







IV. CONFORTER LE RÔLE DES ESSAIS CLINIQUES DANS L'ACCÈS PRÉCOCE DES PATIENTS AUX TRAITEMENTS INNOVANTS
Les essais cliniques précoces, dont le nombre connaît une augmentation régulière dans le monde , sont devenus l'un des moteurs essentiels des innovations thérapeutiques et font désormais souvent partie intégrante des soins. C'est le cas en particulier dans le domaine de la cancérologie, où d'importants besoins médicaux demeurent insatisfaits et où l'avènement de la médecine de précision et la révolution de l'immunothérapie ont ouvert de nouvelles perspectives pour les patients, les soignants et les chercheurs.
Dans ce contexte, la recherche implique une gestion d'études de plus en plus complexes dans un contexte international devenu extrêmement compétitif . Si l'expertise de nos chercheurs est unanimement reconnue et des initiatives structurantes ont vu le jour, la nécessité d'améliorer l'environnement de la recherche clinique française, notamment précoce, par l'identification de priorités stratégiques, une interaction plus étroite de tous les acteurs et le soutien au développement des infrastructures est un constat partagé de tous. Au-delà des enjeux majeurs de financement de la recherche, que le présent rapport n'a pas vocation à aborder, de nombreux freins organisationnels et administratifs doivent en effet être levés pour garantir l'attractivité de la France sans remettre en cause les garanties de sécurité et de déontologie au respect desquelles il convient en toute circonstance de s'astreindre.
A. LA RECHERCHE CLINIQUE COMME VOIE D'ACCÈS PRÉCOCE AUX TRAITEMENTS INNOVANTS
1. Un encadrement juridique étroit, qui a pour principal objectif d'assurer la sécurité des volontaires
a) Les essais cliniques, une étape prometteuse mais encore très incertaine dans le processus de développement d'un médicament
La commercialisation d'une ou de plusieurs molécules sous la forme d'un médicament à usage humain nécessite la mise au jour préalable de connaissances biologiques et médicales acquises dans le cadre d'une recherche biomédicale réalisée sur l'homme. Celle-ci se concrétise par la mise en place d'un essai clinique permettant de tester un médicament « candidat » ou une nouvelle façon d'utiliser un traitement connu. Il s'agit d'établir ou de vérifier, selon la classification opérée à l'article R. 1121-1-1 du code de la santé publique, les données pharmacocinétiques (absorption, distribution, métabolisme et excrétion), pharmacodynamiques (mécanisme d'action) et thérapeutiques (efficacité et tolérance) d'un médicament.
Les essais cliniques s'inscrivent ainsi dans le prolongement d'une phase initiale de recherche exploratoire permettant d'identifier une molécule susceptible de répondre à un besoin médical non pourvu ou d'améliorer les prises en charge existantes, suivie d'une phase obligatoire de recherche préclinique en laboratoire sur des cellules in vitro et/ou sur des animaux. Cette dernière vise à évaluer l'innocuité et l'absence de toxicité de la substance testée. Elle permet en outre de déterminer les doses qui seront administrées à l'homme au cours des essais cliniques.
Une fois ces deux premières phases achevées, la molécule sélectionnée est brevetée et s'ouvre la phase de recherche clinique et industrielle. Réalisés soit chez le volontaire malade, soit chez le volontaire sain, les essais cliniques se décomposent en trois, voire quatre phases successives . Au cours de la phase I, la molécule est évaluée sur un nombre limité de volontaires sains dans l'objectif de déterminer sa tolérance et sa sécurité d'emploi et d'évaluer son évolution dans l'organisme. La phase II consiste à administrer la substance à des patients malades afin d'identifier les doses auxquelles elle est efficace et tolérée. La réalisation d'essais cliniques à plus grande échelle est conditionnée à la preuve de concept établie au cours de cette deuxième phase. Au cours de la phase III, la molécule est administrée à une population significativement plus nombreuse, avec l'objectif de valider à la fois son efficacité et sa tolérance à plus grande échelle et son rapport bénéfice-risque. Si les résultats s'avèrent concluants, le promoteur a la possibilité d'introduire une demande d'AMM en vue d'une éventuelle commercialisation du produit. Enfin, le suivi du médicament en vie réelle se déroule en phase IV dans le cadre de la pharmacovigilance dont l'objectif est d'identifier tout effet secondaire grave et/ou inattendu du médicament.
Les quatre phases des essais cliniques
|
Nombre
|
Durée indicative |
Objectif |
|
|
Phase I |
20-100 |
De plusieurs mois
|
Sécurité et tolérance |
|
Phase II |
jusqu'à 100 |
De plusieurs mois
|
Tolérance à court terme
|
|
Phase III |
100 à plus de 1 000 |
De 1 à 4 ans |
Évaluation et comparaison bénéfice/risque |
|
Phase IV |
Plus de 1 000 |
De 1 à 4 ans |
Tolérance et recherche
|
Source : Ligue nationale contre le cancer
Au total, pour un médicament finalement commercialisé, en fonction des pathologies concernées et du nombre de volontaires participant, environ cinq années pourront s'être écoulées du début de la recherche pharmaceutique à la fin de la phase préclinique et dix au terme de la recherche clinique. Seul un nombre relativement très faible de molécules sont développées jusqu'au terme de ce processus.
b) Un régime juridique contraignant pour protéger les personnes qui acceptent de se prêter à une recherche
La réalisation d'un essai clinique étant empreinte de nombreuses inconnues et incertitudes, il est apparu essentiel de prévoir un encadrement strict, minimisant la prise de risque pour les personnes, quand bien même celles-ci se prêtent volontairement à la recherche sans en attendre de bénéfice individuel direct. Définies pour la première fois par la loi de 1988 relative à la protection des personnes qui se prêtent à des recherches biomédicales 86 ( * ) , les règles applicables ont été régulièrement actualisées tout en reposant sur l'idée constamment réaffirmée que l'intérêt des volontaires qui se prêtent à une recherche clinique doit toujours primer sur l'intérêt de la science et de la société, conformément aux principes éthiques fondamentaux consacrés dès 1964 dans la déclaration d'Helsinki 87 ( * ) .
La loi de 1988, dite « Huriet-Sérusclat » , a ainsi introduit le principe d'une protection obligatoire des patients fondé un devoir d'information des volontaires, la nécessité de recueillir leur consentement écrit et l'obligation pour le promoteur d'une recherche clinique de souscrire un contrat d'assurance spécifique pour couvrir les risques encourus.
Afin d'évaluer le respect de ces principes et les protocoles de recherche, la loi institue dans chaque région un comité consultatif de protection des personnes qui se prêtent à la recherche biomédicale (CCPPRB). Pour chaque projet, elle impose l'identification d'un promoteur - personne morale (laboratoire pharmaceutique, prestataire de service, association, établissement de soins ou de recherche) ou physique -, qui prend l'initiative de l'essai clinique, et d'un investigateur principal, médecin chargé de diriger et de surveiller la réalisation de l'essai. La recherche fait l'objet d'une contractualisation entre le promoteur et le ou les centres d'investigation.
Ce régime a été renforcé par la loi de 2004 relative à la politique de santé publique 88 ( * ) qui permet l'application en France de la directive européenne du 4 avril 2001 89 ( * ) harmonisant les règles de vigilance des essais thérapeutiques entre les Etats membres. Cette loi confère à l'Agence française de sécurité sanitaire des produits de santé (Afssaps) - ancêtre de l'ANSM - la mission de veiller à ce que les conditions d'un consentement éclairé soient réunies ainsi que la possibilité de mener des investigations en cours d'essai par l'intermédiaire d'un comité d'éthique indépendant. Elle a transformé les CCPPRB en comités de protection des personnes (CPP) et rendu leur avis non plus consultatif mais décisionnel.
Le cadre législatif aujourd'hui applicable aux essais cliniques (articles L. 1121-1 à L. 1126-12 du code de la santé publique) est issu de la loi relative aux recherches impliquant la personne humaine de 2012, dite loi « Jardé » 90 ( * ) , qui n'est devenue applicable qu'en 2016 par l'édiction d'une ordonnance prise sur le fondement de la loi de modernisation de notre système de santé du 26 janvier 2016 91 ( * ) , dans l'attente par le Gouvernement de la révision des règles européennes applicables en la matière 92 ( * ) . Le décret d'application de la loi « Jardé » 93 ( * ) précise les modalités de réalisation des recherches impliquant la personne humaine (RIPH) et ajuste les règles relatives au rôle et au fonctionnement des CPP et de l'ANSM. Elle distingue plusieurs catégories de recherche en fonction des niveaux de risque, parmi lesquelles celles requérant le plus haut niveau de vigilance sont dénommées « recherches interventionnelles qui comportent une intervention non justifiée par sa prise en charge habituelle » (1° de l'article L. 1121-1 du code de la santé publique).
Pour ces dernières, le démarrage d'un essai clinique est soumis à la double condition d'un avis favorable d'un CPP et d'une autorisation de l'ANSM (art. L. 1121-4). Le promoteur a le choix d'introduire, simultanément ou non, la demande d'avis et celle d'autorisation.
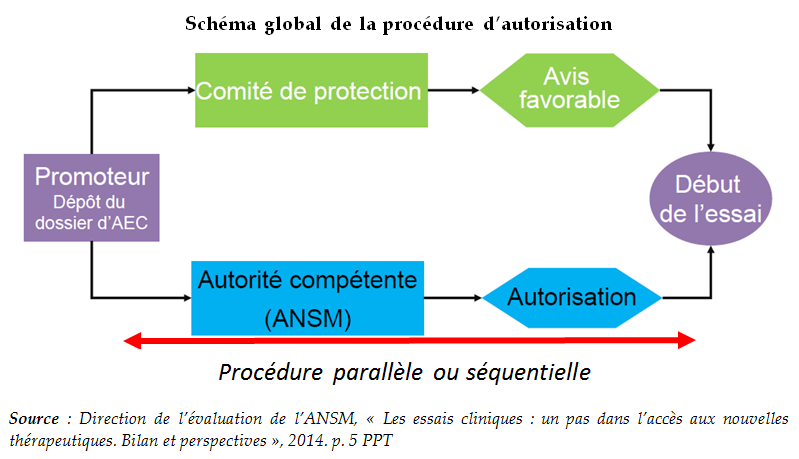
En vertu de l'article L. 1121-11 du code de la santé publique, les volontaires, recensés sur un fichier national, font l'objet d'un examen médical préliminaire et aucune contrepartie financière directe ou indirecte hormis un remboursement des frais exposés ne peut leur être versée. Cette indemnisation est limitée à 4 500 euros pour une période de douze mois consécutifs. Une période d'exclusion entre les recherches doit être respectée. Enfin, les personnes bénéficiant d'une protection renforcée (mineurs, femmes enceintes, majeurs sous tutelle, etc.) ne peuvent être inclus dans les essais sauf en cas de bénéfice majeur direct possible pour leur santé.
Le CPP, dont la mission principale est de garantir la protection des volontaires, rend son avis sur les conditions de validité de la recherche au regard principalement des critères suivants 94 ( * ) :
- la protection des personnes, au premier rang desquelles les participants ;
- l'adéquation, l'exhaustivité et l'intelligibilité des informations écrites à fournir ainsi que la procédure à suivre pour obtenir le consentement éclairé, et la justification de la recherche sur des personnes incapables de donner leur consentement éclairé ;
- la nécessité éventuelle d'un délai de réflexion ;
- la nécessité éventuelle de prévoir, dans le protocole, une interdiction de participer simultanément à une autre recherche ou une période d'exclusion ;
- la pertinence de la recherche, le caractère satisfaisant de l'évaluation des bénéfices et des risques attendus et le bien-fondé des conclusions ;
- l'adéquation entre les objectifs poursuivis et les moyens mis en oeuvre ;
- la qualification du ou des investigateurs ;
- les montants et les modalités d'indemnisation des participants ;
- et enfin les modalités de recrutement des participants.
Le promoteur a l'obligation de soumettre à l'ANSM un protocole de recherche, qui se fonde sur une revue de la littérature scientifique et démontre la nécessité de réaliser une nouvelle recherche. En application de l'article L. 1123-12 du code de la santé publique, l'agence évalue l'objectif, les conditions de réalisation et de déroulement de l'essai, les modalités d'inclusion, d'information, de traitement et de surveillance des volontaires par les médecins investigateurs ainsi que les procédures de recueil des informations sur l'efficacité et la tolérance des médicaments. Une fois l'autorisation octroyée et l'essai débuté, l'ANSM est tenue informée des effets indésirables graves et inattendus pouvant être liés au médicament expérimental et de tout fait nouveau lié à la recherche susceptible de remettre en cause la sécurité des personnes se prêtant à la recherche. Le cas échéant, elle peut prendre toute décision de suspension ou d'interdiction. L'ANSM inspecte en outre certains sites d'essais cliniques.
Par ailleurs, en vertu des bonnes pratiques cliniques (BPC), dont la dernière version date du 24 novembre 2006, toute recherche doit faire l'objet de contrôles de qualité réalisés en début et en cours d'essai sous la responsabilité du promoteur de la recherche. Cette mission échoit en général aux assistants de recherche clinique (ARC) du promoteur.
En mars et juin 2016, à la suite de l'accident survenu à Rennes au cours d'un essai de phase I conduit par la société Biotrial pour le compte du laboratoire Bial chez un volontaire sain avec première administration chez l'homme d'une nouvelle molécule, ces règles de vigilance ont été encore renforcées. En particulier, le promoteur d'un essai doit désormais informer sans délai l'ANSM, le CPP et l'ARS de tout fait nouveau survenant dans le cas d'un essai de première administration ou utilisation d'un médicament expérimental chez des volontaires sains, en suspendre l'administration ou l'utilisation et prendre toutes les mesures de sécurité appropriées dans l'attente de l'adoption de mesures définitives.
2. Les essais cliniques comme voie d'accès précoce à des médicaments innovants
Dans certaines situations, pour les patients qui présentent une résistance aux traitements classiques ou qui demeurent sans solution thérapeutique efficace, des essais de phase précoce peuvent permettre d'accélérer la mise à disposition de nouvelles stratégies thérapeutiques . Ces essais, qui regroupent des caractéristiques d'essais de phases I et II, avec une évaluation concomitante de la sécurité du dosage et de l'efficacité, permettent d'accéder à un nouveau médicament plusieurs années avant sa commercialisation, de 3 à 4 ans selon le Leem.
Ce type d'essai remet en cause la distinction traditionnelle entre la recherche et le soin, alliant les deux approches dans une même démarche. Il soulève de nouveaux enjeux éthiques et juridiques, au regard notamment des conditions d'accès aux soins et de sélection des volontaires. La grande majorité des essais précoces sont aujourd'hui mis en oeuvre en cancérologie où les molécules à l'essai visent très souvent des cibles protéiques anormales caractéristiques d'un type particulier de tumeur dans le cadre d'une médecine de précision. Si ce type d'essais est en plein essor, il ne concerne ainsi qu'un petit nombre de malades. Comme l'indiquent Valérie Gateau et Philippe Amiel, « en cancérologie, les essais précoces sont les premiers tests d'administration d'une molécule, ou combinaison de molécules, chez l'être humain, à la suite des essais précliniques en laboratoires et/ou sur l'animal. Ils concernent environ 2500 malades par an, soit moins de 1 % de ceux qui participent à des essais cliniques en France tous les ans. Ces essais de phase I non randomisés visent à évaluer la dose maximale tolérée. Ils sont conduits sur un petit nombre de patients (de moins d'une dizaine à une trentaine). Contrairement aux essais dans d'autres pathologies, en raison de la toxicité des molécules testées en cancérologie, il n'y a que peu d'études menées sur des volontaires sains. » 95 ( * )
Les autorités ont progressivement pris la mesure de l'importance des essais précoces pour développer l'innovation thérapeutique et des initiatives structurantes ont vu le jour, en particulier pour permettre un accès plus égalitaire des personnes malades . C'est l'un des objectifs poursuivis dans le cadre des plans Cancer avec la mise en place à compter de 2010 des centres d'essais cliniques de phase précoce (Clip) labellisés par l'Institut national du cancer (INCa). Actuellement, 16 Clip existent, dont 6 en pédiatrie, qui sont labellisés jusqu'en 2019. Une attention particulière a ainsi été portée à l'amélioration de la couverture territoriale et au développement d'une activité de phase précoce en cancérologie pédiatrique.
Ainsi que l'a indiqué l'INCa, ce réseau vise tout autant à améliorer l'accès à l'innovation qu'à augmenter la visibilité et l'attractivité internationales des centres français de recherche. La moitié de l'activité de phase I des Clip est réalisée par le département d'innovation thérapeutique et d'essais précoces (DITEP) de l'Institut Gustave Roussy que vos rapporteurs ont pu visiter au cours d'un déplacement.
|
Le département d'innovation thérapeutique
A travers son département d'innovations thérapeutiques et d'essais précoces (DITEP), l'Institut Gustave Roussy a fait des essais précoces un moteur de l'innovation thérapeutique en cancérologie. Son approche combinée des soins et de la recherche, à travers la réalisation d'essais qui répondant de façon concomitante aux objectifs poursuivis au cours des phases I et II, vise à mettre en évidence l'efficacité d'une molécule de manière accélérée. Le délai d'accès aux innovations peut ainsi être ramené à 4-6 ans au lieu de 8 environ . Selon le DITEP, « à l'issue d'un essai clinique de phase I, la maladie régresse ou se stabilise chez près de 50 % des patients . » 96 ( * ) Le DITEP a été créé en 2013 à partir du service des innovations thérapeutiques et des essais précoces, première unité d'hospitalisation française intégralement dédiée aux essais précoces en cancérologie mise en place en 2008. Sa transformation en un département à part entière se fondait sur la volonté d'atteindre une masse critique, de fluidifier les processus et d'optimiser les performances. Aujourd'hui, le DITEP est le plus grand centre d'essais de phase I en France et en Europe et le septième dans le monde. En 2016, 417 recherches biomédicales ont été mises en oeuvre, dont 366 essais thérapeutiques. En 2017, 3 617 patients ont été inclus dans 438 recherches biomédicales, soit 28 % des 13 109 patients traités pour un cancer. L'Institut Gustave Roussy est le promoteur de la moitié des études menées par le DITEP. Le nombre annuel d'essais précoces en cours a connu une hausse constante, passant de 41 en 2010 à 104 en 2017. Les principales difficultés identifiées par les équipes de très haut niveau rencontrées par vos rapporteurs tiennent aux délais d'approbation réglementaire des essais précoces : pour l'année 2017, sur 25 essais précoces, le délai d'autorisation par l'ANSM s'élève à 121 jours en moyenne (la fourchette étant de 32 à 212 jours), et le délai d'avis pour les CPP à 91 jours (de 43 à 234). Le DITEP regrette en outre l'impossibilité pour les coordonnateurs académiques d'interagir avec les autorités réglementaires dans un esprit constructif pour faciliter la mise en place des essais. S'agissant enfin des CPP, ils relèvent l'expertise très variable de leurs membres et l'absence d'harmonisation des pratiques. Au total, une inquiétude existe quant à la défiance croissance des promoteurs industriels à développer leur activité en France. |
* 86 Loi n° 88-1138 du 20 décembre 1988 relative à la protection des personnes qui se prêtent à des recherches biomédicales.
* 87 « Principes éthiques applicables aux recherches médicales sur des sujets humains », Déclaration d'Helsinki de l'Association médicale mondiale (AMM), 1964.
* 88 Loi n° 2004-806 du 9 août 2004 relative à la politique de santé publique.
* 89 Directive 2001/20/CE du Parlement européen et du Conseil du 4 avril 2001 concernant le rapprochement des dispositions législatives, réglementaires et administratives des États membres relatives à l'application de bonnes pratiques cliniques dans la conduite d'essais cliniques de médicaments à usage humain.
* 90 Loi n° 2012-300 du 5 mars 2012 relative aux recherches impliquant la personne humaine.
* 91 Loi n° 2016-41 du 26 janvier 2016 de modernisation de notre système de santé.
* 92 Ordonnance n° 2016-800 du 16 juin 2016 relative aux recherches impliquant la personne humaine.
* 93 Décret n° 2016-1537 du 16 novembre 2016 relatif aux recherches impliquant la personne humaine.
* 94 Article L. 1123-7 du code de la santé publique.
* 95 « Essais précoces en cancérologie, éthique et justice. La lettre du cancérologue, 2012, XXI (10), pp. 514-518.
* 96 https://www.gustaveroussy.fr/fr/essais-precoces (consulté le 13 juin 2018).