







TRAITER ET ACCOMPAGNER LES MALADES : LA DIVERSITÉ DES APPROCHES
M. Jean-Louis Touraine. Je donne maintenant la parole au professeur Philippe Leboulch.
M. Philippe Leboulch, professeur des universités à la faculté de Paris-XI, praticien hospitalier, chef de l'Institut des maladies émergentes et des thérapies innovantes (IMETI) de la direction des sciences du vivant du CEA et d'une unité mixte de recherche INSERM-CEA . Je vais essayer de faire devant vous le point sur les thérapies géniques de ces deux maladies-soeurs que sont la â-thalassémie et la drépanocytose. Ces deux hémoglobinopathies, relativement fréquentes de par le monde, présentent l'intérêt de pouvoir constituer d'intéressants modèles pour la thérapie génique.
Ces maladies sont faussement simples. Bien qu'il s'agisse des premières dans l'histoire de la médecine dont le mécanisme a été identifié au niveau moléculaire, on ne sait pas pour autant les traiter de manière efficace et durable. Beaucoup de temps a été nécessaire pour accomplir les premiers progrès. Ceux-ci s'accélèrent désormais.
Dans notre sang circulent à chaque instant plus de vingt mille milliards de globules rouges. Il n'est pas simple de les traiter quand ils sont, comme dans le cas des hémoglobinopathies, déformés. Si on parvenait à traiter ces maladies par thérapie génique, il y aurait des retombées non seulement pour d'autres maladies rares mais pour l'ensemble de la médecine et de la biologie. L'essai clinique que nous avons mené a permis de mieux comprendre comment les cellules souches hématopoïétiques donnaient naissance à l'ensemble des éléments figurés du sang et conduit à remettre en question les modèles simples qui nous étaient enseignés en faculté de médecine.
La drépanocytose et la â-thalassémie sont surtout répandues en Afrique sub-saharienne et en Asie, sur toute la bande équatoriale et sub-tropicale. Cela tient au fait que le parasite du paludisme, transmis par un moustique, a besoin d'une hémoglobine normale pour pouvoir se reproduire. Les individus hétérozygotes, porteurs sains du gène, se trouvent être résistants aux formes sévères du paludisme, ce qui explique que cette particularité génétique se soit amplifiée dans certaines zones d'Afrique jusqu'à être présente chez 80%, voire 90%, de la population. Les mouvements migratoires expliquent l'essaimage de ces maladies aux Antilles, en Amérique du Nord et en Europe.
On dénombrerait environ 500 000 nouveau-nés par an dans le monde atteints de formes sévères d'hémoglobinopathies. Il n'y en a quasiment pas en France atteints de â-thalassémie grâce à l'efficacité du diagnostic prénatal, mais en Thaïlande, pays de population comparable à la nôtre, on compte environ 3 000 nouveaux cas par an. Les patients aujourd'hui atteints en métropole sont originaires d'Afrique du Nord, du Bassin méditerranéen et d'Asie. On compte en revanche dans notre pays, DOM-TOM inclus, 15 000 patients atteints de drépanocytose, essentiellement antillais ou africains, et ce nombre devrait, avec environ 400 nouvelles naissances d'enfants atteints par an, passer à 20 000 vers 2020. En l'absence de traitement, ces maladies sont mortelles. Il n'existe, hélas, presque que des traitements palliatifs, le seul traitement curatif résidant dans une greffe de moelle osseuse avec donneur compatible.
Ces maladies sont d'origine génétique. Chez les individus malades, chacun des deux chromosomes codant pour la â-globine présente une ou plusieurs mutations - diverses dans le cas de la â-thalassémie, toujours la même dans le cas de la drépanocytose - empêchant le fonctionnement normal des globules rouges. Lorsque chacun des parents possède sur l'un des deux chromosomes concernés le gène muté, si l'enfant n'hérite d'aucun des chromosomes porteurs de la mutation, il est sain et ne risque pas de transmettre la maladie. S'il hérite d'un seul chromosome atteint, celui de son père ou celui de sa mère, il est sain également mais, porteur de la mutation, il peut transmettre la maladie - le seul avantage pour lui est que cela le protège des formes sévères de paludisme. Si par malchance, il hérite de chacun de ses parents du chromosome porteur du gène muté, il est atteint.
La â-globine est l'un des principaux constituants protéiques du tétramère d'hémoglobine, lequel comporte deux molécules de â-globine, une molécule d'á-globine et une molécule de fer. Les globules rouges ne contiennent quasiment rien d'autre que de l'hémoglobine, qui assure tous les échanges gazeux depuis les poumons jusque dans les tissus.
Chez les malades atteints de â-thalassémie, la production de â-globine est insuffisante, que le gène codant pour cette protéine présente une mutation qui l'empêche de s'exprimer ou ait disparu par délétion. En l'absence de traitement, les individus homozygotes, qui ne peuvent fabriquer de globules rouges, meurent dans la petite enfance, alors même qu'ils sont nés normaux. En effet, le foetus est protégé pendant la vie intra-utérine grâce au gène foetal de la globine, dit ã, lequel cesse de s'exprimer dans les deux ou trois premières années de la vie.
Chez les malades atteints de drépanocytose, la production de â-globine est suffisante mais la protéine fabriquée est anormale. Elle polymérise dans les globules rouges qu'elle déforme - ils prennent la forme d'une faucille - et rigidifie, si bien qu'ils adhèrent à la paroi des vaisseaux qui s'obstruent, provoquant d'atroces douleurs, puis des micro-infarctus dans les différents tissus de l'organisme.
Dans la â-thalassémie majeure, on observe un retard de croissance, des problèmes endocriniens, une hypertension pulmonaire, des thromboses veineuses, des atteintes cardiaques, une cirrhose, un diabète, une arthropathie, des déformations osseuses, une hépatosplénomégalie... Bref, tous les organes sont touchés. Non traitée, la maladie entraîne le décès avant l'âge de deux ans. Mal traitée, ce qui est le cas dans beaucoup de pays moins favorisés que le nôtre, elle provoque tous ces symptômes. Et même lorsqu'elle est traitée de la meilleure manière, cela n'empêche pas un décès prématuré, en général entre 30 et 40 ans.
Dans la drépanocytose, les crises vaso-occlusives et hémolytiques détruisent peu à peu les tissus, ce qui, dans les formes sévères, peut raccourcir de beaucoup l'espérance de vie.
Le seul traitement curatif actuellement disponible consiste en une greffe de moelle prélevée chez un donneur compatible, membre de la fratrie possédant les mêmes caractéristiques HLA d'histo-compatibilité. Le taux de succès peut atteindre 80% à long terme si le patient greffé ne développe pas une maladie du greffon contre l'hôte (GVHD), toujours possible même avec un donneur dit compatible. C'est d'ailleurs pourquoi on ne sollicite quasiment jamais de donneurs extra-familiaux, même compatibles. Outre que 75% des patients n'ont pas de donneur compatible, il faut compter avec le risque de réaction du greffon contre l'hôte. Ce traitement est donc loin d'être la panacée.
D'où l'intérêt de la thérapie génique. On prélève des cellules des patients, on y corrige le gène déficient, puis on les leur réinjecte une fois la correction effectuée. Dans la mesure où il s'agit d'une greffe autologue, il n'y a pas de risque de rejet. On réserve pour l'instant cette thérapie, encore balbutiante, aux cas les plus graves et où le patient n'a pas de donneur compatible. Si les succès se confirment, elle pourra être étendue.
On pourrait imaginer de corriger in situ le gène codant de façon erronée pour la â-globine dans les cellules souches hématopoïétiques de la moelle osseuse, capables d'auto-renouvellement. Mais ces cellules sont très difficiles à isoler, à manipuler et à conserver sans qu'elles perdent leurs propriétés spécifiques de cellules souches. Il faudrait en outre s'assurer que le nouveau gène ne s'exprimera bien ultérieurement que dans les globules rouges. On arrive aujourd'hui à corriger in vitro le gène déficient, pas encore in vivo.
L'autre méthode, celle utilisée aujourd'hui, consiste à introduire, grâce à un vecteur, un gène dans les cellules souches hématopoïétiques destiné à s'exprimer dans les globules rouges. Des essais ont déjà eu lieu chez l'animal. J'en profite pour dire que ce serait une folie que de prétendre se passer des expérimentations sur l'animal : le modèle animal demeure indispensable. Les vecteurs qui marchent chez l'animal pour les cellules souches hématopoïétiques sont les lentivirus. Tout l'art de la vectorologie est de ne conserver du rétrovirus que les parties mobilisant le gène jusqu'au chromosome.
Il nous faut mieux comprendre le fonctionnement des cellules souches hématopoïétiques comme des globules rouges pour pouvoir les modifier, savoir comment y insérer un gène, comprendre comment la moelle « corrigée » pourra se substituer à la moelle d'origine, savoir comment fabriquer les gènes-médicaments avec toute la sécurité nécessaire et en juste quantité. Une souris est environ trois mille fois plus petite qu'un homme. Ce qu'on réussit chez elle, il faut être capable de le reproduire en trois mille fois plus grand chez l'homme ! C'est ainsi que pour un seul patient on produit de vingt à quarante litres de vecteur, ensuite concentrés. Les premières souris traitées avec succès par thérapie génique pour des hémoglobinopathies l'ont été en 2000-2001 et le premier patient en 2007. En réalité, contrairement à ce que l'on entend parfois dire, c'est allé très vite pour faire tous les essais nécessaires, prendre toutes les garanties en matière de toxicité, obtenir l'aval des agences sanitaires et fabriquer en grande quantité les vecteurs dans des conditions GMP ( Good manufacturing process) .
Comment cela se passe-t-il en pratique ? On prélève des cellules de moelle osseuse, soit par voie intra-veineuse soit par ponction de l'os sous anesthésie locale. On les mélange pendant quelques heures avec le vecteur, auparavant préparé, congelé puis décongelé. On congèle les cellules puis on les analyse pour s'assurer que tout est normal. Le patient revient plus tard à l'hôpital où lui est administrée une chimiothérapie, beaucoup moins lourde que celle normalement pratiquée lors d'une greffe de moelle, dans la mesure où on n'a pas besoin de déprimer son immunité. Et on lui réinjecte ses propres cellules qui ont été mêlées auparavant au vecteur.
M. le président Claude Birraux. Il faut préparer trente à quarante litres de vecteur pour un seul patient. Mais combien lui en injecte-t-on ?
M. Philippe Leboulch. Quelques dizaines de millilitres seulement. On concentre la préparation jusqu'à n'obtenir plus que cette quantité qu'on mélange avec les cellules.
C'est en juin 2007 que nous avons traité de la sorte un premier patient, après avoir obtenu l'aval de l'Agence française de sécurité sanitaire des produits de santé (AFSSAPS). Il était atteint d'une thalassémie majeure de type â E /â 0 , devait être transfusé tous les mois depuis l'âge de trois ans et prendre des chélateurs du fer pour éliminer la surcharge en fer, inéluctable lorsqu'on est obligé d'être transfusé à un rythme aussi rapproché - jusqu'à l'apparition de formes orales beaucoup mieux tolérées de chélateurs du fer ; ce patient, depuis l'enfance, avait une perfusion avec une pompe branchée dans son abdomen cinq nuits par semaine. Il était arrivé que son taux d'hémoglobine tombe avant transfusion à 4,5 g/dl. Il souffrait d'une hépatomégalie et d'une splénomégalie qui avait nécessité l'ablation de la rate à l'âge de six ans. Les tentatives de traitement à l'hydroxyurée avaient échoué et il n'avait pas de donneur intra-familial compatible qui aurait permis une greffe de moelle. Quatre ans après cette thérapie génique, il n'est pas guéri mais son état s'est très nettement amélioré. Depuis trois ans, il n'a pas eu besoin d'une seule transfusion et son taux d'hémoglobine oscille entre 9 et 10 g/dl. Il travaille à plein temps. Et pour éliminer le fer qui s'est accumulé dans son organisme au cours des années antérieures, il suffit de lui faire régulièrement des saignées, ce qui est beaucoup moins lourd que d'administrer des chélateurs. Ce patient reste suivi de près mais le bénéfice de la thérapie génique a été pour lui considérable.
En conclusion, je voudrais insister sur la nécessité que les politiques et les associations de patients continuent de nous aider.
Même si tout est loin d'être parfait en France et que du retard a été pris dans le domaine de la génomique, notamment par rapport aux Etats-Unis et à la Chine qui ont réalisé des investissements colossaux, notre pays s'est donné les moyens d'être leader en matière de thérapie génique et le demeure aujourd'hui - je pense aux travaux d'Alain Fischer à l'hôpital Necker ou bien encore à ceux de José Sahel sur les pathologies rétiniennes à l'hôpital des Quinze-Vingts. Ces recherches ont besoin de financements pérennes.
On ne peut pas non plus se passer des modèles animaux, y compris avec les grands singes, les seuls vraiment pertinents avant de passer à l'essai clinique chez l'homme.
Enfin, l'AFSSAPS, sans se départir de son indispensable regard critique, doit comme elle l'a fait pour les premiers travaux de thérapie cellulaire et de thérapie génique, accompagner la phase ultérieure du développement de médicaments géniques pour les patients auprès de l'EMA ( European Medicine Agency) .
Il faut aussi nouer des partenariats industriels car il n'est pas envisageable de développer autrement ce type de thérapie jusqu'au stade du médicament. Les petites sociétés de biotechnologies sont le lieu idéal de développement des initiatives aux tous débuts. Les idées, les projets y sont foisonnants mais il faut aider ces sociétés à conclure des partenariats avec les grands laboratoires pharmaceutiques. On a évoqué le rachat de Genzyme par Sanofi. Il faut favoriser ce type d'alliances.
Il pourrait, à première vue, sembler que les traitements classiques de la drépanocytose coûtent moins cher que des thérapies plus innovantes. Il n'en est rien car les malades doivent être soignés durant toute leur vie. Ces thérapies ne sont donc pas réservées aux pays riches. Un malade atteint de la maladie de Gaucher, qui est une mucopolysaccharidose, coûte aux Etats-Unis - le coût est voisin en France - de 250 000 à 800 000 dollars par an pour la seule protéine recombinante qui devra lui être administrée sa vie durant. Si cette maladie pouvait être traitée par thérapie génique, sans effets secondaires majeurs, cela reviendrait au final bien meilleur marché car, même coûteux, le traitement n'aurait à être administré qu'une fois. Si le développement des thérapies géniques a un coût élevé, leur utilisation permettrait ensuite des économies considérables par rapport aux médicaments classiques. En Thaïlande, on dénombre chaque année 3 000 nouveaux cas de â-thalassémie. Depuis plusieurs années, le gouvernement thaïlandais a décidé de prendre en charge le coût des greffes de moelle allogéniques chez les enfants atteints, pour la raison simple que cette technique est au final financièrement plus avantageuse que des transfusions et l'administration de chélateurs du fer à vie.
M. Jean-Louis Touraine. Je donne maintenant la parole à Mme Cécile Martinat qui dirige à I-STEM l'équipe qui travaille sur les maladies du motoneurone.
Mme Cécile Martinat, chargée de recherche à l'INSERM et à l'Institut des cellules souches pour le traitement et l'étude des maladies monogéniques (I-STEM). Je me réjouis de pouvoir vous présenter aujourd'hui une partie de nos travaux sur un thème débattu récemment à l'occasion de la révision des lois de bioéthique, à savoir le potentiel des cellules souches embryonnaires humaines et des cellules pluripotentes induites (iPS) dans le cadre de maladies rares. Je profite de l'occasion pour dire combien je regrette que la législation française n'ait pas évolué vers une autorisation encadrée des recherches sur les cellules souches embryonnaires. Je m'inquiète de ce que pourra être la position de la France sur le plan international dans ce domaine de recherche.
Je vais vous présenter les avancées qui ont pu être obtenues à la suite de travaux menés à I-Stem sur les cellules souches embryonnaires humaines pour la modélisation pathologique de maladies monogéniques et comment ces nouveaux modèles ont pu être utilisés pour identifier de nouvelles thérapeutiques.
Les lignées de cellules souches embryonnaires sont dérivées d'une cellule extraite de la masse interne du blastocyte, aux tous premiers stades du développement embryonnaire -les embryons utilisés sont les embryons surnuméraires des fécondations in vitro . Les cellules pluripotentes induites (iPS), elles, s'obtiennent à partir d'une cellule somatique adulte dédifférenciée puis reprogrammée pour retrouver des propriétés proches de celles d'une cellule souche embryonnaire.
Ces deux types de cellules ont la capacité de s'autorenouveler : elles constituent donc en théorie une source illimitée de matériel de recherche. Elles ont également la capacité de se différencier en tous les types cellulaires de l'organisme - neurones, cellules du muscle, de la peau... Leur premier champ d'application est de remplacer les cellules détruites ou endommagées dans le cas de maladies dégénératives. Une autre application, qui est celle dont je traiterai aujourd'hui, découle de la possibilité d'obtenir des lignées de cellules souches embryonnaires ou de cellules iPS portant la mutation causale d'une maladie monogénique. Il y a deux moyens d'y parvenir. Le diagnostic pré-implantatoire permet d'identifier les embryons porteurs du gène et d'en dériver des lignées de cellules souches embryonnaires porteuses de l'anomalie. Avec les iPS, il est possible de reprogrammer des cellules somatiques adultes de façon qu'elles contiennent le gène en question.
Pourquoi créer de nouveaux modèles ? Tout d'abord, parce que pour mettre au point des traitements adaptés, il faut mieux connaître les mécanismes de certaines pathologies. Dans le cas des maladies monogéniques, il faut comprendre comment la mutation aboutit au développement de la maladie. Ensuite, les modèles animaux utilisés jusqu'à présent, qu'il s'agisse de la souris, de la drosophile ou du ver, présentent l'inconvénient d'une restriction liée à l'espèce. Quant aux modèles cellulaires, qu'il est possible d'élaborer à partir de biopsies humaines, ils présentent eux aussi des difficultés. Il est parfois difficile d'effectuer les prélèvements nécessaires, notamment dans le cas de maladies neurologiques. Il l'est également de maintenir en culture les cellules prélevées. Et je ne m'étendrai pas sur la difficulté intrinsèque d'utiliser des cellules immortalisées par génie génétique, les propriétés de ces cellules se trouvant modifiées du fait de cette immortalité même. Tout cela explique qu'il soit nécessaire de disposer de nouveaux modèles plus pertinents.
Quel est le principe de la modélisation pathologique à partir de cellules souches embryonnaires humaines prélevées au cours d'un DPI ? Notre stratégie a été de faire se différencier ces cellules en progénies d'intérêt pour les pathologies auxquelles nous nous intéressions, de démontrer la relevance pathologique en recherchant la présence de stigma caractéristiques de ces maladies et de voir si, forts de ces résultats, nous pouvions utiliser ces modèles à la fois pour mieux comprendre les mécanismes pathologiques jusqu'au niveau moléculaire et identifier de nouveaux composés potentiellement thérapeutiques. Nous avons travaillé en laboratoire sur des cellules souches embryonnaires humaines portant la mutation causale de la myotonie dystrophique de type I (MD1), maladie neuro-musculaire d'origine génétique.
Nous avons d'abord différencié ces cellules en précurseurs mésenchymateux et recherché la présence des signes caractéristiques de la MD1 : agrégation de transcrits mutés au niveau du noyau, défaut d'expression de l'une des isoformes du gène codant pour le récepteur à l'insuline... Nous nous sommes ensuite demandés si nous pouvions utiliser ces modèles pour mieux comprendre les mécanismes de la maladie et faire du criblage thérapeutique.
Nous avons recherché tous les gènes qui s'exprimaient de manière différente dans les cellules malades et les cellules témoins. Grâce à la transcriptomique, nous avons pu en identifier toute une liste. Deux nous ont paru particulièrement intéressants, qui appartiennent à la famille des SLITRK, connue pour coder des protéines impliquées dans la communication entre neurones et myocytes. La mauvaise communication entre ces deux types de cellules est à l'origine même du syndrome myotonique associée à la DM1.
Exploitant au maximum le potentiel de pluripotence des cellules souches embryonnaires, nous les avons différenciées en neurones de la moelle épinière et avons étudié la capacité des cellules malades à interagir avec des cellules musculaires. Nous avons ainsi pu montrer que le défaut d'expression des gènes SLITRK résultait d'un défaut de communication entre les neurones et leurs cibles musculaires. Ce travail, qui a fait l'objet d'une publication récente dans une revue internationale, a apporté la preuve que des cellules souches embryonnaires humaines porteuses de la mutation causale d'une maladie monogénique pouvaient servir à identifier de nouveaux mécanismes pathologiques.
Nous avons également utilisé les cellules souches embryonnaires humaines pour identifier de nouvelles molécules thérapeutiques. Nous avons développé une plate-forme de criblage thérapeutique pour identifier des composés capables de faire disparaître les agrégats nucléaires et améliorer l'expression du récepteur à l'insuline. Une molécule est actuellement en phase d'étude pré-clinique.
Alors que depuis 1998, une trentaine de pathologies ont pu être « capturées » grâce à des cellules souches embryonnaires humaines obtenues à partir de DPI - les limites tiennent d'une part à la pratique du DPI elle-même, peu de maladies encore en faisant l'objet, d'autre part à la difficulté de dériver des cellules souches embryonnaires à partir des embryons recueillis -, depuis 2007 et la découverte des iPS par le professeur Yamanaka au Japon, plus d'une vingtaine l'ont été avec ces iPS.
Mais des problèmes sont apparus, liés au processus même de la reprogrammation. On force des cellules somatiques adultes à retrouver les propriétés de cellules embryonnaires en leur faisant surexprimer des gènes normalement impliqués dans le contrôle de l'état embryonnaire. On utilise pour cela différents vecteurs, notamment lentiviraux. Plusieurs études ont montré que cette reprogrammation induisait dans les iPS un grand nombre de défauts, notamment dans le contrôle de l'ADN - certaines régions du génome sont, à l'état embryonnaire, éteintes, mais deviennent actives lorsqu'on pousse la cellule à se différencier. Une cellule iPS n'a plus de zones éteintes et garde la mémoire de la cellule somatique adulte dont elle est issue. Une autre conséquence de la reprogrammation est l'apparition de nombreuses mutations dans les chromosomes. Environ 126 ont été identifiées, sur lesquelles 50 affectent des gènes potentiellement impliqués dans la survenue de cancers.
Après la parution d'articles sur le sujet en mars 2011, les mises en garde de la communauté scientifique internationale se sont multipliées sur les dangers de l'utilisation des iPS, invitant en tout cas à la précaution.
Pour terminer, je dresserai un tableau comparatif des avantages et inconvénients respectifs des cellules souches embryonnaires humaines et des cellules iPS.
Ces deux types de cellules présentent l'intérêt de permettre de dériver des lignées à grande échelle, de disposer de phénotypes très divers, et de pouvoir être cultivées de façon standardisée.
Tous deux présentent l'inconvénient d'une certaine instabilité génomique en culture. Se posent également la question de la pertinence du modèle - est-il pertinent d'utiliser des cellules souches embryonnaires pour modéliser par exemple des pathologies dégénératives survenant à l'âge adulte ? - et la difficulté de modéliser des maladies multifactorielles.
Les iPS offrent l'avantage que la reprogrammation permet de choisir les génotypes afin de répliquer exactement les maladies génétiques, dans leur extrême diversité. Elles présentent en revanche l'inconvénient que la reprogrammation entraîne des altérations de l'ADN. Il semble aussi maintenant qu'elles n'aient pas tout à fait la même pluripotence, et donc les mêmes capacités de différenciation que les cellules souches embryonnaires.
Un dernier mot sur I-Stem, premier institut français à avoir obtenu les autorisations nécessaires pour travailler sur des cellules souches embryonnaires humaines. I-Stem a été créé par Marc Peschanski en 2005, sous l'impulsion de l'INSERM, de l'université d'Evry et de l'AFM, l'Association française contre les myopathies. Alors qu'il ne comptait que quinze personnes à son lancement, I-STEM, qui a été labellisé en 2007 et reconnu à la fois par l'INSERM et par l'AFM comme bras armé, compte aujourd'hui 90 chercheurs répartis en onze équipes, dont le seul objectif est d'explorer le potentiel offert par les cellules souches embryonnaires humaines, notamment dans l'espoir de trouver des traitements pour les maladies monogéniques.
M. Jean-Louis Touraine. Je donne enfin la parole au professeur Soumeya Bekri, qui a notamment travaillé sur le traitement enzymatique de la maladie de Fabry.
Mme Soumeya Bekri, professeur des universités, praticien hospitalier, responsable du dépistage et du suivi des maladies métaboliques au CHU de Rouen. J'ai le plaisir de vous présenter aujourd'hui de nouvelles thérapies de maladies monogéniques, notamment celles dues à un déficit enzymatique. Je me focaliserai sur les maladies lysosomales de surcharge. C'est un groupe hétérogène qui comprend plus de cinquante maladies, dont la prévalence combinée est de un sur cinq mille naissances. Elles se caractérisent toutes par un défaut de catabolisme ou de transport d'un substrat.
Leur caractéristique commune est l'accumulation progressive de différentes macromolécules dans les lysosomes. Ce sont des pathologies graves, entraînant des atteintes multiviscérales - hépatosplénomégalie, problèmes osseux et souvent une atteinte neurologique... Ces manifestations cliniques ne sont pas immédiates, n'apparaissant qu'après accumulation suffisante du substrat non dégradé, mais elles sont alors inexorables. La morbi-mortalité est très élevée. Il est important de pouvoir porter un diagnostic devant des signes cliniques évocateurs, non nécessairement spécifiques, car il existe des traitements pour certaines de ces maladies.
Les maladies lysosomales de surcharge les plus connues et les plus fréquentes sont la maladie de Gaucher et la maladie de Fabry.
On dénombre un cas de maladie de Gaucher sur 50 000 naissances. Cette maladie, de transmission autosomique récessive, entraîne des atteintes osseuses et hématologiques, ainsi qu'une hépatosplénomégalie. Dans les types II et III, on trouve également des atteintes neurologiques. Le type I est connu pour être la forme « non neurologique ».
La maladie de Fabry est une pathologie héréditaire du métabolisme des glycosphingolipides, de transmission récessive liée au chromosome X, et due à un déficit en alpha-galactosidase A, enzyme lysosomale. Ce défaut enzymatique conduit à l'accumulation du substrat non dégradé dans les tissus et le plasma. On dénombre un cas sur 80 000 naissances. Dans sa forme classique, la maladie touche plus sévèrement les hommes hémizygotes. Les femmes hétérozygotes, conductrices de la maladie, présentent souvent des symptômes, mais plus variables et généralement moindres que les hommes. La maladie entraîne des atteintes neurovasculaires, avec notamment des accidents vasculaires cérébraux chez le sujet jeune, des atteintes cardiaques avec une hypertrophie ventriculaire gauche, des atteintes du rein aboutissant à une insuffisance rénale, parfois aussi des atteintes ophtalmologiques et dermatologiques.
Les maladies lysosomales de surcharge ont constitué un modèle pour le développement de nouvelles thérapies ces dernières années. Il est possible de remplacer l'enzyme manquante ou déficiente par une enzyme exogène - c'est le principe de l'enzymothérapie substitutive - ; de limiter la production du substrat non dégradé ; de stabiliser l'enzyme mutante et d'en augmenter l'activité résiduelle grâce à des molécules chaperons.
Le lysosome est un compartiment cellulaire au pH acide, dont la principale fonction est de dégrader des macromolécules intra et extracellulaires. C'est une composante du système endosome/lysosome, impliqué dans l'homéostasie générale de la cellule, et qui permet la communication entre milieu intracellulaire et milieu extracellulaire.
Pour comprendre le principe de l'enzymothérapie substitutive, il faut revenir brièvement sur la synthèse et l'adressage des enzymes lysosomales. La plupart d'entre elles sont synthétisées au niveau du réticulum endoplasmique puis migrent vers l'appareil de Golgi, où elles subissent des modifications dites post-traductionnelles qui leur permettent ensuite de fixer le résidu que constitue le mannose-6-phosphate. L'ensemble constitué du mannose-6-phosphate, de son récepteur spécifique et de l'enzyme est adressé au compartiment cellulaire de l'endosome tardif, lequel, en s'acidifiant, devient un lysosome. L'enzyme ainsi adressée est active. Les récepteurs mannose-6-phosphate, eux, sont recyclés soit vers l'appareil de Golgi pour recruter de nouvelles enzymes, soit vers la membrane plasmique.
Dans le cas d'une maladie lysosomale, l'enzyme soit n'est pas synthétisée soit est mal adressée, en tout cas elle n'est pas assez active dans le lysosome. Dans l'enzymothérapie substitutive, on apporte une enzyme exogène, synthétisée in vitro , qui va être reconnue par le récepteur au mannose-6 phosphate, internalisée par endocytose dans une vésicule qui va rejoindre l'endosome précoce puis l'endosome tardif, appelé à devenir lysosome. Une fois parvenue dans le lysosome, cette enzyme exogène pourra y remplacer l'enzyme déficiente.
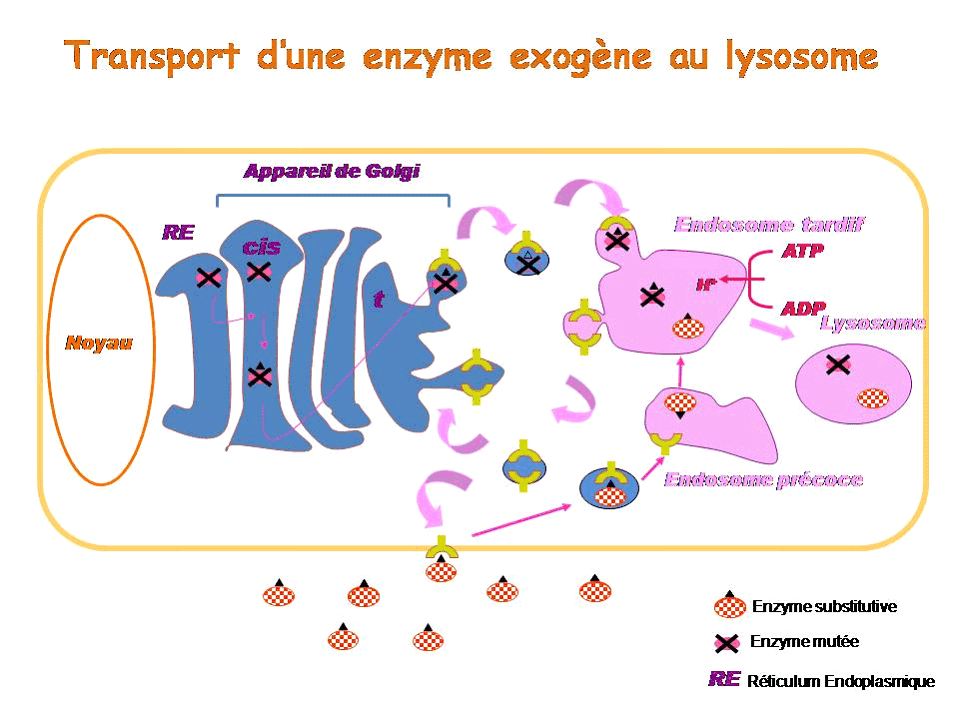
Il existe actuellement une enzymothérapie substitutive pour la maladie de Gaucher, la maladie de Fabry, les mucopolysaccharidose de type I, II et VI, et la maladie de Pompe. Son efficacité est variable selon la pathologie. Les résultats sont très bons pour la maladie de Gaucher, plus discutés pour la maladie de Fabry, et le recul est encore insuffisant pour les mucopolysaccharidoses. L'efficacité varie également selon le stade de la maladie et l'organe atteint. L'enzymothérapie est très efficace sur la viscéralomégalie, beaucoup moins sur les atteintes osseuses. Les enzymes en question ne peuvent pas être administrées pour l'instant par voie orale car elles sont trop volumineuses pour passer la barrière intestinale. Elles le sont donc par voie intra-veineuse et leur administration doit se poursuivre à vie. Une enzyme de substitution, mise au point en Israël sur des cellules de carottes et qui vient d'être approuvée par la Food and drug administration (FDA) américaine, semblerait toutefois pouvoir être administrée par voie orale : une étude clinique est en cours.
Un autre inconvénient de l'enzymothérapie substitutive est que, comme il s'agit de protéines, elle ne passe pas la barrière hémato-encéphalique : les atteintes neurologiques ne sont donc pas accessibles au traitement.
L'administration de ces enzymes peut en outre entraîner une réaction immunologique avec le développement d'anticorps neutralisants, susceptibles de réduire l'efficacité du traitement.
Enfin, le coût de cette thérapie est très élevé, d'au moins 200 000 euros par an et par patient, et ce à vie. Le traitement d'une mucopolysaccharidose de type VI coûte 500 000 euros par an pour un enfant.
En sus de l'enzymothérapie substitutive, il existe d'autres thérapies en cours d'évaluation ou venant d'être approuvées. Elles visent à réduire la quantité de substrat ou font appel à des molécules chaperons. Leur avantage est qu'elles peuvent être administrées par voie orale et qu'elles ont une meilleure distribution tissulaire. Il semble aussi qu'elles passent la barrière hémato-encéphalique et qu'elles n'entraînent pas de réaction immunologique. Leur coût de production serait également moins élevé. Cela étant, on manque encore beaucoup de recul.
Dans un métabolisme normal, un substrat B, synthétisé à partir d'une molécule A, est dégradé en une molécule C. Dans le cas d'une maladie enzymatique, l'enzyme qui permet la dégradation de B en C, est déficiente, si bien que le substrat B s'accumule, alors même que sa synthèse se poursuit normalement. Le principe de la réduction de substrat est de limiter la synthèse.
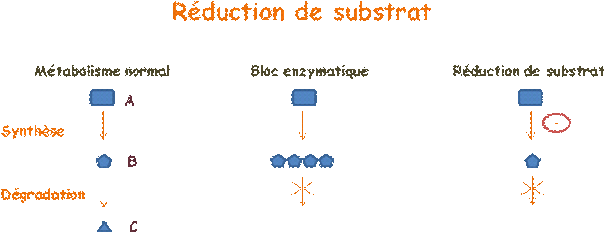
Un médicament, le Miglustat, est aujourd'hui disponible pour la maladie de Gaucher de type I et la maladie de Niemann-Pick de type C. Un autre, l'Eliglustat, commercialisé par Genzyme pour la maladie de Gaucher de type I, est en phase d'essai clinique. La Genistein, destinée à traiter la mucopolysaccharidose de type III, elle aussi en cours d'essai clinique, présente un intérêt tout particulier car les manifestations de cette maladie étant surtout neurologiques, on ne peut faire appel à l'enzymothérapie substitutive.
Un mot des traitements par molécules chaperons. Ces molécules permettent la stabilisation tridimensionnelle des enzymes qui sont synthétisées au niveau du réticulum endoplasmique mais ne se replient pas pour prendre leur conformation normale. Ce sont pour la plupart des analogues de substrat ayant une très forte affinité pour le site catalytique. Elles aident au repliement de l'enzyme et l'adressent au lysosome. En milieu acide, les inhibiteurs se détachent de l'enzyme, laquelle peut alors dégrader le substrat. Ces traitements, porteurs d'espoir, ne peuvent toutefois servir qu'à traiter les maladies résultant d'une mutation induisant un défaut de repliement. Aujourd'hui, on teste in vitro l'efficacité de la molécule chaperon avant de traiter le patient. Deux molécules sont en cours d'essai clinique : le Migalastat dans la maladie de Fabry et l'Isofogamine pour la maladie de Gaucher de type I et de type III.
En conclusion, je voudrais insister sur le caractère trompeur de la dénomination « maladies rares » pour les maladies lysosomales de surcharge car leur incidence globale est loin d'être négligeable : nous rencontrons un grand nombre de cas au quotidien. Ces maladies, connues pour être pédiatriques, concernent un nombre croissant d'adultes. Certaines d'entre elles se manifestent dès la vie in utero. Ces maladies rares font néanmoins l'objet de recherches actives, dont les retombées sont beaucoup plus larges. Grâce à ces recherches, plusieurs approches thérapeutiques innovantes ont pu être mises au point.
Débat
M. Jean-Louis Touraine. Ces nouvelles thérapeutiques, qui se sont développées en peu de temps, sont fascinantes. Dans les années 70, où rien de tout cela ne pouvait même être imaginé, nous pratiquions des greffes de cellules de foie foetal chez des patients atteints de maladie de Fabry ou de Gaucher. Cela permettait d'enrayer la progression de la maladie, sans toutefois permettre d'en faire régresser les symptômes. Mais des malades ainsi traités ont pu « attendre » jusqu'à la mise au point de l'enzymothérapie substitutive, qui a pris le relais. Tous les traitements, même ceux qui n'ont pas encore fait la preuve d'une efficacité totale sur tous les symptômes de la maladie, sont intéressants, ne serait-ce que parce qu'ils peuvent permettre à certains malades de survivre jusqu'à ce qu'un traitement plus efficace soit disponible. Toutes ces thérapies innovantes sont porteuses de beaucoup d'espoirs.
Mme Soumeya Bekri. Être en mesure de proposer une solution partielle ou d'attente change complètement la donne. C'est un espoir considérable pour les malades. J'ai en tête le cas d'une fillette atteinte de mucopolysaccharidose de type VI, dont l'intelligence n'avait pas été atteinte par la maladie, et qui a pu bénéficier de l'une de ces thérapies. Sa vie et son pronostic en ont été révolutionnés.
M. Jean-Louis Touraine. Il est très encourageant que de nouveaux traitements semblent franchir la barrière hémato-encéphalique. En effet, jusqu'à présent, en cas d'atteinte neurologique lourde, on ne disposait d'aucun outil, ne serait-ce que pour stopper, sans parler même de réversibilité, les atteintes cérébrales. Il était très frustrant de ne pas pouvoir protéger le système nerveux.
M. le président Claude Birraux. Je trouve moi aussi tous ces progrès très encourageants. Des résultats ont d'ores et déjà été obtenus, il faut le faire savoir plus largement... y compris à M. Bergé, qui s'en était pris au Téléthon.
Le coût élevé de ces traitements tient, entre autres, à celui des recherches nécessaires à leur mise au point. Quid de la propriété intellectuelle des découvertes ?
Mme Soumeya Bekri. Nous avons acquis une culture du brevet, que nous n'avions pas auparavant. Les universités comme l'INSERM nous incitent de plus à plus à protéger nos découvertes avant de les publier.
M. Philippe Leboulch. Qu'on le déplore ou qu'on s'en réjouisse, force est de constater que s'il n'y avait pas d'application industrielle en aval des découvertes fondamentales, on ne pourrait pas aller très loin. Il faut en effet des moyens matériels et financiers considérables pour en arriver jusqu'aux essais cliniques puis à la mise à disposition de nouveaux médicaments pour les patients. Les étapes-clés des découvertes doivent donc être protégées par des brevets, de façon que les industriels puissent non seulement rentrer dans leurs frais mais faire des bénéfices leur permettant de continuer à investir.
Le prix des médicaments dont on a parlé aujourd'hui est très élevé, peut-être trop élevé. Mais leur coût même de production fait qu'il ne pourra pas s'abaisser au-delà d'un certain seuil. La thérapie génique, elle, marque une rupture car le traitement, dans la plupart des cas, est destiné à n'être administré qu'une seule fois. Une transplantation de moelle avec transfert de gène coûtera nécessairement davantage qu'une transplantation de moelle simple. Mais le surcoût sera au final quasiment négligeable par rapport aux thérapies innovantes non géniques qui nous ont été présentées ou aux thérapies palliatives qui doivent durer toute la vie du patient.
M. Jean-Louis Touraine. Les retombées potentielles de la thérapie génique sont considérables. Elle pourrait être utilisée dans le traitement de maladies comme le cancer, le diabète, le sida. Le coût d'une thérapie génique unique, aussi onéreuse soit-elle, serait bien moindre au final chez les malades du sida, dont l'espérance de vie avoisine désormais la normale, que l'administration à vie des trithérapies actuelles, dont l'observance sur le long terme est par ailleurs quotidienne et imparfaite chez de nombreux malades. Il faut donc encourager la recherche sur les thérapies géniques, quel que soit le montant des investissements nécessaires, car en résulteront des progrès médicaux considérables pour les malades mais aussi, à terme, des économies pour nos systèmes de santé.
M. Philippe Leboulch. Des équipes travaillant sur le sida ont découvert un corécepteur dont l'absence ou la mutation expliquent que certaines personnes soient comme protégées de l'infection par le VIH. En Allemagne, un patient atteint à la fois d'une leucémie et du sida a reçu une greffe de moelle à partir d'un donneur compatible qui se trouvait posséder la mutation de ce corécepteur. Ce patient a non seulement été guéri de sa leucémie mais semble l'avoir été aussi du sida, les cellules qu'on lui a greffées n'étant plus infectables. Un essai clinique est en cours aux Etats-Unis, utilisant le même vecteur que celui utilisé pour les hémoglobinopathies pour exprimer dans les cellules un gène empêchant ce corécepteur d'être présent et ainsi immuniser « de l'intérieur » les cellules du patient contre le VIH. Voilà l'exemple-type d'une retombée d'une recherche conduite initialement pour des maladies génétiques rares dans le traitement d'une maladie fréquente, d'ailleurs infectieuse et non génétique.
M. Nicolas Lévy. Je voudrais abonder dans le sens de mes collègues. Certaines des molécules qui ont fait la preuve d'une certaine efficacité dans le traitement des maladies lysosomales de surcharge - je pense en particulier au Miglustat et à toutes les molécules capables d'induire le repliement de certaines protéines - peuvent trouver des applications dans bien d'autres champs. Elles pourront servir à traiter toutes les maladies liées à une anomalie du « contrôle de qualité » ou de repliement des protéines du fait d'une mutation génétique. Il importe donc de développer la recherche dans ces domaines.
Certains opposent thérapies pharmacologiques classiques et thérapies innovantes, dont la thérapie génique et la thérapie cellulaire. Cette opposition n'a pas de sens. Tout d'abord, les secondes n'excluent pas de recourir aux premières, y compris les plus classiques. Mais, pour certaines maladies du muscle, de la rétine, du sang ..., les thérapies pharmacologiques n'ont pas apporté de solution et sont vraisemblablement vouées à demeurer impuissantes. Il faut donc absolument soutenir les approches innovantes, parmi lesquelles la thérapie génique, par transfert de gène, ou, plus « à la carte », par saut d'hexon ou modification génomique grâce à des molécules anti-sens. Les deux approches s'opposent d'autant moins que certaines formes de thérapie génique sont appelées à être administrées comme un médicament classique. C'est le cas des petites molécules d'ADN anti-sens d'ores et déjà utilisées dans certains essais cliniques.
M. le président Claude Birraux. Si je puis me permettre en conclusion un trait d'humour sur un sujet aussi sérieux, au moins dans ce domaine n'y a-t-il pas de faucheurs volontaires pour ruiner les recherches !