







Rapport d'information n° 653 (2011-2012) de M. Bernard CAZEAU , fait au nom de la MCI portant sur les dispositifs médicaux implantables, déposé le 10 juillet 2012
Disponible au format PDF (1,2 Moctet)
-
INTRODUCTION
-
PREMIÈRE PARTIE - UN ENCADREMENT
INADAPTÉ AUX ÉVOLUTIONS DE SECTEURS
MÉDICO-ÉCONOMIQUES EN PLEINE EXPANSION
-
I. LA RÉGLEMENTATION APPLICABLE AUX
DISPOSITIFS MÉDICAUX IMPLANTABLES : LA LIBRE CIRCULATION
PLUTÔT QUE LA SÉCURITÉ
-
A. UNE RÉGLEMENTATION EUROPÉENNE
ENCORE RÉCENTE, DOMINÉE PAR LA DOCTRINE DE LA
« NOUVELLE APPROCHE »
-
1. Une réglementation européenne qui
entend « laisser aux entreprises la plus grande latitude pour remplir
leurs obligations vis-à-vis du public »
-
2. Le marquage CE matérialise la
conformité du dispositif médical aux « exigences
essentielles » communautaires
-
3. Les responsabilités en matière
d'évaluation, de contrôle et de surveillance des dispositifs
médicaux
-
4. Un marché aux contours difficiles
à appréhender
-
1. Une réglementation européenne qui
entend « laisser aux entreprises la plus grande latitude pour remplir
leurs obligations vis-à-vis du public »
-
B. LE MARQUAGE CE REMIS EN CAUSE
-
1. Une inquiétude croissante quant à
la capacité du système de régulation européen
à garantir la sécurité des patients
-
2. Une trop grande disparité entre les Etats
membres dans les exigences de compétence et de transparence applicables
aux organismes notifiés
-
3. Des exigences en matière d'investigation
clinique limitées
-
4. Un partage des informations insuffisant
-
1. Une inquiétude croissante quant à
la capacité du système de régulation européen
à garantir la sécurité des patients
-
C. UNE COMPARAISON INTERNATIONALE PEU
FAVORABLE
-
A. UNE RÉGLEMENTATION EUROPÉENNE
ENCORE RÉCENTE, DOMINÉE PAR LA DOCTRINE DE LA
« NOUVELLE APPROCHE »
-
II. LES INTERVENTIONS À VISÉE
ESTHÉTIQUE : UN ENCADREMENT TARDIF ET INSUFFISANT
-
A. UN ENJEU DE SOCIÉTÉ À PART
ENTIÈRE
-
B. LA CHIRURGIE ESTHÉTIQUE : UN
ENCADREMENT JURIDIQUE SOLIDE
-
C. LA MÉDECINE ESTHÉTIQUE : UNE
RÉGLEMENTATION QUI COURT APRÈS DES TECHNIQUES EN
PERPÉTUELLE ÉVOLUTION
-
A. UN ENJEU DE SOCIÉTÉ À PART
ENTIÈRE
-
I. LA RÉGLEMENTATION APPLICABLE AUX
DISPOSITIFS MÉDICAUX IMPLANTABLES : LA LIBRE CIRCULATION
PLUTÔT QUE LA SÉCURITÉ
-
DEUXIÈME PARTIE - CRÉER LES
CONDITIONS D'UNE VÉRITABLE SÉCURITÉ
-
I. DE L'IMPLANTABLE À
L'IMPLANTÉ : RENFORCER LE CONTRÔLE DES DISPOSITIFS
MÉDICAUX À TOUTES LES ÉTAPES
-
A. CLARIFIER LES CONDITIONS DE MISE SUR LE
MARCHÉ DES DISPOSITIFS MÉDICAUX
-
1. Préciser le rôle des organismes
notifiés
-
2. Assurer la transparence du processus de
certification
-
3. Rendre plus transparentes les relations entre
professionnels de santé et fabricants
-
4. Repenser le marquage CE
-
a) Inciter la Commission à prendre
pleinement en compte les exigences de sécurité des dispositifs
médicaux
-
b) Créer une liste positive des dispositifs
médicaux les plus risqués au niveau européen
-
c) Interdire l'utilisation de produits
cancérogènes, mutagènes et reprotoxiques dans les
dispositifs médicaux
-
a) Inciter la Commission à prendre
pleinement en compte les exigences de sécurité des dispositifs
médicaux
-
1. Préciser le rôle des organismes
notifiés
-
B. INSTITUER UNE VÉRITABLE SURVEILLANCE DES
DISPOSITIFS MÉDICAUX IMPLANTÉS
-
1. Une évidence : appliquer enfin le
droit existant
-
2. Mettre en oeuvre sans tarder les
avancées de la loi « Médicament » en
matière de dispositifs médicaux
-
3. Remettre la matériovigilance au coeur du
système de sécurité sanitaire
-
4. Améliorer l'identification des
dispositifs médicaux implantables et le partage des
données
-
5. Assurer la pleine traçabilité des
dispositifs médicaux implantés grâce à des registres
exhaustifs
-
a) Respecter des principes de mise en oeuvre
précis
-
b) Lever l'obstacle juridique de la protection des
données personnelles
-
c) Créer en priorité des registres
de dispositifs médicaux à risque, en coopération avec les
parties intéressées
-
d) Parvenir enfin à valoriser les
informations des systèmes d'information hospitaliers
-
a) Respecter des principes de mise en oeuvre
précis
-
6. Revoir les modalités de
l'évaluation et de la surveillance clinique des dispositifs
médicaux implantables avant et après leur
commercialisation
-
7. Garantir la qualité de l'information des
praticiens et des patients
-
1. Une évidence : appliquer enfin le
droit existant
-
A. CLARIFIER LES CONDITIONS DE MISE SUR LE
MARCHÉ DES DISPOSITIFS MÉDICAUX
-
II. RESPONSABILISER LES ACTEURS DE
L'ESTHÉTIQUE POUR MIEUX PRENDRE EN COMPTE SES IMPLICATIONS
SANITAIRES
-
A. POSER LES BASES D'UNE TYPOLOGIE DES PRATIQUES
DE MÉDECINE ESTHÉTIQUE
-
1. Instituer un marquage CE spécifique aux
dispositifs et produits esthétiques afin de garantir leur
innocuité
-
2. Assurer la transparence de l'information sur
les actes à finalité esthétique
-
3. Privilégier la sécurité
-
4. Renforcer les exigences de formation et de
compétence
-
a) Instituer un diplôme national de
médecine esthétique
-
b) Créer un registre qualité des
médecins pratiquant des actes à visée
esthétique
-
c) Encadrer les exigences de qualification des
assistants des médecins esthétiques
-
d) Préciser les obligations de formation
des professions non médicales du secteur de l'esthétique
-
a) Instituer un diplôme national de
médecine esthétique
-
5. Clarifier les conditions de tarification des
actes médicaux à visée esthétique
-
1. Instituer un marquage CE spécifique aux
dispositifs et produits esthétiques afin de garantir leur
innocuité
-
B. METTRE EN PLACE UNE VÉRITABLE
« ESTHÉTICOVIGILANCE »
-
A. POSER LES BASES D'UNE TYPOLOGIE DES PRATIQUES
DE MÉDECINE ESTHÉTIQUE
-
I. DE L'IMPLANTABLE À
L'IMPLANTÉ : RENFORCER LE CONTRÔLE DES DISPOSITIFS
MÉDICAUX À TOUTES LES ÉTAPES
-
CONCLUSION - PRIMUM NON NOCERE
-
LES PRINCIPALES PROPOSITIONS DE LA MISSION
D'INFORMATION
-
COMPTE RENDU DE LA RÉUNION D'EXAMEN DU
RAPPORT
-
ANNEXE - LISTE DES PERSONNES
AUDITIONNÉES
N° 653
SÉNAT
SESSION EXTRAORDINAIRE DE 2011-2012
|
Enregistré à la Présidence du Sénat le 10 juillet 2012 |
RAPPORT D'INFORMATION
FAIT
au nom de la Mission commune d'information portant sur les dispositifs médicaux implantables et les interventions à visée esthétique (1),
Par M. Bernard CAZEAU,
Sénateur.
Tome I : Rapport.
|
(1) Cette commission est composée de : Mme Chantal Jouanno, présidente ; M. Bernard Cazeau, rapporteur ; Mme Aline Archimbaud, M. Gilbert Barbier, Mmes Laurence Cohen, Catherine Deroche, Catherine Génisson, Nathalie Goulet, vice-présidents ; M. Philippe Bas, Mmes Marie-Thérèse Bruguière, Françoise Cartron, Caroline Cayeux, MM. Alain Fauconnier, Michel Fontaine, Mme Christiane Kammermann, MM. Ronan Kerdraon, Jacky Le Menn, Jean Louis Masson, Alain Milon, Jean-Jacques Mirassou, Alain Néri, Mmes Isabelle Pasquet, Gisèle Printz, MM. Gérard Roche, René-Paul Savary, René Teulade et Mme Catherine Troendle. |
INTRODUCTION
Mesdames, Messieurs,
La création par le Sénat d'une mission commune d'information portant sur les dispositifs médicaux implantables et les interventions à visée esthétique répondait à l'émotion suscitée par le scandale des prothèses mammaires PIP. Cette triste affaire, désormais entre les mains de la justice, est venue malheureusement confirmer les craintes que les sénateurs avaient exprimées à l'automne 2011, lors de l'examen de la loi sur la sécurité du médicament et des produits de santé : après le Mediator, le nouveau scandale sanitaire concerne bel et bien un dispositif médical. Il est d'ailleurs regrettable qu'il ait fallu attendre l'émoi suscité dans l'opinion par cette affaire pour que l'attention se porte enfin sur un volet de la politique de santé resté dans l'ombre malgré les trop nombreux incidents enregistrés au cours des années récentes concernant, par exemple des prothèses de hanche ou des sondes cardiaques.
Avant d'aborder le cadre dans lequel la mission a mené à bien ses travaux, il est utile d'effectuer un retour en arrière sur le scandale PIP. Au-delà de l'utilisation d'un gel de silicone à usage industriel dans un produit médical, il est l'illustration la plus choquante des graves dysfonctionnements de la réglementation des dispositifs médicaux.
I. AFFAIRE PIP, RETOUR SUR UN SCANDALE SANITAIRE EXEMPLAIRE
Une technologie longtemps controversée
Les premières prothèses mammaires ont été mises sur le marché aux Etats-Unis en 1962, bien avant l'adoption d'une réglementation régissant les dispositifs médicaux. Du fait d'une suspicion relative au risque de maladies auto-immunes et des cas de ruptures et de fuites observés, l'utilisation des prothèses remplies de gel de silicone y fut interdite en avril 1992, pour n'être autorisée à nouveau qu'en 2006. Dans l'appréciation générale de ces implants qu'elle a menée à bien en juin 2011 1 ( * ) , la Food and Drug Administration (FDA), autorité sanitaire américaine, reconnaît qu'il n'existait alors que peu de données sur les événements rares et les effets à long terme de ces prothèses. En conséquence, elle avait imposé aux fabricants de mener à bien des études après mise sur le marché. Les investigations cliniques menées depuis lors sur ces types d'implants font ressortir un haut niveau de satisfaction des femmes à l'égard de leur image corporelle et de l'aspect, de la taille et de la sensation de leurs implants. En outre, ces études ne démontrent pas d'incidence négative de ces implants sur la fertilité 2 ( * ) .
En France, la commercialisation de ce type de prothèses remplies de gel de silicone a fait l'objet de moratoires successifs à compter de 1995 ; la suspension de leur mise sur le marché n'a été levée qu'en 2001.
Une procédure de certification dévoyée
Créée en 1991, la société Poly Implant Prothèse (PIP) n'a pas fait parler d'elle avant 1995. Aux Etats-Unis, elle n'a jamais demandé l'autorisation de commercialiser ses produits remplis de gel de silicone. En revanche, des prothèses remplies de sérum physiologique ont été utilisées dans ce pays, dans le seul cadre de la reconstruction mammaire. Ceci explique que la FDA ait été conduite à les contrôler. Dès 1996, elle a d'ailleurs envoyé une lettre d'avertissement ( « warning letter » ) à PIP . Ce courrier suivait une visite d'inspection sur place ; il faisait état de non-conformités du niveau le plus élevé (aux exigences de bonne pratique de fabrication).
En 2000, la FDA adresse une nouvelle lettre d'avertissement à PIP . La même année, les échanges de courriers entre la société et l'Agence française de sécurité sanitaire des produits de santé (Afssaps) commencent. En réponse aux demandes de l'agence, PIP transmet notamment les certificats de conformité européenne (dits marquage CE) délivrés par l'organisme qu'elle a choisi pour faire certifier ses produits, TÜV Rheinland, puis, en août 2001, le bilan d'audit « ne faisant pas apparaître de risques particuliers » 3 ( * ) .
A la suite d'un audit sur site, réalisé du 17 au 19 novembre 2003, l'autorité sanitaire australienne (Therapeutic Goods Administration - TGA) relève trois non-conformités majeures (trous dans le sol et les murs d'une salle propre ; ouverture simultanée de portes exposant les installations à contamination depuis l'extérieur ; méthodes utilisées par PIP afin de démontrer l'absence de contamination) ainsi que plusieurs problèmes de moindre importance. L'obtention de la certification européenne délivrée par un organisme habilité permet de lever les restrictions préalables à l'homologation des prothèses.
Entendus par la mission le 9 mai 2012, les représentants des organismes notifiés allemands, notamment de TÜV Rheinland, ont souligné que pour obtenir la certification de ses prothèses, PIP avait choisi la voie de la documentation plutôt que de l'examen du produit type . Au sens des directives européennes, ces deux procédures sont considérées comme équivalentes. Si le fabricant choisit le processus de documentation, il a toute latitude pour réaliser les contrôles de qualité lui-même . Il transmet ensuite le résultat de ces derniers à l'organisme notifié, qui les vérifie sur papier. Interrogés par les membres de la mission, les représentants des organismes notifiés allemands ont expliqué que, si le producteur décide de recourir à la documentation, « l'organisme notifié ne peut travailler que sur le dossier qui lui est fourni, tant que la documentation est conforme aux exigences de production dudit produit. La possibilité de recourir à la demande d'échantillons est une exception. Il faut, pour y avoir recours, des indications montrant que des problèmes existent sur le produit contrôlé » .
Des signaux d'alerte négligés
Pourtant, dès 2001, neuf déclarations de chirurgiens transmises à l'Afssaps relèvent, notamment, des cas de rupture ; les incidents interviennent généralement peu de temps après l'implantation. Dans tous les cas signalés, le chirurgien procède à l'explantation des prothèses. Des événements indésirables de même ampleur sont signalés à l'agence au cours des années suivantes. En mai 2006, vingt-huit plaintes sont déposées simultanément au Royaume-Uni, dans le cadre du Consumer Protection Act ; elles mettent en cause la résistance des enveloppes des prothèses. Les autorités sanitaires britanniques n'ont, semble-t-il, pas été informées de ces procédures contentieuses.
En 2008, les cas de matériovigilance rapportés à l'Afssaps se multiplient . Certains déclarants effectuent plusieurs signalements. Pourtant, en avril 2009, le conseil scientifique de l'agence considère que le taux d'incidents sur les prothèses mammaires en silicone - toutes marques confondues - est globalement stable bien que le volume de vente soit en croissance. Lors d'une réunion à l'Afssaps, le 18 décembre 2009, PIP fournit la même explication pour expliquer l'accroissement du nombre d'anomalies rapportées.
Au cours d'une inspection de la société le 17 mars 2010, la fraude est découverte . Une plainte est déposée auprès du procureur du tribunal de grande instance de Toulon. Le 29 mars, l'Afssaps décide le « retrait et la suspension de la mise sur le marché, de la distribution, de l'exportation et de l'utilisation des implants mammaires préremplis de gel de silicone fabriqués par la société PIP » .
Finalement, le 31 mars 2010, l'Afssaps prévient ses homologues de cette décision. Ce n'est qu'à compter de cette date que les autorités des différents pays échangent véritablement sur cette question. Leur réaction est très différente, d'autant que le sujet ne prend de l'ampleur qu'à la fin de 2011.
Une information trop peu partagée
L'enchaînement des événements montre de graves déficiences dans la circulation de l'information.
Sur le plan européen, comme l'a souligné le représentant de TÜV Rheinland lors de son audition par la mission, « l'audit de qualité en entreprise n'a rien à voir avec la surveillance du marché, qui dépend des autorités compétentes de chaque Etat membre.
« Par exemple, le chiffre relatif au nombre de ruptures de prothèses n'est pas signalé à l'organisme notifié. »
De même, aucune sanction ne semble exister si le fabricant ne signale pas à l'organisme notifié qu'il s'est vu opposer un refus par un autre organisme pour obtenir la certification de son produit.
Sur le plan international, la FDA a multiplié les lettres d'avertissement à PIP (celle de juin 2000 est mentionnée dans le rapport précité de l'Afssaps et de la DGS, remis au ministre de la santé en février 2012). En mars 2000, son comité technique spécialisé ( « General and Plastic Surgery Panel » ) a recommandé à l'unanimité le rejet de la demande d'autorisation de mise sur le marché formulée par PIP pour ses implants mammaires remplis de sérum physiologique. Rien ne dit que cette recommandation ait été communiquée aux autorités françaises, pas plus que les échanges de courrier entre l'agence et la société (mai 2000, lettre de PIP à la FDA l'informant de son intention de renoncer à sa demande d'autorisation de mise sur le marché ; le 15 mai 2000, lettre de la FDA à PIP lui enjoignant de cesser la distribution de ses produits et de tous les retirer du marché ; novembre 2002, lettre de la FDA à PIP exigeant que la société écrive à l'ensemble des 1 175 médecins concernés afin de leur demander de lui retourner les prothèses non implantées).
Les résultats du contrôle sur site réalisé par la TGA australienne en 2003 n'ont vraisemblablement pas été portés à la connaissance des autorités sanitaires d'autres pays ; aucune d'entre elles n'a pris connaissance des procédures en réparation engagées au Royaume-Uni.
PIP, une entreprise qui éprouve des difficultés à s'assurer
La mauvaise circulation de l'information est tout aussi flagrante en matière d'assurance.
La loi du 4 mars 2002 4 ( * ) a mis à la charge des professionnels de santé, y compris des producteurs, exploitants et fournisseurs de produits de santé, l'obligation de souscrire une assurance destinée à les garantir pour leur responsabilité civile ou administrative susceptible d'être engagée en raison de dommages subis par des tiers et résultant d'atteintes à la personne.
Comme en matière de dommage ouvrage, d'assurance automobile ou de catastrophe naturelle, l'assujetti qui a essuyé deux refus d'assurance a la faculté de saisir le Bureau central de tarification (BCT) afin que celui-ci impose à l'assureur choisi par l'entreprise de le prendre en charge. PIP s'est trouvée dans l'obligation de saisir le BCT dès 2004. Rien ne dit que l'entreprise était assurée avant cette date. A l'appui de sa demande, PIP invoque des problèmes financiers, liés à une baisse importante de son activité en France, qui l'empêchent de s'assurer. En l'absence de sinistre au cours des cinq années précédentes, Allianz est désigné comme son assureur , moyennant le paiement d'une surprime égale à 2 % du chiffre d'affaires. La décision du BCT en date du 28 juin 2005 est notifiée à l'assureur et à l'entreprise mais elle n'est pas suivie d'effet car une double faille dans la réglementation apparaît : le fabricant n'est pas tenu d'appliquer la décision du BCT et aucun organe ou administration n'a compétence pour contrôler que l'entreprise a bien rempli ses obligations d'assurance.
En 2008, PIP saisit à nouveau le BCT. Le dossier présenté par l'entreprise fait état de nombreux incidents de matériovigilance. Une nouvelle fois, en l'absence de sinistre durant les cinq années précédentes et malgré des inquiétudes sur la qualité même des produits , rien ne permettait au BCT, dans la limite de ses attributions, de majorer la prime d'assurance pour circonstances aggravantes.
On ne peut néanmoins s'empêcher de penser que ces difficultés récurrentes à s'assurer auraient dû constituer un nouveau signal d'alerte pour les pouvoirs publics, d'autant que les demandes d'intervention du BCT formulées par les fabricants de produits de santé sont très peu nombreuses.
Des victimes durablement traumatisées
Le temps des études scientifiques, comme celui de la justice, n'est pas compatible avec le ressenti de femmes qui souffrent quotidiennement. A ce titre, elles méritent la considération de l'ensemble de la communauté nationale. Dans ce contexte difficile et qui risque de le rester encore longtemps, la reconnaissance de la validité du contrat d'assurance de PIP auprès d'Allianz pourrait constituer un premier élément de satisfaction. En l'espèce, contraint en 2005 puis en 2008 d'être l'assureur en responsabilité civile de la société PIP, Allianz avait estimé avoir été trompé et avait demandé la reconnaissance de la nullité du contrat. L'assureur avait donc porté plainte contre les liquidateurs judiciaires de cette société, le 27 juillet 2010, auprès du tribunal de commerce de Toulon, notamment pour fausse déclaration délibérée. Le 12 juin 2012, le tribunal a débouté Allianz de toutes ses demandes et a constaté « la validité des polices d'assurance délivrées dans le cadre strict de l'obligation édictée par la loi de 2002 » . L'un des conseils de l'assureur s'est immédiatement déclaré surpris « d'avoir à garantir les circonstances d'une fraude manifeste » et a annoncé son intention d' « examiner l'éventualité d'interjeter un appel » .
De même, le 18 juin 2012, Allianz a été condamné par le tribunal de grande instance de Lyon à verser une indemnité de 19 650 euros à une femme porteuse d'implants PIP en réparation des préjudices liés à la défectuosité des produits fabriqués par cette société.
Si elles étaient confirmées, ces décisions pourraient permettre de lever les nombreux obstacles que rencontrent les victimes, au moins en termes d'indemnisation du préjudice qu'elles ont subi.
Dans un communiqué commun publié le 9 juin 2012, les deux principales associations de victimes 5 ( * ) ont marqué leur impatience face à ce qu'elles interprètent comme l'inertie des pouvoirs publics. Sur le plan médical, les explantations se poursuivent ; elles montrent un taux de rupture bien supérieur à celui qui était attendu . Cette découverte contribue à alimenter l'angoisse des victimes, en particulier de celles qui se trouvent dans l'impossibilité de faire face aux coûts des opérations chirurgicales, voire même d'avancer les sommes par la suite prises en charge par la collectivité au titre des seules explantations. Elle devrait également inciter à accélérer l'évaluation scientifique des conséquences ou séquelles physiques et psychologiques pour les victimes.
Une gestion de crise à expertiser
Peu de pays ont fait le choix de recommander l'explantation préventive des prothèses mammaires PIP à toutes les femmes qui en sont porteuses, même en l'absence de signes cliniques de détérioration des implants. La France est l'un d'entre eux. Elle a également décidé de prendre en charge toutes les opérations d'explantation, sans considération des motifs qui avaient conduit à la pose des prothèses.
La plupart des femmes souhaitent, semble-t-il, bénéficier de la réimplantation de nouvelles prothèses. Praticiens et industriels ont été appelés à faire preuve de tact et de mesure, de sorte de ne pas rendre ces opérations financièrement impossibles. Cette recommandation ne paraît pas toujours respectée et des victimes ont dû renoncer à les faire réaliser. La découverte d'un taux de rupture plus élevé que celui qui était anticipé valide pourtant le choix d'une explantation généralisée, ne serait-ce qu'en raison de considérations financières : privilégier le suivi régulier des femmes porteuses de prothèses PIP par rapport à leur explantation immédiate pourrait s'avérer à long terme plus coûteux pour la collectivité.
Il est donc indispensable de disposer de données précises et incontestables sur l'ensemble du processus. La mission souhaite que le Gouvernement demande à l'inspection générale des affaires sociales (Igas) de dresser un bilan détaillé de la gestion de cette crise sanitaire. Ce bilan devra notamment s'attacher à affiner les constatations faites sur les risques spécifiques des prothèses PIP, y compris en termes d'apparition de pathologies associées, à faire le point sur le déroulement des opérations d'explantation et de réimplantation et sur les conditions dans lesquelles les chirurgiens sont appelés à les pratiquer.
*
Par son ampleur, par le caractère systématique et d'ailleurs reconnu des manquements observés 6 ( * ) ainsi que par le nombre des victimes avérées ou potentielles partout dans le monde (au moins quatre cent mille), ce scandale est exceptionnel . La justice mettra sans doute plus de temps à se prononcer sur les infractions commises par PIP et ses dirigeants que les victimes ne sont aujourd'hui prêtes à attendre. Il nous faut dès maintenant en tirer les conséquences, car si le scandale des prothèses PIP représente un problème sanitaire d'une nature tout à fait particulière, il comporte plusieurs éléments récurrents dans ce type d'affaire : l'information circule mal, les personnes qui achètent les produits de la société font confiance à ceux qui sont chargés de son contrôle, les lanceurs d'alerte ne sont pas pris au sérieux, le régulateur agit avec retard.
Le scandale PIP constitue le cas extrême des graves insuffisances de la réglementation des dispositifs médicaux implantables. C'est aujourd'hui celle-ci, dans son ensemble, qui doit faire l'objet d'une investigation approfondie.
II. LES TRAVAUX DE LA MISSION D'INFORMATION
La mission d'information du Sénat est née d'une volonté, celle de faire toute la lumière sur les enchaînements qui n'ont pas permis de détecter rapidement les pratiques de la société PIP. Pour faire la lumière sur cette affaire, elle a étendu ses investigations à l'ensemble des dispositifs médicaux implantables. Depuis qu'elle a commencé ses travaux, des difficultés majeures concernant d'autres dispositifs que les prothèses mammaires sont apparues au grand jour.
Soucieuse d'éviter qu'une fois le système d'autorisation et de contrôle des dispositifs médicaux mis à plat, un nouveau scandale éclate, dans le domaine de la chirurgie ou de la médecine esthétiques, elle a choisi d'étendre le champ de ses investigations aux interventions à visée esthétique, qui constituent désormais un enjeu à part entière.
Depuis sa constitution, le 21 février dernier, la mission a mené trente-deux heures d'auditions, dont six tables rondes. Au total, ce sont plus de cinquante organisations, associations ou entreprises qui lui ont fait part de leurs préoccupations ou suggestions. Conformément à la volonté de transparence décidée dès le départ, ces auditions - à de rares exceptions près - ont été ouvertes au public et à la presse. Elles ont également fait l'objet d'une diffusion simultanée sur le site Internet du Sénat, ainsi que d'un enregistrement vidéo.
Trois déplacements ont permis de compléter les éléments recueillis au cours de ces auditions. Le suivi du processus de fabrication de prothèses en silicone par six membres de la mission ainsi que la présentation d'un centre de formation à la pose de dispositifs implantables utilisés en cardiologie ont fait l'objet d'un reportage audiovisuel disponible en ligne.
Afin d'obtenir des précisions sur la législation applicable dans les deux domaines relevant de son champ d'investigation, la mission s'est rendue aux Etats-Unis, notamment au Maryland, Etat modèle en matière de régulation esthétique, ainsi qu'en Suède et au Danemark, deux pays aux réglementations plus drastiques ayant mené des expérimentations intéressantes sur les registres médicaux. Elle a complété ces informations auprès de l'ambassade d'Australie en France, pays qui lui était apparu en pointe, tant en matière de suivi des dispositifs médicaux que de vigilance par rapport aux pratiques de médecine esthétique.
Elle s'est enfin attachée à appréhender l'état d'avancement de la refonte des directives européennes relatives aux dispositifs médicaux et s'est rendue à Bruxelles , pour s'entretenir avec les représentants de la direction générale de la santé et des consommateurs de la Commission ainsi qu'avec Gilles Pargneaux, député européen, rapporteur de la résolution du Parlement européen sur les prothèses mammaires.
Les dispositifs médicaux :
un secteur
méconnu à l'impact croissant en matière de
santé
Parce qu'il recouvre une multitude de produits, du pansement au pacemaker , de l'abaisse-langue à la prothèse de hanche, de la lentille de contact au drug eluting stent 7 ( * ) , le secteur des dispositifs médicaux reste largement méconnu. Il se situe toujours dans l'ombre du médicament, bien qu'il s'agisse d'un domaine fondamentalement différent .
|
Médicament et dispositif
médical :
|
||
|
Médicament |
Dispositif médical |
|
|
Evolution technologique |
Lente |
Rapide |
|
Mode d'action |
Chimique |
Physique |
|
Industries de base |
Chimie |
Multiples : chimie, électronique, mécanique, textile, plasturgie... |
|
Durée d'action |
Courte |
Longue |
|
Possibilité d'expérimentation in vitro |
Forte |
Faible |
|
Faculté de mettre en oeuvre des traitements à l'aveugle |
Forte |
Faible ou nulle |
|
Faculté de mener des essais sur des populations de patients étendues |
Forte |
Quasi nulle |
|
Source : mission d'information du Sénat sur les dispositifs médicaux implantables et les interventions à visée esthétique |
||
Le nombre de produits regroupés sous cette appellation dépasse de très loin celui des médicaments et la nature des fabricants est très variée, de la multinationale à la micro-entreprise innovante. Pourtant, les dispositifs médicaux concernent chacun d'entre nous, à des degrés divers, quotidiennement ou presque. A l'avenir, l'allongement de la durée de la vie aura un impact fort sur l'utilisation des plus sophistiqués d'entre eux - les dispositifs médicaux implantables - sur lesquels la mission d'information a choisi de faire porter ses investigations en priorité 8 ( * ) . L'évolution des techniques permettra de recourir à des matériels de santé plus personnalisés, plus petits et moins invasifs. Ces transformations auront pour conséquence d'accentuer les difficultés que pose leur évaluation préalable : contrairement aux médicaments, ces produits de santé ne peuvent par définition faire l'objet d'études à l'aveugle, avec groupe témoin, sous placebo. L'étroitesse des populations concernées constitue un frein supplémentaire à la réalisation d'essais touchant des cohortes de malades.
La réflexion ne saurait oublier l'impact économique de ces deux secteurs . La France constitue le quatrième producteur mondial de dispositifs médicaux, filière qui emploie soixante mille personnes. Sa technologie est reconnue en matière d'aide technique, de systèmes de chirurgie mini-invasive, de diagnostic par imagerie, de diagnostic in vitro et, pour ce qui entre dans le champ d'étude de la mission, de prothèses.
Car, ne l'oublions pas, dans tous les cas, ces produits s'adressent à des personnes qui souffrent de problèmes de santé. La mesure du rapport entre le bénéfice attendu et le risque encouru est au coeur de l'analyse qui doit en être faite.
Lorsque la vie même du patient est en cause, un certain niveau de risque doit être accepté. Mais encore faut-il que ce risque soit correctement mesuré avant la commercialisation du produit et fasse l'objet d'un suivi précis après l'implantation .
Les interventions à visée
esthétique :
nouvelles pratiques, nouveaux dangers
La problématique des interventions à visée esthétique est fondamentalement différente : réparer une infirmité, lutter contre la maladie ou prolonger la vie de personnes qui souffrent dans leur chair n'est pas la seule motivation de ceux qui y font appel. Au contraire, beaucoup sont en bonne santé physique ; on ne peut donc admettre que leur vie soit mise en danger et leur intégrité physique menacée.
Il est logique que ces interventions ne présentent aucun danger. Une prise de conscience semble se dessiner, si l'on en croit le recul sensible des techniques les plus invasives. En revanche, de nouvelles techniques de médecine esthétique apparaissent sans cesse. Médecins, dentistes, professionnels de l'esthétique, revendiquent de pouvoir pratiquer ces actes. De nouveaux marchés se créent pour les industriels. L'imagination est sans limite et, si elle n'est pas condamnable en soi, la volonté de nos contemporains de prendre en compte leur image ne doit pas leur faire perdre de vue l'essentiel : ne pas nuire à leur santé. Evidemment, toutes ces interventions ont un coût ; en conséquence, le marché du tourisme esthétique médical est né, et avec lui de nouveaux risques sanitaires.
En matière d'esthétique, la France continue d'attirer les amoureux de la « French touch » , chirurgie esthétique dont les résultats paraissent plus naturels que les interventions réalisées dans d'autres pays. Les soins esthétiques constituent une activité économique en plein développement, pourvoyeuse d'emplois non délocalisables au sein d'un secteur - les services à la personne - en forte croissance.
La mission a pleinement conscience de ces enjeux, sans qu'ils puissent prévaloir sur les implications de santé publique. Il est fondamental de mettre en oeuvre un nouvel équilibre entre innovation et évaluation, entre impact économique et protection des consommateurs, de sorte à placer la sécurité de tous au premier rang des priorités.
PREMIÈRE PARTIE - UN ENCADREMENT INADAPTÉ AUX ÉVOLUTIONS DE SECTEURS MÉDICO-ÉCONOMIQUES EN PLEINE EXPANSION
I. LA RÉGLEMENTATION APPLICABLE AUX DISPOSITIFS MÉDICAUX IMPLANTABLES : LA LIBRE CIRCULATION PLUTÔT QUE LA SÉCURITÉ
Dans le cadre de la finalisation du marché commun européen à partir de 1986, la nécessité de supprimer les entraves aux échanges commerciaux a conduit les institutions communautaires et, en particulier, la Commission européenne, à procéder à une harmonisation des règles techniques applicables à un certain nombre de produits. A l'époque, dans un contexte de renforcement progressif des exigences en matière de santé publique dans plusieurs pays européens, les caractéristiques de sécurité et les procédures de contrôle des dispositifs médicaux différaient sensiblement d'un Etat membre à l'autre.
C'est pourquoi, dans le cadre de la « nouvelle approche » 9 ( * ) poursuivie par les institutions communautaires en matière d'harmonisation technique et de normalisation, celles-ci ont élaboré une réglementation visant à concilier le principe de libre circulation, applicable aux dispositifs médicaux, et l'exigence du maintien d'un niveau élevé de sécurité et de qualité en ce qui concerne les prestations et soins impliquant ces produits de santé.
A. UNE RÉGLEMENTATION EUROPÉENNE ENCORE RÉCENTE, DOMINÉE PAR LA DOCTRINE DE LA « NOUVELLE APPROCHE »
1. Une réglementation européenne qui entend « laisser aux entreprises la plus grande latitude pour remplir leurs obligations vis-à-vis du public »10 ( * )
a) La libre circulation, principe général
Conformément à l'esprit de la « nouvelle approche », la législation d'harmonisation technique communautaire entend soumettre, à partir de la fin des années 1980, des familles de produits à des « exigences essentielles » en vue de leur libre circulation dans le marché unique. Les tentatives précédentes consistant à élaborer, sous forme de directives particulières, des règles techniques applicables à des catégories plus restreintes de biens s'étaient avérées incompatibles avec le libre-échange au sein du marché intérieur.
Dans son Guide relatif à la mise en application des directives élaborées sur la base des dispositions de la nouvelle approche et de l'approche globale , publié en 2000, la Commission européenne souligne que « ces approches complémentaires ont en commun de limiter l'intervention des pouvoirs publics à l'essentiel et de laisser aux entreprises la plus grande latitude pour remplir leurs obligations vis-à-vis du public » .
C'est ainsi que le Conseil de l'Union européenne a adopté, au début des années 1990, deux directives relatives aux dispositifs médicaux 11 ( * ) afin de les soumettre à des exigences essentielles de sécurité compatibles avec un haut niveau de performance des prestations de santé :
- la directive 90/385/CEE du Conseil du 20 juin 1990 concernant le rapprochement des législations des Etats membres relatives aux dispositifs médicaux implantables actifs ;
- la directive 93/42/CEE du Conseil du 14 juin 1993 relative aux dispositifs médicaux.
Leur dernière modification remonte à 2007, avec la directive 2007/47/CE du Parlement européen et du Conseil du 5 septembre 2007 12 ( * ) . Celle-ci a rendu obligatoire l'évaluation clinique des dispositifs médicaux les plus à risque.
b) Le risque potentiel, seul élément de spécificité des dispositifs médicaux
Au fur et à mesure de leurs modifications successives, les directives de 1990 et 1993 ont adopté une définition commune de la notion de dispositif médical afin d'y inclure une très grande variété de produits. Leur champ d'application se fonde sur la destination du produit , c'est-à-dire l'utilisation à laquelle il est destiné. C'est ainsi qu'un logiciel est considéré comme un dispositif médical dès lors qu'il est destiné par le fabricant à être utilisé à des fins diagnostique et/ou thérapeutique.
Dans leur version consolidée à ce jour, les directives précitées définissent comme dispositif médical :
« tout instrument, appareil, équipement, logiciel, matière ou autre article, utilisé seul ou en association, y compris le logiciel destiné par le fabricant à être utilisé spécifiquement à des fins diagnostique et/ou thérapeutique, et nécessaire au bon fonctionnement de celui-ci, destiné par le fabricant à être utilisé chez l'homme à des fins :
« - de diagnostic, de prévention, de contrôle, de traitement ou d'atténuation d'une maladie,
« - de diagnostic, de contrôle, de traitement, d'atténuation ou de compensation d'une blessure ou d'un handicap,
« - d'étude ou de remplacement ou modification de l'anatomie ou d'un processus physiologique,
« - de maîtrise de la conception,
« - et dont l'action principale voulue dans ou sur le corps humain n'est pas obtenue par des moyens pharmacologiques ou immunologiques ni par métabolisme, mais dont la fonction peut être assistée par de tels moyens. »
Le seul élément permettant d'illustrer la spécificité des dispositifs médicaux en tant que produits de santé (par rapport aux autres produits industriels et commerciaux régis par la « nouvelle approche ») consiste , dans la directive de 1993, en une classification en fonction du risque potentiel qu'ils représentent pour la sécurité des utilisateurs, patients et médecins . Inspirée du système américain, cette classification est également proche de celle des autres pays occidentaux (Canada, Australie, Japon) ou émergents (Brésil, Corée, Russie). Les dispositifs médicaux sont, ainsi, répartis en quatre classes : I, II a , II b et III .
|
La classification des dispositifs
médicaux
|
||
|
Classe de dispositif |
Exemples de dispositifs concernés |
|
|
Classe I
|
Dispositifs non invasifs, pansements, abaisse-langue, scalpels, fauteuils roulants, lunettes |
|
|
Classe II
a
|
Instruments de diagnostic, lentilles de contact, appareils d'aide auditive, oxygénateurs, agrafes cutanées |
|
|
Classe II
b
|
Dispositifs contraceptifs, préservatifs, hémodialyseurs, pompes à perfusion, sutures internes |
|
|
Classe III
|
Dispositifs invasifs en contact avec le coeur, le système sanguin ou le système nerveux central, prothèses mammaires, sondes d'aspiration aortique, prothèses articulaires de la hanche, du genou et de l'épaule |
|
|
Source : mission d'information du Sénat sur les dispositifs médicaux implantables et les interventions à visée esthétique |
||
La même directive précise que les procédures d'évaluation de conformité aux exigences essentielles dépendent de la classe à laquelle appartient le dispositif médical concerné :
- les procédures d'évaluation de conformité pour les dispositifs de la classe I peuvent être effectuées, en règle générale, sous la seule responsabilité des fabricants , vu le faible degré de vulnérabilité de ces produits ;
- pour les dispositifs de la classe II a , l' intervention obligatoire d'un organisme notifié doit avoir pour objet le stade de la fabrication , et suppose l'évaluation et le contrôle du système d'assurance qualité mis en place par l'entreprise ;
- pour les dispositifs des classes II b et III qui présentent un potentiel élevé de risque, un contrôle effectué par un organisme notifié s'impose en ce qui concerne la conception des dispositifs ainsi que leur fabrication ;
- la classe III est réservée aux dispositifs les plus critiques pour lesquels la mise sur le marché présuppose « une autorisation préalable explicite sur la conformité » .
2. Le marquage CE matérialise la conformité du dispositif médical aux « exigences essentielles » communautaires
Les directives de 1990 et 1993 soumettent l'introduction sur le marché intérieur des dispositifs médicaux à l'obtention de la marque CE. L'apposition de ce symbole sur un produit est conditionnée à la réalisation, à la charge de son fabricant, des contrôles et essais qui garantissent sa conformité aux exigences essentielles définies par la directive concernée en matière de sécurité, de santé et de protection de l'environnement. Le marquage CE ne constitue pas une marque de certification ou d'appréciation de la qualité du dispositif , ni ne détermine son origine géographique de production, puisqu'il peut être apposé sur un produit fabriqué hors de l'Union européenne .
Les exigences essentielles auxquelles les dispositifs médicaux doivent satisfaire afin d'obtenir le marquage CE sont précisées au sein des annexes I de chacune des deux directives précitées. En l'absence de disposition propre aux dispositifs médicaux implantables dans leur ensemble, c'est celle du 20 juin 1990, relative aux dispositifs médicaux implantables actifs qui se rapproche le plus du champ des exigences qui leur sont applicables. Dans son annexe I, elle prévoit comme première exigence essentielle, que « les dispositifs doivent être conçus et fabriqués de telle manière que leur utilisation ne compromette pas l'état clinique ni la sécurité des patients lorsqu'ils sont implantés dans les conditions et aux fins prévues. Ils ne doivent pas présenter de risques pour les personnes qui les implantent ni, le cas échéant, pour des tiers » .
Dans le même ordre d'idées, l'annexe I de la directive du 14 juin 1993 définit la première exigence essentielle applicable à tout dispositif médical dans les termes suivants : « les dispositifs doivent être conçus et fabriqués de telle manière que, lorsqu'ils sont utilisés dans les conditions et aux fins prévues, leur utilisation ne compromette pas l'état clinique et la sécurité des patients ni la sécurité et la santé des utilisateurs ou, le cas échéant, d'autres personnes, étant entendu que les risques éventuels liés à leur utilisation constituent des risques acceptables au regard du bienfait apporté au patient et compatibles avec un niveau élevé de protection de la santé et de la sécurité » .
L'évaluation de la conformité des dispositifs médicaux aux exigences essentielles des directives est effectuée par un « organisme notifié » chargé de vérifier que les produits satisfont aux dispositions pertinentes de la directive concernée. A titre d'exemple, dans le cadre de la procédure dite de « déclaration CE de conformité », il est précisé que « l'organisme notifié procède périodiquement aux inspections et aux évaluations appropriées afin de s'assurer que le fabricant applique le système de qualité approuvé et fournit un rapport d'évaluation au fabricant » . Il peut, en outre, « faire des visites inopinées au fabricant » .
La directive du 5 septembre 2007 a précisé dans l'article 9 de la directive du 20 juin 1990 et dans l'article 11 de la directive du 14 juin 1993 que « les décisions des organismes notifiés [...] ont une durée de validité de cinq ans maximum . Elles peuvent être reconduites pour des périodes supplémentaires d'une durée maximale de cinq ans, sur demande formulée à la date prévue par le contrat signé par les deux parties » .
3. Les responsabilités en matière d'évaluation, de contrôle et de surveillance des dispositifs médicaux
Les directives de 1990 et 1993 permettent d'identifier quatre acteurs concernés par la mise en oeuvre de la réglementation relative aux dispositifs médicaux :
- les fabricants ;
- les organismes notifiés ;
- les autorités compétentes , communautaires et nationales, chargées de veiller au respect de la réglementation et, le cas échéant, de son adaptation ;
- les utilisateurs (professionnels de santé, patients, tiers).
a) Les organismes notifiés évaluent les niveaux de performance et de sécurité des dispositifs médicaux
L'article 1 er de la directive du 14 juin 1993 définit, pour la première fois, la notion de fabricant d'un dispositif médical. Il s'agit de « la personne physique ou morale responsable de la conception, de la fabrication, du conditionnement et de l'étiquetage d'un dispositif en vue de sa mise sur le marché en son propre nom, que ces opérations soient effectuées par cette même personne ou pour son compte par une tierce personne » .
Cette définition privilégie la responsabilité du fabricant sur l'ensemble du cycle de vie du produit, au-delà de sa seule activité de production.
Il doit veiller à « réduire [...] le risque d'une erreur d'utilisation due aux caractéristiques ergonomiques du dispositif et à l'environnement dans lequel le dispositif doit être utilisé (conception pour la sécurité du patient) » et à « prendre en compte les connaissances techniques, l'expérience, l'éducation et la formation et [...] l'état de santé et la condition physique des utilisateurs auxquels les dispositifs sont destinés (conception pour les utilisateurs profanes, professionnels, handicapés ou autres) » . 13 ( * )
Le fabricant ou son mandataire doit, ainsi, « notifier aux autorités compétentes de l'Etat membre dans lequel il a son siège social, l'adresse de son siège social ainsi que la désignation des dispositifs concernés » 14 ( * ) .
Les Etats membres notifient auprès de la Commission européenne les organismes chargés d'évaluer les performances et le respect des caractéristiques de sécurité par les dispositifs médicaux.
Les organismes notifiés ont pour mission d'évaluer la conformité du dispositif médical sur la base d'un dossier technique établi par le fabricant. Ils sont conduits à mettre en oeuvre, dans le cadre de vérifications sur pièces, des procédures de certification et des audits des systèmes de gestion de la qualité.
Contrairement aux Etats-Unis où la responsabilité de l'ensemble de la régulation du secteur des dispositifs médicaux incombe à un seul organe, la Food and Drug Administration (FDA), l'octroi du marquage CE, condition de la commercialisation sur le marché européen, est réservé en Europe aux organismes notifiés, en règle générale sociétés de droit privé à but lucratif, seuls responsables de l'évaluation des données techniques et cliniques communiquées par le fabricant .
Les Etats membres de l'Union européenne, ainsi que les Etats de l'Espace économique européen et la Turquie, disposent en pratique d'un haut degré de liberté dans la désignation des organismes notifiés. Il existe donc potentiellement plus de vingt-sept systèmes nationaux de désignation et de surveillance de ces organismes. Dans ces conditions, un niveau uniforme d'évaluation et de sécurité des dispositifs médicaux n'est pas garanti au sein de l'Union européenne.
La liste des organismes notifiés comprend également des établissements situés en Suisse et en Turquie, dans la mesure où la réglementation européenne concerne des pays associés au marché commun européen, et en Australie, en application d'accords de reconnaissance mutuelle.
Certains critères communs dans la désignation par les Etats membres des organismes notifiés ont été élaborés à partir des directives relatives aux dispositifs médicaux et de la décision n° 768/2008/CE 15 ( * ) relative à un cadre commun pour la commercialisation des produits. Les critères principaux retenus par les directives sont :
- l'indépendance ;
- l'impartialité ;
- une expertise dans le domaine des dispositifs médicaux ;
- un personnel scientifique en nombre suffisant et doté d'une expérience et de connaissances suffisantes pour évaluer, sur le plan médical, le caractère fonctionnel et les performances des dispositifs ;
- du matériel et des locaux adaptés ;
- une bonne formation professionnelle portant sur l'ensemble des opérations d'évaluation et de vérification pour lesquelles l'organisme est désigné ;
- une connaissance satisfaisante des prescriptions relatives aux contrôles qu'il effectue ;
- une pratique suffisante des contrôles, l'aptitude requise pour rédiger les attestations, procès-verbaux et rapports qui constituent la matérialisation des contrôles effectués et une assurance de responsabilité civile.
La Commission européenne a également développé des lignes directrices (« Guidelines ») concernant la désignation et la surveillance des organismes notifiés qui sont plus complètes que les directives. Elles n'ont pas de caractère contraignant pour les Etats membres . Ces derniers doivent également s'assurer que les organismes notifiés maintiennent un niveau élevé de compétences. Toutefois, il n'existe pas de procédure de contrôle harmonisée au niveau communautaire : il revient aux Etats membres seuls de choisir les moyens et les méthodes des contrôles à mettre en oeuvre .
Lorsqu'il a connaissance d'un problème concernant un organisme notifié qui cesse de remplir les critères précités, l'Etat membre concerné doit retirer ou suspendre la notification de l'organisme après l'en avoir averti. L'information doit alors être communiquée à la Commission européenne ainsi qu'aux autres Etats membres. Le retrait de la notification n'affecte pas les certificats délivrés par l'organisme notifié tant que la preuve n'est pas apportée qu'ils doivent également faire l'objet d'un retrait.
Il n'est pas possible d'établir une carte européenne de la qualité des organismes notifiés : les prothèses mammaires PIP ont été certifiées par l'organisme allemand TÜV Rheinland et les prothèses de hanche métal-métal fabriquées par la société américaine DePuy, aujourd'hui retirées du marché, par l'organisme britannique BSI. Ils jouissent tous deux d'une excellente réputation auprès des régulateurs européens et des fabricants. De plus, ils font partie des huit organismes notifiés ayant obtenu l'homologation du « Canadian Medical Devices Conformity Assessment System » (CMDCAS) permettant d'autoriser l'entrée de dispositifs sur le marché canadien 16 ( * ) .
En France, le seul organisme habilité par l'Agence nationale de sécurité du médicament et des produits de santé (ANSM, ex-Afssaps) et notifié auprès de la Commission européenne est la structure G-Med , créée en 1994 et intégrée au laboratoire national de métrologie et d'essais (LNE). Le LNE, dont l'origine remonte à 1901, dispose du statut d'établissement public à caractère industriel et commercial (Epic).
b) Les autorités compétentes des Etats membres sont chargées de surveiller le marché national et les organismes notifiés
Il appartient aux autorités compétentes des Etats membres de veiller à ce que le marché national soit conforme aux exigences des directives. A ce titre, l'ANSM est chargée, en France :
- d'assurer la mise en oeuvre de la procédure de vigilance visée à l'article 8 de la directive du 20 juin 1990 et à l'article 10 de la directive du 14 juin 1993 prévoyant l'enregistrement dans un système centralisé des informations concernant les incidents liés à un dispositif ;
- de procéder à des enquêtes périodiques ou inopinées , en particulier lorsqu'elle reçoit d'utilisateurs - professionnels de santé ou patients - des informations concernant d'éventuels dysfonctionnements liés à un dispositif médical.
Dans le cadre de la surveillance du marché des dispositifs médicaux, l'action de l'ANSM est :
- soit réactive, en mettant en oeuvre les décisions administratives ou de police sanitaire requises en réponse à un signal d'alerte ;
- soit proactive, en sélectionnant chaque année des types de dispositifs médicaux qu'elle souhaite examiner, sur la base de critères précis. Elle tient compte de l'évolution très rapide des marchés, notamment en cas de saut technologique substantiel nécessitant de vérifier de manière globale la qualité des nouveaux dispositifs. Dans la période récente, l'ANSM s'est ainsi, par exemple, intéressée aux prothèses dentaires dont l'importation s'est considérablement développée.
Dans l'exercice de son contrôle, l'ANSM déploie trois modalités d'intervention susceptibles de se combiner : soit en examinant sur dossier , c'est-à-dire en demandant au fabricant de lui soumettre son dossier de marquage CE ; soit en inspectant l'entreprise sur site ; soit en effectuant un contrôle en laboratoire . Cette dernière possibilité est la plus compliquée à mettre en oeuvre, certains dispositifs (tel le gel de silicone) se prêtant mieux que d'autres (tel le stimulateur cardiaque) à des tests en laboratoire.
Principales étapes du processus de certification des dispositifs médicaux en Europe
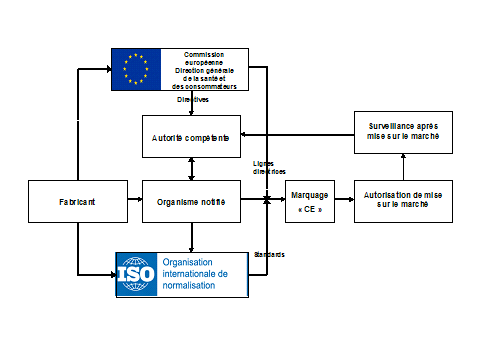
Les flèches pleines représentent les exigences formelles, les flèches en pointillés représentent les lignes directrices ou les conseils prodigués.
Source : « Clinical evaluation of
cardiovascular devices:
principles, problems, and proposals for European
regulatory reform »
in European Heart Journal Advance Access,
14 mai 2011.
4. Un marché aux contours difficiles à appréhender
Il est difficile d'évaluer le marché des dispositifs médicaux, en Europe et en France, en termes de nombre de produits commercialisés et de revenu global généré. Selon la Haute Autorité de santé (HAS), « il n'y a pas moins de quatre mille classes de dispositifs médicaux qui représentent, selon les spécialistes, cent soixante mille à huit cent mille références , des pansements aux valves cardiaques » . Selon d'autres estimations, le nombre de dispositifs médicaux mis à la disposition des professionnels de santé et des patients dans le monde est évalué à près de cinq cent mille 17 ( * ) .
M. Gilles Johanet, président du Comité économique des produits de santé (Ceps), regrettait, pour sa part, la vision très partielle du marché des dispositifs médicaux dont disposent les autorités françaises, comme en témoigne le décalage entre les chiffres livrés par l'Inspection générale des affaires sociales (Igas) en novembre 2010 (21 milliards d'euros) 18 ( * ) , et ceux du Ceps (6,8 milliards d'euros). Une des raisons pour lesquelles il demeure difficile, en France, d'appréhender la globalité de ce marché réside dans l'impossibilité pour notre système d'assurance maladie de connaître tant la valeur que le volume des dispositifs achetés par l'hôpital public : dans la pose d'un dispositif médical, son coût et, a fortiori , son modèle, ne sont pas spécifiquement identifiés au sein des groupes homogènes de séjour (GHS), qui constituent la nomenclature des actes remboursés à l'hôpital.
En privilégiant la libre circulation et en ménageant des marges de manoeuvre substantielles aux fabricants dans le choix des procédures de certification de la conformité de leurs produits aux exigences essentielles des directives, la réglementation européenne a posé les conditions d'un taux de renouvellement particulièrement élevé des dispositifs médicaux en circulation dans le marché unique. Ainsi, M. Jean-Claude Ghislain, directeur de l'évaluation des dispositifs médicaux au sein de l'ANSM, a souligné devant la mission que ses services reçoivent, chaque mois, « soixante communications de nouveaux dispositifs de classe III et cent dix de classe IIb, dont une partie est aussi constituée de dispositifs implantables » . Ce volume significatif doit toutefois être relativisé : les nouveaux dispositifs entrant sur le marché sont moins souvent des créations d'un genre nouveau qu'une évolution d'un précédent produit.
D'une façon générale, l'encadrement réglementaire européen applicable aux dispositifs médicaux est jugé propice au développement de l'innovation thérapeutique . Le processus de marquage CE est décrit comme plus rigoureux pour les dispositifs moins risqués et moins exigeant que le mécanisme d'autorisation américain ou canadien pour les dispositifs les plus critiques. Les patients de l'Union européenne auraient accès aux nouvelles techniques en matière de dispositifs médicaux avec près de trois ans d'avance , en moyenne, sur les patients américains. M. David Gollaher, président-directeur général de l'Institut de santé de Californie, groupe de pression de l'industrie médicale, entendait alerter les pouvoirs publics américains sur le fait que la simplification des procédures d'autorisation de mise sur le marché dans l'Union européenne avait débouché sur « un transfert spectaculaire des essais cliniques et des lancements de produits conduits par des entreprises américaines vers l'Europe » 19 ( * ) . Selon la même source, les dispositifs médicaux ayant obtenu, en 2010, une autorisation de mise sur le marché par la FDA, dans le cadre de la procédure la plus exigeante dite de « Premarket Approval » (PMA), auraient reçu une autorisation de mise sur le marché européen avec, en moyenne, près de quatre ans d'avance 20 ( * ) .
Le système de classification établi par les directives a vocation à tenir compte de l'évolution rapide des techniques. Les autorités françaises sont ainsi intervenues à plusieurs reprises depuis 2000, parfois de concert avec leurs partenaires britanniques, en vue de faire remonter dans la classification un certain nombre d'implants , notamment des implants mammaires. Ils ont été reclassifiés de la classe II b à la classe III par la directive 2003/12/CE de la Commission du 3 février 2003 21 ( * ) . La reclassification a également concerné les prothèses articulaires totales de hanche, d'épaule et de genou. Lors de son audition devant la mission, M. Dominique Maraninchi, directeur général de l'ANSM, n'a pas manqué de souligner que « la classification des dispositifs de I à III n'est apparue que très récemment et qu'elle commence à peine à être mise en oeuvre » , ajoutant qu'il aura « fallu au moins trois ans pour digérer la classification actuelle » .
En outre, il convient de rappeler qu'une des spécificités du marché européen des dispositifs médicaux, par rapport notamment au marché américain, réside dans un taux de pénétration élevé des produits de fabrication étrangère , à quelques exceptions près, comme les prothèses de hanche.
Il existe traditionnellement moins de marques de dispositifs médicaux commercialisées sur le marché américain que sur le marché européen, en raison principalement de la charge financière que représentent la production de données cliniques, leur exploitation et la surveillance du marché, à laquelle s'ajoute la responsabilité induite lors de la délivrance des produits. La réglementation européenne constitue un cadre propice à l'innovation et à la créativité des petites entreprises fabriquant des dispositifs médicaux à destination de publics très ciblés .
B. LE MARQUAGE CE REMIS EN CAUSE
1. Une inquiétude croissante quant à la capacité du système de régulation européen à garantir la sécurité des patients
Interrogé par les membres de la mission sur les différences essentielles entre les réglementations européenne et américaine, M. Dominique Maraninchi a plaidé pour des efforts tendant à « rapprocher notre système de celui d'un processus d'autorisation de mise sur le marché explicite , plutôt que d'essayer de répondre à des exigences essentielles » . Pour l'heure, le considérant 15 de la directive de 1993 se borne à une référence à une « autorisation préalable explicite sur la conformité » conditionnant la mise sur le marché des dispositifs médicaux de la classe III.
Une étude consacrée aux liens entre les rappels de dispositifs médicaux au Royaume-Uni et le système de régulation applicable, publiée en mai 2011 dans le British Medical Journal , a dénoncé l' absence totale de publicité sur les données cliniques transmises par les fabricants aux organismes notifiés . Les auteurs de l'enquête ont ainsi souligné s'être à plusieurs reprises heurtés au refus des organismes notifiés de communiquer les éléments cliniques associés aux dispositifs médicaux de classe III autorisés, ceux-ci ayant répondu que « toutes les données cliniques concernant des dispositifs médicaux qu'ils ont autorisés ne pouvaient être mises à la disposition du public. L'organisme notifié est un client travaillant pour le compte du fabricant et considère les données cliniques comme des informations à caractère commercial sensibles » 22 ( * ) .
Les directives communautaires relatives aux dispositifs médicaux exigent des investigations cliniques qu'elles soient conformes aux principes de la déclaration d'Helsinki , adoptée en 1964 et amendée à plusieurs reprises depuis. Ce document prévoit précisément l' inscription d'un essai dans un registre public avant l'enrôlement du premier patient ainsi que la publication des résultats des études cliniques afin, notamment, de permettre aux médecins de donner une information éclairée.
Cette situation a conduit M. Jeffrey Shuren, directeur du centre des dispositifs médicaux et de la santé radiologique ( « Center for Devices and Radiological Health » - CRDH) de la FDA, à déclarer en juillet 2011, à l'occasion d'une audition devant la commission de l'énergie et du commerce de la Chambre des représentants des Etats-Unis, que « l'Union européenne approuve généralement de façon plus rapide que les Etats-Unis l'entrée sur son marché de dispositifs présentant un niveau élevé de risque dans la mesure où, contrairement à ces derniers, elle ne requiert pas du fabricant qu'il fasse la démonstration que son dispositif présente effectivement un bénéfice pour les patients » 23 ( * ) .
Il est communément admis que la procédure américaine d'autorisation de mise sur le marché des dispositifs les plus critiques requiert des études cliniques préalables plus poussées et implique des délais d'autorisation plus longs. Il n'en demeure pas moins que, selon une étude commandée par l'association européenne des fabricants de dispositifs médicaux Eucomed, « la répartition des rappels de dispositifs d'une certaine gravité est semblable en termes aussi bien de domaines thérapeutiques concernés que de motifs de rappel, suggérant ainsi que les différences entre les deux systèmes [européen et américain] sont sans effet, en dernier ressort, sur leur efficacité respective » 24 ( * ) .
2. Une trop grande disparité entre les Etats membres dans les exigences de compétence et de transparence applicables aux organismes notifiés
Ce n'est qu'à partir de 2007 que les institutions communautaires ont fait le choix de modifier la réglementation applicable aux dispositifs médicaux dans le sens d'un renforcement de l'évaluation clinique qui fait de la mesure du rapport bénéfices-risques un élément central avant mise sur le marché pour les dispositifs médicaux les plus critiques. Parallèlement, la surveillance des dispositifs après leur mise sur le marché passe prioritairement par la réalisation d'études cliniques. Mais, seuls les organismes notifiés et leurs experts continuent de disposer de la capacité à attribuer le marquage CE.
C'est précisément sur cette latitude concédée aux fabricants dans le choix de leur organisme notifié et de la procédure de certification que se concentrent les principales critiques à l'encontre de la réglementation européenne.
Si la France ne possède qu'un seul organisme notifié habilité à certifier les dispositifs médicaux implantables, l'Allemagne en compte quatre, l'Italie cinq et la Belgique trois. Il est aujourd'hui difficile d'apprécier l'uniformité des critères de compétence et de transparence sur la base desquels les organismes de certification sont notifiés par les Etats membres à la Commission européenne.
a) Des procédures de certification laissées au libre choix du fabricant
A cela s'ajoute la possibilité pour le fabricant de choisir la procédure de certification qui lui semble la plus à même de garantir à son dispositif médical l'octroi du marquage CE. En ce qui concerne les dispositifs de classe III, il existe , selon l'article 11 de la directive de 1993, deux procédures de certification distinctes qui font porter le contrôle soit directement sur le produit lui-même, soit sur la documentation de conception du produit :
- la première procédure , dite « examen CE de type » (annexe III de la directive de 1993), repose sur l'examen d'un produit type, sur la base d'un échantillon représentatif de la production . L'organisme notifié procède aux examens et essais appropriés afin de vérifier la conformité du produit aux exigences de la directive. L'organisme notifié examine également la société, son système de contrôle qualité ainsi que les conditions de fabrication du produit. Au cours de la première phase du contrôle, qui porte donc sur le produit en tant que tel, un échantillon est prélevé et vérifié en laboratoire ;
- dans le cadre de la seconde procédure , dite de « déclaration CE de conformité » (annexe II de la directive de 1993, point 4 « Examen de la conception du produit »), le producteur livre la documentation de conception du dispositif médical . Il dépose une présentation d'examen de dossier, où il explique le système de contrôle qualité qu'il souhaite introduire, puis fournit une documentation complète relative au produit, aux conditions de la production et aux matériaux de base utilisés. Enfin, un audit du système de qualité est réalisé de manière périodique. En ce cas, le producteur n'a pas à subir de prélèvement sur son produit et c'est lui qui est chargé de procéder aux contrôles qualité en vue d'en présenter les résultats à l'organisme de certification, qui les vérifie. Dans le cadre de cette procédure, il appartient toutefois à l'organisme notifié d'examiner le système de gestion de la qualité, en s'assurant que la société en question est en mesure de procéder aux différents contrôles nécessaires .
Le fabricant a toute liberté pour choisir la procédure de certification à laquelle il entend soumettre son dispositif. L'organisme notifié n'a, à aucun moment, le moyen de se prononcer sur l'opportunité de la procédure concernée compte tenu des caractéristiques et des finalités du produit. L'entreprise PIP avait choisi la voie de la documentation pour obtenir la certification des prothèses mammaires qu'elle fabriquait, avec les conséquences que l'on connaît.
Dans ces conditions, les fabricants peuvent être tentés de choisir l'organisme notifié le plus abordable en termes de prix, qui audite le plus vite et qui n'est pas réputé pour être excessivement exigeant sur un certain nombre de caractéristiques techniques. Dès lors, plusieurs interlocuteurs de la mission d'information ont souligné la nécessité de s'interroger sur la notion d'un tarif unique proposé par les organismes notifiés, de façon à concilier la concurrence avec un niveau élevé de contrôle de sécurité . Il convient également de réfléchir aux moyens de prévenir les conflits d'intérêts entre les experts engagés par les organismes notifiés et les entreprises contrôlées.
b) Une évaluation du système de qualité
Au-delà de sa composante « produit » qui repose sur l'examen d'un produit type ou de la documentation de conception, la démarche de certification comporte également une composante « système » axée sur l'évaluation par l'organisme notifié du système de qualité mis en place par le fabricant d'un dispositif médical de classe III :
- dans le cas d'une procédure de certification centrée sur le produit, le fabricant veille à l'application du système de qualité approuvé pour la conception, la fabrication et le contrôle final du produit ;
- dans le cas d'une procédure de certification centrée sur l'analyse de la documentation de conception, l'organisme notifié constate et atteste qu'un échantillon représentatif de la production satisfait aux dispositions de la directive de 1993.
Les directives de 1990 et 1993 prévoient que tout changement « important » dans le système de fabrication doit être signalé à l'organisme notifié qui est appelé à le valider. A l'heure actuelle, seuls quelques organismes notifiés procèdent à des audits annuels ou semestriels de surveillance du système de qualité de leurs fabricants. Or, un système de gestion des changements au sein des processus de qualité de production, qui doit faire l'objet d'audits réguliers par l'organisme notifié, semble incontournable, surtout dans un domaine de progrès technologiques rapides.
Lors de son audition, l'organisme notifié allemand TÜV Rheinland a indiqué procéder, au moins une fois par an, à un audit du fabricant, au cours duquel elle vérifie le système de contrôle qualité après sa certification. Tous les cinq ans, elle effectue un nouveau contrôle du produit. Celui-ci peut également être diligenté en cas d'incidents remettant en question la recertification du produit.
Face aux disparités entre Etats membres dans l'observation de critères minimaux de compétence et de transparence des contrôles en vue de la désignation des organismes notifiés, plusieurs des personnalités auditionnées par la mission, en particulier les représentants de l'ANSM et de la HAS, ont plaidé pour la mise en place, au niveau européen, d'un comité d'experts indépendants chargé de l'évaluation des données cliniques cruciales concernant les dispositifs médicaux les plus risqués. Ce comité aurait vocation à garantir le passage d'une certification de conformité avec des référentiels normatifs à un mécanisme d'évaluation véritable.
La question du rôle des autorités sanitaires de chaque Etat membre doit être examinée avec attention, en leur confiant une mission de contrôle de la qualité et de la transparence des évaluations cliniques des dispositifs médicaux : elles ne sont toujours pas, jusqu'ici, associées aux procédures d'évaluation clinique, aussi bien avant qu'après la mise sur le marché des dispositifs, et en sont réduites à n'intervenir qu' a posteriori une fois les dysfonctionnements constatés .
De l'avis des représentants de l'ANSM, cette « centralisation » de l'analyse des données cliniques relatives aux dispositifs médicaux les plus critiques doit permettre, à terme, la définition, au niveau européen, d'une liste positive de dispositifs médicaux invasifs, au sein de la classe III, dont les risques sont jugés acceptables au regard des bénéfices thérapeutiques.
3. Des exigences en matière d'investigation clinique limitées
La notion de « données cliniques » est entendue par les directives européennes de façon suffisamment large pour recouvrir aussi bien des bancs d'essais comparatifs que des essais cliniques conduits sur des sujets humains.
Depuis la directive 2007/47/CE du 5 septembre 2007, l'annexe X de la directive de 1993, relative à l' évaluation clinique, précise que « la confirmation du respect des exigences concernant les caractéristiques et performances [...] dans des conditions normales d'utilisation d'un dispositif ainsi que l' évaluation des effets indésirables et du caractère acceptable du rapport bénéfices-risques [...] doivent être fondées sur des données cliniques ».
La directive précise que, « dans le cas de dispositifs implantables et de dispositifs faisant partie de la classe III, les investigations cliniques doivent être réalisées , sauf si le recours aux données cliniques existantes peut être dûment justifié » .
|
Un fabricant a trois possibilités de satisfaire à l'obligation d'évaluer cliniquement les dispositifs médicaux implantables : - soit une évaluation critique de la littérature scientifique pertinente actuellement disponible concernant la sécurité, les performances, les caractéristiques de conception et de la destination du dispositif démontrant l'équivalence du dispositif avec le dispositif auquel se rapportent les données et le respect des exigences essentielles concernées ; - soit une évaluation critique des résultats de toutes les investigations cliniques réalisées ;
- soit une évaluation critique de la combinaison
des données cliniques visées aux deux points
précédents.
|
La très grande majorité des fabricants de dispositifs médicaux ont recours à la procédure d'équivalence, fondée sur l'analyse de la littérature scientifique disponible . A l'heure actuelle, l'équivalence d'un nouveau dispositif par rapport à un dispositif existant peut être prouvée selon trois modalités :
- l'équivalence de matière : elle est, à l'évidence, la plus facile à établir, en démontrant l'analogie de composition et de conception des produits comparés ;
- l'équivalence de biocompatibilité ;
- l'équivalence de procédé de fabrication : elle est, par définition, plus difficile à justifier dès lors que le secret industriel empêche logiquement la divulgation de l'intégralité des processus de fabrication par les producteurs.
Il n'existe aucune méthodologie précise et harmonisée, acceptée et mise en oeuvre par l'ensemble des organismes notifiés, pour déterminer la pertinence du critère de démonstration de l'équivalence choisi par le fabricant. Seules des lignes directrices ( « guidelines » ) a minima ont été élaborées au niveau européen concernant le respect des critères d'équivalence mais leur caractère non contraignant ne permet pas de garantir une application uniforme de ces critères par les organismes notifiés des différents Etats membres.
Team NB, l'Association européenne des organismes notifiés pour les dispositifs médicaux, a, dès 1998, rappelé les principes à respecter en matière d'investigations cliniques, en soulignant notamment que « le besoin d'effectuer une enquête clinique est le résultat des considérations suivantes :
« - des données cliniques doivent être obtenues dans tous les cas ;
« - les données cliniques peuvent s'appuyer sur l'évaluation de la littérature pertinente, de données pertinentes, de résultats d'investigations cliniques ou toute combinaison de ces informations ;
« - à moins que la sécurité et l'efficacité d'un dispositif puissent être démontrées par d'autres moyens (expérience convaincante résultant d'un usage précédent), l'investigation clinique d'un dispositif médical devrait s'avérer nécessaire en particulier dans les circonstances suivantes :
« 1. lorsqu'un dispositif médical entièrement nouveau est proposé à l'entrée sur le marché, dont les composants, les caractéristiques ou les principes d'action sont inconnus jusqu'ici ;
« ou
« 2. lorsqu'un dispositif existant est modifié et que cette modification est susceptible d'affecter de façon significative la sécurité et l'efficacité cliniques ;
« ou
« 3. lorsqu'un dispositif déjà commercialisé est proposé pour une nouvelle indication ;
« ou
« 4. lorsqu'un dispositif incorpore de nouveaux matériaux, jusqu'ici inconnus, entrant en contact avec le corps humain ou des matériaux existants appliqués sur une zone jusqu'ici non exposée à de tels matériaux, et pour lequel il n'existe pas d'expérience clinique, ou dans le cas où ce dispositif sera utilisé sur une période significativement plus longue. » 25 ( * )
Ces recommandations ne contiennent aucune indication quant aux principes méthodologiques devant être appliqués par le fabricant pour procéder à une investigation clinique convaincante sur le plan statistique et scientifique. C'est à lui seul de déterminer le nombre de sujets, le type de plan d'étude, les paramètres d'évaluation principaux et secondaires, le type et le calendrier des évaluations, ou encore la période minimum de suivi du patient. S'il peut toutefois consulter l'organisme notifié pour obtenir des éclaircissements sur ces différents points, le fabricant dispose donc d'une très grande liberté pour mener à bien les investigations cliniques.
4. Un partage des informations insuffisant
a) Le lent développement d'Eudamed
L'article 14 bis de la directive du 14 juin 1993 a créé une banque de données européenne destinée à centraliser l'ensemble des « données réglementaires conformes à la présente directive » afin de les mettre à la disposition des autorités compétentes.
L'objectif consistait, en particulier, à partager au niveau européen les « informations sur des incidents survenus après la mise des dispositifs sur le marché » , collectées par les autorités nationales compétentes c'est-à-dire en l'espèce :
« a) tout dysfonctionnement ou toute altération des caractéristiques et/ou des performances d'un dispositif ainsi que toute inadéquation de l'étiquetage ou de la notice d'instructions susceptibles d'entraîner ou d'avoir entraîné la mort ou une dégradation grave de l'état de santé d'un patient ou d'un utilisateur ;
« b) toute raison d'ordre technique ou médical liée aux caractéristiques ou aux performances d'un dispositif pour les raisons visées au point a) et ayant entraîné le rappel systématique du marché par le fabricant des dispositifs appartenant au même type. » 26 ( * )
Cette base de données doit ainsi contenir les informations suivantes :
« a) les données relatives à l'enregistrement des fabricants, des mandataires et des dispositifs [...], à l'exclusion des données relatives aux dispositifs sur mesure ;
« b) les données relatives aux certificats délivrés, modifiés, complétés, suspendus, retirés ou refusés [...] ;
« c) les données obtenues conformément à la procédure de vigilance ;
« d) les données relatives aux investigations cliniques [...]. »
Une décision adoptée par la Commission européenne le 19 avril 2010 a rendu l'utilisation de cette banque de données européenne sur les dispositifs médicaux, Eudamed ( European Databank on Medical Devices ), obligatoire à compter du 1er mai 2011 . Elle est présentée comme un « portail web sécurisé visant à l'échange rapide d'informations entre les autorités nationales » 27 ( * ) . Dans ce cadre, les autorités nationales compétentes sont incitées à utiliser la nomenclature globale sur les dispositifs médicaux « GMDN » ( « Global Medical Device Nomenclature » ), afin de remplir leurs fiches de manière harmonisée.
Un an après sa mise en oeuvre, Eudamed est jugée insuffisamment fonctionnelle. Les principales critiques se concentrent autour de :
- l'accessibilité : l'accès à la base de données Eudamed est aujourd'hui restreint aux seules autorités nationales compétentes ainsi qu'à la Commission européenne . Des voix se font entendre pour qu'elle puisse être ouverte, au moins en partie, au public, en particulier à la communauté médicale. Les fabricants y sont, pour l'instant, réticents, alors même que l'article 20 de la directive de 1993 prévoit la non-confidentialité des informations fournies par les fabricants dans les termes suivants : « Ne sont pas considérées comme confidentielles les informations suivantes :
« a) informations relatives à l'enregistrement des personnes responsables pour la mise sur le marché des dispositifs [...] ;
« b) informations aux utilisateurs établies par le fabricant, son mandataire ou un distributeur concernant une mesure au sens de l'article 10, paragraphe 3 ;
« c) informations contenues dans les certificats délivrés, modifiés, complétés, suspendus ou retirés » ;
- la saisie et la confidentialité des données : Eudamed connaît quelques dysfonctionnements liés à sa complexité technique , mais aussi à la charge de travail supplémentaire que l'alimentation de la base représente pour les autorités nationales . En outre, les fabricants semblent réticents à divulguer les informations qu'ils estiment trop sensibles ;
- le rôle des organismes notifiés : les progrès apportés par la base Eudamed ne pourront suffire à résoudre le problème posé par les organismes notifiés, qui font l'objet d'une supervision encore perfectible. Si nombre d'entre eux sont performants et fiables, certains fournissent un travail de qualité insuffisante. L'accréditation des organismes et le suivi de leur travail doivent être améliorés.
b) Une information trop cloisonnée
Pour l'heure, le partage des données au niveau européen se limite aux seules conclusions des autorités compétentes, c'est-à-dire les décisions de police sanitaire prises . Toutes les autres mesures de police plus « administratives », telles que les sanctions à l'égard des entreprises, sont, au mieux, rendues publiques sur les sites Internet des agences de surveillance mais ne donnent pas lieu à alerte .
L'affaire des prothèses mammaires PIP est symptomatique de l'absence de cadre opérationnel en vue d'un travail commun, en Europe, sur les données de vigilance : quand bien même la France avait pris soin d'informer la Commission européenne, les Etats membres et d'autres pays de sa décision de police sanitaire concernant les prothèses PIP, certains d'entre eux ont malgré tout continué à commercialiser ces produits, parfois jusqu'en décembre 2011 (exemples : Chine,...). Dans ces conditions, il apparaît qu'une suspension de commercialisation n'est pas interprétée comme contraignante par les autres Etats. C'est une faiblesse du système des dispositifs médicaux qui ne se retrouve pas dans celui du médicament .
En matière pharmaceutique, la déclaration par une autorité nationale compétente de la suspension d'un produit déclenche une procédure européenne dans le cadre de laquelle l'Agence européenne du médicament est conduite à ouvrir une instruction afin de déterminer s'il est légitime que ce médicament soit suspendu dans les autres Etats membres.
C. UNE COMPARAISON INTERNATIONALE PEU FAVORABLE
1. Les Etats-Unis : un modèle ?
a) Des conditions de mise sur le marché plus strictes, du moins en apparence
La réglementation américaine relative aux dispositifs médicaux est née en 1976, quinze ans avant la première directive communautaire. Elle s'est insérée parmi les missions de certification et de contrôle confiées par le législateur américain à la Food and Drug Administration (FDA), dont le périmètre d'intervention s'étend au quart des produits de consommation courante. Les dispositions relatives aux dispositifs implantables apparaissent plus contraignantes que le marquage CE, d'autant qu'elles sont plus anciennes : les dispositifs médicaux implantables ne sont en théorie autorisés qu'après des essais cliniques approfondis , destinés à démontrer non seulement la qualité de la fabrication et l'innocuité du produit mais aussi son efficacité.
Tout comme les directives européennes, la réglementation américaine distingue plusieurs classes de dispositifs médicaux selon le degré de risque qu'ils présentent. Ceux qui présentent le risque le plus élevé, sont en principe approuvés préalablement à leur mise sur le marché ( Premarket Approval - PMA ). Il s'agit d'un examen très poussé, fondé sur la démonstration de l'utilité du dispositif au terme d'essais cliniques réalisés par répartition aléatoire (« randomisés »). La procédure simplifiée - dite 510(k) - n'est censée s'appliquer qu'aux dispositifs moins risqués .
|
Classe de dispositifs |
Procédure d'autorisation |
Exigences cliniques |
Type d'autorisation |
Temps moyen d'obtention de l'autorisation |
Exemples de dispositifs concernés |
|||
|
Classe I
|
Exemption |
Application des bonnes pratiques de fabrication (1) (emballage et étiquetage adéquats, enregistrement de l'établissement de production et du dispositif auprès de la FDA, production dans le cadre d'un système de qualité) |
Présomption de conformité aux bonnes pratiques de fabrication |
Entrée immédiate sur le marché |
Pansements, abaisse-langue, scalpels |
|||
|
Classe II
|
Notification 510(k) (à l'exception de certains dispositifs appartenant à la liste des produits exemptés) |
En sus des exigences de bonnes pratiques de fabrication applicables aux dispositifs de classe I : contraintes spécifiques en matière d'étiquetage, de données précliniques justifiant le respect de normes d'efficacité obligatoires et éléments de surveillance après mise sur marché |
Décharge ( clearance ) |
3 à 6 mois |
Aiguilles chirurgicales, systèmes à rayons X, pompes |
|||
Essai pilote
|
Données précliniques
|
Autorisation de mise sur le marché |
12 à 24 mois |
Valves cardiaques, stimulateurs cérébraux, pacemakers |
(1) Good manufacturing practices (GMP).
(2) Center for Devices and Radiological Health (Centre pour les dispositifs médicaux et la santé radiologique ; division de la FDA charge des dispositifs médicaux)
Source : David Gollaher, Simon Goodall, California
Health Institute -
Boston Consulting Group, Competitiveness and
regulation :
the FDA and the future of America's biomedical industry,
février 2011
Deux considérations juridiques viennent néanmoins tempérer cette exigence :
- en application de la « clause du grand-père » les dispositifs médicaux présents sur le marché avant 1976 sont exemptés de toute autorisation car considérés comme remplissant les conditions nécessaires ;
- l'acceptation par la FDA de l'équivalence substantielle de dispositifs, à la demande de leurs fabricants, tend singulièrement à restreindre le champ du PMA . Dès lors que la démonstration de ce que le nouveau dispositif est « substantiellement équivalent » à un dispositif déjà commercialisé - le « predicate » - est accepté par la FDA, celui-ci peut être mis sur le marché au terme de la procédure 510(k), moins longue et beaucoup moins coûteuse 28 ( * ) . En cas de refus, le fabricant peut faire appel de cette décision auprès d'un « supervisor » .
On comprend mieux, dans ces conditions, pourquoi moins de 10 % des dispositifs médicaux sont mis sur le marché au terme de la procédure la plus contraignante, et pourquoi seulement 4 % des certifications selon la procédure 510(k) ont été accordées sur la base d'un dossier propre (et non par assimilation à un « predicate » ) au cours des cinq premiers mois de l'exercice budgétaire 2012 29 ( * ) ... L'apparition de défaillances postérieures démontre les conséquences de cette moindre exigence : la prothèse de hanche ASR XL Acetabular, fabriquée par DePuy et retirée en 2010, avait été approuvée selon la procédure 510(k). En outre, des observateurs se sont émus du fait que cette procédure permettait d'utiliser comme dispositifs de référence certains produits retirés du marché en raison de problèmes de fonctionnement ou d'absence de service rendu.
Au nom de la perte de chance liée au retard observé dans l'autorisation des nouveaux dispositifs médicaux de classe III aux Etats-Unis par rapport à l'Union européenne, tant les industriels que les parlementaires qui se font l'écho de leurs préoccupations mettent en avant l'excessive lenteur de la FDA. A leurs yeux, la procédure PMA est trop tournée vers l'impératif d'assurer la sécurité des patients.
Mais la mise à disposition effective des dispositifs plus risqués traduit à la fois le temps mis à autoriser leur mise sur le marché et le délai nécessaire à l'acceptation de la prise en charge de leur remboursement. De l'avis unanime des interlocuteurs de la mission, ce délai est bien moindre aux Etats-Unis qu'en Europe, en particulier en France. Les nouveaux dispositifs seraient en définitive disponibles auprès des patients au terme d'une période de durée équivalente des deux côtés de l'Atlantique.
Les industriels plaident pour un juste équilibre entre impératifs de sécurité d'une part, et diffusion de l'innovation du service rendu aux patients, d'autre part. D'autres intervenants, notamment les associations de consommateurs, sont davantage sensibles aux préoccupations de sécurité et dénoncent la légèreté des règles actuelles : comment accepter qu'une prothèse de hanche puisse être considérée comme un produit présentant un risque modéré ?
L'existence d'une agence fédérale garantit une uniformité des mécanismes d'autorisation sur l'ensemble du territoire des Etats-Unis. Du point de vue américain, c'est la très grande hétérogénéité du système européen qui apparaît la plus frappante : le travail des organismes notifiés serait très inégal et se fonderait sur des procédures internes mal connues. Alliée à l'absence de véritable contrôle de ces organismes notifiés, la procédure européenne contribuerait à faire de l'Union européenne un champ d'expérimentation de la recherche américaine. Les industriels que la mission a rencontrés ont d'ailleurs confirmé une fuite de la recherche vers l'étranger, notamment vers l'Europe.
b) Des mécanismes de surveillance contraignants et bientôt renforcés
Le système de matériovigilance ( Medical Device Reporting ) fait peser une charge importante sur le fabricant. Il doit garantir la traçabilité de son produit, c'est-à-dire pouvoir retrouver la première personne échappant directement à son contrôle, soit le plus souvent le distributeur avec qui il entretient des relations contractuelles. Il doit également pouvoir suivre le dispositif médical jusque chez le patient ( « tracking » ). Le signalement des défauts de fonctionnement et événements indésirables graves voire mortels lui incombe, même si d'autres acteurs peuvent l'effectuer.
Schéma de déclaration des événements indésirables graves aux Etats-Unis
Fabricant
Action corrective
5 jours ouvrables
Déclarations volontaires
Mort, événement indésirable grave, mauvais fonctionnement
Etablissement
de santé
Mort, événement indésirable grave
10 jours ouvrables
Mort, événement indésirable grave, mauvais fonctionnement
30 jours calendaires
Importateur Distributeur
Mort, événement indésirable grave, mauvais fonctionnement
30 jours calendaires
FDA
Source : mission d'information du Sénat sur les dispositifs médicaux implantables et les interventions à visée esthétique
La section 522 du Federal Food, Drug and Cosmetics Act permet à la FDA d'exiger du fabricant la conduite d'études cliniques après mise sur le marché dès lors que le dispositif obéit à l'un des quatre critères suivants :
- sa défaillance pourrait avoir une conséquence importante en termes de santé du patient ;
- le dispositif est beaucoup utilisé en pédiatrie ;
- il est conçu pour être implanté pour plus d'un an ;
- il a une incidence vitale pour le patient.
Ce système va donc bien au-delà d'une simple surveillance passive. Il permet à la FDA de conduire des contrôles sur place inopinés ou non, sous réserve de respecter certains critères, liés par exemple à l'enregistrement de plaintes faisant suspecter un danger grave. Les inspections de routine ainsi que celles réalisées dans le cadre de la PMA ou d'un suivi de conformité après délivrance d'une lettre d'avertissement ( warning letter ) sont toutefois annoncées cinq jours à l'avance.
Les registres constituent l'autre modalité de surveillance après mise sur le marché. Les Etats-Unis ont acquis une certaine expérience en ce domaine. Intermacs ( Interagency Registry for Mechanically Assisted Circulatory Support ) est un registre des patients ayant reçu un système d'assistance circulatoire mécanique après un accident cardiaque 30 ( * ) . Il comporte un grand nombre de données par patient et est parfois présenté comme un modèle du genre. Les Etats-Unis ont d'ailleurs récemment proposé à tous les centres de transplantation cardiaque, au niveau international, de se joindre à cette banque de données, à travers le registre Imacs ( ISHLT Mechanically Assisted Circulatory Support ). Mais tous les registres ne peuvent atteindre ce niveau de détail, ne serait-ce qu'en raison du coût de leur mise en oeuvre. De plus, leur mise en place se heurte à la même difficulté qu'en Europe : en l'absence de standardisation, il est très difficile de collecter les données. Chaque fabricant, chaque spécialité médicale, chaque établissement de soins dispose de son propre système de référencement, sans lien entre eux.
Lier le remboursement des dispositifs médicaux à leur traçabilité constitue une voie intéressante. Mais l'expérience américaine incite à la prudence car les registres sous l'égide de Medicare , qui prend en charge tous les Américains de plus de soixante-cinq ans, sont l'exception. Le système d'assurance maladie, aussi puissant soit-il par le nombre des personnes couvertes, ne peut jouer le même rôle que celui qu'il tient en matière de médicament car les modalités de prise en charge de la dépense sont différentes. Comme en France, l'essentiel des dispositifs sont achetés par le praticien directement ; il se fait ensuite rembourser dans le cadre global qui correspond à l'acte médical accompli.
L'étude de grande ampleur relative aux implants mammaires (elle concerne près de cinquante-sept mille femmes engagées pour dix ans sur une base volontaire), actuellement menée par United BioSource Corporation, montre les difficultés et les limites de la tenue de registres. 1 026 sites sont concernés par cette étude, ce qui confirme la nécessité de l'exhaustivité. Prévue pour durer dix ans, cette étude n'en est qu'à sa troisième année mais moins des deux tiers des femmes ayant subi une augmentation mammaire et s'étant engagées dans ce suivi remplissent encore les données.
|
Le 26 juin, le Congrès a adopté une loi visant notamment à renforcer la sécurité des patients dans le secteur des dispositifs médicaux, à la suite des récents scandales concernant des prothèses de hanche ou encore des défibrillateurs cardiaques. Les éléments les plus déterminants organisent une véritable réforme du système d'évaluation, de contrôle et de surveillance des dispositifs médicaux : - la loi consacre le prolongement, pour la période 2013-2017, de taxes devant être acquittées par les fabricants de dispositifs médicaux ( « medical device user-fee » ), destinées à financer l'examen des demandes d'autorisation de commercialisation des dispositifs médicaux et à garantir leur sécurité et leur efficacité ; - la FDA se voit confier la possibilité d'engager un processus plus rapide de reclassification des dispositifs médicaux, en fonction de l'évolution des techniques , en particulier en ce qui concerne les dispositifs présentant un degré élevé de vulnérabilité pour le patient afin de pouvoir renforcer, dans ce cas, les exigences d'évaluation et de contrôle applicables. Toute reclassification doit être justifiée sur la base d'informations, étayées sur le plan clinique, reçues par la FDA. Elle ne peut intervenir qu'après consultation de l'ensemble des parties prenantes, dont les fabricants. La reclassification peut également, en sens inverse, conduire à l'inscription d'un dispositif médical dans une catégorie présentant un risque plus modéré, ce qui devrait toutefois moins concerner les dispositifs jusqu'ici soumis à une demande d'autorisation de mise sur le marché ( « Premarket Approval » ) et pour laquelle les fabricants ont investi des sommes importantes en termes d'études cliniques préalables ; - la loi a codifié une pratique de la FDA consistant à conditionner l'octroi d'une autorisation de mise sur le marché à la mise en place, par le fabricant, d'études de surveillance après mise sur le marché ( « postmarket study » ). En conséquence, des amendes pourront être infligées aux producteurs ne s'étant pas acquittés dans les délais de leurs obligations de suivi clinique des dispositifs après leur mise sur le marché ; - les dispositifs médicaux sont désormais inclus dans la procédure « Sentinel Initiative » , outil de surveillance après mise sur le marché jusqu'ici essentiellement réservé aux médicaments. Toutes les informations concernant les événements indésirables sérieux liés à des dispositifs médicaux ont désormais vocation à nourrir le système d'identification et d'analyse des risques ; - le suivi des procédures de retrait est considérablement renforcé . La loi exige la collecte d'informations permettant d'identifier les tendances en nombre et type de dispositifs retirés, les types de dispositifs au sein de chaque classe faisant le plus fréquemment l'objet de retraits et les causes des retraits. Des audits devront être conduits en vue de vérifier la mise en oeuvre effective des retraits ;
- les autorités fédérales sont
autorisées à interrompre toute étude clinique susceptible
de présenter des risques disproportionnés pour ses sujets,
donnant ainsi au commanditaire de la recherche le temps nécessaire pour
procéder à une réévaluation et à
d'éventuels ajustements.
|
c) La transparence, instrument de régulation du système
Ce nouveau pas dans la surveillance devrait permettre de remédier aux faiblesses actuelles des règles relatives aux dispositifs médicaux. La transparence générale existant aux Etats-Unis contribue également à pallier la faiblesse des moyens à la disposition de la FDA pour effectuer des contrôles des mécanismes de suivi des dispositifs après leur implantation. La défaillance d'un dispositif médical peut être signalée par un patient mais aussi par un concurrent de son fabricant. Ces difficultés étant intégralement répertoriées sur le site de l'agence, un industriel peut utiliser ces signalements pour s'interroger sur les faiblesses potentielles de ses propres produits et leur apporter préventivement des améliorations. Une surveillance particulière du patient ou une modification du dispositif peuvent s'avérer plus appropriées que son explantation et son remplacement complet.
La question des conflits d'intérêts a également été résolue avec pragmatisme : partant du fait qu'un très petit nombre de professionnels sont spécialistes d'un dispositif spécifique et participent d'ailleurs souvent à son élaboration, les Etats-Unis ont rendu obligatoire la déclaration de ces liens ou conflits ( Sunshine Act ). Dès lors que ces liens sont connus, un spécialiste peut travailler avec qui bon lui semble, notamment dans les domaines dans lesquels tant praticiens que fabricants sont peu nombreux.
|
Disposition du Patient Protection and Affordable Care Act (loi sur la protection des patients et la garantie de soins abordables), le Physician Payment Sunshine Act ou Sunshine Act entend répondre à la question posée par un éditorial du New York Times en date du 20 janvier 2012, intitulé « Who Else is Paying the Doctor ? » (Qui d'autre paie le médecin ?). Il vise à garantir la transparence dans les relations d'intérêts entre médecins et producteurs de médicaments, de dispositifs médicaux ou de fournitures médicales, sachant qu'à l'heure actuelle, aux Etats-Unis, environ un quart des médecins perçoivent des versements de la part de l'industrie pharmaceutique ou des fabricants de dispositifs médicaux et les deux tiers acceptent des invitations à déjeuner. Le Sunshine Act comporte deux volets : les fabricants ont l'obligation de déclarer auprès du ministère de la santé (Department of Health & Human Services) les paiements effectués à des médecins ; cette déclaration est rendue publique. S'il n'effectue pas la déclaration, le fabricant s'expose à de lourdes sanctions financières (jusqu'à un million de dollars en cas d'omission volontaire). Les fabricants concernés doivent indiquer le nom et l'adresse du bénéficiaire ; le numéro d'identification de l'hôpital ou du praticien ainsi que sa spécialisation ; le montant du paiement ou autre transfert de valeur, ainsi que sa date et la nature précise du service fourni par le médecin et le produit qui fait l'objet d'une promotion par le médecin. Ils doivent également expliquer dans quel cadre cette somme a été versée (paiement d'une formation, invitation à un congrès...). Sont également concernés par cette mesure tous les organismes appartenant à l'industrie et intervenant dans la production, la préparation, le développement, l'élaboration, la transformation, la commercialisation, la promotion, la vente ou la distribution de ces produits. Le Sunshine Act est applicable depuis le 1 er janvier 2012 s'agissant des faits à déclarer. Les déclarations elles-mêmes ne seront obligatoires qu'à partir du 31 mars 2013. Désormais , le Centers for Medicare and Medicaid Services (CMS) devra communiquer les informations tirées des déclarations d'intérêts via un site internet, qui, selon la loi, devra être interrogeable et compréhensible (« searchable and understandable ») , afin que le public puisse avoir connaissance des financements existant entre un médecin et un fabricant clairement identifié. Seront mentionnés, le nom du fabricant, le nom du bénéficiaire, l'adresse commerciale, le montant du paiement, la date à laquelle il a été fait au bénéficiaire et sous quelle forme. A partir de 2013 et avant le 1 er avril de chaque année, le Secrétaire à la Santé et aux Services Sociaux (Health & Human Services Secretary, équivalent américain du ministre de la santé ) devra transmettre au Congrès les informations communiquées durant l'année précédente, agrégées pour chaque fabricant, ainsi que toutes les mesures coercitives prises pour exercer cette obligation de déclaration. * Plusieurs Etats ou universités avaient décidé d'encadrer les flux financiers entre l'industrie pharmaceutique ou des dispositifs médicaux et les professionnels de santé avant l'adoption du Sunshine Act. En février 2009, la Harvard Medical School avait fixé à 20 000 dollars (16 000 euros environ) le plafond annuel des sommes reçues par ses professeurs et chercheurs au titre de leur activité de consultant ou dans le cadre du financement d'une recherche. |
Dans le cadre très particulier de l'utilisation de dispositifs médicaux, la transparence doit aussi être entendue comme un vecteur d'éducation à la santé. Car, en ce domaine plus que dans d'autres, les patients attendent la perfection et ne comprennent pas qu'un dispositif puisse ne pas fonctionner dans leur cas, pour des raisons propres, par exemple d'ordre génétique. Définir les moyens d'alerter le public sur les risques potentiels présentés par un dispositif donné est d'autant plus difficile que peu de patients sont capables d'identifier le problème qu'ils rencontrent. Le caractère parfois vital de ces dispositifs impose de bien réfléchir au message transmis au public, surtout si leur explantation n'est pas la seule option possible mais que les défaillances constatées se traduisent simplement par un suivi plus régulier.
Le dernier élément d'équilibre du système réside dans les possibilités de mise en jeu de la responsabilité du fabricant. Les conditions de mise en oeuvre d'une procédure d'action de groupe ( « class action » ) sont néanmoins très difficiles à remplir . D'une manière générale, la conduite de ce type de procédure dans les domaines du médicament et des dispositifs médicaux semble le plus souvent vouée à l'échec en raison de la jurisprudence de la Cour suprême issue des décisions « Amchem » 31 ( * ) et « Ortiz » 32 ( * ) . Le fait que chaque organisme humain réagisse de manière spécifique aux effets d'un dispositif médical rend difficile la démonstration d'un intérêt commun à demander réparation de préjudices dont la nature peut varier selon les plaignants. La justice américaine considère généralement que les « litiges de masse » ( « mass torts » ) concernant des dispositifs médicaux, ne sont pas éligibles à ce type de procédure.
En tout état de cause, la mise en oeuvre d'une action de groupe supposerait la démonstration de la faute du fabricant (par exemple, la communication de données erronées ou l'absence de déclaration d'événements indésirables à la FDA), à l'inverse du médicament, pour lequel un simple défaut d'information suffit. Pour la Cour suprême, l'approbation du dispositif médical par la FDA vaut présomption de bon fonctionnement, ce qui restreint singulièrement les possibilités d'action pour les victimes.
En décembre 2011, dans le cadre de l'examen en première lecture du projet de loi visant à renforcer les droits, la protection et l'information des consommateurs, le Sénat a adopté le principe de la création d'une procédure d'action de groupe « à la française ». Compte tenu des strictes limites existant aux Etats-Unis en matière de responsabilité médicale, ce pays ne pourra pas être cité comme un modèle à suivre en ce domaine, comme sur plusieurs dispositions essentielles du fonctionnement du marché des dispositifs médicaux .
2. Suède, Danemark et Australie : les registres au coeur de la surveillance des dispositifs médicaux après leur mise sur le marché
a) La Suède : une expérience poussée en matière de registres
La Suède se caractérise par une décentralisation particulièrement avancée ainsi que par un niveau élevé de déconcentration de son administration centrale. En effet, à la suite d'une réforme en profondeur de l'administration centrale à la fin des années 1970, les effectifs des onze ministères suédois ont été significativement réduits. Les ministres sont à la tête d' administrations d'« état-major » chargées pour l'essentiel de définir la stratégie et les priorités globales dans leurs domaines de compétences respectifs, et de préparer, en conséquence, les textes législatifs et réglementaires correspondants.
Ce sont les agences , - plus de trois cents dans l'ensemble du pays -, qui sont chargées de la mise en oeuvre des politiques publiques, conformément aux conditions préalables d'opération définies par le gouvernement chaque année, au travers de lignes directrices ( « appropriation directions » ) et d'ordonnances. Ces agences sont dotées de moyens humains importants ; certaines d'entre elles emploient plus de dix mille agents. Une fois son budget et sa lettre de mission arrêtés, chaque agence dispose d'une grande liberté de manoeuvre.
L' Agence des produits médicaux ( « Läkemedelsverket » ) est responsable de la régulation, du développement et du contrôle de la production, de la vente et de la distribution des médicaments, des dispositifs médicaux et des autres produits de santé. Elle est placée sous la tutelle du ministère de la santé et des affaires sociales. En tant qu'agence d'Etat, son activité est principalement financée par l'impôt et ses effectifs s'élèvent à près de six cents agents, dont la plupart sont issus de la communauté médicale, notamment des médecins et des pharmaciens. L' Agence nationale de la santé et de la protection sociale ( « Socialstyrelsen » ), également placée sous l'autorité du ministère de la santé et des affaires sociales, doit, pour sa part, s'assurer que les standards de qualité de service sont effectivement respectés au sein des institutions sociales et de santé suédoises (services sociaux, établissements de santé...) et encadrer, à ce titre, l'utilisation et l'entretien des dispositifs médicaux à l'intérieur du système de santé.
Chaque année, l'Agence des produits médicaux reçoit près de huit cents formulaires de déclaration de renseignements correspondant aux mouvements d'environ onze mille dispositifs médicaux (entrées sur le marché, altérations de dispositifs, arrêts de production...). Pour l'heure, l'agence n'a toujours pas transposé en droit interne les dispositions de l'article 14 de la directive du 14 juin 1993 relatives à l' enregistrement des personnes responsables pour la mise sur le marché des dispositifs médicaux en ce qui concerne ceux de classes II a , II b et III . Les modalités de l'enregistrement des dispositifs médicaux implantables de classe III ne sont, à l'heure actuelle, qu'en cours d'étude.
La Suède a mis en place soixante-treize registres nationaux (vingt-sept autres projets étant en cours d'examen) consacrés au suivi aussi bien de dispositifs médicaux que de nouvelles techniques médicales. Ils sont gérés par sept centres de compétence directement bénéficiaires de financements de l'Etat. Le registre le plus ancien fut créé en 1975, en vue de suivre le parcours des prothèses de genou. Il constituait alors une première mondiale.
En 1976, un registre des prothèses de hanche a été mis en place. Il se distingue aujourd'hui par un taux d'enregistrement de 99,5 % et a permis d'identifier, avec près de trois ans d'avance, les dysfonctionnements survenus sur les prothèses ASR de la marque DePuy. Le registre suédois des prothèses de hanche vise la plus grande exhaustivité des informations relatives à leur parcours, en particulier concernant les départements hospitaliers d'intervention, les dates d'intervention, les données personnelles du patient telles que son numéro de sécurité sociale, son diagnostic, le type d'implant, ou encore l'historique des interventions.
La capacité des registres suédois à détecter très en amont les dysfonctionnements présentés par certains dispositifs explique en partie le nombre limité de marques de dispositifs commercialisées par rapport au reste de l'Europe. Six marques de prothèses de hanche sont aujourd'hui commercialisées en Suède, sensiblement moins que dans d'autres pays.
Sans contrevenir aux règles de libre circulation des biens au sein de l'Union européenne, il est donc possible de ne plus avoir recours à une prothèse, sans prendre de mesures d'interdiction, au nom de la police sanitaire.
 Source : Agence
suédoise des produits médicaux.
Source : Agence
suédoise des produits médicaux.
En Suède, la santé relève des vingt conseils des régions . Les 290 municipalités sont compétentes en matière de services sociaux et de prise en charge des personnes âgées. Depuis 2007, la propriété des registres nationaux de qualité a été transférée aux régions et aux communes qui en ont confié la gestion à l'Association suédoise des communes et des régions ( « Sveriges Kommuner och Landsting » - SKL). Celle-ci a signé avec le gouvernement suédois un accord relatif au développement et au financement des registres nationaux de qualité pour la période 2012-2016.
Cet accord prévoit un financement annuel des registres à hauteur de 35,6 millions d'euros par an pendant cinq ans. Il est assuré à 70 % par le gouvernement et à 30 % par les collectivités territoriales. Un autre accord a été conclu sur les relations entre l'industrie des dispositifs médicaux et l'association SKL, en vue notamment de traiter d'enjeux économiques et de définir, sur le plan juridique, les modalités d'accès aux données des registres.
Un comité de pilotage ( « steering committee » ), composé de représentants du gouvernement, des conseils de région, des autorités locales et de l'association SKL, est chargé de définir la stratégie globale à poursuivre en matière de registres. Il revient à un comité exécutif de déterminer la répartition des moyens financiers entre les registres et les différents « centres de compétence » responsables de leur gestion. Ces deux comités peuvent s'appuyer sur l'expertise et les avis de groupes référents , représentatifs des patients et des consommateurs, des professionnels de santé et des chercheurs. Des groupes d'experts ont également été mis en place afin d'évaluer la qualité des projets de registres.
La Suède a traditionnellement pris soin d'écarter toute représentation des intérêts de l'industrie des dispositifs médicaux au sein du système de gestion des registres médicaux. Cette relation est appelée à être réexaminée afin de trouver un équilibre satisfaisant permettant aussi bien de préserver l'indépendance des organes administratifs responsables de la gestion des registres que de stimuler l'innovation thérapeutique.
Au nombre de sept , les centres de compétence sont chargés d'accompagner la mise en oeuvre des initiatives de registres émanant de la communauté médicale - celle-ci se trouve en effet à l'origine de la mise en place des registres des dispositifs médicaux implantés dans ce pays -, en offrant des fonctions support en matière d'études statistiques, d'épidémiologie, de systèmes d'information et d'analyse juridique. Dans un esprit de synergie, les moyens humains, fonctionnels et budgétaires ainsi que les équipements sont mutualisés entre plusieurs registres au sein d'un même centre de compétence. La Suède se distingue, en Europe, par une très forte implication de la communauté médicale dans le suivi et l'évaluation de la performance des nouvelles technologies médicales, ce qui se traduit par un très fort dynamisme des initiatives visant à mettre en place des registres de dispositifs implantés.
La communauté médicale suédoise est particulièrement attachée à ce que les registres constituent des outils d'apprentissage, de consolidation des techniques et d'amélioration globale de l'efficience du système de soins. Ils n'ont, en aucun cas, vocation à servir d'instrument de supervision et d'encadrement des équipes médicales.
Dans ce contexte, les registres suédois sont à l'origine d'améliorations significatives des techniques médicales : le registre national de la cataracte a conduit à une diminution des infections postopératoires de l'ordre de 80 % et le registre des prothèses de hanche a permis de ramener le taux de ré-intervention à seulement 10 %.
La saisie des données en ligne se développe de plus en plus à partir de formulaires directement mis à la disposition des patients. En outre, la Suède s'efforce de rendre publics les résultats des études conduites à partir des registres sur la sécurité et l'efficacité des différents dispositifs implantés . L'objectif consiste, en 2013, à mettre à la disposition des cliniques, sur Internet, les données de près de 95 % des registres de qualité. A l'heure actuelle, la totalité des registres présentant un taux de remplissage supérieur à 80 % est mise à la disposition du grand public.
|
Un des plus récents registres créés en Suède est le registre national pour la chirurgie des mains (Hakir), mis en place en 2010, géré par la région de Stockholm au sein de l'hôpital Södersjukhuset de la capitale. La Suède dispose désormais de sept centres chirurgicaux spécialisés dans les opérations de la main, soit un volume d'environ vingt mille opérations par an. Le développement, au cours des deux dernières décennies, de nouvelles techniques coûteuses (implants articulaires, chirurgie des fractures de la main ou encore traitement enzymatique de la maladie de Dupuytren), a justifié la création d'un tel registre. Après avoir bénéficié d'un financement public de 20 000 euros en 2008 puis de 50 000 euros en 2010, ce registre est désormais financé à hauteur de 200 000 euros pour la période 2011-2012. Il a été conçu de façon à pouvoir être pleinement opérationnel grâce à la seule saisie des données sur Internet . Les patients sont sollicités au bout de trois mois, puis de douze mois, pour remplir des questionnaires relatifs aux résultats de l'opération, avec des données précises quant à la nature et à la cause des complications observées. Des messages SMS sont envoyés aux patients par l'administrateur du registre afin de leur rappeler la nécessité de renseigner les questionnaires. Chaque département a accès aux informations saisies par ses patients.
Concrètement, la saisie des informations dans la salle
opératoire est effectuée par l'infirmière qui est
chargée d'enregistrer le numéro de sécurité sociale
du patient, le numéro de lot de l'implant ou encore les codes
associés à l'intervention. Le chirurgien vérifie les
informations avant de signer le protocole. C'est ainsi que
10 180 opérations ont été enregistrées
dans ce registre depuis le 1
er
janvier 2010, pour un taux de
réintervention liée à des complications de l'ordre de
2,7 %.
|
b) Le Danemark : des registres cliniques en pleine expansion
Le Danemark a mis en place un système de registres cliniques géré par les régions dans le cadre de leur programme de développement de la qualité du système de soins. Ces registres poursuivent trois missions :
- améliorer la qualité des soins (prévention, diagnostic, traitement et rééducation) ;
- renforcer la transparence des informations médicales mises à la disposition du grand public ;
- développer l'innovation thérapeutique dans le secteur des dispositifs médicaux.
Les registres danois se fondent sur la collecte de données personnelles des patients, qui n'est pas conditionnée à leur consentement. Ils font l'objet d'une évaluation tous les trois ans par l'Agence nationale de la santé et leurs résultats sont publiés chaque année. On dénombre entre soixante et soixante-dix registres cliniques certifiés au Danemark, dont un grand nombre est consacré au suivi du traitement de maladies comme le cancer ou le diabète.
Les premiers registres danois ont été créés au milieu des années 1970 à l'initiative des sociétés savantes, qui en étaient les propriétaires. Ce n'est qu'en 1993 qu'a été élaborée une stratégie nationale en matière de registres cliniques, posant les bases de leur financement public, bien que les sociétés savantes en demeurent propriétaires . En 2003, la législation nationale a pris soin de définir le concept de registre de qualité et d'en déterminer le cadre réglementaire. Les registres sont ainsi gérés par un « conseil politique » au sein duquel sont représentées les régions, les autorités sanitaires et les organisations représentatives des professionnels de santé et des patients .
Le système des registres danois est entièrement financé par les régions , pour un budget global de 6,5 millions d'euros, soit seulement 15 % environ de l'effort financier consacré aux registres en Suède. Le Danemark plaide, de longue date, pour une coopération renforcée en la matière entre pays scandinaves mais également avec le reste des membres de l'Union européenne. En effet, les registres danois se caractérisent traditionnellement par la faiblesse des volumes de patients concernés.
Le Danemark s'est cependant illustré par la mise en place, en 1995, d'un registre de prothèses de hanche, à la suite d'un premier scandale concernant des prothèses défectueuses contenant des composants en ciment n'ayant pas fait l'objet de tests préalables. Ce registre comporte aujourd'hui des données relatives à près de cent mille patients dont neuf mille nouvelles entrées chaque année. De 2005 à 2006, six cents patients danois ont été concernés par la pose de prothèses ASR de la marque DePuy, quarante d'entre eux ayant dû être réopérés par la suite . Malgré une première alerte émanant du Royaume-Uni en 2005, les prothèses ASR n'ont été retirées du marché danois qu'en 2010. L'absence d'information ou d'ordre d'explantation en direction des patients exposés a provoqué un scandale sanitaire de grande ampleur, relayé par la presse dès janvier 2011.
c) L'Australie : une culture des registres qui a fait ses preuves
Tout comme les Etats-Unis et l'Union européenne, l'Australie est membre fondateur de la GHTF 33 ( * ) . Elle a mis en place un système d'autorisation qui repose sur la même typologie que celle ayant cours en Europe. Le marquage CE est d'ailleurs reconnu par équivalence, au terme d'un accord de reconnaissance mutuelle ( Mutual Recognition Agreement - MRA). Ce mécanisme permet aux dispositifs médicaux européens, comme beaucoup d'autres produits, d'entrer sur le marché australien sans vérification supplémentaire. Le certificat CE est admis comme « preuve du fabricant » pour faire figurer le dispositif sur le registre australien des produits thérapeutiques (Australian Register of Therapeutic Goods - ARTG ) ; le système de gestion de la qualité est fondé sur la norme Iso 13485, comme en Europe.
En 2010, le gouvernement australien a néanmoins engagé une réforme de la réglementation applicable aux dispositifs médicaux. Cette volonté de refonte suivait sept ans d'application d'un encadrement fondé sur le modèle défini par la GHTF. Elle s'appuyait notamment sur le souhait de la Commission européenne d'engager une refonte des directives relatives aux dispositifs médicaux. Telle que décrite par le ministère australien de la santé 34 ( * ) , la démarche de la Commission traduisait également ses préoccupations à propos du processus de désignation des organismes notifiés et de leur compétence relative, notamment en matière de dispositifs médicaux les plus risqués. Tenant compte de ce nouvel élément, l'Australie a engagé, en 2009, une révision de l'accord de reconnaissance mutuelle avec l'Union européenne. Un des points essentiels de ce processus concerne les mesures de nature à permettre d'avoir confiance dans le système de désignation des organismes notifiés par chaque partie.
En matière de registres , l'Australie se distingue également par une culture qui vise à s'inspirer des meilleures pratiques internationales. A ce titre, conjointement avec la Nouvelle-Zélande, elle tient un registre des essais cliniques ( Australian New Zealand Clinical Trials Registry ), sur le modèle de celui existant aux Etats-Unis 35 ( * ) . Le ministère australien de la santé garde trace de tous les essais relatifs aux dispositifs qui lui sont notifiés. Ceci inclut l'utilisation d'une nouvelle technologie, d'un nouveau matériau, d'une nouvelle modalité de traitement ou d'une utilisation pour une autre indication que celle décrite à l'appui de la demande d'autorisation de commercialisation.
Outre de nombreux registres de suivi de différentes pathologies, l'Australie a mis sur pied plusieurs registres relatifs aux dispositifs médicaux. Le National Joint Replacement Registry (NJRR) a permis de donner l'alerte, à la suite de la détection des difficultés particulières posées par les prothèses de hanche ASR. Lorsqu'une difficulté semble se dégager à partir des données de ce registre, l'autorité compétente en matière de dispositifs médicaux ( Therapeutic Goods Administration - TGA) au sein du ministère de la santé ( Department of Health and Aging ) saisit un groupe d'experts ( Orthopaedic Expert Working Group ) composé de chirurgiens orthopédiques. Celui-ci recommande au ministère les actions qu'il estime nécessaires. Depuis 2007, l'OEWG a été saisi à neuf reprises et a examiné le cas de soixante-seize implants orthopédiques, qui présentaient des taux de reprise supérieurs à ceux anticipés. Pour cinquante d'entre eux, il a préconisé de ne prendre aucune mesure particulière ou, simplement, de veiller à un suivi précis. Sur les vingt-six prothèses restantes, quinze ont été retirées du marché, quatre restent sous surveillance et sept sont toujours commercialisées, après l'examen par le ministère des actions correctives engagées par les fabricants. Elles font néanmoins encore l'objet d'une vigilance particulière.
Parmi les cas de retrait du marché, celui des prothèses de hanche ASR constitue l'exemple le plus connu du bon fonctionnement du registre australien.
|
La pose de prothèses articulaires est une opération très répandue, qui apporte une qualité de vie et une diminution de la douleur très appréciables pour les patients. Les poses de telles prothèses augmentent d'ailleurs rapidement, d'autant plus que le vieillissement de la population tend à multiplier le nombre des personnes susceptibles de bénéficier de cette technique. Le suivi de ces prothèses fait intervenir de nombreux facteurs, parmi lesquels l'âge du receveur, son sexe, le type de prothèse implantée et la technique de pose. Forte de ces considérations et face au rythme rapide de l'évolution des techniques, l'association orthopédique australienne ( Australian Orthopaedic Association - AOA) a décidé, en 1993, de créer un registre national de l'ensemble des prothèses implantées : hanche, genou, épaule, poignet, coude et disques vertébraux. Lors de l'implantation, les patients sont informés (en quinze langues, dont le français) des conditions de leur participation, des modalités de protection des données personnelles, des risques et avantages du registre pour eux. S'inspirant du modèle suédois, le NJRR a pour vocation d'identifier les meilleures techniques et de permettre de réduire les coûts du système de santé. En 1998, le ministère de la santé a décidé d'apporter un financement au registre. La collecte de données a commencé dans l'Etat d'Australie méridionale en 1999 et a été ensuite étendue à l'ensemble du territoire australien. Le champ des opérations enregistrées s'est également mis en place peu à peu, de sorte que le registre soit pleinement opérationnel en novembre 2007. Depuis cette date, tous les hôpitaux australiens participent au registre. En 2009, la loi a autorisé le ministère de la santé à percevoir une redevance sur les matériels afin de participer au financement du système. Sur le plan pratique, en 2007, l'analyse par la TGA des données du registre a démontré un taux de remplacement anormalement élevé pour les prothèses de hanche ASR Resurfacing , fabriquées par DePuy. Le ministère de la santé australien a notifié cette observation à ses homologues étrangers regroupés au sein de la GHTF et a confié à un comité d'experts indépendants (« Orthopaedic Expert Working Group ») le soin de déterminer les causes de ce dysfonctionnement. Celui-ci a estimé qu'il était probablement lié à la très grande technicité de la pose de la prothèse. Il a recommandé de compléter la formation des chirurgiens concernés.
Malgré la mise en oeuvre d'un programme de
formation, les données du NJRR ont montré, en 2009, que le niveau
de remplacement des prothèses ASR
Resurfacing
demeurait
élevé. La TGA s'est alors retournée vers DePuy afin qu'il
retire ce modèle du marché australien, ce qui fut fait en
décembre 2009, plusieurs mois avant son retrait au niveau
mondial.
Entretemps, la TGA, conjointement avec le fabricant et l'AOA,
s'était assurée de la connaissance du retrait de la part de tous
les chirurgiens orthopédiques australiens ainsi que de la bonne
information des patients.
|
Bien que cela ne soit pas à l'origine de sa mise en place, l'impact financier du NJRR n'est pas négligeable. Après quatre ans de fonctionnement, le taux de réintervention a diminué de 14,8 % à 11,1 % en ce qui concerne la hanche et de 10,4 % à 7,9 % pour le genou, ce qui a permis d'économiser la somme de 44,6 millions de dollars australiens 36 ( * ) . Même s'il reste difficile de mesurer la baisse des dépenses de santé susceptible de découler de la mise en oeuvre de registres, ces éléments peuvent inciter certains intervenants à participer à leur financement, notamment les services qui prennent en charge ces frais.
*
L'étude comparée des systèmes de régulation des dispositifs médicaux montre qu'aucun n'apparaît, en définitive, supérieur à l'autre. Peu ou prou, tous les pays du monde tendent à appliquer des procédures de plus en plus harmonisées au niveau international. Dernier signe en date de ce rapprochement, le 20 avril 2012, l'administration chargée de la réglementation de la santé à Singapour (« Health Services Authority ») a instauré une voie d'évaluation rapide des dispositifs médicaux ainsi qu'une voie d'enregistrement immédiat. Peuvent bénéficier de l'évaluation rapide ceux ayant déjà obtenu l'autorisation d'au moins une des instances des pays de la GHTF (FDA, TGA, ministère de la santé japonais, Santé Canada ou organisme notifié européen) et ayant été utilisés pendant une durée d'au moins trois ans. De même, la FDA s'apprête à lancer un programme pilote d'un an permettant aux fabricants de dispositifs médicaux d'éviter les inspections en soumettant les audits de leurs produits réalisés selon la norme Iso 13485. Ce faisant, la FDA semble faire un pas supplémentaire dans la voie de l'alignement sur les directives de la GHTF.
Le nombre des retraits du marché ainsi que les difficultés à les accomplir pleinement constituent également des motifs de préoccupation dans beaucoup de pays.
II. LES INTERVENTIONS À VISÉE ESTHÉTIQUE : UN ENCADREMENT TARDIF ET INSUFFISANT
A. UN ENJEU DE SOCIÉTÉ À PART ENTIÈRE
La multiplication spectaculaire des recours aux interventions à visée esthétique soulève de nombreux enjeux à la fois sociaux, culturels et économiques .
Le développement du secteur de l'esthétique reflète une préoccupation généralisée d'embellissement des corps qui n'est pas nouvelle. Cependant, compte tenu du poids de l'image et du regard porté sur le vieillissement dans les sociétés contemporaines, les enjeux de cette évolution se posent en des termes nouveaux.
Face à ces inéluctables évolutions et à une appréhension du risque de plus en plus marquée, il ne s'agit pas de moraliser les pratiques mais de réglementer les transactions et les produits dans un contexte où les transformations du corps passent de plus en plus par le marché.
1. Un secteur économique florissant
a) Un marché en forte croissance
En 2008, le marché mondial de la beauté et du bien-être était évalué à 500 milliards de dollars, dont 100 milliards pour les soins de lutte contre le vieillissement. La croissance de cette portion du marché devrait être comprise entre 8 % et 10 % par an au cours des prochaines années.
Globalement, la même image de la femme tend à s'imposer aux populations des grandes métropoles, qu'elles soient situées en Occident ou dans les pays émergents. C'est au sein de la zone Asie-Pacifique que l'accroissement du marché est le plus rapide. La demande de produits anti-âge, de blanchiment des dents, d'éclaircissement de la peau et de protection contre le soleil y est particulièrement forte.
|
Les vingt-cinq pays où le recours aux
interventions esthétiques médicales
|
||
|
1. Etats-Unis |
11. Argentine |
21. Australie |
|
2. Chine |
12. Russie |
22. Venezuela |
|
3. Brésil |
13. Italie |
23. Arabie saoudite |
|
4. Inde |
14. France |
24. Pays-Bas |
|
5. Mexique |
15. Canada |
25. Portugal |
|
6. Japon |
16. Taïwan |
|
|
7. Corée du Sud |
17. Royaume-Uni |
|
|
8. Allemagne |
18. Colombie |
|
|
9. Turquie |
19. Grèce |
|
|
10. Espagne |
20. Thaïlande |
|
|
Source : International Society of Aesthetic Plastic Surgery (Isaps) Biennial Global Survey, 2010 |
||
b) Des interventions de plus en plus diversifiées
Les interventions de chirurgie et médecine esthétiques les plus pratiquées sont globalement les mêmes dans les principaux pays occidentaux. En mars 2012, la société américaine des chirurgiens esthétiques (American Society for Aesthetic Plastic Surgery - Asaps) , a indiqué que près de 9,2 millions d'interventions ont été pratiquées aux Etats-Unis, en 2011. Les interventions les plus couramment accomplies en chirurgie esthétique étaient la liposuccion, et les injections de toxine botulique, au titre de la médecine esthétique.
La chirurgie esthétique représentait 18 % des actes pratiqués (contre 82 % pour les actes de médecine esthétique) mais 63 % du chiffre d'affaires total (contre seulement 37 %) pour la médecine esthétique.
Le marché est essentiellement féminin puisque 8,4 millions d'actes ont été réalisés pour cette clientèle (soit 91 % du total) contre huit cent mille pour les hommes. Les dépenses totales s'établissaient à 10 milliards de dollars dont 6,2 milliards au titre de la chirurgie esthétique, 1,7 milliard pour les injections, 1,6 milliard pour les actes de réjuvénation. Près de 400 millions étaient consacrés aux procédures non invasives, notamment l'utilisation du laser pour la dépilation et le traitement des varices.
Par comparaison, la France représente un marché de faible importance. En 2009, près de trois cent mille actes de chirurgie et de médecine esthétiques ont été réalisés par environ 465 chirurgiens plasticiens, soit 1,7 % des procédures mondiales. La France se classe au quatorzième rang mondial, derrière les Etats-Unis, la Chine et le Brésil qui figurent aux premiers rangs du classement 37 ( * ) .
Quant aux actes de médecine esthétique, dont la croissance annuelle atteint 11 % en moyenne dans le monde et 5 % à 8 % en France, ce sont principalement des injections de toxine botulique, de neuromodulateurs et d'acide hyaluronique, l'épilation par laser, ou encore l'injection de graisse autologue.
Si la part des femmes demeure encore largement majoritaire dans ces interventions (près de 85 %), celle des hommes va croissant.
Aux actes de médecine esthétique viennent s'ajouter les nombreuses prestations proposées dans les divers centres et instituts de beauté. Epilations, stylisme d'ongles et de cils, bronzage artificiel, blanchiment des dents, modelage, spas, soins du corps et du visage, traitements amincissants : plus de onze millions de soins de beauté et de bien-être auraient été réalisés en France en 2011 .
Selon les informations communiquées par la Confédération nationale artisanale des instituts de beauté (Cnaib), la France compterait trente-six mille entreprises et soixante-cinq mille esthéticiennes relevant des chambres de métier et de l'artisanat, pour un chiffre d'affaires de 1,5 milliard d'euros . Quant aux entreprises représentées par la Confédération nationale de l'esthétique parfumerie (instituts, centres de bronzage en cabine, spas, prothésistes d'ongles mais aussi fabricants de cosmétiques, équipementiers, distributeurs), affiliées à 90 % aux chambres de commerce et d'industrie, elles emploieraient près de cinquante mille salariés dans environ quarante mille entreprises, pour un chiffre d'affaires annuel de près de 3 milliards d'euros .
Ces montants démontrent que le secteur de l'esthétique médicale est bien engagé sur la voie de la banalisation. Tandis que les demandes des patients évoluent au fil de l'apparition de nouvelles techniques, le regard sur les interventions a changé . Le recours à la chirurgie et à la médecine esthétiques peut apparaître moins futile et plus avouable.
Dans le même temps, comme en témoigne le recours aujourd'hui plus fréquent à la médecine esthétique qu'à la chirurgie esthétique , les techniques les plus prisées sont celles qui sont les moins invasives . Ainsi qu'en conviennent les professions médicales concernées, les demandes dont elles sont saisies expriment souvent le souhait d'aller vers « quelque chose de naturel », si possible sans effraction cutanée ou période de convalescence. Aux opérations irréversibles sont préférées les interventions ponctuelles mais régulières, qui se résorbent progressivement et permettent, en cas de besoin, de « corriger le tir ».
2. L'apparence, une préoccupation ancestrale amplifiée par l'allongement de la durée de la vie
Nul doute que les entreprises de transformation et d'embellissement du corps ne datent pas d'aujourd'hui. Comme les ethnologues aiment à le rappeler, jamais les êtres humains ne se sont contentés de leur corps biologique.
La construction de l'image du corps a lieu tout au long de la vie par la confrontation au regard d'autrui : d'abord la famille, dont le rôle premier dans l'élaboration d'une bonne ou mauvaise image de soi est démontré, puis l'école et les différentes instances sociales dans lesquelles l'individu est conduit à évoluer.
L'élaboration de l'image du corps revêt également une dimension culturelle car elle a toujours été fonction des modèles véhiculés par le milieu social. Dans l'Antiquité, les Olmèques, société mère des Aztèques et des Mayas, déformaient le crâne des enfants de l'aristocratie pour lui donner une forme oblongue qui puisse ressembler au dieu du maïs.
Une rupture est toutefois intervenue à l'époque des Lumières. Jusqu'alors les transformations avaient été le fait de groupes, le plus souvent dans le cadre de rites. Désormais , la transformation de l'apparence physique et la lutte contre l'inné et l'inéluctable commencent à être vécues comme un choix du libre arbitre, voire comme un facteur de démocratisation. La liberté pour l'individu de disposer de son corps apparaît comme une marque des sociétés modernes.
De ce point de vue, des pratiques aussi diverses que le sport et la musculation, le tatouage, le piercing, la scarification, le maquillage ou encore l'ascèse alimentaire ne seraient que les manifestations modernes d'une tension qui a toujours existé entre la manière d'exister en tant qu'individu et le fait de devoir se raccrocher à un langage partagé par un groupe.
Cependant, avec l'importance accordée à l'image et la difficile acceptation du vieillissement dans nos sociétés contemporaines, les enjeux des interventions à visée esthétique sont renouvelés.
La forte médiatisation et la valorisation de stéréotypes culturels du corps et de l'apparence (minceur, jeunesse, fermeté) sur les multiples écrans (télévision, ordinateurs, cinéma, panneau publicitaire) et dans les magazines rendent plus douloureux tout écart par rapport à la représentation . De manière générale, l'existence connue de techniques esthétiques potentiellement correctrices rend d'autant moins acceptable le maintien de ce que nos contemporains ressentent comme des imperfections. Les nouveautés, dont la connaissance est largement diffusée, créent de nouvelles demandes.
En outre, l'accroissement de l'espérance de vie appelle une nouvelle appréhension des choses. La période qui fait suite à la vie active s'allonge ; chacun d'entre nous voit s'ouvrir devant lui plusieurs décennies de vie à réinventer. Le désir d'aborder ces années en retardant autant que faire se peut les marques de vieillesse corporelle ne parait pas illogique.
Peu à peu s'est instaurée une certaine continuité entre la prévention de la maladie et les actes de maintien de soi . La médecine moderne évolue de plus en plus vers une médecine du mieux-être et le recours croissant aux interventions médicales à visée esthétique contribue à brouiller les frontières entre santé et bien-paraître.
Il existe des liens établis entre la mésestime de son corps et la mauvaise estime de soi-même. La souffrance psychologique à laquelle les candidats aux interventions esthétiques souhaitent mettre un terme est par nature en partie subjective et ses causes très variées : simple douleur psychologique causée par une apparence jugée disgracieuse ou une malformation, trouble psychique qui résulte de la tendance à donner à un élément déplaisant plus d'importance qu'il n'en a réellement, ou, ce que la psychiatrie et les chirurgiens esthétiques qualifient de « trouble de l'image du corps » ou de phénomène de « corps-écran » 38 ( * ) , c'est-à-dire le report sur le corps de difficultés psychologiques ou psychopathologiques . Dans ce dernier cas, il n'est pas certain que la réalisation de l'acte envisagé soit réellement favorable au patient.
Les chirurgiens ou les médecins sont en effet amenés à se poser la question du service rendu au patient. L'opération souhaitée rendrait-elle service au patient à titre individuel ? Le fait d'accéder à sa demande lui donnera-t-il plus d'estime de soi ?
Enfin, dans les représentations collectives, influencées dans une large mesure par les différents supports médiatiques et publicitaires, les gestes de beauté et l'apparence physique sont souvent présentés, au moins implicitement, comme un signe de bonne santé . En témoigne notamment l'utilisation du terme de médecine « anti-âge ».
3. « Ne vieillissez pas trop vite » ou le risque accru d'exclusion sociale par l'apparence
Au regard de l'importance accordée aujourd'hui à l'apparence, il demeure difficile de démêler, dans le cadre d'une intervention à visée esthétique, ce qui relève d'une démarche spontanée de ce qui est du ressort d'une contrainte intégrée . En tout état de cause, la pression sociale exercée par le poids des apparences demeure élevée.
Plusieurs études sociologiques convergentes révèlent la stigmatisation dont font l'objet les personnes dont l'apparence physique ne se situe pas dans la norme. Comme l'a déclaré Jean-François Amadieu lors de son audition, la taille et le poids constitueraient, à égalité avec les noms et prénoms, le premier motif déclaré par les Français de moquerie, de mise à l'écart ou de refus d'un droit.
L'impact discriminatoire de l'apparence physique se manifeste jusque dans la vie professionnelle en général et dans l'accès à l'emploi en particulier, voire pour le maintien dans l'emploi. Dans un sondage réalisé en mai 2003, l'Observatoire des discriminations 39 ( * ) a montré que 82 % des personnes interrogées ont le sentiment que, par rapport au passé, l'apparence personnelle et la façon de se présenter jouent un rôle plus important (dont, pour 46 % d'entre elles un rôle « beaucoup » plus important) dans la vie professionnelle et le déroulement d'une carrière . Cette opinion est partagée par les hommes et les femmes et concerne tout particulièrement les jeunes (plus de 90 % des moins de trente-cinq ans) ainsi que les catégories moyennes et populaires (84 % des employés et 88 % des ouvriers).
En matière d'emploi, une véritable concurrence entre discriminations s'install e : il est difficile de lutter à la fois contre la discrimination par le nom et contre l'exclusion par l'apparence. Comme l'a souligné Jean-François Amadieu lors de son audition par la mission, « s'agissant de l'emploi, la question la plus sensible est celle du recrutement. C'est une hérésie que de valoriser les CV en vidéo ou avec photo. Il convient de tout faire pour que l'aspect physique ne joue pas dans le recrutement » .
Le jeunisme conduit à dévaloriser la vieillesse et alimente des formes de discriminations liées à l'âge . Dans ce contexte, une personne âgée peut être perçue comme s'étant négligée. Elle n'a pas fait le nécessaire pour conserver une apparence ferme et tonique et en est tenue pour responsable. Tout se passe désormais comme si les signes du vieillissement revêtaient une connotation négative. C'est ce phénomène qu'incarne cette campagne de publicité lancée par une grande chaîne de radio, en 2010, au slogan provocateur : « Ne vieillissez pas trop vite » .
En conséquence, au-delà d'une appréhension psychologique accrue du vieillissement, de nouvelles formes d'inégalités apparaissent entre ceux qui ont les moyens financiers de combattre les déterminismes biologiques, y compris en ayant recours à des interventions esthétiques, et les autres.
S'il est difficile de lutter contre cette discrimination par l'argent, on peut noter que les pouvoirs publics ne sont pas restés inactifs afin de sensibiliser l'opinion au développement de cette nouvelle forme d'inégalité. Lors de son audition, Jean-François Amadieu notait d'ailleurs que « les campagnes nationales contre la violence et le harcèlement à l'école ont eu un impact. On y voit justement un garçon moqué parce qu'obèse » .
Lutter contre les discriminations et éduquer nos contemporains à dépasser les stéréotypes n'empêchera jamais certains d'entre eux de vouloir corriger ce qui leur apparait comme la source d'un mal-être, voire une difformité. Sinon pourquoi la presse magazine consacrerait-elle régulièrement des articles, voire des dossiers entiers, à des thèmes tels que « Rajeunir » , « Spécial kilos » ou encore « Cellulite » ?
Comme l'a expliqué Monique Le Dolédec, rédactrice en chef adjointe du magazine Elle, au cours de la table ronde sur la place de l'esthétique dans la ligne éditoriale de la presse magazine organisée par la mission, « tous ces numéros se vendent bien. Par les sujets traités, nous ne faisons que refléter les préoccupations des femmes qui ont des attentes, des espérances, des envies, mais aussi des méfiances et des interrogations. Nous sommes comme des éponges qui intègrent et répondent à ce qui se passe dans la tête et le corps des femmes » ... et, de plus en plus, des hommes.
En présence de titres accrocheurs comme « La chirurgie esthétique a changé ma vie » , il est toutefois possible de douter de la capacité des intéressé(e)s à ne pas laisser libre cours à leur envie de recourir à toutes les techniques disponibles sur le marché. Les personnes en souffrance, ou tous ceux qui aspirent au rajeunissement, continueront d'autant plus à vouloir lutter contre les effets du temps que les préoccupations ancestrales en la matière s'accompagnent aujourd'hui de considérations d'ordre économique.
B. LA CHIRURGIE ESTHÉTIQUE : UN ENCADREMENT JURIDIQUE SOLIDE
Jusqu'en 1981, le Conseil national de l'ordre des médecins attribuait la compétence « chirurgie plastique reconstructrice et esthétique » soit à des chirurgiens, soit à des médecins ayant acquis une spécialité chirurgicale (comme l'ophtalmologie, la stomatologie ou l'oto-rhino-laryngologie) et suivi une formation supplémentaire en chirurgie plastique au cours de leur cursus universitaire. Depuis 1988, la chirurgie plastique reconstructrice et esthétique constitue une spécialité médicale à part entière , à laquelle est associée une formation universitaire spécifique, au même titre que l'ophtalmologie ou la dermatologie.
En France, la loi du 4 mars 2002 précitée , assortie des décrets d'application du 11 juillet 2005 et d'une circulaire en date du 23 décembre 2005, pose les fondements de la réglementation applicable à la chirurgie esthétique en disposant que les actes de chirurgie esthétique ne peuvent être pratiqués que par des médecins spécialistes en chirurgie plastique reconstructrice et esthétique .
1. Un cadre légal précis
a) Une distinction très nette entre les installations de chirurgie esthétique et les établissements de santé
Comme le rappelle une circulaire 40 ( * ) du 23 décembre 2005, les installations dans lesquelles est pratiquée la chirurgie esthétique « reçoivent des personnes non malades, non blessées, pour des interventions qui n'ont pas de motif curatif, quel que soit le bien-être qu'elles entendent procurer aux personnes intéressées » .
Elles ne relèvent donc pas de la catégorie des établissements de santé définie par les articles L. 6111-1 et L. 6111-2 du code de la santé publique qui assurent le diagnostic, la surveillance et le traitement des malades, des blessés et des femmes enceintes et ne sont pas, par conséquent, soumises pour leur création à l'autorisation prévue à l'article L. 6122-1 de ce même code. Leur inscription dans le schéma régional d'organisation sanitaire (Sros), prévue à l'article L. 1434-7, est incertaine.
En outre, la circulaire précitée rappelle la distinction entre les actes chirurgicaux à visée esthétique et les interventions de chirurgie plastique ou reconstructrice réalisées à la suite d'un accident ou d'un traitement, ou pour la correction d'une malformation ou d'un déficit fonctionnel, qui s'inscrivent dans une démarche thérapeutique.
Le décret du 20 juillet 2005 41 ( * ) a codifié un certain nombre de dispositions réglementaires relatives aux actes à visée esthétique. L'article R. 6322-1 du code de la santé publique définit ainsi la chirurgie esthétique comme l'ensemble « des actes chirurgicaux tendant à modifier l'apparence corporelle d'une personne, à sa demande, sans visée thérapeutique ou reconstructrice » .
L'annexe 2 jointe à la circulaire précitée du 23 décembre 2005 rappelle à titre purement indicatif les actes les plus courants :
|
Interventions sur le visage (tête et cou) : - traitement chirurgical de la calvitie par détonsuration ou prélèvements de lambeaux ; - lifting frontal, temporal ou cervico-facial ; - chirurgie esthétique du nez (rhinoplastie) ; - chirurgie esthétique des paupières (blépharoplastie) ; - chirurgie des oreilles décollées ; - chirurgie esthétique des lèvres (épaisses ou fines et minces) ; - chirurgie du cou (cervicoplastie) ; - implants (pommettes, par exemple) ; - liposculpture - lipoaspiration ; - lipostructure (réinjection de graisse autologue). Chirurgie du thorax et de l'abdomen, sans préjudice des indications de chirurgie reconstructrice : - mammoplastie d'augmentation avec pose de prothèse en cas d'aplasie ou d'hypoplasie mammaire ; - mammoplastie pour hypertrophie ; - chirurgie de la ptose mammaire en l'absence d'hypertrophie ; - plastie abdominale ; - liposuccion, lipo-aspiration. Techniques de liposuccion ou lipo-aspiration, notamment : - traitement des surcharges graisseuses : - de la paroi abdominale ; - de la face interne des bras ; - de la région trochantérienne (apophyse située à l'extrémité supérieure du fémur) ; - des cuisses ; - de la face interne des genoux. Dermabrasion :
La dermabrasion mécanique est l'acte qui consiste
à enlever la couche superficielle de la peau avec une meule à
rotation très rapide. L'indication dans le domaine de la chirurgie
esthétique est celle de l'effacement des ridules de la lèvre
supérieure et de la lèvre inférieure.
|
b) Une autorisation administrative préalable des installations
La loi du 4 mars 2002 a inséré, dans le titre II du livre III de la sixième partie du code de la santé publique, un chapitre II intitulé « Chirurgie esthétique » . Les articles L. 6322-1 à L. 6322-3 de ce code soumettent ainsi les installations de chirurgie esthétique à une autorisation spécifique préalable à leur mise en service, délivrée par le directeur général de l'agence régionale de santé territorialement compétente .
Le régime d'autorisation des installations de chirurgie esthétique repose sur une double obligation :
- l'exigence d'une accréditation des installations , au même titre que les établissements de santé publics et privés. Cette « procédure externe d'évaluation dénommée certification » est mise en oeuvre par la Haute Autorité de santé (HAS) en application de l'article L. 6113-3 du code de la santé publique ;
- la délivrance d'une autorisation par le directeur général de l'agence régionale de santé territorialement compétente.
La circulaire du 23 décembre 2005 précise que l'autorisation est valable pour une durée d'exploitation de cinq ans.
Les installations sont, par ailleurs, soumises aux normes de fonctionnement en vigueur pour la chirurgie, la chirurgie ambulatoire et la sécurité anesthésique. Elles ont ainsi l'obligation d'adopter le système prévu à l'article L. 6111-1 du code de la santé publique applicable aux établissements de santé, qui leur impose d'assurer notamment « les examens de diagnostic, la surveillance et le traitement des malades » ainsi que de mener une réflexion sur les questions éthiques posées par l'accueil et la prise en charge des malades.
c) Des actes non pris en charge qui ne peuvent bénéficier de publicité
Aux termes de l'article L. 6322-1 du code de la santé publique, « l'activité, objet de l'autorisation, n'entre pas dans le champ des prestations couvertes par l'assurance maladie au sens de l'article L. 321-1 du code de la sécurité sociale » .
Le même article interdit aux installations autorisées de bénéficier d'une publicité « directe ou indirecte, sous quelque forme que ce soit » . Cette prescription a le même fondement de principe que les interdictions déontologiques faites aux médecins par l'article R. 4127-19 du code de la santé publique, qui dispose que « la médecine ne doit pas être pratiquée comme un commerce » et qu'à ce titre « sont interdits tous procédés directs ou indirects de publicité et notamment tout aménagement ou signalisation donnant aux locaux une apparence commerciale » .
La circulaire du 23 décembre 2005 précise que cette interdiction concerne tous les moyens d'information, Internet compris. En revanche, elle souligne qu' « elle n'empêche aucunement les titulaires de l'autorisation de donner au public, sans employer les procédés de la publicité, des renseignements de fait sur leurs installations, leurs activités et les compétences de leurs praticiens, en les présentant avec sobriété » .
Cette interdiction ne fait aucunement obstacle aux communications de nature scientifique, dans les revues spécialisées par exemple, ni aux ouvrages d'enseignement, dès lors que ces publications ne comportent pas de mentions spécifiques en faveur d'un établissement.
2. Une responsabilité effective des chirurgiens
La responsabilité du praticien esthétique peut être recherchée sur un double fondement : un défaut d'information et une faute médicale.
a) Une exigence accrue en matière d'information
Parallèlement à l'article L. 1111-2 du code de la santé publique, qui régit l'ensemble des dispositions relatives à l'obligation d'information pesant sur tout praticien, le législateur a édicté, à l'article L. 6322-2 du même code, des dispositions spécifiques relatives à l'obligation d'information en matière de chirurgie esthétique. Ce régime spécial, plus exigeant en raison de l'absence de finalité thérapeutique des interventions réalisées, s'est très largement inspiré de la jurisprudence.
Ainsi, préalablement à la loi du 4 mars 2002, la jurisprudence avait déjà soumis l'exercice de la chirurgie esthétique à des obligations d'information renforcées par rapport aux autres spécialités médicales :
- dans un arrêt du 8 janvier 1981, la cour d'appel de Lyon avait estimé que « le chirurgien esthéticien doit, plus que tout autre, informer très exactement son client de tous les risques inhérents à l'opération qu'il conseille et des séquelles pouvant en résulter [...] , le devoir d'information ne cessant pas avec l'achèvement de l'acte opératoire » ;
- dans un arrêt du 13 janvier 1959, la cour d'appel de Paris a considéré que cette obligation particulière d'information devait conduire le praticien à refuser certaines interventions au regard des risques encourus ;
- dans un arrêt du 17 février 1998, la Cour de cassation a imposé au chirurgien d'informer son patient sur les risques graves de l'intervention ainsi que sur tous les inconvénients qui pourraient en résulter. Dans le même ordre d'idées, la cour d'appel de Paris a rappelé, dans un arrêt du 1 er octobre 1998, la nécessité pour le chirurgien esthétique d'informer son patient sur les difficultés de cicatrisation et la survenance de complications postopératoires, et, dans un arrêt du 2 avril 1999, de préciser à son patient toutes les circonstances liées à la cicatrisation d'un lifting et notamment sa durée.
La loi du 4 mars 2002 est venue consacrer l'obligation, pour le chirurgien esthétique, d'informer la personne concernée et, s'il y a lieu, son représentant légal, des conditions de l'intervention, des risques et des éventuelles conséquences et complications (article L. 6322-2 du code de la santé publique).
En regard du régime général d'information des praticiens édicté à l'article L. 1111-2 du code la santé publique, qui prévoit que « l'information porte sur les différentes investigations, traitement ou action de prévention qui sont proposés, leur utilité, leur urgence éventuelle, leur conséquence, les risques fréquents ou graves normalement prévisibles qu'ils comportent ainsi que sur les autres solutions possibles et sur les conséquences prévisibles en cas de refus » , il y a lieu de considérer que le chirurgien esthétique doit également informer le patient des risques exceptionnels encourus .
De plus, la loi impose au praticien de remettre au patient un devis détaillé et d'observer un délai de réflexion obligatoire (article L. 6322-2 précité). Le décret du 11 juillet 2005 42 ( * ) a ainsi fixé à quinze jours le délai minimum entre la remise du devis et l'intervention.
Des inspections et des sanctions en cas d'infraction aux obligations précitées sont prévues par les articles L. 6324-1 et L. 6324-2 du code de la santé publique.
b) Une obligation de moyens renforcée
Toute intervention sur le corps humain comporte une part d'aléa liée aux réactions imprévisibles de ce dernier et la chirurgie esthétique n'échappe pas à de tels risques au prétexte qu'il s'agit d'interventions non curatives. Il apparaît d'ailleurs d'autant plus inconcevable d'attendre d'un chirurgien esthétique de produire avec exactitude les résultats escomptés par le patient que ces souhaits sont par nature subjectifs.
Par conséquent, le chirurgien esthétique ne peut être astreint à une obligation de résultat . A l'image de ce qui vaut pour les autres spécialités médicales, l'obligation porte sur les moyens . C'est ainsi que dans un arrêt du 7 octobre 1992, à l'occasion d'une implantation de prothèses mammaires à visée esthétique dont le résultat ne donnait pas satisfaction à la patiente, la Cour de cassation a estimé qu' « attendu qu'après avoir relevé que selon l'avis du docteur E. (expert), le résultat inesthétique n'était pas dû à une faute du chirurgien mais à la méthode elle-même, le résultat des prothèses mammaires étant encore statistiquement aléatoire [...] , Mme R., dont la décision de subir l'intervention avait été raisonnée, n'apportait pas la preuve d'un manquement du docteur B. à ses obligations » .
Pour autant, la jurisprudence apprécie l'obligation de moyens de façon plus stricte dans le cas de la chirurgie esthétique, d'où l'expression consacrée d' « obligation de moyens renforcée » . En effet, la cour d'appel de Nancy, dans un arrêt du 18 mars 1991, a jugé qu' « en matière de chirurgie esthétique, l'obligation de moyens pesant sur le praticien doit être appréciée beaucoup plus strictement que dans le cadre de la chirurgie classique, dès lors que la chirurgie esthétique vise, non pas à rétablir la santé, mais à apporter une amélioration et un réconfort esthétique à une situation jugée insupportable par le patient » .
Le juge a précisé au fil de ses décisions le contenu de l'obligation de moyens renforcée :
- le risque de l'intervention doit être proportionné à l'importance de la disgrâce constatée. Dans un arrêt du 17 janvier 1999, la cour d'appel de Versailles a ainsi considéré qu' « en matière de chirurgie esthétique, l'atteinte à l'intégrité physique du malade ne peut se justifier que si elle respecte l'existence d'un certain équilibre entre le mal causé par l'intervention et le profit espéré, de sorte que le médecin ne doit pas mettre en oeuvre une thérapeutique dont les inconvénients risqueraient de surpasser la disgrâce qu'il prétend traiter ou dont la gravité serait hors de proportion avec l'embellissement espéré » 43 ( * ) ;
- l'intervention doit être effectuée dans les règles de l'art , de façon rigoureuse, avec prudence et diligence. Dans un arrêt du 28 septembre 1990, la cour d'appel de Paris a estimé qu'à la suite d'une implantation de prothèses mammaires ayant entraîné une asymétrie flagrante, la patiente n'était pas tenue de régler les honoraires du chirurgien 44 ( * ) ;
- au titre de son obligation de non-aggravation de l'état du patient , le praticien doit veiller à ce que les séquelles opératoires ne dépassent pas le défaut esthétique initial. Dans un arrêt du 16 avril 1981, la cour d'appel d'Aix-en-Provence a jugé que « lorsqu'un chirurgien intervient sur une jeune fille « fraîche et belle » parce qu'elle se trouve « un peu joufflue » et « mal dans sa peau », il s'engage à ne pas la défigurer et à ne pas lui apporter de gêne fonctionnelle » 45 ( * ) .
c) Le chirurgien esthétique est soumis au droit pénal
Comme tout praticien médical, le chirurgien esthétique est soumis au droit pénal et peut être sanctionné à ce titre lorsqu'il a commis une infraction. Il peut donc être concerné par deux types d'infractions :
- les infractions pénales générales applicables à l'ensemble des citoyens : atteintes involontaires à l'intégrité de la personne, mise en danger de la vie d'autrui, non-assistance à personne en danger, ou encore atteintes volontaires à l'intégrité corporelle ;
- les infractions propres à la profession médicale : violation du secret professionnel, rédaction de faux certificats médicaux, administration de substance nuisible, consultation dans des locaux commerciaux, défaut d'autorisation d'exercice des activités de chirurgie esthétique.
Le dispositif législatif et réglementaire mis en place par la loi du 4 mars 2002 et par les décrets d'application des 11 et 20 juillet 2005 s'articule autour de deux volets : le premier concerne la qualité des installations requises pour la pratique de la chirurgie esthétique et le second est relatif au renforcement des obligations du praticien envers les patients. Comme le rappelait M. Vorhauer, secrétaire général du Conseil national de l'ordre des médecins, lors de son audition par la mission 46 ( * ) , « la chirurgie réparatrice et esthétique est une activité bien connue et réglementée quant aux compétences et à la formation requises » . De nombreux pays cherchent aujourd'hui à s'inspirer de la réglementation française, qui apparaît comme un exemple à suivre en matière d'information des patients sur les prix, les risques et les qualifications du chirurgien 47 ( * ) .
A l'heure du développement du tourisme médical, qui permet de recourir à la chirurgie esthétique à moindre coût, c'est plutôt à l'étranger que résident les risques de techniques mal maîtrisées. Aucun des interlocuteurs de la mission n'a pu fournir d'éléments statistiques et l'assurance maladie ne tient pas de décompte des actes qu'elle est conduite à prendre en charge au titre du suivi et de la correction des interventions réalisées à l'étranger. Certains chirurgiens esthétiques reconnaissent néanmoins que les interventions de reprise constituent une part non négligeable de leur activité.
A l'inverse, la médecine esthétique constitue un domaine dans lequel règne un certain désarroi face à un vide réglementaire à la fois source de confusion et propice au développement de toutes sortes d'abus.
Dans un domaine qui touche au plus profond de ce que chacun de nous ressent, il sera toujours difficile d'éviter toute insatisfaction quant aux modifications corporelles effectuées.
C. LA MÉDECINE ESTHÉTIQUE : UNE RÉGLEMENTATION QUI COURT APRÈS DES TECHNIQUES EN PERPÉTUELLE ÉVOLUTION
L'encadrement très strict des pratiques de chirurgie esthétique est en partie né de l'assimilation du patient à un consommateur de soins à caractère médical mais à finalité non thérapeutique. C'est donc en contrepartie de l'absence de nécessité thérapeutique que les obligations du chirurgien esthétique ont été renforcées. Le même raisonnement aurait pu prévaloir en matière de médecine esthétique, mais la circulaire du 23 décembre 2005 a précisé que « n'étaient pas concernées les pratiques dites de « médecine esthétique » , telles que l'utilisation de la toxine botulique ou l'injection de matériaux résorbables ou de substances, notamment pour le comblement des rides » .
La médecine esthétique ou « médecine anti-âge 48 ( * ) » ne constitue pas une spécialité médicale référencée par le Conseil national de l'ordre des médecins. Elle peut se définir, selon François Turmel, président de la Fédération française des médecins experts en médecine esthétique et anti-âge, « comme l'ensemble des prescriptions et des actes visant à prévenir, améliorer ou corriger les aspects inesthétiques ou jugés comme tels par un sujet sain, et ce grâce à une approche pluridisciplinaire » 49 ( * ) .
La portée de cette définition est vaste : elle recouvre en fait l'ensemble des actes esthétiques non chirurgicaux. Dans ces conditions, certains actes aux contours mal définis peuvent prêter à confusion et il n'est pas toujours aisé de savoir ce qui relève, d'une part, de la chirurgie ou de la médecine esthétiques, d'autre part, des professionnels de l'esthétique ou des médecins.
La très grande majorité des personnes auditionnées par la mission, que ce soient les représentants de l'ordre des médecins, des enseignants ou des institutions de régulation, a regretté le manque de formation qualifiante en matière de médecine esthétique et l'absence d'un statut clairement défini de médecin esthétique, alors même que l'engouement de nos concitoyens pour ce type de médecine et l'augmentation exponentielle de la demande montrent qu'il s'agit d'un véritable enjeu de société.
La médecine esthétique est une médecine à part entière , qui nécessite une formation universitaire et sur laquelle pèsent les mêmes obligations que pour tout médecin, notamment en termes de tenue du dossier médical ou d'assurance. Elle entre dans le champ de la loi sur les droits des malades du 4 mars 2002 et, en cas de préjudice sans faute, les victimes sont indemnisées par les assureurs privés ou par l'Office national d'indemnisation des accidents médicaux (Oniam) selon le degré de déficit fonctionnel permanent (en-dessous de 25 % d'incapacité, indemnisation par l'assureur ; au-delà, par l'Oniam).
Cependant, devant l'augmentation du nombre d'alertes liées à des pratiques esthétiques non évaluées et donc au risque, pour des personnes en bonne santé, d'avoir à subir « des conséquences graves de pratiques dangereuses ou réalisées par des personnes non qualifiées, voire dans des conditions totalement contraires aux règles élémentaires d'hygiène ou de bonne pratique médicale 50 ( * ) » , le législateur a décidé d'encadrer précisément le domaine des pratiques esthétiques 51 ( * ) .
La loi « HPST » du 21 juillet 2009 52 ( * ) a donc créé les articles L. 1151-2 et L. 1151-3 du code de la santé publique, qui prévoient deux modalités d'encadrement : une première, sur la formation et la qualification des médecins réalisant des actes à visée esthétique, et une seconde sur l'interdiction de certains actes esthétiques jugés dangereux.
L'encadrement de ces actes, complexe à entreprendre, mêle à la fois droit de la santé et droit de la consommation. Le dispositif mis en place au travers de ces deux articles n'est pas sans faille et témoigne de la difficulté à gérer le risque sanitaire dans une matière où les techniques sont en perpétuelle évolution.
3. Des actes aux contours mal définis
La médecine esthétique recouvre un grand nombre d'actes et de techniques , dont les plus courantes peuvent se décliner comme suit :
- les exfoliations et les stimulations dermo-épidermiques : il s'agit des peelings , qu'ils soient réalisés au moyen d'agents chimiques, mécaniques (dermabrasion) ou physiques (laser, lampe à lumière pulsée, cryothérapie de contact) et les stimulations de photorajeunissement par lumière pulsée ;
- la mésothérapie : ce sont des injections de principes actifs dans les tissus superficiels ;
- les implantations dermiques ou hypodermiques : elles visent à corriger des rides profondes ou superficielles et des défauts de volume par des injections d'implants dermiques ou hypodermiques, qu'ils soient temporaires ou définitifs ;
- le comblement des dépressions corporelles innées ou acquises, par les autogreffes sous-cutanées de cellules adipeuses ;
- la prise en charge de la surcharge pondérale : traitement des lipodystrophies localisées par aspiration et par d'autres traitements locaux, chimiques ou mécaniques ;
- le traitement de la calvitie par micro-greffes capillaires ;
- les traitements vasculaires superficiels : le traitement de la couperose, de l'érythrose, des varicosités, par des lasers vasculaires, des lampes flash, de l'électrothérapie ou de la micro-sclérose ;
- le traitement de la pilosité jugée excessive : épilation longue durée par des lasers, des lampes à lumière pulsée ou de l'électrothérapie.
En constante évolution, certains actes ou techniques de médecine esthétique prêtent à confusion et semblent pouvoir relever aussi bien du chirurgien que du médecin esthétiques .
a) Entre médical et esthétique : une frontière difficile à tracer
La frontière traditionnelle entre médecine esthétique et métiers de l'esthétique se définit en fonction du caractère invasif ou non des techniques employées . Les actes non médicaux non invasifs à visée esthétique sur peau saine, n'impliquant pas de modification importante de celle-ci et non susceptibles d'induire des complications requérant des compétences médicales (épilation à la pince ou à la cire, maquillage, modelage, manucure, bronzage UV, conseil et vente de produits cosmétiques...) entrent dans le champ de compétence des esthéticiennes, dont la formation est encadrée par des diplômes nationaux de différents niveaux. A partir du moment où il y a injection, application de produits actifs sur la peau ou utilisation de dispositifs médicaux, c'est au médecin d'agir. L'examen clinique, la discussion du rapport bénéfices-risques et le recueil du consentement éclairé et informé du patient sont les caractéristiques cardinales de l'acte médical que ne peuvent réaliser les professionnels de la beauté. Cependant, les champs de compétence des esthéticiennes et des médecins esthétiques ont beaucoup en commun : l'épilation, l'amincissement et la lutte contre le vieillissement cutané.
Si l'on examine chacun de ces champs de compétences séparément, on comprend combien la détermination de ce qui relève de l'institut de beauté ou du cabinet médical peut être difficile et que l'on puisse se trouver face à des « actes frontières » .
Les lampes à lumière pulsée sont de plus en plus utilisées dans les instituts de beauté pour réaliser des épilations dites « définitives », en contradiction avec les dispositions de l'arrêté du 6 janvier 1962 53 ( * ) qui fixe la liste des actes médicaux ne pouvant être pratiqués que par des médecins, et parmi lesquels on trouve « tout mode d'épilation, sauf les épilations à la pince ou à la cire » . Il est vrai que les autres techniques d'épilation, à l'époque où cet arrêté a été pris, étaient beaucoup plus invasives (à base d'aiguilles) et qu'il était alors impossible de prévoir l'évolution des techniques en matière esthétique. Ces appareils comportent également une dimension médicale en matière d'hirsutisme ou de lutte contre l'acné, par exemple.
Une délégation de la mission s'est rendue dans les locaux du groupe Eurofeedback, dont l'une des activités est la fabrication et la commercialisation d'équipements à vocation médicale et esthétique utilisant une technologie à base de lumière pulsée.
Le département recherche et développement de cette entreprise a évalué les risques potentiels de la photodépilation à base de lumière pulsée en comparant les études cliniques disponibles les plus pertinentes sur cette technologie. Il en ressort que l'efficacité d'une machine dont la puissance serait inférieure à 12 joules par centimètre carré n'est pas garantie, et qu'une puissance supérieure à 38 joules par centimètre carré présente un taux d'incidents élevé (dépigmentation, brûlures...).
|
La lumière pulsée, qui a un spectre plus large que le laser et qui ne présente donc aucun risque de brûlure en cas de rencontre avec la rétine (la lumière pulsée diverge dès la sortie du filtre, à la différence du laser, lumière cohérente et monochromatique), va agir directement sur la mélanine des poils en la chauffant. Cette augmentation de température va permettre de détruire la papille des poils qui sont dans la première de leurs trois phases de croissance (soit environ 20 % de la pilosité totale du corps humain) et donc de les éradiquer définitivement.
Le risque de brûlure est quasi nul dans la mesure
où la peau contient très peu de mélanine, à
l'exception des peaux présentant un phototype élevé
(à savoir les peaux métissées et noires : phototypes
5 et 6). La seule précaution à prendre, en plus d'une juste
évaluation du phototype, est le rasage de la partie destinée
à l'épilation définitive, afin d'éviter que le poil
chauffé ne soit en contact avec la peau et n'occasionne ainsi une
brûlure superficielle.
|
En conséquence, Eurofeedback a décidé de limiter la puissance des machines vendues aux esthéticiennes à 20 joules (en plus d'une limitation aux quatre premiers phototypes de peau) et celle des machines destinées aux médecins à 38 joules par centimètre carré. Pour ses promoteurs, cette décision paraît de nature à garantir à la fois l'efficacité du matériel mis à disposition des instituts de beauté (contrairement aux lampes à lumière pulsée individuelles disponibles dans les circuits de grande distribution et dont la puissance atteint rarement 10 joules par centimètre carré) et sa sécurité.
Rien ne garantit que les concurrents de cette entreprise prennent les mêmes précautions, notamment les importateurs de matériels très souvent fabriqués en Chine.
De plus, et afin d'assurer la meilleure utilisation possible de ses machines, Eurofeedback a mis en place un dispositif de formation préalable : les machines ne sont livrées aux instituts de beauté qu'après que les utilisateurs ont suivi une formation auprès d'un médecin lui-même familier de ces appareils.
Il en va du vieillissement cutané et de l'amincissement comme de l'épilation : les mêmes techniques peuvent être à la fois utilisées par des médecins et par des instituts de beauté.
La photoréjuvénation - ou photorajeunissement - est une technique utilisant la lumière pulsée et certains lasers vasculaires permettant de donner une apparence plus jeune à la peau du visage, du cou et des mains.
En traversant l'épiderme, les rayonnements lumineux émis par la lampe à lumière pulsée 54 ( * ) (seul appareil autorisé dans les instituts de beauté, l'usage des lasers étant réservé aux médecins ou sous leur contrôle) vont cibler trois zones en priorité : les micro-vaisseaux capillaires, pour atténuer les rougeurs et taches rouges ; les pigments, pour effacer les taches brunes ; les firoblastes, cellules fabriquant du nouveau collagène naturel, et le collagène lui-même en favorisant sa contraction.
Alors que l'arrêté de 1962 leur interdit l'usage des appareils à lumière pulsée pour la photodépilation, les esthéticiennes ont la possibilité d'utiliser ces machines pour les soins de photorajeunissement. Voilà qui semble pour le moins paradoxal : médecins esthétiques et esthéticiennes utilisent les mêmes appareils pour les mêmes soins de photoréjuvénation mais seuls les premiers disposent de la compétence pour établir un diagnostic dermatologique.
Nous nous trouvons à nouveau devant un « acte frontière » , dont le caractère médical ou esthétique mériterait d'être précisé et qui contribue à entretenir le flou dans l'esprit de nos concitoyens.
Dans son avis du 13 juin 2001 relatif à l'utilisation des lasers (ou autres sources de puissance) dans le domaine de l'esthétique 55 ( * ) , la Commission de la sécurité des consommateurs recommandait déjà de « procéder à une mise à jour de la réglementation et de la normalisation concernant les lasers ou lampes flash afin de reclassifier les différents dispositifs de lasers médicaux ou paramédicaux en fonction des utilisations thérapeutiques ou esthétiques [...], [d'] exiger de la part des personnes mettant en oeuvre les techniques utilisant les lasers et les lampes flash des connaissances minimales qui pourraient être prodiguées dans le cadre d'une formation faisant l'objet d'une réglementation [...] , [et d'] élaborer un texte réglementaire qui permettrait à différents services de contrôle d'intervenir sur le terrain, tant sur les personnels que sur les matériels » . Ce constat est malheureusement toujours d'actualité.
b) L'interdiction des actes dangereux
En revanche, la loi a expressément prévu la possibilité d'interdire les actes à visée esthétique qui s'avèrent dangereux pour la santé de ceux sur lesquels ils sont pratiqués. En application de l'article L. 1151-3 du code de la santé publique, un décret d'interdiction peut être pris si la mise en oeuvre des actes concernés présente un danger grave ou une suspicion de danger grave pour la santé humaine, après avis de la Haute Autorité de santé. Au sein de celle-ci, c'est la Commission nationale d'évaluation des dispositifs médicaux et des technologies de santé (CNEDiMTS) qui est chargée de se prononcer. La même procédure doit être suivie pour lever une interdiction.
La loi met donc en place l'idée d'une interdiction de principe, montrant bien par là que la sécurité des personnes est placée au coeur du dispositif d'encadrement . Las, en droit public, toute interdiction doit être motivée pour ne pas être entachée d'illégalité dans le cadre d'un recours pour excès de pouvoir. Or, comment démontrer le caractère dangereux d'une pratique esthétique en l'absence de données scientifiques ou statistiques fiables et objectives ?
La contestation des dispositions du décret du 11 avril 2011 56 ( * ) montre toute la difficulté de cet exercice. Le décret en question avait voulu interdire, en application de l'article L. 1151-3 du code de la santé publique, les actes de lyse adipocytaire à visée esthétique, qui consistent en la destruction des cellules graisseuses par des techniques invasives ou non.
Saisi en référé, le Conseil d'Etat s'est prononcé le 17 juin 2011 sur le recours pour excès de pouvoir déposé par plusieurs associations de professionnels et d'industriels du secteur de l'esthétique. Il a annulé l'article 2 de ce texte, lequel visait les techniques non invasives. Il a considéré que les méthodes invasives présentaient non un danger grave mais une suspicion de danger grave pour la santé humaine et pouvaient, à ce seul titre, être interdites, et a estimé qu'il ne ressortait pas de l'avis de la HAS qu'une telle suspicion existât pour celles des techniques utilisant des agents physiques externes, sans effraction cutanée.
L'annulation partielle de ce décret témoigne de la difficulté à établir précisément le caractère dangereux d'une technique innovante en l'absence d'études scientifiques fiables.
Devant ce manque d'éléments probants, le Conseil d'Etat a donc décidé que les complications présentant un certain degré de gravité avancées dans le décret pour qualifier ces techniques de danger grave étaient insuffisantes compte tenu du faible nombre de cas répertoriés et du manque de données disponibles. Il est donc très difficile d'interdire une pratique esthétique sans pouvoir apporter la démonstration scientifique de sa dangerosité.
L'interdiction des pratiques de lyse adipocytaire invasives a néamoins pu être maintenue grâce à la requalification, par le ministère de la santé, du « danger grave » en « suspicion de danger grave ». Le Conseil d'Etat a accepté cette substitution de motifs - il revient aujourd'hui aux promoteurs de ces techniques de démontrer leur innocuité - mais la question de la difficulté d'évaluation scientifique des pratiques esthétiques reste à résoudre.
c) Des exemples étrangers peu concluants
Plusieurs pays industrialisés ont conduit une réflexion approfondie sur le cadre réglementaire le plus pertinent pour les actes à visée esthétique, en se posant, en premier lieu, la question de la définition et du champ de ce type d'interventions. En Australie , dans un rapport de 2011, le groupe de travail sur les interventions de chirurgie et médecine esthétiques mis en place par le conseil consultatif des ministres de la santé des Etats fédérés d'Australie a défini les actes à visée esthétique comme « l'ensemble des opérations et autres procédures qui ont pour objet de corriger ou de modifier l'apparence, la couleur, la texture, la structure ou la position de caractéristiques et éléments corporels normaux avec la seule intention d' atteindre ce que le patient considère comme une apparence plus souhaitable ou de renforcer son estime de soi » 57 ( * ) .
L'Agence nationale suédoise de la santé et de la protection sociale a confié, en septembre 2011, à une personnalité indépendante, Mme Karin Lindell, le soin de rédiger un rapport consacré à l'analyse de la réglementation en vigueur en Suède dans le secteur de l'esthétique, et de réfléchir, le cas échéant, au renforcement de cet encadrement dans le sens d'une plus grande protection des patients.
Les premières analyses conduites dans le cadre de cette mission ont fait apparaître l'absence, en Suède, d'une réglementation claire applicable aux interventions non chirurgicales à visée esthétique, à l'exception de quelques règles spécifiques disséminées entre le code de la santé publique, le code de l'environnement ou encore le code de la consommation. Faute d'une définition réglementaire des actes à visée esthétique, la Suède ne prévoit aucune règle précise concernant les compétences requises, aussi bien des professionnels de santé que des professions non médicales, ou les exigences de sécurité applicables à ce secteur. Dans ces conditions, les patients suédois sont dans la majorité des cas renvoyés au droit des contrats ou au code de la consommation.
Dans son rapport remis, le 20 juin 2012, à l'agence nationale de la santé et de la protection sociale, Mme Karin Lindell propose de réserver l'exécution des interventions à visée esthétique à caractère médical aux seuls médecins disposant d'une licence. Cette licence leur serait octroyée sous réserve qu'ils apportent la preuve d'une formation adéquate et d'une expérience minimale dans la réalisation de ces actes spécifiques. En outre, elle propose d'interdire de pratiquer des actes à visée esthétique sur des sujets de moins de dix-huit ans.
Dans un décret en date du 24 octobre 2007 58 ( * ) , le Danemark a posé les bases d'un encadrement strict des actes à finalité esthétique, en donnant valeur réglementaire, dans son article 1 er , à la définition suivante des traitements à caractère esthétique : « toute procédure correctrice pour laquelle la considération esthétique constitue la principale indication, ou tout traitement destiné principalement à changer ou améliorer l'apparence » .
Devant la multiplication des plaintes déposées par des patients victimes de complications consécutives à des interventions pratiquées par des personnes non qualifiées en dehors d'établissements de soins accrédités, un certain nombre d'Etats fédérés des Etats-Unis ont entrepris des démarches visant à renforcer la réglementation applicable aux actes non invasifs de médecine esthétique.
C'est sous l'impulsion de M. Lawrence J. Green, dermatologue et président du comité d'encadrement des pratiques de la Société américaine de chirurgie dermatologique ( American Society for Dermatologic Surgery - ASDS), que l'Etat du Maryland a rendu exécutoire, le 23 août 2010, une réglementation des interventions à visée esthétique et de l'utilisation des dispositifs médicaux à visée esthétique.
Consignées dans le code du Maryland 59 ( * ) , ces nouvelles dispositions établissent un cadre pour la pratique de procédures dites « médicales à visée esthétique » ( « cosmetic medical procedures » ).
Afin de distinguer le champ de la médecine esthétique de celui des interventions à caractère chirurgical, la législation de l'Etat du Maryland établit une définition précise des procédures médicales à visée esthétique :
« (a) Une procédure médicale à visée esthétique est une procédure utilisant un dispositif médical à visée esthétique ou un produit médical destiné à améliorer l'apparence d'un individu.
« (b) Les procédures médicales à visée esthétique incluent les procédures suivantes :
« (i) les traitements de la peau impliquant l'utilisation de lasers ;
« (ii) les traitements de la peau impliquant l'utilisation de lumière pulsée intense ;
« (iii) les traitements de la peau impliquant l'utilisation de radiofréquences, de rayonnement micrométrique ou d'impulsions électriques ;
« (iv) les peelings en profondeur ;
« (v) les traitements de la peau impliquant le recours à la photothérapie ;
« (vi) la microdermabrasion ;
« (vii) les injections de produits médicaux par voies sous-cutanée, intradermique ou intramusculaire ;
« (viii) les traitements visant à enlever ou détruire de la graisse ; et
« (ix) tout traitement utilisant un dispositif médical à visée esthétique dans le but d'améliorer l'apparence d'un individu » ;
- des dispositifs médicaux à visée esthétique :
« (a) Un dispositif médical à visée esthétique est un dispositif qui altère ou endommage des tissus vivants.
« (b) Les dispositifs médicaux à visée esthétique incluent l'un des dispositifs suivants lorsqu'il est utilisé dans un but esthétique :
« (i) un laser ;
« (ii) un dispositif émettant de la lumière ou de la lumière pulsée intense ;
« (iii) un dispositif émettant des radiofréquences, des impulsions électriques ou des ondes sonores ;
« (iv) un dispositif de microdermabrasion ; et
« (v) des dispositifs utilisées pour l'injection ou l'insertion de substances naturelles ou étrangères dans la peau, la graisse, les tissus du visage, le muscle ou l'os. »
Le droit de l'Etat du Maryland reconnaît explicitement les interventions à visée esthétique, telles que l'épilation laser ou l'injection de toxine botulique, comme des actes médicaux . Le Conseil de l'ordre des médecins de l'Etat est régulièrement interrogé sur la pratique d'actes à visée esthétique au sein de spas dits « médicaux » ( « medical spas » ). A cet égard, le conseil précise que les « spas médicaux » ne peuvent être assimilés à des établissements de soins ( « health care facility » ) et qu'un esthéticien ne peut être assimilé à un professionnel de santé au regard de la législation du Maryland. Il est donc interdit à tout médecin de déléguer l'exécution d'interventions à visée esthétique reconnues comme des actes médicaux à des esthéticiennes.
4. Des compétences à clarifier, une formation à renforcer
a) Une répartition des compétences sujette à interprétation
De nombreux actes esthétiques se trouvent à la frontière entre médecine et chirurgie. Ce qui relevait autrefois uniquement du chirurgien , le lifting par exemple, peut être remplacé aujourd'hui par de simples injections de toxine botulique ou de plasma riche en plaquettes 60 ( * ) dans les zones du visage jugées inesthétiques. Avant leur interdiction, il en allait de même de certaines pratiques invasives de lyse adipocytaire, qui pouvaient se substituer aux liposuccions. D'où une certaine confusion dans l'esprit de patients enclins à croire que les médecins qui pratiquent ces actes à la fois invasifs et non chirurgicaux ont les mêmes capacités qu'un chirurgien esthétique.
De même, qui peut le plus peut le moins , et les actes de médecine esthétique « courante » peuvent également être pratiqués par les chirurgiens esthétiques qui ont l'obligation de proposer à leurs patients la solution la mieux adaptée à leurs attentes en fonction du rapport bénéfices-risques de la technique concernée.
La question de l'habilitation des médecins à pratiquer des soins esthétiques à la frontière de la chirurgie se pose donc en raison de la confusion qui peut s'installer chez leurs patients. Il en va de même d'une autre profession médicale : celle des chirurgiens-dentistes .
Ces derniers ont été récemment au coeur d'une polémique au sujet de leur capacité à injecter des produits visant au comblement des rides (comme l'acide hyaluronique) dans le sillon naso-génien . Le 24 janvier 2012, un courrier conjoint de la direction générale de la santé (DGS) et de la direction générale de l'offre de soins (DGOS), adressé à l'ordre des chirurgiens-dentistes, disposait que l'injection de produits de comblement de rides sur le visage n'était plus autorisé aux chirurgiens-dentistes parce qu'un tel acte les conduisait à intervenir dans une zone anatomique extra-buccale. Par la suite, un courrier signé de la secrétaire d'Etat à la santé les a maintenus dans cette capacité après qu'ils ont fait valoir que l'article L. 4141-1 du code de la santé publique faisait entrer dans le champ de leur capacité professionnelle « la prévention, le diagnostic, le traitement des maladies congénitales ou acquises, réelles ou supposées, de la bouche, des dents, des maxillaires et des tissus attenants ». La difficulté d'appréciation réside dans l'interprétation des mots « tissus attenants » , auxquels appartient le sillon naso-génien. Le comblement de ce canal est un acte à la frontière du thérapeutique (notamment dans le cadre d'un traitement prothétique et d'implants dentaires) et de l'esthétique. Lors de leur audition par la mission, les représentants des chirurgiens-dentistes ont dénoncé avec force ce qu'ils considéraient comme une atteinte à leur capacité professionnelle (en s'appuyant sur l'article L. 4141-1 précité, transposant la directive 78/687/CEE du 25 juillet 1978), bien qu'elle portât sur une activité marginale de leur profession.
b) La formation des médecins esthétiques ou les vertus du diplôme
En 2008, un rapport 61 ( * ) de la direction générale de la santé soulignait qu'il « existe de nombreuses formations complémentaires à la pratique des actes à visée esthétique dont le niveau est très variable : diplôme d'université (DU), diplôme interuniversitaire (DIU), formations par les fabricants d'appareil ». Il proposait donc la mise en place d'un enseignement de niveau national et soulignait également que les chirurgiens spécialisés en chirurgie plastique reconstructrice et esthétique ainsi que les dermatologues disposaient déjà de diplômes adaptés à l'exercice de la médecine esthétique.
La vraie question semble donc porter sur la formation initiale des médecins esthétiques dans la mesure où « aucune spécialisation dans le domaine de l'esthétique n'est actuellement reconnue au plan national ni exigée au plan réglementaire. Or, l'exercice de ces actes nécessite un complément de formation afin que ces médecins acquièrent les compétences indispensables à cet exercice » .
La plupart des personnes entendues par la mission lors d'une table ronde sur la médecine esthétique réunissant représentants du Conseil national de l'ordre des médecins (Cnom), enseignants et médecins esthétiques, se sont accordées pour considérer que la médecine esthétique ne devait pas constituer une spécialité. Elle se caractérise plutôt par la pluridisciplinarité de ses actes qui sont susceptibles de faire appel à des compétences relevant de spécialités médicales diverses : chirurgie esthétique, dermatologie, oto-rhino-laryngologie, médecine générale, gynécologie-obstétrique, chirurgie dentaire, médecine et chirurgie vasculaires. Cette approche pluridisciplinaire de la médecine esthétique semble d'ailleurs faire consensus.
Face au manque de formation qualifiante et à l'augmentation exponentielle de la demande en médecine esthétique, il est apparu nécessaire de mettre en place les premiers outils de reconnaissance d'une qualification professionnelle . De trop nombreux patients risquaient de se trouver face à de soi-disant professionnels, peu compétents sinon incompétents.
En 2004, le Cnom a donc réuni les représentants des chirurgiens et des oto-rhino-laryngologistes (ORL) afin de créer un diplôme interuniversitaire validant l'enseignement de ces techniques et, en 2007, a accordé aux titulaires du DIU « médecine morphologique et anti-âge » le droit de s'en prévaloir sur leurs plaques et leurs ordonnances, permettant aux patients de disposer de listes départementales de ces praticiens.
Enseigné à l'université de Lyon I, le DIU dépend également des universités de Montpellier, de Paris et de Pointe-à-Pitre. Le ministère de l'enseignement supérieur a demandé aux responsables de ce diplôme de continuer à mieux impliquer certaines régions (Est et Sud-ouest de la France) et à respecter le numerus clausus de quatre-vingt-cinq médecins formés chaque année (soit près de six cents médecins depuis la création du diplôme). Le DIU comporte deux cents heures d'enseignement , dispensées par un grand nombre d'intervenants. Y sont enseignés, par exemple, les gestes de prévention, l'endocrinologie, la dissection du visage (huit heures d'enseignement) ou encore les conséquences médico-légales des actes.
Ce diplôme , qui permet d'offrir un apprentissage des actes qui fait cruellement défaut en matière de médecine esthétique, n'est cependant pas obligatoire : s'il n'est pas titulaire du DIU, un médecin peut tout de même pratiquer des actes de médecine esthétique.
La formation et le droit au titre qui l'accompagne doivent être au coeur de toute politique de santé publique en matière de médecine esthétique : il est important que nos concitoyens prennent conscience du fait que ces actes sont d'ordre médical, ce qui suppose examen et diagnostic préalables avant traitement. Dispenser un acte de médecine esthétique revient à faire exercice de la médecine à part entière.
c) Des expériences étrangères hétérogènes
La situation française n'a rien d'unique. La question de la formation des médecins esthétiques se pose également en Espagne, en Italie, en Allemagne ou en Grande-Bretagne . Un rapport du régulateur britannique 62 ( * ) , chargé du contrôle des hôpitaux et des lieux où est pratiquée la médecine, dénonce l'existence de nombreux établissements fournissant des interventions esthétiques non régulées, telles que les techniques d'injection et de remodelage. Ce rapport recommande l'accréditation des praticiens privés opérant dans le champ esthétique, ce qui est rendu difficile par l'absence de formation spécifique reconnue, et préconise donc avec force la mise en place d'une telle formation.
La situation est un peu différente aux Etats-Unis. En l'absence d'encadrement fédéral de l'exercice de la médecine, y compris pour tout ce qui s'apparente à la médecine esthétique, ce sont les Etats fédérés qui sont compétents en la matière.
L'injection de Botox, pourtant défini comme un médicament, illustre bien la grande diversité des pratiques selon les Etats : dans le Maryland, cet acte est considéré comme de la médecine ; dans l'Etat voisin de Virginie, n'importe qui peut le réaliser.
D'une manière générale, la législation et la réglementation n'ont pas suivi les évolutions techniques. Ainsi, plus de vingt Etats, aux Etats-Unis, n'ont aucune disposition relative aux conditions de délégation par les médecins des interventions au laser. Quelques-uns seulement ont pris des dispositions concernant les injections. Surtout, alors que chaque Etat donne sa propre définition de la pratique de la médecine, beaucoup la limitent au traitement de maladies ou d'affections. Lorsque les interventions à visée esthétique ne sont pas considérées comme entrant dans le champ médical, elles ne sont pas soumises aux mêmes protections en matière de formation, de supervision et de contrôle que les procédures « médicalement nécessaires ».
De plus, aux Etats-Unis , les médecins ont la possibilité de déléguer la réalisation de certains actes sous leur contrôle et sous réserve d'un examen préalable du patient ainsi que son information : dans certains Etats, les injections de toxine botulique peuvent donc être exécutées par des infirmières. Il en va d'ailleurs de même en Grande-Bretagne ou en Suède .
L'exemple de ce pays illustre parfaitement la difficulté que peuvent éprouver les pouvoirs publics à réguler les actes de médecine esthétique : alors qu'il a fait l'objet, en 2010, d'un avis motivé (ultime étape avant la saisine de la Cour de justice de l'Union européenne) de la Commission européenne lui enjoignant d'adoucir sa législation sur les produits cosmétiques 63 ( * ) , les produits de comblement (acide hyaluronique ou toxine botulique, entre autres) ont été classés dans la catégorie des dispositifs médicaux sur la base d'une réglementation écrite à une époque où ces produits n'existaient pas encore. Dès lors, n'importe quelle personne travaillant dans un institut de beauté et n'ayant aucune formation médicale peut procéder à de telles injections alors que les risques de ces actes sont aujourd'hui bien connus - paralysie faciale ou percement de veine, par exemple.
Au Danemark, vingt-huit types d'interventions à visée esthétique ne peuvent être pratiquées que par des médecins disposant des compétences requises.
La réglementation danoise couvre l'ensemble des interventions à finalité esthétique : les interventions chirurgicales, l'utilisation de produits de comblement, la sclérothérapie, l'injection de toxine botulique, la lipothérapie, la dermabrasion, l'usage de lasers ou de lumière pulsée et les peelings chimiques. Ces traitements (y compris l'utilisation de dispositifs d'épilation tels que le laser et la lampe à lumière pulsée) ne peuvent être exécutés que par des médecins , le cas échéant assistés par des personnes dûment qualifiées, enregistrés auprès de l'Agence nationale danoise de la santé et des produits médicaux. Certaines interventions sont expressément réservées à des médecins spécialisés dans la partie du corps concernée ou à des médecins apportant la preuve d'une qualification pertinente et reconnue.
A titre d'exemple, les chirurgiens esthétiques sont autorisés à pratiquer l'ensemble des procédures à caractère esthétique mentionnées dans la liste établie par le décret du 24 octobre 2007 précité, à l'exception des traitements par laser ou par lumière pulsée. Les dermatologues ont, pour leur part, la possibilité de réaliser l'intégralité des interventions au niveau de la peau et du cuir chevelu, de même que les actes de lipothérapie. Les ophtalmologues sont susceptibles de pratiquer des interventions chirurgicales dans la région de l'oeil, et sont habilités à procéder à des injections de toxine botulique. Les oto-rhino-laryngologues peuvent intervenir, sur le plan chirurgical, au niveau du nez et des oreilles et les chirurgiens vasculaires sont autorisés à pratiquer de la sclérothérapie par injection.
Le recours à des assistants doit faire l'objet d'une autorisation délivrée par l'Agence nationale de la santé et des produits médicaux. Tout médecin enregistré auprès de l'agence doit ainsi soumettre la documentation relative aux qualifications de ses assistants et à leur expérience dans l'exécution de certains actes. Les médecins demeurent responsables des actes effectués par leurs assistants.
DEUXIÈME PARTIE - CRÉER LES CONDITIONS D'UNE VÉRITABLE SÉCURITÉ
Par les répercussions qu'il a pour les femmes qui en sont victimes, le traumatisme lié au scandale des prothèses PIP sera très long à s'estomper. Les séquelles - physiques et psychologiques - seront malheureusement difficiles à surmonter.
Au-delà de l'utilisation frauduleuse d'un gel interdit pour un usage humain, cette douloureuse affaire revêt un caractère exemplaire. Car aux Etats-Unis, ce n'est pas l'utilisation de ce gel qui a suscité la réaction négative des autorités sanitaires. C'est l'organisation même du processus de fabrication en vigueur dans l'entreprise qui fut à l'origine du refus d'homologation. Les constatations de l'inspection sur site des autorités sanitaires australiennes, en 2003, sont tout aussi accablantes. Les suites qui y furent données sont révélatrices des graves lacunes du système de régulation des dispositifs médicaux : tout repose sur la confiance mise dans l'évaluation accomplie par les organismes notifiés .
Aujourd'hui, il convient évidemment d'éviter la répétition de fraudes comme celle de PIP.
Mais il est tout aussi fondamental de faire en sorte que les drames sanitaires puissent être évités, y compris et surtout lorsqu'ils ne sont pas le résultat d'une fraude.
En matière de santé, chacun d'entre nous attend la perfection et refuse le droit à l'erreur, à l'absence de résultat. Il est important de prendre conscience que nul système d'autorisation, nul mécanisme de régulation ne permettra d'y parvenir.
A l'inverse, il n'est plus possible de se satisfaire d'une situation dans laquelle des matériels de plus en plus complexes, et dont l'impact sur la survie des patients est de plus en plus décisif, présentent des risques mal détectés.
Des défauts propres à la France doivent aussi être corrigés. Si aucun système n'est parfait - les défaillances rencontrées par certains dispositifs médicaux américains le soulignent amplement -, il n'est pas acceptable que la France accuse un retard dans la détection des défaillances par rapport à certains pays soumis aux mêmes règles ou à une réglementation équivalente .
De nombreux dysfonctionnements des procédures censées garantir la sécurité des dispositifs médicaux implantables et des insuffisances du cadre législatif et réglementaire relatif aux interventions à visée esthétique ont été portés à la connaissance de la mission d'information. Par leur ampleur, ils ne peuvent appeler une réponse unique de sa part ; un principe évident, malheureusement trop souvent oublié, doit néanmoins prévaloir : la protection de la santé publique.
Les autorités publiques chargées de la régulation de ces secteurs d'activité et de la préservation de la santé des citoyens doivent être en mesure d'évaluer les innovations et les techniques nouvelles qui apparaissent en continu, que ce soit dans le domaine médical ou dans celui de l'esthétique pour éviter que celles présentant un danger pour la santé des patients ou des consommateurs puissent leur être proposées. Elles doivent également faire preuve d'une plus grande réactivité afin de mettre en oeuvre dans les plus brefs délais les mesures conservatoires qui s'imposent lorsque des doutes crédibles sur la fiabilité d'un dispositif médical apparaissent.
Dès lors, la mission recommande d'agir dans deux directions , qui recoupent le champ de ses investigations :
- renforcer strictement les contrôles effectués sur les dispositifs médicaux implantables dès leur mise sur le marché et durant toute leur durée d'utilisation, afin de restaurer la confiance que les scandales sanitaires qui se sont succédé ces dernières années ont commencé à éroder ;
- reconnaître que le développement rapide qu'ont connu les interventions à visée esthétique , aussi bien par le nombre de personnes concernées que les techniques proposées, constitue une problématique majeure en matière de santé publique . Dans ce contexte, l'absence d'encadrement réglementaire devrait inévitablement aggraver la situation dans un futur proche.
Les propositions que fait la mission d'information pour corriger les carences qu'elle a identifiées ne requièrent pas toutes de profondes réformes législatives. Au contraire, la solution se situe parfois dans l'application correcte, dans son esprit et dans sa lettre, de la législation existante. Le droit ne peut toutefois pas régler l'ensemble des problèmes soulevés. Une action de pédagogie visant à faire évoluer les consciences et à responsabiliser tous les acteurs - professionnels, patients, consommateurs, régulateurs - devrait permettre de corriger des comportements parfois dangereux ou inacceptables.
I. DE L'IMPLANTABLE À L'IMPLANTÉ : RENFORCER LE CONTRÔLE DES DISPOSITIFS MÉDICAUX À TOUTES LES ÉTAPES
Contrairement au médicament, dont la mise en vente est conditionnée à l'obtention d'une autorisation de mise sur le marché (AMM) pour laquelle la réalisation d'importants essais cliniques est obligatoire, le dispositif médical implantable peut être vendu et utilisé même en l'absence de tests in vivo . Qui plus est, son suivi après l'implantation, sur une période longue, reste lacunaire. En l'absence de registres ou d'outils comparables recensant l'ensemble des dispositifs implantés d'un même type, il n'est pas possible d'identifier rapidement des anomalies ou des taux de dysfonctionnement supérieurs à la moyenne. En cela, la sécurité sanitaire des porteurs d'implants n'est pas correctement assurée.
Certaines mesures correctives relèvent du socle commun communautaire . A cet égard, il apparaît urgent de modifier les règles qui définissent la procédure conduisant à la certification et à la commercialisation d'un dispositif médical implantable. Il faut renforcer les contrôles et s'assurer que les organismes notifiés, sur lesquels repose le système du marquage CE, proposent des prestations d'une qualité équivalente et soient supervisés uniformément, quel que soit leur pays d'origine .
Il convient également d'agir au plan national car d'autres pays tirent d'ores et déjà davantage parti des mécanismes existants. L'exemple des registres montre que la surveillance des implants commercialisés, dite post market , peut être réalisée de manière plus efficace. Il faut recueillir plus de données et mieux partager celles déjà en la possession des autorités sanitaires et des fabricants. Il est également nécessaire de rappeler l'importance de la matériovigilance à tous les professionnels de santé et de s'interroger sur les moyens de rendre cette obligation plus compatible avec leur activité quotidienne.
A. CLARIFIER LES CONDITIONS DE MISE SUR LE MARCHÉ DES DISPOSITIFS MÉDICAUX
1. Préciser le rôle des organismes notifiés
La Commission européenne 64 ( * ) recense soixante-dix-sept organismes notifiés dans l'UE et les Etats partenaires. Il est du ressort des autorités compétentes de chaque Etat de désigner à ce titre les organismes qui en font la demande, sur la base de critères minimaux fixés par l'annexe XI de la directive de 1993. Toutefois, celles-ci n'appliquent malheureusement pas le même niveau d'exigence ou de sévérité dans ce domaine ou lors des contrôles qu'elles effectuent ensuite. Un niveau homogène d'application des exigences réglementaires à l'échelle européenne et la définition de règles plus précises concernant l'encadrement des organismes notifiés sont donc indispensables.
a) Revoir les conditions de désignation des organismes notifiés
Il ne s'agit pas d'une problématique nouvelle née des récents scandales sanitaires. Au contraire, elle a été identifiée peu de temps après l'entrée en vigueur de la directive de 1993, conduisant notamment à la création du Groupe opérationnel des organismes notifiés, ou N-Bog, organisme de concertation regroupant la Commission européenne et les autorités sanitaires nationales, en juillet 2000. Celui-ci a, dans chacune des résolutions adoptées après ses sommets annuels (à Vienne en 2006 ou encore à Bonn en 2007), souligné le caractère hétérogène et inégal des procédures suivies par les autorités de chaque Etat concernant les organismes notifiés, malgré la réalisation d'un manuel des autorités notifiantes.
Ces dernières années, des réflexions ont été menées sur le sujet afin d'améliorer la législation. La Commission européenne avait ainsi imaginé la mise en oeuvre d'un système centralisé de désignation finale et de contrôle et de surveillance des organismes notifiés . Cet organisme centralisé de surveillance des organismes notifiés aurait été créé sous la forme d'un département spécifique au sein de l'Agence européenne du médicament (EMA) ou du Centre commun de recherche rattaché directement à la Commission européenne ( Joint Research Centre - JRC).
C'est la logique même de la régulation des organismes notifiés par les autorités sanitaires nationales, de leur enregistrement à leur fonctionnement quotidien, qu'il est nécessaire de modifier. Celle-ci ne doit plus être ponctuelle, ni se concentrer uniquement sur le contrôle sur pièces. Il faut inventer de nouvelles méthodes de contrôle globales, qui prennent pleinement en compte toutes les dimensions des travaux des organismes notifiés et qui font de la sécurité sanitaire la première des priorités.
La première étape consiste en un renforcement des critères d'agrément des organismes notifiés . Dans les conclusions de leur rapport commun de février 2012 65 ( * ) sur l'affaire PIP, l'Afssaps et la direction générale de la santé (DGS) proposent que les critères minimaux qui doivent actuellement être remplis soient remplacés par des « critères maximaux », c'est-à-dire une obligation de disposer de moyens, humains comme matériels, pour effectuer un nombre prédéterminé d'audits chaque année. Il est également suggéré que la notification d'un organisme soit le résultat d'une évaluation menée, conjointement ou séparément, par les régulateurs d'au moins deux Etats. Enfin, les critères d'habilitation devraient être modulés en fonction du type de dispositif médical à certifier.
Il est tout à fait souhaitable que ces propositions de bon sens soient suivies d'effet et que la France les défende à Bruxelles. Il faut toutefois aller plus loin, en s'assurant notamment de la compétence du personnel et en encadrant le recours à la sous-traitance. Les capacités d'expertise interne de ces structures doivent être évaluées et les liens d'intérêts rendus publics.
b) S'assurer des moyens mis en oeuvre pour effectuer les contrôles
Comment les organismes notifiés travaillent-ils ? Selon quels critères évaluent-ils les produits qui leur sont présentés ? Quels sont les moyens qu'ils mettent en oeuvre ? Autant de questions pour lesquelles il est difficile d'obtenir une réponse claire.
Il est donc nécessaire d'agir, au niveau européen, pour obtenir la mise au point d'un cahier des charges commun à tous les organismes notifiés définissant clairement la procédure aboutissant à la délivrance du marquage CE. Cette méthodologie homogène fait aujourd'hui défaut. En conséquence, le niveau d'exigence peut varier selon les organismes et les pays d'origine. Certains fabricants se livrent à du « forum shopping » . Cette pratique consiste, pour ceux-ci, à faire examiner leurs produits par un organisme notifié moins regardant après avoir subi un premier refus de certification.
Proposition n° 1 :
Mettre au point un
cahier des charges commun à tous les organismes
notifiés
Des mesures visant à garantir, au sein des organismes notifiés, un niveau de compétence et des capacités d'expertise suffisants, permettraient donc de corriger les pratiques anticoncurrentielles qui se développent actuellement au bénéfice des structures les plus laxistes et au détriment des citoyens.
Si l'on en croit les témoignages recueillis par la mission, les fabricants ne sont pas opposés à des actions dans ce sens. Auditionné par la mission d'information, M. Eric Le Roy, directeur général du Syndicat national de l'industrie des technologies médicales (Snitem), organisation professionnelle représentant l'industrie du dispositif médical, a ainsi estimé « qu'une meilleure harmonisation éliminerait les moins-disant » . Déplorant le trop grand nombre d'organismes notifiés, il n'en a pas moins souligné l'importance, pour les industriels, d'avoir des interlocuteurs sérieux. Il a également reconnu leur attachement à un marquage CE fiable et incontestable.
Il est donc unanimement reconnu qu'un effort important en matière de transparence du processus de certification des dispositifs médicaux doit être réalisé de la part de tous les acteurs concernés et, en premier lieu, des organismes notifiés. Toutefois, c'est l'insuffisance des règles en vigueur qui contribue largement à la situation actuelle. Il appartient donc au législateur européen, mais aussi aux différents organes de concertation mis en place sur le sujet, comme le N-Bog ou Team-NB, l'association des organismes notifiés européens, de s'emparer du sujet, de faire avancer les bonnes pratiques et d'inciter tous les acteurs à les adopter.
En conséquence, les pouvoirs des organismes notifiés en matière de contrôle et d'inspection doivent être mieux exercés et, dans certains cas, renforcés. Il en va ainsi de la réalisation de visites inopinées au fabricant , déjà prévues par la directive 93/42/CEE mais très peu effectuées en pratique. Pourtant, dans le cadre d'une fraude de grande ampleur comme l'affaire PIP, l'effet de surprise est le seul moyen de découvrir la tromperie et de confondre les coupables. De même, la fréquence des audits des fabricants par les organismes notifiés , qui ne fait l'objet d'aucune harmonisation européenne en dehors du contrôle réalisé, tous les cinq ans, lors de la recertification d'un dispositif médical, devrait être précisément définie.
Proposition n° 2 :
Multiplier les
contrôles inopinés chez les fabricants
2. Assurer la transparence du processus de certification
a) Vérifier périodiquement la qualité des prestations des organismes notifiés
La commercialisation d'un dispositif médical implantable est conditionnée à la délivrance, pour celui-ci, du marquage CE par un organisme notifié. Dès lors, il apparaît indispensable de s'assurer que ce certificat de qualité et de conformité avec les exigences essentielles posées par le droit européen fasse l'objet d'une procédure uniforme sur tout le territoire de l'UE.
Force est malheureusement de constater que ce n'est pas le cas.
En vertu de la réglementation actuelle, la plupart des décisions concernant les modalités de certification et d'évaluation des dispositifs médicaux relèvent du fabricant. Elles ne peuvent être remises en cause par l'organisme notifié, qui ne dispose d'aucune marge de manoeuvre ou de pouvoir d'appréciation à ce sujet. Comme le prévoient les annexes de la directive européenne, le producteur choisit librement s'il souhaite faire porter le contrôle de l'organisme notifié sur son produit ou sur un dossier de documentation technique.
Ainsi que Mme Laurence Dagallier, directrice déléguée du Laboratoire national de métrologie et d'essais (LNE), seul organisme notifié français, l'a précisé lors de son audition, si un organisme notifié peut exiger d'un fabricant la réalisation d'essais supplémentaires lors de l'examen de la conception d'un produit, il ne peut pas décider unilatéralement de mener à bien de tels essais après mise sur le marché en l'absence d'une nouvelle demande de certification.
Pour un observateur extérieur, il est difficile de savoir quelles sont les méthodes de travail des organismes notifiés et donc légitime de s'interroger sur l'équivalence des différentes méthodes d'examen des dispositifs médicaux qu'ils utilisent.
De plus, les relations financières existant entre les organismes notifiés et les fabricants, qui les rémunèrent pour obtenir la certification de leurs produits, constituent un obstacle à leur parfaite indépendance. Sans remettre en cause le professionnalisme de leurs employés, cette situation, à elle seule, devrait être corrigée pour lever le soupçon de partialité vis-à-vis du donneur d'ordre qu'elle est susceptible de créer .
La surveillance des organismes notifiés au niveau communautaire mérite d'être renforcée, au moins pour assurer un pilotage stratégique des contrôles à l'échelle de l'Union. En dotant le N-Bog d'un statut légal et d'un véritable pouvoir en la matière, il serait possible de réagir de manière coordonnée et de mutualiser les ressources à la disposition des Etats membres, favorisant la coopération entre eux au-delà de ce qui existe aujourd'hui.
Proposition n° 3 :
Doter le N-Bog d'un
statut légal dans la réglementation européenne et lui
confier
la mission d'harmoniser les modalités de surveillance des
organismes notifiés
Le fait de vérifier à intervalles réguliers , si possible annuellement, la qualité des prestations et l'expertise des organismes notifiés doit redevenir l'une des priorités des autorités sanitaires partout en Europe. La régulation ne doit pas être un avantage favorisant la concurrence déloyale à laquelle se livrent ceux d'entre eux qui bénéficient des insuffisances des pouvoirs publics nationaux. Il est urgent que la Commission européenne agisse en ce sens.
b) Renforcer le partage d'informations
Par ailleurs, il faut améliorer le partage d'informations entre organismes notifiés ainsi qu'entre ceux-ci et les autorités sanitaires nationales .
L'absence de circulation de l'information a constitué un des ressorts majeurs de l'affaire PIP : les incidents graves constatés par les médecins n'ont pas été portés à la connaissance des différents acteurs ni, a fortiori , les doutes du Bureau central de tarification quant à la qualité des prothèses mammaires fabriquées par cette entreprise.
Les organismes notifiés ont déjà obligation de communiquer à leurs homologues les décisions de refus, retraits ou suspensions de certifications qu'ils sont conduits à prendre. Il est urgent d'étendre cette obligation d'information aux observations de non-conformité majeures. Un tel renforcement corrigerait une des failles du droit en vigueur et constituerait sans doute un moyen de percevoir plus tôt les premiers signaux d'un risque sanitaire majeur.
Proposition n° 4 :
Etendre l'obligation
d'information des organismes notifiés aux observations de
non-conformité majeures
c) Multiplier les inspections croisées des organismes notifiés
Un certain nombre d'associations de praticiens et d'experts du domaine de la santé ont également proposé que les dossiers techniques en vue d'une évaluation des dispositifs médicaux de la classe III soient soumis directement à un département spécifique, qui serait créé en ce sens au sein de l'EMA. Cette dernière émettrait ainsi un avis pour la Commission européenne relatif à la mise sur le marché du produit concerné.
Enfin, la majorité des participants à la consultation publique engagée en 2008 par la Commission européenne sur la refonte de la réglementation relative aux dispositifs médicaux préconisait qu'Eudamed intègre des informations concernant les performances des organismes notifiés, dans un souci de plus grande transparence.
Cette prise de conscience tardive a été suivie de quelques effets, notamment dans la coopération entre les autorités nationales compétentes. Ainsi, elles conduisent désormais des inspections communes d'organismes notifiés afin de partager leurs méthodes de travail et d'échanger sur les meilleures pratiques. L'organisme notifié allemand TüV Süd, par exemple, a récemment été contrôlé par l'autorité espagnole. Le développement de ces évaluations par les pairs constitue une avancée notable qu'il faut saluer. Il reste néanmoins anecdotique par rapport au chemin qui reste à parcourir pour assurer un encadrement des organismes notifiés à la hauteur de leur rôle dans la préservation de la santé publique.
Proposition n° 5 :
Généraliser les inspections communes des organismes
notifiés par les autorités sanitaires de plusieurs pays
européens
3. Rendre plus transparentes les relations entre professionnels de santé et fabricants
L'effort indispensable à réaliser en matière de transparence passe aussi par une plus grande publicité des relations entre les professionnels de santé et les fabricants de dispositifs médicaux.
Les dispositifs médicaux implantables sont, plus que tout autre produit de santé, « opérateur-dépendant », c'est-à-dire qu'ils sont développés par les industriels en coopération avec ceux, médecins et chirurgiens, qui seront amenés à les utiliser. Ainsi, ils peuvent être personnalisés selon des besoins spécifiques et répondre de manière plus adaptée aux besoins des praticiens. Ce mode de fonctionnement a permis des avancées importantes dans le traitement de nombreuses pathologies et a contribué à l'amélioration générale de la qualité des soins ces dernières années.
Néanmoins, des efforts restent à accomplir pour dissiper les soupçons suscités par les liens d'intérêts qui peuvent exister entre médecins, experts ou scientifiques et l'industrie du dispositif médical. Les liens d'intérêts ne sont pas condamnables en eux-mêmes. Pour des dispositifs médicaux qui sont de plus en plus spécialisés, il est par nature difficile de trouver des experts qui n'entretiennent aucun lien d'intérêts avec les fabricants.
Il convient cependant de rendre publics les liens d'intérêts afin qu'ils ne se transforment pas en conflits d'intérêts, c'est-à-dire qu'ils puissent avoir une influence sur la manière dont la personne concernée exerce ses fonctions, ou en donner l'apparence. Comme l'affaire du Mediator l'avait révélé, la collusion entre l'industrie du médicament et certains médecins peut avoir des conséquences graves pour les patients.
La France a tenté de répondre à ce défi avec la loi du 29 décembre 2011 66 ( * ) dont l'objet était, à l'aune du scandale du Mediator, de restaurer la confiance dans le système de sécurité sanitaire. Celle-ci n'a que partiellement tenu compte des travaux de réflexion qui l'ont précédée, notamment du rapport 67 ( * ) de la mission commune d'information que le Sénat avait alors mise en place.
Il faut agir sur plusieurs fronts afin de garantir l'indépendance de tous , des étudiants en médecine aux professeurs des universités - praticiens hospitaliers (PU-PH), face aux entreprises. Des progrès ont été réalisés en matière de publication des liens d'intérêts des personnes qui siègent dans les organismes et commissions publics du champ sanitaire. Après la publication du décret du 9 mai 2012 68 ( * ) , le formulaire de déclaration d'intérêts auprès de l'ANSM a été actualisé et sera disponible par télédéclaration à compter du 18 juillet 2012. Le site de l'agence (consulté le 4 juillet 2012) précise toutefois que « pour des raisons d'ordre technique, les montants des rémunérations perçues et des participations financières détenues [...] ne seront demandés que dans un second temps » .
Sous cette réserve, les dispositions de la loi « Médicament » relatives aux liens d'intérêts entre industries des produits de santé et experts sont maintenant applicables. Il n'en est pas de même de celles qui concernent les avantages consentis par les entreprises aux professionnels de santé.
Il semble ainsi illusoire , et de faible portée concrète, d'imposer 69 ( * ) seulement aux entreprises de rendre publique l'existence de conventions passées avec des praticiens , leurs associations, des établissements de santé ou encore des étudiants en médecine, sans que les flux financiers en cause ne soient révélés. C'est cette voie qu'ont choisie les Etats-Unis avec le Sunshine Act en imposant que les informations relatives au paiement ou autre transfert de valeurs, faits à un bénéficiaire par un fabricant, soient mises à la disposition du public de manière claire et accessible, avant le 31 mars de l'année suivant celle au cours de laquelle ils ont été effectués. La publicité annuelle de ces flux financiers et des liens d'intérêts qu'ils entraînent doit être rendue obligatoire en France .
Afin que les contrats de complaisance puissent être sanctionnés, il est important de doter les instances ordinales des pouvoirs nécessaires. Comme l'a indiqué M. André Deseur, responsable de la section « exercice professionnel » du Conseil national de l'Ordre des médecins (Cnom), lors de son audition, la commission médecins-industrie de celui-ci, ou les instances départementales de l'Ordre, reçoivent tous les contrats passés entre un médecin et un industriel et les examinent. Toutefois, il a déploré que l'avis de cette commission ne s'impose à aucune des parties. La loi de décembre 2011 a exigé de l'industriel qu'il informe l'instance ordinale des suites données à cet avis. La moindre des choses serait de compléter ces dispositions en l'obligeant à apporter une justification au cas où il déciderait de passer outre un avis négatif et que le Cnom puisse s'opposer à cette décision.
Proposition n° 6 :
Rendre publics les
flux financiers et les liens d'intérêts entre fabricants
de
dispositifs médicaux et bénéficiaires de ces
avantages
Les modalités de l' enseignement de la médecine sont un autre aspect important de l'indépendance des professionnels de santé et conditionnent leurs pratiques ultérieures. C'est dès la formation des futurs médecins que l'impartialité dans le jugement et les opinions scientifiques doit être assurée. Il faut donc assurer la publicité des liens entre les entreprises et les établissements qui assurent la formation initiale médicale ou qui y concourent . De plus, afin d'éviter que les industriels ne puissent avoir une influence indue sur eux, il serait souhaitable de leur interdire de passer des conventions d'hospitalité avec des étudiants ou de leur octroyer des avantages . Il ne faut pas donner aux étudiants l'habitude d'entretenir des liens trop étroits avec l'industrie des produits de santé, même s'il n'est pas question non plus de les priver d'une contribution au financement de leurs études. Il s'agit simplement d'écarter la création de liens trop directs entre l'industrie médicale et les professionnels de santé dès le début de leurs études supérieures.
Proposition n° 7 :
Mutualiser les sommes
allouées par l'industrie des produits de santé aux
étudiants dans le cadre des conventions d'hospitalité
Enfin, une fois toutes ces informations rendues publiques, il serait utile de les centraliser afin d'en faciliter l'accès pour tous . C'est pourquoi la création d'un site internet unique et gratuit , dont les contours restent à préciser, constituerait une avancée majeure vers plus de transparence. Comme la mission commune d'information du Sénat consécutive à l'affaire du Mediator 70 ( * ) le préconisait, la mise en place d'un registre public des avantages consentis par l'industrie des produits de santé aux professionnels de santé constituerait la solution optimale, celle qui dissiperait les doutes sur les liens d'intérêts que le secret engendre. Elle devrait évidemment s'accompagner de sanctions pour ceux refusant de se soumettre à ces obligations de transparence.
Il conviendrait également de revoir les modalités de formation continue des médecins . Si cette question complexe dépasse le champ d'investigation de la mission d'information et a fait l'objet de plusieurs réformes ces dernières années, notamment dans la loi « HPST » 71 ( * ) , les personnes auditionnées n'en ont pas moins souligné à plusieurs reprises la place indue qu'y tiennent les industriels. Comme l'a exprimé le professeur Daniel Loisance, membre de l'Académie de médecine, « il n'est pas souhaitable que l'industrie ait le monopole de l'information des médecins dans leur pratique quotidienne » . Les praticiens subissent ensuite d'importantes pressions et voient leur exercice nécessairement influencé par les recommandations et les produits d'un fabricant. Une fois encore, seule une plus grande transparence dans les relations entre l'industrie et le corps médical constituerait une première réponse à cette question, avant d'engager une réflexion sur une plus grande implication de l'université, en coopération avec ceux qui mettent au point et produisent les dispositifs médicaux.
4. Repenser le marquage CE
a) Inciter la Commission à prendre pleinement en compte les exigences de sécurité des dispositifs médicaux
Quelques mois après l'adoption de la directive 2007/47/CE du 5 septembre 2007, la Commission européenne a pris l'initiative de lancer une grande consultation relative aux dispositifs médicaux. Le scandale des prothèses PIP a souligné l'urgence qui s'attache à poursuivre sur la voie du renforcement de la réglementation.
L'idée de départ était d'aboutir à une proposition de révision de la réglementation pour juillet 2012.
Confrontée à ce drame, qui concerne près de quatre-vingt-dix mille personnes dans toute l'Union, la Commission a appelé les Etats membres, le 9 février 2012, à prendre des mesures immédiates pour « assurer la pleine et stricte application de la législation actuelle » . Par la voix de John Dalli, commissaire à la santé et à la politique des consommateurs, elle appelait de ses voeux une action concertée dans trois directions : renforcer les contrôles, accroître la surveillance, restaurer la confiance.
Les mesures préconisées tendent notamment à :
- vérifier les désignations des organismes notifiés, afin de garantir qu'ils ne le sont que pour le champ d'évaluation qui correspond à leurs moyens et compétences ;
- s'assurer de la réalisation d'une véritable évaluation de la conformité, y compris par des inspections inopinées chez les fabricants ;
- renforcer la surveillance du marché ;
- améliorer les modalités de la matériovigilance, en facilitant la déclaration et l'analyse des événements indésirables ;
- mettre au point des outils permettant d'assurer la traçabilité des dispositifs médicaux.
Beaucoup de ces mesures rejoignent les recommandations que la mission sera amenée à formuler : au niveau européen comme au plan national, la première urgence consiste tout simplement à mettre véritablement en oeuvre les dispositions qui existent déjà.
Qu'il s'agisse du fonctionnement des organismes notifiés, de la surveillance du marché, de la coordination des interventions ou de la transparence de la matériovigilance, la Commission partage largement les vues de la mission, qui sont détaillées ci-après.
Sur le plan plus général de la refonte des directives existantes, la Commission a fait le choix de privilégier le renforcement du marquage CE plutôt que de mettre en place une véritable AMM pour les dispositifs les plus risqués . Pour écarter cette assimilation au médicament, la commission se fonde notamment sur des considérations financières : elle estime que la création d'une nouvelle agence dédiée au contrôle des dispositifs médicaux, voire un élargissement des missions de l'Agence européenne du médicament, relèvent de l'utopie car cela supposerait de mobiliser des ressources scientifiques et techniques très variées, pour un coût qu'elle juge inacceptable. Laisser le soin à chaque Etat membre d'autoriser la mise sur le marché de ces dispositifs conduirait à devoir mettre en oeuvre un mécanisme de reconnaissance mutuelle, lui aussi fort onéreux.
Interrogés par la présidente et le rapporteur de la mission, le 6 juin 2012, les services de la Commission ont d'ailleurs indiqué privilégier un renforcement des dispositions existantes. Un « marquage CE renforcé » serait mis en place. Le comité restreint du MDEG 72 ( * ) disposerait d'un pouvoir d'évocation de l'ensemble des dispositifs de classe III et en cas de suspicion de problèmes sanitaires pour les autres. Un tel système permettrait également de vérifier l'homogénéité du travail des organismes notifiés. Il favoriserait l'information des autorités nationales, averties en amont de l'arrivée de nouveaux produits sur le marché.
Certains - les fabricants notamment - objecteront probablement qu'un tel dispositif retarderait la mise sur le marché de dispositifs innovants. D'autres regretteront, à l'inverse, que le comité n'ait que la faculté de délivrer un simple avis, plutôt que de disposer d'un véritable pouvoir de blocage de la certification. Si la question n'est pas encore tranchée, la Commission objecte, en tout état de cause, que l'avis rendu par le comité sera immanquablement suivi d'effet de la part des organismes notifiés.
Par courrier en date du 20 février 2012, la France et cinq autres Etats membres ont fait part à la Commission de leur préoccupation quant au renforcement des exigences de mise sur le marché des dispositifs médicaux.
La révision de la directive a pris du retard. Prévue pour le printemps, elle est aujourd'hui annoncée pour l'automne 2012. Ce délai doit être mis à profit pour prendre en compte cette préoccupation, afin de prévoir une véritable appréciation du rapport bénéfices-risques, de renforcer les pouvoirs de contrôle des organismes notifiés et d'instituer un mécanisme d'autorisation des dispositifs médicaux de classe III les plus risqués.
Proposition n° 8 :
Inciter la Commission,
dans le cadre de la refonte des directives, à prendre pleinement en
compte les exigences de sécurité des dispositifs
médicaux
b) Créer une liste positive des dispositifs médicaux les plus risqués au niveau européen
Parallèlement à la réflexion menée par la Commission, qui devrait déboucher sur un projet de règlement à l'automne, le Parlement européen s'est saisi de l'affaire PIP. Le 14 juin 2012, il a adopté une résolution « sur les implants mammaires en gel de silicone défectueux fabriqués par la société française PIP » .
La résolution pointe du doigt l'échec du système de certification des organismes notifiés et appelle à tirer les leçons de cette fraude. Ce texte invite par conséquent les autorités nationales et communautaires à renforcer la qualité des contrôles exercés par les organismes notifiés et à améliorer la traçabilité de ce type d'implants.
Le Parlement européen élargit la problématique au cas des prothèses de hanche, laquelle, estime-t-il, illustre également « l'échec de l'actuel système de certification de la conformité aux exigences essentielles de santé et de sécurité ainsi que de la surveillance et des contrôles effectués par les organismes notifiés par les autorités nationales compétentes ».
Il considère que « le souhait de garantir au patient un accès rapide à de nouveaux dispositifs médicaux ne doit jamais prévaloir sur la nécessité d'assurer sa sécurité » .
La mission partage cette préoccupation, tout comme la demande d'un renforcement des contrôles des dispositifs médicaux déjà sur le marché.
La résolution appelle la Commission à examiner « la nécessité d'une autorisation de mise sur le marché pour les dispositifs médicaux dangereux, qui soit conforme ou analogue aux obligations à remplir pour les produits médicaux » . Ce parallèle avec le médicament peut être approuvé s'il s'agit d'accroître les exigences requises pour assurer la sécurité des dispositifs médicaux avant leur mise sur le marché. Subordonner la certification d'un nouveau dispositif médical risqué à la réalisation d'essais cliniques préalables permettrait de créer une liste positive , ce que souhaite vivement la mission.
En revanche, il n'est pas possible de transposer au dispositif médical l'ensemble de la procédure d'autorisation de mise sur le marché applicable au médicament, compte tenu des spécificités de ce secteur (multiplicité des classes de produits, rythme de l'évolution technologique, équilibre économique des entreprises innovantes...).
Proposition n° 9 :
Créer une liste
positive des dispositifs médicaux
les plus risqués au niveau
européen
c) Interdire l'utilisation de produits cancérogènes, mutagènes et reprotoxiques dans les dispositifs médicaux
En revanche, on peut regretter que ni la Commission ni le Parlement européen n'aient saisi l'occasion pour prendre en considération la question de l'utilisation de produits cancérogènes, mutagènes et reprotoxiques (CMR) dans les dispositifs médicaux.
Pour certaines substances, comme les phtalates, le débat est déjà ancien. Dans certaines conditions, cette substance, dont la toxicité est établie, est susceptible d'être relarguée par le plastique. C'est pourquoi la directive 2007/47/CE a institué un étiquetage obligatoire pour les dispositifs médicaux contenant des phtalates lorsqu'ils sont « destinés à administrer dans l'organisme et/ou à retirer de l'organisme des médicaments, des liquides biologiques ou autres substances ou des dispositifs destinés au transport et au stockage de ces liquides ou substances » . La directive exige également la justification spécifique de l'utilisation de cette substance et l'indication dans la notice d'information des risques résiduels et des mesures de précaution appropriées dans le cas où les dispositifs médicaux sont spécialement destinés aux enfants ou aux femmes enceintes ou allaitant. Ces dispositions sont entrées en vigueur en mars 2010.
Dans des recommandations formulées en mars 2009, l'Afssaps avait invité ces populations à risque à « rechercher des solutions de substitution » . Aujourd'hui, la révision des directives relatives aux dispositifs médicaux offre l'occasion d'aller plus loin car il n'est pas acceptable qu'un dispositif médical implantable soit plus dangereux qu'un jouet quand il est question de substances cancérogènes, mutagènes et reprotoxiques.
Proposition n° 10 :
Interdire l'ensemble
des CMR de catégorie 2 dans les dispositifs médicaux
destinés aux nourrissons, jeunes enfants et femmes enceintes
B. INSTITUER UNE VÉRITABLE SURVEILLANCE DES DISPOSITIFS MÉDICAUX IMPLANTÉS
1. Une évidence : appliquer enfin le droit existant
Lorsqu'une faille du droit existant apparaît, en particulier dans le domaine de la sécurité sanitaire, le premier réflexe est souvent d'appeler à sa réforme et de critiquer son insuffisance. Les récents scandales relatifs à des dispositifs médicaux implantables ont bien mis en lumière des failles dans la réglementation actuelle, mais la première des choses consiste à s'assurer que toutes les dispositions en vigueur étaient bien appliquées. Or, au cours de ses travaux, la mission a pu constater des réponses contradictoires de la part de ses interlocuteurs, souvent peu au fait des modalités d'organisation de la surveillance des dispositifs médicaux après leur commercialisation.
Pourtant, conformément au droit communautaire, et en particulier à la directive du 5 septembre 2007 73 ( * ) qui est venue modifier celle de 1993 74 ( * ) , tous les dispositifs médicaux implantables doivent désormais faire l'objet d'investigations cliniques , sauf si le recours aux données cliniques existantes peut être dûment justifié 75 ( * ) . Les données recueillies sont ensuite tenues à la disposition de l'ANSM.
En matière de matériovigilance , c'est-à-dire la surveillance du bon fonctionnement des dispositifs médicaux et des incidents dont ils peuvent être la cause une fois sur le marché, des possibilités d'encadrement particulier existent depuis 2004 . La loi du 9 août 2004 76 ( * ) a confié au pouvoir réglementaire la possibilité de fixer « les règles particulières applicables en matière de vigilance » exercée sur des dispositifs médicaux désignés par arrêté du ministre de la santé (article L. 5212-3 du code de la santé publique). L'article R. 5212-36 du même code 77 ( * ) , qui en fait application, prévoit que ces produits sont soumis à des règles de traçabilité « depuis la réception des dispositifs médicaux dans la structure sanitaire ou de chirurgie esthétique où ils seront utilisés jusqu'à leur utilisation chez le patient » .
Le but est bien évidemment de pouvoir identifier le plus rapidement possible, si l'innocuité de dispositifs médicaux venait à être infirmée, les patients chez qui ceux-ci auraient été utilisés ainsi que leurs lots d'origine. Il est ensuite de la responsabilité des établissements de santé, des pharmaciens hospitaliers et des services utilisateurs de dispositifs médicaux, chacun dans leurs compétences respectives, de veiller à ce que l'enregistrement du dispositif médical utilisé soit correctement réalisé et que le dossier médical du patient soit également renseigné (articles R. 5212-37 à R. 5212-40 du code de la santé publique). Enfin, à l'issue des soins, le patient doit se voir remettre un document récapitulant l'identification du dispositif utilisé, le lieu et la date d'utilisation ainsi que le nom du médecin utilisateur (article R. 5212-42 du code de la santé publique) .
L'application de ces dispositions était conditionnée à la publication d'un arrêté listant les dispositifs médicaux qui devaient y être soumis. Un arrêté du 26 janvier 2007 78 ( * ) a notamment prévu leur application immédiate aux valves cardiaques et différée au 31 décembre 2008 pour les autres dispositifs médicaux implantables.
Le « passeport dispositif médical » , présenté par certains des interlocuteurs de la mission comme un pas en avant important à accomplir en matière de matériovigilance, fait donc partie du droit positif depuis près de six ans et constitue une obligation légale depuis plus de trois ans ! Il est à tout le moins curieux de constater que nombreux sont les professionnels de santé qui semblent l'ignorer et que les autorités sanitaires ne font pas de la sensibilisation sur ce sujet l'une des priorités de leur action .
De toute évidence, la bonne mise en oeuvre de ces mesures aurait permis d'éviter les difficultés rencontrées en 2010 et 2011 pour identifier toutes les porteuses d'implants PIP et la confusion qui s'en est suivie . Le fait que de nombreuses femmes ne connaissaient pas le fabricant de leurs prothèses mammaires a pu causer chez elles un sentiment de crainte et de méfiance, d'autant plus fort que les pouvoirs publics ont officiellement recommandé de réaliser, à titre de précaution, l'explantation de tous les implants PIP. A l'avenir, et sans qu'il y ait besoin de modifier le droit existant autrement qu'à la marge, il devrait être possible d'informer individuellement chaque personne concernée du rappel d'un dispositif médical implantable.
2. Mettre en oeuvre sans tarder les avancées de la loi « Médicament » en matière de dispositifs médicaux
a) Tirer profit des avancées de la loi « Médicament »
L'objectif affiché de la loi du 29 décembre 2011 était de corriger les insuffisances du système français de pharmacovigilance, mises en lumière par le scandale du Mediator. Son champ dépassait celui des règles applicables au contrôle et à la surveillance du médicament et concernait également, à travers plusieurs de ses dispositions, les dispositifs médicaux : renforcement des conditions dans lesquelles ceux pris en charge par l'assurance maladie sont contrôlés (article 35), évaluation de ceux utilisés à l'hôpital (article 37), encadrement de la publicité pour les dispositifs médicaux (article 34), création d'un groupement d'intérêt public (Gip) habilité à réaliser des études de vigilance à partir des données de l'assurance maladie (article 33).
Six mois après la promulgation de la loi, les décrets d'application de ces articles ne sont toujours pas parus , bien que l'échéancier de mise en application de la loi, disponible sur le site Légifrance 79 ( * ) , affiche toujours une publication envisagée en avril 2012. Seules les modalités d'application des mesures concernant la publicité ont été fixées par un décret du 9 mai 2012 80 ( * ) .
Une telle situation est regrettable car les dispositions en question répondent à plusieurs critiques communément soulevées à l'encontre de la réglementation des dispositifs médicaux implantables et corrigent certaines de ses lacunes.
Proposition n° 11 :
Publier rapidement
les textes d'application de la loi
« Médicament »
relatifs aux dispositifs
médicaux
b) Contrôler la conformité des dispositifs médicaux aux spécifications requises pour pouvoir être remboursés
L'article 35 donne à l'ANSM le pouvoir d'effectuer un contrôle des dispositifs médicaux inscrits sur la liste des produits et prestations remboursables (LPP), c'est-à-dire ceux remboursés par l'assurance maladie, afin de s'assurer qu'ils respectent les critères techniques sur la base desquels l'inscription sur cette liste est autorisée. Si, en conduisant un tel contrôle, elle constate qu'un fabricant n'a pas respecté ses obligations, l'ANSM le met en demeure de s'y conformer et le comité économique des produits de santé (Ceps) peut lui infliger une pénalité financière dont le montant peut aller jusqu'à 10 % du chiffre d'affaires réalisé par le fabricant en France.
La procédure d'inscription la plus courante sur la LPP, dite par description générique 81 ( * ) , permet à des dispositifs médicaux présentant des caractéristiques techniques similaires à celles de produits faisant déjà partie de la liste d'y figurer après une simple déclaration à l'ANSM. Seuls les dispositifs médicaux innovants ou nécessitant un suivi particulier du fait de leur impact économique ou sanitaire doivent être inscrits dans une ligne spécifique, sous forme de nom de marque. Dans ce cas, une évaluation de ce produit et de son service attendu par la Commission nationale des dispositifs médicaux et des technologies de santé (CNEDiMTS) de la Haute Autorité de santé (HAS) est obligatoire.
Il existe aujourd'hui plus de 3 100 lignes génériques dans la LPP. Cet article permet donc de compenser l'aspect purement administratif de cet enregistrement en ouvrant la voie à des contrôles renforcés et à des sanctions pour les fabricants dont les produits et prestations ne sont pas conformes aux principes de sécurité établis par les législations communautaires et françaises. Il s'agit donc d'un progrès en matière de surveillance du marché des dispositifs médicaux. Il représente également un premier pas vers une maîtrise de la dépense en la matière, car lorsqu'un manquement aux règles d'inscription a entraîné un remboursement indu, l'assurance maladie peut se retourner contre le fabricant ou, le cas échéant, son mandataire et le recouvrer auprès de lui.
c) Etendre l'obligation d'évaluation des dispositifs médicaux utilisés à l'hôpital
De même, l'article 37 modifie profondément les conditions dans lesquelles les établissements de santé peuvent utiliser des dispositifs médicaux et obtenir leur prise en charge par l'assurance maladie. A l'heure actuelle, depuis la mise en place de la tarification à l'activité (T2A), deux modalités de financement cohabitent. La première, celle de droit commun, consiste en l'intégration des dispositifs médicaux dans les tarifs des groupes homogènes de séjour (GHS). Toutefois, comme l'a présenté à la mission M. François-Xavier Selleret, directeur général de l'offre de soins (DGOS) au ministère des affaires sociales et de la santé, certains dispositifs médicaux implantables sont remboursés en sus des GHS, pour des raisons médicales et de coût. D'après ses explications, « il faudrait que leur prix, pour une pathologie donnée, ne varie pas pour qu'ils puissent être inclus dans un séjour. Or, pour les deux grandes familles de dispositifs médicaux implantables, en cardiologie et en orthopédie, la dispersion des coûts est souvent supérieure à 30 % » .
Ces produits sont donc inscrits sur une « liste en sus », selon l'expression consacrée, et bénéficient d'un remboursement intégral de la part prise en charge par l'assurance maladie. Ils doivent obligatoirement figurer, au préalable, sur la LPP. Leur suivi est facilité et ils ont, dans certains cas, déjà fait l'objet d'une évaluation par la CNEDiMTS.
Ce n'est pas le cas des dispositifs médicaux intégrés aux GHS, qui ne sont soumis à aucune étude particulière et dont la connaissance même, tant au niveau de la consommation que du rapport, pour chacun, entre bénéfices et risques, est limitée.
Dans ce contexte, l'article 37 de la loi a limité la prise en charge des dispositifs médicaux, entrant dans des catégories homogènes prédéterminées, achetés et utilisés par les établissements de santé, à ceux inscrits sur une liste établie par les ministres de la santé et de la sécurité sociale, après avis de la CNEDiMTS. Pour y figurer, un dispositif médical devra soit avoir fait la preuve de son efficacité clinique, soit démontrer des spécifications techniques particulières, soit avoir montré son efficience au regard des alternatives thérapeutiques disponibles. La CNEDiMTS sera chargée du traitement des demandes d'inscription au regard de ces critères et pourra exiger que des études complémentaires soient réalisées. Les hôpitaux qui achèteraient des dispositifs médicaux appartenant à ces catégories homogènes mais ne figurant pas sur cette liste s'exposeraient à des sanctions, tout comme les fabricants qui refuseraient de réaliser les éventuelles études cliniques demandées.
La généralisation de règles jusque-là applicables aux seuls dispositifs médicaux innovants constitue donc la concrétisation d'une nouvelle orientation en matière de santé publique , grâce à la prise en compte de critères scientifiques et sanitaires pour déterminer quels produits seront ouverts au remboursement. En l'attente d'un décret d'application, ces dispositions restent encore virtuelles. Une mesure d'une telle ampleur, qui modifie en profondeur les pratiques dans les hôpitaux, ne saurait être mise en place en quelques semaines. La mission émet néanmoins le voeu qu'elle entre en vigueur rapidement car elle constitue assurément un pas dans la bonne direction en matière d'encadrement du marché des dispositifs médicaux.
3. Remettre la matériovigilance au coeur du système de sécurité sanitaire
Les défaillances du système de matériovigilance français ont contribué à ce que l'utilisation de dispositifs médicaux implantables non conformes aux règles de sécurité sanitaire et dangereux pour la santé des patients se poursuive alors que leurs effets néfastes avaient déjà été identifiés, soit isolément par certains médecins, soit à l'étranger. Pourtant, il appartient à tous les acteurs du secteur des dispositifs médicaux de faire remonter les informations dont ils pourraient disposer sur les incidents qu'un de ces produits aurait pu causer.
a) Faire de la matériovigilance une responsabilité mieux partagée, sous l'égide de l'ANSM
Le corps médical n'est pas le seul acteur de la matériovigilance. En effet, en application de l'article L. 5212-2 du code de la santé publique, « le fabricant, les utilisateurs d'un dispositif et les tiers ayant connaissance d'un incident ou d'un risque d'incident mettant en cause un dispositif ayant entraîné ou susceptible d'entraîner la mort ou la dégradation grave de l'état de santé d'un patient, d'un utilisateur ou d'un tiers doivent le signaler sans délai à l'ANSM » . La mise en place et le fonctionnement du système national de matériovigilance sont une des missions de cette agence, au sein de laquelle une commission nationale de sécurité sanitaire des dispositifs médicaux est plus spécialement chargée de ces questions.
C'est surtout au niveau local qu'il importe d'avoir des structures aptes à recueillir des signalements d'incidents. Chaque établissement de santé, ainsi que tout fabricant de dispositifs médicaux, est donc tenu de désigner un correspondant local de matériovigilance, chargé ensuite de déclarer à l'ANSM les incidents dont il aura eu connaissance. Quant aux tiers et, en particulier, aux médecins libéraux, ils sont tenus de faire leur signalement directement à l'ANSM.
Une fois l'ANSM informée d'incidents concernant un dispositif médical, elle peut suspendre sa commercialisation s'il présente un danger ou une suspicion de danger pour la santé humaine et enjoindre le fabricant de prendre les mesures que la situation impose, comme, le cas échéant, la destruction du produit et la mise en garde du public.
b) Rendre plus effective l'obligation de signalement des incidents liés à des dispositifs médicaux
Bien que les règles du code de la santé publique en matière de matériovigilance apparaissent claires, il ressort des auditions menées par la mission d'information que certains professionnels de santé ne satisfont pas correctement à leur obligation de signalement des incidents qu'ils constatent et que, lorsqu'ils le font, l'ANSM ne les tient pas informés des suites qu'elle y donne ni du délai dont elle a besoin pour traiter ces informations .
|
Les découvertes de fraudes sur la qualité d'un dispositif médical ou de problèmes de conception majeurs, susceptibles de porter gravement atteinte à la santé de ses utilisateurs, sont souvent facilitées par l'action de personnes, professionnels médicaux ou non, qui ont porté à la connaissance des pouvoirs publics des informations dont elles disposaient sur le sujet. Le rôle de ces lanceurs d'alerte est reconnu depuis de nombreuses années dans la plupart des pays occidentaux, notamment aux Etats-Unis où les « whistleblowers » bénéficient d'un statut juridique spécifique. Le droit français ne reconnaissait pas juridiquement les lanceurs d'alerte dans le domaine médical jusqu'à la loi « Médicament » du 29 décembre 2011. Désormais, quiconque a alerté de bonne foi son employeur, la justice ou les autorités administratives de faits, découverts dans l'exercice de ses fonctions, concernant la sécurité sanitaire des produits de santé est protégé contre d'éventuelles sanctions, en particulier pour le déroulement de sa carrière 82 ( * ) . De plus, en cas de litige, la charge de la preuve est présumée bénéficier au lanceur d'alerte et c'est à son employeur qu'il appartient, le cas échéant, d'apporter la preuve que les mesures qu'il a prises à l'encontre de son employé lanceur d'alerte sont sans lien avec son initiative.
Un cadre juridique solide est en place afin d'inciter ceux qui
disposent d'informations concernant la sécurité des dispositifs
médicaux à les rendre publiques. Il reste désormais
à voir de quelle façon ces règles seront appliquées
par la justice.
|
Comme l'a reconnu Jean-Yves Grall, directeur général de la santé (DGS) au ministère des affaires sociales et de la santé, lors de son audition par la mission d'information, dans l'affaire PIP « la détection n'a pas été optimale » . Relativement au nombre de porteuses de prothèses, il y a eu très peu de déclarations d'effets indésirables avant l'inspection de l'usine PIP réalisée par l'Afssaps en mars 2010.
Il convient donc de simplifier la procédure de signalement , qui consiste encore à l'heure actuelle en l'envoi par fax à l'ANSM d'un formulaire Cerfa. Il est précisé sur celui-ci que si l'émetteur n'a pas reçu d'accusé de réception dans un délai de dix jours, il doit confirmer son envoi par lettre recommandée avec accusé de réception ! La mise en place d'un portail unique par internet , permettant une télétransmission rapide et sécurisée, serait de nature à répondre à une partie des doléances des professionnels de santé devant le caractère fastidieux de la procédure actuelle. Ceux-ci, ainsi que les patients, devraient bien évidemment être associés à sa conception.
Proposition n° 12 :
Mettre en place un
groupe de travail chargé de simplifier la procédure
de
signalement des incidents de matériovigilance
De plus, en obligeant l'ANSM à prendre contact systématiquement avec le déclarant et à le tenir informé des suites données à son signalement , celui-ci se sentirait plus impliqué, pourrait faire part de manière plus détaillée de ses observations et verrait sa démarche mieux prise en considération.
Proposition n° 13 :
Mieux associer les
déclarants aux suites données aux déclarations
de
matériovigilance
Il pourrait également être envisagé d'utiliser comme relais , ce qui est déjà parfois le cas de manière non formalisée, l'Ordre des médecins , que ce soit ses instances nationales ou départementales. Un effort de pédagogie de la part du Cnom et des pouvoirs publics en direction des médecins, par exemple à travers une campagne nationale de communication commune centrée sur la matériovigilance , ne serait sans doute pas superflu afin d'identifier plus rapidement les dispositifs médicaux qui, à l'usage, présentent un risque particulier pour la santé. Avec un nombre de signalements accru et une plus grande implication des professionnels de santé, il ne fait guère de doute que les cas de fraude avérée, comme celui des prothèses PIP, ne pourraient prospérer dans les mêmes conditions que par le passé et que le nombre de victimes serait bien moins important.
Proposition n° 14 :
Mener une campagne
d'information commune, avec le Cnom,
sur les enjeux de la
matériovigilance
L'ANSM doit donc incontestablement faire des efforts en matière de matériovigilance, aussi bien dans ses procédures internes de traitement des signalements, notamment en matière de délai, que dans la réactivité de son action une fois une situation anormale découverte. Dans le cadre de la signature prochaine, entre l'Etat et cette agence, d'un contrat d'objectifs et de performance visant à remplacer celui qui portait sur la période 2007-2010, il serait tout à fait opportun que le renforcement de la matériovigilance et l'amélioration de son efficacité y tiennent une place centrale et soient consacrés comme des priorités de l'action de l'ANSM . La mise en place d'indicateurs objectifs permettrait, à l'échéance prévue de ce document contractuel, que son action en la matière soit évaluée. De toute évidence, il est dans l'intérêt général que la France dispose enfin d'un système de matériovigilance d'excellence, où tous les acteurs coopèrent efficacement et s'acquittent pleinement de leurs responsabilités.
Proposition n° 15 :
Faire du renforcement
de la matériovigilance l'un des points centraux
du prochain contrat
d'objectifs et de performance Etat-ANSM
4. Améliorer l'identification des dispositifs médicaux implantables et le partage des données
Dans un marché européen où règnent la libre circulation et un marché mondial qui s'ouvre progressivement, avec un rapprochement général des législations applicables en matière de commercialisation des dispositifs médicaux, il n'est plus possible de penser à l'échelle nationale les prochaines étapes de l'évolution de la réglementation qui leur est applicable, en particulier en matière d'identification et de suivi.
Confrontés à cette réalité, les Etats ont mis en place des instances de concertation pour mettre au point des normes communes, au premier rang desquelles l'identifiant unique pour les dispositifs médicaux (UDI ou Unique Device Identifier ). A l'échelle européenne, la base de données Eudamed permet aux autorités nationales d'échanger facilement des informations sur les dispositifs médicaux, en particulier sur les incidents survenus dans chaque pays et la surveillance du marché.
Il est désormais nécessaire d'agir pour que les travaux des organismes en question soient pleinement soutenus par l'Union européenne et par la France au sein de celle-ci. L'accès aux outils développés, en Europe, au profit des régulateurs doit être étendu aux organismes notifiés et, à terme, pour des raisons de transparence, à tous les citoyens.
a) Soutenir les initiatives du Forum mondial des régulateurs de dispositifs médicaux (IMDRF)
L' International Medical Device Regulators' Forum (IMDRF) a pris en 2012 la suite de la Global Harmonization Task Force (GHTF), qui regroupait depuis 1992 les représentants d'organismes nationaux de réglementation des dispositifs médicaux et de l'industrie du secteur. Associant désormais l'UE, les Etats-Unis, le Japon, le Canada, le Brésil, l'Australie, la Chine et l'Organisation mondiale de la santé (OMS), l'IMDRF poursuit les activités de la GHTF et cherche à encourager la convergence entre ses membres des pratiques en matière de régulation afin d'assurer l'innocuité, l'efficacité, le rendement et la qualité des dispositifs médicaux et de promouvoir l'innovation et les échanges internationaux.
La principale activité de la GHTF était la publication et la diffusion de documents d'orientation harmonisés sur les pratiques de réglementation de base. Elaborés par ses cinq groupes d'études, ils pouvaient ensuite être mis en oeuvre dans chacun des pays membres. Son action connaissait néanmoins plusieurs limites, au premier rang desquels la lenteur des membres fondateurs à adapter leur législation aux guides élaborés de manière consensuelle et le peu d'écho rencontré par ses travaux.
La création de l'IMDRF vise à corriger ces insuffisances. Cet organisme a d'ores et déjà créé cinq groupes de travail visant notamment à accompagner la mise en oeuvre de l'identifiant unique et à créer une liste de standards internationaux, reconnus universellement, en matière de régulation des dispositifs médicaux.
Cette nouvelle instance n'en est encore qu'à ses premiers mois d'existence mais dispose d'un potentiel important en matière d'harmonisation des réglementations dans le monde, ce qui ne peut qu'être source d'amélioration de la sécurité des dispositifs médicaux et du partage d'informations relatives à la matériovigilance. Il convient donc de soutenir ses travaux et de mettre en oeuvre ses recommandations, à condition que la volonté de réciprocité de nos partenaires soit avérée et qu'elles n'aboutissent pas à un affaiblissement des normes en vigueur.
b) Accélérer la mise en oeuvre de l'identifiant unique pour les dispositifs médicaux (UDI)
L'UDI est un procédé d'identification visant à faire correspondre un numéro unique à un ensemble d'informations industrielles et médicales relatives à chaque dispositif médical. Ce numéro unique permet d'identifier la classe de dispositif médical à laquelle celui-ci appartient et de recenser les différentes informations de suivi clinique le concernant au sein d'une base de données.
Il présente de nombreux avantages pour la sécurité des patients en garantissant un partage continu des données entre les parties concernées tout au long du cycle de vie des dispositifs médicaux. Ainsi :
- le renforcement de la traçabilité des produits rend la matériovigilance et la surveillance du marché plus efficaces, facilitant en particulier le retrait rapide du marché des dispositifs défectueux et la transmission des informations relatives aux dysfonctionnements, incidents et effets indésirables observés ;
- la réunion des données nécessaires à la constitution de registres médicaux concernant des dispositifs médicaux est facilitée ;
- la collecte de données issues d'observations cliniques permet de mesurer effectivement le rapport bénéfices-risques, afin de servir de support à la certification et à l'autorisation de mise sur le marché de nouveaux produits ;
- il contribue à la prévention de la contrefaçon.
Le Food and Drug Administration Amendments Act du 27 septembre 2007 visait à surmonter les difficultés liées à la multiplicité de systèmes d'identification développés par les fabricants, les distributeurs et les hôpitaux qui ralentissent considérablement la recherche des dispositifs médicaux implantés lorsqu'un produit ou un lot défectueux fait l'objet d'un rappel.
|
Le document ci-dessous est la reproduction d'une carte de porteur d'endoprothèse. Trois stents ont été posés, dont deux actifs identifiés à la main. Chaque fabricant indique les références et le numéro de lot, mais avec son propre système.
|

Le 2 juillet 2012, la FDA a proposé des modifications pratiques de mise en oeuvre de ce système d'identification qui devrait concerner, dans un premier temps, les dispositifs médicaux les plus à risque, de classe III, avant d'être étendu graduellement à l'exception des dispositifs médicaux en vente libre pour lesquels il existe déjà un code d'identification aux Etats-Unis. Il facilitera grandement la collecte d'informations très précises pour le suivi des implants dans des registres. Ce mécanisme permettra d'observer l'évolution dans le temps de leur fonctionnement selon leur marque et leur modèle. Les études cliniques post market pourront se généraliser et le repérage d'anomalies statistiques dans les incidents liés à un dispositif médical spécifique deviendra plus aisé.
L'affectation à un dispositif médical de son code UDI relève de la responsabilité du fabricant : ce dernier doit créer un code UDI en fonction des standards internationaux applicables aux dispositifs médicaux. En effet, à la différence des médicaments pour lesquels chaque pays dispose de son propre système d'identification, les dispositifs médicaux ont vocation à se voir attribuer un identifiant fonctionnel dans tous les pays se soumettant aux standards internationaux élaborés dans le cadre de la GHTF. Le numéro UDI doit recouvrir deux procédés d'identification :
- l' identifiant produit ( partie statique ) : sur le même principe que l'EAN ( « International Code Number » , originellement « European Code Number » ), code-barres à treize chiffres permettant l'identification d'un produit de consommation tout au long de sa chaîne de distribution, cet identifiant devrait intégrer le « Global Trade Item Number » (GTIN), code international permettant d'identifier toute unité commerciale ;
- l' identifiant de production ( partie dynamique ) : il comprend des éléments issus de la production tels que le numéro du lot ou sa date d'expiration. La réglementation n'exigera la sérialisation des dispositifs médicaux, c'est-à-dire l'apposition d'un numéro de série, que pour certains groupes de dispositifs, notamment les dispositifs médicaux implantables.

Source : Actes des journées Euro-Pharmat Lyon (11, 12 et 13 octobre 2011).
Afin de faire correspondre au numéro UDI l'ensemble des informations relatives au dispositif en question, la FDA a prévu la création d'une base de données associée : la « Global Unique Device Identification Database » (Gudid). Cette base de données a vocation à collecter uniquement l'information dite « statique » concernant le dispositif et susceptible de renseigner les questions suivantes : de quel dispositif s'agit-il ? Quel est son modèle de fabrication ? De quelle manière le produit a-t-il été contrôlé ? Est-il possible de trouver un numéro de série ou de lot sur ce produit ? A qui doit-on s'adresser en cas d'incident ? Dans quelles conditions le produit doit-il être conservé ? Le produit est-il stérile ? Il est envisagé que cette base soit accessible à la communauté médicale comme au public.
La Commission européenne a annoncé réfléchir à une recommandation relative aux principes généraux de mise en oeuvre de l'UDI. Ce document devrait être finalisé d'ici la fin de l'année 2012. Il est indispensable que l'Union européenne, qui coordonne les travaux à ce sujet au sein de l'IMDRF, ne prenne pas de retard important sur les Etats-Unis. Tout décalage dans la mise en oeuvre de ce système se ferait alors au détriment de la santé et de la compétitivité en Europe.
Proposition n° 16 :
Accélérer la mise en oeuvre de l'UDI au sein de l'Union
européenne
c) Elargir l'accès à Eudamed
Formellement instituée par une décision de la Commission européenne du 19 avril 2010 83 ( * ) , Eudamed est un outil qui reste réservé aux autorités sanitaires nationales . Elle a été conçue afin d'améliorer le fonctionnement et la surveillance du marché des dispositifs médicaux grâce à un meilleur partage de l'information, notamment sur les certifications refusées ou retirées. L'utilisation d'Eudamed est obligatoire pour tous les Etats membres de l'UE depuis le 1 er mai 2011.
Toutefois, ainsi que l'ont expliqué plusieurs experts auditionnés par la mission d'information, l'accès à Eudamed devrait être étendu afin de tirer pleinement parti de son potentiel et d'assurer que ses données soient à la disposition de tous ceux qui peuvent, par leur activité, en avoir besoin. C'est notamment le cas des organismes notifiés. Ainsi Eric Vicaut, président du groupe de travail « dispositifs médicaux » des Assises du médicament, a déploré qu'Eudamed reste très fermée et a préconisé une plus grande ouverture pour une meilleure information au niveau européen. De même Jean-Yves Grall, directeur général de la santé, a jugé utile, au nom de la transparence, qu'Eudamed soit plus ouverte.
Cette question semble donc faire l'objet d'un consensus. A l'occasion de la refonte de la réglementation européenne sur les dispositifs médicaux, il est important que la Commission propose qu'Eudamed soit, progressivement si une telle mesure comporte des difficultés techniques, ouverte aux organismes notifiés et, à terme, au public. En effet, il serait très utile aux entités qui ont pour mission de garantir la sécurité et la qualité des dispositifs médicaux d'avoir accès aux données de matériovigilance détenues par les autorités nationales.
Proposition n° 17 :
Elargir
l'accès à Eudamed aux organismes notifiés et, à
terme,
au moins partiellement au public
En l'état actuel du droit, les organismes notifiés ne sont tenus informés que des certificats suspendus, retirés ou refusés. N'ayant connaissance des incidents survenus lors de l'utilisation d'un dispositif médical que lorsqu'un fabricant sollicite le renouvellement de son marquage CE, ils ne sont pas en mesure de jouer pleinement le rôle que leur confie la réglementation européenne au sein de la chaîne de sécurité sanitaire de ces produits de santé.
Le même souci de transparence, en vertu duquel il faut imposer la publicité de tous les liens d'intérêts des professionnels de santé, doit conduire à ouvrir Eudamed, ou tout du moins certaines des informations qu'elle contient, au plus grand nombre. Il n'y a rien à gagner à entretenir le secret sur la fiabilité des dispositifs médicaux et à ne communiquer que de manière irrégulière sur le sujet. Aux Etats-Unis, la délivrance de l'autorisation de commercialisation - quelle que soit la procédure suivie - fait l'objet d'une publicité sur le site de la FDA. Plus l'information sera disponible, plus les citoyens pourront avoir confiance dans l'action des structures chargées d'assurer le respect des règles de santé publique relatives aux dispositifs médicaux.
5. Assurer la pleine traçabilité des dispositifs médicaux implantés grâce à des registres exhaustifs
Comme les exemples australiens, suédois ou américains en sont la preuve, la création de registres des dispositifs médicaux implantés permet d'assurer efficacement leur surveillance et de détecter les éventuels dysfonctionnements avant qu'ils ne se transforment en véritables scandales de santé publique.
Dans ce domaine, la France accuse un retard flagrant, notamment dans le domaine orthopédique. Les seules initiatives privées existantes, comme celle de la Société française de chirurgie orthopédique et traumatologique (Sofcot) pour les prothèses totales de hanche, reposent sur le volontariat des praticiens, connaissent des difficultés de financement et ne parviennent pas à atteindre le niveau de développement de leurs modèles anglo-saxons ou scandinaves.
Comme l'avait souligné le professeur Daniel Loisance lors de son audition par la mission d'information, « un registre qui n'est pas complet n'a strictement aucune valeur : il est même trompeur » . C'est la raison pour laquelle il convient de s'inspirer des expériences étrangères afin de développer efficacement cet outil en France et, à long terme, à l'échelle européenne. S'ils ne constituent pas, à eux seuls, la panacée en matière de matériovigilance et de suivi clinique des patients, les registres bien conçus et correctement alimentés par les professionnels de santé n'en restent pas moins l'un des outils les plus utiles pour la réalisation de ces tâches.
a) Respecter des principes de mise en oeuvre précis
Les détails des quelques registres orthopédiques décrits ci-dessous permettent d'identifier les conditions générales de leur réussite :
- maîtrise par la communauté médicale, le registre ne devant pas constituer un moyen de contrôler les pratiques ;
- décentralisation, chaque registre spécialisé étant administré par une équipe différente ;
- financement stable, le plus souvent assuré par l'Etat ;
- participation la plus exhaustive possible, pas toujours obligatoire mais exigeant le consentement éclairé du patient ;
- identification du patient avec des données personnelles permettant de le retrouver rapidement.
Les principes de fonctionnement des principaux registres orthopédiques étrangers
|
Administration |
Financement (1) |
Participation obligatoire (2) |
Numéro de sécurité sociale |
Consentement du patient |
|
|
Angleterre et Pays de Galles |
G |
R |
+ (3) |
+ |
+ |
|
Australie |
A |
G |
- |
+ |
- |
|
Canada |
G |
G |
- |
+ |
+ |
|
Danemark |
|||||
|
hanche |
A |
G |
+ |
+ |
- |
|
genou |
A |
G |
+ |
+ |
+ |
|
Finlande |
G |
G |
+ |
+ |
- |
|
Nouvelle Zélande |
A |
G, A |
- |
+ |
+ |
|
Norvège |
A |
G |
- |
+ |
+ |
|
Roumanie |
A |
G |
- |
+ |
+ |
|
Slovaquie |
A |
G |
+ |
+ |
- |
|
Suède |
|||||
|
genou |
A |
G |
- |
+ |
- |
|
hanche |
A |
G |
- |
+ |
- |
|
coude |
A |
A |
- |
+ |
- |
|
épaule |
A |
G, A |
- |
+ |
- |
|
Suisse |
A |
A |
- |
- |
- |
|
(1) A, associations; G, gouvernement, R : redevance sur les implants |
|||||
|
(2) + : oui ; - : non |
|||||
|
(3) dépend du statut de l'hôpital |
|||||
|
Source : mission d'information du Sénat sur les dispositifs médicaux implantables et les interventions à visée esthétique |
|||||
b) Lever l'obstacle juridique de la protection des données personnelles
Tout registre doit également respecter la législation relative à l'utilisation et à la conservation de données personnelles, en particulier la loi « Informatique et libertés » du 6 janvier 1978 84 ( * ) . Il doit donc être déclaré auprès de la Commission nationale de l'informatique et des libertés (Cnil) et se soumettre aux éventuelles modifications que celle-ci imposerait.
|
En application du chapitre IX de la loi du 6 janvier 1978, la mise en place de « traitements de données à caractère personnel ayant pour fin la recherche dans le domaine de la santé » est soumise à une procédure en plusieurs étapes qui vise à s'assurer de leur sérieux scientifique et de l'adéquation des mesures de protection des données envisagées. Partant du principe que chacun doit pouvoir faire valoir son droit d'opposition à l'utilisation de ses données personnelles, tout projet doit d'abord être soumis pour avis au Comité consultatif sur le traitement de l'information en matière de recherche dans le domaine de la santé (CCTIRS), placé auprès du ministère chargé de la recherche et qui étudie la méthodologie scientifique utilisée. Dès lors que celui-ci s'est prononcé favorablement, la Cnil, bien que n'étant pas juridiquement liée par la position du comité, autorise la mise en oeuvre du traitement de données.
L'avis favorable d'une troisième instance, le
Comité national des registres (CNR), est nécessaire pour obtenir
l'appellation officielle de « registre qualifié ».
Selon l'arrêté du 6 novembre 1995 relatif à ce
comité, un registre est défini comme «
un recueil
continu et exhaustif de données nominatives intéressant un ou
plusieurs événements de santé dans une population
géographiquement définie, à des fins de recherche et de
santé publique, par une équipe ayant les compétences
appropriées
». C'est donc sur la base de ces
critères cumulatifs que le CNR, qui s'appuie sur les services de
l'Institut national de la santé et de la recherche médicale
(Inserm) et de l'Institut de veille sanitaire (InVS) prend ses
décisions. Leur validité est de trois ans pour les registres en
création et de quatre ans pour ceux en fonctionnement.
|
Selon les données communiquées par la Cnil à la mission d'information, il y aurait actuellement en France 135 registres médicaux autorisés, dont 10 portant sur des dispositifs médicaux implantables.
L'articulation de ce dispositif ne favorise pas la création de véritables registres de dispositifs médicaux. Les projets soumis à l'avis du CCTIRS portent parfois le titre d'« observatoire » ou de « base de données ».
Ils ne peuvent se voir attribuer le titre de registre que lorsqu'ils sont qualifiés par le CNR.
La rédaction de l'article 2 de l'arrêté du 6 novembre 1995 relatif au comité national des registres s'oppose à la constitution de registres pour l'enregistrement de poses de dispositifs médicaux implantables et le suivi des patients implantés.
Sous réserve qu'ils contiennent différentes données attendues d'une recherche menée dans le domaine de la santé (exhaustivité, continuité...), les projets soumis à l'avis du CCTIRS peuvent être autorisés lorsqu'ils portent le titre d'« observatoire » ou de « base de données » . Par exemple, le 5 juillet 2012, le CCTIRS devait examiner une demande portant sur une « étude observationnelle prospective multicentrique évaluant la tolérance à long terme » d'un implant sur mesure « au regard du taux d'explantation et du nombre d'infections ».
En revanche, les projets tenus par des sociétés savantes, en collaboration avec des industriels, ne peuvent être qualifiés de registres, surtout si l'on considère qu'ils ne correspondent pas à la définition d'un registre de morbidité dans une zone géographique définie, département ou région. Le « registre des hémopathies malignes de Côte d'Or » correspondrait bien à un registre, contrairement à l'observatoire cité ou à un autre projet soumis à l'avis du CCTIRS le même jour, qui consistait en une « étude observationnelle multicentrique sur l'utilisation » d'une prothèse orthopédique.
Si elle ne portait sur une préoccupation de santé publique, cette controverse pourrait sembler purement sémantique.
Selon beaucoup d'observations, faute de disponibilité, les médecins participent de moins en moins à la mission de recueil de données. Mais la Cnil nous a précisé que leur implication serait plus forte si le recueil portait le nom de registre. Le législateur a légitimement voulu protéger les citoyens en restreignant l'utilisation de données personnelles y compris dans le domaine de la santé. Mais au nom de cet impératif, l'utilisation du terme registre pourrait être étendue aux études observationnelles ou bases de données relatives au suivi de dispositifs médicaux implantés.
Il reste toutefois un obstacle majeur à la création de registres de suivi des patients efficaces : les conditions très restrictives d'utilisation du numéro d'inscription au répertoire national d'identification des personnes physiques, le Nir, qui figure sur la carte Vitale de chaque assuré . Le Nir, souvent appelé numéro Insee, fournit plusieurs données de nature personnelle : sexe, année, mois, département et commune de naissance. A ce titre, il diffère très sensiblement des systèmes étrangers. Selon l'article 27 de la loi de 1978, un traitement de données utilisant le Nir doit être autorisé par décret en Conseil d'Etat.
La solution passe par le développement de l'identifiant national de santé (INS) prévu à l'article L. 1111-8-1 du code de la santé publique. Il n'est pas signifiant, contrairement au Nir, c'est-à-dire que sa connaissance ne permet pas de déduire des informations de nature personnelle sur son titulaire. Cinq ans après sa création, son décret d'application n'est toujours pas paru bien qu'il soit utilisé dans le cadre du déploiement du dossier médical personnel (DMP) et du dossier pharmaceutique. Au vu des difficultés et retards que connaissent ces deux outils, sa généralisation est donc peu probable à court terme.
Une fois ces considérations présentées, il convient d'agir. Lors de son audition par la mission, Dominique Maraninchi, directeur général de l'ANSM, avait résumé la situation en ces termes : « Il y a des avantages et des inconvénients à avoir des registres. Il n'en reste pas moins que nous sommes pénalisés parce que nous n'en avons pas du tout. La France doit se lancer dans ce domaine » .
c) Créer en priorité des registres de dispositifs médicaux à risque, en coopération avec les parties intéressées
Pour toutes ces raisons, la France connaît un retard certain en matière de registres. Fixer comme objectif de constituer d'emblée un registre général des dispositifs médicaux implantables n'aurait guère de sens, d'autant que chaque spécialité médicale présente ses spécificités. Il convient donc d'accorder la priorité aux dispositifs médicaux implantables innovants ou à risque, préalablement identifiés par les autorités sanitaires et les professionnels de santé et nécessitant un suivi très fin. Les deux spécialités principalement concernées sont l'orthopédie et la cardiologie.
Il est tout d'abord nécessaire d'associer au développement d'un registre les sociétés savantes et les médecins intéressés , ou tout du moins de s'assurer de leur collaboration. Ensuite, plusieurs options s'offrent aux pouvoirs publics pour inciter praticiens et hôpitaux à alimenter les registres . Le caractère jusqu'à présent purement volontaire des registres a malheureusement montré ses limites. La seule bonne volonté de leurs promoteurs se montre toujours insuffisante pour assurer leur exhaustivité. C'est pourquoi il faut envisager d'intégrer une éventuelle obligation de renseigner un registre dans les règles de fonctionnement plus générales de notre système de santé .
Proposition n° 18 :
Rendre obligatoire,
sous certaines conditions, le renseignement
des registres par les
médecins
Ainsi, il serait envisageable de prévoir, dans le cadre de la procédure de certification obligatoire des établissements de santé par la HAS, une clause concernant la participation à un registre, pour un ou plusieurs types de dispositifs médicaux implantables utilisés en son sein. Le même type de raisonnement pourrait être adapté au niveau local afin de faire des registres un objet de contractualisation entre les hôpitaux et les agences régionales de santé (ARS). Dans les deux cas, si l'hôpital en question ne tenait pas ses engagements, il perdrait son agrément pour poser l'implant en question.
L'idée, plusieurs fois évoquée devant la mission et adoptée par d'autres pays, comme les Etats-Unis avec Intermacs, de conditionner la prise en charge du dispositif médical implantable et de l'acte chirurgical qui y est associé au renseignement d'un registre, ne semble pas directement transposable à l'heure actuelle à la France. Le codage des procédures et des produits de santé dans le programme de médicalisation des systèmes d'information (PMSI) n'est pas suffisamment précis pour permettre d'identifier précisément les dispositifs médicaux utilisés à l'hôpital, en particulier ceux qui sont intégrés dans les GHS ou qui sont inscrits en ligne générique dans la liste en sus. Il s'agit néanmoins d'une idée séduisante dont il faudrait étudier plus en détail les modalités d'application pour envisager son adoption à moyen terme.
d) Parvenir enfin à valoriser les informations des systèmes d'information hospitaliers
Malgré ces difficultés, les données du PMSI sont une source très riche d'informations dont l'utilité en matière de suivi épidémiologique et sanitaire a été reconnue ces dernières années. Ainsi, le Gip « Etudes en santé publique » créé par l'article 33 de la loi du 29 décembre 2011 a pour but de faciliter leur exploitation.
Toutefois, pour l'instant, comme l'a précisé lors de son audition Thomas Fatome, directeur de la sécurité sociale au ministère des affaires sociales et de la santé, « aucune donnée dans le système d'information de l'assurance maladie n'existe pour les dispositifs médicaux implantables intégrés au tarif des GHS » . La mise au point d'un codage spécifique pour mieux appréhender leur utilisation est envisageable et techniquement possible, sans toutefois régler le problème des lignes génériques, qui ne permettent pas d'identifier un produit spécifique.
Néanmoins, l'enrichissement des données du PMSI et l'adoption d'un codage plus précis semblent être la solution à privilégier pour pouvoir, sur les bases existantes, construire un véritable outil de suivi de l'utilisation des dispositifs médicaux implantables . Cela permettra d'automatiser la remontée de l'information sans modifier en profondeur les pratiques des équipes médicales ni alourdir leur charge de travail. Un travail important de modification des systèmes d'information des hôpitaux sera cependant nécessaire . C'est pourquoi il est important d'engager dès aujourd'hui les travaux préparatoires qui poseront les bases de ce chantier et de déterminer, dans le cadre d'une concertation avec tous les professionnels, la nature précise des données qui devront être récoltées. La mise en oeuvre, dans le même temps, de l'UDI devrait simplifier le suivi des dispositifs médicaux en faisant figurer celui-ci dans le PMSI.
Proposition n° 19 :
Mettre en place un
codage plus fin dans le PMSI
afin de mesurer précisément
l'usage des DMI à l'hôpital
6. Revoir les modalités de l'évaluation et de la surveillance clinique des dispositifs médicaux implantables avant et après leur commercialisation
Alors que le marquage CE constitue avant tout une garantie de conformité des dispositifs médicaux avec des règles de sécurité et de fabrication, tous les acteurs du secteur s'accordent sur la nécessité de renforcer l'évaluation de ces produits de santé par la systématisation des études réalisées après leur mise en vente.
Cette préoccupation n'est pas nouvelle. Comme on l'a vu précédemment, la directive 2007/47/CE du 5 septembre 2007 a complété la directive de 1993 en rendant obligatoires les investigations cliniques pour les dispositifs médicaux implantables ainsi que pour ceux de classe III, « sauf si le recours aux données cliniques existantes peut être dûment justifié » (annexe X de la directive 93/42, §1.1 bis ; article R. 5211-36-2 du code de la santé publique). Malheureusement, le droit en vigueur est souvent méconnu et mal appliqué.
a) Corriger les ambiguïtés de la réglementation européenne
Jean-Luc Harousseau l'a déploré lors de son audition : « les essais cliniques, que la directive 2007/47/CE encourage pour les dispositifs de classe III, se mettent en place lentement, les produits étant souvent fabriqués par des toutes petites entreprises pour des publics très ciblés. [...] Il faudrait inciter, plus que ne le fait la directive, les industriels à lancer des études » . En l'absence de lignes directrices claires, leurs termes sont négociés entre le fabricant et l'organisme notifié qui examine son dossier. Les autorités nationales de régulation du secteur, comme l'ANSM en France, n'ont pas directement accès à ces données et ne peuvent pas s'assurer que les études sont réalisées selon un protocole scientifiquement irréprochable. Elles ne connaissent pas non plus le plan de surveillance établi par le fabricant afin d'obtenir sa certification CE.
Dans ces circonstances, il est regrettable de constater que les dispositions de l'annexe X de la directive 93/42, qui définit les modalités de l'évaluation clinique obligatoire pour les dispositifs médicaux implantables, soient de manière quasi systématique contournées par les fabricants et interprétées de telle sorte qu'ils soient exonérés de leurs obligations. En effet, ce texte prévoit que les investigations cliniques doivent être réalisées « sauf si le recours aux données cliniques existantes peut être dûment justifié » (§ 1.1 bis ). Malheureusement, aucun contrôle n'est effectué sur le recours à cette alternative qui aurait dû, dans l'esprit du texte, rester l'exception . La mauvaise application de cette réglementation lui ôte toute sa force. De plus, l'évaluation clinique a un caractère souvent biaisé lorsqu'elle est réalisée par des experts rémunérés par les industriels.
Il est donc indispensable que, dans le cadre de la refonte de la réglementation européenne qui s'annonce, les obligations en matière d'évaluation clinique soient précisées et rendues plus contraignantes. D'ores et déjà, les organismes notifiés pourraient se montrer plus exigeants au niveau de l'évaluation des données de conception des dispositifs médicaux implantables . La démarche d'évaluation de la performance clinique d'un dispositif médical par son fabricant devrait être soumise au régulateur. Afin de s'assurer de la qualité de l'expertise scientifique, il serait même souhaitable de mettre en place une double validation, par l'organisme notifié puis par l'autorité nationale compétente, des données cliniques avant la délivrance du marquage CE .
La procédure d'évaluation clinique par équivalence, basée sur la littérature scientifique existante, devrait être mieux encadrée afin qu'elle cesse d'être celle choisie dans près de 90 % des cas . L'organisme notifié et le régulateur devraient pouvoir demander plus de détails au fabricant sur la méthodologie utilisée et vérifier qu'elle est applicable à chaque cas d'espèce.
Surtout, il convient de mieux encadrer la notion même d'équivalence, afin d'éviter un « effet domino » qui permet aujourd'hui à des fabricants, par une suite d'équivalences successives, de considérer que la littérature scientifique relative à un dispositif médical existant est pertinente pour évaluer le comportement d'un nouveau produit sensiblement différent. Il faudrait donc définir clairement les critères permettant de l'apprécier et de la restreindre à une équivalence en lien direct entre deux dispositifs médicaux précis. Afin de garantir incontestablement le sérieux scientifique de cette voie d'évaluation et renforcer son bien-fondé, elle devrait être limitée aux cas où le dispositif médical auquel le fabricant prétend que son produit est équivalent a lui-même fait l'objet d'investigations cliniques avant sa commercialisation .
Proposition n° 20 :
Limiter
« l'effet domino » en définissant plus
précisément l'équivalence
pouvant être
acceptée entre deux dispositifs médicaux
pour satisfaire
à l'obligation d'évaluation clinique
Proposition n° 21 :
Imposer que le
dispositif médical auquel un nouveau produit est présenté
comme équivalent ait lui-même fait l'objet d'investigations
cliniques
Les mesures proposées par la mission d'information visent à mettre un terme à des pratiques et des comportements, de la part des fabricants, des organismes notifiés et des autorités nationales, qui ont eu pour conséquence de rendre quasi inopérantes, dans les faits, les dispositions de la réglementation européenne relatives à l'évaluation clinique des dispositifs médicaux implantables.
Profitant des imprécisions du texte et du peu d'empressement des certificateurs et des régulateurs à s'assurer de sa bonne application, les producteurs ont réussi jusqu'à présent à s'affranchir de leurs obligations en ayant massivement recours à la voie la moins contraignante qui, si elle peut être justifiée dans certaines situations spécifiques, devrait néanmoins rester exceptionnelle. C'est pourquoi, pour des raisons qui tiennent à la surveillance du marché, à la protection de la santé publique et au développement de l'expertise scientifique dans le domaine des dispositifs médicaux implantables, il est nécessaire de renforcer les contrôles et de clarifier les textes afin d'affirmer sans aucune ambiguïté que des essais cliniques scientifiquement crédibles et impartiaux doivent constituer une obligation préalable à la commercialisation des dispositifs médicaux implantables.
b) Assurer une meilleure évaluation post-inscription des dispositifs médicaux implantables remboursés en France
Pour l'instant, la principale forme d'évaluation conduite dans notre pays est celle faite par la CNEDiMTS, instance de la HAS, lorsque l'inscription d'un dispositif médical sur la LPP en vue d'obtenir sa prise en charge par l'assurance maladie est demandée. Elle mesure le rapport bénéfices-risques du dispositif en question par comparaison avec les stratégies thérapeutiques existantes, se fondant sur son service médical attendu (SMA). Toutefois, lorsque le dispositif médical doit être enregistré sous son nom de marque, en raison de son caractère innovant ou très spécifique, une étude clinique complète peut être menée à la demande de la CNEDiMTS et conditionner son remboursement.
Le récent accord-cadre entre le Comité économique des produits de santé (Ceps) et les organisations professionnelles des fabricants de dispositifs médicaux, signé le 16 décembre 2011, vient faciliter la réalisation de telles études. Comme l'a expliqué Jean-Luc Harousseau, président de la HAS, à la mission d'information, cet accord prévoit que, pour un dispositif médical, « quand un manque clinique sera constaté, une étude devra être mise en place dès son inscription. La CNEDiMTS devra ensuite réévaluer le produit » . Plus précisément, ses articles 10 à 15 donnent à la CNEDiMTS et au Ceps le droit de demander à un fabricant de mener une évaluation clinique complémentaire, sous l'égide d'un comité scientifique, dont les résultats doivent faire l'objet d'une publication. La même procédure est applicable lors du renouvellement de l'inscription d'un produit sur la LPP. Enfin, des études peuvent également être conduites sur des produits qui y sont inscrits sous description générique, sachant que la CNEDiMTS réévalue la cohérence et la pertinence thérapeutiques des lignes génériques tous les trois à cinq ans.
Entendu par la mission d'information, Jean-Michel Dubernard, président de la CNEDiMTS, a souligné la grande importance de cet accord-cadre, qui marque une nouvelle étape dans les relations entre les fabricants et les autorités chargées de déterminer si un dispositif médical est admissible au remboursement et quel doit être son prix. Ainsi, selon son article 19, « le choix entre l'inscription en nom de marque et l'inscription sous description générique relève exclusivement des ministres chargés de la santé et de la sécurité sociale sur proposition du Ceps après avis de la CNEDiMTS » . L'affirmation de ce principe général donne un pouvoir accru à ces deux organismes pour mettre au point une stratégie réfléchie d'évaluation des dispositifs médicaux implantables, qu'ils soient uniques et innovants ou fondus dans une ligne générique.
Il est évident qu'il ne sera pas possible de procéder, à brève échéance, à l'évaluation post-inscription de tous les dispositifs médicaux implantables présents sur le marché. Néanmoins, les dispositions de cet accord-cadre ainsi que celles de la loi « Médicament » 85 ( * ) sont des compléments bienvenus aux pouvoirs des autorités sanitaires afin qu'elles mettent en oeuvre une politique volontariste, sur le marché français, de suivi clinique des dispositifs médicaux, dans l'attente d'une modification de la réglementation européenne qui reste, sur ce sujet, trop imprécise.
c) Continuer à favoriser l'innovation
Les dispositifs médicaux évoluant au gré des changements technologiques et des avancées scientifiques, il est indispensable de soutenir l'innovation et de mettre en place des procédures spécifiques de financement de la recherche médicale et de prise en charge temporaire des produits les plus innovants. C'est la raison pour laquelle plusieurs programmes d'aide, intervenant aux différentes étapes de développement d'un dispositif médical, ont été mis en place par le législateur et le pouvoir réglementaire, sous l'égide du ministère de la santé.
Le programme de soutien aux techniques innovantes, coûteuses ou non (Pstic) , permet de tester les nouveaux produits de santé contre le standard existant et d'évaluer leur efficience, afin de valider leur utilité clinique et médico-économique. Cette procédure a donc lieu une fois que l'efficacité clinique a été démontrée. Un dispositif médical inscrit à ce programme doit disposer du marquage CE, et les résultats du Pstic ont vocation à faciliter l'évaluation qui doit ensuite être réalisée par la HAS pour permettre sa prise en charge par l'assurance maladie. En pratique, des appels à projet sont lancés annuellement afin de recueillir des candidatures, sélectionnées ensuite par le ministre chargé de la santé, après avis d'un comité d'experts. Les établissements de santé lauréats bénéficient ensuite d'un financement spécifique destiné à compenser le surcoût lié à l'innovation, sur une durée de vingt-quatre mois, dans le cadre des missions d'enseignement, de recherche, de référence et d'innovation (Merri).
A côté du Pstic, le « forfait innovation » prévu à l'article L. 165-1-1 du code de la sécurité sociale, conditionne la prise en charge temporaire, à titre dérogatoire, d'un produit innovant à la réalisation d'études cliniques. Dans le cas des dispositifs médicaux, il concerne ceux d'entre eux pour lesquels la HAS a jugé le service attendu insuffisant, en raison du manque de données disponibles. Les conditions générales de prise en charge par l'assurance maladie ne sont donc pas remplies. Néanmoins, si le dispositif innovant présente un intérêt potentiel, le forfait innovation constitue un moyen d'approfondir les recherches cliniques à son sujet tout en permettant son utilisation dans des établissements de santé prédéterminés. Contrairement au Pstic, il intervient à la suite de l'évaluation réalisée par la HAS. Le forfait innovation apporte un financement global prenant en charge à la fois l'acte, les frais d'hospitalisation et le produit en question. Son but est de démontrer que l'innovation concernée est efficace, sûre et présente une réelle utilité clinique.
Les conditions de recours au forfait innovation
 Source : Direction
générale de l'offre de soins
Source : Direction
générale de l'offre de soins
La France dispose donc d'outils de soutien à l'innovation de nature à couvrir les différents cas de figure qui peuvent se présenter lors du développement d'une technique nouvelle. Toutefois, la question des délais et de la mise à disposition des dispositifs médicaux les plus innovants aux praticiens et aux patients n'est pas encore résolue . Créé par la loi de financement de la sécurité sociale (LFSS) pour 2009 puis modifié par la loi « HPST », le forfait innovation n'a véritablement été opérationnel qu'à partir de 2011. Seulement deux dispositifs médicaux ainsi qu'un acte sont en phase d'examen. Il conviendrait donc d'accélérer sa mise en place et d'en élargir la portée.
7. Garantir la qualité de l'information des praticiens et des patients
Aujourd'hui, les citoyens qui souhaitent obtenir des informations fiables sur les dispositifs médicaux implantables disposent de peu de sources. Il n'existe pas de portail officiel de santé publique. L'information n'est pas unifiée mais dispersée entre plusieurs organismes. L'ANSM diffuse principalement des bulletins d'alerte et des mises en garde lorsqu'un dispositif médical a été identifié comme dangereux pour la santé. De leur côté, l'Inpes, pour l'éducation à la santé, et l'InVS pour la veille sanitaire, communiquent séparément et selon des modalités différentes.
Contrairement à ce qu'on pourrait penser, les professionnels de santé ne bénéficient pas d'une situation réellement plus favorable. En dehors des revues médicales dans lesquelles sont publiés les résultats des principales études cliniques, il leur est parfois difficile d'obtenir une information incontestable ainsi qu'une évaluation impartiale des innovations. Le fait que les fabricants subventionnent une partie des essais réalisés ne permet pas de garantir leur objectivité.
La loi « Médicament » a tenté de répondre à une partie de ces problèmes en introduisant un encadrement de la publicité pour les dispositifs médicaux. Son article 34 prévoit que la publicité « définit de façon objective le produit, le cas échéant ses performances et sa conformité aux exigences essentielles concernant la sécurité et la santé, [...] et favorise son bon usage » (article L. 5213-2 du code de la santé publique). Il pose également la règle générale de l'interdiction de la publicité pour les dispositifs médicaux qui font l'objet d'une prise en charge par l'assurance maladie sauf pour ceux, limitativement énumérés dans un arrêté des ministres chargés de la santé et de la sécurité sociale, qui présentent un faible risque pour la santé. Surtout, cette loi a instauré un mécanisme d'autorisation préalable, délivrée par l'ANSM, pour la publicité en faveur de certains dispositifs médicaux particulièrement risqués. Pour donner un caractère dissuasif à ces dispositions, toute infraction peut être punie de deux ans d'emprisonnement et de 30 000 euros d'amende, auxquels peuvent s'ajouter des peines complémentaires telles que l'interdiction d'exercer dans le domaine des dispositifs médicaux pour une durée maximale de cinq ans.
Le décret du 9 mai 2012 86 ( * ) fixe la date d'entrée en vigueur de ces mesures au 1 er janvier 2013. Elles devraient certainement avoir pour conséquence une amélioration bienvenue de la qualité de l'information délivrée, en permettant d'identifier clairement le caractère publicitaire d'un message et en interdisant toute mention qui serait de nature à induire en erreur sur les effets du produit en question.
Il serait également possible pour le régulateur de tirer parti de ce nouvel outil réglementaire pour s'assurer de la qualité des évaluations cliniques réalisées par le fabricant. En effet, pour les dispositifs médicaux à risque dont la publicité sera soumise à autorisation préalable, le code de la santé publique donne le pouvoir au directeur de l'ANSM « d'exiger la communication de tous les éléments d'information indispensables au contrôle de l'exactitude des caractéristiques et des performances annoncées » 87 ( * ) . Voilà donc, par une voie il est vrai quelque peu détournée, un moyen d'obtenir des informations de la part des fabricants et de déterminer le sérieux des études que ceux-ci ont menées.
Les mesures sur la publicité de la loi « Médicament » constituent une avancée certaine en rendant impossibles les dérives de la publicité, trompeuse ou accrocheuse, qu'il a parfois été possible de constater. Les dispositifs médicaux sont des produits de santé qui peuvent avoir des effets équivalents à ceux des médicaments. Il était donc indispensable que les patients ne puissent pas être indûment influencés par les campagnes de promotion des industriels. Toutefois, elle n'apporte pas de solution au problème plus large de l'accès à une information médicale objective et de qualité en dehors du cabinet d'un professionnel de santé.
C'est pourquoi il convient , dans un premier temps, de rendre plus d'informations disponibles sur le site internet de l'ANSM , sur le modèle de celui de la FDA où sont mises en ligne toutes les déclarations d'incidents et d'événements de matériovigilance. De même, le dernier compte rendu de la commission nationale de sécurité sanitaire des dispositifs médicaux disponible sur le site de l'agence française 88 ( * ) remonte au 8 septembre 2010. Il est regrettable que l'impératif de transparence en la matière, pourtant reconnu à l'article L. 5324-1 du code de la santé publique, qui prévoit la publicité des réunions des commissions de l'ANSM, soit ainsi traité.
Proposition n° 22 :
Enrichir le contenu
du site internet de l'ANSM et assurer la publication rapide
des comptes
rendus de ses commissions et organes de travail internes
A terme, la mission appelle de ses voeux la création d'un portail dédié aux dispositifs médicaux et à la matériovigilance , ou tout du moins à celle d'une page spécifique au sein d'un site internet plus généralement consacré aux différentes formes de vigilance sanitaire. La centralisation des informations disponibles à ce sujet et l'effort de pédagogie ainsi réalisé constitueraient sans nul doute une réponse adaptée à la crise de confiance causée par la succession rapide, ces derniers mois, de scandales liés à des fraudes ou à des dysfonctionnements majeurs de dispositifs médicaux implantables pourtant déclarés conformes à la réglementation en vigueur.
Proposition n° 23 :
Envisager la
création d'un site d'information public sur les dispositifs
médicaux
II. RESPONSABILISER LES ACTEURS DE L'ESTHÉTIQUE POUR MIEUX PRENDRE EN COMPTE SES IMPLICATIONS SANITAIRES
A. POSER LES BASES D'UNE TYPOLOGIE DES PRATIQUES DE MÉDECINE ESTHÉTIQUE
1. Instituer un marquage CE spécifique aux dispositifs et produits esthétiques afin de garantir leur innocuité
La plupart des substances injectables et des dispositifs médicaux utilisés dans le cadre d'interventions à visée esthétique sont soumis au contrôle de l'ANSM, qui dispose de pouvoirs de police sanitaire à l'égard de produits de santé dangereux. A ce titre, dans une décision du 16 août 2011, l'ANSM a restreint, par exemple, la mise sur le marché et l'utilisation des dispositifs médicaux injectables (acide hyaluronique) dans le comblement et l'augmentation des volumes corporels à visée esthétique, en excluant l'indication d'augmentation mammaire à visée esthétique.
Toutefois, le statut des produits à visée exclusivement esthétique continue de faire débat : si l'on s'en tient à une lecture littérale de la définition d'un dispositif médical par les directives communautaires, les dispositifs conçus dans une finalité purement esthétique pourraient apparaître exempts de l'obligation de marquage CE au titre des directives de 1990 et 1993, les fabricants étant libres de se soustraire aux évaluations et contrôles cliniques correspondants.
Certains lasers épilatoires sont considérés comme des dispositifs médicaux dès lors qu'ils sont utilisés dans le traitement d'affections telles que l'hirsutisme. A ce titre, ils sont soumis aux exigences des directives relatives aux dispositifs médicaux et font l'objet d'un contrôle après leur mise sur le marché par l'ANSM. Néanmoins, ces mêmes lasers peuvent être utilisés à des fins exclusivement esthétiques et ne sont pas, dans ces circonstances, assimilés à des dispositifs médicaux, bien qu'ils reposent sur les mêmes principes de fonctionnement que les lasers à usage médical.
Le droit français a anticipé assez tôt la problématique posée par les lasers médicaux. L'article 2 de l' arrêté du 30 janvier 1974 portant réglementation concernant les lasers à usage médical précise que « les lasers à usage médical sont des appareils devant être utilisés par un médecin ou sous sa responsabilité » . En établissant une classification des lasers, qu'ils soient à impulsion ou à émission continue, cet arrêté permet de couvrir l'ensemble des lasers susceptibles d'être utilisés à des fins d'épilation : il en réserve donc l'usage aux seuls médecins, qu'ils interviennent dans un cadre thérapeutique ou dans une perspective exclusivement esthétique .
En revanche, se pose la question de l'autorité nationale compétente dans le contrôle des « produits frontières » , c'est-à-dire les produits à finalité esthétique inclassables, qui ne sont ni des dispositifs médicaux au sens des directives communautaires, ni des produits cosmétiques . Parmi ces produits, on recense aussi bien les cabines de bronzage que les lampes à lumière pulsée utilisées pour l'épilation (« lampes flash ») et les dispositifs de blanchiment des dents.
Pour ces produits, la direction générale de la consommation, de la concurrence et de la répression des fraudes (DGCCRF) dispose d'une compétence générale qui demeure, cependant, difficile à exercer en pratique. En application du principe de la libre circulation des marchandises au sein de l'Union européenne et à la liberté du commerce et de l'industrie, une interdiction requiert un motif de santé publique , lequel ne peut être mis en évidence que par une expertise scientifique . L'expertise est réalisée par les agences sanitaires (l'Agence nationale de sécurité sanitaire de l'alimentation, de l'environnement et du travail (Anses), la HAS, l'ANSM), ou encore des laboratoires d'experts et ne parvient donc qu'indirectement à la DGCCRF.
Plusieurs enquêtes ont pourtant fait apparaître que les appareils mettant en oeuvre la technique de la lumière pulsée, utilisés de façon croissante par les esthéticiennes, ne font pas toujours l'objet des contrôles permettant de garantir le respect des exigences de sécurité, en particulier pour prévenir tout risque de brûlure ou de dépigmentation 89 ( * ) .
Le même type de problème se pose pour les cabines de bronzage à ultraviolets (UV). En juillet 2009, le Centre international de recherche sur le cancer (Circ) a classé « cancérogènes certains pour l'homme (groupe 1) » les rayonnements ultraviolets solaires ainsi que les rayonnements émis par les installations de bronzage artificiel. Une étude publiée par des chercheurs américains en avril 2012 90 ( * ) a conclu à une accentuation du risque de mélanome chez les jeunes , multiplié par huit chez les femmes et par quatre chez les hommes en l'espace de quarante ans, dans le cas d'une exposition répétée aux UV en cabine de bronzage. Selon le docteur Jerry Brewer, directeur de l'étude, « les personnes recourant fréquemment à la lampe à bronzer ont 74 % plus de risques de développer un mélanome » . La France compte plus de quinze mille cabines UV fréquentées par près de 16 % de la population, ce chiffre étant en augmentation constante.
Un grand nombre de produits et dispositifs médicaux sont donc susceptibles d'être utilisés dans un but esthétique. Tant les exigences de sécurité sanitaire que les autorités compétentes en matière de contrôle varient selon les cas :
- les produits à finalité exclusivement esthétique , tels que les cabines UV, sont soumis à un marquage CE non médical, dont l'octroi est conditionné au respect de simples exigences de sécurité de type « horizontal » (comme par exemple la sécurité électrique) ;
- les dispositifs médicaux historiquement destinés à des actes de chirurgie reconstructrice à caractère invasif ont vu leur usage progressivement détourné dans le cadre d'indications exclusivement esthétiques (produits de comblement, prothèses mammaires). Si l'on s'en tient à une lecture stricte de la définition des dispositifs médicaux et du champ d'application des directives de 1990 et 1993, un produit qui ne revendique que des finalités esthétiques et ne poursuit aucune visée thérapeutique n'est pas tenu d'obtenir le marquage CE. Face à cette difficulté, la Commission européenne entend régulariser la situation juridique des dispositifs à visée esthétique et à caractère invasif en les intégrant dans le champ des dispositifs médicaux, comme ce fut le cas lors de la reclassification des prothèses mammaires en dispositifs de classe III ;
- les produits « frontière », moins invasifs mais dérivés de techniques médicales, sont utilisés dans un but exclusivement esthétique : c'est le cas d'un certain nombre de lasers épilatoires, de puissance inférieure aux lasers exclusivement réservés à l'usage médical dans certains pays, ou des lampes « flash » à lumière pulsée. Ces produits ne sont pas formellement soumis à l'obligation du marquage CE préalablement à leur commercialisation. Toutefois, afin de pouvoir attester du niveau de sécurité de leurs matériels, un certain nombre de fabricants recherchent l'obtention de la certification auprès d'organismes notifiés sans que leur classification en dispositifs médicaux soit forcément justifiée.
Il est raisonnable de penser, à la suite du scandale des prothèses mammaires PIP, que les autorités communautaires consacreront le caractère de dispositif médical pour tous les produits invasifs ou implantables à visée esthétique dans le cadre de la révision des directives relatives aux dispositifs médicaux. Néanmoins, l'absence de règles spécifiques de classification concernant les autres dispositifs utilisés à des fins esthétiques, en particulier ceux commercialisés auprès des professionnels non-médecins du secteur de l'esthétique, voire du grand public, ne permet pas de garantir la mise à la disposition des professionnels et des clients d'informations fiables sur l' évaluation de leur rapport bénéfices-risques . Certains de ces produits ne sont pas soumis au marquage CE applicable aux dispositifs médicaux ou sont, au mieux, soumis aux exigences essentielles de sécurité horizontale imposées par d'autres directives européennes dans le cadre de la « nouvelle approche ». L'innocuité des dispositifs autorisés à la commercialisation en vente libre doit pouvoir être étayée par des données cliniques.
Les référentiels traditionnels de l'évaluation du bénéfice thérapeutique organique sur un sujet malade ne sont pas appropriés pour les produits et dispositifs à finalité esthétique. Ils s'en distinguent, d'une part, par un niveau différent d'acceptation du risque par rapport au bénéfice escompté , et, d'autre part, par la très grande diversité de la technique employée, de la conception et de la composition de ces produits . Il n'est pas possible de laisser au fabricant la possibilité d'apprécier seul si son produit mérite ou non un marquage CE. Il doit se soumettre à des tests d'évaluation des bénéfices que l'utilisateur est en droit d'attendre de ce produit et des risques potentiellement encourus.
Dans ces conditions, la mission estime indispensable de prévoir une réglementation européenne spécifique pour l'ensemble des produits et dispositifs à finalité esthétique , qu'ils soient utilisés par des médecins ou par les professions non médicales du secteur de l'esthétique. Conformément à la logique de la « nouvelle approche », ces produits seraient soumis à des exigences de sécurité sanitaire harmonisées et à des principes d'évaluation clinique adaptés à leur nature : une telle réglementation aurait le mérite de clarifier le cadre juridique actuel, en ne laissant plus aux seuls fabricants le choix de soumettre leur produit au processus d'évaluation que suppose l'octroi d'un marquage CE.
Proposition n° 24 :
Instituer un
marquage CE spécifique aux dispositifs et produits
esthétiques
afin de garantir leur innocuité
|
Différences entre un dispositif médical et un dispositif esthétique |
||
|
DM
|
« DE »
|
|
|
Destination |
Médicale |
Esthétique |
|
Marquage CE |
|
|
|
Etude ou investigation clinique |
Obligatoire (avec un résultat de bénéfice sur risque avantageux) |
Facultative |
|
Matériovigilance |
Oui |
Non |
|
Documentation et formation |
Complète, et associée à la sécurité d'utilisation |
Suffisante pour une bonne utilisation |
|
Source : mission d'information du Sénat sur les dispositifs médicaux implantables et les interventions à visée esthétique |
||
2. Assurer la transparence de l'information sur les actes à finalité esthétique
L'importance du thème de l'esthétique dans la ligne éditoriale de la presse magazine ainsi que la multiplication des sites d'information sur les différentes méthodes morpho-esthétiques et anti-âge sur Internet participent d'un vaste mouvement de vulgarisation des connaissances sur les techniques de correction de l'apparence et de rajeunissement, qu'elles soient chirurgicales ou non. Mésothérapie, « vampire lift » , laser fractionné, cryothérapie, dermabrasion, laser-abrasion, lyse adipocytaire, liporéduction, électro-lipolyse, lipocryolyse, carboxythérapie, radiofréquence, micro-courants anti-âge, photoréjuvénation, luxopuncture, éveinage : la liste des interventions à visée esthétique est inépuisable . Une simple recherche en ligne portant sur l'une de ces techniques produit une multitude de résultats parmi lesquels il est difficile, pour le grand public, de faire la part entre information purement commerciale et documentation solide sur le plan scientifique.
L'abondance de l'information relative aux actes à visée esthétique disponible dans la presse et sur Internet tranche avec l'absence de données officielles concernant le nombre de médecins et de professionnels de l'esthétique autorisés à les exécuter. Hormis les chirurgiens esthétiques, il n'existe pas, pour l'heure, de système de déclaration d'activité des médecins pratiquant des interventions à visée esthétique , que ce soit auprès du Conseil national de l'ordre des médecins (Cnom) ou du ministère de la santé. En outre, en l' absence d'une réelle cotation des actes de médecine esthétique , qui échappent logiquement à la nomenclature de l'assurance maladie, il n'est pas possible de recenser les actes effectués par les différents médecins concernés.
Face à la profusion des messages à caractère commercial émanant des professionnels du secteur de l'esthétique et des fabricants de produits et dispositifs à visée esthétique, la mission estime indispensable, dans un premier temps, de créer les conditions d'un accès du grand public à une information indépendante et transparente , diffusée sur une plateforme numérique qui serait cogérée par la HAS (plus particulièrement par la CNEDiMTS, sa commission spécialisée dans les dispositifs médicaux et les techniques médicales) et l'ANSM, destinée à regrouper l'ensemble des informations susceptibles de démontrer la qualité des actes à visée esthétique et de leurs outils et de préciser les risques qu'ils représentent 91 ( * ) .
Dans un contexte de développement du tourisme esthétique, cette plateforme aura également vocation à informer le grand public des précautions et des mises en garde à observer dans le cadre de soins esthétiques effectués à l'étranger.
|
Le développement du tourisme
esthétique
Compte tenu de la réputation de son système de soins hospitaliers et de la qualité de la formation de ses médecins, la France constitue traditionnellement une destination pour le tourisme chirurgical. Depuis quelques années, un flux inverse est apparu. De plus en plus de Français se tournent vers des cliniques situées à l'étranger pour faire réaliser des interventions de chirurgie esthétique. La motivation des intéressés tient essentiellement au prix des interventions pratiquées à l'étranger, parfois trois fois moindre qu'en France. Les charges de structure étant bien moins élevées, la facture finale est donc plus faible. En outre, un chirurgien esthétique travaille, en France, avec toute une équipe et une infrastructure soumises à des conditions strictes d'hygiène et de sécurité, contrôlées dans le cadre d'accréditations et d'inspections sur site. Par ailleurs, les soins postopératoires sont incontournables dans des pays soumis à des réglementations strictes et représentent, potentiellement, des coûts considérables. L'absence de suivi et de correction éventuelle pour les opérations low cost réalisées à l'étranger diminue la facture globale de 30 %. Enfin, des formules « tout compris » permettent de prolonger le séjour au bord de la piscine d'un hôtel attenant à la clinique de chirurgie esthétique, voire à la plage, au mépris des règles de comportement postopératoire. Bien évidemment, une fois rentré en France, le client ne bénéficie d'aucun suivi particulier : il ne peut revenir en consultation après l'opération.
Les démarchages pour des séjours
« esthétiques » à l'étranger,
auprès des demandeurs de soins esthétiques, se multiplient, dans
des formes publicitaires de plus en plus agressives, en particulier sur
Internet. L'interdiction, en France, de la publicité pour la chirurgie
esthétique ne vaut que pour les installations autorisées en
France. Les cliniques situées à l'étranger sont
évidemment libres de toute règle lorsqu'elles diffusent des
publicités sur Internet.
|
Proposition n° 25 :
Mettre en ligne
l'ensemble des informations sur le rapport bénéfices-risques des
produits et dispositifs à finalité exclusivement
esthétique
Lors de son audition par la mission, M. Jean-Luc Harousseau, président de la HAS, regrettait le manque de données et d'essais cliniques concernant les interventions à visée esthétique : « nous ne pouvons nous appuyer que sur quelques publications scientifiques, sur les rapports de sinistralité des assureurs, sur les données éventuelles de cosmétovigilance ou de pharmacovigilance, sur l'expérience internationale, et, éventuellement, sur les réseaux de vigilance des sociétés professionnelles » . Dans ces conditions, lorsqu'elle est conduite à formuler un avis sur certains actes à visée esthétique, sur le fondement de l'article L. 1151-3 du code de la santé publique, la HAS ne peut invoquer, bien souvent, qu'une suspicion de danger grave basée sur le mode d'action potentiel des techniques invasives. Cette difficulté a trouvé une nouvelle illustration lorsque le Conseil d'Etat, appelé à statuer sur la régularité du décret d'avril 2011 visant à interdire les techniques de lyse adipocytaire, a considéré, dans sa décision du 17 février 2012, que la dangerosité des techniques non invasives n'était pas suffisamment établie sur le plan scientifique.
Les actes à visée esthétique doivent répondre aux mêmes exigences que celles applicables aux interventions thérapeutiques. C'est pourquoi la mission propose la mise en place de prérequis expérimentaux et d'études cliniques afin d' évaluer scientifiquement le rapport entre les bénéfices attendus, les risques encourus e t l'incertitude quant au résultat acceptable associé à une technique d'intervention à visée esthétique présentant un danger potentiellement grave pour la santé humaine, au sens de l'article L. 1151-3 du code de la santé publique. Ces données doivent être rendues publiques par la HAS lorsque celle-ci est appelée à rendre des avis d'interdiction ou de levée d'interdiction concernant des actes à visée esthétique, en vertu de ce même article.
Proposition n° 26 :
Rendre publiques
toutes les informations sur le rapport bénéfices-risques
des
interventions esthétiques
Il appartiendra à l'ANSM de garantir la mise à la disposition des professionnels de santé, des professions non médicales du secteur de l'esthétique et du grand public, l'ensemble des informations relatives à l'évaluation des bénéfices et des risques liés à l'utilisation des produits et dispositifs à finalité exclusivement esthétique. L'existence de ces données d'évaluation est bien sûr conditionnée à la mise en oeuvre du marquage CE spécifique - proposé par la mission - pour les produits et dispositifs à visée esthétique, qui suppose la conduite préalable d'études cliniques rigoureuses sur le plan scientifique.
Le développement d'une démarche d'évaluation des bénéfices et des risques qui s'attachent à l'utilisation d'un produit esthétique pose la question de sa composition. Il n'existe aucun référentiel transparent sur les éléments, substances et matériaux autorisés à entrer dans la composition des produits et dispositifs à visée esthétique. Or les professionnels, qui les achètent, doivent pouvoir accéder, au-delà des seules informations publicitaires transmises par les fabricants, à une information indépendante sur les composants autorisés et non autorisés dans leur production. L'ANSM doit donc établir une liste des composants ne présentant pas de danger pour la santé et à même d'entrer dans la fabrication des produits et dispositifs à visée esthétique.
Proposition n° 27 :
Etablir une liste des
matières premières, des substances et des composants
autorisés pour la fabrication de produits et de dispositifs à
visée esthétique
3. Privilégier la sécurité
a) Réserver les actes présentant des risques sérieux aux médecins qualifiés
Le législateur a confié au pouvoir réglementaire le soin de déterminer les règles de formation et de qualification applicables aux personnes habilitées à exécuter des interventions à visée esthétique. Créé par l'article 61 de la loi « HPST » 92 ( * ) , l'article L. 1151-2 du code de la santé publique dispose que « la pratique des actes, procédés, techniques et méthodes à visée esthétique autres que ceux relevant de l'article L. 6322-1 [interventions de chirurgie esthétique] peut, si elle présente des risques sérieux pour la santé des personnes, être soumise à des règles, définies par décret, relatives à la formation et la qualification des professionnels pouvant les mettre en oeuvre, à la déclaration des activités exercées et à des conditions techniques de réalisation » . Il prévoit également que l'exécution de ces actes « peut également être soumise à des règles de bonnes pratiques de sécurité fixées par arrêté du ministre chargé de la santé » .
Le ministère de la santé travaille, à l'heure actuelle, à l'élaboration, en collaboration avec les représentants du secteur, d'un projet de décret relatif aux actes, procédés, techniques et méthodes à visée esthétique présentant des risques sérieux pour la santé, autres que la chirurgie esthétique. Le précédent gouvernement avait envisagé d'insérer dans la partie réglementaire du code de la santé publique un ensemble de dispositions relatives à l'encadrement des « actes à visée esthétique présentant des risques sérieux réservés aux médecins » . Le but de ces dispositions aurait été de réserver aux seuls médecins la pratique des actes, procédés, techniques et méthodes à visée esthétique présentant des dangers sérieux pour la santé dès lors qu'elle a pour objet :
« 1° Le comblement de toute partie du corps par injection de produits ou utilisation de dispositifs, de techniques, de rayonnements électromagnétiques ou d'ultra-sons.
« Le comblement de toute partie du corps concerne toute action qui vise à redonner du volume ou à redessiner des volumes pour corriger l'apparence physique ;
« 2° L'amincissement par utilisation de dispositifs, de techniques, de rayonnements électromagnétiques ou d'ultra-sons.
« L'amincissement concerne toute action qui vise à affiner la silhouette ;
« 3° Le traitement des lésions cutanées par utilisation de produits à l'exclusion des produits cosmétiques mentionnés à l'article L. 5131-1 du code de la santé publique, de dispositifs, techniques, de rayonnements électromagnétiques ou d'ultra-sons.
« Le traitement des lésions cutanées ayant un retentissement esthétique concerne toute action qui vise à effacer ou à atténuer les cicatrices, les lésions vasculaires, les taches pigmentaires, les vergetures ou réaliser un détatouage ;
« 4° L'épilation par utilisation de dispositifs, de techniques, de rayonnements électromagnétiques ou d'ultra-sons.
« L'épilation concerne toute action qui vise à éliminer, temporairement ou définitivement les poils sur un corps humain ;
« 5° Le traitement des rides par injection ou application de produits à l'exclusion des produits cosmétiques mentionnés à l'article L. 5131-1 du code de la santé publique ou utilisation de dispositifs, de techniques, de rayonnements électromagnétiques ou d'ultra-sons.
« Le traitement des rides concerne toute action qui vise à atténuer les sillons à la surface de la peau ;
« 6° Le traitement des calvities ou des alopécies par injection de produits ou utilisation de dispositifs, de techniques, de rayonnements électromagnétiques ou d'ultra-sons.
« Le traitement des calvities ou des alopécies concerne toute action qui vise à freiner la chute, stimuler la repousse ou réimplanter des follicules pileux ;
« 7° Le tatouage médical par utilisation de produits, de dispositifs, de techniques, de rayonnements électromagnétiques ou d'ultra-sons.
« Le tatouage médical concerne toute action qui vise à redonner une pigmentation normale de la peau. »
Il est urgent d'établir une liste d'interventions à visée esthétique dont le degré de dangerosité et la nature des compétences requises pour leur exécution justifient leur pratique par des médecins préalablement formés. Afin de ne pas être rapidement rendue obsolète par l'évolution des techniques, cette liste devra se fonder sur les finalités des interventions et être périodiquement réactualisée.
Proposition n° 28 :
Fixer par
décret la liste des interventions à visée
esthétique ne pouvant être exécutées que par des
médecins dûment qualifiés
b) Encadrer strictement le recours par les esthéticiennes à certains dispositifs à visée esthétique
L'arrêté du ministre chargé de la santé, en date du 6 janvier 1962, a fixé la liste des actes médicaux ne pouvant être pratiqués que par des docteurs en médecine. Cette liste mentionne « tout mode d'épilation » , à l'exception des « épilations à la pince ou à la cire » . Or un grand nombre d'esthéticiennes proposent désormais à leurs clients des séances d'épilation utilisant des lampes à lumière pulsée.
Certains de ces appareils sont désormais en vente libre et si leur puissance est pour l'instant différente de celle des lampes à usage médical, nul ne peut garantir qu'il en sera ainsi à l'avenir. Il serait pour le moins paradoxal de maintenir l'interdiction faite à des personnes formées d'effectuer des actes que tout un chacun peut librement réaliser à son domicile.
A l'inverse, au nom des impératifs de santé publique, la mission considère qu'il convient d'encadrer strictement les conditions dans lesquelles les professionnels de l'esthétique pourraient être autorisés à pratiquer la photodépilation par lumière pulsée.
Proposition n° 29 :
Encadrer
strictement le recours par les esthéticiennes
à certains
dispositifs à visée esthétique
Cette autorisation devrait être assortie de conditions strictes en matière de transparence de l'information du patient, d'hygiène et de sécurité :
- préalablement à toute séance de photodépilation par lumière pulsée au sein d'un centre esthétique, le client doit être en possession d'une attestation dermatologique déclarant sa peau saine ;
- l'esthéticienne doit apporter la preuve d'une formation adéquate à la technologie de la photodépilation par lumière pulsée et d'une connaissance solide des précautions d'emploi des équipements et d'administration des traitements ainsi que des risques et événements indésirables potentiels ;
- l'intensité maximale des appareils de photodépilation destinés à un usage non médical doit être limitée en fixant une norme générale en la matière ;
- les informations relatives aux traitements et aux zones concernées doivent être consignées dans un dossier remis au client afin que celui-ci puisse garder la trace de ces interventions et en informer les professionnels auxquels il s'adressera par la suite, ces informations étant cruciales pour permettre au professionnel de déterminer l'état d'avancement d'un traitement épilatoire à la lumière pulsée et d'évaluer le nombre de séances restantes nécessaires ;
- le client doit manifester son consentement par écrit en signant un formulaire comprenant l'ensemble des explications relatives à la procédure.
c) Interdire les cabines de bronzage hors usage médical pour éviter un futur scandale sanitaire
L'assimilation d'une peau bronzée à la santé est profondément ancrée dans l'esprit de nos contemporains. L'utilisation des lampes UV à des fins esthétiques n'est aujourd'hui interdite qu'au Brésil. Elle le sera en 2014 en Nouvelle-Galles du Sud, l'Etat le plus peuplé d'Australie.
Les tenants du bronzage en cabine expliquent que l'éducation à la santé et la prévention constituent le meilleur remède à la multiplication des pathologies associées au « soleil artificiel ». Et si cet argument ne suffisait pas, ils ajoutent souvent qu'il favorise la synthèse de la vitamine D, dont la carence affecte la majorité de la population française en hiver. Par le passé, ils ont su trouver d'autres arguments pour présenter les bienfaits supposés du bronzage artificiel, par exemple le fait que les lampes utilisées ne délivrent que des UVA, considérés comme moins dangereux.
L'Institut national du cancer (INCa) a récemment consacré plusieurs études au sujet. Toutes confirment la nocivité du bronzage en cabine. Après un premier rapport en 2010 93 ( * ) , l'institut concluait ainsi un rapport de juillet 2011 94 ( * ) réalisé dans le cadre du Plan cancer 2009-2013 : « La pratique du bronzage par UV artificiels présente un risque cancérogène avéré pour la peau. Ainsi, en l'état actuel des connaissances scientifiques, le rapport bénéfices-risques des expositions répétées aux UV artificiels délivrés par les appareils de bronzage UV ne peut donc être que négatif et en défaveur des UV artificiels. »
Le Centre international de recherche sur le cancer (Circ) a établi, sur la base des données épidémiologiques les plus récentes, l'existence d'une relation entre risque de mélanome et exposition aux UV artificiels. En conséquence, il a ajouté, en 2009, les UV artificiels (UVA et UVB) à la liste des cancérogènes pour l'homme, au même titre que le rayonnement solaire.
Selon l'INCa, au vu des mêmes résultats scientifiques, il n'est pas possible de déterminer une fréquence minimum pour laquelle l'exposition aux lampes UV est sans risque. Celle-ci n'entraîne aucun bénéfice pour la santé : elle ne prépare pas la peau au soleil, ne permet pas ou très peu la production de vitamine D et entraîne un vieillissement cutané prématuré . De plus, les mêmes travaux démontrent que l'affirmation, avancée par certaines études américaines, selon laquelle les UV auraient un effet protecteur sur l'incidence de certains cancers non cutanés, ne repose pas sur des bases scientifiques solides.
La mission première des pouvoirs publics est de protéger la santé de nos concitoyens de dangers sanitaires avérés. C'est bien le cas des UV artificiels, en dehors des quelques usages médicaux pour lesquels ils peuvent être prescrits (traitement de certaines formes d'acné ou de psoriasis). Faute de le faire, il leur faudra assumer toutes les conséquences de leur inaction, lorsque, d'ici à vingt ans, le laxisme actuel se traduira par un surcroît de pathologies cutanées.
Proposition n° 30 :
Interdire les
cabines de bronzage hors usage médical
pour éviter un futur
scandale sanitaire
4. Renforcer les exigences de formation et de compétence
Selon le projet de décret soumis à l'avis des professionnels de l'esthétique, il était envisagé de réserver l'exécution des interventions précitées aux catégories de médecins suivantes :
« 1° Les médecins mentionnés aux 1, 2 et 4 de l'article D. 6322-43 95 ( * ) et les médecins spécialistes qualifiés en dermatologie ;
« 2° Les médecins titulaires des qualifications, diplômes, certificats ou autres titres nécessaires à la réalisation de ces actes et fixés par arrêté du ministre chargé de la santé et du ministre chargé de l'enseignement supérieur. »
En outre, il reviendra au ministre chargé de la santé de déterminer, par arrêté, les conditions dans lesquelles pourront être réalisées les interventions à visée esthétique réservées aux seuls médecins.
a) Instituer un diplôme national de médecine esthétique
En raison de la reconnaissance de la chirurgie plastique, reconstructrice et esthétique en tant que spécialité médicale à part entière et du régime déclaratif des installations correspondantes auprès des ARS, il est possible de recenser le nombre de praticiens concernés : 500 établissements publics et privés sont autorisés à pratiquer des interventions de chirurgie esthétique et reconstructrice, dont 350 privés. En outre, certains actes esthétiques peuvent être réalisés par des médecins libéraux dans leur cabinet. Il existe 809 spécialistes en chirurgie plastique reconstructrice et esthétique titulaires d'un diplôme d'études de spécialité (DES) en la matière, dont 70 % exercent à titre libéral exclusivement. En complément, des ORL peuvent accomplir certains actes, tout comme des gynécologues obstétriciens peuvent effectuer de la reconstruction mammaire. Les établissements qui ne sont que des centres autonomes de chirurgie esthétique sont une trentaine, concentrés en Ile-de-France et en région Provence-Alpes-Côte d'Azur.
En revanche, en l'absence de diplôme national, il est difficile de recenser avec exactitude les médecins esthétiques pratiquant en France. Lors de son audition par la mission, M. François-Xavier Selleret, directeur de la DGOS, estimait entre quatre cents et cinq cents le nombre de médecins esthétiques , précisant qu'un travail de recensement était en cours. En l'absence de réglementation précise, la pratique des interventions à visée esthétique est ouverte à tous les médecins.
La très grande majorité des organisations représentatives du secteur de la médecine esthétique n'estime pas souhaitable de faire de la médecine esthétique une spécialité médicale à part entière , au même titre que d'autres spécialités telles que la dermatologie ou la chirurgie plastique reconstructrice et esthétique. Toutefois, devant l'augmentation exponentielle d'actes à visée esthétique non déclarés, le Cnom a demandé aux médecins disposant d'une expérience dans la pratique de ces actes d'élaborer des référentiels techniques et de préparer un enseignement national afin de les valider.
Initiées par des organisations professionnelles regroupant des médecins esthétiques, des formations diplômantes ont été mises en place à la fin des années 1990 au niveau des universités, sous la forme de diplôme universitaire (DU) puis de diplôme interuniversitaire (DIU), afin de pallier l'absence de formation initiale spécifique dans ce domaine.
Malgré ces progrès incontestables, un diplôme interuniversitaire n'offre pas de garantie de quota , ni de pratique homogène, ni d'uniformité dans la qualité des actes. Dans ces conditions, un certain nombre de praticiens plaident pour la mise en place d'une véritable capacité à exercer la médecine esthétique , sous la forme d'un diplôme d'études spécialisées complémentaires (DESC). Ce diplôme permettrait de certifier l'acquisition d'une compétence complémentaire et non exclusive dans le cadre de chaque spécialité médicale.
La mise en place d'un DESC se heurte cependant à plusieurs difficultés tenant à l'absence bien logique de services hospitaliers dédiés à la médecine esthétique. Une solution pourrait être trouvée dans l'insertion de stages pratiques en centres libéraux (cliniques ou cabinets) dans les requis de la maquette d'enseignement qui devraient être fixés par arrêté conjoint des ministres chargés de la santé et de l'enseignement supérieur.
Proposition n° 31 :
Définir
le contenu d'un DESC de médecine morpho-esthétique
et de
traitement des effets du vieillissement
L'article 2 du projet de décret relatif aux actes à visée esthétique présentant des dangers potentiels pour la santé, prévoit un mécanisme transitoire de « validation des acquis de l'expérience » pour les médecins ne disposant pas d'un diplôme reconnu de médecine esthétique mais ayant acquis une expérience solide dans la pratique de ce type de traitement 96 ( * ) .
Dans un contexte d'aggravation des déséquilibres de la démographie médicale , l'augmentation de la demande de soins esthétiques susceptible d'intéresser un nombre croissant de médecins généralistes est perçue comme une cause potentielle d'aggravation du risque de désertification médicale. Dans ces conditions, le ministère de la santé a mis en place un plan d'action sur la médecine de proximité afin de favoriser un exercice attractif et diversifié de la médecine générale.
Pris en application des articles du code de la santé publique relatifs à l'inscription au tableau de l'ordre (L. 4111-1, L. 4112-1 et L. 4112-3), le décret du 3 mai 2012 97 ( * ) ouvre aux médecins, sous conditions, la possibilité d'obtenir un droit d'exercice complémentaire à celui de la spécialité dans laquelle ils sont initialement qualifiés.
Dans le cadre de la loi « HPST » précitée, le développement professionnel continu (DPC) des professions de santé a été créé en vue de regrouper la formation médicale continue (FMC) et celles des autres professions de santé ainsi que l'évaluation des pratiques professionnelles (EPP). Afin de tenir compte du renforcement de la formation médicale continue ainsi que de la diversification des compétences des médecins, le décret du 3 mai 2012 vise à permettre aux médecins spécialistes d'exercer une spécialité dite « non qualifiante » relevant du groupe I des diplômes d'études spécialisées complémentaires de médecine (telles que l'allergologie, la nutrition, l'addictologie, la toxicologie, la dermatopathologie, la médecine d'urgence, la médecine du sport ou la médecine du travail). Ces spécialités n'ouvrent pas droit à la qualification de spécialiste correspondant à l'intitulé du diplôme.
Afin de bénéficier de ce droit d'exercice complémentaire, les médecins doivent justifier d'une formation et d'une expérience qui assurent tout ou partie des compétences requises pour l'exercice des spécialités correspondantes. L'obtention de ce droit relève de la compétence de l'Ordre national des médecins, les décisions étant prises par le conseil départemental de l'ordre après avis d'une commission constituée par spécialité.
L'article 5 de ce décret prévoit qu'en fonction de l'évolution de la démographie médicale constatée au cours d'une année civile et au regard des besoins de prise en charge des patients, le nombre maximum de médecins pouvant en bénéficier sera fixé chaque année par arrêté du ministre chargé de la santé, pour chaque région et spécialité.
Dans le cas où la médecine esthétique ferait l'objet d'un diplôme d'études spécialisées complémentaires relevant du groupe I, elle serait reconnue comme une « spécialité non qualifiante » : un médecin justifiant des compétences requises par l'Ordre national des médecins pourrait se voir attribuer un droit d'exercice dans les conditions prévues par ce décret.
La reconnaissance au niveau national d'une formation diplômante rigoureuse en matière de médecine esthétique permettra de mettre de l'ordre parmi les formations sponsorisées et alimentées par des représentants de fabricants de dispositifs esthétiques dans le cadre, bien souvent, de conférences ou colloques internationaux qui n'offrent aucune assurance sérieuse de qualité.
b) Créer un registre qualité des médecins pratiquant des actes à visée esthétique
L'institut indépendant britannique de conseil en matière de soins de santé 98 ( * ) a lancé, le 13 avril 2010, avec le soutien du gouvernement et le financement du ministère de la santé, un registre d'assurance-qualité des professionnels de santé pratiquant des injections de produits dans un but esthétique, le « IHAS Register of Injectable Cosmetic Providers » . Ce registre vise à mettre à la disposition du grand public des informations relatives aux formations, à l'expérience et aux antécédents des professionnels de santé pratiquant des actes de médecine esthétique (médecins, infirmières et dentistes) qui auront procédé à leur enregistrement ainsi qu'à celui de leurs installations et sites en ligne 99 ( * ) .
Au cours de leur enregistrement, les professionnels de santé doivent remplir une documentation d'auto-évaluation et soumettre les pièces justificatives correspondantes. L'inscription, d'une durée d'un an à renouveler, suppose le paiement de 50 livres 100 ( * ) de frais par les organismes au sein desquels les praticiens proposent des traitements par injection de produits cosmétiques ou par les praticiens exerçant seuls en cabinet. On estime à cinq mille le nombre d'installations dans lesquelles sont pratiqués près de deux cent mille actes à visée esthétique chaque année au Royaume-Uni. Jusqu'à la mise en place du registre précité, ces actes échappaient au contrôle de la Commission de la qualité des soins ( « Care Quality Commission » ).
Les standards définis par l'IHAS comprennent des règles concernant :
- la sécurité sanitaire : des modes opératoires normalisés doivent être mis en oeuvre en vue de garantir la sécurité et l'efficacité de la sélection, de l'achat, de l'approvisionnement, du stockage, de la prescription, de l'administration, de l'élimination et du contrôle des produits médicaux ;
- la consultation en personne : tous les patients doivent effectuer une consultation directe avec le prescripteur préalablement à l'administration ou à l'usage d'un produit prescrit ;
- le consentement : un consentement éclairé sous forme écrite doit être obtenu préalablement à l'administration de tout traitement à visée esthétique par injection de produit et le formulaire de consentement doit être conservé dans le dossier médical du patient ;
- la démonstration de la qualité des soins dispensés au patient et les qualifications du prestataire : le prestataire doit démontrer qu'il dispose des qualifications cliniques nécessaires et qu'il est en mesure de fournir le meilleur niveau de soin et d'établir un diagnostic permettant de sélectionner le traitement le plus approprié en fonction des antécédents médicaux du patient.
La mission estime nécessaire la mise en place d'un tel registre des professionnels de santé pratiquant des actes à visée esthétique, une fois qu'un diplôme de valeur nationale aura été reconnu pour les médecins esthétiques : ce registre devrait pouvoir être géré par le Cnom, moyennant le paiement de frais d'inscription par les professionnels concernés qui devront s'engager à respecter un guide des bonnes pratiques en matière d'actes à visée esthétique. Il devra comprendre des informations relatives à leurs qualifications et à leur expérience ainsi qu'aux installations dans lesquelles sont réalisés ces actes.
Proposition n° 32 :
Créer un
registre qualité des médecins pratiquant des actes à
visée esthétique
c) Encadrer les exigences de qualification des assistants des médecins esthétiques
La plupart des assurances contractées par les médecins esthétiques exigent que leurs assistants soient des professionnels de santé, en grande majorité, des infirmières. A l'heure actuelle, il existe un diplôme interuniversitaire « dermatologie esthétique, lasers dermatologiques et cosmétologie » comportant un module spécifiquement destiné aux infirmiers diplômés d'Etat souhaitant accéder au statut d'« aide-lasériste » conformément aux avis du Cnom, des assurances professionnelles et de la Société française de dermatologie. Ce module a pour objectif d'assurer une formation dans le domaine des lasers à usages médical et esthétique et de délimiter le cadre d'exercice et les compétences des aides-laséristes intervenant dans les cabinets médicaux. Ce DIU est dispensé dans cinq universités et son module « aide-lasériste » est coordonné par le professeur Béatrice Crickx, enseignant à l'université Paris Diderot-Paris VII (hôpital Bichat).
En tout état de cause, les assistants ne peuvent se voir déléguer l'exécution d'un acte à visée esthétique en lieu et place d'un médecin : dans ce cas, le délégué serait seul pleinement responsable de son acte, ce qui correspondrait, en l'absence de toute supervision par un médecin, à un exercice illégal de la médecine . En revanche, il est possible à un médecin esthétique de transférer à son assistant des compétences dans l'exécution de certains actes nécessaires à la réalisation d'une intervention à visée esthétique, à la condition que ces actes soient effectués sous sa supervision . De manière plus générale, les consultations des patients et le recrutement, la formation et la supervision des assistants doivent être assurés par les médecins responsables de l'installation.
Il est indispensable de limiter la dimension entrepreneuriale de certains centres ou cabinets au sein desquels des actes médicaux à visée esthétique sont de plus en plus accomplis par de simples auxiliaires paramédicaux en l'absence de toute supervision directe par des médecins. Aux Etats-Unis, où l'exercice de la médecine est peu réglementé, la société américaine de chirurgie dermatologique a souligné auprès de la mission la nécessité d' interdire sur le plan réglementaire la pratique de la médecine dans un cadre entrepreneurial ( « corporate practice of medicine » ) afin d'empêcher tout médecin de n'être qu'un simple « directeur médical fantôme » ( « ghost medical director » ). Il convient d'interdire tout arrangement permettant à un médecin de prêter son nom à un centre médical dans lequel il n'intervient pas directement lors des consultations avec les patients et ne joue pas un rôle déterminant dans le recrutement, la formation et la supervision des personnes chargées d'exécuter les procédures à caractère médical. En l'absence de contrôle effectif assuré par un médecin qualifié, ces cabinets esthétiques n'ont de médical que le titre du médecin qui les « sponsorise » et sont assimilables, en réalité, à des centres de spas dans lesquels sont pratiquées illégalement des interventions à caractère médical.
Proposition n° 33 :
Encadrer les
centres et cabinets médicaux spécialisés
dans les
interventions à visée esthétique
La mission considère que tout assistant d'un médecin esthétique (auxiliaire médical ou paramédical, infirmière) doit pouvoir justifier de compétences et d'expériences pratiques dans l'exécution d'interventions à visée esthétique sous supervision d'un médecin. Dans ces conditions, le médecin esthétique responsable doit renseigner, dans le registre précité proposé par la mission, l'ensemble des informations relatives aux qualifications de ses assistants.
Proposition n° 34 :
Imposer aux
médecins de communiquer au registre du Cnom l'ensemble des informations
relatives aux qualifications et aux compétences
d'exécution
transférées à leurs assistants
Proposition n° 35 :
N'autoriser la
participation des assistants à l'exécution d'un acte à
visée esthétique que sous la supervision du médecin
responsable
d) Préciser les obligations de formation des professions non médicales du secteur de l'esthétique
Le secteur de l'esthétique à caractère non médical emploie des diplômés des niveaux V, IV et III selon la nomenclature des niveaux de formation. Le certificat d'aptitude professionnelle (CAP, niveau V) d'esthétique-cosmétique-parfumerie est le premier diplôme de la branche et obligatoire pour exercer. Les études menant à ce diplôme peuvent être poursuivies dans le cadre du brevet professionnel, de niveau IV, qui s'obtient exclusivement en alternance. Il correspond à un baccalauréat sans les matières générales.
En 2005, dans le cadre de la réforme du lycée sous l'égide du ministre délégué à l'enseignement scolaire, M. Xavier Darcos, la Confédération nationale de l'esthétique-parfumerie (Cnep) a oeuvré à la mise en place d'un baccalauréat professionnel en trois ans. La réforme du brevet de technicien supérieur (BTS), diplôme à bac + 2 (niveau III) s'est achevée récemment.
Au niveau du CAP, les esthéticiennes sont qualifiées pour assurer les soins du visage, des mains et des pieds, les épilations et le maquillage. Pour les soins du corps, le brevet professionnel est nécessaire, avec une formation aussi bien technique que juridique et réglementaire. Le BTS est susceptible de conduire à des métiers de laboratoire, de formation, de cadre ou de manager .
Une étude du centre de recherche pour l'étude et l'observation des conditions de vie (Credoc) a mis en lumière, en 2010, les axes d'amélioration possibles en matière de formation continue des professionnels de l'esthétique. Ce sont la gestion, l'anglais et les nouvelles technologies, qui évoluent plus rapidement que les diplômes du secteur.
5. Clarifier les conditions de tarification des actes médicaux à visée esthétique
Le 10 avril 2012, la direction générale des finances publiques (DGFiP) a publié un rescrit fiscal 101 ( * ) mettant un terme à une exonération fiscale bénéficiant aux médecins spécialisés dans l'esthétique, datant d'une directive européenne de 2003. Désormais, les actes de chirurgie, de médecine et de dermatologie à visée strictement esthétique sont soumis à la TVA à 19,6 %. Seuls les actes à caractère esthétique mais s'inscrivant dans une démarche thérapeutique ou reconstructrice, pratiqués par des médecins, sont éligibles à l'exonération de TVA sur le fondement de l'article 261 du code général des impôts : il s'agit des « actes pris en charge totalement ou partiellement par l'assurance-maladie, c'est-à-dire notamment les actes de chirurgie réparatrice et certains actes de chirurgie esthétique justifiés par un risque pour la santé du patient ou liés à la reconnaissance d'un grave préjudice psychologique ou social » .
Toutefois, la DGFiP indique qu'une consultation est en cours sur le critère déterminant de la finalité thérapeutique des actes réalisés, au sein d'un groupe de travail avec les organisations représentatives du secteur de la santé, piloté par la direction de la législation fiscale. Dans l'attente des résultats de ces travaux, le droit en vigueur reste applicable.
Conformément à l'article 132 de la directive 2006/112/CE du 28 novembre 2006, « les prestations de soins à la personne effectuées dans le cadre de l'exercice des professions médicales et paramédicales telles qu'elles sont définies par l'Etat membre concerné » sont exonérées de la TVA. Cette disposition a été transposée, en droit français, à l'article 261 du code général des impôts, dont le 1° du point 4 dispose que les soins dispensés aux personnes, notamment par les membres des professions médicales et paramédicales réglementées, sont exonérés de TVA.
Concernant la condition tenant à la nature des soins, la Cour de justice de l'Union européenne (CJUE) a précisé que seules les prestations à finalité thérapeutique, entendues comme celles menées dans le but de prévenir, de diagnostiquer, de soigner et, dans la mesure du possible, de guérir des maladies ou anomalies de santé sont susceptibles de bénéficier de l'exonération de TVA 102 ( * ) .
S'agissant de la condition tenant à la qualification du praticien, seuls les membres des professions médicales et paramédicales réglementées par une disposition législative ou par un texte pris en application d'une telle disposition sont susceptibles d'entrer dans le champ de cette exonération.
Il s'agit essentiellement des professions visées dans la quatrième partie du code de la santé publique mais également de professions 103 ( * ) qui, bien que non visées par ces dispositions, fournissent des prestations reconnues comme de qualité identique qui doivent, à ce titre, bénéficier également de l'exonération pour respecter le principe de neutralité de la TVA 104 ( * ) .
En matière de médecine esthétique par conséquent, les actes pratiqués par les médecins ne sont éligibles à l'exonération que dans la mesure où ils sont considérés comme poursuivant une finalité thérapeutique. Aussi, les actes à visée purement esthétique, qui ne peuvent être considérés comme poursuivant un tel but, doivent être soumis à la TVA.
Le gain potentiel pour l'Etat, résultant de la sujétion à la TVA des actes à finalité exclusivement esthétique, est évalué à 23 millions d'euros, pour un marché français dont le revenu global est estimé, en 2012, à 120 millions d'euros. La suppression de cette exonération fiscale correspondrait à une dépense fiscale additionnelle de 40 000 euros de TVA en moyenne par cabinet. La presse estime que huit cents chirurgiens, un millier de médecins et près de mille deux cents dermatologues seraient concernés 105 ( * ) .
La mission estime que cette clarification du statut fiscal des actes à visée exclusivement esthétique va dans le bon sens car elle permettra d'établir une base déclarative de ce type d'interventions . Elle préconise une classification des différents actes médicaux n'ouvrant pas droit à l'exonération de TVA afin de pouvoir recenser au mieux, parmi ces actes non exonérés déclarés, ceux qui poursuivent une finalité esthétique.
B. METTRE EN PLACE UNE VÉRITABLE « ESTHÉTICOVIGILANCE »
L'ensemble des produits et des dispositifs à finalité esthétique doivent faire l'objet d'une « esthéticovigilance », à l'image de la matériovigilance à laquelle sont soumis les dispositifs médicaux .
La surveillance des incidents ou des risques d'incidents résultant de l'utilisation des produits et dispositifs à visée esthétique suppose la mise en place d'un mécanisme d'alerte à partir des signalements obtenus des différents acteurs du secteur.
Pour être efficace, ce dispositif d'alerte ne saurait se concevoir sans une amélioration du traçage des produits et des dispositifs concernés.
1. Renforcer le dispositif d'alerte sur les interventions à visée esthétique
Il convient de mettre un terme à la confusion des responsabilités entre la DGCCRF et l'ANSM dans le contrôle des produits à la frontière du médical et de l'esthétique pure , dont l'utilisation peut avoir des conséquences notables sur la santé des utilisateurs. C'est pourquoi la mission recommande de clarifier la situation juridique actuelle, en confiant à l'ANSM (qui est déjà chargée de contrôler les produits cosmétiques et de tatouage) une compétence de police spéciale dans le contrôle des produits et dispositifs utilisés à des fins exclusivement esthétiques.
En conséquence, la mission propose de modifier l'article L. 5311-1 du code de la santé publique détaillant les compétences de l'ANSM afin de confier à cette dernière des fonctions de police sanitaire sur l'ensemble des produits et dispositifs à finalité esthétique.
Proposition n° 36 :
Confier à
l'ANSM une compétence de police sanitaire sur l'ensemble des
produits
et dispositifs à finalité exclusivement
esthétique
Au titre de sa compétence de police sanitaire, l'ANSM serait chargée de recueillir toutes les informations relatives aux signalements d'effets indésirables ou d'incidents survenus sur les clients à l'occasion ou à la suite de l'utilisation d'un dispositif ou d'un produit à visée esthétique. L'obligation de signalement de la part des praticiens ferait partie des bonnes pratiques médicales surveillées par le Cnom. Devraient obligatoirement donner lieu à signalement :
- toute réaction nocive et non voulue se produisant lors de l'utilisation d'un produit ou dispositif à visée esthétique, que celle-ci ait été conforme ou non aux instructions du fabricant ;
- tout dysfonctionnement ou toute altération des caractéristiques ou des performances du produit ou du dispositif médical utilisé ;
- toute insuffisance (erreur ou omission) figurant dans le mode d'emploi ou manuel de maintenance des produits ou dispositifs concernés.
L'obligation de signalement vaudrait également pour les fabricants ou leurs mandataires et pour les distributeurs des produits et dispositifs à visée esthétique, lorsqu'ils feraient la constatation ou auraient connaissance d'incidents ou de risques d'incidents mettant en cause un produit et/ou un dispositif à visée esthétique.
Les patients seraient fortement incités, par l'intermédiaire de formulaires ad hoc disponibles sur une plateforme numérique de recueil des données de vigilance, à communiquer à l'ANSM tout effet secondaire ou incident dont ils seraient victimes. L'ANSM mettrait à disposition de tous une base de données centralisant l'ensemble des informations obtenues et figurant sur cette plateforme dédiée.
L'ANSM serait chargée de superviser les travaux d'évaluation et l'exploitation des informations obtenues dans un but de prévention et, le cas échéant, de faire usage de ses pouvoirs de police sanitaire.
Proposition n° 37
:
Confier
à l'ANSM le soin de centraliser tous les signalements relatifs aux
effets indésirables ou incidents consécutifs à une
intervention à visée esthétique
2. Sécuriser le parcours de soins esthétiques
L'objectif d'une meilleure traçabilité des interventions à visée esthétique est double. Il s'agit, d'une part, de sécuriser le parcours de soins , en particulier dans le contexte du nomadisme esthétique. Celui-ci peut en effet conduire à une répétition potentiellement nocive d'actes similaires. Il s'agit, d'autre part, de faciliter le repérage des produits et des dispositifs ayant causé des réactions secondaires tardives ou faisant l'objet de signalements indésirables graves et de pouvoir en avertir rapidement les patients concernés .
L'exigence de traçabilité a été mise en lumière par l'Afssaps notamment, dans ses recommandations aux praticiens relatives à l'usage de produits injectables de comblement de rides 106 ( * ) . Elle y rappelle en particulier qu' « il est recommandé au praticien de questionner le candidat sur les traitements esthétiques antérieurs notamment sur la nature du produit préalablement injecté. Car il est fortement déconseillé d'avoir recours à un produit résorbable après l'utilisation d'un produit non résorbable sur le même site d'injection » . En outre, comme l'indique l'agence, certains effets secondaires associés notamment aux produits lentement et non résorbables peuvent survenir plusieurs mois voire plusieurs années après l'implantation du produit . Une illustration de certains effets à retardement observés après l'injection de produits de comblement des rides est fournie dans le tableau ci-après :
|
Réactions |
Effets secondaires |
Durée estimée |
|
Immédiate (1j-1 sem.) |
• Hématome, érythème, oedème |
8 j |
|
Semi-retardée (1-3 sem.) |
• Infection (relative aux conditions d'asepsie) • Nécrose • Inflammation non spécifique |
1-6 mois 3 mois |
|
Retardée (3-24 mois) |
• Allergie, érythème • Pigmentation |
1-12 mois |
|
Retardée (rare) (> 3 mois - x années) |
• Granulome |
x mois - définitif |
|
Source : ANSM |
||
Dans ces conditions, les praticiens sont encouragés à consigner les actes effectués (nombre d'injections, délai entre deux injections), les produits et les volumes utilisés ainsi que les zones corporelles visées. Pour l'agence, il faut « conserver les données sur une durée de quinze ans » et « fournir toutes ces informations dans un « carnet esthétique » » .
La mission estime que la mise en place d'un « carnet du parcours de soins esthétiques » devrait être rendue obligatoire pour l'ensemble des dispositifs et des produits à finalité esthétique utilisés par les professions médicales.
La délivrance et la mise à jour du carnet de soins esthétiques ainsi que la sensibilisation du patient à l'importance de cet outil feraient partie des obligations incombant aux praticiens sous le contrôle du Cnom .
Il serait fait obligation au praticien d'inscrire dans ce carnet tous les renseignements relatifs à l'identification du dispositif ou du produit utilisé au cours de l'intervention (dénomination, numéro de lot, nom du fabricant ou de son mandataire), la date d'utilisation, les zones corporelles concernées et le nom du praticien ainsi que la mention du lieu d'utilisation . Ces mêmes renseignements seraient obligatoirement conservés par le praticien sans limitation de durée au sein du dossier médical personnel du patient ou de tout autre dossier de suivi . Ils permettraient de garantir un traçage précis des dispositifs en question ainsi que de leurs conditions d'utilisation.
Proposition n° 38 :
Mettre en place un
carnet de suivi des interventions à visée
esthétique
CONCLUSION - PRIMUM NON NOCERE
Au terme de ses travaux, la mission d'information a acquis la conviction que le scandale des prothèses mammaires revêt un caractère plus large que ce que l'opinion en a retenu : l'attention s'est quasi exclusivement portée sur la rupture des implants remplis de gel de silicone et sur les conséquences qui en résultent pour les femmes à qui ils avaient été posés. Cette situation est dramatique mais elle n'est que le signe le plus tangible d'une affaire plus vaste : pourquoi et comment une entreprise qui présentait autant d'anomalies et de manquements graves à la réglementation a-t-elle pu continuer à vendre ses produits pendant tant d'années ?
Poser cette question revient à s'interroger sur les graves lacunes du système européen de contrôle des dispositifs médicaux , notamment les plus critiques. Le scandale PIP démontre également que ces produits ne peuvent plus rester les grands oubliés de la politique de santé . Chaque jour, les dispositifs médicaux les plus complexes contribuent à prolonger l'existence de patients pour lesquels il n'existait pas d'alternative thérapeutique et à améliorer la qualité de vie de beaucoup d'autres. Il est temps de comprendre que les dispositifs médicaux ne constituent pas une sorte de « médicament au rabais » mais bel et bien un outil sanitaire indispensable.
Les investigations de la mission la conduisent à remettre en cause d'autres idées reçues :
- penser que les Etats-Unis constituent un modèle indépassable , tant sont nombreux les aménagements qui permettent d'échapper à la réglementation la plus exigeante. Le nombre de retraits de dispositifs et les difficultés récurrentes rencontrées par des matériels américains illustrent bien cette réalité : les principaux pays appliquent tous plus ou moins le même système de classification en fonction du degré de risque présenté par le produit ;
- croire que l'institution d'une procédure d'autorisation de mise sur le marché pour les dispositifs médicaux les plus risqués permettrait d'éviter toute défaillance à l'avenir relève de la même logique : un dispositif médical ne sera jamais un médicament. En la matière, il est impossible de demander à des volontaires sains de tester les produits, il n'existe pas de placebo, les études cliniques préalables sont difficiles à mettre en oeuvre.
En revanche, force est de constater les graves dangers qui découlent des modalités d'application de la réglementation européenne : le marquage CE ne peut plus dépendre du bon vouloir des fabricants, avec la passivité des organismes notifiés . Ce qui devait rester l'exception - l'assimilation « dûment justifiée » à un produit existant - est devenue la règle. Il est urgent d'inverser cette logique car la volonté de promouvoir l'innovation thérapeutique et d'offrir aux malades toutes les possibilités issues du développement de technologies nouvelles ne doit pas faire oublier le but premier de tout dispositif médical : améliorer l'état de santé du patient.
La première réforme consiste donc à appliquer rigoureusement la réglementation existante , d'autant que la révision des textes communautaires n'entrera en vigueur que dans deux ans au mieux ; la mise en oeuvre des mesures d'harmonisation décidées au niveau international, notamment l'institution d'un système d'identifiant unique, prendra nécessairement du temps. Sans attendre, notre pays peut faire le choix de la sécurité en modifiant en profondeur la réglementation nationale. Car la France se trouve dans une position plus inconfortable que ses principaux partenaires :
- le délai mis à admettre au remboursement les dispositifs médicaux innovants est le plus long de l'Union européenne , de sorte que les patients ne peuvent bénéficier du bon côté de la procédure de certification européenne, sa rapidité. Ainsi, le dispositif Watchman TM dont la mission a eu la démonstration 107 ( * ) , est disponible dans plusieurs pays européens mais pas encore en France. Les patients du nord de la France n'ont pour seule solution que de se le faire implanter en Allemagne ;
- les défaillances des mécanismes d'alerte sont flagrantes . La protection des données personnelles constitue une pierre angulaire de notre système juridique. En matière de santé, elle doit pouvoir être aménagée afin de mettre en place des registres de patients pour chaque spécialité ou acte clairement identifié (prothèse de hanche ou de genou) ;
- l'accès à une information simple et exhaustive reste un objectif à atteindre car la transparence constitue le seul moyen pour restaurer la confiance.
La création d'un portail d'information unique permettra également d'assurer une éducation à la santé , qui devra concerner aussi bien les dispositifs médicaux que les interventions à visée esthétique. Par exemple, qui sait, aujourd'hui, que la prothèse qui lui est implantée a une durée de vie limitée et qu'il sera peut-être nécessaire de procéder à son remplacement ?
On pourrait s'interroger longtemps pour connaître et analyser les motivations et ressorts psychologiques de la demande d'actes ayant pour finalité de parfaire l'apparence de personnes a priori en bonne santé. En tout état de cause, le législateur ne peut assister sans réagir à la multiplication des pratiques à visée esthétique. Il est urgent d'agir dans trois directions :
- définir précisément ce qui relève de la médecine et ce qui appartient à l'esthétique , sans se fonder sur les actes proprement dits car ceux-ci évoluent sans cesse ;
- clarifier les compétences des différentes professions et mettre en place un véritable plan d'action en matière de formation ;
- instituer un véritable parcours de soins esthétiques de nature à alerter les consommateurs sur la nécessaire prudence qui s'impose en la matière.
Il n'existe pas de fatalité à voir se répéter les scandales sanitaires ; il importe seulement de remettre la sécurité au premier rang des priorités de l'action publique en ce domaine.
LES PRINCIPALES PROPOSITIONS DE LA MISSION D'INFORMATION
|
1) Mettre au point un cahier des charges commun à tous les organismes notifiés 2) Multiplier les contrôles inopinés chez les fabricants 3) Doter le N-Bog d'un statut légal dans la réglementation européenne et lui confier la mission d'harmoniser les modalités de surveillance des organismes notifiés 4) Etendre l'obligation d'information des organismes notifiés aux observations de non-conformité majeures 5) Généraliser les inspections communes des organismes notifiés par les autorités sanitaires de plusieurs pays européens 6) Rendre publics les flux financiers et les liens d'intérêts entre fabricants de dispositifs médicaux et bénéficiaires de ces avantages 7) Mutualiser les sommes allouées par l'industrie des produits de santé aux étudiants dans le cadre des conventions d'hospitalité 8) Inciter la Commission, dans le cadre de la refonte des directives, à prendre pleinement en compte les exigences de sécurité des dispositifs médicaux 9) Créer une liste positive des dispositifs médicaux les plus risqués au niveau européen 10) Interdire l'ensemble des CMR de catégorie 2 dans les dispositifs médicaux destinés aux nourrissons, jeunes enfants et femmes enceintes 11) Publier rapidement les textes d'application de la loi « Médicament » relatifs aux dispositifs médicaux 12) Mettre en place un groupe de travail chargé de simplifier la procédure de signalement des incidents de matériovigilance 13) Mieux associer les déclarants aux suites données aux déclarations de matériovigilance 14) Mener une campagne d'information commune, avec le Cnom, sur les enjeux de la matériovigilance 15) Faire du renforcement de la matériovigilance l'un des points centraux du prochain contrat d'objectifs et de performance Etat-ANSM 16) Accélérer la mise en oeuvre de l'UDI au sein de l'Union européenne 17) Elargir l'accès à Eudamed aux organismes notifiés et, à terme, au moins partiellement au public
18)
Rendre obligatoire, sous certaines
conditions, le renseignement des registres par les médecins
19) Mettre en place un codage plus fin dans le PMSI afin de mesurer précisément l'usage des dispositifs médicaux implantables à l'hôpital 20) Limiter « l'effet domino » en définissant plus précisément l'équivalence pouvant être acceptée entre deux dispositifs médicaux pour satisfaire à l'obligation d'évaluation clinique 21) Imposer que le dispositif médical auquel un nouveau produit est présenté comme équivalent ait lui-même fait l'objet d'investigations cliniques 22) Enrichir le contenu du site internet de l'ANSM et assurer la publication rapide des comptes rendus de ses commissions et organes de travail internes 23) Envisager la création d'un site d'information public sur les dispositifs médicaux 24) Instituer un marquage CE spécifique aux dispositifs et produits esthétiques afin de garantir leur innocuité 25) Mettre en ligne l'ensemble des informations sur le rapport bénéfices-risques des produits et dispositifs à finalité exclusivement esthétique 26) Rendre publiques toutes les informations sur le rapport bénéfices-risques des interventions esthétiques 27) Etablir une liste des matières premières, des substances et des composants autorisés pour la fabrication de produits et de dispositifs à visée esthétique 28) Fixer par décret la liste des interventions à visée esthétique ne pouvant être exécutées que par des médecins dûment qualifiés 29) Encadrer strictement le recours par les esthéticiennes à certains dispositifs à visée esthétique 30) Interdire les cabines de bronzage hors usage médical pour éviter un futur scandale sanitaire 31) Définir le contenu d'un DESC de médecine morpho-esthétique et de traitement des effets du vieillissement 32) Créer un registre qualité des médecins pratiquant des actes à visée esthétique 33) Encadrer les centres et cabinets médicaux spécialisés dans les interventions à visée esthétique 34) Imposer aux médecins de communiquer au registre du Cnom l'ensemble des informations relatives aux qualifications et aux compétences d'exécution transférées à leurs assistants 35) N'autoriser la participation des assistants à l'exécution d'un acte à visée esthétique que sous la supervision du médecin responsable 36) Confier à l'ANSM une compétence de police sanitaire sur l'ensemble des produits et dispositifs à finalité exclusivement esthétique 37) Confier à l'ANSM le soin de centraliser tous les signalements relatifs aux effets indésirables ou incidents consécutifs à une intervention à visée esthétique
38)
Mettre en place un carnet de suivi
des interventions à visée esthétique
|
COMPTE RENDU DE LA RÉUNION D'EXAMEN DU RAPPORT
Réunie le mardi 10 juillet 2012 , sous la présidence de Mme Chantal Jouanno, présidente, la mission commune d'information a procédé à l' examen du rapport de M. Bernard Cazeau, rapporteur .
Mme Chantal Jouanno, présidente . - Avec l'examen du rapport, notre mission d'information arrive à son terme, après un point d'étape le 5 juin dernier. Nous avons mené une cinquantaine d'auditions, visité trois entreprises, étudié quatre pays : Etats-Unis, Danemark, Suède et Australie.
A Bruxelles, nous avons rencontré le cabinet du commissaire à la santé et aux consommateurs. La Commission n'a nullement l'intention de substituer au système actuel de certification un mécanisme d'approbation préalable plus exigeant, même pour une liste limitée de dispositifs médicaux. Elle plaide, au contraire, pour un renforcement du marquage CE, avec un pouvoir d'évocation confié aux autorités nationales en cas de doutes sur la sécurité d'un dispositif médical en particulier. Outre que le processus serait fort lourd, le mécanisme resterait non contraignant et le marquage CE pourrait être délivré malgré l'avis négatif de l'instance regroupant les autorités nationales. Après avoir envisagé une révision des directives relatives aux dispositifs, la Commission s'oriente désormais vers une proposition de règlement, dont le texte serait disponible à l'automne et adopté, au mieux, dans deux ans. Dans cet intervalle, à nous de peser sur la Commission pour amender ce projet.
Un mot sur un sujet qui me tient à coeur, celui des perturbateurs endocriniens. Il y a un an, notre collègue Gilbert Barbier a consacré un rapport à ce sujet, au nom de l'office parlementaire d'évaluation des choix scientifiques et technologiques (Opecst), intitulé « Perturbateurs endocriniens, le temps de la précaution ». On a coutume de dire que les dispositifs médicaux ne sont pas mieux encadrés que les jouets. Les substances cancérogènes, mutagènes et reprotoxiques sont interdites dans les jouets - et les sextoys... - mais entrent toujours dans la fabrication des dispositifs médicaux ! Les règles de la libre circulation des marchandises nous empêchent d'agir seuls au niveau national. Raison de plus pour proposer l'interdiction de ces substances pour les personnes les plus exposées afin que la future réglementation européenne soit amendée en ce sens. Je compte sur le soutien du Parlement européen, qui a récemment adopté une résolution sur les prothèses PIP, preuve que nos préoccupations sont partagées.
Avec M. Cazeau, nous nous sommes rendus à Villepinte à l'invitation de la société Boston Scientific pour visiter son centre européen de formation dans le domaine de la cardiologie interventionnelle. Nous avons découvert comment les médecins sont formés à l'implantation de dispositifs médicaux. Et cette présentation nous a renforcés dans la conviction que le dispositif médical n'était pas un médicament au rabais. Instituer des règles identiques pour ces deux catégories de produits de santé n'aurait donc pas de sens.
Un dernier mot sur l'esthétique. Ce que nous avons découvert tout au long de nos travaux est effrayant. Sans adopter une posture moralisatrice sur les raisons du recours à ce type d'intervention - qui va se développer avec le vieillissement de la population - nous devons avant tout faire progresser la sécurité.
M. Bernard Cazeau, rapporteur . - Cette mission a été créée en écho à l'émotion suscitée par le scandale PIP. Bien plus qu'une simple fraude au gel de silicone, cette triste affaire illustre les dysfonctionnements de l'encadrement juridique des dispositifs médicaux. Les signaux d'alerte - lettres d'avertissement de la Food and Drug Administration (FDA), mission d'inspection de l'autorité sanitaire australienne, plaintes déposées en Angleterre, jusqu'aux interrogations du bureau central de tarification sur la qualité des produits de PIP - montraient pourtant que la chaîne de fabrication et l'organisation même de l'entreprise posaient problème. Le Gouvernement doit demander à l'inspection générale des affaires sociales (Igas) un bilan exhaustif de la gestion de cette crise. Il faut y voir clair, en termes de taux de rupture et de pathologies associées, ainsi que sur les conditions, y compris financières, des opérations d'explantation et de réimplantation. C'est le moins que nous puissions faire pour les trop nombreuses victimes.
Mais cette affaire est l'arbre qui cache la forêt. Au début des années 1990, la volonté de mettre en place le grand marché unique européen a conduit à une nouvelle approche réglementaire, faisant primer la libre circulation des produits sur la sécurité des patients.
Je reviens sur le rôle des organismes notifiés dans la certification des dispositifs médicaux. Je regrette que la possibilité offerte aux fabricants de faire certifier leurs produits par la voie dite bibliographique, qui devait rester l'exception, soit devenue la règle. Dans la plupart des cas, ils se contentent donc de présenter les études déjà disponibles, avec un effet domino : un dispositif peut être assimilé à un produit déjà existant, qui a lui-même été certifié par la voie bibliographique, et ainsi de suite. D'aucuns prétendent qu'un dispositif peut même être certifié par assimilation avec un produit retiré du marché. L'évaluation clinique est insuffisante, même pour les dispositifs les plus risqués. Il faudrait que la certification par équivalence ne puisse jouer qu'une fois ; j'espère que le Gouvernement reprendra cette proposition à son compte.
Pour ce qui est de la chirurgie esthétique, le cadre légal existe et la France est plutôt citée en exemple. Notre attention s'est donc portée sur la médecine esthétique, qui souffre d'une réglementation inadaptée, voire inexistante : c'est une vraie jungle. Il n'existe pas de véritable formation en médecine esthétique et chaque spécialité médicale revendique le droit de pratiquer un certain nombre d'actes, sans compter les dentistes au nom de leur capacité à traiter le sillon naso-génien. Les techniques et les produits évoluent très rapidement et de nouveaux marchés apparaissent - blanchiment des dents, épilation par lumière pulsée, etc. - sans que leur innocuité ait été établie.
Fort de ces constatations parfois dramatiques, souvent désolantes, je voudrais aujourd'hui formuler un ensemble de propositions. Mais avant cela, je souhaite dissiper quelques idées reçues. La première est que les règles régissant le médicament pourraient être transposées sans peine aux dispositifs médicaux. Ce sont deux produits de santé très différents. Nous pouvons nous inspirer du système du médicament mais en l'adaptant aux spécificités du secteur des dispositifs médicaux, je pense par exemple à l'institution d'une éventuelle autorisation de mise sur le marché.
Autre idée reçue : il suffirait d'adopter un modèle existant. Certains de nos interlocuteurs ont présenté la réglementation américaine comme la panacée alors que ses dispositions les plus contraignantes peuvent être contournées dans beaucoup de cas. Très voisin de ce qui existe en Europe, le système de reconnaissance par équivalence substantielle est pratiqué dans 90 % des cas.
Troisième idée reçue, fonder le suivi des dispositifs médicaux sur leur remboursement. Or la pose d'un dispositif médical s'appuie à la fois sur un acte et sur un matériel. Il faut disposer de données précises sur la dépense liée à l'utilisation de dispositifs médicaux dans les établissements de santé en général et à l'hôpital en particulier. Je serais étonné que la Cour des comptes ne se saisisse pas du sujet un jour ou l'autre. Fonder le suivi médical des dispositifs médicaux implantés sur le codage du PMSI est une tout autre affaire.
Dernière idée reçue : croire que tout se joue à Bruxelles. Notre mission en Suède et au Danemark ainsi que les éléments que nous avons recueillis sur le système australien montrent bien que la tenue de registres exhaustifs suppose des conditions précises et se heurte à une réticence quasi culturelle dans notre pays. Mais ce n'est pas la réglementation européenne qui nous empêche d'agir.
J'en viens aux propositions du rapport. Tout d'abord, il faut repenser en profondeur le mécanisme de contrôle des dispositifs médicaux, notamment au niveau européen. La Commission a annoncé vouloir réformer le système des organismes notifiés plutôt que mettre en oeuvre une véritable autorisation de mise sur le marché, sur le modèle du médicament, pour les dispositifs les plus risqués. Nous pensons pour notre part que la certification de ces produits doit se fonder sur des études cliniques poussées, de sorte d'établir une véritable liste positive. Il faut également revoir le mode de fonctionnement des organismes notifiés, aussi bien leur désignation, leurs méthodes de travail ou leur contrôle par les autorités compétentes. Nous proposons d'agir sans délai au niveau national. Il faut commencer par appliquer les dispositions existantes, par prendre les textes d'application de la loi « Médicament ». Deux domaines sont plus particulièrement concernés : l'évaluation des dispositifs médicaux utilisés à l'hôpital et les liens d'intérêts. Les Etats-Unis accordent une très grande importance à la transparence : en complément du Sunshine Act, plusieurs Etats ou universités ont plafonné les sommes susceptibles d'être perçues par les médecins. En l'absence de données précises, nous pourrions dans un premier temps suggérer la mutualisation des sommes perçues et demander la publication en ligne des flux financiers concernés.
Il faut par ailleurs simplifier un certain nombre de procédures afin de rendre la matériovigilance plus réactive : création d'un portail unique de déclaration des incidents, meilleure association des médecins et des professionnels de santé.
Nous souhaitons instituer de véritables registres ; ces instruments de suivi existent dans plusieurs pays et ont fait la preuve de leur efficacité. En Australie, ils ont permis la détection précoce des problèmes liés aux prothèses de hanche DePuy. Il faut encourager les initiatives des sociétés savantes et inciter les médecins à intégrer cette obligation de recueil des données. Au besoin, il nous faudra revoir les règles de protection des données personnelles, quand il y a un impératif de santé publique. La précision du numéro de sécurité sociale fait obstacle à la tenue de registres par les personnes de droit privé. A l'inverse, pour assurer un suivi efficace, il faut pouvoir contacter rapidement les patients et donc les identifier facilement. Nous sommes encore loin du système du registre suédois de la chirurgie de la main où les patients peuvent être contactés par SMS, mais cela ne doit pas nous empêcher de faire évoluer les mentalités.
Enfin, le public doit pouvoir accéder à une information claire et précise sur l'ensemble des incidents car la transparence est le seul moyen de restaurer la confiance.
J'en viens aux interventions à visée esthétique. Il est important de clarifier les compétences et les actes qui relèvent de la médecine esthétique. Pour éviter d'être dépassé par l'évolution des techniques, c'est à la finalité des interventions qu'il faut s'attacher plutôt qu'aux actes eux-mêmes. Une fois cette classification établie, il faut déterminer clairement les compétences des uns et des autres : on ne peut laisser les actes ou matériels dangereux, lampes à lumière pulsée ou cabines de bronzage, entre les mains de personnes peu qualifiées. Non pas que tous les professionnels de l'esthétique soient des irresponsables. Au contraire : l'entreprise Eurofeedback, que nous avons visitée, limite volontairement la puissance des lampes à lumière pulsée. Cela pourrait constituer une des bases de l'encadrement de leur utilisation par les esthéticiennes.
Avec les cabines de bronzage, le danger n'est plus potentiel mais avéré. Je vous propose donc une mesure radicale : leur interdiction pure et simple, hors usage médical. Lorsqu'un risque sanitaire est avéré, nous ne pouvons rester inactifs. Je préfère être accusé aujourd'hui d'excès de précaution que demain d'inertie ! Prenez l'exemple de l'Islande : avant l'apparition des cabines de bronzage dans les années 1980, ce pays ne connaissait quasiment pas de cancers de la peau. Toutes les études montrent que ces cabines ont contribué à la brusque augmentation de l'incidence du mélanome du tronc qui y a été observée depuis 1992.
Il est également urgent de réserver certains actes aux médecins et de doter ceux qui les pratiquent d'une véritable capacité, à travers la création d'un diplôme d'études spécialisées complémentaires (DESC), plutôt que d'ériger la médecine esthétique en spécialité médicale à part entière.
Il faut créer un parcours de soins esthétiques, mettre en place une véritable esthéticovigilance. Les Français doivent être alertés sur les dangers qu'ils courent, sur les risques du tourisme esthétique. Dans un premier temps, il faut créer un site internet regroupant l'ensemble des incidents liés aux interventions à visée esthétique. J'espère que cette meilleure information ouvrira les yeux des consommateurs. La création d'un carnet de soins esthétiques constituerait un autre élément utile.
Voici le socle des propositions concrètes, réalistes et réalisables, formulées dans le rapport. Sans perdre de vue l'incidence économique des secteurs concernés ainsi que la nécessité de ne pas freiner l'innovation, je pense qu'elles sont de nature à replacer la sécurité au premier rang des priorités car, selon le principe d'Hippocrate, Primum non nocere.
Mme Gisèle Printz . - Les cabines de bronzage présentent un danger potentiel, dites-vous. Mais comment s'y prendre pour les interdire ? Faut-il une proposition de loi ?
M. Bernard Cazeau, rapporteur . - L'interdiction se ferait par décret.
Mme Catherine Génisson . - Je remercie et félicite la présidente et le rapporteur pour la qualité du rapport, en regrettant de n'avoir pu assister à toutes les auditions. Le rapport comporte beaucoup de propositions fondamentales, qui visent à encadrer sans pour autant empêcher.
Je crains toutefois que si les cabines de bronzage sont interdites dans les instituts esthétiques, elles ne se retrouvent chez les médecins. Certes, l'acte ne sera pas remboursé, et l'encadrement médical sera sans doute bienvenu, mais on ne fait que déplacer le problème. La réglementation des cabines de bronzage a déjà bien progressé, il ne faudrait pas que cette proposition soit inopérante.
M. Bernard Cazeau, rapporteur . - Les UVA dispensés dans les cabines de bronzage ne font pas bronzer et ne préparent pas à l'exposition au soleil, ils ne font que donner bonne mine. Ces cabines, qui sont fréquentées par beaucoup de jeunes, sont souvent mal utilisées, en durée ou en intensité, au risque de brûlures. On a constaté depuis quelques années une multiplication des tumeurs de la peau, dont la plus grave est le mélanome, mortel s'il n'est pas détecté précocement.
Je sais que cette proposition d'interdiction suscitera des protestations, mais les arguments scientifiques sont irréfutables. Les officines de bronzage ne disent pas la vérité aux clients !
Mme Chantal Jouanno, présidente . - Le rapport avance des chiffres : 16 % de la population a eu recours à ces cabines.
M. Gilbert Barbier . - A mon tour de féliciter la présidente et le rapporteur. Je vous trouve toutefois très gentils avec les pouvoirs publics de notre pays. Ceux-ci n'ont pas davantage réagi pour les prothèses PIP que pour le Mediator ! Peut-on parler de faute ? Je ne sais pas. Il faut toutefois s'interroger sur les procédures qui ont permis à cette entreprise de commercialiser pendant des années ses prothèses défectueuses.
Le rapport formule trente-huit propositions. Ne pourrait-on pas les hiérarchiser, afin de faire ressortir celles qui sont vraiment prioritaires, comme l'évaluation des organismes de certification ?
Au niveau européen, les choses évoluent lentement, et le libéralisme est la règle. En termes de matériovigilance, la Société française de chirurgie orthopédique et traumatologique (Sofcot) gère un registre des prothèses implantées : c'est un bon exemple de surveillance des patients, qui mériterait d'être officialisé au niveau du ministère de la santé. Même chose pour l'organisme associatif qui assure le suivi des pacemakers. Il manque toutefois un contrôle par les pouvoirs publics.
On ne peut totalement dédouaner les autorités sanitaires de notre pays, qui doivent pouvoir agir plus efficacement en matière de suivi et de contrôle.
M. René-Paul Savary . - La proposition choc du rapport est l'interdiction des cabines de bronzage. Outre l'aspect économique, ne craignez-vous pas que cette interdiction n'entraîne des dérives, un développement du tourisme esthétique ? Etant donné la législation européenne, ne pourrait-on, dans un premier temps, conditionner l'accès aux cabines de bronzage à la présentation d'un certificat médical ? Avez-vous envisagé une phase intermédiaire avant l'interdiction totale ?
M. Jean-Jacques Mirassou . - Sans condamner a priori tout ce qui tend à améliorer la perception esthétique que les gens ont d'eux-mêmes, nous sommes face à une question de société : comment développer l'esprit critique face aux canons esthétiques ? L'engouement pour les cabines de bronzage résulte bien d'un phénomène de société, d'une mode qui n'existait pas il y a vingt ans et passera peut-être dans vingt ans. Reste que, dans l'intervalle, elles peuvent être à l'origine de pathologies gravissimes.
Je plaide pour une information renforcée, afin que les gens qui ont recours à ces pratiques le fassent en pleine connaissance de cause. C'est un impératif. Dans une autre vie, je me suis frotté aux soins dentaires : dans ce domaine aussi, il existe des phénomènes de mode, et j'ai souvent refusé des opérations que je ne jugeais pas nécessaires. Pour le comblement des caries dentaires, certains pays ont disqualifié les amalgames, alors que le matériau de substitution - une résine qui se révèle n'être pas dénuée de risques - vaut avant tout pour son aspect esthétique, les laboratoires en ont d'ailleurs fait l'ardente promotion. Malgré une quantité d'études attestant de l'innocuité des amalgames - qui ne sont dangereux qu'au moment où on les retire - la question revient sur la table.
Comment développer l'esprit critique des gens ? La médecine esthétique est un moindre mal, mais que dire des autres pratiques, comme le blanchiment des dents ?
Je suis pour l'interdiction des cabines de bronzage : on n'a pas le droit de causer des mélanomes par pure coquetterie.
Mme Chantal Jouanno, présidente . - Notre mission n'a pas voulu traiter directement des amalgames dentaires, même si le sujet a été abordé via celui des perturbateurs endocriniens.
M. Bernard Cazeau, rapporteur . - Monsieur Savary, la position de l'Europe est ambiguë. Pour Bruxelles, le système est bon dans son principe, seule la pratique est mauvaise. Il faut donc mieux encadrer les organismes notifiés, renforcer le suivi, se doter d'une commission de contrôle. Mais il n'est pas question d'aller vers un système d'autorisation de mise sur le marché qui se rapproche de celui du médicament. Les députés européens, notamment le rapporteur de la résolution sur les prothèses PIP, Gilles Pargneaux, vont certes plus loin, mais c'est souvent le point de vue de la Commission qui l'emporte.
Les avancées attendues d'ici un ou deux ans au niveau européen ne changeront pas le système. Rien n'empêche d'agir au niveau national en matière de matériovigilance, par décret, par exemple en confiant à la nouvelle Agence nationale de sécurité du médicament et des produits de santé (ANSM) un certain nombre de nouvelles prérogatives.
Jamais Bruxelles n'interdira les cabines de bronzage. Pourtant, l'exemple de l'Islande est édifiant : depuis l'introduction des cabines dans les années 1980, les mélanomes se multiplient. Mais en Europe, c'est la loi de la concurrence qui prime. Il faut informer les gens sur le danger qu'ils courent !
M. René-Paul Savary . - Ils iront ailleurs...
M. Bernard Cazeau, rapporteur . - Tant pis pour eux. Il y en a bien qui vont se faire refaire le nez ou les seins en Tunisie, dans des conditions effarantes...
J'invite M. Barbier à se reporter aux pages 9 à 15 du rapport : on ne peut pas dire que nous soyons « gentils » avec les autorités nationales dans l'affaire PIP. Nous en démontrons les manquements. Les Américains, parfois laxistes, ont fini par interdire les prothèses PIP ; quant aux Australiens, ils étaient édifiés après avoir visité l'usine où elles étaient fabriquées.
Il est difficile de hiérarchiser les propositions, qui sont toutes importantes, d'autant que beaucoup découlent les unes des autres.
En matière d'esthétique, nous proposons que l'ANSM ait une compétence de police sanitaire, qu'elle centralise tous les signalements, qu'elle crée des sites internet ou des banques de données pour mieux informer la population. Nous demandons bien aux instances françaises de faire leur travail !
M. Mirassou a soulevé une question de société, celle du standard esthétique. Il faut d'abord renforcer l'information, sur Internet, à la télévision, et mettre en garde, à l'instar de ce qui existe sur les paquets de cigarette ou pour les accidents de la route. Nous faisons des propositions ; il appartient au Gouvernement et au législateur de décider. Si l'on dit clairement que les cabines de bronzage ne font pas bronzer mais favorisent en revanche le mélanome, cela devrait faire réfléchir.
Mme Chantal Jouanno, présidente . - S'agissant de l'affaire PIP, précisons que la procédure judiciaire en cours nous oblige à adopter un ton neutre dans le rapport.
Mme Aline Archimbaud . - A mon tour de remercier la présidente et le rapporteur. Les propositions du rapport sont intéressantes. Beaucoup reprennent des sujets qui ont été débattus dans le cadre de l'examen de la loi « Médicament », à commencer par la nécessité de mettre fin à l'opacité des relations financières entre les fabricants et les médecins.
Sur la question de la vigilance, j'ai été frappée, lors de notre déplacement en Suède, par le récit de cette chirurgienne, spécialisée dans la médecine de la main, qui a décidé de monter, seule avec son équipe, un registre ensuite étendu au niveau régional.
C'est une piste à explorer pour donner une suite concrète à ce rapport, pour faire évoluer les esprits. En Suède, les médecins se sont organisés face à l'inertie des autorités nationales et à la grande faiblesse de la réglementation européenne. Cela fonctionne. Les chirurgiens y utilisent seulement trois ou quatre marques sur les six cents existantes. Ne peut-on pas confier aux régions ou aux agences régionales de santé le soin de monter des expériences pilotes sur les registres avec des médecins volontaires ?
M. Jacky Le Menn . - Je salue le travail considérable mené par la présidente et le rapporteur. Grâce à la précision des questions posées lors des auditions, nous sommes allés au plus profond des thèmes abordés. Ce rapport est donc très riche. Contrairement à M. Barbier, il ne me semble pas insuffisamment sévère à l'égard des pouvoirs publics. Le marquage CE existe ; à nous de le renforcer pour éviter le laxisme, l'indifférence ou, pire, la complaisance.
Le triptyque santé, beauté, sécurité, dans le titre du rapport est bien trouvé. Il met le doigt sur l'émergence de l'attention portée au corps et à la recherche de la beauté dans notre société tout en insistant sur la priorité de la sécurité.
La clarification des compétences entre chirurgie esthétique, médecine esthétique et « simple » esthétique est un vrai sujet. La médecine esthétique appartient à un ensemble plus global qui doit être considéré dans son entier. Nous devons faire des choix clairs sur les compétences des uns et des autres sans accabler les professionnels de l'esthétique qui croient souvent bien faire. Quelles compétences doivent rester du ressort du médecin ? Lesquelles transférer aux autres professions médicales et sous quelles conditions de formation complémentaire ?
Les trente-huit propositions du rapport sont toutes pertinentes, le personnel médical doit se les approprier. En tout cas, elles doivent servir à faire avancer la réflexion européenne sur les dispositifs médicaux et esthétiques.
Mme Catherine Deroche . - A mon tour de saluer la qualité de ce travail engagé après le scandale des prothèses PIP. La frontière entre médecine et esthétique est difficile à tracer, vous avez su la marquer clairement.
Les propositions sur la formation et les diplômes qualifiants sont particulièrement intéressantes. Il faut combler le manque actuel.
Quant aux registres, la Sofcot en a bien créé un, mais il est peu renseigné par les chirurgiens sans que l'on ne sache très bien pourquoi.
Je suis favorable à l'interdiction des cabines de bronzage. D'autant que leur utilisation, a priori, ne dépend pas d'indications thérapeutiques, sauf pour...
Mme Catherine Génisson . - ...le psoriasis. Néanmoins, le détournement des actes médicaux n'est pas rare, notamment chez les kinésithérapeutes.
Mme Catherine Deroche . - L'idée que les cabines de bronzage préparent la peau au soleil est encore très présente dans les esprits. Pour autant, cette proposition choc ne doit pas occulter le reste du travail entrepris, qui est considérable.
Mme Marie-Thérèse Bruguière . - Effectivement ! La plupart des gens pensent que, grâce ces séances, ils bronzeront mieux. Tout un pan de l'économie est concerné. L'interdiction des cabines de bronzage fera l'effet d'une bombe.
Dans le même ordre d'idées, je pense aux officines de blanchiment des dents. C'est une activité juteuse pratiquée par des gens qui, contrairement aux dentistes, n'ont pas de compétences médicales. Nous allons au devant d'une autre catastrophe sanitaire.
Les complications liées au tourisme esthétique, par exemple une septicémie qui peut entraîner une incapacité, coûtent cher à la sécurité sociale. Ne peut-on pas demander aux médecins de déclarer que l'infection est la conséquence d'une opération à visée esthétique pratiquée à l'étranger ?
Mme Catherine Deroche . - C'est impossible car cela équivaudrait à un refus de prise en charge.
Mme Marie-Thérèse Bruguière . - La personne sera soignée, mais on progressera dans la connaissance du nombre de malades.
Mme Catherine Génisson . - C'est une proposition forte à élargir au suivi de toutes les opérations, qu'elles soient pratiquées en France ou à l'étranger, par des études randomisées. Il faut comparer les taux de complication des interventions réalisées en France de celles pratiquées à l'étranger.
Ce rapport soulève des problèmes sociétaux qui seraient à considérer sous un angle philosophique. Au XIX e siècle, une belle femme devait avoir la peau blanche et laiteuse, des formes avantageuses. Aujourd'hui, elle se doit d'avoir la peau hâlée et d'être mince. Les images de mannequins contribuent, en partie, à la progression de l'anorexie chez les adolescentes.
Ce rapport doit maintenant déboucher sur des propositions concrètes : des études randomisées par type d'intervention, par exemple la pose de prothèses mammaires, et un registre obligatoire pour les prothèses de hanche. Nous avancerons ainsi dans la connaissance objective des dispositifs et des complications que leur implantation peut entraîner. Sans oublier que ces complications sont parfois le fait du matériel, du médecin ou de l'état général du patient.
M. Alain Néri . - Le rapport est très intéressant, il met l'accent sur l'action pédagogique. Cela dit, des gens continuent de fumer bien que leur père soit mort d'un cancer du poumon ou qu'ils lisent tous les jours « fumer tue » sur leur paquet de cigarettes. Dans ce domaine, il est difficile d'avoir une action efficace. Lorsque j'ai travaillé sur le dopage, nous avions pris des précautions oratoires. Notre proposition de loi était relative à la « santé publique et à la protection des sportifs ». Cela n'a pas suffi à persuader les sportifs d'une vingtaine d'années de ne pas prendre des produits qui leur détruiraient la santé trente ans plus tard. Dans notre monde de plus en plus virtuel, tout le monde veut être beau et fort. Si, en prime, tout le monde pouvait être riche, ce serait encore mieux !
Comment traduire concrètement les propositions de ce rapport sans tomber dans des mesures si précises et sévères qu'elles deviendraient inapplicables ? Pourquoi vouloir appliquer aux pays du Sud de l'Europe les solutions du Nord et réciproquement ? Ce n'est pas forcément la solution. Nous sommes des Latins, et nous ne partageons pas forcément la vision des Anglo-Saxons.
Le rapport est achevé, il faut maintenant que tout ce travail ne se résume à l'interdiction des cabines de bronzage.
Mme Gisèle Printz . - Après le scandale des prothèses PIP, les implantations de prothèses mammaires ont-elles régressé ?
M. Bernard Cazeau, rapporteur . - Je vais aborder les différents sujets les uns après les autres. Les registres ? Notre proposition n° 18 est de les rendre obligatoires, sous les conditions posées par la Cnil, en y associant les médecins et le Conseil national de l'ordre des médecins. Sans eux, ils ne fonctionneront pas. L'exemple suédois est intéressant, mais ce pays compte neuf millions d'habitants, et la France plus de soixante-quatre millions. Il faudrait atteindre le seuil représentatif de 70 % mais notre culture est bien différente.
Les compétences sont à clarifier entre chirurgie esthétique, médecine esthétique et esthéticiennes. Le premier secteur est bien encadré ; pour les autres, il reste des marges de progression. Prenons l'épilation : les lampes à lumière pulsée d'une puissance inférieure à dix joules que les esthéticiennes ont le droit d'utiliser ou celles que l'on trouve dans le commerce, ne brûlent pas les poils à la racine. Au contraire, elles accéléreraient la repousse.
Les officines de blanchiment des dents ? Leur toxicité est moindre que celle des cabines de bronzage : les gens s'en sortent avec une bonne gingivite, leur vie n'est pas en danger.
Sur les complications, Mme Génisson a raison. En France, si elles sont plutôt rares et moyennes, elles existent aussi. Elles sont fréquentes à l'étranger pour une raison simple : les gens sont opérés puis se reposent trois jours à l'hôtel sans aucun suivi. Le risque d'infection est maximal durant cette période. Les gens consultent leur médecin généraliste en rentrant en France et la sécurité sociale paie. Comment l'éviter ? Je n'ai pas la solution.
La comparaison que M. Néri a établie avec le dopage est assez juste. Nous ne sommes pas sous une dictature, nous ne pouvons pas faire le bonheur des gens contre leur gré !
Mme Chantal Jouanno, présidente . - Si personne ne demande plus la parole, nous allons pouvoir passer au vote.
Mais avant cela, je vous propose le titre de notre rapport, qui pourrait être « Santé, beauté, une priorité : la sécurité ».
Quelqu'un a-t-il des observations ?
La mission commune d'information adopte cette rédaction.
Mme Chantal Jouanno, présidente . - Nous passons maintenant au vote du rapport. Qui est pour ? Qui est contre ? Qui s'abstient ?
Le rapport est adopté à l'unanimité.
Mme Chantal Jouanno, présidente . - Conformément aux règles que nous avions définies au début de nos travaux, je vous propose d'autoriser la publication du rapport que nous venons d'adopter.
Il en est ainsi décidé.
ANNEXE - LISTE DES PERSONNES AUDITIONNÉES
Mardi 28 février 2012
Dominique Maraninchi , directeur général, et Jean-Claude Ghislain , directeur de l'évaluation des dispositifs médicaux, de l'Agence française de sécurité sanitaire des produits de santé (Afssaps)
Mercredi 7 mars 2012
Gilles Johanet , président du Comité économique des produits de santé (Ceps)
Xavier Bertrand , ministre du travail, de l'emploi et de la santé
Mardi 13 mars 2012
Jean-Luc Harousseau , président de la Haute Autorité de santé (HAS), et Jean-Michel Dubernard , président de la Commission nationale d'évaluation des dispositifs médicaux et des technologies de santé (CNEDiMTS)
Edouard Couty , président du conseil d'administration, Erik Rance , directeur, et Sabine Gibert , directrice juridique, de l'Office national d'indemnisation des accidents médicaux, des affections iatrogènes et des infections nosocomiales (Oniam)
Alain Sautet , secrétaire général, et Christian Delaunay, responsable du registre, de la Société française de chirurgie orthopédique et traumatologique (Sofcot)
Mardi 20 mars 2012
Docteurs Walter Vorhauer , secrétaire général, Xavier Deau, responsable de l'enseignement, et André Deseur , président de la section « Exercice Professionnel », du Conseil national de l'ordre des médecins (Cnom)
Laurence Dagallier , directrice de la certification et de la formation, Thierry Thomas , responsable du pôle G-MED, et Corinne Delorme , responsable des affaires réglementaires, du Laboratoire national de métrologie et d'essais (LNE)
Table ronde sur les enjeux des interventions à visée esthétique dans les sociétés contemporaines : Jean-François Amadieu , sociologue, professeur à l'université Paris 1 Panthéon-Sorbonne, Elisabeth Azoulay , ethnologue, enseignante à Sciences-Po, et le professeur Maurice Mimoun , chef du service de chirurgie plastique, reconstructrice et esthétique et traitement chirurgical des brûlés à l'Hôpital Saint-Louis
Mardi 27 mars 2012
Professeur Eric Vicaut , vice-président de la chaire santé de Sciences-Po, membre de la Commission d'évaluation des produits et prestations (CEPP) de la Haute Autorité de santé (HAS)
Stéphane Pénet , directeur des assurances de biens et de responsabilité, et Anne-Marie Papeix , chargée de mission, de la Fédération française des sociétés d'assurance (FFSA)
Eric Le Roy , directeur général, et François-Régis Moulines , directeur des affaires publiques, juridiques et de la communication, du Syndicat national de l'industrie des technologies médicales (Snitem) ; Antoine Audry , président, et Timothé de Romance , en charge du secrétariat général, de l'Association pour la promotion de l'innovation des dispositifs médicaux (Apidim)
François-Xavier Selleret , directeur général de l'offre de soins (DGOS)
Mardi 3 avril 2012
Table ronde consacrée à la chirurgie esthétique : docteur Bruno Alfandari , président du Syndicat national de chirurgie plastique reconstructrice et esthétique (SNCPRE) ; docteur Michel Rouif , secrétaire général de la Société française des chirurgiens esthétiques plasticiens (Sofcep)
Marianick Lambert , secrétaire générale du Collectif interassociatif sur la santé (CISS) et Claude Rambaud , présidente du LIEN, association d'aide aux victimes d'infections contractées dans une clinique ou un hôpital
Murielle Ajello , présidente du Mouvement de défense des femmes porteuses d'implants et de prothèses (MDFPIP)
Mardi 10 avril 2012
Alain Griset , président, et Béatrice Saillard , directrice du département des relations institutionnelles, de l'Assemblée permanente des chambres de métiers et de l'artisanat (APCMA) ; Monique Amoros , coprésidente, et Martine Berenguel , membre, de la Confédération nationale artisanale des instituts de beauté (CNAIB)
Christian Couzinou , président du Conseil national de l'ordre des chirurgiens-dentistes ; Jacques Levoyer , vice-président de l'Union des jeunes chirurgiens-dentistes - Union dentaire (UJCD-UD)
Table ronde consacrée à la médecine esthétique : docteur Luc Sulimovic , président du Syndicat national des dermatologues et vénérologues ; docteur François Turmel , président de la Fédération française des médecins experts en médecine esthétique et anti-âge (FFMEAA) et vice-président de l'Union nationale des médecins à exercice particulier; docteur Catherine de Goursac , membre du conseil d'administration de l'Association française de médecine morpho-esthétique et anti-âge (AFME) ; docteur Lydia Houri , présidente de la Société française de médecine morphologique et anti-âge (SOFMMAA) et responsable du DIU médecine morphologique et anti-âge ; professeur Christian Dubreuil , coresponsable du diplôme universitaire de médecine morphologique et anti-âge à l'université Lyon 1 ; docteur Xavier Deau , vice-président du Conseil national de l'ordre des médecins
Mardi 24 avril 2012
Jean-Yves Grall , directeur général de la santé (DGS)
Stanislas Martin , chef du service de la protection des consommateurs et de la régulation des marchés, Axel Thonier , sous-directeur en charge du secteur industrie, santé et logement, Alain Boulanger , chef du bureau produits et prestations de santé et services à la personne, et Daniel Miles , membre du bureau produits et prestations de santé et services à la personne, de la direction générale de la concurrence, de la consommation et de la répression des fraudes (DGCCRF)
Table ronde avec des représentants de la presse magazine concernant la place de l'esthétique dans leur ligne éditoriale : Julie Lasterade , chef de service santé/forme, Grazia ; Ariane Goldet , rédactrice en chef beauté forme santé, Marie Claire ; Monique Le Dolédec , rédactrice en chef adjointe beauté, et Isabelle Sansonetti , journaliste spécialisée esthétique, Elle
Mercredi 25 avril 2012
Pierre Chirac , membre de la rédaction de la revue Prescrire, et Mathieu Escot , chargé d'études santé à l'UFC-Que Choisir, au nom du collectif Europe et Médicament
Jean-Luc Besse , président de l'Association française de chirurgie du pied (AFCP)
Mercredi 9 mai 2012
Représentants d'organismes notifiés allemands : Daniel Pflumm (Verband der TÜV), Hans-Heiner Junker (TÜV Süd) et Björn Clüsserath (TÜV Rheinland)
Thomas Fatome , directeur de la sécurité sociale
Maître Georges Lacoeuilhe , avocat au barreau de Paris
Professeur Daniel Loisance , membre de l'Académie nationale de médecine
Mardi 22 mai 2012
Laure Lechertier , responsable du département Politique des produits de santé, et Vincent Figureau , responsable du département Relations institutionnelles, de la Mutualité française
Confédération nationale de l'esthétique parfumerie (Cnep) : Régine Ferrère , présidente, représentant l'Union des professionnels de la beauté et du bien-être (UPB) et la Fédération française des écoles de l'esthétique et de la parfumerie (FFEEP), Jean-Yves Martin , trésorier de l'Union des marques du matériel (UMM), Jean-Claude Sirop , président de l'Union des marques de l'esthétique (UME), et Hervé Corlay , vice-président du Syndicat national des professionnels du bronzage en cabine (SNPBC)
*
Déplacement à Washington - 14 au 17 mai 2012
François Delattre , Ambassadeur de France
Cyril Cosme , conseiller pour les affaires sociales
Frédéric Badey, Sanofi, et Stanislas Vilgrain , Cuisine solutions, Conseillers du Commerce extérieur de l'Ambassade
Kimberly A. Trautman , associate director for international affairs, office of the center director, U.S. department of health and human services
Jack Mitchell , Chief of oversight and investigations, Sarah Levin , senior health policy advisor, for the Senate Special Committee on Aging chaired by Senator Herb Kohl (Démocrate-Wisconsin)
Docteur Harvey Fineberg , president of the Institute of Medicine
Docteur Timothy Baldwin , program director, Marissa A. Miller , branch chief, National Heart, Lung and Blood Institute (NHLBI)
Docteur Roderic Pettigrew , directeur du National Institute of Biomedical Imaging and Bioengineering (Nibib)
Docteur Diana Zuckerman , présidente du National Research Center for Women & Families, fondatrice du Cancer prevention and Treatment Fund
Michael B. Gropp , Vice President, Medtronic (Global Regulatory Strategy), April Veoukas , Abott (Regulatory Affairs), Ralph F. Ives , Executive Vice President, Janet E. Trunzo , Executive Vice President, Abby Pratt , Associate Vice President, Advanced Medical Technology Association (AdvaMed)
Nabil Mouline, senior director, international business development, Medco Health Services
United Biosource Corporation (conférence téléphonique)
Docteur Lawrence J. Green , American Board of Dermatology, ainsi que des représentants de l'association nationale des dermatologistes et de l'académie de médecine du Maryland
Déplacement à Boston - 17 et 18 mai 2012
Christophe Guilhou , Consul général de France
Docteur Frederick J. Schoen , director, BWH Biomedical Research Institute (BRI) technology innovation programm ;
Docteurs Aaron Kesselheim , Daniel Kramer («Experts in medical device regulation» en Europe et aux Etats-Unis) et Gregory Curfman , executive editor, New England Journal of Medicine
Cary Hagan , President and CEO of Intrinsic Therapeutics, Inc., Greg Lambrecht , Founder and Executive Director of Instrinsi Therapeutics, Inc., Stephen A. Hull , Principal and Founder of Hull Associates MedTech Consulting, et Thomas J. Sommer , President of Massachusetts Medical Device Industry Council
Déplacement en Suède - 22 et 23 mai 2012
Olivier Guérot , Premier conseiller, et Fabrice Perrin , conseiller pour les affaires sociales et la santé, Ambassade de France en Suède
Rencontre avec des représentants de l'Agence nationale de la santé et de la protection sociale et de l'Agence des produits médicaux
Karin Johansson , secrétaire d'Etat au ministère de la santé et des affaires sociales
Bodil Klintberg , directrice de la division pour les registres médicaux auprès de SKL, association des communes et régions de Suède
Marianne Arner , directrice du Centre de gestion du registre de la main
Déplacement au Danemark - 23 et 24 mai 2012
Véronique Bujon-Barré , Ambassadeur de France au Danemark
Docteur Paul Daniels Bartels , médecin en chef et directeur du programme de développement de la qualité des régions danoises
Julie Marie Cederholm , responsable de la division de la politique médicale et des affaires juridiques, ministère de la santé, Anne Mette Dons , médecin en chef et directrice à l'Agence de la santé, et Carlo V. Andersen , représentant de l'ancienne Agence des produits médicaux, désormais intégrée à l'actuelle Agence de la santé
Docteur Lisbet Rosenkrantz Hölmich , médecin en chef à la division de chirurgie plastique de l'hôpital d'Herlev, chercheuse dans le domaine des opérations mammaires
Entretien avec une association danoise de patients
Déplacement à Bruxelles - 6 juin 2012
Nils Behrndt , chef de cabinet adjoint de M. John Dalli, commissaire européen en charge de la santé et des consommateurs
Sabine Lecrenier et Aurélien Perez , unité technologies de la santé et cosmétiques à la direction générale de la santé et des consommateurs de la Commission européenne
Vincent Houdry , conseiller santé à la représentation permanente de la France auprès de l'Union européenne
Gilles Pargneaux , député européen, rapporteur de la résolution du Parlement européen sur les implants mammaires en gel de silicone défectueux produits par la société française PIP
* 1 Food and Drug Administration ; Center for Devices and Radiological Health; FDA Update on the Safety of Silicone Gel-Filled Breast Implants, June 2011.
* 2 En revanche, selon un autre document publié par l'agence (Food and Drug Administration ; Center for Devices and Radiological Health ; Anaplastic Large Cell Lymphoma in Women with Breast Implants : Preliminary FDA Findings and analyses, January 2011), si les porteuses d'implants mammaires sont plus susceptibles de développer une forme très rare de lymphome à grandes cellules, ce risque n'est pas plus élevé pour celles qui portent des prothèses à base de gel de silicone.
* 3 Afssaps - Direction générale de la santé - Etat des lieux des contrôles opérés par les autorités sanitaires sur la société Poly Implant Prothèse, février 2012.
* 4 Loi n° 2002-303 du 4 mars 2002 relative aux droits des malades et à la qualité du système de santé.
* 5 L'association de défense des porteuses de prothèses de la marque PIP (PPP) et le Mouvement de défense des femmes porteuses d'implants et de prothèses (MDFPIP).
* 6 Si l'on en croit une dépêche AFP du 6 janvier 2012, le fondateur de PIP, Jean-Claude Mas, incarcéré depuis, a reconnu, lors d'un interrogatoire réalisé en octobre 2011, avoir produit et utilisé pour ses prothèses un gel non homologué « moins cher [...] et de bien meilleure qualité ».
* 7 Dispositif médical implantable actif de cardiologie utilisé dans le traitement de l'athérosclérose.
* 8 Elle a toutefois exclu de son champ d'étude trois domaines qui relèvent de problématiques particulières : le tatouage et le piercing, régis par une réglementation qui date de 2008 et n'est pas contestée ; la chirurgie de changement de genre, qui dépasse de loin la préoccupation esthétique ; les implants dentaires, qui justifient une investigation propre.
* 9 Résolution du Conseil 85/C 136/01 du 7 mai 1985 concernant une nouvelle approche en matière d'harmonisation technique et de normalisation.
* 10 Guide relatif à la mise en application des directives élaborées sur la base des dispositions de la nouvelle approche et de l'approche globale, publié en 2000 par la Commission européenne.
* 11 La directive 98/79/CE du Parlement européen et du Conseil du 27 octobre 1998 concerne, quant à elle, les dispositifs médicaux de diagnostic in vitro.
* 12 Directive 2007/47/CE du Parlement européen et du Conseil du 5 septembre 2007 modifiant la directive 90/385/CEE du Conseil concernant le rapprochement des législations des Etats membres relatives aux dispositifs médicaux implantables actifs, la directive 93/42/CEE du Conseil relative aux dispositifs médicaux et la directive 98/8/CE concernant la mise sur le marché des produits biocides.
* 13 Annexe 1 de la directive 1993/42/CEE du Conseil du 14 juin 1993.
* 14 L'article 14 de cette même directive prévoit l'« enregistrement des personnes responsables pour la mise des dispositifs sur le marché ».
* 15 Décision n° 768/2008/CE du Parlement européen et du Conseil du 9 juillet 2008 relative à un cadre commun pour la commercialisation des produits et abrogeant la décision 93/465/CEE du Conseil.
* 16 Ces huit organismes sont : BSI (Royaume-Uni), Kema (Pays-Bas), LGA Intercert (Allemagne), LNE/G-Med (France), NSAI (Irlande), TÜV Rheinland (Allemagne), TÜV Süd (Allemagne), et TÜV Nord (Allemagne).
* 17 Eucomed, What Medical Technology Exactly is ( http://www.eucomed.org/medical-technology ), cité dans Heneghan, et al., « Medical-device recalls in the UK and the device-regulation process: retrospective review of safety notices and alerts », in British Medical Journal , mai 2011.
* 18 Igas, Evolution et maîtrise de la dépense des dispositifs médicaux, rapport n° RM2010-154, novembre 2010.
* 19 David Gollaher, Simon Goodall, California Health Institute - Boston Consulting Group, Competitiveness and regulation : the FDA and the future of America's biomedical industry , février 2011 ( http://www.bcg.com/documents/file72060.pdf ).
* 20 Ibidem.
* 21 Directive 2003/12/CE du 3 février 2003 concernant la reclassification des implants mammaires dans le cadre de la directive 93/42/CEE relative aux dispositifs médicaux.
* 22 Heneghan, et al., « Medical-device recalls in the UK and the device-regulation process: retrospective review of safety notices and alerts », in British Medical Journal, mai 2011.
* 23 Déclaration de M. Shuren devant la sous-commission de surveillance et d'enquête de la commission de l'énergie et du commerce de la Chambre des représentants. ( http://democrats.energycommerce.house.gov/sites/default/files/image_uploads/Testimony_07.20.11_Shuren.pdf ).
* 24 http://www.eucomed.org/uploads/Press%20Releases/BCG%20study%20report.pdf .
* 25 Recommendation NB-Med/2.7/Rec1, Co-ordination of Notified Bodies Medical Devices (NB-Med) on Council Directives 90/385/EEC, 93/42/EEC and 98/79/EC, «Guidance on clinicals».
* 26 Article 10 de la directive de 1993.
* 27 Communiqué de la Commission européenne, 19 avril 2010.
* 28 220 050 $ pour la procédure PMA et 4 049 $ pour le régime 510(k) (respectivement 55 013 $ et 2 024 $ pour les petites entreprises, c'est-à-dire réalisant moins de 100 millions de dollars de chiffre d'affaires) ; les dispositifs à usage pédiatrique exclusif sont exonérés.
* 29 Selon la déclaration de Jeffrey Shuren devant la commission de l'énergie et du commerce (sous-commission de la santé) de la Chambre des Représentants, « FDA user fees 2012 : How innovation helps patients and jobs ».
* 30 Intermacs distingue événements majeurs (mort, transplantation, guérison et changement de dispositif) et événements indésirables (mauvais fonctionnement du dispositif, infection, saignement, dysfonctionnement neurologique, problème respiratoire...). Il permet, entre autres, d'évaluer plus rapidement les nouveaux dispositifs.
* 1 Charles Grassley, sénateur de l'Iowa, un des deux principaux auteurs et promoteurs du Sunshine Act.
* 31 Amchem Products, Inc. v. Windsor, 521 U.S. 591 (1997).
* 32 Ortiz v. Fibreboard Corp., 527 U.S. 815 (1999).
* 33 Les deux autres membres fondateurs sont le Canada et le Japon.
* 34 Australian Government ; Department of Health and Aging; Therapeutic Goods Administration: Reforms in the Medical Devices Regulatory Framework (25 october 2010).
* 35 http://clinicaltrials.gov/
* 36 Environ 36,1 millions d'euros.
* 37 Isaps Biennial Global Survey.
* 38 Pour reprendre l'expression employée par Maurice Mimoun dans « L'Impossible limite, Carnets d'un chirurgien », Albin Michel, 1996.
* 39 « Les discriminations sur l'apparence dans la vie professionnelle et sociale », étude réalisée pour Adia par l'Observatoire des discriminations en collaboration avec la Sofres, mai 2003.
* 40 Circulaire n° DGS/SD2B/DHOS/O4/2005/576 du 23 décembre 2005 relative à l'autorisation et au fonctionnement des installations de chirurgie esthétique.
* 41 Décret n° 2005-840 du 20 juillet 2005 relatif à la sixième partie (dispositions réglementaires) du code de la santé publique.
* 42 Décret n° 2005-77 du 11 juillet 2005 relatif à la durée de réflexion prévue à l'article L. 6322-2 du code de la santé publique ainsi qu'aux conditions techniques de fonctionnement des installations de chirurgie esthétique.
* 43 David Picovschi, « Le chirurgien plasticien et la justice - Responsabilités, prévention et conduite à tenir en cas de litige » , thèse présentée pour le diplôme de docteur en médecine, faculté de médecine de Strasbourg, 2002, n° 72.
* 44 Ibidem.
* 45 Ibidem.
* 46 Audition du 20 mars 2012.
* 47 Dans sa résolution du 14 juin 2012 sur les implants mammaires en gel de silicone défectueux produits par la société française PIP (n° 2012/2621(RSP)), le Parlement européen « invite les Etats membres à mieux sensibiliser les patients aux risques potentiels associés à la chirurgie esthétique et à mieux réglementer la publicité dans ce domaine pour veiller à ce que les intéressés soient pleinement conscients des risques et des avantages ». La France fait donc figure de précurseur en ayant mis en place un tel dispositif dès 2002.
* 48 L'expression « anti-âge » est la transcription littérale de la terminologie utilisée aux Etats-Unis pour dénommer cette pratique. En France, on ne peut utiliser le même mot pour désigner des qualifications dans des spécialités différentes. L'adjectif « esthétique » étant réservé à la « chirurgie plastique, reconstructrice et esthétique », il a fallu trouver un autre titre pour les médecins exerçant dans ce domaine. En Italie et en Espagne on parle bien de « médecine esthétique ».
* 49 Auditions du 10 avril 2012, dans le cadre d'une table ronde sur la médecine esthétique.
* 50 Rapport n° 380 (2008-2009), tome I, fait par Alain Milon au nom de la commission des affaires sociales du Sénat sur le projet de loi portant réforme de l'hôpital et relatif aux patients, à la santé et aux territoires, page 217.
* 51 L'article 52 de la loi du 4 mars 2002 ne visait en effet que les interventions de chirurgie esthétique dans le nouvel article L. 6322-1 qu'elle a inséré dans le code de la santé publique.
* 52 Loi n° 2009-879 du 21 juillet 2009 portant réforme de l'hôpital et relative aux patients, à la santé et aux territoires.
* 53 Arrêté du 6 janvier 1962 fixant la liste des actes médicaux ne pouvant être pratiqués que par des médecins ou pouvant être pratiqués également par des auxiliaires médicaux ou par des directeurs de laboratoires d'analyses médicales non-médecins.
* 54 Aussi dénommée « lampe flash ».
* 55 BOCCRF n° 13 du 23 septembre 2001.
* 56 Décret n° 2011-382 du 11 avril 2011 relatif à l'interdiction de la pratique d'actes de lyse adipocytaire à visée esthétique.
* 57 « Cosmetic medical and surgical procedures - A national framework », rapport final du « I nter-jurisdictional cosmetic surgery working group », « Australian Health Ministers' Advisory Council », 2011.
* 58 Statutory order n° 1245, 24 octobre 2007, « Statutory order regarding cosmetic treatment ».
* 59 Titre X « Département de la santé et de l'hygiène mentale », sous-titre 32 « Conseil de l'ordre des médecins », chapitre 9 « Délégation et attribution de l'exécution de procédures médicales à visée esthétique et utilisation de dispositifs médicaux à visée esthétique ».
* 60 Il s'agit de la technique dite du « vampire lift » : le médecin prélève 5 à 10 ml de sang qu'il va passer à la centrifugeuse afin de séparer les cellules, puis réinjecte dans le visage du patient ce concentré riche en plaquettes censé activer le renouvellement cellulaire et stimuler la production de fibres de collagène et d'élastine. A ce jour, aucune étude scientifique n'a démontré son efficacité.
* 61 Direction générale de la santé, « Rapport sur les actes à visée esthétique », décembre 2008.
* 62 « Provision of cosmetic surgery in England », Healthcare Commission (devenue « Care Quality Commission » en 2009).
* 63 Le régulateur suédois obligeait les distributeurs de produits cosmétiques importés à suivre une procédure de notification spécifique alors même que ces produits avaient déjà fait l'objet d'une telle procédure dans un autre Etat membre. En outre, l'agence de santé suédoise demandait des informations précises aux fabricants des produits importés alors que la directive cosmétique n° 76/768/EEC impose seulement, pour la surveillance du marché, de déclarer l'adresse de l'usine de fabrication des produits visés ou le lieu de leur première importation au sein de l'Union.
* 64 Dans sa base de données Nando, à la date du 10 juillet 2012.
* 65 Afssaps - DGS, Etat des lieux des contrôles opérés par les autorités sanitaires sur la société Poly Implant Prothèse, février 2012.
* 66 Loi n° 2011-2012 du 29 décembre 2011 relative au renforcement de la sécurité sanitaire du médicament et des produits de santé.
* 67 « La réforme du système du médicament, enfin », rapport d'information n° 675 de Marie-Thérèse Hermange (2010-2011).
* 68 Décret n° 2012-745 du 9 mai 2012 relatif à la déclaration publique d'intérêts et à la transparence en matière de santé publique et de sécurité sanitaire.
* 69 Article L. 1453-1 du code de la santé publique.
* 70 Rapport précité ; proposition n° 6.
* 71 Loi n° 2009-879 du 21 juillet 2009 portant réforme de l'hôpital et relative aux patients, à la santé et aux territoires.
* 72 Le « Medical Device Expert Group » regroupe les autorités compétentes des Etats membres, les fabricants et les autres parties prenantes du secteur des dispositifs médicaux intéressés à la mise en oeuvre des directives les concernant. Il peut se réunir en comité restreint, ne comprenant que les autorités compétentes.
* 73 Directive 2007/47/CE du Parlement européen et du Conseil du 5 septembre 2007 précitée.
* 74 Directive 93/42/CEE du Conseil du 14 juin 1993 précitée.
* 75 Dispositions transposées en droit interne à l'article R. 5211-36-2 du code de la santé publique.
* 76 Loi n° 2004-806 du 9 août 2004 relative à la politique de santé publique.
* 77 Créé, comme les articles R. 5212-37 à R. 5212-41 du code de la santé publique, par le décret n° 2006-1497 du 29 novembre 2006.
* 78 Arrêté du 26 janvier 2007 relatif aux règles particulières de la matériovigilance exercée sur certains dispositifs médicaux, pris en application de l'article L. 5212-3 du code de la santé publique ; JO du 10 février 2007, page 2567.
* 79 http://www.legifrance.gouv.fr ; voir le dossier législatif de la loi du 29 décembre 2011, consulté le 10 juillet 2012.
* 80 Décret n° 2012-743 du 9 mai 2012 relatif à la publicité pour les dispositifs médicaux.
* 81 Article R. 165-3 du code de la sécurité sociale.
* 82 L'article L. 5312-4-2 du code de la santé publique définit précisément l'étendue de la protection offerte à un lanceur d'alerte. Il « ne peut faire l'objet d'une mesure discriminatoire, être écarté d'une procédure de recrutement ou de l'accès à un stage ou à une période de formation professionnelle, ni être sanctionné ou faire l'objet d'une mesure discriminatoire, directe ou indirecte, notamment en matière de rémunération, de traitement, de formation, de reclassement, d'affectation, de qualification, de classification, de promotion professionnelle, de mutation ou de renouvellement de contrat ».
* 83 Décision de la Commission 2010/227/UE du 19 avril 2010 relative à la banque de données européenne sur les dispositifs médicaux.
* 84 Loi n° 78-17 du 6 janvier 1978 relative à l'informatique, aux fichiers et aux libertés.
* 85 Articles 35 et 37 de la loi n° 2011-2012 du 29 décembre 2011 relative au renforcement de la sécurité sanitaire du médicament et des produits de santé.
* 86 Décret n° 2012-743 du 9 mai 2012 relatif à la publicité pour les dispositifs médicaux.
* 87 Article R. 5213-6 du code de la santé publique.
* 88 Sur http://ansm.sante.fr ; site consulté le 10 juillet 2012.
* 89 Système de refroidissement, filtre efficace pour neutraliser les sections indésirables du spectre lumineux des infrarouges et des ultraviolets...
* 90 Mayo Clinic Proceedings, volume 87, issue 4, pages 328-334, avril 2012.
* 91 A titre d'exemple, l'Etat de Victoria, en Australie, a mis en place, à partir de 2008, une rubrique spécifiquement consacrée aux interventions à visée esthétique au sein du site d'information indépendant « Better Health Channel » : elle permet l'accès à un ensemble d'informations dont la qualité est certifiée par un comité d'experts indépendants .
* 92 Loi n° 2009-879 du 21 juillet 2009 portant réforme de l'hôpital et relative aux patients, à la santé et aux territoires, précitée.
* 93 INCa, « Installations de bronzage UV, état des lieux des connaissances sur les risques de cancer », avril 2010.
* 94 INCa, « UV (artificiels et solaires), vitamine D et cancers non cutanés », juillet 2011.
* 95 Soit les médecins spécialistes, à l'exception des anesthésistes-réanimateurs, autorisés à faire partie d'équipes médicales réalisant des interventions de chirurgie esthétique.
* 96 Selon ce projet, « les médecins qui, à la date de publication du présent décret, justifient de trois ans de pratique dans le domaine des actes, procédés, techniques et méthodes médicaux à visée esthétique, peuvent continuer à les pratiquer, sans remplir les conditions de formation et de diplômes prévues [par le code de la santé publique] , sous réserve d'une validation par le Conseil national de l'Ordre des médecins, après avis de la commission nationale de validation de la pratique des actes à visée esthétique placée auprès du Cnom [...] . Cette validation intervient au plus tard avant le 31 décembre 2013, après examen d'un dossier présenté par les candidats ».
* 97 Décret n° 2012-637 du 3 mai 2012 relatif aux conditions dans lesquelles les docteurs en médecine peuvent obtenir une extension de leur droit d'exercice dans une spécialité non qualifiante.
* 98 Independent Healthcare Advisory Services (IHAS).
* 99 Sur www.treatmentsyoucantrust.co.uk (« les traitements dans lesquels vous pouvez avoir confiance »)
* 100 Soit environ 62 euros.
* 101 Rescrit du 10 avril 2012, n° 2012/25.
* 102 Arrêts du 20 novembre 2003, aff. C-212/01 « Margarete Unterpertinger » et aff. C-307/01 « Peter d'Ambrumenil ».
* 103 Notamment les praticiens autorisés à faire usage légalement du titre d'ostéopathe et les psychologues, psychanalystes et psychothérapeutes titulaires d'un des diplômes requis, à la date de sa délivrance, pour être recrutés comme psychologues dans la fonction publique hospitalière.
* 104 En application duquel un traitement identique doit être appliqué à des opérations identiques.
* 105 « La chirurgie esthétique rattrapée par le fisc », www.leparisien.fr , 7 juin 2012.
* 106 « Produits injectables de comblement de rides - Recommandations aux patients », juillet 2010.
* 107 Watchman TM est un petit dispositif en forme d'ombrelle implanté à l'entrée de l'auricule afin de l'obturer et d'éviter ainsi le passage d'éventuels caillots dans la circulation artérielle. Il s'adresse aux patients qui ne peuvent suivre un traitement anticoagulant.