







QUATRIÈME PARTIE
DISPOSITIONS RELATIVES AUX
DÉPENSES
POUR L'EXERCICE 2020
TITRE Ier
POURSUIVRE LA
TRANSFORMATION
DU SYSTÈME DE SOINS
CHAPITRE
Ier
RÉFORMER LE FINANCEMENT
DE NOTRE SYSTÈME DE
SANTÉ
Article 24 A
Visibilité
pluriannuelle sur les ressources des établissements de santé
Objet : Cet article, inséré par l'Assemblée nationale, ouvre la possibilité pour l'État et les fédérations hospitalières de conclure un protocole fixant, pour une période maximale de trois ans, les trajectoires relatives au montant des ressources pluriannuelles des établissements de santé publics et privés, ainsi que les engagements réciproques afférents.
I - Le dispositif adopté par l'Assemblée nationale
L'Assemblée nationale a adopté, à l'initiative du Gouvernement, un amendement traduisant l'engagement de la ministre des solidarités et de la santé, exprimé lors de son audition devant la commission des affaires sociales, en faveur d'une meilleure visibilité pluriannuelle sur les financements dédiés aux établissements de santé .
À cet effet, cet article prévoit la possibilité d'établissement d'un « protocole » entre l'État et les fédérations hospitalières visant à définir les trajectoires relatives au montant des ressources pluriannuelles des établissements de santé publics et privés et les engagements réciproques afférents, portant sur une période maximale de trois ans.
Ce protocole serait élaboré au sein de l'observatoire économique de l'hospitalisation publique et privée, transformé par le même article en « comité » économique de l'hospitalisation publique et privée.
Ce comité se voit également confier le suivi et l'application du protocole.
|
L'observatoire économique de l'hospitalisation publique et privée Institué par la loi de financement de la sécurité sociale pour 2007, d'abord à titre temporaire pour cinq ans, l'observatoire économique de l'hospitalisation publique et privée a été pérennisé par la loi de financement de la sécurité sociale pour 2012 (article L. 162-21-3 du code de la sécurité sociale). Créé auprès des ministres chargés de la santé et de la sécurité sociale, il est chargé du suivi des dépenses d'assurance maladie relatives aux frais d'hospitalisation et de la situation financière des établissements de santé publics et privés. L'observatoire est composé : - de représentants des services de l'État ; - de représentants des organisations nationales les plus représentatives des établissements de santé publics et privés ; - de représentants des organismes nationaux de l'assurance maladie. Le code de la sécurité sociale prévoit en outre que : - cet observatoire remet au Gouvernement et au Parlement un rapport semestriel sur l'évolution des dépenses d'assurance maladie relatives aux frais d'hospitalisation. Le dernier rapport publié date toutefois d'octobre 2015 ; - le Gouvernement le consulte préalablement à la mise en oeuvre de la procédure de modification des tarifs hospitaliers prévue en cas de risque sérieux de dépassement de l'Ondam hospitalier identifié par le Comité d'alerte de l'Ondam. |
II - La position de la commission
Votre rapporteur salue le signal donné par cet article en faveur d'une meilleure visibilité pluriannuelle des établissements de santé sur leurs ressources, qui répond à des attentes fortes des acteurs hospitaliers.
Le récent rapport sur l'Ondam présenté au nom de la Mecss 94 ( * ) a notamment préconisé, en ce sens, la mise en place d'une construction pluriannuelle de la tarification des établissements de santé afin de sortir d'un pilotage à courte vue qui obère la capacité des établissements de santé à porter des projets de long terme ou décider de choix d'investissements.
Cette avancée ne lève pas, cependant, toutes les inquiétudes sur la situation de l'hôpital qui accompagnent l'examen du PLFSS.
Sur la proposition de son rapporteur, la commission a adopté un amendement rédactionnel (amendement n° 164) et un autre tendant à préciser que les trajectoires pluriannuelles portent non seulement sur le montant mais également la répartition des ressources des établissements de santé (amendement n° 145), de manière à avoir une visibilité a minima sur quelques grandes catégories de dotations , en particulier en matière d'innovation ou d'investissement.
La commission vous demande d'adopter cet article ainsi modifié.
Article 24
Réforme
du financement des hôpitaux de proximité
Objet : Cet article modifie le mode de financement dérogatoire des hôpitaux de proximité dont la loi relative à l'organisation et à la transformation du système de santé a actualisé les missions, en le fondant sur une garantie pluriannuelle de financement et une dotation de responsabilité territoriale.
I - Le dispositif proposé
A. Une réforme destinée à accompagner la rénovation en cours du modèle des hôpitaux de proximité
• L'article 35 de la loi n° 2019-774 du 24 juillet 2019 relative à l'organisation et à la transformation du système de santé a actualisé la définition des missions des hôpitaux de proximité , issue de la loi de financement de la sécurité sociale pour 2015, dans l'objectif, affiché par le plan « Ma Santé 2022 », d'en faire le premier niveau de la gradation des soins hospitaliers et le lieu de coopérations renforcées avec la médecine de ville.
L'évolution de ces missions et activités (définies à l'article L. 6111-3-1 du code de la santé publique) est retracée dans le tableau ci-après.
L'entrée en vigueur de ces dispositions a été fixée à une date définie par décret et au plus tard le 1 er janvier 2021. Cette réforme devrait s'accompagner d'une montée en charge du modèle des hôpitaux de proximité, qui concerne à l'heure actuelle 241 établissements 95 ( * ) .
L'évolution des missions et activités des hôpitaux de proximité
|
Ancien modèle (LFSS pour 2016) |
Modèle rénové (Loi « santé » de juillet 2019) |
|
|
Statuts |
Établissements de santé publics ou privés |
Établissements de santé publics ou privés ou sites identifiés de ces établissements |
|
Missions |
- Contribuent par des coopérations avec les professionnels de santé du territoire à l'offre de soins de premier recours - Participent à la coordination et à la continuité des parcours de santé |
- Apportent un appui aux professionnels de santé de ville et autres acteurs notamment quand la prise en charge de leurs patients nécessite un cadre hospitalier |
|
- Contribuent à la permanence des soins et à la continuité des prises en charge en complémentarité avec les structures et les professionnels de la médecine ambulatoire - Favorisent la prise en charge des personnes en situation de vulnérabilité et leur maintien dans leur lieu de vie - Participent à la prévention et à des actions de promotion de la santé |
||
|
- Assurent au besoin l'orientation des patients vers des structures de second recours, avec lesquelles ils développent des partenariats |
- Orientent les patients qui le nécessitent vers les établissements de recours ou de référence ou vers les autres structures adaptées à leurs besoins |
|
|
Activités |
Médecine ou soins de suite et de réadaptation (SSR), dans la limite d'un seuil réglementaire |
Activités obligatoires : - médecine (et actes techniques) - consultations de plusieurs spécialités en complémentarité avec l'offre libérale - présence ou accès à des plateaux techniques d'imagerie, de biologie et à des équipements de télésanté |
|
Accès à des consultations spécialisées dans le cadre de coopérations |
||
|
Pas de chirurgie ou d'obstétrique |
Pas de chirurgie ou d'obstétrique. Mais dérogation possible pour une liste limitative d'actes chirurgicaux programmés (sur décision ARS) |
|
|
Missions optionnelles (en fonction des besoins et de l'offre de soins) dont : - médecine d'urgence - activités pré et post-natales - SSR - soins palliatifs - équipes mobiles |
• Les autres éléments de cette réforme - à savoir les critères de labellisation des futurs hôpitaux de proximité ainsi que les modalités de leur organisation et de leur gouvernance - ont été renvoyés à une prochaine ordonnance qui devrait être publiée dans le courant de l'année 2020 .
D'après les premières propositions concertées avec les acteurs, l'adhésion au label pourrait se faire sur la base du volontariat des établissements de santé.
B. La confirmation d'un modèle de financement dérogatoire et hybride
• La loi de financement de la sécurité sociale pour 2015 qui a défini le cadre des hôpitaux de proximité leur a adossé un financement mixte dérogatoire à la tarification à l'activité , « sous la forme de recettes issues de leur activité et d'une dotation forfaitaire » (article L. 162-23-16 du code de la sécurité sociale).
Ces modalités ont alors eu vocation à rectifier les dotations annuelles des anciens hôpitaux locaux, qui apparaissaient déconnectées de l'activité réelle de ces établissements, tout en évitant de leur appliquer strictement la tarification à l'activité afin de garantir une certaine stabilité de recettes.
Ces modalités, précisées par un décret du 20 mai 2016 96 ( * ) , conduisent à allouer à chaque établissement une dotation forfaitaire annuelle garantie qui comporte :
- une part « socle » correspondant à un pourcentage des recettes « historiques », à savoir les recettes d'hospitalisation perçues par l'établissement au cours des deux années précédentes (environ 70 %) ;
- une part modulable par l'agence régionale de santé (ARS), la dotation organisationnelle et populationnelle, en fonction des caractéristiques du territoire (part de la population âgée de plus de 75 ans ou en dessous du seuil de pauvreté, densité de population, nombre de médecins généralistes pour 100 000 habitants), ainsi que des engagements de coopération, de partenariat et de coordination pris par l'établissement.
D'après les indications transmises par la DGOS, ce modèle conduit à garantir le financement de l'activité de médecine , au niveau macro-économique, à hauteur de 90 % de la moyenne des recettes des deux années antérieures. En pratique, le niveau de la garantie est variable selon les établissements et s'échelonne entre 75 et 103 %.
Si la valorisation de l'activité est supérieure au niveau de la garantie, l'établissement perçoit un complément de recettes . D'après un bilan réalisé par l'ATIH en 2018, 60 % des établissements labellisés hôpitaux de proximité ont perçu un complément de recettes issu de leur activité.
• La réforme proposée conserve le caractère dérogatoire et mixte de ce financement mais en redéfinit les modalités .
Cette réforme répond à plusieurs objectifs. Il s'agit d'abord, en cohérence avec la nouvelle définition des missions de ces établissements, de faire de leur mode de financement un levier de décloisonnement des pratiques, de sécuriser les activités socles, de financer leurs missions élargies et d'éviter, comme le note l'étude d'impact, une « logique de « production » d'actes » en incitant plutôt à des actions de prévention et à la coordination.
D'après les indications transmises à votre rapporteur par la DGOS, la réforme proposée vise, en ce sens, à remédier aux limites du modèle actuel :
- en dépit du principe de garantie de financement, le modèle actuel ne permet pas aux établissements de disposer d'une visibilité suffisante sur leurs recettes d'une année sur l'autre, alors qu'elles sont soumises à plus de volatilité du fait d'un volume d'activité souvent plus réduit ;
- une autre limite est son caractère insuffisamment protecteur et peu encourageant au développement d'activités de coordination avec les acteurs du territoire : le niveau de fixation de la garantie à 90 % en moyenne des recettes antérieures fragilise les établissements confrontés à des baisses continues d'activité ;
- enfin, selon la DGOS, le modèle actuel repose de façon trop importante sur le volume de l'activité et insuffisamment sur les besoins concrets de la population du territoire.
Le modèle proposé vise, en réponse à ces limites, à introduire une part de pluriannualité dans la garantie (contre une réévaluation annuelle dans le modèle actuel), à rehausser le niveau de la garantie sur la base de 100 % de recettes historiques sur deux ou trois exercices (au lieu de 90 % aujourd'hui en moyenne sur la base des deux derniers exercices) et à financer, via une dotation spécifique, les missions conduites avec les acteurs du territoire.
À cet effet, il combine deux volets ( I ) :
- une garantie pluriannuelle de financement de l'activité de médecine obligatoire, dont le montant est fondé sur plusieurs critères : le volume d'activité, les recettes perçues « antérieurement » au titre de l'activité, les besoins de santé du territoire et la qualité des prises en charge. Comme à l'heure actuelle, un complément de recettes issues de l'activité pourra être alloué si celles-ci dépassent le montant de la garantie pour l'année donnée.
Des travaux se poursuivent en concertation avec les acteurs pour déterminer les modalités de fixation et de révision de cette garantie (dont le niveau devrait être déterminé a priori pour trois ans), ainsi que la part respective des différents critères. D'après les indications de la DGOS à votre rapporteur, le nombre d'années de référence des recettes pris en compte sera ainsi fixé par voie réglementaire afin de « trouver un juste équilibre entre le lissage des effets liés à la volatilité de l'activité et la juste prise en compte des dynamiques territoriales (à la hausse comme à la baisse) ». Quant aux critères de qualité, ils seraient spécifiques aux hôpitaux de proximité et distincts de ceux relevant du dispositif IFAQ d'incitation financière des établissements de santé à la qualité et à la sécurité des soins auquel les hôpitaux de proximité resteront éligibles ;
- une dotation de responsabilité territoriale , ayant vocation à accompagner les autres missions des hôpitaux de proximité (consultations de spécialités, plateaux techniques d'imagerie, de biologie et équipements de télésanté). Son montant sera déterminé en fonction « de l'organisation et de la réalisation » de ces missions et de la qualité des prises en charge. La dotation serait allouée par les ARS sur la base d'une contractualisation. D'après la DGOS, sous réserve des concertations qui se poursuivent avec les acteurs, les indicateurs de qualité seraient distincts de ceux pris en compte dans la garantie de financement et le dispositif IFAQ, afin qu'un établissement ne soit pas pénalisé plusieurs fois au titre d'un même critère.
Cette dotation de responsabilité territoriale inclura une indemnité allouée aux professionnels de santé libéraux concourant à l'exercice des missions des hôpitaux de proximité, y compris dans les établissements privés ; le III en pose le principe à l'article L. 6146-2 du code de la santé publique, au bénéfice des médecins, des sages-femmes et des odontologistes, concernés par le contrat d'exercice libéral ouvert à tous les établissements publics de santé.
Cette indemnisation, qui s'ajoutera aux honoraires conventionnels, vise, d'après l'étude d'impact, à « rémunérer les professionnels libéraux pour leurs activités hospitalières « non cliniques » nécessaires à l'exercice de la mission commune à tout hôpital de proximité » , par exemple les réunions d'équipe et temps de coordination avec les équipes soignantes, la participation aux instances médicales, l'organisation des admissions directes ou la préparation des sorties d'hospitalisation. Selon les indications transmises par la DGOS, plusieurs options sont à ce stade envisagées : soit une rémunération pour chaque type d'acte soit, de façon privilégiée, une rémunération plus forfaitaire, qui devra nécessairement s'adapter à la participation effective des professionnels dans la vie institutionnelle de l'hôpital de proximité.
• Le Gouvernement évalue le surcoût global du modèle cible de financement des hôpitaux de proximité à 100 millions d'euros à compter de 2021. A titre de comparaison, le surcoût du mode de financement actuel par rapport à un financement T2A est estimé par la DGOS à 21 millions d'euros 97 ( * ) .
La montée en charge serait progressive dès 2020, avec un surcoût de 40 millions d'euros anticipé pour les établissements qui entreront dans le dispositif cette année-là, qui inclut : 10 millions d'euros permettant d'offrir une garantie de recettes de 100 % (au lieu de 90 %), 15 millions d'euros en supposant les établissements qui seront labellisés plus nombreux et de taille plus importante et enfin 15 millions d'euros pour le versement dès 2020 de la dotation de responsabilité territoriale.
Lors de son audition, la DGOS a indiqué que la labellisation serait engagée à compter du second semestre 2020, une fois que l'ordonnance relative à la gouvernance et à l'organisation des hôpitaux de proximité aura été prise, avec une cible évaluée à 600 établissements supplémentaires.
II - Les modifications adoptées par l'Assemblée nationale
Outre des modifications rédactionnelles, l'Assemblée nationale a adopté, avec l'avis favorable du Gouvernement, des amendements de sa commission des affaires sociales précisant à la marge la rédaction de l'article :
- à l'initiative de Brahim Hammouche et de membres du Modem, afin de spécifier que les besoins de santé pris en compte pour la détermination de la garantie pluriannuelle de financement sont bien évidemment ceux de la « population » du territoire ;
- à l'initiative de Martine Wonner et de membres du groupe LREM, pour indiquer que les besoins de santé pris en compte sont ceux définis par le projet régional de santé et ses déclinaisons territoriales.
L'Assemblée nationale a complété en outre cet article d'un IV demandant un rapport au Parlement sur le financement des établissements hospitaliers dans les collectivités ultra-marines , afin d'évaluer les coefficients spécifiques de ces territoires et les différents modes de financement dont ils font l'objet (amendement de Justine Benin, députée du Modem, adopté avec l'avis favorable de la commission et du Gouvernement).
L'Assemblée nationale a adopté cet article ainsi modifié.
III - La position de la commission
• Votre rapporteur souscrit aux objectifs de cette réforme de sécuriser le financement des hôpitaux de proximité en leur donnant notamment une plus grande visibilité sur leurs ressources.
Cette réforme s'inscrit dans le prolongement de la rénovation, par la loi « santé » du 24 juillet 2019, du modèle et des missions des hôpitaux de proximité, que la commission avait accueillie favorablement en dépit des craintes légitimes qu'elle suscite, notamment chez des élus locaux, d'un déclassement de certains établissements périphériques. La commission avait considéré que ce modèle, à la condition d'être conçu comme réellement innovant, pouvait contribuer à la nécessité de repenser l'offre de soins à l'échelle des territoires selon une logique de complémentarité et de subsidiarité d'une part, et selon un principe de coordination des soins et de continuité des parcours de santé axé sur les besoins des patients d'autre part.
• Il est toutefois regrettable que cette réforme, dont un autre volet essentiel a été renvoyé à une ordonnance ultérieure, soit abordée par étapes et non de façon globale : les auditions conduites par votre rapporteur ont mis en avant les fortes attentes des médecins libéraux quant au rôle qui leur sera effectivement dévolu dans la gouvernance de ces établissements, et la crainte que ceux-ci, intégrés aux groupements hospitaliers de territoire, restent trop hospitalo-centrés. La volonté initiale d'en faire « l'hôpital de la médecine de ville » répond à un réel besoin de décloisonnement des prises en charge qui devra se traduire dans les ordonnances à venir.
Votre rapporteur note enfin qu'au-delà des principes généraux posés par cet article, qui reçoivent globalement l'approbation des fédérations hospitalières, ce sont les modalités pratiques de mise en oeuvre qui seront déterminantes , notamment pour prendre en compte la diversité des établissements et des dynamiques de territoire et ne pas fragiliser plus encore ceux situés dans un bassin de vie à la démographie peu dynamique.
Sous réserve de ces observations, la commission vous demande d'adopter cet article sans modification.
Article 25
Réforme
du financement de la psychiatrie
et évolution du modèle-cible
de financement des SSR
Objet : Cet article propose de simplifier le modèle-cible de financement des établissements de soins de suite et de réadaptation (SSR), dont l'application est reportée à fin 2020, et de faire évoluer le modèle de financement des établissements de soins psychiatriques vers un objectif de dépenses globalisé.
I - Le dispositif proposé
A. Soins psychiatriques et soins de suite et de réadaptation : des modalités de financement dérogatoires
Le financement des établissements de soins psychiatriques et des établissements de soins de suite et de réadaptation (SSR) fait l'objet de dispositions particulières , qui s'expliquent en grande partie par la spécificité des activités de ces secteurs. Contrairement aux activités de médicine, chirurgie et obstétrique (MCO), le financement fait encore intervenir un versement de dotations de fonctionnement , et ne s'intègre pas dans un objectif global de dépenses.
Concrètement, là où les établissements de MCO voient leur financement principalement couverts par des tarifs, les établissements de soins psychiatriques publics et privés non lucratifs bénéficient d'une dotation globale de fonctionnement et demeurent contraints de facturer les prestations d'hospitalisation et les spécialités pharmaceutiques auxquels ils ont recours à la sécurité sociale . Cette dichotomie de financement, en plus d'alourdir les modalités de gestion, ne favorise pas la responsabilisation des gestionnaires.
Le secteur des SSR, historiquement financé de la même manière, a connu en 2016 une réforme importante de son financement sur le modèle d'un objectif global de dépenses, dont l'application demeure toutefois différée.
1. Soins psychiatriques
Concernant les établissements de santé psychiatrique publics ou privés à but non lucratif , qui assurent environ 75 % des prestations de soins, leurs frais de fonctionnement sont financés par une dotation annuelle de financement (DAF) mentionnée à l'article L. 174-1 du code de la sécurité sociale (CSS), dite « DAF psy ». Il s'agit d'une enveloppe budgétaire fermée .
Cette dotation constitue la part principale de l'objectif des dépenses d'assurance maladie (ODAM) mentionné à l'article L. 174-1-1 du même code, lequel est arrêté chaque année par l'État et réparti en dotations régionales limitatives en fonction de critères très largement définis (« l'activité des établissements, des orientations des schémas régionaux ou interrégionaux de santé et des priorités nationales ou locales en matière de politique sanitaires »).
La « DAF psy » intègre par ailleurs l'ensemble des frais relatifs aux actes et consultations externes pratiqués en établissement psychiatrique, dans la limite de leurs tarifs. Les prestations hospitalières et les dépenses de médicaments sont, pour leur part, remboursées par la sécurité sociale aux établissements sur la base des tarifs arrêtés.
Concernant les établissements de santé psychiatrique privés à but commercial , qui sont exclus du champ de la « DAF psy », les dépenses remboursables de soins (prestations hospitalières et spécialités pharmaceutiques) réalisées sont encadrées par un objectif quantifié national (OQN) dont le montant est arrêté chaque année par l'État.
Enfin, de façon générale, les établissements de soins psychiatriques publics, privés à but non lucratif et privés à but commercial signataires de Cpom bénéficient d'une dotation nationale de financement des missions d'intérêt général et d'aide à la contractualisation (Migac), dite « Migac psy », pour leurs actions de formation, de recherche, d'amélioration de la qualité des soins ou de réponse aux priorités définies par les schémas nationaux ou régionaux.
2. Soins de suite et de réadaptation
a) Un nouveau modèle de financement qui prévoit la couverture globalisée des dépenses de soins
Aux termes de l'article L. 162-23 du CSS, créé par la loi de financement de la sécurité sociale (LFSS) pour 2016 98 ( * ) , les activités de soins de suite et de réadaptation (SSR) ne sont plus financées sous le régime de la dotation annuelle mais bénéficient d'une couverture intégrale assurée par un ODAM (dit « ODAM SSR »), sans distinction de statut public ou privé . Cet ODAM doit désormais couvrir l'ensemble des dépenses engagées par le secteur.
Ainsi, l'ODAM SSR distingue deux composantes, une principale et une complémentaire . La composante principale, qui substitue à la DAF une dotation modulée à l'activité (DMA), intègre des recettes issues de l'activité de soins , et rassemble :
- une dotation calculée chaque année sur la base de l'activité antérieure et valorisée par une fraction des tarifs appliqués aux prestations hospitalières remboursées par les régimes obligatoires de sécurité sociale. Divers coefficients concourant à l'équité géographique et au respect de l'Ondam par ailleurs peuvent également lui être appliqués ;
- un montant forfaitaire pour chaque séjour, auquel s'appliquent les mêmes règles de valorisation et de coefficient que précédemment exposé.
Pour sa part, la composante complémentaire finance, le cas échéant, la prise en charge des médicaments et des plateaux techniques spécialisés .
Par ailleurs, les établissements de SSR publics, privés à but non lucratif et privés à but commercial signataires de Cpom bénéficient, de façon similaire aux établissements de soins psychiatriques, d'une dotation nationale de financement Migac (dite « Migac SSR »). Contrairement à la dotation « Migac psy », celle-ci n'est cependant pas directement prélevée sur l'Ondam mais intégrée à l'ODAM SSR .
b) Des dispositions transitoires qui maintiennent pour une large part un financement à la dotation annuelle de financement
Ce mode de financement, dont l'objectif est de faire entrer le secteur des SSR dans le droit commun du financement hospitalier, n'est pour l'heure que très partiellement appliqué, en raison de plusieurs dispositions transitoires introduites par la LFSS pour 2016, puis la LFSS pour 2018 99 ( * ) :
- les établissements de SSR sont régis, jusqu'au 31 décembre 2019, par un mode de financement composite . Leur dotation comprend deux montants cumulatifs correspondant ( pour 90 % ) aux recettes telles que calculées d'après les modalités antérieures de la DAF et ( pour 10 % ) aux recettes calculées d'après les modalités de la DMA en vigueur. Par ailleurs la composante complémentaire du nouveau financement des SSR, couvrant les spécialités pharmaceutiques et les plateaux techniques spécialisés, ne sera versée qu'à partir du 1 er janvier 2020 pour les premières, et au plus tard au 1 er janvier 2020 pour les seconds ;
- jusqu'au 31 décembre 2019, l'ODAM SSR ne désigne pas, comme le prévoit le CSS, le financement des SSR selon ses modalités nouvelles, mais le financement versé selon ses modalités transitoires. Ainsi, une part importante de l'ODAM SSR tel qu'actuellement fixé par arrêté continue d'être constituée de dotations annuelles de financement ;
- en conséquence, du 1 er janvier 2020 jusqu'au 1 er mars 2022 au plus tard, les prestations d'hospitalisation en SSR demeurent , indépendamment du calcul de la DMA, remboursées par la sécurité sociale mais seulement pour une part, affectée d'un coefficient de transition.
Ces dispositions transitoires dessinent un cadre de financement particulièrement complexe dont le tableau ci-après tente de rendre compte.
Financement actuel des établissements
de
soins psychiatriques et de SSR
|
Soins psychiatriques |
SSR |
|||||
|
Public/Privé à but non lucratif |
Privé à but commercial |
Public/Privé à but non lucratif |
Privé à but commercial |
|||
|
90 % |
10 % |
90 % |
10 % |
|||
|
Frais de fonctionnement |
DAF psy |
DAF SSR |
ODAM SSR |
ODAM SSR |
||
|
Prestations hospitalières |
Droit commun |
OQN psy |
Droit commun |
« OQN SSR » |
||
|
Spécialités pharmaceutiques |
||||||
|
Spécialités pharmaceutiques « en sus » 100 ( * ) |
||||||
|
Actes et consultations externes |
DAF psy |
Droit commun |
||||
|
Formation, recherche, amélioration de la qualité |
Migac psy (au sein de l'enveloppe Migac) |
Migac SSR (au sein de l'ODAM SSR) (art. L. 162-23-8 CSS) |
Idem
|
|||
Source : Commission des affaires sociales
B. Bilan des campagnes budgétaires de 2018 et 2019
Plusieurs mesures budgétaires spécifiques ont été récemment prises par le ministère des solidarités et de la santé 101 ( * ) .
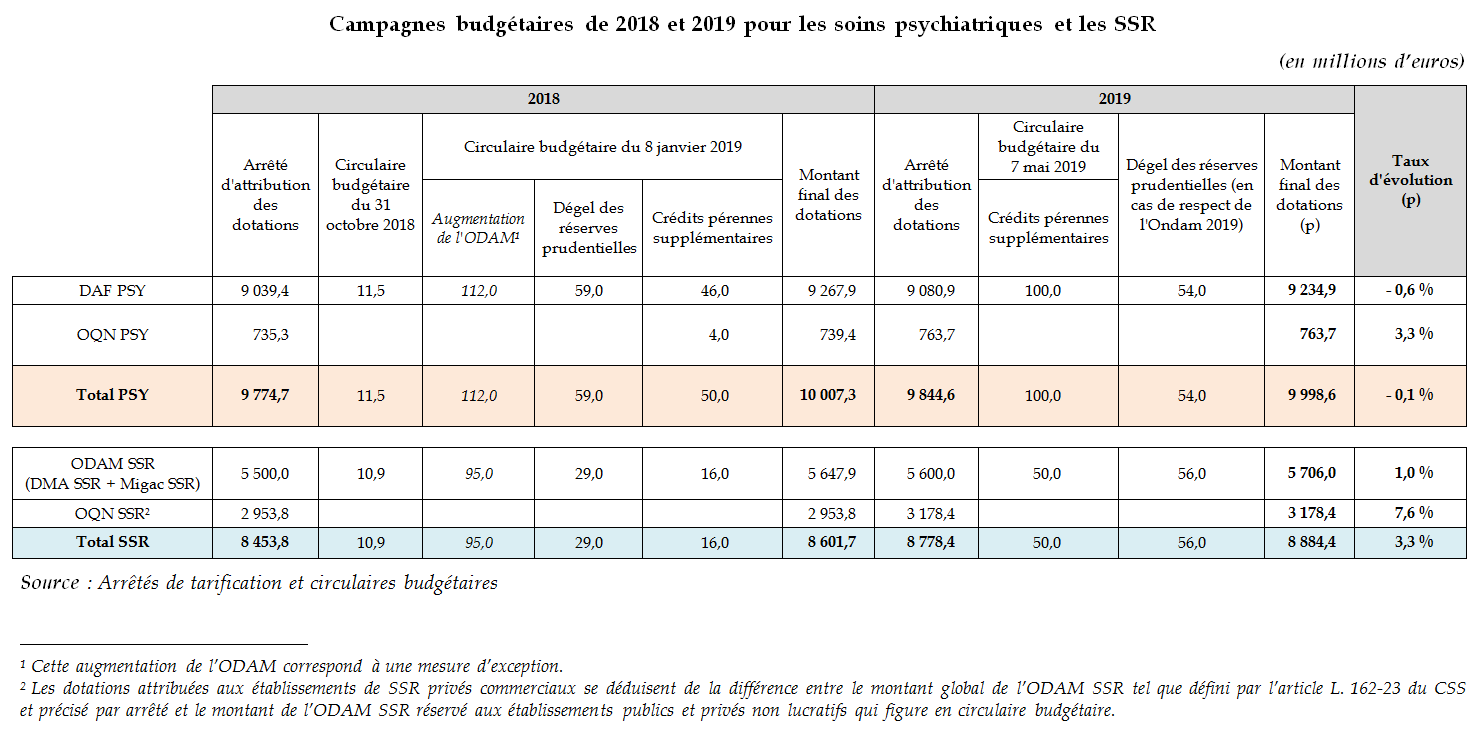
Malgré l'abondement notable de crédits intervenu en janvier 2018 (plus de 230 millions d'euros), dont certains ont été reconduits au cours de l'exercice 2019, les taux d'évolution des dotations des secteurs des soins psychiatriques et des SSR se maintiennent à des niveaux préoccupants, et permettent deux constats :
- même en neutralisant l'évolution des crédits de l'apport exceptionnel de 112 millions d'euros pour 2018, la progression du financement du secteur psychiatrique ( - 0,1 % en tenant compte de l'apport, 1 % sans en tenir compte ) évolue à un rythme significativement moins élevé que l'Ondam hospitalier (2 %) ;
- dans les deux secteurs des soins psychiatriques et des SSR, les taux de progression des dotations pour le secteur privé commercial sont beaucoup plus élevés que pour le secteur public et le secteur privé non lucratif .
C. Le dispositif proposé par l'article 25
1. Financement des SSR : une simplification du modèle et une prorogation des délais d'application
Le I de l'article 25 procède à plusieurs simplifications du modèle de financement des SSR.
Le 2° du I réécrit la base législative de la composante principale de la dotation attribuée aux établissements de SSR . Ses deux éléments sont désormais fondus dans un « financement mixte » associant des recettes directement issues de l'activité, avec un maintien des divers coefficients susceptibles de l'ajuster et d'une dotation forfaitaire visant à sécuriser de manière pluriannuelle le financement de ces activités, dont les modalités seront précisées par décret. L'ancien élément du montant forfaitaire par séjour se trouve donc en quelque sorte absorbé par la DMA , dont l'article 25 entend à terme faire la principale clef de financement des établissements de SSR.
Le 3° du I abroge les modalités de calcul de la DMA (dans ses contours actuels), qui faisaient intervenir une fraction des tarifs nationaux des prestations hospitalières pratiquées en établissements de SSR et le taux moyen de prise en charge par l'assurance maladie au titre de ces prestations. La nouvelle rédaction simplifie considérablement le calcul par un renvoi direct au tarif plein de la prestation. Il est néanmoins conservé la possibilité de minorer la nouvelle DMA d'un coefficient géographique, d'un coefficient visant à concourir au respect de l'Ondam et d'un coefficient visant à concourir au respect de l'ODAM SSR pour la part qu'il consacre au remboursement des spécialités pharmaceutiques.
Le 5° du I prévoit la possibilité pour les établissements de SSR, à partir du 1 er janvier 2021 ( V ), de se voir rembourser par la sécurité sociale, au titre de la liste en sus, les médicaments qui bénéficient d'une autorisation temporaire d'utilisation (ATU), à la condition que ces derniers aient été prescrits au sein d'un établissement de santé.
Le 4°, le 6° et le 7° du I procèdent à des coordinations.
Le II proroge les délais d'application de la réforme du financement des SSR. Il s'agit principalement de prolonger d'un an (jusqu'au 31 décembre 2020) les modalités transitoires de financement .
2. La création d'un « ODAM psychiatrie »
Le 8° du III prévoit une réforme en profondeur des modalités de financement des activités de soins psychiatriques. Est créée une sous-section spécifique composée de deux articles qui prévoient :
- la création d'un ODAM spécifique aux activités de psychiatrie . À l'instar du secteur des SSR, il s'agit d'intégrer l'ensemble des activités de soins psychiatriques au sein d'un unique objectif de dépenses (et d'ainsi fondre la « DAF psy » et les remboursements de la sécurité sociale au sein d'un système de financement unique) ;
- que cet objectif national sera composé de trois dotations distinctes :
• une dotation populationnelle , dont le montant tiendra principalement compte des besoins de la population concernée. Cette dotation globale sera ensuite répartie en dotations régionales, qui seront versées aux établissements par les ARS, en fonction de critères définis au niveau régional ;
• des dotations complémentaires , dont les montants tiendront compte de l'activité des établissements et qui seront versées en fonction de critères définis par arrêté ministériel ;
• d'une dotation accordée sur des critères de qualité et d'amélioration des soins.
Ces trois dotations ne seront pas attribuées aux établissements selon des modalités identiques. Seule la dotation populationnelle fera l'objet d'une répartition préalable entre régions, « dans le but de réduire progressivement les inégalités » subsistant actuellement entre ces dernières. Leur distribution aux établissements par les ARS, selon des critères propres à chaque territoire, s'effectuera dans un second temps. Les autres dotations seront versées uniformément par les ARS aux établissements selon des critères établis par arrêté ministériel.
Le texte indique que les médicaments bénéficiant d'une ATU ou ayant bénéficié d'une telle autorisation peuvent être inscrits sur la liste en sus de tout établissement de santé financé par dotation globale, dont feront désormais partie les établissements de soins psychiatriques ( 1°du I ).
Il est en outre précisé que les actes et consultations externes, actuellement financés par la « DAF psy », demeureront couverts par l'ODAM psychiatrie ( 11° du III ).
Par coordination, le financement des transports sanitaires , qui reste à la charge de l'établissement prescripteur, est inclus dans le nouveau mode de financement des établissements psychiatriques ( b) du 2° du III ).
La création de l'ODAM psychiatrie se traduisant par une uniformité du financement pour tous les établissements, de statut public ou privé, l'article 25 procède à l'abrogation des dispositifs spécifiques au secteur psychiatrique privé ( 1°, a) du 2°, 3°, 4°, 5°, 6° et 12° du III ).
Elle se traduit également par une redéfinition du modèle de financement des établissements dispensant des soins psychiatriques aux personnes incarcérées, dont le financement par DAF est maintenu mais dont le calcul prendra désormais en compte les nouvelles modalités d'attribution de la DMA ( 7° du III ).
Enfin, le 9° du III renomme la sous-section rassemblant les dispositions communes aux différents établissements financés par objectif global de dépenses en y intégrant les activités psychiatriques, et le IV intègre les coordinations nécessaires au code de la santé publique (CSP).
À noter que, bien que l'application de ce nouveau mode de financement soit prévu pour le 1 er janvier 2021 ( V ), il n'est pas prévu de dispositif de financement transitoire .
3. Activités continuant de faire l'objet d'un versement en dotation annuelle de financement (DAF)
L'article 25 tire les conséquences du retrait des activités de soins psychiatriques de l'ODAM défini à l'article L. 174-1-1 du CSS rassemblant les financements des établissements maintenus dans le système de la DAF. Le 13° du III redéfinit ainsi les contours de cet ODAM spécifique pour les activités de soins d'établissements limitativement énumérés (établissements de soins pour personnes incarcérées, unités de soins de longue durée, Institution nationale des invalides, établissement public de santé de Mayotte...).
À ce titre, les paragraphes 14° à 22° du III procèdent aux coordinations qu'appelle cette réécriture.
II - Les modifications adoptées par l'Assemblée nationale
Outre onze amendements rédactionnels, l'Assemblée nationale a adopté treize amendements à l'article 25, dont la grande majorité apporte des clarifications à la définition et à la répartition des dotations qui structurent le nouveau mode de financement des soins psychiatriques :
- un amendement du rapporteur général visant à intégrer à la future couverture des soins psychiatriques assurée par l' ODAM les frais de « prise en charge » en plus des « frais d'hospitalisation », afin d'acter la pleine participation du soin psychiatrique au virage ambulatoire ;
- un amendement du rapporteur général précisant que les dotations régionales composant l'ODAM devront tenir compte du projet régional de santé (PRS) ;
- quatre amendements du rapporteur général et de plusieurs députés du groupe Mouvement démocrate (Modem) sont venus préciser les critères de définition de la dotation populationnelle et de la dotation complémentaire qui composent l'ODAM , afin que ces dernières tiennent également compte du virage ambulatoire ;
- les critères de répartition entre régions de la dotation populationnelle qui compose l'ODAM ont par ailleurs fait l'objet de trois amendements spécifiques : l'un déposé par Mme Wonner visant à y intégrer la spécificité sociologique et démographique des territoires et les deux autres déposés par le rapporteur général visant à tenir compte du maillage de l'offre médico-sociale ainsi que des indications du projet régional de santé (PRS) ;
- trois amendements du rapporteur général et un amendement de plusieurs députés du groupe Les Républicains ont apporté des précisions aux critères de répartition par les ARS des dotations populationnelles régionales entre établissements, qui devront tenir compte des projets territoriaux de santé mentale (PTSM) ainsi que des avis des associations d'usagers et de représentants des familles ;
- un amendement du rapporteur général a affiné les critères de répartition par les ARS des dotations complémentaires régionales , qui pourront tenir compte des activités auxquelles participe l'établissement destinataire et non plus seulement des activités qu'il assure .
L'Assemblée nationale a adopté cet article ainsi modifié.
III - La position de la commission
Votre commission ne peut que se réjouir du dispositif prévu à l'article 25, qui marque la fin pour le secteur psychiatrique d'un système de financement inadapté . Elle tient toutefois à inviter le Gouvernement à la plus grande prudence lorsqu'arrivera le temps de la déclinaison de ce modèle par voie réglementaire.
La réforme du financement des établissements d'hébergement pour personnes âgées dépendantes (Ehpad), déployée depuis 2016, s'était appuyée sur un principe similaire d'une meilleure visibilité des financements et d'une convergence des modèles tarifaires entre les différents établissements, quel que soit leur statut public ou privé. Pour autant, l'application d'un modèle tarifaire uniforme s'était immanquablement traduite par des « gagnants » et des « perdants », sans que les pertes n'aient été préalablement objectivées et anticipées.
Cette remarque formulée, et compte tenu des rythmes actuels de progression des dotations dans le secteur psychiatrique, largement favorables au secteur privé commercial, on peut néanmoins s'attendre à ce que la convergence tarifaire se traduira par un rééquilibrage au bénéfice du secteur public . Votre rapporteure s'y montrera d'autant plus attentive que la direction générale de l'offre de soins (DGOS) lui a confirmé que « s'agissant plus spécifiquement de l'évolution des crédits alloués à chaque secteur, il n'y aura pas de convergence à proprement parler, mais le financement de l'ensemble des établissements à travers un modèle de financement commun permettra de rapprocher sensiblement les dynamiques de progression des enveloppes allouées à chacun des secteurs ».
Les orientations actuelles du nouveau financement du secteur psychiatrique, largement inspirées des résultats de la mission « Réforme des modes de financement et de régulation » conduite par Jean-Marc Aubert, dessinent un compromis dont l'ensemble des acteurs semble se satisfaire. La dotation populationnelle devrait normalement représenter 80 à 85 % de l'ONDAM psychiatrie avec, à l'issue des travaux de l'Assemblée nationale, une pondération spécifique par des éléments relatifs à la précarité et à la couverture médico-sociale. Votre commission a souhaité, par l'amendement n° 195 , y ajouter la prise en compte des soins pédopsychiatriques .
Votre commission se montrera également attentive aux effets de l'introduction d'une dotation liée à l'activité, que le secteur aura à s'approprier . La DGOS a indiqué à votre rapporteure que « la répartition entre les deux dotations pour les différents secteurs pourra être différente afin de garantir de la souplesse et de la progressivité dans la montée en charge de la réforme ». Un amendement n° 196 vise ainsi à pallier les effets potentiellement délétères que pourra provoquer l'introduction d'une dotation liée à l'activité pour les établissements assurant des soins psychiatriques. En effet, les établissements montrant la file active la plus faible sont souvent les établissements les moins dotés en personnel médical, sans être ceux dont les besoins sont pour autant les moins élevés. La transition vers le nouveau modèle de financement ne doit pas se traduire, pour ces établissements en particulier, souvent situés en zone sous-dotée, par une baisse de leur dotation.
Enfin, bien que l'Assemblée nationale se soit montrée soucieuse de préciser les critères de définition et de répartition des dotations régionales par les ARS aux établissements, votre commission a été fortement sensibilisée aux risques de maintien d'inégalités infrarégionales que le silence du législateur permettrait de perpétuer . La ministre des solidarités et de la santé a certes assuré, lors de son audition par votre commission le 19 octobre 2019, que « la répartition territoriale [...] sera assurée par les agences régionales de santé, qui connaissent les besoins et les indicateurs de santé des territoires », mais il conviendrait de préciser ces critères de répartition en y ajoutant les spécificités des territoires en matière de présence de professionnels de santé. C'est l'objet de l'amendement n° 197 .
Votre commission vous demande d'adopter cet article ainsi modifié.
Article 26
Réforme du
ticket modérateur à l'hôpital
Objet : Cet article modifie le mode de calcul du ticket modérateur à l'hôpital.
I - Le dispositif proposé
A. Le droit en vigueur : un dispositif censément transitoire, peu transparent, et qui accroît les restes à charge
1. Un dispositif initialement transitoire
Depuis la tarification à l'activité (T2A), instaurée par la loi du 18 décembre 2003 de financement de la sécurité sociale pour 2004, les établissements de santé sont financés selon le principe de tarifs correspondant à un paiement forfaitaire par type de séjour donné. Les ressources tirées antérieurement de la dotation globale ne pouvant correspondre aux tarifs nouveaux, le législateur a mis en place des dispositifs transitoires, notamment s'agissant de la part restant à la charge de l'assuré.
Avant la réforme de la T2A, l'assuré participait à l'ensemble des charges supportées par l'établissement au moyen du ticket modérateur, cette part de 20 % appliquée aux tarifs journaliers de prestations (TJP) calculés dans chaque établissement , ainsi que par le paiement du forfait journalier hospitalier fixé nationalement.
Maintenir le même niveau de participation des assurés exigeait, une fois passé à la T2A, de choisir entre asseoir le ticket modérateur sur les seules prestations d'hospitalisation mais en en augmentant le taux, ou maintenir son taux à 20 % mais en l'asseyant également sur les autres composantes du financement des établissements - dotations finançant les missions d'intérêt général et d'aide à la contractualisation (Migac), forfaits annuels d'urgence et de greffe, etc.
La deuxième solution a été choisie par l'article 33 de la loi du 18 décembre 2003, en sorte que le ticket modérateur non seulement fait participer l'assuré au financement des Migac et des fonds annuels d'urgence, mais surtout dépend toujours des TJP, établis pour chaque établissement. Pour les établissements privés, en revanche, le ticket modérateur est calculé sur l'assiette des groupes homogènes de séjour, c'est-à-dire sur la base des tarifs de l'assurance maladie pour la pathologie dont relève le patient hospitalisé.
Conçu pour être transitoire, ce mécanisme n'en a pas moins été prorogé jusqu'au 31 décembre 2019 par la LFSS pour 2016, après l'avoir été jusqu'au 31 décembre 2015 par la LFSS pour 2013 et jusqu'au 31 décembre 2012 par la LFSS pour 2009. Le rapporteur général pour le Sénat du PLFSS pour 2016, Jean-Marie Vanlerenberghe, en a donné dans son rapport sur la branche assurance maladie la double raison :
• d'une part, « l'impossibilité, pour les établissements anciennement sous dotation globale, d'établir une facturation individuelle sur la base des tarifs nationaux » ;
• d'autre part, l'effet financier qu'aurait eu la modification du mode de calcul du ticket modérateur, compte tenu du moindre niveau des tarifs nouvellement définis par rapport au financement global des séjours.
Ces deux raisons sont restées d'actualité. Dans les observations soumises au Conseil constitutionnel à l'occasion de sa saisine par soixante députés sur l'article 77 du PLFSS pour 2016, le gouvernement de l'époque assurait que sa fin provoquerait une « perte nette de recettes pour les établissements d'environ 2,5 Md€ par an puisque ces derniers sont dans l'incapacité technique d'établir une facture patient sur la base des groupes homogènes de séjour. Cette perte représenterait environ 5 % des recettes des établissements concernés, ce qui mettrait ces structures dans une situation financière extrêmement difficile voire insoutenable et poserait la question de la continuité des soins » .
2. Un dispositif complexe
Le TJP est calculé en divisant les charges d'exploitation engagées par l'hôpital au cours des séjours des malades par le nombre de journées prévisionnel. Il est fixé tous les ans par les ARS sur proposition du directeur d'établissement et doit suivre des modalités de calcul précisées par voie réglementaire.
Aux termes du décret n° 2009-213 du 23 février 2009, « Pour les activités de médecine, chirurgie, obstétrique et odontologie, les tarifs de prestations servant de base au calcul de la participation des patients [...] sont établis pour au moins chacune des catégories suivantes :
1° L'hospitalisation complète en régime commun en distinguant :
a) Services spécialisés ou non ;
b) Services de spécialités coûteuses ;
c) Services de spécialités très coûteuses ;
2° L'hospitalisation à temps partiel ;
3° La chirurgie ambulatoire ;
4° L'hospitalisation à domicile ;
5° Les interventions de la structure mobile d'urgence et de réanimation ».
3. Un dispositif qui a contribué à alourdir le reste à charge des patients
Dans son rapport sur l'application des lois de financement de la sécurité sociale de 2016, la Cour des comptes relève que « les valeurs retenues ont largement correspondu, au moins jusqu'en 2012, aux demandes de chaque établissement en fonction de ses conditions d'exploitation, de ses coûts et de sa situation financière propre, sans toujours respecter les modalités de calcul réglementaires ».
Il en a résulté une grande hétérogénéité des TJP, à types de soins identiques, selon les établissements. Selon l'Observatoire citoyen des restes à charge en santé, l'éventail des TJP pratiqués en médecine par les CHU allait en 2012 de 862 euros par jour à l'AP-HP à 1 476 euros par jour au CHU de Rouen ; en dehors des CHU, certains TJP étaient inférieurs à 300 euros. Des travaux effectués par la Cour des comptes en 2013 sur des données 2011 ont mis en évidence des écarts similaires : de 140 à 1 832 euros en médecine, de 429 à 2 243 euros en chirurgie et de 146 à 1 318 euros en psychiatrie.
La Cour des comptes, dans le même rapport, estimait en outre que « l'augmentation de la valeur des TJP entre 2004 et 2012 à un rythme nettement supérieur à celle de la dépense hospitalière a[vait] augmenté de près de 1 Md€ la part relative de financement des ménages au sein de la CSBM ».
Depuis 2012 et à l'exception de 2018, la circulaire ministérielle annuelle relative à la campagne tarifaire des établissements de santé reconnaît bien volontiers que « le niveau des tarifs journaliers de prestation (TJP) entre établissements est très hétérogène et entraîne une inégalité dans le reste à charge des patients qu'il est nécessaire de modérer », mais n'appelle les gestionnaires qu'au strict respect des règles de calcul prévues par le décret de 2009 et à la « baisse progressive des TJP supérieurs de plus de 15 % au niveau auquel ils devraient être » en application de ces règles. En 2019 ainsi, la diminution des TJP de ces établissements devait atteindre 3 %.
Depuis la LFSS pour 2016, les établissements non concernés par cette consigne sont certes tenus de respecter un plafond d'augmentation des TJP, fixé par le décret n° 2016-650 du 20 mai 2016 au niveau du sous-objectif « Dépenses relatives aux établissements de santé tarifés à l'activité » déterminé par la LFSS de l'année en cours. Ainsi, en 2019, l'augmentation des TJP ne pouvait-elle dépasser 2,4 %.
Reste donc que les hôpitaux publics peuvent depuis 2005 laisser aux patients des restes à charges importants. Leur niveau est d'autant moins sensible depuis que la loi n° 2013-504 du 16 juin 2013 a rendu obligatoire la prise en charge du ticket modérateur par les contrats collectifs que les employeurs sont tenus de souscrire au profit de leurs employés.
D'après Santéclair, le TJP représentait en 2016 pour de grandes mutuelles 54 % des dépenses hospitalières prises en charge en MCO, charge en hausse de 31 % sur trois ans 102 ( * ) . Les complémentaires couvrent ainsi une part de marché de plus en plus large des frais de santé des patients hospitalisés.
B. Une réforme nécessaire, des modalités à surveiller
L'article 26 maintient le principe des TJP mais les rationalise selon une nomenclature nationale.
Le I 1° dispose que la participation de l'assuré aux frais d'hospitalisation est proportionnelle aux bases de calcul mentionnées à l'article L. 162-20-1 du code de la sécurité sociale, qui instaure ( I 2° ) une « tarification nationale journalière des prestations » établie par voie réglementaire en fonction des soins donnés et du niveau de l'activité de l'établissement où ils sont donnés, et ne concerne que les établissements des secteurs public, privé non lucratif participant au service public hospitalier, et privé non lucratif ayant opté pour la dotation globale de financement.
Pour les autres établissements privés, les GHS continueront à servir de base au calcul de la participation de l'assuré pour les activités de médecine, chirurgie, gynécologie-obstétrique et odontologie. Les activités de psychiatrie et les soins de suite et de réadaptation en revanche reposeront elles aussi sur la nouvelle tarification nationale journalière des prestations.
Le II précise que la tarification nationale journalière et les GHS continueront à servir également à l'exercice de recours contre des tiers, à la facturation de soins des patients relevant d'un autre système de sécurité sociale coordonné avec le système français, et à la facturation des soins et de l'hébergement des patients qui ne sont pas couvertes par un régime d'assurance maladie.
Le III dispose que les GHS restent applicables aux patients affiliés au régime d'assurance maladie, maternité, invalidité et décès de Mayotte, relevant d'un des régimes de la protection sociale généralisée de la Polynésie française, bénéficiant de l'aide médicale d'État ou bénéficiant de la prise en charge des soins urgents.
Les I 3° et 4° suppriment des dispositions devenues inutiles. Les 5° , 6° et 7° sont de coordination.
Le II procède à des coordinations dans le code de la santé publique. Son 2° retire notamment au directeur d'établissement le pouvoir de fixer les « propositions de tarifs » des prestations de soin.
Le III modifie la LFSS pour 2003 pour repousser au 31 décembre 2020 l'extinction du régime actuel.
Le IV précise que le nouveau dispositif entrera en vigueur le 1 er janvier 2021 pour les activités de MCO, et à la date prévue au V de l'article 25 du projet de loi pour les activités de psychiatrie et de SSR, soit le 1 er janvier 2021.
Le V prévoit une période transitoire : à compter du 1 er janvier 2021 et jusqu'au 31 décembre 2023 au plus tard, les montants annuels de la dotation populationnelle de chaque établissement, prévue par l'article 25 du présent PLFSS pour les activités de psychiatrie et de SSR, ainsi que de la dotation Migac pour les autres établissements, seront modulés selon les modalités fixées par arrêté afin de limiter l'effet de la mise en oeuvre de la tarification nationale journalière des prestations sur les recettes des établissements.
D'après l'étude d'impact, les centres hospitaliers non régionaux et les établissements à but non lucratif seront les plus nombreux à bénéficier favorablement de la réforme ; les proportions sont inversées pour les centres hospitaliers régionaux et les centres de lutte contre le cancer. Les niveaux de gains et de pertes restent cependant limités : plus ou moins 0,9 % des recettes totales, la catégorie la plus touchée étant celle des centres hospitaliers non régionaux, avec une variation possible de plus ou moins 1,3 % des recettes totales. Ces effets seront par ailleurs lissés sur trois ans comme le prévoit le V du dispositif.
II - Les modifications adoptées par l'Assemblée nationale
L'Assemblée nationale a adopté quatre amendements rédactionnels à l'initiative du rapporteur général.
Un amendement déposé par des députés du groupe LREM a également été adopté, avec un avis favorable du Gouvernement, qui inclut les services de santé des armées dans le champ d'application de cet article.
L'Assemblée nationale a adopté l'article ainsi modifié.
III - La position de la commission
Cet article est le seul du PLFSS à s'attaquer, si l'on peut dire, au reste à charge des patients à l'hôpital, dont de nombreuses études, telles celles du Haut conseil pour le financement de l'assurance maladie de 2013, celle de l'Irdes de 2016 ou encore l'Observatoire de la Mutualité française de 2019, ont montré le caractère inéquitable.
Même si l'on peut regretter qu'il ne soit pas explicitement mis fin à un régime initialement transitoire et que le ticket modérateur ne soit pas enfin assis sur les GHS, cette réforme semble à votre rapporteure de nature à remédier aux inconvénients du système actuel, source d'importants restes à charge et d'inégalités de traitement des assurés selon les établissements.
Certains acteurs auditionnés s'inquiètent néanmoins de la concomitance d'une telle réforme, pour les activités de SSR et de psychiatrie du secteur privé, avec celle de leur financement, opérée par l'article 25, craignant notamment un transfert de charges au détriment des patients. La période transitoire prévue par le V semble toutefois de nature à absorber l'impact du nouveau régime sur la situation financière de ces établissements.
L'essentiel de la mise en oeuvre de cette réforme dépend cependant de mesures réglementaires qui devront être prises. Compte tenu de leur impact sur les assurés, il conviendra de surveiller de près leur élaboration.
La commission vous demande d'adopter cet article sans modification.
Article 26 bis
(nouveau)
Réforme du financement des services d'urgence
Objet : Cet article, inséré par l'Assemblée nationale, vise à réformer le mode de financement des services d'urgence en combinant une dotation populationnelle, un financement à l'activité et une dotation complémentaire liée notamment à la qualité des prises en charge.
I - Le dispositif adopté par l'Assemblée nationale
L'Assemblée nationale a adopté, avec l'avis « très » favorable du Gouvernement, un amendement de la commission des affaires sociales issu d'une initiative de Thomas Mesnier et des membres du groupe La République en Marche, réformant le financement des services d'urgence.
Cet amendement est issu du travail mené par Thomas Mesnier et Pierre Carli, président du conseil national de l'urgence hospitalière, dans le cadre de la mission sur l'amélioration de la situation dans les services d'urgences que leur a confiée la ministre des solidarités et de la santé en juin 2019. Il traduit en outre des annonces faites par la ministre dans le « pacte de refondation des urgences » présenté le 9 septembre dernier.
• Le financement des structures d'urgence comporte à l'heure actuelle plusieurs éléments essentiellement liés à l'activité , dont les modalités sont fixées par voie réglementaire :
- un forfait « accueil et traitement des urgences » (ATU) d'un montant de 25,32 euros dû pour chaque passage aux urgences non suivi d'une hospitalisation dans un service de MCO (médecine, chirurgie, obstétrique) ou dans une unité d'hospitalisation de courte durée (UHCD) du même établissement ;
- un « forfait annuel urgences » (FAU) qui s'élève depuis 2016 à 730 000 euros pour les 9 000 premiers passages, avec des suppléments par tranche de 2 500 passages au-delà de 9 000.
À ces financements s'ajoutent ceux liés à la facturation des consultations et examens de biologie et d'imagerie, ainsi que des recettes de séjour en cas d'hospitalisation en UHCD.
D'après la Cour des comptes 103 ( * ) , les services d'urgence des établissements de santé ont accueilli 21,2 millions de passages en 2016 pour un coût de 3,1 milliards d'euros à la charge de l'assurance maladie et des autres financeurs (complémentaires santé et ménages). Ces dépenses ont connu une hausse moyenne annuelle de 4 % depuis 2013, pour une progression de l'activité de l'ordre de 5 % en moyenne annuelle pour les passages non suivis d'hospitalisation.
• Le présent article rétablit au sein du code de la sécurité sociale un article L. 162-22-8-2 réformant, tout en l'érigeant au niveau législatif, le mode de financement de l'activité de soins de médecine d'urgence au sein des établissements de santé publics comme privés.
Le modèle de financement mixte et dérogatoire proposé ( I ) s'appuie sur trois composantes :
- une dotation populationnelle qui aurait vocation à être majoritaire (de l'ordre de 66 %) dont le montant serait arrêté par région en fonction des besoins de la population - en fonction notamment de la démographie - et des caractéristiques de l'offre de soins, puis répartis entre les établissements selon des critères définis au niveau régional ;
- des recettes liées à l'activité , dans lesquelles est introduit le principe d'une modulation selon l'intensité des prises en charge des patients. Les modalités restent à travailler en concertation avec les acteurs mais d'après les indications transmises à votre rapporteur, ces critères pourraient notamment prendre en compte l'âge des patients, étroitement corrélée à la complexité des prises en charge ;
- enfin, un financement complémentaire à la qualité , sur la base de critères liés à « l'amélioration de la qualité et de l'organisation des prises en charge de cette activité » , dont les modalités sont renvoyées à un décret en Conseil d'État. Cette dotation est distincte du dispositif IFAQ de financement à la qualité des soins auquel les établissements concernés sont par ailleurs éligibles et les critères (qui pourraient par exemple prendre en compte le temps de passage) devraient être également distincts.
• L'entrée en vigueur de cette réforme est prévue à compter du 1 er janvier 2021 ( II ).
Ce nouveau modèle de financement concernerait également les SMUR (services mobiles d'urgence et de réanimation) qui sont actuellement financés sur la base d'une dotation MIG (mission d'intérêt général), dans l'objectif, d'après les indications transmises à votre rapporteur, de favoriser une plus grande mutualisation entre les SMUR et les services d'urgence. En revanche, les services d'aide médicale urgente (SAMU ou centre 15) en sont explicitement exclus.
II - La position de la commission
• Le caractère inadapté du mode actuel de financement des services d'urgence a été relevé par de nombreux rapports.
La commission des affaires sociales l'a ainsi souligné dans un rapport de juillet 2017 sur les urgences hospitalières 104 ( * ) , qui mettait en évidence le paradoxe selon lequel ce mode de financement « incite à l'inflation de l'activité sans pour autant couvrir les besoins des établissements. » Ce rapport préconisait un mode de financement incitant à une meilleure coopération entre les acteurs de la prise en charge des soins non programmés, en conservant un financement mixte (forfaitaire et à l'activité) et en modulant le montant du financement à l'activité en fonction de la gravité des pathologies et de la technicité des actes réalisés.
La Cour des comptes, dans le rapport précité de février 2019, relève, dans le même sens, un financement désincitatif à la coopération avec les autres acteurs et à la réorganisation des prises en charge : « dans le cadre actuel du financement des structures d'urgence, la croissance du nombre de passages permet aux établissements de dynamiser leurs recettes alors que le report d'une partie des passages évitables sur la médecine de ville, porteur d'économies pour l'assurance maladie, est financièrement pénalisant pour eux. En effet, la tarification au forfait opère une péréquation entre les cas légers, qui sont « sur-financés », et les cas lourds, qui sont « sous-financés ». »
• Votre rapporteur accueille donc favorablement cette réforme dont les modalités restent encore cependant à discuter avec les acteurs concernés. Il conviendra notamment de prendre en compte le besoin de rééquilibrage des inégalités territoriales dans la répartition des moyens tout en prenant en compte les besoins spécifiques de certains territoires.
Sous réserve d'un amendement du rapporteur portant des clarifications rédactionnelles (amendement n° 146), la commission vous demande d'adopter cet article ainsi modifié.
Article 26 ter
(nouveau)
Rapport au Parlement sur le financement et l'évolution
du financement des missions de recherche
et d'innovation des
établissements de santé
Objet : Cet article, inséré par l'Assemblée nationale, demande la remise d'un rapport au Parlement faisant le point sur le financement des missions de recherche et d'innovation des établissements publics de santé et leur évolution.
I - Le dispositif adopté par l'Assemblée nationale
Cet article résulte d'un amendement présenté par Jean-Louis Touraine et des membres du groupe La République en Marche, approuvé par la commission des affaires sociales de l'Assemblée nationale en dépit des réserves exprimées par son rapporteur général, et adopté avec l'avis de sagesse du Gouvernement.
Il demande la remise au Parlement, dans un délai de six mois, d'un rapport sur « le financement et l'évolution des missions de recherche et d'innovation au sein des établissements publics de santé » .
Il s'agit comme le soulignent les auteurs de l'amendement dans son exposé sommaire, de réaliser un bilan permettant d'évaluer l'utilisation des dotations au titre des MERRI (missions d'enseignement, de recherche, de recours et d'innovation) afin que ces crédits ne servent pas de variable d'ajustement, « d'en mesurer les impacts concrets dans l'organisation et le fonctionnement des établissements publics de santé et d'envisager une évolution du financement des missions de recherche et d'innovation » .
II - La position de la commission
Votre rapporteur partage les inquiétudes exprimées par les auteurs de l'amendement quant au financement de la recherche et de l'innovation au sein des établissements de santé .
Destinés à compenser, lors de la mise en place de la tarification à l'activité, les surcoûts liés notamment aux activités de recherche et d'enseignement, les crédits MERRI constituent une sous-catégorie de l'enveloppe MIGAC 105 ( * ) , dont ils représentent 54 % soit 3,5 milliards d'euros en 2016 106 ( * ) . Le rapport sur les centres hospitaliers universitaires (CHU) établi par la Cour des comptes à la demande de la commission des affaires sociales 107 ( * ) appelait à réformer le mode d'allocation des crédits MERRI, notant leur dilution sur un plus grand nombre d'établissements éligibles, leur caractère volatile et imprévisible et un « défaut de pilotage stratégique » .
Votre rapporteur a par ailleurs déjà attiré l'attention sur des modes de financement inadaptés en matière d'accès à l'innovation, à travers notamment l'enveloppe fermée du RIHN (répertoire des actes innovants hors nomenclature) en matière de biologie médicale, qui ne permet pas de couvrir l'ensemble des dépenses engagées par les établissements de santé 108 ( * ) .
Quelle que soit l'importance de ces enjeux, votre rapporteur doute qu'une demande de rapport sur le sujet - si tant est que ce rapport soit effectivement transmis au Parlement - soit une solution efficace.
L'article L. 162-22-19 du code de la sécurité sociale prévoit déjà la présentation au Parlement, avant le 15 septembre de chaque année, d'un rapport sur les actions menées sur le champ du financement des établissements de santé, dont un volet porte explicitement sur les dotations MIGAC, leur évolution et critères d'attribution .
L'analyse des crédits MERRI s'inscrit pleinement dans ce cadre. Il serait cependant souhaitable que ce rapport soit effectivement transmis au Parlement, dans les délais prévus, ce qui est en pratique rarement le cas. La dernière version transmise date en effet de 2017.
C'est la raison pour laquelle votre rapporteur propose la suppression de cet article (amendement n° 147).
La commission vous demande de supprimer cet article.
Article 27
Refonte de la
nomenclature des actes médicaux et paramédicaux de ville
Objet : Cet article engage la refonte de la nomenclature des actes médicaux et paramédicaux relevant de la médecine de ville.
I - Le dispositif proposé
A. Le droit existant : une procédure longue et complexe, préjudiciable à la pertinence et à l'efficience des soins
1. Une procédure longue et complexe
Depuis la loi n° 2004-810 du 13 août 2004, les actes pris en charge par l'assurance maladie doivent, pour être remboursés aux professionnels, être inscrits sur une liste des actes et prestations prévue par l'article L. 162-1-7 du code de la sécurité sociale.
La décision de l'Union des caisses d'assurance maladie (Uncam) du 11 mars 2005 a distingué deux parties dans cette liste : la classification commune aux actes médicaux (CCAM), qui regroupe les actes techniques réalisés par les médecins, et la nomenclature générale des actes professionnels (NGAP), qui regroupe les actes des chirurgiens-dentistes, des sages-femmes et des auxiliaires médicaux. À cela s'ajoute la nomenclature des actes de biologie médicale (NABM), mentionnée à l'article L. 162-1-7-1.
Ces trois nomenclatures représentent plus de 12 000 catégories d'actes et redistribuent un montant de l'ordre de 27,4 milliards d'euros, soit environ 14 % de la dépense de santé nationale et 16,5 % des dépenses à la charge de l'assurance maladie.
L'inscription d'un acte sur cette liste implique de suivre la procédure législative et réglementaire prévue par les articles L. 162-1-7, R. 162-52 et R. 162-52-1 du code de la sécurité sociale, qui se recompose comme expliqué ci-après.
|
1. La demande d'inscription peut émaner de sociétés savantes ou de professionnels de santé ou, plus fréquemment, de l'Uncam, qui fait part de son intention au ministre compétent ainsi qu'aux organisations représentatives des professionnels de santé autorisés à pratiquer cet acte, et saisit la Haute autorité de santé (HAS) pour avis. 2. L'évaluation médicale de l'acte est effectuée, au sein de la HAS, à la demande de son collège, par la commission nationale d'évaluation des dispositifs médicaux et des technologies de santé (Cnedimts), qui propose un avis formellement rendu, ensuite, par le collège de la HAS, au plus tard à la fin du sixième mois qui suit sa saisine par l'Uncam - délai renouvelable une fois. 3. L'évaluation scientifique et technique est ensuite effectuée par l'Uncam : l'acte est hiérarchisé par des experts choisis par elle, dont les propositions sont transmises à une instance de cohérence composée d'autres experts, qui présente le score obtenu par l'acte à une des commissions créées pour chacune des professions dont les rapports avec les organismes d'assurance maladie sont régis par une convention - ou « commissions de hiérarchisation des actes et prestations » (CHAP). 4. L'évaluation médico-économique et la tarification sont aussi de la compétence de l'Uncam. L'article R.162-52 indique qu'elle « définit le tarif de l'acte ou de la prestation dans le respect des règles de hiérarchisation », et que « lorsque l'acte constitue une alternative à des traitements thérapeutiques déjà inscrits (...) l'Uncam évalue l'opportunité de l'inscription de l'acte et définit, le cas échéant, son tarif au regard des coûts de mise en oeuvre comparés de ces différents traitements ». 5. L'information et la consultation . L'Uncam consulte ensuite l'Union nationale des organismes d'assurance maladie complémentaire (Unocam) sur l'inscription ou le remboursement des actes et prestations ; elle dispose de six mois pour rendre son avis. 6. L'inscription de l'acte sur la liste des actes et prestations pris en charge par l'assurance maladie relève du collège des directeurs de l'Uncam, qui peut ne pas donner suite à un avis favorable de la CHAP. 7. L'approbation par les ministres compétents doit être recueillie sous 45 jours. 8. La publication au Journal officiel a lieu dans un délai de 30 jours en l'absence d'opposition des ministres. La révision de la hiérarchisation ou de la tarification d'un acte implique de suivre à nouveau les étapes 3 à 8. |
Cette procédure se voit fréquemment reprocher sa longueur. Le Snitem estimait en 2012 à 1 000 jours le délai moyen d'examen des dossiers, dont 500 jours pour la HAS, et 581 jours pour l'Uncam. Dans le dernier de ses rapports disponibles en ligne faisant figurer cette information, la Cnedimts indique qu'en 2017, trois catégories de produits concernant des descriptions génériques avaient été évaluées, pour une durée moyenne de 18 mois.
2. Une complexité préjudiciable à la pertinence et à l'efficience des soins
La procédure décrite ci-dessus se heurte à deux grandes catégories de critiques, bien analysées par le rapport de l'Igas de 2012 et celui de la task force « Réforme du financement du système de santé », rendu en janvier 2019 109 ( * ) :
• d'une part, elle est mal comprise et mal acceptée. « La procédure actuelle est vécue par les acteurs concernés autres que l'Uncam comme longue, peu lisible, génératrice de dysfonctionnements et d'incompréhension des arbitrages rendus » , écrit l'Igas en 2012 110 ( * ) .
• d'autre part, elle manque d'objectivité. Comme le résume l'Igas, la procédure « est pilotée de fait par la CNAM. La CNAM prend l'initiative de soumettre un acte nouveau à la HAS, elle anime les travaux nécessaires à la hiérarchisation qui seront soumis à la Commission de hiérarchisation des actes professionnels (CHAP), elle décide de l'inscription » [...] « Le fait que l'organisme en charge de la maîtrise des dépenses ait la maîtrise entière du processus peut, à tort ou à raison, faire craindre un conflit d'intérêt ».
Les conséquences de telles caractéristiques sont celles que l'on peut attendre d'une nomenclature mal calibrée sur l'état de l'art médical : frein à l'intégration des innovations, maintien d'activités ne correspondant plus aux pratiques en cours, création de rentes économiques pour certaines professions ou certaines activités sans rapport avec l'utilité collective des actes réalisés, etc.
Dans le rapport précité, l'Igas conclut ainsi : « ces constats militent pour qu'une équipe dédiée, indépendante de la CNAM, prenne en charge la procédure d'actualisation de la nomenclature. Il faut en effet que les moyens consacrés à la gestion de l'introduction des actes nouveaux soient préservés et que l'objectif des personnels qui s'y consacrent soit exclusivement la qualité descriptive de la classification ».
« Il convient donc de constituer une équipe indépendante de la CNAM en charge d'actualiser la nomenclature, de conduire les opérations de réévaluation des points travail et de maintenir les coûts de la pratique [...] La CHAP pourrait dans cette hypothèse conserver le rôle qu'elle joue aujourd'hui en matière d'orientation générale des travaux méthodologiques, de définition des priorités de travail et d'instance de validation des travaux techniques effectués. »
Une révision de la méthode d'inscription des actes à la liste des actes et prestations devrait ainsi se donner pour principes directeurs la simplification d'une part, et d'autre part une distinction plus nette entre la hiérarchisation médicale et technique des actes et leur tarification.
B. Le dispositif proposé
L'article 27, dans sa version issue du texte déposé par le Gouvernement sur le bureau de l'Assemblée nationale, disposait que l'ensemble des actes et prestations inscrits sur une liste mentionnée à l'article L.162-1-7 du code de la sécurité sociale feraient l'objet, à la date d'entrée en vigueur de la présente loi de financement, d'un examen en vue d'une nouvelle hiérarchisation dans un délai de cinq ans, selon des modalités précisées par décret en Conseil d'État.
Votre rapporteure accueillait initialement avec circonspection la méthode retenue initialement, consistant à autoriser un décret en Conseil d'État à déroger, pour engager la réforme de la nomenclature, aux règles de concertation avec les professionnels fixés par le code de la sécurité sociale.
II - Les modifications adoptées par l'Assemblée nationale
L'Assemblée nationale a adopté en séance publique trois amendements rédactionnels, ainsi que l'amendement n° 1958 rectifié du Gouvernement, qui réécrit entièrement l'article 27.
Le 1° du I dispose que l'inscription sur la liste peut être provisoire pour les actes innovants dans des conditions fixées par décret et faire l'objet d'une révision en respectant une durée minimale de trois ans renouvelable une fois.
La procédure prévue par l'article L. 162-1-7 est ensuite réécrite de la manière suivante, et ne concerne que les actes faisant partie de la CCAM :
1. La demande d'inscription est adressée par l'Uncam ou par les ministres chargés de la santé et de la sécurité sociale pour avis à la HAS. La demande peut aussi émaner, outre de conseils nationaux professionnels, d'associations d'usagers agréées, ce qui est nouveau. Dans ce cas de figure, c'est la HAS qui fixe le délai dans lequel elle rend son avis.
2. L'évaluation médicale de l'acte est effectuée par la HAS. Elle porte sur l'évaluation du service attendu ou du service rendu de l'acte ou la prestation qui lui est soumis ainsi que, le cas échéant, sur les actes existants dont l'évaluation pourrait être modifiée en conséquence. À la demande du collège, l'avis de la HAS peut être préparé par une commission spécialisée - rôle actuellement tenu par la Cnedimts. Cet avis est transmis à l'Uncam dans un délai de six mois suivant le dépôt de la demande, renouvelable une fois pour les évaluations complexes.
3. L'évaluation scientifique et technique est de la compétence d'un nouvel organe, le Haut conseil des nomenclatures (HCN). Le HCN établit son rapport en tenant compte des enjeux de pertinence médicale. Ce rapport est ensuite remis, dans un délai de six mois , renouvelable une fois pour les évaluations complexes, à l'Uncam, après avis simple de la commission professionnelle compétente pour la profession de médecin.
Le HCN, chargé de proposer une méthodologie de description et de hiérarchisation des actes et prestations et d'étudier à cette fin ceux qui lui sont soumis, est composé d'un nombre égal de médecins libéraux et de praticiens hospitaliers, ainsi que de personnes qualifiées nommées dans des conditions fixées par décret. Un représentant de la HAS, un représentant des patients ainsi que le président de la commission professionnelle compétente pour la profession de médecins assistent à ses travaux. Il remet chaque année un rapport annuel d'activité après consultation de l'ensemble des acteurs impliqués dans la hiérarchisation, qui est rendu public. Son secrétariat est assuré par l'Uncam.
Les commissions professionnelles compétentes pour chacune des professions sont des commissions dont les rapports avec les organismes d'assurance maladie sont régis par une convention, à l'instar des CHAP actuelles. Elles sont chargées du suivi de l'activité de hiérarchisation. Présidées par une personnalité désignée d'un commun accord par leurs membres, elles sont composées de représentants des syndicats représentatifs des professionnels de santé et de représentants de l'Uncam. Un représentant de l'État assiste à leurs travaux.
La commission compétente pour la profession des médecins est tenue informée des travaux du HCN qui lui adresse ses rapports. Elle valide la proposition de méthodologie de description et de hiérarchisation des actes et prestations du HCN. Elle émet également un avis sur les rapports du HCN relatifs à la description et à la hiérarchisation de l'acte ou de la prestation dans un délai défini par décret.
Les actes cliniques et les actes effectués par les biologistes-responsable et biologistes coresponsables mentionnés à l'article L. 162-14 sont inscrits par l'Uncam après avis de la commission compétente pour leur profession.
4. La phase de tarification n'apparaît pas dans le dispositif - elle est actuellement régie par la partie réglementaire du code - mais, d'après la CNAM, resterait inchangée, c'est-à-dire de la compétence de l'Uncam en fonction de la hiérarchisation.
5. Consultation. L'Uncam sollicite l'avis de l'Unocam et, le cas échéant, de la HAS lorsque la décision porte sur l'évaluation du service attendu ou du service rendu d'un acte ou d'une prestation.
6. Inscription. Les conditions d'inscription d'un acte ou d'une prestation, leur inscription et leur radiation sont décidées par l'Uncam. Les décisions d'inscription de l'Uncam sont réputées approuvées sauf opposition motivée des ministres chargés de la santé et de la sécurité sociale.
Le VIII dispose que tout acte ou prestation inscrit fait l'objet d'un examen en vue d'une nouvelle hiérarchisation, dans les conditions prévues aux alinéas précédents, dans les cinq ans qui suivent l'entrée en vigueur de la décision de l'Union nationale des caisses d'assurance maladie.
Le IX dispose que les conditions d'application du présent article sont précisées par décret en Conseil d'État.
Le 2° abroge, par cohérence, l'article L. 162-1-7-2 du code de la sécurité sociale, relatif à la nomenclature des actes de biologie médicale.
Le 3° modifie l'article L. 162-1-8 pour disposer que :
a) En l'absence de hiérarchisation par la commission professionnelle compétente, dans un délai qui ne peut être supérieur à cinq mois à compter de la transmission à l'Uncam de l'avis de la HAS mentionné au troisième alinéa du même article et de l'évaluation mentionnée au deuxième alinéa du présent article, l'Uncam peut procéder à la hiérarchisation d'un acte dont le service attendu est suffisant sous réserve qu'il entre dans l'une des quatre catégories précisée par le b) :
• actes présentant un niveau d'amélioration du service attendu déterminé et dont l'inscription sur la liste est nécessaire à l'utilisation ou à la prise en charge par l'assurance maladie
• actes pratiqués uniquement au sein d'un établissement de santé et ayant ou étant susceptibles d'avoir un impact significatif sur l'organisation des soins et les dépenses de l'assurance maladie
• actes ayant fait l'objet d'une tarification provisoire dans le cadre d'une expérimentation, et présentant un niveau d'amélioration du service attendu déterminé, ou étant susceptibles d'avoir un impact significatif sur l'organisation des soins et les dépenses de l'assurance maladie.
• actes inscrits dans un protocole de coopération ayant fait l'objet d'une proposition par le comité national des coopérations interprofessionnelles
Les d) , e) et f) procèdent à des coordinations.
Le g) ajoute un alinéa à l'article L. 162-1-8 pour disposer que le ministre de la santé peut procéder d'office à l'inscription ou à la radiation d'un acte ou d'une prestation pour des raisons de santé publique par arrêté pris après avis de l'HAS. Dans ce cas, il fixe la hiérarchisation de l'acte ou de la prestation dans le respect des règles ci-dessus. Les tarifs de ces actes et prestations sont publiés au JO.
Le 4° ajoute une phrase à l'article L. 162-14-1 précisant que la ou les conventions déterminent pour les actes techniques la trajectoire de convergence vers le prix de l'acte établi à partir de la hiérarchisation déterminée par le HCN.
Le 5° précise, ainsi que le I 1° , que l'Uncam assure le secrétariat du HCN.
Le II précise que la liste fait l'objet d'une révision, dont les modalités seront précisées par un décret en Conseil d'État. Ce dernier précisera notamment l'organisation des travaux du HCN, chargé de cette révision.
Le III indique que le présent article entre en vigueur le 1 er avril 2020.
III - La position de la commission
Votre rapporteure observe que ce nouveau dispositif permettra a priori de mieux distinguer les phases de description et de tarification, et donc de tenir compte des critiques récurrentes relatives au manque d'impartialité de la procédure actuelle, et donc de rendre la nomenclature plus adéquate à l'état de l'art médical.
La représentation à parité des médecins libéraux et hospitaliers au sein du Haut conseil des nomenclatures est également de nature à rassurer les professionnels de santé, que votre rapporteure a auditionnés sur ce point.
Il n'est en revanche pas manifeste que l'inscription d'actes nouveaux gagnera en célérité, sauf à imaginer que la procédure de révision emprunte une forme beaucoup plus simplifiée. Donner au HCN la compétence d'élaborer la procédure de révision devrait permettre d'assurer la qualité des travaux.
En toute hypothèse, le décret précisant la composition exacte et le mode d'organisation du HCN et le décret en Conseil d'État prévu pour préciser la procédure de révision devront faire l'objet d'une attention particulière.
La commission vous demande d'adopter cet article sans modification.
Article 28
Réforme
de la prise en charge des dispositifs médicaux
Objet : Cet article porte une réforme importante de la prise en charge de certains dispositifs médicaux. Il précise les obligations incombant à chaque acteur de la mise sur le marché, prévoit une procédure de référencement afin de mieux réguler la couverture et la prise en charge de certains dispositifs et instaure la possibilité d'une remise en bon état d'usage pour les fauteuils roulants.
I - Le dispositif proposé
A. Des précisions apportées au circuit de distribution
L'attribution individuelle d'un dispositif médical 111 ( * ) fait potentiellement intervenir plusieurs acteurs, dont chacun est défini au sein d'un règlement européen dédié 112 ( * ) .
|
Définitions des acteurs
Fabricant : personne physique ou morale qui fabrique ou remet à neuf un dispositif ou fait concevoir, fabriquer ou remettre à neuf un dispositif, et commercialise ce dispositif sous son nom ou sous sa marque ; Mandataire : personne physique ou morale établie dans l'Union européenne ayant reçu et accepté un mandat écrit d'un fabricant, situé hors de l'Union, pour agir pour le compte du fabricant aux fins de l'accomplissement des tâches déterminées liées aux obligations incombant à ce dernier ; Importateur : personne physique ou morale établie dans l'Union qui met un dispositif provenant d'un pays tiers sur le marché de l'Union ; Distributeur : personne physique ou morale faisant partie de la chaîne d'approvisionnement, autre que le fabricant ou l'importateur, qui met un dispositif à disposition sur le marché jusqu'au stade de sa mise en service. Par ailleurs, l'article 15 du PLFSS pour 2020 insère un nouvel article L. 165-1-1-1 au code de la sécurité sociale qui introduit la qualité générique d' exploitant d'un produit de santé , défini comme « le fabricant ou le distributeur de ce produit, en assurant l'exploitation. L'exploitation comprend la commercialisation, ou la cession à titre gratuit, sur le marché français. [...] Lorsqu'un mandataire agit pour le compte d'un fabricant, le mandataire est regardé comme étant exploitant ». |
Il a été précisé à votre rapporteure au cours de ses auditions que la mise sur le marché des dispositifs médicaux pouvait faire intervenir deux types de distributeurs : les distributeurs « en gros » , assimilables aux grossistes-répartiteurs du secteur du médicament, et les distributeurs « au détail » , désignant les prestataires (pharmacies ou dépôts de prestataires spécialisés) en lien direct avec le patient.
Les articles du code de la sécurité sociale (CSS) relatifs aux obligations incombant aux acteurs de la filière des dispositifs médicaux au moment de leur mise sur le marché ne visent actuellement que les « fabricants ou distributeurs ». Or, malgré la définition précise qu'en donne le règlement européen précité, cette désignation semble maintenir une ambiguïté dommageable sur l'application de ces obligations aux détaillants, qui sont les premiers concernés par la qualité et le suivi du service rendu.
C'est pourquoi l'article 28 substitue la rédaction « exploitant ou distributeur au détail » , la qualité d'exploitant comprenant celle de distributeur en gros, à celle actuellement en vigueur ( A, B, 1°, a) et b) du 3° du D, 1° du F, G, I, 2° à 4° du J, K à Q du I ).
Inversement, la même ambiguïté se retrouve pour les obligations incombant aux acteurs de la filière au moment de l'inscription du dispositif médical sur la liste des produits et prestations remboursables (LPPR) . La mention des « fabricants ou distributeurs » fait actuellement courir le risque aux distributeurs de détail de voir leur responsabilité engagée pour manquement à des obligations relatives aux qualités techniques de fabrication, qui pourtant ne serait imputable qu'à l'exploitant. C'est pourquoi le C du I protège les distributeurs de détail des sanctions que l'agence nationale de sécurité des médicaments et des produits de santé (ANSM) pourrait prendre quand un produit ne respecte pas les spécifications techniques qui ont motivé son inscription sur la LPPR.
B. Le contingentement du marché des dispositifs médicaux par une nouvelle procédure de référencement
Le B du I prévoit, sur initiative ministérielle, le conditionnement possible de l'inscription d'un dispositif médical sur la LPPR à une procédure de référencement visant à limiter la distribution des produits et, le cas échéant, des prestations associées, selon plusieurs critères. Ces derniers, que le projet de loi ne mentionne que de manière générique, s'articuleront toutefois autour de trois types de leviers :
- la qualité intrinsèque , à savoir le respect des spécifications techniques et la qualité générale des produits et prestations ;
- le nombre , à savoir le volume nécessaire pour garantir un approvisionnement suffisant du marché ;
- le prix , à savoir les conditions tarifaires au regard de l'objectif d' efficience 113 ( * ) des dépenses d'assurance maladie.
Le référencement des produits et prestations sélectionnés ne pourra dépasser une durée de deux ans prorogeable un an . Il pourra également, à la condition de ne pas engendrer une situation de monopole , exclure de la LPPR les produits ou prestations les moins avantageux au regard des critères de sélection.
Il s'agit, en droit comme en fait, d'une faculté discrétionnaire accordée au pouvoir réglementaire d'administrer, pour une durée maximale de trois ans, un ou plusieurs segments du marché des dispositifs médicaux remboursables .
Le E du I précise par ailleurs que la mise en oeuvre de la procédure de référencement pourra impliquer, de la part des exploitants ou des distributeurs au détail des produits et prestations sélectionnés, un engagement à « fournir des quantités minimales de produits et prestations sur le marché français en cas de sélection » et à « garantir une couverture suffisante du territoire français ». L'article prévoit l'application d'un régime de sanctions administratives ou financières en cas de non-respect de ces engagements.
C. La possibilité nouvelle d'une remise en bon état d'usage
1. Un contexte propice au recyclage de certains dispositifs médicaux : l'important reste à charge lié aux fauteuils roulants
La couverture financière des dispositifs médicaux fait intervenir un nombre importants de financeurs publics et privés. L'inscription de ces dispositifs médicaux sur la liste des produits et prestations remboursables (LPPR) des régimes obligatoires de sécurité sociale ouvre droit pour l'assuré à une couverture partielle par l'assurance maladie. Pour le cas des accidentés de la vie ou des accidents du travail, il convient également de tenir compte de la participation des assureurs agissant conséquemment à l'engagement de la responsabilité civile du commettant.
Les dispositifs médicaux implicitement visés par la possibilité de remise en bon état d'usage prévue par l'article 28 recouvrent principalement la catégorie des fauteuils roulants (catégorie IV de la LPPR), qui sont également revêtus de la qualité d' aides techniques et à ce titre attribuables spécifiquement aux personnes en situation de handicap. Le conseil départemental est dans ce cas appelé à intervenir, de façon subsidiaire par rapport aux financeurs précédents 114 ( * ) , à travers le versement de la prestation de compensation du handicap (PCH) et par l'intervention des fonds départementaux de compensation (FDC).
Enfin, vient éventuellement s'ajouter la participation facultative d'autres financeurs publics (Carsat, collectivités territoriales) et des assureurs complémentaires individuels ou collectifs , auxquels les assurés ont souscrit par ailleurs.
À l'issue de ces financements, le reste à charge net assuré directement par la personne handicapée se situe en moyenne autour de 12 % du coût total de l'aide 115 ( * ) . Des études plus récentes ont porté cette moyenne à 16 % , en montrant qu'une majorité de départements parvenait à contenir le reste à charge net à un niveau inférieur à 15 % , mais qu'il pouvait être porté à plus de 20 % pour une petite quinzaine d'entre eux 116 ( * ) .
Ce niveau moyen de prise en charge, a priori relativement contrôlé, cache des réalités très disparates, non seulement entre départements, mais également entre natures d'aides techniques financées. Ce sont les dépenses de fauteuils roulants verticalisateurs ou à propulsion électrique qui sont les plus difficilement absorbables par les personnes. Une étude menée en 2014 par l'association AFM-Téléthon a montré l'importance de ces restes à charge spécifiques.
Reste à charge pour le financement
d'un
fauteuil roulant non manuel
|
Montant moyen (en euros) |
|
|
Coût initial |
22 895 |
|
Reste à charge après financements légaux sans conditions de ressources (assurance maladie, PCH, FDC) |
9 837 |
|
Reste à charge après financements complémentaires sous conditions de ressources (autres collectivités et caisses de sécurité sociale) |
5 458 |
|
Reste à charge après financements privés |
1 850 |
Source : AFM-Téléthon
Votre rapporteure tient à souligner que, bien que le reste à charge moyen puisse être « réduit » à 1 850 euros, le reste à charge « légal » moyen s'élève quant à lui à 9 837 euros. Les réductions consécutives dépendent de conditions « extra-légales », qui varient selon l'implantation géographique ou la situation personnelle de l'intéressé.
2. L'innovation proposée par l'article 28
a) Un nouveau circuit de distribution
L'une des principales innovations de l'article 28 réside dans l'inscription au sein du CSS de la possibilité d'une remise en bon état d'usage d'un dispositif médical . Bien qu'ils ne soient pas explicitement désignés, la mesure concernera essentiellement les fauteuils roulants et autres dispositifs médicaux ayant la qualité d'aides techniques à destination des personnes en situation de handicap.
La remise en bon état d'usage d'un dispositif médical doit être soigneusement distinguée de son renouvellement , quand bien même ce dernier implique ponctuellement une opération de réparation. En effet, le renouvellement d'un dispositif inscrit sur la LPPR constitue un droit du seul bénéficiaire , ouvert et pris en charge par l'assurance maladie dans les cas où le produit initialement prescrit est hors d'usage, irréparable ou inadapté, ou encore lorsque son utilisation doit se prolonger au-delà de sa durée normale d'utilisation au profit du même bénéficiaire (article R. 165-24 du CSP).
Or la remise en bon état d'usage, telle que décrite par l'article 28 du projet de loi, vise un dispositif médical dont l'usage, une fois sa première durée d'utilisation écoulée, peut être prolongé en faveur d' un autre bénéficiaire . Ne figurant pas tel quel au sein de l'article R. 165-24, il n'ouvre actuellement aucun droit à la prise en charge par l'assurance maladie. Autrement dit, l'exemplaire d'un produit spécifique inscrit sur la LPPR ne peut voir sa prise en charge prolongée qu'au profit unique de son premier attributaire (en cas de besoin) .
Le II de l'article 28 instaure donc un nouvel article au sein du code de la santé publique (CSP) disposant que certains dispositifs médicaux à usage individuel figurant sur une liste spécifique établie par arrêté ministériel pourront faire l'objet d'une remise en bon état d'usage en vue d'une réutilisation par un patient différent .
Cet article prévoit également que la réalisation de cette remise en bon état usage puisse être subordonnée au respect de certains critères de qualité et sécurité sanitaire ainsi qu'à une procédure d'homologation dispensée par des centres ou des professionnels spécifiquement autorisés.
b) Un nouveau régime d'obligations
L'introduction de ce nouveau régime de distribution de dispositifs médicaux emporte deux types d'obligations ( E du I ), les unes à la charge du distributeur et les autres à la charge des bénéficiaires :
- nouvelles obligations incombant au distributeur : l'article prévoit la possibilité d'adjoindre aux règles de distribution des dispositifs médicaux concernés l'obligation, à la charge du distributeur au détail, d' informer le patient de la disponibilité d'un exemplaire remis en bon état d'usage. Le non-respect de cette obligation pourra se voir sanctionné d'une pénalité financière d'un montant maximal du 5 % du chiffre d'affaires hors taxes total réalisé en France par le distributeur ;
- nouvelles obligations incombant aux bénéficiaires : l'éligibilité d'un dispositif à sa remise en bon état d'usage pourra se traduire, si un arrêté ministériel le prévoit, par la double obligation pour le bénéficiaire de s'engager à restituer le dispositif à un centre autorisé lorsqu'il n'en a plus l'usage ou lorsqu'il ne correspond plus à son besoin médical et de s'acquitter d'une consigne .
Il est par ailleurs précisé que la consigne ne pourra donner lieu, au moment de son versement, à aucune prise en charge au titre de la sécurité sociale, qu'elle soit assurée par les régimes obligatoires ou par les régimes complémentaires ( R du I ) ou de la protection sociale. Elle sera rétrocédée au bénéficiaire au moment de la restitution du dispositif.
c) Un nouveau régime de fixation des prix
En outre, le CEPS acquiert la possibilité de moduler à la baisse, par convention avec l'exploitant ou le distributeur au détail ou par décision unilatérale, le tarif de responsabilité associé à un dispositif médical, en fonction de sa remise en bon état ( 2° du F du I ). Aux termes de l'article R. 165-15 du CSS, le CEPS peut déjà, par convention ou par décision unilatérale , modifier le prix des produis ou des prestations figurant sur la LPPR .
L' impact sur le reste à charge lié aux fauteuils roulants, bien que non explicitement évoqué par l'article 28, ne manquera pas d'être sensible. Le CEPS, lors de son audition par votre rapporteure, a assuré à cette dernière qu'un dispositif médical remis en bon état d'usage bénéficiera d'un tarif révisé .
d) Un nouveau système d'informations
Enfin, l'article prévoit la création d'un référentiel nommé « Enregistrement relatif à la circulation officielle des dispositifs médicaux » rassemblant les informations relatives à la mise en circulation du produit, à l'identification du patient bénéficiaire et aux opérations de réparation et de maintenance.
D. Une obligation de déclaration des prix des dispositifs médicaux au CEPS
Le H du I insère un nouvel article au CSS visant à obliger tout exploitant ou fournisseur de distributeur au détail de dispositif médical à déclarer au CEPS le prix auquel a été vendu chaque produit ou prestation . L'inapplication de cette disposition pourra se traduire par une pénalité fixée par le CEPS ne pouvant être supérieure à 5 % du chiffre d'affaires hors taxes de l'exploitant ou du fournisseur intéressé.
E. Un renforcement du contrôle de la qualité de la prescription
Le 2° et le c) du 3° du D du I visent à pouvoir soumettre le prescripteur du dispositif médical à un dispositif d'évaluation visant à établir la qualité de la prise en charge ainsi que la satisfaction du patient, qui pour l'heure ne concernait que le distributeur. Cette possibilité nouvelle est par ailleurs assortie d'une sanction financière pouvant aller jusqu'à 10 000 euros par an en cas de méconnaissance par le prescripteur des obligations afférentes.
II - Les modifications adoptées par l'Assemblée nationale
Outre cinq amendements rédactionnels du rapporteur général, l'Assemblée nationale a apporté trois modifications importantes à l'article 28 .
Un amendement de plusieurs députés du groupe Mouvement démocrate (Modem) vise à qualifier le centre chargé de la remise en bon état d'usage de « centre homologué », afin de garantir la sécurité du dispositif médical qui y sera reconditionné.
Un amendement identique déposé par l'ensemble des groupes politiques de l'Assemblée nationale a supprimé le dispositif de la consigne , considérant que l'engagement du bénéficiaire à restituer le dispositif médical suffisait à sécuriser son cycle de renouvellement.
Enfin, un amendement identique déposé par plusieurs groupes politiques de l'Assemblée nationale a rendu obligatoire la procédure d'homologation du dispositif médical remis en bon état d'usage, alors que le texte initial s'était limité à une procédure facultative.
L'Assemblée nationale a adopté cet article ainsi modifié.
III - La position de la commission
Les dispositions diverses qui figurent à l'article 28, présentées comme formant un ensemble cohérent visant à faciliter l'accès des patients aux dispositifs médicaux, n'emportent pas toutes l'adhésion de votre commission.
A. Sur la procédure de référencement
Votre commission se montre particulièrement réservée quant à l'instauration d'une « procédure de référencement », dont le terme déguise mal la possibilité ouverte au pouvoir réglementaire de contingenter, de façon unilatérale, tout un segment du marché des dispositifs médicaux . En l'occurrence, bien que le texte actuel ne prévoie pas de restriction particulière, la direction de la sécurité sociale a indiqué que cette procédure serait limitée aux fauteuils roulants.
Votre commission entrevoit trois effets principaux .
En premier lieu , comme les représentants des prestataires de santé à domicile en ont fait part à votre rapporteure, le référencement ne pourrait avoir de sens que pour un produit dont les prix actuellement pratiqués entraînent un reste à charge important pour le bénéficiaire. Or le prix d'un fauteuil roulant n'est pas tant le fait de l'objet considéré strictement comme dispositif médical inscrit à la LPPR, mais peut être accompagné de certaines aides techniques qui, n'y étant pas inscrites, ne pourraient être concernées par le référencement.
Un rapport récent de l'IGAS mentionne que « sur environ 10 000 aides techniques [attribuables aux personnes en situation de handicap ou en perte d'autonomie et donc susceptibles d'engendrer un reste à charge] , 250 sont aussi inscrites en LPPR [donc seraient éligibles à la remise en bon état d'usage] » 117 ( * ) . L'IGAS indique par ailleurs que le « fondement de la distinction entre aides techniques et dispositifs médicaux n'apparaît pas d'évidence et ce d'autant moins que les informations objectives qui pourraient fonder cette discrimination sont rares ou absentes ».
Ainsi, bien qu'un fauteuil roulant soit en soi inclus au titre IV de la LPPR, les personnes auditionnées par votre rapporteure lui ont signalé que le reste à charge lié au fauteuil résidait essentiellement dans cet appareillage technique , constitué de pièces indépendantes, qui ne figure pas, pour sa part, sur la LPPR. Telle qu'actuellement décrite, cette procédure de référencement court donc le risque de manquer sa cible, faute d'englober les éléments réellement facteurs de reste à charge .
En second lieu , votre rapporteure s'est montrée particulièrement sensible aux avertissements formulés par le CEPS, selon lequel les éventuels effets bénéfiques d'un référencement sélectif à court terme pouvaient à long terme se montrer nocifs. Il est en effet certain que, lors d'un premier appel d'offre, un référencement permettra d'obtenir des prix plus faibles et des restes à charge plus limités. Cependant, en empêchant pendant deux ou trois ans tout nouvel entrant de pénétrer le marché et en entraînant de facto la disparition des entreprises concurrentes qui ne seront pas sélectionnées, des oligopoles ne manqueront pas de se former , ce qui engendrera à long terme une hausse des prix.
Enfin , l'injonction qui pourra être faite aux opérateurs (exploitants ou distributeurs détaillants) sélectionnés à l'issue de la procédure de référencement de « fournir des quantités minimales de produits et de prestations sur le marché français » ne paraît pas réaliste au vu du maillage actuel de la filière du dispositif médical, composé à 84 % de très petites entreprises (TPE).
Pour ces raisons, la procédure de référencement telle que proposée par le texte présente les caractères d'un dispositif mal abouti aux effets trop incertains . Il serait, aux yeux de votre commission, beaucoup plus opportun de déterminer au préalable les facteurs véritables du reste à charge et de viser ces derniers à l'aide de procédures moins contraignantes. Par conséquent, votre commission a adopté un amendement n° 198 visant à supprimer l'introduction de la procédure de référencement .
B. Sur la procédure de remise en bon état d'usage
La procédure de remise en bon état d'usage, qui vise explicitement (d'après l'étude d'impact) les fauteuils roulants, s'inscrit parfaitement dans la ligne du rapport de notre collègue Philippe Mouiller relatif au financement des politiques du handicap 118 ( * ) . Votre commission, soucieuse de favoriser la diminution du reste à charge des personnes en situation de handicap, ne peut donc que se montrer favorable aux mesures visant à diminuer le coût final des dispositifs médicaux qui leur sont spécifiquement destinés .
Elle considère toutefois le dispositif proposé comme perfectible à plusieurs égards .
• L'article 28 ne dit rien des modalités d'attribution du dispositif médical remis en bon état d'usage. Il précise certes que l'assuré bénéficiaire doit s'engager à la restitution du dispositif médical pouvant faire l'objet d'une remise en état mais reste muet quant aux conditions déterminant son attribution.
N'est par ailleurs mentionnée qu'une obligation d'information du distributeur détaillant à son égard. Il en résulterait, aux yeux de votre commission, le maintien d'une liberté de choix laissée au patient quant au bénéfice d'un dispositif neuf ou usagé, qui pourra ainsi décider selon le reste à charge .
• Le dispositif pourrait être étendu aux aides techniques : les représentants des prestataires de santé à domicile ont fait remarquer à votre rapporteure que, pour viser les fauteuils roulants à fort reste à charge, la LPPR à elle seule n'était pas une cible pertinente, puisqu' elle n'incluait pas les aides techniques qui, tout en contribuant à la compensation de la perte d'autonomie ou du handicap, pouvaient ne pas disposer de la qualité de dispositif médical et n'étaient donc pas inscrites à la LPPR. Votre commission a donc souhaité préciser, par un amendement n° 200 , le champ de la remise en bon état d'usage en y incluant les aides techniques en tant que dispositifs contribuant à la compensation d'un handicap ou de la perte d'autonomie.
• Le dispositif pourrait également être davantage sécurisé au profit du patient .
En premier lieu, l'obligation faite au distributeur d'indiquer au patient l'existence d'un dispositif médical remis en bon état d'usage ne semble pas explicitement porter sur le même dispositif médical que celui initialement prescrit. Or la mise à disposition d'un produit usagé, à laquelle on ne peut qu'adhérer, ne doit pas pour autant permettre des substitutions de produits, qui seraient dommageables à la prise en charge. C'est pourquoi votre commission a adopté un amendement n° 199 visant à préciser l'identité générique du dispositif prescrit et du dispositif usagé.
Par ailleurs, votre commission a adopté un amendement n° 201 substituant le « patient » à l' « assuré », afin d'homogénéiser la rédaction de l'article.
En outre, il n'est actuellement prévu qu'une possibilité ouverte au CEPS de baisser, par convention ou décision unilatérale, le tarif de responsabilité d'un dispositif médical remis en bon état d'usage. Votre commission regrette qu'il ne s'agisse pas d'une obligation, qui seule aurait assuré la pleine effectivité de la baisse du reste à charge mais également évité les ruptures potentielles d'égalité entre un bénéficiaire de dispositif neuf et un bénéficiaire de dispositif usagé.
Votre commission vous propose d'adopter cet article ainsi modifié.
Article 28 bis
(nouveau)
Accès précoce aux dispositifs médicaux
Objet : Cet article, inséré par l'Assemblée nationale à l'initiative du Gouvernement, redéfinit le régime de prise en charge temporaire des dispositifs médicaux qui ne sont pas encore inscrits sur la liste des produits et prestations remboursables.
I - Le dispositif proposé par l'Assemblée nationale
A. Le droit existant
L'article 28 bis , inséré à l'initiative du Gouvernement, procède à une réécriture intégrale de l'article L. 165-1-5 du code de la sécurité sociale (CSS). Cet article traite de l'ouverture d'une prise en charge temporaire par l'assurance maladie d'un dispositif médical dont la demande d'inscription sur la liste des produits et prestations remboursables (LPPR), au titre d'une indication thérapeutique particulière, est en cours d'instruction .
Cette prise en charge temporaire peut être décidée par arrêté ministériel après avis de la commission nationale d'évaluation des dispositifs médicaux et des technologies de santé (Cnedimts) de la Haute Autorité de santé (HAS). La distribution des dispositifs médicaux temporairement pris en charge ne pourra alors être assurée que par un nombre limité d'établissements de santé. Elle est alors financée par une compensation versée à chaque entreprise et fixée par arrêté ministériel .
À l'instar du secteur du médicament, l'article 65 de la loi de financement de la sécurité sociale (LFSS) pour 2019 119 ( * ) a prévu un mécanisme de régulation financière .
Ce mécanisme obéit au principe suivant : le fabricant ou le distributeur du produit ou de la prestation reverse aux organismes de sécurité sociale, chaque année de la période de la distribution précoce, sous forme de remises, la différence entre le chiffre d'affaires facturé et le montant qui aurait résulté de la valorisation des unités vendues selon la compensation .
Une fois le produit ou la prescription inscrit sur la LPPR, il fait l'objet d'un prix ou d'un tarif fixé par convention entre le fabricant et le comité économique des produits de santé (CEPS). S'il résulte de ce prix ou de ce tarif que le fabricant ou le distributeur aurait réalisé un chiffre d'affaires supérieur à celui effectivement facturé aux établissements de santé, minoré des remises, la différence entre ces deux montants lui est alors restituée . Cette différence ne peut excéder le montant des remises préalablement versées.
B. Le dispositif proposé
• Une nouvelle dénomination des acteurs
En cohérence avec l'article 16 du présent projet de loi, qui porte une nouvelle définition de l'« exploitant d'un produit de santé » , l'article 28 bis substitue ce terme à la dénomination moins précise actuellement en vigueur d'« entreprise commercialisant le produit ou la prestation ». Le renvoi à la notion d'exploitant permet ainsi d'étendre la distribution précoce des produits de santé aux fabricants et aux distributeurs en gros. Compte tenu du caractère limité de la distribution précoce à certains établissements de santé, le dispositif exclut les distributeurs détaillants.
• La demande de prise en charge transitoire
Le a) du 1° de l'article 28 bis redéfinit le champ et les conditions de la prise en charge temporaire, en renommant cette dernière « prise en charge transitoire ». Cette dernière n'est plus conditionnée au dépôt préalable par l'exploitant d'une demande d'inscription sur la LPPR . Néanmoins, le dispositif indique explicitement que la prise en charge transitoire, lorsqu'elle est accordée, est suspendue si aucune demande d'inscription sur la LPPR n'a été déposée dans un délai de 12 mois suivant la demande de prise en charge transitoire.
Par ailleurs, le texte précise que, dans le cas où la demande de prise en charge transitoire d'un dispositif médical, ce dernier doit disposer d'un marquage « CE » , qui prouve sa conformité aux normes européennes afférentes 120 ( * ) .
Le texte maintient par ailleurs la décision par arrêté ministériel prise après avis de la Cnedimts, ainsi que la limitation de la distribution précoce à certains établissements de santé.
• Le renouvellement de la demande de prise en charge en cas de suspension
Le texte ouvre par ailleurs la faculté à l'exploitant dont le produit de santé a fait l'objet d'une prise en charge transitoire suspendue, de renouveler sa demande de prise en charge temporaire dans un délai de douze mois après la suspension . Au-delà de cette période, le droit au renouvellement ne s'applique plus.
• La redéfinition du mécanisme de la compensation
Le b) du 1° de l'article 28 bis modifie le régime de fixation de la compensation accordée à l'exploitant . Cette dernière sera désormais fixée à l'issue d'un « dialogue » entre ce dernier et les ministres compétents : après une première indication par l'exploitant du niveau maximal de la compensation qu'il réclame, les ministres peuvent lui communiquer, par une décision motivée, une proposition alternative. En cas de refus par l'exploitant, la demande de prise en charge transitoire est réputée abandonnée.
Il est implicitement prévu que cette compensation, en tant qu'elle peut être « réclamée » par l'exploitant aux établissements de santé, définit le plafond en-deçà duquel l'exploitant est autorisé à leur facturer le produit en distribution précoce .
C'est donc une modification notable par rapport au droit existant, qui permet la libre détermination initiale du prix par l'exploitant, conditionnée toutefois au reversement à l'assurance maladie de la différence entre ce montant et le montant de la compensation accordée, une fois le prix fixé.
• La redéfinition du circuit de la régulation financière
En conséquence des nouvelles modalités de fixation de la compensation, l'article 28 bis modifie le régime de la rétrocession financière une fois le produit ou la prestation inscrit sur la LPPR. La compensation définissant le plafond du chiffre d'affaires facturé à l'établissement de santé, il n'est par conséquent pas envisagé que la négociation conventionnelle avec le CEPS aboutisse à la fixation d'un prix ou d'un tarif de référence qui lui sera inférieur.
Ainsi, l'article 28 bis ne prévoit que l'hypothèse où l'exploitant, dans le seul cas d'un prix ou un tarif négocié inférieur à la compensation, sera redevable à l'égard de l'assurance maladie de la différence entre le chiffre d'affaires facturé aux établissements de santé et celui qui aurait résulté de l'application de ce prix ou tarif. Dans ce cas, il sera tenu de lui reverser, en une seule fois , cette différence sous la forme d'une remise.
• La possibilité laissée au CEPS d'une modulation du prix de référence
L'article 28 bis reconnaît par ailleurs au CEPS la possibilité de faire ultérieurement évoluer le prix ou le tarif de référence d'un produit de santé admis à la distribution précoce dans le cas général où ce dernier aurait fait l'objet d'une inscription sur la LPPR, mais également dans les deux cas particuliers où les ministres compétents lui auraient refusé cette inscription et où aucune demande d'inscription sur la LPPR n'aurait été déposée dans un délai de trente mois.
Cette possibilité ouverte au CEPS ne pourra intervenir que dans les cas énumérés aux articles L. 165-2 et L. 165-3 du CSS, qui décrivent pour une large part des situations où la couverture financière des produits de santé par l'assurance maladie serait exposée à des menaces liées à leur coût.
• Une obligation de continuité des traitements à la charge de l'exploitant
Le 2° prévoit l'obligation pour l'exploitant d'un produit de santé qui fait l'objet d'une distribution précoce de s'engager à assurer la continuité des traitements initiés pendant toute la durée de la prise en charge transitoire et jusqu'à un an après cette dernière, mais aussi, le cas échéant, pendant la durée de la suspension de cette prise en charge. Le délai d'un an est raccourci à 45 jours en cas de refus d'inscription sur la LPPR.
Tout manquement à cette obligation de continuité des traitements pourra entraîner la prononciation par le CEPS d'une pénalité financière pouvant aller jusqu'à 30 % du chiffre d'affaires hors taxes.
II - La position de la commission
Votre commission reconnaît plusieurs avantages à l'article 28 bis, dont il convient de mentionner que l'examen, compte tenu de l'importance de la matière traitée, aurait mérité des délais supérieurs à ceux qui s'imposent en cas d'adoption d'un dispositif en cours de navette.
Elle reconnaît dans la mention obligatoire du marquage « CE » , dans la possibilité ouverte à l'exploitant de renouveler sa demande de prise en charge temporaire en cas de suspension et dans l'énonciation d'une obligation de continuité des traitements des avancées notables dans l'accès précoce aux produits de santé.
Pour autant, par cohérence avec la position qu'elle a exprimée lors de l'examen de l'article 28 et qu'elle réitérera lors de l'examen des articles 29 et 30, elle maintient ses interrogations face à la remise en cause de la fixation du prix d'un dispositif médical par négociation conventionnelle. Elle propose en conséquence, par un amendement n° 212 , d'introduire un délai maximal pour la contre-proposition formulée par le ministère concernant la compensation maximale.
Votre commission vous propose d'adopter cet article ainsi modifié.
Article 28 ter
(nouveau)
Rapport au Parlement sur la prise en charge des dispositifs
médicaux
Objet : Cet article, inséré par l'Assemblée nationale à l'initiative de plusieurs députés du groupe Gauche démocrate et républicaine (GDR), prévoit la remise au Parlement d'un rapport sur la dépense d'assurance maladie relative aux dispositifs médicaux.
I - Le dispositif proposé par l'Assemblée nationale
L'article 28 ter prévoit que, dans un délai de six mois à compter de la promulgation de la présente loi, le Gouvernement remet au Parlement un rapport sur le montant consolidé de l'ensemble des dépenses d'assurance maladie résultant du remboursement des dispositifs médicaux, ventilé selon les différentes modalités de remboursement.
II - La position de la commission
Bien que votre commission se montre habituellement réservée sur l'adoption d'articles contenant des demandes de rapport, le contenu spécifique de cet article lui paraît pertinent.
L'Assemblée nationale a en effet évoqué, au moment de son adoption, le caractère insuffisamment étayé de l'annexe 7 du PLFSS qui « se contente d'indiquer le volumes d'économies attendues sur les dispositifs médicaux pour l'année à venir » 121 ( * ) , sans distinguer les modalités de leur remboursement (liste en sus ou liste intra GHS). Or ces éléments pourraient être de nature à éclairer utilement le législateur sur la spécificité des dispositifs médicaux (plus ou moins intensifs en technologie) visés par les mesures d'économies .
Aussi, la présente demande de rapport pourra-t-elle être considérée comme satisfaite si l'annexe 7 est enrichie des éléments nécessaires.
Votre commission vous propose d'adopter cet article sans modification.
Article 29
Prise en charge
et régulation des prix
de certains médicaments
particuliers
Objet : Cet article comporte de nombreuses dispositions relatives à la prise en charge et à la régulation des prix de certains médicaments particuliers : les médicaments faisant l'objet d'importation ou distribution parallèle sont intégrés au droit commun de la régulation des spécialités pharmaceutiques, le recours aux médicaments biosimilaires est restreint et les prix de certains médicaments distribués aux établissements de santé et des médicaments de nutrition parentérale font l'objet d'une fixation unilatérale par la puissance publique.
I - Le dispositif proposé
A. Une précision relative à l'autorisation de mise sur le marché des spécialités hybrides
L'article L. 5121-10-2 du code de la santé publique (CSP) prévoit, dans sa version actuelle, qu'une autorisation de mise sur le marché (AMM) puisse être délivrée, avant l'expiration des droits de propriété intellectuelle attachés au médicament de référence, à tout médicament présentant des caractéristiques communes avec ce dernier sans pour autant en être un générique .
L'article 66 de la loi de financement de la sécurité sociale pour 2019 122 ( * ) a précisé le statut de ces médicaments spécifiques, les désignant comme « spécialités hybrides », et a posé comme obligation préalable à leur AMM la réalisation d'essais précliniques et cliniques déterminés en fonction des différences qu'elles présentent avec leur spécialité de référence. Par conséquent, la délivrance de l'AMM d'une spécialité hybride n'est plus décidée en fonction des caractéristiques communes qu'elle présente avec la spécialité de référence, ce qui contredit la disposition de l'article L. 5121-10-2 précitée.
Le 1° du I procède par conséquent à sa suppression.
B. La réglementation applicable aux médicaments faisant l'objet d'une importation ou d'une distribution parallèle
1. Un cadre réglementaire lacunaire
La distribution parallèle de médicaments est informellement définie par le ministère des solidarités et de la santé comme « le fait pour un opérateur économique (communément appelé distributeur parallèle), étranger au circuit de distribution officiel du titulaire de l'autorisation de mise sur le marché (AMM) , d'acquérir une spécialité faisant l'objet d'une AMM communautaire [délivrée par l'agence européenne des médicaments] obtenue par le biais de la procédure centralisée (règlement (CE) n° 726/2004 du Parlement européen et du Conseil du 31 mars 2004), en vue de sa commercialisation en France » 123 ( * ) .
Elle est à distinguer de l' importation parallèle , qui concerne les spécialités bénéficiant d'une AMM délivrée par l'État membre de provenance et d'une AMM délivrée par l'État membre de destination.
Le CSP détaille les conditions dans lesquelles un médicament peut être importé sur le territoire français, mais limite singulièrement son champ d'application aux spécialités ayant reçu une AMM délivrée par un État membre de l'Union européenne (l'importation parallèle), et non à celles ayant reçu une AMM communautaire (la distribution parallèle). En outre, alors qu'est précisément défini le cadre réglementaire de l'importation parallèle (articles R. 5121-108 et suivants), aucune précision similaire n'existe pour la distribution parallèle.
Par ailleurs, le distributeur parallèle, en raison de la nature spécifique de l'AMM communautaire, n'est pas tenu d'être titulaire ou mandataire de son exploitation . En effet, la distribution parallèle d'un médicament s'applique au médicament qui a obtenu une AMM selon la procédure dite « centralisée ». Dans le cadre de cette procédure, l'AMM est délivrée par l'agence européenne du médicament et autorise la libre distribution du produit dans tous les États membres de l'Union européenne , en dehors du réseau de distribution mis en place par le fabricant ou son distributeur agréé, sans autre contrainte juridique que de répondre aux conditions fixées dans l'autorisation pour le marché de distribution considéré 124 ( * ) .
Or l'article L. 162-16-4 du code de la sécurité sociale (CSS) dispose explicitement que le prix de vente au public des médicaments remboursables est fixé par convention entre l'entreprise exploitant le médicament et le comité économique des produits de santé (CEPS). Dans les termes actuels de la loi, le distributeur parallèle n'est donc pas intégré au champ conventionnel du CEPS. En conséquence, l'ensemble des prérogatives exercées par ce dernier, parmi lesquelles figure notamment celle de contrôler le prix de vente d'un produit au public ou aux établissements lorsque ce dernier menace l'équilibre financier des comptes de la sécurité sociale, ne lui est pas applicable.
On peut à ce titre utilement signaler que la procédure centralisée au niveau européen est obligatoire pour les médicaments de thérapie innovante , ceux issus des biotechnologies , les médicaments contenant une nouvelle substance active et dont l'indication thérapeutique est le traitement de certaines affections (SIDA, cancer, maladie neurodégénératives, diabète, maladies auto-immunes et maladies virales) ainsi que les médicaments orphelins indiqués dans le traitement des maladies rares 125 ( * ) .
|
Annexe du règlement (CE) n° 726/2004
du Parlement européen
Aux termes de l'article 3 du règlement, aucun médicament de la liste suivante ne peut être mis sur le marché dans la Communauté sans qu'une autorisation de mise sur le marché n'ait été délivrée par la Communauté : 1) médicaments issus de l'un des procédés biotechnologiques suivants : technologie de l'acide désoxyribonucléique recombinant, expression contrôlée de gènes codant pour des protéines biologiquement actives dans des procaryotes et des eucaryotes, y compris des cellules transformées de mammifères, méthodes à base d'hybridomes et d'anticorps monoclonaux ; 2) médicaments à usage vétérinaire destinés principalement à être utilisés comme améliorateurs de performance pour accélérer la croissance ou pour augmenter la productivité des animaux traités ; 3) médicaments à usage humain contenant une nouvelle substance active qui, à la date d'entrée en vigueur du présent règlement, n'était pas autorisée dans la Communauté et dont l'indication thérapeutique est le traitement d'une des affections suivantes : syndrome d'immunodéficience acquise, cancer, maladie neurodégénérative, diabète et, à compter du 20 mai 2008, maladies auto-immunes et autres dysfonctionnements immunitaires ainsi que les maladies virales ; 4) médicaments désignés comme des médicaments orphelins. |
Ainsi, la distribution parallèle de médicaments, bien que fondamentalement dérogatoire au schéma classique de la distribution des produits de santé (le distributeur est titulaire de l'exploitation ou mandataire de l'exploitant) ne fait l'objet d' aucun régime juridique spécifique . La seule formalité requise figure (incidemment) à l'article R. 5121-132-1 du CSP et prévoit que l'établissement pharmaceutique distributeur parallèle notifie son intention au fabricant ou à l'exploitant titulaire de l'AMM, à l'ANSM ainsi qu'à l'agence européenne des médicaments.
2. L'article 29 pose une définition de la distribution parallèle et l'intègre au droit commun de la distribution du médicament en France
a) Une définition de rang législatif
La définition de la distribution parallèle, qui n'était visée à ce jour par aucune disposition juridique au sein du CSP 126 ( * ) , fait désormais l'objet d'un nouvel article ( 3° du I ), qui rappelle la réunion nécessaire des deux conditions suivantes :
- la spécialité doit bénéficier d'une AMM délivrée par l'Union européenne ;
- elle est importée d'un autre État membre ou partie à l'Espace économique européen par un établissement pharmaceutique autre que le titulaire de l'AMM .
b) L'application des mécanismes de régulation de droit commun
L'article 29 prévoit la complète application des différents leviers de régulation du marché des médicaments aux spécialités issues de la distribution parallèle. Ces leviers agissent concurremment sur les volumes et sur les prix des produits distribués.
La régulation par le volume est ainsi assurée par un triple mécanisme :
- un encadrement des fournitures aux collectivités publiques demandeuses et des possibilités de remboursement par les caisses de sécurité sociale : elles ne seront désormais possibles qu'à la condition d'une inscription des médicaments distribués sur la liste des indications thérapeutiques ouvrant droit à une prise en charge ( 2° du I et 10° du II ) ;
- une obligation de définir ultérieurement, par décret en Conseil d'État, les obligations des distributeurs parallèles de médicaments ainsi que les conditions de leur commercialisation ( 4° du I ) ;
- l'intégration du chiffre d'affaires hors taxes résultant de l'activité des distributeurs et importateurs parallèles de médicaments à l'assiette des différentes contributions auxquelles sont assujettis les grossistes et industriels du médicament :
• la contribution grossistes-répartiteurs ( 1° du II ) ;
• la contribution sur le montant M (dite « clause de sauvegarde ») ( 2°et 3° du II ) ;
• la contribution sur les dépenses de publicité ( 18° du II ) ;
• la contribution sur le chiffre d'affaires ( 20° du II ).
La régulation par le prix est réalisée, quant à elle, par l'intégration des distributeurs parallèles de médicaments au champ conventionnel du CEPS, jusqu'alors limité au seul exploitant :
- la possibilité est ouverte aux importateurs et distributeurs parallèles de médicaments de définir, au sein d'une convention avec le CEPS, des remises conventionnelles sur les spécialités concernées ( 4° et 16° du II ) ;
- le a) du 6° et le 8° du II disposent que le prix de cession au public des médicaments ayant fait l'objet d'importation ou de distribution parallèle sera désormais fixé par convention entre l'importateur ou le distributeur d'une part, et le CEPS d'autre part. En cohérence, le 9° du II intègre le tarif de responsabilité et le prix limite de vente aux établissements des médicaments issus de l'importation ou de la distribution parallèle au champ conventionnel du CEPS ;
- par ailleurs, le b) du 6° du II prévoit la possibilité, par avenant à une convention signée avec le CEPS ou par décision unilatérale de ce dernier, d'une baisse du prix de vente de médicaments spécialisés non titulaires d'une AMM européenne et ayant fait l'objet d'une importation ou d'une distribution parallèle 127 ( * ) ;
- de façon plus large, les médicaments diffusés par les importateurs et distributeurs parallèles pourront également voir leur prix ou leur tarif de responsabilité modulé par le CEPS en cas de dépassement d'un certain seuil de remboursement par la sécurité sociale, avec la possibilité d'éviter cette modulation par le versement d'une remise conventionnelle à l'assurance maladie ( 14° du II ) ;
- les distributeurs parallèles pourront se voir assujettis à la pénalité financière définie en cas de rétention d'information ( 15° du II ).
Enfin, les spécialités issues de la distribution parallèle pourront être inscrites, à la demande des distributeurs ou sur décision de l'État, sur la liste des médicaments remboursables en sus des prestations hospitalières ( 17° du II ).
C. La fin de la possibilité d'une substitution de médicaments biosimilaires dispensés en officine
La publication en mai 2016 par l'agence nationale de sécurité du médicament et des produits de santé (ANSM) d'un rapport général sur les médicaments biosimilaires 128 ( * ) a permis de mieux identifier les enjeux spécifiques liés à ces spécialités pharmaceutiques.
Il s'agit moins d'apporter une modification aux médicaments biologiques de référence existants, dont les propriétés suffisent à couvrir le besoin médical correspondant, que d'élargir la couverture des besoins par une offre pharmaceutique quasi-bioéquivalente et qu' une concurrence par les prix rendrait plus accessible . Par ailleurs, en raison des difficultés d'approvisionnement qu'engendre la production délicate des médicaments biologiques, la promotion des médicaments biosimilaires rend le marché « moins sensible aux tensions, accidents de production et/ou aux éventuelles ruptures de stock ».
Les articles L. 5125-23-2 et L. 5125-23-3 du CSP déclinent le cadre dans lequel le pharmacien peut, au moment de la dispensation en officine, substituer un médicament biosimilaire à un médicament biologique de référence :
- l'article L. 5125-23-2 pose le principe de substitution d'un biosimilaire à un médicament biologique de référence, auquel il ne peut être fait exception qu'en cas de mention expresse de non-substitution apposée manuellement sur la prescription par le médecin (mention « non-substituable ») ;
- l'article L. 5125-23-3 précise les conditions dans lesquelles le pharmacien d'officine peut, sans accord exprès et préalable du médecin, délivrer un biosimilaire par substitution au médicament biologique : appartenance au même groupe biologique similaire, substitution obligatoirement réalisée en initiation de traitement, lorsque le prescripteur n'a pas exclu la possibilité d'une substitution et remboursement selon les modalités de droit commun. La mise en oeuvre de cette possibilité de substitution ainsi que de l'information obligatoire du prescripteur, devaient être précisées par un décret en Conseil d'État, qui n'a jamais été pris .
Le 5° du I de l'article 29 abroge ces deux articles , avec pour effet de réintégrer la substitution d'un produit biosimilaire à un produit biologique de référence dans le droit commun de la dispensation décrit au premier alinéa de l'article L. 5125-23 : l'impossibilité pour le pharmacien d'officine de dispenser un autre médicament que celui qui a été prescrit sans l'accord exprès et préalable du médecin . Le 5° du II opère une coordination.
On rappelle à cet égard que le principe général régissant la dispensation est en effet celui d'une subordination du produit dispensé au produit prescrit, ou ayant la même dénomination commune que le produit prescrit . C'est en vertu de ce principe que le pharmacien est autorisé à dispenser un médicament générique ou hybride dans le cas d'une prescription libellée en dénomination commune . Si la spécialité est nommément désignée dans la prescription, la dispensation d'une spécialité générique ou hybride est également possible, à la condition que le prescripteur n'ait pas expressément exclu cette possibilité dans l'ordonnance.
D. De nouveaux modes de fixation des prix
Le 7° du II de l'article 29 crée deux nouveaux mécanismes de régulation des prix, en ouvrant la possibilité d'une fixation directe par arrêté ministériel , pour certaines catégories de médicaments.
Est tout d'abord instaurée la fixation automatique du prix de cession des préparations magistrales et des préparations hospitalières pour la nutrition parentérale , lorsqu'elles sont délivrées par des établissements de santé, afin de permettre, selon l'étude d'impact, la définition de « règles homogènes de facturation de cette activité pour les établissements hospitaliers sur l'ensemble du territoire ».
Par ailleurs, est également prévue la possibilité pour les ministres de la santé et de la sécurité sociale de fixer un prix maximal de vente pour « certains » médicaments fournis aux établissements de santé ou pour « certains » produits de santé financés au titre des prestations hospitalières dans au moins l'une des deux situations suivantes :
- risque de dépenses injustifiées , notamment au regard d'une augmentation significative des prix de vente constatés, ou au regard des produits de santé comparables ;
- caractère particulièrement coûteux pour certains établissements.
Selon le CEPS, il pourrait s'agir de médicaments en situation de monopole qui, à l'occasion d'un rachat, voient leur prix facial augmenté par le nouvel exploitant.
Notons que le 13° du II retire le prix de tous les médicaments vendus aux établissements de santé (et non les seuls médicaments visés par le 7°) du champ des conventions tarifaires conclues entre le CEPS et les entreprises pharmaceutiques.
E. Un nouveau « bulletin officiel des produits de santé »
Le 11° et le 12° du II prévoient que les informations relatives au remboursement, à la prise en charge, aux prix, aux tarifs et à l'encadrement de la prescription et de la dispensation des médicaments, des dispositifs médicaux et des autres produits de santé soient désormais publiés au sein d'un document officiel spécifique : le Bulletin officiel des produits de santé.
II - Les modifications adoptées par l'Assemblée nationale
Outre quatre amendements rédactionnels du rapporteur général, l'Assemblée nationale a adopté quatre modifications au dispositif de l'article 29.
Un amendement du rapporteur général est venu utilement clarifier la définition d'une spécialité pharmaceutique faisant l'objet d'une distribution parallèle, en précisant que cette dernière pouvait également être importée par un établissement autre que l'entreprise qui en assure l'exploitation au vue de sa commercialisation en France.
Un amendement du Gouvernement précise le régime applicable aux substitutions que le pharmacien peut opérer au moment de la dispensation. Il précise que le pharmacien, même lorsque l'ordonnance ne l'exclut pas , peut être tenu, pour certaines situations médicales précisées par arrêté, de ne pas appliquer de substitution entre la spécialité prescrite et une spécialité générique ou hybride et d'obligatoirement dispenser le médicament princeps .
Un amendement du rapporteur général clarifie les conditions de fixation du prix maximal de cession aux établissements de santé de certains médicaments ou produits de santé, en précisant que cette fixation se fera en-dehors de la négociation conventionnelle entre les exploitants et le CEPS, mais tiendra tout de même compte des éléments qui interviennent habituellement au cours de cette dernière.
Enfin, deux amendements identiques du Gouvernement et des membres du groupe La République en marche modifient substantiellement le dispositif de « tiers payant contre générique » , qui consiste à consentir aux assurés une dispense d'avance de frais totale ou partielle au moment de la facturation, à la condition que ces derniers consentent à la dispensation d'un médicament générique.
Ce dispositif ne s'applique toutefois pas lorsque le remboursement de ces génériques est soumis à un tarif de responsabilité défini par le CEPS ou lorsque le prix du générique est supérieur ou égal à celui du princeps . Autrement dit, le tiers payant peut toujours être applicable au patient à qui est dispensé un princeps dont le prix est inférieur ou égal à celui de son générique.
L'amendement supprime l'application du tiers payant en cas d'égalité de ces deux prix et le limite au seul cas où le princeps est moins coûteux strictement que le générique. Dans le cas contraire, le patient devra opter pour le générique avec tiers payant, ou conserver le princeps sans bénéfice du tiers payant.
L'Assemblée nationale a adopté cet article ainsi modifié.
III - La position de la commission
Bien que l'article 29 soit porteur de plusieurs avancées en matière de régulation (notamment pour ce qui regarde les médicaments faisant l'objet d'une distribution parallèle), votre commission se montre circonspecte quant à trois de ses dispositions principales : le régime de substitution des biosimilaires , l'avènement d'un régime de fixation unilatéral des prix des produits de santé et l'application du mécanisme de tiers payant aux patients auxquels sont dispensés des génériques .
A. La fin de la substitution des biosimilaires : un dispositif a priori incohérent avec l'ambition affichée par le Gouvernement
L'abrogation des articles L. 5125-23-2 et L. 5125-23-3 du CSP semble de prime abord contradictoire avec un discours politique favorable à la pénétration du marché par les médicaments biosimilaires , porté sans ambiguïté par la ministre lors de son audition par votre commission le 19 octobre 2019 129 ( * ) et par ailleurs confirmé par le dispositif de l'article 43 du présent projet de loi.
Initialement exclus de toute interchangeabilité ou substitution , les médicaments biosimilaires ont fait l'objet, en 2016, d'une importante modification de doctrine de la part de l'ANSM.
|
Les recommandations de l'ANSM
« Bien que les médicaments biosimilaires soient en principe autorisés pour traiter les mêmes maladies que le médicament de référence, un médicament biosimilaire peut avoir moins d'indications que le médicament de référence , le plus souvent faute d'études probantes d'efficacité et de sécurité dans l'indication concernée. » « Si le choix entre deux médicaments biologiques (de référence ou biosimilaire) reste libre en l'absence de traitement antérieur identifié, il n'est cependant pas souhaitable, pour des raisons de sécurité et de traçabilité, de modifier la prescription initiale , en remplaçant une spécialité par une autre, sans garantie. » « Au vu de l'évolution des connaissances et de l'analyse continue des données d'efficacité et de sécurité des médicaments biosimilaires au sein de l'Union européenne, il ressort qu' une position excluant formellement toute interchangeabilité en cours de traitement ne parait plus justifiée . » Ainsi, si tout échange non contrôlé entre médicaments biologiques de référence et biosimilaires doit être évité, une interchangeabilité peut être envisagée si : - le patient traité est informé et donne son accord ; - il reçoit une surveillance clinique appropriée lors du traitement ; - une traçabilité sur les produits concernés est assurée. |
La position de l'ANSM est donc la suivante : la substitution d'un biosimilaire à un médicament biologique de référence, même en cours de traitement, ne présente pas de danger particulier, pour autant que cette substitution soit exclusivement de l'initiative du médecin (« interchangeabilité ») et non de celle du pharmacien d'officine sans accord exprès et préalable du prescripteur (« échange non contrôlé »).
C'est en ce sens que les dispositions de l'article 29, qui abrogent la possibilité pour le pharmacien d'opérer la substitution sans accord préalable du prescripteur, se font l'écho des recommandations de l'ANSM. Votre commission le regrette d'autant plus que :
- le contrôle par le prescripteur était exercé ex ante par la possibilité qui lui était ouverte de spécifier le caractère non substituable du médicament biologique de référence (par l'apposition de la mention NS) ;
- l'article L. 5125-23-3 prévoyait bel et bien l'information du prescripteur en cas de substitution par le pharmacien, mais n'a pu être mis en oeuvre en l'absence du décret requis.
Il a été objecté à votre rapporteure que les impacts de cette abrogation seraient de toute façon mineurs, étant donné que les substitutions entre médicament biologique de référence et médicament biosimilaire se faisaient dans un cadre essentiellement hospitalier, sans intervention du pharmacien d'officine.
Deux constats militent néanmoins pour le maintien de ces dispositions :
- le signal très négatif qui serait envoyé aux fabricants de biosimilaires alors que l'on exige d'eux des décotes tarifaires significatives ;
- l'utilité pour certaines pathologies (notamment le diabète) d'une substitution d'un biosimilaire (notamment de l'insuline glargine) par le pharmacien d'officine.
C'est pourquoi votre commission a adopté un amendement n° 202 visant à rétablir la possibilité, sous certaines conditions, de la substitution par le pharmacien d'un biosimilaire à un biologique de référence .
B. L'attachement à la détermination du prix par négociation conventionnelle
Cette disposition de l'article 29, manifestement disproportionnée, a suscité l'incompréhension de votre commission, pour deux raisons principales :
- en ouvrant la possibilité d'une fixation unilatérale par le pouvoir réglementaire d'un prix de vente maximal de médicaments ou de produits de santé, elle introduit une dérogation notable et dommageable au principe équilibré de la négociation conventionnelle du prix entre l'industriel et le CEPS ;
- les critères susceptibles de provoquer cette fixation unilatérale sont décrits de façon à être à tout moment opposables aux industriels, au mépris de la loyauté qui régit normalement les conditions de la commande publique : il suffirait en effet que le ministère des solidarités et de la santé se prévale du « risque » de dépenses injustifiées ou du coût « prévisible » de certains produits.
En conséquence, votre commission a adopté un amendement n° 203 qui renvoie à la négociation conventionnelle entre l'industriel et le CEPS les deux cas évoqués par l'article 29, tout en précisant leurs contours. Elle a également supprimé, par le même amendement, la disposition de l'article 29 qui ôte la négociation du prix des médicaments rétrocédés aux établissements de santé du champ de la négociation conventionnelle 130 ( * ) .
C. Tiers payant et génériques : une disposition insuffisamment concertée
Votre commission déplore enfin qu'une disposition aussi importante que l'impossibilité pour un patient de se voir appliquer le tiers payant lorsqu'il privilégie le princeps en cas d'égalité de prix entre ce dernier et son générique ait été introduite par un simple amendement du Gouvernement en séance publique à l'Assemblée nationale. Elle a en conséquence adopté un amendement n° 210 de suppression de cette disposition.
Sous réserve de l'adoption de l'amendement n° 215 rédactionnel, votre commission vous propose d'adopter cet article ainsi modifié.
Article 29 bis
(nouveau)
Expérimentation de l'usage médical du cannabis
Objet : Cet article, introduit par l'Assemblée nationale par l'adoption d'un amendement du rapporteur général, ouvre la possibilité d'expérimenter, pour une durée de deux ans, l'usage médical du cannabis.
I - Le dispositif proposé
• À la demande de la ministre des solidarités et de la santé, l'ANSM a mis en place, le 18 septembre 2018, un comité scientifique spécialisé temporaire (CSST) chargé d'évaluer, sur une durée d'un an, la pertinence et la faisabilité de la mise à disposition du cannabis thérapeutique en France. Après avoir jugé, en décembre 2018, qu'il était pertinent d'autoriser l'usage du cannabis dans le traitement de certaines situations cliniques, le CSST a proposé à l'ANSM, en juin 2019, un cadre pour l'expérimentation du cannabis thérapeutique. À la suite de la consultation des différentes parties sur cette question, dont le président de votre commission qui a transmis au CSST un avis écrit, l'ANSM a validé en juillet 2019 le cadre expérimental proposé par le comité.
En octobre 2019 131 ( * ) , le directeur général a ainsi institué un comité scientifique temporaire dédié à la mise en oeuvre de l'expérimentation du cannabis médical en France, chargé de collaborer à la rédaction du cahier des charges de cette expérimentation (médicaments utilisés, contenu des formations des professionnels de santé et contenu du registre de suivi des patients) et des recommandations à destination des prescripteurs. Pour mémoire, les situations thérapeutiques retenues par le CSST pour l'administration du cannabis thérapeutique sont les suivantes :
- les douleurs réfractaires aux thérapies (médicamenteuses ou non) accessibles ;
- certaines formes d'épilepsie sévères et pharmacorésistantes ;
- les soins de support en oncologie ;
- les situations palliatives ;
- la spasticité douloureuse de la sclérose en plaques.
Ce comité scientifique devrait rendre ses conclusions dans un délai de six mois. L'ANSM et les services de l'État se sont d'ores et déjà engagés à déployer cette expérimentation à l'issue de ce délai.
• Adopté par l'Assemblée nationale par la voie d'un amendement du rapporteur général Olivier Véran, l'article 29 bis du PLFSS pour 2020 vise à poser un cadre législatif pour l'expérimentation de l'usage thérapeutique du cannabis qui est autorisée pour une durée de deux ans. Il précise ainsi que l'usage médical du cannabis doit être envisagé « sous la forme de produits répondant aux standards pharmaceutiques, dans certaines indications ou situations cliniques réfractaires aux traitements indiqués et accessibles » ( I ). Il prévoit, par ailleurs, que les conditions de mise en oeuvre de cette expérimentation seront définies par voie réglementaire, notamment les conditions de prise en charge, le nombre de patients concernés, les modalités d'importation, de production, d'approvisionnement, de prescription et de délivrance par les pharmacies hospitalières et d'officine, ainsi que les conditions d'information, de suivi des patients et de formation des professionnels de santé ( II ).
Enfin, dans un délai de six mois avant le terme de l'expérimentation, un rapport sur sa mise en oeuvre devra être adressé par le Gouvernement au Parlement. Ce rapport a vocation à examiner la pertinence du maintien du cadre de l'usage médical du cannabis à l'issue de l'expérimentation ( III ).
II - La position de la commission
• Dans son avis adressé à l'ANSM, le président de votre commission avait souligné que le succès et la crédibilité de cette expérimentation étaient conditionnés à la pleine adhésion des professionnels de santé et des patients concernés à la démarche de suivi qui lui était associée. À cet égard, le caractère obligatoire de la participation tant du prescripteur que du patient au registre national électronique de suivi de l'expérimentation constitue une exigence incontournable.
Ce registre n'aura pas vocation à collecter des données cliniques à proprement parler, l'expérimentation n'étant pas un essai clinique destiné à évaluer le service médical rendu du cannabis thérapeutique. La pertinence de son utilisation dans un certain nombre d'indications a déjà été validée par le CSST à partir de la littérature scientifique existante. D'autres essais cliniques sont du reste en cours pour déterminer l'efficacité clinique de plusieurs médicaments à base de cannabis.
L'objectif de l'expérimentation du cannabis thérapeutique en France étant de tester la pertinence du cadre d'administration validé par l'ANSM, le registre de suivi doit, selon le président de votre commission, avoir vocation à collecter des données de vie réelle. Ces données observationnelles peuvent être collectées par le médecin prescripteur ou l'équipe médicale de suivi à l'occasion de points routiniers dont la périodicité pourrait être précisée par l'ANSM. Il s'agit de pouvoir non seulement recenser les effets indésirables ressentis ou constatés mais également évaluer l'observance et la tolérance du traitement, identifier les éventuelles comorbidités, les interactions avec d'autres médicaments ou encore les facteurs de risque (par exemple, pour les personnes insuffisantes rénales ou hépatiques, immunodéprimées, schizophrènes ou bipolaires...).
Il recommandait, par ailleurs, de s'appuyer sur les actuels centres d'évaluation et de traitement de la douleur labellisés par les agences régionales de santé, déjà répartis sur le territoire 132 ( * ) , qui pourraient alimenter la réflexion sur le cahier des charges des centres de référence habilités à prescrire du cannabis médical et sur la formation des professionnels de santé.
Dans la mesure où la quasi -totalité des produits susceptibles d'être utilisés dans le cadre de l'expérimentation devrait provenir d'entreprises étrangères, l'avis du président de votre commission attirait l'attention des pouvoirs publics sur la nécessité d'assurer une traçabilité solide de ces produits, non encore commercialisés en France, tout en permettant aux pharmaciens hospitaliers d'adapter la posologie ou la forme galénique des produits, en particulier dans le cadre d'un usage pédiatrique.
Enfin, le président de votre commission insistait sur la délivrance d'une information complète et loyale au patient candidat, réactualisée tout au long de l'expérimentation afin de tenir compte d'éventuelles nouvelles contre-indications. En l'état des connaissances scientifiques sur les effets du cannabis pour la santé, ne peuvent être écartés les soupçons de lien entre la consommation de cannabis et l'aggravation de troubles psychotiques, notamment par des phénomènes de décompensation pour certaines pathologies psychiques (schizophrénie, bipolarité...), la prolifération tumorale ou encore le risque de réduction de l'efficacité de certains traitements anticancéreux.
• Afin de garantir que les données cliniques et de vie réelle collectées lors de l'expérimentation soient complètes et couvrent un temps suffisamment long, votre commission a adopté un amendement tendant à :
- inscrire dans le cadre de l'expérimentation du cannabis thérapeutique le caractère obligatoire d'un registre de suivi des patients qui devra permettre de collecter des données de vie réelle afin d'évaluer la tolérance du cannabis thérapeutique et de pouvoir, le cas échéant, déterminer le lien entre l'administration du cannabis médical et d'éventuelles comorbidités, de possibles interactions avec d'autres médicaments et traitements et des facteurs de risque ;
- réduire de six à deux mois avant le terme de l'expérimentation le délai dans lequel un rapport de bilan devra être transmis au Parlement, afin de permettre à l'ANSM de disposer du maximum de données dans son évaluation du cadre d'administration du cannabis thérapeutique (amendement n° 173).
Votre commission vous propose d'adopter cet article ainsi modifié.
Article 30
Accès
précoce et soutenabilité financière des ATU
Objet : Cet article apporte d'importantes modifications au régime d'accès ainsi qu'au régime financier des médicaments auxquels a été attribuée une autorisation temporaire d'utilisation (ATU). Il restreint de façon notable les conditions d'accès aux ATU nominatives et revoit en profondeur les modalités de remboursement par les laboratoires des remises dues au titre de médicaments en post-ATU non encore inscrits au remboursement de droit commun.
I - Le dispositif proposé
A. Conditions d'accès précoce dans le cas d'autorisations temporaires d'utilisation nominatives
1. Un régime d'accès particulièrement conditionné
L'article L. 5121-12 du code de la santé publique (CSP) précise le régime juridique applicable aux médicaments bénéficiaires d'une autorisation temporaire d'utilisation (ATU). Il s'agit de « certains médicaments, dans des indications thérapeutiques précises, destinés à traiter des maladies graves ou rares, en l'absence de traitement approprié », auxquels n'a pas encore été attribuée d'autorisation de mise sur le marché (AMM).
L'attribution d'une ATU se fait à plusieurs conditions : une condition générale et plusieurs conditions complémentaires, pour le cas spécifique de l'ATU nominative.
La condition générale consiste à :
- pour une ATU de cohorte , qui s'adresse à des groupes de patients, présenter une présomption forte d'efficacité et de sécurité de ces médicaments au vu d' essais thérapeutiques auxquels il a été procédé dans le cadre d'une demande d'AMM (qui, si elle n'a pas encore été déposée, doit l'être au maximum un an après l'octroi de l'ATU). Les conditions d'octroi de l'ATU de cohorte concernent donc des produits dont le développement clinique est avancé et dont les effets sont solidement documentés ;
- pour une ATU nominative , délivrée pour un patient nommément désigné qui ne peut participer à une recherche impliquant la personne humaine, présenter un « bénéfice » individuel pour son état de santé et d'une présomption simple d'efficacité et de sécurité au vu des connaissances scientifiques disponibles. L'octroi se fait alors sous la responsabilité du médecin prescripteur.
La demande d'ATU nominative n'est en outre valable qu' à l' une des conditions complémentaires suivantes :
- l'industriel a déposé ou s'engage à déposer, pour le même médicament, une demande d'ATU de cohorte ;
- l'industriel a déposé ou s'engage à déposer, pour le même médicament, une demande d'AMM nationale ou européenne ;
- des essais cliniques sont conduits en France ou ont été demandés.
Ces conditions complémentaires ont vocation à s'assurer que la demande d'ATU nominative formulée par l'industriel ne se fait pas au détriment d'une mise ultérieure sur le marché .
Ces conditions complémentaires d'attribution de l'ATU nominative peuvent toutefois être contournées dans l'un des cas suivants :
- en l'état des thérapeutiques disponibles, l'état du patient est susceptible de fortement s'aggraver ;
- le médicament est commercialisé pour une autre indication thérapeutique et il existe de fortes présomptions d'efficacité et de sécurité dans l'indication sollicitée ;
- l'entreprise s'est vue refuser une ATU de cohorte pour l'indication thérapeutique considérée, mais le médicament est susceptible de présenter un bénéfice individuel pour le patient.
Ce régime d'exception permet qu'une ATU nominative soit accordée, sans considération particulière de sa commercialisation ultérieure, mais sous réserve que l'état personnel du patient soit sensiblement amélioré.
2. Un resserrement important des conditions d'octroi prévu par l'article 30
Le I de l'article 30 du projet de loi entend resserrer les conditions d'octroi de l'ATU nominative .
Il redéfinit en premier lieu la condition générale d'attribution de l'ATU nominative en :
- substituant un « une efficacité cliniquement pertinente et un effet important » au seul « bénéfice » individuel du patient ( a) du 1° du A ) ;
- en adjoignant la condition de conséquences graves pour l'état du patient dans l'état des thérapeutiques disponibles ( b) du 1° du A ) ;
- en substituant une présomption forte d'efficacité et de sécurité à la présomption simple ( c) du 1° du A ).
Il étoffe en second lieu la liste des conditions complémentaires en :
- précisant qu'aucune des demandes d'ATU de cohorte ou d'AMM ne doit avoir reçu de réponse (positive ou négative) pour que l'ATU nominative soit accordée ( b) du 2° du A ) ;
- supprimant le simple dépôt de demande d'essai clinique et en exigeant la réalisation de ces derniers ( c) du 2° du A ) ;
- limitant dans le temps l'engagement de l'industriel de déposer l'une des deux demandes précitées ( d) du 2° du A ) ;
- prévoyant une condition complémentaire supplémentaire d'urgence vitale pour le patient, pour le seul cas des maladies aiguës sans alternative thérapeutique possible ( e) du 2° du A ) ;
- ajoutant un deuxième ensemble de conditions complémentaires obligatoires consistant pour le médicament à ne pas dépasser un certain nombre total d'ATU nominatives et à ne pas déjà disposer d'une première AMM ou d'une ATU de cohorte ( f) du 2° du A ). Cette mesure explique la coordination du B du II .
Enfin , il réduit considérablement les possibilités de dérogation aux conditions complémentaires en les limitant au seul cas d'un médicament autorisé pour une autre indication thérapeutique mais pour lequel il existe de fortes présomptions de sécurité et d'efficacité sur l'état de santé du patient ( 3° du A ).
B. Prise en charge financière des médicaments bénéficiant d'une ATU et dispensés en établissement de santé
1. La chaîne d'autorisation du médicament innovant expose le patient à certains risques de rupture de la prise en charge
La prise en charge financière d'un médicament innovant bénéficiant de l'ATU soulève deux principaux problèmes, comme l'a récemment montré un rapport 133 ( * ) présenté à la commission des affaires sociales du Sénat :
- un problème de viabilité financière , lié au caractère particulièrement coûteux de nouveaux traitements contre l'hépatite C et de nouvelles molécules anticancéreuses ;
- un problème de continuité de la prise en charge financière , lié à un manque de coordination entre la fin du dispositif de l'ATU, qui marque théoriquement la fin de la couverture intégrale de son prix par l'assurance maladie (ce dernier ayant été librement fixé par les laboratoires), et son entrée dans le droit commun du remboursement des produits de santé (le prix est fixé par le CEPS à l'issue d'une négociation). Il peut concrètement s'écouler une période dite « post-ATU » (fixée à 7 mois maximum par le législateur) au cours de laquelle le médicament anciennement sous ATU, auquel l'AMM a bien été délivrée, n'est pas encore inscrit au remboursement et continue de bénéficier de la couverture financière ATU .
Comme l'a souligné le rapport précité, la délivrance de plus en plus précoce des AMM a récemment abouti à une « compression de la phase d'ATU proprement dite et à une extension concomitante de la séquence post-ATU », entraînant la concentration des enjeux - financier et de continuité de la prise en charge - sur cette phase de post-ATU .
2. Une régulation financière du dispositif ATU qui repose sur un partage des coûts
L'article 97 de la LFSS pour 2017 134 ( * ) , puis l'article 65 de la LFSS pour 2019 135 ( * ) , ont été adoptés dans l'objectif affiché par le précédent gouvernement de « préserver » les dispositifs d'ATU et de post-ATU en assurant leur soutenabilité financière pour l'assurance maladie. Il s'agissait de mettre en place un « accès précoce plus régulé », reposant sur « un partage des coûts entre l'industriel et la collectivité ».
A été introduit un plafonnement de la dépense moyenne annuelle par patient au titre des produits sous ATU : pour tout produit dont le chiffre d'affaires hors taxes global (tous industriels confondus) excède 30 millions d'euros par an, le coût annuel par patient est limité à 10 000 euros.
Le cas échéant, ce plafonnement se traduit par un remboursement rétroactif annuel à l'assurance maladie, par le laboratoire pharmaceutique, de la part de son chiffre d'affaires en dépassement de ces seuils , sous la forme d'une remise. Ce montant est calculé par rapport au « prix net de référence » (prix net des remises conventionnellement consenties par le laboratoire) pour le produit concerné 136 ( * ) .
3. Un dispositif particulier de prise en charge pour les extensions d'indications de médicaments en post-ATU
Le rapport sénatorial précité s'est longuement penché sur certaines rigidités liées au séquençage de la chaîne financière de prise en charge (faisant se succéder les régimes financiers de l'ATU, du post-ATU une fois l'AMM délivrée et du remboursement de droit commun). Il souligne notamment que « passée la délivrance de l'AMM [donc en post-ATU] , le périmètre de l'ATU se fige et se trouve limité au produit employé dans les indications ayant fait ou faisant l'objet de la demande d'AMM » 137 ( * ) , ce qui restreint le public éligible au médicament post-ATU à deux catégories seulement : « les patients répondant strictement aux indications de l'AMM et ceux relevant d'extensions d'indications demandées par l'industriel et en cours d'évaluation par l'agence européenne des médicaments ».
L'extension d'indication non concernée par la procédure d'AMM européenne tombait par conséquent dans le champ du droit commun de la négociation conventionnelle avec le CEPS, ce qui ne présentait pour le laboratoire exploitant qu'une faible incitation et exposait de nombreux patients à d'importantes pertes de chances. C'est pourquoi l'article 65 de la LFSS pour 2019 a introduit une disposition spécifique prévoyant que lorsqu'une spécialité pharmaceutique dispose d'une AMM pour au moins l'une de ses indications tout en bénéficiant du régime financier du post-ATU, l'autorité ministérielle fixe « la compensation accordée à l'entreprise exploitant la spécialité pour sa mise à disposition dans le cadre de l'indication 138 ( * ) pour laquelle une prise en charge est autorisée ». Ce régime de la compensation se trouve néanmoins limité aux spécialités bénéficiant d'une ATU de cohorte .
Par ailleurs, ce même article soumet l'entreprise pharmaceutique, pour les spécialités bénéficiant du versement de cette compensation, à un régime financier de remboursement à l'assurance maladie de la différence entre le chiffre d'affaires facturé au titre de l'indication spécifique hors AMM (pour toutes les spécialités commercialisées par l'entreprise) et le montant qui aurait résulté de la valorisation des unités vendues et utilisées dans le cadre de cette indication et selon la compensation (donc comme si le médicament considéré avait effectivement bénéficié du régime financier post-ATU).
4. Le dispositif proposé par l'article 30
L'extrême complexité des régimes financiers des médicaments non-inscrits au remboursement, variable selon leur entrée dans le circuit de financement (ATU ou post-ATU) et selon les indications thérapeutiques pour lesquelles ils sont prescrits, est depuis longtemps fortement critiquée, notamment par les laboratoires pharmaceutiques qui dénoncent leurs effets désincitatifs et l'aberration comptable du caractère rétroactif du remboursement (qui les contraint à des provisionnements importants).
a) Fin du remboursement rétroactif et aménagement des modalités de versement des remises
Le II de l'article 30 comprend plusieurs mesures de redéfinition et d'assouplissement du régime général de ce remboursement auxquels les laboratoires sont soumis pour les spécialités sous ATU ou post-ATU.
Le b) du 1° du A prévoit que l'intégralité des remises 139 ( * ) dues par le laboratoire devra être versée en une seule fois au titre de l'année au cours de laquelle l'inscription au remboursement a eu lieu. L'intégralité des remises s'apprécie à l'aune de l'indication, et non du produit. Le remboursement cesse donc d'être rétroactif , pour ne désormais intervenir qu'au moment de l'inscription du produit dans le droit commun de la prise en charge .
Le laboratoire pourra par ailleurs bénéficier de modalités dérogatoires de versement de ces remises s'il signe avec le CEPS une convention prévoyant :
- soit le versement sur deux années successives d'un montant de remises ne pouvant être inférieur à celui qu'il doit (le laboratoire y bénéficie donc d'un étalement de la période de versement des remises, postérieure à l'inscription au remboursement) ;
- soit le versement en une seule fois, au titre de l'année de l'inscription au remboursement, d'un montant de remise égal à celui prévu hors convention, auquel cas pourra être appliquée une décote maximale de 3 % . Il est manifeste, dans ce cas-ci, que le laboratoire devra, pour bénéficier de la décote, adapter en conséquence sa négociation avec le CEPS pour la définition du prix du médicament mis sur le marché .
Il est également prévu que le ministre chargé de la sécurité sociale communique au laboratoire exploitant le médicament en ATU ou en post-ATU, uniquement pour l'indication pour laquelle l'AMM du produit a été demandé , un montant prévisionnel auquel l'assurance maladie pourra prendre en charge cette indication.
b) Prise en charge financière pour une extension d'indication de médicament en post-ATU dans le cas d'une ATU nominative
Le 2° du A du II étend le bénéfice du mécanisme de la compensation (qui joue, pour rappel, dans le cas d'une demande d'extension d'indication d'un médicament en post-ATU) aux laboratoires exploitant des médicaments auxquels a été attribuée une ATU nominative , mais uniquement pour ceux d'entre eux auxquels il a également été attribué une ATU de cohorte .
C. Application de l'article 30
Le A du III prévoit une application à partir du 1 er mars 2020 des dispositions de l'article 30 relatives à :
- la restriction des conditions d'accès aux ATU nominatives ;
- l'éligibilité de certaines ATU nominatives au bénéfice de la compensation en cas d'extension d'indication thérapeutique en cours de phase post-ATU.
Le B du III prévoit une application des dispositions de l'article 30 relatives aux nouveaux modes de versement des remises liées à la phase post-ATU à compter d'une date postérieure à l'entrée en vigueur du présent projet de loi ou pour lesquelles la prise en charge a pris fin au cours de l'année 2019.
Le C du III exclut les ATU nominatives de la disposition de l'article 30 prévoyant la communication obligatoire par le ministre de la sécurité sociale aux laboratoires d'un montant prévisionnel indiquant la prise en charge après inscription au remboursement.
Le D du III garantit le maintien de l'engagement des laboratoires exploitants à garantir la continuité des traitements ATU ou post ATU initiés, malgré les modifications apportées par le présent article 30.
II - Les modifications adoptées par l'Assemblée nationale
En plus de huit amendements rédactionnels, le rapporteur général a déposé un amendement de précision visant à soustraire l'ensemble des ATU nominatives déposées (et non délivrées) avant le 1 er mars 2020 du champ d'application du présent article.
L'Assemblée nationale a adopté cet article ainsi modifié.
III - La position de la commission
L'article 30, dont l'objectif est de garantir la soutenabilité financière du modèle de l'ATU, contient plusieurs mesures visant concurremment à faciliter le pilotage financier des ATU nominatives pour les entreprises pharmaceutiques tout en restreignant leur accès. Or votre commission, à travers de nombreux travaux, s'alarme depuis plusieurs années des restrictions croissantes que connaît le régime des ATU en termes d'accès précoce aux médicaments innovants.
A. Des dispositions favorables au pilotage des coûts pour les laboratoires
Votre commission salue les dispositions de l'article 30 qui clarifient le régime de financement des ATU par l'assurance maladie, notamment :
- la fin du caractère rétroactif du remboursement après la délivrance de l'AMM ;
- la possibilité de négocier l'étalement de ce remboursement avec le CEPS ;
- l'extension du mécanisme de la compensation aux ATU nominatives ayant déjà bénéficié d'une ATU de cohorte ;
- l'obligation faite au ministère de la sécurité sociale de communiquer au laboratoire un montant prévisionnel auquel l'assurance maladie pourrait prendre en charge la spécialité sous ATU pour l'indication ayant fait l'objet de l'AMM.
Ces mesures sont de nature à permettre aux exploitants d' adapter au mieux leurs prévisions et d'assurer un pilotage financier plus prospectif des spécialités qui ne sont pas encore ouvertes au remboursement.
B. La restriction des conditions d'accès aux ATU nominatives
Pour autant, votre commission s'inquiète vivement des nouveaux critères de délivrance des ATU nominatives, qui font l'objet d'un resserrement d'autant moins compréhensible que leur financement est de toute façon rétrocédé à l'assurance maladie en cas de chiffre d'affaires prévisionnel annuel de plus de 30 millions d'euros, pour la part supérieure à 10 000 euros par patient traité.
Elle déplore la mesure prévoyant que le nombre total d'ATU nominatives par médicament sera désormais limité par un plafond fixé par arrêté ministériel, qui ne manquera pas de drastiquement limiter l'accès des patients à l'innovation thérapeutique .
Elle interroge par ailleurs la cohérence du dispositif retenu, qui refuse à un médicament l'ATU nominative s'il dispose d'une première AMM, indépendamment de l'indication pour laquelle la demande est effectuée, mais qui autorise tout de même son attribution si « le médicament a fait l'objet d'un arrêt de commercialisation, si l'indication thérapeutique sollicitée est différente de celle de l'autorisation du médicament ayant fait l'objet de cet arrêt et qu'il existe de fortes présomptions d'efficacité et de sécurité du médicament dans l'indication thérapeutique sollicitée ».
Elle a donc adopté un amendement n° 204 proposant une réécriture moins restrictive des conditions d'accès aux ATU nominatives. Elle a également, par un amendement n° 213 , précisé le cas d'éligibilité du patient à l'ATU nominative : l'absence d'alternative thérapeutique doit s'apprécier au regard de la poursuite efficace du traitement.
Votre commission vous propose d'adopter cet article ainsi modifié.
Article additionnel après
l'article 30
Expérimentation d'une évaluation et d'une prise
en charge temporaires de certains médicaments sur la base de leur valeur
thérapeutique relative
Objet : Cet article, adopté par la commission à l'initiative de son rapporteur, met en place à titre expérimental une évaluation de certains médicaments sur la base de leur valeur thérapeutique relative et une prise en charge conditionnée à la production de nouvelles données de vie réelle.
L'amendement n° 149 fait suite, notamment, aux travaux engagés par la Mecss sur l'accès précoce aux médicaments innovants 140 ( * ) .
Ceux-ci ont mis en évidence la nécessité d'adapter les procédures de droit commun au changement de paradigme en matière d'innovation thérapeutique, afin de fluidifier l'accès des patients aux médicaments innovants après leur autorisation de mise sur le marché.
Les conclusions, en juillet 2018, du 8 ème conseil stratégique des industries de santé (CSIS) ont confirmé cet objectif et ont envisagé, dans un objectif de lisibilité, une réforme de l'évaluation du médicament sur la base de sa valeur thérapeutique relative (VTR), telle que préconisée par le rapport remis en 2015 par Dominique Polton. Cette réforme n'est pas encore engagée.
Or, l'évaluation de l'amélioration du service médical rendu (ASMR) par la commission de transparence de la Haute Autorité de santé est rendue difficile dans certaines situations, lorsque l'autorisation de mise sur le marché est délivrée par l'agence européenne du médicament sur la base de données précoces, jugées peu robustes sur le plan scientifique.
Il est donc proposé, à titre expérimental , de pouvoir fonder le prix du médicament sur un autre critère, la VTR, qui serait soumis à des réévaluations périodiques en fonction des données de vie réelle collectées après l'autorisation de mise sur le marché .
La prise en charge de ces médicaments, y compris sur la liste en sus à l'hôpital, serait conditionnée à la collecte de ces données. Afin de rendre ce dispositif opérationnel, un mécanisme de récupération du différentiel de prix est envisagé en cas de révision du prix.
Il s'agit ainsi d'une proposition équilibrée entre la volonté de favoriser l'accès des patients à une innovation thérapeutique porteuse d'espoir, les exigences indispensables en termes d'efficacité et de sécurité ainsi que les enjeux de maîtrise du prix des médicaments par une réévaluation plus dynamique.
La commission vous demande d'adopter cet article additionnel ainsi rédigé.
Article 31
Transfert du
financement
de l'ANSP et de l'ANSM vers l'Ondam
Objet : Cet article procède au transfert du financement de l'agence nationale de sécurité du médicament et des produits de santé et de l'agence nationale de santé publique de l'État vers l'assurance maladie.
I - Le dispositif proposé
• L'agence nationale de sécurité du médicament et des produits de santé (ANSM) et l'agence nationale de santé publique (ANSP), plus communément connue sous le nom de « Santé publique France », sont aujourd'hui financées sur le budget de l'État, par le programme 204 « Prévention, sécurité sanitaire et offre de soins » de la mission « Santé ». En loi de finances initiale pour 2019, les crédits de paiement consentis à ces deux opérateurs se sont établis à :
- 118 millions d'euros pour l'ANSM ;
- 153,74 millions d'euros pour Santé publique France.
Deux autres opérateurs sanitaires sont financés par le programme 204 : l'institut national du cancer (INCa) et, pour partie, l'agence nationale de sécurité sanitaire de l'alimentation, de l'environnement et du travail (ANSéS).
• Comme l'a décrit notre collègue Corinne Imbert dans le rapport pour avis de votre commission sur les crédits de la mission « Santé » dans le projet de loi de finances pour 2019, plusieurs modifications du périmètre du programme 204 sont intervenues dans la période récente dans un souci de simplification des circuits de financement des politiques publiques de santé afin de mettre en oeuvre les préconisations de la Cour des comptes pour un décroisement des financements des opérateurs sanitaires par l'État et l'assurance maladie :
Ø dans la loi de finances initiale pour 2015 : ont été transférées à l'assurance maladie les parts de financement par l'État :
- de l'agence technique de l'information et de l'hospitalisation (ATIH) (3,3 millions d'euros de la part de l'État en 2014) ;
- du centre national de gestion des praticiens hospitaliers et des personnels de direction de la fonction publique hospitalière (CNG) (3,7 millions d'euros de la part de l'État en 2014) ;
- de la Haute Autorité de santé (HAS) (14,8 millions d'euros de la part de l'État en 2014) ;
- de la formation médicale initiale 141 ( * ) (139 millions d'euros de la part de l'État en 2014) ;
Ø dans la loi de finances initiale pour 2017 :
- a été transférée à l'assurance maladie la part de financement par l'État du fonds d'intervention régional (FIR) 142 ( * ) (116 millions d'euros de la part de l'État en 2016) ;
- a été transférée à l'État la part de financement par l'assurance maladie de Santé publique France (ANSP) (65 millions d'euros de la part de l'assurance maladie en 2016) ;
Ø dans la loi de finances initiale pour 2018 : ont été transférées à l'assurance maladie les parts de financement par l'État :
- de l'agence de la biomédecine (ABM) (14 millions d'euros de la part de l'État en 2017) ;
- de l'école des hautes études en santé publique (EHESP) (9 millions d'euros de la part de l'État en 2017).
• Afin de mettre en oeuvre le transfert de financement envisagé, l' article 31 du PLFSS pour 2020 procède aux modifications suivantes au sein du code de la santé publique :
- le I rétablit à l'article L. 1413-12 du code de la santé publique un 2° prévoyant le financement de Santé publique France par une dotation des régimes obligatoires d'assurance maladie versée et répartie dans des conditions fixées par décret ;
- le II ajoute à l'article L. 5321-2 du code de la santé publique un 5° prévoyant, parmi les ressources de l'ANSM, une dotation des régimes obligatoires d'assurance maladie versée et répartie dans des conditions fixées par décret.
Il est à noter que l'article 31 du PLFSS pour 2020 n'exclut pas définitivement un éventuel financement complémentaire de ces opérateurs par l'État dès lors que sont maintenues les dispositions permettant, aux articles L. 1413-12 et L. 5321-2 précités, un financement par une subvention de l'État.
• L' article 17 du PLFSS pour 2020 prévoit une compensation à l'euro près de ces transferts par une majoration, au bénéfice de l'assurance maladie, du transfert des recettes de TVA de l'État aux régimes de sécurité sociale, à hauteur de 268,6 millions d'euros, dont 156,1 millions d'euros pour le financement de Santé publique France et 112,5 millions d'euros pour le financement de l'ANSM 143 ( * ) .
II - Les modifications adoptées par l'Assemblée nationale
L'Assemblée nationale a adopté cet article sans modification.
III - La position de la commission
Votre commission partage le souci de simplifier les modalités de financement des opérateurs sanitaires, à la condition que tout décroisement de ce financement soit opéré dans le respect des missions de chaque opérateur et de l'exigence d'un pilotage national efficient.
Le financement intégral de l'ANSM par l'assurance maladie apparaît ainsi justifié au regard de la place centrale que cette agence occupe dans la mise en oeuvre de notre politique de sécurité du médicament et d'encadrement des pratiques médicales, principalement financée par l'assurance maladie. Deux autres opérateurs intervenant dans la mise en oeuvre de cette politique sont du reste d'ores et déjà financés intégralement par l'assurance maladie : la HAS et l'agence de la biomédecine 144 ( * ) .
En revanche, le transfert du financement de Santé publique France de l'État vers l'assurance maladie intervient seulement trois ans après que le Gouvernement eut précisément fait le choix inverse en loi de finances initiale pour 2017 145 ( * ) . Il était alors apparu au Gouvernement légitime d'assurer un financement intégral par l'État de cet opérateur chargé principalement de missions de surveillance épidémiologique, de prévention et de promotion de la santé, à l'instar de l'ANSéS, autre agence intervenant en matière de veille sanitaire et de surveillance épidémiologique et financée par l'État 146 ( * ) et par une part du produit de la taxe sur les ventes de pesticides.
Dans un contexte de multiplication des risques sanitaires qui mobilise fortement les agences de surveillance épidémiologique 147 ( * ) , votre commission craint que le désengagement financier de l'État n'affaiblisse le pilotage national de notre politique de veille sanitaire. En outre, la disparition de tout financement de Santé publique France par l'État conduirait à dépouiller encore plus la mission « Santé » des opérateurs chargés de mettre en oeuvre ses deux principaux objectifs : « améliorer l'état de santé de la population et réduire les inégalités territoriales et sociales de santé » et « prévenir et maîtriser les risques sanitaires ».
Actant le transfert envisagé par l'article 31 du PLFSS pour 2020, le projet annuel de performances de la mission « Santé » annexé au projet de loi de finances pour 2020 n'intègre plus désormais que le financement et les emplois de l'INCa, l'ANSéS étant financée en premier lieu par le programme 206 de la mission « Agriculture, alimentation, forêt et affaires rurales ». Or l'INCa est un groupement d'intérêt public (GIP) chargé d'élaborer, dans le domaine de l'oncologie, des recommandations de bonne pratique à destination des professionnels et des établissements de santé, et d'accompagner les patients et leurs proches dans leur parcours de prise en charge : la question du transfert de son financement de l'État vers l'assurance maladie se posera donc vraisemblablement à l'avenir. Compte tenu de ses missions étroitement liées à l'organisation de l'offre de soins en oncologie, le transfert de son financement apparaît d'ailleurs plus légitime que celui de Santé publique France, d'autant que la CNAM est membre de ce GIP. Dans cette éventualité, la mission « Santé » se retrouverait dépourvue de tout opérateur sanitaire à part entière.
Par conséquent, votre commission a adopté un amendement tendant à supprimer le I de l'article 31 afin que le financement de Santé publique France reste intégralement assuré par le budget de l'État (amendement n° 174).
La commission vous demande d'adopter cet article ainsi modifié.
* 94 « Pilotage de la dépense de santé : redonner du sens à l'Ondam », rapport d'information n° 40 (2019-2020) de Catherine DEROCHE et René-Paul SAVARY, au nom de la mission d'évaluation et de contrôle de la sécurité sociale et de la commission des affaires sociales, Sénat, 9 octobre 2019.
* 95 D'après la liste fixée par l'arrêté ministériel du 23 juin 2016, révisé le 18 avril 2018.
* 96 Décret n° 2016-658 du 20 mai 2016 relatif aux hôpitaux de proximité et à leur financement.
* 97 Ce montant correspond au différentiel entre le montant de la garantie lorsqu'elle est effectivement perçue (soit quand elle est supérieure à la seule activité valorisée) et ce qu'auraient perçu les établissements en l'absence de garantie. Il est à rapprocher de la masse financière globale d'environ 500 millions d'euros que représentent à l'heure actuelle les 243 hôpitaux de proximité.
* 98 Loi n° 2015-1702 du 21 décembre 2015 de financement de la sécurité sociale pour 2016, article 78.
* 99 Loi n° 2017-1836 du 30 décembre 2017 de financement de la sécurité sociale pour 2018, article 68.
* 100 Correspondent aux spécialités pharmaceutiques qui peuvent être prises en charge, sur présentation des factures, en sus des prestations d'hospitalisation remboursables par la sécurité sociale, dont la liste est fixée par arrêté.
* 101 La circulaire du 4 mai 2018 prévoyant notamment que « les dotations de la psychiatrie doivent faire l'objet d'une attention particulière pour garantir le développement de la spécialité et permettre les transformations attendues dans les territoires pour répondre aux besoins de la population ».
* 102 L'Argus de l'assurance, le 21 janvier 2016.
* 103 « Les urgences hospitalières : des services toujours trop sollicités », Cour des comptes, rapport public annuel 2019, février 2019.
* 104 « Les urgences hospitalières, miroir des dysfonctionnements de notre système de santé », rapport d'information n° 685 (2016-2017), présenté par Laurence COHEN, Catherine GÉNISSON et René-Paul SAVARY, au nom de la commission des affaires sociales, Sénat, 26 juillet 2017.
* 105 Missions d'intérêt général et d'aide à la contractualisation.
* 106 Source : Cour des comptes, rapport cité ci-après.
* 107 « Le rôle des CHU dans l'enseignement supérieur et la recherche médicale », rapport d'information n° 228 (2017-2018), Sénat, 17 janvier 2018.
* 108 Sur ce sujet, cf. rapport d'information n° 569 (2017-2018), « Médicaments innovants : consolider le modèle français d'accès précoce ».
* 109 « Réforme des modes de financement et de régulation », janvier 2019.
* 110 Igas, « Évaluation de la tarification des soins hospitaliers et des actes médicaux », mars 2012.
* 111 On désigne par « dispositif médical » tout produit ou prestation couvert par la définition qu'en donne l'article L. 165-1 du code de la sécurité sociale : « dispositifs médicaux à usage individuel, tissus et cellules issus du corps humain quel qu'en soit le degré de transformation et leurs dérivés, produits de santé autres que les médicaments et prestations de services et d'adaptation associées ».
* 112 Règlement (UE) n° 2017/745 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux, modifiant la directive 2001/83/CE, le règlement (CE) n° 178/2002 et le règlement (CE) n° 1223/2009 et abrogeant les directives du Conseil 90/385/CEE et 93/42/CEE.
* 113 À noter que, contrairement aux médicaments, les mécanismes de régulation de l'offre des dispositifs médicaux ne tiendront pas compte du respect quantitatif de l'Ondam, mais d'un respect qualitatif de l'objectif d'efficience des dépenses d'assurance maladie.
* 114 Il subsiste un débat relatif au droit au recours subrogatoire dont les conseils départementaux s'estiment titulaires à l'égard des assureurs des tiers dans le cas d'une aide technique financée suite à un accident de la vie.
* 115 IGAS, Évaluation de la prise en charge des aides techniques pour les personnes âgées dépendantes et les personnes handicapées , avril 2013.
* 116 IGAS, Évolution de la prestation de compensation du handicap , op. cit.
* 117 IGAS, Évaluation de la prise en charge des aides techniques pour les personnes âgées dépendantes et les personnes handicapées , avril 2013.
* 118 Repenser le financement du handicap pour accompagner la société inclusive , rapport d'information de M. Philippe MOUILLER, fait au nom de la commission des affaires sociales, n° 35 (2018-2019), 10 octobre 2018.
* 119 Loi n° 2018-1203 du 22 décembre 2018 de financement de la sécurité sociale pour 2019.
* 120 Pour les dispositifs médicaux, il s'agit de s'assurer de leur conformité aux dispositions du règlement (UE) 2017/745 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux, modifiant la directive 2001/83/CE, le règlement (CE) n° 178/2002 et le règlement (CE) n° 1223/2009 et abrogeant les directives du Conseil 90/385/CEE et 93/42/CEE.
* 121 Chiffré à 200 millions d'euros pour l'exercice 2020.
* 122 Loi n° 2018-1203 du 22 décembre 2018 de financement de la sécurité sociale pour 2019.
* 123 Site internet du ministère.
* 124 Jean LORENZI, « Les nouveaux facteurs de la distribution du médicament », LSJEA , n° 35, 2007.
* 125 Médicaments innovants : consolider le modèle français d'accès précoce , rapport d'information de M. Yves DAUDIGNY, Mmes Catherine DEROCHE et Véronique GUILLOTIN, présenté à la mission d'évaluation et de contrôle de la Sécurité sociale (Mecss) de la commission des affaires sociales du Sénat (n° 159, 2017-2018).
* 126 À la différence de l'importation parallèle, définie à l'article R. 5121-115 du CSP (simplement au niveau réglementaire).
* 127 On notera que le cas d'une distribution parallèle d'un médicament non titulaire d'une AMM européenne n'est juridiquement pas réalisable, l'AMM européenne définissant précisément la distribution parallèle.
* 128 ANSM, État des lieux sur les médicaments biosimilaires , mai 2016.
* 129 « Les médicaments biosimilaires contribuent à sécuriser la disponibilité de certaines classes de médicaments et à dégager des marges financières par la mise en concurrence. Le PLFSS complète les outils existants pour accompagner le développement des bio-similaires, en encourageant les établissements de santé à acheter ces médicaments. Une marge de progrès existe qui devrait permettre des gains considérables pour nos hôpitaux publics. »
* 130 Cette disposition de l'article 29 était fort mal coordonnée avec l'article L. 162-16-5 du CSS qui maintenait le principe général de la négociation conventionnelle des prix de cession des médicaments au public.
* 131 Décision DG n° 2019-365 du 15 octobre 20019 portant création d'un comité scientifique temporaire « mise en oeuvre de l'expérimentation du cannabis médical en France » à l'agence nationale de sécurité du médicament et des produits de santé.
* 132 Notamment auprès des centres de lutte contre le cancer.
* 133 Médicaments innovants : consolider le modèle français d'accès précoce , rapport d'information de M. Yves DAUDIGNY, Mmes Catherine DEROCHE et Véronique GUILLOTIN, fait au nom de la mission d'évaluation et de contrôle de la sécurité sociale et de la commission des affaires sociales, n° 569 (2017-2018).
* 134 Loi n° 2016-1827 du 23 décembre 2016 de financement de la sécurité sociale pour 2017.
* 135 Loi n° 2018-1203 du 22 décembre 2018 de financement de la sécurité sociale pour 2019.
* 136 Cette disposition figure au III de l'article L. 162-16-5-1 du CSS : lors d'une première inscription au remboursement, si le prix net d'une spécialité est inférieur au montant de l'indemnité ATU déclarée au CEPS, le laboratoire reverse sous forme de remise la différence entre le chiffre d'affaires facturé aux établissements de santé, au titre de la période s'entendant de l'obtention de l'ATU à la première date d'inscription au remboursement, et le chiffre d'affaires qui aurait résulté de la valorisation des unités vendues au prix net de référence.
* 137 Ce qui résulte des dispositions combinées du deuxième alinéa de l'article L. 162-16-5-1-1 et du premier alinéa de l'article L. 162-16-5-2 du CSS.
* 138 Qui n'est pas celle pour laquelle l'AMM a été accordée.
* 139 On rappelle que les remises désignent la part de son chiffre d'affaires facturé au titre de la spécialité en dépassement du seuil de 10 000 euros par patient.
* 140 « Médicaments innovants : consolider le modèle français d'accès précoce », rapport d'information n° 569 (2017-2018) de Yves DAUDIGNY, Catherine DEROCHE et Véronique GUILLOTIN, au nom de la mission d'évaluation et de contrôle de la sécurité sociale et de la commission des affaires sociales, Sénat, 13 juin 2018.
* 141 Essentiellement l'indemnisation des internes de médecine générale et de certaines spécialités en dehors de leur centre hospitalier universitaire de rattachement.
* 142 Qui subventionne des actions et expérimentations territoriales dans le domaine de la santé validées par les agences régionales de santé (ARS).
* 143 Montants des dotations précisés par le projet annuel de performances de la mission « Santé » annexé au projet de loi de finances pour 2020.
* 144 Qui encadre notamment les pratiques médicales et thérapeutiques associées à l'assistance médicale à la procréation, à l'examen des caractéristiques génétiques humaines ou encore aux dons d'organes, de tissus et de cellules.
* 145 Loi n° 2016-1917 du 29 décembre 2016 de finances pour 2017.
* 146 La subvention pour charges de service public globale de l'ANSéS comprend également des dotations de l'État au titre des programmes 206 « Sécurité et qualité sanitaires de l'alimentation », 111 « Amélioration de la qualité de l'emploi et des relations du travail », 181 « Prévention des risques » et 190 « Recherches dans les domaines de l'énergie, du développement et de la mobilité durables ».
* 147 Maladie de Lyme, affaire Lactalis, agénésies transverses des membres supérieurs de nouveaux-nés, émissions toxiques de l'usine de Mourenx, pollution au plomb à la suite de l'incendie de Notre-Dame, conséquences de l'incendie de l'usine Lubrizol...