SYNTHÈSE : LE PARCOURS D'ACCÈS AUX MÉDICAMENTS
|
Du médicament au patient : synthèse
de la procédure de mise à disposition
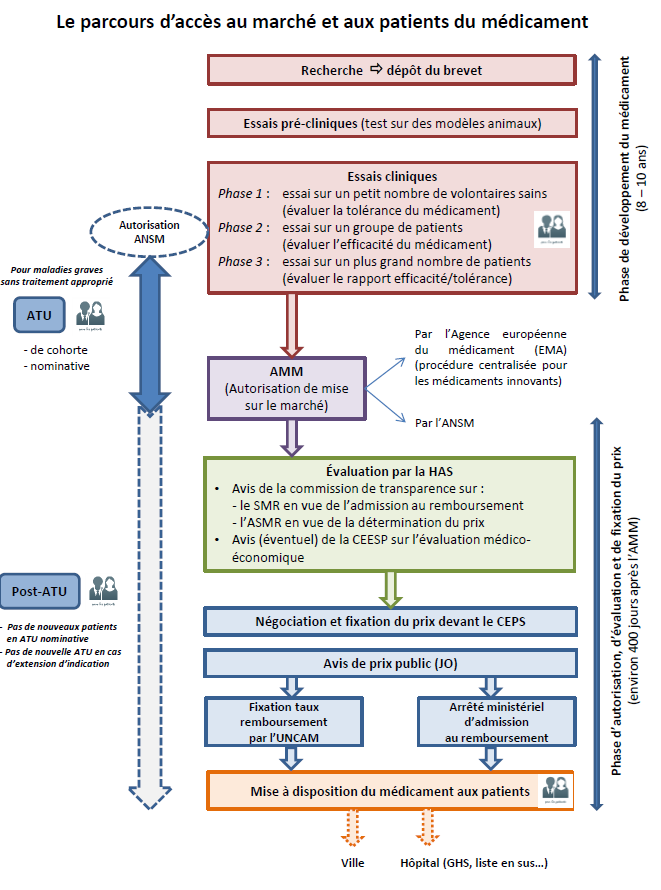
Comme l'illustre le schéma ci-après, les patients français peuvent avoir accès à des molécules nouvelles et susceptibles de constituer un progrès thérapeutique à plusieurs étapes de la chaîne de développement et de mise sur le marché du médicament, explicitées ci-après. Les essais cliniques À la suite de l'identification d'une molécule susceptible de répondre à un besoin médical non pourvu et d'une phase de recherche préclinique en laboratoire suivie d'un dépôt de brevet, la réalisation d'un essai clinique vise à évaluer successivement la tolérance du médicament-candidat sur un petit nombre de volontaires sains (phase I), son efficacité sur des volontaires malades (phase II) et son rapport efficacité/tolérance à plus grande échelle (phase III). Lorsqu'il s'agit d' essais dits « précoces », la tolérance et l'efficacité sont évaluées concomitamment, sur des patients sans alternative thérapeutique, dans une démarche qui peut permettre d'allier recherche et accès aux soins . Si les résultats de l'essai clinique sont convaincants, le promoteur peut engager une procédure en vue de l'éventuelle commercialisation du médicament. La procédure « de droit commun » d'accès au marché des médicaments - L'autorisation de mise sur le marché (AMM) , sollicitée par l'industriel, constitue le point de départ de cette procédure. Celle-ci est accordée sur la base d'une évaluation scientifique de l'efficacité, de la sécurité et du bénéfice/risque du médicament dans la ou les indications pour lesquelles l'AMM est demandée. Elle est délivrée, pour les médicaments innovants, essentiellement au niveau européen (selon une procédure dite centralisée), après avis de l'Agence européenne du médicament (EMA). Dans le cas d'une procédure nationale, c'est en France l'Agence nationale de sécurité du médicament et des produits de santé (ANSM) qui procède à cette évaluation et à l'examen du dossier. - À partir de l'octroi de l'AMM, l'industriel qui commercialise un médicament peut demander son inscription sur la liste des produits remboursables : s'engage alors, au niveau national (et selon des modalités différentes d'un pays européen à l'autre) une procédure en vue de l'inscription du médicament au remboursement et de la fixation de son prix. Elle doit en théorie s'inscrire dans un délai de 180 jours, fixé par une directive européenne. - L'évaluation par la Haute Autorité de santé (HAS), autorité indépendante, en constitue la première étape. Il s'agit dans tous les cas d'une évaluation clinique , à laquelle procède la commission de la transparence de la HAS sur la base du dossier remis par l'industriel. L'objectif est de déterminer : si le produit doit être remboursé et à quel taux, sur la base de son « service médical rendu » (SMR), c'est-à-dire de son intérêt clinique, si le produit apporte un progrès thérapeutique par rapport aux traitements existants, sur la base de l'évaluation de son « amélioration du service médical rendu » (ASMR), qui sert ensuite pour la détermination du prix. Cette évaluation clinique peut être couplée, dans certains cas encore rares, d'une évaluation médico-économique à laquelle procède la commission d'évaluation économique et de santé publique (CEESP) de la HAS. Cette évaluation a vocation à être prise en compte pour la fixation du prix. - La phase de fixation du prix s'engage ensuite sur la base des résultats de l'évaluation, dans le cadre d'une démarche conventionnelle entre les industriels et le Comité économique des produits de santé (CEPS), organisme interministériel. En cas d'accord, cette procédure aboutit à la publication d'un prix public au Journal Officiel, sachant que des remises conventionnelles protégées par le secret des affaires sont négociées entre les industriels et le CEPS (ce prix net de remises n'est pas public). - Ce n'est qu'au terme de ces deux séquences - et après la publication de l'arrêté ministériel d'admission au remboursement du produit et la fixation de son taux de remboursement par le directeur général de l'Uncam (Union nationale des caisses d'assurance maladie) - que le médicament entre dans le circuit de commercialisation « de droit commun » . Il peut alors être distribué en ville (officine) ou dans les établissements de santé selon différents modes de prise en charge : dans le cadre de la rétrocession (médicaments délivrés à l'hôpital mais destinés aux patients en ambulatoire), des GHS (groupes homogènes de séjour), ou encore de la « liste en sus » qui permet la prise en charge de médicaments onéreux en sus des GHS. • Le dispositif des autorisations temporaires d'utilisation (ATU) : une voie d'accès précoce spécifique et dérogatoire La France a prévu que dans certaines conditions, un médicament considéré comme innovant puisse être mis à disposition des patients selon un mode dérogatoire et anticipé, qui se situe en quelque sorte entre les essais cliniques et la commercialisation « de droit commun » : il s'agit d'une autorisation d'utiliser, pour une durée limitée éventuellement renouvelable et « à titre exceptionnel, certains médicaments destinés à traiter des maladies graves ou rares en l'absence de traitement approprié, lorsque la mise en oeuvre du traitement ne peut pas être différée ». Ce dispositif d'autorisation temporaire d'utilisation (ATU) est sollicité soit par l'industriel (ATU de cohorte concernant un groupe de patients dans le cadre d'un protocole déterminé), soit par le médecin prescripteur (ATU nominative pour un patient donné). L'examen de la demande et l'autorisation relèvent de l' ANSM . A ce stade, le produit n'a pas encore fait l'objet d'une évaluation par la HAS ; son prix (appelé indemnité) est par ailleurs fixé librement par le laboratoire . L'ATU est sollicitée en général en fin de période de développement et d'essais cliniques, toujours en amont de l'AMM . Les laboratoires ont l'obligation, quand ils sollicitent une ATU de cohorte, de présenter une demande d'AMM. L'ATU se déroule donc parallèlement à la phase d'examen de l'AMM par l'EMA. Elle prend fin théoriquement au moment de l'obtention de l'AMM . Toutefois, suspendre l'ATU à ce moment, alors que le médicament n'est pas encore distribué car s'engage alors la phase décrite ci-dessus, conduirait à priver les patients, pendant plusieurs mois, de leur traitement. Aussi, une phase dite de « post-ATU », toujours dérogatoire, prend le relais , pendant que se déroule la procédure d'évaluation par la HAS puis celle de fixation du prix. On voit donc que l'intérêt du dispositif d'ATU est non seulement d'anticiper l'AMM mais également, et de plus en plus, d'anticiper la commercialisation effective du médicament selon le circuit de droit commun après son AMM. Les difficultés qui se posent aujourd'hui au dispositif d'ATU, en particulier pour traiter des extensions d'indication d'un médicament sous ATU ou des modalités de régulation financière du dispositif, se comprennent donc à la lumière de l'analyse de cette procédure de « droit commun » dont les délais ont tendance à s'allonger dans un contexte de forte tension sur les prix. |