circuits, donc des accès sécurisés et surtout des systèmes d'anonymisation fiables. Est-on suffisamment avancé dans ces techniques pour pouvoir les garantir ?
Manipulant des quantités phénoménales de données, des algorithmes extrêmement spécifiques et des informations personnelles sensibles, la génomique et la biochimie requièrent des supercalculateurs hautement performants et adaptés aux tâches qu'ils auront à accomplir. En génomique, par exemple, de nombreux programmes de recherche s'intéressent à la comparaison de deux populations, l'une malade et l'autre saine, pour confronter leurs génomes et tenter d'isoler le ou les points qui les différencient.
Seuls des supercalculateurs de dernière génération couplés à d'énormes capacités mémoire sont en mesure d'accomplir ces calculs dans des délais acceptables. Modéliser les molécules, découvrir les règles qui déterminent leur géométrie, évaluer le jeu des influences croisées, tels sont les objectifs d'un domaine d'une complexité extrême, dont les progrès sont directement tributaires des capacités de calcul disponibles, de la maîtrise et de la fiabilité des outils informatiques.
De nombreux problèmes techniques se posent en pratique. En France on constate un déficit de biostatisticiens et bio-informaticiens. Or, la demande des chercheurs est très forte pour mobiliser des capacités par thème, par type de maladie, assortie d'une interopérabilité. Cette vision des choses implique la création d'organismes spécialisés, d'un répertoire mondial fiable de toute l'information trouvée, car aujourd'hui une bonne partie de l'information est perdue.
Cela requiert aussi une normalisation, une standardisation des conditions de prélèvement et d'analyse, une nouvelle conception des banques de données. Il faudra également créer des fichiers d'interopérabilité, ce qui entre dans le domaine de compétence de la CNIL, mais on s'interroge alors à juste titre sur l'insuffisance de ses moyens financiers et humains pour accomplir ces missions. Qui va gérer l'organisation du stockage des données : une ou plusieurs institutions ? « Une sorte de CNIL de la santé ? Une institution unique n'acquerrait-elle pas trop de puissance ? À l'inverse, une pluralité d'institutions ne se ferait-elle pas au détriment de l'interopérabilité ? » s'interroge Christian Byk (217 ( * )) , magistrat.
La dimension européenne s'impose pour trouver des solutions. Au sein de l'Union européenne, on constate une prise de conscience sur ces questions et une nécessité de renforcer les textes actuels, d'aller vers plus de transversalité et d'articuler les différents niveaux, nationaux, européens et internationaux. La directive 95/46 relative à la protection des données à caractère personnel est en phase de réécriture. Il existe aussi une réflexion avancée sur le « cloud », sur la création éventuelle d'un « cloud » européen souverain pour limiter la dépendance à l'égard des stockages nord-américains.
b. L'accès aux bio-banques et la propriété des données
i. L'accès aux bio-banques : un enjeu d'importance
La question de la gestion et de l'accès aux bio-banques constitue aussi un enjeu considérable, eu égard à leur importance pour la recherche. « Les personnes ayant participé à une cohorte demandent à être informées sur ce que devient la cohorte. Elles ont signé un consentement, elles savent qu'elles font partie d'un échantillon, qu'elles sont dans une étude avec une puissance statistique sans forcément de valeur pour elles-mêmes. On peut discuter dans certains cas qu'il puisse y avoir un retour, mais la première demande des personnes est : qu'est devenue cette cohorte ? Surtout quand elle dure des années. Elles ont participé à quelque chose et voudraient savoir si elles ont rendu service à la recherche, à la société » remarque le Pr Hervé Chneiweiss (218 ( * )) .
Plus crucial encore sera de déterminer « q ui aura obligation de fournir au patient les données dans quinze ou vingt ans, quand il pourra en avoir besoin pour une pathologie qui se déclarera ? À partir d'un génome entier, qui donnera des informations gigantesques et pourra offrir une liste de pathologies auxquelles le patient serait exposé, qui va décider du choix des informations à lui donner ? L'industriel qui aura fait le travail ? En outre, lorsque des progrès scientifiques interviendront, de nouveaux facteurs de risque auront été déterminés, qui aura obligation de prévenir le patient ? » s'interroge M. Jean-Marc Grognet (219 ( * )) .
C'est toute la question de la durée de l'archivage des données et de la propriété de ces données. Il faudra bien retraiter périodiquement l'information, si un nouveau traitement existe. Qui informera le patient et quand ? Dès lors qui paiera pour obtenir la donnée, pour la confirmer ? Qui en est propriétaire ? Le patient, celui qui a réalisé l'examen, la collectivité qui a payé l'examen ? Il existe en principe une règle aujourd'hui en vertu de laquelle chacun est propriétaire de ses propres données médicales, mais l'Islande a vendu les données génétiques de sa population à une firme privée.
ii. Les risques d'appropriation des données
Ce type de projet, vise à séquencer une population entière et ainsi alimenter une banque de données biologiques nationale ou internationale.
• Le cas de l'Islande
Un projet de séquençage de la population avait été initié en Islande à la fin des années quatre-vingt-dix ; et en décembre 1998, les parlementaires, après des débats assez houleux, ont confié la gestion et l'exploitation exclusive de la banque de données génétiques à une société privée DeCode genetics au terme d'une loi intitulée Health Sector Database (HSD ). DeCode Genetics , société qui avait été fondée en 1996 par un Islandais, le neurologue Kari Stefansson, avait son siège aux États-Unis et était cotée en bourse.
Le Pr Kari Stefanson était convaincu qu'il y avait là un gisement de connaissances et aussi des possibilités commerciales. Il en allait de même du gouvernement islandais qui avait autorisé cette société à exploiter commercialement le génome collectif des Islandais, l'État islandais obtenant une participation sur les bénéfices éventuels futurs de la société. Certains s'étaient indignés à l'époque que l'on puisse de la sorte exploiter commercialement le patrimoine génétique d'une population. Au sein de la classe politique islandaise, la question avait fait débat.
Cependant ce projet de cession a été abandonné à la suite d'une décision de la Cour suprême islandaise en 2003 fondée sur des lacunes dans la protection des données et de la vie privée. DeCode a déposé son bilan en 2009, et a été rachetée en 2010 par Saga Investments , un groupe dirigé par deux sociétés de capital-risque. DeCode n'a jamais vraiment eu les moyens financiers de développer des médicaments.
Amgen, une des plus grandes sociétés mondiales de biotechnologie, aurait versé 415 millions de dollars pour acquérir DeCode Genetics , en décembre 2012 Interrogée par les rapporteurs, Mme Corinne Blachier-Poisson (220 ( * )) est restée évasive sur la teneur de l'accord Amgen/DeCode. Amgen espère capitaliser sur les découvertes futures de DeCode.
Vos rapporteurs avaient formé le projet de se rendre en Islande et de rencontrer le Pr Kari Stefanson, mais la mission n'a pu avoir lieu et, en tout état de cause, le Pr Kari Stefanson ne souhaitait pas, semble-t-il, s'entretenir avec des parlementaires, quels qu'ils soient. À cette occasion et grâce à l'aide de notre ambassade en Islande, nous avons appris que l'Icelandic Heart Association avait collaboré avec DeCode durant plusieurs années. Leurs différentes approches de la recherche génomique sur le fond ont mis un terme à cette collaboration car les deux centres de recherches n'avaient pas les mêmes conceptions quant à l'utilisation de la base de données.
DeCode , essentiellement financé par des fonds privés, visait surtout à constituer une carte génétique islandaise alors que Icelandic Heart Association , financée par des fonds publics, souhaitait mettre en place des calculateurs de risques avec des systèmes de contrôle des données par les patients eux-mêmes. Le facteur de l'indépendance dans la recherche génomique est donc au coeur de la comparaison entre ces deux centres.
• Le projet FarGen aux Iles Féroé
D'après Mme Mathilde Reynaudi (221 ( * )) , les Iles Féroé souhaitent entreprendre un séquençage génétique complet de tous leurs habitants et créer un précédent en offrant à tous les habitants de l'archipel (environ 50 000 personnes) un séquençage complet de leur génome dans les cinq prochaines années. Selon ce projet, les données génétiques seraient ensuite ajoutées au dossier médical informatisé dont dispose d'ores et déjà chaque habitant. Ces données seraient accessibles à l'usager s'il le souhaite et à son médecin, si celui-ci considérait ces données utiles dans la prise en charge de son patient. Les ADN séquencés seraient également mis à profit par la recherche.
c. Le partage scientifique des grandes banques de données
L'avenir de la médecine personnalisée implique un partage public de larges collections de données et donc de résoudre le défi de la gestion de ces énormes banques de données et du risque de leur mésusage. Une fois rendues publiques afin de permettre à d'autres chercheurs de réaliser des analyses complémentaires dans le même cadre, ou dans un cadre non prévu au départ, ces informations sont censées pouvoir circuler librement pour nourrir la recherche. Or, les usages qui pourront être faits par d'autres, des données mises en commun dans ces grandes banques, peuvent rarement être prévus à l'avance. Il est probable, et même espéré que les usages effectifs de ces données dépasseront ceux initialement présentés aux personnes qui ont accepté d'y participer. On ne peut non plus écarter un risque de détournement d'usage, qui reste particulièrement préoccupant, s'agissant d'informations médicales personnelles touchant à la vie privée de la personne et de sa famille comme le montre l'article de la revue Science précité.
Le Dr Catherine Bourgain (222 ( * )) a constaté au sein de la littérature scientifique internationale, une tendance, notamment américaine, à penser que la notion de vie privée serait en train de changer, que les individus auraient de moins en moins peur de la transparence sur leurs données personnelles, compte tenu des bénéfices sociaux susceptibles d'être obtenus grâce au large partage des données. En juin 2013 a été lancée, sur une base de volontariat, une Alliance globale pour le partage des données génomiques (223 ( * )) qui regroupe maintenant une soixantaine d'institutions issues d'une quarantaine de pays.
Pour la France on y trouve l'INCa et le Consortium international de la recherche sur les maladies rares ( International Rare Diseases Research Consortium IRDiRC), la majorité de ces institutions restant toutefois américaine. Cette Alliance ambitionne de mettre en commun les données génétiques et aussi cliniques de ses partenaires et de prendre les mesures nécessaires pour encourager leur partage en définissant des standards techniques et de procédure harmonisés pour le respect de la vie privée et de l'éthique.
Il s'agit de créer les conditions d'un écosystème ouvert sur le modèle Internet, permettant l'innovation. « Disposer de bases communes ou interactives permettra d'atteindre plus rapidement des volumes conséquents de données, indispensables pour mieux caractériser les cancers, notamment les plus rares » indique le Pr Fabien Calvo, directeur-adjoint de l'INCa. Dans son texte fondateur, l'Alliance insiste à de nombreuses reprises sur la nécessité de prendre en compte les spécificités nationales. Néanmoins, l'esprit général qui se dégage du texte fondateur d'une cinquantaine de pages, d'inspiration très largement américaine, reprend nombre des arguments des tenants de l'évolution vers une moindre protection de la vie privée.
Une réflexion s'impose donc sur les moyens de concilier d'une part, l'indispensable partage scientifique des données à partir de larges tailles d'échantillons permettant des méta-analyses et d'autre part le respect de la confidentialité et de la sécurité de l'accès aux données génétiques des personnes. Comme le souligne M. Jean-François Deleuze (224 ( * )) , il faut arriver à ce que « le spectre pernicieux de la génétique un peu diabolique se change en un cercle vertueux où l'individu participe à la collectivité en mettant ses données au service de l'analyse globale et qu'avec beaucoup de chances il lui revienne quelque chose » .
C. GARANTIR UN ACCÈS ÉQUITABLE DE TOUS AUX SOINS ET LIMITER LA MÉDICO-SURVEILLANCE
Un risque de discrimination surgit quant à l'égalité d'accès à des technologies qui ne seront pas toujours accessibles à tous financièrement ou territorialement. Certains patients plus riches pourront à leur frais faire séquencer leur tumeur, le plus souvent à l'étranger, alors que les autres continueront à être traités par des méthodes et des médicaments conventionnels moins efficaces. Certains se trouveront trop éloignés géographiquement des plateformes de thérapies ciblées ou bien, mal informés, iront trop tardivement dans les centres spécialisés.
On peut légitimement s'interroger sur les risques de discriminations potentielles inhérents à une forme de médecine concevant des traitements pour de petits groupes de populations différenciés en fonction de leurs caractéristiques génétiques et de leur propension à mettre en oeuvre ou non les changements de mode de vie recommandés en fonction des risques identifiés de survenance de telle ou telle maladie. Médecine de riches, médecine de pauvres ? La question peut en effet se poser, en termes d'information éclairée des patients, de niveau social, de localisation plus ou moins proche de centres médicaux performants.
1. Lutter contre l'exclusion due aux inégalités sociales
Le Pr Jean-Claude Ameisen (225 ( * )) faisait remarquer que « dans la dernière étude de l'INSEE, le premier facteur de mortalité de cancer, ce n'est pas la séquence génétique particulière de la tumeur, mais le niveau socio-économique des personnes atteintes de cancer, car il entraîne un retard d'accès au diagnostic, donc un retard de traitement. Or, cela ne se lit pas dans le génome. On a donc un risque de dire : vous savez tout sur vous, vous êtes responsable. Mais pour les deux millions d'enfants qui vivent sous le seuil de la pauvreté, ce n'est pas dans l'analyse de leur corps aujourd'hui que l'on peut imaginer et prédire les conséquences en termes de santé, dues à un environnement socio-économique ».
De même, selon le degré de proximité d'un grand hôpital et centre de recherche médicale, on a sans doute plus ou moins de chances d'être bien pris en charge et de ne pas risquer une erreur de diagnostic. Aux États-Unis, on évoque souvent à propos de la médecine personnalisée la notion de « customized medicine », celle qui se vend. Un effort de réflexion doit être fait pour déterminer ce vers quoi on se dirige, quel nouveau système de santé on se doit construire.
a. Les paradoxes de la stratification
Paradoxalement la médecine personnalisée, réinsère la personne dans un groupe et cette démarche de la ranger dans un groupe pour pouvoir mieux traiter, possède un pouvoir potentiel de discrimination. Un des aspects inégalitaires de cette médecine porte sur les groupes de patients qui ne seront pas traités, parce qu'ils sont non-répondeurs au traitement ou qu'ils risquent de pâtir d'effets secondaires insupportables. On ne peut pas leur donner pas le traitement parce qu'il n'est pas assez efficace d'un point de vue statistique et engendre trop d'effets secondaires.
Á un moment, on exclut les personnes pour lesquelles le résultat serait moins bon et qui devront se contenter des méthodes traditionnelles de soins plus lourdes et moins performantes. Aujourd'hui trop peu de cancers peuvent être soignés par thérapies ciblées et il faut au préalable améliorer l'offre de soins pour cette majorité de malades. L'offre médicale a besoin de concentration, de concertation et d'échanges inter-hospitaliers et entre centres de traitements. Aussi est-il nécessaire d'agir au niveau européen, où se trouve une masse critique de patients offrant des données suffisantes et une mutualisation des expertises pour réaliser les évaluations.
Á cet égard, vos rapporteurs souhaitent reprendre, à l'échelon national, une suggestion du Pr Axel Kahn (226 ( * )) : « par rapport aux inégalités territoriales, on observe que la médecine personnalisée génère des inégalités dans l'information et il serait intéressant de favoriser la création d'un site de références avec un accès mis à jour pour confronter les sollicitations dont le public est l'objet et les allégations réelles de santé. C'est un gros travail et les autorités publiques devraient en assurer la diffusion. On ne peut en effet éviter que la population soit démarchée, bien qu'un très petit nombre de tests soit réellement utile ». Un site public d'informations sur les avancées et les risques des nouvelles formes de thérapies proposées en clinique et aussi sur Internet serait fort profitable à tous nos concitoyens. Mme Emmanuelle Prada-Bordenave (227 ( * )) a d'ailleurs annoncé l'ouverture, en 2014, d'un site Internet à destination du grand public intitulé www.genetique-medicale.fr , construit en liaison étroite avec le Groupe stratégie en génétique et en diagnostic, qui permettra de transmettre un certain nombre d'informations sur les examens génétiques. C'est une excellente initiative.
b. Promouvoir l'éducation à la santé
Dans son rapport annuel pour 2012, l'Agence de la biomédecine indique qu'une enquête menée par ses soins auprès du grand public et des professionnels de santé sur les représentations de la génétique a montré le fort intérêt du public pour cette discipline, mais aussi une méconnaissance et des inquiétudes. Le niveau de connaissance est faible, la génétique étant perçue comme une discipline extrêmement complexe. Un renforcement de l'éducation à la santé s'avère donc nécessaire.
Cette politique peut prendre toutes sortes de formes et devra passer par les instances publiques, (telle que l'Agence de la biomédecine), ou par les associations. Une information, éventuellement très ciblée sur certaines populations défavorisées, une formation à l'école sur la protection de sa propre santé, dans les clubs sportifs, artistiques, culturels sont souvent beaucoup plus efficaces que des spots à la télévision. Il a été prouvé qu'un travail sur les menus à la cantine avait un impact important à la maison sur les familles. C'est un moyen pour les communes de toucher les populations les plus sensibles.
De nombreux chercheurs rencontrés par vos rapporteurs ont aussi insisté sur la nécessité d'une meilleure personnalisation des approches de prévention. Dans le cas de certains cancers où des facteurs environnementaux et comportementaux ont clairement été mis en évidence, le Pr Agnès Buzyn (228 ( * )) s'inquiète du mauvais résultat des dépistages nationaux organisés, malgré les campagnes de publicité lancées pour les encourager : 52 % de participation pour le cancer du sein, 30 % pour le cancer colorectal et cela n'augmente pas. Elle déplore également les nombreuses polémiques sur l'efficacité des dépistages et vaccins (hépatite B). Pour le vaccin contre le cancer du col de l'utérus (virus du papillome humain HPV) chez les jeunes filles, on ne dépasse pas les 10 %.
Notre pays semble très en retard et la défiance à l'égard des messages de prévention reste difficile à dépasser. Le taux de prévalence du tabagisme est à moins de 20 % au Royaume-Uni alors qu'il est à 33 % en France et qu'il ne baisse pas, contrairement à ce qui se passe chez nos voisins. Les facteurs socio-culturels sont nombreux et majeurs. Il faut travailler sur les comportements à risques et sur l'observance des traitements.
Il appartient à l'Institut de veille sanitaire (InVS) d'y veiller et de réfléchir aux moyens sociaux d'accompagnement des comportements de nos concitoyens qui ne sont pas en mesure socialement, économiquement et éthiquement de changer leur mode de vie. La médecine personnalisée , au-delà des thérapies ciblées, c'est une prise en charge personnalisée de l'individu. Elle doit aller des messages personnalisés de prévention primaire et secondaire à la personnalisation du parcours de soins en fonction de l'âge, du terrain, des niveaux de risques particuliers, et ensuite du traitement selon les aspects génétiques de la maladie.
Mme Catherine Vergely (229 ( * )) a suggéré lors du débat avec les associations de malades : « Il faut former le citoyen, et pas seulement dans la connaissance scientifique, mais aussi dans son esprit critique afin qu'il soit inclus dans les différentes étapes de décision, et qu'il ait une vision pertinente de son acceptation en tant que malade dans une société et dans des choix qu'on lui offre. Il faut respecter le citoyen et ses décisions. Il est absolument indispensable de l'intégrer dans l'ensemble des instances de stratégie et de décision de médecine et de recherche d'une façon globale, et de la médecine personnalisée plus particulièrement » .
c. Limiter la médico-surveillance
Responsabiliser les citoyens, leur transmettre un maximum d'informations sur leur propre santé, leurs comportements à risques, mieux accompagner le plus fragile, certes, mais encore faut-il aussi respecter leur liberté et leur choix de vie. On attend en effet une participation accrue des patients dans la prise en charge de leur propre santé. On leur explique qu'ils seront mieux soignés dès lors qu'on séquence son génome, qu'on cible leurs modes de vie plutôt que de donner un médicament standard qui ne convient peut-être pas. En contrepartie, le patient doit être encore plus coopératif et donner des informations sur lui-même et son environnement intime, et, en plus, une fois qu'on a trouvé le traitement approprié, il devra s'y conformer strictement. On voit ainsi poindre l'engrenage d'une société hygiéniste.
M. Didier Tabuteau (230 ( * )) met en garde contre le risque d'excessive surveillance : « cette médecine peut engendrer, cette fois en toute légalité, un véritable Big Brother médico-génétique. Les connaissances et données médicales produiront un dispositif par lui-même potentiellement dangereux. La médico-surveillance, avec ses avantages thérapeutiques, est un système de surveillance supplémentaire dans une société, et mérite attention en termes de préservation des libertés publiques et individuelles. Il vaut mieux s'organiser, juridiquement, administrativement, politiquement, au début du processus, que de se lancer tardivement à sa poursuite ».
Comment donc ajuster la prise en charge par l'assurance-maladie en respectant le libre arbitre des patients ? Il ne faudrait pas que l'assurance-maladie devienne une police des moeurs. Il existe une vraie tentation. La France avait jusqu'à présent échappé, au contraire du Royaume Uni, au débat sur les comportements de vie qui ont des conséquences sur la couverture par l'assurance-maladie (tabac, alcoolisme, sports et conduites à risques...). Or, le principe de fonctionnement de l'assurance-maladie française, qui était de prendre en charge dans tous les cas, vient de connaître une première exception : un arrêté du 9 janvier 2014 conditionne le remboursement du port d'un masque de traitement du syndrome de l'apnée du sommeil à la durée et à la régularité du port du masque par le patient, ce contrôle étant au surplus désormais effectué par télésurveillance.
Quelle sera la place du médecin découvrant que le résultat de la surveillance électronique à distance se soldera par un déremboursement ? C'est toute la question de la responsabilisation financière des patients, et même des médecins, qui est ainsi posée et la tendance déjà bien amorcée visant à un meilleur remboursement des malades vertueux. Ce débat, certes loin d'être illégitime puisqu'il est motivé par un souci d'économies pour la sécurité sociale sur des dépenses considérées comme non justifiées, impose de fixer des limites à la médico-surveillance et aux restrictions éventuelles de prise en charge des traitements médicaux.
Si la télésurveillance contribue certainement à améliorer l'ajustement des traitements, il ne faudrait pas pour autant qu'elle se transforme en une pénalisation de malades éprouvant des difficultés à les respecter et en une intrusion excessive dans leur vie intime. Il convient de rester vigilant dans la politique de prise en charge des traitements médicaux et de fixer des limites à la médico-surveillance. Il faudra donc veiller à maintenir un juste équilibre entre la pression collective engendrée par l'acceptation de traitements ciblés et le maintien de la solidarité de notre système de santé publique. Il faudra veiller à maintenir un juste équilibre entre la pression collective engendrée par l'acceptation de traitements ciblés et le maintien de la solidarité de notre système de santé publique.
2. Informer les citoyens
Si l'on souhaite que les changements de notre système de santé engendrés par les progrès technologiques s'effectuent sans heurts, il faut les accompagner d'une politique d'éducation à la santé, de débats-citoyens. La réussite des innovations thérapeutiques de la médecine personnalisée ne viendra pas seulement des scientifiques ou des industriels ; elle ne deviendra massive que grâce à une appropriation par les citoyens et une demande de leur part d'y participer totalement.
La prise en compte du patient dans sa singularité doit être remise au centre de notre système médical. La dimension européenne, la mise en garde contre les pratiques mercantilistes sont essentielles. Les malades ont un rôle à jouer ; c'est absolument évident, Il en va de même des associations de malades, qui ont souvent été de véritables moteurs de la recherche sur le sida ou sur les maladies rares, notamment. La parole des malades et la façon d'orienter cette prise de parole par les patients sont d'une grande importance.
a. Organiser des débats publics
Compte tenu des bouleversements induits par la médecine personnalisée, il faut organiser rapidement des débats publics pour en expliquer les avancées, les risques et les enjeux, et ce sur plusieurs années pour préparer l'opinion et éviter une, importante déconvenue éthique, populationnelle et même en termes de santé publique. L'opinion publique n'est pas prête aujourd'hui à voir prescrire, à 25 ans par exemple, toute une batterie de tests sur les risques de maladies les plus graves. Il est important de savoir ce qui est acceptable par le corps social afin d'éviter des effets délétères.
L'Agence de la biomédecine constitue certainement le rouage public le mieux adapté pour lancer cette communication et elle a d'ailleurs bien pris la mesure du manque d'information du public sur la génétique. Dans son dernier rapport annuel, elle expose les diverses initiatives entamées pour y remédier : « Pour la première fois en 2012, l'Agence a présenté au public, lors d'une conférence de presse institutionnelle, les enjeux de la génétique médicale en termes de santé publique et de prise en charge des patients confrontés à une maladie génétique. Elle a également mené deux actions préparatoires à la définition des axes d'une communication grand public sur la génétique. D'une part, une analyse des productions de la presse sur le thème de la génétique postnatale et prénatale de 2010 à 2011, notamment lors de la mise en place du nouveau dépistage de la trisomie 21, afin de comprendre quelles images les journalistes en forgent ».
Ces différentes initiatives, tout comme la mise en place d'un site Internet grand public dédié à la génétique, sont à porter au crédit de l'Agence et sont un excellent exemple de ce qui devrait être complété dans d'autres arènes grand public en élargissant les débats à l'ensemble des enjeux éthiques et socio-économiques de la médecine personnalisée énumérés dans le présent rapport.
De même le Comité consultatif national d'éthique pour les sciences de la vie et de la santé a un rôle important à jouer car aux termes de l'article L.1412-1-1 du code de la santé publique introduit par la loi relative à la bioéthique de 2011, il organise un débat public sous forme d'états généraux sur les projets de réforme sur les problèmes éthiques et les questions de société soulevés par les progrès de la connaissance dans les domaines de la biologie, de la médecine et de la santé.
On peut reprendre à cet égard les propositions faites par MM. Jean-Yves Le Déaut et Claude Birraux, députés, membres de l'OPECST dans leur rapport de janvier 2012 sur « L'innovation à l'épreuve des peurs et des risques » (231 ( * )) . Ils préconisaient l'organisation de débats publics par la Commission nationale du débat public, au travers de conférences de citoyens et de comités d'instance spécialisés où sont représentés associations et syndicats. Parmi les recommandations du rapport, on retiendra plus particulièrement les suivantes :
« - s'inspirer des exemples de débat public mis en place à l'étranger avec notamment un usage massif des NTIC, par la création de sites Internet thématiques participatifs, mis à jour régulièrement et effectuant un suivi des actualités scientifiques, par la mise en place de conférences de citoyens, ... ;
- développer au sein des structures concernées une cellule de veille des réseaux sociaux (Facebook, Twitter, blogs,...) afin de prendre le pouls de la société et de pouvoir répondre aux interrogations dès qu'elles apparaissent ;
- encourager les émissions scientifiques sur les chaînes publiques de télévision et créer, suite à l'appel d'offre du CSA, une chaîne de télévision destinée à promouvoir la science et la culture scientifique, par exemple par la présentation des grands enjeux scientifiques d'aujourd'hui dans un langage clair et accessible et dans un format interactif ;
- créer, au sein des universités et des organismes de recherche, des équipes de liaison avec les associations comme celles de patients dans le domaine médical, afin de leur proposer des services d'expertise et de conseil sur les thématiques sociétales ».
Les associations de malades ont bien évidemment un rôle-clé à jouer, qu'elles remplissent déjà fort bien sur les maladies objet de leur domaine d'intervention, mais qu'il faut aussi déborder pour qu'elles participent à des débats plus larges concernant toutes les autres pathologies et sur les enjeux éthico-économiques des bouleversements annoncés de notre système actuel de santé.
Tous ces débats ont besoin des medias qui doivent être correctement informés pour se les approprier et en faire ressortir les aspects positifs et négatifs plus clairement possible et ce à destination de différents publics.
On peut conclure avec M. Didier Tabuteau (232 ( * )) que « selon que le débat public sera bien porté, la formation du public bien effectuée, que les connaissances seront bien appropriées par une bonne partie de la population, et aussi d'ailleurs par les professionnels de santé, cette évolution de la médecine peut être le pire ou le meilleur. Il existe un risque de basculement. Cela peut conduire à une rationalisation, une amélioration du système ou au contraire à une dérive hypocondriaque, une sélectivité et une inégalité croissante. Et ce débat n'est pas simple à mener, or c'est une échéance importante ».
L'éducation du citoyen reste la solution, il doit savoir ce qu'il en est et comprendre ce que tout cela veut dire. M. Didier Tabuteau précise : « il serait par ailleurs intéressant de favoriser la création d'un site de références avec un accès mis à jour pour confronter les sollicitations dont le public est l'objet et les allégations réelles de santé. C'est un gros travail et les autorités publiques devraient en assurer la diffusion. On ne peut en effet éviter que la population soit démarchée, bien qu'un très petit nombre de tests soit réellement utile ».
b. Accompagner les patients : l'importance du conseil génétique
Lors de l'audition publique du 25 juin 2013 les associations de malades, ont toutes insisté sur la nécessité de recentrer notre système médical sur l'accompagnement personnalisé du malade et l'approche globale de la personne. Ce n'est pas seulement un parcours de soins qu'il faut mettre en oeuvre, mais un parcours de santé qui offre une approche globale allant du médical au social, avec des référents parcours de santé afin que le patient se sente en confiance. Il faut qu'il dispose de relais médicaux accessibles et disponibles et qu'il n'ait jamais l'impression d'être exclu de son propre parcours de santé.
Ce nouveau modèle d'organisation des soins, allant du dépistage aux séquelles des traitements administrés, est d'autant plus nécessaire que certains patients ne sont pas éligibles aux thérapies ciblées. Lors de l'annonce d'un diagnostic, il faut que le praticien soit capable de délivrer le message qui accompagnera la découverte d'une mutation délétère et explique à la personne le suivi indispensable.
Le professionnel de santé concerné par la génétique deviendra un conseiller dans des situations extrêmement variées. Il lui faudra gérer de l'information produite à un temps T sans savoir si elle sera utile un jour. Les associations de patients pourraient apporter une contribution importante sur la façon de prendre en charge cette question et notamment celle des limites du « droit de ne pas savoir ».
La relation médecin-patient dans le cas d'un diagnostic de maladie génétique exige un processus d'accompagnement en plusieurs phases, qui a été encadré par les recommandations de l'Agence de la biomédecine et dans lequel l'écoute et le conseil par le généticien, le spécialiste de la maladie, le psychologue ont un rôle important afin de nourrir la réflexion du patient. Puis une fois la décision prise en fonction des informations reçues, il reste l'étape délicate du rendu du résultat, et enfin le suivi personnalisé. Ce processus d'accompagnement exige du temps et des compétences spécifiques de la part des professionnels de santé, comme l'a expliqué le Pr Florent Soubrier (233 ( * )) .
3. Impliquer les associations de malades dans les décisions
L'avenir de la médecine personnalisée passe forcément d'abord par l'implication des patients, leur participation à des essais thérapeutiques et l'application des nouvelles thérapies et combinaisons de médicaments. Le meilleur exemple de réussite d'une médecine innovante initiée et prise en mains par les malades et leurs associations est celui des trithérapies qui ont ouvert la voie de la guérison aux malades du sida après tant d'années sans espoir.
Le Pr Thomas Tursz (234 ( * )) l'a rappelé : « ce sont les patients, qui l'ont orienté contrairement aux intérêts des laboratoires, qui voulaient pousser leurs molécules individuellement sans tester de combinaisons de molécules et luttaient pour des prix relativement élevés...Ce sont les patients qui savaient s'organiser en groupes de pression qui ont changé la situation, et les trithérapies actuelles sont donc arrivées sur le marché relativement vite. C'est une leçon très forte sur la façon dont la médecine personnalisée peut se développer en France avec rapidité, efficacité, équité, et pour un surcoût acceptable pour la collectivité » .
Les exemples de succès des nouvelles thérapies largement portés par les associations de patients, souvent contre l'avis des industriels, sont nombreux. On peut encore citer le cas de l'oncologie en pédiatrie, avec 20 % de guérison il y a trente ans, 80 % maintenant, obtenus aussi parce que les parents se sont organisés très vite avec les médecins pour demander des essais thérapeutiques, très tôt dans le cours de la maladie, et non à la fin quand tout avait échoué. Comme l'a souligné le Pr Thomas Tursz, l'action déterminante de l'UNAPECLE (235 ( * )) et de sa secrétaire générale, Mme Catherine Vergely, a été déterminante vis-à-vis des industriels effrayés à l'idée d'utiliser un nouveau médicament chez l'enfant parce qu'ils pensaient le marché petit et peu rentable. Ils craignaient qu'une toxicité grave empêche le développement d'un blockbuster utilisable et très rentable chez l'adulte.
Mme Laurence Thiennot-Herment (236 ( * )) a rappelé le rôle atypique et majeur joué par son association dans le domaine de la recherche, et aussi sur la partie médicale et sociale, pour les maladies rares. Pour faire avancer la recherche de thérapeutiques innovantes dans ce secteur délaissé par les géants de l'industrie, l'AFM est allée jusqu'à créer son propre site de production de thérapie génique : « nous avons pu avec les moyens du Téléthon engager un milliard d'euros dans la recherche depuis sa première édition, donc en 27 ans, et 660 millions d'euros dans l'aide aux malades. Cela nous a fait jouer un rôle important notamment à travers les laboratoires que nous avons pu créer, Généthon, I-Stem, Atlantic Gene Therapies, l'Institut de myologie, laboratoires dans lesquels la gouvernance majoritaire est donnée aux malades et aux parents de malades » .
C'est pourquoi, vos rapporteurs soutiennent entièrement son souhait « qu'à tous les niveaux dans cette chaîne de santé, du diagnostic jusqu'au prix du médicament, les patients soient présents en tant que ?partenaires? » et en font une des recommandations prioritaires du présent rapport.
C'est bien en effet l'AFM, qui a lancé ce chantier immense de la carte du génome humain, financé grâce au Téléthon, et prouvé que la thérapie génique était la seule réponse possible à l'absence de médicaments sur le marché pour traiter les maladies rares. Les activités génomiques de Généthon ont été reprises en 1998 par le Centre national de génotypage au Génopole d'Evry, pionnier de cette d'activité en France.
Ce sont encore les associations qui ont pris conscience les premières de l'indispensable prise en compte de la dimension européenne, car elles sont largement présentes dans les consortiums et réunions européens. Ainsi, M. Yann Le Cam, directeur général d'EURODIS (237 ( * )) et vice-président d'EUCERD (238 ( * )) , rappelle-t-il : « a ujourd'hui la question n'est pas celle de la valeur du médicament, mais plutôt celle de la capacité à financer, pour le système de santé, ces nouveaux traitements ciblés, qu'ils soient personnalisés ou stratifiés. Pour cela, notre réponse est de promouvoir une nouvelle approche : plutôt plus d'Europe que moins d'Europe. Pas de repli national pour réfléchir sur de toutes petites populations, très souvent sans avoir l'expertise sur ces maladies ou sur la valeur de ces produits, mais plutôt de travailler au niveau européen, où l'on trouve une masse critique de patients pour avoir des données suffisantes, et une mutualisation des expertises pour réaliser les évaluations collectives » .
Il est d'ailleurs bien regrettable qu'en France les associations de malades ne soient pas aussi fortes qu'aux États-Unis, où elles disposent d'une puissance suffisante pour faire évoluer l'innovation thérapeutique, aussi bien contre le monde industriel que les pouvoirs publics. L'apport des nouvelles connaissances issues de la génétique montre aussi le grand intérêt qu'elles auraient à élargir leur champ d'intervention et à dépasser le regroupement autour d'une ou deux maladies spécifiques. Á l'heure où l'on commence à reconnaître une origine génétique à de nombreuses maladies, particulièrement en oncologie avec la nouvelle nosologie des cancers, elles ont un rôle déterminant à jouer pour entrainer la recherche industrielle dans la voie des combinaisons de médicaments, notamment grâce à la coordination de l'action de leurs membres.
On peut conclure avec les propos de Mme Martine Bungener (239 ( * )) : « cette nouvelle médecine va s'exprimer sous forme de probabilité. Qui est apte aujourd'hui pour traiter ce type d'information ? Comment feront les médecins eux-mêmes, quelle capacité auront-ils à traiter cette information ? Les associations de malades ont un rôle à jouer, et on l'a vu dans d'autres situations, pour justement aider à traiter cette information et à la diffuser. On est bien là dans une dimension qui doit mobiliser l'ensemble des associations, qui s'inscrit dans leurs fonctions. Informer les malades, elles en ont les compétences, elles savent le faire... Le GRAM milite depuis sa création pour montrer que le dialogue avec les associations est vraiment une partie prenante de la reconstruction de ces mécanismes de formation de la confiance collective vis-à-vis de la recherche et de la médecine, et nous sommes aujourd'hui face à un enjeu majeur avec les nouveaux traitements et ces nouvelles formes de médecine ».
CONCLUSION
La médecine personnalisée bouleverse la conception traditionnelle de la médecine. Elle opère un véritable changement de paradigme qui influe considérablement sur l'évolution de notre système de santé, sur la façon dont nous l'organiserons dans l'avenir. Il faut s'y préparer sans tarder. Induisant une véritable révolution sociétale, cette nouvelle approche de la médecine doit être accompagnée d'une politique d'éducation à la santé, de débats publics permettant aux citoyens d'en comprendre les apports et d'en apprécier les bienfaits et les contraintes.
Ce nouveau mode de prise en charge du patient présente des avantages dans la mesure où il s'adapte à la personne et met la singularité de l'individu au coeur du système de santé. Pour autant, la médecine personnalisée, en grande partie fondée sur une forme de « transparence de l'homme », pourrait vite apparaître déshumanisée et, en quelque sorte, dépersonnalisée, confiant à des outils techniques performants le soin de tout voir, de tout mesurer, donc, de tout prévoir, tout dépister, tout prévenir, tout traiter ou tout remplacer. Verra-t-on émerger « un corps numérique à côté du corps réel » se demandait le Pr Hervé Chneiweiss ? Qu'en sera-t-il de la singularité de la personne par rapport à un profil type dans lequel la technologie l'a rangé ? « Sera-elle réduite à un point dans un nuage de points ? » s'interrogeait le Pr Jean-Claude Ameisen.
Si de grandes avancées scientifiques ont déjà été obtenues dans les champs de la recherche génomique et protéomique ainsi que de la biologie moléculaire, de nombreux obstacles restent à lever. L'enjeu majeur auquel les médecins et les scientifiques devront répondre portera sur la capacité de savoir quels gènes ou quels mécanismes biologiques prédisposent un patient à développer telle ou telle pathologie et sur la possibilité de transformer cette compréhension en stratégie de prévention ou de traitement. L'analyse et la classification des pathologies devront s'adapter au développement de la connaissance génomique.
Des ensembles de symptômes ou de données cliniques attribués aujourd'hui à une seule et même maladie pourraient se révéler être, en fait, le résultat de variations génétiques différentes, requérant des approches thérapeutiques elles aussi différentes.
La nouvelle classification des maladies en fonction de leurs aspects génétiques et moléculaires, en cancérologie notamment, constitue un tournant et un défi majeurs pour notre système de santé et invite à s'interroger sur son impact organisationnel ou économique. Selon que les nouvelles thérapeutiques resteront à la marge, très spécialisées, localisées ou qu'elles concerneront progressivement de 5, 10 ou 30 % du système de santé, elles produiront des bouleversements plus ou moins considérables sur notre système solidaire de santé publique. Il ne faudrait pas oublier qu'aujourd'hui encore l'innovation ne concerne qu'une très petite minorité de patients, que trop peu de cancers peuvent être soignés par thérapies ciblées : moins de 5 % des patients français ont actuellement accès à un essai thérapeutique ou à une thérapie ciblée au cours de leur vie.
De nouveaux métiers, de nouveaux acteurs majeurs de la lutte contre des pathologies incurables, les biologistes du génome et les bio-informaticiens, sont apparus indispensables pour présenter les résultats multiples des analyses génomiques sous forme visible, lisible et interprétable pour les médecins. Aucun médecin n'est capable d'interpréter un résultat génomique seul si on ne lui explique pas quel médicament il faut utiliser en fonction des anomalies génétiques et moléculaires qui s'accumulent au sein de la même cellule. On a longuement souligné la nécessaire adaptation à cette évolution des études médicales vers un travail en équipes pluridisciplinaires.
Une utilisation optimale des nouvelles technologies de l'imagerie et des outils de diagnostic fait également partie de ce défi. Alors que de nouveaux biomarqueurs sont découverts, le rythme auquel ils sont qualifiés et validés est encore lent. L'avenir sera à la combinaison de drogues entre elles pour contourner les résistances et aussi à une mise en commun des savoirs des grands laboratoires de l'industrie pharmaceutique.
Des normes de gestion des données génétiques et des dossiers médicaux personnels devront être définies pour permettre le déploiement de cette technologie et garantir un recueil et une interprétation standardisés des données cliniques. Aussi, l'accès sécurisé à ces données personnelles et identifiantes et leur protection est-il un enjeu majeur, tant au plan éthique et sociétal qu'au plan économique. La participation active de la France à l'élaboration et à l'application d'instruments internationaux visant à protéger ces données d'une exploitation en dehors de la sphère scientifique et médicale, est essentielle.
La médecine personnalisée transforme et transformera inéluctablement le champ de la prévention et du soin. Au-delà de l'oncologie, elle devrait être plus utilisée dans la prévention des effets des traitements en adaptant mieux les posologies au métabolisme des patients, quelles que soient les pathologies. Si elle place le patient au centre du dispositif de prévention et de soin comme le préconise la « Stratégie nationale de santé » et le rapport de la commission « Innovation 2030 », elle accroît le nombre et la technicité des intervenants sur la santé, les informations dont le patient doit disposer non seulement sur lui-même, mais aussi sur les membres de sa famille.
La France dispose d'un système centralisé de santé, que beaucoup de pays lui envient, avec des plateformes de génétique moléculaire sur tout le territoire offrant un égal accès gratuit à tous, mais elle souffre de grandes faiblesses au niveau informatique en termes de personnels formés, d'outils d'analyses, de capacités de calcul et de stockage qui nécessitent des efforts d'investissement majeurs, si elle entend espérer rester compétitive. Notre pays a donc tout intérêt à accroître sa participation aux initiatives européennes et internationales ; la coopération internationale sera un facteur d'accélération, notamment quant aux progrès à accomplir sur les maladies rares et en oncologie.
La réussite du passage de l'ère actuelle d'une médecine semi-stratifiée à celle d'une réelle médecine personnalisée impliquera un travail considérable d'information des citoyens, une mise en place de mesures de prévention ciblées, expliquées et comprises, une adaptation du monde médical, afin de veiller à éviter toute panique ou tout espoir irraisonné, tout syndrome du Dr Knock ou encore toutes fausses promesses de guérison. La médecine est certes une science, mais avant tout un art de la relation avec le patient, et elle doit à la fois intégrer et humaniser la technologie.
C'est pourquoi l'information des citoyens et l'organisation de débats-citoyens sont importantes, et l'OPECST ne manquera pas d'y participer aux côtés de la communauté scientifique. La médecine personnalisée ne sera un véritable facteur de progrès que si elle réduit les inégalités d'accès aux soins, territoriales, sociales et culturelles, que si elle accroît l'intégration au sein de la société des personnes qui souffrent de handicap. Elle n'atteindra ses objectifs d'optimisation de notre système de santé que si elle permet au malade de prendre en charge sa propre pathologie, de se retrouver en possession de toute l'information médicale qui le concerne. Elle doit avant tout parvenir à reconstruire un parcours de confiance humanisé, équilibré, sans examens médicaux redondants, entre, d'un côté, la médecine de ville et, de l'autre, l'hôpital et ses plateformes spécialisées d'analyses biologiques.
RECOMMANDATIONS
Préparer les institutions françaises au changement de paradigme induit par la médecine personnalisée dans l'approche de la maladie et du traitement :
- Intégrer ce changement dans la stratégie nationale de santé.
- Définir une méthodologie et une stratégie globale pour s'y préparer incluant l'ensemble des acteurs de la politique de santé notamment les personnels de santé et les associations de malades.
- Préciser les parcours de santé et le rôle du médecin référent afin de mieux articuler les liens entre les généralistes et les centres pratiquant cette médecine personnalisée.
- Généraliser l'informatisation des dossiers médicaux et en accélérant la mise en service du Dossier Médical Personnel (DMP).
Encourager la recherche et le développement :
- Reconduire les financements des projets de recherche dans le cadre des investissements d'avenir, notamment en oncologie.
- Créer une plateforme nationale de séquençage à très haut débit de recherche et de diagnostic qui centralise les cas spécifiques pour éviter « l'errance diagnostique » et qui conserve les variants génétiques pertinents identifiés par toutes les plateformes.
- Réactiver le développement de nouvelles molécules.
- Évaluer les potentialités des anciennes molécules en recourant aux nouvelles techniques de criblage de masse ( screening ).
- Favoriser le développement de la nano-médecine.
- Veiller à ce que la convergence des technologies Nano-Bio-Info-Cogno (NBIC) conduise à la création de nouveaux cursus.
Réformer la formation des personnels de santé :
- Appliquer les dispositions des articles 39 à 41 de la loi n° 2013-660 relative à l'enseignement supérieur et à la recherche du 22 juillet 2013 pour accélérer la réforme des études de santé.
- Poursuivre la réforme des études de médecine afin de mieux préparer les futurs professionnels à un exercice en équipes, à une approche pluridisciplinaire mettant l'accent sur les relations humaines et sociétales.
- Encourager le décloisonnement des disciplines scientifiques dans les cursus universitaires.
- Diversifier les parcours pour favoriser les doubles cursus sciences « dures » ou sciences humaines et sociales et médecine-biologie, en intégrant différentes disciplines des sciences et des mathématiques et biostatistiques notamment.
- Créer des cursus formant aux nouveaux métiers de biomédecine : bio-informaticiens, biostatisticiens, biochimistes, ingénieurs et techniciens pour analyser, interpréter et savoir conserver les données de séquençage.
- Proposer une formation professionnelle continue aux personnels de santé mieux adaptée aux évolutions induites par la médecine personnalisée.
- Assurer une formation en informatique permettant à tous les soignants de trouver et d'extraire la bonne donnée pour effectuer leurs choix en connaissance de cause.
Aider au développement des traitements ciblés :
- Maîtriser le coût des tests génomiques et des nouvelles thérapies ciblées, afin de conserver l'égal accès de tous à un coût supportable pour l'assurance-maladie.
- Intégrer les analyses coût-efficacité au cours des premières phases des essais cliniques et du développement.
- S'inspirer du modèle maladies rares pour les essais cliniques tout en assurant la sécurité des patients.
- Harmoniser le processus d'assemblage du test compagnon et de la molécule.
- Encourager une concertation entre les industriels et les agences de régulation pour permettre le développement et l'évaluation conjointe des molécules et des tests compagnons.
- Diminuer la durée des essais préalables à l'autorisation de mise sur le marché (AMM) pour enregistrer plus rapidement les nouveaux médicaments tout en privilégiant les AMM conditionnelles.
- Favoriser la création de regroupements ( pools ) de brevets pour réduire les coûts et encourager les coopérations entre les différents partenaires de la recherche.
Informer sur la valeur prédictive des tests génétiques :
- Créer rapidement le site Internet grand public dédié à la génétique prévu par l'Agence de la biomédecine pour confronter les sollicitations dont le public est l'objet et les exigences réelles de la santé.
- Renforcer l'information du public sur les modalités légales d'accès aux tests génétiques en France (240 ( * )) .
- Assurer une veille institutionnelle sur le recours aux tests génétiques en libre accès sur Internet.
- Effectuer des mises en garde par les instances habilitées tant sur la fiabilité des tests proposés via Internet que sur l'usage possible des résultats de ces tests, qui ne sont pas couverts par le secret, voire l'anonymat.
- Informer sur les risques d'erreurs, voire de piratage des données lors de l'achat de tests par Internet.
- Sensibiliser les citoyens par des campagnes d'informations adaptées sur les limites de la fiabilité et de la valeur prédictive des tests génétiques proposés par des sociétés commerciales.
- Insister auprès du public sur la nécessité de recourir à des consultations génétiques spécialisées auprès de professionnels médicaux pour toute demande de test génétique dès le premier entretien, afin qu'il puisse être pleinement informé et conseillé sur les implications et le résultat attendus.
- Ratifier le protocole additionnel à la Convention sur les Droits de l'Homme et la biomédecine relatif aux tests génétiques à des fins médicales, signé par la France en décembre 2011.
Assurer un égal accès de tous les citoyens aux nouvelles thérapies :
- Placer le patient au centre du dispositif de soins et en veillant à sa prise en charge complète dans un parcours de soins allant du diagnostic au suivi médical terminal.
- Lutter contre les discriminations et les inégalités territoriales pour garantir un égal accès aux nouvelles thérapies.
- Fixer des limites à une médico surveillance notamment par voie de télémédecine, dès qu'elle constitue une intrusion dans la vie personnelle des patients.
- Conserver un système de santé publique fondé sur la solidarité et limitant strictement les restrictions de prise en charge financière des traitements.
- Intégrer les associations de malades, en tant que « partenaires », à tous les niveaux de la chaîne de santé, du diagnostic au prix du médicament.
Protéger les données personnelles de santé :
- Renforcer la confidentialité des données de santé et en sécurisant le futur dossier médical personnalisé.
- Sensibiliser les personnels de santé à cette protection et en vérifiant les consentements donnés pour toutes utilisations.
- Renforcer les moyens de la CNIL pour assurer la confidentialité et la circulation des données de santé.
- Encourager la création d'un répertoire européen public fiable des mutations de l'ADN.
- Rendre accessibles aux chercheurs les grandes banques de données anonymisées et en favorisant le partage scientifique des données.
- Encadrer la création de biobanques privées réunissant les données et analyses génétiques de malades, dont la divulgation et la vente doivent être interdites.
REPRISE DE RECOMMANDATIONS ANTÉRIEUREMENT PROPOSÉES PAR L'OPECST (241 ( * ))
- Organiser une veille conjointe de l'Agence de la biomédecine et de la CNIL, sur l'utilisation de données de santé dans le but de définir par ordinateur les caractéristiques d'une personne.
- Renforcer les procédures de codage et de sécurisation des bases de données de l'assurance-maladie et des autres banques de données médicales.
- Assurer la traçabilité de l'accès des personnels habilités à connaître ces données.
- Améliorer la formation et la sensibilisation des personnels médicaux au respect du secret médical et à la délivrance de données médicales.
- Assurer la sécurité de l'hébergement et du transfert des données médicales.
- Soumettre à des conditions strictes d'agrément les hébergeurs de données de recherches sur de grandes cohortes.
- Participer activement à la négociation et à l'adoption d'une convention internationale pour encadrer la circulation des informations médicales.
Assurer l'information des citoyens sur la médecine personnalisée
- Organiser des débats publics sur les enjeux et risques de la médecine personnalisée : débats, conférences de citoyens avec des professionnels de santé, des chercheurs, des associations de malades.
- Mettre en place une éducation à la santé, des formations sur la protection de la santé dans les collèges et lycées ainsi que dans les clubs sportifs, artistiques et culturels.
- Pallier les insuffisances actuelles de la médecine préventive par des campagnes médiatiques plus adaptées et mieux ciblées sur les groupes de personnes à risques.
- Faciliter l'action des associations de malades auprès du grand public et en les encourageant à faire connaître leurs travaux au-delà de leurs membres.
EXAMEN DU RAPPORT PAR L'OFFICE
L'OPECST s'est réuni le 21 janvier 2014, sous la présidence de M. Bruno Sido, président, pour examiner le présent rapport d'information.
« M. Jean-Yves Le Déaut, député, premier vice-président de l'OPECST . Le sujet de ce rapport démontre encore une fois que l'OPECST remplit bien sa mission d'exploration des sujets scientifiques en avant-garde pour identifier et analyser les problèmes d'ordre juridique ou éthique. Alors que la notion de « médecine personnalisée » était encore mystérieuse au moment du lancement du rapport, elle s'est imposée au cours des derniers mois dans le champ des préoccupations des pouvoirs publics, notamment, en octobre dernier, avec le rapport de Mme Anne Lauvergeon, « Innovation 2030 », qui fait de cette médecine l'un des sept axes stratégiques d'innovation pour la France. L'étude de l'Office était donc prémonitoire.
Avec le terme «personnalisée», on est confronté à la difficulté d'une traduction de l'anglais, avec la tendance des scientifiques à simplement franciser les termes plutôt que de les traduire. Le rapport de Mme Anne Lauvergeon évoque plus judicieusement la notion de médecine « individualisée », terme plus correct en français, mais qui pose la question de ce que serait la médecine si elle ne visait pas justement depuis toujours à soigner chaque personne pour les maux dont elle souffre. Il s'agit en fait aujourd'hui, d'une médecine «stratifiée » plus que d'une médecine véritablement «individualisée» pour des raisons évidentes d'économie d'échelle dans la production des tests et des médicaments.
M. Alain Claeys, député, rapporteur. La Commission des affaires sociales de l'Assemblée nationale a saisi l'OPECST d'une demande d'étude sur les enjeux scientifiques, technologiques, éthiques de la médecine dite personnalisée. Dans l'étude de faisabilité, nous indiquions que la définition de cette médecine était malaisée. Toute bonne pratique médicale est, par essence, « personnalisée. Chaque expert a sa définition de cette nouvelle notion. Pour le Pr Axel Kahn, mieux vaudrait parler de médecine de prévision. Quant à la Commission « Innovation 2030 », elle évoque le concept de médecine individualisée car la manière de se soigner en 2025 sera différente de l'actuelle.
Nous avons organisé le rapport en trois parties : d'abord l'étude du changement de paradigme dans l'approche de la maladie et du traitement, puis les implications pour le secteur de la santé, et, enfin, les garanties à donner aux citoyens face aux enjeux éthiques de la médecine personnalisée.
Le changement considérable de paradigme dans l'approche de la maladie et du traitement résulte de progrès scientifiques et technologiques importants. L'analyse biologique expérimentale et appliquée est passée récemment à une approche globale. Les techniques de séquençage du génome ont connu une évolution de grande ampleur en termes de rapidité et de coûts. La première version du génome entier a été publiée en 2001 et, à partir de 2006, de nouvelles techniques de séquençage plus rapides apparaissent. Elles permettent aujourd'hui de séquencer plusieurs génomes en 24 ou 48 heures. La baisse des coûts est drastique : d'environ 2,7 milliards de dollars sur 15 ans pour le premier génome humain à un montant compris entre 1 000 et 3 000 dollars aujourd'hui.
En France, ces dernières années, on constate une accélération dans l'évolution des équipements des hôpitaux, grâce au financement des projets « équipements d'excellence » (EQUIPEX) dans le cadre du grand emprunt ; s'y ajoutent vingt-huit plateformes de l'INCa, dédiées aux traitements du cancer. Les infrastructures françaises en génomique sont variées et réparties sur tout le territoire. Cependant se pose la question de la création d'un centre de référence d'intérêt national capable d'effectuer une analyse génétique globale grâce à des séquenceurs à très haut débit de troisième génération, avec des moyens d'analyse et de stockage importants. Il s'agit là d'un problème de souveraineté nationale pour éviter de sous-traiter l'analyse de génomes entiers à l'étranger et sauvegarder en France les données y afférentes. Certains experts ont explicitement fait référence à une extension possible, à l'horizon 2015, des capacités de l'Institut de génomique du CEA, qui travaille en coopération avec l'INSERM et participe à l'effort de recherche dans les sciences du vivant dans le cadre de l'Alliance pour les sciences de la vie et de la santé (AVIESAN).
M. Jean-Sébastien Vialatte, député, rapporteur. S'agissant des autres techniques, on observe une extension de l'usage des puces à ADN qui, pour celles de dernière génération, ont la capacité de contenir et d'analyser environ 50 000 polymorphismes. Ces puces sont développées en production industrielle et le passage du laboratoire à l'industrie s'est accompagné d'une amélioration de la qualité ; ce sont des dispositifs simples. Dans un avenir proche, un projet de carte d'identité métabolique permettra d'adapter la posologie des médicaments aux patients. La diversité des thérapeutiques personnalisées est grande, telles la thérapie génique et la thérapie cellulaire ; cette dernière bénéficie désormais, en France, de la modification de la loi de bioéthique de 2011, car la loi du 6 août 2013 pose le principe d'autorisation de la recherche sur les cellules souches embryonnaires sous le contrôle de l'Agence de la biomédecine.
C'est en oncologie que la médecine personnalisée se développe depuis la fin des années 1990 avec l'émergence des thérapies ciblées et l'usage de l'Herceptine dans le cancer du sein. En France, l'INCa joue un rôle moteur dans le développement des thérapies ciblée, grâce aux 28 plateformes et aux 17 thérapies ciblées qui sont accessibles à tous les patients. La structuration française est unique au monde, puisque tous les patients du territoire, où qu'ils soient traités, ont leur tumeur testée et séquencée pour détecter les anomalies génétiques. Cela concerne les cancers colorectaux, ceux du poumon, les leucémies, les cancers du sein. On estime qu'entre 90 ou 100 nouveaux médicaments ciblant des anomalies spécifiques seront disponibles dans les années à venir. Cependant, selon les experts, si l'on entre bien dans l'ère de la médecine personnalisée, on pratique actuellement une médecine stratifiée. On se dirige vers une nouvelle nosologie fondée sur le génome tumoral. Désormais certains oncologues se demandent si donner le traitement ciblé correspondant à la détermination du profil moléculaire de la tumeur chez chacun des patients atteints d'un cancer, est une stratégie plus efficace que la détermination des traitements à partir de la localisation organique de la tumeur, comme cela se fait actuellement à l'hôpital.
Par ailleurs, des tentatives de modélisation des traitements par l'utilisation d'un avatar personnalisé de cancer offre la possibilité d'essayer plusieurs combinaisons de molécules et même de faire des erreurs, avant de commencer véritablement le traitement. C'est la direction que prennent plusieurs équipes de recherche. Cette technique du double biologique, en général un rongeur, ne se limite pas au cancer, elle est exploitée pour analyser in vivo le cas de personnes souffrant ou risquant de souffrir de diabète de type 1 .
Les techniques de médecine personnalisée sont également utilisées pour comprendre les inégalités devant les maladies infectieuses ou les raisons de non réponse à un vaccin. Pour le Sida, on constate que chaque mutation du virus signifie une résistance à une molécule ; on personnalise alors le traitement en fonction du virus et du patient.
S'agissant des maladies chroniques, de nombreux gènes responsables du diabète et de l'obésité ont été identifiés. Il est possible d'adapter le traitement en fonction des caractéristiques génétiques du patient. Cela a abouti à définir des profils dont les particularités permettent de mieux savoir ce que seront les réponses au traitement et les préconisations. On est donc conduit à des soins plus personnalisés du diabète.
Des évolutions relevant de la médecine personnalisée sont à l'oeuvre dans les maladies rares qui représentent d'importants enjeux de recherche, car elles servent de modèle de développement de médicaments. Environ 80 % de ces maladies sont d'origine génétique, et l'explosion actuelle des techniques diagnostiques permet une meilleure approche de leur origine, et parfois de leur traitement.
Les maladies rares sont graves, puisque le pronostic vital est en jeu dans la moitié des cas, et elles sont à l'origine d'un tiers de la mortalité infantile, de 10 % de la mortalité entre un et cinq ans et de 12 % entre cinq et quinze ans. Les maladies rares ont donné naissance, avec ces premières thérapies ciblées, à de vraies innovations de rupture, à partir de thérapies géniques, de l'ARN, cellulaires. Dans ce contexte, la médecine personnalisée modifiera la différence entre les maladies rares et les maladies dites fréquentes, en faisant de ces dernières l'équivalent d'une myriade de maladies rares pour ce qui concerne les traitements.
Aussi un bouleversement socio-économique est-il à l'oeuvre dans le secteur de la santé. Les nouveaux outils technologiques utilisés par la médecine personnalisée génèrent une quantité d'informations considérable, désormais largement en possession du patient lui-même. Tout un corps numérique est en train d'apparaître à côté du corps réel ce qui induira une évolution de la relation médecin-malade, soit parce que le médecin en saura trop par rapport à un patient qui souhaite ne pas savoir, soit parce que le patient viendra le consulter avec les résultats d'un test dans son téléphone portable. Le praticien se voit également contraint de respecter un modus operandi et des protocoles de soins plus ou moins rigides, ce qui ne lui laisse qu'une marge réduite d'appréciation. La relation médecin-malade deviendra un colloque singulier avec une augmentation colossale du volume d'informations, et l'intervention fréquente de tiers scientifiques, mathématiciens, physiciens, d'ingénieurs, bio informaticiens spécialisés. Le patient se trouvera face à de multiples interlocuteurs. L'intervention de la médecine se fera plus tôt dans la vie des personnes, car les possibilités médicales permettront d'anticiper une pathologie susceptible d'apparaître cinq ou dix ans plus tard. La limite entre le normal et le pathologique deviendra de plus en plus délicate à cerner : l'avenir sera sombre pour les hypocondriaques.
La multiplicité des informations dont disposera le patient induira une responsabilité accrue de sa part dans la prise en charge de sa propre santé ; la tentation de lier l'assurance-maladie au comportement du patient sera forte. La médecine personnalisée déchire le voile d'ignorance de l'avenir pour le patient, sa famille, voire pour les tiers, ce qui n'est pas sans conséquences éthiques et juridiques.
M. Alain Claeys . Cela appelle une réforme de la formation des personnels de santé ; tous les experts rencontrés ont insisté sur la nécessité d'un décloisonnement des disciplines scientifiques entre elles, sur l'importance d'un enseignement en informatique adapté et sur la nécessité d'introduire des sciences humaines et sociales dans ce contexte évolutif. La question de la formation des futurs professionnels de santé apparaît cruciale. C'est d'ailleurs l'un des cinq piliers de la stratégie nationale de santé. Les études de santé ne répondent pas à ces attentes, car l'un des écueils de la formation médicale aujourd'hui réside dans l'explosion énorme du corpus de connaissances. L'élaboration de la loi relative à l'enseignement supérieur et à la recherche de juillet 2013 a été l'occasion d'en débattre . Les propositions vont toutes dans le sens d'une réforme en profondeur. Le rapport de notre collègue, M. Jean-Yves Le Déaut recommande une spécialisation plus progressive au cours des trois années de licence. Le Pr Patrick Berche, doyen de l'Université de Paris-Descartes propose la suppression du concours de la première année d'étude de santé (PACES) et de nouveaux cursus. La loi sur la recherche et l'enseignement supérieur prévoit la mise en oeuvre, dès 2014, de dispositions expérimentales pendant six ans dans toutes les formations de santé. La loi vise à diversifier le recrutement des professions médicales et pharmaceutiques en développant des passerelles. De plus, il faudra renforcer et adapter le système de formation continue.
Par ailleurs, on s'oriente vers un nouveau modèle de recherche et développement (R&D) pour l'industrie pharmaceutique. La subdivision de tout champ pathologique en un très grand nombre de sous-domaines, chacun justifiable d'un traitement particulier, marque la fin presque généralisée du modèle de produit à forte valeur ajoutée ( blockbuster ) sur lequel l'industrie pharmaceutique reposait jusqu'à présent . Or, parallèlement, on observe une baisse de l'innovation et des bénéfices de l'industrie pharmaceutique. En effet, la plupart des médicaments les plus vendus tombent ou tomberont dans le domaine public à court ou moyen terme.
La mutation du monde pharmaceutique a commencé en 2004-2005, indépendamment de la médecine personnalisée, à la suite du scandale autour du médicament Vioxx. Merck, l'un des premiers laboratoires pharmaceutiques, a été mis en péril par les actions collectives ( class actions ) et autres procès intentés contre lui. D'après le Pr Laurent Degos, cette affaire a conduit différents laboratoires à arrêter ou limiter la recherche de traitements innovants pour de grandes populations, et à s'en tenir à des traitements pour des populations de 1 000 à 2 000 personnes dans chaque pays, car si un accident survient une fois sur 20 000 patients, il faudra dix ans pour qu'il survienne une fois. En application de ce raisonnement, le risque d'être poursuivi pour un accident causé par un médicament devient quasi nul. Dès lors, l'industrie pharmaceutique a considéré que, pour le moment, les vieilles molécules, souvent des génériques, étaient suffisantes pour traiter de grandes populations et des maladies fréquentes. De plus, elle y voit une opportunité de ressortir certains médicaments qui ne semblaient guère efficaces statistiquement, dénommés « les anges déchus ».
Désormais, l'industrie pharmaceutique s'intéresse non plus au seul médicament, mais au couple médicament-test diagnostique (le test compagnon). Cette évolution exige une diversification de compétences allant du diagnostic in vitro au dispositif médical. Pour un médicament donné, il s'agit, par ces tests, d'identifier les sujets répondeurs et non répondeurs ainsi que ceux qui pourraient provoquer un effet indésirable afin d'adapter la dose à chaque individu. La médecine personnalisée implique alors le développement simultané d'un test compagnon, un deuxième produit qui permet de déterminer si le médicament sera efficace ou non sur tel ou tel patient. Il faut, dans l'intérêt des malades, arriver à un compromis entre le désir d'une mise à disposition rapide des nouveaux produits, reposant parfois sur des dossiers insuffisamment étayés, et le désir d'une mise à disposition la plus sûre possible, sans transiger sur la sécurité des patients.
L'incitation à la recherche et au développement de tests génétiques prédictifs ou diagnostiques assortis à une thérapie ciblée dépend des solutions qui seront trouvées pour résoudre les questions de brevetabilité et de propriété industrielle liées à ces textes. Au-delà de la méthode de traitement, la médecine personnalisée doit prendre une forme concrète pour que les spécialistes de la propriété industrielle puissent lui assurer une protection juridique. L'Office européen des brevets ne permet pas aujourd'hui de breveter de telles méthodes.
Cette problématique induit une tension depuis vingt ans entre, d'une part, les sociétés de médecine moléculaire, alliées aux associations de patients et, d'autre part, les entreprises qui détiennent des brevets larges de gènes. Aux États-Unis, plusieurs procédures judiciaires sont emblématiques, notamment l'affaire Myriad Genetics qui a mis aux prises, d'un côté, des représentants de la communauté scientifique et des associations de citoyens et, de l'autre, Myriad Genetics ; elle s'est terminée, après une longue procédure émaillée de décisions contradictoires, par une décision de juin 2013, réduisant largement la possibilité de breveter un gène. Pour éviter des brevets très larges portant sur des gènes, certaines solutions comme le recours à des « communs » sont envisageables. Ce sont des entités juridiques proposant de nouveaux usages du droit de la propriété intellectuelle, en fractionnant les différents attributs de cette propriété pour les partager entre différentes parties prenantes.
M. Jean Sébastien Vialatte. Il faudra garantir l'équité et l'information des citoyens face aux enjeux éthiques de la médecine personnalisée. Quand on disposera de tests prédictifs ou de marqueurs biologiques suffisamment convaincants au niveau scientifique, nombre de questions devront être soulevées. Quand effectuer les tests ? À quel âge ? Avec quelle périodicité ? Pour quelle partie de la population et avec quelle prise en charge ? Quel sera l'accès à la prévention et pour quel coût, sachant que la prévention précoce pourrait être efficace, mais qu'il ne faudrait pas pour autant inquiéter les patients inutilement ? Or, d'ores et déjà, une partie de ces questions se pose, car le rôle des tests génétiques à valeur prédictive s'accroît.
En effet, la médecine personnalisée inclut largement la médecine prédictive ou de prévision dans son champ. Largement fondée sur l'analyse génétique, elle conduit à un recours large aux tests génétiques, ce qui induit des interrogations d'ordre éthique et sociétal que nous avions abordées en 2008, dans le cadre de l'OPECST, lors de l'évaluation de la loi relative à la bioéthique, et au cours de l'audition publique organisée, en 2011, par M. Claude Birraux et M. Jean-Louis Touraine sur les maladies monogéniques. Les interrogations, observations et recommandations émises alors restent encore d'actualité alors même que l'usage des tests s'étend. À mesure que l'accès au séquençage à haut débit, voire à très haut débit se développe, le recours aux tests génétiques tendra à s'accroitre.
D'après l'Agence de la biomédecine, on constate une augmentation vertigineuse du nombre de tests effectués entre 2009 et 2012. La loi règlemente le recours aux examens génétiques, leur prescription, le rendu des résultats. Ces dispositions introduites pour la plupart d'entre elles par la loi relative à la bioéthique de 2004 ont été peu modifiées par la loi de 2011, sauf en ce qui concerne l'information de la parentèle. L'Agence de la biomédecine a élaboré, en liaison avec la Haute autorité de santé, un arrêté en mai 2013 définissant les règles de bonnes pratiques applicables à l'examen des caractéristiques génétiques d'une personne à des fins médicales.
L'évolution rapide des technologies s'accompagne de la volonté d'assurer aux patients une information fiable sur les bénéfices attendus des examens génétiques pour leur permettre de distinguer entre des examens génétiques prescrits dans un contexte médical, et cette sorte de génétique récréative que suscitent les tests en libre accès sur Internet, accessibles sur une quarantaine de sites. Ces tests proposent, pour 100 ou 200 dollars, de rechercher les mutations les plus fréquentes, ce qui est finalement assez rapide, des machines étant disponibles à l'étranger pour traiter l'information en quantité. Ces tests, qui sont généralement des tests de susceptibilités faussement rassurants ou inquiétants peuvent être dangereux, car, en dehors de tout cadre protecteur, on tend à faire croire aux personnes qui les utilisent qu'elles ne risquent pas de développer telle ou telle maladie ou, à l'inverse, on les angoisse à tort en focalisant leur attention sur d'autres pathologies dont elles ne seront pas atteintes. La régulation est difficile à mettre en oeuvre.
Quant aux aspects éthiques relatifs aux tests génétiques, le seul texte juridique international contraignant est la Convention sur les Droits de l'Homme et la biomédecine du Conseil de l'Europe, dite Convention d'Oviedo, qui limite l'utilisation des tests génétiques prédictifs de maladie aux seules fins médicales ou de recherche médicale, et interdit toute forme de discrimination à l'encontre d'une personne sur la base de son patrimoine génétique. Sur la base de ces principes, un nouvel instrument juridique, complétant les dispositions de la Convention dans le domaine des tests génétiques à des fins médicales, a été élaboré : c'est le Protocole additionnel à la Convention sur les Droits de l'Homme et la biomédecine, relatif aux tests génétiques à des fins médicales.
La valeur prédictive des tests et son impact est débattue de manière récurrente. Le développement des tests pose des questions sur la précision technologique et la fiabilité des analyses. Aujourd'hui, les diagnostics prénataux (DPN) et préimplantatoires (DPI) sont strictement encadrés en France. Or le séquençage à très haut débit et les possibilités nouvelles de prélever l'ADN du foetus dans le sang maternel changent la donne. Plusieurs sortes de tests se développent, des tests génétiques prénuptiaux visent principalement à lutter contre la transmission de maladies génétiques à forte prévalence dans certaines populations. Progressivement, ces tests se diversifient et couvrent de plus en plus de pathologies génétiques. Ils se diffusent notamment aux États-Unis, en Israël, en Asie et dans les pays du Golfe. La notion de risque d'apparition d'une maladie commence à être prise en compte dans le cadre du diagnostic préimplantatoire (DPI), avec des risques de dérive eugéniste vers la quête de l'enfant parfait.
La médecine prédictive pourrait être détournée de sa fonction sanitaire et être exploitée à des fins non médicales, dans le domaine des assurances et du travail. L'individualisation des risques pourrait avoir un impact important sur les mécanismes d'assurance. La prédiction lèverait le « voile d'ignorance » sur lequel repose le principe de la mutualisation du risque. Certes, actuellement, l'état de santé est déjà pris en compte dans les contrats d'assurance. En effet, contrevenir au principe de bonne foi en cachant une pathologie ou des antécédents médicaux et familiaux à un assureur peut provoquer une annulation du contrat. De plus, la détermination de risques de santé pourrait aussi intéresser les employeurs. Elle risquerait alors d'être utilisée soit pour introduire une sélection à l'embauche, soit pour gérer les ressources humaines.
La protection et la gestion des données personnelles issues des tests est complexe. Il s'agit de données personnelles, identifiantes, et pour partie invariantes tout au long de la vie. En France, il existe un cadre juridique protecteur des données génétiques que la Commission nationale de l'informatique et des libertés (CNIL) est chargée de faire respecter. Cependant, vis-à-vis de ces vastes bases de données qui se constituent aujourd'hui grâce aux outils technologiques puissants dont on dispose, de nombreuses règles de la législation sur l'informatique et les libertés sont remises en cause, telle la durée de conservation des données limitée à la finalité de la recherche. En effet, comment assurer le respect du droit des personnes, quand la recherche est appelée à durer très longtemps, et à évoluer ? On ne pourra pas redemander un consentement à chaque étape et, de plus, certaines personnes disparaîtront avant le terme de la recherche. Des exemples de ré-identification, à partir de données minimales anonymisées contenues dans des bases à accès ouvert, ont été récemment décrits. Il faudra donc trouver un système qui garantisse à la fois une qualité de la recherche, une certaine réactivité de celle-ci, et en même temps la confidentialité des données du patient . Toutes les données personnelles du dossier médical doivent être sauvegardées, et protégées de la même façon. Le dossier médical personnel (DMP), en cours d'informatisation, sera concerné au premier chef, car il devra inclure les données génétiques.
Le stockage et le partage d'énormes banques de données constituent un problème important . Les outils de stockage sont d'une dimension nouvelle, et seront difficiles à contrôler. Comment collecter, rassembler et archiver les informations ? Qui aura obligation de stocker les données, alors que celles susceptibles de circuler exigent des espaces considérables ? Pour être utiles, interprétables, ces données doivent pouvoir transiter, ce qui suppose une fiabilité extrême des circuits, donc des accès sécurisés et, surtout, des systèmes d'anonymisation fiables. Est-on suffisamment avancé dans ces techniques pour pouvoir les garantir ? Qui va gérer l'organisation du stockage des données : une ou plusieurs institutions ? Un e sorte de CNIL de la santé ?
L'accès aux bio-banques et la propriété des données sont un enjeu essentiel eu égard à leur importance pour la recherche. Qui en est propriétaire ? Le patient, celui qui a réalisé l'examen ou la collectivité qui a payé l'examen ? Il existe, en principe, une règle aujourd'hui en vertu de laquelle chacun est propriétaire de ses propres données médicales, mais l'Islande a vendu les données génétiques de sa population à une firme privée. Les risques d'appropriation des données existent donc. Cependant, l'avenir de la médecine personnalisée implique un partage public de larges collections de données.
M. Alain Claeys . Il faut veiller à garantir un accès équitable de tous aux soins, limiter la médico-surveillance , et lutter contre les risques d'exclusion des groupes de patients qui ne seront pas traités parce qu'ils seront classés non-répondeurs au traitement. C'est un des aspects inégalitaires de cette médecine. Il faut, en tout état de cause, promouvoir l'éducation à la santé par une information très ciblée sur certaines populations défavorisées. Il faudra donc veiller à maintenir un juste équilibre entre la pression collective engendrée par la demande de traitements ciblés et le maintien de la solidarité du système de santé publique. Compte tenu des bouleversements engendrés par la médecine personnalisée, il faut organiser rapidement des débats publics pour en expliquer les avancées, et les risques.
Ainsi, du point de vue de l'OPECST, cinq sujets sont à suivre. Premièrement, la médecine personnalisée doit s'inscrire dans les thèmes de recherche et représenter une priorité pour la France, qui doit être présente dans ce secteur ; c'est pourquoi il faut appuyer la création d'un Centre national de séquençage. Deuxièmement, la relation singulière médecin-malade doit s'inscrire dans un parcours de soins, ce qui, au niveau de la formation des personnels de santé, est insuffisamment pris en compte, alors que cela doit devenir une priorité. Troisièmement, les enjeux sont importants au niveau de la recherche, du développement et de l'innovation, car il s'agit de faire entrer, dans le cadre de la propriété intellectuelle, des méthodes de traitement sans breveter les acquis de la connaissance ; la France doit prendre position dans ce débat européen qui nous ramène vingt ans en arrière au moment de la transposition de la directive européenne sur la brevetabilité du vivant ; notre action n'avait pas abouti alors. Quatrièmement, la protection des données doit susciter un regain de réflexions : la loi de bioéthique suffit-elle ? Exige-t-elle des aménagements ? Cinquièmement, il conviendra de concilier le financement de la médecine en partie stratifiée, personnalisée et la solidarité ; cela nécessite des débats publics incluant les associations de malades pour éviter les contre-sens et les espoirs injustifiés.
M. Jean-Sébastien Vialatte. Voici les principales préconisations que nous avons regroupées et détaillées en sept points : la préparation des institutions françaises au changement de paradigme induit par la médecine personnalisée, la réforme de la formation des personnels de santé, l'aide au développement des traitements ciblés, l'information de la valeur prédictive des tests génétiques, la garantie d'un égal accès de tous les citoyens aux nouvelles thérapies, la protection des données personnelles de santé et l'information des citoyens sur la médecine personnalisée. À ces nouvelles préconisations s'ajoute la reprise de recommandations antérieures figurant dans d'autres rapports de l'OPECST
M. Bruno Sido. Je vous félicite et vous remercie d'avoir mis en valeur l'apport des rapports précédents et repris des recommandations antérieures de l'Office. Je m'interroge sur la diminution de la durée des essais préalables aux autorisations de mises sur le marché (AMM) : avez-vous des raisons particulières pour estimer qu'une accélération à ce niveau est exempte de risques sérieux pour le malade ? S'agissant de l'idée de renforcer la sécurité des données de santé, notamment en sécurisant le futur dossier médical personnalisé, quelles seront les garanties techniques ? Une formation en informatique donnée à tous soignants est certes très souhaitable, mais, connaissant la situation des établissements de santé, leurs difficultés financières et le manque de temps des soignants, n'est-ce pas une recommandation déconnectée de la réalité ? Dans l'ensemble, vos recommandations très techniques n'excèderaient-elles pas la capacité moyenne d'implication du malade quel que soit d'ailleurs son niveau d'instruction ? Que pensez-vous de la nouvelle lentille de contact mise au point par Google pour permettre aux diabétiques de disposer d'une détection immédiate de leur taux de glucose dans le sang ? Cette information étant transmise immédiatement au téléphone portable de l'intéressé, quelle est la fiabilité d'un tel procédé alors que cette donnée essentielle peut-être interceptée, voire modifiée ? Êtes-vous informés de ce développement technique effectué par un acteur nouveau dans le domaine de la santé ? Partagez-vous mes réserves à cet égard ? Par ailleurs, dans la logique de votre analyse sémantique initiale, le titre du rapport ne devrait-il pas être précisé ?
M. Alain Claeys . On manque de bio-informaticiens et de structures capables d'en faire émerger, car ce n'est pas aisé dans notre organisation et avec notre structuration de la recherche. C'est un sujet important auquel nous avons été sensibilisés par de nombreux experts, particulièrement lors de notre visite au Génopole d'Évry. On manque de passerelles entre les différentes disciplines scientifiques.
M. Jean-Sébastien Vialatte . L'autorisation de mise sur le marché (AMM), telle qu'on la conçoit en médecine personnalisée, s'inspire de ce qui existe pour les maladies rares et le diabète. En effet, on génère des groupes et des sous-groupes de plus en plus étroits, et on s'oriente vers des mises sur le marché conditionnelles impliquant une grande vigilance.
Google s'intéresse de très près à la santé et à la médecine personnalisée qui exige des moyens de stockage des données importants. Or Google, comme d'autres opérateurs de l'Internet, dispose de moyens de stockage et d'analyse des données considérables. Ces entreprises deviennent, et deviendront, des acteurs incontournables du secteur de la santé sauf si les États s'impliquent. Quant au piratage, on constate qu'actuellement tout est connecté et les données de santé pourraient l'être en tout premier lieu, compte-tenu de l'importance qu'elles revêtent.
M. Gérard Bapt, député . Raccourcir les délais d'autorisation de mise sur le marché (AMM), ou faire des essais post-AMM, si cela n'entre pas dans le renforcement d'un plan de pharmacovigilance ou une autorisation temporaire d'utilisation, c'est un sujet explosif au plan des concepts ; mieux vaudrait éviter une telle préconisation. Comment maintenir la solidarité et la protection sociale quand, déjà actuellement, on observe une tentative de s'extraire du système public et d'aller vers des organismes privés moins onéreux ? La médecine personnalisée renforcera ces tendances, et cela ouvre un champ immense de discussions.
M. Jean-Yves Le Déaut. Je me félicite de ce rapport. J'observe la pertinence de décrire la médecine sur mesure ou de précision par rapport aux « 4P » : Personnalisée, Préventive, Prédictive, Participative.
Les connaissances sur le génome permettent aujourd'hui de brosser le portrait moléculaire de chaque patient, mais le coût en reste élevé. On peut désormais être caractérisé par ses gènes, et les premières conclusions ont été que chacun d'entre nous réagit de manière différente quand on lui administre un médicament notamment en oncologie. Demain, on pourra, grâce à ces techniques, trouver et décider du médicament le mieux adapté pour chacun. De même, grâce à de nouvelles techniques de screening, on pourra réhabiliter d'anciennes molécules. En effet, à propos des « anges déchus », ces anciennes molécules que l'on pourrait utiliser en ciblant mieux leurs potentialités, j'ai visité récemment une start-up nancéienne, Harmonic Pharma ; grâce au criblage moléculaire, elle vérifie très vite si une molécule est active. Le rôle considérable de l'informatique dans la modélisation de nouveaux médicaments aura un impact sur les essais cliniques. C'est une véritable révolution que connaîtra la médecine. Cela est d'ailleurs très bien analysé dans le rapport, qui traite également de la nécessité de la recherche.
Aussi, aux sept groupes de recommandations proposées, j'en suggère un huitième qui portera sur la recherche et l'innovation, car ces points sont largement abordés dans le rapport. Je propose de regrouper ces recommandations relatives à ces deux thèmes sous l'intitulé : « Encourager la recherche et le développement ». Il s'agira de reconduire les financements des projets de recherche dans le cadre des investissements d'avenir, notamment en oncologie ; de créer une plateforme de séquençage à très haut débit de recherche et de diagnostic qui se focalise sur les cas spécifiques ; de réactiver le développement de nouvelles molécules ; d'évaluer les potentialités des anciennes molécules en recourant aux nouvelles techniques de criblage de masse ( screening ) ; de favoriser le développement de la nano-médecine, et de veiller à ce que la convergence des technologies Nano-Bio-Info-Cogno (NBIC) conduise à la création de nouveaux cursus.
Des questions se posent sur le coût élevé du ciblage par de nouveaux bio-marqueurs et des tests : comment y remédier ? Comment gérer les risques des tests à bas coûts en accès libre sur Internet ? Ne faut-il pas intégrer des cours sur la protection des données personnelles au cursus de tous les soignants ? Quel seront le contrôle et la gestion des tests prénataux et néonataux, dès lors qu'il est possible de les effectuer à partir du sang de la mère ? Faudra-t-il modifier les règles concernant les essais cliniques ?
M. Jean-Sébastien Vialatte. Certains tests génétiques sont en vente libre sur Internet, sans protection des données, sans avertissement quant à leur fiabilité ; les données qu'ils génèrent peuvent être utilisées par les sociétés qui les proposent en ligne. On assiste à la naissance d'une forme de tourisme prédictif, à l'instar de ce qui s'est développé pour la gestation pour autrui (GPA). En effet, actuellement, une entreprise américaine opérant en France propose, en violation de la loi, d'organiser une GPA à l'étranger et de faciliter les démarches, le choix de la mère porteuse, le choix du sexe de l'enfant, etc . Avec les tests en libre accès, on se trouve face à des risques de dérives eugénistes difficiles à contrôler. Ainsi, l'Internet ouvre la possibilité de démarcher une large clientèle internationale pour promouvoir des techniques interdites.
S'agissant de l'évaluation économique,
l'industrie pharmaceutique défend son
pré-carré,
prétend que l'administration des bonnes molécules aux bons
patients permettra des économies. Il y existe peu d'études
coût-efficacité.
L'anonymisation des données est possible, mais elle est malaisée à maintenir et à garantir. Plusieurs publications ont récemment démontré qu'en croisant différentes données avec une géolocalisation, on parvient à retrouver le nom de la personne concernée. Par ailleurs, concernant la protection des banques de données, l'Islande, sous la pression de ses problèmes budgétaires, a fini par vendre à une société privée les données génétiques de sa population. L'entreprise qui a mené cette opération refuse d'ailleurs de recevoir les parlementaires d'où qu'ils soient.
M. Alain Claeys. Les pouvoirs publics islandais ont croisé les données génétiques et les dossiers médicaux de leur population ; c'est après avoir vendu ces fichiers qu'ils ont tenté de revenir en arrière.
Mme Anne-Yvonne Le Dain, députée . Je m'interroge sur l'intitulé du rapport. La médecine personnalisée porte-t-elle sur le soin de la personne en tant que ce qu'elle est véritablement, compte tenu à la fois de son environnement, de sa vie, de son passé, de son histoire, de ses conditions de vie ? À l'inverse, dans le cadre de cette médecine, il semble que la personne soit définie par sa génétique, ce qui est extrêmement réducteur. Car les données de génétique primaire nient l'adaptation de la personne à l'environnement. Le code génétique évolue au cours de l'existence avec les processus de vieillissement. C'est l'une des raisons pour lesquelles le clonage ne fonctionne pas. De fait, seules certaines maladies monogéniques sont concernées. Il faut donc relativiser la puissance des vendeurs de mirage. L'attention devrait être davantage portée sur le patient qui vient se faire soigner avec des pathologies polygéniques. C'est pourquoi l'intitulé devrait être précisé.
M. Alain Claeys. Comme c'est l'usage, nous avons repris l'intitulé figurant dans la saisine, mais je vous rejoins. Il y a une ambiguïté que Mme Fagot-Largeault a clairement exprimée. Je cite « La publicité faite pour la médecine dite personnalisée tombe dans un contexte où l'on constate une aspiration à un contact plus personnel, voire à une empathie du médecin, qu'on ne trouve pas, qu'on ne rencontre plus. On s'abrite derrière une formule attirante, rassurante, qui donne une image attractive, mais fausse et c'est très préoccupant ».
On a perdu la bataille de la communication, on se trouve face à une médecine stratifiée, voire une médecine de précision.
M. Jean-Sébastien Vialatte. Je considère aussi que la médecine personnalisée est une médecine plutôt dépersonnalisante. Dans ce type de médecine, l'individu risque d'être réduit à sa génétique, à une somme de données, à des milliers de mutations ; il devient un point dans un nuage de points.
M. Jean-Yves Le Déaut . On se trouve en réalité devant une approche génétique de la médecine. Il faudrait en effet changer l'intitulé du rapport.
M. Gilbert Barbier, sénateur. Comment s'articule la recherche public-privé dans ce domaine ? Comment orienter et encourager les laboratoires de recherche ? Faut-il encourager certaines recherches ? Quel serait le rôle de l'Agence de la biomédecine ?
M. Alain Claeys. Cette distinction public-privé n'a plus vraiment de réalité concrète. Il s'agit de savoir sur quoi porteront les investissements publics, comme les investissements d'avenir, par rapport aux recommandations du rapport de Mme Anne Lauvergeon, et de veiller à une concordance entre les investissements publics et les investissements privés. Il faudra peut-être élargir les compétences de l'Agence de la biomédecine, qui joue déjà ce rôle coordinateur dans certains autres domaines de la recherche.
M. Jean-Sébastien Vialatte . Certaines institutions ou groupes privés joueront un rôle, car un séquenceur Illumina est à la portée d'un laboratoire de taille moyenne ; mais son utilisation n'est pas à la portée de tous. En France, les laboratoires d'analyses génétiques doivent obtenir un agrément de l'Agence de la biomédecine ; il faut maintenir cet encadrement et les règles de confidentialité qu'il implique.
M. Jean-Louis Touraine, député. Je félicite les rapporteurs et adhère aux recommandations. La limite de cette médecine dite personnalisée est épigénétique. Or l'acquis importe. L'utilisation de la génétique permet à la médecine de cibler un groupe de personnes en fonction de mutations génétiques communes. On assiste à une modification des paradigmes : la proximité génétique des tumeurs est prise en compte pour un groupe de patients. Ainsi le cancer du poumon se décline maintenant en 150 maladies. On reste cependant dans une démarche de médecine hippocratique, pour laquelle les mêmes causes produisent les mêmes effets.
La médecine dite personnalisée pour la population est, en fait, une médecine individualisée avec ses particularités. Je pose l'hypothèse qu'il faudra reprendre la réflexion d'avant Hippocrate on assistera à un changement radical avec l'avènement d'une médecine individualisée. Cela existe déjà avec le système des antigènes des leucocytes humains (HLA) qui caractérise un individu et qui commande sa réaction immunitaire aux maladies infectieuses. Ce système a permis la survie de l'espèce car il produit des réponses distinctes aux grandes épidémies. Si on intègre les données du système HLA dans la médecine personnalisée, les groupes de patients seront de plus en plus petits ; on en arrivera à une médecine totalement individualisée, notamment dans les biothérapies cellulaires et l'usage des cellules souches ; on modifiera les cellules du malade lui-même ; chaque malade disposera alors d'un traitement qui lui est propre.
M. Jean-Yves Le Déaut. Je suggère, avec l'accord des rapporteurs, un nouvel intitulé : « Progrès de la génétique : vers une médecine de précision ? » en conservant l'intitulé actuel comme sous-titre.
M. Bruno Sido. Le rapport est adopté à l'unanimité, avec les différentes modifications évoquées. »
COMPOSITION DU COMITÉ DE PILOTAGE
- Mme Simone BATEMAN, sociologue, directrice de recherche au CNRS ;
- Dr Catherine BOURGAIN, chargée de recherche en génétique humaine et statistiques à l'INSERM ;
- Pr Anne FAGOT-LARGEAUT, professeur émérite, Collège de France, Chaire de philosophie des sciences biologiques et médicales, membre de l'Académie des sciences ;
- Mme Hélène GAUMONT-PRAT, professeure des Universités, directrice du Laboratoire Droit de la santé, Université Paris VIII, ancien membre du CCNE ;
- Pr. Jean-Marc GROGNET, directeur de l'Institut de biologie et technologies du CEA (l'iBiTec-S), membre de l'Académie de pharmacie ;
- Pr Axel KAHN, ancien directeur de l'Institut Cochin, ancien président de l'Université Paris-Descartes, membre de l'Académie des sciences ;
- M. Didier TABUTEAU, conseiller d'État, responsable de la chaire santé de l'Institut d'études politiques
LISTE DES PERSONNES ENTENDUES PAR LES RAPPORTEURS
20 novembre 2012 :
- Pr Anne FAGOT-LARGEAULT, professeur émérite, Collège de France, Chaire de philosophie des sciences biologiques et médicales, membre de l'Académie des sciences.
28 novembre 2012 :
- Pr Axel KAHN, médecin généticien, ancien président de l'Université Paris-Descartes, membre de l'Académie des sciences et du Conseil scientifique de l'OPECST.
11 décembre 2012 :
- M. Maurice CASSIER, sociologue au Centre de recherche médecine, sciences, santé et société (CERMES).
18 décembre 2012 :
- Mme Hélène GAUMONT-PRAT, professeur des universités, directeur du Laboratoire Droit de la santé, Université Paris VIII, ancien membre du CCNE.
19 décembre 2012 :
- M. Didier TABUTEAU, conseiller d'État, responsable de la chaire santé de l'Institut d'études politiques (IEP).
15 janvier 2013 :
- Mme Catherine BOURGAIN, chargée de recherche en génétique humaine et statistiques à l'INSERM ;
- Mme Simone BATEMAN, sociologue, directrice de recherche au CNRS.
5 février 2013 :
- Pr Claude HURIET, président de l'Institut Curie, ancien sénateur ;
- Pr Dominique STOPPA-LYONNET, professeur de génétique ;
- Dr Christophe LE TOURNEAU, oncologue, responsable de l'essai SHIVA.
6 février 2013 :
- M. Jean-Marc GROGNET, docteur es-pharmacie, directeur de l'institut de Biologie et technologies du CEA (l'iBiTec-S).
12 février 2012 :
- Pr Bertrand JORDAN, biologiste moléculaire, fondateur de la Génopole de Marseille.
13 février 2013 :
- Dr. Jean-François DELEUZE, directeur du Centre national de génotypage (CNG).
20 février 2013 :
- Mme Corinne BLACHIER-POISSON, directrice des affaires gouvernementales et économiques d'Amgen.
20 février 2013 :
- Pr Laurent DEGOS, ancien Président de la Haute Autorité de Santé HAS.
26 février 2013 :
- Pr Philippe MONTEYNE, vice-président R&D France à Sanofi ;
- Mme Isabelle DIAZ, directeur Biotechnologies et Recherche au LEEM (Les Entreprises du Médicament).
12 mars 2013 :
- Mme Frédérique LESAULNIER, coordinatrice du pôle santé du service des affaires juridiques ;
- M. Geoffroy SIGRIST, attaché parlementaire à la Commission nationale de l'informatique et des libertés (CNIL).
18 mars 2013 :
- Pr Agnès BUZYN, hématologue, présidente de l'Institut National du Cancer (INca).
20 mars 2013, à l'Institut Curie :
- Pr Claude HURIET, président de l'Institut ;
- Pr Pierre TEILLAC, directeur de l'ensemble hospitalier ;
- Pr Dominique STOPPA-LYONNET, professeur de génétique ;
- Dr Christophe LE TOURNEAU, oncologue, responsable de l'essai SHIVA ;
- Dr Sergio ROMAN-ROMAN, chef du Département de transfert ;
- Dr Olivier DELATTRE, directeur de l'Unité de génétique et biologie des cancers ;
- M. Thomas RIO-FRIO, responsable de la plateforme de séquençage haut débit ;
- Dr Emmanuel BARILLOT, directeur bio-informatique et systèmes d'information ;
- Dr Claude HOUDOYER, responsable du laboratoire du département de biologie des tumeurs.
13 avril 2013, à l'hôpital St Louis :
- Pr Emmanuel RAISON, directeur de l'hôpital ;
- M. Philippe SUDREAU, directeur du groupe hospitalier des Hôpitaux universitaires St Louis-Lariboisière-Fernand Vidal ;
- Pr Hugues de THÉ, chef du service de biochimie/biologie moléculaire ;
- Pr François SIGAUX, directeur de l'Institut Fédératif de Recherche 105 « St-Louis Institut d'Hématologie » ;
- Mmes Valérie LALLEMAND, Morgane LEBRAS et Dominique VITOUX et MM Nicolas SETTERBLAD, Omar FERHI et Marika PLA, chercheurs à l'Institut universitaire d'hématologie de l'hôpital.
28 mai 2013 :
- M. Christian BYK, magistrat, conseiller à la Cour d'appel de Paris, secrétaire général de l'Association internationale droit, éthique et science.
19 juin 2013 :
- Pr Thomas TURSZ, cancérologue, directeur honoraire de l'Institut Gustave Roussy.
17 juillet 2013 :
- Dr Jean-Marc EGLY, directeur de recherche à l'INSERM à l'Institut de génétique et de biologie moléculaire (IGBMC), membre du Conseil scientifique de l'OPECST.
1 er octobre 2013, à Bruxelles :
- Mmes Olga SOLOMON et Agnès MATTHIEU, responsables de l'Unité Produits pharmaceutiques-Autorisation à l'Agence européenne des médicaments, DG Santé et consommateurs de la Commission européenne ;
- M. Vincent HOUDRY, conseiller santé, médicaments et dispositifs médicaux à la Représentation permanente auprès de l'Union européenne ;
- Mme Irène NORSTEDT et M. Jean-Luc SANNE, respectivement chef d'unité et responsable de l'Unité Médecine personnalisée à la DG Recherche de la Commission européenne ;
- M. Laurent ALEXANDRE, Président de DNA Vision.
8 octobre 2013, au Génopole de Marseille :
- Pr Georges LÉONETTI, doyen de la Faculté de médecine de la Timone ;
- Pr Nicolas LÉVY, directeur du Laboratoire U910, service de génétique médicale ;
- Pr Christian CHABANNON, responsable de l'Unité de thérapie cellulaire et du Centre de ressources biologiques de l'Institut Paoli Calmettes ;
- Mme Fabienne HERMITTE, directrice R&D et affaires réglementaires, Ipsogen/Quiagen ;
- Mme Marie MALISSEN, responsable Immunophénotypage de la plateforme du Centre d'ImmunoPhénomique (CIPHE) ;
- Dr. Jean IMBERT, responsable de la plateforme Transcriptomique et Génomique ;
- Pr Bertrand JORDAN, biologiste moléculaire, fondateur de la Génopole de Marseille, directeur de recherche émérite au CNRS.
6 novembre 2013 :
- Pr. Jean-Paul MOATTI, directeur de l'ITMO Institut de santé publique de l'Alliance nationale pour les sciences de la vie et de la santé (AVIESAN).
12 novembre 2013 :
-Participation à la séance commune de l'Académie des sciences et de l'Académie de médecine sur « Génome personnel et exercice de la médecine » à l'Académie de médecine
20 novembre 2013 :
- Mme Mathilde REYNAUDI, chargée de mission au Commissariat général à la stratégie et à la prospective (CGSP).
27 novembre 2013, au Génopole d'Évry :
- M. Pierre TAMBOURIN, directeur général du Génopole ;
- M. Jean WEISSENBACH, directeur du Génoscope- Centre national de séquençage (CNS) ;
- Dr. Jean-François DELEUZE, directeur du Centre national de génotypage (CNG) ;
- M. François GENDRE, conseiller à la direction des sciences de la vie du CEA, directeur général d'AMAbiotics ;
- M. Yves CHAMPEY, conseiller auprès du directeur général du Génopole ;
- M. Pierre LE BER, directeur-adjoint de l'Institut de génomique/CEA ;
- M. Claude SCARPELLI, directeur informatique du Génoscope ;
- M. François ARTIGUENAVE, directeur bio-informatique du Génoscope.
29 novembre 2013 :
- Pr Hugues de THÉ, chef du service de biochimie/biologie moléculaire ;
- Mme Valérie LALLEMAND, chargée de recherche à l'INSERM
3 décembre 2013 :
- Pr Philippe FROGUEL, endocrinologue, professeur de biologie moléculaire à l'université Lille 2.
4 décembre 2013 :
- Pr Thierry FRÉBOURG, directeur-adjoint de l'ITMO génétique, génomique et bio-informatique AVIESAN, coordinateur de l'Institut de recherche biomédicale de Rouen ;
- Pr Hélène DOLFUS, professeur de génétique moléculaire, service de génétique médicale des hôpitaux universitaires de Strasbourg ;
- Pr Soumeya BEKRI, professeur des universités, responsable du dépistage et du suivi des maladies métaboliques au CHU de Rouen.
4 décembre 2013, à Gustave Roussy Cancer Campus :
- Pr Alexander EGGERMONT, directeur général de Gustave Roussy Cancer Campus ;
- Pr Eric SOLARY, directeur de la recherche ;
- Dr Fabrice ANDRÉ, directeur del'UMP981 biomarqueurs prédictifs et nouvelles stratégies moléculaires en thérapeutique anticancéreuse ;
- Dr Ludovic LACROIX, responsable du laboratoire de recherche translationnelle.
5 décembre 2013 :
-Participation au 2 ème colloque de l'ITMO santé publique sur « Médecine personnalisée et innovations biomédicales : enjeux de santé publique, économique, éthiques et sociaux » au Grand auditorium de la Bibliothèque nationale de France (BNF)
7 janvier2014 :
- Pr Marie-Christine FAVROT, adjointe au directeur général de la santé du ministère des affaires sociales et de la santé, chef des politiques de santé.
ANNEXES
ANNEXE N° 1 :
COMPTE RENDU DE
L'AUDITION PUBLIQUE DU 27 MARS 2013
« MÉDECINE PERSONNALISÉE : QUELS DÉFIS SCIENTIFIQUES, TECHNIQUES ET MÉDICAUX ? »
OUVERTURE
PAR MM. ALAIN CLAEYS ET JEAN-SÉBASTIEN
VIALATTE,
DÉPUTÉS, RAPPORTEURS
M. Alain Claeys, député de la Vienne, rapporteur. Je vous remercie d'avoir répondu à l'invitation de Jean-Sébastien Vialatte et de moi-même, pour cette audition publique concernant la médecine personnalisée. C'est la Commission des affaires sociales qui a saisi l'Office d'une demande d'étude qui concerne les enjeux scientifiques, technologiques, éthiques et juridiques de la médecine personnalisée.
Cette saisine s'inscrit dans la lignée des travaux de l'Office sur les sciences du vivant, de la santé, et plus généralement de la bioéthique. Nous avons eu l'occasion de mener des études et d'entendre certains d'entre vous au moment de l'évaluation et l'élaboration de la loi de bioéthique, votée il y a un an. Et donc, nous avons déjà abordé, indirectement, certains aspects de la médecine personnalisée.
Sur ce sujet, qu'attendons-nous de vous ? Cela vous étonnera peut-être, mais nous avons un problème de définition. Dans ce terme de médecine personnalisée, un thème récent et ancien à la fois, on met bien des éléments parfois contradictoires. En second lieu, il s'agit d'analyser toutes les conséquences éthiques, sociétales, scientifiques, qui se poseront très concrètement à travers le développement de ce type de médecine.
Nos débats seront organisés autour de quatre tables rondes. La première sera médicale : quelles sont les pathologies concernées ? La deuxième table sera technologique : quels sont les outils utilisés ? Ensuite, cet après-midi, toute une série d'interventions traiteront des thèmes de la recherche, ce qui pose nombre de questions sur les transferts, les brevets. Enfin, la quatrième table ronde traitera des modèles de R & D (recherche-développement).Ce qui sous-tend cette réflexion scientifique, c'est la question bien plus vaste de notre modèle social de solidarité et de prise en charge de la maladie. C'est un sujet qui va devenir de plus en plus prégnant.
Je remercie le Pr Axel Kahn, qui fait partie de notre conseil scientifique, et le Pr André Syrota, d'avoir accepté de clarifier le sujet dans leurs propos introductifs. Je remercie chacune et chacun d'entre vous.
M. Jean-Sébastien Vialatte, député du Var, vice-président de l'OPECST, rapporteur. Je m'associe aux remerciements d'Alain Claeys et me félicite de votre présence. La saisine de la Commission des affaires sociales sur le thème de la médecine personnalisée nous offre la possibilité de débattre d'un concept assez difficile à cerner et à définir. De tout temps, les médecins se sont efforcés d'adapter les traitements à leurs patients et ont considéré, à juste titre, que c'était là l'élément essentiel de la relation médecin-malade et de la qualité de leurs soins. Ils disposaient d'outils de diagnostic qui se perfectionnaient progressivement à mesure des avancées scientifiques et techniques pour les aider dans l'accomplissement de cette mission. Or, nous assistons depuis une vingtaine d'années à une évolution ultra-rapide des méthodes d'investigation et de diagnostic cliniques. Ces méthodes se sont diversifiées et améliorées en termes de précision et de rapidité.
Parallèlement, des spécialistes bio-informaticiens, physiciens et autres, s'interposent dans le dialogue singulier entre le médecin et son patient. La médecine personnalisée exige une interdisciplinarité enrichissante pour les équipes à condition qu'elles disposent de personnels formés. Elle peut apparaître déstabilisante pour les patients qui subissent de multiples examens, dont le caractère parfois anxiogène ne doit pas être sous-estimé. Aussi, en tant que parlementaires de l'Office, sommes-nous attachés à l'information du public quant aux avancées scientifiques et technologiques nécessaires à la personnalisation des traitements, pour éviter qu'elles ne suscitent des craintes infondées ou des espoirs inconsidérés.
En présentant l'étude de faisabilité devant nos collègues de l'Office, nous nous sommes attachés à déterminer avec eux les principales questions que soulève la médecine personnalisée.
Sur la définition du concept, qui n'est pas simple, et sur laquelle nous allons entendre le Pr Axel Kahn et le Président André Syrota, nous espérons que les débats d'aujourd'hui apporteront des éclaircissements, tant il apparaît que les conceptions sont variables.
C'est la raison pour laquelle il nous est apparu intéressant de cerner les diverses pathologies, pour lesquelles les traitements ciblés et personnalisés avaient permis des progrès notables, ou pouvaient susciter des espoirs à court ou moyen terme, et d'interroger à cet égard les institutions qui sont dédiées à ces pathologies. Tel sera l'objet de notre première table ronde. Nous savons que nous ne couvrirons pas tout le spectre des applications possibles de la médecine personnalisée et des traitements ciblés, mais il nous a paru important de tenter cette première approche que nous complèterons par des visites sur le terrain.
Nous avons observé combien la médecine personnalisée, qu'elle soit qualifiée de médecine génomique personnalisée ou de médecine stratifiée, voire de médecine prédictive, dépendait largement de l'interaction entre les avancées technologiques, notamment en bio-informatique, et des progrès de la recherche fondamentale et translationnelle. S'agissant par exemple du temps de séquençage d'un génome entier comme du coût de ce séquençage, chacun constate une réduction drastique de la durée de l'opération qui prend aujourd'hui moins de deux jours pour plusieurs génomes ; ceci s'accompagne d'une baisse des coûts ces dernières années, plus rapide que ne le permet la loi de Moore. Ainsi, il était de 500 millions de dollars il y a une douzaine d'années, de quelques centaines de milliers de dollars en 2004 ou 2005 et il est aujourd'hui à environ 1 000 dollars, et ce coût s'apprête à descendre encore plus bas.
D'ailleurs, le coût du traitement numérique de ces données pourrait devenir supérieur à celui du séquençage proprement dit, car il est extrêmement complexe. Loin d'être un aboutissement, la démocratisation du séquençage apparaît de plus en plus comme un point de départ. Désormais, pouvoir obtenir, pour un coût relativement modique, le génome de vastes populations, autorisera des analyses à grande échelle qui, seules, permettront de comprendre le comportement et le rôle des gènes. À condition toutefois de disposer des ressources informatiques adéquates, de savoir gérer, protéger et interpréter les masses de données ainsi générées. Nous évoquerons cela lors de la deuxième table ronde.
Comme vous le savez, les questions liées à la formation et à la recherche dans toutes ses composantes sont essentielles au développement de la médecine personnalisée. Ces sujets seront abordés au cours de l'après-midi. Ils sont cruciaux pour préparer l'avenir.
La troisième table ronde sera consacrée à la valorisation de la recherche et au problème de la brevetabilité du vivant.
Pour anticiper les évolutions du secteur, l'industrie pharmaceutique cherche un certain nombre de pistes pour refonder son modèle de développement à court terme. Il s'agirait, pour surmonter la crise de l'innovation médicamenteuse, de changer de paradigme, d'abandonner le modèle du médicament, pour développer des traitements ciblés et les tests compagnons qui permettent de les prescrire. Nous débattrons de ce modèle de R&D avec les représentants des industries pharmaceutiques.
Le développement de la médecine personnalisée aura des implications à court et moyen terme pour notre système de santé. Il pose des questions d'égalité d'accès et de traitement, de coût et de protection des personnes contre toutes discriminations. Nous les aborderons de manière plus approfondie lors d'une prochaine audition publique.
PROPOS INTRODUCTIFS
Pr Axel Kahn, directeur de recherche à l'INSERM, membre de l'Académie des sciences et du Conseil scientifique de l'OPECST. Comme l'a dit M. le vice-président Jean-Sébastien Vialatte, la médecine a toujours tenté d'être personnalisée, c'est-à-dire de s'adapter à la nature du malade, et cela fait bien longtemps que l'on prend en compte l'âge du malade, sa situation. Est-ce une femme enceinte ? Un vieillard malade ? Y a-t-il une pathologie particulière ? On prend en compte l'insuffisance rénale pour les doses de médicaments, des incompatibilités, le diabète, etc.
Par conséquent, ce qui nous intéresse aujourd'hui, c'est de voir en quoi cette personnalisation a changé, non point de nature, mais de possibilité d'efficacité, ce qui a modifié la nature, avec l'introduction de l'outil génétique.
Effectivement, il est possible de donner plusieurs définitions de la « médecine personnalisée ». Je commencerai par le sens le plus strict du mot, le traitement le plus personnalisé possible. Et à ce stade, il peut lui-même être divisé en deux grandes catégories de propos. Premièrement, il s'agit de proposer le traitement qui est le mieux adapté aux mécanismes d'une maladie donnée, et deuxièmement, en dehors de la personnalisation, il faut essayer de prévoir les effets, bénéfiques ou maléfiques du médicament.
Tout d'abord, le traitement lié aux mécanismes des maladies. On sait depuis très longtemps qu'une maladie donnée, un cadre pathologique, peuvent résulter d'un grand nombre de mécanismes biochimiques, et il paraît de ce fait parfaitement logique que chacun de ces mécanismes puisse bénéficier, si le traitement n'est pas symptomatique, d'un apport médicamenteux tout à fait idoine et spécialisé. Le domaine dans lequel cela est pratiqué depuis fort longtemps est celui du cancer. Les proliférations tumorales peuvent être liées à des mutations de gènes multiples, et toucher, suivant la nature du phénomène déclenchant, soit la tumeur, soit son aggravation ; le traitement doit être naturellement modifié. Cela fait très longtemps qu'on a pris en compte les récepteurs hormonaux, et aujourd'hui les études sont très diversifiées.
De même, plusieurs mécanismes peuvent entraîner des maladies cardiovasculaires : une artériosclérose, une hypertension artérielle, des insuffisances rénales, et chacune d'entre elles mérite d'être traitée de manière idoine. En ce sens, tout ce qui permet de mieux adapter un traitement à la nature, aux mécanismes de la maladie, est un incontestable progrès de la médecine.
De la même manière, dans le champ des progrès, on peut émettre la possibilité de bien mieux prévoir qu'auparavant les effets délétères des médicaments, mais également des mécanismes qui joueront sur la dose efficace. Il existe des métaboliseurs plus ou moins rapides des médicaments, et par exemple, connaître ces déterminants va permettre d'adapter la dose du médicament à la rapidité de son élimination, de son métabolisme, de sa modification, etc. En ce domaine, la médecine personnalisée se situe dans la continuité de la volonté d'être de mieux en mieux adaptée à la réalité d'un malade, toujours pris dans sa globalité. Cela peut être considéré comme un très grand progrès médical.
Il existe une autre définition, que vous avez abordée, qui est de considérer que la médecine personnalisée, c'est la prédiction de l'effet des médicaments, et que de la prédiction de l'effet des médicaments on peut plus généralement s'orienter vers la prédiction de la survenue des maladies, c'est-à-dire passer de la médecine personnalisée à proprement parler à la médecine prédictive. Je n'aime pas le terme « prédictive » qui évoque Madame soleil, mais c'est le terme consacré. J'aurais préféré la médecine de « prévision », puisqu'on essaie de prévoir la survenue des maladies.
Cette médecine prédictive, il en a été fait état plusieurs fois dans ces lieux, dans les travaux de l'Office parlementaire, quand il s'est agi de traiter des tests génétiques. Mais bien entendu, et cela vient d'être souligné, les perspectives offertes aujourd'hui par la généralisation à un faible coût du séquençage du génome, ou au moins de la partie la plus significative du génome, l'exome -sans doute pour une centaine ou quelques centaines d'euros d'ici quelques années-, modifie très certainement le paysage, et mérite qu'on s'y arrête.
Je me focaliserai sur le phénomène de généralisation, voire de très grande diffusion, de la connaissance par les personnes de leurs déterminants génétiques individuels. Nous sommes à peu près tous persuadés que dans une dizaine d'années, cela fera partie des informations qui seront chargées sur le téléphone portable, et pourra ainsi être présenté dans les consultations médicales pour un traitement informatique idoine pour les paramètres qui n'auront pas été déjà analysés.
Le séquençage du génome va incontestablement permettre d'affiner considérablement, les déterminants génétiques des maladies en général, comme outil de recherche, comme outil d'épidémiologie moléculaire, comme outil de meilleure compréhension des différents mécanismes pouvant entraîner une maladie. Il s'agit là d'un outil extraordinairement précieux et nous en attendons tous des progrès.
Je voudrais surtout m'attarder sur les conséquences en termes de santé individuelle et de santé publique de la diffusion de cette méthode. Il faut être franc : il est très difficile aujourd'hui de se faire une idée claire de ce que cela donnera, tant en termes d'amélioration des paramètres de santé, de possibilités de mieux prendre en compte les maladies, qu'en termes d'impact sur l'évolution des dépenses de santé.
Chaque fois que le séquençage du génome permettra de déterminer des anomalies génétiques, qui sont des déterminants forts de la susceptibilité à une maladie qu'il est possible de traiter préventivement ou d'éviter facilement, on peut imaginer que ce sera un progrès incontestable, et tel est le cas de plusieurs situations. Je pense par exemple à l'hémochromatose. Déterminer une hémochromatose permet tout simplement de proposer à quelqu'un d'être donneur de sang régulier et d'éviter que le fer ne s'accumule, et par conséquent de tomber malade.
Mais en réalité, dans l'immense majorité des cas, les déterminants génétiques ne sont pas des déterminants de pénétrance forte, mais de pénétrance extrêmement faible. Il n'empêche que ce sont des renseignements extrêmement importants d'un point de vue scientifique, pour autant le bénéfice individuel en est aujourd'hui incertain. Quel bénéfice une personne, individuellement, peut-elle tirer de la notion qu'elle a un déterminant qui intervient dans 3 %, 4 % ou 10 % de la pathologie ? C'est évidemment incertain. La question qui se pose et que vous discuterez est la suivante : cela pourra-t-il dans le futur être utilisé pour déterminer des agrégats de risques assurantiels ? J'étais il y a quelques jours devant une assemblée d'assureurs justement pour traiter ce point qui est d'une extraordinaire importance pour le futur, et notamment, la place de la solidarité dans les politiques publiques.
Dans certains cas, l'apport est incontestable, dans de très nombreux cas, le renseignement scientifique est tout à fait évident et précieux, et l'avantage individuel est incertain. Quand bien même l'avantage serait tout à fait évident, parce qu'il serait possible de proposer des traitements préventifs, ou en tout cas une hygiène de vie permettant de diminuer un risque auquel on est sensible, il reste une question majeure qu'il ne faut jamais oublier : entre la connaissance de la certitude d'un risque et la modification des pratiques pour éviter la manifestation de ce risque, se trouve un immense écart. On sait que la voiture est extrêmement dangereuse, il n'empêche, on roule en voiture et on a des accidents. On sait que fumer donne le cancer de la vessie, du poumon et bien d'autres maladies, et on a énormément de difficultés à diminuer l'usage du tabac. On sait que manger trop, notamment certains aliments qui rendent obèses, provoquent des maladies cardiovasculaires, il n'empêche que dans nos pays, un problème de cet ordre demeure.
Ainsi l'idée selon laquelle une multiplication extraordinaire des connaissances des susceptibilités sur une base génétique, aboutira très facilement à une modification très généralisée des modes de vie, est sans doute illusoire, quand bien même cette information sera importante. Cela mérite qu'on y travaille plus, mais reste sans doute totalement illusoire.
En termes de dépenses de santé, de la même manière, il est tout à fait impossible de prédire ce que seront les conséquences de la généralisation de ces pratiques, puisque d'un côté, il peut être beaucoup moins onéreux en effet d'éviter qu'une maladie ne survienne par des dispositions ad hoc plutôt que de la soigner. D'un autre côté, médicaliser des risques relatifs de manière tout à fait généralisée peut représenter un surcoût de santé.
En outre, vous l'avez dit, et c'est tout à fait important, généralement la médecine prédictive et la médecine personnalisée amèneront dans le champ économique l'industrie pharmaceutique à se reposer la question de ses modèles, puisque, à terme, la subdivision de tout champ pathologique en un très grand nombre de sous-domaines, chacun justifiable d'un traitement particulier, marque la fin presque généralisée du modèle de blockbuster sur lequel l'industrie pharmaceutique reposait jusqu'à présent. D'un côté, cela permet de réhabiliter des médicaments qui n'auraient pas démontré leur efficacité dans le champ pathologique qui est global, de l'autre, cela diminue le champ d'utilisation d'une drogue donnée, et par conséquent, pose des problèmes de rentabilité généralisée. C'est une question sur laquelle il faudra se pencher. Nous n'avons pas de réponse aujourd'hui.
En définitive, je crois en effet que le champ ouvert aujourd'hui, par cette possibilité totalement bénéfique d'améliorer l'abord thérapeutique des patients, en déterminant le ou les médicaments les mieux adaptés à un mécanisme donné, en évitant leurs effets délétères, en disposant dans un bon nombre de cas de la possibilité d'intervenir avant que la maladie ne survienne, sont réellement des progrès sinon des triomphes de la médecine. Comme toujours, il faut se garder des idéologies et des illusions. L'illusion selon laquelle on pourra connaître tout dans l'avenir est tout à fait folle.
En réalité, et je terminerai par ce point : il est bon de se rappeler qu'aujourd'hui 30 % de la morbidité est liée à des conduites à risques, lesquelles sont liées à une réaction psychologique à un environnement psychologique, socio-économique, etc. Certains pensent que les conduites à risques sont également génétiquement déterminées. Tel n'est point mon avis très largement. De ce point de vue, je termine par une affirmation qu'un généticien est en situation de rappeler : un gène ne code jamais un destin. L'idée selon laquelle un gène code un destin est une idée fausse, qui procède d'une méconnaissance du gène. Un gène code des propriétés d'une cellule, ou, si l'on connaît parfaitement le jeu des gènes travaillant ensemble, les propriétés d'un organisme. Aussi le destin est-il la résultante d'un être vivant qui dispose de propriétés données, confrontées à un environnement qui lui n'est pas génétiquement déterminé.
Pr André Syrota, président-directeur général de l'Institut national de la santé et de la recherche médicale (INSERM), président de l'Alliance nationale pour les sciences de la vie et de la santé (AVIESAN). Le concept de médecine personnalisée est d'abord apparu aux États-Unis dans les années 2000, mais en fait, la réalité qu'il recouvre est déjà plus ancienne. Cette médecine personnalisée, qu'on appelle aussi maintenant plutôt la médecine stratifiée, la taylor-made medicine ou la médecine sur mesure, est issue du transfert progressif vers la clinique des progrès scientifiques et technologiques qui ont été observés à partir des années quatre-vingts. Ces progrès sont issus de la recherche académique, et pas de la recherche provenant des grandes compagnies pharmaceutiques.
Parmi ces progrès des connaissances, des méthodes et des outils, on peut citer les techniques d'analyse biologique, le séquençage déjà évoqué, mais aussi l'immuno-analyse, l'hybridation moléculaire, la spectrométrie de masse, qui ont engendré d'abord la génomique, puis dans son sillage toutes les omiques liées à la lecture, de plus en plus rapide, de plus en plus complète, du vivant à l'échelle atomique, à l'échelle moléculaire, à l'échelle cellulaire. On a eu aussi les évolutions d'imagerie in vitro, in vivo , avec les résolutions spatiales, temporelles qui n'ont fait que croître. S'y sont ajoutées les avancées de la numérisation, de la bio-informatique, les micro-nanotechnologies, qui permettent de réaliser des analyses biologiques de plus en plus détaillées sur des surfaces de plus en plus réduites, les bio-puces, mais aussi d'enregistrer, de conserver, de transférer, de croiser, de grandes quantités biomédicales, le big data , ou encore de cibler, avec une extrême précision la thérapie, nano-médicaments, etc. Puis, sont venus ce qu'on a appelé autrefois le génie génétique, la biologie de synthèse, nourrissent les biothérapies géniques, les biothérapies cellulaires, la production de tissus, d'organes, de biomatériaux, dans une approche régénérative.
Les conséquences de toutes ces évolutions scientifiques et technologiques sur la médecine ont été évidemment extrêmement importantes. On a découvert la complexité intrinsèque du vivant qui a fait sortir, maintenant depuis longtemps, du paradigme « un gène - une protéine - une maladie ». Plus on comprend le fonctionnement normal et pathologique des gènes, des cellules, des tissus, des organes, plus on constate la très grande diversité de leurs altérations, et donc de leur protection ou de leur réparation. En d'autres termes, la médecine devient individualisée parce que le vivant est individualisé. Le corollaire de cela, est que la découverte de nouvelles cibles, de nouvelles voies de signalisation, de leur modification au cours de telle ou telle pathologie, ouvre chaque fois des perspectives nouvelles pour les malades. Cette recherche, c'est essentiellement une recherche académique.
Chaque patient possède un patrimoine génétique particulier qui peut le prédisposer à certaines maladies. On a évoqué la baisse importante du coût du séquençage, (pour le génome entier on atteindra moins de 1 000 euros par patient très rapidement. On pourra identifier pour chaque individu des gènes, ou des combinaisons de gènes, susceptibles d'augmenter ou de diminuer des facteurs de risques pour des pathologies fréquentes ou des pathologies rares. Ces tests sont techniquement possibles, dès la naissance, mais également au stade foetal, voire avant, si on est capable de détecter les cellules embryonnaires qui circulent précocement dans le sang maternel.
Chaque patient possède un environnement spécifique, lequel, croisé à son génotype, présentera ou non une augmentation des facteurs de risques dans le déclenchement des pathologies. On sait bien que les maladies les plus fréquentes, les maladies inflammatoires, cardiovasculaires, tumorales, neurologiques, émergent à la rencontre d'un terrain génétique favorable et d'influences externes du milieu liées aux pratiques de vie : l'alimentation, la sédentarité, les addictions, la profession, etc. Donc la personnalisation implique la prise en compte de cette dimension environnementale. Aujourd'hui, on parle de nutrigénomique, qui analyse l'alimentation sur la santé via le métabolisme des macronutriments. Un autre sujet se développe aussi, notamment aux États-Unis, l'étude de l'exposome, qui consiste à prendre en compte le maximum de données environnementales, qu'on croise à la génétique.
Enfin, les pathologies elles-mêmes sont mieux caractérisées qu'autrefois. Il existe aujourd'hui de nombreux sous-types qui n'empruntent pas les mêmes voies de signalisation moléculaire et cellulaire ; autrement dit ce sont des maladies différentes. Des consortiums internationaux réunissent désormais des cohortes de grande taille sur chaque type de maladie, qui mettent en évidence des dizaines, voire des centaines de profils
Il existe aussi une nouvelle nosographie qui se dessine, même pour des maladies considérées apparemment comme simples, je pense par exemple à l'asthme. Il en va de même pour les traitements. Tous les patients, le Pr Axel Kahn l'a expliqué, ne répondent pas de la même manière à un médicament, que ce soit en terme d'efficacité ou d'effets secondaires, et les traitements ne sont pas forcément adaptés à tous les sous-groupes de leurs pathologies cibles.
La pharmaco-génomique est aussi en train de bouleverser les analyses d'innocuité, de tolérabilité, d'efficacité, là encore, dans le sens d'une plus grande précision et de personnalisation, de même que le pronostic de réponse au traitement est considérablement affiné pour un patient donné.
Outre la conception du médicament, son administration est aussi en train de changer. C'est le principe du ciblage thérapeutique, qui permet d'administrer le médicament au bon endroit, au bon moment, avec les bonnes doses, chez le bon malade. Or ces paramètres sont variables selon la personne et sa pathologie.
Les débats et les échanges de cette journée vont permettre de préciser les enjeux scientifiques, technologiques et cliniques que je viens d'esquisser. Et l'on comprend que la médecine personnalisée évoluera toujours à la frontière entre les connaissances les plus récemment acquises et leurs applications cliniques dans le système de soins.
J'aimerais souligner quelques défis et enjeux de cette médecine personnalisée sur les plans éthique, économique et sociétal. Au niveau éthique, il ne s'agit pas seulement de comprendre avec une précision croissante les mécanismes du vivant, puisqu'on peut désormais intervenir sur eux. L'humanité gagne en savoir, elle gagne en pouvoir : corriger des gènes, produire des tissus, restaurer des organes, poursuivre ou interrompre un développement embryonnaire.
Le vivant est de plus en plus scruté, maîtrisé, artificialisé, et la personne humaine n'échappe pas à cette tendance d'ores et déjà avec le diagnostic prénatal, le diagnostic préimplantatoire, le Pr Axel Kahn a fait allusion aux tests génétiques, tout cela pose de nombreux problèmes. Les patients sont-ils correctement informés de la signification des biomarqueurs, notamment lorsque leur interprétation est probabiliste ? Existe-t-il des limites à l'intervention selon la nature des maladies, ou des handicaps, dont on identifie un risque ? Les données médicalisées personnelles, qui sont désormais faciles à stocker et à transférer numériquement, disposent-elles de garanties suffisantes en matière de confidentialité et de respect de la vie privée ? Les systèmes de protection publique, la sécurité sociale, ou privés, les assurances et les mutuelles, risquent-ils d'exercer des pressions pour vérifier l'existence du diagnostic ou du pronostic sur la santé de l'individu, et cela parfois même avant sa naissance ? On voit que chaque médaille a son revers, et que la médecine personnalisée ne peut être réduite à sa promesse de bien-être.
Sur le plan économique, les progrès de la médecine expérimentale moderne ont notamment été fondés sur la découverte de médicaments à très large spectre. Par exemple, autrefois les vaccins, les antibiotiques ont été à leur manière les premiers blockbusters de l'industrie pharmaceutique. Le développement de nouveaux traitements coûte de plus en plus cher, avec un délai de mise sur le marché de plus en plus long, et une probabilité de succès de plus en plus difficile à estimer d'avance. Le blockbuster assurait un certain équilibre dans le portefeuille des molécules détenues par un industriel, et pour les systèmes de santé publique, les traitements à large spectre permettaient de faire facilement accéder tous les citoyens à un même niveau de soins. La médecine personnalisée va à l'encontre de la logique du grand nombre. Elle consiste au contraire à subdiviser la population générale de patients en groupes, en sous-groupes, de plus en plus réduits, afin de choisir les traitements les plus adaptés à chaque profil. Quel sera dès lors le modèle de retour sur investissement, notamment pour les sociétés pharmaceutiques et biotechnologiques, qui travaillent sur des traitements de niche ? Les thérapies les plus avancées coûtent des dizaines, des centaines de milliers d'euros par an. Comment financera-t-on leur accès ?
L'ascension de la médecine personnalisée n'est donc pas séparable d'un questionnement de fond sur l'économie de la recherche industrielle et de la santé publique, et du rapport entre la recherche académique et la recherche industrielle. Je ne vais pas m'attarder sur les fermetures de centres de recherche d'industries pharmaceutiques. AstraZeneca vient encore d'annoncer il y a deux jours 5 000 suppressions d'emplois, sans parler d'autres sociétés dans le même cas.
Sur les plans sociaux et sociétaux, la médecine personnalisée soulève d'immenses espoirs chez les patients. Pour soi, pour les proches, chacun souhaite évidemment bénéficier d'une médecine de pointe, où la qualité de la prévention, la précision du diagnostic, l'efficacité du traitement, seront optimisés. Le niveau d'information du grand public est désormais élevé. Les médias ne manquent pas de couvrir les percées médicales et les découvertes scientifiques. L'INSERM d'ailleurs y concourt, je crois, largement.
Les associations sont très investies aux côtés de la recherche, et parfaitement au fait des promesses cliniques que recèlent les avancées biomédicales. Ainsi on ne compte pas moins de 475 associations de malades au sein du Groupe de réflexion avec les associations de malades de l'INSERM, (GRAM), intéressé par cette médecine personnalisée. Les attentes sont donc fortes, chez les patients et leurs familles. Cela pose un autre problème celui de la démocratisation de la médecine personnalisée dans des systèmes de soins qui, théoriquement, ont des objectifs égalitaires. La médecine dite de masse n'était pas parvenue à réduire toutes les inégalités d'accès aux soins, mais ne court-on pas le risque, avec la médecine personnalisée, de voir l'émergence d'une médecine plus dédiées aux riches qu'aux autres ?
Toujours au plan sociétal se pose la question de la frontière entre le soin et le confort. Les pathologies les plus invalidantes font évidemment l'unanimité, mais la personnalisation, et la précision du diagnostic génétique, par exemple, pourront concerner, non pas seulement de grandes maladies, mais aussi de simples caractères du phénotype, pas forcément considérés comme des anomalies ou des maladies. Par exemple, un trait physique ou un trait cognitif. La médecine personnalisée risque-t-elle d'opérer un changement de paradigme, en ajoutant aux vocations préventives et curatives de la médecine déjà évoquées, des dimensions régénératives, ou, pourrait-on dire, « amélioratrices » ? Comment s'articulera l'équilibre entre l'offre et la demande d'intervention sur ces plans ?
Il nous faut aujourd'hui anticiper toutes ces questions éthiques, économiques, sociétales. C'est la condition pour ne pas briser l'élan prometteur de la médecine personnalisée, et pour s'assurer qu'elle deviendra progressivement la routine médicale des sociétés de demain. Les percées scientifiques ont toujours perturbé, voire déstabilisé nos systèmes de pensée et leurs modes d'action. C'est probablement nécessaire pour les faire évoluer, mais la responsabilité nous revient de préserver, au cours de cette mutation nécessaire, un équilibre collectif à long terme.
Pour conclure, du point de vue d'un responsable d'un organisme public, jamais autant de perspectives ne se sont ouvertes dans la recherche biomédicale. Le concept de médecine personnalisée s'est imposé par les avancées de la recherche fondamentale. Jamais autant de possibilités de création d'entreprises ne se sont offertes. Or tous les pays avancés font le même constat : à nous de saisir les opportunités, c'est-à-dire d'être aussi capables de financer ces recherches et de continuer à être attractifs pour les jeunes chercheurs, notamment, les post-doctorants. Ce n'est pas simple. Nous étions lundi à Lyon pour la mise en place du Comité stratégique de filières des industries et technologies de santé. Il y a des espoirs ; il faut qu'on soit à même d'y répondre.
PREMIÈRE TABLE RONDE :
POUR QUELLES
PATHOLOGIES ?
|
Point de vue :
|
J'adresse d'abord un grand merci aux orateurs précédents. Ils ont très bien posé cette question moderne, assez complexe, et aux contours un peu flous, qu'est la médecine personnalisée. Il s'agit d'une attente majeure de la part de nos concitoyens, attente qui est audible, nécessaire, et parfois excessive d'une personnalisation complète de la médecine. En effet, chaque individu a un peu tendance à vouloir être considéré comme unique, singulier. Évidemment, il est légitime que l'on substitue au traitement des maladies, le traitement des malades en particulier. Pour autant, tous les progrès du diagnostic, de la thérapeutique, ont exigé de définir des lois générales basées sur l'étude de groupes de patients. L'individualisation de la médecine a des limites, même si elle est nécessaire, pour tenir compte de chacune des caractéristiques de chaque malade particulier.
La médecine personnalisée a connu un essor que vous connaissez tous, notamment en oncologie et dans l'industrie pharmaceutique. En oncologie, une nouvelle approche s'est substituée à l'approche précédente qui consistait à donner par exemple, une première ligne de chimiothérapie à des patients qui devaient en subir les effets adverses éventuels, mais pas toujours le bénéfice. Quand cette première ligne de chimiothérapie n'avait pas d'efficacité sur la tumeur, on passait alors à une autre variété de chimiothérapie, jusqu'à ce qu'on trouve le traitement qui empiriquement s'avérait d'une relative ou d'une grande efficacité. Aujourd'hui cela ne se fait plus de cette façon. Dans un nombre croissant de cancers, on dispose de la possibilité de définir d'emblée, grâce aux biomarqueurs, le choix de la chimiothérapie efficace. Cela est également vrai pour divers autres traitements. Il y a là effectivement une stratification, déjà évoquée, et qui est d'une grande importance.
L'industrie pharmaceutique a développé cela, en évoquant un nouveau paradigme, et aussi un nouveau modèle économique. En effet cela aboutit à définir des traitements pour des groupes plus restreints de patients, ce qui exige tout autant d'investissements pour l'étude et le développement de chaque traitement, lequel aura un champ d'application plus restreint. Peut-être s'agit-il là d'une stratification de la médecine plutôt que d'une authentique personnalisation, mais cela rentre à l'évidence dans le champ de ce que nous traitons aujourd'hui.
La médecine personnalisée concerne aussi nombre d'autres maladies chroniques qui selon la Haute autorité de santé (HAS), touchent aujourd'hui au moins 15 millions de Français. C'est donc un champ immense. On peut citer le diabète de type 1 et encore plus, peut-être celui de type 2. Là encore une approche stratifiée est apparue opportune, permettant d'adapter le traitement au patient en fonction de ses caractéristiques biologiques, mais aussi de son comportement, de ses habitudes, de son habitus. Cela a abouti à définir des profils dont les particularités permettent de mieux savoir quelles seront les réponses au traitement et les préconisations. On est donc conduit à des soins plus personnalisés du diabète que ce qui était traditionnel.
Il en va de même dans le champ vaste des maladies infectieuses. Le séquençage d'une variété de virus ou de bactéries permet de progresser dans l'adaptation du traitement. Mais depuis très longtemps la seule pratique de l'antibiogramme permettait de définir l'antibiotique adapté contre telle ou telle maladie infectieuse. Actuellement, dans le cas du Sida par exemple, dont traitera sans doute le Pr Jean-François Delfraissy, on dispose maintenant pour chacun des patients du génotypage du VIH. Cela permet de choisir la trithérapie adaptée hors des résistances à l'un ou l'autre des médicaments du fait de mutations d'un virus qui a malheureusement une propension très importante à muter et à développer par ces mutations des résistances à certains des antirétroviraux. Donc on a là la définition d'une adaptation très précise du traitement en fonction des caractéristiques, cette fois du virus, bien plus que du patient.
Quant aux maladies dites rares ou orphelines, elles ne sont pas si rares que cela à l'échelle mondiale, mais le nombre de patients est limité, et donc leur traitement par « un médicament orphelin », est compliqué à développer, restant cher à produire pour un groupe restreint de patients. Cela oblige à définir des traitements extrêmement ciblés. Le Pr Odile Kremp dressera sans doute un état des lieux des approches thérapeutiques dans ce domaine.
La médecine personnalisée s'applique également à la procréation. Avec le développement des diagnostics prénataux et plus récemment préimplantatoires, on pourra arriver à cibler ou à personnaliser les décisions opportunes. De nouvelles techniques permettent maintenant d'extraire l'ADN foetal dans le sang maternel, et donc d'aller encore plus loin dans la personnalisation et le choix de la décision souhaitée par les familles et par les équipes compétentes.
Pour terminer, je voudrais évoquer la question des frontières, qui restent floues concernant la médecine personnalisée. Quand se trouve-t-on vraiment dans le champ des traitements ciblés, d'une médecine stratifiée, d'une médecine sur mesure, comme cela a été indiqué, ou dans celui d'un traitement parfaitement individualisé ? Ce dernier existe, par exemple dans le domaine de la thérapie cellulaire, mais c'est à peu près le seul cas où le traitement est vraiment individualisé. Le Pr Marina Cavazzana l'explicitera. C'est le cas où l'on prélève des cellules à un patient, on les traite in vitro pour leur conférer des propriétés additionnelles, et on réinjecte au patient ses propres cellules. C'est un système de médecine totalement personnalisée, voire individualisée. Entre cet extrême-là et la médecine stratifiée que j'ai évoquée au début, il y a tous les intermédiaires. On le comprend, le champ de la médecine personnalisée est vaste, important, et il définit une authentique mutation dans nos pratiques médicales.
*
* *
Pr Jean François Delfraissy, directeur de l'Agence nationale de recherches sur le sida et les hépatites virales (ANRS). Médecin à l'hôpital Bicêtre, je dirige l'ANRS et l'Institut thématique multi-organismes (ITMO) « Microbiologie et maladies infectieuses » de l'AVIESAN. Dans le cadre de l'ensemble des maladies infectieuses nous avons du retard en matière de médecine personnalisée, d'après les différentes définitions qui ont été données. On dispose de beaucoup d'informations sur la génétique des bactéries, des virus, sur les diagnostics de résistance, de sensibilité, mais comme le rappelait judicieusement le Pr Axel Kahn, les bactéries ne sont pas encore des personnes. Et finalement, ce qui est lié aux personnes est lié à l'hôte, à la réponse de l'hôte, et à l'interaction entre l'hôte et le virus, ou la bactérie par exemple.
La communauté que je représente est en retard sur ce sujet, et par rapport à ce qui se passe en oncologie, en métabolisme, on a encore un certain nombre de questions. Cette communauté a vécu avec les antibiotiques. Un agent pathogène responsable d'une maladie est quelque chose d'aigu. On n'a pas trop le temps de réfléchir, l'attaquer avec un antibiotique. Il faut disposer de l'antibiotique le plus adapté à la bactérie. La puissance des antibiotiques a été longtemps importante, même si elle l'est moins maintenant. Dans les maladies virales chroniques que sont l'infection VIH ou les hépatites, la puissance s des antiviraux, après des débuts difficiles, est maintenant importante. Face à cette puissance, fallait-il vraiment s'intéresser à la personnalisation ?
On sait maintenant qu'on a affaire à des maladies infectieuses chroniques, et la réflexion vis-à-vis des biomarqueurs se modifie. Je rappelle que les maladies infectieuses, au sens très large du terme, demeurent la deuxième grande cause de mortalité dans le monde. Je conseille à tout le monde la lecture de l'éditorial du New England Journal of Medicine : « Preparing for Precision Medicine » daté du 13 mars 2012. Il pose véritablement les grandes questions, indépendamment des maladies infectieuses, en termes de société, de conséquences, et pour tous les décideurs. D'ailleurs à l'expression « médecine personnalisée », il préfère « Precision Medicine », et là, on est au coeur du débat pour un médecin.
En ce qui concerne les maladies infectieuses, je soulèverai quatre points. Après vous avoir expliqué qu'on n'était pas en avance, je reconnais que des avancées ont quand même été faites.
La première grande approche, concerne les marqueurs d'évolutivité et de pronostic des maladies infectieuses. Nous sommes inégaux devant les maladies infectieuses : une petite fraction de la population va avoir une forme très grave de l'infection, alors que la majorité n'aura qu'une forme intermédiaire, quand d'autres n'auront rien du tout. Une première grande question se pose : qui fait des formes graves ? Est-on capable de trouver un profil, avec les techniques d'omique qui ont déjà été évoquées ? Dans le cas d'un sepsis grave par exemple, le projet génétique est-il le même que pour une maladie virale x ou y ? Qu'en est-il des cas de certaines personnes qui développent des tuberculoses graves ou des réactions très particulières au BCG ?
La France est très en pointe sur certains domaines de la médecine personnalisée. L'équipe du Pr Jean-Daniel Casanova (Hôpital Necker) par exemple, est finalement partie d'une observation très simple sur les enfants qui développent une réaction très particulière après un BCG. Elle a commencé à se demander qui faisait une tuberculose grave et a trouvé un certain nombre de marqueurs, en particulier de récepteurs de cytokines, ou de relations entre récepteurs et différents types de cytokines, montrant l'existence de mécanismes intimes de la réponse cellulaire pouvant être prévus et qui faisaient qu'on développait ou non, une tuberculose grave, à la fois au nord et au sud.
En France, nous disposons d'un réseau de réanimateurs qui s'intéressent au sepsis grave. Pourquoi quelqu'un en pleine forme développe-t-il tout d'un coup, à l'occasion d'une infection, un choc septique et se retrouve en réanimation, avec une défaillance poly-viscérale dans les heures qui suivent, et a un sepsis grave ? Quels sont les marqueurs qui vont mettre en évidence un sepsis grave, cet orage de cytokines ? Nous disposons d'une étude sur ce sujet qui démontre d'abord l'intérêt des grandes cohortes. Avec tout l'omique possible, on peut essayer de mettre en évidence certains marqueurs. Ce sont des marqueurs d'évolutivité et de pronostic, et ce n'est pas tout à fait de la médecine personnalisée. On se trouve encore dans la compréhension, dans la physiopathologie. Mais voilà une approche en maladies infectieuses.
Le dernier exemple, ce sont les vaccins. Pour des raisons qu'on ne comprend pas bien, environ 3 % des personnes ne répondent pas à un vaccin. Ces non-répondeurs, on a beau les vacciner, les revacciner, ils ne répondent pas à un vaccin. Ils peuvent répondre à certains vaccins, mais pas à d'autres. On constate qu'il existe une relation entre la génétique de l'hôte et les peptides des bactéries ou virus contre lesquels on veut vacciner. L'approche des non-répondeurs vaccinaux est également très intéressante. Dans le cadre de l'ITMO « Microbiologie et maladies infectieuses », on est en train de monter une banque de données sur les non-répondeurs pour essayer d'expliquer pourquoi certaines personnes ne répondent pas aux vaccins. À partir du moment où l'on est capable de comprendre ce type de phénomènes, on pourra éventuellement développer des vaccins du futur.
La deuxième grande approche en matière de maladies infectieuses est à nos portes. C'est le « point-of-care », le lieu d'intervention des soins, ou comment faire de la microbiologie sans culture, avec la possibilité, au lit du patient, en moins d'une heure, d'obtenir des conditions de diagnostic. S'agit-il d'une mycobactérie ou d'une mycobactérie atypique par exemple ? Est-elle sensible ou résistante ? Cela prend du temps. Le « point-of-care » n'est pas encore très courant, ni en France ni même aux États-Unis, y compris dans les très grands centres où on n'en dispose pas encore au lit du patient. La bactériologie sans culture est à la fois loin et très proche !
En France par exemple, la société bioMérieux s'est beaucoup investie dans ce domaine qui compte aussi d'autres grandes entreprises. Nous sommes en pointe sur ce sujet. Des biomarqueurs de sensibilité à différentes molécules, d'identification et de résistance, devraient être disponibles d'ici deux à trois ans, et disponibles dans les hôpitaux d'ici cinq ans. Cela changera complètement la donne en termes de diagnostic et de prise en charge. C'est un peu en marge, mais pas si loin que cela de la médecine personnalisée, ou en tout cas de la « Precision Medicine ».
Un des exemples, qui ne vaut pas seulement pour le nord, mais aussi pour le sud -parce qu'en matière de maladies infectieuses, il faut en permanence se préoccuper de ce qui se passe au sud-, est la nouvelle technique dite « GeneXpert». Elle a commencé à des coûts très impressionnants, mais aujourd'hui elle est de l'ordre de 10 à 15 dollars. Elle permet d'effectuer le diagnostic de la tuberculose et de la sensibilité de la tuberculose en moins d'une demi-heure. Avant, il fallait six semaines de culture. Cela change complètement la donne, que ce soit au nord ou au sud. Voici un exemple précis : dans les prisons françaises, l'incidence de la tuberculose est particulièrement importante et si l'on n'a pas un diagnostic très rapide, imaginez ce qui adviendra dans une cellule de huit prisonniers. Voilà une conséquence sociétale.
En troisième point, je reviendrai au coeur du débat, sur la réponse au traitement et sur la notion de bio-médicament, et de cible thérapeutique. En matière de maladies infectieuses, nous sommes beaucoup moins avancés que dans d'autres domaines, comme l'oncologie et le métabolisme. On progressera sur les facteurs liés à l'hôte, on enregistre déjà un certain nombre de grands succès. Par exemple, sur la réponse dans le traitement des hépatites, en particulier des hépatites C. Jusqu'à maintenant, le traitement des hépatites C comprenait de l'Interféron pégylé plus d'autres molécules, auxquelles certaines personnes répondaient bien et d'autres moins bien.
Il existe un marqueur de réponse à l'Interféron, avec une expression génique, et ensuite protéique, qui est liée à l'expression de IL28. Selon que l'on a ou pas ce marqueur, la réponse au traitement dans les hépatites est complètement différente. Ce grand succès est probablement celui pour lequel on a trouvé la corrélation la plus forte, hors cancérologie bien sûr, entre une sensibilité à un traitement et ce marqueur. On n'avait pas du tout obtenu cela dans l'infection HIV. Cela vous montre aussi la relativité des choses. Associées à ce traitement à l'Interféron, arrivent depuis deux ans, d'autres molécules, plus ciblées, et qui surpassent l'efficacité de l'Interféron. Et là, le marqueur IL28 ne joue plus de rôle, parce qu'il est surpassé par la puissance du traitement. C'est ce que j'évoquais précédemment au sujet des maladies infectieuses au sens large du terme. Selon la puissance des agents infectieux dont on dispose au départ, le marqueur de sensibilité aura un rôle plus ou moins important. Il s'agit là d'un très grand succès issu de la recherche fondamentale, et non pas de l'industrie, et qui ensuite est passé en recherche translationnelle dans un délai très court. Tout le monde utilise ce marqueur pour le suivi des patients. Dans trois ans, on ne l'utilisera probablement plus, parce que des médicaments complètement nouveaux l'auront surpassé. En ce qui concerne la tuberculose multi-résistante, on dispose également d'un certain nombre de marqueurs de ce type, et de sensibilité en fonction de l'hôte. Cela a été évoqué sur le tropisme viral dans l'HIV, où l'on est capable de mieux cibler. Mais là aussi, ce n'est pas tout à fait lié à l'hôte, c'est plus lié à l'agent pathogène, et donc on se trouve à la frontière de la médecine personnalisée.
Enfin, sur les effets secondaires des médicaments, je vous cite un exemple pour en montrer l'intérêt et avoir une réflexion plus globale. Il existe une molécule qui est utilisée dans le traitement du VIH et qui entraîne des effets secondaires chez certains patients. C'est l'Abacavir. Chez certains patients qui ont un HLA ( Human Leukocyte Antigene , antigène de leucocyte humain) particulier, l'Abacavir entraîne des intolérances qui peuvent être graves, voire quasi mortelles. D'où le rôle d'un marqueur permettant de ne pas recourir à l'Abacavir, utilisé en pratique courante, pour éviter les effets secondaires. Il est intéressant de constater que ce marqueur n'est pas universel. L'expression de ce sous-type de HLA particulier est fréquente dans le nord de l'Europe, intermédiaire dans nos pays, et n'existe pratiquement pas dans les pays du sud. Donc voilà un médicament qui nécessite un biomarqueur pour le nord, et qui, pour l'Afrique par exemple, peut être utilisé sans biomarqueur sans aucun problème.
En quatrième point, j'aborderai la nouvelle classification des maladies et les sous-classes de patients, évoquées tout à l'heure, pour lesquelles la « Precision Medicine » s'adapte parfaitement bien. Il s'agit de patients déjà traités pour des maladies virales chroniques, par exemple l'hépatite B ou le VIH. Ils sont traités, vont bien, mais les effets secondaires de ces traitements sont lourds, par exemple l'adhésion à la trithérapie pour les patients VIH. Comment peut-on avancer pour une personne qui a une charge virale indétectable sous trithérapie ? Est-ce un bloc homogène ? Non, c'est un bloc très hétérogène. À partir de là, il nous faut une certaine forme de sous classification de ces patients, soit parce qu'ils peuvent être éventuellement de futurs contrôleurs induits par le traitement (vous avez vu qu'on a commencé à décrire que c'était une des approches dans l'éradication fonctionnelle : comment peut-on les repérer ?), soit parce qu'on peut simplifier le traitement, en ayant moins de traitement. L' International AIDS Society a d'ailleurs tenu une réunion organisée sur ce thème des biomarqueurs pour essayer de sortir de la notion de maladie chronique et arriver à avoir des éléments plus ciblés autour de tel ou tel type de médicament. C'est vraiment une problématique pour ces deux grandes infections chroniques que sont l'hépatite B et le VIH.
Les conséquences de ces réflexions dépassent les maladies infectieuses. En termes de recherche, en particulier pour la recherche translationnelle, pour aborder ce type de problèmes, on n'aura peut-être plus besoin non plus, de grands essais randomisés pendant une très longue période. Il s'agit d'une vraie réflexion car ces grands essais randomisés sont longs et coûteux et c'est ce qui explique aussi le coût d'un certain nombre de molécules. À partir du moment où l'on sélectionne les patients au départ, on peut progresser bien plus vite avec des essais de plus petite taille.
La deuxième conséquence concerne la relation avec l'industrie, mais aussi avec les autorités de santé, les autorités d'enregistrement. Les critères d'enregistrement des médicaments peuvent être profondément bouleversés, en particulier pour les autorisations de mise sur le marché (AMM) et les équivalents.
Enfin, les associations de patients doivent aussi s'interroger sur ce que signifie la médecine personnalisée. Elles sont en général les représentantes d'une pathologie dans le cadre du VIH ou des hépatites. À présent, on leur demandera de mener une réflexion qui ne soit plus celle d'un bloc social complet concernant une pathologie donnée, mais de s'interroger sur leur hétérogénéité aussi à l'intérieur d'elles-mêmes afin d'avoir une réflexion autour de sous-groupes personnalisés. Je pense que cela remodifiera un peu, probablement dans le bon sens, la relation entre les académiques, les chercheurs et le milieu associatif.
En conclusion, on évolue dans le domaine de l'infectieux. On est un peu en retard par rapport à la cancérologie, mais j'ai tenté de vous expliquer pourquoi et comment la dynamique était en train de se mettre en place.
M. Alain Claeys. Je vous remercie et nous essaierons de revenir sur l'implication des associations de malades.
Pr Odile Kremp, directrice d'Orphanet (portail d'information sur les maladies rares et les médicaments orphelins). Les maladies rares posent un problème par rapport à l'ensemble des maladies chroniques déjà évoquées. Comme vous le savez sans doute, elles sont définies par leur prévalence. La définition retenue en France est celle d'une prévalence inférieure à 1/2000. Ce taux est différent dans d'autres pays (1/1000 aux États-Unis). Ainsi, en France, moins de 30 000 personnes sont concernées pour une maladie donnée. On estime que 2 millions de Français en sont atteints, mais on n'en est pas sûr, parce que le recueil de données est difficile. Au niveau européen, cela concernerait 30 millions de patients. Certaines de ces maladies sont connues : drépanocytose, sclérose latérale amyotrophique, mucoviscidose,... Et il y a des maladies beaucoup plus rares, comme la progéria sur laquelle travaille le Pr Nicolas Levy à Marseille.
Ces maladies soulèvent de multiples enjeux, en termes de recherche, de connaissance de diagnostic, et donc de santé publique. En termes de recherche, elles sont de plus en plus prises en compte. Un consortium international de recherche sur les maladies rares a été mis en place récemment, l'IRDiRC (International Rare Diseases Research Consortium), dont l'ANR (Agence Nationale de la Recherche) fait partie. En effet, ces pathologies peuvent servir de modèle pour des maladies plus fréquentes, que ce soit en termes de physiopathologie ou de développement de médicaments. Elles ont aussi une dimension européenne très importante pour l'organisation des soins et de la recherche.
Avant tout, ces maladies constituent un enjeu de santé publique. Très souvent elles sont graves et invalidantes et démarrent tôt dans la vie. Nombre d'entre elles sont des maladies chroniques, avec des déficits sensoriels, intellectuels et moteurs associés. Elles sont graves, puisque le pronostic vital est en jeu dans la moitié des cas, et expliquent un tiers de la mortalité infantile (35 % des décès avant l'âge d'1 an), 10 % de la mortalité entre 1 et 5 ans, 12 % entre 5 et 15 ans. Pour toutes ces raisons, les maladies rares exigent une prise en charge globale qui s'est organisée à partir de la prise en compte du médicament orphelin.
Elles représentent un enjeu de connaissance important : 6 000 à 7 000 maladies rares sont identifiées à ce jour. On décrit de nouveaux syndromes chaque semaine dans la littérature internationale. Environ 80 % sont d'origine génétique, et l'explosion actuelle des techniques diagnostiques font qu'elles sont de mieux en mieux connues. Cependant, on constate une rareté de l'expertise des professionnels, un déficit de connaissance et d'information des professionnels de santé et des patients, à l'origine d'une « errance diagnostique » importante. Cela est générateur d'inégalités dans la prise en charge (remboursements, indemnisations et accès aux produits de santé). Les malades sont en outre mal repérés dans le système de soins. Orphanet a développé à l'INSERM, avec le soutien de la Direction générale de la santé (DGS), une plateforme d'information destinée à la fois aux professionnels, aux malades et aux institutions.
D'après les données de la base Orphanet, une grande partie des maladies rares est liée à des anomalies du développement (16,40 %), dont des anomalies morphologiques et troubles intellectuels ; il y a des maladies oncologiques (10,87 %), neurologiques (10,78 %) et tous les grands champs pathologiques sont concernés. La France a été leader dans la prise en charge des maladies rares, et elle le reste en Europe, à partir d'une réflexion commencée autour des médicaments orphelins (1995 : mission des médicaments orphelins au ministère de la santé menée par Annie Wolf), puis les deux Plans nationaux maladies rares (2005-2008, 2011-2014), et la réflexion sur la recherche.
Un autre enjeu est celui du diagnostic puisque pour pouvoir participer à la recherche, il faut que le malade soit diagnostiqué. Le premier Plan national maladies rares a mis en place un réseau de centres de référence et de compétences qui a permis aux patients d'avoir accès au diagnostic. En termes de traitement, on a encore beaucoup de lacunes. Au niveau de la HAS (Haute Autorité de Santé), seuls 50 protocoles nationaux de diagnostic et de soins ont été mis en place avec les centres de référence. C'est peu pour 6 000 maladies rares.
Le Plan national maladies rares 2011-2014 a mis aussi l'accent sur la recherche, avec la création de la Fondation maladies rares début 2012. Plusieurs appels à projet ont été lancés en recherche fondamentale sur des modèles thérapeutiques sur les souris. Un appel à projets a également été lancé en recherche en sciences sociales, où il y a très peu de données sur les maladies rares. Par ailleurs, toute une réflexion se poursuit sur l'amélioration de l'accès aux soins et sur la recherche, notamment avec le soutien du ministère chargé de la santé au développement des plateformes de génétique moléculaire.
En Europe de nombreuses recherches se développent : plus de 4 500 projets de recherche et près de 1800 essais cliniques sont répertoriés dans Orphanet. On y trouve aussi de nombreux tests diagnostiques et les laboratoires experts, avec leur répartition dans différents pays. Dans certains pays européens, plus de 500 gènes peuvent être testés pour permettre de diagnostiquer de plus en plus de maladies (fin 2011 : 1 129 en France, 1 449 en Allemagne). Un des objectifs du consortium IRDiRC est que la plupart des maladies rares puissent être diagnostiquées d'ici 2020.
En ce qui concerne les traitements, on observe que de nombreuses maladies n'en n'ont pas encore : sur le marché européen, en 2012 il existait 68 médicaments orphelins, dont 9 ont eu l'autorisation de mise sur le marché (AMM) cette année-là, et 75 médicaments avaient au moins une indication pour une maladie rare ou un groupe de maladies. L'autre objectif de l'IRDiRC est de développer 200 nouveaux médicaments d'ici 2020 à l'échelle internationale.
La prise en compte européenne est extrêmement importante, à travers des recommandations pour disposer de stratégies sur les maladies rares dans les différents pays. Celles-ci se mettent en place progressivement, avec aussi la volonté de constituer des réseaux européens d'experts de référence. Certains existent déjà, mais il faut mieux les afficher pour que les malades puissent être pris en charge et participer à l'ensemble des essais.
Actuellement, une grande inquiétude se développe chez les patients et les associations autour de l'application de la « loi médicament », avec les Recommandations Temporaires d'Utilisation (RTU). En effet, de nombreux médicaments utilisés dans les maladies rares n'ont pas d'AMM, notamment en pédiatrie. Comme il a été indiqué précédemment, les associations de patients jouent un rôle majeur dans les maladies rares, notamment l'Association française contre les myopathies (AFM), mais aussi l'Alliance maladies rares, qui fédère plus de 200 associations de malades en France, et Eurordis, qui fédère plus de 560 associations membres, dont plusieurs Alliances nationales maladies rares dans 51 pays. Elles participent aux essais thérapeutiques et aux réflexions en cours sur les maladies rares.
Même si la connaissance génétique des maladies rares se développe, qui permet de mieux les caractériser, de comprendre leur physiopathologie et de chercher de nouvelles thérapeutiques, le défi reste celui du faible nombre de traitements disponibles.
Pr Marina Cavazzana, directrice du département de biothérapie de l'hôpital Necker-Enfants malades, prix Irène Joliot-Curie 2012 . Je vais présenter un parcours des soins en prenant comme exemple les maladies génétiques et la thérapie cellulaire au service de la médecine personnalisée. Il s'agit d'abord de relever un défi scientifique, celui du diagnostic moléculaire, dont les impacts immédiats sont la médecine prédictive et le diagnostic prénatal ou préimplantatoire qui en découle.
L'éradication des maladies génétiques par leur prévention reste encore la forme la plus efficace de médecine personnalisée. Ensuite, la connaissance de la base moléculaire d'une maladie héréditaire permet de comprendre sa physiopathologie. Autrement dit, comment relier un certain nombre de symptômes à la mutation d'un gène ? A ce stade des connaissances d'une maladie, le défi technique consiste à modéliser la pathologie. Dans un certain nombre de cas, les modèles animaux (petits rongeurs) dont nous disposons ne reproduisent pas les signes cliniques et les manifestations pathologiques dont souffrent les malades. On peut alors avoir recours à des modèles cellulaires : les cellules souches pluripotentes induites (iPS) ou les cellules souches embryonnaires obtenues directement des malades, les deux modèles ayant un apport irremplaçable.
La découverte d'un gène et de sa physiopathologie nous amène aussi à envisager une thérapeutique personnalisée, qui peut relever de la pharmacologie, ou de la thérapie cellulaire. Dans le champ de la thérapie cellulaire, la thérapie génique reste une source d'espoir grandissante pour toutes les maladies génétiques, monogéniques et autosomiques récessives. Elle a fait ses preuves pour les maladies des systèmes hématopoïétiques, pour les pathologies de la rétine et pour la production des facteurs de coagulation pour le foie.
La thérapie génique représente non seulement un exemple extraordinaire de médecine personnalisée, mais également une approche thérapeutique importante pour les maladies acquises et les tumeurs. En effet, ces dernières années, l'apport des connaissances provenant des maladies héréditaires a également permis des progrès pour les maladies acquises. On attend également des avancées pour les malades atteints du sida, il s'agit là de rendre leurs cellules résistantes à l'infection.
La thérapie génique appliquée aux maladies héréditaires des systèmes hématopoïétiques consiste à introduire une copie normale du gène muté dans les cellules souches du patient. Théoriquement on pourrait penser que toutes les pathologies héréditaires des systèmes hématopoïétiques curables par greffe de cellules souches hématopoïétiques pourraient bénéficier de l'utilisation de cellules souches autologues (du patient lui-même) génétiquement modifiées. Cela résoudrait le manque de donneurs d'organes, les problèmes immunologiques liés à la greffe seraient évités, et cela pourrait permettre d'atteindre un taux de survie avec guérison sans complications très élevé.
Dans ce champ nouveau qu'est la thérapie génique, les problèmes de sécurité de l'utilisation des rétrovirus demeurent. À cet égard, on distingue deux périodes en thérapie génique. De 1997 à 2006, l'utilisation de rétrovirus a apporté quelques bénéfices indiscutables, des inefficacités et un certain degré de toxicité. De 2007 à 2013, on a démontré l'efficacité en absence de toute toxicité liée aux vecteurs modifiés utilisés. Ces résultats nous permettent de poser ouvertement la question : quand pourrons-nous remplacer par la thérapie génique la greffe non apparentée faite à partir de donneurs volontaires du registre international, ou les greffes intrafamiliales partiellement compatibles ? Tel est notre futur défi technique.
Par ailleurs, ce type de médecine personnalisée présente un troisième défi qui est médico-économique. Je voudrais donner deux exemples pour illustrer les répercussions de la médecine personnalisée sur les dépenses de santé ou du système de sécurité sanitaire national.
Le premier exemple concerne le déficit en adénosine désaminase, qui est un déficit immunitaire combiné grave, et pour lequel le pronostic est sévère à courte échéance. Les patients peuvent décéder dans les mois qui suivent la naissance. L'enzymothérapie de substitution, introduite à la fin des années soixante-dix, a un coût par flacon de 5 200 euros. Il faut un flacon par semaine pour traiter un nourrisson atteint de cette maladie, soit 270 000 euros par an. Si le patient est plus grand, on utilise deux flacons par semaine, soit un coût de plus de 500 000 euros par an. Devenu adulte, il lui faudra trois flacons, soit un coût de plus d'1 million d'euros par an. Le coût moyen d'une greffe allogénique est de 120 000 euros, et dans ce cas, receveur et donneur doivent être HLA compatibles. Le coût de la thérapie génique est inférieur à celui de la greffe allogénique si on exclut le coût de la phase de développement.
Le deuxième exemple d'une greffe de cellules souches allogénique pour la drépanocytose est encore plus parlant. La drépanocytose est une maladie génétique qui touche les globules rouges. On estime à 120 millions les porteurs d'un trait drépanocytaire ou de l'hémoglobine S, soit 1,7 % de la population mondiale. En France, 15 000 patients sont atteints d'un syndrome drépanocytaire majeur, dont 50 % d'enfants. C'est la première maladie génétique en France. Elle touche chaque année 300 nouvelles naissances. Et sa fréquence en Ile-de-France est de 1/1100. En 2014, on prévoit que 30 000 adultes seront atteints de drépanocytose. Actuellement, l'âge moyen de décès est de 55 ans, hors mortalité pédiatrique.
Trois traitements existent. Le coût moyen de l'hydroxyurée, un produit destiné à augmenter chez l'adulte la production d'hémoglobine foetale, est de 10 000 euros par an. Si l'hydroxyurée échoue, le coût d'un programme transfusionnel est de 50 000 euros par an. Une variable entre en ligne en compte pour la mise en place d'un programme transfusionnel : l'approvisionnement des produits sanguins labiles. Cela posera un problème de santé publique dans les années qui viennent, surtout pour des populations immigrées à groupe sanguin rare.
Le coût moyen d'une greffe allogénique est toujours identique, 100 000 euros l'année de sa réalisation, puis 1 700 euros par année de suivi post-transplantation. Le coût d'une greffe la première année correspond au coût d'une transfusion pendant un an et demi et à huit années de traitement par l'hydroxyurée. Le coût d'une greffe post-transplantation correspond au coût de douze jours de transfusion et de 64 jours de traitement à l'hydroxyurée. Les coûts de la thérapie génique après sa phase de développement sont bien inférieurs à ceux de la greffe allogénique et le coût sociétal de la morbidité et de la mortalité de ces individus deviendrait extrêmement inférieur à ce qu'il est actuellement.
À travers ces deux exemples, la médecine personnalisée montre son intérêt scientifique et économique. L'extension de l'utilisation de la thérapie génique est une promesse de changement radical pour le pronostic d'un groupe important de maladies héréditaires, que ce soit en termes d'efficacité (nombre de jours d'hospitalisation) ou d'offre de soins (possibilité d'étendre les soins en dehors des centres de référence).
Pr Florent Soubrier, responsable du département de génétique du groupe hospitalier de la Pitié-Salpêtrière-Charles Foix . En réponse au titre de la table ronde qui vise à définir les pathologies qui doivent bénéficier de la médecine personnalisée, je vous livrerai un certain nombre d'éléments plus pour essayer de cerner le périmètre de ces pathologies, que pour les définir de façon précise. Je suis délégué par l'Académie nationale de médecine, mais ce que je vais expliquer ne reflète pas forcément ce que pense l'Académie.
Pour reprendre ce qu'a dit le Pr Jean-François Delfraissy, historiquement, nous passons progressivement de l'« evidence-based medicine » à la « precision-based medicine ». Mon internat était bercé d'« evidence-based medicine », toute la pratique médicale était basée sur des données publiées concernant des biomarqueurs, des traitements, des investigations, qui permettaient d'avoir un jugement fondé sur des statistiques irréprochables pour effectuer des choix médicaux.
Progressivement, on est passé à la « precision-based medicine » qui fait appel à la multiplication des paramètres d'analyse du patient, et surtout l'utilisation des omiques en clinique, que je détaillerai après. C'est un saut technologique. Il permet l'analyse globale du patient. L'étape la plus difficile concernera l'intégration de ces données, qui sont multidimensionnelles, pour une analyse globale de l'état clinico-biologique du patient. Médecine personnalisée ou médecine de précision, je crois que ce sont de bons synonymes.
En mars 2012, un article de l'équipe de Michael Snyder fait la couverture du prestigieux journal Cell. Cette équipe a suivi un patient de façon longitudinale pendant 400 jours, en s'aidant de quantité de données d'omiques permettant de suivre une infection virale ou une susceptibilité au diabète. On peut utiliser les cellules, les tissus, et les liquides biologiques, et on verra l'importance des fèces sur différents aspects. Maintenant on peut obtenir sans problème la séquence complète du génome, mais aussi les modifications épi génétiques qui surviendront sur ce génome et en modifier la fonction ; les modifications de l'expression des gènes, leur produit (protéome) dans les différents milieux biologiques, le métabolome, c'est-à-dire l'ensemble des métabolites ou des auto-anticorps, et la séquence des microbes peuplent notre tube digestif et qui forment le microbiote intestinal. On peut analyser ces données dans leur globalité et obtenir ce que Michael Snyder avait appelé un profil intégré des omiques personnels l'iPOP, Integrative Personal Omics Profile ).
Je pense qu'il faut revenir à des fondamentaux et se souvenir que l'analyse intégrée d'un patient, d'un sujet, doit comprendre tous les différents déterminants du phénotype. Il y a l'influence des gènes, qui restent tout au long de la vie, mais aussi l'influence de l'environnement, qui ira croissant à tous les âges de la vie, et il faut prendre en compte l'ensemble des données pour obtenir une vision globale, une vision intégrée qui est difficile à obtenir, de façon à pouvoir juger de l'état clinique de ce patient.
Par rapport à la maladie, on peut se situer avant sa survenue. C'est le domaine du diagnostic pré-symptomatique, par exemple le diagnostic d'une maladie génétique, une prédisposition héréditaire au cancer qui va survenir plus tard dans la vie et donc nécessiter une surveillance particulière. Ce peut être la prise en compte d'un facteur de risque pour des maladies vasculaires ou d'autres, sur lesquelles on va pouvoir mettre en place des actions de prévention. Cette prise en compte du risque apportera effectivement un bénéfice au patient, si tant est que celui-ci utilise les données qu'on lui fournit sur ce risque de pathologies.
La médecine de précision peut aussi intervenir pendant la maladie. Pour effectuer une évaluation globale du patient, on utilisera des biomarqueurs, des omiques quand cela est nécessaire, et en particulier la pharmacogénétique pour adapter le bon traitement à la bonne dose, et ainsi intégrer l'ensemble de ces données pour justement choisir le bon traitement au bon moment.
Enfin, la médecine de précision peut se situer après la maladie pour son suivi : utilisation des biomarqueurs, utilisation des omiques, ces données intégrées qui adapteront le traitement, de façon à évaluer le processus pathologique en cours : est-il toujours en cours, quelle est l'amélioration obtenue par le traitement ?
Un point me frappe. Nous connaissons des sauts technologiques très brutaux : sur les omiques par exemple, l'arrivée du séquençage massivement parallèle a complètement changé la donne en quelques années. Les méthodes d'analyse bénéficient très vite ces sauts technologiques, et en même temps, pour la médecine, ces progrès sont incrémentaux, car il faut que toute la chaîne soit impliquée, depuis l'imagerie, les paramètres biologiques, jusqu'aux traitements. C'est pourquoi les progrès sont plus lents dans les applications médicales.
On a parlé de changement de paradigme. Ma vision est un peu différente. On se trouve dans l'approche probabiliste, qui consiste à définir un groupe de patients par un paramètre, un biomarqueur, et à partir de là à pouvoir définir une probabilité pour le patient d'être guéri par un traitement. Progressivement, on passera à une approche beaucoup plus déterministe. Grâce à toute une série de mesures, de paramètres, d'omiques, à l'intégration de ces données, on va pouvoir définir le traitement adapté pour chaque patient. C'est le grand bénéfice de cette médecine de précision : donner un traitement efficace, et éviter le traitement inutile. Je pense que les cancérologues ici présents traiteront de ces chimiothérapies ciblées que l'on sait inutiles, voire délétères en fonction de la présence de certaines mutations dans les tumeurs. Il y aura aussi moins d'effets indésirables grâce à la pharmacogénétique, le rapport coût /bénéfice sera très favorable, mais dépendra des maladies.
Autre point important, il faut adapter le degré de précision à atteindre selon la maladie, parce que les enjeux sont différents. Je prends des exemples très variés. Pour une fracture de jambe, a-t-on vraiment besoin d'une médecine de précision ? On posera une broche, une plaque, et je considère qu'il y a un bénéfice en termes de santé publique et de coût, si par exemple on adapte, grâce à un bilan précis de l'hémostase, le traitement anti-thrombotique nécessaire pour ce patient donné, ce qui lui évitera la thrombose, l'embolie pulmonaire, avec toutes les complications que cela peut entraîner. Pour le cas d'une infection grave, la médecine de précision peut déterminer l'état immunitaire du patient, la bonne dose et le bon médicament. Dans le cas d'un cancer, on ira jusqu'à séquencer le génome tumoral, en partie ou totalement pour adapter le traitement car l'enjeu thérapeutique est majeur.
La médecine personnalisée trouve ses limites, d'abord dans la complexité de l'environnement et des signes cliniques, bien plus difficiles, bien plus imprécis que la détermination d'un génome qu'on sait faire de façon très précise. S'y ajoute la nécessité de créer des traitements spécifiques pour chaque cible pouvant être en cause. On a rappelé la fin des blockbusters . Enfin, au niveau sociétal, le problème des coûts demeure.
Je conclurai sur ces trois remarques. Les progrès de la médecine personnalisée/de précision sont incrémentaux, alors que les sauts technologiques qui permettent de la mettre en place sont bien plus rapides. Par ailleurs, la mise en place de chaque progrès doit tenir compte des enjeux de la maladie, et donc du bénéfice qui peut être tiré. Enfin, l'intégration de l'ensemble des données constitue un défi : le défi de la « biologie de système » permettant d'intégrer toutes ces données.
Pr Hugues de Thé, chef du service de biochimie/biologie moléculaire à l'hôpital Saint-Louis . Mon approche sera un peu différente, elle est ciblée sur un exemple qui représente probablement le succès le plus éloquent de la médecine personnalisée, au niveau clinique, et peut-être aussi l'exemple du cancer le mieux traité par des traitements individualisés. Vous me pardonnerez de ne parler que d'une maladie sur laquelle je travaille depuis vingt-cinq ans, mais c'est probablement pour cela que j'ai été invité. Cet exposé illustrera aussi ce qu'expliquait le Pr André Syrota sur les apports du monde académique dans la médecine personnalisée, puisque tous les progrès concernant cette leucémie sont venus du monde académique.
Leucémie signifie le « sang blanc », c'est une prolifération incontrôlée des cellules du sang. Les premiers progrès dans la classification de ces maladies ont été réalisés dans les années soixante-dix avec la classification FAB (Française Américaine Britannique), fondée sur la morphologie des cellules et sur l'expression des marqueurs de surface. Vingt ans plus tard, cette classification a été progressivement remplacée par une classification moléculaire qui se fondait en particulier sur des anomalies chromosomiques, et plus récemment sur la séquence génomique complète d'un assez grand nombre de types de leucémies. Au niveau thérapeutique, l'introduction de la chimiothérapie a obtenu les premiers succès dans les années soixante.
Un tableau tiré d'une revue parue à la fin des années quatre-vingt-dix montre la diversité génétique de deux maladies qui étaient précédemment considérées comme relativement homogènes : la leucémie aiguë lymphoblastique et la leucémie aiguë myéloïde. Sans entrer dans le détail, on voit qu'elles concernent des anomalies chromosomiques très variées qui vont chacune définir un type de maladie spécifique.
La leucémie aiguë promyélocytaire est une maladie rare, une maladie orpheline (100 cas par an en France). Elle est grave du fait de l'existence de troubles de la coagulation qui mettent en péril la vie du malade dès les premières heures du diagnostic. Un taux de guérison de 25 % avait été obtenu par des chimiothérapies conventionnelles proposées par le Pr Jean Bernard en 1967. Deux thérapeutiques empiriques ont été décrites, et là on arrive à l'apport du monde académique : une hormone, l'acide rétinoïque (1988, Wang) et un toxique notable, l'arsenic (1995, Chen). L'un et l'autre auront des effets quasi miraculeux sur cette maladie, conduisant à la guérison de tous les malades par l'association des deux médicaments.
L'acide rétinoïque in vivo provoquera un changement de la morphologie des cellules et la disparition progressive de celles-ci. La mise en évidence de la sensibilité de cette leucémie à cette hormone a donc permis la caractérisation de la translocation chromosomique à l'origine de cette maladie. Cette pathologie induira la fusion de deux protéines pour créer une protéine de fusion PML/RARA, qui est le marqueur moléculaire de cette maladie, responsable de son déclenchement. Puisque cette protéine comprend l'un des récepteurs de l'acide rétinoïque, l'acide rétinoïque se trouve en fait être un traitement ciblé.
Les travaux de plusieurs équipes ont montré que ces deux médicaments, partagent la propriété de dégrader la protéine à l'origine de la maladie. On se trouve donc face à ce que les théoriciens chinois appelaient « combattre l'ennemi dans ses plans », en ciblant la protéine responsable du déclenchement de cette maladie, et ce de manière intelligente. Il a été démontré que ces deux médicaments induiront la dégradation de cette protéine en ciblant chacune de ses extrémités constitutives. On se trouve dans une situation où l'empirisme avait permis de trouver une manière très efficace pour contrecarrer l'action de cette protéine, et donc détruire la maladie.
Des critiques ont été émises sur l'importance des souris, des modèles animaux dans la compréhension des pathologies humaines. Dans cette maladie, les modèles de souris ont été au contraire extrêmement bénéfiques. Des modèles très efficaces ont été obtenus et reproduisent parfaitement la physiopathologie, l'évolution, la réponse thérapeutique de cette maladie. Ils ont permis de montrer la synergie formidable de l'association de ces deux médicaments, permettant de détruire complètement la maladie et de guérir toutes les souris dans un temps record. Alors que d'autres tests, non fondés sur des souris, avaient conclu à l'antagonisme fonctionnel de ces médicaments, conduisant à ne pas les associer chez les patients.
Aujourd'hui, cliniquement, dans tous les pays, au nord comme au sud, l'arsenic remplace progressivement la chimiothérapie en première ligne. En l'absence de chimiothérapie, on arrive à guérir tous les patients ayant une forme simple. Et ceci représente un succès éclatant de cette thérapeutique ciblée, peut-être l'un des modèles les plus achevés de la guérison par un traitement personnalisé.
Peut-on généraliser ce modèle particulier à l'ensemble des leucémies ? Pour l'acide rétinoïque et l'arsenic, on a une efficacité de 100 % dans cette maladie spécifique, définie par une translocation chromosomique, mais aucune efficacité sur toutes les autres leucémies. On se trouve bien dans le contexte de ces traitements ciblés, personnalisés.
Il existe des biomarqueurs permettant d'effectuer un diagnostic quasi immédiat par imagerie cellulaire. En quelques heures, ils permettent de poser l'indication thérapeutique. Cela nécessite la formation des équipes pour une maladie rare, la disponibilité des produits (acide rétinoïque, arsenic). On a également évoqué le problème du prix prohibitif de certains de ces produits. Néanmoins, on arrive à la guérison de la quasi-totalité des malades, et ceci représente des économies très importantes dans la prise en charge de cette maladie. Un article tiré de Nature Medicine (vol. 13, n° 9, septembre 2007) a contesté le prix prohibitif auquel l'arsenic est mis à disposition dans la plupart des pays, environ 50 000 dollars. C'est un composé dont la fabrication est extrêmement simple. Et il n'a pas demandé beaucoup d'efforts de R&D.
En conclusion, ce modèle est assez extraordinaire au niveau des résultats cliniques. De plus, le degré de compréhension des mécanismes moléculaires de cette maladie sont, je crois, inégalés dans le domaine du cancer. Malheureusement, ce modèle n'est pas généralisable. Certains cancers ou leucémies ont un mécanisme simple : une translocation chromosomique induira la maladie. Cependant, la plupart des maladies cancéreuses possèdent une physiopathologie qui répond à des altérations complexes, multiples, et posent des problèmes de hiérarchie, de compréhension du rôle respectif des différentes anomalies. Ils commencent à être abordés par les techniques de séquençage complet, mais il reste encore beaucoup de mystère, que ce soit au niveau de la biologie ou de la prise en charge.
Ce modèle illustre la façon dont on peut obtenir des bénéfices cliniques éloquents grâce au traitement ciblé. Il n'y a pas de raison pour que ce type d'approche ne soit pas étendu au moins à certaines autres maladies cancéreuses.
Pr Pierre Laurent-Puig, médecin au service de biochimie de l'hôpital européen Georges Pompidou et directeur de recherche INSERM UMR-S775, université Paris V Descartes . Je traiterai des cancers colorectaux qui représentent un modèle du développement de la médecine personnalisée au cours de ces dernières années. Depuis le début de l'année, je dirige le Cancer Research and Personalized Medicine (CARPEM), qui est au centre de nos débats. C'est un site de recherche intégrée en cancérologie, financé par l'Institut national du cancer (INCa), et qui réunit deux hôpitaux de l'assistance publique, l'hôpital Georges Pompidou et l'hôpital Cochin, ainsi que 22 équipes de recherche. Il comprend trois axes de recherche : un axe pharmaco-génomique, un axe immunitaire qui étudie la réponse de l'hôte à sa tumeur - c'est extrêmement important dans la détermination du pronostic des tumeurs -, et un axe éthique avec en particulier une recherche sur les pratiques médicales induites par la médecine personnalisée.
Les cancers colorectaux sont l'un des trois cancers les plus fréquents, avec 40 000 nouveaux cas par an en France. Pendant très longtemps, cette maladie a été considérée comme une maladie homogène, en particulier sur le plan histologique, puisqu'on considère que 95 % des cancers sont des adénocarcinomes. C'est une particularité des cancers colorectaux, alors que d'autres cancers ont des types histologiques différents. Mais en fait, c'est une maladie qui est hétérogène sur le plan moléculaire.
Si l'on applique des omiques aux cancers colorectaux, on est capable de distinguer au moins six groupes complètement différents de cancers colorectaux, alors que sur le plan histologique rien ne les distinguait. Ces types moléculaires sont pour certains d'entre eux extrêmement distants les uns des autres, et on peut attribuer à chacun de ces groupes moléculaires particuliers des caractéristiques spécifiques. Par exemple, le groupe C4 est le plus distant, et on peut retrouver une signature de cellules souches. On peut donc dire que ce groupe-là dérive probablement de cellules souches. On peut aussi leur attribuer des origines de transformation différentes, en particulier, deux groupes ont été identifiés comme dérivant soit d'adénomes, soit de lésions prenéoplasiques de type festonné.
Ces groupes moléculaires ont des diagnostics différents. Les groupes C4 et C6 ont un mauvais pronostic comparés aux autres groupes. Ceci a été montré maintenant sur plusieurs milliers de malades. De plus ces tumeurs ont un paysage thérapeutique compliqué. On dispose de plusieurs types de molécules efficaces et dont l'efficacité est identique. Ces molécules sont de plus en plus chères, et jusqu'à maintenant, on ne disposait d'aucun biomarqueur permettant de choisir le traitement face à un patient qui a un cancer colorectal.
En résumé pour les stratégies thérapeutiques, on dispose de trois molécules classiques de chimiothérapie (5-FU, Irinotecan, Oxaliplatin), de deux thérapies ciblées (un anti-angiogénique, le Bevacizumab, et de deux anticorps anti-EGFR : Cetuximab et Panitumumab), et on mélange, en particulier pour les lignes métastatiques, ces différents composés sans avoir aucun biomarqueur permettant de choisir son traitement face à un patient. Cela a changé, au moins pour l'une des thérapies ciblées, qui est le Cetuximab et le Panitumumab.
Depuis une vingtaine d'années, on a fait des progrès en termes de survie. Par exemple, pour un patient atteint d'un cancer du côlon métastatique, on est passé de 6 mois de survie pour des soins de support à plus de 24 mois pour une trithérapie associant deux chimiothérapies classiques à une thérapie ciblée. Parallèlement, l'évolution des coûts est de quelques centaines d'euros pour un traitement de support à plusieurs milliers ou dizaines de milliers d'euros pour un traitement associant des thérapies ciblées.
L'ensemble des thérapies ciblées qui sont développées dans le cancer sont centrées sur des voies de signalisation qui transmettent le signal du milieu extracellulaire au noyau de la cellule tumorale. Cette thérapie ciblée vise à interrompre la communication entre le récepteur, situé à la membrane, et le noyau. Les cellules tumorales ont la particularité d'activer ces voies de signalisation, soit par des mutations activatrices d'un certain nombre de partenaires, soit par des mutations inactivatrices sur un certain nombre de partenaires.
Si l'on donne un anticorps qui cible le récepteur à l'EGF, c'est-à-dire situé à la membrane, et que l'on est en présence d'une tumeur dont la voie de signalisation est activée en dessous du récepteur, on peut imaginer que le traitement sera inefficace. Malgré l'inhibition de l'activation du récepteur, de toute façon il y aura une activation de la voie de signalisation. C'est ce qui se passe avec les anticorps anti-EGFR qui ciblent le récepteur à l'EGF. Lorsqu'on a une mutation du gène KRAS fréquente dans les tumeurs colorectales (près de 40 % des tumeurs colorectales possèdent cette mutation du gène KRAS activée) le traitement est inefficace. Nous avons montré avec d'autre qu'on observe aucune réponse chez des malades à qui on donne un anticorps anti-EGFR, lorsque leur tumeur est mutée KRAS.
En 2007 et 2008, il y a eu une modification d'autorisation de mise sur le marché (AMM) pour ces molécules qui ne sont maintenant réservées qu'à des patients dont la tumeur possède un gène KRAS sauvage. Cela se traduit aussi en termes de survie sans progression plus longue pour les patients qui n'ont pas de mutation KRAS au sein de leur tumeur.
Cette découverte a été appliquée par l'INCa au niveau des plateformes de biologie moléculaire. Des études médico-économiques ont démontré, en tout cas pour la première fois dans les tumeurs solides, qu'en ciblant le traitement chez un sous-groupe de patients, qui représente 60 % de l'ensemble des patients atteints d'un cancer du côlon métastatique, on pouvait faire des économies. Le coût du test KRAS a été évalué à 2 millions d'euros, ce qui représente environ 20 000 tests KRAS/ an en France, et cela permet d'éviter un traitement inefficace chez 40 % des patients atteints d'un cancer du côlon métastatique, ce qui représente une économie de 39 millions d'euros.
Dr Christophe Le Tourneau, médecin oncologue à l'Institut Curie . Où en est la médecine personnalisée en cancérologie ? L'Institut national du cancer américain définit la médecine personnalisée en cancérologie par l'utilisation des données biologiques des patients, afin de prévenir, diagnostiquer ou traiter. En réalité, le terme de médecine personnalisée est né en cancérologie avec l'émergence des thérapies ciblées à la fin des années quatre-vingt-dix.
La chimiothérapie des médicaments qu'on utilise depuis longtemps en cancérologie détruit les cellules en division. C'est pour cela qu'ils ont des toxicités importantes, car ils détruisent également des cellules saines en division. Les patients perdent leurs cheveux, ils ont des toxicités sur leurs globules, et donc de la fièvre, de l'aplasie, etc.
À la fin des années quatre-vingt-dix, sont apparues les thérapies ciblées qui sont des médicaments synthétisés de novo par les industriels pour inhiber une cible thérapeutique bien précise dans la cellule tumorale ou dans son environnement. Cette cible peut être un ligand, un récepteur ou une voie de signalisation intracellulaire. Les toxicités de ces thérapies ciblées sont différentes des toxicités des chimiothérapies. Leur mode d'administration peut être intraveineux, mais également oral.
Aujourd'hui, et M. Jean-Louis Touraine l'a bien expliqué, en cancérologie, on pratique une médecine stratifiée. Le premier niveau de décision pour donner un traitement à un patient atteint d'un cancer, est la localisation de son cancer. Pour les patients qui ont un cancer du poumon, nous avons des chimiothérapies, et également des thérapeutiques ciblées que nous prescrivons dans certains cas. De même, pour les patients qui ont un cancer du côlon, comme l'a montré le Pr Pierre Laurent-Puig, nous possédons certaines chimiothérapies et certaines thérapeutiques ciblées que nous leur prescrivons. Mais là encore, le premier niveau de décision reste la localisation de la tumeur dans le corps pour décider du traitement.
Je prends deux exemples. Pour les patientes qui ont un cancer du sein, on recherchera systématiquement s'il existe une anomalie sur le récepteur HER-2. 20 % des patientes ont cette anomalie, on leur donne un traitement avec un anticorps qui cible ce HER-2, pendant un an après l'opération, et cela diminue le risque de récidive par deux. De la même manière, pour des patients qui ont un cancer du poumon, on va rechercher systématiquement s'il existe une mutation sur le gène de l'EGFR. Cela représente 10 % des cas. Nous avons pour ces patients un traitement qui inhibera l'EGFR et qui est particulièrement efficace.
Il existe donc un certain nombre d'anomalies moléculaires pour lesquelles nous avons des traitements ciblés qui correspondent à chacune de ces anomalies moléculaires. Leur incidence diffère selon les cancers. 20 % des patients atteints d'un cancer du sein ont une anomalie sur HER-2 ; moins de 2 % des patients atteints d'un cancer du poumon ont une anomalie sur HER-2.
Aujourd'hui, la communauté scientifique s'interroge. Faut-il toujours donner les traitements, en particulier les thérapies ciblées, en fonction de la localisation de la tumeur des patients ? Ou faut-il faire abstraction de la localisation tumorale et uniquement prendre en compte le profil moléculaire de la tumeur ? Cela implique de rechercher l'ensemble des anomalies moléculaires connues, prédictives de réponses à des thérapies ciblées, chez tous les patients, quel que soit leur cancer, afin de déterminer s'il existe une thérapie ciblée qu'on pourrait leur proposer.
Cette approche, envisageable aujourd'hui, ne l'était pas il y a deux ans, pour la simple et bonne raison que la technologie n'était pas prête. On ne pouvait pas encore regarder en un seul coup d'oeil toutes les anomalies moléculaires d'une tumeur d'un patient, non seulement parce que c'était trop coûteux, mais également parce que c'était trop long, et donc pas compatible avec la prise en charge des patients à l'hôpital.
Effectuer un profil moléculaire de la tumeur chez chacun des patients atteints d'un cancer et donner le traitement ciblé qui correspond, est-ce une stratégie finalement plus efficace que ce qu'on fait tous les jours à l'hôpital, c'est-à-dire déterminer les traitements en premier lieu à partir de la localisation tumorale ? Cette question est ouverte. La démonstration de la médecine personnalisée n'a pas encore été faite. Aujourd'hui, on ne peut pas dire s'il est plus efficace d'effectuer un profil moléculaire de la tumeur des patients pour leur donner un traitement ciblé qui correspond, par rapport à ce qui se fait tous les jours en pratique clinique.
À l'hôpital, aujourd'hui encore, les patients sont traités en premier lieu en fonction de la localisation de leur tumeur. Cette question, nous l'avons soulevée dans un essai intitulé SHIVA en octobre 2012, que j'ai initié à l'Institut Curie et dans six autres centres en France. Il consiste à faire la carte génétique simplifiée, sans regarder le génome dans son entier, de la tumeur des patients, et s'il y a une anomalie moléculaire, nous proposons au patient le traitement ciblé qui correspond. Il s'agit d'un essai comparatif. Nous comparons cette approche-là à l'approche de tous les jours à l'hôpital, c'est-à-dire aux traitements que nous donnerons au patient si jamais il n'était pas inclus dans cette étude. Depuis le mois d'octobre, un peu plus de 130 patients ont été inclus, et les inclusions continuent.
Comment cela se passe-t-il dans la pratique ? Nous avons besoin de nouveaux métiers. Dans cet essai, environ 10 personnes travaillent pour rendre la carte génétique. Ces 10 personnes d'habitude ne travaillent pas en routine pour traiter les patients à l'hôpital. Qui sont-elles ? Premièrement, il faut prélever la tumeur. Nous avons donc besoin des radiologues et des chirurgiens pour faire des prélèvements supplémentaires que nous ne faisons pas d'habitude en routine. Nous avons besoin de l'anatomo-pathologie, c'est-à-dire de personnels qui vont regarder au microscope pour bien vérifier qu'il s'agit de cellules cancéreuses, et savoir quelle est la proportion de cellules cancéreuses. Ces mêmes personnes extraient l'ADN des cellules cancéreuses. Ce sont des manipulations que nous ne faisons pas d'habitude en routine.
L'ADN est envoyé à des plateformes de séquençage, lesquelles réalisent la carte génétique de la tumeur, mais produisent des données ininterprétables par des médecins. Ces données sont donc ensuite transférées à la bio-informatique. Plusieurs bio-informaticiens travaillent pour traiter ces données et faire un compte-rendu lisible et compréhensible par les médecins et interprété par les biologistes. Enfin, nous disposons, nous, les médecins, d'un compte rendu de la carte génétique qui nous permet de savoir si oui ou non, il existe une anomalie moléculaire pertinente pour laquelle nous pouvons donner un traitement.
Par rapport à la routine de l'hôpital au quotidien, un certain nombre de personnes entrent dans le jeu pour déterminer la carte génétique. D'habitude, ces personnes ne travaillent pas à cela.
L'essai SHIVA, aussi simple soit-il, nécessite un flux des données qui part des données cliniques des patients jusqu'aux données de la carte génétique, puis jusqu'à la décision. Ces données doivent être envoyées à certains moments de façon automatisée. C'est un réel défi, car à la fin, il faut que le compte rendu de la carte génétique du patient, non seulement corresponde bien au bon patient, mais encore que celle-ci soit validée et soit de qualité. Tout cela nécessite des compétences que nous n'utilisons pas d'habitude en pratique à l'hôpital.
Pour conclure, j'estime que finalement, aujourd'hui en cancérologie, nous pratiquons surtout une médecine stratifiée. Nous utilisons des données biologiques pour décider de traitements, et nous catégorisons les cancers en des sous-groupes de cancers. Ces sous-groupes sont encore relativement importants. Nous ne sommes pas encore parvenus à la médecine personnalisée qui permettrait de dire : « faisons une carte génétique de la tumeur d'un patient, quel que soit son cancer, pour lui donner le traitement qui correspond ». Cette démonstration est probablement en cours avec des essais comme SHIVA qui se déroule actuellement, ou d'autres essais qui démarreront en France ou à l'étranger. Très probablement dans les années à venir, la communauté scientifique en est persuadée, nous arriverons à une stratégie qui consistera effectivement à adapter les traitements en fonction de la carte génétique des cancers des patients, et non plus en fonction de la localisation tumorale primitive.
M. Alain Claeys. Je remercie chacun des orateurs de ces exposés et j'ouvre le débat.
|
Débat |
M. Jean-Louis Touraine. J'ai deux questions. L'une n'entre pas tout à fait dans le cadre des pathologies mais s'en rapproche, et elle est à l'ordre du jour. Elle porte sur le développement bien nécessaire d'une contraception personnalisée. Ce n'est pas de la pathologie, mais c'est indispensable.
Ma deuxième question s'adresse au Pr Jean-François Delfraissy qui a indiqué qu'actuellement il y avait un relatif retard en pathologie infectieuse par rapport à la cancérologie dans le développement de la médecine personnalisée. Pour autant, la nature a développé cette infinie diversité dans la réponse aux infections. Chacun connaît bien l'immense diversité de notre système d'histocompatibilité. D'ailleurs celui-ci existe dans les autres espèces de mammifères. Ce système HLA est complexe et bien entendu important dans l'ontogénie de systèmes immunitaires, notamment intra-thymiques. Depuis Rolf Zinkernagel et Peter Doherty, on sait que le lymphocyte T reconnaît l'antigène de concert avec la molécule d'histocompatibilité propre. Ainsi chacun d'entre nous reconnaît différemment les différents antigènes infectieux par exemple, ce qui détermine une immunité différente de chacun vis-à-vis de ces infections. Et donc la probabilité de survie est différente. Tout se passe comme si la sélection naturelle avait privilégié la survie de l'espèce humaine plutôt que celle de l'individu, acceptant de passer par pertes et profits dans chaque épidémie une partie de l'humanité, mais sélectionnant une partie de l'humanité pour être résistante à chaque épidémie.
Alors je voudrais savoir s'il existe une voie de recherche qui pourrait utiliser cette réflexion. Certes, les traitements les plus puissants passent par-dessus ces différences de réponse individuelle, mais il reste que par exemple pour la plupart des maladies virales, il n'y a pas actuellement de traitement suffisamment efficace. C'est donc bien encore l'immunité individuelle de chacun d'entre nous qui nous permet de guérir de nombreuses maladies virales. Cette immunité est très différente d'un sujet à l'autre. Pourra-t-on personnaliser cette approche pour tirer parti de cette immense diversité d'immunité ?
Pr Jean-François Delfraissy. Cela soulève une question générale. La médecine personnalisée ne doit pas uniquement s'intéresser à la médecine de soins des patients, mais aussi beaucoup plus en amont à la prévention. Les systèmes de prévention, ou par exemple les nouveaux outils biomédicaux de la prévention sur un certain nombre de maladies, doivent évidemment se poser la question de la personnalisation de cette nouvelle approche de la prévention. On ne doit pas l'oublier, car il est vrai que nous sommes tous dans la prise en charge et dans le traitement.
Pr Claude Huriet, président de l'Institut Curie. Le Pr Jean-François Delfraissy a expliqué, je cite « on n'a plus besoin de grands essais randomisés » ; et le Pr Odile Kremp, à propos des maladies orphelines, a déploré le fait qu'un certain nombre de médicaments utilisés dans le cadre des maladies rares soient prescrits hors AMM pour des raisons qui, je pense, sont liées à la méthodologie des essais cliniques. C'est sur ce point que porte ma question.
La méthodologie des essais cliniques s'appuie sur des analyses statistiques, ce qui signifie que si les données des statistiques ne sont pas suffisamment élaborées, on a tendance à ne pas retenir le protocole. Or pour les traitements personnalisés, par nature, il est très difficile de réaliser des cohortes comportant des centaines, voire des milliers de patients, alors même que les exigences en termes d'appréciation du bénéfice-risque vont dans le sens de l'allongement des délais d'observation et de l'accroissement du nombre de personnes participant aux essais.
Autrement dit, il y a une contradiction sur laquelle il est important que l'Office se prononce. En effet, les organismes qui financent les essais cliniques sont amenés à dire : ce protocole ne répond pas aux exigences des analyses statistiques. En conséquence, ce projet ne peut pas être financé. C'est la démarche même du développement des essais cliniques qui se heurte à une antinomie profonde entre le terme même de « personnalisé » et l'importance des analyses statistiques.
Jean-François Delfraissy. C'est tellement important que je suis obligé de réagir. Le concept même de grand essai randomisé n'est pas remis en question, loin de là, mais en tout cas, il doit être revisité, y compris en recherche de la science, de la méthodologie des essais. Ma génération vient de vivre vingt ans, où l'on nous a appris l'importance de l'essai randomisé en médecine, sous l'égide du Pr Laurent Schwartz, père fondateur en France de cette méthodologie.
Cette méthodologie permet en effet de lever toute une série de biais, qui sont maintenant très connus, mais a aussi ses contraintes. Contraintes d'effectifs, contraintes d'un certain nombre de patients qui ne veulent pas rentrer dans des essais en double aveugle par exemple. L'essai randomisé permet de répondre à un certain nombre de questions. La philosophie générale est de considérer que si on compare A versus B, ensuite on pourra d'une part, regarder à l'intérieur de cet essai randomisé ce qui relève véritablement de l'efficacité de telle ou telle molécule, ou ce qui relève d'autres biais, et d'autre part, on trouvera des facteurs explicatifs d'une réponse ou d'une non-réponse à l'intérieur de cela. C'est donc l'essai qui lui-même génère l'explication de la réponse ou de la non-réponse .
Dans le cadre de la médecine personnalisée, on part dans le sens inverse. À la base, on dispose d'un certain nombre de marqueurs qu'on connaît en partie, peut-être pas totalement, mais sur lesquels on possède un certain nombre de pistes. Au lieu de faire un essai randomisé, on peut prendre une population très spécifique, peut-être une population de l'extrême, qui a tous les marqueurs en l'occurrence. Les cancérologues commencent à le faire, et c'est vrai dans d'autres pathologies, avec un marqueur prédictif d'une réponse ou au contraire d'une non-réponse, qu'on en soit sûr ou qu'on le suppose.
Si je sélectionne cette population avec des marqueurs, je démontre plus rapidement la limite sur un plus petit nombre de patients, si finalement la molécule que j'apporte va avoir un bénéfice ou pas. C'est une vraie question, qui fait débat en ce moment. Toute une part de la science s'est bâtie pendant des années et des années autour de l'essai randomisé en double aveugle. Et je fais partie des gens qui admettent que cela a construit leur vie, mais la vie est faite pour évoluer. On doit être extrêmement plastique en recherche. Posons-nous des questions sur ce qui doit se monter pour les années qui viennent, maintenant qu'on a des biomarqueurs et une technologie des biomarqueurs qu'on n'avait pas il y a vingt ans.
Donc je nuance. Je ne suis pas en train de démolir aujourd'hui les essais randomisés, sinon vous allez me faire assassiner dans les jours qui viennent.
Pr Claude Huriet. Pardon Monsieur le président. Ma remarque ne portait pas du tout sur la randomisation. Elle porte exclusivement sur la notion même de cohorte et l'exigence statistique d'avoir des cohortes les plus nombreuses possibles, ce qui est incompatible avec le concept même de traitement personnalisé.
Pr Philippe Amouyel, directeur général de la Fondation Plan Alzheimer. Le Pr Jean-François Delfraissy a pris l'exemple tout à l'heure de l'Abacavir qui est un excellent exemple pour comprendre comment on est arrivé à la médecine personnalisée pour essayer d'éliminer les effets indésirables de l'Abacavir.
Je crois qu'il n'existe pas de confusion. Quand on évoque les cohortes, on confond nombre de patients, nombre de sujets nécessaires. Lorsque les effets sont faibles et qu'on veut montrer qu'avec un nouveau médicament il y a une amélioration de quelques pourcents dans une maladie extrêmement fréquente comme une maladie cardiovasculaire par exemple, on est obligé de recourir à de très grandes populations. Dans cet exemple on ne se trouve pas dans cette situation, on espère obtenir un maximum de bénéfices pour un minimum de patients. Cela signifie que l'effet sera extrêmement fort. L'effet se mesure en termes de risques relatifs ou de hazard ratio , et dans le cas de l'Abacavir, les patients qui ont le fameux génotype HLA, ont un hazard ratio de l'ordre de500 et 1 000. La moyenne habituelle des risques relatifs dans la plupart des essais thérapeutiques, c'est 1,2 - 1,3 - 1,4.
On n'est donc pas du tout sur les mêmes effets, mais on utilise toujours les mêmes méthodologies. Et quand on a démontré un effet pharmacogénétique pour l'Abacavir, on est parti en effet d'un groupe de patients spécifiques, et ensuite on a refait un essai randomisé, pour démontrer que cet effet était bien retrouvé en conditions expérimentales. Parce qu'aujourd'hui, on dispose d'informations descriptives de recherche, on associe les éléments, mais après, il faut refaire l'analyse inverse, un essai clinique ad hoc, pour savoir si on a un bénéfice et si l'on obtient bien l'effet qu'on attend de ce produit ou de ce biomarqueur.
Pr Odile Kremp. J'ai voulu dire dans mon propos que compte tenu du nombre de maladies rares, du faible nombre de malades pour une maladie donnée, et du peu de médicaments actuellement disponibles, personnaliser la médecine semblait encore difficile, même si la connaissance de la génétique de ces maladies progresse vite. Ce n'est pas le cas pour des maladies rares qui sont déjà bien connues, et l'on a eu d'excellents exemples avec la drépanocytose et la leucémie.
Par ailleurs, une réflexion est menée à l'échelle européenne. La valeur ajoutée de l'Europe compte, parce que cela permet d'inclure plus de patients. En ce qui concerne la modification de stratégie des essais, il y a eu des recommandations du Comité européen d'experts sur les maladies rares pour commencer les traitements, et continuer à suivre très précisément les effets secondaires et les effets bénéfiques, mais plus tôt dans le cours de la vie d'un médicament orphelin que pour des médicaments qui sont utilisés à beaucoup plus grande échelle ( http://www.eucerd.eu/?post_type=document&p=2137 ). Cela rejoint la réflexion sur les autorisations temporaires d'utilisation (ATU) ou les recommandations temporaires d'utilisation (RTU). C'est sûrement une piste, avec bien sûr les garanties de méthodologie qu'il faut adopter.
Mme Anne-Yvonne Le Dain, députée de l'Hérault, vice-présidente de l'OPECST. Je ne suis ni chercheur ni médecin en biologie santé. Nous sommes ici à des niveaux de travail et de précision qui sont considérables et d'une immense valeur. Cependant, j'ai eu un peu le sentiment qu'on était très orienté sur le cancer, un peu sur l'infectiologie, mais pas trop sur l'immense majorité des maladies que vivent les hommes. Le cancer, tout le monde le vit, et c'est lié en partie au vieillissement de la population. C'est une réalité objective dans nos pays développés. On sauve de plus en plus de vie, de la naissance à la mort. On étend de manière considérable la vie, c'est essentiel et formidable. Ensuite, cela a été explicité reste la question du bénéfice médical, en termes de soins, de santé ou de vie de la personne, et du coût sociétal. Ce dernier point a été évoqué, mais finalement on l'évoque assez peu.
Ma question porte donc sur le lien entre ce travail exceptionnel et la médecine de ville, et le lien avec les cancérologues que les malades rencontrent. Les patients ne sont pas tous soignés dans le système hospitalier, public ou privé. Ils sont souvent soignés en ambulatoire et se pose le problème du lien entre médecine de ville et s progrès scientifiques.
Cette problématique nous éloigne certes quelque peu de la randomisation des essais ou des cibles plus précises. Nous sommes en train de lutter contre la sélection naturelle, et il est bon que chacun puisse en bénéficier. L'approche individualisée est aussi une lutte contre cette sélection naturelle, il faut le rappeler. Or sur la médecine personnalisée, je pose la question : comment, par rapport aux maladies banales que nous vivons tous, le travail scientifique se fait-il aujourd'hui ? Entre les grands cancers, qui sont nombreux, et les maladies orphelines, existe l'espace pour le quotidien de chacun et le lien avec la médecine de ville individuelle. Ce lien me paraît compliqué et malaisé à construire. En tant que parlementaires, notre ambition est de construire un projet de société autour de cette question de la médecine personnalisée. Comment va-t-on le construire, et comment vous, les scientifiques et les médecins, allez-vous nous aider à le construire ?
M. Alain Claeys. Cette interpellation est très importante, pour nous, parlementaires. Qui veut répondre ?
Pr Pierre Laurent Puig. Je serais bien incapable de répondre à l'ensemble de vos interrogations, mais j'estime qu'un effort de formation doit être accompli pour expliquer la médecine personnalisée. Actuellement, pour les pathologies graves le ciblage thérapeutique et l'accès à certains médicaments, uniquement si la tumeur possède certaines caractéristiques, cela ressemble à une double peine. C'est-à-dire que vous avez le cancer, mais vous ne pouvez pas accéder au traitement. C'est une phase transitoire tant qu'on ne dispose pas pour tous les types tumoraux, tous les sous-types pour un type tumoral donné, de l'accès à une thérapeutique qui permettra de pouvoir traiter le patient. C'est relativement compliqué de faire comprendre cela, et je pense que cela exige un effort de formation. Le bénéfice est qu'on évite des effets secondaires inutiles.
En ce qui concerne la médecine de ville, je ne pense pas que la médecine personnalisée change particulièrement les choses. C'est plus un effort d'explication. Si on a accès à la détermination du phénotype tumoral, de manière égalitaire, ce qui est le cas pour l'instant, il n'y a pas de difficulté sur ce point dans l'immédiat. Grâce à l'effort fourni par l'INCa sur les plateformes de génétique, tout le monde a accès à la détermination des altérations génétiques ciblables de sa tumeur. Peut-être que l'introduction de l'exome ou du génome modifiera cet aspect.
M. Jean-François Deleuze, directeur du Centre national de génotypage au CEA. Je vais apporter une autre dimension. Effectivement, ces avancées donnent l'impression d'être très techniques, mais je pense qu'elles sortiront aussi du champ de l'hôpital et de la recherche, et que le patient aura accès, de par lui-même, à son information génétique. C'est le cas aujourd'hui à l'étranger et ce le sera peut-être en France demain. Pour quelques centaines d'euros, on peut déjà l'acheter ailleurs. Le patient viendra avec son information génétique, et il aura accès à des logiciels d'analyse, qui se trouveront certainement aussi sur des bases de données publiques, permettant de décrire déjà un ensemble de prédispositions. Il sera donc déjà en possession de son information génétique. Par rapport au lien entre médecine de ville et hôpital, l'information appartiendra au patient.
Pr Philippe Armouyel. Aujourd'hui, vous pouvez avoir accès à cette information. De toute façon, les médecins de ville y seront confrontés, puisque les patients viendront avec cette information dans leur cabinet.
En réponse à la première question : applique-t-on ces principes pour les maladies autres que le cancer ? La réponse est positive, mais ce qui est particulier dans le domaine du cancer, c'est qu'on est plus avancé et qu'on dispose de réelles preuves d'intérêt thérapeutique. Dans tous les domaines, on pratique le même type d'approche, et comme le soulignait le Pr André Syrota, c'est la recherche qui nous fait avancer dans ce domaine-là.
À l'autre bout de la chaîne, on a bien sûr la formation, mais aussi l'intégration des informations, comme dans l'essai SHIVA. Quand on mesure l'importance du transfert et la complexité des informations, on constate qu'il est impossible de faire tout cela seul, que l'on soit un universitaire, un chercheur ou un médecin généraliste. D'où le besoin d'outils d'informations et d'informatique, de la bioinformatique, de la biostatistique nécessaires pour intégrer cela et produire des éléments exploitables par le médecin généraliste.
M. Jean-Louis Touraine. Pour le médecin de ville, je ne suis pas sûr que ces avancées soient une authentique révolution. C'est l'évolution, plus scientifique, plus rigoureuse, de ce qu'il faisait précédemment. Comme vous l'avez expliqué, c'est effectivement le rôle de la médecine, depuis toujours de lutter contre la sélection impitoyable de la nature, qui élimine les faibles, généralement avant l'âge de reproduction, écartant ainsi les gènes de susceptibilité, et ne laisse suivre que les plus forts d'entre nous qui survivront jusqu'à l'âge adulte. Donc le médecin généraliste, le pédiatre, tous les médecins, de tout temps dans leur pratique, ont toujours lutté et favorisé la possibilité aussi bien pour les personnes vulnérables que pour les autres, d'atteindre un âge plus avancé.
Par le passé, on avait déjà cette pratique, mais avec des notions moins rigoureuses. L'interrogatoire des comportements des personnes, mais aussi de leurs antécédents, apportait au médecin la précision. Les femmes dont les mères ou les grand-mères avaient eu un cancer du sein, on le savait depuis longtemps, avaient plus de risques d'avoir un tel cancer. C'était également le cas pour les maladies cardiovasculaires, ce qui imposait davantage de propension à une alimentation mieux contrôlée et un exercice physique plus important. Tout cela était déjà établi.
Aujourd'hui, ce qu'on apporte en plus au médecin, ce sont des données scientifiques, rigoureuses, qui lui permettent de pouvoir étayer son argumentation sur des éléments très précis. Et cela a été souligné par le Pr Axel Kahn, ce n'est pas pour autant que cela aboutira à une translation immédiate. On sait déjà tous les méfaits du tabagisme et on a beaucoup de peine à lutter contre, à obtenir une réduction de ce fléau dans la population générale.
Dr Catherine Bourgain, chargée de recherche en génétique humaine et statistiques à l'INSERM. Je citerai un éditorial paru récemment, le 28 mars 2013, dans Science , qui posait la question du « risque de dépersonnalisation de la médecine », avec la montée en puissance de toutes les technologies qui permettront certains progrès. Cela a été mentionné à propos du recrutement de personnel dans l'essai SHIVA, on voit que les compétences ne sont pas les mêmes pour ce type de médecine. Cela pose la question de la répartition des moyens : doit-on payer des informaticiens, des bio-informaticiens, des personnels capables de traiter tous ces flux de données mais qui à l'évidence seront moins au contact des patients. Une tension se joue là. Il ne faut pas la négliger, car elle fait partie de la réflexion autour des promesses de la médecine personnalisée.
Pr Jean-François Delfraissy. Je ferai trois remarques très rapides. Il est vrai que la cancérologie est en avance. On a évoqué l'infectiologie mais en neurosciences, dans le domaine des maladies cardiovasculaires et du métabolisme, des avancées en médecine personnalisée sont vraiment en cours. Les applications sont beaucoup plus larges que celles présentées ce matin.
Deuxièmement, j'estime qu'il faut faire un effort considérable, et cela rejoint les médecins généralistes, sur la formation, y compris celle des étudiants. Cela dépend des facultés, selon qu'une personnalité s'intéresse particulièrement à ce domaine, mais globalement on constate un retard autour de cela. Et donc la transmission par rapport aux médecins généralistes est en jeu.
Troisièmement, sur la place du milieu associatif, je partage ce qui a été évoqué - on trouve énormément d'informations sur Internet et il y en aura de plus en plus -. On peut s'interroger sur l'interaction avec le milieu associatif. Personnellement je l'ai vécu il y a quelques années dans le domaine du VIH où l'on a joué l'interaction avec le milieu associatif qui a contribué à nous stimuler et qu'on a contribué à former. Le bilan vingt ans plus tard est solide, le milieu associatif continue de nous interpeller, et il nous faut d'une certaine façon continuer à les former. Je considère qu'en ce moment, sur une série de pathologies, il faut que le citoyen puisse s'emparer de cette expérience et qu'on contribue, nous scientifiques et médecins, à les amener et à les former à cette réflexion. C'est un enjeu sociétal particulièrement important.
Mme Anne-Yvonne Le Dain. Je vous remercie de la diversité de vos réponses. C'est très éclairant, non seulement sur ce que vous faites les uns et les autres, mais aussi sur l'humanité avec laquelle vous le faites. Je crois que le public a besoin d'être rassuré là-dessus aussi et de savoir que nous ne sommes pas dans une médecine en train de se déshumaniser.
Je m'interroge sur cette information très juste, selon laquelle bientôt les patients arriveront avec leur carte génétique chez le médecin. Ils peuvent acheter cela pour pas très cher sur Internet. C'est interdit mais on peut le faire.
M. Alain Claeys. Il faut peut-être nuancer cela.
Mme Anne-Yvonne Le Dain. Certes mais malheureusement, il faut quand même le souligner, et mon propos est de rappeler que la vie humaine n'est pas qu'une affaire de gènes. On n'a pas un gène à la naissance qui amènera à avoir un cancer à 57 ans ou à 12 ans, ou un accident vasculaire cérébral à 43 ans. Cela n'est vrai qu'en partie, pour un certain nombre de pathologies, peut-être identifiables, et peut-être même déjà identifiées. Cependant dans la plupart des cas, les caractères sont polygéniques, certainement liés à l'environnement, les conditions de vie et les lieux de vie. Tous les travaux d'épi-génétique, et de modélisation sur les conditions de vie, sont absolument indispensables à conduire et à mener. Il faut rappeler que nous ne sommes pas déterminés parce que le monde n'est pas déterministe.
M. Jean-François Deleuze. Je suis assez d'accord mais il existe quand même des dizaines de marqueurs monogéniques et très déterministes. Rien que pour ceux-là, il faut continuer. Tous les autres ne constituent que des facteurs de risques. Il faut donc effectivement, moduler l'analyse.
M. Alain Claeys. Je vous remercie et nous allons entamer la deuxième table ronde.
DEUXIÈME TABLE RONDE :
AVEC QUELS
OUTILS ?
M. Jean-Sébastien Vialatte. Nous allons entamer cette deuxième table ronde. La première, comme nous l'avons vu, a soulevé un certain nombre d'interrogations sur la formation médicale et sur la formation des métiers accompagnant les médecins, notamment les bio-informaticiens. Ont également été posées la question de la protection des données et de leur stockage dans le cadre du décryptage total du génome, et la question de l'égal accès à ces techniques pour tous nos concitoyens.
Il existe une autre problématique à examiner, celle des outils dont nous avons besoin pour mettre en oeuvre cette médecine personnalisée. C'est l'objet de cette table ronde. Je vous donne la parole, professeur Patrick Boisseau.
Pr. Patrick Boisseau, responsable du programme nanomédecine au CEA-Leti, président de l'European Technology Platform on Nanomedicine . Je tiens tout d'abord à me présenter. Je travaille au CEA-Leti à Grenoble Nous faisons de la recherche technologique et transférons ensuite nos technologies vers l'industrie. Je représente également la plateforme technologique européenne Nanomédecine, une structure européenne qui rassemble depuis 2005 les cliniciens, industriels, chercheurs académiques et agences concernés par le développement de la nanomédecine.
Ma présentation portera sur une boîte à outils, actuellement en pleine évolution, que l'on appelle « les nanotechnologies appliquées à la médecine » ou « nanomédecine ». Les nanotechnologies, c'est tellement petit que l'on a du mal à imaginer ce que c'est ! Pour éviter que mon exposé ne soit trop obscur pour certains d'entre vous, je vais donc essayer d'illustrer mon propos avec le plus grand nombre d'exemples possible.
Nos collègues américains de l' Alliance for Nanotechnology in Cancer ( NCI) ont présenté un schéma extrêmement intéressant qui résume les différentes étapes qui ont d'ailleurs été évoquées ce matin au travers des exposés de mes confrères cliniciens. C'est comme si, en France, l'Institut national du cancer (INCa) engageait une action spécifique pour identifier toutes les situations dans lesquelles les nanotechnologies peuvent permettre d'appréhender le cancer, et ce dans les phases de diagnostic, de traitement et de suivi thérapeutique. On s'aperçoit que la nano-médecine peut apporter - elle n'est bien sûr pas la seule à le faire - des solutions, ou des réponses, en tout cas des outils à chacune de ces étapes.
Le terme de nano-médecine désigne le recours aux nanotechnologies en médecine. Que sont les nanotechnologies ? C'est le fait d'utiliser des nanomatériaux qui ont, à l'échelle nanométrique, des propriétés physiques, chimiques et biologiques particulières. Il faut tout de même souligner que le préfixe « nano » représente 10 -9 unités de base, ici des mètres. Cela ne vous dit rien du tout. Prenez un cheveu, coupez son diamètre en 100 000 et cela vous donnera un ordre de grandeur de ce qu'est un nanomètre. C'est vraiment très petit. C'est 1 000 fois plus petit qu'une cellule.
Quand on fait des nano dispositifs, des nano assemblages moléculaires, on travaille à peu près, dans la même échelle de taille que les biomolécules, c'est-à-dire toutes les molécules intervenant dans des processus pathologiques. Par conséquent, dès lors que l'on réussit à fabriquer des entités d'une taille à peu près équivalente à celle de la molécule que l'on veut capter, suivre, détruire ou neutraliser, dès lors que l'on se situe dans le même rapport de taille, on parvient à le faire. Il y a des exemples de la reconnaissance entre un nano assemblage et une cible thérapeutique, par exemple un antigène ou des récepteurs surexprimés sur la surface d'une cellule. En étant 1 000 fois plus petit qu'une cellule, on se situe dans un rapport de taille correct pour pénétrer dans une cellule et effectuer un traitement ou de l'imagerie à l'intérieur de celle-ci.
S'agissant notamment de la détection de ce qui a déjà été mentionné en matière de prédispositions génétiques, de fond génétique, et plus largement de l'analyse des « omics », on a utilisé les puces à ADN, que j'appelle les « dinosaures de la nano-médecine », et on dispose désormais de dispositifs un peu plus sophistiqués, comme les nano-pores, permettant de faire du séquençage à haut débit. En matière de détection et donc de diagnostic, interviennent les capteurs, voire les biocapteurs. Ce diagnostic pouvant être fait in vitro ou in vivo , on trouve là aussi tout le domaine de l'imagerie moléculaire, dans lequel les nanotechnologies apportent de nouvelles fonctionnalités.
Puis, dans la phase de thérapie, citons les thérapies ciblées, notamment grâce aux nano vecteurs que j'évoquerai ultérieurement. C'est un domaine important et sur le plan industriel, aujourd'hui, le plus large domaine d'application des nanotechnologies. Enfin, au niveau du suivi thérapeutique, on retrouve la grande diversité des capteurs : capteurs portés sur la personne, capteurs implantés ou capteurs d'ambiance.
Il y a trois grands domaines dans lesquels les nanotechnologies apportent de l'innovation. Le premier d'entre eux est le diagnostic personnalisé. C'est le diagnostic in vitro qui se fait grâce à des microsystèmes pouvant être multi-paramétrés, plus rapides, capables de traiter un plus grand nombre d'échantillons ou permettant de travailler sur des échantillons de bien plus petite taille. C'est accessible soit dans les laboratoires centraux, soit, comme cela a déjà été souligné, à travers le point-of-care : au lit du patient, au cabinet du médecin traitant ou dans d'autres configurations.
Mais il y a aussi tout le domaine de l'imagerie moléculaire. On peut amener à peu près n'importe quel dispositif d'imagerie -IRM, imagerie nucléaire, imagerie de fluorescence, opto acoustique, photo acoustique- au plus près de la cible thérapeutique, pourvu que l'on possède sur cette cible, une modification de surface suffisamment spécifique pour qu'on puisse la reconnaître dans le grand brouhaha du reste de l'organisme. Par exemple, la société française Fluoptics propose un dispositif d'imagerie moléculaire permettant de guider le geste chirurgical dans l'exérèse des tumeurs abdominales. On rend les tumeurs et, surtout leurs marges, fluorescentes, afin de permettre au chirurgien d'enlever, avec d'autant plus d'efficacité, toutes les cellules tumorales et ainsi éviter qu'il puisse y avoir une reprise du cancer quelques mois plus tard.
Le deuxième grand domaine concerne les thérapies ciblées. On a coutume de considérer que la nano-médecine, même si elle intervient à l'échelle nanométrique, l'échelle moléculaire, ne recouvre pas toutes les molécules thérapeutiques. La molécule « simple », ou éventuellement couplée à un anticorps pour cibler son adressage, relève de la chimie ou de la biochimie. Dans le cas des thérapies ciblées, le grand domaine d'application des nanotechnologies, c'est bien sûr l'utilisation des nano-vecteurs, que vous avez peut-être connus sous des noms tels que liposomes, micelles, particules polymériques de l'épidote. De la même manière que l'on transporte des agents d'imagerie, ces particules servent de transporteurs à de petites molécules médicamenteuses. Après une administration veineuse, nasale ou orale - là encore, toutes les formulations sont possibles - , elles amènent le traitement thérapeutique au bon endroit dans l'individu.
Quelqu'un évoquait ce matin la réduction des effets secondaires adverses : si elle n'est pas la seule solution possible, l'encapsulation dans ces nanoparticules peut permettre de transporter des drogues extrêmement toxiques jusqu'au point souhaité, et pas à côté. On évoque en règle générale de « nanotherapeutics », car on peut transporter des drogues, mais aussi encapsuler des cellules - dès lors, on ne se situe plus à l'échelle nanométrique -, comme c'est le cas dans certaines transplantations.
Le troisième domaine est celui du suivi thérapeutique, où les technologies font oeuvre d'une grande créativité pour développer des capteurs. Les capteurs peuvent être portés sur la personne, implantés dans la personne, sachant qu'à l'échelle nanométrique ou micrométrique et nanométrique, on arrive à réduire d'une manière assez extrême les capteurs comme l'indiquait le Pr André Syrota. À partir du moment où le capteur est très petit, on peut alors l'implanter assez intimement dans l'individu pour mesurer des biomarqueurs ou la variation de paramètres physiques ou physico-chimiques.
Les nano assemblages ont la même taille que les particules biologiques. L'intérêt de détecter des phénomènes à l'échelle moléculaire, est de pouvoir le faire à un stade plus précoce que celui du symptôme global, au niveau d'un tissu. Par ailleurs, si le capteur que l'on va placer sur une personne n'est pas nanométrique, il y a ce qu'on appelle du « nano inside », c'est-à-dire qu'une partie du capteur est de taille nanométrique.
La médecine dite « asymptomatique » ou « préventive » peut être envisagée avec l'utilisation des nanotechnologies. En détectant d'une manière précoce, et en effectuant un diagnostic précoce, on peut prendre en charge un patient avant qu'il ne développe des symptômes cliniques. C'est une évolution dans la manière d'appréhender cette prise en charge, sachant qu'il s'agit là d'un concept vers lequel nous tendons, mais qui n'est pas encore mis en oeuvre à l'hôpital. On ne trouve pas les nanotechnologies seulement dans les laboratoires de recherche.
Il existe un ensemble d'applications médicales qui concerne des pathologies très variées et différentes phases : diagnostic ou thérapie. Aujourd'hui, 44 produits utilisent des nano-vecteurs, dont 18 produits nano-pharmaceutiques qui sont déjà mis sur le marché. Il existe 15 agents d'imagerie utilisés en nanotechnologie et actuellement 70 essais cliniques à travers le monde utilisent des nanotechnologies. Je voudrais citer un exemple, le plus proche géographiquement. Il s'agit de l'essai clinique de phase 1 qui est mené actuellement, à l'Institut Gustave Roussy, par la société Nanobiotix : des nanoparticules métalliques sont implantées en intra-tumoral, en l'occurrence dans des sarcomes, et permettent de magnifier l'effet de la radiothérapie. À terme, un tel procédé pourrait permettre de diminuer les doses de radiothérapie.
Aujourd'hui, non seulement les produits sont disponibles sur le marché mais, selon les dernières études, celui-ci représenterait un peu moins de 100 milliards de dollars au niveau mondial, avec des perspectives de croissance assez importantes. Ces perspectives sont corroborées par tous les essais cliniques en cours : ce sont des produits qui arriveront demain sur le marché.
Pour montrer que la nano-médecine est une réalité y compris chez nous, je voudrais conclure en présentant quelques exemples de sociétés françaises positionnées sur le secteur. Ces entités représentent la partie visible de l'iceberg, celui-ci étant constitué de toute une activité académique, qui est d'ailleurs de très bonne qualité en France, comparée à celle de nos confrères européens, voire d'Amérique du Nord.
Les entreprises sont souvent le fruit de développements issus de la recherche académique, plus en amont. À partir du moment où des technologies ont été développées, elles sont transférées parce qu'un nano-médicament est un médicament avant tout, un nano-dispositif est un dispositif médical et, en ce sens, tous deux suivent les mêmes voies réglementaires que les autres médicaments ou dispositifs, avec notamment ce temps d'accès au marché qui est un peu long. Cela nécessite que ces entreprises gèrent des essais cliniques pendant un certain temps.
En parvenant à contrôler la construction de nano-assemblages à l'échelle nanométrique, on parvient à développer de nouveaux outils pour le diagnostic et la thérapie. Ces outils peuvent être appliqués dans le cadre de la médecine personnalisée. Ils permettent même de mettre en place une stratégie de médecine personnalisée parce que l'on va cibler un tissu spécifique, particulier à l'individu. Ce n'est pas que du rêve : aujourd'hui, des produits sont sur le marché. Je ne sais pas s'il faut employer le terme « tsunami », mais on voit déferler actuellement, dans les laboratoires et au niveau des essais cliniques, une grosse vague de produits très innovants. Ces évolutions se feront au bénéfice du patient en Europe, mais aussi de notre industrie et de notre économie.
M. Jean-François Deleuze, directeur du Centre national de génotypage au CEA . Comme je viens du Centre national de génotypage, je vais donc essentiellement présenter les technologies et d'ADN. J'ai un profil un peu atypique dans le monde académique car, pendant quinze ans, j'ai été responsable de la recherche en génétique chez Sanofi, société que j'ai décidée de quitter voilà quelques mois. J'ai pu apprécier combien il est difficile de faire entrer ces concepts dans le monde industriel de façon assez pratique. Peut-être pourrons-nous en débattre plus tard...
Ma présentation concernera donc l'ADN, la génomique, et même plus, la génétique. Je n'ai pas volontairement choisi de la focaliser sur cet axe : nous pourrions aborder bien plus de sujets, mais cela prendrait beaucoup plus de temps et concernerait des aspects un peu plus techniques. Mon propos sera également très redondant puisque des généticiens, bien plus éminents que moi, se sont déjà exprimés et m'ont « enlevé » tous mes scoops.
Pourquoi personnaliser ? Je pense que la réponse à cette question est claire : parce que nous sommes tous différents ! Nous le sommes déjà en apparence. Il suffit de regarder autour de la table : nous ne sommes pas deux à être pareils ! Ces différences, qu'elles soient déterministes ou pas, sont tout de même pour beaucoup d'entre elles, inscrites dans notre séquence d'ADN. Nous sommes différents en apparence, évidemment en sexe, mais aussi face à la maladie ou en termes de réponse aux médicaments.
Le Pr Axel Kahn a évoqué le problème de la réponse aux traitements. Certaines personnes n'ont pas de traitement du tout. On doit donc personnaliser car au sein d'un groupe, on trouve ceux qui répondent au traitement, ceux qui ne répondent pas, mais aussi des populations qui sont dans des impasses médicales totales et pour lesquelles les traitements n'existent pas. On a signalé que, dans le cas des maladies rares, les populations sont trop petites pour que l'incitation au développement de thérapies soit suffisamment forte. Cependant bien d'autres raisons entrent en jeu et, effectivement la question de la personnalisation peut revêtir de nombreux aspects.
Par ailleurs, comme cela a aussi été mentionné, ces pratiques ne sont pas très nouvelles. Aujourd'hui on a tendance, peut-être trop rapidement, à associer personnalisation à génomique. À cet égard, j'ai beaucoup apprécié l'intervention de du Pr Patrick Boisseau : la personnalisation passera aussi par des dispositifs , comme les pompes à insuline qui, implantées demain dans le bras d'un patient, pourront permettre de réguler la façon dont celui-ci doit prendre l'insuline. Ce genre d'avancées constituera déjà un effort majeur, sans qu'il soit nécessaire d'aller tripoter le génome.
On observera donc différentes époques dans la médecine personnalisée, qui ne se réduira pas à une médecine génétique. Ce sera aussi une médecine de dispositifs, une médecine de phénotypage, une médecine de caractérisation. Ainsi une prise de sang est une personnalisation puisque l'on ne vous transfusera pas un sang ne correspondant pas à votre groupe sanguin. Elle est peut-être partielle, mais c'est bien une personnalisation. Avec l'examen cytobactériologique des urines, (ECBU), on cherche en général à donner l'antibiothérapie correspondant à la bactérie qui peut infecter le patient.
En avançant dans des domaines plus moléculaires, on trouve la greffe avec le système HLA et les questions de compatibilité. Dans le cas d'une greffe allogénique, le patient reçoit l'organe d'un tiers, et on ne le lui transplantera pas si cet organe n'est pas a minima compatible avec le système immunitaire de ce receveur. Cette personnalisation est certes partielle, mais je pense que personne ne reviendrait aujourd'hui sur le bénéfice à la fois thérapeutique et économique de ces tests. Où a-t-on progressé ? On utilise de plus en plus du phénotypique et du moléculaire et, effectivement ce qui nous pousse à vouloir personnaliser, peut-être plus que nous ne le devrions, est l'accès au génome humain ce qui constitue une révolution.
Faisons un bref rappel sur le génome humain, il représente 3 milliards de paires de bases et il ne se passe pas une semaine sans qu'on en apprenne des sur le sujet. Comme nous sommes à l'Assemblée nationale, je n'ai pas résisté à cette petite phrase tirée de la Déclaration des droits de l'homme et du citoyen : « nous naissons et demeurons libres et égaux en droits »... mais nos génomes sont différents ! Malheureusement, nous ne sommes pas égaux devant la maladie et devant la réponse aux médicaments. J'ignore comment cela doit être pris en compte au niveau sociétal, mais c'est une réalité.
Le séquençage d'ADN humain représente 3 milliards d'éléments groupés en 46 chromosomes, et sur ces 3 milliards d'éléments, on comptabilisera 3 millions de différences entre les génomes de deux individus. Ces différences sont à l'origine de notre apparence - notre phénotype extérieur, mais aussi nos maladies -, de notre réponse aux médicaments et de nombreux autres phénomènes.
Effectivement, comme le rappelait précédemment
une intervenante, l'environnement se surimpose à cela. Or
l'environnement sans le patrimoine génétique et ce que celui-ci
exprime n'aurait pas non plus d'influence. Il s'agit en fait d'un mariage entre
les deux. Pour compliquer un peu la situation, 200 types cellulaires existent.
Il faut en effet sortir d'un certain dogme qui consiste à croire
- c'est un peu moins vrai aujourd'hui qu'il y a quelques
années
-
que l'ADN est exactement le même
dans toutes les cellules. Quand on évoque l'ADN, il s'agit le plus
souvent de l'ADN issu du sang, car le sang est le plus accessible de
façon la moins invasive. Notre ADN, qui est différent selon les
200 types cellulaires, subit aussi des modifications chimiques : on
appelle cela l'épigénome, qui peut également être un
révélateur de maladies, d'influences à l'environnement,
etc
. La question est donc bien plus complexe que le simple fait de
regarder l'ADN dans le sang.
Les différences sont de multiples types. Dans le cas des mutations ponctuelles, une lettre est changée par une autre. Un « A » devient un « C » par exemple. Cela peut être absolument neutre ou totalement délétère. Il y a également des délétions - des morceaux entiers sont enlevés -, des inversions - l'ordre dans lequel sont placées les bases est modifié - ou des amplifications.
Voici deux exemples d'implications fonctionnelles d'une mutation par rapport à un phénotype. La mucoviscidose, tout d'abord, est une des maladies monogéniques les plus fréquentes. Si, dans la séquence normale 3 bases « CTT » sautent - 3 bases sur 3 milliards ! -, on passe à la forme suivante. Si malheureusement, on a deux copies de cette forme obtenue par cette fameuse mutation, extrêmement commune, qui s'appelle ÄF508 - delta pour « délétion » -, on est atteint de mucoviscidose. Au regard de la réflexion précédente sur la question du déterminisme, je me permets d'insister : malheureusement, avec deux copies de ÄF508, on est atteint de mucoviscidose dans 99 % des cas, même si les formes peuvent varier. C'est extrêmement déterministe. Une variation génétique peut donc conduire à l'introduction d'une maladie.
Le deuxième exemple aborde la réponse au médicament. Le Pr Axel Kahn et le Pr André Syrota ont traité ce matin des effets toxiques. Le cas qui nous intéresse concerne un cytochrome, le cytochrome 2D6, qui est un enzyme hépatique prenant en charge de nombreux médicaments pour les détoxifier. Ce cytochrome est malheureusement très polymorphe et prend des formes très variables selon les individus. Certaines personnes n'en ont aucune copie et accumulent les drogues sans les métaboliser. D'autres en ont de nombreuses copies et vont donc métaboliser très rapidement. On comprend immédiatement que la posologie d'une drogue devrait être adaptée à notre contribution génétique par rapport à ce polymorphisme. Et sur ce point, c'est assez déterministe. C'est vrai pour une grande partie des enzymes du métabolisme des médicaments.
Un article assez ancien de Nature sur le sujet, dans lequel il est question d'adaptation s'y réfère, et j'estime que c'est acceptable en termes de stratification. On n'en est pas à dire qu'il faut tel ou tel médicament. Mais, de façon intermédiaire, on pourrait imaginer adapter la posologie au profil génétique du patient. La question du business model sera évoquée ultérieurement. De nouveau, nous discuterons des dispositifs certainement acceptables.
Quels sont les outils ? Comme cela a été expliqué, et pour brosser la situation de manière un peu simpliste, il existe deux outils majeurs pour observer l'ADN : le génotypage et le séquençage. Le génotypage consiste à observer des endroits précis : plutôt que d'examiner tout le génome, ce qui coûte cher, je regarde des zones décidées à l'avance, des marqueurs. Le séquençage, de façon manichéenne, consiste à tout regarder.
Un point probablement important pour vos débats futurs, Messieurs les députés, réside dans la question de l'utilisation de ces technologies pour la découverte ou pour le diagnostic, car ces finalités ne recouvrent pas du tout le même monde réglementaire. Les thèmes discutés ce matin au sujet des hôpitaux concernaient, pour beaucoup, le domaine de la recherche clinique, et pas franchement celui du diagnostic, avec une réglementation posée derrière. La façon dont on réglementera le séquençage total, l'utilisation du génome humain, est déjà en soi un débat majeur. On dispose de deux grandes techniques, et de robots nombreux et variés pour effectuer ensuite l'analyse génétique.
Un exemple concerne le laboratoire Bayer qui a développé une statine ayant entraîné un certain nombre de cas, assez rares, de rhabdomyolyses. Cette situation est bien sûr très préjudiciable au patient, mais elle a également causé beaucoup de tort à l'industrie pharmaceutique, qui a failli ne pas s'en relever. Ceci est un exemple d'analyse génétique par génotypage qui a donc été faite, en partie en France, en collaboration avec Merck, pour tenter de répondre à la question suivante : est-ce que ces personnes ont des rhabdomyolyses parce qu'ils ont une susceptibilité génétique ?
Il n'y avait pas tant de cas que cela moins de 90, me semble-t-il, sur 3 000 contrôles effectués dans cette étude, et, pour ces 90 personnes ayant fait une rhabdomyolyse sous statines, on a interrogé environ 300 000 positions du génome humain pour savoir si l'une d'entre elles était liée à l'apparition de la rhabdomyolyse. Il s'est avéré qu'un seul signal fonctionne : c'est un gène candidat. Sur les 387 000 marqueurs, il y en a un qui, de façon statistique, répond à la question : oui, ce marqueur est associé à l'apparition de la rhabdomyolyse en cas de prise de statines. Qu'en est-il de ce marqueur ? Il s'agit d'un variant du transporteur hépatique des statines. C'est donc logique. Malheureusement, quand on a le mauvais variant, la mauvaise forme du transporteur hépatique des statines, et qu'on prend cette statine-là, on a une plus grande chance de développer une rhabdomyolyse.
Cette étude a donc un impact immédiat : éviter de donner éventuellement des statines aux personnes ayant le mauvais génotype CC. Mais elle ouvre aussi une autre voie pour l'industrie, qui consiste à trouver une statine qui n'est pas sensible à ce phénotype. On parle de personnalisation, mais le modèle peut être aussi d'éviter de trouver des cibles thérapeutiques qui sont polymorphes dans la population, en évitant justement d'être dans des systèmes trop polymorphes. C'est une autre stratégie possible !
L'exemple suivant, cité par le Pr Pierre Laurent-Puig, concerne le cancer. À ce propos, on a oublié de rappeler qu'il y a des tumeurs que l'on peut réséquer, prendre et étudier. Quand on veut étudier le pancréas d'un patient, on ne le lui enlève pas ; on n'enlève pas le coeur d'un individu. On peut donc extraire une tumeur en morceaux et l'étudier de façon moléculaire. Or les altérations individuelles moléculaires du cancer sont très fortes. C'est une maladie très fréquente, mais aussi une maladie sur laquelle il était envisageable de progresser.
Un certain nombre de thérapies ciblées représentent des succès de médecine stratifiée. Il y a tout d'abord eu, pour le cancer du sein métastasé, l'Herceptin - Christophe Le Tourneau a évoqué la question du récepteur HER-2 - qui a apporté un réel bénéfice thérapeutique. Et les solutions ne cessent d'augmenter, avec de nombreux anticorps.
Je crois que l'on dispose d'environ 17 ou 20 couples d'altérations génétiques pour lesquelles, aujourd'hui, on peut attribuer une thérapeutique dite « cliniquement actionnable ». Quand on peut associer à une altération une intervention thérapeutique, on aboutit à des procédés puissants.
Le Centre national de génotypage (CNG) que je pilote depuis le mois de septembre dispose, en capacité machines de 11 séquenceurs, ce qui en fait probablement le deuxième centre européen de séquençage. Il faut néanmoins y mettre un bémol : en tant que séquenceurs, nous intervenons dans le domaine de la recherche et n'avons pas, à l'heure actuelle, de capacité en termes de diagnostic. Nous sommes capables de séquencer quasiment trois génomes humains par jour. Ce résultat peut être mis en parallèle avec les efforts qu'il a fallu déployer, voilà dix ans, pour séquencer un génome : cela a pris plusieurs années et coûté des centaines de milliers de dollars. Si l'on raisonne en consommable, le prix d'un génome avoisine désormais 3 000 euros et ce coût continuera de baisser. Il existe donc une force de frappe en France, elle sert beaucoup la communauté scientifique nationale essentiellement sur des activités de recherche et c'est d'ailleurs sa mission. Par ailleurs, nous sommes certainement le leader européen en matière de génotypage, c'est-à-dire pour l'examen d'altérations spécifiques.
Qu'en est-il de l'évolution ? Nous avons grosso modo trois solutions. Selon l'argent dont nous disposons et l'évolution des coûts, nous pouvons regarder la séquence totale. Comme je l'ai indiqué, le prix est de 3 000 euros environ et, si nous ne disposons pas actuellement des outils d'analyse, une évolution se profile dans les mois et années à venir. Nous sommes déjà capables d'analyser de façon localisée des marqueurs, ce qui revient beaucoup moins cher. Enfin, nous pouvons examiner des gènes ou l'ensemble des gènes, et ce que l'on considère être aujourd'hui l'endroit comprenant le plus d'information du génome.
Tous ces concepts sont en train d'évoluer et, aujourd'hui, en 2013, les coûts ne sont plus les mêmes que par le passé. Nous ne séquençons pas tout le monde massivement, mais il est évident que le prix du whole genome sequencing sera, dans les deux ans qui viennent, équivalent au prix de l'exome ou du génotypage que l'on faisait avant. Se posera alors la question de savoir comment on réagit. Il me semble que personne ne pourra contrecarrer ces évolutions, et que tout le monde est d'accord sur ce point.
J'en viens aux défis. Premier défi, il manque une filière en France en bio-informatique et bio-statistique. Nous générons beaucoup plus d'informations que nous sommes capables d'en traiter. Il faudra donc des bio-informaticiens, des bio-analystes, des biostatisticiens en nombre beaucoup plus important que les effectifs actuels. Le fait d'être capable de générer des giga-bases de données ou des centaines de giga-bases de données chaque jour sans être en mesure d'analyser un tel volume de données est un vrai défi, aussi bien en termes d'outils d'analyse qu'en termes de filière et de personnel.
Deuxième défi, il y a un problème au niveau de l'informatique, ce point, me semble-t-il, a été soulevé par le Pr Axel Kahn ou le Pr André Syrota - dans la mesure où le stockage des données coûtera pratiquement plus cher dans le futur que leur génération. Des efforts majeurs doivent donc être fournis dans le domaine informatique, en matière de stockage et de capacité de calcul.
Troisième défi, la génomique fonctionnelle. C'est aussi une science qu'il ne faut pas oublier. Nous sommes capables de décrire beaucoup de marqueurs, mais nous ne comprenons pas vraiment comment ils fonctionnent. Si nous voulons être capables de personnaliser, pour reprendre les exemples donnés par Axel Kahn, au niveau des cibles thérapeutiques, nous devons être capables de comprendre l'intérêt de nos cibles.
Quatrième défi, l'accès aux échantillons biologiques. Il faut arrêter de penser que le sang est le seul tissu pertinent pour l'analyse. Dans le cas de nombreuses pathologies, il faudra aller chercher les échantillons dans les tissus car il faudra regarder l'épigénome. Des technologies sont à développer pour permettre cet accès. Si, par exemple, on pense « épigénome » et « cerveau », on perçoit immédiatement la complexité de travailler, en situation « non post mortem », sur le cerveau d'un individu. Ce n'est pas possible et soulève effectivement beaucoup de problèmes.
Dr Catherine Bourgain, chargée de recherche en génétique humaine et statistiques à l'INSERM . Je poursuivrais la réflexion sur la génétique, puisque je suis généticienne moi aussi. J'ai choisi de me concentrer sur un point qui a été évoqué ce matin et qui constitue un des enjeux de la médecine personnalisée, à savoir la problématique de la prédiction. En quelques minutes, j'essayerais de faire le tour de ce que l'on peut, ou ce que l'on pourra prédire, à partir de l'analyse de l'ADN.
Le premier domaine dont il a été question est celui des maladies monogéniques. Actuellement, les progrès réalisés portent sur les nouvelles méthodes d'analyse par génotypage ou par séquençage, lesquelles permettent, dans une même analyse, de rechercher de façon simultanée un très grand nombre de mutations impliquées dans de nombreuses maladies génétiques pédiatriques différentes.
Dans un article sorti en 2011, qui a eu un grand retentissement, il était question d'effectuer des recherches systématiques de mutations concernant près de 500 maladies pédiatriques différentes. En faisant ces recherches sur des personnes non malades, on arrivait à la conclusion qu'en moyenne, chacun des individus analysés était porteur de 3 mutations pour une de ces maladies, ce nombre de mutations variant entre 0 et 7 à peu près. Ces nouvelles technologies permettant de faire des analyses concomitantes d'un grand nombre de mutations peuvent être couplées avec d'autres progrès, en particulier ceux qui sont liés à la capacité d'analyser le génome des foetus quand ceux-ci se trouvent encore dans le ventre de leur mère.
On a récemment parlé de la mise au point de la technologie permettant le dépistage de la trisomie 21 dès la dixième semaine de grossesse à partir d'une simple prise de sang chez la mère. Depuis, cela a progressé, l'année dernière, un article a été publié, montrant qu'il avait été possible de procéder à un séquençage intégral d'un génome de foetus à 19 semaines de grossesse, à partir d'une simple prise de sang chez la mère. Cela illustre bien comment différentes technologies peuvent se combiner pour engendrer de nouvelles capacités.
Ces évolutions ont des conséquences en termes de diagnostic prénatal, de diagnostic préimplantatoire, mais également sur le développement de ce que l'on appelle les tests génétiques préconceptionnels. Il s'agit de proposer aux membres d'un couple, avant la procréation, de se soumettre à ce type d'analyses pour voir si, par hasard, ils seraient tous les deux porteurs d'une mutation impliquée dans une même maladie. Le cas échéant, des analyses de diagnostic prénatal, voire préimplantatoire, pourraient leur être proposées. Du fait du développement des tests préconceptionnels, le nombre de personnes potentiellement concernées par ces tests peut être considérablement élargi, puisque le public auquel on s'adresserait serait, non plus uniquement des enfants malades ou des couples ayant déjà eu un enfant atteint, mais potentiellement tous les couples désireux de procréer.
Je perçois au moins deux enjeux autour de ce type
d'analyses. Le premier enjeu porte sur la précision technologique. Dans
quelle mesure les outils disponibles permettent-ils de détecter avec une
bonne fiabilité la présence de mutations ? Dans le cadre du
fort développement actuel des technologies, des interrogations se posent
véritablement. Est-on sûr de ce que l'on détecte ?
N'y a-t-il pas des erreurs de technique ? On sait effectivement que
celles-ci sont assez nombreuses.
Le deuxième enjeu est de savoir si les valeurs de pénétrance que l'on a employées jusqu'ici, c'est-à-dire la probabilité de tomber malade pour les personnes porteuses de ces mutations, sont si bonnes que cela. En effet, en analysant un nombre croissant de personnes qui ne sont pas malades, on se rend compte que, dans certains cas, des individus porteurs de mutations, parfois en double dose, auraient dû être considérés comme malades alors qu'ils n'ont pas développé la maladie. L'essor de ces technologies interroge aussi notre capacité à bien mesurer les risques, même dans le cas de maladies censées avoir un très fort déterminisme du point de vue génétique.
Venons-en à la deuxième catégorie de maladies. Ce sont toutes les « maladies multifactorielles », parce qu'elles résultent d'une combinaison de facteurs génétiques et de facteurs d'environnement, au sens le plus large de ce terme, c'est-à-dire à la fois la vie foetale et tous les facteurs non génétiques. Des mesures de composante génétique sont souvent mises en avant dans les débats pour justifier les mesures de risque basées sur la seule génétique. C'est la mesure d'héritabilité. On estime que, pour telle maladie, cela vaut la peine de rechercher des facteurs de risque génétique, car l'héritabilité de cette maladie atteint 30 % ou 40 %.
Or cette notion d'héritabilité est extrêmement discutée dans la communauté scientifique, parce qu'elle présuppose l'absence d'interaction entre les effets génétiques et les effets d'environnement. On sait pertinemment que c'est faux. Un même génome d'un individu, selon l'environnement dans lequel celui-ci sera plongé tout au long de sa vie, aura un risque très différent d'induire telle ou telle pathologie.
Les mesures censées décrire la part de la génétique dans les maladies complexes sont donc à prendre avec beaucoup de précautions. Pour autant, depuis trente ans, de nombreux travaux ont été menés pour essayer d'identifier les facteurs de risque génétiques de ces maladies multifactorielles et certains l'ont été, comme ceux dont il a été question dans la présentation précédente. Ces risques qui sont associés à tel ou tel facteur génétique sont des valeurs moyennes sur l'ensemble des individus porteurs du variant génétique identifié. Il s'agit bien d'une valeur en population. Or si l'on veut faire de la prédiction, il faut se mettre au niveau de l'individu, et à ce niveau, les risques calculés uniquement sur l'ADN sont en définitive de très mauvais déterminants, et ce pour au moins deux raisons.
Premièrement, les risques mesurés sont toujours des risques qui ne sont absolument pas déterministes. Dans le cas de la maladie d'Alzheimer, par exemple, pour laquelle une personne aura un risque moyen, si l'on ne fait pas d'analyse génétique, de 10 % environ, son risque variera de 8 % ou de 51 % selon son génotype pour le gène ApoE. Cela signifie qu'en prenant en compte l'information génétique, on fait varier la mesure du risque, mais en restant dans des échelles de risque qui ne sont pas du tout celles des maladies monogéniques. Ce n'est absolument pas du déterminisme, comme cela a été évoqué précédemment.
Deuxièmement, pour toutes ces mesures, il y a en pratique très peu de recommandations efficaces, voire aucune, qui soient applicables à ces individus plutôt qu'à d'autres. La plupart du temps, ce qui va être suggéré est de faire attention à son alimentation, à sa pratique sportive, etc. , autant de recommandations applicables à pratiquement toute la population, puisque nous sommes tous à risque.
La faiblesse de cette information génétique pour prédire l'apparition de maladies multifactorielles a été illustrée par un exemple. En 2007, aux États-Unis de nombreuses de compagnies se sont développées pour vendre des mesures de risques individualisés pour les maladies multifactorielles, directement au consommateur, sur Internet. Deux des plus emblématiques entreprises ont fermé en 2012 et ont été rachetées par d'autres sociétés. Les services de mise en ligne de mesures de risque ont quant à eux été fermés, l'objectif des industries ayant racheté ces compagnies étant de mettre la main sur les bases de données qui avaient été constituées. Ces mesures de risque individuel n'ont effectivement pas d'utilité. En revanche, le fait de disposer d'importantes bases de données peut être utile pour le développement ultérieur de traitements plus ciblés.
Je voudrais évoquer rapidement un troisième point. D'aucuns pensent qu'il faut aujourd'hui intégrer les données génétiques avec les données environnementales pour peut-être obtenir des améliorations notables dans la précision des prédictions. Il a été question, dans les interventions précédentes, d'un nombre considérable de données additionnelles que l'on peut envisager d'intégrer aux données génétiques. Les biocapteurs ont été évoqués, ainsi que la possibilité d'effectuer des mesures régulières au cours d'une journée, au cours de la vie, et le fait qu'il y a plusieurs génomes et plusieurs épigénomes. Cela forme un ensemble d'informations pouvant se cumuler, ce qui aboutit à des modèles de plus en plus complexes.
Il me semble que la question de savoir s'il suffira d'avoir le bon nombre de bio-informaticiens et la bonne puissance de calcul pour intégrer réellement toutes ces données est ouverte. En effet, il existe aussi des difficultés conceptuelles à modéliser des systèmes aussi complexes, dans lesquels les paramètres ne cessent d'être toujours plus nombreux et qui, en plus, sont en interaction les uns avec les autres, avec des effets « système », si je puis dire, créant des interactions supplémentaires.
En définitive, on ne peut absolument pas savoir si, demain, même avec toutes ces données, on sera en mesure de faire des prédictions ayant une valeur certaine, parce que l'on sait qu'au-delà d'un certain niveau de complexité, on a souvent des difficultés à trouver des concepts permettant de traiter toutes ces informations. J'ajoute qu'il s'agit souvent de données issues de multiples sources et dont le traitement et l'analyse, de façon concomitante, posent des problèmes.
Sur ces enjeux de prédiction à partir de l'ADN, beaucoup de promesses importantes ont été faites, de nombreux discours surfent sur le sensationnel, mais je crois qu'il est extrêmement important d'aller dans le détail pour savoir comment, déjà à l'heure actuelle, nous utilisons ces informations, et cerner ce que nous sommes vraiment capables de réaliser aujourd'hui et ce que nous pensons avoir de bonnes chances de réaliser demain. Quand on interroge les scientifiques, on est souvent capable de faire la différence entre ce qui a de bonnes chances d'advenir et ce qui est de l'ordre de la promesse totalement ouverte. S'agissant de la constitution de toutes ces grandes bases de données, de nombreuses questions demeurent.
Le problème du bénéfice-risque doit donc être abordé avec une mesure la plus fine possible des bénéfices, tels qu'ils sont aujourd'hui, et la problématique de la prédiction, en tout cas pour les maladies multifactorielles, reste tout de même très ouverte.
Dr. Anne Cambon-Thomsen, directrice de recherche au CNRS UMR U 1027 « Épidémiologie et analyses en santé publique » de l'INSERM, et de l'Université de Toulouse 3 Paul Sabatier, responsable de l'équipe « Génomique, biothérapies et santé publique ». Je souscris à bien des points évoqués par le Dr Catherine Bourgain. Mon propos sera orienté sur les biomarqueurs. Pour commencer, je vais citer une phrase tirée d'une assemblée australienne similaire à celle que nous tenons aujourd'hui, préalablement à l'examen d'une législation concernant la génétique dans ce pays : « La rapidité des changements a produit deux réactions sociales puissantes, mais opposées. On constate d'un côté, une très forte attente du public pour des avancées prometteuses en termes de diagnostic et de traitement, et de l'autre, des craintes concernant la perte grandissante de la sphère privée, le risque de discrimination génétique, et la possibilité de réguler la science génétique pour l'intérêt public. » C'est sur ce dernier point que je vais insister. Je crois en effet que la dernière partie de cette phrase se trouve au coeur de ce dont nous discutons aujourd'hui.
Nous avons déjà parlé des définitions que l'on peut donner de la « médecine personnalisée » et de l'importance de ces termes. Je veux souligner que l'expression peut devenir un slogan. Je ne peux plus entendre des phrases telles que « Le bon traitement, au bon moment, pour le bon patient ». Cela a été totalement galvaudé. Cela donne une fausse idée de ce qu'est la réalité. Ce qui est vrai dans certains cas, ne l'est pas pour d'autres. En d'autres termes, il faut susciter l'espoir, mais sans fausses promesses. Or de fausses promesses sont faites tout le temps ! L'espoir et des réalisations existent aussi. Il faut donc faire la part des choses et c'est là toute la difficulté de l'exercice.
S'agissant des biomarqueurs, nous nous sommes déjà arrêtés sur les caractéristiques des sauts technologiques et sur la plus grande lenteur de leur application dans la médecine. Cela est évidemment dû au fait qu'on ne prend pas en compte les seules capacités techniques lorsque l'on met en application un certain nombre d'avancées dans un système de santé. La problématique du niveau de preuves à établir peut donc se résumer ainsi : on se trouve face à des biomarqueurs extrêmement variés, apportant des messages plus ou moins validés et des degrés d'information différents, et cet ensemble est très difficile à appréhender.
Ces enjeux sont aussi liés à la définition du niveau de preuve avant la mise en oeuvre. Il n'existe pas forcément d'éléments précis pour tous les types de marqueurs que l'on peut générer. Ils sont bien sûr liés aux conséquences au niveau individuel et collectif. Il y a des conclusions au niveau individuel dans certains cas et des enjeux à l'échelle de populations dans d'autres cas, la frontière n'étant pas toujours tranchée entre les deux. Enfin, les enjeux sont liés aux relations entre médecins et patients, au rôle joué par les patients et leurs associations, et aux stratégies industrielles.
On se retrouve avec un paysage complexe, dans lequel les biomarqueurs sont des éléments centraux. On en dénombre plusieurs catégories : ceux que j'appellerai « constitutionnels », comme les marqueurs génétiques ; ceux qui ont un rapport avec l'évolution de la maladie ou le développement de la maladie elle-même, comme les marqueurs de tumeur ; ceux qui sont liés aux capacités de réponse à des molécules, donc au traitement et à l'évolution de ce dernier. Outre l'environnement, cet ensemble, composé d'éléments assez variés, forme l'ensemble des paramètres à prendre en compte dans la médecine personnalisée.
Comme cela a été souligné dans les propos introductifs, il existe une sorte de paradoxe, car on parle de personnalisation pour des démarches consistant à définir plus finement des groupes. On se trouve tout de même dans la stratification et la constitution de groupes. Or le fait d'employer le terme « personnalisée » génère une autre attente. C'est pourquoi, je préfère utiliser les notions de « médecine de précision », ou de « stratification », parce que cela correspond davantage à la réalité.
Par ailleurs, la personnalisation vue à travers la caractérisation biologique, fine, peut aussi être perçue comme réductrice, d'où l'importance du dialogue. Le dialogue avec les professionnels de santé reste donc au coeur de la médecine. Ces professionnels, certes aidés par des outils informatiques, doivent en outre intégrer une quantité importante d'informations de différents types, et il n'est pas toujours facile d'apporter une information claire. De ce fait, la question de l'éducation est extrêmement importante, on l'a mentionnée pour les étudiants en médecine, mais c'est également vrai pour les professionnels de santé, au fil de l'apparition des nouveaux outils, dans le cadre de la formation continue.
J'en viens au sujet de la prédictivité, déjà abordé. S'agissant de génétique, on se situe dans cette idée de prédictivité, derrière laquelle se profile la question de l'interprétation. Face à un génome entier en séquence, ou bien un exome, on dispose de capacités très variées d'interprétation des différents points de ce génome ou exome. Ce constat n'est pas toujours mis en avant. On sait générer toute une série d'informations sans savoir les interpréter.
À propos de cette prédictivité, je citerai une phrase du Pr Didier Sicard, ancien président du Comité consultatif national d'éthique ( CCNE) : « La médecine s'engouffre dans cette prédiction en pensant que plus on prédira un risque, moins il aura de chances de survenir. Il existe une confusion permanente entre l'usage que l'on fera de cette prédiction et la prédiction elle-même ». Ce point, me semble-t-il, a été rappelé dans les propos du Dr Catherine Bourgain.
J'en viens à cette question : peut-on considérer, réguler, expliquer tout type de test génétique, de biomarqueur, de la même façon? La réponse est négative. Mais quand on en vient à la régulation et à l'organisation au niveau d'un système de santé, on se trouve en face de certaines réalités et, dans ce cadre, il est commode d'avoir des éléments clairs et des catégories. Or l'information génétique peut être classée dans une catégorie un jour donné et, parce qu'on aura un peu plus d'informations sur l'environnement, sur les interactions, parce qu'on aura progressé dans la capacité à interpréter certains domaines du génome, elle se retrouvera le lendemain dans une autre catégorie, au niveau des capacités productives. Comment gérerons-nous cela ? L'information génétique est évidemment à considérer parmi d'autres biomarqueurs. La dimension familiale de cette information, ainsi que la dimension de catégorie ou de groupe, doivent être prises en compte dans ce cadre. Je reviendrai sur les associations en terminant mon propos.
Par ailleurs, la masse d'informations générées par les tests et examens variés, d'informations génétiques notamment, est bien supérieure à l'information utile dont on se servira au temps T. Dès lors, que faire des informations qui deviennent disponibles, mais sont a priori non recherchées ? Jusqu'où faut-il fouiller la masse d'informations générées quand on a tout un génome ? Que faut-il systématiquement analyser ? Autour de ces enjeux, d'importants débats sont engagés au sein du collège des généticiens aux États-Unis, et une réflexion est entamée sur le sujet à la Société européenne de génétique humaine. Il s'agit bien de déterminer ce qu'il faut faire des découvertes incidentes et s'il doit exister une obligation à regarder systématiquement certains aspects du génome. Évidemment, derrière ces problématiques, se profile tout un pan de l'éducation professionnelle et de l'accompagnement.
Nous avons évoqué le sujet économique. En définitive, au-delà de la question du coût, se pose aussi celle de l'accès équitable. Par ailleurs, les patients sont de plus en plus « catégorisés », peut-être jusqu'à l'individu, c'est le cas dans les thérapies cellulaires, mais, pour le reste, on travaille par groupe, et ce n'est pas toujours à leur avantage. Il est sans doute nécessaire d'envisager des formations particulières pour faciliter la gestion et le partage de certaines informations, par exemple lorsque l'on doit expliquer à un patient qu'il n'est pas dans le groupe que l'on sait traiter, mais que l'on espère que cette situation évoluera. La multiplication des outils et des informations rend ces cas plus fréquents que par le passé. Auparavant, au moins, on gardait espoir même s'il fallait parfois gérer, par la suite, effets secondaires et problèmes thérapeutiques. Il est important de ne pas tuer l'espoir par l'information et la qualité du dialogue est un élément majeur qui ne se résout pas par la bio-informatique.
Je considère en outre, qu'en santé publique il y a une grande nécessité à mener des études d'impact. Nous avons entendu un certain nombre de propos, notamment en matière d'économie de la santé, mais il me semble que des points sont à développer, plus largement, dans le domaine de la santé publique, autour de la question de l'impact de ce qu'on appelle la « médecine personnalisée ». À cet égard, l'Institut thématique multi-organismes (ITMO) de santé publique organise un colloque annuel à l'automne, le 5 décembre 2013. Cette année, le thème en sera la médecine personnalisée. Il serait très intéressant que certaines des personnes ici présentes participent aussi à cet événement et puissent confronter ce qui se dit dans ces auditions publiques et ce qui se dira dans ce colloque scientifique.
Les enjeux sont variés. Ils sont tout d'abord liés au concept, la question étant la suivante : la personnalisation d'une médecine est-elle plus grande parce qu'elle est « biologiquement fondée » ? Le fait de détenir plus de précisions biologiques conduit-il automatiquement à plus personnalisation, au sens de la considération centrale de la personne et non de « ses données biologiques »? Cette interrogation a été soulevée précédemment et je crois qu'elle est essentielle.
Je conclurai en indiquant que nous avons à différencier plusieurs éléments. Tout d'abord, il y a l'application d'un degré de précision nouveau à l'exercice médical, enjeu assez classique et standard. Des exemples ont été donnés sur ce point. Puis vient l'aspect lié à la médecine de prévision ou médecine prédictive, qui constitue une partie de la personnalisation ou de la stratification. On a ensuite ce que l'on appelle « la médecine basée sur les données ». Ces informations sont générées à grande échelle, parce que cela coûte (ou coûtera bientôt) moins cher que leur acquisition en plusieurs étapes successives, parce que la technologie nous pousse à le faire, parce que, finalement, il est logique d'aller dans ce sens, et ce, avec des niveaux d'utilité variés. C'est un sujet que l'on ne sait pas véritablement gérer et il n'y a pas encore de recommandations consensuelles dans ce domaine qui change la logique intrinsèque de l'exercice médical.
Enfin, il faut considérer l'individualisation de l'accès aux données biologiques, notamment les aspects abordés par le Dr Catherine Bourgain sur la mise à disposition publique d'informations. On ne se trouve pas tout à fait dans une situation où c'est systématiquement le médecin qui ordonnera la génération d'informations. Dans certains cas, les patients pourront, et ils le font déjà, se présenter avec des informations sur lesquelles ils n'auront pas toujours obtenu une explication précise, exacte, prenant en compte les hypothèses qui sous-tendent les modèles donnant naissance aux calculs de risques tels qu'on les connaît aujourd'hui.
Dr. Laurent Alexandre, président de DNAVision, membre de l'Académie des technologies . Je voudrais apporter un témoignage personnel de ce que l'on observe au travers de la médecine personnalisée. En effet, j'ai effectué le séquençage de la totalité de mon propre génome à deux reprises, à très haut coverage , je le précise à l'attention des techniciens, donc sans faux-positifs. Entendons-nous bien, mais c'est là un débat de spécialistes, je veux dire qu'à un coverage de 200 ou 300, au lieu d'avoir quelques centaines de milliers de faux-positifs sur les 3 milliards de paires de bases, on n'en aura que quelques-uns. Il y en a tout de même peu !
Cette expérience m'a interpelé par rapport aux discours théoriques, aux discours d'industriels que je tenais sur le sujet, mais également par rapport à l'approche que j'ai développée, voilà quinze ans, en tant que fondateur du site Doctissimo. Cette approche pouvait se résumer ainsi : il faut tout dire au patient en direct ; le patient peut tout entendre, même sans l'intermédiaire du médecin.
Même si l'univers dans lequel nous nous trouvons n'est pas toujours déterministe, la masse d'informations auxquelles j'ai eu accès en me séquençant m'a interpelé, et je suis personnellement opposé à ce que les individus accèdent à leur séquençage sans un intermédiaire médical. On ne peut pas apprendre que l'on a des variants graves sans un intermédiaire médical. Les résultats sont très difficiles à interpréter, y compris pour un spécialiste de la génomique, pour un médecin comme je le suis. On imagine donc les difficultés que rencontreraient des personnes n'ayant pas de culture génomique pour comprendre ce qu'est un variant sur une maladie récessive ou pour interpréter des variants évalués sur des « SNP flags » (Single Nucleotide Polymorphisms ) qui ne sont pas fonctionnels. C'est très difficile.
Bien évidemment, on rencontre dans ce domaine un phénomène de « surpromesses ». Des compagnies comme 23andMe, avec des technologies de génotypage et de microarray relativement rudimentaires, ont proposé des offres grand public. Il faut réfléchir à cette question, car elle soulève de vrais problèmes, que l'on perçoit encore mieux après s'être soi-même séquencé et avoir découvert que l'on était porteur de variants, dont certains sont de mauvais variants. À ce titre, il est important de noter que plus on vieillit et moins le séquençage est informatif. À 90 ans, on peut être certain de ne pas avoir eu de maladie de Huntington, de ne pas être porteur d'une mutation du gène LRRK2, qui donne une forte probabilité de développer la maladie de Parkinson, de ne pas avoir de myopathie.
C'est donc sur le foetus que le séquençage est, par définition, le plus informatif, puisque l'on connaît alors peu de choses. A contrario , plus on vieillit, moins ce caractère informatif est important. Savoir que j'ai les deux principaux variants de protection contre la calvitie ne m'apporte pas grand-chose : je me suis bien rendu compte que je n'étais pas chauve ! Si j'avais été séquencé à cinq ans, cela aurait été plus informatif.
Par ailleurs, il est très difficile de prévoir ce que l'on fera avec ces technologies. Il faut rappeler, à l'attention de ceux qui n'ont pas l'historique du séquençage en tête, que l'on assiste tout de même à un véritable tsunami technologique. Cela ne veut pas dire que l'on va découvrir un univers génomique déterministe, cela signifie simplement que personne n'avait prévu ce qui nous arrive. En 1990, on avait un consensus mondial dans la communauté des généticiens sur le fait qu'on ne saurait jamais séquencer intégralement les 3 milliards de paires de bases. À l'époque, deux ou trois iconoclastes avaient déclaré qu'on y arriverait. D'après eux, il fallait trois à cinq siècles. Or nous y sommes arrivés !
La première fois, cela a coûté 3 milliards de dollars, mobilisé 20 000 personnes et duré treize ans. La deuxième fois, il a fallu six mois et 1,5 milliard de dollars. Aujourd'hui, un séquençage vaut entre 1 000 et 2 000 dollars, et tombera à 100 dollars. Le coût baisse de 50 % tous les cinq mois. Personne n'avait imaginé cette évolution. Nous avons donc beaucoup de mal à déduire les conséquences médicales, industrielles, technologiques et éthiques de cette évolution du séquençage, parce que personne ne l'avait prévue.
Au-delà de l'effondrement des coûts du séquençage, nous avons aussi découvert que ce que nous avons longtemps appelé le junk DNA ou « ADN poubelle » - pardon pour les spécialistes - n'existe pas. Depuis 1971, on était persuadé que 98,5 % de l'ADN ne servait à rien. On avait appelé cette zone ADN poubelle, le « junk DNA » . Or on se rend compte que ces 98,5 % de l'ADN contiennent en réalité de nombreuses zones régulatrices très importantes, des long ncRNAs , des enhancers , des régulateurs à distance, etc . Les 3 milliards de paires de bases servent à quelque chose, et une bonne partie de cette zone sombre influence notamment le câblage neuronal, la neuro-embryogénèse et l'immunité. Ainsi nous découvrons que nous devons analyser les 3 milliards de paires de bases, alors que nous pensions ne séquencer que quelques petits morceaux de nos gènes, et il nous faut un peu de temps pour en comprendre toutes les implications.
Enfin, nous avons longtemps pensé « monovariants », c'est-à-dire que nous recherchions le gène du diabète, le gène de la schizophrénie,... Or, non seulement nous découvrons que les zones régulatrices, en dehors des gènes, jouent un grand rôle, mais nous comprenons aussi, que, hormis les maladies monogéniques comme la mucoviscidose, la plupart des pathologies sont un mélange complexe de facteurs environnementaux et de multiples variants génomiques. Il n'y a pas un variant, mais il y en existe plein, et ils sont différents d'un individu à l'autre.
En matière de médecine personnalisée, on commence à voir deux grands sujets. Le premier, c'est la personnalisation des traitements cancérologiques, c'est-à-dire le souci de donner le bon traitement, au bon malade, au bon moment. Contrairement à ce que l'on a pu espérer après l'expérience très favorable du Glivec, il n'y a pas, dans la plupart des cancers, un biomarqueur qui permet de savoir quelle chimiothérapie donner. En réalité, de nombreux marqueurs doivent être examinés et il faudrait faire des cocktails de drogues. C'est très compliqué. La stratégie utilisée jusqu'à présent, qui consiste à aller chercher un test compagnon, un biomarqueur unique, avec une chimiothérapie, a échoué. Cela ne donne que quelques mois de survie supplémentaire, à l'exception heureuse du Glivec sur lequel la rémission est de plusieurs années, mais c'est un cas très particulier du point de vue génomique.
En France, on a généralisé les analyses mono-marqueurs en cancérologie. C'est comme si l'on avait donné le minitel pour tous, et c'est une très bonne chose. Mais dans les grands centres aux États-Unis, c'est Internet que l'on commence à proposer, à savoir l'analyse complète du génome tumoral. Nous allons donc vers une vraie problématique éthique et médico-économique. Faut-il continuer avec le minitel pour tous ? Peut-on donner l'Internet, soit une analyse globale de l'ADN tumoral, à tous ?
Autre sujet brûlant, déjà évoqué, on parvient désormais à faire ce qui était inimaginable voilà seulement cinq ans : séquencer le génome du foetus sans amniocentèse autour de 18 à 20 semaines. Et il est probable qu'avec l'amélioration des techniques d'amplification, on puisse le faire à partir de 11 à 12 semaines ; ce qui signifie que l'on pourra séquencer le génome des foetus alors même que l'avortement pour convenance personnelle est libre dans notre pays. Dès lors, on risque d'avoir un peu de mal à empêcher certaines personnes d'opter pour un foetus à la carte et de choisir d'avorter. À moins d'interdire l'avortement, cette question se posera rapidement. C'est donc un sujet très important, auquel on n'a pas beaucoup réfléchi.
Quand le premier séquençage intégral du génome d'un foetus a été annoncé dans un article publié dans la revue Science , voilà environ deux ans, cela semblait impensable. D'ailleurs au début, on n'y a pas cru. Il a fallu attendre un deuxième article pour que l'on croie que cela fonctionnait. Et effectivement, cela fonctionne ! Le problème est important. Quelqu'un mentionnait tout à l'heure la possibilité de séquencer le conjoint d'une personne chez qui l'on a découvert un mauvais variant. C'est un vrai sujet. Moi, j'ai par exemple trois mauvais variants sur des pathologies graves, mais je n'ai qu'une fois l'allèle. Étant hétérozygote, je ne suis bien sûr pas touché. J'ai eu mes enfants avant d'être séquencé. Si je devais les avoir aujourd'hui, je pense que je ferai séquencer ma femme. Au vu de la gravité des variants concernés, elle accepterait sans aucun doute. Voilà pourquoi il y a là un vrai sujet et des possibilités de développement de ce domaine. Quand on connaît ses variants graves hétérozygotes, on se demande s'il est raisonnable de prendre le risque que le conjoint ait le même variant et que l'on ait un enfant très gravement malade.
S'agissant des maladies multigéniques,
multifactorielles, multivariants, telles que le diabète, les maladies
cardiovasculaires, la maladie d'Alzheimer,
etc
., on se trouve en
présence d'un mélange entre environnement,
épigénome
-
épigénome
étant presque synonyme d'environnement - et de multiples variants
génomiques. C'est tellement compliqué que nous n'allons pas
avancer très vite sur ces pathologies. Par exemple sur la
génomique du diabète, pour que l'on soit capable de
connaître le degré de déterminisme et d'analyser
l'épigénome, on parle de 2020, plutôt que 2014
ou 2015. On est donc face à plusieurs problématiques : le
diagnostic prénatal et le diagnostic préimplantatoire, le
traitement du cancer, et puis bien sûr la question des maladies
monogéniques traditionnelles. Ces technologies ne sont pas encore
complètement banalisées, mais elles le seront bientôt, et
le coût du séquençage baissera beaucoup.
Je suis en désaccord sur un point de stratégie avec l'évolution actuelle : il concerne la bio-informatique. Celle-ci est évidemment nécessaire, mais nous devons faire attention car, à l'heure actuelle, on est en train de réinventer le fil à couper le beurre dans chaque centre de séquençage. Chaque centre refait son aligneur, son logiciel d'analyse de variants, etc . C'est un peu comme si en 1985, dans chaque PME française, on avait cherché à réinventer Word et Excel. Il est clair que ce n'est pas tenable. Je pense donc que nous avons besoin de moins de bio-informaticiens qu'il n'y en a. En revanche, nous avons besoin de plus de concepteurs de logiciels bio-informatiques.
Le principal problème auquel nous allons nous heurter est de prévoir une stratégie politique et médicale sur ces sujets-là, car nous avons une assez mauvaise connaissance des évolutions qui vont survenir d'un point de vue technologique et nous ne savons pas très bien comment nous allons nous servir des nouvelles technologies.
Enfin, en tant que médecin, permettez-moi de vous dire que le corps médical est, globalement, complètement dépassé. La connaissance génomique des médecins est nulle. La moyenne d'âge du corps médical en France est très élevée puisqu'après les années 1969 à 1973 pendant lesquelles les portes des facultés de médecine ont été laissées ouvertes, un numerus clausus a été mis en place. Pour une grande partie, les médecins sont donc de cette génération 1969-1973. Leur moyenne d'âge est l'une des plus élevées au monde. Or, apprendre la génomique à 55 ans, n'est pas facile. Apprendre un gène, c'est faisable, mais apprendre à gérer 3 milliards de paires de bases, c'est un peu compliqué. Cette question constituera également un vrai sujet dans le cadre du déploiement de la médecine personnalisée.
Je terminerai par une remarque d'éthique. Certains d'entre vous ont probablement noté que les Chinois qui possèdent le plus grand centre de séquençage au monde - il est un peu plus grand que le Centre national de génotypage puisqu'il compte 200 séquenceurs - viennent de lancer un programme très controversé. Ce programme vise à séquencer le génome de 2 000 surdoués ayant un quotient intellectuel supérieur à 160, surdoués que les Chinois sont allés chercher dans le monde entier, y compris chez Google, à la recherche de variants expliquant le surdoué. On ne sait pas s'ils vont trouver des variants, mais le sujet est très délicat. Chez nous, cela semble tabou et impensable. Selon le Wall Street Journal , le gamin qui gère le programme et qui est lui-même un surdoué - le chef de projet a 21 ans - a dit : « c'est un problème pour vous en occident, mais pour nous, le fait de rechercher les gènes permettant d'être surdoué n'est pas du tout un problème ». On comprend ainsi que nos principes éthiques, humanistes, ne seront pas forcément partagés. La science génomique étant mondiale, il n'est pas certain que les conséquences éthiques et médicales du séquençage complet du génome aillent dans le sens que nous souhaitons.
J'ajoute que nous ne maîtrisons pas les technologies sous-jacentes. Que ce soit au niveau du Centre national de génotypage ou au niveau de mon propre centre de séquençage, qui est plus petit, nous utilisons des machines qui viennent d'ailleurs. Ce n'est donc pas nous qui déterminons la technologie, et compte tenu de notre retard technologique, il n'est pas certain que nous soyons capables de déterminer les bornes éthiques - les lignes Maginot éthiques - correspondant aux valeurs que nous partageons.
M. Alain Claeys, rapporteur. Je remercie tous les participants à cette table ronde et j'ouvre le débat.
|
Débat |
M. Hervé Chneiweiss, directeur de recherche
groupe « Plasticité gliale et tumeurs
cérébrales » à l'université
Paris-Descartes, membre du conseil scientifique de l'OPECST.
J'ai
entendu des propos extrêmement intéressants, auxquels je peux
adhérer, et je poserai aussi quelques questions. Prenons justement le
risque d'idéologie de la promesse. Quand on sait qu'on évalue
aujourd'hui à environ 15 millions les types de variations existant entre
le jeu de gènes que l'on reçoit de son père et celui que
l'on reçoit de sa mère, c'est-à-dire simplement des
variants
-
soit dans les changements de l'être,
soit dans des répétitions de certains duplex, soit dans des trous
que l'on a au milieu du
génome
-
, la prise en
considération de la combinatoire et de l'interaction de cette
combinatoire avec l'environnement amène tout de même à
garder un certain degré de modestie par rapport à l'ampleur de
notre ignorance, y compris de notre ignorance sur le plan du génome
lui-même.
J'analysais, il y a quinze jours, un certain nombre de programmes européens dans lesquels cet épigénome s'étend aujourd'hui sur des modifications des ARN. Comme la plupart des stratégies utilisent la copie inverse du transcriptome, si l'on a de nouvelles modifications, de type méthylation, sur différentes bases de ces ARN, alors un grand nombre des données du transcriptome telles que nous les avons actuellement sont fausses. C'est simplement lié au fait que nous sommes encore ignorants d'une grande partie des modes d'expression de nos gènes.
Je suis aussi médecin et je m'occupe aussi de tumeurs. J'adhère donc à certains éléments forts qui ont été évoqués. Je suis très heureux de pouvoir diagnostiquer, peut-être prédire et, désormais, éventuellement cibler un certain nombre de modifications dans des tumeurs. Mais je pense que l'étendue de notre ignorance, qui comprend aussi ce que nous ignorons ignorer, doit nous conduire à une certaine modestie.
Dr. Christelle Besnard-Charvet, gynécologue. Je suis gynécologue-obstétricienne et j'ai travaillé en centre anticancéreux. Je souhaiterais vous faire réfléchir quelques instants sur la question du dépistage des mutations des gènes BRCA1 et BRCA2 car je connais les femmes concernées par ce type de démarches au quotidien. On a essayé de mettre en place un procédé personnalisé. On a donc dépisté une mutation chez des femmes ne se plaignant absolument de rien, mais ayant des antécédents familiaux. On se sert d'une technique individualisée pour leur proposer, comme solution thérapeutique face à un risque de cancer, des ovariectomies et des mastectomies bilatérales. Or, il n'est pas du tout certain que le risque identifié se concrétise finalement.
D'une part, en tant que médecin, je ne suis pas persuadée de rendre service à ces femmes. D'autre part, on s'est aperçu que chez ces femmes qu'on surveillait par le biais de mammographies, celles-ci étaient dangereuses, comme on aurait pu le prévoir. En effet, ces personnes ont forcément des seins beaucoup plus sensibles aux rayons, puisqu'elles sont porteuses de la mutation. Non seulement nous n'avons pas rendu service à ces femmes mais en plus, notre action a été délétère pour elles, puisque nous avons probablement aggravé leur risque de cancer.
C'est une réflexion que je livre. J'ai beaucoup de mal à gérer ces femmes au quotidien, des femmes se retrouvant ménopausées pour un risque qui n'est pas connu. Je me demande donc ce qui se passerait si on commençait à tester les mutations BRCA1 et BRCA2 dans toute la population. Combien de femmes se retrouveraient avec une mastectomie bilatérale ?
Pr. Florent Soubrier. Je me permets d'intervenir en tant qu'onco-généticien. Nous avons monté à l'hôpital Tenon le premier centre de suivi des femmes à haut risque en France. Je ne peux pas ne pas m'émouvoir des propos qui viennent d'être tenus : cela signifierait qu'il ne faut pas dépister ces femmes porteuses de mutations BRCA1 et BRCA2, alors que différentes études ont prouvé que par le dépistage et en adaptant les mesures de surveillance à leur risque, on améliore leur survie. Vous faites allusion, madame, à un certain nombre de mesures drastiques telles que les mastectomies, les ovariectomies, les femmes mutilées, mais je pense qu'il faut raison garder. Ces mesures sont adaptées en fonction de l'âge de la femme, de sa vie génitale, des enfants qu'elle a eus ou qu'elle souhaite avoir dans l'avenir. Il faut donc vraiment décrire les choses telles qu'elles sont. Aujourd'hui, le risque lié aux mutations BRCA1 et BRCA2 a été largement dépisté en France, grâce au soutien de l'INCa, et ce faisant, il me semble que nous avons fait gagner de nombreuses années de vie à ces femmes en dépistant leur risque.
Je crois que vous allez très loin en émettant des doutes sur le fait que vous leur rendez service. Cela voudrait dire qu'il faudrait laisser ces femmes dans la nature, ne plus les dépister et que finalement, on surveillera de la même façon ces femmes présentent un risque de 50 % ou 60 % de développer un cancer du sein ou de 40 % de développer un cancer de l'ovaire, qu'une femme qui a moins de 20 % ou 10 % de développer ces pathologies. Il faut être très prudent avec ce type de discours.
Dr. Christelle Besnard-Charvet. C'est certainement mon ressenti, lié à des épisodes difficiles que j'ai vécus avec ces patientes. Mais je suis aussi très amie avec de nombreux onco-généticiens, qui m'ont donné les mêmes explications que celles qui viennent d'être avancées. Je les comprends parfaitement. Ce qui me gêne, c'est plus la « non-personnalisation » actuelle. Certes, les choses évoluent, mais il y a tout de même des protocoles que les centres anticancéreux appliquent, dans ma région en tout cas.
Évidemment, cela se fait toujours avec l'accord des patientes. Nous ne leur imposons jamais rien et les décisions sont systématiquement prises en discutant avec elles. J'ai eu, par exemple, deux cas de patientes qui ont fait la recherche et ne sont pas allées chercher leurs résultats. C'est ennuyeux, car on se retrouve avec des patientes qui, malgré les explications, n'ont peut-être pas compris jusqu'où la démarche les engageait.
À l'avenir, il serait peut-être souhaitable que nous ayons une réflexion éthique dans le cadre de la mise en place de programmes de dépistage de ce type et qu'une meilleure formation nous soit délivrée, à nous, praticiens de terrain, pour pouvoir suivre correctement ces femmes et leur donner des explications claires. S'agissant de la surveillance par radio, je voudrais justement avoir votre avis sur le sujet. On commence à entendre qu'il faudrait peut-être éviter de faire des mammographies à ces femmes à risque parce qu'elles ont des seins plus sensibles.
Pr. Florent Soubrier. C'est un débat très spécialisé. Des données montrent effectivement aujourd'hui que le fait de pratiquer des mammographies sur ces femmes, avant l'âge de 30 ans, peut augmenter le risque de cancer. Immédiatement, tous les centres qui suivent cette population ont réadapté leur mode de surveillance, en diminuant au maximum les mammographies, en recourant aux échographies, à l'IRM, aux mammographies sur une seule incidence en oblique tout au long de la vie, pour limiter les radiations. Tous les onco-généticiens et les cancérologues sont extrêmement sensibilisés à ce problème.
Au sujet de ces femmes qui ne viennent pas chercher leurs résultats, quand on fait ce type de diagnostic pré-symptomatique, il faut faire très attention à l'environnement psychologique. Un véritable centre de suivi qui fait ce type de diagnostic proposera l'intervention d'un ou d'une psychologue spécialisée et une vraie prise en charge, la préparation pouvant prendre plusieurs mois avant que le test ne soit réalisé et le résultat rendu.
Dr. Laurent Alexandre. On voit bien que notre discipline est encore récente. Les gènes BRCA1 et BRCA2 sont impliqués dans ce que l'on appelle la réparation, l'intégrité de l'ADN. Les personnes ayant une mutation de ces gènes ont une plus grande fragilité des systèmes de réparation de l'ADN et de maintien de son intégrité. Il était évident que les rayons ne seraient pas très bons pour les individus ayant une telle faiblesse. Pourtant, on n'y a pas pensé, et il est invraisemblable qu'il ait fallu attendre 2012 pour se poser cette question. Alors que c'était évident, sauf qu'on n'y avait pas pensé et qu'on n'avait pas fait les études épidémiologiques permettant de trouver une corrélation entre mammographie et augmentation de la mortalité chez ces personnes à risque. Nous sommes donc au début de la connaissance des mécanismes génomiques et des évidences nous échappent encore.
Sur BRCA1 et BRCA2, il y a tout de même un espoir que l'on puisse à l'avenir mieux cibler les femmes ayant réellement un risque important. On a découvert, voilà quelques mois que d'autres variants que BRCA1 et BRCA2 sont impliqués On a découvert une quinzaine de gènes et de variants, et on en découvrira encore, ce qui permettra d'améliorer la prédictibilité. Cela ne signifie pas qu'il faudra encore plus s'orienter vers la mammographie systématique dans le futur, mais on va pouvoir affiner la probabilité d'avoir un cancer.
Ce qui me choque personnellement, c'est que, sur le fondement d'un diagnostic prénatal, on a commencé à pratiquer des avortements pour des foetus atteints de mutations BRCA1 et BRCA2. Ces cas ne sont pas fréquents, mais ils existent, notamment dans les pays anglo-saxons. Je trouve choquant de pratiquer avortement sur un foetus en 2012 ou 2013 au motif qu'il est porteur de telles variations BRCA1 et BRCA2, alors même que le risque de développer un cancer ne surviendra que vers 2035, 2040 ou 2045. On peut espérer que, d'ici là, les méthodes de dépistage et de traitement se seront améliorées. Il me paraît peu raisonnable de pratiquer un avortement sur la base d'une pathologie qui va apparaître aussi loin de nous dans le futur.
M. Jean-François Deleuze. Je pense qu'il faut tout de même que nous dissocions, dans notre discussion, les aspects liés à la recherche des aspects liés au diagnostic. Je suis assez d'accord avec de nombreux points évoqués, même si, bien évidemment, je ne partage pas tout. Il y a aujourd'hui des études populationnelles en génomique qui sont absolument fondamentales pour permettre des avancées thérapeutiques. Par exemple, pour la maladie d'Alzheimer ou d'autres, on a découvert des dizaines de gènes, mais on est encore en retard dans ce domaine et, malgré les nombreuses découvertes, on ne change pas vraiment la prise en charge thérapeutique du patient. Il faut donc continuer ces analyses génomiques pour trouver de nouvelles cibles qui permettront d'ouvrir de nouveaux accès thérapeutiques.
Je partage les remarques concernant la faiblesse de la prédiction dans de nombreux cas de maladies multifactorielles, mais il est vraiment nécessaire de dissocier les deux discussions. Il ne faudrait pas que reconnaître ne pas savoir nombre de choses sur le génome humain et avoir une capacité de prédiction faible, nous empêche de développer les grandes analyses génomiques dont nous avons besoin pour comprendre les pathologies.
Il existe énormément de pathologies que nous ne comprenons tout simplement pas : la schizophrénie, la maladie bipolaire, etc... Nous avons pratiqué toutes les analyses phénotypiques, intelligentes, biologiques, et utilisé la génomique au sens large, l'épi-génomique apparaît aujourd'hui comme une voie possible que nous voulons creuser pour essayer de détecter ces réseaux. Cela se fera au travers d'études populationnelles, sur la base desquelles on espère, à un moment donné, revenir pour un bénéfice individuel. Quoi qu'il en soit, selon moi, il faut vraiment distinguer deux parties : une partie recherche et une partie diagnostic.
M. Alain Claeys, rapporteur. Je vous remercie et vous propose d'en rester là pour cette deuxième table ronde.
APRÈS-MIDI
PROPOS INTRODUCTIFS
Pr Agnès Buzyn, présidente de l'Institut national du cancer (INCa). Je remercie l'OPECST de m'avoir donné l'opportunité de participer à cette réflexion et d'anticiper les problèmes à venir, et surtout de présenter le point de vue de l'INCa. Historiquement, la médecine personnalisée a été un axe majeur du deuxième Plan Cancer 2009-2013. C'était probablement l'un des premiers volets de la « médecine 4P » telle qu'on l'envisageait à l'époque : prédictive, préventive, personnalisée, participative.
L'objectif affiché de ce deuxième Plan Cancer était double : d'une part, favoriser l'accès aux thérapeutiques ciblées, qui étaient développées en premier dans le cancer, en particulier dès les années 2000 avec l'arrivée de l'Imatinib ; d'autre part, favoriser une meilleure compréhension de l'oncogenèse des tumeurs, et puis apporter des marqueurs pronostiques ou prédictifs de certaines tumeurs. Grâce à ce Plan Cancer, à l'INCa, nous avons décliné cette médecine personnalisée par la création de plateformes dédiées, dont une partie est financée par la Direction générale de l'offre de soins (DGOS) du Ministère de la santé, une autre partie par l'INCa, et certaines le sont avec une participation de l'industrie pharmaceutique.
Les premiers types de plateformes ont été dédiés à l'oncogénétique, avec des consultations d'oncogénétique associées, ciblant les facteurs de risque du cancer du sein et du cancer colorectal, afin de mieux identifier les familles à risque et donc les individus à haut risque de cancer.
Le deuxième type de plateformes a été dédié à la génétique somatique. 28 plateformes de génétique moléculaire permettant de donner une signature génétique de la tumeur de chaque patient, et de rationnaliser la prescription face à des traitements innovants et très coûteux. L'idée était de rationaliser la prescription des médecins sur cette signature génétique, et d'améliorer l'accès aux thérapies ciblées.
Actuellement, nous avons 17 thérapies ciblées dans le cancer, et donc sur ces 28 plateformes labellisées INCa, nous effectuons les tests compagnons nécessaires à ces 17 thérapies ciblées. Le principe repose sur une égalité d'accès à ces tests sur tout le territoire français. Tous les patients atteints de tumeur en France, qu'ils soient pris en charge dans le privé ou dans le public, ont accès à ces tests. Ils sont fiables, reposent sur un contrôle qualité, et ces plateformes ont un objectif de réactivité, c'est-à-dire que les tests compagnons doivent être prêts dès qu'une nouvelle thérapeutique est sur le point d'obtenir une autorisation de mise sur le marché (AMM) dans une indication donnée.
Pour l'instant en 2011, nous assistons à une montée en charge qui continue sur ces plateformes, avec plus de 55 000 patients testés au diagnostic. Il faut monter jusqu'à 155 000 patients testés chaque année si l'on considère à la fois le diagnostic et le suivi moléculaire de certaines hémopathies.
A ce stade, nous pensons cependant que le mot « médecine personnalisée » est un peu dévoyé. Pour nous, il s'agit pour l'instant d'une médecine stratifiée, c'est-à-dire basée sur le démembrement des cancers fréquents en cancers rares, qui ont une signature génétique particulière. Par exemple, 2 % des cancers du poumon vont avoir une mutation d'ALK donnant accès au crizotinib. Les enjeux sont nombreux pour l'avenir. En ce qui concerne la génétique somatique, nous sommes actuellement en train de pratiquer des monotests, un test compagnon est développé par thérapie ciblée. Avec l'arrivée du séquençage à haut débit, et la diminution du coût du séquençage, on peut imaginer pouvoir effectuer des multitests pour une tumeur donnée, et cesser de tester spécifiquement chaque anomalie génétique. Ce séquençage à haut débit pourrait cibler des zones de mutation, ou plus largement s'adresser à la totalité du génome ( whole genome sequencing ). On pourrait également imaginer ne séquencer que les exomes.
Mais nous nous demandons si rechercher simplement des mutations sur des voies de signalisation suffit pour interpréter la vie moléculaire d'une tumeur. Faut-il également réaliser systématiquement un transcriptome pour observer l'impact de ces mutations sur d'autres voies de signalisation et des hyper expressions de gènes ? Et puis quelle place aura l'épigénétique dans tout cela ? Très vraisemblablement, il faudra s'intéresser également à ce domaine.
Une autre question que nous nous posons : à quel rythme implémenter ces plateformes de génétique moléculaire avec du séquençage haut débit, s'il le faut réellement ? Est-ce qu'il faut s'intéresser uniquement à la tumeur, au diagnostic ou suivre l'évolution des métastases qui peuvent présenter des évolutions clonales avec d'autres anomalies génétiques apparaissant au fur et à mesure de la « vie de la tumeur » ? Quelle sera la soutenabilité à la fois technique et en ressources humaines de bio-informatique de ces plateformes ? Quelle sera la capacité des professionnels de santé à comprendre ces signatures ? Quel sera le niveau de formation à atteindre pour interpréter ces données ? Je pense que le Doyen Patrick Berche en traitera. Quelle sera la soutenabilité financière, à la fois pour l'INCa en termes d'équipements, et pour la société en termes de coût ? Enfin, cela générera énormément de données, de connaissances. Quelles capacités aurons-nous, collectivement, à stocker l'ensemble de ces données pour favoriser la recherche française ? Cela nécessitera des équipements extrêmement lourds, et aura aussi un coût.
En ce qui concerne la génétique constitutionnelle, d'autres enjeux m'apparaissent. Celui de la pharmacogénétique, qui est encore relativement peu implémentée. Peut-on imaginer des développements de la connaissance de la pharmacogénétique pour chaque individu et de l'adaptation des drogues à ces profils ? Y a-t-il un impact de la génétique constitutionnelle sur l'addiction à certaines drogues, telles que le tabac ? Ceci nécessiterait pour nous de repenser autrement les messages de prévention vis-à-vis de l'addiction au tabac ou à l'alcool ce qui est également un champ d'action de l'INCa. Quel sera l'impact de la génétique dans la prévention et dans les dépistages ? Peut-on imaginer des dépistages plus à la carte, et pas uniquement ciblés sur des tranches d'âge, comme c'est le cas actuellement pour les dépistages organisés du cancer du sein ou du cancer colorectal ?
Enfin, en ce qui concerne les biomarqueurs, les marqueurs pronostiques, on sait bien qu'ils émergent actuellement mais qu'ils sont étroitement liés à des protocoles cliniques, à des usages de certains médicaments. En outre, ils ne sont pas forcément reproductibles d'une population à l'autre. À l'INCa, nous restons extrêmement vigilants sur l'implémentation de ces facteurs pronostiques en routine, - que ce soit des tests génétiques, ou biomarqueurs -, et dans l'appréciation des facteurs de risque des patients face à la maladie.
Ainsi pour l'INCa, les enjeux sont extrêmement nombreux. Nous nous posons beaucoup de questions, et nous considérons que nous n'en sommes qu'aux balbutiements de la médecine personnalisée. Aujourd'hui, ce terme reste un abus de langage. Il s'agit pour l'instant d'une médecine stratifiée. Les systèmes deviendront de plus en plus complexes, pour aller vers une vraie personnalisation de la prise en charge, fondée à la fois sur la génétique constitutionnelle et sur la génétique somatique. De plus, nous considérons qu'actuellement on confond médecine personnalisée et médecine basée sur la génétique. Cela nous semble un peu réducteur. Du point de vue de l'INCa, la médecine personnalisée doit s'envisager plus largement, et doit transcender la signature génétique. Un individu, une personne, demeure au-delà de sa signature génétique.
Nous souhaitons vraiment replacer l'individu et son parcours de vie, puis de santé, au coeur de la réflexion. C'est la raison pour laquelle nous essayons également de personnaliser les parcours de santé sur des caractéristiques de l'individu qui ne sont plus seulement des caractéristiques génétiques. Par exemple, nous organisons des parcours de santé autour des tranches d'âge, gériatrie ou pédiatrie, autour de certains types de cancer. Nous avons par exemple organisé des réseaux de cancers rares pour s'adapter à des prises en charge très complexes et spécifiquement adaptés à des cancers extrêmement rares.
Nous souhaiterions également personnaliser les messages de prévention. Actuellement, ces messages sont très généraux et mal entendus par la population. Par exemple, quand on s'adresse à des adolescents, il est clair que pour leur éviter de rentrer dans le tabagisme, le message ne sera pas le même que celui face à un chômeur qui fume depuis 30 ans. Je rappelle que plus de 50 % des chômeurs sont tabagiques. Il faut donc personnaliser la prévention primaire et les préventions secondaires, c'est-à-dire les dépistages organisés. Nous envisageons, sous le terme de médecine personnalisée, un concept qui va bien au-delà de la simple génétique.
Le troisième point que je souhaite soulever, c'est qu'il faut développer la médecine personnalisée pour le bénéfice des individus. Nous sommes attentifs aux risques de stigmatisation face à une signature génétique de haut risque de cancer par exemple, ou de haut risque d'addiction. Cela pose des problèmes éthiques que nous ne sommes pas capables à l'INCa de résoudre seuls. Une réflexion comme celle d'aujourd'hui participe à la nécessité de réfléchir collectivement, et bien en amont, à toutes ces problématiques, en particulier éthiques, qui se posent autour de la personnalisation.
*
* *
Pr Patrick Berche, doyen de la faculté de médecine de Paris-V Descartes . La médecine est en transition. Elle connaît actuellement une phase d'explosion de connaissances qui va poser beaucoup de problèmes à la formation médicale dont je vais vous faire un bref exposé. Moi non plus je n'aime pas ce terme de « médecine personnalisée », puisque par définition la médecine est personnalisée, tout du moins il faut l'espérer. Je l'appellerai plutôt « médecine prédictive ». Mais « médecine stratifiée » me convient assez bien.
La médecine prédictive a un caractère dual. D'un côté, prédire l'avenir d'un bébé à la naissance en disant qu'il aura un risque de diabète ou de cancer, cela pose un problème difficile de sciences humaines et sociales. C'est la possibilité d'une dérive vers l'eugénisme ou le mercantilisme. Mais d'un autre côté, prédire qu'une tumeur va être sensible à telle chimiothérapie comme on le fait pour les antibiotiques, ou prédire la toxicité d'un médicament par la pharmacogénétique sera un apport considérable.
Il faut replacer les progrès continuels de la médecine, comme d'autres sciences, en tenant compte de l'acceptation et de la compréhension par les citoyens de ces avancées. La population les ressent d'une façon ambiguë, entre la crainte et l'espoir. Aussi faut-il absolument remettre tous les progrès médicaux dans le contexte économique et sociétal pour essayer de faire mieux comprendre les enjeux à la population.
L'un des écueils de la formation médicale aujourd'hui est donc cette explosion énorme du corpus de connaissances. Personne n'est plus capable d'appréhender l'ensemble des connaissances médicales. Cela a deux conséquences. D'une part, il y a une nécessité pour l'enseignement multidisciplinaire de la médecine. D'autre part il y a une nécessité de faire des choix pour adapter les programmes. Mais quels choix ? Qu'est-ce qui est essentiel ?
Quelles sont les forces et les faiblesses de l'enseignement médical en France ? Il y a indubitablement un certain nombre de forces. La qualité de la formation clinique est excellente : trois ans de formation à mi-temps pour les externes, et quatre ou cinq ans de formation à plein temps à l'hôpital. L'apprentissage par compagnonnage est, je crois, le point fort des études médicales en France. Et puis les facultés acceptent volontiers les nouvelles techniques de communication : Internet, simulations sur mannequin, jeux électroniques, E-learning, cours en ligne...
Mais faiblesses sont nombreuses. C'est d'abord l'obsolescence des programmes actuels. L'approche est très souvent mono-disciplinaire. La formation scientifique est médiocre, confinée souvent à la première année commune aux études de santé (PACES), et encore. L'absence des Sciences Humaines et Sociales (SHS) est absolument aveuglante : quelques 30 ou 40 heures dans le programme officielles pour les six ans de premier et deuxième cycles. On peut aussi relever une faiblesse de l'enseignement concernant certains domaines de la médecine qui sont très importants ; par exemple la médecine préventive, la douleur, les soins palliatifs, les addictions, les maladies chroniques, le handicap et le vieillissement ne sont pas réellement bien traités au cours des études de médecine.
De plus, ces études sont encadrées par deux concours archaïques. On a d'abord le concours de la PACES suivi du concours de l'examen commun national (ECN). Pour sélectionner les futurs médecins dont les qualités essentielles pour moi sont l'empathie et la capacité à travailler en équipe, le concours initial n'est pas satisfaisant. Quelques 50 000 étudiants se présentent au PACES, et 7 000 sont admis à poursuivre des études de médecine. Les autres ont perdu leur temps. L'ECN est un héritage du consulat. Il a été créé en 1801. C'est un classement de 7 000 étudiants sur un concours national au mérite, c'est-à-dire que pour quelques centièmes de points, on impose le choix d'une spécialité à un étudiant pour le reste de la vie, parfois au hasard. Le cursus actuel des études médicales en France, encadré par deux concours apprend aux étudiants la compétition, sûrement pas l'empathie, ni l'esprit d'équipe.
Pour moi, il est évident que ce système doit être réformé. En s'inspirant partiellement du système américain, on pourrait proposer la mise en place d'un premier cycle sous forme d'une licence de santé de haut niveau de trois ans, incluant des sciences médicales (anatomie, physiologie, biochimie, biophysique...), des sciences fondamentales (physique, chimie, statistiques...) et l'introduction des SHS pour au moins un tiers du programme. Ensuite, on pourrait classer les étudiants par les notes cumulées pendant les trois ans de cursus. Et les meilleurs d'entre eux seraient soumis à des interviews l'admission par des jurys d'enseignants, d'étudiants et de psychologues. Nous sommes le seul pays en Occident où il n'y a pas d'interviews des étudiants en médecine. Étant chargé de les gérer pendant tout leur cursus, je peux vous dire que l'interview serait fort utile. Exit le concours de la PACES.
Le 2
ème
cycle des études
médicales serait de quatre ans d'externat
(D1-D2-D3-D4), ce qui
rallonge les études d'un an, mais je rappelle que 85 % des
étudiants redoublent la PACES. Avec cet externat de quatre ans à
mi-temps (tous les matins à l'hôpital), on leur apprendrait un
programme minimum, essentiellement clinique, axé sur les pathologies et
les urgences courantes, et la façon de les prendre en charge. Un tel
programme laisserait du temps de libre à chaque étudiant pour
construire, s'il le désire, un cursus personnalisé, en fonction
de sa motivation. Le déroulement du nouveau cursus des études
médicales en France serait le suivant : trois ans de licence,
avec une approche multidisciplinaire, notamment avec l'addition de SHS, mais
aussi de sciences médicales et sciences fondamentales (renforçant
la formation scientifique des étudiants français), un externat de
quatre ans, et un internat de quatre ans, comme c'est le cas actuellement. S'y
ajouteraient des enseignements modulaires complémentaires, puisqu'en
étalant l'enseignement clinique sur quatre ans, les étudiants
auront plus de temps à leur consacrer. Ces enseignements modulaires
complémentaires optionnels (diplômes universitaires, master,
thèse d'université...) seraient validés par
l'Université ou le Ministère et comptés sous forme de
bonus.
La construction d'un cursus personnalisé est une idée inspirée directement de l'École de médecine de Harvard. C'est un enseignement parallèle précoce, orientant vers les filières. Vous voulez être médecin, chirurgien, biologiste, santé publique, psychiatre, pourquoi vous imposer d'apprendre la psychiatrie pour être chirurgien, ou vice-versa ? La validation de modules d'enseignement selon l'orientation souhaitée aurait lieu pendant le temps libre, les modules pouvant être acquis au sein des facultés (de médecine, des sciences, de SHS, psychologie, droit, sciences po, etc. ) ou d'Instituts de recherche (psychologie, sociologie, santé publique, économie, recherche...).
On pourrait même créer des pôles pédagogiques (virtuels) qui dépasseraient les institutions sur des problématiques particulières. Par exemple, on pourrait imaginer mettre en place un enseignement sur la maladie d'Alzheimer, ou sur le diabète ou sur le cancer, qui rassemblerait sur l'ensemble de la France ou d'une région, des neurologues, des psychiatres, des gériatres des ergothérapeutes, des infirmières, des spécialistes de santé publique, des économistes de la santé, des sociologues, etc . Ces « instituts virtuels multidisciplinaires » délivreraient une attestation ou un diplôme à travers leurs Universités. Ce melting-pot améliorerait les études de médecine et donc la qualité de l'enseignement. Cet enseignement pourrait être multi-université dans les TICE ou sous forme d'un enseignement en présentiel pendant de courtes périodes.
Ainsi pourrait-on supprimer le concours de l'internat et le remplacer par un examen régional, associé à un numerus clausus pour la région. Cet examen régional anonyme serait segmenté par discipline (médecine, chirurgie, psychiatrie, santé publique, biologie...), chaque discipline ayant son numerus clausus défini par le Ministère de la santé en fonction des besoins régionaux. Au terme de l'examen, les étudiants auraient, disons, une note sur 20. Si le numerus clausus pour accéder à une discipline est par exemple de 14, les étudiants ayant validé un bonus pourraient incrémenter leur note, se hissant ainsi au-dessus de la barre. Les étudiants pourraient se présenter à plusieurs examens régionaux et à plusieurs disciplines. Exit le concours de l'internat.
Quelles sont les leçons de l'histoire ? Au regard des différents systèmes qui fonctionnent dans les pays en Occident depuis des décennies, aux États-Unis, en Allemagne, en Angleterre et en France, on s'aperçoit de l'importance de l'enseignement clinique par apprentissage, ce qui est habituellement bien fait en France. La formation de médecins de qualité, capables de compléter toute leur vie leurs connaissances, nécessite un enseignement scientifique de bon niveau et conjointement une approche plus approfondie des sciences humaines et sociales, deux domaines qu'il faut renforcer dans le système français. Ceci implique bien sûr une forte intégration des facultés de médecine dans l'université pluridisciplinaire. Il faut enfin insister sur l'importance de la recherche clinique et fondamentale comme moteur à la fois de la qualité des soins, de la pédagogie et du progrès médical.
Enfin, pourquoi ne pas affirmer la nécessité d'une sélection précoce et rigoureuse des étudiants, tenant compte de leurs qualités et de leurs mérites ? De même que cela devrait être le cas pour les enseignants. Est-ce une utopie que de supprimer les deux concours qui encadrent les études de médecine ?
M. Jean-Sébastien Vialatte, député, rapporteur. Depuis ce matin on entend parler des tumeurs, des mutations génétiques et des traitements spécialisés. Peut-on considérer que les tumeurs sont homogènes ? Au sein d'une même tumeur, ne peut-on trouver des cellules tumorales mutées et d'autres qui ne le sont pas ? Et le fait de proposer un traitement ciblé sur une certaine mutation ne risque-t-il pas au contraire de présélectionner d'autres types de cellules ?
Pr Agnès Buzyn. La réponse était dans votre question. Les tumeurs sont extrêmement hétérogènes. Elles ont déjà une hétérogénéité intra-clonale au moment du diagnostic. En fonction de la pression que vont exercer certaines thérapies ciblées, des clones résistants via des activations de nouvelles voies de signalisation émergeront, et c'est en cela que la médecine personnalisée va se compliquer. Il faudra non seulement étudier la tumeur au moment du diagnostic, son hétérogénéité clonale, analyser ce qui est vraiment prépondérant, mais aussi être capable de suivre l'évolution de ces clones qui vont émerger, sous pression, et c'est une pression sélective, ce qui a un coût et est extrêmement complexe à interpréter. C'est pourquoi, j'estime que nous n'en sommes qu'aux balbutiements. Aujourd'hui, on effectue un mono-test pour rechercher une anomalie génétique prépondérante dans une tumeur, ce qui est excessivement réducteur par rapport à l'univers qu'on pourrait découvrir par le séquençage à haut débit de la totalité du génome.
TROISIÈME TABLE RONDE :
AVEC QUELLE VALORISATION
DE LA RECHERCHE ?
Mme Cécile Tharaud, présidente du directoire d'INSERM Transfert . Pour nous, professionnels du transfert de technologie, la médecine personnalisée est un enjeu important, tout autant qu'une énigme à ce jour. J'étais assez contente d'entendre le Pr Agnès Buzyn énumérer ses nombreuses questions. C'est assez rassurant. Nous ne sommes pas les seuls à ne pas savoir comment progresser. Je commencerais par des généralités sur le transfert de technologie. Je crois qu'elles sont importantes et s'appliquent aussi, et grandement, au domaine de la médecine personnalisée.
Le premier grand mouvement auquel on assiste aujourd'hui, et qui rend le transfert de technologie particulièrement crucial actuellement dans le domaine de la santé, est une ouverture des industriels petits, moyens, et grands, vers plus petit que soi quand il s'agit de partenariat entre une grande industrie pharmaceutique et une petite société de biotechnologie, ou d'un partenariat entre tous les acteurs privés et la recherche académique.
Pourquoi ce mouvement est-il si important ? La complexification des technologies est devenue le grand problème du développement en matière de santé en termes d'efficacité, plutôt que de rentabilité. Il en résulte qu'aucun industriel aujourd'hui ne peut prétendre s'en sortir tout seul et développer tout seul. Aussi existe-t-il un grand mouvement de recherche public-privé, privé-privé, en réseau, à l'échelle nationale, internationale, européenne, à tous niveaux. Ce mouvement d'innovation ouverte s'est formalisé en 2003 et commence maintenant à prendre forme, au-delà du concept, dans les industries de santé, bien qu'il soit plus développé dans d'autres secteurs industriels. Il représente pour la recherche académique et pour les offices de transfert de technologie une opportunité et un défi très importants.
Ce défi consiste à repenser de façon stratégique le métier du transfert de technologie, de façon à en faire un métier d'apport de valeur, c'est-à-dire transformer une découverte en un potentiel innovant, ce qui n'est pas tout à fait la même chose. Cela nécessite que les professionnels du transfert de technologie comprennent la demande industrielle, et la demande sociétale, les contraintes ou demandes ou nécessités réglementaires et de propriété intellectuelle ; cela fait de ce métier aujourd'hui, un métier stratégique assez complexe, mais qui se développe beaucoup. La France l'a compris, puisqu'elle a investi, notamment au travers des investissements d'avenir, plus d'un milliard d'euros dans le seul secteur du transfert de technologie. Cette généralité étant posée, je reviendrais à des tendances industrielles plus spécifiques pertinentes pour le sujet qui nous anime aujourd'hui.
En premier lieu, l'industrie, de la même façon que la recherche translationnelle, redéfinit ses efforts de R&D autour du patient. L'innovation est au coeur des questions que se pose l'industrie, depuis la compréhension de la maladie, vers la compréhension de la question posée en matière de diagnostic, de prévention, d'autres thérapeutiques ou de vaccin. Cette question, directement celle que soulève la médecine personnalisée, implique que le développement d'un produit se fasse avec accès au patient, et donc à des cohortes plus ou moins importantes. Depuis la définition et la compréhension des maladies de manière stratifiée, il n'existe plus maintenant un diabète où l'on ne cherche à comprendre les segments du diabète jusqu'à la définition de nouvelles cibles thérapeutiques ou de diagnostic.
Ainsi tout au long du développement du médicament, y compris en post-inscription d'AMM et donc en période de commercialisation, il y a un retour incessant aux populations de patients, de façon à répondre aux questions qu'ils se posent tout au long du développement et de l'utilisation du médicament. On en arrive au point où des structures comme l'INSERM aujourd'hui, -puisque nous sommes la structure de transfert de technologie de l'INSERM-, créent des partenariats avec leurs collaborateurs industriels tout au long du développement du médicament. Ce n'est plus seulement quand il s'agit de trouver de façon caricaturale un gène de maladie, mais bien tout au long de la chaîne de développement que nous tissons des partenariats, aussi bien en découverte qu'en développement, et aujourd'hui en post-inscription. Le transfert de technologie s'adresse aussi à la santé publique, et cela me paraît tout à fait pertinent quand on s'en réfère à la médecine personnalisée.
La demande que nous fait l'industrie est très claire : c'est la compréhension de la maladie, c'est-à-dire la stratification des maladies, puis la stratification des populations, de façon à dessiner des développements cliniques. Mais la première demande, est la compréhension fine des grandes pathologies qui sont les défis sociétaux aujourd'hui : la maladie d'Alzheimer, l'obésité, le diabète, la polyarthrite rhumatoïde. En transfert de technologie, nous ne sommes pas seulement challengés sur l'oncologie, où le sujet de la médecine personnalisée est assez avancé. Mais selon les mêmes pratiques, les mêmes concepts et les mêmes outils, l'un des derniers accords qu'on a par exemple signé consistait à passer une licence sur trois marqueurs génétiques a priori pertinents sur une particularité de réponse à un médicament sur la polyarthrite rhumatoïde. Ainsi, cela ne relève pas seulement du cancer, et c'est important.
L'offre académique est pléthorique. Quand un industriel vient nous voir aujourd'hui en médecine personnalisée, notre offre est pléthorique. Je dois dire qu'en médecine personnalisée, le biomarqueur est probablement le sujet sur lequel nous observons le plus grand nombre d'inventions sortir de nos laboratoires. C'est aussi le sujet sur lequel nous « tuons » le plus. Très peu de biomarqueurs sont d'un réel intérêt pour l'industrie, dans la mesure où l'on n'a pas répondu sur ces biomarqueurs à la série des 56 questions que nous a posées le Pr Agnès Buzyn. Le potentiel industriel est donc énorme, mais aujourd'hui on constate une grande immaturité sur l'offre académique.
J'en viens à ce qui paraît nécessaire de développer, et les points importants sur lesquels nous nous focalisons en transfert de technologie. C'est la preuve de concept, un mécanisme général sur tous les segments thérapeutiques, mais particulièrement en médecine personnalisée, puisque tous ces biomarqueurs n'ont pas fait la preuve de leur utilité et de leur robustesse.
M. Alain Claeys, député de la Vienne, rapporteur. Ce tri que vous faites au niveau de la recherche académique se fait-il en fonction des attentes des industries pharmaceutiques ou bien sur d'autres critères ?
Mme Cécile Tharaud. Quand je parle de tri, je considère qu'une invention dispose d'un potentiel intéressant pour entrer en collaboration de développement avec un partenaire ou en licence, quelle que soit la modalité finalement. Un industriel peut-il se préoccuper de valoriser, de déployer un effort de recherche sur ce biomarqueur à un instant T ? Il s'agit de décider si l'on dépose un brevet ou pas ou même s'il n'y aura pas brevet. Sur les biomarqueurs, on ne dépose pas forcément tout de suite, car la recherche sera très longue, et on peut rester sur du secret le temps d'avancer. Très peu de ces biomarqueurs sont assez signifiants, et assez proches dans leur utilisation, pour que l'industrie s'en occupe vraiment immédiatement. Donc quand j'explique que le biomarqueur est le sujet sur lequel on « tue » le plus, cela signifie gentiment qu'on a peu de potentiel de collaboration industrielle ou de type capital-risque à un instant T0.
Cela ne signifie pas qu'on ne s'occupe pas du marqueur. Le transfert de technologie aujourd'hui s'applique à trouver les modalités de développement supplémentaire pour obtenir une meilleure valeur sur cette hypothèse. Si cette hypothèse n'est pas prête, si elle présente encore trop de risque, ou une nécessité d'investissement trop forte, l'industrie ne s'en occupera pas, ce qui ne signifie pas que le secteur public ne doit pas s'en occuper. D'où l'investissement de l'État en preuve de concept de façon générale en valorisation, et d'où une nécessité de mettre un fort accent notamment sur les cohortes, puisque valider un marqueur implique très vite la nécessité de tester l'hypothèse sur quelques milliers de patients. Cela coûte bien plus cher que ce que l'on fait classiquement dans le cadre d'une recherche académique, ce qui n'est pas nécessaire pour montrer l'intérêt du marqueur.
Avant d'avancer dans un développement, il faut réunir nombre de preuve de concept pour valider les marqueurs, or on manque de financement de plateformes de prototypage. Ce manque est encore plus important que sur les cohortes, puisque les investissements d'avenir ont bien financé cette demande, qui était une demande conjointe du secteur académique et du secteur privé. Le financement de plateformes de prototypage, peut-être un peu moins financé, est pourtant très nécessaire aujourd'hui. Contrairement à l'Allemagne, la France n'est pas un pays bien équipé pour prototyper des tests de diagnostic. Pourtant cela permet d'avancer plus rapidement dans le développement et le transfert industriel de certains tests, quand le marqueur ou la série de marqueurs, est validée et en vaut la peine. Il faut donc encore financer les recherches amont du secteur industriel.
Sur la propriété intellectuelle, je laisserai la parole aux spécialistes, mais aujourd'hui, nous observons plusieurs écueils. La médecine personnalisée, est par essence un mélange d'éléments centrés sur le patient, ce qui soulève une première question de propriété intellectuelle qui ne sait pas tout à fait définir une revendication de propriété intellectuelle incluant le patient. Il faudra donc réussir à phraser l'objet de propriété intellectuelle sans attirer le patient dans cette définition, ce qui n'est pas encore très clair pour les praticiens.
Deuxièmement, les méthodes de traitement sont non brevetables en Europe. La médecine personnalisée est essentiellement comprise comme une méthode de traitement. Donc il faudra à nouveau que la jurisprudence évolue, de façon à ce que la médecine personnalisée prenne une forme concrète que les spécialistes du droit de la propriété intellectuelle arrivent à décrire selon leur arsenal juridique.
Troisièmement, une jurisprudence bien plus récente qui vient des États-Unis semble montrer que les États-Unis s'orientent vers le refus de breveter des méthodes de diagnostic qui seraient issues de l'observation de phénomènes naturels. Or dans le domaine des marqueurs on se trouve typiquement en train de développer du diagnostic qui reproduit, ce qui est issu de l'observation de phénomènes naturels. Cette jurisprudence est récente.
M. Alain Claeys. Que dit l'Office européen des brevets (OEB) sur cette question ?
Mme Cécile Tharaud. À ma connaissance, l'OEB n'a pas encore réagi sur ce point. En revanche, l'OEB contrairement à celui des États-Unis, ne permet pas aujourd'hui de breveter des méthodes de traitement. Or en médecine personnalisée, a priori le recours au test compagnon relève de la décision de prescrire un médicament basé sur un test de diagnostic. Au sens de la propriété intellectuelle européenne, on est encore face à une interrogation.
Mme Elisabeth Thouret-Lemaitre, consultante, spécialiste de propriété industrielle. Les méthodes de traitement en tant que telles ne sont pas brevetables.
Mme Cécile Tharaud. De plus, le test compagnon, qui est une portion de ce que peut être la médecine personnalisée, n'a pas non plus, à ma connaissance, d'existence réglementaire très claire, au sens de la réglementation pharmaceutique. De nombreux industriels expliquent qu'aujourd'hui la route réglementaire est encore très aléatoire sur le principe du compagnonnage. Apparemment, c'est une forte demande pour eux.
Aujourd'hui en matière de développement industriel, quand des biomarqueurs ont montré la preuve de leur utilité, les premiers industriels à venir faire leurs courses sont malheureusement des industriels étrangers, pour lesquels les industries de service du diagnostic sont très développées. On se retrouve alors en amont, à donner une licence à de grands laboratoires qui vendent sous forme de services, un diagnostic relativement peu réglementé, avant que ne soit possible la commercialisation de tests sous une forme plus commerciale, -plus produits, plus kits-, par des industriels qui répondent à des contraintes réglementaires plus aiguës. Les industriels présents pourront commenter cela, mais il s'agit d'une vraie question en matière d'optimisation et d'enrichissement du tissu industriel français et européen, à partir d'une science foisonnante. Je soumets cette question à la sagacité des industriels ici présents.
Pr Dominique Stoppa-Lyonnet, chef du service de génétique de l'Institut Curie, professeur de génétique médicale à l'université Paris-Descartes, membre du Comité national consultatif d'éthique (CCNE). Mon propos portera en particulier sur la médecine génomique personnalisée. Je m'arrêterai aux tests génétiques. Pour moi, la médecine génomique personnalisée repose sur le fait que les caractéristiques génétiques permettront d'identifier des sous-groupes de malades atteints d'une maladie donnée, qui répondront mieux à un traitement, ou à l'inverse n'y répondront pas, ou présenteront une intolérance à ce traitement.
L'une des premières applications de la médecine génomique personnalisée concerne, on l'a souligné, le traitement des cancers, pour lesquels le génome étudié n'est pas le génome natif de la personne, mais celui de sa tumeur, la tumeur étant alors le siège d'altérations génétiques acquises, dont certaines lui confèrent son caractère malin.
La médecine génomique personnalisée concerne aussi la médecine prédictive, terme qui n'est pas très heureux, car il s'agit plutôt d'une médecine de prévision, et donc d'estimer le risque de survenue de telle ou telle maladie. Cependant, l'hypothèse fondatrice de la médecine prédictive repose sur le fait que le savoir permet de mettre en oeuvre des mesures de prévention. Nous savons que nous avons des capacités de prévision des maladies génétiques, sans pour autant toujours disposer de capacités de prévention, et cela peut générer un certain nombre de problèmes éthiques qui seront sans doute rediscutés plus tard. Un autre domaine est celui des tests sur le foetus. Il s'agit rarement de perspectives de traitement, puisque l'enjeu est plus souvent l'interruption médicale de grossesse. Les tests génétiques foetaux font-ils partie de vos préoccupations dans le cadre de la médecine personnalisée ? La question reste posée.
Depuis 2003, date du dépôt dans des bases de données internationales des 3 milliards de paires de bases du génome humain, à aujourd'hui, les capacités d'analyse des séquences ont augmenté d'un facteur 50 000. Le séquençage à très haut débit ou NGS ( Next-Generation Sequencing ) permet ainsi d'analyser, soit un grand nombre de gènes chez un grand nombre de personnes, soit le génome entier d'une seule personne, et de plus en plus le génome d'un grand nombre de personnes. Il est d'ailleurs probable qu'il sera bientôt plus facile de séquencer un génome entier plutôt que des gènes sélectionnés, ou des séquences ciblées, parce que la capture de ces gènes ou séquences est en fait complexe. Et il vaut mieux séquencer l'ensemble de ce qui se trouve dans le « tube », c'est-à-dire l'ADN entier de la personne.
Or ceci générera un très grand nombre de données dont beaucoup seront non interprétables. Le NGS possède une capacité formidable de génération des données, mais il y a un fossé, un saut, entre l'obtention facile de ces données et leur interprétation. Je ne voudrais cependant pas tout remettre en cause et dire que rien n'est interprétable, car notamment pour les maladies génétiques mendéliennes, ou mono-géniques, dont le déterminisme est relativement simple, les interprétations sont souvent solides. Il n'empêche, la génération de ce grand nombre de séquences soulève de nombreuses interrogations.
On pourrait identifier quatre situations. D'abord il existe des régions chromosomiques que nous possédons, en plus ou en moins. On parle alors de copy number variation, dont l'interprétation est complexe aujourd'hui, mais elle se simplifiera plus tard.
Deuxièmement, on trouve des mutations aux variations qui sont inactivatrices, qui introduisent un codon stop dans un gène. Mais aujourd'hui, ce gène n'est pas associé à une maladie particulière. Que penser du retentissement de cette protéine absente sur l'économie d'un individu ?
Troisième point, il existe des variations dans des gènes qui sont associées à une maladie connue, mais on ne sait pas que penser de cette variation. Est-t-elle associée elle-même à la maladie ? Ce problème occupe de nombreux généticiens aujourd'hui dans les laboratoires. Quel est le retentissement biologique de la substitution d'un acide aminé par un autre ? Ce changement a-t-il un rôle dans l'apparition de la maladie ?
Je rajouterais un quatrième point. En cancérologie, les cellules tumorales ont un génome très instable et donc génèrent un très grand nombre de mutations. Le cancer repose sur un modèle darwinien : de très nombreuses mutations surviennent, mais seulement quelques-unes sont sélectionnées par le processus tumoral car conférant un avantage sélectif à la cellule. Il s'agira de séparer les mutations driver , qui jouent un rôle direct dans le processus tumoral, et qui font l'objet de recherches intenses car ce sont des cibles potentielles de traitement des très nombreuses mutations passenger qui sont en quelque sorte le bruit de fond de l'instabilité du génome tumoral.
Voilà donc quelques exemples de la complexité de l'interprétation des données. Et c'est véritablement un défi qu'il faudra relever. En la matière, on dispose de deux grands outils : d'une part, la génomique fonctionnelle, une approche expérimentale qui examine in vitro puis in vivo le retentissement biologique de telle ou telle variation, et d'autre part, la génétique épidémiologique, qui est sûrement un enjeu très important, à travers notamment les études de cohortes de patients.
On a évoqué l'importance d'études de populations, de cohortes de patients. Cela signifie disposer non seulement d'un accès au génome de ces patients, mais surtout au phénotype, c'est-à-dire aux caractéristiques cliniques. De plus, on a également besoin de cohortes de sujets « normaux », en ayant leur phénotype. Or qui dit cohorte, dit suivi de ces personnes sur de nombreuses années.
Je crois qu'il faut faire un effort très important. Il a été fait en partie par le Grand emprunt. Mais il faut savoir que pour des maladies rares, un seul laboratoire, un seul pays, ne suffira pas pour avoir assez de puissance pour conclure à la signification de tel ou tel variant, à l'identification de facteurs génétiques modificateurs de telle ou telle maladie. Aussi voit-on naître des consortiums internationaux qui sont indispensables à la meilleure connaissance de ces maladies. Et puis se mettent en place des consortiums de consortiums, notamment un très beau projet international, le Human Variome Project , dont la vocation est de réunir toutes les données sur de nombreuses maladies génétiques.
Je citerai également des consortiums en cancérologie : TGCA ( The Cancer Genome Atlas ) aux États-Unis, ICGC en Europe ( International Cancer Genome Consortium ). Ces consortiums ont pour vocation de séquencer un très grand nombre de tumeurs. Ils ont un rôle majeur à jouer notamment dans la génération de nouveaux savoirs, et par là dans la génération d'une nouvelle organisation de la recherche, et donc dans l'élaboration de nouveaux modèles de propriété intellectuelle.
Nous en sommes, je crois, à l'ère du Data-Sharing, du partage des données. La généticienne que je suis espère sincèrement que le retour de la science et des nouvelles connaissances vers la société sera à un coût moindre que celui des monopoles et licences exclusives. À mon sens, la nécessité de se mettre à plusieurs pour générer ces nouveaux savoirs remet en question le système des brevets. Celui-ci repose sur un équilibre finalement fragile de l'investissement et du retour sur investissement, d'un inventeur ou d'un groupe d'inventeurs. Or dans ce domaine, le groupe d'inventeurs est énorme. Il est aussi important que celui vers lesquels vont se retourner les connaissances.
Le temps des brevets me semble donc compté. En 2010, un juge fédéral américain, Robert Sweet, a révoqué, après une plainte d'associations de citoyens, les brevets sur les gènes BRCA1 et BRCA2 . Cette décision a été contrariée en appel. Elle est actuellement examinée par la Cour suprême. Il me semble que la tendance va vers une révocation des brevets. Maurice Cassier nous en dira plus. Les brevets individuels, sur les 23 000 gènes de notre génome, rendraient impossibles la médecine génomique personnalisée. De mon point de vue, c'est plutôt rassurant, mais tel n'est vraisemblablement pas le point de vue des industriels.
Pour conclure, même si on se situe actuellement vers bien plus de brevets sur les gènes, on voit se développer des formes insidieuses de dépendance vis-à-vis de la connaissance du génome. En fait, je voudrais évoquer les tests compagnons de la prescription de médicaments. Actuellement, dans le cadre d'essais cliniques utilisant par exemple en cancérologie un traitement ciblé, l'éligibilité à un essai peut reposer dans certains cas sur la mise en évidence de telle ou telle anomalie. Une société pharmaceutique a pu développer elle-même, ou faire une alliance avec une société de biotechnologie qui réalise le test. On l'a évoqué. Ce n'est pas avec un kit, c'est avec l'envoi du prélèvement dans cette société. Le coût du test vient s'associer à celui du médicament.
Tant qu'on se situe dans un essai clinique, cela me semble assez cohérent, car on a besoin d'une très grande homogénéité dans la réalisation des essais, dans la désignation des personnes porteuses de telle ou telle anomalie ou dans la caractérisation de telle ou telle lésion tumorale. Mais je m'interroge : qu'en sera-t-il plus tard, lorsque le médicament aura une AMM, l'efficacité du médicament sera-t-elle garantie par la modalité du test ? Si c'est le cas et que tests compagnons et médicaments sont intrinsèquement liés, alors aucune concurrence aucune perspective d'amélioration, et de baisse des coûts ne sera possible. Je m'interroge donc sur le développement des tests compagnons, car je crains qu'ils ne brident le développement de la médecine personnalisée.
Mme Elisabeth Thouret-Lemaitre, consultante, spécialiste de propriété industrielle . Je traiterai de propriété industrielle, et particulièrement de brevets, matière difficile s'il en est, après les considérations scientifiques, éthiques, sociétales.
Je souhaitais vous faire part des revendications particulières à la médecine personnalisée, mais je ferai d'abord le point de la situation du diagnostic en Europe et aux États-Unis.
Les brevets européens sont délivrés pour des inventions brevetables, c'est-à-dire nouvelles, impliquant une activité inventive, et susceptibles d'applications industrielles. Ce sont des grands principes qu'il faut ensuite mettre en application.
Dans la nouvelle convention européenne sur les brevets, il y a un article important concernant les exceptions à la brevetabilité. Il est bien précisé que les brevets ne sont pas délivrés pour les méthodes de diagnostic appliquées au corps humain ou animal, cette disposition ne s'appliquant pas au produit pour la mise en oeuvre d'une de ces méthodes. Donc ces méthodes en tant que telles sont non brevetables. Les produits pour la mise en oeuvre des méthodes peuvent être brevetés à condition bien sûr, d'être nouveaux et d'avoir une activité inventive.
En Europe, les choses sont à peu près claires : les méthodes de diagnostic sont non brevetables. Différentes décisions l'ont déjà précisé, et l'on peut dresser le bilan suivant. Les méthodes de diagnostic in vitro sont brevetables si elles apportent la nouveauté et l'activité inventive. Les réactifs sont brevetables. Les méthodes, dont le résultat permet directement de prendre une décision au sujet du traitement médical, ne sont pas brevetables, parce que c'est une analyse de données. Mais la méthode visant à obtenir des résultats ou valeurs intermédiaires a été considérée comme brevetable. Ces décisions ne sont pas issues de la grande chambre de recours pour le moment, et sont donc peut-être sujettes à interprétation ou aux différents points de vue des chambres de recours technique.
Aux États-Unis, dans une récente décision, les méthodes de diagnostic utilisant des phénomènes naturels n'ont pas été acceptées comme brevetables, considérant qu'une suite de phénomènes naturels conduisant à une décision ou à un constat, n'est pas brevetable.
Dans le domaine de la médecine personnalisée, plusieurs décisions au niveau de l'Office européen des brevets (OEB) ont été prises. Une revendication typique de ce domaine est la suivante : une substance X pour l'utilisation dans une méthode de traitement de la maladie Y, chez un individu ou un groupe d'individus possédant le biomarqueur Z.
La question principale pour les déposants et les offices de brevets est la nouveauté de l'invention. Car très souvent, le traitement du groupe de patients possédant le biomarqueur Z, dans cette nouvelle revendication, représente une limitation par rapport au traitement déjà connu dans l'art antérieur pour tous les patients, ceux qui possèdent le biomarqueur Z et ceux qui ne le possèdent pas. Y a-t-il nouveauté ?
L'OEB a décidé que, dans un test en deux étapes, s'il y a recouvrement en partie des groupes de patients, et que le nouveau groupe est arbitraire, il n'y a pas brevetabilité. La revendication n'est pas nouvelle. Il faut donc arriver à une solution qui permette de faire en sorte que le groupe de patients nouvellement traités avec ce biomarqueur n'est pas compris dans ou ne recoupe pas le groupe précédent.
Une décision de 1986 T19/86 appelée « Cochons 2 » portait sur un traitement prophylactique des animaux ; la brevetabilité a été reconnue car la population traitée était différente sur le plan immunologique.
La décision essentielle est la décision T 233/96. Elle a conclu que le groupe de patients n'était pas distinctif, et donc de ce fait, la revendication n'était pas acceptable. Le groupe de patients ne peut pas être arbitraire... L'OEB a proposé un test en deux parties pour juger de la nouveauté : les groupes de patients ne doivent pas se recouvrir et il doit y avoir une relation fonctionnelle entre le biomarqueur et le traitement thérapeutique. A ce moment-là, la brevetabilité est acceptée.
Trois décisions de 2001, 2004 et 2006 : T83/01, T1399/04 et T1642/06 concluent à la brevetabilité de ces traitements pour certains groupes de patients, parce qu'il y a une relation fonctionnelle entre le biomarqueur et le traitement thérapeutique. Ces décisions ne sont que des décisions de chambres de recours techniques. Nous restons donc pour l'instant un peu dans l'incertitude. Et nous attendons sans doute d'autres décisions à venir.
On peut faire un parallèle avec les décisions relatives au régime de dosage. Il a été accepté au niveau de l'OEB, dans une décision de la Grande chambre de recours, qu'une posologie particulière, avec une administration particulière, était brevetable. Malheureusement, les tribunaux nationaux en France, en Allemagne et en Grande-Bretagne qui ont eu à traiter de brevets relatifs à telles posologies, n'ont pas tous pris les mêmes décisions. La France n'a malheureusement pas accepté ce brevet.
Pour ce qui est de la médecine personnalisée, il y a peu de décisions.
M. Maurice Cassier, sociologue au Centre de recherche médecine, sciences, santé et société (CERMES). De manière liminaire, je rappellerai que l'application du droit des brevets dans le champ de la médecine, dans le champ médical, a toujours été problématique et difficile. Le droit des brevets lui-même se limite, on vient d'en avoir un exemple quand le droit européen déclare que les méthodes de diagnostic ou les méthodes thérapeutiques appliquées au corps humain ne sont pas brevetables.
Le fondement de cette exclusion, était d'éviter des restrictions de circulation des technologies à l'intérieur du corps médical, pour que l'offre médicale ne soit pas restreinte. Les restrictions que peuvent faire courir les brevets sur l'offre de soins, sur l'offre de service médical, est à la racine des contestations qu'on observe depuis vingt-cinq ans, le Pr Dominique Stoppa-Lyonnet et l'Institut Curie y ayant contribué en France et en Europe. Cela a fondé ce que la revue Nature a appelé « la révolte des généticiens européens », c'était le refus de restriction de l'offre médicale et de la pratique médicale dû à l'existence de brevets qui gênaient l'exercice des médecins.
Depuis vingt ans, on se trouve face à une continuité de ces interrogations et contestations. La particularité est que des discussions juridiques ont lieu à la fois à l'intérieur de l'Office européen des brevets et aujourd'hui aux États-Unis, où se déroule un procès sur la brevetabilité des gènes qui arrive à la Cour suprême ce printemps 2013. Ce qui est intéressant, c'est aussi l'engagement, que ce soit en Europe ou aux États-Unis, des associations médicales, des médecins, dans la discussion de ces brevets et de leurs effets. Lors du procès américain sur les brevets de gènes, ce qui est étonnant, c'est la mobilisation de toutes les grandes associations médicales américaines. Toutes les sociétés de médecine moléculaire, alliées aux associations de patients, puisqu'il s'agit de brevets sur les gènes de prédisposition au cancer du sein, se sont portées plaignantes contre ces brevets pour demander leur annulation.
La question de la brevetabilité est donc à la fois une question juridique, mais aussi de politique de santé. Je rappelle qu'au début des années 2000, la question des brevets sur les gènes, et sur ces brevets qui arrivent devant la Cour suprême, a été évoquée à plusieurs reprises à l'Assemblée nationale, en 2002, en 2003, dans des questions écrites. Lors de la révision de la loi de bioéthique, elle a même fait l'objet d'un amendement particulier. La licence obligatoire est en effet une sorte de dispositif de régulation du droit des brevets. La loi de bioéthique de 2004 prévoit une extension des mesures de licence obligatoire pour les tests biologiques.
M. Alain Claeys. Tout était parti de la transposition de la directive européenne.
M. Maurice Cassier. C'est cela. Mais c'est la conjonction, je pense, de la mobilisation des médecins en France et en Europe à cette époque, de plusieurs États qui se sont portés opposants contre ces brevets, et de la discussion sur la transposition qui a conduit à cet amendement.
M. Alain Claeys. Tout à fait.
M. Maurice Cassier. Je voudrais lister, de manière rapide, les moyens de régulation, ou éventuellement d'émancipation, du droit des brevets, qu'on peut utiliser, ou qui sont utilisés, dans le domaine de la génétique et des innovations médicales.
J'ai rappelé qu'en Europe, ce qui est fréquemment utilisé dans le domaine de la biologie, ce sont les procédures d'opposition. On l'a vu aussi pour des thérapies cellulaires. Des médecins se portent opposants pour demander l'annulation de brevets. Ils sont rejoints parfois par des ministères de la santé inquiets des conséquences à la fois sur le prix et sur la disponibilité de ces technologies. Ces procédures d'opposition ont été utilisées par les généticiens, par les médecins. C'est un moyen de régulation. Evidemment, la limite de ces dispositifs, est qu'on fait une opposition contre un brevet particulier, sur une technologie particulière. Il faudrait multiplier les actes d'opposition pour arriver à des régulations plus stables et plus durables.
Un autre moyen qui a été utilisé en Europe, c'est une fondation médicale qui prend un brevet sur un des gènes, et qui distribue gratuitement des licences à tous les laboratoires médicaux susceptibles de pratiquer des tests génétiques. Dans ce cas, les institutions médicales utilisent le brevet, en quelque sorte pour contrarier et empêcher ses effets nocifs.
Aux États-Unis, le procès en cours, est engagé depuis trois ans par les grandes associations médicales, et a connu plusieurs rebondissements. Il est devant la Cour suprême. On peut en tirer deux enseignements principaux. Ce procès vise en fait à mettre dans le domaine public les gènes en tant que produits de la nature. Le raisonnement est le suivant : dès lors qu'ils sont simplement isolés de leur environnement naturel, les gènes ne sont pas des inventions brevetables. Ce point a été acté par la décision d'un juge fédéral en 2009. La Cour d'appel du circuit fédéral, qui était plutôt favorable au brevet au cours de ces vingt dernières années, est revenue sur cette décision, mais avec un vote de deux juges contre trois. Or quand on lit les arguments des juges, la position de l'un des juges est extrêmement mitigée. Il pense effectivement que les gènes sont plutôt des entités naturelles non brevetables, mais en raison de l'histoire des biotechnologies aux États-Unis, de ces trente ans d'investissements passés, il se rallie à ces brevets. C'est donc un argument plus économique que juridique qui l'emporte.
On a fait référence à une méthode de dosage, où la Cour suprême a affirmé la non-brevetabilité des lois naturelles, des corrélations naturelles. La montée à la Cour suprême pourrait traduire ce mouvement. Il n'est pas impossible que la Cour suprême reconnaisse la non-brevetabilité des gènes comme entités naturelles.
En conclusion, je reviendrai sur une situation particulière. D'un côté on est face à la situation des monopoles, de l'autre face à la fragmentation de la propriété industrielle, c'est-à-dire qu'on a un très grand nombre de brevets sur un très grand nombre de marqueurs biologiques. Cette fragmentation et cette dispersion est susceptible de contrarier les nouvelles générations de tests évoqués à plusieurs reprises, c'est-à-dire des tests multi-marqueurs, multi-gènes, ou des tests de séquençage du génome complet. Dans ce cas-là, même certaines sociétés de biotechnologies aux États-Unis qui sont les plus engagées dans ces outils-là s'avèrent ouvertement opposées aux brevets sur les marqueurs biologiques, et font valoir que cela freine l'innovation dans ce domaine, alors que d'autres sociétés, qui sont également engagées, soutiennent les brevets en reconnaissant que cela leur pose des difficultés considérables.
Deux dispositifs sont proposés et expérimentés. Soit la Cour suprême annule les revendications de brevets sur les gènes, et dans ce cas-là, les gènes tombent dans le domaine public et les sociétés qui veulent développer des tests multiplex multi-marqueurs, pourront le faire librement. Soit on va vers un dispositif qui est en cours d'expérimentation aux États-Unis, à l'initiative des National Institutes of Health (NIH), et qui consiste à regrouper des pools de brevets à l'échelle de plusieurs grandes universités et d'instituts anti-cancéreux. Un pool a été monté à l'automne 2012. Au départ, il a rassemblé 500 brevets, pour favoriser justement, et avec le principe de licence non exclusive, la réunion, la collecte, la collection, d'un grand nombre de brevets et de marqueurs génétiques.
M. Alain Claeys. Y a-t-il beaucoup de brevets actuellement qui associent un gène ?
M. Maurice Cassier. Des comptes divers ont été effectués. On estime qu'entre 4 000 et 5 000 gènes sont brevetés, soit environ 20 % du génome humain.
M. Alain Claeys. Donc ces entreprises propriétaires de ces brevets peuvent revendiquer toute une série de brevets dépendants.
M. Maurice Cassier. C'est exact et c'est ce qui ennuie les industriels les plus engagés dans les tests multiplex et multi marqueurs. Il me semble qu'en France, ou en Europe peut-être, on pourrait réfléchir à la constitution de tels pools, pour autant qu'on ait des brevets sur des marqueurs et au niveau des Alliances, l'AVIESAN par exemple, on pourrait favoriser le regroupement de pools sur des marqueurs qui seraient mis en guichet unique pour des sociétés ?
M. Alain Claeys. Le ministère y travaille-t-il ?
Mme Cécile Tharaud. Non l'INSERM n'y travaille pas. Je pense qu'on a beaucoup appris, suite au séquençage du génome humain. Je veux rappeler que sur cette bagarre des brevets que M. Maurice Cassier mentionnait tout à l'heure -un gène est-il brevetable ou non ?- un beau jour les Américains ont mis l'intégralité de la séquence du génome en libre accès sur leur base de donnée ce qui rend le brevetage beaucoup plus complexe, c'est-à-dire qu'on a arrêté de déposer des séquences, des listings de séquences dans un carton. Personnellement, j'en ai déposé beaucoup, des cartons et des kilos de séquences d'ADN. J'étais chez Genset à l'époque. On avait développé une industrie qui était basée sur le pari que la jurisprudence évoluerait. La génomique fonctionnelle a eu son temps. Il me paraît important de comprendre, que les jurisprudences mettent vingt à vingt-cinq ans à évoluer, mais enfin, elles évoluent.
On n'est pas en train d`expliquer qu'on brevète une séquence aujourd'hui. Les brevets de gènes d'aujourd'hui ne sont ceux qu'on déposait en 2000, mais un non spécialiste de ces brevets ne s'en rend pas bien compte. On a l'impression qu'un brevet de gènes, c'est un simple brevet de gènes, alors qu'il exige une montagne d'expérimentations, de preuves, de descriptions de l'utilité de l'application qui en sortira, qui sont de plus en plus exigeantes.
On peut envisager plusieurs stratégies. Soit on dépose dans le domaine public pour forcer la jurisprudence à avancer d'un grand coup. C'est comme cela que les Américains ont réagi en mettant toute la séquence du génome en accès public, ce qui n'était pas le cas jusqu'au jour où ils l'ont fait. Soit on adopte la stratégie du patent pool groupement de brevets, mais elle ne sera jamais parfaite. On n'a jamais le bon marqueur dans l'hôpital d'à côté. Il faudrait créer une base de données sur un pool mondial, ce qui revient finalement à mettre l'ensemble sur une base publique. Le pool, c'est très empirique, et ça marche une fois sur on ignore combien...
M. Maurice Cassier. Pour terminer, et pour rester un peu dans la même logique, dans le domaine des marqueurs biologiques, on a l'expérience de consortiums qui ont marché au début des années 2000. Toute une série de consortiums sur des marqueurs biologiques ont adopté des licences open source , et qui ont mis également, au bout d'un certain temps, les données dans le domaine public. Ces consortiums-là ont effectivement été utilisés. Ils ont permis le développement d'outils de diagnostic mis au point par les sociétés de biotechnologies.
Dans ce cas-là, on va vers des stratégies de mutualisation via les consortiums, via les Patent pools , précurseurs éventuellement à des mises dans le domaine public. Les incertitudes sont grandes, avec plusieurs types d'initiatives. Mais le principe est plutôt la mutualisation et la mise dans le domaine public.
Mme Audrey Aboukrat, doctorante à l'école de droit de l'université Paris I Panthéon-Sorbonne . Ma présentation portera sur les rapports entre médecine personnalisée et brevetabilité des gènes humains aux États-Unis. Comme cela a été évoqué, c'est une question d'actualité. Je vais retenir une acception large des brevets sur les gènes humains, comprenant d'une part les brevets qui portent sur les séquences d'ADN isolées et d'autre part les brevets qui portent sur les méthodes d'analyse ou de comparaison de ces séquences aux fins de diagnostic car ces deux types de brevets sont essentiels pour le sujet de la médecine personnalisée. L'un des champs privilégiés de développement de la médecine personnalisée est en effet celui de la pharmaco-génomique qui a pour fonction de déterminer les variations de réactions des individus par rapport à des médicaments en fonction de leurs différences génétiques. Pour déterminer ces variations, il est nécessaire de procéder à des tests génétiques permettant aux médecins et aux chercheurs d'analyser et de comparer les séquences génétiques des individus concernés. Or, si ces tests et/ou les gènes impliqués dans l'affection ou la susceptibilité recherchée sont brevetés, alors l'utilisation des tests sans autorisation (licence d'exploitation) est de nature à constituer un acte de contrefaçon.
J'envisagerai les rapports entre médecine personnalisée et brevetabilité des gènes humains selon deux questions. D'une part, celle de la brevetabilité des « gènes » humains aux États-Unis, puisque telle est la question que vous m'avez posée. Je m'interrogerai d'autre part sur le risque de blocage que les brevets sur les gènes peuvent constituer pour le développement d'une médecine personnalisée.
Posée à la section 101 du Patent Act américain, la brevetabilité d'une invention est définie en termes très généraux. C'est la jurisprudence américaine qui a façonné au fil du temps les éléments de définition du caractère brevetable ou non d'une création.
Dans le temps qui m'est imparti, il ne m'est pas possible de retracer toute l'évolution jurisprudentielle américaine relative à la question de la brevetabilité des gènes humains depuis le célèbre arrêt Diamond v. Chakrabarthy rendu par la Cour Suprême des États-Unis en 1980 et qui a constitué le « terreau » de ce que l'on appelle généralement la « brevetabilité du vivant ». Car déjà dans cette décision existait en germe le principe de la brevetabilité des gènes humains. La Cour Suprême y faisait de l'intervention humaine un critère important de la brevetabilité d'une création, permettant ce faisant la brevetabilité de séquences génétiques isolées ou modifiées par l'homme et cela, même si les séquences génétiques considérées existaient telles quelles dans la nature, dans la mesure où une intervention humaine était nécessaire pour les obtenir.
Alors même que les États-Unis avaient été parmi les initiateurs de ce principe de brevetabilité des gènes humains, c'est contre toute attente qu'une affaire actuellement pendante devant la Cour Suprême de ce même pays pourrait être le siège d'un revirement de jurisprudence important sur la question. Il s'agit de l'affaire dite Myriad Genetics qui oppose l'AMP (Association for molecular pathology) parmi nombre de demandeurs et l'Office américain des brevets et des marques (USPTO) ensemble avec Myriad Genetics , défendeurs.
Comment en est-on arrivés là ?
Myriad Genetics détient sept brevets aux États-Unis, couvrant un grand nombre de revendications relatives aux gènes BRCA1 et BRCA2. Sont notamment revendiquées les séquences d'ADN isolées correspondant à ces gènes et à certaines altérations ou mutations qui seraient associées à une prédisposition aux cancers du sein et des ovaires. Sont également revendiquées des méthodes d'analyse ou de comparaison de ces séquences aux fins de diagnostic d'une telle prédisposition. Parmi les nombreuses revendications couvertes par ces sept brevets, trois types d'entre elles ont été mis en cause. Tout d'abord, des revendications portant sur les gènes humains isolés BRCA 1 et BRCA 2 ainsi que certaines altérations ou mutations de ces gènes qui pourraient être associées à une prédisposition aux cancers du sein et des ovaires. Ensuite, une revendication de procédé portant sur les méthodes d'analyse ou de comparaison des séquences BRCA 1 et BRCA 2 du patient avec les séquences d'un patient sain dans l'objectif d'identifier la présence d'altérations ou de mutations prédisposant le patient aux cancers du sein et des ovaires. Enfin, une revendication portant sur une méthode d'identification de médicaments et de leur efficacité potentielle dans le traitement des cancers du sein et des ovaires.
En mars 2010, en première instance, le juge Robert Sweet invalide l'ensemble des revendications des brevets de Myriad Genetics sur le fondement de la section 101 du Patent Act 242 ( * ). Pour ce faire, il distingue entre d'une part les revendications qui portent sur les séquences d'ADN isolées et d'autre part les revendications qui portent sur les méthodes de diagnostic.
S'agissant des séquences d'ADN, le juge Sweet a retenu qu'elles n'étaient pas suffisamment distinctes des séquences existant dans la nature. Se référant notamment à des affaires décidées au 19 ème siècle, le juge explique que « la seule purification d'un produit de la nature ne suffit guère à transformer ce produit en invention brevetable ». Cette position est intéressante en ce qu'elle diffère de la position classique retenue en droit américain des brevets. La position classique consiste à analyser le gène comme un élément chimique et donc le seul fait de l'isoler du corps humain suffirait à le rendre brevetable, sous réserve du respect des autres conditions de brevetabilité. Le juge Sweet s'oppose à cette interprétation et considère que c'est la fonction biologique du gène qui est à prendre en considération pour juger de la brevetabilité, soit sa fonction en tant que porteur de l'information. Or, en l'espèce, la version isolée et la version « naturelle » des gènes BRCA 1 et BRCA 2 portaient toutes deux la même information génétique. Par conséquent, le juge Sweet a retenu que les revendications associées portaient sur des produits de la nature, exclus en tant que tels de la brevetabilité.
Sur les revendications relatives aux méthodes d'analyse et de comparaison des séquences d'ADN aux fins de diagnostic, ces méthodes ont également été jugées non brevetables. Le juge a retenu que celles-ci consistaient simplement en des opérations mentales abstraites n'impliquant aucune transformation physique, ne constituant rien de plus qu'une étape préparatoire de collecte des données. En ce sens, ces méthodes se trouvaient exclues de la brevetabilité.
La décision était donc plutôt sévère contre Myriad Genetics dont l'ensemble des revendications contestées a été invalidé. En particulier, la position du juge Sweet de retenir que les gènes humains et plus précisément les séquences d'ADN isolées des gènes BRCA 1 et BRCA 2 n'étaient pas brevetables en tant que telles aurait pu sembler être un combat perdu d'avance, tant le principe de brevetabilité des gènes humains est ancré dans la jurisprudence américaine. De fait, dans un arrêt rendu en juillet 2011, la Cour d'appel fédérale a cassé la décision rendue en première instance sur ce point. Si la Cour d'appel s'aligne sur la décision du juge Sweet en ce qui concerne les méthodes d'analyse et de comparaison des séquences d'ADN aux fins de diagnostic (retenant que ces méthodes constituent une opération mentale abstraite et sont donc exclues de la brevetabilité), elle revient en revanche à la position classique en ce qui concerne la brevetabilité des gènes, analysant ces derniers comme des éléments chimiques et concluant à la brevetabilité des séquences d'ADN isolées revendiquées par Myriad Genetics .
Toutefois, l'hésitation jurisprudentielle qui s'est fait jour a été de nature à faire resurgir comme une question de principe, aux plans juridique et politique, la question de la brevetabilité des gènes humains aux États-Unis. En décembre 2011, l'Association pour les libertés civiles aux États-Unis ( American Civil Liberties Union ) et la Fondation Publique des Brevets ( Public Patent Foundation ) ont donc adressé une demande à la Cour Suprême visant à ce que la Cour d'appel fédérale revoit sa décision. Le 26 mars 2012, la Cour Suprême, soit la plus haute institution juridictionnelle américaine, a favorablement accueilli leur demande et a annulé la décision rendue par la Cour d'appel fédérale, lui ordonnant de revoir sa décision à la lumière d'une autre décision rendue par la juridiction suprême en mars 2012 dans une affaire Mayo Collaborative Services v. Prometheus Laboratories 243 ( * ).
Pourquoi, à la lumière de cette décision, s'interroger une troisième fois sur la brevetabilité des gènes humains ? Dans l'affaire Mayo Collaborative Services v. Prometheus Laboratories , la Cour suprême s'était prononcée sur la validité de revendications portant sur une méthode d'optimisation de l'efficacité thérapeutique et de réduction de la toxicité associées à un traitement de troubles gastro-intestinaux. Pour trancher cette affaire, le juge Breyer, qui a écrit l'opinion majoritaire de la Cour suprême, a distingué entre une loi de la nature, non brevetable car elle constitue un phénomène naturel, et une application d'une loi de la nature qui, elle, est brevetable. En l'espèce, la méthode revendiquée visait à établir des corrélations entre les concentrations de certains métabolites dans le sang et la probabilité que le dosage du médicament se révèle inefficace ou trop toxique pour le patient. Le juge a déduit que de telles revendications ne suffisaient pas à transformer ces relations en application d'une loi de la nature. Ces simples corrélations tombaient ainsi dans la catégorie des lois de la nature.
Cette distinction entre une loi de la nature et l'application d'une loi de la nature est certes intéressante au plan de la brevetabilité, mais extrêmement complexe à mettre en oeuvre. On comprend mieux pourquoi la Cour suprême a voulu que la décision de la Cour d'appel fédérale soit revue à la lumière de cette distinction pour trancher la question de la brevetabilité des gènes humains. En pratique cependant et comme cela était prévisible, l'impact de l'affaire Mayo Collaborative Services sur l'affaire Myriad Genetics a été assez faible. En effet, dans l'affaire Mayo Collaborative Services v. Prometheus Laboratories , les brevets en cause portaient sur une méthode de diagnostic ; il s'agissait donc de brevets de procédé. Or, dans l'affaire Myriad Genetics , les revendications qui posaient vraiment problème et dont l'appréciation opposait la première instance et la Cour d'appel fédérale portaient sur les séquences isolées d'ADN, soit un produit, et non sur les méthodes de diagnostic. La solution retenue dans l'affaire Mayo Collaborative Services v. Prometheus Laboratories ne contrôlait donc pas la question de la brevetabilité des revendications relatives aux séquences d'ADN isolées dans l'affaire Myriad Genetics . Aussi la Cour d'appel fédérale a-t-elle retenu la même solution qu'initialement et confirmé que l'arrêt Diamond v. Chakrabarthy servait de référence pour décider de la brevetabilité des séquences d'ADN associées aux gènes BRCA 1 et BRCA 2 244 ( * ) . Plus précisément, la Cour d'appel fédérale maintient que les séquences d'ADN revendiquées ne constituent pas un produit de la nature mais plutôt résultent d'une intervention humaine. En effet, l'isolement des séquences d'ADN ne se limite pas à leur séparation de corps étrangers mais consiste également en une transformation des séquences en une entité moléculaire différente de celle qui existe dans la nature. En ce sens, la Cour d'appel fédérale a retenu que les séquences d'ADN revendiquées, quoiqu'obtenues à partir de produits existant dans la nature, résultaient de l'ingéniosité humaine et étaient donc brevetables.
En septembre 2012, la Fondation publique des brevets et l'Association pour les libertés civiles aux États-Unis ont demandé à la Cour suprême de se saisir de l'affaire véritablement pour réviser la solution retenue par la Cour d'appel fédérale. L'affaire Myriad Genetics est actuellement pendante devant la Cour Suprême qui entendra, le 15 avril prochain, les arguments oraux des multiples parties avant de rendre sa décision dans les mois qui suivent. Notons que le Gouvernement américain a pris position tout au long de l'affaire Myriad Genetics contre la brevetabilité des gènes humains. S'il est difficile de prédire la solution que retiendra la Cour Suprême, au moins est-il intéressant de constater que le monde judiciaire se ressaisit de la validité d'un principe - la brevetabilité des gènes humains - depuis vingt ans assis sur une construction politico-administrative, à savoir la pratique de l'USPTO (United States Patent and Trademark Office) qui est responsable de la délivrance de brevets dans ce domaine.
La deuxième question que j'annonçais concerne les risques de blocage, par les brevets sur les gènes humains, de la recherche en médecine personnalisée. En tant que juriste, je ne suis pas forcément la mieux outillée pour me prononcer, d'autant qu'en l'absence d'études de terrain sur cette question, il est difficile de trancher. Mais au moins peut-on dresser quelques constats.
Premier constat : aux États-Unis, les avis les plus divers se côtoient à propos de cette question. Certains considèrent qu'il existe un risque réel de blocage de la recherche par les brevets. On peut citer Michael A. Heller et Rebecca S. Eisenberg qui ont théorisé, à travers l'expression d'anti-communs, ce qu'ils estiment être un vrai risque de blocage de la recherche biomédicale par les brevets : appliquée au cas des gènes humains, cette position met en relief les dangers de brevets revendiquant de nombreuses fonctions de gènes et verrouillant de ce fait l'exploitation de ces fonctions par d'autres. Dans ce cas de figure, les incidences seraient néfastes pour les chercheurs en médecine personnalisée, dans le cadre notamment du séquençage du génome humain entier (lequel leur permet d'avoir accès à un plus grand spectre de l'information génétique d'un individu et de ses éventuelles variations pour déterminer avec plus de précision les relations entre la génétique et les réactions au traitement pour le personnaliser, l'adapter à l'individu). Si des brevets couvrent en effet l'ensemble des fonctions d'un gène humain, l'analyse et l'exploitation des variations du gène risque de constituer un acte de contrefaçon.
En contrepoint, d'autres auteurs estiment qu'un tel blocage n'est en rien écrit. Ils défendent la thèse selon laquelle de nombreux domaines (notamment celui des nouvelles technologies de l'information et de la communication) se caractérisent par un enchevêtrement de brevets conférant à leurs titulaires des monopoles étendus, ce qui n'empêche pas les concurrents d'avancer, notamment par le biais d'accords de licences croisées.
Si c'est effectivement bien le cas, on observera néanmoins que la multiplication des brevets sur les gènes peut entraîner d'importants coûts de transaction pour les opérateurs (s'informer des brevets délivrés et de l'étendue des monopoles conférés, déterminer la liberté d'exploitation laissée aux concurrents) et une fragilité importante pour tous ceux qui ne bénéficient pas de portefeuilles de brevets leur permettant de conclure des accords de licence leur permettant de continuer à travailler.
Deuxième constat : il est important de rappeler qu'aux États-Unis, comme en Europe, l'obtention d'un brevet est conditionnée à la vérification de plusieurs conditions : outre la mise en évidence d'une invention, il faut remplir un certain nombre de critères de brevetabilité, parmi lesquels le critère de l'applicabilité industrielle. Sur ce point, en 2001, l'Office américain des brevets et des marques (USPTO) a développé des directives pour l'examen des demandes de brevets sur des gènes humains, ajoutant des exigences pour la démonstration d'une applicabilité industrielle, laquelle doit désormais être spécifique, substantielle et crédible. Par exemple, il faut qu'aucune fonction du gène humain n'ait été identifiée auparavant pour que l'invention associée puisse être protégée par un brevet. Si davantage de recherches sont exigées pour identifier l'applicabilité industrielle de la séquence d'ADN identifiée, le brevet ne sera pas délivré. L'USPTO a donc ajouté des critères pour les seuls brevets sur les gènes humains, afin d'éviter que les brevets délivrés ne soient trop larges. Cette position permet d'éviter que l'étude puis l'exploitation d'une autre fonction du même gène, et même sa brevetabilité, tombent dans la dépendance d'un premier brevet. C'est un garde-fou important.
Toujours dans cette voie, notons que dans le cadre de la réforme très récente du droit américain des brevets qui a eu lieu en 2011, le Congrès a demandé à l'Office américain de mener une étude sur les tests génétiques de diagnostic pour formuler des recommandations concernant des moyens efficaces de garantir l'indépendance de l'activité de diagnostic en présence de brevets sur les gènes et de licences exclusives. Cette étude, en cours de réalisation, permettra de mieux comprendre la relation entre les brevets sur les gènes et les licences exclusives sur des tests génétiques d'une part et l'activité de diagnostic destinée à permettre au patient d'obtenir un second avis, indépendant de celui fourni par le test génétique protégé, d'autre part. Cette étude apportera sans doute des éléments de réflexion sur la question de savoir si les brevets sur les gènes ou les licences exclusives associées à des tests génétiques sont susceptibles de limiter l'accès des patients aux activités de diagnostic protégées.
|
Débat |
Pr Philippe Amouyel. J'ai une remarque et une question. On n'a parlé que de la brevetabilité du génome. Notre séance est consacrée à la médecine personnalisée. Donc qu'en est-il de tous les diagnostics d'autre nature ? Si la séquence complète du génome se diffuse et se démocratise, parce que le prix baissera de façon absolument drastique, et que chacun pourra obtenir une séquence complète, et si les patients utilisent cette séquence avec leur médecin pour savoir si on trouve telle ou telle mutation, comment peut-on défendre un brevet ? Comment peut-on réclamer les droits du brevet ?
Mme Elisabeth Thouret-Lemaître. Je ne comprends pas votre deuxième point.
Pr Philippe Amouyel. Si l'on prend l'exemple de Myriad Genetics , aujourd'hui des sociétés interdites en France, mais qui ont pignon sur rue sur le web et pratiquent des diagnostics de BRCA1, BRCA2, ne me semblent pas payer quelque redevance que ce soit à Myriad Genetics .
Mme Elisabeth Thouret-Lemaître. J'ignore si Myriad Genetics exerce une surveillance totale de ce qui se passe.
M. Philippe Amouyel. C'est un exemple, mais il y en a des centaines d'autres. Ainsi sur le diabète existe-t-il plus de 60 gènes testables et sur Alzheimer on arrive à 25. Il y en a vraiment beaucoup. Cela va nécessiter une surveillance globale. Comment protéger ces découvertes ?
Mme Elisabeth Thouret-Lemaître. Si Myriad Genetics le juge utile, je pense qu'elle poursuivra en contrefaçon. Mais elle attend surtout une décision très importante aux États-Unis, pour voir ce qu'elle peut faire. En Europe, il y a déjà eu une limitation très importante de ces revendications de brevets. Je pense que la surveillance est en cours et que peut-être on attend un peu. Il existe peut-être des accords qu'on ignore, des licences, même pas chères.
M. Maurice Cassier. Je pense qu'en Europe, Myriad Genetics s'est vu rétablir partiellement ses brevets en 2008 par l'Office européen des brevets. Mais ce rétablissement est très problématique. L'application des revendications est très difficile à faire, puisque c'est un certain type de mutations qui a été reconnu, et que pour pouvoir reconnaître ce type de mutation, il faut déjà pratiquer le test. Il faut voir aussi qu'en Europe, Myriad Genetics n'a rien réclamé, n'a poursuivi personne. Pourquoi ? Parce qu'il y a eu un mouvement social extrêmement important de la part des professionnels de santé, des instituts anti-cancéreux, des hôpitaux, des laboratoires publics, des sociétés de génétique qui ont refusé ces brevets. Et donc Myriad Genetics ne poursuivra personne en Europe. En plus, il y a la menace d'une éventuelle licence obligatoire. Donc ils ne feront rien.
Aux États-Unis, s'il s'avère qu'un des acteurs concentrait un grand nombre de tests, des industriels pourraient éventuellement décider d'aller le voir. Ils ne s'en sont pas privés dans le passé, en allant voir un certain nombre de laboratoires hospitaliers pour demander la fermeture de leur service de tests ou pour leur imposer des licences.
Mme Audrey Aboukrat. Je souhaiterais répondre à la première question du Pr Philippe Amouyel. Il est vrai que notre propos concernait essentiellement les brevets sur les gènes, mais effectivement il y a d'autres types de brevets qui peuvent intéresser la médecine personnalisée. Aux États-Unis, des brevets ont été délivrés pour des médicaments « ethniques » et « raciaux ». C'est notamment le cas du BiDil, un médicament censé traiter l'insuffisance cardiaque et spécifiquement destiné à la population afro-américaine aux États-Unis. D'autres brevets sont donc susceptibles d'être associés à la médecine personnalisée, en sus des seuls brevets sur les gènes humains.
Mme Cécile Tharaud. Pour répondre à la question du Pr Philippe Amouyel, on demande nombre de licences à des sociétés américaines de services de diagnostic sur des séquences de gènes. Ces sociétés affichent, y compris dans la documentation financière, que 30 % à 60 % de leur marché potentiel, notamment européen, est « cannibalisé » par les structures hospitalières. Elles l'affichent d'autant plus tranquillement qu'il leur faudrait attaquer en contrefaçon, si tant est qu'une telle approche soit éthiquement recevable, autant de centres hospitaliers que de services pratiquant le diagnostic. Donc économiquement, ce n'est pas possible. Un lien s'est fait naturellement entre les équilibres économiques et un certain nombre de questions éthiques.
M. Vincent Fert, directeur général de Qiagen Marseille . En tant que représentant de l'industrie, je voudrais défendre les brevets sur le diagnostic. Pour un industriel, un brevet, c'est une façon de maîtriser son retour sur investissement. C'est le principe, que ce soit pour un médicament, les technologies de l'information, ou dans d'autres secteurs industriels. En ce qui concerne un test de diagnostic, la question est de savoir s'il nécessite un investissement particulier pour être utile au patient. Le débat ne portait pas sur ce point-là et donc et je voudrais le recentrer. On a l'impression qu'un test de diagnostic, une séquence par exemple, s'apparente à une suite de mots, et on imagine que Gutenberg aurait breveté l'impression. Mais en réalité ce n'est pas du tout cela. On ne peut pas breveter une suite de signes pour ensuite récupérer des royalties sur le dos des systèmes de santé.
En tant qu'industriels, nous développons des tests, nous démontrons leur validité clinique, nous explorons leur sensibilité, leur spécificité, nous regardons quel est le taux de faux positif et de faux négatif. Un faut négatif pour des mutations EGFR, c'est un manque de chance pour un patient. Nous insérons ces tests dans des systèmes automatisés de façon à ce qu'ils puissent être utilisés à des coûts raisonnables par le système de santé. On nous demande, à juste titre, notamment aux États-Unis, de démontrer ces performances dans des essais prospectifs. On se rapproche du coût de développement du médicament. C'est pourquoi je pense qu'attaquer le droit des brevets sur le diagnostic est dangereux pour l'innovation et pour le patient.
Pr. Philippe Amouyel. Les points qui ont été évoqués ne sont pas de cette nature. Aujourd'hui, vous pouvez envoyer de l'ADN au Beijing Institute of Technology à Pékin, ils le séquenceront, moyennant un coût qui n'est pas encore raisonnable, mais qui le sera bientôt. Je l'aurai donc à ma disposition, en tant qu'individu, et j'irai consulter mon médecin. Donc l'acte du diagnostic que vous avez évoqué sera finalement dilué. Quel sera le poids du brevet déposé sur la maladie d'Alzheimer par exemple, par rapport à quelqu'un qui aura sa séquence et qui ira directement lire cela sur son ADN pour 100 dollars et un micro-ordinateur ?
M. Vincent Fert. Est-ce nouveau ? Est-ce inventif ? Cela peut-il être appliqué industriellement ? Je crois que le droit des brevets est extrêmement bien fait de ce point de vue-là.
M. Alain Claeys. L'interprétation du droit des brevets sur le vivant est assez complexe. C'est pour la raison de tous ces débats aux États-Unis ou en Europe. Quand vous analysez les alinéas dans la directive européenne, de nombreux spécialistes expliquent qu'on peut y trouver une contradiction entre un alinéa et un autre. Ce n'est pas si évident que cela, et je comprends votre préoccupation.
Mme Cécile Tharaud. D'une façon générale, la valeur du brevet correspond à la valeur ajoutée que le brevet apporte. Le brevet de gène de la maladie d'Alzheimer est intéressant, parce qu'on a une potentialité de développer de la thérapeutique derrière ; ce n'est pas seulement intéressant parce que la séquence est jolie à regarder.
Mme Elisabeth Thouret-Lemaître. Ce sont les revendications dont j'ai parlé dans la deuxième partie de mon exposé : les substances pour le traitement avec biomarqueur. Comment distingue-t-on le groupe de patients par rapport au groupe de patients antérieur, s'il y a eu un groupe de patients antérieur ?
QUATRIÈME TABLE RONDE :
AVEC QUELS MODELES DE
RECHERCHE-DEVELOPPEMENT ?
Pr Gilles Bouvenot, président de la commission de la transparence, membre du collège de la Haute Autorité de Santé (HAS). Je vais m'exprimer au nom de la Haute Autorité de Santé (HAS), mais également au nom de l'Académie nationale de médecine comme prévu. Je crains d'avoir à donner pour cette dernière table ronde quelques petits coups de canif dans le conte de fée dont on nous parle depuis le début de cette audition. Malheureusement, je n'ai pas pu entendre les préliminaires.
M. Alain Claeys . On a aussi été critique ! (à l'assistance) Avez-vous eu l'impression d'entendre un conte de fée, pour ceux qui étaient là ce matin ? (réprobation) Non.
Pr Gilles Bouvenot. Je vais peut-être requalifier mon sujet en parlant de l'expérience de l'évaluation des thérapies ciblées, et uniquement des thérapies ciblées, par la HAS, entre 2002 et 2013. Cette expérience porte sur 18 produits 245 ( * ) . À propos de ces 18 produits, la commission de transparence n'a donné d'avis défavorable au remboursement en matière de thérapie ciblée qu'une seule fois, pour l'un de ces produits dans le cadre d'un traitement du cancer métastasé du pancréas, au motif que la performance thérapeutique ne consistait qu'en une prolongation de la survie moyenne de trois semaines par rapport au comparateur. Ce produit a d'ailleurs été admis dans une autre indication.
La commission de transparence de la HAS n'effectue qu'une l'évaluation médicotechnique. D'une certaine manière, il faut se réjouir de l'indépendance dans notre pays, entre l'évaluation médicotechnique pure, que je représente aujourd'hui, et l'évaluation médico-économique. Ce n'est pas le cas dans d'autres pays. Le NICE ( National Institute for Health and Clinical Excellence), pour ce qui concerne l'Angleterre et le Pays de Galles, a par exemple refusé le Xalkori au motif qu'il n'est pas coût-efficace. Ce n'est pas encore le cas dans notre pays où il n'y a pas de rationnement, mais seulement des efforts, pas toujours efficaces, de rationalisation.
Tableau récapitulatif des spécialités dites « thérapies ciblées » évaluées par la CT de janvier 2002 à mars 2013
|
Spécialité (dci) |
Indication |
cible |
SMR |
ASMR |
Population cible |
|
ADCETRIS (Brentuximab vedotin) |
Traitement du lymphome hodgkinien (LH) CD30 positif récidivant ou réfractaire chez l'adulte : 1. après greffe autologue de cellules souches (ASCT) ou 2. après au moins deux traitements antérieurs quand l'ASCT ou une poly-chimiothérapie n'est pas une option de traitement. ADCETRIS est indiqué dans le traitement du lymphome anaplasique à grandes cellules systémique (LAGCs) récidivant ou réfractaire chez l'adulte. |
Antigène CD 30 |
Important |
ASMR III dans la prise en charge du lymphome hodgkinien CD30 positif |
245 à 275 patients par an |
|
CAPRELSA vandetanib |
cancer médullaire de la thyroïde agressif et symptomatique. |
Mutation RET positive |
Important |
IV |
70 à 130 patients par an |
|
ERBITUX (cetuximab) |
Traitement des patients présentant un cancer colorectal métastatique avec gène KRAS de type sauvage exprimant le récepteur du facteur de croissance épidermique (EGFR)
|
Gène KRAS |
Important |
- V en 1ère et 2éme ligne métastatique - IV en 3ème ligne métastatique |
7 400 à 12 300 patients par an pour toutes les lignes |
|
GLIVEC (imatinib) |
- Leucémie myéloïde chronique (LMC) à Chromosome Philadelphie positif - Tumeur stromale gastro-intestinale GIST Kit (CD117) positive |
- le Chromosome Philadelphie positif dans LMC - présence du KIT (CD117) pour les GIST |
Important dans toutes les indications |
- I dans LMC phase chronique et II dans les autres phases - I GIST métastatique - III dans le cadre du traitement adjuvant du GIST |
LMC : 500 patients par an GIST : 500 à 600 patients par an |
|
HERCEPTIN (trastuzumab) |
- traitement adjuvant du cancer du sein avec HER positif - traitement néo-adjuvant du cancer du sein avec HER positif |
Surexpression du gène HER2 |
important |
- I dans le traitement adjuvant - IV en traitement néo-adjuvant |
735 à 1 712 patients par an |
|
IRESSA (géfitinib) |
cancer bronchique non à petites cellules (CBNPC) localement avancé ou métastatique avec mutations activatrices de l'EGFR-TK |
Mutation du gène EGFR |
Important |
- V en 1ère ligne métastatique - IV en 2ème ou 3ème ligne métastatique |
5 600 patients par an. |
|
MABTHERA (rituximab) |
Lymphome non-hodgkinien CD20 positif |
Gène CD 20 |
Important |
ASMR I |
5 900 patients par an |
|
TARCEVA (erlotinib) |
Cancer du poumon avec mutation du gène de l'EGFR |
Gène EGFR |
important |
IV en première ligne de traitement du cancer pulmonaire métastatique |
2 300 à 3 200 patients par an |
|
Spécialité (dci) |
Indication |
cible |
SMR |
ASMR |
Population cible |
|
SPRYCEL (dasatinib) |
LMC à chromosome Philadelphie positif |
Chromosome Philadephie |
Important en seconde intention SMR insuffisant en 1ère intention (tolérance : cancers secondaires) |
II en phase chronique de la LMC I en phase accélérée ou blastique Sans objet |
150 à 200 patients par an Sans objet |
|
TARCEVA (erlotinib) |
Cancer du poumon avec mutation du gène de l'EGFR |
Gène EGFR |
important |
IV en première ligne de traitement du cancer pulmonaire métastatique |
2 300 à 3 200 patients par an. |
|
TASIGNA (nilotinib) |
LMC à chromosome Philadelphie positif |
Chromosome Philadephie |
Important en 2ème intention SMR important en 1ère intention |
II en phase chronique de la LMC I en phase accélérée ou blastique ASMR IV |
100 patients par an. 600 à 1200 patients par an. |
|
TYVERB |
- cancer du sein, avec surexpression des récepteurs HER2 (ErbB2) en échec au trastuzumab - cancer du sein avec surexpression des récepteurs HER2 (ErbB2) sans échec au trastuzumab |
Surexpression du gène HER2 |
Important dans les 2 indications |
ASMR III en cas d'échec au trastuzumab ASMR V en situation de non échec au trastuzumab |
4 000 et 4 800 patientes par an. 100 à 500 patientes par an |
|
VECTIBIX (Panitumumab) |
cancer colorectal métastatique exprimant le gène KRAS non muté (type sauvage) |
gène KRAS |
ASMR V |
11700 à 14000 patientes par an |
|
|
ZELBORAF (vemurafenib) |
Mélanome métastatique avec mutation BRAF V600 |
Gène BRAF |
Important |
ASMR III |
1 200 patients par an |
|
XALKORI (crizotinib) |
Cancer du poumon non à petites cellules anaplastic lymphoma kinase (ALK) positif |
Mutation ALK |
Important |
ASMR III |
630 patients par an. |
|
KALYDECO (ivacaftor) |
Traitement de la mucoviscidose chez les patients âgés de 6 ans et plus, porteurs de la mutation G551D du gène CFTR (mutation CFTR-G551D |
Mutation G551D du gène CFTR |
Important |
ASMR II |
Maximum 80 patients |
|
NEXAVAR (sorafenib) |
- Carcinome hépatocellulaire - Carcinome rénal |
Inhibiteur tyrosine kinase et anti angiogène |
Important Important |
ASMR IV ASMR II |
1 500 patients 3 400 patients par an |
|
AVASTIN (bevacizumab) |
- Cancer colorectal - Cancer du sein - Cancer du rein - Cancer Poumon petites cellules avancé ou métastatique - Cancer de l'ovaire |
Anti VEGF |
Important Faible Important Important Important |
ASMR II ASMR IV ASMR V ASMR IV ASMR V ASMR IV |
18 000 patients par an 3 300 patients par an --- 5 500 patients par an 6 500 à 6 900 patients par an 3 460 patients par an |
|
JAKAVI (ruxolitinib) |
Splénomégalie myéloïde |
Inhibiteur JAK / SAT |
Important |
ASMR III |
500 patients par an |
|
SUTENT (sunitinib) |
- Tumeur neuroendocrine pancréas - Cancer du rein avancé - Tumeur stromale G.I. |
Anti angiogène |
modéré Important Important |
ASMR V ASMR III ASMR II |
170 patients par an 5 440 patients par an 230 patients par an |
Les 18 produits représentant concernant des thérapies ciblées ne sont pas tous accompagnés. D'ailleurs le mot « test compagnon » m'a toujours posé problème. Le test compagnon, pour la commission de la transparence de la HAS, c'est le test quasiment fourni par l'industriel dans le cadre d'un kit, afin de permettre la bonne identification du patient présentant la cible. Or ce que la commission observe dans les dossiers, c'est l'existence d'une cible, et par conséquent l'existence d'un produit qui a été mis au point pour toucher la cible. Il ne m'appartient pas de faire cette différence, peut-être byzantine, entre le test compagnon et tout simplement l'existence d'un biomarqueur mis en évidence et qui est utilisé.
Quel est le conte de fée ? On présume d'une meilleure efficacité de ces produits, et on comprend très bien pourquoi. On présume de leur meilleure tolérance, ce qui reste à discuter, j'y reviendrai. On présume éviter grâce à eux de prescrire des traitements inutiles, et alors là on en est à peu près certain, et par conséquent, d'imposer un certain nombre d'effets indésirables, superfétatoires, à des patients. Mais il faut nuancer, en rappelant que tant que ces produits ne seront pas des produits de première ligne, -à supposer qu'ils le deviennent rapidement-, ils interviennent soit en deuxième ligne chez des patients ayant déjà subi les méfaits des chimiothérapies, ou en association avec des chimiothérapies, et on aboutit alors à une sommation des effets indésirables.
Enfin, probablement parce que le traitement est bien ciblé, et que ne reçoivent le traitement que ceux qui sont susceptibles d'y répondre, on arrive probablement, dit-on, et c'est l'élément essentiel du conte de fée, à un coût diminué pour la solidarité nationale. En tant que citoyen, je n'en suis pas persuadé. Pour l'instant, l'objectif est que les thérapies ciblées entrent dans le cadre de ce que nous appelons très improprement la « soutenabilité », c'est-à-dire la possibilité de s'offrir ces produits, et ce, de manière pérenne. J'évoque évidemment la solidarité nationale.
Quelques notions en termes d'efficacité. La commission de transparence a pour mission de noter les produits. Elle note leurs progrès thérapeutiques de 1 à 5 : 1 pour un progrès majeur. 5 pur une absence de progrès. Sur les 18 produits que nous avons évalués, seuls 3 d'entre eux ont eu la note 1, c'est-à-dire progrès majeur. Ainsi pour les 15 autres produits, le progrès, le bénéfice, est assez minime. Il est quelquefois très notable, mais enfin, il n'est pas toujours très important. La prolongation de survie est de l'ordre de deux à trois mois. Notez bien que nous sommes confrontés à un problème : la plupart des dossiers ne nous donnent pas d'idée sur le gain véritable de prolongation de la vie des patients, mais seulement de la survie sans progression.
Ces produits ont reçu une autorisation de mise sur le marché (AMM) de caractère un peu mixte, à la fois une AMM d'organe et une AMM de cible. Deviendra-t-elle un jour une AMM ubiquitaire de cible ? C'est possible, mais nous n'en sommes pas là. Nous en sommes encore à la mixité. Par ailleurs, ces dossiers montrent que la plupart des essais qui ont conduit ces produits à l'AMM ont été interrompus par des comités scientifiques ou des comités d'éthique, considérant qu'à partir d'un certain moment, il n'y avait pas lieu de poursuivre l'essai, et que les patients qui n'étaient pas dans le bon groupe, c'est-à-dire dans le groupe thérapie ciblée, auraient une perte de chance en poursuivant leur participation à l'essai. Pour nous, évaluateurs, ceci obère de manière très notable ce que nous pensons de la qualité des produits et très probablement, sous-estime, on peut l'imaginer et on l'espère, leur efficacité. Cette efficacité est loin d'être aussi importante que le concept pourrait le laisser présager. Nous en sommes absolument désolés, mais c'est ainsi.
Et puis il y a des résistances. Nous les observons aussi bien en cours d'essais que dans la vie réelle. Nos amis scientifiques, des sciences fondamentales, de la génétique, de la génomique, savent bien quelles sont les grandes causes de ces résistances, pas seulement l'existence de cellules stromales qui entourent la cellule cancéreuse, mais aussi peut-être, et c'est là le problème, l'existence d'autres cibles, sur lesquelles on n'intervient pas, et qui pourraient interférer avec la cible sur laquelle on intervient. Soyons humbles, nous observons un certain nombre de biomarqueurs prédictifs, mais la plupart du temps, nous le savons bien, il faudrait des « polymarqueurs », et on n'en dispose pas. Outre qu'on n'intervient pas sur eux, il est probable que certains interfèrent dans le mauvais sens avec ceux sur lesquels on intervient.
Une autre réflexion qu'on peut faire quand on évalue des thérapies ciblées, c'est qu'on aimerait que pour ces thérapies ciblées, comme pour tout traitement onéreux, on dispose de règles d'arrêt précises. Quand doit-on arrêter le traitement ? Quand doit-on penser qu'il n'y a pas lieu de le poursuivre et que le patient n'en tire plus de bénéfices, mais seulement des effets indésirables non négligeables ? On aurait pu croire que les effets indésirables des thérapies ciblées seraient moins importants, moins graves, que les effets indésirables des chimiothérapies, mais ce n'est pas le cas.
Mes sources dans le domaine sont aussi celles dont sont issues les publications de l'INCa, et l'INCa, à juste titre, comme nous, se préoccupe des effets indésirables encore inconnus. Il n'est probablement pas anodin d'introduire dans l'organisme des substances qui agissent sur un récepteur, sur une cible que nous connaissons, mais probablement aussi sur d'autres que nous n'avons pas identifiées, et dont les effets à long terme pourraient être préoccupants. Je ne parlerais pas de bombe à retardement. Ce ne serait pas un coup de canif, ce serait un coup de poignard, et je n'en suis pas là. Mais méfions-nous. Il faut exercer une surveillance, probablement prolongée si tant est que ce type de patients puisse bénéficier une surveillance prolongée quand on prescrit ces thérapeutiques.
Tout n'est donc pas rose dans le domaine des résultats des thérapies ciblées, parce que le profilage des patients est encore insuffisant. Mais il est permis de penser que des progrès arriveront, que des associations de thérapies ciblées verront le jour et se posera le problème de la prise en charge par la solidarité nationale de traitements ciblés associés. On n'en restera pas seulement au traitement avec son test compagnon. On en viendra à un ou deux traitements ciblés associés, avec évidemment les éventuels tests compagnons.
Telle est l'expérience de l'évaluation de ces médicaments. Nous la prenons très au sérieux, nous tentons, autant que faire se peut, de permettre la mise à la disposition aux patients de ces médicaments. Il n'y a pas, dans les avis de la commission de transparence, la moindre arrière-pensée médico-économique. Non pas qu'il ne faille pas en avoir une, mais il n'appartient pas à la commission de transparence de la HAS d'en avoir. Ainsi pour ce qui concerne la France, nous n'avons pas à rougir : 18 médicaments présentés, 18 acceptations. Alors bien sûr, comme les notes que nous attribuons ne sont pas toujours très valorisantes, il en résulte qu'en aval de nous, les négociations de prix par le comité économique des produits de santé peuvent être difficiles, pour ne pas dire très difficiles.
En tout cas, l'objectif de l'évaluation par la commission de transparence est de donner à ces produits la note qu'ils méritent, ni plus, ni moins, sans se préoccuper, en aucune manière, des conséquences économiques, ou comptables, de nos avis.
M. Alain Claeys. Cette intervention est importante. Certains veulent-ils réagir ? Non. Alors continuons.
Pr François Ballet, président du comité R&D de Medicen Paris région . Je traiterai des modèles d'organisation qu'on peut proposer pour favoriser le développement de la médecine personnalisée. Mon propos sera orienté du point de vue des industriels. Il est important de rappeler que le champ de la médecine personnalisée est extrêmement vaste. Il y a l'aspect médicament mais aussi l'aspect détermination de facteurs de risque chez le sujet sain pour permettre de mettre au point des stratégies de prévention adaptées et individualisées, et de pouvoir faire un diagnostic précoce, voire ultra-précoce dans les populations à risque qu'on aura ciblées. On a également la possibilité de stratifier les malades pour mieux déterminer leurs facteurs de risque, leurs facteurs de gravité et enfin la possibilité de déterminer la meilleure molécule pour le patient, en termes d'efficacité et de toxicité.
Deuxièmement, j'évoquerai les bases de la susceptibilité individuelle, qui expliquent mieux les différents types d'activité qu'on doit inclure pour développer cette activité. Prenons par exemple le problème de la toxicité du médicament, les effets secondaires. On sait que l'un des problèmes majeurs actuellement dans le cadre du développement des médicaments est la toxicité idiosyncratique, la toxicité rare, qu'on n'est pas capable de détecter, ou très difficilement, dans les essais cliniques. Sur l'animal, c'est impossible à détecter. Très souvent, cette toxicité, compte tenu de sa très faible incidence, n'apparaît que lorsque la molécule est sur le marché.
Il est extrêmement frustrant, pour un laboratoire pharmaceutique qui développe une molécule très active, d'être obligé d'arrêter son développement en raison d'un effet indésirable chez 1 % ou 2 % des patients, alors que chez 98 % des patients, la réponse est très bonne et la tolérance est parfaite. On perçoit bien l'intérêt de détecter à l'avance les patients à risque, afin de les exclure du traitement. On connaît mieux les bases de cette susceptibilité. Dans un premier temps, on a pensé que les facteurs génétiques pouvaient l'expliquer. On s'aperçoit maintenant que ce n'est pas le cas. Les polymorphismes génétiques peuvent expliquer une partie très faible de cette susceptibilité, mais le reste de la susceptibilité dépend de facteurs externes, environnementaux, particuliers à l'hôte, la maladie sous-jacente, son immunité, son micro-biote intestinal, etc .
Pour pouvoir prédire le risque in fine , il faudrait non seulement des tests biologiques, mais également d'autres paramètres, bien plus difficiles à maîtriser, et aussi des algorithmes permettant réellement de prédire le risque. Ce qui paraissait relativement simple au début, quand on a pu séquencer le génome, est en réalité bien plus complexe. La toxicité des médicaments est un bon exemple. C'est exactement la même chose pour la susceptibilité des maladies. La maladie apparaît lorsqu'il y a une convergence de facteurs individuels, génétiques, mais également environnementaux, externes, etc.
Ces données permettent de comprendre deux notions importantes. Si l'on veut encourager le développement de la médecine personnalisée, premièrement, il faut favoriser les approches multidisciplinaires, rapprocher de nombreuses compétences permettant de développer vraiment une approche de médecine translationnelle, c'est-à-dire multidisciplinaire, centrée sur le malade, en rapprochant les chercheurs, les cliniciens et les industriels. C'est un élément extrêmement important pour aborder des problématiques aussi complexes que celle-là.
Deuxièmement, on s'aperçoit que le champ industriel et les industries qu'il est nécessaire de mobiliser pour développer des projets de médecine translationnelle, sont finalement extrêmement divers. Sans diagnostic, on ne peut pas faire de médecine personnalisée, mais il est des domaines industriels auxquels on ne pense pas forcément, et qui sont également extrêmement importants.
Par exemple, au niveau du dispositif médical, l'une des formes particulières de la médecine personnalisée, est d'être capable d'adapter un les doses d'un traitement, aux paramètres de logique du patient. Le pancréas artificiel, qui est un système permettant de délivrer des doses d'insuline parfaitement adaptées à la glycémie du patient, est une approche thérapeutique de médecine personnalisée. Les industries qui développent ces systèmes ne sont ni les laboratoires pharmaceutiques traditionnels, ni les laboratoires de diagnostic, mais les laboratoires de dispositifs médicaux. Les technologies de l'information et de la communication, et l'e-santé sont aussi très importantes pour le développement de la médecine personnalisée. Il en va de même pour les d'algorithmes prédictifs de risques, c'est encore une autre industrie qui devra les développer.
La chirurgie personnalisée est en train de se développer, c'est un domaine auquel on ne se réfère pas. À l'IRCAD (Institut de recherche contre les cancers de l'appareil digestif à Strasbourg), on développe des méthodes d'imagerie virtuelle permettant d'étudier, avant d'opérer l'anatomie spécifique, la vascularisation de du foie par exemple du patient, de façon à pratiquer des hépatectomies réglées bien plus précises qu'avant. L'imagerie médicale, et l'industrie du numérique, sont extrêmement importantes pour développer ces approches.
Un autre domaine industriel auquel on ne pense pas est celui de l'industrie agroalimentaire. Un concept assez révolutionnaire est en train de se développer actuellement, c'est le micro-biote intestinal comme facteur prédictif de maladie et en même temps comme paramètre important susceptible de moduler l'immunité. La flore intestinale de chaque personne est différente. On s'aperçoit qu'en fonction de sa composition, on peut être sensible à telle ou telle maladie. Cela peut donc constituer non seulement un biomarqueur de risque, mais on peut imaginer qu'en modulant ce micro-biote intestinal, on pourra éventuellement prévenir l'apparition de certaines maladies, voire éventuellement les traiter.
Or le développement de pro-biotiques, c'est-à-dire d'agents alimentaires susceptibles de modifier la flore intestinale, particulière à chaque malade, est une approche qui est actuellement développée par Danone Research , dont le centre de recherche est en région parisienne, en collaboration avec l'Institut national de la recherche agronomique (INRA), dans le cadre de grands projets européens. Ce sont également des approches de médecine personnalisée extrêmement intéressantes.
Je représente le pôle Medicen Paris région, un pôle de compétitivité qui a pour objectif de fédérer les différents acteurs de l'innovation dans une région donnée. Un pôle de compétitivité est un acteur très important pour développer la médecine personnalisée, parce qu'il est précisément capable, et c'est son objectif, de rapprocher les différents acteurs de la recherche, les chercheurs académiques, les cliniciens, les industriels grands et petits, et également de rapprocher différents champs industriels qui n'ont pas forcément l'habitude d'échanger.
Au moment où la médecine personnalisée s'est développée, il n'était pas si simple de faire discuter le monde du diagnostic avec le monde de la pharmacie, le monde de la pharmacie avec l'industrie du dispositif médical, ou des technologies de l'information et de la communication, ou avec l'industrie agroalimentaire. En réalité, on s'aperçoit que c'est la mise en réseau de tous ces mondes qui permettra de parvenir à des solutions innovantes, pour vraiment réaliser la révolution de la médecine personnalisée. Ils représentent des opportunités de business , de création et développement d'entreprises extrêmement importants, très largement au-delà du cercle classique de la médecine personnalisée qui est celui du médicament et de la pharmacogénétique.
M. Alain Claeys. Pr Gilles Bouvenot, on voudrait avoir une précision sur le problème de notation. Quel est l'objet de la notation ? Vous avez dit de 1 à 5. Mais lorsque vous analysez un test, soit il est efficace, soit il ne l'est pas, soit il comporte des risques, soit il n'en comporte pas, soit on le met sur le marché, soit on ne peut pas. Cela ne sert pas uniquement à fixer un prix.
Pr Gilles Bouvenot. Nous ne revenons jamais sur les autorisations de mise sur le marché (AMM) qui ont été octroyées à ces produits, quelques mois auparavant, par l'EMA (Agence européenne des médicaments). Il n'en est pas question et ce n'est pas notre mission. Il nous est demandé essentiellement trois choses. La première, c'est de préciser l'intérêt thérapeutique du nouveau médicament, en le classant dans l'une des catégories suivantes : intérêt thérapeutique important, modéré, faible ou insuffisant. On ne peut proposer au remboursement des produits d'intérêt thérapeutique insuffisant.
Nous quantifions donc cet intérêt thérapeutique, en sachant que le taux de remboursement est corrélé au niveau d'intérêt thérapeutique attribué (important, modéré, etc. ) Á noter toutefois qu'en ce qui concerne les thérapies ciblées, sur les 18 produits, 17 produits tiennent au cancer et aux hémopathies malignes et un tient à la mucoviscidose. Il s'agit donc d'affections de longue durée dont la prise en charge par la solidarité nationale est de 100 %.
Notre deuxième mission est au moins aussi importante, en particulier pour les thérapies ciblées, puisqu'il nous revient de dire quels progrès ces nouveaux produits apportent par rapport à ceux dont on dispose déjà. Pour ce faire, nous attribuons une note de progrès thérapeutique allant de 1 à 5. 1 : c'est un progrès majeur, une vraie révolution thérapeutique comme, par exemple l'a été le Glivec dans la leucémie myéloïde chronique. Et quand nous attribuons 5, c'est qu'il n'y a pas de progrès thérapeutique.
La fixation du prix, à la suite de négociations entre les firmes et le comité économique des produits de santé (CEPS), dépend d'un certain nombre de facteurs, dont le niveau de note que nous avons attribué. Le Comité économique des produits de santé est le seul à pouvoir vous dire la pondération qu'il accorde à la note donnée par la commission de la transparence dans le contexte général de fixation des prix. En matière de thérapies ciblées, seuls 3 produits sur les 18 examinés, ont été qualifiées de progrès majeurs, avec la meilleure note.
Enfin nous avons une troisième mission, non négligeable elle aussi. Les pouvoirs publics nous demandent en effet de déterminer la population cible des patients justiciables des nouveaux traitements. Il nous revient donc de déterminer si tel nouveau produit va intéresser 200, 500, 1 000 ou 10 000 patients. À la lecture de la liste des populations cibles, on voit qu'elles dépassent rarement quelques milliers de patients. Dans le cadre d'accord prix-volume entre les pouvoirs publics et les firmes pharmaceutiques, cette estimation a son importance sur la fixation du prix.
M. Alain Claeys. Quand vous proposez la note 5, proposez-vous aussi le déremboursement ?
Pr Gilles Bouvenot. Non : en matière de progrès thérapeutique (ASMR), la note 5 indique seulement que le produit n'apporte pas de progrès et ne doit donc pas être à l'origine de surcoût par rapport aux produits qui existent déjà. Encore une fois, j'ai occulté délibérément la première partie de notre mission liée à l'intérêt thérapeutique, considérant qu'elle n'est pas vraiment concernée dans le cadre des traitements ciblés, qui pour l'instant, touchent les malades ayant un cancer ou une mucoviscidose. Ces produits, il les leur faut, sauf si évidemment ils avaient obtenu des AMM indues, ce qui n'est jamais arrivé. C'est la note de progrès thérapeutique, - celle que vous lisez quelquefois dans la presse sous le nom d'amélioration du service médical rendu (ASMR) - qui compte. C'est elle qui a de l'influence sur la fixation du prix et influe aussi ceux qui lisent les avis de la HAS. Quand un produit a obtenu la note 1, cela signifie qu'il est un progrès majeur. Quand il a eu 5, cela signifie qu'il n'est sûrement pas le meilleur.
M. Pierre Attali, directeur général délégué, stratégie & affaires médicales, BioalliancePharma. Combien de produits ont eu les ASMR notées 5, quel en est le pourcentage ? Finalement, comme vous l'avez expliqué fort justement, l'ASMR est très fortement reliée au prix. Pour faire simple, avec une ASMR 5, vous n'avez pas de prix, et tout le coût de votre développement tombe à l'eau.
Pr Gilles Bouvenot. Il est extrêmement difficile de répondre à la question que vous me posez. Pourquoi ? Je vais être bref, mais j'aimerais être clair. Il est fait de nos avis un certain nombre d'usages que nous ne contrôlons pas. Je vais prendre un exemple trivial : en fonction des notes que nous attribuons, les produits peuvent être ou pas inscrits sur la liste en sus. Mais il nous faut absolument résister à l'idée que si nous ne donnons pas une très bonne note à tous ces produits, il y aura quelques difficultés dans les négociations de prix ultérieurement. Nous devons rendre des avis purement scientifiques.
Une mission médico-économique a été attribuée à la HAS. Le Parlement nous a fixé le seuil du 1 er septembre 2013 pour rendre désormais un double avis pour ce type de produits : un avis de la commission de la transparence qui ne sera que médicotechnique et un avis d'une commission médico-économique. Les deux avis iront conjointement au comité économique des produits de santé.
Votre question c'est probablement : quand la commission de la transparence attribue une ASMR de niveau 5, le produit est-il mort ? Je ne le crois pas. Quand on donne une ASMR 5, c'est-à-dire pas de progrès thérapeutique, le code de la sécurité sociale est clair. Quand la commission de transparence n`attribue pas une note de progrès, autrement dit quand elle donne 5, l'introduction du médicament est possible, mais elle doit induire des économies pour l'assurance maladie. Pour ce type de produits, quand nous donnons 5, cela signifie que l'industriel ne pourra pas exiger un surcoût par rapport à la concurrence, mais je ne suis pas sûr que le prix fixé l'empêche de le mettre à la disposition des patients.
M. Jean-Sébastien Vialatte. Nous allons poursuivre avec le Pr Philippe Monteyne, vice-président R&D France de Sanofi, qui intervient pour Les entreprises du médicament (LEEM).
Pr Philippe Monteyne, pour Les Entreprises du Médicament (LEEM), vice-président R&D France de Sanofi . En préambule, j`indiquerai qu'en vous donnant le point de vue de l'industrie, je vous décevrai peut-être, ou vous rassurerai, je l'espère, dans le sens où je ne vais pas vous apporter des éléments très nouveaux ou très différents de ce que vous avez pu déjà entendre... En particulier, je vous confirmerai plutôt que le point de vue de l'industrie n'est pas si loin du monde académique ou du monde de la science.
Voltaire disait que « les médecins administrent des médicaments dont ils savent très peu, à des malades dont ils savent moins, pour guérir des maladies dont ils ne savent rien ». Ce à quoi l'on veut parvenir aujourd'hui, c'est exactement l'inverse, c'est comprendre beaucoup mieux les maladies, les malades, et bien entendu les médicaments qu'on administre, très tôt dans le développement. Nous prônons de plus en plus ce que nous appelons chez Sanofi une médecine des « 4P » : préventive, prédictive, personnalisée, participative. C'est une vision à long terme qui couvre tout le modèle R&D, et c'est ce vers quoi nous voulons aller.
Une médecine préventive, car on a déjà pu prévenir de nombreuses maladies infectieuses, mais progressivement on doit aussi pouvoir, dans l'industrie, participer à la prévention de maladies non infectieuses. Certaines maladies font le lien entre les deux. Par exemple, le vaccin anti-HPV protège contre des lésions précancéreuses, et de façon ultime, contre le cancer de l'utérus. Il y a donc des liens entre l'infectieux et le non-infectieux, et progressivement on doit pouvoir, y compris pour un industriel du médicament, se diriger vers une prévention de pathologie non infectieuse.
Une médecine prédictive, parce que c'est très important de prédire le risque et d'intervenir en fonction du risque prédit. Une médecine personnalisée, et vous l'avez déjà entendu, avant d'arriver à la médecine personnalisée, beaucoup d'entre nous pensent qu'il y a une étape intermédiaire, celle de la médecine stratifiée. Nous allons tous dans le sens d'une médecine personnalisée, qui est probablement l'étape ultime. Une médecine participative, parce que cela ne peut se faire qu'avec la participation du malade à la prise en charge de sa santé.
En ce qui concerne la médecine stratifiée, nous travaillons de plus en plus avec ce qu'on appelle la médecine translationnelle, c'est-à-dire en ayant accès à du matériel humain, en rapprochant le lit du malade de la paillasse, et inversement. Depuis les années 90, sans doute dans le monde scientifique en général, y compris dans l'industrie, on a attaché beaucoup trop d'importance aux modèles animaux. On a cru que les modèles de souris résoudraient tous les problèmes et on s'est lancé dans de grands développements, en sorte que parfois on se retrouvait en phase 3 en ne connaissant pas assez le mode d'action du médicament, parce qu'une trop grande partie de cette connaissance était basée sur des modèles animaux. De plus en plus, on se rend compte qu'il faut aller vers du matériel humain, il faut beaucoup plus tôt comprendre la maladie chez l'homme.
On ira progressivement vers une médecine stratifiée, de l'aval, c'est-à-dire du traitement symptomatique, vers l'amont, c'est-à-dire vers le mode d'action des maladies, et finalement vers la génétique quand il y a une base génétique. En agissant de la sorte, on doit découper les maladies. C'est de cette manière qu'on s'aperçoit que des maladies comme le diabète par exemple, ne sont probablement pas une seule maladie ; dans le cancer il y a probablement plus de deux cents maladies, et c'est aussi le cas des maladies rares. Par exemple, un traitement contre la mucoviscidose a été découvert l'année dernière, mais il y a plus de 500 mutations, et ce traitement ne s'adresse qu'à 4 % des 70 000 patients atteints de mucoviscidose dans le monde.
On perçoit bien qu'il faudra aller vers une médecine fortement stratifiée, avec des outils de médecine translationnelle, avec un accès à des matériels humains, et avec des biomarqueurs. Pour les industriels, l'accès à des matériels humains signifie qu'ils doivent se rapprocher beaucoup plus du monde académique et du monde hospitalo-universitaire, en particulier pour accéder à du matériel humain, mais aussi à l'innovation. C'est pourquoi, on évoque de plus en plus l'innovation ouverte, mais certaines entreprises, ont historiquement un système d'innovation beaucoup trop fermé. Cela n'implique pas que nous voulons externaliser notre recherche. Nous continuerons à faire de la recherche en interne, mais nous devons être beaucoup plus ouverts sur le monde académique et sur le monde hospitalo-universitaire, en particulier très tôt dans le développement.
D'où l'intérêt de l'écosystème, des regroupements. En France, les regroupements sont plus que bienvenus. On vient d'avoir l'exemple avec le Pr François Ballet, de Medicen Paris région. Les pôles de compétitivité sont un aspect très important de l'écosystème. Au niveau des regroupements hospitalo-universitaires, en région parisienne, on a la chance d'avoir l'Assistance Publique- Hôpitaux de Paris (AP-HP), qui est un bel exemple.
Quant à Sanofi, où nous sommes en train de réorganiser notre recherche /développement (R&D),a première raison de cette réorganisation tient à la baisse de productivité de la R&D, dans la santé et dans l'industrie en général. Nous considérons que pour un dollar investi, nous avons 70 cents de retour sur investissement. Évidemment, ce modèle n'est pas viable à long terme. Face à cette réalité, on peut améliorer la productivité en diminuant l'attrition. C'est ce qui explique notre intérêt pour la médecine translationnelle. Nous voulons mieux comprendre les malades et les maladies, et avoir accès à du matériel humain pour mieux percevoir les risques dès le début du développement.
Dans l'équation de la productivité, cela jouera essentiellement sur les chances de succès, car ce sont tous les échecs qui pèsent énormément sur les retours sur investissement, et coûtent très cher. Si on n'a moins d'attrition, par définition le retour sur investissement va augmenter. Il nous faut donc diminuer l'attrition, être plus efficaces et nous regrouper. Sanofi est très ancré en France et nous avons la volonté de le rester. La moitié de la R&D de Sanofi regroupe 6 000 collaborateurs en France. Nos unités thérapeutiques sont regroupées sur un minimum de sites en fonction de l'écosystème. Donc dans l'industrie, on s'adapte à ce modèle.
Pour conclure, je citerais sept défis majeurs, et éventuellement obstacles, que nous avons identifiés pour ce modèle R&D à long terme, en particulier pour la médecine stratifiée et la médecine personnalisée.
Premier défi, il faut effectuer des études cliniques dès la phase zéro, avant même d'avoir un médicament en développement, pour mieux comprendre le malade, pour avoir accès à des échantillons. Et il faut d'ailleurs que le patient donne son autorisation pour qu'on utilise le matériel de façon assez large.
Deuxième défi, il faut une épidémiologie, une bonne connaissance des populations de malades. Le Pr Gilles Bouvenot parlait de profilage des patients. C'est exactement ce à quoi je fais allusion.
Le troisième défi, c'est la propriété intellectuelle. Vous en avez largement débattu dans la troisième table ronde. La propriété intellectuelle doit couvrir des aspects beaucoup globaux que ce qui est couvert aujourd'hui, si l'on veut une approche globale.
Le quatrième défi, ce sont les difficultés pour un grand industriel de faire face à des exigences réglementaires qui sont malheureusement trop souvent différentes entre la Food and Drug Administration (FDA) et l' European Medicines Agency (EMA), pour ne citer que celles-là, malgré toutes les communications qui sont mises en place entre les deux agences.
Cinquième défi, il y a un modèle économique qui n'existe pas aujourd'hui. Si l'on veut faire une médecine plus personnalisée, avec une prise en charge globale, qui ira de la prévention, au diagnostic, au traitement, et dans ce traitement une partie classique sur prescription, une partie prise en charge du malade, avec des aspects de santé grand public hors prescription, cela doit former un ensemble devant faire partie d'un modèle économique qui n'existe pas aujourd'hui. Il faudra sûrement y travailler.
Le sixième défi est très français, c'est notre fameux « principe de précaution » qui constitue un défi majeur. Il faut absolument construire et bâtir la confiance dans le médicament, et avoir une confiance dans les traitements et les vaccins qui sont développés. Nous connaissons tous l'exemple typiquement français du vaccin contre l'hépatite B, dont la réputation souffre encore aujourd'hui de ce qui s'est passé dans les années quatre-vingts dix.
Le septième défi, provient des systèmes de remboursement dans le monde entier. Même si le président de la commission de la transparence de la HAS a bien expliqué que cela n'entrait pas dans sa fonction, on y a fait beaucoup allusion. Les systèmes de remboursement dans le monde entier datent de l'après-Seconde Guerre mondiale, et ne sont sûrement pas adaptés à une médecine du futur, laquelle doit être une prise en charge plus globale du malade, où le malade fait partie du modèle économique. Il faudra repenser la façon dont on rembourse par une approche globale, sur la base d'une performance et d'une qualité, plutôt que sur un médicament unique. L'industriel, nous l'espérons, aura alors un apport bien plus global.
Mme Corinne Blachier-Poisson, directrice de l'accès au marché et des affaires publiques d'Amgen . Je représente un laboratoire américain leader des biotechnologies et un des rares à avoir mis sur le marché un exemple de médicament de médecine personnalisée.
Je commencerai par donner un exemple pratique illustrant à la fois le succès et les échecs qu'on peut rencontrer dans le développement de ces médicaments. Cette molécule, dont le nom ne vous intéressera sans doute pas, appartient à une classe d'anti-cancéreux. Comme il a été expliqué, la grande majorité des médicaments utilisés dans la médecine personnalisée le sont uniquement pour le cancer, à une exception près. La molécule dont je souhaite vous parler, a commencé à être développée dans le cancer colorectal métastatique. L'essai clinique laissait apparaître une réponse très variable des patients. À la fin de l'analyse, les statisticiens se sont rendu compte qu'il y avait vraiment de grandes différences entre les répondeurs et les non-répondeurs. A posteriori, ils ont analysé le lien entre certains biomarqueurs et la réponse au traitement, et se sont alors aperçus qu'on pouvait stratifier les patients en deux groupes : ceux dont le gène KRAS était non muté étaient répondeurs, et ceux dont le gène KRAS était muté ne répondaient pas du tout au traitement, n'avaient que des effets secondaires et ne tiraient aucun bénéfice de ce médicament.
Ces résultats ont conduit à l'enregistrement d'un essai clinique basé sur des données d'analyse a posteriori , ce qui n'est pas classique dans le développement des médicaments. Habituellement, on part d'une hypothèse, que l'on veut tester et si l'on ne vérifie pas cette hypothèse, on n'a pas le droit de travailler les données a posteriori . Cela soulève déjà la question de l'analyse statistique des essais de ces médicaments dans la médecine personnalisée. On est obligé de tester plusieurs biomarqueurs pour savoir quel est celui qui va a posteriori avoir un lien.
Dans un deuxième temps, on a suivi la recommandation de cliniciens spécialistes de cancers de la tête et du cou, où il existe très peu de médicaments disponibles, et qui avaient testé en phase 2 ce médicament, considérant qu'il avait une efficacité. De nouveau, on a effectué une analyse des statistiques des essais cliniques disponibles. En fait, il s'est avéré impossible de trouver un biomarqueur qui soit prédictif et permette d'obtenir une réponse cliniquement significative. On a donc arrêté le développement dans cette indication, à la grande déception de certains cliniciens. Clairement, un besoin existait, et il semblait qu'il y avait un bénéfice, mais on n'était pas capable de trouver le gène permettant d'identifier les répondeurs. C'était un exemple pour montrer qu'en effet, l'histoire n'est pas un conte de fée, pour reprendre les propos du président Gilles Bouvenot.
La médecine personnalisée peut certainement bien nous aider et faire évoluer notre modèle de R&D, mais elle ne règlera certainement pas tous les problèmes, les principaux, étant les coûts. Le coût de développement d'un médicament a énormément augmenté ces deux dernières décennies. Il y a vingt ans, on disait qu'il fallait en moyenne 250 millions de dollars pour mettre un médicament sur le marché. Aujourd'hui on parle d'1 milliard de dollars. Ce sont toujours des calculs assez difficiles à établir, mais cela donne à peu près une idée des proportions.
Naturellement, au sein des laboratoires, toutes les directions R&D cherchent à diminuer ce coût. Cela passe par une accélération des essais cliniques, par une diminution de l'attrition comme cela a été expliqué en s'adressant tout de suite à la population chez laquelle on trouvera le plus grand bénéfice.
La médecine personnalisée paraît être la réponse idéale pour faire évoluer le modèle. D'une part, elle permettra de s'adresser à des pathologies souvent rares, ou peu fréquentes, pour lesquelles il existe un fort besoin médical et peu de solutions thérapeutiques. C'est vrai qu'en général on préfère arriver sur un marché dans lequel on trouve peu de comparateurs, car on a plus de chances de démontrer un plus grand bénéfice. D'autre part, cette médecine permettra de s'adresser à des prescripteurs et des médecins moins nombreux, pour lesquels on pourra mieux encadrer l'information et la pharmacovigilance. C'est important car on maîtrise certainement plus la pharmacovigilance d'un médicament quand il est prescrit par 1 000 médecins que par 60 000 médecins généralistes. C'est donc aussi pour nous, une façon de diminuer le risque d'une mauvaise utilisation, et de minimiser la toxicité.
Cependant la médecine personnalisée n'a pas que des avantages. Elle soulève aussi certaines difficultés que j'ai listées, sans être exhaustive. Au niveau du recrutement des médecins qui vont mener les études, il faut bien voir que ces médecins ne seront pas seuls, ils seront dans des centres. Comme il a été dit, ces centres regroupent de nombreuses compétences et doivent donc être multidisciplinaires. La recherche clinique sera donc restreinte à certains centres apportant toutes les garanties en termes de respect des protocoles de recrutement des essais, lesquels sont extrêmement stricts. En cas de non-respect de ces protocoles, les patients ne sont pas inclus dans les essais, et même, les centres peuvent être exclus des essais cliniques à venir.
Au niveau du recrutement des patients, on se trouvera avec des pathologies où il y a peu de patients à recruter. Il est plus facile de faire des essais dans l'hypertension où, en France, dix millions de personnes sont concernées, que dans le cancer de l'ovaire par exemple, qui concerne 9 000 personnes sur une année, réparties dans différents sous-groupes. De plus, nous sommes plusieurs industriels à chercher dans ces cancers assez rares. Il existe donc une concurrence dans le recrutement de ces patients.
L'aspect du co-développement en parallèle avec le test diagnostic est très nouveau pour nous. Nous sommes un peu dans une situation de dépendance par rapport aux tests. Il faut que le test soit bien fait si l'on veut que le bon traitement soit donné au bon patient. On le savait déjà avec certains médicaments basés sur les tests, mais les biomarqueurs demandent une certaine compétence. En France, dans le domaine du cancer, on a la chance que tout cela soit bien géré par l'INCa. C'est fait par des plateformes agréées et contrôlées.
Par exemple, dans le test du gène KRAS, on n'a eu aucun problème de qualité en France. En revanche, dans d'autres pays européens, c'est un vrai souci et cela peut être un obstacle à notre AMM. Les autorités européennes, quand elles ont évalué notre médicament, étaient très réticentes à donner une AMM, parce que certains États membres faisaient état d'une mauvaise qualité des tests dans leur pays. Finalement, l'autorisation de mise sur le marché d'un médicament devient liée à celle d'un test diagnostic. La majorité des laboratoires, à de rares exceptions près, ne conduisent pas à la fois une activité diagnostique et une activité pharmaceutique. Ce n'est pas du tout le même mode de fonctionnement. Comme l'a expliqué le Pr Philippe Monteyne, ce nouveau mode de fonctionnement nous oblige à travailler de manière pluridisciplinaire, ce qui est très nouveau pour l'industrie pharmaceutique.
Ensuite, se pose la question de la nécessité d'identifier les bons gènes qui demande de faire de l'épi-génétique et donc d'avoir accès à de larges cohortes de patients. Il n'est pas toujours facile de disposer de bonnes qualités de données. Il se trouve que mon laboratoire a décidé d'acquérir une base de données islandaise qui réunit tous les génotypes et les phénotypes de toute la population islandaise. L'objectif est de pratiquer toutes les analyses de liens entre la génétique et la maladie. Clairement, il faut donc accéder à certains types de données de l'épi-génétique qui ne sont pas si nombreuses, ni si faciles d'accès.
Je terminerai sur un point déjà été mentionné. En matière de thérapie ciblée, une fois que nous obtenons l'AMM pour ces médicaments et que nous sommes passés par la commission de la transparence, le sort du médicament est lié à celui du test. Or, le remboursement et le prix de ces tests ne suivent pas du tout le même circuit administratif. Aujourd'hui, les pouvoirs publics ont une vision qu'il faudra très certainement revoir en tenant compte de cette nouvelle approche.
La commission de la transparence, le Comité économique des produits de santé (CEPS) ont des règles de fonctionnement et des délais auxquels les industriels tiennent beaucoup. Mais le remboursement d'un test dépend de la nomenclature de la Caisse nationale d'assurance maladie (CNAM), laquelle n'a absolument aucune obligation légale en matière de délai administratif. La CNAM inscrit les tests sur la liste dans un délai qui peut aller de un à trois ans, voire jamais ce qui pose un problème de cohérence. Il y a certainement à travail à faire du côté des administrations pour s'assurer que les deux, et le test et le médicament, arrivent en même temps sur le marché, sinon les patients français ne pourront pas bénéficier de la médecine personnalisée, même si tout a été accompli du côté du médicament.
Dr Frédéric Eberlé, responsable médical de Roche Diagnostics France . Je vous présenterai le modèle de développement du Groupe Roche dans la médecine personnalisée. Le groupe Roche est l'une des rares industries de santé qui possède deux divisions Roche Pharma et Roche Diagnostics. Chacune a son indépendance, mais comme nous le verrons, elles ont de très fortes synergies, notamment dans le cadre de la médecine personnalisée. La division Pharma est largement représentée dans le monde, avec des centres d'expertise essentiellement en Europe et aux États-Unis. C'est également le cas de la division Diagnostics. Le siège du groupe Roche est à Bâle.
Comme le Dr Christophe Le Tourneau l'a exposé, le Trastuzumab a été une révolution il y a une dizaine d'années. Après le Glivec en oncohématologie, l'Herceptin a fondé la médecine personnalisée dans les tumeurs solides. Pour mémoire, dans 20 % des cancers du sein, il y a une amplification du gène HER2 au niveau du noyau, qui se répercute au niveau membranaire par une surexpression du récepteur. L'Herceptin est un anticorps monoclonal qui bloque le récepteur et les voies de signalisation, en déclenchant également un phénomène de réaction immunitaire appelé ADCC ( Antibody Dependent Cellular Cytotoxicity ).
Le médicament peut être prescrit lorsqu'il y a une forte surexpression (3+) de la protéine à la surface de la cellule. Ce sont les anatomopathologistes qui réalisent ces tests. Dans les cas de surexpression (2+), une deuxième technique devra être mise en oeuvre : l'hybridation in situ par fluorescence ou par chromo génie. Il y a une dizaine d'années, l'Herceptin a donc été lancé avec des tests compagnons réalisés par une autre société que Roche.
Il existe deux nouveaux développements de médicaments chez Roche pour bloquer HER-2. Le premier concerne le Pertuzumab qui empêche la dimérisation du récepteur. Le deuxième (T-DM1) consiste à greffer une chimiothérapie sur le corps de l'anticorps monoclonal thérapeutique qu'est l'Herceptin. Ce faisant, le Pertuzumab d'une part ciblera la molécule tumorale HER-2 positive (comme l'Herceptin), et en outre délivrera la chimiothérapie dans la cellule. L'an dernier, un accord a été passé entre le groupe Roche et le groupe Areva pour greffer sur le corps de l'Herceptin, non pas une chimiothérapie, mais un radionucléide (plomb 212 d'Areva), ce qui permet de faire une irradiation ciblée intracellulaire des cellules tumorales.
Trois modes de développement de médecine personnalisée peuvent être décrits. Le premier (en 1998) fut l'Herceptin avec un test compagnon qui n'était pas celui de Roche. Le deuxième (années 2004-2008) décrit par Mme Blachier-Poisson, portait sur le Panitumab qui a obtenu une AMM avant qu'un test compagnon (KRAS) ne devienne obligatoire. Enfin, le troisième exemple, le plus récent, concerne le mélanome métastatique, où le Zelboraf (molécule de Roche Pharma), a été développé en parallèle avec le test compagnon cobas BRAF (de Roche diagnostics). Les études cliniques conduites en vue de l'obtention de l'AMM du Zelboraf l'ont été avec ce seul test, ce qui permet de dire que le test cobas BRAF est cliniquement validé. Il est aussi marqué CE in vitro diagnostic (CE-IVD), c'est-à-dire conforme à la directive européenne pour répondre aux « exigences essentielles » pour garantir la sécurité des patients et des utilisateurs.
Le Zelboraf est absolument révolutionnaire. Il fait partie des médicaments qui ciblent une mutation précise (« driver mutation » dont a parlé le Pr Dominique Stoppa-Lyonnet), c'est-à-dire une mutation causale nécessaire et suffisante au développement du cancer. L'article de G. Bollag publié dans Nature 2010 a montré des résultats spectaculaires entre J0 et J15. En général, les thérapies ciblant une « driver mutation » présentent des résultats initiaux très impressionnants. La photo de l'article de N. Wagle publié dans le Journal of clinical oncology en 2011 montre un patient sur lequel on observe énormément de métastases à J0. En seulement 15 jours, le Zelboraf a « nettoyé », expression d'usage chez les médecins, les métastases. Malheureusement ce patient va rechuter en semaine 23, à cause d'une mutation acquise de la protéine MEK située en aval de BRAF. Donc le modèle peut être résumé comme suit : un test diagnostic identifiant une « driver mutation » confère l'éligibilité des patients à une thérapie ciblée efficace. Cela n'exclut pas l'apparition ultérieure de mutations de résistance et sans doute faudra-t-il aller vers des bithérapies.
Roche Diagnostics, dispose d'un large panel d'instruments et de réactifs. Roche Diagnostics est très connu des biologistes dans les disciplines de l'immunologie, la biochimie et la biologie moléculaire, avec les tests PCR (Polymerase Chain Reaction), tel le test cobas BRAF marqué CE-IVD précédemment décrit. Nous avons également des instruments de séquençage à haut débit. Pour la cancérologie où les tests réalisés par les anatomopathologistes sont fondamentaux (hybridation in situ et immunohistochimie), Roche Diagnostics apporte aussi des techniques automatisées de coloration, des techniques d'imagerie (scanner de lames).
Les deux divisions de Roche travaillent donc en grande synergie. Les biomarqueurs peuvent être identifiés par des experts académiques ou par nos propres chercheurs. Mais le véritable savoir-faire de l'industrie pharmaceutique, est de développer le test compagnon en tant que réactif de laboratoire standardisé détectant le biomarqueur d'intérêt accompagnant la thérapie ciblée. Roche Diagnostic vise à obtenir un marquage CE-IVD conjointement à l'AMM du médicament (comme cela a été fait pour le Zelboraf).
Roche comprend la société Genenteche en tant qu'entité travaillant avec une grande autonomie. La R&D de Genentech porte sur de nombreux médicaments, notamment en oncologie, dont plus de la moitié sont développés conjointement avec un test diagnostic compagnon de type moléculaire ou histologique ou biochimique ou immunologique. Du côté de la R&D intrinsèque à Roche Pharma, 80 % des molécules sont accompagnées du développement d'un test diagnostic compagnon. Nous venons de décrire les molécules en phase précoce de développement.
S'agissant des molécules lancées d'ici deux ans, six nouveaux traitements sont accompagnés d'un test compagnon sortiront. Je ne reviens pas sur le Pertuzumab et le T-DM1 en oncologie. Pour les cancers bronchiques non à petites cellules, le MetMAb est un anticorps monoclonal anti-cMET pour lequel la surexpression de la protéine MET sur les prélèvements bronchiques sera recherchée par un test Roche Diagnostics. Pour le traitement de l'hépatite virale C, le Danoprevir est une anti-protéase et la Mericitabine une anti-polymérase. Les patients concernés sont suivis par les mesures de leur charge virale (qui sont donc de véritables tests personnalisés). Ces tests ont été développés chez Roche Diagnostics il y a une vingtaine d'années.
L'interleukine 13 (IL-13) peut être particulièrement impliquée dans l'asthme sévère et le Lebrikizumab développé par Roche est une thérapie ciblée ant-IL-13 présentant un potentiel très intéressant. Administré à des patients non sélectionnés, la réponse au Lebrikizumab peut apparaître comme modérée (je cite un article de J. Corren du New England Journal of Medicine de septembre 2011). En revanche, administré à des patients présentant une forte implication de l'IL-13, le traitement apparaît comme efficace. Un moyen indirect d'évaluation de l'implication de l'IL-13 est de doser le taux sanguin de périostine sur un instrument de type cobas. L'article cité confirme la preuve de concept d'une stratification des patients asthmatiques par leur taux sanguin de périostine comme critère prédictif de réponse au traitement.
Roche présente une politique volontariste de partenariats de R&D. En France, 56 partenariats sont en cours, avec des partenaires académiques. 22 millions d'euros ont été investis depuis 2009 dans ces partenariats, avec un modèle collaboratif en vue de bénéfices mutuels.
Pour conclure, l'accès des patients aux traitements très innovants de médecine personnalisée que nous avons présentés est fondamental au vu des résultats cliniques observés, mais on ne peut nier que la sélection des patients par un test compagnon complique la stratégie thérapeutique pour les industriels, les cliniciens et les autorités de santé. L'industriel a la charge d'évaluer simultanément le test-compagnon et le médicament pour viser l'inscription du test aux nomenclatures des actes de biologie médicale ou des actes d'anatomopathologie et l'AMM du médicament. Il doit également s'enquérir de la bonne maîtrise de la réalisation des tests par les centres de prise en charge des patients. Roche Diagnostics accompagne très fortement ses clients au niveau de la détection de HER-2. L'an dernier, nous avons soutenu le contrôle de qualité (CQE) des tests BRAF effectués en France. C'était une préoccupation majeure de Roche en vue d'une dispensation juste du Zelboraf. Cette préoccupation est aussi partagée par l'INCa, qui en 2013 met en place des contrôles de qualité au niveau de l'EGFR, BRAF et KRAS.
Dr Jérôme Garnier, directeur en oncologie de Roche France . Évidemment, je m'inscris complètement dans la ligne de présentation du Dr Frédéric Éberlé. Je voudrais insister sur la nécessaire qualité, exercice difficile, mais qui est absolument fondamental dans la recherche de tests. J'en veux pour exemple le MetMAb, anticorps qui cible la protéine cMET exprimée à la surface des cellules de cancers bronchiques ou de cancers gastriques, ainsi que dans un grand nombre de tumeurs. Des résultats d'études de phase 2 ont été présentés il y a bientôt deux ans dans un congrès américain. Ils ont clairement montré que lorsque le médicament est administré à la population de patients qui expriment fortement cMET, le bénéfice est vraiment significatif en survie globale et en survie sans progression. À l'inverse, lorsque le médicament est administré à une population atteinte des mêmes cancers bronchiques et qui n'expriment pas la protéine cMET à la surface des cellules, le résultat est totalement inverse, décrémental par rapport au standard of care .
Cela montre bien qu'on est vraiment là dans une recherche quasi absolue de la perfection dans la présence de cette protéine, de sorte qu'on l'administre qu'à la population qui en bénéficiera, et bien sûr pas à la population qui n'exprime pas la protéine. Dans ce cas, celle-ci serait non seulement exposée aux effets indésirables du médicament, mais de surcroît, on lui rajouterait un effet délétère. Cet exemple très précis est d'autant plus important à prendre en considération qu'il ne s'agit pas de biologie moléculaire, à savoir de réalisation des tests sur les plateformes de génétique moléculaire des cancers de l'INCa, mais de réalisation de test dans tous les laboratoires d'anatomie pathologique sur le territoire. L'enjeu de la qualité du test est forcément très fort quand on l'associe aux médicaments.
En outre, lorsqu'on développe un médicament en vue d'un enregistrement, en tant qu'industriel on ne s'arrête pas là dans la production de connaissances. Il est absolument crucial d'accompagner les prescripteurs dans la meilleure compréhension possible des mécanismes associés, telle que la résistance primaire ou secondaire aux médicaments, et de mieux comprendre en vie réelle ce qui se passe chez les malades. Il y a une nécessaire collaboration entre le monde industriel et le monde académique après le lancement des médicaments.
Cela rejoint ce que mon collègue précisait précédemment à propos de la médecine translationnelle. Finalement, ce sont ces allers-retours entre le malade et la paillasse qui permettront de mieux comprendre les mécanismes des médicaments. La publication très importante du New England Journal of Medicine daté d'avril 2012 a montré très clairement l'hétérogénéité tumorale et l'instabilité génétique des tumeurs. Cela signifie qu'il peut y avoir des écarts entre un prélèvement réalisé sur un cancer dans l'organe primitif et la situation mutationnelle dans les métastases. Donc se fier à des résultats obtenus sur l'organe primitif pour pouvoir ensuite administrer un traitement dit ciblé, c'est probablement faire une erreur.
L'instabilité génétique des tumeurs a été illustrée par le Dr Frédéric Éberlé. Quand on expose un patient porteur d'un mélanome métastatique BRAF muté à un inhibiteur BRAF, il y a des voies de contournement de ce blocage de BRAF, avec entre autres l'expression par exemple de MEK, ou l'amplification de BRAF. La tumeur a un grand nombre de possibilités pour contourner ce blocage, et donc il est absolument nécessaire de poursuivre encore la recherche, pour mieux comprendre ce qu'il faut faire. Il faut peut-être associer des médicaments ou mieux les utiliser au cours du cursus du malade.
Mme Christine Guillen, directrice du programme ADNA « Avancées Diagnostiques pour de Nouvelles Approches thérapeutiques », Institut Mérieux . Je vous remercie de nous donner l'opportunité de présenter un programme fédéré par l'Institut Mérieux. Ce programme, dédié à la médecine personnalisée, a été initié par Christophe Mérieux en 2005 avec la volonté de promouvoir et d'améliorer la médecine personnalisée dans le domaine des maladies infectieuses mais également des cancers et des maladies génétiques rares. Ce programme s'appelle ADNA : Avancées Diagnostiques pour de Nouvelles Approches thérapeutiques. Il fédère des partenaires d'horizons différents et en cela il illustre ce qui vient d'être discuté. Il réunit des industriels qui développent des produits thérapeutiques ou des tests de diagnostic tout en s'appuyant sur l'expertise de nombreux cliniciens. Le programme, centré sur le patient, a pour principal objectif de réaliser le meilleur diagnostic et le meilleur suivi de la pathologie, mais également d'identifier des biomarqueurs permettant d'établir les traitements les mieux adaptés aux patients.
En 2013, ADNA est entré dans sa septième année. Il bénéficie du soutien de l'État français (par l'intermédiaire d'OSEO) dans le cadre des Programmes Mobilisateurs à l'Innovation Industrielle. Il a permis de mettre autour de la table des partenaires scientifiques de cultures différentes qui n'avaient pas l'habitude de travailler ensemble. Ils ont appris des uns et des autres pour aboutir à la mise sur le marché de produits diagnostiques et thérapeutiques innovants.
En complément à ce qui a été mentionné précédemment sur l'identification d'un biomarqueur, je prendrai l'exemple de notre partenaire Transgene qui développe un vaccin thérapeutique dans le traitement du cancer du poumon « non à petites cellules ». La société dispose déjà d'un test immuno-histochimique pour identifier si l'antigène d'intérêt est exprimé par les cellules tumorales chez les patients à traiter. Transgene a réalisé une étude de phase 2B à l'issue de laquelle il a identifié de manière rétrospective l'existence d'un marqueur complémentaire. Il s'agit d'un marqueur lié à des cellules immunitaires, et plus précisément à la présence d'un taux de cellules TrPAL (Triple positive activated lymphocytes) prédictif de la réponse au traitement des patients. Cela a permis de poser l'hypothèse que ce biomarqueur permettrait de sélectionner les patients avant l'administration du traitement. Comme l'hypothèse de départ de l'étude clinique n'intégrait pas la sélection des patients en fonction du niveau d'activation de ces cellules, Transgene a donc consulté différentes agences réglementaires afin de définir le design de la phase clinique suivante. Les réponses ont été différentes suivant les agences, mais il est apparu clairement qu'il fallait mener une nouvelle étude clinique combinant non plus seulement un test compagnon mais deux tests compagnons avec des technologies extrêmement différentes, afin de valider ces biomarqueurs.
Le premier test compagnon est un test immuno-histochimique robuste et standardisable, facile à développer, qui a pu être mis au point dans le cadre des essais cliniques de Transgene sans trop de difficultés. En revanche, le deuxième test compagnon repose sur une technologie de cytométrie en flux, très difficilement automatisable et surtout très variable selon le centre clinique qui la met en oeuvre. Le partenaire Transgene a donc été amené à travailler très tôt avec des spécialistes du domaine car ce type de test ne pouvait pas être développé en interne. Ils se sont alors heurtés à deux difficultés : premièrement, la durée de développement de ce genre de test est différente de celle d'une phase clinique et deuxièmement, les discussions réglementaires liées à l'enregistrement de ce test compagnon sont à mener en parallèle à celles du produit.
Ce deuxième test permet la stratification des patients, suivant le taux de cellules NK activées. Il permettrait de distinguer deux populations : celle ne tirant malheureusement pas de bénéfice du traitement en raison d'un taux élevé de cellules NK activées, et celle en tirant un bénéfice. La phase 2B en cours doit valider la pertinence de ce biomarqueur pour la sélection des patients.
Dans le cas de notre partenaire bioMérieux et de ses partenaires (CEA/Leti, ST Microelectronics, Hospices Civils de Lyon et CNRS), ceux-ci prévoyaient, lorsque ce programme a démarré, la mise sur le marché d'un certain nombre de biomarqueurs en oncologie et dans les maladies infectieuses. Sept ans plus tard, un taux d'attrition important a été observé, en cohérence avec les résultats de l'ensemble des acteurs du domaine, en raison en particulier des performances cliniques insuffisantes des marqueurs candidats. Nous savons aujourd'hui que la recherche et la validation de biomarqueurs ayant une vraie valeur médicale ajoutée sont extrêmement longues et difficiles. BioMérieux prévoit néanmoins la validation de biomarqueurs pour le dépistage du cancer du foie, le pronostic et le diagnostic du sepsis, et pour la détection des maladies nosocomiales liées, entre autres, à la ventilation artificielle. Chez le partenaire bioMérieux, la plateforme de diagnostic automatisée initialement envisagée a été revue en raison de l'évolution rapide des technologies.
Par ailleurs, dans le cas des maladies rares, notre partenaire Généthon lance un essai clinique pour l'évaluation d'un traitement de thérapie génique pour la myopathie de Duchenne. L'ensemble des patients atteints par cette pathologie portent des mutations dans le gène de la dystrophine et dans un premier temps le produit développé par Généthon s'adresse à une sous-population de patients porteurs d'une mutation précise. Son objectif à terme toutefois est de développer des produits pouvant s'adresser au plus grand nombre. Ce développement pionnier d'une biothérapie innovante pour une maladie mortelle chez l'enfant et l'adolescent présente de nombreux défis, dont celui de produire selon des normes pharmaceutiques et à grande échelle un traitement particulièrement complexe. Ainsi, des méthodes de production ont été mises au point, « inventées » par Généthon, à l'occasion de ce projet, et pourront servir pour d'autres produits en particulier pour le traitement d'autres maladies rares mais aussi fréquentes. En parallèle, une réflexion doit être menée concernant la prise en charge par la société de ce type de traitement dans un souci de maîtrise des dépenses de santé. Aujourd'hui, nous réfléchissons aux problématiques à la fois réglementaires, économiques et de remboursement.
En conclusion, l'identification et la validation des biomarqueurs sont les points communs de tous les partenaires du programme dans leurs domaines respectifs : maladies infectieuses, diagnostics de cancers et leur traitement ciblé, maladies génétiques rares. Certaines études ou activités se sont avérées prometteuses comme dans le cas de la société GenoSafe avec une pérennisation de ses tests d'immuno-monitoring qui répondent aux besoins de développement de produits de médecine personnalisée.
S'il fallait tirer les grandes leçons de ce programme, sur le fond, nous avons été confrontés aux difficultés propres aux approches innovantes, sur la forme, nous avons assisté à de nombreux échanges de compétences, de savoir-faire entre des partenaires de cultures et d'expertises différentes mais tous mobilisés autour d'un objectif partagé.
Pour terminer, il faut réfléchir au modèle de propriété industrielle. Aujourd'hui, il est extrêmement compliqué de gérer le modèle standard de copropriété. Dans ADNA, nous avons réussi à faire émerger un modèle adapté aux projets issus des synergies nées entre les partenaires. Nous apprenons à gérer cette question de propriété industrielle afin qu'elle ne soit pas un facteur de blocage pour la valorisation, qu'elle aboutisse à un bénéfice pour les patients et qu'elle valide ainsi l'approche de médecine personnalisée.
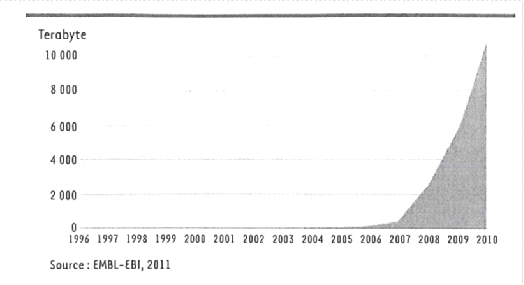
M. Vincent Fert, directeur général de Qiagen Marseille . De même que le Dr Fréderic Éberlé, je représente l'industrie. Le groupe Qiagen, basé en Allemagne, est une société de taille mondiale, dont le chiffre d'affaires est d'1,2 milliard d'euros. Il est représenté aux États-Unis, en Europe, en Asie. Ses centres de recherche sont répartis dans le monde entier. C'est l'un des acteurs importants de la médecine personnalisée.
On a entendu aujourd'hui que sans tests de diagnostic, il n'y avait pas de médecine personnalisée. Qiagen est l'une de ces sociétés engagées de façon très volontariste dans ce domaine via des outils essentiellement d'analyse des acides nucléiques, d'extraction d'échantillons, d'automatisation de ces extractions, d'analyse des séquences d'ADN via des technologies PCR ou de pyro- séquençage. Et puis récemment, nous avons investi dans les technologies de séquençage de nouvelle génération qui permettent d'analyser un génome complet en quelques jours.
Je vous décrirai comment Qiagen aborde le domaine de la médecine personnalisée. Nous proposons aux industriels de la pharmacie des outils et des solutions pour le développement de leurs molécules. À ce jour, Qiagen a une quinzaine de partenariats avec les plus grands groupes pharmaceutiques. Qiagen a notamment lancé l'an dernier un test KRAS dans le cancer du côlon, en collaboration avec Bristol-Myers-Squibb et Lilly pour le cetuximab. C'était le premier test KRAS enregistré. Cette année, nous allons probablement obtenir le label de la Food and Drug Admnistration (FDA) pour le test EGFR dans le cancer du poumon, en collaboration avec une autre société pharmaceutique.
Pour une société de diagnostic, les collaborations avec l'industrie pharmaceutique sont vraiment un domaine émergent. Comme on l'a entendu, que ce soit au niveau réglementaire ou à celui de l'accès au marché, beaucoup reste à faire. Plusieurs éléments sont importants pour une société pharmaceutique qui développe une thérapie ciblée. D'abord le diagnostic ne doit pas peser sur le développement en termes de délais. À l'exception des molécules faites rétrospectivement dans le cancer du côlon (notamment le cetuximab), tous les développements, sont effectués de façon synchrone avec le médicament. On collabore à partir des phases 2 pour définir les prérequis du test. On fige un test à l'issue des phases 2, de façon à ce qu'en phase 3 du médicament, on puisse valider ce test et l'enregistrer de façon conjointe. Ce sont évidemment les prérequis appliqués dans le système américain au niveau de la FDA. Mais c'est un système dominant qu'en général les sociétés pharmaceutiques appliquent pour les autres pays.
L'industrie pharmaceutique veut parvenir à un développement de test compagnon abordable qui au final, ne doit pas être un frein au niveau de la commercialisation en termes de prix. Le bras de fer est assez inégal entre l'industrie pharmaceutique et l'industrie du diagnostic, néanmoins, nous sommes engagés dans ces discussions et des collaborations. Du coup, et c'est très nouveau pour nous, l'industrie du diagnostic subit de plein fouet, les taux d'attrition de ces molécules. Les industries pharmaceutiques adoptent les biomarqueurs et les projets tests compagnons diagnostiques de plus en plus tôt dans le développement, à partir de la phase 1. Nous sommes donc associés à des projets de phase 1, et nous savons très bien que le taux d'attrition est important et que nous le subirons.
Je prendrai deux exemples particuliers par rapport au sujet de cette table ronde consacrée aux modèles de R&D. Le premier exemple est un test développé à partir d'une découverte faite à l'Institut Gustave-Roussy. Il s'agit d'une mutation dans le gène JAK2 qui permet de porter un diagnostic des syndromes myélo-prolifératifs qui est un excellent exemple de cercle vertueux de l'innovation. Notre partenaire l'Institut Gustave-Roussy a été un très bon partenaire, mais cela aurait aussi bien pu être l'INSERM.
En 2005, une publication dans Nature de l'équipe du Pr William Vainchenker au sein de l'Institut Gustave-Roussy démontre l'existence de cette mutation qui permet de classer ces leucémies pour lesquelles on ne disposait pas réellement d'outils de diagnostic. En 2006, nous avons fait l'acquisition d'une licence exclusive et mondiale sur ce test. En 2007, un test marqué CE est lancé sur le marché. Ce test a représenté 50 % des revenus de la société qui était à l'époque Ipsogen et qui est aujourd'hui Qiagen Marseille, société toujours en activité et en croissance. Cette licence a été exploitée sous forme de produits qui ont été diffusés, et qui le sont encore, mondialement. Nous avons un projet pour enregistrer ce test à la FDA et des licenciés l'utilisent dans d'autres pays. Chaque année, je verse à l'Institut Gustave-Roussy (IGR) une somme qui représente le montant des redevances payées sur les revenus de ce test. Ce montant est partagé, puisque c'est une découverte collégiale qui implique l'INSERM et l'AP-HP.
J'ai fait un petit calcul que j'avais présenté il y a deux ans pour le dixième anniversaire de la structure de valorisation de l'IGR. Il montre que les redevances que nous reversons aux instituts de recherche pour ce test financent largement les tests JAK2 réalisés en France. Il faut savoir que ces redevances proviennent à 75 % des États-Unis, où l'on respecte le droit des brevets sur les diagnostics, jusqu'à aujourd'hui.
C'est donc un bon exemple de cercle vertueux de l'innovation. Cet argent sert à financer la recherche publique. Mais comme je le disais tout à l'heure de façon un peu volontariste, je pense que le diagnostic, et notamment le compagnon diagnostic, nécessite des investissements importants en termes de développement, d'essais cliniques, et d'une certaine façon, le droit des brevets est utile dans notre domaine.
Le deuxième exemple est un test que nous avons développé parallèlement au succès du Glivec qui est l'archétype des médecines personnalisées, pour lequel l'amélioration du service médical rendu (ASMR) n'est pas équivoque. En 2003, nous avons développé un test en collaboration avec un réseau européen d'experts académiques, avec qui il faut dans notre domaine, collaborer de façon très large. Nous sommes le leader pour ce genre de test. Il consiste à mesurer assez précisément le taux de transcrits, d'anomalies moléculaires dans le sang, de façon à suivre l'efficacité du Glivec qui maintenant devient un traitement chronique. Certains patients sont sous traitement au Glivec depuis dix ans. C'est finalement le même modèle que les anti-HIV.
Avec l'arrivée des molécules de deuxième génération, plus efficaces, le taux de cette anomalie dans le sang chute encore, et on arrive aux limites de sensibilité. Par ailleurs, on se demande si ces patients peuvent soutenir l'arrêt du traitement. Cela permettrait d'éviter au système de santé de payer des médicaments inutiles et pour les patients des toxicités. Des essais cliniques sont en cours, avec des résultats encourageants. Mais pour que ces essais cliniques puissent être conduits, il faut un test plus sensible que nous développons actuellement.
Je conclurai sur le problème de l'accès au marché. En ce qui concerne la réglementation, la directive CE en Europe est extrêmement différente en termes de prérequis par rapport à ce que nous demande la FDA. Ces marchés de médecine personnalisée sont mondiaux. La France représente seulement 10 % du chiffre d'affaires d'un test comme JAK2 ou BRCA. L'harmonisation des réglementations est donc extrêmement nécessaire, en particulier dans le domaine du test compagnon diagnostic, puisque les exigences de la FDA sont celles que j'ai citées tout à l'heure.
En ce qui concerne l'évaluation, il n'y a pas d'ASMR dans le diagnostic. Elle existe pour le médicament, mais pas pour le diagnostic. Il serait nécessaire qu'on en ait une pour le diagnostic. C'est une demande que j'adresse formellement. Si nous avons des ASMR pour le diagnostic, nous pourrons peut-être être acteurs du remboursement. Aujourd'hui, un industriel du diagnostic n'est pas qualifié pour soumettre un dossier de remboursement à la CNAM ou ailleurs. C'est un point extrêmement important.
Dr Pierre Attali, directeur général délégué stratégies et affaires médicales de BioalliancePharma . Chez BioAlliance, nous sommes dans une situation qui est un peu différente de celles décrites dans cette table ronde. Nous sommes une petite société de biotechnologie française, cotée en bourse. Pour développer nos produits, nous n'avons pas du tout les mêmes revenus et donc les mêmes facilités que les grands groupes et les multinationales.
Quelques rappels très brefs déjà été évoqués : qu'entend par « médecine personnalisée » ? Des modalités diagnostiques adaptées à chaque maladie, avec un traitement adapté à chaque malade, ce qui veut dire théoriquement un diagnostic précoce, une efficacité améliorée, pas ou peu d'effets indésirables. Ce n'est pas du tout ce qui passe aujourd'hui. Cela a des implications pour la R&D dans les domaines du diagnostic et pharmaceutique en raison de la complexité liée à au moins deux « facteurs » : le malade lui-même et la maladie .
En ce qui concerne les malades, chaque malade a son propre profil génétique individuel et cela implique des conséquences thérapeutiques. En tant que praticien à l'Assistance publique, je vais donner des exemples. En hépatologie, on connaît bien le problème des hépatites virales C chroniques G1 et l'on sait que la réponse thérapeutique varie de 25 à 80 % en fonction du polymorphisme génétique des patients. Il y a donc vraiment une efficacité qui dépend du profil génétique propre. Il existe aussi une tolérance qui dépend du profil génétique, avec une adaptation des doses selon le métabolisme individuel. Par exemple, l'anémie liée à la ribavirine en fonction du métabolisme individuel.
En ce qui concerne les maladies, pour les agents infectieux par exemple, on sait qu'il existe des sous-types viraux ou bactériens qui vont répondre plus ou moins aux traitements, avec des résistances innées (Anti-protéases et HCV G3) ou acquises (pour les agents bactériens).En cancérologie, il y a des récepteurs et des voies de signalisation intracellulaires qui diffèrent selon les cancers (sein, poumon, mélanome. Elles diffèrent même au sein d'un même type de cancer (poumon, etc .), avec une ou plusieurs voies de signalisation surexprimées ou inhibées, avec des modifications de profil de ces maladies, en fonction de la pression thérapeutique.
Cela intervient de façon importante pour la R&D, car cette complexité est accrue par cumul des facteurs intrinsèques et extrinsèques. Finalement, à force de restreindre et d'individualiser les traitements, on individualise les malades, mais on individualise aussi les maladies orphelines. On risque de rentrer très rapidement dans les maladies orphelines et ultra-orphelines. Pour les maladies orphelines, la définition est bien connue : en Europe, une prévalence 5/10 000 sujets, avec une contribution majeure à la santé publique. Pour les maladies ultra-orphelines, c'est-à-dire finalement un nombre de patients extrêmement restreint, il n'y a pas de définition. Certaines définitions courent « sous le manteau » : moins de 20 000 patients dans le monde et une prévalence en Europe inférieure à 1/50 000 sujets. En Grande-Bretagne, le National Institute for Health and Care Excellence (NICE) les définit à moins de 1 000 patients. Si vous reportez ce chiffre à la population européenne, vous obtenez une prévalence inférieure à 1/50 000 sujets.
On se situe dans des systèmes de R&D où l'on développe des médicaments pour des populations extrêmement restreintes. C'est complètement différent de tout ce qui a déjà été fait en développement. Jusqu'à présent, on développait des produits pour de larges populations, avec accès à des marchés colossaux, un nombre de malades colossal. Actuellement c'est l'inverse qui se passe. On restreint de façon considérable le nombre de patients à traiter pour des coûts qui ne sont pas diminués.
Sur l'aspect diagnostic, le modèle de R&D vis-à-vis du malade doit faire intervenir le secteur académique et le secteur industriel, avec une collaboration indispensable entre les deux. Celle-ci n'existe pas toujours. Vis-à-vis des maladies, les collaborations sont importantes. Comme le faisait remarquer le Pr François Ballet, elles ne sont pas uniquement nécessaires entre le secteur académique et le secteur industriel, mais aussi avec un ensemble d'autres acteurs, en particulier l'imagerie médicale, les produits de contraste, toutes ces méthodes vraiment très importants dans ces modèles de R&D en train de changer avec la médecine personnalisée.
Sur l'aspect thérapeutique, le développement de thérapeutiques ciblées intervient dans les produits classiques des entités chimiques, des protéines, des acides nucléiques. Mais on rentre aussi dans la thérapie génique, qui est vraiment de la médecine personnalisée, et qui pose un certain nombre de problèmes en termes éthiques et de développement pour l'industrie pharmaceutique. On rentre dans les domaines de transplantation de cellules souches, de nano médicaments, de vaccins thérapeutiques.
Cet ensemble est beaucoup plus compliqué, parce que les modèles expérimentaux n'existent pas aujourd'hui. Il faut parfois en créer, avec des animaux transgéniques, des bi-transgéniques, et j'en passe. Ces développements doivent être adaptés, aussi bien en préclinique qu'en clinique à des maladies orphelines ou ultra-orphelines, avec des « médicaments » qui sont innovants ou ultra-innovants, et où tout est à faire.
Nous sommes donc en train d'explorer des voies, ou de créer des chemins qui ne sont pas bien connus. Si c'est déjà compliqué pour de grandes sociétés, vous imaginez les difficultés que rencontrent des petites sociétés comme la nôtre de 55 personnes, et pourtant, elles font l'innovation de demain.
Première difficulté : les durées de développement sont longues. Il ne faut pas croire que dans les maladies orphelines ou ultra-orphelines, la mise au point des modèles se fera très rapidement. Par définition, il n'y a pas beaucoup de malades, il faut les chercher un peu partout, en Europe et aux États-Unis, en Asie. Deuxième difficulté : les coûts sont très élevés (essais mondiaux, durée de vie prolongée, ...).Troisième difficulté : nous sommes parfois obligés de recourir à des combinaisons de thérapeutiques ciblées (ex : mélanome, ...). Pour cela, nous devons faire appel à d'autres acteurs, à d'autres sociétés pharmaceutiques pour co-développer le produit, ce qui complique encore le système.
Enfin, en écho à ce qu' expliquait le Pr. Gilles Bouvenot, nous avons un petit nombre de malades et donc nous sommes obligés de prendre des engagements, pour l'enregistrement et en post enregistrement, de suivi, d'études particulières de pharmacovigilance, etc. Quand on a l'enregistrement, et qu'en plus on a la chance d'avoir un prix, on n'est même pas arrivé au bout de nos peines.
Le système est complexifié par la multiplicité des acteurs. On fait régulièrement appel aux acteurs académiques (instituts de recherche, hôpitaux) et parmi les acteurs industriels, il y a la biotechnologie ( drug discovery ), les sociétés de service (formulation, préclinique, clinique), les sociétés pharmaceutiques. Les pouvoirs publics sont également importants pour nous. D'une part, ils peuvent nous faciliter les communications entre les différents acteurs publics ou privés. D'autre part, l'ensemble des agences qui évaluent nos produits, que ce soit en France, en Europe et dans le Monde (USA, Japon, BRICS et pays émergents), doivent nécessairement être à peu près d'accord sur ce qu'il faut faire. Par exemple, dans le domaine de la pédiatrie, on a des demandes européennes et américaines d'études pour tous les nouveaux médicaments. Pour une société comme la nôtre, développer un médicament, c'est déjà difficile. Les développer en pédiatrie, c'est compliqué. Et quand les agences ne vous demandent pas les mêmes choses, c'est encore plus compliqué.
Pour conclure, nous avons besoin d'établir des partenariats stratégiques de long terme, avec des rôles et des responsabilités de chacun des acteurs bien définis. Le cadre de la propriété intellectuelle doit être clarifié. C'est important, car sans propriété intellectuelle, il n'y a pas de rentabilité. Il faut arrêter de penser qu'on peut demander 10 % de royalties à des produits qu'on prendra dans les hôpitaux ou dans les instituts de recherche, parce que cela obère tout accord de licence pour les petites sociétés de biotechnologie, donc être trop gourmand, cela tue les projets
En ce qui concerne les financements, nous avons besoin de subventions. à ce titre, la France fait bien son travail. J'en profite pour remercier tous ces acteurs : Oseo, FSI, auxquels nous faisons largement appel. Les subventions européennes sont également les bienvenues, mais c'est beaucoup plus difficile pour les PME d'y avoir accès. L'administration de ces consortiums européens est très compliquée. Je vous garantis, pour y avoir participé, que ce n'est pas commode à gérer et à manager au quotidien.
Dernier point, et c'était l'objet de ma question au Pr. Gilles Bouvenot, c'est le problème de la rentabilité. Il est nécessaire pour nous d'avoir des prix élevés car nos investissements, à ce niveau, sont très importants. Et il est nécessaire aussi d'avoir des remboursements. Ainsi dans le domaine anti-infectieux, à force de vouloir obtenir des prix peu élevés, de saboter les remboursements, on a abouti aujourd'hui à l'absence d'antibiotiques nouveaux, et d'antituberculeux. Je tire la sonnette d'alarme. On finit par détruire totalement des pans de recherche entiers dans des domaines où existent des besoins. Les agents anti-infectieux en sont une démonstration formelle. Le soutien des pouvoirs publics est nécessaire dans un certain nombre de domaines qui ne font plus aujourd'hui l'objet de la R&D. On a certes beaucoup évoqué le cancer aujourd'hui, mais il n'y a pas que cette pathologie, même si BioAlliance est aussi largement positionné en cancérologie.
CONCLUSION
M. Jean-Sébastien Vialatte. Nous remercions tous les intervenants de cette journée. Ce terme de médecine personnalisée reste encore très difficile à définir. Aujourd'hui, on a surtout traité de médecine personnalisée génomique, en laissant de côté tous les autres aspects.
On a vu poindre un certain nombre de problèmes, en particulier des problèmes éthiques, en premier lieu la possibilité de faire un dépistage du génome complet du foetus à partir des cellules circulant dans le sang maternel. On perçoit bien tous les risques de dérive eugénique qui risquent d'en découler. C'est un des sujets dont le législateur devra à un moment se saisir.
On a aussi constaté que cette médecine a un coût. La dernière intervention le montre. Jusqu'où ce coût est-il soutenable par la solidarité nationale ? Ne risque-t-on pas de se diriger vers une médecine destinée à une catégorie de personnes ayant les moyens de la financer ? Que va pouvoir prendre en compte la solidarité nationale ?
D'une façon générale, à la fin de ces journées d'audition, plus de questions que de réponses sont apparues par rapport à celles qu'on se posait précédemment.
M. Alain Claeys. Nous vous remercions chaleureusement. Nous serons peut-être amenés à vous recontacter dans le cadre de nos travaux. Le mérite de ces auditions, c'est aussi d'ouvrir de nouveaux champs à notre réflexion. À un moment ou à un autre, le législateur aura des recommandations à faire. C'est comme cela que nous concevons notre travail.
Je remercie chacune et chacun d'entre vous, en espérant qu'on ne se sera pas trop raconté de contes de fée, et qu'on aura été le plus concret possible.
ANNEXE N° 2 :
COMPTE RENDU DE
L'AUDITION PUBLIQUE DU 25 JUIN 2013
« MÉDECINE
PERSONNALISÉE :
ENJEUX ÉTHIQUES ET
SOCIÉTAUX »
SOMMAIRE
___
Pages
OUVERTURE PAR MM. ALAIN CLAEYS ET JEAN-SÉBASTIEN VIALATTE, DÉPUTÉS, RAPPORTEURS 3
PROPOS INTRODUCTIFS 7
Pr Jean-Claude Ameisen, immunologiste, président du Comité consultatif national d'éthique pour les sciences de la vie et de la santé (CCNE). 7
Pr Hervé Chneiweiss, président du comité d'éthique de l'Institut national de la santé et de la recherche médicale (INSERM), directeur du laboratoire de neuroscience de l'institut de biologie Paris-Seine, membre du conseil scientifique de l'OPECST 11
PREMIÈRE TABLE RONDE : MÉDECINE DE PRÉVENTION OU DE PRÉDICTION : QUELLE RELATION MÉDECIN-MALADE ? 15
Mme Emmanuelle Prada-Bordenave, directrice générale de l'Agence de la biomédecine. 15
Pr Anne Fagot-Largeault, professeure au Collège de France, membre de l'Académie des sciences. 18
Pr Philippe Amouyel, professeur d'épidémiologie et de santé publique au Centre hospitalier et universitaire de Lille, directeur général de la fondation Plan Alzheimer. 20
Dr Anne Cambon-Thomsen, directrice de recherche au CNRS, UMR 1027 « Épidémiologie et analyses en santé publique » de l'INSERM et de l'Université de Toulouse 3 Paul Sabatier, responsable de l'équipe « Génomique, biothérapies et santé publique ». 25
Pr Florent Soubrier, responsable du département de génétique du groupe hospitalier de la Pitié-Salpêtrière-Charles Foix 28
Mme Simone Bateman, sociologue, directrice de recherche au CNRS, Centre de recherche médecine, sciences, santé, santé mentale, société (CERMES3) - CNRS UMR8211/Université Paris Descartes/EHESS/ Inserm U988. 31
DÉBAT 35
DEUXIÈME TABLE RONDE : QUELLE PROTECTION DES PERSONNES, DE LEUR VIE PRIVÉE ET DES DONNÉES PERSONNELLES ? 43
POINT DE VUE : M. Jean-Louis Touraine, député du Rhône, membre de l'OPECST 43
Pr. Dominique Stoppa-Lyonnet, chef du service de génétique de l'Institut Curie, professeur de génétique médicale à l'université Paris-Descartes, membre du CCNE 45
Dr. Catherine Bourgain, chargée de recherche en génétique humaine et statistiques à l'INSERM. 47
Pr Marc Delpech, chef du service de biochimie et génétique moléculaire de l'hôpital Cochin 50
Dr Jean-François Deleuze, directeur du Centre national de génotypage au CEA 54
Mme Audrey Aboukrat, doctorante à l'école de droit de l'université Paris I Panthéon Sorbonne 57
Mme Frédérique Lesaulnier, coordonnatrice du pôle santé de la Commission nationale de l'informatique et des libertés (CNIL). 64
DÉBAT 69
APRÈS-MIDI 75
PROPOS INTRODUCTIFS 75
Pr Agnès Buzyn, présidente de l'Institut national du cancer (INCa). 75
M. Didier Tabuteau, conseiller d'État, responsable de la chaire santé de l'Institut d'études politiques de Paris (IEP). 77
TROISIÈME TABLE RONDE : QUELLE ORGANISATION DU SYSTÈME DE SANTÉ ? 81
Pr Gilles Bouvenot, président de la Commission de la transparence, membre du collège de la Haute autorité de santé (HAS), membre de l'Académie nationale de médecine 81
Pr Véronique Trillet-Lenoir, professeur de cancérologie, université Claude Bernard Lyon 1, chef du service d'oncologie médicale au Centre Hospitalo-Universitaire de Lyon, présidente du Cancéropôle Lyon-Rhône-Alpes-Auvergne 84
Pr Thomas Tursz, cancérologue, directeur honoraire de l'Institut Gustave Roussy 87
Mme Valérie Seror, économiste, chargée de recherche INSERM-IRD. Université Aix-Marseille 92
Mme Elisabeth Thouret-Lemaitre, consultante, spécialiste de propriété industrielle 94
Pr Laurent Degos, ancien président de la Haute autorité de santé (HAS), membre de l'Académie de médecine, et membre correspondant de l'Académie des sciences 96
M. Olivier Perche, (LEEM) pour les entreprises du médicament, responsable du développement chez Roche Diagnostics 99
DÉBAT 101
QUATRIÈME TABLE RONDE : QUELS REGARDS DES PATIENTS ET DES ASSOCIATIONS DE MALADES ? 105
Mme Martine Bungener, directrice de recherche au CNRS, économiste, présidente du GRAM, groupe de réflexion avec les associations de malades de l'INSERM 105
Mme Catherine Vergely, secrétaire générale de l'Union des parents d'enfants atteints de cancer et de leucémie (UNAPECLE). 107
Mme Laurence Tiennot-Herment, présidente de l'AFM-Téléthon (Association Française contre les Myopathies). 110
M. Yann Le Cam, directeur général d'EURORDIS (Organisation européenne des maladies rares), vice-président du Comité d'experts sur les maladies rares (EUCERD). 113
OUVERTURE
PAR MM. ALAIN
CLAEYS ET JEAN-SÉBASTIEN VIALATTE,
DÉPUTÉS,
RAPPORTEURS
M. Alain Claeys, député de la Vienne, rapporteur. Je vous remercie d'être présents aujourd'hui pour cette deuxième audition publique consacrée à la médecine personnalisée.
Je rappellerai rapidement le contexte de notre étude que certains d'entre vous connaissent déjà. L'Office a été saisi par la commission des affaires sociales d'une étude sur les enjeux scientifiques, technologiques et éthiques de la médecine personnalisée, la lettre de saisine spécifiant notamment que cette médecine exigeait des investissements coûteux ainsi que la nécessité de définir des règles précises de conservation des données.
Notre première audition publique du 27 mars dernier visait à définir la médecine personnalisée à travers les pathologies ciblées, les outils utilisés, et à mesurer son impact en termes de recherche-développement (R&D). Des évolutions scientifiques et technologiques considérables ont fait découvrir la complexité extrême du vivant et sa profonde diversité. Les outils comme le séquençage à haut débit, la bio-informatique, les nanotechnologies, ont permis de progresser dans la compréhension du fonctionnement normal et pathologique des gènes, des cellules, des tissus, des organes. Comme le constatait le Pr André Syrota, la médecine devient individualisée parce que le vivant est individualisé. La découverte de nouvelles cibles, de nouvelles voies de signalisation, de leur modification au cours de telle ou telle pathologie, ouvre des perspectives nouvelles pour les malades. Ainsi le vivant est-il scruté, analysé voire réparé, ce qui suscite chez nos concitoyens des espoirs mais aussi des craintes fondées et infondées, qu'il nous appartient de prendre en considération.
L'audition publique d'aujourd'hui tend à mesurer les défis éthiques et sociétaux que la personnalisation des traitements implique. Nous avons constaté lors de nos visites et auditions que le concept de médecine personnalisée faisait débat, induisant un questionnement subtil non seulement sur la relation médecin-malade, mais également sur la place du patient et sur celle des différents intervenants impliqués dans cette relation. Comment ce colloque singulier entre le médecin et son patient évoluera-t-il à l'heure de la génomique ? Comment le patient lui-même gérera-t-il les informations qui lui parviendront sur sa propre santé présente, voire future, et sur celle des membres de sa famille, informations qui vaudront pour le présent et parfois pour l'avenir ? L'intervention de la médecine se fera-t-elle plus tôt dans la vie des personnes ? Même en étant bien portant, sans signe clinique apparent, le patient aura noué une relation avec le médecin, car les possibilités médicales permettront d'anticiper une pathologie susceptible d'apparaître dans cinq ou dix ans. Quel sera l'impact éthique et sociétal de la baisse drastique des coûts de séquençage ultra rapide du génome, sur laquelle chacun s'accorde ?
Voilà autant de questions que nous devons nous poser, et pour lesquelles il nous faut essayer d'apporter des réponses. Une autre interrogation est également importante : qui aura accès à cette nouvelle médecine et comment les systèmes de santé fondés sur la solidarité comme le nôtre, pourront-ils fonctionner ? Je pourrais multiplier les interrogations, mais c'est l'objet des tables rondes d'aujourd'hui de tenter de donner des éléments de réponse.
En vous renouvelant nos remerciements pour votre participation, je passe la parole à Jean-Sébastien Vialatte
M. Jean-Sébastien Vialatte, député du Var, vice-président de l'OPECST. Je m'associe aux remerciements d'Alain Claeys. Comme je le constatais en conclusion de la première audition publique, qui fut très instructive, le caractère flou du concept de médecine personnalisée induit plus de questions que de réponses tranchées. Nous sommes en quelque sorte à la croisée des chemins, les outils technologiques désormais à notre disposition ont accru très rapidement nos connaissances. Les pathologies sont mieux caractérisées qu'autrefois. Une nouvelle nosographie se dessine. Il en va de même des traitements. Parallèlement, les personnes tendent de plus en plus à connaître leurs déterminants de santé, qu'ils soient génétiques ou autres. Cependant, malgré la possibilité qu'offre la médecine personnalisée de préciser et de quantifier des facteurs de risques individuels, ces derniers ne se transformeront jamais en certitudes. Un risque restera toujours un risque, donc source d'incertitude malgré l'apport de la science. Les médecins devront décider du traitement à offrir à leur patient selon des résultats qui confèrent certes plus de certitude, mais qui possèdent aussi une incertitude inhérente. Comment par exemple, prendre la décision clinique de donner tel ou tel traitement à un patient dont le profil génétique implique qu'il a 30 % de chance de subir des effets secondaires ? Dès lors, quelles seront les attentes des patients surinformés de leurs éventuelles prédispositions ? Que préviendra-t-on ? Que soignera-t-on ? Comment évoluera la relation médecin-malade dans ce contexte ? Ce sera le sujet de notre première table ronde, ce qui sera sans doute l'occasion de débattre de la définition de la médecine personnalisée.
Il est probable que dans quelques années, les patients se présenteront avec une série de données de santé incluses dans leur portable. Une autre partie pourra être conservée dans telle ou telle banque de données au risque de diffusion malencontreuse, voire volontaire, s'il s'agit de piratage. Quel sera le niveau de protection de ces données très personnelles ? Est-on en mesure de garantir cette protection à nos concitoyens ?
En tant que biologiste, je constate que la médecine personnalisée suscite d'immenses espoirs car elle est perçue comme une médecine de pointe. Le niveau d'information du grand public est désormais élevé. Mais quelles sont les informations retenues et privilégiées par lui ? Il est probable que l'on n'échappe pas totalement à un certain goût du sensationnel, générateur de déception, car si la connaissance des mécanismes pathologiques a progressé, le transfert et l'application de ces découvertes au lit du malade se révèlent bien lents. Lors de la première audition publique, nous avons eu l'occasion d'en débattre pour constater que la médecine personnalisée induit un changement de paradigme dans le modèle de R&D des médicaments, car elle va à l'encontre de la logique d'application d'un même traitement à un grand nombre de patients. Les blockbusters assuraient un certain équilibre dans le portefeuille des molécules détenues par un industriel, et pour les systèmes de santé publique, les traitements à large spectre permettaient de faire facilement accéder tous les citoyens à un même niveau de soins. Or la médecine personnalisée est une médecine stratifiée qui oblige à reconsidérer le modèle médicaments et induit l'utilisation d'un test compagnon du traitement. Comment financera-t-on l'accès équitable à ces nouveaux traitements ?
Quelle sera la réaction des grands groupes pharmaceutiques après la récente décision de la Cour suprême américaine dans le litige médiatisé opposant la société Myriad Genetics et deux associations, l'Union américaine pour la défense des libertés et la Public Patent Foundation ? L'ADN humain est un produit de la nature et ne peut donc être breveté a décidé très récemment la Cour Suprême. Dès lors quel sera l'impact de ces changements de paradigme sur l'accès aux soins ? Comment améliorer l'organisation de notre système de santé pour garantir un égal accès aux traitements et une égale qualité des soins ?
Nous savons combien les associations de patients sont investies aux côtés de la recherche, et parfaitement au fait des promesses cliniques que recèlent les avancées biomédicales. On mesure d'ailleurs leur impact car aux États-Unis elles se sont impliquées dans le procès contre Myriad Genetics . Les attentes sont donc fortes, chez les malades et leurs familles. Il est fort important pour nous parlementaires de mieux connaître la perception, le rôle qu'entendent jouer les associations de patients, car elles représentent les premiers concernés, à savoir les malades. Quelle est leur vision du système de santé ? Comment l'adapter ? Ce sera l'objet de notre quatrième table ronde.
La médecine personnalisée reste un concept paradoxal pour la médecine contemporaine. Très scientifique et objective, fondée sur une masse impressionnante de données numériques dont l'interprétation est dévolue à des tiers à la relation médecin-malade, elle cherche à prendre en compte tous les paramètres de la situation unique de chaque patient.
PROPOS INTRODUCTIFS
Pr Jean-Claude Ameisen, immunologiste, président du Comité consultatif national d'éthique pour les sciences de la vie et de la santé (CCNE). Comme vous le disiez, la notion de médecine personnalisée pose autant de questions qu'elle apporte de réponses possibles, et un premier point est qu'au fond cette démarche d'individualisation, d'adaptation de la médecine à la singularité de la personne, est ancienne. Traiter par antibiotique une personne infectée en vérifiant que la bactérie est sensible à cet antibiotique, est une démarche ancienne. Il en va de même, de l'adaptation au fonctionnement rénal de la personne, de la dose d'un médicament éliminé par le rein. La découverte des groupes sanguins et leur application dans les transfusions, la découverte des groupes HLA et leur application dans les greffes, sont ainsi des démarches de ce que l'on appelle aujourd'hui une médecine personnalisée.
Le terme donne l'impression qu'il s'agit d'une médecine sur mesure, s'adressant spécifiquement à la personne en tant que telle, c'est l'une des ambigüités. Or il s'agit comme pour toutes les démarches de la médecine, -et même si c'est un changement d'échelle et peut-être pas de paradigme-, de rattacher le patient à un sous-groupe abstrait de personnes qui sur les caractéristiques nous intéressant lui ressemblent le plus. D'une certaine façon, la médecine personnalisée, si l'on veut évaluer son efficacité par une démarche de médecine fondée sur les preuves, donc sur les statistiques, n'est pas une démarche sur mesure, mais plutôt d'appariement le plus congruent d'un patient à d'autres groupes de patients et à des croisements de ces groupes. C'est la congruence de cet appariement à d'autres sous-groupes qui définit la notion même de médecine personnalisée.
En outre, ce concept fait appel de manière peut-être plus frappante à des croisements de sous-groupes où chaque personne est une mosaïque, même si cela a toujours été le cas. C'est plus visible en cancérologie, puisque le séquençage du génome de la tumeur permettra d'apparier la tumeur du patient à des tumeurs d'autres personnes, et le séquençage du génome du patient permettra de l'apparier pour les effets secondaires et la tolérance aux traitements à d'autres personnes. Donc, on dissocie dans ces appariements, différents éléments qui constituent cette personne ; il y a ainsi un effort de formalisation mathématique, encore débutant qui vise à considérer les personnes comme des réseaux de molécules, de gènes, de fonctionnements. On est devant un paradoxe : ce que l'on appelle « médecine personnalisée », revient au fond à l'étude et à l'appariement de points précis dans des nuages de points. Comme le traitement lui-même repose sur la recherche et donc sur la validation statistique, cette médecine, ce traitement personnalisé, devient un sujet de recherche qui rétrospectivement, déplacera les points que l'on avait inscrits dans d'autres nuages de points. C'est mouvant et d'autant plus efficace que la singularité de la personne peut être décomposée en éléments non singuliers. C'est donc un peu différent de la vision habituelle que la société se fait de la démarche.
On met l'accent justifié la génomique, mais elle n'est pas, et de loin, la seule à prendre en considération. Le généticien Richard Lewontin disait que l'intérieur et l'extérieur s'interpénètrent, que chaque être vivant est à la fois le lieu et le produit de ses interactions. Ainsi l'environnement, dans la façon dont il déterminera la manière dont les gènes sont utilisés par les cellules et les corps, détermine en retour la manière dont les gènes sont utilisés par les cellules et les corps. L'une des approches de la médecine personnalisée, très différente des approches habituelles, consiste à équiper des personnes de capteurs qui les suivent dans leur environnement au fur et à mesure de leurs déplacements, et révèlent toutes les molécules, éventuellement toxiques, avec lesquelles cette personne singulière est en contact. Il y a, en outre, le microbiome, les centaines de milliers de milliards de bactéries que nous hébergeons, et dont des études montrent aujourd'hui la singularité de la composition chez chaque personne. Le microbiome a un effet sur notre métabolisme, nos réactions inflammatoires et sur la manière dont fonctionne notre système immunitaire. Les éléments de l'environnement, les éléments constitutifs de la personne et les microbes que nous hébergeons, constituent une singularité évolutive et multiple.
Cela produit une masse extraordinaire de données, suivant les aspects singuliers de la constitution biologique et de l'environnement que l'on étudie. Cela pose en effet un problème de protection de la vie privée, de la confidentialité, car au fond ces données ne sont utiles que si elles sont recueillies sur un nombre suffisant de personnes, et si elles sont centralisées ou circulent d'un laboratoire à un autre. Comment les protéger, d'autant que des travaux récents indiquent que l'idée de protéger l'anonymat de données génétiques est sans doute illusoire ? C'est donc un véritable problème à prendre en considération. L'interprétation de ces données pose un deuxième problème : faire la part de variations singulières et de ce qui peut avoir une traduction significative en termes de maladie ou de santé.
Le troisième souci réside dans la discordance entre ce que l'on peut interpréter, et ce que l'on peut changer en intervenant. Comment communiquer cette information ? Que faire de cette information ? Dans l'avis n°120 récent du Comité consultatif national d'éthique (CCNE) sur les « questions éthiques associées au développement des tests génétiques foetaux sur sang maternel, le CCNE a réfléchi sur l'évolution des outils de séquençage, et une question se pose. Faut-il tout lire et choisir ensuite ce que l'on va dire, faire le tri entre le signifiant et non signifiant ? Faut-il ne pas tout lire ? Ce serait peut-être la première fois dans la médecine que des médecins s'interdiraient d'acquérir une information par peur de ne pas savoir quoi en faire. Cette masse d'informations et la difficulté de l'interpréter engendre des problèmes nouveaux particulièrement en matière d'organisation et de réflexion sur le conseil génétique. On se trouve face à un décalage de plus en plus grand entre ce à quoi la technique donne accès, les règles mises en place sur le choix libre et informé, et ce que la personne peut faire de l'information.
Une autre implication, vous l'évoquiez, est que la médecine personnalisée va modifier la différence entre les maladies rares et les maladies dites fréquentes en faisant de ces dernières une myriade de maladies rares pour les traitements. Sur le plan économique, il faudra repenser la manière dont on recherchera les traitements adaptés. Nous disposons d'une expérience dans ce domaine avec les maladies rares, les nouveaux modèles de R&D de médicaments mis en place dans le cadre de cette particularité, pourront devenir le nouveau modèle.
Paradoxalement, il existe aussi me semble-t-il, un risque d'exclusion. En effet, si on estime que mieux vaut donner à un sous-groupe de patients un traitement efficace avec des effets secondaires rares, que fait-on avec les sous-groupes de patients pour lesquels on ne dispose pas de médicaments efficaces et bien tolérés ? Si l'on ne donne pas le traitement parce qu'il n'est pas assez efficace d'un point de vue statistique et engendre des effets secondaires ; il marche, mais un peu, quel sera le seuil ? À un moment, on exclut les personnes pour lesquelles le résultat serait moins bon. Or si on donne ce médicament à tout le monde, à ce moment-là il n'est guère utile de séparer les sous-groupes. C'est donc un véritable problème, sauf si, pour chaque sous-groupe, le développement thérapeutique donnait des résultats équivalents, ce qui est a priori peu probable, en tout cas dans une même durée.
La prédiction n'est jamais qu'une extrapolation du passé. Il n'existe pas de boule de cristal dans laquelle les chercheurs lisent l'avenir. Si les conditions changent, les mêmes relations de causalité n'induiront pas les mêmes conséquences. Encore une fois, la prédiction consiste à inclure le patient dans un groupe, sans prendre spécifiquement en considération son avenir. On écrase l'avenir, dans la mesure où on donne un poids démesuré à une forme de prévision, par rapport à l'ensemble des autres évènements imprévisibles. Dès lors, libère-t-on, augmente-t-on le degré de liberté de la personne, ou risquons-nous de l'enfermer ? Que fait-on d'une information quand la prédiction en particulier ne peut pas se traduire en prévention, et qu'elle est simplement un dire de l'avenir ? Arnold Munnich disait que « prédire sans pouvoir prévenir, c'est médire ». Cela interroge sur le service que l'on rend éventuellement à prédire l'avenir.
Un article est paru la semaine dernière sur la version Internet de la revue Science, qui corrèle des profils génétiques sur un grand nombre de personnes, à la capacité de poursuivre des études et au niveau d'études que l'on peut poursuivre. Dans ce domaine éloigné de la santé, cela va-t-il aider les personnes, ou les instrumentaliser, les enfermer dans un avenir prédit ? C'est une question importante.
Un des paradoxes de la médecine personnalisée, ce qui montre que ce terme de personnalisé est étrange, consiste à réinsérer une personne dans un groupe. C'est-à-dire : vous, personne singulière, vous avez un diabète, un cancer, un risque de maladie d'Alzheimer, de maladie psychiatrique. La démarche tout à fait naturelle de vous ranger dans un groupe pour pouvoir mieux définir, mieux traiter, possède un pouvoir ou un risque potentiel de discrimination. Comme toutes les démarches médicales, la médecine personnalisée comporte un risque de discrimination, car elle range la personne dans un groupe. Cela amène à réfléchir profondément.
Comme on l'évoquait dans l'avis n° 120 précité, cela interroge sur le rôle que la société veut voir jouer au médecin. C'est un problème de formation. Comment interpréter et pouvoir transmettre cette masse énorme de données, comment faire ré-émerger la personne derrière cette approche médicale qui paradoxalement en fait une identité absolument abstraite, un point dans un nuage de points ? Faut-il repenser le rôle de la médecine en faisant du médecin un médiateur, un interlocuteur parmi tous les spécialistes qui recueillent et interprètent les données ? Doit-il être un homme-orchestre qui sait à la fois interpréter les résultats du séquençage du génome et de toutes les données, et en plus parler à la personne ? Cela amène à s'interroger sur un risque paradoxal, pas si étonnant que cela, de dépersonnalisation lié à l'approche de la médecine personnalisée. Au fond, la véritable singularité de la personne, son histoire, ses espoirs, ses craintes, apparaissent comme une gêne, une interférence avec un processus qui justement a essayé d'établir un profil-type par rapport aux groupes où on l'a rangé.
Longtemps la médecine s'est définie comme un art dans la mesure où elle n'était pas une science, et désormais, il s'agit peut-être de savoir si à mesure qu'elle devient une science, elle peut également rester ou redevenir un art, celui de prendre en compte le patient dans sa singularité, en tant que personne humaine. Le rôle des sciences humaines et sociales, de la réflexion éthique, des associations de patients et des autres secteurs de la société est sans doute important, non pas comme demandeurs, mais comme partenaires à part entière dans cette réflexion. Le terme de médecine personnalisée est sans doute trop ambigu et pas parfaitement adéquat, car il semble dire que la personne est prise en considération alors qu'en fait, elle doit l'être en plus de cette démarche.
Cette approche exige, comme toutes les avancées en médecine, de se garder du triomphalisme, risque principal. En effet, il empêche une réflexion critique, court-circuite toutes les manières de réfléchir avec l'impression d'être devant la solution à tous les problèmes, comme il y a douze ans, avec le séquençage du génome humain qui pendant un temps relativement bref a donné l'impression que toutes les maladies allaient être guéries, et que la médecine allait radicalement changer.
Pr Hervé Chneiweiss, président du comité d'éthique de l'Institut national de la santé et de la recherche médicale (INSERM), directeur du laboratoire de neuroscience de l'institut de biologie Paris-Seine, membre du conseil scientifique de l'OPECST . Vous avez tous les trois déjà très bien posé les questions. Je vais les reprendre simplement pour les présenter d'une autre façon. Qu'est-ce que la médecine personnalisée ? En tant que médecin, la première question est : pourrait-il exister une médecine qui ne soit pas personnalisée ? Le colloque singulier patient-médecin, caractéristique de la médecine, est évidemment toujours une démarche personnalisée. Les hasards de la traduction automatique m'ont fait trouver une piste. Si vous regardez « personalized medicine » par un traducteur de type Google, cela donne "médecine orientée vers le patient". Cela ressemble beaucoup aux conclusions que Jean-Claude Ameisen présentait : on peut se demander si la technicité ou l'illusion de la technicité ne nous aurait pas conduits à perdre de vue le patient derrière la maladie. Effectivement, quand j'étais étudiant en médecine, un de mes maîtres répétait sans cesse « la médecine est un art qui meurt et une science qui naît ». Le modèle anatomo-clinique fondé en France par Magendie, puis Bichat et Broca, les progrès de la biologie, ont permis les succès de cette médecine basée sur les preuves, permettant l'élaboration de recommandations de plus en plus précises. Grâce aux progrès des biotechnologies, nous pourrions la dépasser, c'est ce que l'on imagine possible dans un avenir proche, obéissant ainsi à une certaine idéologie de la promesse. C'est dès demain que nous pourrions lire la séquence de l'ADN ou enregistrer les paramètres de ce corps numérique avec les 400 mesures déjà possibles : la pression artérielle, la glycémie, l'imagerie de différents organes, etc .
De fait, tout un corps numérique est en train d'apparaître à côté du corps réel. Allons-nous céder à la croyance que nous pourrions tout voir/mesurer, donc tout prévoir, tout dépister, tout prévenir, tout traiter ou tout remplacer ? Axel Kahn, dès les années quatre-vingt-dix évoquait une médecine instrumentale unique, et Claude Le Pen, l'émergence d'un médecin ingénieur qui gère une maladie dont les malades ne sont que les supports. Il ne s'agirait à coup sûr pas d'une médecine de la personne mais d'une technologie médicale adaptée à chaque individu.
Mais que pourrait-on raisonnablement reprocher à cet objectif de délivrer le bon traitement au bon sujet et au bon moment ? Cet objectif final doit offrir une meilleure santé, réduire les coûts globaux en sortant du système non efficace du « one fits all » , c'est-à-dire un même médicament pour la même maladie pour tous. Le raisonnement est en effet simple : des médicaments dangereux et inefficaces provoquent des décès évitables, des réactions indésirables, les fameux effets secondaires dont certains entraînent des hospitalisations coûteuses, et pour le moins un gaspillage en médicaments qui ne fonctionnent pas. La médecine adaptée à chaque individu suppose toutefois une infrastructure de soins qui n'existe pas. Dès aujourd'hui, l'accès aux soins reste très souvent difficile pour les patients ; elle sera un fardeau plus lourd encore pour le système de santé « d'une médecine à la carte ». Déjà les coûts explosent au fur et à mesure des nouvelles techniques « pour tous ». Quelle que soit l'orientation prise, on constate que la maîtrise de ces coûts semble inaccessible pour la recherche, pour un usage clinique en particulier, et pour les nouvelles technologies qu'exige la médecine personnalisée.
En conséquence, on observe d'ailleurs que les différents acteurs, au lieu de s'engager sur la voie royale de cette médecine de demain, ont tendance à ne pas se précipiter. L'industrie pharmaceutique est réticente à s'adapter car la médecine personnalisée réduit la taille du marché et les bénéfices liés à ses médicaments vedettes, car derrière les « one fits all » , il y a aussi les blockbusters. Le système prudentiel résiste aux changements car le retour sur investissement des diagnostics et des thérapies adaptées à chaque individu n'est tout simplement pas là. Au contraire dans un premier temps, cette démarche, y compris la démarche de prévention coûte cher, et la perspective de rentabilité est éloignée. Les patients pourraient être une force motrice, mais à part quelques groupements autour de maladies rares ou du VIH, les associations de patients pour des maladies plus générales, comme le diabète, ne sont pas présentes. Les organismes de règlementation continuent de débattre sur la meilleure façon de gérer la nouvelle complexité, et l'on peut par ailleurs se demander dans quelle mesure ces agences de régulation ont le pouvoir légal de créer de nouvelles règles.
La médecine personnalisée oblige à tout un ensemble de tests-compagnons du traitement : génétiques, d'imagerie médicale, de biologie, et d'autres aspects encore. Il est éthiquement important de déterminer quelle entité doit contrôler l'accès à ces différents tests. Est-ce la loi du marché comme dans certains pays, les payeurs, le médecin ou les ordres médicaux, le Gouvernement ? Devons-nous continuer à faire confiance à l'industrie pour la mise sur le marché de tests qui répondent à des normes nécessairement plus strictes ?
On se demande finalement si ce corps numérique n'est pas aujourd'hui l'objet d'une vaste dérèglementation, avec des outils ou des procédés de traitements qui apparaissent, alors que la preuve n'a pas été totalement faite. Lors d'une réunion scientifique récente il était fait allusion à une société de biotechnologies récemment rachetée, qui a mis au point un appareil permettant de détruire certaines petites ramifications nerveuses autour de l'artère rénale pour traiter l'hypertension. Il semble que les fondements médicaux sur laquelle repose cette technologie soient extrêmement minces, en termes « d'evidence-based médicine » . Mais comme c'est un procédé de soin, il échappe à toute une partie de la règlementation. Comme techniquement il fait dans de bonnes conditions de sécurité ce qu'il annonce, à savoir détruire les ramifications nerveuses visées, il peut obtenir un label CE et apparaître dès lors comme un procédé certifié.
Nous devons également considérer que nous sommes devant une société ouverte dans laquelle les patients disposent d'un droit à l'information sur leur santé, et peuvent aussi exiger que des tests soient disponibles. Nous sommes dans une société de l'Internet et donc si ce n'est pas disponible en France ou s'il existe une régulation française, il n'est pas nécessaire d'aller très loin pour arriver à obtenir des avis ou des possibilités d'accès supplémentaires.
En 2010, le Nuffield Council en Grande-Bretagne a rendu un avis sur le « medical profiling » et la médecine en ligne, et cela s'appelait l'éthique de la médecine personnalisée, « ethics of personalized care in a consumer age ». Le Nuffield Council a identifié cinq grands chapitres de tension, où s'opposent à chaque fois dans ce droit à l'information, les avantages et les inconvénients de la mise à la disposition du grand public de ces tests. Il s'agit de tests génétiques, mais aussi d'autres tests. Ainsi on trouve en ligne de nombreux tests pour auto-diagnostiquer ou non un début de maladie d'Alzheimer. On peut s'interroger sur la validité de ces tests et sur leur intérêt. D'un côté les avantages sont une meilleure information, une meilleure prise en charge, un meilleur respect de l'autonomie de la personne, un meilleur usage des deniers publics, de l'autre côté les risques sont liés aux difficultés de compréhension des résultats. Ce qui est vrai pour les variants génétiques, l'est tout aussi pour l'imagerie médicale, et pour d'autres paramètres de la personne, qui peuvent varier au cours du temps (pression artérielle, glycémie, etc .). Le risque est également un éloignement, voire la perte de la relation médecin-malade. Si tout passe par Internet, il n'y a plus de relation personnelle, on risque l'atteinte à la vie privée et le mercantilisme dans les pays où l'on peut faire de la publicité directe par rapport à un certain nombre de tests. Je pense que Dominique Stoppa-Lyonnet évoquera ces questions de susceptibilité liées au diagnostic. Par exemple pour le cancer du sein : la publicité de Myriad Genetics était « vous avez toujours voulu savoir, maintenant, vous pouvez savoir », par rapport à une mutation qui portait sur 2 à 3 % des cancers du sein...
Trois grandes questions éthiques sont dès lors ouvertes :
- en premier lieu l'individualisation permet un traitement sur mesure, pas toujours nécessaire, et n'évitant pas toujours tous les effets secondaires. Il existe un risque d'idéalisation de cette personnalisation, dans la tendance générale du zéro défaut, ou du zéro risque ;
- en second lieu, elle permet la responsabilisation, avec les limites de la responsabilité individuelle. Si l'on vous informe sur tous vos facteurs de risques, comment cette responsabilisation entrera-t-elle en conflit éventuel avec le principe de solidarité de l'assurance maladie ? Un assureur remboursera-t-il les soins d'un diabétique qui n'a pas arrêté de manger du sucre ?
- en troisième lieu les personnes orientées par des groupes de pression, par la publicité ou le marché, voudront avoir systématiquement accès au dernier test ou à la dernière molécule. Dans ce consumérisme le médecin n'est pas forcément celui qui est le plus écouté.
L'essentiel est d'intégrer les bénéfices de la biomédecine adaptée à une histoire de la personne dans un questionnement éthique, de ne pas confondre le corps numérique avec le corps réel, de ne pas confondre le corps avec la personne, d'être capable de combiner la technique et la considération de l'être, qui ne se résume pas à la maladie, ni même à la personne malade.
Pour reprendre une formulation d'André Grimaldi, il faut créer la médecine intégrée plus que la médecine personnalisée, une médecine intégrée qui associe à la composante biomédicale adaptée au corps numérique, la composante psychosociale, adaptée au sujet, et une composante pédagogique essentielle adaptée à la personne. Il faudra former des personnels à la prise en charge des différents éléments de ces corps numériques. Cela est nécessaire pour donner à l'individu les moyens au quotidien de prendre réellement en charge soit sa pathologie, soit le potentiel d'une pathologie, et surtout bien vivre avec.
PREMIÈRE TABLE
RONDE :
MÉDECINE DE PRÉVENTION OU DE
PRÉDICTION : QUELLE RELATION MÉDECIN-MALADE ?
Mme Emmanuelle Prada-Bordenave, directrice générale de l'Agence de la biomédecine. Merci beaucoup M. le président d'avoir permis à l'Agence de la biomédecine de participer à cette table ronde et de vous présenter son rôle. Cette Agence publique est une agence sanitaire qui dépend de la Direction générale de la santé du Ministère chargé de la santé. Dans le cadre de la réunion d'aujourd'hui, c'est-à-dire de la génétique et de la médecine personnalisée, je parlerai uniquement de la relation médecin-malade. Quel rôle, agence de l'État, pouvons-nous jouer ?
Le Parlement nous a demandé de nous intéresser à cette question. Il a confié à l'Agence un rôle général en matière de génétique (article L.1418-1 du code de la santé publique), et un rôle particulier sur la réflexion en amont et l'élaboration d'un arrêté de « bonnes pratiques applicables à la prescription et la réalisation de l'examen des caractéristiques génétiques d'une personne et de son identification par empreintes génétiques à des fins médicales » (article L. 1131-2 du même code). C'est donc en liaison étroite avec un groupe de professionnels que nous avons réfléchi pendant plusieurs mois à l'élaboration de cette proposition, qui a fait l'objet finalement d'un arrêté en date du 27 mai dernier, publié au Journal officiel du 7 juin 2013.
Nous ne sommes pas les seuls à intervenir aux côtés des patients et des professionnels en tant qu'instance publique et à nous préoccuper de cette question, puisque, selon les sujets et les domaines, d'autres autorités publiques interviennent : la Haute autorité de santé (HAS), l'Institut national du cancer (INCa) pour les sujets relatifs au cancer, l'Agence nationale de sécurité du médicament et des produits de santé ( ANSM ) pour le contrôle qualité des dispositifs médicaux car les tests génétiques sont des dispositifs médicaux de diagnostic in-vitro au sens des textes.
L'Agence de la biomédecine n'intervient pas dans le domaine de la génétique somatique, c'est-à-dire l'étude des tumeurs, mais se concentre sur la réalisation des bonnes pratiques, et plus généralement dans son champ de compétence, sur la génétique constitutionnelle, c'est-à-dire l'étude des caractéristiques génétiques de la personne elle-même. Notre champ de compétences couvre tout le champ de l'histoire de la vie de l'homme, depuis presque avant son commencement, puisque le diagnostic préimplantatoire est fait avant toute grossesse, et éventuellement ensuite un diagnostic prénatal. Arrive ensuite la génétique constitutionnelle post-natale et c'est ce dont il sera question aujourd'hui.
Je voudrais vous donner quelques chiffres, car les laboratoires qui travaillent dans ce domaine sont tenus d'adresser un rapport annuel d'activité à l'Agence. Ce sera dans le rapport à venir : il y a eu en 2012 dans notre pays 82 756 tests de cytogénétique et 416 767 tests de génétique moléculaire, qui couvrent aujourd'hui plus de 1 500 maladies. Il est intéressant de se reporter en 2009, car ces tests couvraient simplement à peu près 1 000 maladies. Il s'en est rajouté un peu plus de 500, et surtout les tests de génétique moléculaire étaient seulement 271 330. On voit donc une augmentation vertigineuse du nombre de tests entre 2009 et 2012. 228 laboratoires réalisent ces tests, et nous adressent régulièrement leurs rapports d'activité.
Les règles de bonnes pratiques ont été élaborées avec énormément de soin car, au sein de la médecine, et cela a été rappelé tout à l'heure par les orateurs précédents, cette génétique tient une place très particulière et le diagnostic génétique a des implications et une nature particulière. Le résultat d'un examen de génétique pour un patient est définitif. Il parle de ses caractéristiques génétiques, acquises à un stade précoce de son développement, ou transmises. Á partir de sa naissance ou de la fin de ce stade précoce, elles sont définitivement acquises et peuvent avoir des conséquences sur l'ensemble de sa famille. Aussi, les patients sont-t-ils très différents de ceux rencontrés habituellement dans un cabinet médical.
Pour des personnes malades, bien évidemment, une analyse génétique amènera un diagnostic. Cependant il existe aussi toute une catégorie de personnes, que l'on appellera patients, mais qui ne sont pas vraiment malades, ou en tout cas ne présentent pas les signes de la maladie. Ce sont par exemple, les membres de la famille de la personne qui s'est présentée spontanément pour une maladie : toutes ces personnes ne sont pas vraiment malades, ne présentent pas de symptômes et pour eux, la relation médecin-malade sera complètement différente. Dans cette catégorie, se trouvent d'abord des personnes asymptomatiques, que l'on dit porteurs sains. On en connaît l'existence depuis des siècles. Une analyse génétique révélera un risque de transmission à la descendance et pourra déboucher, s'il y a un projet parental, sur du conseil génétique. Puis on trouve des personnes encore asymptomatiques, mais que l'on peut dire pré-symptomatiques : elles ont dans leur génome la trace d'une maladie déjà présente, mais qui se révèlera plus tardivement. C'est là que sera posé un diagnostic pour le futur, comme l'expliquait Jean-Claude Ameisen. Cette maladie se révèlera dans le futur : aujourd'hui, on en est sûr.
Vient ensuite la prédiction avec les tests de prédisposition. Une personne totalement asymptomatique est soumise à un test de prédisposition, et l'on va déterminer un facteur de risque de développer une maladie avec une forte probabilité.
En dernier lieu, il y a les tests de susceptibilité qui finalement occupent en grande partie nos sujets de réflexion ; cela concerne bien plus de monde. Cela touche des personnes asymptomatiques, qui vont très bien pour lesquelles on fera une analyse génétique et déterminera un facteur de risque de développer une maladie. Pour eux, le risque est faible, parfois très faible. Nous avons tous des variants génétiques, parmi lesquels beaucoup sont identifiés ; or l'on sait qu'ils ne modifient que très faiblement les risques de maladie, et que le risque de développer la maladie est bien inférieur à celui de la prédisposition. Il est très important de souligner que l'anomalie génétique n'est ni nécessaire, ni suffisante pour développer la maladie. Alors que dire à ces patients, comment les prendre en charge ? Sont-ils vraiment des patients, ou simplement monsieur et madame tout le monde ?
Un autre point a été évoqué et porte sur les tests compagnons. Ils concernent des patients non symptomatiques d'une maladie génétique, mais sans doute atteints d'une pathologie, et auxquels on s'apprête à prescrire ou pas un médicament. Il s'agira alors de prédire la toxicité ou l'efficacité de ce médicament par ces tests. Ce sont bien sûr des analyses de pharmacogénétique.
Ce que nous essayons de faire passer, chaque fois que nous avons la parole en tant qu'agence de l'État, c'est l'idée que l'homme n'est pas tout génétique, et que bien sûr ces différents patients et les différents examens et diagnostics montrent une combinaison avec des pondérations variables entre les facteurs environnementaux et génétiques. Il faut que progressivement les Français comprennent cette complexité et se l'approprient, car ils sont sans doute encore restés sur l'idée qu'un variant génétique détermine avec certitude une maladie, très grave.
Nous avons élaboré cet arrêté qui définit les règles de bonne pratique en liaison avec la Haute Autorité de santé. Il insiste sur le fait que les caractéristiques de génétique constitutionnelle sont définitives, que les résultats ont des conséquences non seulement pour la personne mais aussi pour sa famille. Cet arrêté a vocation à être diffusé très largement, bien au-delà du cercle un peu restreint des spécialistes de la génétique, pour toucher les prescripteurs. Il n'y a pas aujourd'hui, sauf exceptions très rares, de restriction à la prescription des tests génétiques. Il est donc rappelé dans l'arrêté que l'individu doit rester au centre des préoccupations des acteurs. Une importance particulière doit être donnée à l'information, au consentement, aux modalités de rendu d'un résultat. Les travaux des généticiens qui se sont réunis pendant plusieurs mois au sein de l'Agence, dont certains sont présents ici aujourd'hui, ont tourné en grande partie autour de ces questions d'information, de consentement et de rendu du résultat, notamment pour toutes les découvertes incidentes, et pour tous les variants dont on sait qu'ils n'ont pas vraiment de portée, ou dont on ignore quelle portée ils ont.
Les examens génétiques ne doivent être prescrits que lorsqu'ils ont une utilité clinique et sont souhaités par la personne. Le seul fait qu'un examen soit disponible et réalisable ne justifie ni de sa prescription, ni sa réalisation. Ces règles seront une aide, je pense, pour les prescripteurs, pour la retenue, pour les patients, et pour les finances publiques.
On a évoqué l'augmentation fulgurante du nombre de tests réalisés. Dès qu'un test est inscrit à la nomenclature - il y en a très peu aujourd'hui -, cela se traduit presque immédiatement par une augmentation au décuple du nombre de tests prescrits, et donc de la charge pour l'assurance maladie. La prescription n'est pas soumise à des restrictions, mais dans les règles de bonnes pratiques, il est prévu qu'en principe elle doit être effectuée par quelqu'un qui connaît la maladie et ses aspects génétiques, éventuellement un médecin spécialiste.
Enfin on a formulé une mise en garde sur les tests de susceptibilité. L'attention des médecins qui vont prendre connaissance de ces règles de bonnes pratiques est attirée, sur le fait que la contribution d'un test de susceptibilité est variable et généralement mineure, et contribue très peu, aujourd'hui en tout cas, à la prise en charge de la personne. On attire l'attention des prescripteurs sur la retenue dont ils doivent faire preuve.
La transmission du résultat est très importante, elle doit être faite par le prescripteur et non pas être envoyée par courrier. C'est d'ailleurs dans les textes, le Code de la santé le prévoit. Enfin l'on doit toujours prévoir un accompagnement psychologique. C'est toute la différence d'avec les tests par Internet où vous recevez un résultat non-rendu par un médecin, et non-accompagné d'une explication, et notamment sans accompagnement psychologique, alors qu'il peut être souhaité.
Nous allons à l'automne prochain ouvrir un site Internet à destination du grand public. Il n'est pas encore accessible, mais est en train de se construire en liaison étroite avec le Groupe stratégie en génétique et en diagnostic, dont l'adresse sera www.genetique-medicale.fr , qui permettra de transmettre un certain nombre de ces notions, en tout cas je l'espère.
Pr Anne Fagot-Largeault, professeure au Collège de France, membre de l'Académie des sciences. Je vais essayer de distinguer deux contextes dans lesquels on peut comprendre la notion de médecine personnalisée
Le premier sera celui de la médecine curative, essentiellement le traitement du cancer, qui est à l'origine de la notion. Je rappelle que cette notion est très récente. Elle a été lancée par l'industrie pharmaceutique dans les années 90, il y a donc seulement une petite vingtaine d'années, à propos d'un médicament du cancer du sein. Ici, il s'agit d'une médecine moléculaire adaptée à certains traits du génome de l'individu, et il est très difficile de penser dans quel sens c'est personnel, puisque, comme cela a été très bien dit précédemment, c'est une caractéristique de groupe ou de sous-groupe que l'on cherche chez le malade. L'avantage promis à travers cette attitude de recherche à quel groupe vous appartenez, c'est que d'une part nous serons plus efficaces en ne prescrivant qu'aux bons répondeurs, et d'autre part que nous ferons faire des économies au système de santé, car le prix du séquençage du génome baisse, en prescrivant moins, à juste titre, et même en intervenant de façon préventive si l'on sait détecter les prédispositions sur votre génome. Le côté noir de cette médecine, habituellement passé sous silence, est le problème de ceux qui ne seront pas traités, parce qu'ils seront classés non-répondeurs au traitement. On voit apparaître à travers cette perspective l'aspect inégalitaire de cette médecine.
Le second contexte dans lequel on peut penser la médecine personnalisée, est celui de la médecine régénérative, celle qui fait des greffes, qui répare des tissus, qui vous redonne des parties de votre corps, voire une amélioration esthétique ou de performance. Ici, littéralement, on se rapproche d'une médecine personnelle, sur mesure. Il vaut peut-être mieux dire individualisée plutôt que personnalisée. Puisqu'en raison de la complexité de nos systèmes immunitaires nous sommes tous différents, une médecine pertinente sera adaptée à notre propre système. Bien sûr, ce n'est pas encore tout à fait la réalité : on greffe encore des tissus ou des organes d'autrui en combattant la réaction immunitaire par un traitement immunosuppresseur, mais on commence déjà à voir la possibilité demain, de recourir à des cellules souches venant de la personne malade pour lui reconstruire une peau neuve, des muscles abîmés, un coeur abîmé. Il est exprimé à ce propos des espoirs de rajeunissement ou d'amélioration de l'état de la personne. Cette médecine-là serait réellement adaptée à des besoins particuliers ou des demandes de personnes particulières. Le problème est qu'elle aurait un coût très élevé puisqu'il faudrait une médecine par personne.
On peut distinguer trois niveaux dans cette médecine :
- s'il s'agit de réparer une peau brulée, ou de greffer un rein, on se dit que ce sera pris en charge par le système de santé, ce sera un bon traitement ;
- s'il s'agit de chirurgie esthétique, en principe le système de santé ne le prendra pas sous sa coupe sauf cas spéciaux ;
- mais s'il s'agit d'une médecine d'amélioration, elle n'aura cours que pour les personnes pouvant se l'offrir. Il est clair que l'accès à cette médecine coûteuse serait inégalitaire, car on imagine difficilement que son coût soit supporté par la solidarité.
Je me place maintenant dans la position du malade, du patient, et je fais la remarque générale suivante : il y a souvent à l'heure actuelle une expression des patients qui se plaignent d'une frustration à l'égard de la médecine, qui souffrent d'être pris pour des machines et d'être traités comme tels. Vous téléphonez à l'hôpital, c'est une machine qui répond, vous devez discuter avec une machine. Vos données personnelles, vos paramètres individuels sont numérisés. Ils sont une colonne de chiffres. La pratique des médecins est standardisée dans une médecine « evidence based ». Il y a donc chez beaucoup de patients la revendication d'une médecine qui les comprendrait mieux, personnellement, d'un abord des médecins ou des services médicaux qui prendrait toutes leurs dimensions en compte.
La publicité pour la médecine dite personnalisée tombe dans ce contexte où il y a l'aspiration à un contact personnel, à une empathie du médecin qu'on ne trouve pas, qu'on a le sentiment de ne plus rencontrer. Et l'on croit tout naturellement que la médecine personnalisée va remplir cette attente. C'est faux, bien sûr. Ce qu'on appelle la médecine personnalisée n'a aucun rapport avec cela. Dans la manière dont elle a été lancée, cette médecine est de la publicité. Mais c'est de la publicité à l'occasion de quelque chose de très sérieux, à savoir un tournant difficile à prendre à la fois par l'industrie pharmaceutique, le monde médical, le système de santé. La notion de médecine personnalisée donne une image attractive mais fausse, d'une vérité à la fois attirante et préoccupante : nous avons été habitués à croire que notre médecine, notre système de santé, sont les mêmes pour tous. Or, l'époque de la même médecine pour tous est révolue. Elle devient personnalisée, c'est-à-dire différente pour chacun, « customised », comme on dit, et on le craint, proportionnelle aux ressources de l'individu qui sera capable ou non de se l'offrir.
Pr Philippe Amouyel, professeur d'épidémiologie et de santé publique au Centre hospitalier et universitaire de Lille, directeur général de la fondation Plan Alzheimer. J'aimerais tenter de rendre le plus concret possible cette médecine personnalisée pour pouvoir ensuite analyser son impact sur la relation médecin-malade. Pour ce faire, j'ai essayé de trouver trois exemples publiés dans la littérature, qui donnent des visages à cette médecine personnalisée.
La consultation médicale n'a pas changé, cela reste la relation entre un médecin et son patient. Elle a une particularité tout de même : en général elle est réalisée pour un motif de consultation. On peut espérer que cela ne changera pas et nous verrons qu'indirectement cela aura des implications sur cette consultation médicale. La consultation médicale comprend un interrogatoire, un examen clinique et des examens para-cliniques. Ces informations élargies à la génomique et aux omics en général, existent déjà, certes en quantité moindre. Il y a en général un peu de prévention envers les enfants, essentiellement autour des vaccins et de certains dépistages.
L'apport de ce que l'on appelle la médecine personnalisée, on conviendra tous que c'est un mauvais terme, reste l'étude de vastes ensembles de molécules biologiques et de signatures moléculaires appelées habituellement biomarqueurs. Cela a commencé avec les gènes et le génome, mais cela ne fait que commencer. Les scientifiques développent d'autres systèmes d'information avec le transcriptome, le métabolome, le protéome et tous ces omics qui se déclinent quotidiennement aujourd'hui. On passe d'un mode d'information linéaire, un dosage biologique avec une réponse, à un mode d'information parallèle avec, sur un même sujet, des milliers d'informations que ne peut pas spontanément appréhender un cerveau humain. Il faudra des outils.
Il convient de distinguer deux éléments :
À la consultation du spécialiste, on obtiendra une meilleure description de chaque patient. C'est important, cela répond à une demande. Les cancérologues sont très en pointe dans le domaine, les microbiologistes également. Ces nouveaux examens complexes ajoutent à la précision du diagnostic, à l'amélioration de la prise en charge. Ainsi, nous avons une utilisation quasiment directe. On la qualifie de médecine de précision. Je crois que c'est le bon terme, et l'on peut développer des thérapies ciblées dans le cancer. On est vraiment toujours dans la même relation, avec un apport d'information supplémentaire.
Pour ce qui est de la consultation du généraliste, on change complètement de modèle. Ces sources d'informations vont être rémanentes, codées et accessibles. Une fois la séquence effectuée, on ne va pas la refaire dix fois. Toute l'information sera disponible. En ce qui concerne les tests spécifiques moléculaires, ils sont faits pour une raison particulière en général, pour une cible, une mutation particulière. En une fois, on a toute l'information et elle va être stockée, gardée quelque part. Cela signifie que pour un individu donné il y aura un énorme potentiel d'informations, qui fournira des probabilités plus ou moins importantes de survenue d'évènements. Nous ne sommes plus dans une consultation motivée par un motif de consultation, mais dans des situations absolument non sollicitées, non voulues par le patient, ni même par le médecin. Tout cela décrira un ensemble de déterminants et de situations potentielles, qui soit surviendront dans la vie du patient, soit ne surviendront pas, sans parfois savoir lesquels vont influencer. Une évolution de l'environnement, pour tout un tas de raisons, fera que cette probabilité changera complètement par rapport aux études mises en oeuvre dans le passé, comme le disait très bien Jean-Claude Ameisen.
Ces informations bénéficieront pour certaines, de réponses thérapeutiques et d'actions à mettre en oeuvre, même si ce n'est pas nécessairement un médicament, mais pour beaucoup ce ne sera pas le cas. Le coût pour un million de paires de bases de la séquence va baisser bien plus rapidement que la loi bien connue de Moore, qui a démocratisé complètement l'ordinateur. Cela veut dire que ces éléments vont s'imposer, à un prix relativement accessible. On n'empêchera pas le système de se mettre en place, donc il faut vraiment essayer d'anticiper.
L'autre élément sur lequel j'aimerais insister concerne les conséquences « massives », à cause de la masse d'informations que cela va générer, sur tous les problèmes d'interprétation de ces données liés à leur nature probabiliste. Ce n'est pas parce qu'on connaît l'alphabet cyrillique qu'on est capable de lire la Bible en russe. Il va falloir former les médecins à l'accès à ces informations, informer le patient sur l'accès à ces informations, discuter de l'accès de tiers à ces informations. On sort de la relation médecin-malade et tout ce qui va en découler, en particulier la responsabilité du médecin. Sur des échographies nous avons des responsabilités quant au bon diagnostic, mais cela va être décuplé pour une information génétique. Enfin, il va falloir assurer l'information du patient, et de l'individu qui n'a pas nécessairement une maladie, et surtout la protection de tout cela, avec un accès extrêmement large est très difficile à mettre en oeuvre.
J'ai donc pris trois exemples de la littérature, avec juste une petite information au départ pour vous donner une idée du volume des données.
962 génomes humains disponibles sur Internet ont été analysés. Un génome représente en gros 2 téraoctets par patient ; un millier de génomes représentent 1 pétaoctet ; on commence à entrer dans des chiffres dont il faudra tenir compte. Il y a plus de 25 millions de variations de l'ADN dans l'ensemble de ces à peu près 1 000 génomes. La moitié de ces variations sont individuelles, c'est-à-dire uniques ou simples. On les trouve chez un ou deux individus maximum. Les variants fréquents, plus de 1 %, représentent à peu près 35 % de ces variations, soit à peu près 9 millions. Pour essayer de vous donner une idée du vertige de ce que représente ce volume d'informations : un mégaoctet, c'est un million d'octets, une photo numérique : 4 millions d'octets, un gigaoctet, c'est un milliard d'octets, 20 gigaoctets, c'est l'enregistrement complet des oeuvres de Beethoven. Avec 10 téraoctets on a tous les ouvrages d'une bibliothèque universitaire ; 200 téraoctets, tous les imprimés de la Bibliothèque nationale du Congrès ; un pétaoctet, un million de milliards d'octets, à peu près mille génomes, et 5 120 pétaoctets, c'est le web en 2011. On estime que pour le web, une loi de Moore s'applique tous les 5,3 ans par le doublement du niveau d'informations. 5 éxaoctets, ce sont tous les mots prononcés depuis le début de l'humanité. Je ne sais pas comment ils l'ont calculé, mais c'est ce que j'ai trouvé dans la littérature.
1) Premier exemple : les Beery twins. Les jumeaux Alexis et Noah Beery, (un article est sorti dans Sciences Transnational Medicine en 2011) sont diagnostiqués à deux ans, en 1998, pour une infirmité motrice cérébrale. La Maman, Reeta, observe des fluctuations en particulier au cours de la journée, et on pose à ce moment-là le diagnostic de dystonie dopa sensible. On les met sous L-dopa, et on constate une amélioration spectaculaire de la vie de ces enfants. Mais certains problèmes persistent qui ne sont pas classiques, des tremblements des mains, des maladresses, des troubles de l'attention, et douze ans plus tard Noah commence à présenter des spasmes du larynx violents et des troubles respiratoires graves justifiant des réanimations respiratoires régulières. Entre temps la science a évolué et dans la dystonie dopa sensible on ne cherche que les mutations que l'on connaît à l'époque, c'est-à-dire dans les gènes Tyrosine Hydroxylase et GCH1. Et chez les jumeaux on n'en trouve aucune. Donc question : si ce n'est pas une dystonie dopa sensible, de quoi s'agit-il ? Or il se trouve que le Papa est cadre chez Life Technologies , qui produit les appareils de séquençage de chez Affymétrix en particulier, et il décide avec des chercheurs du Baylor College de réaliser un séquençage complet du génome des deux enfants, du frère ainé, du père, de la mère, et des grands parents. Résultat, on trouve une nouvelle mutation non décrite, qui correspond bien au syndrome, mais elle n'est pas dans les gènes que l'on avait anticipés. Donc un nouveau diagnostic intervient qui permet une modification du traitement compensateur, et on ajoute non seulement un complément en L-dopa, mais aussi en 5 hydroxytryptophane, et un inhibiteur de la capture de la serotonine. Aujourd'hui Noah va bien, il arrive à écrire, il se concentre au collège. Petite conséquence non prévue sur la famille, dans laquelle des fibromyalgies avaient été diagnostiquées chez la mère et la grand-mère : on s'aperçoit qu'elles ont à l'état hétérozygote les mutations des enfants à l'état homozygote ; on les met sous inhibiteurs de la recapture de la sérotonine, et du coup elles vont beaucoup mieux. Voilà un premier exemple des conséquences directes et indirectes de ce type d'information.
2) Deuxième exemple, voici un arbre généalogique familial chargé . Le sujet, Stephen Quake, a 40 ans, un de ses petits neveux est décédé de mort subite, des grands parents et un père morts d'accidents vasculaires cérébraux et d'accidents cardiaques. Il a un peu d'embonpoint mais rien de particulier. Il se pose des questions parce qu'il est ingénieur en bio-informatique dans un laboratoire à Stanford. Il fait faire le séquençage complet de son génome. On lui trouve 2,6 millions de mutations. On fait différents systèmes de calculs de risques ; il se trouve que son risque est élevé pour l'infarctus, le diabète de type 2 et certains cancers. On lui trouve des variants dans trois gènes associés à la mort subite, probablement les mêmes que ceux de son petit neveu qui vient de décéder de mort subite ; des variants associés à l'athérosclérose, et quelques indicateurs de variation de sensibilité aux médicaments. On commence à développer des programmes d'analyse à partir de toute cette information issue des variants, qui permettent d'avoir des visions d'ensemble et de fournir des calculs de risques sur l'ensemble de la vie. Stephen Quake a un risque d'obésité et de maladies coronaires de l'ordre de 40 à 50 % ; cela veut dire que dans toute son existence, il aura 4 à 5 chances sur 10 de développer ce type de maladie. Pour arriver à ce résultat, on a créé des programmes bio-informatiques qui essaient de lier automatiquement mutations, informations associées dans la littérature et risque. On appelle cela de la « bibliomique », cela existe effectivement. Stephen Quake est ingénieur et a publié les résultats de sa recherche avec Euan A. Ashley dans le Lancet en 2010 .
3) Le dernier exemple est celui du patient, Michael Snyder, qui a été suivi pendant plus d'un an et demi. On a pu détecter pendant cette surveillance une infection à HRV, une infection à RSV, et également des changements dans sa vie. Durant cette surveillance, on lui a fait un bilan omics complet, on a effectué bien sûr la séquence complète de son génome, du transcriptome sur ses cellules sanguines, du protéome dans son sérum, bref il a eu droit à tout. Ensuite cela a été intégré dans des logiciels complexes. On fait des calculs de risques tout au long de la vie, et on lui trouve 80 % de risques de faire un glaucome à angle ouvert, par exemple, et toute une autre série de risques, surtout un risque de diabète de type 2 un peu plus élevé. Il y a là l'analyse complète de tous les polymorphismes associés à ce risque. Ce qui est intéressant, c'est que pendant l'année et demie de surveillance, il a eu des glycémies régulières, car cela faisait partie du bilan complet, y compris clinique. Totalement par hasard, c'est pendant cette période que sa courbe de glycémie s'élève brutalement juste après une infection à RSV. Cette dérégulation de sa glycémie persiste, il décide alors de changer énergiquement son style de vie, ce lui permet de ramener sa glycémie à un niveau normal. Donc, il a un diabète, il le découvre complètement par hasard concomitamment à la survenue d'une infection, et il se prend en charge. Toutes ces informations et ces cynétiques sont intégrées dans des profils complexes qui sont maintenant associés à l'individu, et il a même créé un concept qu'il appelle IPOP, ce qui veut dire Integrative Personal Omics Profiling , qu'il considère comme intégrable à son dossier médical, et qui pourrait être utilisé dans le cadre du suivi.
Si on revient à la relation médecin-malade, c'est un colloque singulier, où il y aura une augmentation colossale du volume d'informations, essentiellement sous forme de valeurs probabilistes. Il va falloir modifier la façon dont on aborde les questions. On passe vraiment de la notion d'art « médical » comme la voyait Auguste Comte à celle de science. Il va falloir commencer à utiliser des outils scientifiques, ce qui pose la question de l'accès aux données du patient par le médecin, du dossier numérique et des coûts générés par le stockage. Aujourd'hui, stocker et accéder à un téraoctet coûte extrêmement cher. Se pose alors la question du niveau de formation du médecin, du niveau d'information du patient, du lieu où l'on va réaliser ces études : nos hôpitaux, nos laboratoires, les laboratoires de biologie médicale. Des sociétés sont en train de se positionner sur ce terrain, Google qui gère à la fois l'information et ce genre de données ( Société 23andMe ) est en train de le faire. C'est intéressant d'aller regarder ce qu'ils font, et cela pose d'abord le problème de la protection des individus.
Je vais conclure par une phrase de Michael Snyder parue dans Cell. C'est le scientifique qui a voulu tester ce qu'est la médecine personnalisée dans les trois exemples précités. Il explique deux choses : « je veux savoir, je n'ai pas de raison de ne pas connaître l'information, parce que si cela peut me permettre de prévoir, je veux le savoir ». Donc, on a une vraie volonté de l'individu qui s'exprime. Puis il prend son côté médecin, et l'idée qui émerge est que vraiment il va y avoir un niveau et un volume d'informations tel que le médecin généraliste ne pourra pas le gérer seul. Il faudra qu'il le gère en coordination et cela va changer complètement les pratiques.
Dr Anne Cambon-Thomsen, directrice de recherche au CNRS, UMR 1027 « Épidémiologie et analyses en santé publique » de l'INSERM et de l'Université de Toulouse 3 Paul Sabatier, responsable de l'équipe « Génomique, biothérapies et santé publique ». Je suis généticienne, chercheuse et responsable d'une plate-forme génétique et société à Toulouse. Mon propos sera moins illustratif que celui qui vient d'être présenté. C'est évidemment sur la question du rôle du dialogue médecin-patient dans la prise de décision que je m'attarderai pour traiter de la prédictivité. Elle est liée à la question de l'interprétation des données générées et à celle de la responsabilité de la personne vis-à-vis de la gestion de sa santé. Nous venons de voir un exemple très exigeant à ce niveau ainsi que sur la question de la disponibilité des informations, de l'accès aux informations, qui sont également à considérer hors du contexte médical.
On vient de parler de séquences avec des chiffres éloquents. Que change la large échelle à laquelle on s'expose en faisant notamment du séquençage ? D'une part il y a la constitution de bases de données au long cours, issues à la fois de données de recherche et de données de clinique, qui convergent, de façon anonymisée, vers les mêmes bases de données. On doit informer les personnes de cette possibilité de réutilisation de leurs données mais aussi de l'éventualité de ré-identification à partir de données de séquences croisées avec d'autres types de données accessibles publiquement. Cela concerne peut-être plus la recherche, mais l'inquiétude existe parmi les personnes qui font l'objet de tests génétiques à cette échelle, quel que soit le contexte. En effet des exemples de ré-identification, à partir de données minimales contenues dans des bases à accès ouvert ont été décrits. Un article qui commence à être célèbre, est paru dans Science au début de cette année sur ce sujet (246 ( * )) .
Les questions qui se posent au niveau du dialogue, concernent la validité et les limites du consentement classique, tel que décrit même au sein des textes de régulation des tests génétiques dans le contexte français en particulier. La nature du séquençage est-elle différente de celle d'autres tests génétiques, et doit-il y avoir un consentement managé, opéré d'une façon différente, spécifique, concernant en particulier l'étendue des informations qu'il recouvre ? Ces différentes facettes de l'information génétique massive sont à considérer aussi du côté des patients ou quand on parle de recherche, des participants. Quel est le droit à l'information et concerne-t-il toute information générée ? On sait qu'on génère plus d'informations en faisant une séquence, que lorsqu'on réalise des tests spécifiques ciblés. Quelles sont les mesures d'accompagnement adéquates ? Nous venons d'entendre les mesures de bonne pratique qui viennent d'être édictées en France.
Au-delà des patients, au niveau des professionnels de santé, beaucoup de questions se posent. Un professionnel de santé concerné par la génétique va devenir un conseiller dans des situations extrêmement variées. Il va devoir gérer de l'information produite à un moment où l'on ne sait pas si elle sera utile un jour. Á quelles conditions prescrire la production d'information génétique à large échelle, compte tenu du contexte économique de ces tests dont on vient de parler et qui montre que la production massive d'information par séquençage à large échelle va être économiquement plus avantageuse que la production successive d'informations génétiques ciblées ?
Parmi ces enjeux du séquençage à large échelle, je vais parler, d'une part de ce qui concerne le consentement, dont j'ai déjà évoqué les spécificités, et d'autre part des découvertes non sollicitées ou dite incidentes, en particulier lorsque de telles informations peuvent permettre de façon certaine de prédire une maladie grave qui peut être traitée ou améliorée par une prise en charge précoce ; et aussi qu'en est-il si une telle maladie ne peut être ni prévenue, ni traitée ? Je ne parlerai pas des susceptibilités. Mon propos consiste non pas à décrire ce que sont les bonnes pratiques en France, cela a déjà été fait, mais ce qui se passe ailleurs, et d'élargir au débat international dans ce domaine.
Je vais vous présenter quelques-unes des recommandations de la Société européenne de génétique humaine , parues au mois de mai 2013, qui disent en préambule que des protocoles doivent être établis concernant comment et quand une information de type séquençage du génome devrait être produite, partagée, conservée, et pour combien de temps. Étant donné la rapidité de ces nouvelles découvertes, il est important que des bio-banques et des bases de données soient créées, avec des informations à jour sur les génotypes et de l'information phénotypique sur les variants et les patients. Il ne faut pas oublier cette notion : on constitue des bases en même temps que l'on traite des patients. Les ressources nécessaires pour ces bases doivent évidemment être trouvées quelque part.
J'ai sélectionné quelques-unes de ces recommandations parmi les 11 existantes (247 ( * )) :
- La n° 3 dit que l'utilisation des puces sur génome entier ou l'analyse du génome complet, demandent une justification. Nous nous trouvons dans la même logique que celle décrite tout à l'heure dans le cadre des pratiques en termes de nécessité : besoin de résoudre un problème clinique et de proportionnalité : l'équilibre des avantages et des inconvénients, pour le patient. Donc le simple fait que la technique existe et qu'elle est économiquement intéressante n'est pas suffisant pour poser son indication.
- Quand l'utilisation de ces techniques est envisagée, un protocole doit être établi avant, pour guider la conduite à tenir devant les découvertes non sollicitées. Si la détection de variants génétiques non sollicités indique un problème de santé sérieux que l'on peut traiter ou prévenir, un professionnel de santé devrait donner l'information, ce qui n'est souvent pas le cas hors du système de santé.
- La recommandation suivante dit que des lignes directrices pour le consentement éclairé dans le cadre du diagnostic doivent être développées. Cela veut bien dire qu'on n'en est pas là pour le moment, et il y a effectivement de larges débats et de nombreux travaux et études pilotes pour en parler et essayer différents types et contextes de consentement pour ce genre de tests dans la pratique clinique.
« Le droit de ne pas savoir d'un patient » ne prend pas automatiquement le pas sur la responsabilité propre du professionnel quand la santé du patient ou celle de ses proches parents est en jeu. Cette phrase fait débat. Les associations de patients pourraient apporter une contribution importante sur la façon de prendre en main cette question, et il serait intéressant de revenir sur cette phrase, dite par des professionnels, au sujet de l'avis des patients. Ici effectivement le droit de ne pas savoir commence à connaître certaines limites.
Cela tombe en plein dans un débat qui a lieu aux États-Unis, puisque le Collège américain de génétique et génomique médicale a fait paraître des recommandations au mois de mars de cette année :
- Quand le séquençage est pratiqué dans le cadre clinique, quelle qu'en soit la raison médicale, les laboratoires doivent examiner systématiquement 57 gènes spécifiques. Il y a une liste établie dans ce texte et le but est de déceler des informations additionnelles considérées comme cliniquement utiles. Donc le non-sollicité devient obligatoire.
- Ces découvertes doivent être communiquées aux cliniciens et au patient. Le 25 avril, après plusieurs débats, une clarification a été apportée par ce Collège qui précise que ne pas rapporter de telles découvertes serait considéré comme non éthique.
- Dans ce contexte, le patient n'a pas la possibilité de refuser l'analyse sur ces 57 gènes s'il accepte le test du génome entier.
- Ces recommandations s'appliquent aussi pour les enfants. Évidemment, cela fait débat, et tout le monde n'est pas d'accord même aux États-Unis.
Le 16 mai 2013 dans Science , deux articles sont parus, dont un qui argumente contre ces recommandations, signé exclusivement par des auteurs américains. Il dit que le consentement éclairé est la colonne vertébrale du soin aux patients, que les tests génétiques ont depuis longtemps requis le consentement des patients, qui ont le droit de ne pas connaître leurs résultats. Toutefois la médecine du XXI ème siècle commence à utiliser les outils du séquençage du génome, et un énorme débat surgit sur la question du maintien de ces droits du patient à l'ère de la médecine génomique. Cette publication, signée par Wolf et ses collaborateurs, argumente donc contre les recommandations de l'American College of Medical Genetics and Genomics. Ces auteurs disent que révéler des découvertes incidentes sans le consentement du patient est une méconduite. Dans le même numéro de Science , il y a un autre article dont une partie des auteurs font partie de l'American College , mais ils s'expriment à titre individuel. Ils disent que les laboratoires ont une obligation de révéler les découvertes incidentes qui apportent un bénéfice clinique. Á cet égard, la Commission consultative d'éthique des États-Unis auprès de la Présidence, a lancé une consultation publique sur la question des découvertes incidentes et la date limite pour répondre à cette consultation publique est le 5 juillet.
Ce n'est pas une question facile et il existe effectivement un débat. Je conclurai en ouvrant sur d'autres débats actuels. Le British Medical Journal de mai 2013 se demande s'il faut séquencer le génome de tout un chacun : un article dit oui, un autre dit non, qui se font face, avec des arguments dans les deux sens. Derrière cela, finalement, quelles sont les garanties pour les personnes et les utilisations ? On voit bien que l'on s'éloigne, avec ces questions, de la relation médecin-patient, qui est le coeur de notre table ronde. Mais il ne faut pas ignorer que l'accès aux données génétiques sort aussi de cette relation, et on ne peut pas faire fi de cette réalité.
Pr Florent Soubrier, responsable du département de génétique du groupe hospitalier de la Pitié-Salpêtrière-Charles Foix . J'ai consacré mon exposé à la relation médecin-patient, et il s'agit bien d'un patient et non d'un malade, puisque je vais parler du diagnostic pré-symptomatique dans les maladies génétiques, dans un cadre bioéthique et dans la relation qui s'installe entre le médecin et le patient.
Les différentes circonstances dans lesquelles on peut réaliser des tests génétiques sont les suivantes :
- Des tests de confirmation de la maladie, chez une personne qui a déjà des symptômes, chez qui on va découvrir une maladie héréditaire et l'identifier, par identification de la maladie génétique.
- Des tests prédictifs chez un apparenté, ou des tests prénataux, pour une maladie génétique de l'enfant avec des symptômes particulièrement sévères. Les tests pré-symptomatiques, ceux dont je vais parler, concernent des maladies génétiques de révélation plus ou moins tardive, à l'âge adulte la plupart du temps, mais aussi, disons, au milieu de l'enfance. Ils posent des problèmes particuliers.
- Des dépistages néonataux, mais je n'entrerai pas dans cette considération.
Les diagnostics pré-symptomatiques dans les maladies héréditaires à révélation tardive :
La demande de test est faite pour la personne qui veut savoir si elle est porteuse d'une anomalie génétique.
Il existe un cadre juridique bien précis pour ces analyses : L'article R 1131-4 du code de la santé publique prévoit que : « Préalablement à l'expression écrite de son consentement, la personne est informée des caractéristiques de la maladie recherchée, des moyens de la détecter, du degré de fiabilité des analyses ainsi que des possibilités de prévention et de traitement. En outre, elle est informée des modalités de transmission génétique de la maladie recherchée et de leurs possibles conséquences chez d'autres membres de sa famille » . Le code fixe les conditions de prescription et de réalisation des examens des caractéristiques génétiques d'une personne à des fins médicales. Ces tests pré-symptomatiques sont réalisés au sein d'une équipe pluridisciplinaire, dans le cadre d'un protocole. L'équipe est déclarée, et le médecin délivre une attestation certifiant qu'il a délivré des informations à la personne concernée et a recueilli son consentement. Les enjeux de ce test vont être fonction de la gravité de l'affection dépistée et surtout du risque associé à l'anomalie génétique.
Je ne parlerai que de maladies ayant une certaine sévérité, et surtout des mutations associées à un très fort risque. Je ne parlerai pas des tests de prédisposition et de susceptibilité. L'importance de l'enjeu vient de la possibilité de surveillance, de prévention ou de traitement curatif. On peut voir sur une échelle les différentes proportions de sujets qui vont développer la maladie parmi les porteurs de la mutation. Nous avons des maladies comme l'hypertension artérielle pulmonaire où seulement 15 à 20 % des sujets porteurs d'une mutation du gène BMPR2 vont développer la maladie. A l'inverse, pour BRCA1, BRCA2, on trouve des proportions qui peuvent atteindre 80 % de développer un cancer, et dans la maladie de Huntington, il existe une pénétrance complète, c'est-à-dire que 100 % des sujets porteurs de l'anomalie génétique vont développer la maladie. Donc les enjeux vont être très différents selon les cas, même si dans tous les cas, les maladies sont graves. Cette annonce potentielle d'une mauvaise nouvelle doit se faire dans une relation médecin-patient tout à fait particulière, et justement Emmanuelle Prada-Bordenave faisait allusion à ces bonnes pratiques.
Je soulignerai des points qui me semblent importants, que j'ai pu tirer de la pratique médicale :
- Le premier point est la prise en compte de l'étape de la vie où se situe le patient, et je prendrai un exemple précis, l'annonce d'une mutation BRCA1, BRCA 2. Si cette annonce est faite à une femme de 25 ans, les conséquences ne sont pas les mêmes que pour une femme de 60 ans ou plus. Il faut bien expliquer à la personne, patient, malade, la différence entre être à risque et être atteint. C'est parfois bien difficile à faire assimiler par le patient, mais c'est une notion extrêmement importante pour gérer le quotidien lorsque l'annonce a été faite.
- Dans cette relation médecin-patient, il faut se donner du temps, être à l'écoute, être attentif aux mots choisis qui ont des répercussions majeures même si le patient entend ce qu'il veut entendre la plupart du temps.
- Assurer un suivi de l'annonce est essentiel à la démarche. Ce n'est donc pas une rencontre unique mais il y aura tout un processus d'accompagnement dans lequel les psychologues, les conseillers en génétique notamment, ont un rôle important. Tout cela a été parfaitement cadré par les recommandations de l'Agence de la biomédecine.
M. Alain Claeys. Vous dites, tout cela a été cadré, mais le médecin est-il formé aujourd'hui pour cela ?
Pr Florent Soubrier . Le généticien l'est, mais il est vrai que ces recommandations vont aller vers un cadrage plus strict, et le médecin généraliste n'est pas formé à cela. C'est pour cela qu'il me semble que ce genre de tests doit rester dans le cadre de médecins formés spécialement, car, on l'a vu, les conséquences en fonction des pathologies sont très spécifiques.
Dans la prise en charge il y a des phases importantes : l'écoute, le conseil par le généticien, par le spécialiste de la maladie, une phase de réflexion ensuite où le patient peut avoir le choix de décider ou non de faire le test en fonction des informations qu'il aura reçues ; enfin, la décision est prise et il y aura le rendu du résultat, puis, le dernier point est celui du suivi spécifique.
Les conséquences de la prédiction sont les suivantes :
- Quand le test n'est pas fait, le résultat n'est pas connu, la personne se trouve dans l'état d'être à risque. Ce doute est vécu au quotidien. Il n'est pas toujours facile à gérer, et il peut paralyser l'action, tout projet, et expliquer aussi tous les symptômes que le patient peut éprouver avec un risque subjectif souvent autour de 50 %.
- Le résultat favorable contrairement à ce que l'on peut penser peut entraîner des réactions surprenantes bien qu'explicables. C'est la culpabilité du non-porteur vis-à-vis de son frère ou de sa soeur, qui eux sont porteurs. Cela met le non porteur dans une situation un peu à part. Et quelquefois le risque écarté d'une pathologie sévère a des conséquences tellement importantes que cela va pouvoir changer la façon dont il gère sa vie, c'est-à-dire, avec cette expression très belle d'Alexandra Dürr, « guérir du risque ». Ce n'est pas neutre, parce qu'il va pouvoir complètement changer son cadre de vie, ses projets, quelquefois changer sa vie de couple. Il y a parfois beaucoup de choses qui vont être décidées à la suite d'un résultat favorable.
- Dans les résultats défavorables, il va falloir distinguer porteurs et atteints, gérer le temps qui sépare le résultat de la déclaration de la pathologie quand celle-ci est inéluctable. Il y a une incertitude sur l'âge de début ou de sévérité, et également une adaptation familiale et sociale à faire.
Quel est le pouvoir prédictif du test, avec quelle est la prise en charge ? Il existe une difficulté à prédire la survenue quand la pénétrance n'est pas complète. On ne peut pas aller au-delà d'une certaine information vers le patient, encore moins pour l'âge de début de la pathologie. Mais savoir peut permettre, selon le type de la prédisposition, un dépistage précoce. Je voudrais insister sur l'intérêt des centres de suivi spécialisés que par exemple l'INCa a mis en place pour les personnes porteuses de mutations prédisposant aux cancers, qui permettent une vraie prise en charge et assurent le meilleur suivi. Ce pouvoir prédictif permet également les mesures préventives chirurgicales, qui sont parfois lourdes, mais peuvent apporter des gains importants en termes d'années de vie

Mme Simone Bateman, sociologue,
directrice de recherche au CNRS, Centre de recherche médecine, sciences,
santé, santé mentale, société (CERMES3) - CNRS
UMR8211/Université Paris Descartes/EHESS/
Inserm U988. Pour
répondre à la question posée par la table ronde,
j'aimerais pouvoir me référer à une définition
consensuelle de la médecine personnalisée. La littérature
est abondante ; cependant, une revue de cette littérature m'a
conduit à constater qu'il n'y a de consensus ni sur une
définition, ni sur le champ que le terme recouvre, ni sur le point
d'entrée qu'il convient de privilégier pour comprendre son
développement. Toutefois quelques idées reviennent avec
insistance.
La première est qu'il s'agit d'une médecine faite sur mesure (en anglais, customised, ou tailored , par analogie avec les tailleurs, l'habillement). C'est-à-dire que l'on va tenir compte des caractéristiques individuelles du patient, principalement ses caractéristiques biologiques, notamment génétiques, mais aussi des données concernant son environnement et son mode de vie. Ce profil est défini de manière objective, car l'individu est saisi à un niveau élémentaire de son existence biologique.
Cette personnalisation ne peut se confondre avec des conceptions plus philosophiques de la personne, par exemple un autrui incarné engagé dans une relation, un sujet de droit responsable de ses actes, voire avec l'idée d'une médecine holistique attentive aux besoins de chaque personne au moment où elle se fait soigner. On la retrouve dans la notion de colloque singulier, mais également dans un programme de l'INCa qui personnalise des soins en cancérologie. Ce programme n'a rien à voir avec la médecine personnalisée dont nous parlons, puisqu'il s'agit de mettre en place une meilleure coordination et un accompagnement du patient dans son parcours de soin.
La notion de médecine personnalisée renvoie donc, soit à un système de santé, soit à des soins de santé. Le terme anglais comporte le mot medicine mais aussi parfois le mot health care , donc soins de santé au sens le plus large, non forcément associés à une pathologie. Ce système est censé s'accommoder, à tous les stades de la prise en charge, de ces différences individuelles. Il s'éloigne d'une médecine dite à « taille unique », one size fits all , mais nous savons que cette vision est un idéal puisque dans la pratique, il s'agit d'identifier des sous-groupes de personnes susceptibles de bénéficier de traitements ciblés en fonction de leur profil.
La seconde idée qui revient dans la littérature est celle-ci : des technologies rendent possibles cette nouvelle vision d'une médecine sur mesure. Le terme en anglais est enabling technology . En effet, la médecine personnalisée s'appuie sur un ensemble de connaissances et d'outils techniques qui rendent possibles la caractérisation du profil biologique ou génétique de chaque patient et donc la mise en oeuvre de soins adaptés. Outre les avancées dans les domaines de la génomique, de la bio-informatique, de la pharmacogénétique et de la pharmaco-génomique, il s'agit notamment de la nouvelle génération de machines de séquençage, plus puissantes et donc capables de faire le travail en moins de temps et à moindre coût. Certains rapports privilégient l'analyse des enjeux et des risques associés à l'utilisation de ces nouveaux outils techniques : par exemple, le rapport de la Commission présidentielle américaine pour l'étude des questions de bioéthique, W hole Genome Sequencing : Privacy and Progress , met en avant le séquençage du génome entier ; un rapport du Nuffield Council, Emerging Biotechnologies , consacre une petite partie de son analyse à la médecine personnalisée et à ce qu'ils appellent la médecine génomique; un autre rapport du Nuffield Council mentionné par Hervé Chneiweiss, M edical profiling and online medicine : the ethics of personalised healthcare in a consumer age , identifie Internet comme une technologie importante dans ce champ.
Une troisième idée s'impose également dans le débat : cette médecine dite personnalisée a commencé dans un domaine spécifique, l'oncologie. En effet, le recours aux outils de la génomique a pu s'inscrire dans des pratiques déjà bien établies d'examen et de classification des tumeurs. Dans ce cas, il s'agissait de caractériser non pas le profil du patient, comme il est dit dans les définitions que j'ai pu trouver dans certains rapports, mais les altérations génétiques de la tumeur avec pour but d'affiner le diagnostic, le pronostic du cancer, mais aussi de choisir le traitement le plus efficace ou le mieux toléré, voire d'éviter un traitement inefficace aux effets secondaires toxiques. En d'autres termes, la médecine personnalisée vient renforcer les moyens que l'oncologie met déjà en oeuvre dans sa mission thérapeutique proprement dite.
Certes la rhétorique voudrait qu'on « personnalise » le diagnostic et le traitement, qu'on « prédise » l'efficacité d'un traitement, qu'on « prévoit » la tolérance ou l'intolérance à un traitement, mais nous sommes dans ces cas loin de ce que nous entendons habituellement par médecine préventive ou médecine prédictive. Il s'agit plutôt d'une médecine de précision, qui pour le moment s'applique à un nombre limité de personnes, chez qui l'on a détecté les quelques marqueurs associés actuellement à des thérapies dites ciblées, ce qui a été rappelé également par Anne Fagot-Largeault. Toutefois, au long terme, l'oncologie aspire à une nouvelle classification des cancers fondée sur des caractéristiques génétiques, avec l'espoir d'une efficacité accrue. La traduction de cette aspiration dans la pratique passera également par des essais cliniques dont les modalités, notamment la phase 3, doivent être pensées en fonction du fait que les cibles concernent des groupes de patients de plus en plus petits.
Le cancer peut-il servir de paradigme pour le développement de cette médecine sur mesure dans d'autres spécialités ? Alors que le cancer est aujourd'hui compris par certains comme une maladie du génome, ce n'est probablement pas le cas pour bien d'autres maladies comme par exemple les maladies cardio-vasculaires. Cela limite peut-être l'extension du paradigme d'une médecine individualisée à d'autres spécialités. Sans doute faut-il garder à l'esprit l'idée que la médecine personnalisée est en développement et a encore à faire ses preuves.
Cette insistance sur le mot « médecine », comme sur le mot « personnalisée », laisse dans l'ombre un enjeu essentiel. Son extension repose, comme je l'ai rappelé, sur l'identification de nouveaux variants génétiques associés à des thérapies spécifiques. Or cela ne peut être établi que par la collecte de données à des fins de recherche sur de très grandes populations de patients. Outre les recherches fondamentales déjà menées dans ce domaine, le rapport Toward Precision Medicine du National Research Council américain de la National Academy of Sciences préconisait déjà en 2010 la collecte de données observationnelles en situation clinique en vue d'alimenter un réseau commun de données. La récente initiative de mise en commun de données de recherches par un réseau d'institutions, réunies sous le nom de Global Alliance, va tout à fait dans le sens préconisé. Le schéma présenté dans le rapport a été élaboré voici trois ans, et déjà l'une des pièces commence à se mettre en place. Même les personnes qui dans une démarche privée souhaitent faire établir leur profil génétique par l'un des services directs aux consommateurs existant aux États-Unis, ont elles-mêmes alimenté de telles bases de données. Aujourd'hui certains de ces services sont fermés, et ont vendu leurs bases à d'autres entreprises, dont par exemple des industries pharmaceutiques.
Les retombées de ces recherches ne seront pas immédiates, car la simple production de données n'équivaut pas à l'élaboration d'une information scientifiquement rigoureuse, pertinente et cliniquement utile. Cependant, toute production de données engage le patient ou le client dans une nouvelle forme de contribution à la recherche dont il convient de préciser les termes de consentement à la participation. On assiste là à une reconfiguration du cadre de la relation entre le médecin et son patient, qui dépasse largement celui plus restreint auquel renvoie la notion de colloque singulier.
On peut enfin s'intéresser à l'absence du mot génomique dans l'expression médecine personnalisée, alors que c'est cet ensemble de connaissances et de technologies associées qui rend possible cette nouvelle démarche thérapeutique. Ce fait va de pair avec le constat qu'hormis le débat sur l'intérêt pour l'individu des données relatives aux gènes de susceptibilité fournies dans le cadre des services directs aux consommateurs, l'impact de la génomique sur l'évolution de la génétique médicale comme une forme de médecine personnalisée est plus rarement abordé dans les rapports mentionnés. Cependant, beaucoup d'articles récents sont consacrés aux découvertes fortuites qui pourraient être faites dans le cadre de stratégies de recherches fondées sur l'analyse des génomes. Ces découvertes suscitent des controverses - auxquelles a déjà fait allusion Anne Cambon-Thomsen - sur l'obligation d'informer sur le droit de ne pas savoir, notamment lorsque la médecine ne peut rien proposer en matière thérapeutique. Ces controverses montrent bien que les enjeux de la médecine dite personnalisée se définiront nécessairement au-delà des frontières du seul champ d'une prise en charge thérapeutique mieux ciblée de certaines affections.
La génomique commence à faire ses preuves comme outil pertinent dans le diagnostic prénatal, notamment dans les tests non-invasifs récents faits sur sang maternel. Il s'agit là d'un champ de pratiques et de problèmes distincts, relevant plutôt d'une médecine dite prédictive, au sens où elle détermine des probabilités d'être atteint d'une maladie ou d'une affection. Elle peut être éventuellement préventive lorsqu'il est possible mettre en place une surveillance, voire des mesures prophylactiques. Le fait que les deux champs de pratiques mobilisent un même ensemble de connaissances et de techniques suggère que la médecine personnalisée entendue comme une médecine de précision, ne pourra échapper à certaines des interrogations qui depuis longtemps traversent la génétique médicale. Dans ces circonstances, ce ne sera plus l'individu, défini dans son rapport à une population, qui sera au centre de cette médecine, mais la personne dans son rapport à ses apparentés, et dans ses aspirations à une descendance en bonne santé.
|
Débat |
M. Alain Claeys, député, rapporteur . Je souhaiterais que l'on revienne sur cette reconfiguration de la relation malade-médecin. Pour nous, en tant que parlementaires, c'est un sujet important.
Pr Thomas Tursz, cancérologue, directeur honoraire de l'Institut Gustave Roussy. Je parlerai cet après-midi plus précisément des problèmes posés par le traitement personnalisé du cancer. Mais après ce débat un peu général, j'aurais voulu intervenir sur certains points qui me paraissent plus globaux et importants, et en particulier devant des élus de la République.
On peut spéculer à perte de vue sur l'importance de cette médecine, sur le temps qu'elle mettra à exister, sur les avantages et les inconvénients apportés aux patients et sur son coût pour la société.
Le premier point est que c'est inéluctable, incontournable. C'est un saut de la connaissance, on ne reviendra pas en arrière. Et si dans un pays comme la France, dans une structure comme l'Europe, on ne se donne pas les moyens de le faire bien, intelligemment, de partager ces connaissances et ces résultats, cela durera longtemps, ce sera mal fait, sans contrôle de qualité, et à nouveau ces progrès apparaitront lents, coûteux et peu efficaces.
L'autre risque que je perçois est réellement celui d'une médecine à deux vitesses, une médecine de classe, où les patients riches pourront à leur frais faire séquencer leur tumeur, le plus souvent à l'étranger, et bénéficier de traitements personnalisés, alors que les autres continueront à être traités par les méthodes et les médicaments conventionnels, dont nous voyons aujourd'hui les limites et les inconvénients.
Je crois que la notion de pluridisciplinarité, essentielle en cancérologie, doit maintenant s'élargir vers un réel pluri-professionnalisme. C'est vraiment dans la mise au point stratégique de consortia, de spécialistes différents travaillant avec des technologies différentes, mais partageant l'ensemble de leurs données, l'ensemble de leur savoir, de leurs connaissances, que l'on progressera.
Enfin, contrairement à ce que j'ai entendu, les malades, dans ma spécialité, le cancer, sont très demandeurs de ce type de médecine. Autant l'essai thérapeutique a été longtemps considéré comme le dernier recours quand tout le reste des traitements avait raté, la dernière voie avant le miracle ou avant Lourdes, autant il est en train de devenir l'offre précoce, dans le cours de la maladie, d'une chance réelle de traitement. Ceci change aussi beaucoup la place de la recherche clinique, qui devient de plus en plus importante, et va de plus en plus être réclamée par les malades.
J'en parlerai dans le domaine du cancer, mais ceci concerne d'autres domaines. Je voudrais insister sur le changement de classification des maladies. Ce point crucial va plus loin que la cancérologie. Le Parkinson est probablement un mélange de 30 maladies, l'Alzheimer de 50, et une nouvelle nosologie des tumeurs doit être faite. L'intérêt général, public, social, éthique, scientifique est qu'elle soit faite de la façon la plus courte, rentable, équitable, et avec le plus de critères de qualité.
Je voudrais enfin dire combien ces notions nouvelles vont modifier la relation médecin-malade. Bien sûr elle doit continuer à exister, c'est un médecin qui doit être le référent du malade, l'orienter, le guider, l'appuyer, l'éclairer, mais c'est une équipe de gens partageant leurs connaissances, leurs technologies, leurs savoirs qui orienteront les décisions thérapeutiques, en fonction des résultats d'ordinateurs géants, et non pas de ce qu'ils ont pu lire dans tel ou tel journal, ou dans leur expérience passée. C'est vraiment, pour paraphraser Edgar Morin, un enjeu de gestion collective de la complexité, un changement de paradigme. Et la pire des choses serait de ne rien faire maintenant en pensant que de toute façon cela se fera tout seul, ou dans tellement longtemps que ce n'est pas la peine de prendre des décisions tout de suite. Il faut essayer que cela ne coûte pas trop cher, que ce soit supportable par la société, que cela se fasse bien.
M. Alain Claeys. Par rapport à l'ordinateur, qu'elle est la place du médecin référent ?
Pr Hervé Chneiweiss . Nous avons assisté à deux présentations, non pas conflictuelles, mais tout de même... D'un côté Philippe Amouyel nous a présenté des programmes développés par des personnes qui ont accès aux technologies informatiques et aux technologies omiques, et de l'autre, je suis à peu près d'accord avec ce que vient de dire Thomas Tursz.
Mais il n'y a pas que la génétique, il y a tous les omiques qu'il va falloir intégrer. Un ingénieur de santé va certainement être nécessaire, il va falloir ouvrir la relation médecin-malade. Et va se poser immédiatement l'inégalité d'accès à l'information ou à cette médecine intégrée. En effet, nous l'avons vu sur un des schémas : l' Integrative Personal Omics Profiling (IPOP) qu'a montré Philippe Amouyel intègre les différents domaines. Je ne veux pas minimiser les choses, mais je dirais que le domaine du cancer, c'est-à-dire l'identification de mutations dans les cellules tumorales, le suivi de ces mutations et le ciblage de la tumeur, est presque le cas le plus facile par rapport aux multiplicités du schéma IPOP, qui a montré l'interaction entre les différents facteurs de risques et d'environnement par rapport à la personne.
Donc, comment les pouvoirs publics vont-ils ouvrir la relation médecin-malade ? Florent Soubrier a semblé vouloir garder le conseil génétique au médecin qui prescrit le test. Mais comment va-t-on ouvrir cela aux nouvelles connaissances qu'ont les patients eux-mêmes, à cet ingénieur de santé, à ces nouveaux métiers du conseil génétique, en tenant compte également des nouvelles pratiques des médecins généralistes, qui eux n'y comprennent vraiment pas grand-chose aujourd'hui ? Il y a vraiment des enjeux de santé publique et d'ouverture des données privées à de nouveaux professionnels, auxquels il faut réfléchir.
Pr Philippe Amouyel. Ce que dit Hervé Chneiweiss est tout à fait juste. Par hasard, dans le cadre de programmes de recherches j'ai eu à interagir avec les personnes de l'entreprise 23andM e sur des programmes neuro-dégénératifs Parkinson. Vous savez tous comment cela a commencé. La femme de Sergey Brin, un des patrons de Google, était biologiste moléculaire. Il lui a offert un laboratoire, et avec leur exceptionnel esprit d'entreprise, ils ont monté 23andMe , qui offrait au début la possibilité d'avoir des informations sur la généalogie. C'est ce qui a permis le fameux article de Science entre autres : vous donnez votre génome, et à partir de petits bouts de séquences vous dites, sans le savoir, qui vous êtes, où vous habitez, quels sont vos parents, et votre nom. On peut le faire aujourd'hui sur Internet dans près de 10 % des cas. Et puis Sergey Brin s'est trouvé une mutation dans le Parkinson quand il a fait le test. Cela a complètement changé son approche intellectuelle. Il a mis de l'argent dans le système pour la recherche sur la maladie de Parkinson. 23andMe perd encore beaucoup d'argent, mais il continue, et c'est un fabuleux laboratoire expérimental de ce que l'avenir pourra être.
23andMe est un acteur majeur dans les consortiums de recherche sur les maladies neuro-dégénératives. Leurs bases de données sont composées uniquement de volontaires recrutés sur Internet. Ils ont pu ainsi constituer ainsi une des grosses bases de données génomiques sur Parkinson. Ils font un appel par internet, proposent des génotypes pour trois fois moins cher, et construisent des bases colossales, hors de tout système scientifique ou légal. L'information est simplement issue de questionnaires et de la bonne volonté des gens. On peut se dire que ce n'est pas scientifique. Ils sortent des documents sur des caractéristiques peut-être inutiles, des gènes qui vont gouverner des cheveux frisés ou des choses comme cela. Mais cela veut dire qu'il y a vraiment un enjeu, une population très demandeuse et des outils très puissants.
En effet, quand c'est très ciblé, quand il s'agit du diagnostic qu'évoquait Florent Soubrier ou Thomas Tursz en matière de cancer, on est déjà dans la médecine de précision. Simplement c'est un plus pour le médecin, un nouvel examen qui vient enrichir vraiment une consultation de spécialistes. Dans les cas précédents, nous sommes sur des données qui peuvent générer des informations telles que : « je vais chez un généraliste, il peut m'annoncer que mon père n'est pas mon père, par exemple ». Doit-il le dire ou non ? Il est important que cette relation médecin-malade soit conservée, même s'il y a tout cet environnement en réseaux qui ne manquera pas d'intervenir. Il faudra un « intercesseur » car nous ne sommes que des hommes : « je n'ai peut-être pas envie de savoir si je vais avoir telle maladie, si je vais décéder dans cinq ans. J'aimerais pouvoir discuter avec mon médecin traitant d'abord ».
Pr Jean-Claude Ameisen . Je crois que c'est important ce qu'Anne Cambon-Thomsen évoquait, ce droit de ne pas savoir. Et cette pression qui consiste à penser que toute information est forcément utile, et que l'on est forcé de savoir qui est son père, quel risque on a etc ., est un vrai problème. Si c'est l'ordinateur qui pilote, c'est comme les tests sur le net : on pose une question, on a des réponses, y compris à celles qu'on n'a pas posées. Cela demande non-seulement une formation, une pluridisciplinarité avec des ingénieurs de santé, mais aussi du temps. Or, dans une époque de tarification à l'activité, le temps du dialogue n'est pas considéré comme une activité. Ce paradoxe, la dépersonnalisation de la médecine personnalisée, contient la seule chose dont on va faire l'économie : ce ne sont pas les séquenceurs, ni les ordinateurs, mais le temps.
Comment reconstruire de manière ouverte cette relation entre une personne et un interlocuteur médecin, qui n'est pas forcément celui qui a fait le test, séquencé, analysé ? Il y a quelque chose à repenser en partenariat encore une fois, avec les associations de patients. Le fait de pouvoir identifier la personne sur une séquence, et que les séquences circulent, posent un véritable problème de confidentialité et de risque de discrimination, à une époque où l'on s'aperçoit que celle de nos conversations et de nos échanges de méls n'est peut-être pas aussi grande qu'on le croit.
Un point que l'on n'a pas évoqué, il est un peu éloigné, mais il est très simple : le fait d'envoyer quelques cheveux et d'avoir un séquençage permet non seulement de séquencer son génome, mais aussi celui de n'importe quel voisin. Il n'y a donc pas simplement le problème de confidentialité dans la manière dont les données sont échangées ou recueillies, il y a aussi la question de savoir de qui l'on séquence le génome.
Ce sont des problèmes de fond. Le temps, la réflexion, sont nécessaires, sans encore une fois se laisser entraîner par les côtés très intéressants d'une démarche qui avance à pas forcés.
On a parlé de la validité...Quelque chose me préoccupe depuis quelques années. Contrairement à ce qui se passe aux États-Unis avec la Food and Drug Administration , le marquage CE des dispositifs médicaux de diagnostic ne prend pas en compte l'utilité clinique, mais simplement les caractéristiques techniques. Et si l'on se met à rentrer dans une analyse du génome, du métabolome, du microbiome avec des instruments mis sur le marché, sans que leur utilité clinique et leur intérêt aient été évalués, on a un problème auquel il faut réfléchir au niveau européen.
Un dernier point : nous l'avons vu, il y a non seulement insistance sur ce qu'on peut lire de précis dans le corps, mais de plus un déplacement vers le génome. Comme le plus facile à interpréter d'un point de vue informatique est actuellement l'information génétique, on a l'air de dire que c'est la plus importante. En gros, ce qu'on sait le mieux analyser devrait être le plus important. Or ce n'est pas le plus important. Comment ne pas faire basculer ce qu'on appelle la médecine de précision vers simplement une médecine d'étude génétique ? Dans l'aspect intéressant de responsabilisation de la personne (vous en savez beaucoup plus sur vous si vous le voulez, et vous pouvez changer votre mode de vie), il y a un risque en termes de solidarité, qui n'est pas seulement la difficulté de prendre en charge économiquement ces analyses, mais d'oublier les contraintes d'ordre général, qui jouent un rôle extrêmement important sur la santé.
Il me semble me rappeler que dans la dernière étude de l'INSEE, le premier facteur de mortalité de cancer, ce n'est pas la séquence génétique particulière de la tumeur, mais le niveau socio-économique des personnes atteintes de cancer, car il entraîne un retard d'accès au diagnostic, donc un retard de traitement. Or, cela ne se lit pas dans le génome. On a donc un risque de dire : vous savez tout sur vous, vous êtes responsable. Mais pour les deux millions d'enfants qui vivent sous le seuil de la pauvreté, ce n'est pas dans l'analyse de leur corps aujourd'hui que l'on peut imaginer et prédire les conséquences en termes de santé, dues à un environnement socio-économique.
Il faut donc avancer dans la réflexion sur cette démarche, mais ne pas oublier le rôle des facteurs que nous construisons ou non, de l'ordre de l'environnement au sens très large du terme : les études de l'OMS ou de Michael Marmot sur les déterminants socio-économiques de la santé ne se lisent pas dans le corps du nouveau-né, du foetus, de l'enfant ou de l'adulte, avant que les déterminants de leur environnement n'aient commencé à produire leurs effets.
Mme Simone Bateman . Juste un petit commentaire en réponse à votre question sur le médecin référent. Je suis tout à fait d'accord sur le fait que les tests génétiques doivent être pris en charge par un spécialiste puisque c'est lui ou elle qui connaît le mieux le problème pour lequel consulte la personne. Mais une enquête que j'ai menée auprès de personnes ayant une mutation pour deux types de cancers héréditaires mentionnés par Florent Soubrier, montre que le médecin traitant est très important, car il peut, par méconnaissance, ne pas aiguiller le patient suffisamment rapidement et à bon escient chez le spécialiste. Il peut y avoir une perte de chances liée au fait de perdre du temps dans un parcours de soin inapproprié. Mon sentiment est qu'il est urgent de proposer une meilleure formation en génétique notamment pour le généraliste, médecin que le patient voit le plus souvent et qui joue un rôle clé dans le réseau de soins.
Mme Emmanuelle Prada-Bordenave . Je voudrais juste revenir sur deux points. L'aspect financier, et le gros plus apporté par ces techniques de génétique et la possibilité d'y accéder rapidement : la limitation de l'errance diagnostique. Elle a un coût considérable pour nos systèmes de santé. Il ne faut jamais oublier que si ces technologies apparaissent coûteuses aujourd'hui, elles apportent une économie relative, qui est l'économie de l'errance diagnostique et de son coût, sans parler de la pharmacogénétique, de tout ce qu'on a dit ce matin sur les effets toxiques en cas de mauvaise prescription.
Sur le point qui vient d'être soulevé par Mme Bateman, nous Agence, en liaison avec la Haute Autorité de Santé, et l'ANSM, sommes positionnés dans cette idée que cette médecine devait servir à soigner des malades. Il est vrai, des personnes se présentent sans être malades, et vont se faire prescrire une analyse : autant pour la prédisposition, pour les personnes pré-symptomatiques, il s'agit vraiment d'une démarche clinique, autant pour les tests de susceptibilité il faut vraiment faire preuve d'une très grande prudence. C'est là que les médecins les plus généralistes, les plus proches des patients vont être le plus impactés. Bien sûr, dans le cas du Pr Florent Soubrier, les patients ont déjà été triés, car les pathologies sont graves. Mais le médecin généraliste, ou le gynécologue, devant des troubles de la coagulation, vont être directement confrontés à la nécessité ou non de prescrire des tests qui dans la plupart des cas vont être de susceptibilité. Et là nous devons faire preuve de pédagogie. C'est pour cela que ces règles de bonne pratique vont être diffusées aux généticiens, qui n'en ont pas vraiment besoin puisque ce sont eux qui les ont construites, mais surtout aux prescripteurs non spécialisés dans la maladie génétique, voire non spécialisés dans la maladie très grave.
Il va falloir une sorte d'apprentissage de nos professionnels pour ces nouvelles techniques, et tant mieux si les outils sont moins coûteux et plus puissants. Mais ils doivent toujours être utilisés pour soigner, et non pas pour faire de la génétique récréative. Car la génétique récréative est quand même de la dépense de santé. Entendez-le au sens de la comptabilité nationale. Quand est installé chez nous quelque chose qui fait du séquençage du génome, c'est de la dépense de santé et c'est un poids pour notre société. Si on installe ces grands séquenceurs, et la direction générale de l'offre de soins s'est engagée à le faire de manière déterminée, il faut qu'ils servent à soigner et non pas à savoir si l'on a les cheveux frisés.
Mme Catherine Vergely, secrétaire générale de l'Union des parents d'enfants atteints de cancer et de leucémie (UNAPECLE). Je représente l'Union nationale des associations de parents d'enfants atteints de cancers ou de leucémies. J'interviens ce matin sur un autre sujet.
Vous êtes très éloignés des préoccupations du citoyen moyen, car s'il a envie de faire son génome sur Internet, même s'il n'est pas remboursé, il le fera. Par ailleurs la personne actuellement au SMIC n'a absolument rien à faire des tests que vous avez montrés. Ils sont faits par des gens supérieurement intelligents, avec des moyens pour décoder leurs génomes ou décoder leurs enfants, mais ne sont pas accessibles à la majorité.
J'ai une question importante. On parle bien de la relation médecin-malade, de médecine de prévention, dont on sait qu'elle n'a pas atteint son zénith en France. Nous avons devant nous cette obligation de la mettre en place, c'est un enjeu citoyen, qu'elle soit pour la médecine personnalisée ou pour les autres. Il me semble que la médecine du travail, la médecine de prévention dans les institutions, celle chez nos enfants dans l'éducation nationale, ne sont pas suffisantes.
Il me semble important de replacer la médecine personnalisée, et pas uniquement la génétique, dans la perspective du bien-être du citoyen, sans être uniquement de la médecine de haut vol. Par exemple le diagnostic de la tuberculose, maladie qui remonte en France, est important. Je voudrais remettre le débat à ce niveau. Nous avons peut-être des professionnels hyper-pointus à former, mais déjà, les médecins de prévention et du travail ne sont pas reconnus par nos institutions. Ils auront également, vu leur expérience avec des personnes, malades ou non, des leçons à nous donner pour la médecine personnalisée.
DEUXIÈME TABLE
RONDE :
QUELLE PROTECTION DES PERSONNES, DE LEUR VIE
PRIVÉE
ET DES DONNÉES PERSONNELLES ?
|
Point
de vue :
|
M. Jean-Louis Touraine, député du Rhône, membre de l'OPECST . Je voulais faire le lien avec ce qui a déjà été abordé dans ces questions d'actualité, importantes dans les prochaines décennies, sur la médecine personnalisée. En effet, si la définition est encore un peu incomplètement précise dans ce qu'elle englobe, il n'empêche que des grandes lignes se détachent dès maintenant. Malgré la terminologie de médecine personnalisée, cette médecine n'est clairement pas individualisée. Il est important de le dire à la population générale car certains croient que l'on s'achemine vers une médecine d'individu, de façon très singulière. Il faut bien rappeler que le progrès médical continuera encore pendant de nombreuses années à nécessiter des observations et des thérapeutiques s'appliquant à des groupes de patients.
Par contre, la médecine personnalisée inclut la médecine ciblée, l'usage des biomarqueurs, la prédiction génétique, les médecines de précision et de prévention. Dans tous les cas, elle éloigne encore un peu plus de la réduction traditionnelle au seul colloque singulier médecin-malade. En effet, elle introduit de nombreux tiers supplémentaires dans la relation entre le patient et son médecin, dont certains sont connus, et d'autres inconnus. Elle exige et génère une masse de données de santé, sensibles, qui valent pour le présent mais aussi dans une certaine mesure pour l'avenir des personnes.
Ces données sont personnelles, identifiantes, et pour partie invariantes tout au long de la vie. Elles constituent un dossier qui portera sur les possibilités d'évolution de l'état de santé d'une personne pendant toute sa vie. En outre, même si la médecine personnalisée cible l'individu, la famille sera au courant en cas de maladie génétiquement transmissible. Le risque qui pèse donc sur le secret médical prend une dimension encore plus importante que dans le passé, et les risques d'obtention indue de données sont eux aussi accrus d'autant plus qu'il ne s'agit plus des fiches cartonnées du généraliste, mais d'un système informatisé, donc fragile, consultable à distance, intégrant durablement des données personnelles. Ceci est donc préoccupant, car l'anonymisation de ces données doit pouvoir être garantie pour assurer une protection rigoureuse de ce secret médical, éviter des discriminations préjudiciables aux personnes ou aux familles.
En avons-nous les capacités techniques à l'heure actuelle, surtout à un moment où l'on découvre que la surveillance numérique s'effectue à notre insu dans de très nombreuses circonstances ? Nos concitoyens sont-ils informés de ces risques ? Se posent-ils suffisamment la question de leur intimité numérique ? Par ailleurs, le coût du traitement numérique des données de séquençage, par exemple, risque de devenir aussi important, voire supérieur, à celui du séquençage lui-même. Sa complexité dépendra de la connaissance préalable que l'on a de l'ADN séquencé.
Ainsi, loin d'être un aboutissement, la démocratisation du séquençage apparaît de plus en plus comme un point de départ. Désormais, pouvoir obtenir pour un coût relativement modique le génome de vastes populations autorisera des analyses à grande échelle, qui permettront de comprendre le comportement et le rôle de certains gènes, à condition de disposer des ressources informatiques adéquates. Utiles aux chercheurs et aux patients, ces analyses à grande échelle vont générer des masses de données. Dès lors la gestion, le contrôle, la protection des bases de données, deviennent un enjeu crucial.
Qui aura obligation de stocker les données, alors que celles susceptibles de circuler exigent des espaces considérables ? La Commission nationale de l'informatique et des libertés aura-t-elle les moyens d'assurer leur protection ? Qu'en sera-t-il au niveau international ? Ceci conduit à s'interroger sur la capacité de protéger les masses de données ainsi produites. Pour être utiles, interprétables, ces données transitent, ce qui suppose une fiabilité extrême des circuits, donc des accès sécurisés et surtout des systèmes d'anonymisation fiables. Est-on suffisamment avancé dans ces techniques pour pouvoir les garantir ?
La gestion et l'accès aux bio-banques seront aussi un enjeu considérable eu égard à leur importance pour la recherche. L'impact de leur privatisation par de grands groupes pharmaceutiques inquiète à juste titre. En effet, on constate une tension entre la demande des chercheurs qui souhaitent accéder facilement à des banques de données anonymisées, et les détenteurs privés. Ces tensions conduisent à des interrogations sur la protection des personnes contre un mésusage de ces données, car toute faille dans le système risque d'être préjudiciable aux patients que ce soit en termes d'erreur matérielle, de perte de données ou bien de rupture dans l'anonymisation.
Quelles sont les garanties actuelles ? Où en sont les réflexions sur ces sujets très importants pour chacun d'entre nous ? Il s'agit de notre vie privée, et à partir de là, du regard porté par nos concitoyens sur les avancées de la science. Il importe de progresser dans tous ces domaines à mesure que l'accès à un nombre plus important de données numérisées de caractéristiques génétiques devient disponible.
*
* *
Pr. Dominique Stoppa-Lyonnet, chef du service de génétique de l'Institut Curie, professeur de génétique médicale à l'université Paris-Descartes, membre du CCNE . Merci monsieur le président de me donner l'occasion de m'exprimer sur un sujet qui m'est cher puisque c'est mon quotidien que de prescrire des tests génétiques.
Sur la question posée, très ciblée, la protection des données personnelles et de la vie privée, je propose de voir deux aspects.
Le premier est la « dure vie » de la personne qui fait un test génétique en cas de maladie dominante ou liée à l'X, c'est-à-dire ces situations où le résultat de son test va avoir un impact majeur pour ses apparentés. L'archétype en est les prédispositions aux cancers, puisque l'essentiel de ces prédispositions obéissent à un modèle dominant : dans ces familles, souvent plusieurs personnes ont été atteintes d'un cancer et appartiennent à la même branche parentale. Je voudrais évoquer le cas index, dont la vie privée et les données personnelles risquent d'être mises à mal du fait du retentissement de sa démarche sur ses apparentés.
Le deuxième point est celui de la confidentialité des données de la recherche, données qui ne peuvent être anonymisées de façon définitive du fait du lien indispensable entre données expérimentales et données cliniques et du fait du retour possible des résultats de la recherche dans le dossier du patient. C'est aussi la confidentialité des données du dossier médical, dossier médical informatisé, que l'on attend avec impatience car devant faciliter la prise en charge des patients.
La démarche en oncogénétique débute en général par celle d'une femme ou d'un homme qui va bien, mais s'interroge sur ses risques tumoraux, alerté par une histoire familiale de cancers. Il souhaite avoir des recommandations de prise en charge. L'état des connaissances et la complexité des analyses génétiques conduisent à mener une étude génétique en deux temps. En effet, pour donner une réponse de qualité et en particulier pouvoir rassurer cette personne qui s'interroge, il est nécessaire dans un premier temps de comprendre l'origine de l'histoire familiale. Comprendre l'origine de l'histoire familiale, c'est identifier un facteur génétique de prédisposition, l'altération - ou mutation - d'un gène donné. Aujourd'hui, on est loin de comprendre toutes les formes familiales de cancer - qui ne sont d'ailleurs pas toujours le reflet d'un facteur génétique de prédisposition - du fait de connaissances encore incomplètes. C'est ainsi qu'afin d'être dans la meilleure situation possible pour repérer un éventuel facteur génétique, la première étude dans la famille va être conduite chez l'une des personnes les plus susceptibles d'être prédisposées, en l'occurrence une personne qui a déjà été traitée pour un cancer : le fameux cas index. Ainsi, souvent à une personne qui s'interroge sur ses risques, on explique que ce n'est pas tant chez elle qu'un premier test va être réalisé mais chez l'un de ses apparentés : un frère, une soeur, voire une cousine, une mère, un père qui a été déjà atteint d'un cancer et dont le jeune âge au moment du diagnostic est indicateur d'une prédisposition. Ainsi, un apparenté qui n'a pas fait initialement cette démarche peut se trouver sollicité afin de donner son autorisation d'accès à son dossier médical et pour venir en consultation de génétique.
Admettons que l'histoire personnelle et familiale de cet apparenté, devenu cas index, soit validée, que l'indication d'un test génétique soit retenue. Les enjeux du test pour lui-même et ses apparentés sont exposés. Les enjeux médicaux sont essentiellement en cas de résultat positif - un facteur de risque identifié - des recommandations de prise en charge. Dans certains cas, une intervention chirurgicale de prévention peut être retenue. L'autre enjeu à anticiper est celui de l'information des apparentés en cas de résultat positif. Le cas index aurait alors la mission d'informer ses apparentés potentiellement concernés et non seulement la personne qui aurait initié l'étude familiale. Le cas index en a en fait le devoir selon les lois de bioéthique de 2004 et de 2011, et le décret d'information de la parentèle qui a été publié le 20 juin 2013. À partir de ces trois étapes, demande du dossier médical, réalisation du test, diffusion de l'information des apparentés en cas de résultat positif, une personne qui n'a pas initié une démarche de génétique, est ainsi très sollicitée.
Je ne veux pas vous dire que je dénonce ces dispositions - elles reflètent la limite actuelle des tests génétiques et elles ont une vocation de prévention. Je voudrais souligner cependant combien nous devons être prudents et respecter les choix des personnes de réaliser ou non un test génétique, ici de prédisposition aux cancers. Nous mettons en avant le droit de ne pas savoir. Mais il faut arriver, dans cet exercice difficile, à conjuguer le souhait de la personne que l'on vient chercher, de faire ou de ne pas faire quelque chose, et ce que veulent faire les apparentés.
Le deuxième point dont je voudrais vous faire part concerne la confidentialité des informations, notamment au cours des recherches. Les travaux de recherche sont inhérents aux tests génétiques.
L'essentiel des tests génétiques entrés dans la pratique médicale aujourd'hui correspond à l'identification de maladies mendéliennes, c'est-à-dire obéissant à un mode de transmission simple (un seul gène) et souvent associées à une pénétrance élevée, c'est-à-dire à une probabilité de survenue élevée. Il existe cependant une certaine variabilité individuelle de la pénétrance, et c'est un enjeu majeur que de repérer des facteurs modificateurs de cette variabilité, facteurs pouvant être eux-mêmes génétiques. Il y a un souci d'intégrer dans la prédiction des caractéristiques cliniques, biologiques, mais aussi l'exposition à des facteurs environnementaux ou liés au mode de vie, car, on le sait, le tout-génétique n'existe pas.
Nous avons donc besoin de faire des travaux de recherche. Ils sont extrêmement importants, et je rajouterais même, Thomas Tursz l'a très bien dit, ils sont essentiels, car il ne faudrait pas qu'aujourd'hui, s'ils ne sont pas faits, les tests génétiques soient figés par des intérêts économiques. C'est valable autant pour la génétique constitutionnelle que pour la génétique somatique.
Il n'empêche qu'il va falloir transférer des données génétiques du patient mais aussi des données cliniques, biologiques, anatomopathologiques, à des laboratoires de recherche. Ces données vont être anonymisées de façon non irréversible, pour deux raisons. D'une part, les données de l'évolution clinique du patient sont essentielles à la recherche elle-même et d'autre part, les résultats de la recherche, s'ils ont un intérêt démontré pour la prise en charge médicale, tendent à être restitués au patient, sous réserve que celui-ci le souhaite. J'inclus parmi les résultats de la recherche les données incidentes, c'est-à-dire celles obtenues sans les avoir
Il faut trouver un système, M. Jean-Louis Touraine l'a évoqué, qui garantisse à la fois une qualité de la recherche, une certaine réactivité de la recherche, et en même temps la confidentialité des données du patient.
Un autre point que je voudrais soulever est la place des données génétiques dans le dossier médical. Les données génétiques, si elles ont un retentissement dans la prise en charge du patient, doivent figurer dans le dossier. Je souligne ce point car pour il ne fait pas l'objet d'un consensus. Pour certains, tout résultat d'études génétiques ne doit pas figurer dans le dossier. D'une façon générale, toutes les données personnelles du dossier médical doivent être sauvegardées, protégées de la même façon. Ce serpent de mer du dossier médical personnel, bientôt informatisé, va avoir en plus comme gageure d'inclure ces données génétiques. Je crois qu'un dossier médical personnalisé n° 2 est en cours d'élaboration. Ces données doivent être intégrées, avec un ensemble de protections, nécessaires et efficaces.
Dr. Catherine Bourgain, chargée de recherche en génétique humaine et statistiques à l'INSERM. J'ai décidé de cibler mon intervention sur un point assez précis dont il a été plusieurs fois fait mention par les intervenants de la première table ronde et par Dominique Stoppa-Lyonnet : la question des enjeux de la protection de la vie privée à l'heure du séquençage haut débit et du partage à grande échelle des données.
Pour commencer je voudrais rappeler une idée importante : donner un sens clinique, faire parler les données issues du séquençage haut débit, est une opération très complexe qui repose en grande partie sur la comparaison avec des données similaires récoltées auprès d'autres patients ou d'individus non malades. On cherchera par exemple, après avoir identifié chez un patient un variant particulier de séquence sur l'ADN, si ce même variant est retrouvé chez d'autres patients, ou chez d'autres personnes non malades. Recueillir de telles données fait l'objet de très nombreux projets de recherches, mais lorsque l'effet des variations de séquence étudiées est faible ou lorsque les variations étudiées sont relativement rares, des jeux de données de très grande taille sont nécessaires. C'est la raison pour laquelle les pratiques de partage de données entre groupes travaillant sur des pathologies similaires, ou de plus en plus la constitution dès le départ de gros consortiums de recherche, sont de plus en plus fréquentes.
Sous l'impulsion de certains financeurs de ces études, ces larges collections de données sont souvent rendues publiques une fois les premières analyses réalisées. L'idée est de permettre à d'autres chercheurs de réutiliser ces données afin de réaliser des analyses complémentaires dans le même cadre, ou dans un cadre non prévu au départ. Comme les idées ou les découvertes, ces données rendues publiques sont censées pouvoir circuler librement pour irriguer, et polliniser, d'une certaine façon, la recherche.
Pour être utile à ces différents travaux à venir, ces données doivent être constituées de plusieurs éléments, comme Dominique Stoppa-Lyonnet l'a évoqué. Il y a les informations sur la séquence de l'ADN, mais il faut aussi des informations cliniques sur la personne autant que possible, et des informations susceptibles d'être utiles et disponibles, comme l'âge, le sexe, le lieu de vie, les expositions environnementales ou les activités professionnelles. Cela a été largement répété, dans un souci de protection des personnes, ces données sont systématiquement anonymisées.
Le passage de ces pratiques d'échanges entre collègues, ou au sein de consortiums, aux pratiques de grandes collections ou bases de données publiques, a plusieurs conséquences. J'en soulignerai simplement deux. Les usages qui pourront être faits par d'autres des données mises en commun dans ces bases de données, peuvent rarement être prévus à l'avance. Il est probable, et même espéré que les usages effectifs de ces données dépasseront les usages initialement présentés aux personnes qui ont accepté de participer à ces études.
Parmi ces usages, il en est un qui pourrait plutôt s'apparenter à un détournement d'usage, puisqu'il consiste à essayer de désanonymiser les données, autrement dit à retrouver l'identité des participants. Un article dont il a été plusieurs fois fait mention aujourd'hui, paru dans la revue Science au mois de janvier 2013 écrit par le groupe de Yaniv Erlich, un ancien hacker informatique reconverti dans la génomique, a fait grand bruit. A partir de données publiques du projet 1000 génomes, dont Philippe Amouyel a parlé tout à l'heure, et en utilisant d'autres bases de données publiques, en particulier sur les noms de famille et les lieux de résidence, Erlich et son groupe ont pu identifier 5 hommes inclus dans le projet 1 000 génomes, et une fois ces cinq hommes identifiés, un total de cinquante membres de leurs familles, également présents dans la base de données 1 000 génomes, ont pu être identifiés.
Les spécialistes du domaine s'accordent à dire que, je cite : « notre capacité à protéger l'anonymat des données à l'ère du big data, est pour le moins limitée ». Je cite là les termes employés par Eric Schadt, une figure emblématique de la génomique haut débit, actuellement directeur de l'Institut de génomique et de biologie multi-échelle du Mont Sinai Hospital de New York , dans un éditorial de 2012 intitulé « Le paysage changeant de la vie privée à l'ère du big data ».
Face à ce constat, quelles sont les options ?
En matière d'usage imprévu, une solution de plus en plus mise en avant, en particulier aux États-Unis et au Royaume-Uni est celle du consentement légal portable, en anglais, Portable Legal Consent : une personne qui participe à une étude peut décider que les informations personnelles qu'elle communique dans le cadre de cette étude sont libres de droit. Autrement dit, elles peuvent être partagées et utilisées dans d'autres études à condition que les résultats obtenus à partir de ces données soient eux-mêmes libres de droits et non appropriables. Le sujet accepte de renoncer à son droit de regard sur ses données, en échange d'une assurance qu'elles seront utilisées uniquement dans un objectif de bien public, pour le plus grand nombre de personnes.
En matière de garantie de l'anonymat, il y a au moins deux positions exprimées dans la littérature scientifique internationale. :
- Pour régler ce problème de la désanonymisation des données il faut travailler sur la gouvernance des bases de données, et mieux encadrer leur accès. Par exemple, un usage pourrait être restreint aux seuls utilisateurs jugés fiables, sur la base de leur affiliation académique par exemple, ou de déclarations sur l'honneur.
- D'autres considèrent que, je cite : « les attentes du public en matière de confidentialité sont en évolution ». Je reprends là les termes d'un commentaire publié dans la revue Science , en réaction à la fameuse publication de ce hacker dont je parlais, de responsables de l'Institut national américain de la recherche sur le génome humain. Autrement dit, la notion de vie privée serait en train de changer, les gens auraient de moins en moins peur de la transparence sur leurs données personnelles. En conclusion de leur commentaire, ces responsables de l'Institut américain invitent à réexaminer la façon d'équilibrer la protection des participants aux recherches avec les bénéfices sociaux susceptibles d'être obtenus grâce au partage large des données. Autrement dit les bénéfices escomptés de ces partages valent peut-être de revenir en arrière sur la protection a priori des participants.
Pour accompagner ces changements, les tenants de cette vision soutiennent les initiatives centrées sur les patients dont il a été question tout à l'heure, à propos des travaux en particulier de 23andMe , qui permettront aux patients de suivre l'avancée de travaux de recherches, d'être tenus au courant des évolutions par le biais de réseaux sociaux dédiés. Cette implication plus grande du public serait une façon d'augmenter sa confiance.
Le corollaire de cette position est résumé par Eric Schadt, le bio-informaticien-généticien, qui dit : « il me semble que l'éducation et la législation devraient être moins focalisées sur la protection de la vie privée et plus sur la prévention de la discrimination sur la base des informations génétiques personnelles ». Cette évolution n'est pas anodine puisqu'il s'agit au nom des progrès attendus pour la santé et pour l'économie, de considérer qu'une protection en aval, condamner la discrimination, peut désormais remplacer la protection en amont, pour empêcher les usages non désirés.
Dans cette discussion, la question des bénéfices attendus est centrale. La Commission de bioéthique auprès du Président américain a publié à la fin de l'année 2012 un rapport dont Simone Bateman a parlé tout à l'heure sur les enjeux de la vie privée associés aux techniques de séquençage à haut débit, « Privacy and Progress ». Ce rapport commence par une description des bénéfices attendus du séquençage. En un mot, la Commission considère qu'ils sont gigantesques en matière de santé comme d'économie. Le projet génome humain est présenté comme un investissement de 3,8 milliards de dollars, ayant eu un impact économique de 796 milliards de dollars, et qui aurait créé 310 000 emplois.
Je tiens à préciser que je ne compte pas moi-même parmi les tenants de cette ouverture rapide des données personnelles au prix d'une moindre protection de la vie privée, mais cette façon de voir les choses est très présente dans les discussions de la communauté scientifique internationale, et il convient de les avoir en tête au moment de réfléchir à ces questions.
En particulier, je voulais finir en disant quelques mots à propos de cette Alliance globale dont il a été question, lancée en juin 2013, qui agrège une cinquantaine d'institutions issues de huit pays. Pour la France il y a l'INCa et le Consortium maladies rares, mais la majorité de ces institutions sont toutefois américaines. Cette Alliance ambitionne de prendre les mesures nécessaires pour encourager le partage de données en définissant des standards techniques et de procédure harmonisés, pour le respect de la vie privée et de l'éthique. Il s'agit de créer les conditions d'un écosystème ouvert sur le modèle Internet, permettant l'innovation. Dans son texte fondateur, l'Alliance insiste à de nombreuses reprises sur la nécessité de prendre en compte les spécificités nationales. Néanmoins, l'esprit général qui se dégage de ce texte fondateur d'une cinquantaine de pages, d'inspiration très largement américaine, reprend nombre des arguments des tenants de l'évolution vers une moindre protection de la vie privée.
Pr Marc Delpech, chef du service de biochimie et génétique moléculaire de l'hôpital Cochin. Comme Dominique Stoppa-Lyonnet, je vais rester dans mon domaine qui est celui du diagnostic génétique. Il est en fait dans la réalité un peu éloigné du discours ambiant.
Tout est beaucoup plus compliqué que ce que l'on dit à propos des gènes et l'information n'est pas si simple que cela à extraire. Il faut remonter un peu en arrière. En 1944, année où il a été démontré que la molécule d'ADN contient l'information génétique et que tout notre patrimoine génétique est dans cette molécule. Depuis lors, la croyance dans le public a été que la connaissance de la séquence de cette molécule allait conduire à la connaissance de la totalité de l'individu. La conclusion pour chacun est que lorsque l'on donne son ADN, on a tout donné, on est totalement nu, et celui qui le détient va tout savoir sur vous. Ce fantasme reste dans la tête de beaucoup. Cela est faux, et chaque jour on se rend compte de plus en plus à quel point c'est faux. Il existe de nombreux facteurs en dehors qui interviennent aussi.
La capacité de prévoir qui sera atteint ou pas est réelle pour les maladies dont a parlé Dominique Stoppa-Lyonnet, des maladies où un seul gène est en cause, où la pénétrance (probabilité d'être malade lorsqu'on possède la mutation délétère) est pratiquement totale. Lorsque l'on possède la mutation, il est certain que l'on sera malade, mais fort heureusement ces maladies sont rares. Ce dont on parle à côté, et beaucoup depuis ce matin, c'est de toutes les maladies communes, le diabète, l'asthme, l'obésité, les maladies cardiovasculaires. Dans ces maladies, la part que tient la génétique est relativement limitée, et aujourd'hui, le plus souvent, nous sommes totalement incapables de l'analyser.
Jusqu'au dernier quart du XX ème siècle, les biologistes ne pouvaient rien étudier. Le problème de l'accès à l'information génétique ne s'est pas posé, car nous étions incapables de lire l'information génétique. Tout a changé en 1975 où l'on a commencé, indirectement, à avoir quelques petits renseignements, et puis surtout à partie de 1977 où il a été possible de commencer à lire le génome. Mais c'était vraiment de tous petits bouts. Pendant quelques années encore cela n'a pas posé de gros problèmes. La technologie évoluant, entre autres grâce au Projet génome humain, on a été capable de lire de plus en plus de données au sein de la molécule d'ADN. Cela a relancé dans le public l'inquiétude de cette information : qu'allait-on en faire, qui allait en disposer ?
Cela a conduit en 1994 à la première loi de bioéthique. Cette loi n'était pas parfaite, une des preuves en est que le décret d'application qui a prévu l'encadrement de l'activité quotidienne de génétique au laboratoire a mis six ans à sortir, alors que le législateur, très prudemment, avait prévu une révision de la loi dans les cinq ans. En fait la loi a été révisée dans les dix ans. Le fait qu'il ait fallu six ans pour sortir le décret, loin d'être parfait, montrait bien que ce n'était pas si simple et que tous les problèmes n'étaient pas résolus. Mais cela a été tout de même une avancée considérable, et la France a joué un rôle de pionnier puisque les grands principes de cette loi ont été repris par la majorité des pays européens et occidentaux. Également, les recommandations de l'OCDE pour les tests génétiques lui correspondent tout à fait, avec la nécessité de transmettre l'information à un médecin, des qualifications particulières pour la réalisation, etc .
Cette loi a été un progrès formidable. Sa philosophie offre une très grande sécurité pour le citoyen car elle n'autorise, c'est un des premiers principes, l'utilisation des tests génétiques que dans un but médical, de recherche, ou judiciaire. Cela exclut les diables que sont les assureurs, les employeurs, etc ., et globalement on avait une très bonne protection.
Cette loi a été révisée comme prévu en 2004, puis en 2011, et je suis un peu inquiet de certaines évolutions. Dans la loi de 1994, la parentèle évoquée par Dominique Stoppa-Lyonnet était totalement, ou presque oubliée. C'est pourtant un point important car la différence majeure entre un test génétique et tout autre examen de biologie médicale, est que le résultat de ce test a des conséquences pour toute la famille du patient et pas seulement pour lui-même comme habituellement. Cet élément devait être pris en compte : il l'a été dans la révision de la loi de 2004 où il était précisé que le médecin devait faire tout son possible pour convaincre le patient d'informer sa famille s'il était porteur d'une anomalie génétique susceptible de provoquer une maladie grave alors qu'il est possible de prendre des mesures pour prévenir ses effets. Il n'empêche que plus loin dans la loi, il était dit que nul ne peut être poursuivi pour ne pas avoir transmis ses caractéristiques génétiques à sa famille.
Quand on regarde la révision de la loi de 2011, ce n'est plus cela du tout. Il y a obligation au patient de transmettre l'information génétique à sa famille, suivant des modalités pouvant inclure le fait que lui ne sera pas au courant : à ce moment-là il demande à ce qu'un médecin informe sa famille sans dire ce qu'il en est pour lui. En 2004 il avait été prévu toute une série de mesures pour permettre la transmission de l'information aux membres de la famille à la demande du patient, mais cela était tellement complexe qu'elles n'ont jamais été appliquées, et c'est pour cela que la loi de 2011 a un peu nettoyé le dispositif. Mais il n'empêche que cette nécessité absolue d'informer la famille est une remise en cause du secret médical, compréhensible, mais qui m'inquiète, car c'est un changement majeur.
En France, nous adorons les lois, nous les accumulons. Le problème est que de temps en temps elles se télescopent. Il vient de sortir un guide de bonnes pratiques des tests génétiques, évoqué par Mme Emmanuelle Prada-Bordenave. Il a été rédigé avec des professionnels par l'Agence, a été validé par le Comité d'orientation de l'Agence et par la Haute autorité de santé. Il est sorti début juin.
Dedans, on y trouve des phrases qui n'y étaient pas à l'origine. Le Ministère a modifié le texte adopté par l'Agence de la biomédecine et la HAS. Et ce ne sont pas des modifications anodines. Il est par exemple indiqué que le résultat doit être rendu au laboratoire ayant effectué le prélèvement. C'est un changement absolument fondamental, même s'il est précisé que le laboratoire préleveur ne doit pas transmettre le résultat au patient, mais au médecin prescripteur. Le fait que le laboratoire réalisateur n'adresse pas directement le résultat à celui qui a réalisé la prescription n'est pas satisfaisant. En effet ces examens sont en général excessivement complexes et nécessitent des connaissances que n'ont pas la majorité des biologistes. Seuls les quelques laboratoires en France qui les réalisent les ont. Cette nécessité de dialogue entre celui qui a réalisé l'examen et celui qui va le rendre est ignorée. De plus, en rendant l'examen au laboratoire préleveur, cela fait transmettre un résultat par quelqu'un qui théoriquement n'a pas le droit de le faire, puisque la loi impose que le laboratoire soit autorisé, et que le praticien soit agréé. Or, le laboratoire préleveur en général n'est ni autorisé, ni agréé. Donc c'est un coup de canif majeur dans la loi. Cette modification a été faite simplement pour être conforme à la loi « Hôpital, patients, santé et territoires » (HPST) de 2009, et à l'ordonnance Ballereau de 2010 qui imposent l'obligation au laboratoire préleveur d'être jusqu'au bout responsable de la transmission des examens et cette ordonnance ne prend pas en compte la spécificité de la génétique et les problèmes éthiques sous-jacents. Il y a donc de temps en temps des dérives, et c'est un peu inquiétant.
Depuis cinq à sept ans, les choses ont terriblement évolué. La technologie est devenue d'une puissance inimaginable. Voici quelques années, la détermination de la séquence du génome humain coûtait plus de 3 milliards de dollars. Aujourd'hui le grand centre de séquençage de Pékin peut séquencer plusieurs individus par jour, pour un coût de moins de 1 000 euros. L'information génétique globale va être accessible de manière très étendue, et se posent maintenant tous les problèmes de protection évoqués tout à l'heure.
Il ne faut pas pour autant s'inquiéter. Franchement, si on mettait sur Internet 99 % de mon patrimoine génétique avec sa séquence, je m'en fiche complètement, car cela ne sert absolument à rien. Mais il y a quand même quelques régions extrêmement sensibles, et je me rappelle le souhait de la deuxième personne dont on a déterminé la séquence au monde : c'était James Watson. C'est lui qui a découvert la structure de la double hélice d'ADN. Il a exigé qu'une région du génome, celui de l'apolipoprotéine E dont l'allèle å4 est lié à la maladie d'Alzheimer, ne soit pas accessible, même pas à lui-même.
Cette quantité d'informations extraordinaire va poser des problèmes pour chacun, car nous allons nous retrouver inondés d'informations (si nous le souhaitons), que notre médecin sera totalement incapable d'interpréter. Même si le résultat doit être transmis par un médecin prescripteur compétent, comme il est dit dans la loi, le résultat est souvent « ésotérique » et difficile à comprendre.
Quand il ne s'agit plus de maladies monogéniques, mais de maladies communes, ce qui est rendu n'est pas un diagnostic, mais un risque. Pour l'immense majorité des individus, le risque est une notion assez abstraite. Je peux donner un exemple qui m'a été fourni par Pierre Corvol et que je trouve excellent. Si vous fumez deux paquets de cigarettes par jour, vous multipliez par 25 votre risque d'avoir un cancer du poumon, et à peu près 80-90 % des cancers du poumon sont dus au tabac. Les Français considèrent que ce risque est dérisoire et leur immense majorité fume. Si on prend un autre exemple, la probabilité de gagner le gros lot au loto est de 1 sur 13 millions. Les Français considèrent que c'est absolument gigantesque, et certains jouent plusieurs fois par semaine. Et ce sont les mêmes qui considèrent qu'augmenter 25 fois son risque est sans importance Dans un domaine aussi complexe que l'information génétique, si l'on vous explique que vous avez un risque de 1,05 de développer une maladie cardio-vasculaire, cela peut affoler alors que cela ne veut rien dire. Des entreprises comme 23andMe proposent ce genre de test et sortent des listings entiers de risques possibles, et régulièrement remis à jour, dont la très grande majorité n'a aucune valeur réelle. Je pense qu'il y a un risque non négligeable pour l'individu d'être traumatisé par de tels résultats et de développer des pathologies psychosomatiques, alors qu'il est parfaitement bien portant.
Malheureusement, malgré ce que pensent certains, on ne peut rien faire dans la mesure où le monde est ouvert et où tout est accessible par Internet. Il suffit de prendre un petit écouvillon, de se brosser l'intérieur des joues, d'envoyer cela en Espagne ou en Australie ou ailleurs, et l'on a tout ce qu'on veut. La loi peut faire ce qu'elle veut, le phénomène ne s'arrête pas aux frontières. En France on n'a pas le droit de faire une recherche de paternité, mais il suffit d'aller en Espagne, en Italie ou en Suisse, et c'est possible. On ne peut donc pas compter sur la loi ou alors il faudra montrer beaucoup d'imagination. Pour moi, le facteur principal, fondamental, est l'éducation du citoyen. Il faut qu'il sache ce qu'il en est, qu'il comprenne ce que cela veut dire. Là uniquement se trouve, à mon avis, une solution.
Dr Jean-François Deleuze, directeur du Centre national de génotypage au CEA . Si j'en crois ce que j'ai entendu ce matin il faut former les médecins en génétique et la population en maths et en statistiques, et nous nous en trouverons mieux... Je me sens comme un technicien après tous ces débats. Je ne suis pas un médecin, mais un scientifique qui produit de la séquence. Je vais vous donner quelques éléments dans cette fonction-là. Que se passe-t-il dans un centre de séquençage, comment cela rentre-t-il, qu'en sort-il et quelles sont les précautions prises ?
Je vais aborder deux points : le consentement éclairé, et le côté confidentialité-sécurité des données.
Au CEA, le Centre national de génotypage (CNG), est un mot un peu misleading comme disent les anglais, car on y fait essentiellement du séquençage. Donc CNG est une appellation historique. C'est en gros le grand centre de séquençage du génome humain en France, une entité du CEA. Vous avez entendu parler tout à l'heure du centre de Pékin que tout le monde connaît. En France c'est le CEA qui gère ce groupe, qui n'a pas les capacités du BGI de Pékin qui a 120 ou 150 séquenceurs, mais en dispose d'environ 11. Avec cela nous pouvons faire trois génomes humains par jour, donc à Pékin, en multipliant par 10, ils peuvent en faire probablement 30 ou 50. C'est incommensurable quand vous vous rappelez qu'il a fallu dix ans, dans les années 90 à 2000 pour en faire un. Voilà l'évolution et l'explosion de la recherche. Le CNG est un opérateur, il n'est pas initiateur, ou promoteur au point de vue juridique du terme, de projets. Il opère pour la communauté scientifique. Nous sommes un centre de ressources, essentiellement académique, pour l'INSERM en particulier.
En termes juridiques et de consentement éclairé, cette responsabilité est associée aux collaborateurs avec qui nous travaillons, qui nous garantissent que les projets et les cohortes d'ADN qu'ils nous soumettent, ont été recensés et sécurisés avec les consentements éclairés idoines.
Quand l'ADN arrive au CNG, nous faisons trois types de choses :
On parle de big data, de génome. Il faut peut-être démystifier. Ces dix dernières années, nous regardions entre un et cinq million de positions, parmi les 3 milliards qui existent dans le génome humain, soit donc environ 0,1% de l'information génétique, non fonctionnelle. En fait nous faisions de l'association. Ce sont des études de populations qui permettent, avec de gros échantillons, de trouver de temps en temps des effets faibles ou rares, qui ne renseignent pas énormément au niveau de l'individu.
Aujourd'hui nous faisons de l'exome. Nous sommes passés d'une approche d'association à une approche plus fonctionnelle. Nous regardons ce que l'on considérait encore il y a peu, les éléments fonctionnels du génome, qui représentent 2 %. Tout le reste est à considérer comme de l'ADN poubelle, notion aujourd'hui remise en question. Cet exome ne représente que 2% de la totalité du génome humain. C'est ce qu'on fait aujourd'hui en routine, et nous trouvons des pénétrances fortes pour les maladies rares. Nous avons là la chance probablement de trouver un gène ou une mutation vraiment causale par rapport à la maladie.
L'émergent est le génome entier, les 3 milliards de paires de bases, qu'on sait faire techniquement. Il coûte beaucoup plus cher, et renseigne sur la totalité de l'information présente dans les gènes, non seulement les éléments fonctionnels, mais les éléments régulateurs de l'expression des gènes. On est capable de le produire, mais non de l'analyser, ou de faire des prédictions définitives. D'où l'appellation de big data, car on produit des giga, des tera, des pétaflop de données.
On a produit au CNG des milliers d'exomes, qui occupent, comme le disait Jean-Louis Touraine, des quantités impressionnantes de stockage informatique. Aucune de ces données n'est exposée dans le domaine public. Toutes ces données que nous produisons proviennent d'échantillons totalement anonymes. Ils sont anonymisés et codés par nos collaborateurs. Quand un projet arrive, nous savons que nous avons un échantillon de x patients. Nous recevons des collaborateurs, des tubes et des codes barre, et nous ne sommes pas capables de revenir en arrière. Nous les utilisons comme du matériel biologique. Le collaborateur est celui qui a l'adéquation entre le code barre, et les structures familiales.
Ces données générées, extrêmement massives, sont déposées, sur nos serveurs au CNG, et accessibles pour nos collaborateurs, par des sites web sécurisés avec des mots de passe. Ce n'est pas que nous ne sommes pas intéressés à les mettre dans des bases publiques, mais ce n'est pas fait. Il reste la question de mettre ces données à la disposition, non pas du public, mais d'une communauté intelligente et respectueuse, qui pourra s'en servir à la fois dans le respect de la confidentialité et pour l'intérêt de la science. Il y a un intérêt à partager ces données. On génère des pétaflop de séquences, qui sont envoyés à nos collaborateurs dans des formes très sécurisées.
Sur le côté éthique et consentement éclairé, nous ne sommes pas promoteurs, nous ne sommes pas les gens qui décidons les consentements éclairés. Mais nous demandons qu'ils soient revus, à la fois pour nous assurer que ce qui se passe est en adéquation avec le consentement éclairé, et pour comprendre quelle évolution il va falloir lui apporter.
En gros on a deux types de consentements éclairés :
- Il y a des consentements très larges, pour étudier la génétique de la maladie, qui sont pour le scientifique très confortables, car ils couvrent quasiment toutes les applications.
- Il existe des consentements très restrictifs, comme il y a pu avoir voici quelques années, où il fallait mentionner des listes de gènes, consistant par exemple à autoriser à regarder si des polymorphismes dans un gène précis sont impliqués dans une maladie.
La science va beaucoup plus vite que les réunions des conseils d'éthique. Vous avez dit tout à l'heure qu'il avait fallu dix ans pour faire un décret. Or la science, la technologie avancent au bénéfice de la médecine, et il est extrêmement compliqué de revenir vers un patient, qui peut être mort, avoir changé de pays, pour lui refaire signer quelque chose. Nous sommes dans un dilemme. Le consentement éclairé très large permet de travailler et de faire bénéficier la science et peut-être le patient, des avancées technologiques qui arrivent, c'est le cas du Whole genome . Avec le consentement éclairé très précis, on dit exactement à la personne ce que l'on va faire : un exome, par exemple. Quand le consentement est très large, par exemple génétique du diabète, tout est couvert. Mais c'est très hétérogène, on trouve de tout, des consentements très larges, des très spécifiques, et il faut aussi reconnaître que souvent nous sommes confrontés à ces questions ; puisque le consentement éclairé a une valeur dans le temps et que la technologie va plus vite, il faudrait faire re-consentir toute la cohorte. C'est souvent impossible.
Il faut probablement que nous soyons proactifs. Je ne parle pas seulement des découvertes fortuites, qui sont rarement mentionnées. Pour le retour vers le patient certains consentements sont très clairs : il n'y aura pas de bénéfice direct, et aucun retour. Et pour d'autres la personne peut demander accès à ses données si elle le veut. Je ne dis pas qu'il n'en faut qu'un, mais nous devons probablement gagner un peu d'homogénéité dans ces consentements éclairés.
En matière de confidentialité, le CNG ne reçoit aucun échantillon identifiant par données sociodémographiques. Nous n'avons que des codes barre. Prenons un exemple : nous envoyons des données pour des collaborations avec Philippe Amouyel : il reçoit avec un mot de passe à la clef sur des sites web sécurisés toutes ces data.
Mais il y a quand même un intérêt évident à les combiner et à les partager. Encore une fois je répète ce qu'a dit Catherine Bourgain, il faut certainement trouver une façon de le faire. Il y a des raisons scientifiques de le faire : obtenir les tailles d'échantillons dont nous avons besoin pour la puissance statistique, faire des méta-analyses. Il faut donc réfléchir au meilleur moyen de trouver un forum ou une façon de le faire pour que le spectre pernicieux de la génétique un peu diabolique se change en un cercle vertueux où l'individu participe à la collectivité en mettant ses données au service de l'analyse globale et qu'avec beaucoup de chances il lui revienne quelque chose.
Mme Audrey Aboukrat, doctorante à l'école de droit de l'université Paris I Panthéon Sorbonne. Comme son nom l'indique, la médecine personnalisée propose une adaptation du diagnostic et des traitements proposés au patient, accroissant ainsi l'efficacité de ceux-ci par leur action ciblée. La personnalisation de la médecine peut en ce sens entraîner des distinctions au sein de la population générale si des traitements et diagnostics individuels se dégage un constat collectif. Cependant, si toute distinction entre groupes d'individus au sein d'une population générale n'implique pas nécessairement une stigmatisation des individus, il existe des risques de rupture d'égalité entre les patients, voire des risques de discrimination dans certains cas (248 ( * )) .
Imaginons par exemple un test de diagnostic ou un médicament qui ciblerait des groupes d'individus en fonction de leur « race » ou de leur « ethnie » et que ce ciblage soit en plus figé dans les revendications d'un brevet, alors le risque de discrimination s'intensifie.
Le ciblage de groupes d'individus au sein de populations générales en fonction de leur « race » ou de leur « ethnie » constitue-t-il une première étape légitime, voire décisive, vers une médecine personnalisée ? Ou s'agit-il au contraire, en l'absence de véritables fondements scientifiques, d'un engrenage possiblement discriminatoire pour les « races » ou les « ethnies » ainsi visées ? C'est à cette problématique que je consacrerai ma présentation, qui, il faut le préciser, prend très largement appui sur un article que nous avons écrit, Mme Christine Noiville et moi-même dans un ouvrage publié en 2011 (249 ( * )) .
Pour structurer cette réflexion, je prendrai deux cas d'études : d'une part le BiDil, qui est un médicament destiné au traitement de l'insuffisance cardiaque au sein de la population africaine américaine exclusivement, et d'autre part, le test génétique commercialisé par l'entreprise Myriad Genetics pour le dépistage d'une prédisposition aux cancers du sein et des ovaires chez les femmes juives ashkénazes exclusivement.
L'histoire du BiDil est celle d'un médicament contre l'insuffisance cardiaque, qui sur le point de tomber dans le domaine public après avoir été protégé par un brevet d'invention, a retrouvé la voie de la rentabilité. Pour comprendre comment, il faut resituer ce médicament dans son contexte médical d'abord, administratif ensuite.
Aux États-Unis, environ cinq millions d'individus souffrent d'insuffisance cardiaque et 500 000 nouveaux cas sont diagnostiqués chaque année, ce qui conduit à d'importantes dépenses de santé pour les patients concernés (250 ( * )) et crée un véritable marché pour l'industrie pharmaceutique. Si les bêtabloquants commercialisés réduisent sensiblement le taux de mortalité au sein de la population « blanche », ceux-ci n'ont qu'un effet limité sur les patients dits « Noirs » (251 ( * )) . Dans une société comme la société américaine où le gouvernement prend peu en charge les dépenses de santé, de telles statistiques apportent un éclairage intéressant sur la commercialisation d'un médicament susceptible d'apporter un traitement adapté aux patients Noirs souffrant d'insuffisance cardiaque.
Le BiDil est un médicament qui résulte de la combinaison de deux molécules : l'Isosorbide dinitrate et l'Hydralazine. Destinée au traitement de l'insuffisance cardiaque, cette combinaison de molécules a fait l'objet d'un brevet en 1989 revendiquant « une méthode permettant de réduire le risque de mortalité associé à l'insuffisance cardiaque chronique chez un patient souffrant d'une déficience de la fonction cardiaque en concomitance avec une tolérance réduite à l'exercice » (252 ( * )) . La Food and Drug Administration (FDA), agence américaine chargée d'autoriser la mise sur le marché des médicaments, avait toutefois refusé en 1997 d'autoriser la commercialisation de la combinaison de molécules.
En 2000, alors que le premier brevet relatif à cette combinaison tendait à expirer, NitroMed a cherché à obtenir un nouveau titre portant sur la même combinaison de molécules, mais visant cette fois une nouvelle application thérapeutique : l'utilisation du même médicament de manière ciblée chez les seuls patients tantôt définis comme « Noirs », tantôt comme afro-américains ou africains américains (253 ( * )) . Le brevet fût délivré en 2002 et assigné à l'entreprise NitroMed (254 ( * )) .
Cette fois, la FDA autorise la commercialisation du BiDil en 2005, pour le traitement de l'insuffisance cardiaque chez des patients identifiés comme Noirs ou Africains américains (255 ( * )) . Notons qu'avec le BiDil, c'est la première fois (256 ( * )) que la FDA autorise la commercialisation d'un médicament qui vise des patients identifiés en fonction de leur « race » au sens des recommandations de la FDA (257 ( * )) . Pour les essais cliniques, quant aux données collectées eu égard à la « race » et « l'ethnie » des participants, la FDA recommande en effet de leur proposer, deux « ethnies » (hispanique ou latino et non-hispanique ou latino) et cinq catégories pour la « race », parmi lesquelles la « race » « noire ou africaine américaine » (258 ( * )) . En conclusion, si l'action ciblée d'un médicament pour le traitement d'une affection au sein d'une population déterminée pour laquelle les médicaments existant ne se révèlent pas efficaces nous semble une grande avancée en faveur du développement d'une médecine personnalisée, le fait que les patients soient tantôt présentés comme Africains américains, tantôt comme Noirs, révèle une imprécision terminologique importante quant à la population véritablement visée. En dépit d'un effort de la FDA pour distinguer « race » et « ethnie », ces notions demeurent en pratique le plus souvent indistinctement utilisées pour identifier un même groupe de population ; les Hispaniques, par exemple, sont tantôt regardés comme une « ethnie » (aux États-Unis), tantôt comme une « race » (en Europe) (259 ( * )) . Par conséquent, si la « race » et « l'ethnie » peuvent être envisagées par les chercheurs comme un moyen utile et rapide de classer les patients soumis à un test ou souffrant d'une maladie, les implications d'un tel usage en droit génèrent un tout autre écho, porteur d'un potentiel discriminatoire.
Si dans le cas du BiDil la population visée correspondait à l'une des « races » mises en évidence par la taxonomie de la FDA, ce n'est pas le cas de la population visée par le test proposé par l'entreprise Myriad Genetics , destiné à identifier une prédisposition au cancer du sein chez les seules femmes juives ashkénazes.
L'entreprise Myriad Genetics a obtenu aux États-Unis un brevet (260 ( * )) revendiquant des méthodes et matériels utilisés pour isoler et détecter le gène BRCA2 dont certains allèles mutants causeraient une prédisposition au cancer du sein. En parallèle, Myriad Genetics a déposé une demande de brevet auprès de l'Office européen des brevets (OEB) pour la même invention (261 ( * )) , entrant alors en compétition avec le Centre anglais de recherche sur le cancer (CRUK (262 ( * )) qui avait lui-même déposé des demandes de brevets comparables au Royaume Uni (263 ( * )) avant que Myriad Genetics ne dépose sa propre demande de brevet aux États-Unis (264 ( * )) . C'est le Centre anglais qui s'est finalement vu délivrer en 2004 un brevet européen (265 ( * )) revendiquant une invention portant sur « l'identification et le séquençage du gène BRCA2 de susceptibilité au cancer, et sur le matériel et les méthodes dérivant des résultats obtenus » (266 ( * )) Myriad Genetics a en conséquence décidé d'amender les revendications de sa demande de brevet en précisant ses revendications par la référence à l'usage d'un acide nucléique particulier portant une mutation du gène BRCA2 associée à une prédisposition au cancer du sein, pour un diagnostic in vitro d'une telle prédisposition « chez les femmes juives ashkénazes ». Objet d'une opposition, cette demande de brevet a ensuite été approuvée en 2006 dans sa version modifiée par la Division d'opposition de l'OEB. Les opposants au brevet ont notamment fait valoir son caractère discriminatoire expliquant que l'application de l'invention brevetée aux seules femmes juives ashkénazes était contraire à l'ordre public (267 ( * )) . Rappelons que l'article 53 de la Convention sur le brevet européen interdit la délivrance de brevets pour des inventions contraires à l'ordre public et aux bonnes moeurs. La Division d'opposition de l'Office européen des brevets a répondu que le test breveté se révèlerait bénéfique aux femmes juives ashkénazes et donc que, si discrimination il y avait, celle-ci ne pouvait être qu'en faveur des femmes concernées (268 ( * )) . Plus encore, la Division d'opposition a considéré que ce serait davantage le choix de ne pas développer le test génétique qui pourrait être discriminatoire pour ces femmes souffrant d'une prédisposition particulière.
Si les arguments avancés par la Division d'opposition ont un sens en eux-mêmes, ils perdent en pertinence lorsque l'on compare le test génétique offert par Myriad Genetics à celui du Centre anglais, lequel couvre tous les types de mutations dans la recherche d'une prédisposition au cancer du sein, y compris donc la mutation expressément visée par le test proposé par Myriad Generics. En outre, le Centre anglais accorde des licences libres (c'est-à-dire qu'il n'est pas nécessaire de payer des droits de licence pour exploiter le test) à toutes les institutions médicales qui le demandent, alors que Myriad Genetics monnaie ses licences, ce qui accroît le prix à payer par les patients concernés aux détenteurs de la licence (269 ( * )) . Ainsi, un test pour le diagnostic des mutations associées aux gènes BRCA 1 et 2 coûte entre 2 500 et 3 000 dollars lorsque celui-ci est acheté directement à Myriad Genetics , 5 000 dollars lorsque celui-ci est acquis auprès de l'entreprise allemande Bioscientia qui a obtenu de Myriad une licence (270 ( * )) .
Comme le remarque l'Institut Curie dans son opposition au brevet européen, le test proposé par Myriad Genetics impose aux seules femmes juives ashkénazes le paiement d'une prime élevée pour effectuer le diagnostic (271 ( * )) . D'un point de vue pratique, cela signifie que les femmes qui s'identifient comme juives ashkénazes souhaitant faire dépister une éventuelle prédisposition au cancer du sein et des ovaires se trouvent face à l'alternative suivante : ou bien s'adresser directement à Myriad Genetics et payer le surcoût, ou bien nier leur identité auprès des médecins utilisant le test développé par le CRUK (272 ( * )) , ceux-ci étant contraints de respecter le brevet plus spécifique détenu par Myriad Genetics et donc de ne pas faire le test aux femmes qui se déclarent juives ashkénazes, au risque d'être déclarés contrefacteurs. A la lumière de ces éléments, il semble plus difficile de discerner les avantages du test proposé par Myriad pour les femmes juives ashkénazes.
Face à cette situation, nous avons proposé la réaffirmation de trois grands principes (273 ( * )) :
Tout d'abord, plutôt qu'une « race » ou une « ethnie », les médicaments mis sur le marché devraient continuer à cibler une maladie et les tests génétiques une mutation. En effet, les indications qui fleurissent aujourd'hui tendent à dessiner d'une façon scientifiquement fragile des traits identiques par « races » ou « ethnies » (274 ( * )) et de nombreuses questions restent en suspens, parmi lesquelles les suivantes : le médicament « racial » pourra-t-il être prescrit au patient ne relevant pas de la « race » en question, même s'il est affecté par la pathologie concernée ? Comment déterminer l'appartenance au groupe de population visé ? Le système d'auto-identification des patients est-il suffisant ?
Ensuite, la « race » ou l' « ethnie », ou plutôt le groupe d'ascendance, devraient pouvoir être pris en considération pour la détermination des indications et contre-indications des produits médicaux dès lors que ces catégories sont posées comme étant scientifiquement pertinentes, mais aussi dynamiques et flexibles. Cette réflexion s'aligne avec l'observation faite ce matin sur l'importance d'inclure à la définition de la médecine personnalisée son caractère mouvant. Prenons l'exemple du Crestor, médicament approuvé en mars 2003 par l'Agence nationale de sécurité du médicament et des produits de santé, qui prévoit dans ses contre-indications que la dose de 40 milligrammes ne doit jamais être prescrite pour les personnes présentant certains facteurs de risques, dont les personnes d'ethnie japonaise ou chinoise (275 ( * )) . Si la connaissance de l'ascendance des patients est essentielle dans la recherche biomédicale et ne pose pas de problème à cet égard, il nous semble que les notions de « race » et d' « ethnie » demeurent trop imprécises à elles seules pour décrire les variations génétiques existant au sein de l'espèce humaine (276 ( * )) . Afin d'éviter des confusions entre, d'une part, les notions de « races » et d'« ethnies » et, d'autre part, la génétique et la biologie, il convient d'intégrer ces notions dans des catégories flexibles et dynamiques. Cela signifie que si les critères sous-jacents à la constitution de groupes de populations étaient susceptibles d'évoluer en fonction de l'environnement et du contexte dans lesquels ils sont sollicités, et s'ils étaient étayés par de solides arguments scientifiques, les risques de discrimination sur le long terme s'en trouveraient fortement réduits. Prenons le cas de la maladie périodique, affection héréditaire qui se traduit notamment par des accès récurrents de fièvre, accompagnés de douleurs abdominales et thoraciques. Le groupe de population identifié dans le traitement de cette affection est défini premièrement par une origine géographique commune : le pourtour méditerranéen. Pourquoi ensuite sous-diviser cette étude et figer dans des revendications de brevet des « races » ou « ethnies » particulières, en distinguant les Arméniens, les Grecs, les Juifs, les Sépharades, les Turcs, etc . ? Il nous semble que l'origine géographique est un critère pertinent en tant que tel sans qu'il soit nécessaire, à moins que cela soit étayé par des critères scientifiques pertinents, de sous-diviser ensuite ces populations en sous-groupes.
Enfin, la démonstration d'un lien net s'impose selon nous entre la maladie ou l'exposition à la maladie d'une part, l'« ethnie », la « race » ou le groupe d'ascendance d'autre part, l'intérêt et les effets du médicament ou du test enfin. Il convient selon nous d'éviter à l'avenir que les offices de brevets ne cautionnent trop rapidement un rattachement et un enfermement des variations génétiques ou de certaines populations dans des catégories raciales ou ethniques qui seraient rigides. On peut noter en ce sens trois décisions importantes des chambres de recours de l'Office européen des brevets qui n'acceptent qu'à de strictes conditions la brevetabilité de nouvelles applications thérapeutiques pour le traitement d'une maladie comme affectant un groupe de sujets particuliers (277 ( * )) . Dans ces trois décisions, l'Office impose des exigences strictes, quelle que soit la catégorie proposée au titre de la nouvelle application thérapeutique. Il faut que le groupe de sujets en question soit distinct du groupe antérieurement visé sur les plans physiologique et pathologique ; on est là sur des critères scientifiques, et non pas liés à la « race » ou l' « ethnie », polysémiques et difficiles à définir en droit des brevets. Il faut également que soit prouvée une relation fonctionnelle entre l'état physiologique et pathologique des patients visés d'un côté, et l'effet thérapeutique obtenu de l'autre, de sorte que soit justifiée la distinction opérée entre plusieurs sous-groupes au sein de la population générale (278 ( * )) .
Mme Frédérique Lesaulnier, coordonnatrice du pôle santé de la Commission nationale de l'informatique et des libertés (CNIL). Aujourd'hui les biotechnologies connaissent une véritable révolution, un gigantesque changement d'échelle dans le temps et dans l'espace dû au progrès des sciences et des techniques. Il existe de grands projets fortement médiatisés, internationaux. On a parlé d'Alliance globale tout à l'heure, 70 institutions, 40 pays, qui se met en place. Mais il existe aussi des projets d'envergure nationale à l'échelle d'une population entière, comme DeCODE Genetics en Islande.
Ces bases de données gigantesques ont vocation à être conservées très longtemps et mises à disposition de la communauté des chercheurs à travers une plateforme et à permettre la mise en oeuvre d'études spécifiques ultérieures. Ce phénomène n'est d'ailleurs pas propre aux biothèques. Cette nouvelle organisation de la recherche lance aux autorités de protection des données de nouveaux défis et conduisent à une évaluation des régimes de protection et sans doute une adaptation de ceux-ci. La recherche médicale évolue, de façon globale, vers une mutualisation des moyens, la création de vastes bases de données à des fins de recherches ultérieures, et la réutilisation de bases de données déjà existantes, médico-administratives en particulier, à des fins de recherche, qui appellent probablement de nouvelles modalités d'encadrement
Il est vrai que la génétique a sans doute joué un rôle moteur à la suite du séquençage du génome humain compte tenu de la valorisation des données qui en a résulté. Sans doute faut-il dire que ces vastes bases de données et leur partage présentent un intérêt scientifique majeur. Elles constituent un atout considérable pour la recherche médicale, diagnostique ou thérapeutique, la recherche en santé publique, la génétique des populations, et sont utiles à l'humanité.
D'ailleurs la CNIL autorise nombre de ces traitements. Elle a autorisé l'INSERM, le 8 novembre 2012, à mettre en oeuvre une étude génétique de la population d'origine arménienne, visant à déterminer la structure génétique de cette population et à connaître, ainsi, l'ancienneté de la population étudiée, l'origine et l'histoire du peuplement des différentes régions et faciliter, par la suite, dans un intérêt de santé publique, l'identification de gènes de susceptibilité de maladies polygéniques dans cette population, dans le cadre d'études dites « d'association cas témoins ». Il y a donc un intérêt collectif et individuel majeur.
Pour autant, ces avancées ne doivent pas faire oublier que les données génétiques sont des données de santé, et, à ce titre, ce sont relatives à l'intimité de la vie privée. Elles portent en elles des potentialités discriminatoires. Ce ne sont pas des données médicales comme les autres, et c'est pourquoi elles appellent une protection renforcée. Vous le savez, l'information génétique est une information « à sujets multiples », puisqu'elle concerne une personne, mais aussi toute la lignée de ses apparentés génétiques. Elle présente un caractère unique et permanent pour chaque individu. Enfin, elle présente un caractère prédictif et est susceptible de révéler l'état de santé présent et futur d'une personne, de sorte que lorsque l'on délivre l'information à la personne intéressée, on s'adresse à une personne en bonne santé et on lui révèle la maladie qui la frappera, bien avant qu'elle ne s'exprime. Il faut d'ailleurs être très vigilant sur la prévision et la force attachée à cette prévision en cas de maladie multifactorielle. Et même lorsque l'on a des certitudes, les possibilités thérapeutiques n'évoluent pas parallèlement à la capacité prédictive, de sorte que la personne ne peut tirer aucun bénéfice pour elle-même d'une révélation dotée, en outre, d'une très grosse charge affective. Se pose dès lors la question de l'intérêt de s'entendre annoncer un destin biologique auquel on ne peut échapper par la prévention ou le traitement.
Á tous les titres, la donnée génétique n'est pas une information comme les autres et c'est si vrai que le futur projet de règlement lui reconnaît un caractère sensible.
En raison de ces caractéristiques particulières, le droit français d'une manière générale confère aux données génétiques un statut particulièrement protecteur. En outre, les biotechnologies évoluent dans un réseau d'obligations législatives et réglementaires qui se superposent. Le corps humain, ses éléments, sont des attributs de la personne humaine et soumis à ce titre à un principe d'indisponibilité et de non commercialisation. L'étude des caractéristiques génétiques n'est autorisée que pour certaines fins déterminées par la loi, et à l'inverse, certaines utilisations sont interdites : la discrimination génétique en matière d'emploi, ou d'assurances en particulier, même si on dispose du consentement de la personne. La CNIL a réaffirmé son attachement à ce principe de non-discrimination dans le cadre de la consultation publique lancée sur ce thème par le Comité bioéthique du Conseil de l'Europe en 2012.
En outre les collections d'échantillons biologiques donnent lieu à une déclaration, une demande d'autorisation auprès du Ministère de l'Enseignement supérieur et de la Recherche selon qu'elles sont vouées à une utilisation par l'organisme gestionnaire, ou à être cédées. Enfin les échantillons biologiques ne doivent pas être considérés indépendamment des données à caractère personnel qui les accompagnent, et qui sont en constante interaction. De plus, les échantillons biologiques sont eux-mêmes des vecteurs d'information. C'est la raison pour laquelle la CNIL trouve à en connaitre, et la loi « Informatique et Libertés » a vocation à s'appliquer.
Cette loi pose un principe d'interdiction de traitement des données sensibles, qui connaît des dérogations en cas de consentement de la personne, à des fins de suivi médical individuel, à des fins d'intérêt public, de recherche médicale et d'évaluation de pratiques de soins, selon des procédures certainement à simplifier, compte tenu du parcours du combattant auquel sont confrontés aujourd'hui les chercheurs. Un autre fondement est susceptible de lever cette interdiction, l'anonymisation des données à bref délai
Une fois que l'on entre dans l'une de ces exceptions, qui permettent le traitement des données génétiques, il existe des garanties au respect desquelles la CNIL veille : premièrement, un traitement est mis en place pour une finalité précise, déterminée, légitime, et conforme aux missions de l'organisme ; deuxième principe, les données collectées doivent être pertinentes au regard de cette finalité, et non excessives ; troisième principe, la durée de conservation des données doit être limitée et fonction de cette finalité. Je les mentionne pour montrer qu'ensuite, les modalités nouvelles de la recherche lancent des défis à tous ces principes.
La durée de conservation limitée des données en fonction de la finalité de l'étude, le respect du droit des patients, le consentement des personnes, et bien sûr l'information préalable sont fondamentaux. De ces droits dépendent tous les autres, le consentement des personnes quand il y a des prélèvements biologiques identifiants, et, une fois les données collectées, traitées, le droit d'accès des personnes à des fins de rectification le cas échéant
Le dernier principe est celui de la sécurité des données. Il faut en effet veiller à ce que les données soient conservées par le responsable du traitement dans des conditions de nature à garantir leur confidentialité, mais aussi leur intégrité.
Voilà les différents principes posés.
La législation française offre donc un cadre juridique très présent. Les règles existent, ou plutôt elles coexistent. Les problèmes sont abordés selon des approches différentes qui souvent s'ignorent, et c'est dommage puisque les échantillons biologiques, comme les données à caractère personnel, sont pareillement des attributs de la personne humaine, des émanations de celle-ci, dont elles gardent une empreinte. Ce sont en quelque sorte des choses personnalisées, et elles devraient être soumises à un régime largement commun. En particulier il importe, et c'est l'objet de notre tâche, de faire prévaloir l'approche européenne de la protection des données, largement personnaliste, et de tenir la bride à la logique marchande qui va de pair avec la mondialisation.
Donc les règles existent, même si une approche globale fait défaut. Mais elles sont complètement mises à mal par le gigantesque changement de dimension et aussi le comportement des personnes. Il existe, en effet, un mouvement en faveur de l'autonomie des citoyens qui souhaitent prendre en charge leur santé, leur bien-être, être titulaires de leurs données, avoir accès à leurs données médicales, être titulaires de supports sur lesquels figurent ces données médicales, et qui diffusent sur internet dans le cadre d'une génétique « récréative », des données d'une sensibilité extrême, ce qui, là encore, lance des défis aux autorités de protection des données. Il faut responsabiliser les citoyens et appeler leur attention sur la nécessaire confidentialité qui s'attache à ces données.
Pour revenir aux principes dont je vous ai parlé, je vais prendre pour exemple de la constitution de ces vastes bases de données, la cohorte « Elfe », cohorte longitudinale, constituée par l'INED. Le projet consiste à suivre des enfants depuis la naissance jusqu'à l'âge adulte dans toutes les dimensions de leur vie pour identifier les indicateurs ou les éléments qui conditionnent l'évolution d'un individu.
Cette base, d'ailleurs soutenue et financée par les pouvoirs publics a vocation à être mise à disposition de la communauté des chercheurs à travers une plateforme. C'est la même problématique que pour les bases de données génétiques, et, d'ailleurs, elle en contient. Dans ce cas, le contrôle de la pertinence des données est difficile, compte tenu de la finalité très large de l'étude. Nombre de données particulièrement sensibles peuvent être considérées comme pertinentes à l'égard de cette finalité. La base de données ne saurait être soumise à une durée de conservation limitée dans la mesure où précisément ses éléments ont vocation à être mis à disposition des chercheurs. Au départ de cette cohorte en 2011, dont le lancement a été autorisé par la CNIL, le consentement exprès de l'un des parents à l'entrée de l'enfant dans la cohorte a été nécessaire. Se posera également à terme la question du consentement des enfants, qui n'ont aujourd'hui que deux ans. Indépendamment de cela, la conservation dans le temps de la cohorte est difficilement conciliable avec le recueil du consentement à chaque nouvelle utilisation
Ces vastes bases de données, conservées sur de longues périodes, constamment enrichies et leur mutualisation appellent une réévaluation des dispositifs de protection, des règles applicables en la matière et des règles de gouvernance.
La responsabilité doit être renforcée en matière de sécurité pour les gestionnaires de bases de données. En effet, l'article 34 de la loi « Informatique et Libertés » fait obligation aux responsables de traitements de prendre les mesures nécessaires en fonction du risque pour garantir la confidentialité des données. Mais si elles sont nécessaires à des fins de santé publique, il faut aussi assurer l'intégrité des données, leur disponibilité, leur pérennité.
Les gestionnaires de bases vont avoir une responsabilité renforcée, qui va nécessiter une évaluation du niveau de sécurité nécessaire au moment de la constitution de la base, mais aussi des réévaluations régulières en fonction des risques évoluant avec le temps et les évolutions technologiques. Un audit régulier du niveau de sécurité est nécessaire, une adaptation des mesures mises en bouclier face au risque.
Est-il nécessaire de prévoir en plus de la loi « Informatique et Liberté » une procédure particulière de labellisation, de certification, d'homologation, comme il en existe pour les hébergeurs de données de santé ? En effet, quand un professionnel de santé, un établissement ou un patient externalise les données auprès d'un industriel, ce dernier doit obtenir un agrément du ministre de la santé après avis de la CNIL et d'un comité d'agrément des hébergeurs. Cela a conduit à mettre ces hébergeurs sous contrôle, à garantir un très haut niveau de sécurité. Il est probable que la procédure ainsi mise en place n'est pas adaptée à la recherche ou aux vastes bases de données médico-administratives utilisées à des fins de recherche, et qu'il faudra sans doute la faire évoluer. Elle date de la loi Kouchner de 2002 et du décret de 2006. Elle répondait à un réel besoin, sous réserve d'une simplification et d'une adaptation de cette procédure. Mais en tout état de cause la question se pose de garantir un très haut niveau de sécurité de ces vastes bases de données et de leur encadrement. Peut-être faudra-t-il aussi, c'est une question, que l'activité de gestion de bases fasse l'objet d'un encadrement spécifique.
Voilà la première règle : le niveau de sécurité doit être garanti. Il doit être effectif, et réévalué régulièrement. Une fois qu'on a constitué la base, se pose la question de sa mise à disposition. Le droit actuel suffit-il en la matière ? Une réflexion est à mener sur la gouvernance de ces bases. Y a-t-il besoin d'un contrôle juridique, éthique, de la demande d'extraction, et qui va effectuer ce contrôle ?
Il existe plusieurs modèles possibles. On peut imaginer un modèle de gouvernance centralisé. Pour la cohorte Elfe, chaque organisme gestionnaire de la base fixe des règles explicites d'accès et met en place un guichet unique pour traiter les demandes. Cela s'appelle le Comité d'accès de la cohorte Elfe. Ou alors ce peut être un dispositif décentralisé, une structure centrale indépendante qui gère un guichet unique et fait office d'interface entre les demandeurs et les organismes gestionnaires. C'est le cas notamment du groupement d'intérêt public chargé d'autoriser les études de santé publique portant sur le médicament, et qui nécessite un accès au Système national inter-régime de l'assurance maladie (SNIRAM).
Évidemment on pourrait imaginer, d'anonymiser les données, c'est-à-dire de rendre impossible, directement ou indirectement, de retrouver les personnes. Mais dès lors que le traitement nécessite un suivi des personnes dans le temps, comme c'est le cas pour les cohortes, ou d'un suivi médical, on a besoin de garder le lien avec l'identité des personnes pour pouvoir le cas échéant revenir vers elles et engager une nouvelle collecte.
De la même manière, on a parlé de la volonté des personnes qui participent à des recherches d'être bénéficiaires de retours, d'être informées. Si l'on veut les informer, là encore, il va falloir pouvoir les ré-identifier et leur permettre d'exercer leur droit d'accès le cas échéant. Cela correspond à l'idée de nouveaux droits pour les personnes qui souhaitent adhérer à la démarche de la recherche à laquelle elles participent.
Tout cela nécessite encore une fois une sécurité de très haut niveau puisque des données d'identification vont être gardées. On va devoir les séparer des données de l'étude et les conserver dans des conditions très particulières d'accès
Pour les conditions de mise à disposition garantissant la confidentialité des données, il existe aujourd'hui des solutions. Elles sont par exemple les centres d'accès sécurisés à distance (CASD) qui permettent aux chercheurs de pouvoir travailler sur les données de l'INSEE couvertes par le secret statistique. Ils utilisent une base de travail sans pouvoir les copier, et ensuite l'on veille à ce que l'extraction effectuée ne permette pas une identification des personnes. On pourrait aussi réfléchir à des dispositifs de tiers de confiance, de centres d'accès sécurisés.
Pour la CNIL, ces sujets font l'objet d'une réflexion permanente et de préoccupations majeures. Elle les a inscrits à l'ordre du jour du programme de sa direction des Études, de l'Innovation et de la Prospective, puisqu'il est évident qu'il faut réfléchir à une adaptation de notre cadre juridique et technique, pour que ces dispositifs puissent permettre dans l'intérêt individuel et collectif la recherche, sans porter atteinte aux droits et libertés du citoyen.
|
Débat |
Pr Philippe Amouyel . Ce n'est pas une question, mais juste un témoignage sur le partage de bases de données. Je mène un vaste programme international sur la maladie d'Alzheimer, qui est la plus grosse base de données actuellement existante sur cette affection, puisque elle est la réunion de quatre grands consortiums internationaux avec près de 100 000 individus. S'est posée la question du partage public des données.
Quand ils sont financés par des organismes publics, les Anglais comme les Américains doivent mettre leurs données en ligne au bout d'un certain temps, de l'ordre de deux ans, mais en France il n'existe pas de législation particulière. Nous avons publié en 2009 à peu près tous en même temps nos informations, et avec le Centre national de génotypage (CNG) j'ai mis en ligne des informations détaillées sur certaines données anonymisées de notre base. Pour l'instant les Anglais n'ont rien mis et les Américains non plus. Il y a de grandes différences entre ce qui est annoncé et la réalité. Quand nous avons monté le consortium où chacun a partagé les données, nous nous sommes beaucoup opposés, moi-même et les Anglais, à une mise en ligne des données brutes regroupées dans un seul fichier, puis analysées globalement. Nous avons fait des méta-analyses, c'est à dire que chacun analyse localement ses propres données et en sort des résumés. Ensuite ces résumés sont regroupés. Aucune identification de l'individu n'est alors possible. Cela a finalement satisfait tout le monde, et à ce moment il s'est trouvé que la France a été choisie comme hébergeur.
Autre exemple de l'intérêt majeur suscité par ces données. Nous avons des réunions régulières dans le cadre de ce consortium, toutes les semaines, des conférences téléphoniques à 20 ou 30 personnes. Un représentant du gouvernement américain est toujours présent à ces réunions. Il note et enregistre tout. Il est au coeur de l'information et ses questions sont très orientées. Par exemple : « pensez-vous que l'on puisse développer un médicament ? » J'ai donc proposé à des représentants français de venir également, car il y a un déséquilibre très net entre les pays quant à l'implication gouvernementale. Ce partage de données doit donc être rééquilibré entre les nations.
Autre point. Sur les derniers travaux effectués, les Américains ont créé une base dans laquelle ils mettent tout, y compris des données brutes. Ils m'ont généreusement proposé d'y mettre les données du consortium. Je m'y suis personnellement opposé. Puis, quelques mois après la publication du fameux article de Science, nous avons eu une nouvelle réunion. Les Américains ont commencé, à la lumière de cet article, à se poser des questions en s'interrogeant sur les possibilités de récupérer ces informations avec la génétique récréative... Ils sont donc revenus en arrière.
Je crois que le phantasme : « on est tous des gens très sympathiques, des chercheurs qui veulent le bien de l'humanité, qui vont mettre toutes leurs données ensemble pour que cela puisse avancer... » doit tout de même être considéré à l'aune des enjeux économiques de la recherche, et de la protection individuelle.
La première fois que j'ai fait une analyse, c'était au CNG. Vous savez qu'il y a des différences entre populations, et l'on essaie d'effacer ces différences par des ajustements mathématiques, avec des analyses en composantes principales. (NDLR : méthode statistique permettant de classer des individus sur des similitudes mathématiques sans a priori ). Cela utilise environ 100 000 mutations prises au hasard dans le génome. Dans les premières dimensions de ces analyses en composantes principales, émergent spontanément trois grands groupes de populations : européennes, asiatiques, et africaines. Ensuite vous poursuivez les analyses sur les autres dimensions et arrivez à des cartes de répartition des individus en fonction de leur origine géographique, avec une précision telle qu'en France je peux identifier ceux nés en dessous de la Loire ou au-dessus, voire dans certaines régions bien particulières, simplement avec 100 000 polymorphismes. Il n'est pas besoin de toute l'information du génome pour identifier les individus.
Techniquement, pour vous donner une idée, un génome fait 1 téraoctet. Pour l'exploiter, il faut le transférer vers un ordinateur. Tout moyen de transfert sur Internet aujourd'hui, a un débit moyen de 40 mégaoctets par seconde qui vient limiter la vitesse de transfert de ces informations et en ralentir l'utilisation. Il y a des principes de réalité à prendre en compte pour les grands éléments de séquence. Mais également, il n'est besoin que de très peu d'informations pour assurer l'identification. L'article de Science a été fait à partir de séquences répétées du chromosome Y, trois fois rien. Nous n'avons pas besoin de tout le génome pour rompre la protection de l'identité.
Pr Hervé Chneiweiss . Quand Marc Delpech dit que cela ne le gêne pas du tout que 99 % de son génome soit sur Internet, je le comprends très bien, cela ne me gênerais pas non plus. Mais on ne sait pas exactement ce que représente le 1 %. On sait qu'il comprendra ApoE4 ou des choses comme cela, mais la preuve des séquences répétées du chromosome Y ne compte pas beaucoup, sauf si l'on veut tracer des populations.
La question est : quel est le niveau de protection par rapport à quoi ? Il y a une certaine ambigüité de la loi quand on parle des caractères génétiques du sujet. Parle-t-on de ce qui va avoir du sens quand cela revient vers la personne, ou de ce qui n'a plus de sens, car par exemple dans certaines cohortes les personnes sont décédées ? Or, une très grande difficulté sur les données génétiques est que l'on peut obtenir des échantillons, par exemple l'anatomopathologie, conservés à long terme, et on ne peut plus ensuite retrouver les gens. Pourtant, il n'y a plus de problème si l'on analyse certaines caractéristiques de tumeur ou du tissu environnant, puisqu'il n'y a pas de retour possible vers la personne. Mais il pourrait éventuellement y avoir des conséquences sur la descendance.
Autre point sur la question des cohortes, les personnes demandent à être informées sur ce que devient la cohorte. Elles ont signé un consentement, elles savent qu'elles font partie d'un échantillon, qu'elles sont dans une étude avec une puissance statistique sans forcément de valeur pour elles-mêmes. On peut discuter dans certains cas qu'il puisse y avoir un retour, mais la première demande des personnes est : qu'est devenue cette cohorte ? Surtout quand elle dure des années. Elles ont participé à quelque chose et voudraient savoir si elles ont rendu service à la recherche, à la société.
Dr Anne Cambon-Thomsen . Sur la question de savoir ce qui a été fait avec les cohortes : dans mon équipe à Toulouse nous travaillons sur le concept de mesure de l'impact de ce qu'on appelle les bio-ressources, qui sont les collections d'échantillons, les bases de données, etc . Mettre à disposition d'une façon utilisable des ressources, des données ou éventuellement des échantillons, représente tout un travail. Ce n'est pas seulement donner accès et va chercher qui pourra. Penser qu'il suffit de mettre quelque chose à disposition à l'état brut pour que cela soit utilisable et soit effectivement utilisé est naïf. Ce n'est pas toujours fonctionnellement utilisable par d'autres scientifiques ou médecins et il est nécessaire d'organiser, annoter et structurer des bases de données sur des variations de séquences de certains gènes pour que cette information devienne largement utile.
Ce travail est très important pour la communauté scientifique et médicale, car une description correcte des variants génétiques peut être utilisée immédiatement par des médecins. Il n'est cependant reconnu nulle part dans les évaluations des chercheurs, ou des travaux des médecins. Je ne vais pas décrire plus avant le projet sur lequel nous travaillons, avec la philosophie qu'il faut retourner la situation actuelle où il y a une certaine résistance au partage pour des raisons parfois techniques, ou de protection et de sentiment de perte de l'information et de perte de contrôle sur ce qui en est fait. Si l'on avait moyen de mesurer et de donner une valeur au fait qu'une ressource soit utilisée, on pourrait imaginer mettre en place une espèce de cercle vertueux qui serait : plus quelque chose est utilisé, plus il prend de la valeur, et plus cela apporte de la reconnaissance à ceux qui ont mis en place cette ressource. On a dit l'importance des bases de données, de la protection, du partage. Mais il faut aussi trouver un moyen de reconnaître le travail de mise en place des ressources effectivement et efficacement partageables.
Pr Philippe Amouyel. Pour aller dans le sens de ce que dit Anne Cambon-Thomsen, si jamais il y a une décision comme en Angleterre ou aux États Unis de mise à disposition, il faut prévoir les fonds qui vont la faciliter, car cela représente un coût, répercuté sur l'unité de recherche, sur ses fonds propres. Ouvrir une base de données n'est pas simple. Nous sommes sur des choses qui demandent un travail en soi pour la normalisation de ces informations. C'est un peu le sens de Global Alliance , c'est cela qu'ils veulent faire.
Mme Frédérique Lesaulnier . Surtout qu'il y a un travail, pour mettre à disposition ces données, de granularité de la donnée en fonction du besoin, par identification de lots que l'on soumet à un régime particulier d'accès. Certains, plus sensibles, répondent à un autre régime. C'est une activité à part entière qui demande beaucoup de temps, et sur laquelle nous travaillons dans la définition de lots dits "anonymes" qui pourraient être mis à disposition sans véritable contrôle a priori car leur anonymat a été évalué. C'est un énorme travail préalable, qui nécessite des réévaluations régulières. Quand la donnée est agrégée, il y a moins de risque, mais quand elle est individuelle, sa confrontation au Big data , conduit à des ré-identifications, ne serait-ce que par rapprochement à d'autres données. Il faut réfléchir aussi à la durée de mise à disposition.
M. Jean-Louis Touraine . Les différents aspects soulevés sont loin d'être l'objet d'une conclusion définitive. Nous sommes au milieu du gué. Les questions offrent l'affrontement entre les souhaits formulés par la recherche, la nécessité de protections. L'équilibre est délicat à trouver et probablement différent d'une période à l'autre. Donc il faut se garder de définir des règles absolues valables dans le temps et dans l'espace. Il y aura sûrement des adaptations à développer dans le futur au fur et à mesure de la progression des connaissances, des besoins, des techniques, des nécessités de protection, tout en ayant la contrainte supplémentaire du monde ouvert. Nous ne pouvons être ignorants de règles un peu différentes d'un continent à l'autre. Il nous faut naviguer entre tous ces écueils pour assurer sur notre propre continent les meilleures conditions de progrès et de protection. Nous aurons donc l'occasion probablement de nous réunir à nouveau et de réfléchir en voyant dans quelques années le progrès développé dans ces réflexions.
APRÈS-MIDI
PROPOS INTRODUCTIFS
Pr Agnès Buzyn, présidente de l'Institut national du cancer (INCa). Merci de nous avoir donné l'occasion d'exprimer le point de vue de la cancérologie, qui peut être légèrement différent de celui de la génétique constitutionnelle, longuement débattu ce matin, et qui a peut-être été anticipé par le deuxième Plan cancer.
Le deuxième Plan cancer avait en effet choisi de mettre comme axe majeur de travail la médecine personnalisée. C'était à l'époque, dans l'idée des rédacteurs, que la génétique somatique des cancers allait guider les traitements, allait les personnaliser et donc qu'il fallait s'organiser pour rendre accessible à tous les patients du territoire cette médecine personnalisée. Je vous remercie de m'avoir donné l'opportunité d'ouvrir cette table ronde sur l'organisation des soins puisque cela a été pensé dès le deuxième Plan cancer en 2007, tant en termes d'organisation que de structures. Nous pouvons être fiers collectivement de cette initiative qui nous est maintenant enviée dans le monde entier. Elle a en effet permis de structurer l'accès aux thérapeutiques ciblées via ce qu'on a appelé les plateformes de génétique moléculaire.
Pour ceux qui ne sont pas familiers de l'organisation de la cancérologie, le Plan cancer avait bien vu venir le fait que des traitements allaient cibler des anomalies moléculaires spécifiques de la tumeur de patients. Il est vrai que le terme de médecine personnalisée est probablement trop fort. Nous en sommes plutôt au stade de la médecine de précision, stratifiée, puisque des patients ayant des tumeurs qui présentent les mêmes types d'anomalies sont accessibles aux mêmes traitements. Mais pour qu'ils puissent bénéficier de ces traitements ciblés, il fallait mettre en place des tests compagnons accessibles sur des plateformes et de qualité.
L'Institut national du cancer en 2007 a commencé à identifier 28 centres ou laboratoires de génétique somatique, souvent des laboratoires qui faisaient de la génétique constitutionnelle, ce n'est pas forcément séparé. Il a organisé via des financements dédiés, le fait que tous les patients dont la tumeur pouvait bénéficier d'un traitement ciblé, puissent faire l'objet d'un envoi de leur prélèvement sur la plate-forme de génétique moléculaire, par le laboratoire d'anatomopathologie où se situerait leur prélèvement. Que ces patients soient traités dans le secteur privé ou public, l'INCa finance donc avec un budget PLFSS (279 ( * )) les tests compagnons et l'envoi des prélèvements par les anatomopathologistes. Il les rétribue pour l'organisation que représente le transport des fragments de biopsie.
Actuellement ces plateformes fonctionnent à un niveau accéléré. Il y a actuellement 17 thérapeutiques ciblées accessibles sur le territoire, qui ont une autorisation de mise sur le marché pour certains cancers exprimant certaines anomalies moléculaires, et ont donc besoin de tests compagnons. En 2012, plus de 60 000 nouveaux patients ont eu au moins un test de génétique moléculaire de leur tumeur sur ces plateformes, et ce nombre est en constante augmentation puisqu'on identifie régulièrement de nouvelles anomalies moléculaires spécifiques. Il faut donc implémenter les plateformes avec ces nouveaux tests, en anticipation des traitements en développement et sur le point d'obtenir une autorisation de mise sur le marché. C'est un phénomène d'amélioration continu, dynamique, et en constante expansion.
La structuration française est unique au monde, puisque tous les patients du territoire, où qu'ils soient traités, ont leur tumeur testée pour la présence d'une éventuelle anomalie génétique. Cela concerne les cancers colorectaux, du poumon, les leucémies, les cancers du sein. On prévoit, à l'évidence, une augmentation de ces tests dans les années à venir.
Nous avons cependant plusieurs enjeux pour le développement de cette médecine personnalisée. Les tests les plus anciens vont devoir à un moment donné être considérés comme des tests de routine et rentrer dans le droit commun de la une tarification. Actuellement il s'agit d'une tarification via une Mission d'intérêt général (MIG), ou par l'INCa quand les tests sont très nouveaux et s'implémentent sur les plateformes. Il va falloir que des tests rentrent dans le droit commun, soient évalués, bénéficient d'un tarif et puissent être accessibles dans des laboratoires de ville. En parallèle, il faut que nous gardions notre capacité à implémenter la nouveauté, l'innovation sur ces plateformes, et la difficulté devant laquelle nous nous trouvons est le séquençage et en particulier l'apport du séquençage à haut débit.
En effet, jusqu'à présent, l'organisation permet de réaliser un, deux, trois tests par tumeur, des tests unitaires, alors que des tumeurs vont nécessiter la recherche de X anomalies, peut-être dix ou vingt anomalies à terme, pour accéder aux médicaments à venir. Et bien évidemment il faut être en capacité de séquencer le génome des tumeurs de ces patients qui en auront besoin. Nous sommes en train d'organiser ce changement d'échelle stratégique, qui va nécessiter une organisation en termes de bio-informaticiens, et nous accompagnons à l'Institut national du cancer cette reconversion, cette évolution obligatoire des plateformes de génétique moléculaire, de façon à pouvoir absorber, je dirais ce « tsunami » d'identification de nouvelles cibles d'anomalies moléculaires, et ce « tsunami » de nouvelles molécules que nous voyons arriver : au dernier congrès américain de l'Asco , American society of clinical oncology , on a repéré plus de 900 molécules de thérapies ciblées en développement au niveau des essais de phase 1 ou 2. Ce qui veut dire que si 10% de ces molécules arrivent sur le marché, nous allons devoir absorber 90 ou 100 nouveaux médicaments ciblant des anomalies spécifiques dans les années à venir. Nous accompagnons cette structuration en équipant les plateformes en capacité de séquençage. Nous sommes le seul pays au monde à disposer d'une telle organisation et cela est cité en exemple dans tous les congrès.
Nous devons ne pas rater le tournant du séquençage haut débit, et nous devons également maintenant, et je pense que ce sera l'un des objectifs du 3ème Plan cancer, adapter la façon dont nous faisons de la recherche clinique. Jusqu'à présent celle-ci était adaptée à une ou deux nouvelles molécules de chimiothérapie qui arrivaient sur le marché, et obligeaient à faire des essais de phase 3 randomisés sur des milliers de patients pour juger de l'efficacité d'un traitement. Si nous avons à absorber l'arrivée de 90 nouvelles molécules en cancérologie dans les cinq ans qui viennent ciblées sur de petites populations de patients, il va falloir repenser notre modèle d'essais cliniques et d'évaluation du médicament.
La médecine personnalisée, que j'appelle la médecine stratifiée ou de précision, car nous sommes encore loin de l'individualisation des traitements et nous nous adressons à des sous-groupes de patients, nécessite un accompagnement et une évolution très dynamique de la façon dont nous structurons le paysage des soins, pour adapter ce paysage à l'innovation. Au-delà de ces traitements personnalisés, ou de précision, nous avons la sensation à l'INCa que la médecine devant être appelée personnalisée, doit être beaucoup plus vaste que le fait simplement d'accéder à des thérapeutiques ciblées. Nous pensons qu'elle doit être une prise en charge individualisée, prenant en compte des facteurs de génétique constitutionnelle pour ce qui concerne la prévention, l'addiction à certains toxiques comme le tabac ou l'alcool, et que cette génétique constitutionnelle guide, via des niveaux de risque particuliers, les dépistages organisés. Au-delà d'une individualisation des traitements ciblés, la médecine personnalisée de demain, que nous souhaitons tous, va vers une individualisation des prises en charge et de la prévention au traitement. Je pense que le 3ème Plan cancer proposera cette mise en oeuvre.
M. Didier Tabuteau, conseiller d'État, responsable de la chaire santé de l'Institut d'études politiques de Paris (IEP). Je vais dans ce propos rapide repartir des travaux d'une commission que nous avions réunie à l'Office de prospective en santé qui s'était en partie interrogée, travaillant à l'horizon 2025, sur les conséquences de la médecine personnalisée. Je me bornerai à présenter quatre séries de réflexions sur la possible évolution du système de santé, qui sont apparues à l'occasion de ces échanges.
La médecine personnalisée, cela rejoint ce qui a été dit en conclusion de l'intervention précédente, comprend bien sûr les traitements ciblés, mais également toute la médecine anticipée, les possibilités de prévention ou de médecine prédictive dans leurs applications. Mais la médecine peut être également regardée comme personnalisée par la connexion, la capacité de suivre en permanence et d'individualiser le traitement ou le suivi d'un patient, notamment à travers le télé-suivi ou la télésurveillance. Nous avions considéré l'ensemble des mécanismes faisant que la prévention ou la prise en charge peuvent être totalement adaptées à la personne, à l'individu.
Cette médecine ou cette façon de prendre en charge a bien des avantages, puisqu'elle s'adapte à la personne et met la singularité de l'individu au coeur du système de santé, ce qui, au-delà des effets médicaux, s'inscrit dans le processus d'affirmation du droit des personnes.
Elle invite à s'interroger sur quatre impacts possibles pour le système de santé :
Le premier est l'impact organisationnel ou économique. Si ces thérapeutiques restent à la marge, très spécialisées, localisées, elles ne modifieront pas le fonctionnement du système quand elles s'y intègreront. En revanche, si cette approche de la médecine concerne progressivement de 5, 10, 30 % du système de santé, elle produira des bouleversements considérables. Avec l'inquiétude de certains économistes de la santé sur ses coûts. Un modèle de médecine orpheline aux hyper-coûts justifiés par son application à un nombre restreint de patients va-t-elle se développer ? Ou au contraire, va-t-il permettre de partir du modèle économique des maladies rares et le transformer en modèle économique de droit commun, l'ensemble du système fonctionnant sur cette base avec des conséquences importantes sur les essais cliniques, la tarification des produits et des prestations ?
Cette première interrogation conduit à se demander, et je crois que c'est aujourd'hui qu'il faut le faire, si l'assurance maladie doit continuer à tarifer systématiquement des produits, ou si elle doit entrer dans une réflexion sur des tarifications de protocoles, ou même de résultats thérapeutiques. Cette interrogation est en germe. Elle n'est pas encore très concrète aujourd'hui, mais je crois qu'elle mérite attention. On peut également s'interroger sur l'équilibre entre les surcoûts liés à ces thérapeutiques, et les gains qu'elles permettront, puisqu'elles ont un effet de sélectivité leur permettant d'être efficientes pour le système de santé.
La deuxième conséquence touche l'organisation du système elle-même. Cette médecine se traduit inévitablement par une hyperspécialisation de l'offre de soins et incite à la création de centres nationaux, ou en tout cas très spécialisés. Elle pose la question de l'évolution des plateaux techniques et du système hospitalier dans son ensemble. Là encore, si elle devient une part significative de l'activité médicale, elle peut conduire à une restructuration considérable du système hospitalier et même du système de soins.
Le parcours de soins sera infiniment plus complexe dans une médecine hyperspécialisée du fait de la personnalisation des thérapeutiques ou des processus de prise en charge. Cela se traduira par l'émergence d'un besoin accru de GPS médical, difficile à organiser dans notre système peu régulé, mais aussi par la tentation d'autres acteurs plus ou moins bien intentionnés de tenir ce rôle d'accompagnement par le coaching ou le conseil à des fins purement économiques. On voit des initiatives fleurir dans ce domaine.
La troisième série d'interrogations concerne l'évolution des acteurs du système de santé. Il y aura émergence de pôles de compétences, comme les centres d'analyses de génétique cités tout à l'heure, mais aussi bien d'autres systèmes imaginables. Pour les thérapeutiques les plus complexes et les plus liées à la personne, on peut s'interroger sur la façon dont la séquence prise en charge-soins-élaboration des produits- administration au patient, s'organisera. Respectera-t-on toujours une séparation franche entre les entreprises pharmaceutiques, celles de dispositifs médicaux, et les prestataires de soins ? Cela mérite attention.
Dans cette réflexion sur les acteurs, il faut prendre en compte l'indispensable éclosion de nouveaux métiers de la santé. Des techniciens, ingénieurs, informaticiens du vivant, généticiens non-médecins, devront voir le jour. Déjà, certains centres universitaires témoignent de leur intérêt pour des cursus de formation de ce type. L'évolution de la formation en santé est nécessaire pour adapter le système aux transformations que cette médecine personnalisée pourrait susciter.
Enfin, sur un plan plus juridique ou administratif, cette médecine remet en cause des frontières traditionnelles, les distinctions prévention-soins, actes-produits, médicaments-dispositif médical. Ces distinctions vont voler en éclat si j'en crois les perspectives scientifiques et médicales ouvertes par ces thérapeutiques et les pratiques qu'elles permettront. Or, ce sont ces frontières qui structurent notre organisation administrative et juridique. Le code de la santé publique ou de la sécurité sociale, s'appuient sur ces concepts traditionnels pour les définitions, les processus d'autorisation, de contrôle, d'évaluation. Il y aura un effort à faire pour dépasser ces clivages, ces frontières traditionnelles. Ce ne sera pas simple car nous y sommes habitués. Il faudra donner un statut juridique ou administratif à des pratiques beaucoup plus complexes que celles auxquelles nous étions habitués, et c'est toujours difficile.
J'évoquerai pour terminer quatre risques, car ces processus sont à la fois porteurs d'espoirs thérapeutiques, d'avancées en termes d'efficience pour le système de santé, mais ils comportent des risques dont certains ont été évoqués ce matin. Ils nous étaient également apparus, et je les reprends rapidement.
Le premier est le risque de l'illégalité. Ces processus vont conduire à la banalisation et la dissémination des centres utilisateurs et détenteurs de données sensibles. Les transferts d'informations multiplieront les risques d'atteinte au secret des données médicales. L'évolution globale du système conduit à cette profusion des risques d'atteinte au secret médical. Cela n'est pas propre à la médecine personnalisée, mais compte tenu de la nature particulière de ces données, ce risque mérite d'être encore souligné.
Le deuxième risque est celui de la surveillance. Cette médecine peut engendrer, cette fois en toute légalité, un véritable Big Brother médico-génétique. Les connaissances et données médicales produiront un dispositif par lui-même potentiellement dangereux. La médico-surveillance, avec ses avantages thérapeutiques, est un système de surveillance supplémentaire dans une société, et mérite attention en termes de préservation des libertés publiques et individuelles. Il vaut mieux s'organiser, juridiquement, administrativement, politiquement, au début du processus, que de se lancer tardivement à sa poursuite.
Le troisième enjeu est celui de l'anxiété. Nous sommes dans un système où notamment les perspectives fantasmées ou réelles de la génétique génèrent une espèce d'hypocondrie collective dangereuse pour le bien être de l'individu, mais également pour le fonctionnement de la société, le fonctionnement du système d'assurance maladie et du système de santé. Dans le rapport précité, nous nous étions posé la question de la façon dont le suivi des personnes pourrait retentir sur l'évolution des prises en charge, et nous voyons d'ores et déjà poindre une « automédication génétique » suscitée par des sites internet, qui est inquiétante.
Le dernier risque est la tentation de développer une assurance maladie conditionnelle. Plus le comportement individuel, le suivi personnel en temps réel de la prévention sera possible, étayé scientifiquement, diffusé publiquement, plus la tentation pourra exister dans un contexte économique toujours difficile, de lier la prise en charge aux risques connus et aux comportements des patients. Cette assurance maladie conditionnelle, qui n'est pas inexistante, était jusqu'à présent extrêmement restreinte. Le droit des affections de longue durée (ALD) permet depuis longtemps de subordonner la prise en charge à 100 % à des conditions d'observance et de comportement et de la supprimer si elles ne sont pas respectées. Mais en pratique ce n'est pas utilisé. La médecine personnalisée et la médecine génétique font renaître avec une acuité nouvelle ce risque. Or dans un contexte économique difficile, on commence à voir dans le monde des initiatives encore isolées de systèmes d'assurance maladie introduisant le lien entre le comportement et la prise en charge, ce qui est porteur de très profondes inégalités sociales et socioculturelles. Si elles ne sont pas prohibées, ces pratiques se développeront.
Ces questions, compte tenu de leur impact sur le système de santé, supposent une pédagogie, des décisions publiques, collectives, anticipées. Et je me réjouis tout particulièrement des journées de travail organisées sous votre égide.
TROISIÈME TABLE
RONDE :
QUELLE ORGANISATION DU SYSTÈME DE
SANTÉ ?
Pr Gilles Bouvenot, président de la Commission de la transparence, membre du collège de la Haute autorité de santé (HAS), membre de l'Académie nationale de médecine. Je voudrais être très concret dans mes propos et, bénéficiant de l'expérience de l'audition publique de mars où cours de laquelle vous m'avez demandé des éclaircissements sur certains des termes que j'utilisais, essayer de définir d'abord le contexte général de l'évaluation des médicaments par la HAS avant d'évoquer les évolutions souhaitables pour ce qui concerne les évaluations des traitements ciblés.
Il existe, dans notre pays, un pacte de solidarité nationale qui fonctionne bien et qui fait que l'accès des patients à l'innovation est facile, sinon systématique, même en cas de médicaments particulièrement onéreux. Il y a en particulier les Affections de longue durée (ALD) qu'évoquait à l'instant Didier Tabuteau et d'autre part il y a, pour les établissements de soins, la liste en sus, sur laquelle je reviendrai. La Commission de transparence (CT) de la Haute autorité de santé est d'abord chargée de se prononcer sur le service médical rendu des médicaments, c'est-à-dire sur le bien-fondé de leur prise en charge ou non par la solidarité nationale. Ses avis, consultatifs, constituent une aide à la décision pour le ministre. La CT se prononce aussi sur l'amélioration du service médical rendu (ASMR) par les nouveaux médicaments, c'est-à-dire sur la quantité de progrès que leur utilisation est susceptible d'induire ; le niveau d'ASMR qui leur est octroyé concourt à la fixation de leur prix.
Un point très important à bien considérer est qu'on demande à cette commission de se prononcer sur les performances des médicaments sans se préoccuper en aucune manière des conséquences économiques (disons comptables) de ses avis qui ne peuvent donc être suspectés d'être sous-tendus par des arrières pensées de coût. Par ailleurs, la CT n'a pas à se préoccuper non plus de l'usage que certaines instances peuvent faire, en aval, de ses avis. Prenons l'exemple du Conseil de l'hospitalisation qui propose au directeur général de l'offre de soins et au ministre, d'inscrire ou non tel médicament coûteux sur la liste en sus. Or, nous savons tous qu'un médicament innovant et coûteux non inscrit sur la liste en sus n'est pas accessible aux patients, car à la charge des établissements. La décision d'inscrire ou non se fait dans des conditions que nous respectons, mais sur lesquelles nous n'avons jamais été consultés. C'est ainsi qu'un produit ayant obtenu une note d'amélioration du service médical rendu (ASMR) de niveau IV, ce qui veut dire progrès minime et non pas nul, risque donc fort de ne jamais être inscrit sur la liste en sus, alors qu'un produit avec une ASMR de niveaux I ou II ou III, a beaucoup de chances d'être inscrit. Or, certains médicaments d'ASMR IV peuvent être très utiles aux patients, en particulier dans le domaine du cancer.
Dans le même ordre d'idées, certaines pharmacies centrales ou certaines commissions médicales des établissements de soins ont décidé de ne pas acheter les produits ayant obtenu une ASMR de type V, qui signifie absence de progrès. Pourtant, pour la CT, absence de progrès ne veut pas nécessairement dire inutilité. Nous avions, à ce propos, dans un passé récent et dans l'intérêt des patients, créé l'expression "alternative utile" qui correspondait à des produits n'apportant manifestement pas de progrès au sens réglementaire du terme, mais dont nous pensions qu'ils pouvaient rendre service à certains patients résistants et/ou intolérants aux produits déjà disponibles. Nous avons été censurés par la Cour des comptes qui a considéré que le terme « alternative utile » faisait le lit de l'attribution d'une ASMR indue par le Comité économique des produits de santé, au moment de la fixation du prix. Il nous est donc interdit désormais de parler d'alternative utile, mais nous continuons d'y penser. Car la censure de la Cour des comptes est loin de résoudre le problème posé.
À la Haute autorité de santé comme à l'Académie de médecine, nous partageons l'idée que les traitements ciblés ont beaucoup plus de chance d'être efficaces que les traitements standards et sont même susceptibles d'être source d'économie pour l'assurance maladie dès lors qu'on ne les prescrit qu'à des patients qui, a priori, ont toutes les chances d'y être répondeurs. Néanmoins et même en cas de grande innovation et de traitement ciblé, nous nous devons de nous poser les questions essentielles suivantes. La première est : est-ce que cela marche ? La deuxième est : de combien cela marche-t-il ? La troisième, à ne pas occulter : pendant combien de temps cela va-t-il marcher ? Car si les traitements ciblés peuvent être source d'économies dès lors que leur emploi rationnel évite tout gaspillage, encore faut-il disposer à leur endroit de règles d'arrêt, afin de ne pas prolonger inutilement leur utilisation une fois que le patient leur est devenu résistant.
Nous savons que la recherche et le développement de ce type de produits sont coûteux. Mais les prix demandés en retour sur investissement par les industriels, sont-ils vraiment explicités et justifiés ? Ce n'est pas du ressort de la commission de la transparence de la HAS d'en débattre mais, en démocratie sanitaire, se poser cette question est de la légitimité de tout citoyen. Bien entendu, les traitements ciblés sont des produits de niche et la population cible des patients qui en sont justiciables est généralement très limitée, d'où les exigences des firmes en matière de fixation de prix. Le problème de la fixation des prix est d'autant plus difficile à résoudre que les prix sont fondés en grande partie sur une démonstration de l'efficacité et de la quantité d'effet dans les essais thérapeutiques réalisés avant l'AMM et non sur l'observation des performances de ces produits en vie réelle. C'est tout l'intérêt des études post-inscription, sur lesquelles je ne m'appesantirai pas.
Face à l'émergence de ces thérapies ciblées, quelles évolutions, quelles adaptations nécessaires la HAS doit-elle envisager dans le cadre de ses évaluations réglementaires ?
D'abord, et Didier Tabuteau l'a très bien dit, nous aurons de plus en plus à faire appel à la transversalité de la HAS, dès lors qu'un test compagnon sera associé à l'emploi du médicament dans le but de définir précisément les seuls patients qui en sont justiciables. Dans ces conditions, deux évaluations conjointes s'imposeront : celle du médicament par la CT et celle du test compagnon par la Commission nationale d'évaluation des dispositifs médicaux et des technologies de santé (CNEDiMTS) ce qui ne pose pas de problème particulier, la HAS étant déjà coutumière de ce type d'évaluation transversale.
D'autre part, la CT va devoir réfléchir à l'évaluation spécifique de ces thérapies ciblées qui ne pourra plus être la simple déclinaison de l'évaluation standard des produits « traditionnels ». Certains critères d'évaluation, mieux adaptés, devront être pris davantage en compte qu'ils ne l'étaient jusqu'à présent. La CT n'est pas du tout arcboutée sur l'utilisation systématique de la survie globale comme critère principal d'évaluation en cancérologie. Nous sommes et serons attentifs à l'évolution des consensus internationaux, une évaluation standard et univoque de tous les médicaments du cancer n'étant pas, dans bien des cas, la plus appropriée. Nos évaluations et les critères pris en compte pour la réaliser dépendront du type de cancer et de sa rareté, du type de cible et, même dans certains cas, la survie en fonction de l'évolution spontanée de la maladie ou sous traitement habituel pourra être retenue. Pour autant, nos critères de jugement devront toujours répondre aux deux exigences actuelles retenues par la Commission Européenne : être objectifs et vérifiables.
Un autre élément important d'évolution que la présidente de l'INca appelle de ses voeux, consiste à mieux discriminer les types d'essais cliniques considérés comme pertinents pour démontrer l'efficacité de ce type de médicament. Les thérapies ciblées sont des traitements de niche qui ne concernent que des effectifs restreints de patients. Aussi, ne nous attendons-nous pas à ce que les industriels nous soumettent, dans leurs dossiers de demande de prise en charge, de grands essais de phase 3 ayant inclus quelques dizaines de milliers de patients, comme c'est le cas pour dans le traitement de l'hypertension artérielle, du diabète, ou de la prévention des accidents vasculaires cérébraux dans une fibrillation auriculaire. Nous accepterons des essais d'effectifs limités. L'important est de trouver, dans l'intérêt des malades, une sorte de compromis entre le désir d'une mise à disposition rapide des nouveaux produits (qui suppose parfois des dossiers insuffisamment étayés) et le désir d'une mise à disposition la plus sécuritaire possible. Toutefois, dans l'incertitude, nous ne transigerons pas sur la sécurité des patients face à des AMM un peu trop précipitées. En revanche, nous ferons tout notre possible pour raccourcir nos délais d'évaluation, bien au-dessous des 90 jours imposés par la directive européenne. Encore faut-il que la durée de la négociation pour la fixation du prix par le comité économique des produits de santé (CEPS) n'allonge pas trop, du fait des revendications des firmes, cette mise à disposition.
Enfin, très attentifs aux nouvelles modalités d'utilisation qu'imposent ces nouveaux médicaments, nous savons que leur évaluation ne pourra plus être systématiquement celle, in abstracto , d'un médicament pris isolément versus son placebo ou même versus le médicament de référence. Il s'agira davantage d'estimer les performances d'un nouveau produit dans le cadre d'un parcours séquentiel ou en association à d'autres produits et pour une durée de temps limitée, au vu des résistances qui ne manqueront pas de survenir et de l'arrivée de produits encore plus nouveaux à lui associer ou à lui substituer. C'est du même coup rappeler combien la connaissance des règles d'arrêt de ces traitements est indispensable.
Pr Véronique Trillet-Lenoir, professeur de cancérologie, université Claude Bernard Lyon 1, chef du service d'oncologie médicale au Centre Hospitalo-Universitaire de Lyon, présidente du Cancéropôle Lyon-Rhône-Alpes-Auvergne . Je voudrais vous remercier de me donner l'occasion de m'exprimer ici, et tout particulièrement après Gilles Bouvenot, ce qui est toujours un challenge. Tout d'abord comme on l'a rappelé ce matin, la cancérologie aime bien la terminologie et les mots-clés, puisque c'est elle qui a inventé la médecine personnalisée. Il a également été fait allusion ce matin au programme personnalisé de soins, sur lequel je voudrais revenir.
La médecine personnalisée, telle que la comprennent les cancérologues, est très loin des débats de ce matin, celle vue par la génétique. Nous nous attaquons à des mutations somatiques et le génome que nous étudions est mutant, mouvant. Il n'a finalement plus grand-chose à voir avec le patrimoine génétique initial, objet des préoccupations de ce matin, que les cancérologues ne partagent qu'à travers les prédispositions génétiques au cancer, sur lesquelles je ne reviendrai pas puisque Dominique Stoppa-Lyonnet a longuement évoqué ce problème.
En revanche, la médecine personnalisée des cancérologues est très loin de la génétique récréative, puisqu'il s'agit vraiment de médecine et de thérapies moléculaires, ciblées, de précision a-t-on dit ce matin, et Agnès Buzyn a ajouté tout à l'heure, stratifiées. Dans tous les cas de figure, elle est curative, même si elle est également prédictive. Elle repose donc sur l'identification de cibles par un test compagnon, et l'utilisation de médicaments, qui pour être efficaces, n'en sont pas moins souvent toxiques, et presque toujours coûteux.
Dans les changements d'organisation et de paradigme auxquels vous avez fait allusion, Didier Tabuteau, le premier, déjà signalé par Thomas Tursz ce matin, on trouve le démantèlement nosologique des tumeurs. A l'heure actuelle, rien ne peut ressembler autant à un cancer du sein qu'un cancer du poumon ou de la prostate, pour peu qu'ils partagent les mêmes anomalies moléculaires. On n'assiste donc pas tant à l'émergence de tumeurs rares et de niches, mais plutôt à une juxtaposition de tumeurs rares. Le cancer du poumon est actuellement la juxtaposition de 150 formes rares de cancers du poumon. Un des changements dans l'organisation de notre médecine est un décloisonnement extraordinaire entre les spécialités, et la nécessité absolue de renforcer la médecine transversale. Celle-ci se préoccupe de la biologie moléculaire des cancers plus que de l'organe d'origine qui finalement n'entre plus beaucoup en ligne de compte. C'est un premier mouvement.
Gilles Bouvenot nous a brillamment expliqué l'intérêt de ces thérapies ciblées pour sélectionner les malades candidats, c'est à dire les bons répondeurs, et ainsi faire des économies entre guillemets, ou du moins éviter le gaspillage. Je voudrais tout de même rappeler qu'à mon sens, ce qui coûte le plus cher à une société, ce sont des malades qui vont mal, ou qui ne reçoivent pas au bon moment le bon traitement, ou reçoivent un traitement coûteux prescrit de n'importe quelle manière. Autrement dit, Monsieur Bouvenot, je continue à trouver que dans votre dénonciation du système, vous n'insistez peut-être pas encore assez sur le cloisonnement entre l'autorisation de mise sur le marché (AMM), qui reconnaît l'intérêt d'une molécule, la commission dont vous assurez la présidence qui reconnaît ses performances, la troisième ou quatrième commission qui en déduisent le prix. À aucun moment je n'entends le ratio entre l'investissement d'une société sur la santé de ses citoyens et le retour sur investissement, et cela me donne en permanence le sentiment que le prix du médicament n'est jamais fixé en fonction du seul indicateur qui me paraisse intéressant, l'allongement ou le sauvetage de vies humaines. J'y reviendrai tout à l'heure car il me semble que la recherche en sciences humaines et sociales doit mettre en priorité cette problématique... Mais encore une fois ce « séquençage à haut débit » entre les différentes problématiques continue à me poser problème.
J'ai réagi tout à l'heure à l'apparition de résistances aux thérapies ciblées, car en fait il s'agit moins de résistance que de sélection de cellules qui, ne portant pas l'anomalie moléculaire, continuent à se développer malgré la thérapie ciblée, qui ne les cible pas. Quoi qu'il en soit, merci d'avoir rappelé que la survie globale des patients est un bel objectif. Mais c'est bien grâce à la survie sans progression que nous avons fait passer le cancer d'une maladie aigüe à une maladie chronique avec toutes les problématiques organisationnelles et économiques que cela comporte.
Quant aux thérapies ciblées, il faudrait dans l'avenir, et cela a déjà été longuement dit, booster la recherche les concernant, dans les conditions déjà définies. Elles ne feront probablement pas la différence en termes de guérison, mais en revanche, le dernier congrès auquel Agnès Buzyn a fait allusion a montré que l'immunothérapie des cancers est en train de devenir un outil majeur de ciblage intelligent par la mise à disposition du système immunitaire du patient, et que la complexité biomathématique de ces traitements va probablement faire appel de plus en plus à des modélisations, c'est-à-dire à de nouveaux métiers. Il a été dit à plusieurs reprises que nous ne ferions pas l'économie de plates-formes dans lesquelles les biomathématiciens, les bio-informaticiens, les biologistes des systèmes, devraient interagir en pluridisciplinarité avec les équipes de recherche et les équipes cliniques.
La gradation des soins sera probablement une question nouvelle, puisque nous ne mettrons pas des plates-formes partout, et d'ailleurs les patients n'ont pas besoin qu'elles soient partout. L'organisation des plates-formes de biologie moléculaire qu'a décrite Agnès Buzyn me paraît exemplaire. Il y en a une par région. Il faut que nous travaillions encore plus à l'accès des patients à ces plates-formes, sans pour autant négliger les aspects de proximité.
Je voudrais également attirer l'attention sur le fait que la médecine personnalisée est une médecine dans laquelle on individualise des traitements. Cela ne signifie pas que l'on n'est pas rigoureux vis-à-vis de ce que la science nous a appris, et en particulier des recommandations de pratiques qu'il faut continuer à suivre. Je trouverais particulièrement inquiétant que le terme de « personnalisé », fasse revenir sans garde-fous à des décisions individuelles de médecins brusquement inspirés par une fantaisie qui les éloigne des référentiels, très solides en cancérologie. Médecine personnalisée, oui, mais rigoureuse. Le service à la carte n'est pas aussi bien en médecine qu'au restaurant. Le programme personnalisé de soins est une quadrature du cercle insoluble, puisqu'il s'agit d'adapter à un individu, en le personnalisant pour lui, un programme issu de recommandations de pratiques qui par définition, issues de l'Evidence based médicine , sont universelles. C'est une des raisons pour lesquelles les cancérologues ont jusqu'ici, soyons humbles, échoué à mettre en place, le programme personnalisé de soins.
En revanche, grâce aux deux plans cancer précédents, nous avons appris à travailler en pluridisciplinarité. La réunion de concertation pluridisciplinaire est un modèle. Nous avons appris à sortir du carcan du colloque singulier, tout en le renforçant par la décision pluridisciplinaire. C'est une façon de personnaliser la médecine. Nous avons à côté des nouveaux métiers liés aux biotechnologies et à la progression de la science, personnalisé les soins de support et l'accompagnement des patients par des spécialistes de la douleur, des psychologues, ergothérapeutes, nutritionnistes, esthéticiennes. Ce sont des soignants non-médicaux, accompagnant le geste médical, et personnalisant la prise en charge.
Nous avons encore beaucoup de progrès à faire pour améliorer cette personnalisation de la prise en charge. Nous ne sommes pas bons dans la prévention et le dépistage, les associations de patients l'ont dit. Nous sommes dans un pays latin où la culture de la prévention et du dépistage ne sont pas dans les moeurs, où les météorologues nous annoncent quelques éclaircies mêlées de quelques averses, alors qu'aux États Unis il y a vingt ans qu'on annonce 30 % de risques de pluie et 20 % de risques d'humidité. C'est pour cela que nous avons du mal à convaincre les patients d'entrer dans une culture de prévention et de dépistage. Nous avons des freins liés à des croyances, des crispations de tous ordres, et nous devons progresser en tenant compte de ces spécificités culturelles, en arrêtant d'être dans la coercition, qui a fait la preuve d'une certaine inefficacité en matière de prévention et de dépistage.
Á côté des biomarqueurs biologiques, nous devons être attentifs à des biomarqueurs cliniques. La personnalisation de la médecine, y compris par les thérapies ciblées, dont la plupart sont orales, c'est la prise en compte de la typologie des patients en termes de comorbidité, de co-médication, de grand âge, d'adaptation aux traitements, qui sont autant d'éléments majeurs dans la réussite ou l'échec d'un traitement, dans son bénéfice/risque positif ou non. Nous avons tendance à les négliger au profit d'autres marqueurs beaucoup plus coûteux, mais infiniment moins pertinents.
La plupart des thérapies ciblées sont disponibles sous forme orale, et leur avènement bouleverse l'organisation, puisqu'elles nécessitent une acculturation à un parcours de soins presque exclusivement extrahospitalier. Dans l'environnement très hospitalo-centré où nous sommes, l'implication des médecins généralistes et des structures de ville non médicales est absolument essentiel pour assurer une coordination territoriale de qualité, génératrice d'économies de soins, de qualité, du désengorgement des services d'urgence embolisés par des malades dont la prise en charge n'est pas coordonnée, pour éviter la rupture de la continuité des soins, etc .
Enfin les sciences humaines, sociales, et la recherche interventionnelle doivent être développées. Les patients doivent bénéficier d'éducation thérapeutique, devenir des patients experts. La recherche médico-économique doit être faite si possible en amont de la mise à disposition d'un médicament.
Pour finir, ces thérapies orales mettent l'accent sur la perversité du système de T2A, qui ne reconnait pas l'acte intellectuel mais uniquement l'acte technique. Son bon fonctionnement en matière de thérapie ciblée orale, passera par la définition d'un parcours de soins qui ne sera plus une nomenclature à l'acte.
Pr Thomas Tursz, cancérologue, directeur honoraire de l'Institut Gustave Roussy. Nous avons parlé jusqu'à maintenant d'un petit nombre de médicaments, actifs sur un petit nombre de malades, pour peu de temps, pour des prix très importants et qui nécessitent des tests compliqués. Si nous nous limitons à cela, je crois que nous passons à côté des énormes enjeux et des défis majeurs pour notre système de santé que représente la révolution autour de la nouvelle classification des maladies en fonction de leurs aspects génétiques et moléculaires.
Même si le problème des quelques nouveaux médicaments issus de ces concepts est bien sur important, et j'y reviendrai, il est capital de dire que le plus important aujourd'hui, c'est bien le changement de paradigme. Il influera considérablement dans l'évolution du système de santé, dans la façon dont nous le préparons, le pensons, et dont nous l'organiserons dans l'avenir. En cancérologie, le point-clé est le bouleversement de la classification des tumeurs, qui jusqu'ici reposait avant tout sur l'examen anatomo-pathologique des échantillons tumoraux au microscope, et donc sur des aspects purement morphologiques. Cette classification histologique des tumeurs a été bien sûr très utile, et a permis d'établir des protocoles thérapeutiques permettant de guérir aujourd'hui environ 50 % des cancers. Mais nous en atteignons maintenant les limites. Cette classification ne permet pas de prédire quels seront les malades qui vont ou non rechuter après un traitement, ceux qui vont développer des métastases, et surtout ceux qui vont répondre à tel ou tel médicament ou à la radiothérapie. C'est bien pourquoi en recevant le même traitement, certains malades vont guérir et d'autres vont rechuter. Plutôt que distinguer simplement entre les malades qui « ont de la chance » et « ceux qui n'en ont pas », les progrès de la biologie et des technologies nous montrent que l'hétérogénéité des tumeurs est bien plus grande que ce que nous pensions, et que beaucoup des entités telles que les cancers du sein, du poumon, ne sont pas une maladie unique, mais un mélange de 20 ou 30 maladies différentes (voire plus), liées à des mécanismes génétiques et moléculaires différents, et qu'il faudra donc traiter de façon différente. Si certains malades répondent et vivent, et certains ne répondent pas et meurent, c'est parce qu'ils n'ont pas la même maladie, et qu'ils n'auraient pas dû être soignés de la même façon. Ce démembrement des maladies, cette nouvelle nosologie du cancer est en elle-même une révolution qu'il va falloir assimiler, et qui aura des conséquences lourdes sur les concepts, mais aussi sur les technologies, l'organisation des soins et, bien sûr, les traitements dont les premiers médicaments ciblés, apparus depuis quelques années, ne sont que les précurseurs .
On a parlé du tsunami des nouveaux médicaments anti-cancéreux en développement. Je ne crois pas en ce tsunami de médicaments, et surtout je ne pense pas que l'arrivée de médicaments efficaces chez certains malades soit en soi une mauvaise chose qu'il faille redouter et contenir, pour de pseudo-raisons d'économie. L'enjeu n'est pas le moratoire ou le rationnement de l'innovation, c'est bien son organisation intelligente et efficace qui doit permettre d'administrer rapidement ces nouveaux médicaments aux quelques malades qui sont susceptibles d'en bénéficier, et seulement à eux. Je voudrais rappeler que ce « tsunami » arrive après une pénurie, une disette, un Sahel de médicaments pendant 40 ans en cancérologie. Il n'y a pas eu de médicament nouveau important entre le Cisplatine à la fin des années 60 et les taxanes découverts par un Français, Pierre Potier, à la toute fin du XX ème siècle. Mais ce sont les concepts et les procédés de découverte de ces nouveaux médicaments qui ont été bouleversés par nos nouvelles connaissances biologiques. Les drogues nouvelles étaient autrefois créées d'abord par des chimistes qui jouaient avec les molécules naturelles ou non, et savaient brancher merveilleusement des radicaux sur ces molécules, sans pouvoir préjuger à l'avance de leur éventuel effet anti-tumoral. Ils livraient ainsi des millions de nouveaux composés à des biologistes, pour qu'on les teste et les crible sur de complexes systèmes de lignées tumorales en culture et de modèles de cancers animaux, dont on ne sait toujours pas s'ils sont bien représentatifs des tumeurs humaines, que ces candidats-médicaments étaient éventuellement censés soigner. Il fallait aussi passer par la longue série d'essais cliniques de phases I, II, III pour qu'ils soient finalement enregistrés et mis sur le marché.
Cette stratégie a été utile puisqu'elle a permis de mettre sur le marché 30 à 40 médicaments qui constituent la base de la chimiothérapie conventionnelle. On peut quand même dire qu'elle est extraordinairement peu efficace, longue, et très coûteuse. Il y a de toute façon un problème de fond de la cancérologie, et peut-être de la médecine en général. On ne peut pas continuer comme cela. Actuellement, la découverte du génome et du produit des gènes change tout à fait la donne. La découverte des oncogènes et de leurs mutations permet de comprendre les mécanismes mêmes de la carciniogénèse, c'est-à-dire de la transformation d'une cellule normale en cellule cancéreuse. Les techniques nouvelles de séquençage du génome permettent d'analyser rapidement les anomalies génétiques dont les cellules cancéreuses d'un malade précis sont porteuses, et de prédire, dans une certaine mesure, quelles devraient être la structure et les propriétés d'un médicament potentiellement efficace pour contrecarrer l'effet de ces anomalies génétiques. On peut voir la structure tridimensionnelle d'une molécule, la partie altéré dans cette molécule, et ce sont maintenant les biologistes du cancer qui demandent aux chimistes de leur préparer « à façon » un médicament dont les cibles moléculaires précises et les propriétés seront ainsi prédéfinies. Certes, ce seront bien toujours des chimistes qui synthétiseront ces médicaments du futur, mais non plus en fabricant « à l'aveugle » des milliers de molécules. Ils fabriqueront à la demande une molécule qui ira se fixer exactement à tel endroit, dans telle poche, dans tel repli de telle molécule. Ce tsunami donc, on n'y pourra rien, c'est une avancée inéluctable de la recherche et de la technologie. C'est ainsi que l'on inventera maintenant des médicaments, en nombre, dont peut-être certains seront intéressants. Il faut se préparer à ce changement.
Pour faire une comparaison rapide, ce n'est pas la première fois qu'il existe un tel décalage entre un progrès faramineux des connaissances biologiques, et une lenteur des progrès thérapeutiques. La cancérologie au début du XXI ème siècle est dans la même situation que les maladies infectieuses au début du XX ème , en 1900, c'est 20 à 30 ans après les découvertes fondamentales de Pasteur, qui a introduit la théorie microbienne. Il n'a pas trouvé tous les microbes, mais il a complètement changé la façon de classer, de penser, de réfléchir et même de prendre en charge les maladies infectieuses, même si on connaissait celles-ci avant Pasteur, avec la notion d'épidémie, d'isolement. Il a totalement changé le regard sur ces maladies, comme notre regard sur le cancer est en train de changer totalement. Il a fallu 60 ans entre Pasteur et Fleming, pour que le premier antibiotique, la pénicilline, soit introduite en pratique courante. Comment faire, puisque nous sommes quelque part entre Pasteur et Fleming, pour que ceci ne dure pas 60 ans ?
Or 30 ans se sont déjà passés depuis les découvertes fondamentales, et quelques médicaments plus ou moins intelligents sont en train d'arriver et pourraient être les premiers représentants de ces nouveaux médicaments « intelligents » du cancer, l'équivalent des antibiotiques. L'enjeu est maintenant d'accélérer l'histoire, il ne faut pas que ce processus prenne à nouveau 60 ans. De toutes façons, la recherche ne procède plus de la même façon. Là est l'importance des masses critiques, de la vraie pluridisciplinarité, qui n'est plus simplement la réunion de quelques médecins dans une pièce ou se parlant au téléphone, mais des professionnels d'horizons différents, de métiers différents, partageant leur savoir, leurs technologies, leurs connaissances au service des mêmes malades.
Didier Tabuteau l'a très bien dit en parlant des nouveaux métiers, des nouveaux acteurs majeurs de la lutte contre le cancer que seront les biologistes du génome et les bio-informaticiens, car il va falloir rendre ces résultats multiples sous forme visible, lisible et interprétable pour les médecins. Et aucun médecin n'est capable d'interpréter un résultat génomique seul, si on ne lui explique pas quel médicament il faut utiliser en fonction des anomalies génétiques et moléculaires, qui s'accumulent au sein de la même cellule. Le cancer est bien une maladie génétique, une maladie de plusieurs gènes, et non pas d'un seul, qui accumule des mutations génétiques, car l'instabilité génétique est une des constantes de cette maladie.
Sommes-nous en si bonne situation que cela pour que le monde entier nous admire, comme il a été dit ? Je n'en suis pas sûr du tout. L'innovation ne concerne qu'une très petite minorité des patients. Moins de 5% des patients français, quoi qu'en disent les Plans cancer, ont accès à un essai thérapeutique au cours de leur vie et auront accès à l'un de ces médicaments. Et encore, la plupart de ces essais thérapeutiques ne posent pas ce genre de question, mais sont issus de marketing industriel et servent plutôt à mettre en place des équivalents de médicaments existant déjà, ou à créer des habitudes de prescription chez les médecins. Cela se traduit par le fait que les grandes compagnies pharmaceutiques, ont déjà tendance à déplacer leur recherche fondamentale et translationnelle vers les États-Unis (y compris les firmes françaises ou ex-françaises comme Sanofi-Aventis). Les grands essais de Phase III, qui étaient un peu notre spécialité, où il s'agissait d'inclure un très grand nombre de malades ne seront peut-être plus utiles et de toutes façons, ne se feront plus en Europe, mais pour des raisons de coût, en Europe de l'Est, en Chine et en Inde.
Nous faisons actuellement, en fonction de ce que nous dit l'anatomopathologiste, le même traitement à tout le monde : on surtraite certainement un bon nombre de malades, dont le cancer n'évoluerait pas, avec trop de radio et de chimiothérapie, mais on ne sait pas dire lesquels à l'avance. Et c'est une des principales conséquences attendues de la nouvelle médecine : éviter les traitements inutiles ou inefficaces. Même si nous sommes encore loin des traitements réellement individualisés pour chaque malade, de la haute couture ou du sur-mesure, il nous faut quitter l'ère du prêt-à-porter et de la taille unique pour tous et toutes.
Quelles que soient les critiques qu'on peut aujourd'hui faire au concept de médecine personnalisée, la cancérologie traditionnelle est dans une impasse grave : avons-nous fait des progrès dans les quarante dernières années ? Le cancer du poumon avancé, que l'on n'a pas pu opérer, métastatique, reste un très mauvais cancer. Ne fumez pas. En quarante ans on peut dire, en montrant les meilleures séries, que l'on a augmenté la survie moyenne des malades de huit mois. Pendant le même temps, le coût moyen du traitement a augmenté entre 100 et 1 000 fois. Sans cynisme, on peut se poser la question de savoir si ces huit mois de vie supplémentaires valent vraiment de tels surcoûts. D'autre part la façon dont se font les essais thérapeutiques est l'addition. On montre que A + B est mieux que A, que A+B+C est mieux que A+B, etc . L'on cherche à ajouter des médicaments qui sont en général coûteux, à des thérapeutiques qui existent déjà. Cela ne fait qu'alourdir le traitement, le rendre plus toxique, plus coûteux, et pas beaucoup plus efficace. Il serait beaucoup plus rationnel et avantageux de réserver le médicament A aux malades susceptibles de répondre à A, le médicament B aux seuls malades potentiellement répondeurs à B, etc .
On parle beaucoup des coûts des médicaments, mais on ne parle pas assez du coût des nouvelles biotechnologies. Or, comme il a été dit ce matin, il va probablement s'effondrer, d'autant plus qu'il y aura une concurrence et un marché. Le séquençage complet du génome va devenir rapide et est déjà à moins de 100 dollars. Ainsi, nos modèles médico-économiques ne devront pas prendre en compte seulement le prix des traitements, mais aussi celui de ces technologies. De plus, les progrès et les évolutions de ces technologies, comme dans le cas de la téléphonie mobile ou de la micro-informatique, seront sources de développement économique pour de petites firmes de biotechnologies, et seront à l'origine de nouveaux métiers. Il s'agit là de réels investissements d'avenir, qui devraient être pris en compte dans l'évaluation de ce nouveau type de médecine.
Les cancers vont être démembrés en un certain nombre de maladies qu'on pourrait qualifier d'orphelines, et je peux donner deux exemples qui montrent à la fois la beauté et les inconvénients de cette médecine. Le premier concerne le cancer du poumon, dont je vous ai dit que c'est un mauvais cancer. Des Japonais ont montré que 3 % des malades atteints de ce cancer ont une anomalie d'un gène appelé ALK. Or il y avait dans le pipeline de Pfizer un médicament anti-Alk, que Pfizer ne voulait pas développer et était prêt à jeter à la poubelle. Quand cette publication est sortie, ils ont commencé un essai. Pour traiter 80 malades ils ont génotypé 3 000 personnes pour savoir si elles avaient cette anomalie. On parle des tests compagnons en disant qu'on va les développer. Est-il raisonnable de considérer 3 000 malades pour en traiter 80, et dire à 97 % d'entre eux qu'on ne peut pas les traiter ? N'est-il pas plus raisonnable d'essayer de trouver d'autres anomalies permettant ainsi peut-être un autre traitement ? L'intérêt des techniques pangénomiques me paraît être évident. Par ailleurs je ne suis pas sûr que ce soit à l'État de payer pour donner un médicament de Pfizer ou de Roche. Il y a là une question qui me paraît importante. En tous cas chez ces 3 % de malades ayant cette amplification d'ALK, les résultats sont très spectaculaires avec ce médicament. Certains patients sont encore vivants. C'est la première fois que la FDA a accepté un médicament en phase 2, sans phase 3.
Enfin, je voudrais terminer sur l'importance de nouveaux types d'essais thérapeutiques. Traditionnellement les essais de phase 1 sont des essais de toxicité menés chez des malades ayant épuisé les traitements conventionnels. On y recherche des frémissements de réponse clinique, et 2 à 3 % de ces malades bénéficiant de ces phases 1 ont la chance d'avoir un médicament efficace. Les traitements sont donnés à l'aveugle. Actuellement nous essayons d'adapter les traitements aux anomalies moléculaires présentes, et nous arrivons à des taux de réponses en phase 1 de 30 à 40 %. C'est déjà considérable, et cela montre le potentiel de ces techniques. Il s'agit de malades qui arrivent à ces essais très tardivement après avoir reçus 5, 6, 7 traitements inefficaces, coûteux et toxiques, qui les ont mis en mauvais état général. Essayer de ne plus donner ces traitements serait un élément tout à fait important. L'un des messages est que l'essai ne doit plus être présenté comme un dernier recours, une dernière chance, mais très tôt dans la maladie, comme une chance supplémentaire de survie. Ils peuvent se faire en France même de façon regroupée comme nous l'avons fait dans le cancer du sein. Les malades sont demandeurs. Nous avons inclus en six mois plus de 400 malades pour des essais thérapeutiques dans 18 centres. Certains de ces malades ont eu des réponses alors qu'ils avaient résisté à 3, 4, 5, 7 lignes de chimiothérapie, et ont des anomalies moléculaires multiples. Cependant, dans le mélanome, il arrive que toute la maladie disparaisse, mais qu'au bout de six mois, elle rechute. Ce sont les résistances, qui sont liées à la sélection de sous-clones de cellules tumorales, présentant de nouvelles anomalies génétiques. Ainsi, le médicament a de fait exercé une sélection darwinienne des clones résistants. L'avenir des médicaments ciblés sera ainsi paradoxalement de les combiner entre eux pour contourner ces résistances, à l'image des poly-antibiothérapies de nombreuses infections, ou des trithérapies du sida.
Mme Valérie Seror, économiste, chargée de recherche INSERM-IRD. Université Aix-Marseille . Je vais vous proposer une perspective économique.
Un premier point d'entrée est technique, où il s'agit de considérer le développement des bio-marqueurs, des tests de génomique, qui conduisent à une médecine plus stratifiée, caractérisée par des arbres de décisions médicales plus complexes. Un autre point d'entrée est sociétal, et dans ce cas le développement de la médecine personnalisée peut être une réponse aux demandes des patients pour plus d'implication dans leur prise en charge médicale. Également, comme il vient d'être plus ou moins montré sur le cancer du poumon, l'économique peut être un point d'entrée, le développement de ces tests et bio-marqueurs pouvant être une réponse au rendement décroissant du progrès biomédical. Enfin, le développement de ces tests pourrait être une réponse à la crise de l'innovation pharmaceutique.
En adoptant une perspective sociétale large, beaucoup de travaux ont montré une augmentation de l'individualisme, constatée tous dans nos pays, et également de façon un peu contrastée une montée en charge des mobilisations collectives des patients. On pourrait dire que la médecine personnalisée pourrait contribuer à satisfaire les demandes des patients pour plus de transparence dans l'information médicale, à plus d'implication dans la décision médicale. En revanche, les bio-marqueurs et les tests génomiques ne vont pas faciliter la communication sur les risques.
Cette réflexion un peu large s'inscrit dans un contexte caractérisé par une croissance forte des dépenses de santé. Le cadre du cancer est exemplaire, puisque entre 2005 et 2009, l'histogramme montre une augmentation des dépenses en médicaments de 220 % environ. Les plates-formes, indiquées par Agnès Buzyn, ont été mises en place avec une montée en charge assez rapide des tests, un doublement de leur nombre au bout de deux ans. Ces éléments sont forts et arrivent aujourd'hui un nombre croissant de bio-marqueurs, de thérapies ciblées. Une question se pose alors : le surcoût des tests est-il compensé par un meilleur ratio cout/efficacité ? Des travaux ont été menés dans la littérature internationale. La réponse est oui dans certains cancers colorectaux, dans certains cancers du poumon. Qu'en est-il d'autres indications, d'autres organes atteints, d'autres bio-marqueurs, et que traduisent ces réponses positives sur les relations entre le prix des médicaments et la cotation des tests ?
Pour illustrer, Je vais m'appuyer sur une étude publiée que nous avions menée. Elle évalue l'apport d'un test génomique, qui vise, car il y a une étude prospective en cours, à guider la décision médicale de chimiothérapie dans certaines formes de cancer du sein, avec envahissement ganglionnaire mais sans métastases, c'est-à-dire environ 30 % des cancers du sein. Deux stratégies étaient comparées. La prise en charge standard de ces femmes par chimiothérapie incluant anthracyclines et taxanes; et une autre reposant sur un test génomique. Selon le résultat du test, des patientes identifiées comme de bons pronostics se voyaient proposer une chimiothérapie avec seulement des anthracyclines, alors que celles identifiées comme de mauvais pronostics avaient la prise en charge standard. Sur le plan des efficacités, il n'y a pas de différence significative de survie sans rechute à cinq ans parmi les patients de bon pronostic, ce qui suggère qu'il n'y a pas de bénéfice clair lié à l'adjonction de taxanes dans leur chimiothérapie. En revanche pour les patients de mauvais pronostic, le bénéfice de l'adjonction des taxanes était significatif sur un plan statistique, et absolument flagrant. À l'époque où l'étude avait été menée, le docetaxel, une molécule de la classe thérapeutique des taxanes, n'était pas encore un médicament générique, et le test génomique représentait une option-coût efficace, à condition que son coût reste inférieur à 2 100 euros. Mais si on considérait une baisse de 30%, cohérente avec le passage du médicament au générique, on voyait qu'un coût du test maximal à 1140 euros permettait une option-cout efficace.
Cette analyse coût-efficacité, et en réalité tous les travaux publiés, montrent qu'il y a une interdépendance entre la cotation des tests et le prix des médicaments, une complémentarité entre tests et thérapies ciblées, puisque l'utilisation conjointe du test et du médicament conduit à un bénéfice clinique supérieur à leur utilisation séparée. Il en ressort de mon point de vue, que les enjeux de cotation dépassent la question de l'équilibre des comptes de l'assurance maladie. Celle-ci est soumise à des arbitrages entre faciliter l'adoption d'une amélioration thérapeutique, et avoir les moyens de l'offrir à ses patients.
D'un autre coté les décisions de cotation ont un impact direct sur le marché des médicaments et des tests. Pour aller un peu vite, on peut dire qu'une cotation d'un test trop basse peut être comprise comme une désincitation au développement d'autres tests, alors que si elle est trop élevée, pourrait conduire à des rentes de situation payées par les adhérents de l'assurance maladie.
Par ailleurs, la complémentarité entre les tests et les thérapies met en lumière des barrières organisationnelles, qui ne sont pas propres à la France. Plusieurs travaux publiés en Europe et aux États Unis montrent la même chose. Il y a besoin de davantage de coopération dans l'évaluation de la valeur ajoutée des tests et dans les décisions de prix et de cotation, dans la mesure où cette complémentarité clinique de ces deux éléments, devrait se traduire dans une évaluation conjointe de leur valeur ajoutée, ou du prix.
Le développement de ces tests et de ces thérapies ciblées, pourraient permettre de prescrire certains traitements aux seuls patients susceptibles d'en bénéficier, d'éviter des traitements inutiles, coûteux, toxiques, dans un contexte, en tous cas dans le cancer, d'augmentation très rapide des dépenses de santé et de médicaments. Cela pourrait freiner ou atténuer l'escalade thérapeutique et les coûts associés. Cependant ces travaux militent dans le sens d'analyses coût-efficacité systématiquement intégrés aux essais cliniques, et non pas effectués après.
Mme Elisabeth Thouret-Lemaitre, consultante, spécialiste de propriété industrielle . Il m'a été demandé d'intervenir pour vous présenter la décision de la Cour Suprême Myriad Genetics/Association for Molecular Pathology , qui vient d'intervenir le 13 juin 2013 aux États-Unis et aura peut-être un impact sur l'organisation du système de santé lui-même, ce qui dépasse largement mes compétences. Elle arrive après la présentation des différentes décisions relatives aux mêmes sujets, faite lors de la première audition publique de votre Office au mois de mars.
Cette décision était attendue et a fait l'objet de commentaires nombreux, de manchettes de journaux, qui se sont empressés d'en faire une conclusion rapide, alors qu'elle nécessite à mon avis une lecture approfondie, une réflexion beaucoup plus longue. Il faudra suivre l'application de cette décision aux États-Unis même, puisque ses conséquences interviendront rapidement, l'Office des brevets américain ayant déjà présenté un mémorandum à ses examinateurs pour qu'ils en tiennent compte.
Vous vous souvenez de l'affaire Myriad et je n'en rappelle pas les données. Une décision en première instance a annulé les brevets de Myriad, puis la Cour d'appel les a au contraire remis en vigueur, et à la demande de différents mouvements et de différentes associations qui se sont constituées, la Cour Suprême s'est penchée sur cette affaire. Je vais vous donner une traduction libre et personnelle de l'opinion de la Cour, car il est difficile d'interpréter complètement ce qui a été dit. Le résumé a été communiqué par le rapporteur de la décision.
Myriad a découvert la localisation précise et la séquence de deux gènes humains, dont les mutations peuvent augmenter considérablement les risques du cancer du sein ou de l'ovaire. Myriad a obtenu des brevets pour cette découverte. Ce cas, traité par l'opinion et la Cour suprême, concerne les revendications de trois brevets, et nous demande de résoudre le problème de savoir si un segment d'ADN existant dans la nature est brevetable au sens de l'article 35 USC, paragraphe 101, de la loi américaine, si ce segment est isolé du reste du génome humain. Nous donnons également notre avis sur la brevetabilité de l'ADN créé de manière synthétique, connu sous le nom d'ADN complémentaire, qui contient la même information codante pour la protéine que celle du segment d'ADN naturel, mais ne contient pas de portion d'ADN ne codant pas pour les protéines. « Nous considérons que le segment d'ADN existant dans la nature, est un produit de la nature et n'est pas brevetable simplement parce qu'il a été isolé. En revanche l'ADN complémentaire est brevetable car il n'existe pas dans la nature » dit la Cour.
La décision de la Cour d'appel est donc en partie confirmée, et en partie annulée.
Voilà le résumé de l'opinion de la Cour Suprême. Il suit un long raisonnement de plusieurs pages qui expliquent le pourquoi de cette décision. Si on l'examine complètement, et que l'on connaît la lettre amicus curiae briefs qui a été présentée par le Département de Justice des États-Unis, on s'aperçoit que les arguments sont à peu près les mêmes. D'ailleurs l'opinion et le juge précisent qu'il est important de noter que la décision n'a pas examiné les revendications de méthode, ni les brevets relatifs aux nouvelles applications de la connaissance des gènes BRCA 1 et 2. Cette partie est très importante, partie 3 de l'opinion. Et la Cour n'a pas non plus considéré la brevetabilité d'un ADN dans lequel l'ordre des nucléotides naturels aurait été modifié. Donc les juges et en particulier le rapporteur, ont bien mentionné ce qui n'avait pas été traité. Il s'agit d'une décision limitée aux gènes existant dans la nature.
Il y a eu différents commentaires, certains rapides, et puis des commentaires émanant du domaine de la propriété industrielle, qui soulignent le fait que la décision est limitée aux gènes eux-mêmes, que les revendications d'application n'ont pas été examinées et n'ont pas fait l'objet de la décision. Différentes analyses sont un peu succinctes, mais il y en a une très intéressante sur le blog YP Talk, que je vous engage à lire car elle serait un peu longue à vous expliquer. Il est clair qu'il faut attendre et voir comment cela peut évoluer dans le temps. Comme je vous l'ai dit, l'Office des brevets américains a immédiatement présenté un mémorandum à ses examinateurs pour limiter la délivrance de brevets à des gènes existants.
Il faudrait rappeler qu'en Europe, la protection conférée par un brevet à une séquence de gènes est limitée à la fonction technique de cette séquence, telle que décrite dans la demande de brevet. Il y aurait peut-être un certain rapprochement entre la jurisprudence américaine qui vient de sortir, mais qui n'a pas été traitée complètement, et la jurisprudence européenne. Voilà ce que je pouvais vous dire ce jour.
Pr Laurent Degos, ancien président de la Haute autorité de santé (HAS), membre de l'Académie de médecine, et membre correspondant de l'Académie des sciences. Je me cantonnerai à parler du bémol de Gilles Bouvenot, de ce qu'a dit Didier Tabuteau sur les difficultés économiques en rapports avec les maladies orphelines ou les médicaments orphelins, et ce que n'a pas pu traiter notre ami Thomas Tursz en raison du temps. Il est vrai que ce problème du prix des médicaments dits ciblés ou personnalisés est devenu un problème.
J'ai participé activement à la première découverte d'une médecine personnalisée pour une leucémie, la plus grave, puisqu'elle tuait en huit jours, et qui maintenant est guérie pour 90 % des patients. À l'époque, le médicament que nous avions trouvé, un dérivé de la vitamine A, ne coûtait rien et ne coûte toujours rien. Et petit à petit un autre médicament est venu traiter cette maladie, l'arsenic, un produit relativement naturel également, mais 400 fois plus cher que le premier traitement. Et si l'on considère les traitements personnalisés actuels, le Soliris représente quand même 350 000 euros par an par personne, une somme importante.
Ma question est de savoir pourquoi y a-t-il eu ce grand changement brutal, dans une décennie, du prix de ces médicaments dits personnalisés ?
Tout d'abord, j'aimerais faire un rappel sur certaines confusions. Vous avez vu ce matin, on parle de médicaments personnalisés, de médecine personnalisée, de sous-groupes, de médecine proche... On ne sait pas très bien si c'est le médicament qui est ciblé, ou si c'est la petite population-cible : à ce moment-là nous nous approchons du médicament orphelin. Ces médicaments personnalisés ont besoin de tests, de biomarqueurs. Ils sont souvent faits par des biotechnologies, et s'approchent donc des biothérapies, dans une certaine confusion entre médecine ciblée, personnalisée, de biothérapie. Cette limite est un peu floue, et lorsqu'on parle de médecine personnalisée, ciblée, souvent il est question de médecine de petites populations.
Les prix de ces médicaments de petite population montrent une constante. Si l'on traite 300 personnes, cela revient à 350 000 euros par an par personne en France pour le Soliris. Si l'on traite 3 000 personnes comme le Glivec, c'est 30 000 euros par an par personne. Si l'on traite 30 000 personnes, ce sera 3 000 euros, etc . Quand on multiplie le prix et le volume, on obtient le même nombre. Il y a bien la recherche d'obtenir quelque chose qui s'approche du milliard d'euros en France dans cette dépense.
n peut se poser la question pourquoi il y a eu ce changement. Tout d'abord, les laboratoires Sanofi depuis un an cherchent à arrêter leurs recherches, pas seulement en France, mais leur recherche fondamentale propre. En effet on trouve les médicaments ciblés au hasard de certains mécanismes défectueux. Et l'on ne peut pas avoir dans les laboratoires d'une société pharmaceutique tous les mécanismes, et toute la recherche biologique. Si bien que l'on fait appel aux laboratoires universitaires, académiques, pour pouvoir trouver ces petits mécanismes. Ces laboratoires académiques font émerger des start-up qui vivent ou ne vivent pas, et les entreprises pharmaceutiques reprennent celles qui ont des produits efficaces.
Nous assistons à un changement de la vision de ce qu'est la recherche, et même j'irais plus loin: quelquefois on trouve un produit contre un mécanisme, mais qui n'agira pas du tout sur cette cible, et en trouvera une autre. Par exemple, un anti récepteur contre le virus HIV n'a pas eu d'effet sur le sida, mais par contre est un très bon médicament pour mobiliser les cellules médullaires. Cela n'avait rien à voir. Il y a de plus en plus de molécules pour lesquelles une cible est cherchée, et le jour où elle est trouvée, tant mieux, il est possible de s'en servir.
Une deuxième voie de réflexion des laboratoires pharmaceutiques s'est ouverte après l'affaire Viox, qui a failli mettre à plat le numéro un mondial, Merck, en raison de class actions , car il était insupportable pour ce laboratoire de suivre tous ces procès. Une réflexion a été menée par les différents laboratoires, séparément. On s'aperçoit maintenant que tous ont abouti à la même conclusion. Pour un risque égal, si vous traitez une centaine ou un millier de personnes, mettons que le risque soit de 1 pour 10 000, il faudra au moins dix ans pour voir un cas, et cent ans pour voir dix cas. Par compte si vous traitez 100 000 personnes, ou un million, au bout de quelques années, vous voyez des effets délétères, et au bout de cinq ans, vous avez des procès. Tout le monde s'est dit qu'il était bien préférable de chercher des médicaments pour traiter des petites populations, plutôt que des traitements pour de grandes populations.
Il y a conjonction entre le milieu académique qui trouve des mécanismes assez restreints, et donc des cibles et des médicaments pour de petites populations, et une réflexion industrielle qui se comprend très bien, de ne pas prendre trop de risques pour ne pas mettre toute la maison en danger. De plus, puisque la découverte est faite au niveau académique, il n'y a plus besoin de recherche fondamentale pour ces médicaments orphelins. La mise sur le marché est plus simple, le remboursement est en général accepté et il n'y a pas de taxes car il s'agit de traiter une maladie orpheline. Il n'y a pas besoin de publicité, ni d'aller voir les généralistes car si cette maladie concerne cent personnes, les généralistes n'en verront jamais une seule. Cela permet d'alléger le budget. On y trouve beaucoup d'avantages. Donc tout le monde recherche ces médicaments ciblés pour de petites populations.
C'est ainsi que le cancer, dont beaucoup de mécanismes sont connus, pour lequel nous possédons beaucoup de moyens pour pouvoir réagir, a beaucoup de médicaments ciblés. Mais à chaque fois ils deviennent des médicaments orphelins : la moitié des 100 médicaments orphelins sont des médicaments anticancéreux. Il y a donc un mouvement général auquel nous devons réfléchir, et ma réflexion emporte des conséquences.
L'assurance maladie, est concernée. Avec des médicaments aussi coûteux, le Soliris à 350 000 euros par an par personne, si simplement 0,8% de la population se voyait prescrire ce type de médicament, toute la sécurité sociale y passerait. On ne pourrait plus payer ni un hôpital, ni un médecin, une infirmière... Il y a un vrai sujet. Ces médicaments pour l'instant sont peu nombreux, avec très peu de patients. Mais la montée en charge est grande : 220 % en cinq ans, a-t-il été dit, mais si l'on considère les médicaments oraux anti cancéreux, c'est 400 % d'augmentation. Il y a donc là un mouvement qui pourrait être dangereux pour l'assurance maladie : ne couvrir que les grands risques, et laisser le reste à charge, les petits risques, c'est-à-dire tout ce qui est générique. On peut se poser la question de savoir si la sécurité sociale est faite pour très peu de personnes, un très faible pourcentage de la population.
Au sein de l'Europe, l'Europe de l'Est ne veut pas ce genre de médicaments, et de fait va demander à ce que l'Europe de l'Ouest les paie. Autre conséquence : le Glivec, un modèle, est devenu un blockbuster qui a rapporté énormément au laboratoire Novartis. Cet exemple pose la question de savoir si en fin de compte l'on va garder ce terme de maladie orpheline, destiné à l'origine à protéger la recherche de nouveaux médicaments dans un contexte difficile. L'industrie cherche de progressive blockbusters , c'est-à-dire cherche une petite population, et étend l'indication progressivement. Alors ce médicament rapportera plus.
Comment va-t-on endiguer ces phénomènes ?
En France, nous jouons vraiment le jeu du volume. Avec les plates-formes, nous cherchons l'indication précise, et ne donnons le médicament qu'à ceux qui en ont besoin. En Angleterre, ils cherchent l'utilité, et les médicaments anticancéreux sont très peu mis sur le marché, car cela apporte assez peu de survie, ou de vie augmentée, pour le patient. Les Allemands travaillent sur le prix lui-même. Ils mettent un plafond de prix. Donc chacun des pays cherche à pouvoir endiguer cette montée en charge de ces nouveaux médicaments.
Les laboratoires pharmaceutiques font des médicaments pour des petites populations, et mettent des prix assez élevés pour pouvoir survivre, car ils ont une entreprise mondiale à faire vivre. Pourquoi ne chercherait-on pas à promouvoir de nouveau des médicaments de grandes populations, qui permettraient en fin de compte aux laboratoires de vivre ? Aux États Unis, les industriels ont décidé d'arrêter de fabriquer les vaccins car les risques d'effets adverses, et ensuite de procès, étaient très importants. Et l'Etat fédéral a pris en charge toutes les indemnités de tous les accidents liés aux vaccins.
Certes il faut promouvoir, et je suis tout à fait pour, cette médecine personnalisée, mais aussi s'interroger sur le prix qu'elle représente, et essayer de conforter les laboratoires pharmaceutiques à ne pas abandonner les traitements des grandes populations.
Mr Alain Claeys L'intervention de M. Olivier Perche arrive au bon moment. Nous allons lui demander sa réaction sur l'intervention de M. Laurent Degos.
M. Olivier Perche, (LEEM) pour les entreprises du médicament, responsable du développement chez Roche Diagnostics . J'interviens donc au nom des sociétés pharmaceutiques et de l'industrie du diagnostic. J'aurais du mal à dépasser le mandat qui m'a été confié et de faire un commentaire à chaud de ce que je viens d'entendre.
M.Alain Claeys . Dans le cadre de votre mandat vous aurez, quelques éléments de réflexions, j'en suis sûr.
M. Olivier Perche . J'aurais quelques éléments de réponse. J'espère vous les donner tout au moins.
Dans la médecine personnalisée et l'organisation des soins, les enjeux pour le patient, les professionnels, les autorités ou les industriels, qu'ils soient du diagnostic ou de l'industrie thérapeutique, ont été évoqués au cours des différentes interventions précédentes. L'objet est de pouvoir accéder à l'innovation en coordonnant l'activité de l'ensemble de ces partenaires.
L'activité diagnostic intervient dans le parcours de soins et à ses différentes étapes, pour apporter des réponses sur des facteurs de risques, ou dans le contexte de dépistage ou d'identification de pathologies, pour la prévention, pour l'éligibilité de traitements. Elle s'attache à une molécule, ou au suivi. De nombreux acteurs interviennent et donnent des réponses aux cliniciens : le biologiste, le pathologiste, le radiologue, ou le biologiste moléculaire. Apparait la notion de diagnostic intégratif pour laquelle il n'y a pas encore une coordination complète. Ce peut être un axe de réflexion pour le futur.
L'évolution du diagnostic dans la médecine personnalisée a été évoquée par Mme Agnès Buzyn pour les phases 1 et 2. Le travail bibliographique mené par le LEEM montre que sur des phases 2 et 3, l'on est à plus de 400 molécules pour lesquelles il y a un couple molécule et test prospectif, dont à peu près 170 molécules pour des tests de phase 3. Donc des avancées seront effectivement proposées dans les temps futurs.
Pour autant, des problématiques demeurent, notamment pour la mise sur le marché. En ce qui concerne le médicament, la HAS et le processus sont bien banalisés, bien organisés et tout à fait aptes à répondre aux questions qui sont posés pour mettre des molécules sur le marché. Il en est de même concernant le test de diagnostic in vitro , néanmoins avec des paramètres qui restent bloquants dans le processus, et je mentionne là les délais. L'assemblage des deux dans le contexte d'une médecine personnalisée, est complexe.
Contrairement à ce qui se passe pour un médicament, un industriel ne peut pas demander la mise sur le marché d'un test diagnostic et des désynchronisations s'en suivent. M. Gilles Bouvenot évoquait les critères d'évaluation. L'évaluation technique et médicale du médicament est séparée et désynchronisée du test. Pour le test, ces critères ne sont pas publics, ni connus des industriels eux-mêmes. De même pour les délais. Il n'y a pas à ce jour d'évaluation économique du couple médicament-test diagnostic, et les acteurs sont différents.
M. Gilles Bouvenot évoquait la notion de transversalité à développer dans l'évaluation commune. Des exemples récents montrent que le processus d'un médicament est bien analysé et est arrivé à la publication pour sa mise sur le marché, alors que le test diagnostic associé, malgré une demande antérieure, n'a toujours pas été accordé ou n'est toujours pas évalué pour sa mise sur le marché. Il y a donc une désynchronisation complète des deux aspects, ce qui occasionne une perte de chance pour le patient d'obtenir un traitement lié à son test préalable, des retards pour la commercialisation, un frein à l'innovation. L'évaluation du test reste donc à définir.
Des propositions peuvent être apportées sur l'harmonisation du processus d'assemblage du test et de la molécule : par la saisine des industriels du diagnostic désirant une évaluation simultanée de la molécule et du test, le processus restant évidemment à construire autour de cette synchronisation. L'évaluation technique et économique du test et de la molécule en association pourraient être faite conjointement, ce qui représenterait une nouveauté en particulier pour les industriels du diagnostic, et pourrait amener à la construction d'un acte comprenant une partie test et une partie activité médicale. Ce processus aurait l'opportunité de pouvoir être encadré dans une notion de délai. Par l'utilisation d'un test diagnostic marqué CE, obligatoire, ce qui n'est pas forcément le cas aujourd'hui.
Ce matin a été évoquée la notion d'utilité clinique du test lui-même: un règlement européen est en cours de préparation et sera publié dans les prochains mois. Il devrait apporter ce critère d'évaluation pour la médecine personnalisée. Un remboursement temporaire du test associé à sa molécule, pourrait être un autre paramètre.
L'idée est de pouvoir apporter de la valeur ajoutée au patient, aux autorités par une prise en charge et une meilleure connaissance, pour une sécurité d'évaluation, pour les professionnels de santé dans le rapport entre les bénéfices et la toxicité, et pour les autorités en charge de la définition de son remboursement.
|
Débat |
M. Alain Claeys . Les intervenants ont pu faire leurs exposés. Le temps est compté. Mais y a-t-il quelques questions ?
M. Didier Tabuteau . Je voudrais réagir sur la question du modèle économique. On sent qu'elle est cruciale pour l'évolution de cette thérapeutique, et je crois que l'on doit admettre être dans une phase de changement de paradigme. Cela restera-t-il marginal, ou cela va-t-il s'étendre ?
Il ne faut pas faire une fatalité du fait qu'aujourd'hui des médicaments de niche ont des tarifs très élevés. Cela peut s'expliquer pour des raisons historiques. Laurent Degos en a avancé une explication, il y en a sans doute d'autres. Si des médicaments de niche, ou très spécialisés, ou en soins personnalisés, se développent, la façon de les tarifer va aussi complètement se transformer. On n'est pas du tout enfermé dans un système où la règle de la division ou de la multiplication par 10, prenant en compte le nombre de patients et le coût du traitement, va se poursuivre. Je crois au contraire qu'il faut préparer la modification des modes de tarification. Pour ma part je suis convaincu que dans dix ou quinze ans, si ces médecines se sont développées, les modes de tarification rendront ces évolutions tout à fait compatibles avec des assurances maladies, même sous contrainte. Il ne faut pas raisonner à modèle économique constant. L'enjeu est de le transformer à travers cette évolution.
Pr Dominique Stoppa-Lyonnet . Je m'adresse à Mme Thouret-Lemaître, dont je remercie l'analyse de la décision de la Cour Suprême. Ce que j'ai retenu est que la décision concernant la brevetabilité des ADN complémentaires fait couler beaucoup d'encre, du moins sur les conséquences sur la brevetabilité, notamment des protéines recombinantes, qui seraient un peu modifiées par rapport à ce que l'on trouve dans la nature. Mais concernant la première partie, c'est-à-dire la non-brevetabilité des gènes dits naturels, même s'ils sont le siège de mutations délétères, j'aurais voulu avoir votre point de vue sur les conséquences que cela pourrait avoir précisément sur ce dont on parle, c'est-à-dire sur la brevetabilité des tests compagnons, qui sont associés à une mutation pour une thérapie particulière.
Mme Elisabeth Thouret-Lemaître . C'est une décision importante, qui n'a pas fini de faire couler de l'encre. Elle est très limitée. Pour ce qui est de votre question, je pense que l'on va pouvoir continuer à trouver un intérêt dans ce qui va être fait. Je ne suis pas sûre que tous les commentaires soient terminés. La décision est assez limitée, il reste beaucoup de points à voir.
Pr Dominique Stoppa-Lyonnet. Il me semble que la décision est assez claire concernant l'identification de gènes. En gros, la séquence de gènes, qu'ils soient mutés ou pas, ne peut plus entrer dans le domaine privé, ne peut plus être brevetée. Donc ne peut-on pas considérer que cela ouvre l'ensemble du marché des tests génétiques, que ce soit en génétique constitutionnelle ou somatique ? D'ailleurs, il ne faut pas se leurrer, cette décision arrive à point nommé en même temps que le séquençage très haut débit. Il y a eu, j'imagine, une pression extrêmement forte des sociétés de biotechnologies qui développent le séquençage très haut débit, devenu impossible si plus de 20 % des gènes sont brevetés.
Mme Elisabeth Thouret-Lemaître. Vous avez tout à fait raison, et il faudrait voir la suite et les conséquences, et surtout les commentaires qui vont être faits, parce que la décision américaine est très limitée.
Mme Valérie Seror . Il y a autour de la table des personnes qui sont plus dans la technique, donc ils pourront dire ce qu'il en est véritablement. Il semblerait d'après ce que je lis que pour l'instant que les tests compagnons sont principalement développés sur des médicaments existants, et qu'il ne faut pas s'aveugler derrière le Glivec et l'Herceptin, même si les laboratoires pharmaceutiques commenceraient, semble-t-il à intégrer ce virage de développement conjoint. Du coup les aspects économiques sur les économies d'échelles ont du sens.
M. Yann Le Cam, directeur général d'EURORDIS (organisation européenne des maladies rares), vice-président du Comité européen d'experts sur les maladies rares (EUCERD). Sur l'aspect économique, je ne voudrais pas laisser une trace d'informations erronées de notre point de vue, sur un sujet aussi essentiel pour les malades, notamment en générant des peurs irrationnelles, non fondées sur des faits.
Deux informations fausses ont été apportées dans cette première partie de table ronde :
On parle de tsunami de médicaments orphelins. C'est faux. En 2006 au cours d'un groupe de travail, un colloque européen y a été consacré. On y a présenté un modèle mathématique fondé sur vingt ans d'expérience aux États-Unis et dix ans en Europe, montrant qu'il n'y en avait pas, qu'il ne fallait pas confondre mise sur le marché et désignation orpheline. Ce modèle fonctionne. En 2013 et pour les prochaines années, dans un contexte de fort progrès scientifique, il y a 8 à 12 médicaments, soit 10 médicaments orphelins en moyenne mis sur le marché en Europe par an, pas plus.
Deuxièmement, on ne peut pas faire l'amalgame entre le nombre de malades à traiter et l'augmentation du prix qui serait linéaire. C'est une contre-vérité. Sur la base des 100 premiers médicaments, le prix augmente 10 fois quand la maladie est 100 fois plus rare : donc il n'augmente pas 100 fois.
Voilà, pour restaurer des chiffres et des faits qui permettent un débat mieux éclairé.
Dr Frédéric Staroz, médecin pathologiste, président du syndicat des médecins pathologistes français . J'avais juste deux remarques assez brèves. Le Pr Agnès Buzyn parlait des plateformes qui à l'évidence ont permis un accès très large aux tests compagnons et aux thérapeutiques innovantes. Le fait est que la continuation de ce modèle capte l'information, et en particulier l'accès à cette formation que chacun a jugée indispensable, l'appropriation de ces réalisations de tests.
La deuxième remarque concernait ce que disait le Pr Laurent Degos. Il a été répété que le cancer était extrêmement hétérogène. Je ne sais pas si chacun s'imagine à quel point il l'est. Chaque cellule d'un même cancer va avoir un profil génomique différent, et le Pr. Thomas Tursz parlait d'un phénomène où il y avait un accélérateur à fond et des freins cassés... Il n'est pas toujours nécessaire de démonter totalement la voiture pour savoir ce qui se passe dedans, et de temps en temps, justement, l'observer au microscope permet directement de savoir. Malgré tout, les nosologies actuelles ne sont pas obsolètes, mais tous ces tests moléculaires sont complémentaires et doivent s'y intégrer. Je ne suis pas sûr qu'il faille jeter totalement le bébé avec l'eau du bain dans ces circonstances.
M. Laurent Degos. Pour répondre à Didier Tabuteau tout à l'heure : le modèle économique doit être vu, certainement, mais si l'on baisse le prix des médicaments actuels, alors que les industriels ont surtout ces médicaments, et très peu de grande populations qui deviennent génériques, ils rechercheront soit des fusions-acquisition pour essayer d'augmenter le portefeuille, soit des acquisition de start-up pour la même raison, ou alors il faudra conjointement les aider à avoir des médicaments de grande population, avec tout ce que cela comporte comme difficultés. Si l'on baisse les prix, avec un modèle économique fondé sur de toutes petites populations, ils ne pourront pas avoir d'actionnaires.
Pr Thomas Tursz. Il y a un point très important dans l'évaluation de ce futur modèle économique, c'est encore une fois qu'aucune de ces drogues seules ne guérira le cancer. L'avenir est qu'il y ait combinaison de drogues, car il y aura plusieurs anomalies, des résistances, où émergeront des clones sélectionnés. Or, le schéma actuel de mise sur le marché, comme le schéma que proposent les industriels, vise à mettre un médicament sur le marché tout seul, avant de permettre de faire des associations, en particulier avec des médicaments issus de firmes concurrentes. Ceci est une catastrophe scientifique et économique et amène à de gros problèmes industriels : sur dix substances qui entrent en phase 1, une seule deviendra réellement un médicament. Les industriels jettent donc à la poubelle neuf médicaments sur dix, sur lesquels ils ont déjà énormément investi sur les plans recherche et essais précliniques.
Peut-être certaines de ces drogues, comme celles qui ont été jetées du temps de ces screening que j'évoquais tout à l'heure, ne seraient jamais devenues des médicaments seules, mais auraient pu être intéressantes en association ou en combinaison, parce qu'elles auraient levé ou contourné des résistances. Un système serait intéressant, qui permettrait très précocement de tester sur un fonds particulier, je pense que ce devrait être un fonds européen, des combinaisons de médicament, en fonction des anomalies du génome. En effet on peut réaliser 3, 4 mais pas 10 000 tests compagnons. C'est le gros argument qui amènera le séquençage complet du génome comme une étape nécessaire, et qui finira par économiser de l'argent.
La réalité du cancer quotidien, est de donner à 90 % de la population des médicaments qui ne marchent pas, coûtent cher, et sont toxiques pour tout le monde ; d'en essayer au hasard un deuxième, un troisième... un huitième, ayant de moins en moins de chances de marcher. Si l'on pouvait s'attaquer à ce problème comme à l'un des problèmes majeurs de la médecine personnalisée, dans ce modèle économique nouveau, il y aurait un élément de réflexion très important.
QUATRIÈME TABLE
RONDE :
QUELS REGARDS DES PATIENTS ET DES ASSOCIATIONS DE
MALADES ?
M. Alain Claeys, député. Nous abordons cette quatrième table ronde. Le regard des patients et des associations de malades est un élément important par rapport à tous les sujets que nous avons pu aborder. Avant d'en débattre, nous avons cinq interventions.
Mme Martine Bungener, directrice de recherche au CNRS, économiste, présidente du GRAM, groupe de réflexion avec les associations de malades de l'INSERM . Je voudrais remercier l'Office parlementaire d'avoir demandé au GRAM et à l'INSERM de l'aider à monter cette table ronde.
C'est absolument essentiel d'avoir du temps pour donner la parole aux patients et à leurs associations, d'autant que même si depuis ce matin j'ai entendu, et nous avons entendu, plusieurs fois et avec une grande compétence évoquer les patients et leurs besoins, il me semble que c'était avec le filtre de chacun des intervenants et de sa propre position. On constate des décalages sur lesquels il est important de revenir.
Je suis moi-même chercheur en sciences sociales, et très contente de présider le GRAM, lieu de débats. Je veux que ce temps de parole soit d'abord pour les associations, et je me permets juste d'introduire cinq thématiques très rapidement. Elles sont importantes non seulement pour les patients, mais aussi plus largement pour l'ensemble de la société, et elles réclament une certaine vigilance sociale. On a beaucoup évoqué ce matin des transformations, des changements de paradigme, de modèle économique, et il est donc absolument important que l'ensemble des citoyens soit partie-prenante de ces changements. Une des fonctions des associations de patients sur ce type de thème est de pouvoir exercer cette vigilance sociale dont elles sont en partie les garantes, mais aussi de les associer pour imaginer et mettre en place les formes nécessaires de régulation et de transparence dont nous allons avoir besoin.
Juste avant de revenir très rapidement sur ces cinq thèmes, je vais me permettre cette fois en tant que chercheur, à titre personnel, d'évoquer deux points concernant les propos que j'ai écoutés ce matin.
Un premier point concerne cet appel à la nouveauté que je viens moi-même de reprendre. Il me semble que sur un certain nombre d'éléments comme la protection des données, les transformations des pratiques ou de l'organisation des soins, il faut se garder d'être dans un modèle de "tout est nouveau", de rupture totale. Nous ne sommes pas vraiment dans un modèle de rupture, mais au contraire on observe des permanences à propos des questions posées, des enseignements à tirer et des éléments, certes exacerbés, mais sur lesquels il existe déjà des réflexions. Il ne faudrait pas faire table rase de tout cela, tout recommencer, mais regarder un peu en arrière. Il y a des choses qui existent et doivent être améliorées, des choses qui ont déjà été mises en place, sur lesquelles il faudrait sans doute faire un bilan critique. Prenons quand même le temps de le faire, et je ne vais pas le prendre là, mais il faudra sans doute en rediscuter. En tout cas, des orateurs et des intervenants, d'autres acteurs sociaux disent : « oui, il y a un certain nombre de choses qui existent déjà dans notre pays, et sur lesquelles on peut bâtir cette nouveauté », sans nécessairement repenser, remettre sur le chantier des acquis, en perdant sans doute beaucoup de temps. C'est vrai sur les enjeux économiques notamment, mais aussi sur les enjeux sociaux et politiques.
Le deuxième point était pour évoquer l'histoire que Philippe Amouyel nous a racontée ce matin au sujet des dirigeants, qui eux-mêmes ont eu l'occasion de faire faire le séquençage de leur génome et de se rendre compte qu'ils avaient éventuellement une prédisposition à une maladie. Á un moment il a très précisément évoqué un de ces dirigeants, qui disait "mais moi, je veux savoir". Donc ce dirigeant plaidait pour ce droit au savoir, et le Pr Amouyel nous a expliqué que la personne en question avait entrepris de changer son mode de vie pour tenir compte des prédispositions que son génome lui avait révélées. Mais je voulais demander en tant qu'économiste et sociologue : aujourd'hui face à cette « révolution » médicale qui se dessine, qui parmi nos concitoyens a vraiment la possibilité de changer fondamentalement son mode de vie ? Nous sommes face à des inégalités de positions. Il ne suffit pas simplement de changer un comportement. Cela a été dit plusieurs fois mais je pense qu'il faut le redire, nos concitoyens, dans leur grande majorité, ne sont pas nécessairement en position de changer fondamentalement leur mode de vie, même s'ils apprennent quelque chose sur un risque potentiel. Politiquement, socialement, éthiquement, c'est un point qu'il ne faut absolument pas perdre de vue. Et il faudra penser aussi aux moyens sociaux d'accompagnement de ce changement s'il est nécessaire.
Très rapidement je vais juste repointer les cinq enjeux que j'ai évoqués tout à l'heure, qui je pense vont être repris par l'ensemble des associations.
Le premier enjeu, on l'a évoqué plusieurs fois, est celui qui concerne finalement ces questions de nom ou de mot. On a parlé de médecine personnalisée, aussi de médecine de prévision, de tout ce qui gravite autour de cette notion de médecine personnalisée et de tous les malentendus qu'elle peut générer. Mais à l'inverse, je veux aussi pointer l'enjeu, et cela a aussi été beaucoup discuté, que de toute façon cette médecine va s'exprimer sous forme de probabilité. Qui est apte aujourd'hui pour traiter ce type d'information ? Comment feront les médecins eux-mêmes, quelle capacité auront-ils à traiter cette information ? Les associations de malades ont un rôle à jouer, et on l'a vu dans d'autres situations, pour justement aider à traiter cette information et à la diffuser. On est bien là dans une dimension qui doit mobiliser l'ensemble des associations, qui s'inscrit dans leurs fonctions. Informer les malades, elles en ont les compétences, elles savent le faire.
Le deuxième enjeu est double. Selon les pathologies et leurs formes, il concerne la précision individuelle ou par sous-groupe du diagnostic et du pronostic d'une part, et l'adaptation plus fine des traitements pharmacologiques, des thérapies ciblées notamment pour les cancers, d'autre part. Non seulement les malades ont des choses à dire sur ces aspects, mais également les associations ont véritablement une fonction et des capacités pour socialement répondre à ces questions, qui incluent celle de la cohésion des groupes de malades ou celle de leur division potentielle. Des patients qui, avant, travaillaient ensemble, discutaient ensemble, vont se trouver peut-être dissociés. Certains auront des traitements, d'autres pas, d'autre des pronostics différents. C'est un enjeu important, il faut y réfléchir.
Bien entendu, il est important de reprendre aussi l'enjeu des coûts et de ces nouveaux marchés de recherche de médicaments. Il ne faut pas oublier que les associations sont des groupes de pression, et elles se feront entendre. On l'a redit tout à l'heure à la fin de la table ronde précédente, oui, le modèle d'aujourd'hui n'est sûrement pas le modèle de demain, et il va bouger, parce que les citoyens auront aussi des volontés de le faire bouger.
Le quatrième enjeu est celui des nouvelles dispositions et des nouvelles procédures que ces avancées entraînent pour la recherche clinique, et notamment bien sûr pour l'implication des patients dans ces processus de recherche, avec la rédaction de protocoles et de consentements. C'est l'enjeu de l'alimentation de grosses bases de données. Là aussi le nombre de patients à inclure sera différent et là aussi les associations vont avoir une fonction à jouer.
Enfin, dernier enjeu, on l'a beaucoup évoqué, je me permets juste de le reprendre. C'est bien sûr la question de la protection de toutes ces données. Dans le contexte actuel, on le sait tous, la façon de faire confiance à la médecine et à la recherche est déjà assez ébranlée, a subi beaucoup de coups ces dernières décennies. Il y a eu des crises sanitaires, des crises de recherche.
Le GRAM milite depuis sa création pour montrer que le dialogue avec les associations est vraiment une partie prenante de la reconstruction de ces mécanismes de formation de la confiance collective vis-à-vis de la recherche et de la médecine, et nous sommes aujourd'hui face à un enjeu majeur avec les nouveaux traitements et ces nouvelles formes de médecine.
Mme Catherine Vergely, secrétaire générale de l'Union des parents d'enfants atteints de cancer et de leucémie (UNAPECLE). Merci de nous permettre à nous association de parents de prendre la parole aujourd'hui. Comme je suis la première à parler en tant qu'association de patients, je souhaite à ce titre tenter de vous faire part de la perception sociétale de tous les propos très savants et des quelques erreurs que nous avons entendus depuis ce matin, et je me permettrai de faire des recommandations très pratico-pratiques, ce qui changera un peu.
Tout d'abord l'Union nationale des parents d'enfants atteints de cancer ou de leucémie se préoccupe depuis plus de vingt ans de l'amélioration des traitements, et notre évolution à nous a eu lieu, puisque nous sommes passés de 25 % de guérison à plus de 80 % pour nos enfants, ce qui pour nous a été, parents et associations impliquées, quelque chose de très important. Nous avions anticipé en tant que parents, puisque nous sollicitions le plus souvent les médecins, non seulement sur les avancées, mais aussi la qualité de vie de nos enfants pendant et après les traitements, ce que nous retrouvons aujourd'hui probablement dans la médecine personnalisée.
Une étude récente de l'INSERM montre que ces enfants vivant aujourd'hui, guéris depuis plus de vingt ans quelquefois, rencontrent des difficultés importantes dans leur vie sociale et dans l'acceptation de leur guérison par la société. C'est par ce biais que je vais entrer dans la médecine personnalisée.
En effet depuis de nombreuses années nous connaissons l'impact de l'information, traitée mal ou bien, sur la vie de nos enfants. Dans le carnet de santé il suffit d'avoir quelques indications de maladies, pour s'apercevoir que ces enfants ne sont pas intégrés dans certains systèmes collectifs, qu'ils ne sont pas pris en charge par certaines écoles, même si les lois existent, et que certains postes professionnels ne leurs sont pas accordés. De plus, Il y a encore des surtaxes dans les assurances, malgré toutes les lois et les procédures mises en place par nos élus.
Comment se présente aujourd'hui la médecine personnalisée pour ces enfants atteints de cancer, et surtout quelles sont ses spécificités par rapport à ce que l'on a entendu sur le cancer de l'adulte ?
Pour les enfants atteints de cancer et pour leurs parents, la médecine personnalisée est un grand espoir, comme pour tous les parents dont les enfants sont malades, et qui n'ont pas forcément un traitement qui marche à 100 %. Cet espoir ne se situe pas du tout sur l'avancée ou l'efficacité des traitements, que nous avons déjà en partie résolues, mais surtout sur la qualité de vie durant la maladie et après la maladie. C'est cela qui est essentiel pour nous. Lorsque les parents évoquent cette efficacité, c'est toujours pour les 15 ou 20 % d'enfants qui n'ont pas encore de traitement. Là, nous rejoignons d'autres parties de la pédiatrie où il n'y a pas de traitement efficace.
Dans le cancer de l'enfant, la médecine personnalisée est déjà en place. Comme on vous l'a rabâché depuis ce matin, les tumeurs sont maintenant typées. Mais il faut considérer également la protection et la qualité de vie. Il faut que vous sachiez qu'un garçon et une fille avec la même maladie ne sont pas traités de la même façon tout simplement parce qu'ils sont un garçon et une fille, et je n'ai pas besoin d'un test génétique pour le savoir. La qualité de vie de ce garçon ou de cette fille va être modifiée par le traitement, notamment dans sa stérilité, ou au contraire sa possibilité de procréer. C'est important déjà de penser que la médecine personnalisée est en place, qu'elle est essentielle, car elle va déterminer tout l'avenir de nos enfants.
Si l'on pousse un peu plus loin la médecine personnalisée dans ce qui reste toujours le cancer de l'enfant, avec une étude plus précise du génome, alors les grandes inquiétudes pour les familles apparaissent.
Inquiétude immédiate, d'abord, sur les répercussions directes sur les parents et la fratrie, avec une culpabilité exacerbée pour les familles. Les parents ont peur de ce qui va se passer, car malgré tout ce que vous pensez, ils sont suffisamment pertinents pour penser que le gène n'est pas seul en cause et que peut-être des comportements, voire de l'épigénétique, sont importants dans l'arrivée de la maladie. Ils sont tout à fait conscients que le génome en lui-même ne suffit pas. Il existe ensuite une inquiétude à long terme, sur l'utilisation des données et leur protection. Les parents ont une expérience négative dans ce domaine, mais surtout, et je ne l'ai pas beaucoup entendu aujourd'hui, ils n'ont pas systématiquement confiance dans les institutions de recherche, qui sont très avides de données et d'accumulation de résultats, ni dans les institutions de contrôle dont ils ont déjà perçu les failles à différents niveaux, et où le citoyen n'a pas l'opportunité de jouer toujours un contre-pouvoir. Nous avons vu tout à l'heure l'assurance maladie conditionnelle. C'est un élément essentiel. L'on sait très bien que des enfants ayant été malades, ou des enfants de fratrie dont une soeur ou un frère ont déjà été malades, ont une épée de Damoclès sur leur vie sociale puisqu'on les met dans des catégories à risque.
J'en viens à mes quatre recommandations pratico-pratiques, comme je l'ai déjà dit.
D'abord nous ne réinventons pas la roue. Il existe des modèles adaptables. Utilisons par exemple les avancées des sciences humaines et sociales concernant certaines maladies, et aussi la médecine personnalisée existante destinée à des patients qui ne sont pas malades génétiquement, et appliquons-les dans les maladies génétiques.
Ensuite utilisons les Plans maladies rares dans l'application de la médecine personnalisée, ce qui permettrait peut-être d'inventer des modèles économiques pertinents où l'ensemble des acteurs seraient mis autour d'une table en tant que citoyens et professionnels, pour en discuter.
Il ne faut pas informer le malade, il faut former le citoyen, et pas seulement dans la connaissance scientifique, mais aussi dans son esprit critique afin qu'il soit inclus dans les différentes étapes de décision, et qu'il ait une vision pertinente de son acceptation en tant que malade dans une société et dans des choix qu'on lui offre. Il faut respecter le citoyen et ses décisions. Il est absolument indispensable de l'intégrer dans l'ensemble des instances de stratégie et de décision de médecine et de recherche d'une façon globale, et de la médecine personnalisée plus particulièrement. J'ai entendu parler des bonnes pratiques depuis ce matin. Je n'ai pas perçu la présence de patients, bien qu'ils aient des représentants, dans l'acceptation de ces bonnes pratiques, alors que toutes les institutions se sont cependant concertées.
Enfin, il faut sûrement repenser plus précisément les conséquences sociétales des données acquises dans la médecine personnalisée, notamment pour les enfants. Il ne faut pas multiplier les risques de perturber leur futur, leur vie sociale. Nous demandons donc que dans ce cadre, les enfants devenus adultes soient consultés sur l'utilisation des données accumulées pendant leur enfance, qui leur reviennent. Ce n'est pas parce que des parents ont pris à un moment des décisions pour eux, que ces enfants n'ont pas le droit à l'oubli dans un certain nombre d'éléments, qui sont finalement leur vie personnelle.
Je vous rappelle que la loi Huriet-Sérusclat, mais également la déclaration d'Helsinki, précisent que l'intérêt général ne doit jamais dans la recherche prendre le pas sur l'intérêt individuel.
Mme Laurence Tiennot-Herment, présidente de l'AFM-Téléthon (Association Française contre les Myopathies). Je suis présidente de l'AFM-Téléthon. De par le rôle atypique que notre association a joué dans le domaine de la recherche, et aussi sur la partie médicale et sociale, j'apporterai sans doute une vision particulière. En effet, nous avons pu avec les moyens du Téléthon engager un milliard d'euros dans la recherche depuis sa première édition, donc en 27 ans, et 660 millions d'euros dans l'aide aux malades. Cela nous a fait jouer un rôle important notamment à travers les laboratoires que nous avons pu créer, Généthon, I-Stem, Atlantic Gene Therapies, Institut de myologie, laboratoires dans lesquels la gouvernance majoritaire est donnée aux malades et aux parents de malades.
Les maladies rares rentrent complètement dans la thématique de la thérapie ciblée. Elles sont des maladies modèles à différents niveaux :
car les malades et les parents sont des experts d'expérience de ces maladies. Puisqu'il y avait assez peu de connaissances dans le domaine, ils sont allés les chercher. L'expérience est venue de leurs propres enfants et de leurs propres malades ;
car elles ont nécessité un décloisonnement, un changement de paradigme et un changement de modèle. II a fallu innover. Comme il y a 27 ans on disposait de très peu de connaissances dans le domaine des maladies rares, il a fallu construire des solutions. Nous ne pouvions pas mettre nos pas dans ceux de quelqu'un d'autre.
Les maladies rares sont au nombre de 6 à 8 000, et concernent trois millions de personnes en France, je le rappelle. Elles sont modèles sur plusieurs axes : la génétique, le diagnostic, l'organisation des soins, une prise en charge médicale adaptée, un accompagnement personnalisé, le développement clinique et les premières thérapies ciblées. Elles sont à plus de 80 % d'origine génétique et cela a donné lieu à des découvertes nombreuses. En 1986 on connaissait six gènes à l'origine de maladies rares, et aujourd'hui plus de 3 800. Un vrai bouleversement s'est opéré en ce domaine. Au cours de cette période sont apparus le séquençage à grande échelle, à haut débit et les bio-banques. On voit par ailleurs émerger depuis ces dernières années des bases de données cliniques/génétiques, extrêmement importantes, notamment pour identifier les bonnes cohortes de patients pour les thérapies ciblées qui commencent à naître aujourd'hui.
Sur l'aspect génétique, je donnerai juste une alerte. Il est important que la France retrouve son leadership dans le domaine du séquençage à haut débit. Cela a été largement évoqué par les experts pendant cette journée.
J'évoquais le diagnostic, car pour les maladies génétiques il est important d'avoir un bon diagnostic, et qu'il y ait transfert du diagnostic recherche vers le diagnostic hospitalier. C'est crucial, indispensable. Il faut connaître la maladie très précisément pour avoir une prise en charge médicale adaptée et une médecine préventive adaptée dans certaines maladies.
Par exemple, il y a 25 ans on connaissait environ 20 myopathies. Maintenant on sait qu'il existe 250 formes différentes, autant de gènes différents en cause, et alors qu'on parlait à l'époque d'une myopathie des ceintures, on a identifié aujourd'hui près de 30 sortes de myopathies des ceintures. Dans ces dernières, certaines nécessitent une surveillance cardiaque particulière, et il faut connaître le gène très exactement en cause pour l'assurer. Le diagnostic est donc crucial. Mais pour bénéficier des thérapies ciblées, il faut non seulement savoir quel type de maladie est en cause, mais il faut aussi connaître le type de mutation ou de délétion dans la maladie. Même dans une maladie rare il y a des sous-groupes de maladies. Par exemple dans la myopathie de Duchenne, on sait qu'on trouve des patients candidats au saut de codon stop ou au saut d'exon, etc. Dans ces maladies on doit connaître la bonne mutation. L'alerte sur la partie diagnostic concerne l'accès au diagnostic pour que les actes soient correctement codifiés et les patients bien remboursés. C'est un problème que nous rencontrons.
Une meilleure organisation des soins a permis d'augmenter l'espérance de vie de 15 à 20 ans dans certains cas chez les patients atteints de maladies rares. Dans les consultations pluridisciplinaires, le patient est au centre, et les professionnels exerçant différentes compétences médicales sont autour. Il ne faut pas que le patient aille voir d'un côté l'orthopédiste, de l'autre le neurologue, le cardiologue, etc. Cela a nécessité forcément une nouvelle organisation des soins, la mise en place de consultations pluridisciplinaires, qui ont été reconnues. Ces consultations pluridisciplinaires ont donné lieu à la naissance des premiers centres de référence et des centres de compétences maladies rares. Dans ces centres de référence, coexistent les trois axes indispensables que sont le diagnostic, la prise en charge et la recherche. Après les centres de références on espère demain, dans le cadre du Plan national maladies rares n° 2, la création des premières filières de santé maladies rares par grands groupes de maladies. Cette organisation des soins qui place le malade au centre avec les compétences autour implique une organisation des soins particulière. Et dans l'alerte que je donne, au-delà des filières de santé qui doivent se mettre en place, des guides de bonnes pratiques sont nécessaires.
Un autre point concerne l'accompagnement personnalisé et l'approche globale de la personne. À plusieurs reprises j'entendais cet après-midi l'évocation du parcours de soins. Je dis qu'il ne faut pas que ce soit seulement un parcours de soins, mais un parcours de santé avec une approche globale de la personne, du médical au social, avec des référents parcours de santé pour qu'il n'y ait jamais d'exclusion du patient de son parcours de santé. Cela peut avoir bien sûr un intérêt au-delà des seules maladies rares. Á l'heure des déserts médicaux, si l'on est dans cette logique de réorganisation du parcours de santé avec un accompagnement personnalisé et un référent parcours de santé, c'est aussi une solution pour bon nombre de maladies chroniques.
L'alerte que je donne concerne un nouveau modèle fondé sur l'organisation des soins, un meilleur accompagnement personnalisé, avec une approche globale, d'autant plus, comme le disait Catherine Vergely tout à l'heure, que l'on voit certains patients exclus de ces thérapies ciblées, ou victimes de la problématique de l'annonce du diagnostic. Il faut vraiment qu'il y ait cet accompagnement personnalisé.
Le développement clinique pour les maladies rares montre l'exemple, car leurs thérapies ciblées doivent être efficaces. Je reprends ce que disait le Professeur Thomas Tursz tout à l'heure, quand il évoquait le fait que pour les maladies fréquentes, le ratio de succès des essais clinique est de seulement 10 %. C'est une réalité. Pour les maladies rares, 10 % seulement de ratio de succès est impossible car nous n'avons pas assez de patients. Dans certaines de ces maladies, si dix essais sont lancés et qu'un seul débouche sur un traitement, ce ne sera pas possible. Donc il faut vraiment que ce développement clinique soit particulier, spécifique, adapté, efficace ce qu'il commence à être dans le cadre des maladies rares. Cela nécessite un développement préclinique adapté de la qualité du développement clinique « traditionnel », c'est-à-dire intégrant l'objectif d'efficacité dès le début de l'essai clinique.
Les maladies rares donnent naissance, avec ces premières thérapies ciblées, à de vraies innovations de rupture, à partir de thérapies géniques, de l'ARN, cellulaires. Á chaque fois on le constate, ces innovations de rupture, bien sûr sont efficaces pour des petites populations dans des maladies rares, mais peuvent également s'appliquer à différentes maladies rares, et aussi être des innovations pour des maladies fréquentes.
Je prends un exemple. Le saut de codon stop est une stratégie thérapeutique qui va permettre de sauter une mutation stop dans un gène et rétablir ainsi le cadre de lecture. Je ne suis pas scientifique et les scientifiques me contrediront éventuellement. En tout cas, cette approche thérapeutique est intéressante pour tous les patients qui ont une myopathie de Duchenne avec un codon stop. Mais elle peut l'être parfois aussi dans la mucoviscidose où certains patients ont également une mutation à codon stop. On y voit aussi un intérêt en oncologie, et dans les maladies infectieuses. Ces innovations de rupture sont intéressantes pour des petites catégories de patients dans les maladies rares mais elles auront un intérêt pour plusieurs maladies rares, voire pour des maladies fréquentes, et l'on assiste à l'émergence d'une nouvelle génération de médicaments à partir de ces thérapies ciblées, innovantes nécessitant aussi la construction d'un nouveau modèle économique pour ces médicaments innovants.
Le prix du médicament a été beaucoup évoqué tout à l'heure, avec parfois une confusion entre la règlementation autour du médicament orphelin et le prix du médicament. Le prix du médicament orphelin n'a rien à voir avec la règlementation européenne sur ce type de médicament. Il est fixé, comme pour tout médicament, après discussion entre l'industrie pharmaceutique et les autorités compétentes.
En tant qu'association participant à la gouvernance d'un laboratoire associatif, Généthon, pour lequel nous pensons que, dans les jours qui viennent, nous allons pouvoir annoncer publiquement son statut d'établissement pharmaceutique, car maintenant la loi le permet, nous pensons construire le prix du médicament innovant sur son coût : le coût de développement, de production.... C'est sans doute différent des pratiques actuelles. Cela doit être envisagé, un prix juste et maîtrisé du médicament, davantage basé sur le coût, et en toute transparence, plutôt que sur les seuls critères sur lesquels il est basé actuellement.
En tant que représentante des malades et des familles, je souhaite qu'à tous les niveaux dans cette chaîne de santé, du diagnostic jusqu'au prix du médicament, les patients soient présents en tant que « partenaires ».
M. Yann Le Cam, directeur général d'EURORDIS (Organisation européenne des maladies rares), vice-président du Comité d'experts sur les maladies rares (EUCERD). J'ai le plaisir de présenter la perspective des associations et des patients au niveau européen. EURORDIS représente 585 associations et considère représenter 25 millions de personnes atteintes par une des 4 000 maladies rares, parmi les 6 000 qu'évoquait Laurence Tiennot-Herment tout à l'heure. J'essaierai aussi de partager avec vous la perspective des comités experts européens et notamment de l'Agence Européenne des Médicaments et du Comité Experts sur les Maladies Rares en charge de l'organisation des soins européens, auquel j'ai le plaisir d'appartenir.
Mon exposé contiendra trois parties. J'aimerais contribuer à la clarification des concepts, qui ne se révèlent pas si clairs au cours de cette audition, entre médecine personnalisée, médecine stratifiée, et médicaments orphelins, car nous intervenons souvent dans le cadre de débats comme celui-ci, mais au niveau européen. Il existe même une plate-forme européenne sur les thérapies personnalisées dont EURORDIS fait partie.
De notre point de vue, la médecine personnalisée est prise au sens d'un produit de santé spécifique à une personne. Nous serons dans cette situation extrêmement rarement, dans le cadre d'une thérapie génique ou cellulaire dans un avenir proche, mais cela concernera peu de situations scientifiques et médicales et ne suppose pas une grande adaptation du système de soins en général. Généralement, ces traitements seront curatifs. Cette médecine personnalisée n'aura pas un grand impact budgétaire sur les budgets de santé. Elle en a un, mais généralement en situation curative.
En revanche dans la médecine stratifiée, nous sommes dans une tendance lourde, issue des progrès de la génomique et des thérapies ciblées sur des sous-populations de patients, avec une fragmentation de certaines maladies fréquentes, à commencer par les cancers, mais pas seulement. En font partie également les troubles de comportement et de l'apprentissage. La médecine stratifiée peut concerner autant les maladies fréquentes que rares.
C'est cette contre vérité que je voudrais aborder et aimerais que l'on ne mélange pas les deux. Dans les maladies rares, Laurence Tiennot-Herment l'a dit, si l'on prend la mucoviscidose, un médicament comme le Kalydeco concerne une mutation spécifique (ESSID) dans cette maladie qui comprend 1 800 mutations. On est déjà dans le cadre d'un traitement stratifié de la mucoviscidose, qui ne s'applique pas à une autre maladie pour l'instant, en l'état de la connaissance.
La médecine stratifiée peut concerner un nombre de patients parfois élevé. Pour prendre un autre exemple, pour les mutations non-sens, des médicaments sont en cours de développement. Si je prends l'exemple de l'Ateluren, si la preuve de concept est faite dans la myopathie de Duchenne et dans la mucoviscidose, il est vraisemblable qu'il s'appliquera sur 50 ou 60 maladies, car cette petite molécule agit selon le même mécanisme d'action dans ces différentes maladies. Il n'y aura pas besoin vraisemblablement d'aller jusqu'à l'AMM pour chaque nouvelle maladie, mais plutôt de procéder à la démonstration par des essais cliniques progressifs transversalement pour chacune de ces 60 indications. Il est actuellement discuté au sein de la Food and Drug Administration américaine (FDA) et de l'Agence européenne de pouvoir le faire en abordant globalement cette population sur une mutation non-sens, concernant différentes maladies. Nous sommes sur des concepts vraiment nouveaux, et nous ne parlons plus de populations rares, mais de l'agrégat de populations rares qui devient alors plus large, presque fréquent.
Dans le binôme produit de santé-population cible qu'évoquait tout à l'heure Laurent Degos, le lien est le mécanisme d'action plutôt que la maladie ciblée. Il faut bien faire ces différences.
Dans le médicament orphelin en revanche, le point de départ est une maladie, que l'on appelle une condition dans les textes européens, c'est-à-dire un ensemble de symptômes avec une histoire naturelle de cette maladie, en se plaçant du point de vue du patient et de la clinique. Ce n'est vraiment pas la même chose que de considérer le mécanisme d'action comme dans la situation précédente du stratifié. Nous sommes là sur des prévalences bien définies, 5 sur 10 000 au niveau européen, c'est-à-dire 1 sur 2 000, ou moins de 30 000 personnes en France.
La conclusion sur ces trois concepts est qu'ils ne sont pas équivalents, et je voudrais insister sur le fait qu'ils ne se superposent pas, mais qu'en revanche ils ont des intersections.
Je voudrais aussi apporter une précision supplémentaire, c'est qu'aux États-Unis l'an dernier, en 2012, 39 médicaments ont eu une autorisation de mise sur le marché. Deux-tiers de ces médicaments étaient soit orphelins, soit stratifiés, en cancérologie. On n'est donc pas dans la science-fiction ni dans des problèmes de demain, mais dans des situations que l'on sait traiter aujourd'hui, et sur lesquels existent des concepts assez aiguisés.
Le deuxième aspect de ma présentation est de parler de l'impact sur le règlement européen des médicaments orphelins, puisqu'on a tendance à dire que l'arrivée de ces médecines personnalisées ou stratifiées va le faire exploser. Nous ne le pensons absolument pas, d'abord grâce aux clarifications de concepts que j'espère vous avoir apportées précédemment, mais aussi parce que l'enjeu n'est pas l'existence de ce règlement, mais la façon dont on l'applique. Et le fait d'être bien centré sur la façon dont on définit une condition, et de ne pas partir uniquement sur des mécanismes d'action, peut être travaillé dans le cadre du règlement actuel.
J'ai déjà dit tout à l'heure dans le débat que nous ne sommes pas non plus face au tsunami de médicaments orphelins que certains annoncent. Des éléments chiffrés et validés le montrent à partir de l'analyse des désignations orphelines et des AMM depuis le début des législations américaines et européennes ; cette analyse a été réalisée par EURORDIS avec la FDA et l'Agence européenne des médicaments (EMA). Depuis huit ans, ce modèle mathématique fonctionne aux États-Unis comme en Europe, nous sommes sur un schéma prédictif pour les cinq à dix ans qui viennent, de 8 à 12 médicaments orphelins par an, puis sur le marché si les conditions restent constantes.
Pour conclure sur cet aspect-là, nous pouvons considérer qu'il faut ajuster la politique des médicaments orphelins en fonction de l'évolution de la connaissance scientifique, dans la pratique notamment du Comité des médicaments orphelins. Mais cela ne justifie pas de remettre en cause le règlement lui-même.
Les vrais enjeux sont plutôt en amont et en aval de ce règlement, et ce sera ma dernière partie. J'insisterai sur trois principaux enjeux.
Le premier, en amont, est celui de concrétiser pour les personnes malades l'espoir issu des progrès scientifiques. Nous ne refusons pas d'avancer, nous ne pensons pas à toutes les raisons de ne pas développer ces médecines personnalisées ou stratifiées, par peur de leur énorme coût, mais nous cherchons plutôt les conditions d'une translation de la connaissance scientifique vers les applications thérapeutiques, c'est-à-dire que nous créons les conditions d'une accélération de la durée de la recherche scientifique. Cela peut être fait notamment grâce aux outils informatiques, mais aussi avec les nouveaux design dits adaptifs d'essais cliniques, des approches statistiques différentes, adaptives elles aussi avec les méthodes séquentielles, bayésiennes, etc., qui sont encouragées par l'Agence Européenne des Médicaments, et encore sous-utilisés. Il est également possible de réduire les coûts du développement clinique, notamment en faisant appel à des alternatives de modèles animaux ou aux biomarqueurs.
Dans une conférence récente qui s'est tenue à Dublin sous la présidence de l'Union européenne, première conférence du Consortium international de recherche sur les maladies rares, plusieurs interventions ont démontré que les coûts de développement, en utilisant ces différentes méthodologies existantes, sont 10 à 30 fois inférieurs à ce qu'ils étaient ces dix dernières années, et que l'on entre dans une toute autre économie de ces médicaments.
L'idée est d'offrir au malade et à la société une autre perspective que celle proposée aujourd'hui. Cette dernière soutient que chaque nouveau médicament coûte de plus en plus cher à cause des règlementations qui rendent plus coûteux son développement, mais aussi implique un nombre décroissant entre les candidats au moment de la preuve de concept et le nombre de produits atteignant la mise sur le marché. Il faut plutôt envisager de faire différemment dans les vingt prochaines années que dans les 40 dernières, et plein d'opportunités existent pour offrir un espoir aux malades.
Le deuxième enjeu principal, qui va au-delà du règlement sur les médicaments orphelins, concerne les modalités d'autorisation de ces nouveaux traitements.
Je voudrais insister sur une notion, celle de la prise de risque. Nous considérons que la façon dont les agences réglementaires de médicaments fonctionnent, au niveau européen comme au niveau national, tend à protéger leurs responsabilités ainsi que les élus politiques, plutôt que de servir les patients et la santé publique. Bien sûr, on me l'opposera, cela dépend de quel médicament l'on parle, dans quelle maladie. Effectivement, si l'on est au dixième produit pour l'hypertension, certainement les risques pour la population doivent être regardés de très près, la prise de risque doit tendre vers zéro. En revanche, dans le cas de maladies rares ou non rares, mais sévères, débilitantes, avec une menace pour la survie, l'appréciation n'est pas la même, et les patients sont prêts à prendre plus de risques que ces agences. Et c'est leur droit à la vie. Ce n'est pas iconoclaste de le dire, c'est une tendance lourde des discussions au niveau européen, et c'est le sens de mon propos.
Notamment, une nouvelle perspective s'ouvre, celle de la mise sur le marché progressive « l'accès progressif aux patients », à la fin de phases 2, à partir d'essais cliniques sur un petit nombre de patients. Elle ferait l'objet d'un encadrement très strict de la prescription, en milieu hospitalier, où progressivement serait augmenté le nombre de patients recevant ce traitement. Les conditions économiques de remboursement par les systèmes de santé nationaux s'appliquent dans ce cas dès cette mise sur le marché précoce, ce qui décroît tout de même le coût de développement, et la prise de risque pour l'industriel. Cela génère en échange une collecte de données beaucoup plus importante sur la sécurité et l'efficacité de ces traitements en situations de vie réelle et permet de définir leur véritable valeur ajoutée et leur place dans les stratégies thérapeutiques. Nous plaidons vraiment en faveur de cette approche qui nous semble être parfaitement adaptée non seulement à la problématique des médicaments orphelins actuels mais plus globalement à celle des médecines stratifiées ou personnalisées.
J'ajoute que c'est un domaine au niveau européen très actif actuellement. Il y a des réunions pratiquement chaque mois à l'Agence européenne des médicaments ou ailleurs pour mettre en place ces nouveaux dispositifs. Et EURORDIS est très engagé sur ce dossier.
Sur cette question de l'AMM, je voudrais ajouter qu'il existe trois enjeux.
L'un concerne l'appréciation du bénéfice-risque au stade de l'AMM. Nous sommes en faveur de la participation des patients à cette évaluation, car ils sont les premiers concernés, notamment dans les maladies sévères. Elle doit être rigoureusement encadrée, en pensant d'abord à l'intérêt des personnes malades.
Aux États-Unis les lois ont complètement changé, on n'en a pas encore pris suffisamment conscience en Europe et notamment en France. Les lois adoptées en 2012 changent complètement le paradigme de la prise de risque à la FDA, et l'on attend d'en voir l'impact en Europe.
Un autre enjeu concerne la continuité de collecte des données de sécurité et d'efficacité avant et après l'AMM. Nous sommes à présent dans un continuum. On ne peut plus continuer à opposer avant et après l'AMM. Il faut collecter les données en situation de vie réelle, et avoir une approche plus globale du cycle de vie d'un médicament. Enfin, de manière à mieux répondre aux besoins de santé non satisfaits et à réduire les incertitudes sur ces traitements, le dialogue continu est nécessaire entre les agences réglementaires du médicament, la Haute autorité de santé (HAS) et les payeurs, tout au long du développement du produit.
Le troisième et dernier enjeu que l'on identifie est celui de l'accès au traitement. Aujourd'hui la question n'est pas celle de la valeur du médicament, mais plutôt celle de la capacité à financer, pour le système de santé, ces nouveaux traitements ciblés, qu'ils soient personnalisés ou stratifiés. Pour cela, notre réponse est de promouvoir une nouvelle approche : plutôt plus d'Europe que moins d'Europe. Pas de repli national pour réfléchir sur de toutes petites populations, très souvent sans avoir l'expertise sur ces maladies ou sur la valeur de ces produits, mais plutôt de travailler au niveau européen, où l'on trouve une masse critique de patients pour avoir des données suffisantes, et une mutualisation des expertises pour réaliser les évaluations collectives.
La suite logique de cette coordination au niveau européen est d'avoir des discussions entre la valeur du médicament, le nombre de patients à traiter dans une indication précise - certainement plus restreintes que les indications thérapeutiques actuelles - de parler de volumes, et de lier le prix à la collecte des données post-AMM, c'est-à-dire à la création de connaissances et de niveau de preuves pour réduire les incertitudes. À ce moment-là nous aurons un nouveau modèle économique qui permettra de baisser le prix du médicament, et donc de le rendre plus finançable par les systèmes de santé. Ce que l'on veut offrir c'est le bon traitement, au bon patient, au bon moment, et à un prix plus juste et durablement finançable par la société
Mme Christine Pezel, secrétaire générale de l'association Vaincre la mucoviscidose. Mon intervention va être assez courte car au fur et à mesure des heures, elle a été un peu vidée de son contenu. Beaucoup de choses ont été dites, et surtout au moment des dernières interventions. J'ai une réflexion toute personnelle sur la place qui est accordée à cette table ronde, en fin de journée, intitulée : « quel regard des patients et des associations de malades sur la médecine personnalisée ? »
Une première audition avait eu lieu en mars, qui a réuni des scientifiques autour des pathologies, des technologies, etc . Et depuis ce matin, beaucoup de scientifiques se sont exprimés, laissant peu de place pour les patients, et en l'occurrence pour les associations.
Ce que j'exprime présente beaucoup de similitudes avec ce qui a été dit par Laurence Tiennot-Herment sur les maladies rares. Comme les myopathies, la mucoviscidose est un modèle pour l'expérimentation de la médecine ciblée. C'est une maladie génétique avec un dysfonctionnement de la protéine CFTR, où le génotype du malade permet de prévoir ce que sera le dysfonctionnement de la cellule. Il existe un registre national de la mucoviscidose qui existe depuis 20 ans aujourd'hui et recense 95 % des patients atteints de cette maladie, ce qui était un atout majeur pour une thérapie ciblée. La preuve en a été faite tout récemment.
L'organisation de soins spécifiques est nécessaire. Les centres de compétence avec leur caractère multidisciplinaire permettent une prise en charge globale et un parcours de soins facilité pour le patient. Sans aucun doute, cela a été dit par Didier Tabuteau tout à l'heure, des évolutions des structures de soins seront nécessaires. Il a parlé d'évolution technique, mais également de nouveaux métiers. Il faudra augmenter la formation des médecins, veiller à ce qu'il y ait une place réelle pour les sciences humaines et sociales et également une prise en compte de l'aspect psychologique.
En ce qui concerne la recherche j'adhère à ce qui vient d'être dit. C'est ce qui est réalisé par l'association « Vaincre la mucoviscidose » : à savoir être partie prenante d'une recherche tant au niveau français qu'européen et international. L'intérêt d'une telle démarche est une évidence aujourd'hui.
Pour notre part, nous préférons le terme de médecine adaptée à celui de médecine personnalisée, adaptée au patient, avec un traitement ciblé, même si l'individu est inclus dans un groupe.
Bien sûr pour les patients et les familles concernés par la mucoviscidose, cette médecine personnalisée représente un espoir : cela a été indiqué par l'orateur précédent, tout récemment en 2012 une molécule appelée Kalydeco a été mise sur le marché et profite à des patients atteints de mucoviscidose pour une mutation bien précise, qui concerne assez peu de malades, mais constitue un espoir important, d'autant plus que des essais cliniques vont démarrer pour une autre mutation beaucoup plus répandue.
Une remarque sur le petit nombre de patients et la petite population : c'est parfois difficile à entendre, et les deux orateurs précédents l'on dit, parfois des pathologies peu répandues peuvent profiter à d'autres qui sont plus importantes. Il ne faut pas cloisonner des objectifs communs.
Ces nouvelles molécules coûtent cher, la médecine personnalisée va peut-être coûter plus cher dans un premier temps. Mais il faut savoir dépenser davantage pour, à terme, réaliser des économies.
* (1) http://www.assemblee-nationale.fr/11/dossiers/991871.asp.
* (2) Les biomarqueurs sont des molécules biologiques présentes dans le sang, dans les liquides corporels et les tissus organiques ou dans les tissus malades eux-mêmes.
* (3) Professeur au Collège de France, membre de l'Académie des sciences, audition des rapporteurs du 20 novembre 2012.
* (4) Personalized Medicine for the European Citizen towards more precise medicine for the diagnosis, treatment and prevention of disease, October 2012 (iPM).
* (5) Directeur de recherches à l'INSERM, ancien membre du conseil scientifique de l'OPECST ; audition des rapporteurs du 28 novembre 2012.
* (6) Biologiste moléculaire, fondateur de la Génopole de Marseille ; audition des rapporteurs du 12 février 2013.
* (7) Conseiller d'État, responsable de la Chaire santé de l'Institut d'études politiques de Paris ; audition des rapporteurs du 19 décembre 2012.
* (8) Hématologue, présidente de l'Institut national du cancer (INCa) ; audition des rapporteurs du 19 mars 2013.
* (9) Sociologue, directrice de recherche au CNRS, Centre de recherche médecine, sciences, santé, santé mentale, société (CERMES3) - CNRS UMR8211/Université Paris Descartes/EHESS/Inserm U988.
* (10) Stratégie nationale de santé, feuille de route http://www.social-sante.gouv.fr/IMG/pdf/SNS-version-longue.pdf.
* (11) Président directeur général de l'INSERM Institut national de la santé et de la recherche médicale - Audition publique du 25 mars 2013.
* (12) Professeur de biochimie ; rapport « Techniques d'analyse du génome et de son expression : applications médicales » (Académie nationale de médecine).
* (13) La molécule d'ADN est composée d'éléments de base, les nucléotides de quatre types différents : l'adénine (A), la thymine (T), la cytosine (C) et la guanine (G). Cette molécule est constituée de deux brins complémentaires, un A se trouvant toujours face à un T et un G face à un C.
* (14) Intervention lors de la réunion conjointe Académie des sciences-Académie nationale de médecine le 12 novembre 2013 « génome personnel ».
* (15) Une molécule d'ADN, extraite d'un échantillon est clonée et de nombreuses molécules d'ADN simple brin sont produites. Un petit oligonucléotide (amorce) complémentaire à une partie du fragment d'ADN à séquencer est ajouté et sert de point de départ à la synthèse du brin d'ADN complémentaire. L'ajout d'ADN polymérase et des nucléotides A, T, G et C (les dNTP) permet de générer le brin complémentaire. À ce stade on ajoute en faible concentration quatre nucléotides analogues appelés didésoxynucléotides (les ddNTP) qui ressemblent aux nucléotides normaux, leur incorporation a pour conséquence l'arrêt de la polymérisation et la formation de nouveaux fragments d'ADN de taille variable, qui sont ensuite séparés par électrophorèse.
* (16) Séquençage de l'ADN : les dernières (r)évolutions.
http://www.bulletins-electroniques.com/actualites/69283.htm
* (17) Idem.
* (18) Président de DNAVision, audition publique du 27 mars 2013.
* (19) Chef du service de génétique de l'Institut Curie, professeur de génétique médicale à l'Université Paris-Descartes, audition des rapporteurs du 5 février 2013.
* (20) Responsable du département de génétique du groupe hospitalier de La Pitié Salpêtrière-Charles Foix - Audition publique du 27 mars 2013.
* (21) Directeur du centre national de génotypage- Audition publique du 27 mars 2013.
* (22) Biologiste moléculaire, fondateur de la Génopole de Marseille - Audition des rapporteurs du 12 février 2013.
* (23) Directeur de recherche au CNRS, professeur à l'Université Lille 2, et à l'Imperial College l'Université de Londres - Audition des rapporteurs le 3 décembre 2013.
* (24) Chargée de recherche en génétique humaine et statistiques à l'INSERM, audition des rapporteurs le 15 janvier 2013.
* (25) Professeur d'économie de la santé Université d'Aix-Marseille UMR 912 SESTIM AMU/INSERM/IRD directeur de l'ITM0 Santé Publique de l'AVIESAN, audition des rapporteurs du 25 novembre 2012
* (26) Directeur-adjoint de l'ITMO génétique, génomique et bio-informatique AVIESAN, coordinateur de l'Institut de recherche biomédicale de Rouen, audition des rapporteur du 4 décembre 2013.
* (27) Professeur de génétique moléculaire, service de génétique médicale des hôpitaux universitaires de Strasbourg, audition des rapporteurs du 4 décembre 2013.
* (28) Professeur des universités, responsable du dépistage et du suivi des maladies métaboliques au CHU de Rouen, audition des rapporteurs du 4 décembre 2013.
* (29) Le 28 novembre 2013.
* (30) Audition des rapporteurs du 13 février 2013 et visite des rapporteurs au Génopole d'Évry le 28 novembre 2013.
* (31) Le 28 novembre 2013
* (32) Médecin, oncologue à l'institut Curie, responsable de l'essai SHIVA - Audition publique du 27 mars2013.
* (33) « Médecine personnalisée et innovation médicale : enjeux de santé publique », le 5 décembre 2013.
* (34) Innovation et valorisation de la recherche La nouvelle ambition de l'« Institut Broad » : un vaste programme de recherche en biologie moléculaire http://www.bulletins-electroniques.com/actualites/69103. 2 octobre 2012.
* (35) Innovation et valorisation de la recherche Le centre génomique de New York, un nouvel acteur incontournable du séquençage ? http://www.bulletins-electroniques.com/actualites/70833, 31 aout 2012.
* (36) Président de DNAvision audition publique du 27 mars 2013, visite des rapporteurs à Bruxelles le 1 er octobre 2013, Le Monde des sciences et techniques du 30 septembre 2013
* (37) Les SNP constituent la forme la plus abondante de variations génétiques dans le génome humain. Ils représentent plus de 90 % de toutes les différences entre individus. C'est un type de polymorphisme de l'ADN dans lequel deux chromosomes diffèrent sur un segment donné par une seule paire de bases. Dans deux génomes humains tirés au hasard, 99,9 % de la séquence d'ADN est identique. Le 0,1 % restant contient des variations de séquence dont le type le plus commun est le polymorphisme pour un nucléotide (SNP). Les SNP sont stables, très abondants et distribués uniformément dans tout le génome. Ces variations sont associées à de la diversité entre populations ou individus, une différence de sensibilité à des maladies et la réponse individuelle aux médicaments.
* (38) Professeur de biologie moléculaire, fondateur du Génopole de Marseille, membre de l'Académie des sciences, audition des rapporteurs du 12 février 2013.
* (39) Responsable du programme nano-médecine au CEA-Leti, président de l'European Technology Platform on Nanomedicine, audition publique du 25 mars 2013.
* (40) Directeur du Centre national de génotypage du CEA, audition publique du 27 mars.2013, visite des rapporteurs au Génopole d'Évry le28 novembre 2013.
* (41) Directeur de l'Institut de biologie et technologies du CEA à Saclay IBITECs, audition des rapporteurs du 6 février 2013.
* (42) Bioactif CEA, mars 2012.
* (43) Ensemble des protéines synthétisées par une cellule, aux 25 000 gènes humains correspondent environ un million de protéines différentes.
* (44) Techniques d'analyse du génome et de son expression : applications médicales Bull. Acad. Nationale Méd., 2012, 196, no 1, 151-171, séance du 17 janvier 2012.
* (45) Directrice du département de biothérapie de l'Hôpital Necker-Enfants Malades, audition publique du 27 mars 2013.
* (46) Rapport d'information au Parlement et au Gouvernement de l'ABM, novembre 2013.
* (47) Rapport d'information au Parlement et au Gouvernement de l'ABM, novembre 2013.
* (48) Rapport d'information au Parlement et au Gouvernement de l'ABM, novembre 2013.
* (49) Loi n° 2013-715 du 6 août 2013 tendant à modifier la loi n° 2011-814 du 7 juillet 2011 relative à la bioéthique en autorisant sous certaines conditions la recherche sur l'embryon et les cellules souches embryonnaires.
* (50) Rapport de MM. Alain Claeys et Jean-Sébastien Vialatte, députés, sur la recherche sur les cellules souches (n° 2718 AN - n° 652 Sénat) du 8 juillet 2010.
* (51) Dominique Stoppa-Lyonnet, professeur de génétique, audition des rapporteurs du 5 février 2013.
* (52) Médecin oncologue à l'Institut Curie, responsable de l'essai Shiva, audition publique du 27 mars 2013.
* (53) Médecin généticien, ancien directeur de l'Institut Cochin, ancien président de l'Université Paris-Descartes, audition publique du 27 mars 2013.
* (54) Hématologue, présidente de l'Institut national du cancer (INCa)-Audition publique du 27 mars 2013.
* (55) Hématologue, présidente de l'INCa - Auditions publiques du 27 mars et du 25 juin 2013.
* (56) Voir le site Internet de l'INCa.
* (57) Hématologue, présidente de l'INCa, audition publique du 25 juin 2013.
* (58) Hématologue, Présidente de l'INCa, audition publique du 27 mars 2013.
* (59) Idem.
* (60) Rapport au Président de la République sur le 3 ème Plan cancer.
* (61) Rapport au Président de la République sur le 3 ème Plan cancer.
* (62) Chef du service de biochimie/biologie moléculaire à l'hôpital St Louis, audition publique du 25 juin 2013.
* (63) Chef du service de biochimie/biologie moléculaire à l'hôpital St Louis et Valérie Lallemand, chargée de recherche à l'INSERM, à l'Institut universitaire d'Hématologie de l'Hôpital Saint-Louis visite à l'Hôpital Saint-Louis, le 13 avril 2013, et à Toulon, le 29 novembre 2013.
* (64) Directeur de recherche au CNRS, professeur à l'Université Lille 2 et à l'Imperial College-Université de Londres, audition des rapporteurs le3 décembre 2013.
* (65) Visite des rapporteurs à Gustave Roussy le 4 décembre 2013.
* (66) Cancérologue, directeur honoraire de Gustave Roussy, audition publique du 25 juin 2013.
* (67), Pr Dominique Stoppa- Lyonnet, chef du service de génétique à l'Institut Curie, professeur de génétique et Dr Christophe Le Tourneau, oncologue, responsable de l'essai SHIVA, audition des rapporteurs le 5 février 2013.
* (68) Audition publique du 27 mars 2013 et visite à l'Institut Curie le 20 mars 2013.
* (69) , Médecin oncologue à L'institut Curie, responsable de l'essai SHIVA, audition publique du 27 mars 2013.
* (70) Professeur de cancérologie à l'Institut Claude Bernard Lyon 1, chef du service d'oncologie médicale au Centre hospitalo-universitaire de Lyon, présidente de Lyon Rhône-Alpes Auvergne, audition publique du 25 juin 2013.
* (71) Visite à Gustave Roussy Cancer Campus Grand Paris le 4 décembre 2013.
* (72) Site Internet de Gustave Roussy.
* (73) Cancérologue, directeur honoraire de Gustave Roussy, audition publique du 25 juin 2013.
* (74) Visite des rapporteurs à Marseille le 8 octobre 2013.
* (75) Cancérologue, directeur honoraire de Gustave Roussy, audition publique du 25 juin 2013.
* (76) Ancien président de la Haute autorité de santé (HAS), audition publique du 25 juin 2013.
* (77) Directeur de l'Agence nationale de recherches sur le sida et les hépatites virales (ANRS), audition publique du 27 mars 2013.
* (78) Pr Jean-François Delfraissy, colloque ITMO santé publique du 5 décembre 2013.
* (79) Biologiste moléculaire, fondateur de la Génopole de Marseille, membre de l'Académie des sciences-Audition des rapporteurs du 12 février 2012, audition des rapporteurs du 12 février 2013.
* (80) Biologiste moléculaire, fondateur de la Génopole de Marseille, membre de l'Académie des sciences, audition des rapporteurs du 12 février 2013 et visite des rapporteurs à Marseille le 8 octobre 2013.
* (81) Colloque ITMO santé publique du 5 décembre 2013.
* (82) Directeur de l'Agence nationale de recherches sur le sida et les hépatites virales (ANRS, audition publique du 27 mars 2013.
* (83) Endocrinologue, professeur de biologie moléculaire à l'université Lille 2 et à l'Imperial College-Université de Londres, audition des rapporteurs du 3 décembre 2013.
* (84) Endocrinologue, professeur de biologie moléculaire à l'université Lille 2 et à l'Imperial College-Université de Londres, audition des rapporteurs du 3 décembre 2013.
* (85) Présidente de l'AFM-Téléthon (Association française contre les myopathies) - Audition publique du 25 juin 2013.
* (86) Directeur du Laboratoire U910, service de génétique médicale de l'Institut Paoli Calmettes, visite des rapporteurs à Marseille le 8 octobre 2013.
* (87) Professeur de génétique médicale, praticien hospitalier, Hôpitaux universitaires de Strasbourg, service de génétique médicale - Pôle de biologie, audition des rapporteurs du 4 décembre 2013.
* (88) Professeur des universités, praticien hospitalier, responsable du dépistage et du suivi des maladies métaboliques au CHU de Rouen, audition des rapporteurs du 4 décembre 2013.
* (89) Directeur du département de génétique, Hôpital universitaire de Rouen, directeur de l'Inserm U1079, coordinateur de l'Institut de recherche biomédicale de Rouen, directeur adjoint de l'ITMO Génétique, Génomique et Bio-informatique (GGB), audition des rapporteurs du 4 décembre 2013.
* (90) Directrice d'Orphanet, portail d'information sur les maladies rares et les médicaments orphelins, audition publique du 27 mars 2013.
* (91) Rapport d'information AN n° 4484 et Sénat n° 49 de MM. Claude Birraux et Jean-Louis Touraine, députés, mars 2012.
* (92) Directeur du Laboratoire U910, service de génétique médicale de l'Institut Paoli Calmettes, visite des rapporteurs à Marseille le 8 octobre 2013.
* (93) Professeur de génétique médicale, praticien hospitalier, Hôpitaux universitaires de Strasbourg, service de génétique médicale, Pôle de biologie, audition des rapporteurs du 4 décembre 2013.
* (94) Professeur des universités, praticien hospitalier, responsable du dépistage et du suivi des maladies métaboliques au CHU de Rouen, audition des rapporteurs du 4 décembre 2013.
* (95) Secrétaire générale de l'association « Vaincre la mucoviscidose », audition publique du 25 juin 2013.
* (96) Président du Comité d'éthique de l'INSERM, directeur du laboratoire de neurosciences de l'Institut de biologie de Paris-Seine, membre du Conseil scientifique de l'OPECST, audition publique du 25 juin 2013.
* (97) Immunologiste, président du Comité consultatif national d'éthique pour les sciences de la vie et de la santé (CCNE), audition publique du 25 juin 2013.
* (98) Professeur d'épidémiologie et de santé publique au Centre hospitalier et universitaire de Lille, directeur général de la fondation Plan Alzheimer, audition publique du 25 juin 2013.
* (99) Députée de l'Hérault, vice-présidente de l'OPECST, audition publique du 27 mars 2013.
* (100) Professeure au Collège de France, membre de l'Académie des sciences, audition publique du 25 juin 2013.
* (101) Chargée de recherche en génétique humaine et statistiques à l'INSERM, audition publique du 25 juin 2013.
* (102) Responsable de la Chaire santé de l'Institut d'études politiques (IEP). Audition privée du 19 décembre 2012, audition publique du 25 juin 2013.
* (103) Adjointe au directeur général de la santé, chef des politiques de santé, audition des rapporteurs du 7 janvier 2014
* (104) Professeure au Collège de France, membre de l'académie des sciences, audition publique du 25 juin 2013.
* (105) Doyen honoraire de l'Université Paris V Descartes, audition publique du 27 mars 2013.
* (106) Conseiller d'État, responsable de la Chaire de santé de l'IEP, audition publique du 25 juin 2013.
* (107) Conseiller à la Cour d'appel de Paris, secrétaire général de l'Association internationale Droit, éthique et science, audition des rapporteurs du 28 mai 2013.
* (108) Doctorante à l'École de droit de l'Université Paris I Panthéon-Sorbonne, audition publique du 25 juin 2013.
* (109) http://www.social-sante.gouv.fr/actualite-presse,42/communiques,2322/strategie-nationale-de-sante-vers,16258.html.
* (110) Doyen la faculté de médecine de Paris V Descartes, audition publique du 27 mars 2013.
* (111) Doyen de la faculté de médecine de l'Université d'Aix Marseille, visite des rapporteurs à Marseille le 8 octobre 2013.
* (112) Doyen de l'Université de Paris-Descartes, audition publique du 27 mars 2013.
* (113) Article 41 de la loi n°2013-660 du 22 juillet 2013 relative à l'enseignement supérieur et à la recherche.
* (114) Nouveaux métiers du secteur des technologies de la santé février 2010.
* (115) Membre de l'Académie de pharmacie, directeur de l'Institut de biologie et technologies (iBiTecS) du CEA, audition des rapporteurs du mercredi 6 février 2013.
* (116) Directeur de recherche à l'INSERM, à l'Institut de génétique et de biologie moléculaire (IGBMC), membre du Conseil scientifique de l'OPECST, audition publique du 17 juillet 2013.
* (117) Stratégie Emploi/Formation 2015 Études Compétences/Métiers pour les Industries de Santé en Île-de-France LEEM, Medicen Genopole 2011.
* (118) Directeur général du Génopole d'Évry, visite des rapporteurs du 28 novembre 2013.
* (119) Conseiller d'État, responsable de la Chaire santé de l'Institut d'études politiques de Paris (IEP), audition des rapporteurs du 19 décembre 2012 et audition publique du 25 juin 2013.
* (120) www.social-sante.gouv.fr/...42/.../presentation-de-la-strategie,16141.htmlý.
* (121) Directeur général du Génopole d'Évry, visite des rapporteurs du 28 novembre 2013.
* (122) Directeur de recherche à l'INSERM, membre de l'Académie des sciences et du Conseil scientifique de l'OPECS,- audition publique du 27 mars 2013.
* (123) Présidente du directoire d'INSERM transfert, audition publique du 27 mars 2013.
* (124) Idem.
* (125) Intervenant pour Les Entreprises du médicament (LEEM), vice-président R&D France de Sanofi, audition publique du 27 mars 2013.
* (126) Présidente du directoire d'INSERM transfert, audition publique du 27 mars 2013.
* (127) Président du comité d'éthique de l'Institut national de la santé et de la recherche médicale (INSERM), directeur du laboratoire de neuroscience de l'institut de biologie Paris-Seine, membre du conseil scientifique de l'OPECST, audition publique du 25 juin 2013.
* (128) Député du Rhône, membre de l'OPECST, audition publique du 27 mars 2013.
* (129) Président directeur général de l'INSERM, audition publique du 27 mars 2013.
* (130) Ancien président de la Haute autorité de santé (HAS), membre de l'Académie de médecine, et membre correspondant de l'Académie des sciences, audition publique du 25 juin 2013.
* (131) Intervenant pour les entreprises du médicament LEEM, responsable du développement Roche Diagnostics, audition publique du 25 juin 2013.
* (132) http://www.leem.org/les-biomarqueurs-compagnons.
* (133) Directrice accès au Marché et affaires publiques du laboratoire Amgen, audition des rapporteurs du 23 février 2013.
* (134) Directeur général délégué stratégies et affaires médicales de BioalliancePharma, audition publique du 27 mars 2013.
* (135) Ancien président de la Haute autorité de santé (HAS), membre de l'Académie de médecine et membre correspondant de l'Académie des sciences, audition publique du 25 juin 2013.
* (136) Professeur des universités, directeur du Laboratoire droit de la santé - université Paris VIII, Réflexions sur l'étude « les enjeux scientifiques, technologiques et éthiques de la médecine personnalisée » de l'OPECST, Les petites affiches, 3 octobre 2013, n° 198.
* (137) Le Monde des sciences et techniques, 9 décembre 2013.
* (138) Conseiller d'État, responsable de la Chaire santé de l'Institut d'études politiques de Paris (IEP), audition des rapporteurs du 19 décembre 2012.
* (139) Directrice du programme ADNA « Avancées diagnostiques pour de nouvelles approches thérapeutiques», Institut Mérieux, audition publique du 27 mars 2013.
* (140) http://www.leem.org/les-biomarqueurs-compagnons.
* (141) Directrice de l'accès au marché et des affaires publiques d'Amgen, audition publique du 2 mars 2013.
* (142) Intervenant) pour les entreprises du médicament pour (LEEM,) responsable du développement chez Roche Diagnostics, audition publique du 25 juin 2013.
* (143) Président de la Commission de la transparence de la HAS, membre du collège de la HAS et de l'Académie nationale de médecine, audition publique du 25 juin 2013.
* (144) Oncologue, directeur honoraire de Gustave Roussy, audition publique du 25 juin 2013.
* (145) Présidente du directoire d'INSERM transfert, audition publique du 25 juin 2013.
* (146) Sociologue au Centre de recherche médecine, sciences, santé et société (CERMES), audition publique du 27 mars 2013.
* (147) United States Federal Circuit, Aff. Association for Molecular Pathology contre Myriad Genetics.
* (148) Consultante spécialiste de propriété industrielle, audition publique du 25 juin 2013.
* (149) Consultante spécialiste de propriété industrielle- Audition publique du 25 juin 2013.
* (150) Hélène Gaumont-Prat, professeur des universités, directeur du laboratoire de droit médical, Université de Paris VII, Réflexions sur l'étude « Les enjeux scientifiques, technologiques et éthiques de la médecine personnalisée » de l'OPECST, Les petites affiches, 3 octobre 2013, n 198.
* (151) Chef du service de génétique à l'Institut Curie, professeur de génétique, audition publique du 25 juin 2013.
* (152) Consultante spécialiste de propriété industrielle, audition publique du 27 mars 2013.
* (153) Hélène Gaumont-Prat, professeur des universités, directeur du laboratoire de droit médical, Université de Paris VII, contribution écrite à l'audition publique du 27 mars2013.
* (154) Sociologue au Centre de recherche médecine, sciences, santé et société (CERMES, audition publique du 27 mars 2013.
* (155) Alain Claeys, rapport de l'OPECST, AN n° 1487 - Sénat n° 235.
* (156) Sociologue au Centre de recherche médecine, sciences, santé et société (CERMES), audition publique du 27 mars 2013.
* (157) La construction de communs comme alternative à la privatisation de la connaissance in Médecine/Sciences hors-série n° 2, juin-juillet 2012.
* (158) M. Cassier, D. Stoppa-Lyonnet in Médecine/Sciences hors-série n° 2, juin-juillet 2012.
* (159) Sociologue au Centre de recherche médecine, sciences, santé et société (CERMES), audition publique du 27 mars 2013.
* (160) Professeur de droit privé, Université Paris VIII, directeur du Master Propriété industrielle et industries de santé, Intervention écrite pour l'audition publique du 27 mars 2013.
* (161) Doctorante à l'école de droit de l'Université Paris I Panthéon-Sorbonne, audition publique du 27 mars 2013.
* (162) Visite des rapporteurs à la Commission européenne à Bruxelles, le 1 er octobre 2013.
* (163 Économiste, chargée de recherche INSEM-IRD Université d'Aix-Marseille, audition publique du 25 juin 2013.
* (164) Ancien président de la Haute autorité de santé (HAS), membre de l'Académie de médecine et membre correspondant de l'Académie des sciences, audition publique du 25 juin 2013.
* (165) Professeur d'économie de la santé Université d'Aix-Marseille UMR 912 SESTIM AMU/INSERM/IRD directeur de l'ITM0 Santé publique de l'AVIESAN, audition des rapporteurs du 25 novembre 2012.
* (166) Oncologue, directeur honoraire de Gustave Roussy, audition publique du 25 juin 2013.
* (167) Professeur d'économie de la santé Université d'Aix-Marseille UMR 912 SESTIM AMU/INSERM/IRD directeur de l'ITM0 Santé Publique de l'AVIESAN audition des rapporteurs du 25 novembre 2012.
* (168) Économiste, chargée de recherche INSEM- IRD Université d'Aix-Marseille, audition publique du 25 juin 2013, audition publique du 25 juin 2013
* (169) Président de la Commission de la transparence, membre du collège de la Haute autorité de santé (HAS), membre de l'Académie nationale de médecine, audition publique du 25 juin 2013.
* (170) Président de la Commission de la transparence, membre du collège de la Haute autorité de santé (HAS), membre de l'Académie nationale de médecine, audition publique du 27 mars 2013.
* (171) Professeur de droit privé, Université Paris VIII, directeur du Master Propriété industrielle et industries de santé, Réflexions sur l'étude « les enjeux scientifiques, technologiques et éthiques de la médecine personnalisée » de l'OPECST Petites Affiches, 3 Octobre 2013, n° 198.
* (172) Président de la Commission de la transparence, membre du collège de la Haute autorité de santé (HAS), membre de l'Académie nationale de médecine, audition publique du 25 juin 2013.
* (173) Oncologue, directeur honoraire de Gustave Roussy, audition publique du 25 juin 2013.
* (174) Conseiller d'État, responsable de la chaire de politique de santé de l'IEP, audition publique du 25 juin 2013.
* (175) Ancien président de la HAS, membre de l'Académie des Sciences, audition publique du 25 juin 2013.
* (176) Conseiller d'État, responsable de la chaire de politique de santé de l'IEP, audition publique du 25 juin 2013.
* (177) Professeur de droit privé, Université Paris VIII, directeur du Master Propriété industrielle et industries de santé, Réflexions sur l'étude « les enjeux scientifiques, technologiques et éthiques de la médecine personnalisée » de l'OPECST Petites Affiches, 3 Octobre 2013, n° 198.
* (178) Conseiller d'État, responsable de la chaire de politique de santé de l'IEP, audition publique du 25 juin 2013.
* (179) Conseiller d'État, responsable de la chaire de politique de santé de l'IEP, audition publique du 25 juin 2013.
* (180) Conseiller d'État, responsable de la Chaire de politique de santé de l'IEP, audition publique du 25 juin 2013.
* (181) Rapport d'évaluation de MM. Alain Claeys et Jean-Sébastien Vialatte, députés, la loi n° 2004-800 du 6 août 2004 relative à la bioéthique, AN n° 1325 et Sénat n° 37, janvier 2008.
* (182) Rapport de MM. Claude Birraux et Jean-Louis Touraine, députés, AN n° 4484 et Sénat n° 490, Les maladies monogéniques : état des lieux, compte-rendu de l'audition publique du 7 juin 2011 et de la présentation des conclusions le 25 janvier 2012
* (183) Directeur de recherche à l'INSERM, membre de l'Académie des sciences et du Conseil scientifique de l'OPECST, audition publique du 25 mars 2013
* (184) Directrice générale de l'Agence de la biomédecine, audition publique du 25 juin 2013.
* (185) Responsable du département de génétique du groupe hospitalier de la Pitié- Salpetrière- Charles Foix, audition publique du 25 juin 2013.
* (186) MM Alain Claeys et Jean-Sébastien Vialatte, députés, rapport d'évaluation de la loi relative à la bioéthique, AN n° 1325 - Sénat n° 407.
* (187) Arrêté du 27 mai 2013, Journal officiel du 7 juin 2013 - JORF n° 0130 du 7 juin 2013, page 9469.
* (188) Tests génétiques en accès libre sur Internet - Stratégies commerciales et enjeux éthiques et sociétaux, Pascal Ducournau, Pierre-Antoine Gourraud, Emmanuelle Rial-Sebbag, Alexandre Bulle et Anne Cambon-Thomsen, médecine/sciences 27 (1) 95 (2011).
* (189) Chargée de recherché en génétique humaine et statistique à l'INSERM, audition publique du 25 juin 2013.
* (190) Conseiller d'État, responsable de la chaire de santé publique de l'IEP, audition publique du 25 juin 2013.
* (191) Ancien président de l'Université Paris-Descartes, membre de l'Académie des sciences et du Conseil scientifique de l'OPECST, audition publique du 27 mars 2013
* (192) Sources Médecine prédictive : les balbutiements d'un concept aux enjeux considérables : note n° 289 du centre d'analyse stratégique, octobre 2012.
* (193) Sociologue, directrice de recherche au CNRS, Centre de recherche médecine, sciences, santé, santé mentale, société (CERMES3) CNRS UMR8211/Université Paris-Descartes/EHESS/Inserm U988, audition publique du 25 juin 2013.
* (194) Chargée de mission au Commissariat général à la stratégie et à la prospective (CGSP), audition des rapporteurs du 20 décembre 2013.
* (195) http://conventions.coe.int/Treaty/fr/Treaties/html/203.htm
* (196) Chargée de recherche en génétique humaine et statistiques à l'INSERM, audition publique du 25 juin 2013,
* (197) Directrice de recherche au CNRS UMR U 1027 « Épidémiologie et analyses en santé publique » de l'INSERM, et de l'Université de Toulouse 3 Paul Sabatier, responsable de l'équipe Génomique, biothérapies et santé publique, audition publique du 25 juin 2013.
* (198) Président du CCNE, audition publique du 25 juin 2013.
* (199) Note n° 289, octobre 2012, Médecine prédictives les balbutiements d'un concept aux enjeux considérables.
* (200) In Médecine/Sciences Volume 29, Numéro 6-7, juin-juillet 2013.
* (201) Rapport d'information au Parlement et au Gouvernement, septembre 2013.
* (202) Rapport d'évaluation de MM. Alain Claeys et Jean-Sébastien Vialatte, députés, la loi n° 2004-800 du 6 août 2004 relative à la bioéthique, AN n° 1325 et Sénat n° 37, janvier 2008.
* (203) In Médecine/Sciences Volume 29, numéro 6-7, juin-juillet 2013.
* (204) Sociologue, directrice de recherche au CNRS, audition des rapporteurs du 15 janvier2013.
* (205) Président du CCNE, audition publique du 25 juin 2013.
* (206) Note du centre d'analyses stratégiques n° 289 d'octobre 2012 : « Médecine prédictive : les balbutiements d'un concept aux enjeux considérables ».
* (207) http://www.coe.int/t/dg3/healthbioethic/Source/Final%20F%20consult%20doc.pd.
* (208) Biologie à haut débit et organisation de la recherche, une économie des données ? Médecine/Sciences, décembre 2012, Hors-Série n° 2 Volume 28.
* (209) Gymrek M, McGuire AL, Golan D, Halperin E, Erlich Y. Identifying personal genomes by surname inference. Science. 2013 Jan 18 ; 339(6117):321-4).
* (210) Coordonnatrice du pôle santé de la CNIL, audition des rapporteurs du 12 mars 2013.
* (211) Voir l'audition publique organisée au nom de l'OPECST par M. Pierre Lasbordes, député, le 30 avril 2009 : « Le DMP : quel bilan d'étape pour quelles perspectives ? » http://www.assemblee-nationale.fr/13/dossiers/dmp_bilan.asp.
* (212) ( ) Adjointe au directeur général de la santé et chef des politiques de santé, audition des rapporteurs du 7 janvier 2014.
* (213) Directeur de recherche à l'INSERM à l'Institut de génétique et de biologie moléculaire (IGBMC), membre du Conseil scientifique de l'OPECST, audition des rapporteurs du 17 juillet 2013.
* (214) Directeur de recherche à l'INSERM à l'Institut de génétique et de biologie moléculaire (IGBMC), membre du Conseil scientifique de l'OPECST-Audition des rapporteurs du 17 juillet 2013.
* (215) Directeur de recherche à l'Inserm à l'Institut de génétique et de biologie moléculaire (IGBMC) et membre du conseil scientifique de l'OPECST- Audition des rapporteurs du 17 juillet 2013.
* (216) Audition publique du25 juin 2013
* (217) Conseiller à la cour d'appel de Paris, secrétaire général de l'Association internationale droit, éthique et science, audition des rapporteurs du 28 mai 2013.
* (218) Président du comité d'éthique de l'INSERM, audition publique du 25 juin 2013 précitée.
* (219) Directeur de l'Institut de Biologie et technologies du CEA (iBiTec-S), audition des rapporteurs du 6 février 2013.
* (220) Directrice de l'accès au marché et des affaires publique Amgen, audition des rapporteurs du 20 février 2013.
* (221) Chargée de mission au Commissariat général à la stratégie et à la prospective (CGSP), audition des rapporteurs du 20 novembre 2013.
* (222) Chargée de recherche en génétique humaine et statistiques à l'INSERM, audition publique du 25 juin 2013.
* (223) Global Alliance for genomic and clinical data sharing.
* (224) Directeur du Centre de génotypage au Commissariat à l'énergie atomique (CEA), audition publique du 25 juin 2013.
* (225) Président du CCNE, audition publique du 25 juin 2013.
* (226) Directeur de recherche à l'INSERM, membre de l'Académie des sciences et du Conseil scientifique de l'OPECST, audition des rapporteurs du 28 novembre 2012.
* (227) Directrice générale de l'Agence de la biomédecine, audition publique du 25 juin 2013.
* (228) Présidente de l'INCa, audition publique du 25 juin 2013.
* (229) Secrétaire générale de l'Union des parents d'enfants atteints de cancer et de leucémie (UNAPECLE), audition publique du 25 juin 2013.
* (230) Conseiller d'État, responsable de la Chaire santé publique à l'IEP, audition publique du 25 juin 2013.
* (231) http://www.assemblee-nationale.fr/13/dossiers/innovation_epreuve_peurs_risques.asp .
* (232) Conseiller d'État, responsable de la Chaire de santé publique à l'IEP, audition des rapporteurs du 19 décembre 2012.
* (233) Responsable du département de génétique du groupe hospitalier de la Pitié-Salpêtrière, audition publique du 25 juin 2013.
* (234) Cancérologue, directeur honoraire de Gustave Roussy, audition publique du 25 juin 2013.
* (235) Union des parents d'enfants atteints de cancer et de leucémie, audition publique du 25 juin 2013.
* (236) Présidente de l'AFM-Téléthon Association française contre les myopathies, audition publique du 25 juin 2013.
* (237) Organisation européenne des maladies rares, audition publique du 25 juin 2013.
* (238) Comité européen d'experts sur les maladies rares.
* (239) Chercheur en sciences sociales, présidente du GRAM de l'INSERM, Groupe de réflexion avec les associations de malades, audition publique du 25 juin 2013.
* ( 240 ) MM. Alain Claeys et Jean-Sébastien Vialatte, rapport AN n°1325 - Sénat n°107 sur l'évaluation de la loi de bioéthique du 6 aout 2004 décembre 2008.
* ( 241 ) MM. Alain Claeys et Jean-Sébastien Vialatte, rapport AN n° 4469 - Sénat n° 476 sur l'impact impact et les enjeux des nouvelles technologies d'exploration et de thérapie du cerveau de mars 2012.
* 242 Association for Molecular Biology, et al. v. United States Patent and Trademark Office, et al . (U.S. District Court - Southern District of New York, 29 mars 2010)
* 243 Mayo Collaborative Services v. Prometheus Laboratories, Inc. (U.S. Supreme Court, 20 mars 2012)
* 244 Association for Molecular Biology, et al. v. United States Patent and Trademark Office, et al. (U.S. Court of Appeals for the Federal Circuit, 16 août 2012)
* 245 Voir tableau ci-après.
* ( 246 ) Gymrek M, McGuire AL, Golan D, Halperin E, Erlich Y. Identifying personal genomes by surname inference. Science, 2013 Jan 18;339(6117):321-4).
* ( 247 ) VAN EL CG, et al. Whole-genome sequencing in health care. Eur J Hum Genet . 2013 June;21(6):580-4.
* ( 248 ) Voir J. Kahn, « How a drug becomes «Ethnic»: Law, Commerce, and the Production of Racial Categories in Medicine », Yale Journal of Health Policy, Law & Ethics, n° IV, 2004, p. 13.
* ( 249 ) A. Aboukrat et Ch. Noiville, « Les médicaments «raciaux» ou «ethniques» : manoeuvre commerciale ou enjeu de santé publique ? » dans Les catégories ethno-raciales à l'ère des biotechnologies - Droit, sciences et médecine face à la diversité humaine , Société de législation comparée, UMR de droit comparé de Paris, Vol. n°24 (2011), pp. 55s.
* ( 250 ) Statistiques de 2004 - voir H. J. Wellens, « Tailoring Heart Failure Therapy », New England Journal of Medicine (11 novembre 2004), p. 351.
* ( 251 ) Agency for Healthcare Research and Quality, U.S. Department of Health and Human Services, « Pharmacologic Management of Heart Failure and Left Ventricular Systolic Dysfunction: Effect in Female, Black, and Diabetic Patients, and Cost-Effectiveness », Evidence Report/Technology Assessment n° 82 (juillet 2003).
* ( 252 ) Voir U.S. Patent n° 4 868 179 (19 septembre 1989), en particulier la première revendication.
* ( 253 ) La nouvelle demande de brevet revendiquait en effet « une méthode de réduction de la mortalité associée à l'insuffisance cardiaque, pour améliorer la consommation d'oxygène, pour améliorer la qualité de vie ou pour améliorer la tolérance à l'exercice chez un patient noir comprenant l'administration à ce dernier d'une quantité efficace au plan thérapeutique d'au moins un composé d'Hydralazine (...), et d'au moins un composé d'Isosorbide dinitrate et d'Isosorbide mononitrate » - voir la revendication n° 1 du brevet U.S. Patent n° 6, 465, 463 (délivré le 15 octobre 2002). Voir J. KAHN, « Beyond BiDil: The expanding embrace of race in biomedical research and product development », Saint Louis University Journal of Health Law & Policy, Vol. 3 (2009).
* ( 254 ) U.S. Patent n° 6, 465, 463 (délivré le 15 octobre 2002).
*
(
255
) U.S. Food and
Drug Administration, « FDA Approves BiDil Heart Failure for Black
Patients » (23 juin 2005) :
http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2005/ucm1
08445.htm.
* ( 256 ) J. Kahn, « Beyond BiDil », art. cit. , p. 61.
*
(
257
) FDA,
« Guidance for Industry: Collection of Race and Ethnicity Data in
Clinical Trials » (septembre 2005), p. 3 - note n° 8 :
http://www.fda.gov/downloads/RegulatoryInformation/Guidances/ucm
126396.pdf.
* ( 258 ) Id. , pp. 4-5.
* ( 259 ) Voir et comparer : Id. , p. 5 et European Medicines Agency « Assessment Report for Rebetol » (20 janvier 2010), p. 5, http://www.ema.europa.eu/humandocs/PDFs/EPAR/Rebetol/Rebeto l-H-C-246-II-48-AR.pdf.
* ( 260 ) Brevet européen EP785216 (déposé le 17 novembre 1998) : le brevet portait sur des méthodes et matériel utilisés pour isoler et détecter un cancer du sein chez l'humain prédisposant le gène BRCA2 ainsi que certains allèles mutants qui causent la susceptibilité au cancer, au cancer du sein en particulier.
* ( 261 ) Brevet européen EP785216 (déposé le 17 novembre 1998) : le brevet portait sur des méthodes et matériel utilisés pour isoler et détecter un cancer du sein chez l'humain prédisposant le gène BRCA2 ainsi que certains allèles mutants qui causent la susceptibilité au cancer, au cancer du sein en particulier.
* ( 262 ) « Cancer Research UK ».
* ( 263 ) G.B. Patent Application No. 9523959 (délivré le 23 novembre 1995) et G.B. Patent Application n° 9525555 (délivré le 14 décembre 1995).
* ( 264 ) U.S. Patent n° 5, 837, 492 (demande de brevet déposée le 29 avril 1996).
* ( 265 ) Brevet européen EP0858467 (délivré le 11 février 2004).
* ( 266 ) Voir la description et les revendications du brevet européen EP0858467 sur le site Internet de l'Office européen des brevets : http://v3.espacenet.com/publicationDetails/description?CC=EP&NR=0858467B1&KC=B1&FT=D&date=20040211&DB=EPODOC&locale=en_EP
* ( 267 ) OEB, Division d'opposition, décision du 12 septembre 2005 relative au brevet EP-B1-0 785 216.
* ( 268 ) Voir OEB, Motifs de la décision (Annexe) - Opposition (12 septembre 2005) : https://register.epo.org/espacenet/application?lng=fr&number=EP96309211&tab=doclist
* ( 269 ) W. Nicholson Price II, « Patenting race: the problems of ethnic genetic testing patents », The Columbia Science and Technology Law Review, Vol. VIII (2007) p.128.
* ( 270 ) S. Steimle, « Critics question BRCA2 patent decision in Europe », Journal of The National Cancer Institute, p. 97 (2005).
* ( 271 ) Lettre de F. Faivre Petit & J. Warcoin à l'Office européen des brevets (29 avril 2005), https://register.epo.org/espacenet/advancedSearch?lng=fr (entrer le numéro de publication « EP785216 » ; suivre ensuite le lien « Tous les documents », puis le lien « Lettre relative à la procédure d'opposition »).
* ( 272 ) Voir en ce sens J. Kahn, «Race-ing Patents/Patenting Race: An Emerging Political Geography of Intellectual Property in Biotechnology», 92 Iowa Law Review (2007), p. 382.
* ( 273 ) Voir A. Aboukrat et Ch. Noiville, art. cit.
* ( 274 ) J. Kahn, « Beyond BiDil », art. cit. , p. 76.
* ( 275 ) Voir « CRESTOR (rosuvastatine) et toxicité musculaire : renforcement des précautions d'emploi » : http://www.afssaps.fr/Infos-de-securite/Communiques-Points-presse/CRESTOR-R-rosuvastatine-et-toxicite-musculaire-renforcement-des-precautions-d-emploi/(language)/fre-FR
* ( 276 ) Voir S. A. Tishkoff & K. K. Kidd, « Implications of biogeography of human populations for «race» and medicine », Nature Genetics Supplement, Vol. 36, n° 11 (novembre 2004).
* ( 277 ) En l'espèce, les chambres de recours se prononçaient non sur la brevetabilité d'une nouvelle utilité trouvée à un médicament existant, mais sur une utilité accrue d'un médicament pour un groupe de patients donné. Voir les décisions T_19/86, T_893/90 et T_233/96. www.epo.org. Voir A. Aboukrat et Ch. Noiville, art. cit.
* ( 278 ) Décision T_19/86 ; décision T_893/90 ; décision T_233/96.
* ( 279 ) Projet de loi de financement de la sécurité sociale.