







Rapport d'information n° 739 (2015-2016) de MM. Gilbert BARBIER et Yves DAUDIGNY , fait au nom de la commission des affaires sociales, déposé le 29 juin 2016
Disponible au format PDF (975 Koctets)
-
LISTE DES PRINCIPALES PROPOSITIONS
-
AVANT-PROPOS
-
EXPOSÉ GÉNÉRAL
-
I. LE MÉDICAMENT, PRODUIT D'UNE INDUSTRIE EN
MUTATION AU PLAN MONDIAL, EN DÉCLIN EN FRANCE
-
A. L'INDUSTRIE DU MÉDICAMENT EN
CHIFFRES : UNE PRODUCTION STRATÉGIQUE À LA
COMPÉTITIVITÉ DÉGRADÉE, UNE CONSOMMATION DE MOINS
EN MOINS ATYPIQUE
-
1. Les industries pharmaceutiques, une part
importante de l'économie nationale aujourd'hui en relatif
déclin
-
2. Une dépense maîtrisée, mais
une consommation de médicaments toujours supérieure à
celle des pays comparables
-
3. Une administration des prix du médicament
obéissant à des objectifs contradictoires et incluant une part de
soutien à l'industrie
-
1. Les industries pharmaceutiques, une part
importante de l'économie nationale aujourd'hui en relatif
déclin
-
B. UNE INDUSTRIE EN PROIE À DE PROFONDES
MUTATIONS
-
1. Un secteur industriel en recomposition
-
a) Un phénomène de concentration des
acteurs hors de France
-
b) L'attractivité du territoire
français en question
-
(1) Un environnement réglementaire
perçu comme complexe et imprévisible
-
(2) Des atouts à préserver
-
c) Un enjeu pour la place et l'influence
françaises dans le secteur pharmaceutique
-
d) La gestion des pénuries et des ruptures
de production, un problème récurrent
-
a) Un phénomène de concentration des
acteurs hors de France
-
2. Un changement de stratégie dans
l'innovation pharmaceutique
-
1. Un secteur industriel en recomposition
-
A. L'INDUSTRIE DU MÉDICAMENT EN
CHIFFRES : UNE PRODUCTION STRATÉGIQUE À LA
COMPÉTITIVITÉ DÉGRADÉE, UNE CONSOMMATION DE MOINS
EN MOINS ATYPIQUE
-
II. QUEL PÉRIMÈTRE DE PRISE EN
CHARGE POUR QUEL USAGE ?
-
A. LE MÉDICAMENT NON PRIS EN CHARGE
-
1. L'automédication : liberté
des prix et choix du patient
-
2. L'usage hors AMM, un mal
nécessaire ?
-
3. Le médicament non pris en charge,
excepté dans des indications thérapeutiques
spécifiques
-
4. Des déremboursements mal perçus
par l'opinion publique et parfois difficiles à intégrer
à une stratégie médicale.
-
1. L'automédication : liberté
des prix et choix du patient
-
B. LES MOYENS D'ACTION SUR LE COÛT DU
MÉDICAMENT
-
A. LE MÉDICAMENT NON PRIS EN CHARGE
-
III. QUEL MÉCANISME INSTITUTIONNEL POUR LA
MEILLEURE PRISE EN CHARGE DU MÉDICAMENT ?
-
A. UN MÉCANISME DE MISE SUR LE
MARCHÉ ACCUSÉ D'OPACITÉ
-
B. QUELLE PRISE EN CHARGE DE
L'INNOVATION ?
-
1. Une réforme de l'évaluation
nécessaire, une gestation contrariée
-
2. Faut-il remplacer le système actuel de
fixation des prix pour les médicaments innovants ?
-
a) La rémunération du
médicament en fonction de l'apport en années de vie et en
qualité de vie
-
b) Rémunérer les médicaments
en fonction des « économies »
réalisées par l'assurance maladie
-
c) Adapter la rémunération à
l'efficacité en vie réelle du médicament
-
d) Quel bilan pour le système actuel de
rémunération et d'accès des patients à
l'innovation ?
-
a) La rémunération du
médicament en fonction de l'apport en années de vie et en
qualité de vie
-
1. Une réforme de l'évaluation
nécessaire, une gestation contrariée
-
A. UN MÉCANISME DE MISE SUR LE
MARCHÉ ACCUSÉ D'OPACITÉ
-
I. LE MÉDICAMENT, PRODUIT D'UNE INDUSTRIE EN
MUTATION AU PLAN MONDIAL, EN DÉCLIN EN FRANCE
-
EXAMEN EN COMMISSION
-
LISTE DES PERSONNES AUDITIONNÉES
N° 739
SÉNAT
SESSION ORDINAIRE DE 2015-2016
|
Enregistré à la Présidence du Sénat le 29 juin 2016 |
RAPPORT D'INFORMATION
FAIT
au nom de la commission des affaires sociales (1) sur la politique du médicament ,
Par MM. Gilbert BARBIER et Yves DAUDIGNY,
Sénateurs.
|
(1) Cette commission est composée de : M. Alain Milon , président ; M. Jean-Marie Vanlerenberghe , rapporteur général ; M. Gérard Dériot, Mmes Colette Giudicelli, Caroline Cayeux, M. Yves Daudigny, Mme Catherine Génisson, MM. Jean-Pierre Godefroy, Gérard Roche, Mme Laurence Cohen, M. Gilbert Barbier, Mme Aline Archimbaud , vice-présidents ; Mme Agnès Canayer, M. René-Paul Savary, Mme Michelle Meunier, M. Jean-Louis Tourenne, Mme Élisabeth Doineau , secrétaires ; M. Michel Amiel, Mme Nicole Bricq, MM. Olivier Cadic, Jean-Pierre Caffet, Mme Claire-Lise Campion, MM. Jean-Noël Cardoux, Daniel Chasseing, Olivier Cigolotti, Mmes Karine Claireaux, Annie David, Isabelle Debré, Catherine Deroche, M. Jean Desessard, Mme Chantal Deseyne, M. Jérôme Durain, Mmes Anne Emery-Dumas, Corinne Féret, MM. Michel Forissier, François Fortassin, Jean-Marc Gabouty, Mme Françoise Gatel, M. Bruno Gilles, Mmes Pascale Gruny, Corinne Imbert, MM. Éric Jeansannetas, Georges Labazée, Jean-Baptiste Lemoyne, Mmes Hermeline Malherbe, Brigitte Micouleau, Patricia Morhet-Richaud, MM. Jean-Marie Morisset, Philippe Mouiller, Louis Pinton, Mmes Catherine Procaccia, Stéphanie Riocreux, M. Didier Robert, Mme Patricia Schillinger, MM. Michel Vergoz, Dominique Watrin, Mme Evelyne Yonnet . |
LISTE DES PRINCIPALES PROPOSITIONS
__________
1. Mieux valoriser les innovations liées à la recherche publique française en renforçant les moyens de la recherche fondamentale et en menant une politique plus active en matière de brevets.
2. Agir au niveau européen pour renforcer les exigences relatives à l'évaluation du médicament pour l'autorisation de mise sur le marché
3. Combiner les avis de la commission de la transparence en cas de déremboursement avec des recommandations permettant d'assurer la meilleure prise en charge du patient tout en limitant le report de prescription
4. Définir une politique de santé publique relative au développement de l'automédication
5. Développer des partenariats entre le Ceps et la Cnam afin de favoriser le bon usage du médicament et d'agir sur les comportements de promotion, de prescription et d'usage
6. Mener une action intergouvernementale avec nos principaux partenaires européens afin de définir un cadre commun de négociation du prix des médicaments les plus onéreux
7. Clarifier la notion de secret des affaires en transposant rapidement la directive européenne
8. Proscrire la mise en place de mesures fiscales ponctuelles
9. Prévoir une audition publique annuelle du Ceps devant les commissions des affaires sociales présentant les résultats de la négociation avec les industriels et la comparaison entre les prix du médicament en France et dans les pays voisins
10. Renforcer la place de l'assurance maladie au sein du Ceps
11. Mettre en place selon les préconisations du rapport Polton un critère unique d'évaluation comparative des médicaments, la valeur thérapeutique relative (VTR)
12. Fusionner en un seul les taux de prise en charge à 15 %, 30 % et 65 % en s'appuyant sur les évaluations conduites par le rapport Polton
AVANT-PROPOS
Mesdames, Messieurs,
En ce qui concerne le médicament, on ne peut que partager l'affirmation de la caisse nationale d'assurance maladie, selon laquelle « la situation de la France en Europe reste singulière » 1 ( * ) .
En effet, « sur les huit principales classes de médecine générale [antibiotiques, antidépresseurs, antidiabétiques oraux, antihypertenseurs, inhibiteurs de la pompe à protons (IPP), hypolipémiants, antiasthmatiques, anxiolytiques et hypnotiques], notre pays se situe en deuxième position en volume, mais largement en tête en termes de dépenses par habitant. Ainsi, bien que les dépenses aient décru entre 2013 et 2014, en France (- 6 %) plus que chez nos voisins européens (+ 7,6 % au Royaume-Uni, - 0,9 % en Allemagne), les écarts en 2014 restent significatifs, avec un montant par habitant de 30 % supérieur à celui observé en Allemagne, de + 50 % par rapport au Royaume-Uni et de + 80 % par rapport aux Pays-Bas ».
Les Français restent donc de forts consommateurs de médicaments avec en moyenne 48 boîtes consommées par personne et par an .
Mais leur confiance dans les médicaments qu'ils consomment connaît de fortes variations en fonction des scandales sanitaires (- 12 points entre 2013 et 2014 avant de remonter à 85 % de confiance en 2015 selon le baromètre Ipsos pour le syndicat des industries du médicament, le Leem). La confiance majoritaire des Français dans le médicament s'appuie sur une vision de l'industrie pharmaceutique considérée comme innovante. Le médicament est donc perçu comme un des porteurs des progrès de la médecine. Toutefois, seuls 49 % des Français font confiance aux informations données par les firmes sur leurs produits, ce qui signifie que tant le bénéfice d'un médicament que ses risques doivent faire l'objet d'une analyse extérieure à l'industrie.
L'industrie pharmaceutique est parfois même dénoncée comme un « monstre » assurant ses intérêts financiers au détriment des objectifs de santé publique. Cette dénonciation s'appuie sur deux arguments. D'une part, la mise sur le marché de médicaments peu efficaces dont les effets secondaires dépasseraient leur intérêt si on les compare aux traitements existants et qui pèseraient inutilement sur les dépenses publiques. D'autre part, le prix disproportionné des traitements innovants arrivés récemment sur le marché, qui conduirait à un rationnement des soins. Les appels portés par des associations, des professionnels de santé, des représentants politiques et des journalistes demandent à renforcer la lutte contre les liens d'intérêts entre experts, autorités sanitaires et industrie, et à limiter les profits de l'industrie à un niveau raisonnable. Ils s'appuient sur la volonté d'une évaluation plus juste des risques liés au médicament et de leur meilleure appréhension par les malades . Mais l'impératif de sécurité peut entrer en contradiction avec la demande d'accès rapide aux traitements innovants auxquels sont légitimement attachées les associations de patients.
Le régime des autorisations temporaires d'utilisation (ATU) est au coeur des contradictions qui caractérisent les attentes des Français en matière de médicament. Il est parfois dénoncé comme étant trop souple au regard de l'évaluation du médicament, mais sert de modèle à plusieurs autres pays pour permettre l'accès rapide des malades à l'innovation. Lors de son audition par la commission des affaires sociales le 20 janvier 2016, Jean-Sébastien Bordes, du Formindep, a ainsi relevé qu'en décembre 2014, les six membres du comité antiviraux contre l'hépatite C, saisi par le directeur général de l'Agence nationale de sécurité du médicament et des produits de santé (ANSM) pour donner un avis sur une demande d'autorisation temporaire d'utilisation pour une association d'antiviraux, ont considéré que celle-ci était moins bien tolérée, moins évaluée que celle déjà utilisée et qu'elle entraînait davantage d'effets secondaires. Ils ont donc refusé la demande. Malgré cette prise de position unanime des membres du comité, le directeur général de l'ANSM « a décidé de mettre en place une ATU de cohorte pour les antiviraux d'Abbvie au même titre que l'ATU de cohorte d'Harvoni. La décision de l'agence s'inscrit dans le cadre de sa stratégie d'offrir un accès large, équitable et précoce aux traitements innovants . »
Les propos du Pr Bruno Guerci professeur de diabétologie au CHU de Nancy, publiés dans un communiqué « spécial diabète » inséré dans le quotidien Le Monde en date du mercredi 23 mars 2016 font un constat inverse et sans doute excessif : « En France, dans le domaine de la santé et notamment du traitement du diabète, l'accès à l'innovation thérapeutique est difficile et par conséquent retardé. Pour le médicament comme pour le dispositif médical, certains produits ne sont pas sur le marché français alors qu'ils sont commercialisés depuis plusieurs années dans d'autres pays européens, tandis que d'autres ont un accès restreint. (...) L'accès aux innovations est donc moins fluide que dans d'autres pays européens ou qu'aux Etats-Unis, et ce, au détriment des patients. »
Les autorités régulatrices sont prises entre les demandes contradictoires de ceux qui voient d'abord l'espoir offert par une nouvelle molécule et refusent des délais de procédure vus comme une perte de chance en matière de traitement, et de ceux qui insistent sur la nécessité de distinguer innovations réelles et fausses innovations et de mesurer pleinement les risques qui s'attachent nécessairement à la prise d'une substance active 2 ( * ) . Ces derniers soulignent que les scandales liés au médicament au cours des dernières années ont reposé en partie sur une notification insuffisante des effets indésirables graves liés à des médicaments et sur un défaut de traitement des signaux remontés jusqu'aux autorités régulatrices. Cette question est liée à celle des conditions de mise sur le marché des médicaments car, comme le souligne Catherine Hill, responsable du service d'épidémiologie de l'institut Gustave-Roussy, « Aujourd'hui, on voit bien que l'industriel s'abrite derrière l'ANSM, qui s'abrite derrière les prescripteurs, qui s'abritent derrière l'efficacité du médicament et leur respect des règles édictées par l'ANSM et l'industriel, bouclant ainsi la boucle » 3 ( * ) .
L'affaire du Mediator a entraîné une augmentation très importante de l'exigence publique en matière d'indépendance des agences sanitaires et des experts auxquels elles ont recours. La ressource fiscale prélevée sur les laboratoires et affectée à l'Agence française de sécurité sanitaire des produits de santé (Afssaps, ancien nom de l'ANSM), qui avait longtemps été perçue comme le gage de l'autonomie de cette agence et la garantie de son développement pour assurer les missions qui lui étaient confiées, fut désormais perçue comme un financement direct par l'industrie. Cette affaire qui concerne un médicament commercialisé pendant plus de trente ans a aussi mis en lumière la nécessité d'un meilleur suivi du médicament tout au long de son utilisation par la population.
La meilleure gestion du médicament, de ses bénéfices et de ses risques, des opportunités qu'il offre et de son coût, par les autorités sanitaires et de sécurité sociale est ainsi un enjeu récurrent, que renforce d'un côté la bonne nouvelle du retour de l'innovation thérapeutique après une longue phase de plateau, et de l'autre la contrainte durable qui pèse sur nos finances sociales .
Il a donc paru nécessaire à la commission des affaires sociales de se pencher sur la place du médicament aujourd'hui dans notre système de santé , et particulièrement d'assurance maladie. Produit d'une industrie en mutation qui a changé sa manière d'innover, le médicament est soumis à la contrainte financière qui pèse sur notre système d'assurance maladie et doit faire l'objet d'une évaluation rénovée.
EXPOSÉ GÉNÉRAL
I. LE MÉDICAMENT, PRODUIT D'UNE INDUSTRIE EN MUTATION AU PLAN MONDIAL, EN DÉCLIN EN FRANCE
L'industrie du médicament constitue un secteur stratégique à la fois par la nature de sa production, qui constitue un outil indispensable au service de la santé des populations, mais aussi par son poids économique . Aux termes du rapport du conseil stratégique des industries de santé (CSIS) du 11 avril dernier, elle contribue en outre « à la fois au rayonnement international de la recherche et de l'industrie françaises et à la souveraineté sanitaire du pays » .
Sur le plan économique et à l'échelle mondiale, le dynamisme et la compétitivité de l'industrie pharmaceutique en font un secteur particulièrement porteur pour l'avenir . Cette position s'explique à la fois par la croissance continue de la demande - en raison du vieillissement global de la population, de l'ouverture de nouveaux marchés dans les pays en développement ainsi que de la progression des maladies chroniques et des maladies rares -, et par le caractère très innovant du secteur - que l'on pense au développement des biotechnologies, des thérapies ciblées et de la médecine personnalisée, ou encore aux nouveaux usages du numérique dans le domaine de la santé.
Dans ce contexte, les entreprises françaises du secteur, traditionnellement figures de proue de l'industrie nationale, affichent une érosion continue de leur compétitivité depuis les dernières années . Selon un document préparatoire 4 ( * ) au CSIS du 11 avril dernier, cette tendance s'expliquerait « notamment par l'essoufflement du dispositif de financement et d'innovation et une recomposition difficile du secteur face à la concurrence ».
Vos rapporteurs ont dès lors souhaité entendre plusieurs acteurs de la sphère économique (représentants de l'OCDE et de la DGE, notamment) dont les contributions ont permis d'établir un état des lieux chiffré de la situation fragile de l'industrie pharmaceutique sur le territoire français (A). Les auditions conduites ont par ailleurs permis d'explorer les enjeux liés aux changements de modèle à l'oeuvre dans une industrie récemment entrée dans une nouvelle phase d'innovation (B).
Vos rapporteurs ont ainsi souhaité établir un constat objectif et neutre, qui ne préjuge pas de considérations de politique industrielle - qui ne relèvent d'ailleurs pas du champ de la commission des affaires sociales. Ils soulignent cependant que l'industrie pharmaceutique, aujourd'hui largement financiarisée, ne saurait être considérée comme une industrie tout à fait comme une autre , dans la mesure où son chiffre d'affaires et sa forte rentabilité sont largement financés par la collectivité. Pour l'ensemble de ces raisons, ils n'ont pas souhaité formuler de propositions relatives à la dimension industrielle de la politique de médicament, et ont concentré leurs préconisations sur la question de la recherche publique.
A. L'INDUSTRIE DU MÉDICAMENT EN CHIFFRES : UNE PRODUCTION STRATÉGIQUE À LA COMPÉTITIVITÉ DÉGRADÉE, UNE CONSOMMATION DE MOINS EN MOINS ATYPIQUE
L'industrie du médicament constitue traditionnellement un secteur stratégique pour la France, tout au long de la chaîne de commercialisation du produit. Du point de vue de la production, les industries de santé, en dépit de leur place importante dans l'économie française et de leur résilience face à la crise de 2008, sont entrées au cours de la dernière décennie dans une phase de décroissance. Du point de vue de la consommation, l'enjeu est bien évidemment aujourd'hui celui de la maîtrise de la dépense de médicaments remboursables, qui constituent la majeure partie des produits commercialisés. Dans ce contexte, l'administration des prix du médicament obéit à des objectifs contradictoires qui brouillent sa lisibilité.
1. Les industries pharmaceutiques, une part importante de l'économie nationale aujourd'hui en relatif déclin
a) Un secteur-clé de l'économie française...
Avec 28,7 milliards d'euros d'exportations et une contribution positive au solde commercial de la France de 3,3 milliards d'euros en 2015 5 ( * ) , les produits de santé 6 ( * ) représentent 10 % des exportations industrielles nationales (hors énergie et aéronautique) et constituent le troisième secteur des exportations françaises .
Selon le Leem, les seuls médicaments ont permis de dégager en 2014 un excédent commercial de 6 milliards d'euros , pour une part de la France dans le marché mondial du médicament de 3,9 %.
Décomposition du chiffre d'affaires de l'industrie du médicament en 2014
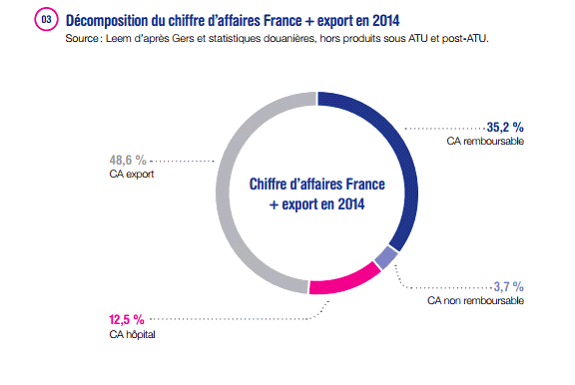
Source : Leem d'après Gers et statistiques douanières, hors produits sous ATU et post-ATU
Le poids des industries de santé dans l'économie nationale est par ailleurs plus important en France que dans les pays comparables : la part du chiffre d'affaires de ces industries (qui représentait au total 70 milliards d'euros en 2014) dans l'industrie manufacturière est en effet de 5,5 % en France, contre 4,1 % au Royaume-Uni et 3,6 % en Allemagne.
Du point de vue de leur contribution à l'emploi, les industries de santé employaient 130 875 salariés en 2013 7 ( * ) , soit 6 % de l'emploi industriel en France, dont 99 715 emplois directs pour le seul médicament à usage humain 8 ( * ) .
Ces chiffres absolus doivent être mis au regard de la capacité d'entraînement de ce domaine industriel sur les autres secteurs de l'économie française. Selon les indications fournies par la DGE, le médicament représenterait au total - en comptant les emplois induits - 200 000 à 300 000 salariés , dont 40 000 pour la seule production industrielle 9 ( * ) . Le rapport préparatoire au CSIS précité relève par ailleurs un potentiel d'entraînement très élevé du secteur des produits de santé , qui se caractérise à la fois par un coefficient multiplicateur sur les industries connexes élevé (2,49) et par une croissance remarquable de ce coefficient entre 2000 et 2010 (de l'ordre de 18 %).
Ce rapport souligne par ailleurs la capacité de résilience de l'industrie pharmaceutique française, en relevant qu'elle a globalement mieux résisté à la crise financière de 2008 que le reste de l'industrie manufacturière.
b) ... mais un avantage compétitif qui se dégrade
Pour autant, l'érosion de ses parts de marché sur le plan international ainsi que la baisse des investissements associés traduisent une tendance au recul des industries de santé françaises .
Le rapport du CSIS du 11 avril dernier relève ainsi « une baisse tendancielle des parts de marché françaises dans les exportations pharmaceutiques de la zone euro », qui ont chuté de plus de 20 % en 2000 à 12 % en 2015, soit une perte de valeur d'environ 10 milliards d'euros. D'après le rapport économique du Leem pour 2015, les exportations de médicaments sont en repli de 5 % en 2014, soit un montant global de 25 milliards d'euros (après cependant une hausse de 4 % en 2013).
Les investissements des entreprises du secteur se sont par ailleurs contractés de 4,5 % en moyenne chaque année entre 2010 et 2013 10 ( * ) , 80 % d'entre eux concernant en outre des sites déjà existants. Ce recul, qui intervient après une phase de croissance du début des années 2000 jusqu'à la crise de 2008 (augmentation de 2,5 milliards d'euros en 2000 à 4,5 milliards en 2008), s'expliquerait notamment par les faibles taux de marge des industries pharmaceutiques françaises par rapport à celles des pays européens comparables.
Enfin, d'après le Leem, l'année 2014 a marqué la troisième année de baisse consécutive du chiffre d'affaires des médicaments en ville (en prix fabricant hors taxes) : celui-ci s'élevait à 20 milliards d'euros, soit - 2 % par rapport à 2013, après - 2,4 % en 2013 et - 2,5 % en 2012.
2. Une dépense maîtrisée, mais une consommation de médicaments toujours supérieure à celle des pays comparables
La France se voit traditionnellement caractérisée par une consommation de médicaments supérieure à celle constatée dans les autres pays comparables . Le problème existerait à la fois du point de vue de la quantité et de la qualité de la consommation ; plusieurs des acteurs auditionnés ont ainsi rappelé que certains produits dangereux, tel que le Mediator, ont continué d'être commercialisés en France plusieurs années après leur interdiction dans d'autres pays.
Selon les acteurs entendus par vos rapporteurs, cette consommation atypique résulterait à la fois de l'attitude des patients et de celle de leurs prescripteurs, dans le cadre d'une culture générale de la « magie du médicament » : il serait ainsi admis que chaque symptôme trouverait sa solution dans la prise médicamenteuse - une simple fatigue passagère pouvant ainsi donner lieu à la prescription d'un sédatif.
Pour autant, ce phénomène d'hyperprescription et d'hyperconsommation n'est pas nécessairement facile à établir objectivement, et se trouve en lien avec une multitude de facteurs . Les travaux menés par vos rapporteurs ont ainsi permis de mettre en évidence plusieurs éléments venant nuancer ce constat, qui demeure cependant en partie vérifié.
a) Une stabilisation de la dépense de médicaments principalement due à la baisse des prix au cours des dernières années
Selon les comptes de la santé, la dépense relative à la consommation de médicaments de ville s'est élevée à 33,9 milliards d'euros en 2014 , dont 30,2 milliards pour le médicament remboursable, en augmentation de 2,7 % par rapport à l'année précédente .
Cette croissance, qui rompt avec le mouvement de baisse amorcé au cours des deux années précédentes (respectivement - 1,3 % et - 1,7 % en 2012 et 2013), s'explique principalement par la hausse très importante de la rétrocession hospitalière (+ 83 %), en raison de l'arrivée des nouveaux traitements contre le virus de l'hépatite C.
Si l'on neutralise les effets de la rétrocession, la consommation globale s'inscrit dans la tendance constatée au cours des années précédentes, avec un recul de 1,4 % en valeur et une hausse des volumes de 2,5 %.
Évolution de la consommation de
médicaments en ville
(y compris rétrocession
hospitalière) entre 2007 et 2014
|
2007 |
2008 |
2009 |
2010 |
2011 |
2012 |
2013 |
2014 |
|
|
Ensemble (en M€) |
32 249 |
32 972 |
33 505 |
33 661 |
34 013 |
33 586 |
33 021 |
33 903 |
|
Remboursables |
29 284 |
29 547 |
29 952 |
30 127 |
30 406 |
29 752 |
29 197 |
30 196 |
|
dont rétrocédés |
1 135 |
1 218 |
1 309 |
1 399 |
1 516 |
1 663 |
1 600 |
2 932* |
|
Non remboursables |
2 965 |
3 425 |
3 553 |
3 534 |
3 608 |
3 834 |
3 824 |
3 707 |
|
Évolution (%) valeur |
3,5 |
2,2 |
1,6 |
0,5 |
1,0 |
- 1,3 |
- 1,7 |
2,7 |
|
prix |
- 2,5 |
- 2,3 |
- 2,6 |
- 2,2 |
- 2,0 |
- 3,2 |
- 4,4 |
- 3,7 |
|
volume |
6,2 |
4,6 |
4,3 |
2,7 |
3,1 |
2,0 |
2,8 |
6,6 |
* Estimation.
Note : La consommation de médicaments non remboursables a été révisée sur les années 2010 à 2013 à la suite de l'intégration de données définitives du Leem.
Source : Drees - Comptes de la santé
Au total, on a ainsi observé au cours des dernières années une certaine stabilisation de la dépense de médicaments - aujourd'hui remise en question dans le contexte de retour de l'innovation, avec notamment l'arrivée sur le marché des nouveaux traitements contre l'hépatite C, qui ont donné lieu à une consommation supérieure de 1,1 milliard d'euros en 2014 (avant dispositifs supplémentaires de remises conventionnelles ad hoc ).
La décomposition de l'évolution de la dépense, qui figure dans le tableau ci-dessus, montre que cette stabilisation est principalement liée à la baisse des prix . Les comptes de la santé relèvent en effet que, calculé à qualité constante par l'INSEE 11 ( * ) , le prix des médicaments diminue de 3,7 % en 2014 (-4,1 % pour les spécialités remboursables).
Cette décrue est en premier lieu due aux actions de baisse des prix, qu'il s'agisse de baisses directes, de la mise en place des tarifs forfaitaires de responsabilité (TFR), du renforcement de la mesure « tiers-payant contre génériques » 12 ( * ) , ou encore de l'augmentation du nombre de spécialités généricables. Si l'on compare d'ailleurs l'évolution de la consommation de médicaments en France et en Allemagne au cours de la décennie 2000-2011, on observe en France une croissance inférieure de 14 % en chiffre d'affaires hors taxes (CAHT), bien que supérieure de 21 % en volume 13 ( * ) .
Ces actions sont complétées par divers dispositifs de maîtrise médicalisée, qui agissent tant sur la baisse des prix que sur celle des volumes de consommation - qui progressent moins vite depuis 2010, avant le rebond de 2014. Il s'agit notamment des campagnes de l'assurance maladie relatives à la consommation d'antibiotiques, de la généralisation de la rémunération sur objectifs de santé publique (ROSP), ou encore des déremboursements de médicaments dont le service médical rendu (SMR) est jugé insuffisant.
Entendus par vos rapporteurs, les professionnels du secteur, représentés par le Leem, ont estimé que l'ensemble de ces actions fait peser un poids trop important sur ces entreprises dans l'effort global de réduction des dépenses de santé, comparativement à leur contribution à ces dépenses - de l'ordre de 15 %. Ils ont par ailleurs relevé que la stabilisation des dépenses de médicament résultait bien davantage des actions de baisse des prix que de la mise en oeuvre de la maîtrise médicalisée.
b) Vers la fin de l'exception
française en matière de consommation
de
médicaments ?
Les comptes de la santé relèvent que la France est toujours « l'un des tous premiers consommateurs européens de médicaments », en volume par habitant comme en chiffre d'affaires hors taxes (CAHT) par habitant . En unités standards 14 ( * ) , la consommation y est ainsi supérieure de plus d'un quart (27 %) à la moyenne constatée pour un groupe de huit pays européens comparables 15 ( * ) .
Cette surconsommation apparente se doublerait d'un phénomène de « malprescription », évoqué devant vos rapporteurs par l'association UFC Que Choisir, dont elle a rappelé qu'elle constituait un enjeu particulièrement important pour les personnes âgées - dont l'organisme, plus fragile, élimine par ailleurs plus difficilement les substances ingérées, qui restent de ce fait plus longtemps dans l'organisme. Une étude de février 2015, publiée dans le numéro 91 de la revue Que choisir Santé , a ainsi mis en évidence, à partir de l'analyse de 327 ordonnances comportant 9 médicaments en moyenne et 21 produits au maximum, un phénomène de surprescription auprès des personnes âgées : sur 40 % de ces ordonnances figurait au moins un médicament jugé inadapté.
Le développement d'une éducation à la santé comme alternative à la consommation médicamenteuse, considéré comme l'une des grandes lacunes de la loi Bertrand de 2011, est ainsi défendu de longue date par plusieurs des acteurs entendus par vos rapporteurs, notamment la revue Prescrire. Il a également été rappelé que toute solution à une pathologie n'est pas nécessairement médicamenteuse : face à la maladie d'Alzheimer, il faudrait ainsi travailler autant sur des solutions thérapeutiques que sur le développement de l'accompagnement social.
Une étude publiée en juin 2012 par l'association des laboratoires de recherche internationaux (LIR) et la chaire santé de l'Essec, et portant sur la période 2000-2011, conclut cependant à « la fin de l'exception française en matière de consommation de médicaments ». Elle relève en effet que, si la France était bien en tête, en 2000, du classement européen de la consommation pour les huit classes thérapeutiques les plus utilisées 16 ( * ) , elle se situe en 2011 dans la moyenne européenne. Cette évolution s'expliquerait par deux facteurs : la modération relative de la consommation de médicaments en France , qui se traduit par de faibles taux d'évolution, tandis qu' un phénomène de rattrapage a été observé dans les autres pays européens .
Évolution de la consommation
médicamenteuse
par classes thérapeutiques en Europe entre 2000
et 2013
(taux d'évolution moyen annuel en
volume)
|
Anti
|
Anti
|
Anti
|
Anxio-
|
Anti
|
Anti
|
Anti
|
Hypo
|
|
|
All |
10,9 |
- 0,1 |
9,5 |
- 3,5 |
0,1 |
5,1 |
7,0 |
9,9 |
|
Be |
11,9 |
0,2 |
5,1 |
- 1,3 |
- 1,0 |
5,3 |
5,2 |
9,3 |
|
Es |
10,2 |
- 0,5 |
6,0 |
1,0 |
0,4 |
4,4 |
4,7 |
12 ,4 |
|
Fr |
8,1 |
- 1,0 |
2,1 |
1,3 |
2,2 |
5,2 |
3,8 |
4,1 |
|
It |
13,1 |
0,7 |
8,8 |
- 0,5 |
0,6 |
3,6 |
5,2 |
15 |
|
RU |
8,6 |
2,3 |
6,3 |
- 1,4 |
1,6 |
7,6 |
4,5 |
17,7 |
Au total, au terme des auditions conduites par vos rapporteurs, il semblerait que « l'exception culturelle » française en matière de consommation de médicaments demeure principalement notable sous trois aspects :
- en premier lieu, la consommation d' antibiotiques , pour laquelle, malgré une diminution de 15 % entre 2000 et 2011, on observe toujours une surconsommation par rapport aux autres pays européens ;
- en second lieu, selon les informations transmises par l'OCDE, un recours plus marqué en France aux médicaments les plus chers ;
- enfin, un plus faible taux de recours aux médicaments génériques , malgré un taux de substitution satisfaisant (de l'ordre de 80 %) mais qui ne porte que sur 32 % des produits disponibles. Cet aspect devrait cependant se normaliser du fait de la généralisation de la prescription en dénomination commune internationale (DCI) et de l'arrivée de nouvelles générations de prescripteurs mieux formés aux génériques.
Plusieurs facteurs doivent par ailleurs être pris en compte pour nuancer le constat d'une consommation atypique en France.
En premier lieu, les données relatives à la consommation devraient sans doute être pondérées en fonction de la prévalence des maladies.
En second lieu, la longueur de certaines ordonnances, notamment à destination des personnes âgées, résulte sans doute en partie de la plus grande prévalence des polypathologies.
Au total, le véritable profil de la consommation française inclurait sans doute un mélange de surconsommation sur certains produits, de sous-consommation sur d'autres, et de mésusages.
3. Une administration des prix du médicament obéissant à des objectifs contradictoires et incluant une part de soutien à l'industrie
Les politiques publiques menées en matière de médicament répondent à des considérations multiples et souvent contradictoires, entre objectifs de santé publique, soutien à l'industrie et contraintes budgétaires. Elles doivent en effet à la fois permettre le plus large accès possible de la population aux innovations en matière de santé et offrir un environnement favorable au développement des industries sur le territoire français, tout en maîtrisant la dépense associée pour garantir l'équilibre du régime d'assurance maladie.
Le dialogue entre l'Etat et les industriels est formalisé de longue date en France . En 2004 a ainsi été créé le conseil stratégique des industries de santé (CSIS) , dont l'objectif est d' « oeuvrer de façon partenariale pour l'attractivité du pays dans le secteur des biens de santé, pour attirer ou maintenir sur son territoire des activités de recherche ou de production de biens de santé ». Cet organisme, dans ses derniers travaux en date d'avril 2016, a réaffirmé les principes du dialogue entre l'Etat et l'industrie et de la préférence conventionnelle en matière de régulation.
Le comité économique des produits de santé (Ceps) , placé sous la tutelle des ministres chargés de la santé, du budget et de l'économie et chargé de négocier avec les industriels le prix des produits de santé remboursés, s'inscrit dans la même logique.
De manière plus récente et dans le but d'anticiper l'arrivée des innovations thérapeutiques coûteuses, un comité d'interface informel entre l'État et l'industrie du médicament a par ailleurs été mis en place dans le cadre de la préparation des projets de loi de financement de la sécurité sociale pour 2015 et 2016.
Selon plusieurs des acteurs entendus par vos rapporteurs, qui voient dans cette organisation une forme d'opacité, il en résulterait en particulier une certaine illisibilité du système d'administration des prix à la française .
Le modèle traditionnel de soutien à l'industrie nationale est en outre soupçonné de favoriser la politique des industries, en France davantage que dans les autres pays de l'OCDE. Ainsi, du fait que les pouvoirs publics n'affirmeraient pas suffisamment leurs exigences face aux laboratoires résulterait l'abandon de certains pans de la production pharmaceutique en raison de leur faible rentabilité, en dépit de leur fort intérêt pour la santé publique. Ce serait le cas s'agissant, notamment, de certains antibiotiques, ou encore du vaccin contre la coqueluche.
Le récent avis rendu par l'autorité de régulation professionnelle de la publicité (ARPP) sur la campagne d'affichage de Médecins du monde, qui dénonce le coût jugé exorbitant de différents traitements pharmaceutiques, est encore venu alimenter ce soupçon.
Pour autant, plusieurs éléments permettent de nuancer ce soupçon.
En premier lieu, l'OCDE a rappelé devant vos rapporteurs la difficulté de procéder à des comparaisons internationales sur le prix des médicaments , en raison à la fois des obstacles à l'établissement d'un panier commun, de la confidentialité des rabais obtenus par les administrations de santé, et des difficultés à prendre en compte les différentes taxations touchant au champ du médicament.
En second lieu, il apparaît que l'enjeu majeur pour les administrations françaises de santé est aujourd'hui celui de la maîtrise du coût des médicaments . Cet objectif est d'ailleurs pris en compte dès le stade de l'admission au remboursement : par exemple, les produits présentant une faible amélioration du service médical rendu (ASMR 5) ne sont plus admis au remboursement que s'ils permettent de réaliser une économie dans le coût du traitement. Cette économie peut résulter du coût facial du produit comme de la forme de traitement proposée : le passage d'un traitement par intraveineuse à un traitement par voie orale permet ainsi de réaliser une moindre dépense. Il arrive ainsi, selon les indications fournies par le Ceps, que certains produits classés en ASMR 5 ne sortent pas sur le marché français.
En sens inverse, la prise en compte des contraintes de l'industrie pharmaceutique peut être opérée dans le sens de la préservation des impératifs de santé publique . Des hausses de prix peuvent ainsi être ponctuellement consenties pour des produits très anciens, dont le prix n'a pas été revalorisé depuis longtemps et dont l'exploitation devient de ce fait déficitaire, mais dont l'intérêt thérapeutique est majeur.
B. UNE INDUSTRIE EN PROIE À DE PROFONDES MUTATIONS
1. Un secteur industriel en recomposition
a) Un phénomène de concentration des acteurs hors de France
Le secteur pharmaceutique français se compose d' un tissu dense de PME , surplombé par quelques entreprises de taille mondiale, et à côté duquel se développe un tissu de jeunes TPE innovantes, spécialisées notamment dans les biotechnologies et les technologies médicales. Outre les laboratoires français tels que Sanofi, Pierre Fabre, Servier ou encore Ipsen, certains laboratoires étrangers sont très implantés sur le territoire, comme par exemple Novartis et GSK. Le marché français est dominé par les groupes Sanofi et Novartis, qui détenaient respectivement 7,8 % et 7 % des parts de marché en 2014.
Cette organisation est cependant remise en question, depuis plusieurs années, par un phénomène de délocalisation de la production et de la recherche hors du territoire français .
Selon les indications transmises par la direction générale des entreprises (DGE), ce mouvement ne se traduit pas, la plupart du temps, par un arrêt brutal de la production en France : le plus souvent, les laboratoires procèdent à un désengagement progressif en cédant leur exploitation, au moins pour un temps, à des sous-traitants . Ce désengagement porte notamment sur des produits anciens , dont la France est fortement productrice. Au total, l'on recense aujourd'hui davantage de ventes d'usines que d'ouvertures d'usines spécialisées dans le médicament sur le territoire français.
Le modèle de production du laboratoire Pfizer, entendu par vos rapporteurs, illustre la réalité de ce phénomène. L'entreprise ne dispose plus aujourd'hui de sites de production en propre en France, la production sur le territoire ayant été cédée à neuf façonniers - qui travaillent pour plusieurs laboratoires, et qui ne sont pas tous français. Elle ne dispose plus davantage de sites de recherche en France ; il a toutefois été indiqué que Pfizer poursuivait un effort d'investissement dans la recherche en France sous la forme de partenariats public-privé, notamment avec l'Inca, pour un montant de l'ordre de 81 millions d'euros par an.
Cette évolution s'accompagne d'un phénomène relatif de concentration des acteurs, dont le rapprochement entre Sanofi et le laboratoire américain Merck, qui a donné lieu à la co-entreprise Sanofi Pasteur-MSD dans le domaine des vaccins constitue un illustre exemple - sa disparition avant la fin de l'année 2016 a cependant été annoncée en mars dernier. En raison de la grande variété des produits, des techniques et des marchés dans le secteur, l'industrie pharmaceutique demeure cependant peu concentrée au niveau mondial 17 ( * ) .
b) L'attractivité du territoire français en question
(1) Un environnement réglementaire perçu comme complexe et imprévisible
Selon les représentants du laboratoire Pfizer, l'évolution de son modèle de production résulterait principalement d'une logique générale d'externalisation , liée à la perte des brevets sur les anciens blockbusters , plus qu'au contexte français lui-même.
Le Leem a cependant fait valoir un phénomène de perte d'attractivité de la France au regard des industriels comme des investisseurs, qui l'empêcherait de bénéficier des investissements internationaux et du mouvement de retour de l'innovation actuellement en cours.
La DGE a souligné que la concurrence entre les industriels du médicament, qui prend davantage place à l'échelle européenne qu'à l'échelle mondiale, se traduit par des choix d'implantation résultant principalement des conditions de compétitivité de chaque pays, notamment en fonction des infrastructures et du coût du travail.
Selon le Leem, la plus faible attractivité de la France résulterait :
- du caractère imprévisible de l'environnement réglementaire français , notamment s'agissant de la fiscalité du médicament : le taux K a ainsi fait l'objet d'une profonde réforme en 2014 pour devenir le taux L, tandis que le mécanisme W a été introduit sur les seuls médicaments contre le virus de l'hépatite C. Cet aspect a également été mis en avant par les représentants du laboratoire Pfizer - qui saluent par ailleurs le modèle français. Les industriels réclament, depuis plusieurs années déjà, l'inscription du taux L dans un cadre pluriannuel , qui permettrait selon eux aux professionnels de gagner de la visibilité et un pouvoir d'anticipation, tandis qu'il permettrait aux pouvoirs publics d'inciter à des réformes visant à réaliser des économies sur le médicament de manière structurelle plutôt qu'à court terme.
- de la complexité des circuits administratifs et de la longueur des délais nécessaires au développement et à la commercialisation des produits de santé, singulièrement des vaccins. Selon le rapport d'activité du Ceps pour l'année 2014, l'ensemble de la procédure d'inscription d'un médicament de ville, du passage devant la commission de transparence à la publication des textes nécessaires au journal officiel (JO), est de 80 jours pour les médicaments génériques, mais de 237 jours pour les non génériques . Selon le rapport préparatoire au CSIS précité, le délai pour la mise en place d' essais cliniques dans le domaine pharmaceutique est de 30 à 40 jours en France, contre 15 jours seulement en Belgique . Serait ainsi en particulier découragée l'innovation sur les petits marchés, ou « niches thérapeutiques », les laboratoires étant réticents à engager des démarches longues et compliquées sur des segments à l'intérêt financier faible.
S'agissant en particulier de la production des médicaments génériques, la moindre attractivité du territoire français ne s'expliquerait pas, selon les informations transmises à vos rapporteurs, par le différentiel de coût de la main-d'oeuvre, qui ne serait que de 5 à 7 centimes d'euros par rapport à une spécialité extraterritoriale.
Il serait davantage le résultat des contraintes réglementaires et juridiques perçues comme trop importantes par les entreprises - quoique des accords de façonnage aient permis de maintenir la production de plusieurs blockbusters sur le territoire français.
Il a cependant été précisé que la pression exercée sur le prix des génériques, qui rogne la profitabilité des entreprises du secteur, aboutirait à la précarisation de ce modèle.
(2) Des atouts à préserver
Pour autant, les atouts importants du modèle français pour les décisions d'investissement des entreprises du secteur ont également été soulignés par les acteurs entendus par vos rapporteurs, y compris par les représentants des laboratoires pharmaceutique.
En premier lieu, la France dispose d'un environnement scientifique et technique particulièrement attractif pour les activités de recherche : l'excellence de ses équipes médicales et de recherche en sciences de la vie, ainsi que la qualité de ses formations, sont ainsi largement reconnues.
Selon la DGE, le crédit impôt-recherche (CIR) renforcé en 2008 constitue un outil crucial pour les entreprises, de même que le crédit d'impôt innovation mis en place en 2013 pour les PME. Selon le rapport du CSIS précité, « la France est reconnue comme l'un des pays qui offrent le traitement fiscal de R&D le plus incitatif pour les entreprises ». L'industrie pharmaceutique qui, avec 3 milliards d'euros, est la 3 e branche industrielle en matière de R&D (après l'automobile et l'aérospatiale), bénéficie d'environ 10 % des crédits du CIR.
Le même rapport du CSIS place également l'engagement des pouvoirs publics en faveur de la recherche dans les sciences du vivant parmi les facteurs d'attractivité de la France.
Il rappelle ainsi que la santé et les biotechnologies sont identifiées comme l'un des six axes stratégiques du programme des investissements d'avenir (PIA), 3 milliards d'euros ayant déjà été mobilisés à ce titre.
Il renvoie par ailleurs à « un ensemble de dispositifs nationaux [...] mis en place afin de soutenir la recherche partenariale et de renforcer les transferts de technologie » : les instituts de recherche technologique (IRT), les six instituts hospitalo-universitaires (IHU), la création de l'alliance nationale pour les sciences de la vie et de la santé (Aviesan) et de l'alliance pour la recherche et l'innovation des industries de santé (Ariis), les conventions industrielles de formation par la recherche (Cifre), ou encore le lancement du fonds d'accélération des biotechnologies en santé avec une dotation de 340 millions d'euros
En matière d' innovation , le mécanisme des autorisations temporaires d'utilisation (ATU) , qui permet une diffusion rapide et sous protocole sécurisé des nouveaux médicaments, avant obtention de leur AMM, est unanimement salué.
La France dispose par ailleurs d'infrastructures industrielles de qualité et d'un savoir-faire reconnu en la matière. La France dispose en particulier de positions solides notamment dans les domaines de l'oncologie et du vaccin.
Selon les indications générales transmises par la DGE, le crédit d'impôt pour la compétitivité et l'emploi (Cice) n'aurait cependant qu'un faible effet sur ce secteur industriel, dans la mesure où il bénéficie principalement aux bas salaires. Le salaire mensuel moyen brut était en effet de 4 408 euros en 2013 dans l'industrie pharmaceutique, tandis que le Cice concerne les salaires allant jusqu'à 2,5 Smic.
L' étendue de sa couverture maladie , pour un pays qui représente 4 % du marché mondial du médicament, ainsi que le mécanisme de la négociation conventionnelle constituent également des facteurs importants.
c) Un enjeu pour la place et l'influence françaises dans le secteur pharmaceutique
Selon les éléments transmis par la DGE, le mouvement de délocalisation pourrait s'intensifier au cours des prochaines années . En dépit du développement de plusieurs entreprises spécialisées dans les biotechnologies, comme le groupe LFB, ou du rapprochement entre Sanofi et la société Genzyme, les entreprises françaises ne semblent en effet pas avoir réellement pris le virage du médicament biologique 18 ( * ) , qui est aujourd'hui le plus porteur d'innovation et de valeur ajoutée.
Les médicaments produits en France sont en effet principalement des produits anciens, dont la marge diminue avec le temps, sous l'effet des pertes de brevets. 40 000 des emplois français du secteur pharmaceutique portent ainsi sur des médicaments chimiques d'ancienne génération ; ils seront donc potentiellement touchés de plein fouet par les baisses de prix qui continueront d'affecter l'industrie française au cours des prochaines années. La DGE a par ailleurs relevé que seulement 40 % des investissements productifs réalisés en 2013 l'avaient été sur des sites de production de médicaments biologiques.
Selon le Ceps, ce phénomène de délocalisation hors de France se traduit très concrètement par le fait que les décisions qui auparavant se prenaient en France, dans le cadre des négociations sur les prix, se prennent désormais à l'étranger - y compris pour le laboratoire Sanofi.
D'une manière plus globale, plusieurs des acteurs entendus par vos rapporteurs ont souligné la nécessité de préserver l'influence française dans ce domaine, autant par l'attractivité économique du territoire que par la sauvegarde de la place de la France dans les institutions du secteur .
Plusieurs acteurs ont ainsi insisté sur la nécessité de conserver une agence délivrant des AMM françaises , ce qui permettrait à la fois de garantir une certaine indépendance de la France par rapport à l'échelle européenne et de conserver la place stratégique du territoire au regard notamment de la conduite d'essais cliniques. Il est ainsi à noter que, si le laboratoire Gilead ne produit pas le traitement contre le virus de l'hépatite C en France, il y conduit cependant des essais cliniques.
Au niveau européen, c'est la qualité de son système de pharmacovigilance qui permet à la France de peser dans les décisions d'AMM . Mesurée par le nombre de positions de rapporteur confiées à chaque pays, la place de la France au sein des instances de décision européenne avait ainsi fortement chuté, jusqu'à la 24 e place, après le scandale du Mediator.
Il est à noter que l'influence française est toujours très forte en matière de fixation des prix : le prix facial déterminé en France constitue en effet un point de référence pour les autres pays, notamment s'agissant des vaccins.
S'agissant enfin de la production de génériques , la question de l'attractivité française se pose davantage en termes de garantie de la qualité de la production . Le maintien d'usines de génériques en France, et plus généralement en Europe, apparaît en effet comme un facteur-clé du maintien de la confiance des consommateurs, alors que la pénétration de ces médicaments a pu être limitée par quelques polémiques récurrentes.
En particulier, la question de la qualité des matières premières employées dans le processus de production avait déjà été évoquée par un rapport de l'Igas de 2012 19 ( * ) comme par un récent rapport de la commission des affaires sociales du Sénat 20 ( * ) sur les médicaments génériques : selon ce dernier, « entre 60 % et 80 % des matières premières seraient fabriquées dans des pays hors Europe, principalement en Asie (Inde et Chine), et les inspections de sites de production, peu nombreuses, mettent en évidence de graves dysfonctionnements ».
d) La gestion des pénuries et des ruptures de production, un problème récurrent
La concentration des acteurs industriels hors du territoire français, voire hors du territoire européen, accroît le risque pesant sur la continuité de la production et de l'approvisionnement en médicaments . Il s'agit là à la fois d'un enjeu de stratégie industrielle et de santé publique : cette situation pose en effet particulièrement problème lorsqu'elle concerne des médicaments d'intérêt thérapeutique majeur, et pour lesquels il n'existe aucune alternative thérapeutique.
En particulier, l'utilisation de matières premières provenant largement de pays situés en dehors de l'Union européenne 21 ( * ) -et ce, que la production intervienne ou non sur le sol français- constitue une vulnérabilité importante du système de production pharmaceutique. Les insuffisances en matière de qualité des produits constituent également un risque, du fait du renvoi de lots par les autorités sanitaires.
Vos rapporteurs soulignent cependant que si ce phénomène de délocalisation contribue à renforcer le risque de rupture d'approvisionnement, il ne l'explique pas à lui seul. Il semble qu'il résulte plus largement des modes de gestion de leur production retenus par les industries pharmaceutiques, qui opèrent souvent à flux tendus, pour des raisons de rentabilité économique principalement.
En tout état de cause, la conjugaison de ces deux logiques contribue à faire en sorte que le moindre imprévu survenant dans la chaîne de production et de commercialisation aboutisse à un complet déséquilibre du système, qui peut entraîner des ruptures d'approvisionnement pouvant durer plusieurs mois.
Le Haut Conseil de la santé publique (HCSP) a souligné devant vos rapporteurs que s'agissant des vaccins, pour lesquels des ruptures de stock ont été constatées à plusieurs reprises au cours des dernières années (notamment sur le BCG, le vaccin contre le méningocoque ou encore contre la coqueluche), ce sont des pans entiers de la politique vaccinale menée auprès des jeunes enfants qui peuvent ainsi se trouver remis en cause.
Afin de répondre à cet enjeu majeur, et dans la continuité de la mesure n° 27 adoptée par le CSIS du 5 juillet 2013, l'article 151 de la loi de modernisation de notre système de santé (LMSS) a prévu divers aménagement visant à mieux anticiper la réponse en cas de rupture d'approvisionnement des molécules les plus indispensables. Selon les informations transmises à vos rapporteurs, plusieurs textes d'application devraient par ailleurs intervenir prochainement.
D'autres ruptures de stock résultent en revanche d'une décision de politique industrielle , par laquelle les entreprises pharmaceutiques mettent fin à la commercialisation d'un produit devenu insuffisamment rentable.
À cet égard, il convient de rappeler que l'accord cadre du Ceps du 9 février 2016 prévoit des pénalités financières pour les laboratoires en cas de cessation de la commercialisation de certains produits, dès lors que cet arrêt aboutit à ce qu'un besoin thérapeutique majeur ne se trouve plus couvert. Il ne s'agit cependant pas nécessairement d'une solution durable, dans la mesure où les industriels peuvent toujours faire le choix d'acquitter cette pénalité plutôt que de relancer leur production.
Plusieurs des acteurs auditionnés par vos rapporteurs ont insisté sur la nécessité d'une revalorisation périodique du prix des médicaments les plus anciens afin d'inciter les laboratoires à poursuivre leur production. Selon les informations transmises, des hausses de prix sont effectivement opérées sur certains produits anciens afin d'éviter leur retrait du marché, comme par exemple en 2015 pour l'adrénaline injectable.
2. Un changement de stratégie dans l'innovation pharmaceutique
a) De la fin des blockbusters au retour de l'innovation
Le modèle économique dans le secteur du médicament a été longtemps fondé sur l'innovation en même temps que le développement de ce que l'on a appelé les produits « blockbusters » , susceptibles de dégager un chiffre d'affaires de plus d'un milliard de dollars par an. La fin de cette période s'est caractérisée par un faible nombre d'innovations thérapeutiques majeures, qui a conduit à une certaine stabilisation du marché du médicament en France.
Ce modèle traditionnel est cependant profondément remis en cause depuis quelques années, sous l'effet de deux facteurs.
En premier lieu, de nombreux brevets sont aujourd'hui tombés , sans que l'innovation n'ait toujours pris le relais dans tous les laboratoires pour remplacer ces médicaments princeps . Entendue par vos rapporteurs, la filiale française du groupe Pfizer 22 ( * ) a ainsi indiqué la perte de nombreux brevets sur des médicaments blockbusters , tel par exemple que le Lipitor. L'entreprise est de ce fait entrée dans une phase de décroissance .
En deuxième lieu, la période actuelle se caractérise, depuis quelques années, par un retour de l'innovation . Selon les indications fournies par le Leem, plus de 180 molécules seraient à l'heure actuelle en développement, et il est probable que plusieurs maladies qui n'ont actuellement pas de solution thérapeutique pourraient en trouver au cours des prochaines années. La HAS a cependant tempéré ces propos devant vos rapporteurs en indiquant que seulement 100 des 650 médicaments mis sur le marché en 2014 étaient réellement innovants.
En tout état de cause, ce mouvement devrait se poursuivre au cours des prochaines années. Dans le champ de la chimiothérapie par exemple, des perspectives importantes sont ainsi ouvertes par le séquençage génétique des tumeurs - la France disposant d'ailleurs de ressources importantes dans ce domaine grâce à ses plateformes de génétique moléculaire.
Ce retour de l'innovation se fait selon un modèle très différent de celui qui avait prévalu dans les dernières décennies. D'une part, le développement des nouvelles molécules se fait fréquemment de manière externalisée , dans le cadre de petites start-ups ensuite rachetées par les grands laboratoires pharmaceutiques. D'autre part, la nouvelle génération de médicaments ainsi développée augmente en technologie par rapport aux précédentes, ce qui entraîne un temps de mise au point plus long - et ce qui justifierait selon les industriels leur caractère extrêmement coûteux.
Selon les éléments transmis à vos rapporteurs, on constaterait ainsi aujourd'hui une très forte segmentation du marché du médicament entre trois types de produits : des médicaments anciens à l'efficacité éprouvée, comme le paracétamol, et au prix peu élevé ; des médicaments destinés au traitement de pathologies mal soignées et pour lesquelles l'innovation est faible, comme la maladie d'Alzheimer, ce qui peut entraîner des prix élevés ; des produits très innovants, comme le Sovaldi, qui ont de ce fait un coût exorbitant.
Si cette dernière catégorie de médicaments pourra se trouver davantage exposée à la concurrence après leurs premières années d'exploitation, leur irruption soudaine sur des cohortes importantes de malades est susceptible de déstabiliser profondément le fonctionnement et le financement de notre système de santé .
Selon plusieurs des acteurs entendus, les médicaments génériques pourront quant à eux permettre de favoriser indirectement l'innovation : les économies dégagées pour la sécurité sociale peuvent en effet permettre de financer les médicaments innovants.
Les auditions conduites par vos rapporteurs ont en ce sens permis de constater que les pouvoirs publics menaient plusieurs actions envers les agissements anticoncurrentiels des laboratoires exploitant les blockbusters associés. L'Autorité de la concurrence a ainsi sanctionné Sanofi-Aventis, en 2013, à hauteur de 40,6 millions d'euros pour avoir mis en place une stratégie de dénigrement à l'encontre des génériques de Plavix®, l'un des médicaments les plus vendus dans le monde. On peut également citer en ce sens la décision de 2014 de la Commission européenne, par laquelle celle-ci a infligé des amendes à Servier et à cinq fabricants de génériques pour avoir freiné l'entrée sur le marché de versions moins chères du Périndopril (pratique dite du « pay for delay »). D'autres pratiques encore ont été signalées à vos rapporteurs, telles que le consentement de remises aux pharmaciens de remises au-delà du niveau légal afin de saturer leurs linéaires de produits princeps .
b) Un retard français en matière d'investissement dans l'innovation
(1) Des budgets de R&D en stagnation
Selon le rapport préparatoire au CSIS précité, le système d'innovation français, évalué au travers des crédits consacrés à la R&D, est en stagnation face à des concurrents actuellement en phase de montée en puissance, et qui consacrent de ce fait des moyens considérables à la recherche.
Ce faible investissement relatif résulterait en particulier de la structure du tissu industriel innovant en France : la plupart des entreprises innovantes, notamment en matière de biotechnologies, sont des PME constituées autour d'une innovation moléculaire spécifique, dont le développement nécessite des capitaux très importants et selon un modèle d'affaires par nature risqué.
Il apparaît dès lors indispensable de développer des initiatives, notamment publiques, pour favoriser le financement de type capital-risque de ce secteur - ce que fait, partiellement et de manière insuffisante, la banque publique d'investissement (BPI).
Cette situation est d'autant plus problématique que, dans le secteur pharmaceutique, « la concurrence [...] est notamment fortement conditionnée par les enjeux de propriété intellectuelle et d'efficacité du système d'innovation ».
Avec 5 900 dossiers, les biotechnologies figuraient ainsi en 2014 au huitième rang des demandes de brevets auprès de l'office européen des brevets (OEB), en hausse de 12,1 % par rapport à l'année précédente - soit la plus forte hausse parmi les grands secteurs.
De ce point de vue, la position française apparaît cependant plus favorable qu'en termes de budgets de R&D, avec un acteur public (l'Inserm) et un acteur privé (Sanofi) présents respectivement à la 5 e place et à la 2 e place en nombre de demandes de brevets déposées auprès de l'OEB. Cette mixité des innovateurs français signe en outre « un bon fonctionnement de l'écosystème d'innovation français dans la pharmacie ».
Entendu par vos rapporteurs, le Professeur Hugues de Thé a cependant insisté sur la nécessité pour les acteurs publics de s'inscrire pleinement dans cet écosystème d'innovation . Il a cité à ce titre le cas d'un traitement contre la leucémie aiguë promyélocytaire, qui permet d'obtenir une guérison quasi systématique. Développé par des équipes de recherche publiques française et chinoise, le traitement, dont la découverte est largement due à l'effet de la chance et du hasard, associe deux molécules à bas coût, l'arsenic et l'acide rétinoïque. Un brevet a cependant été pris sur ce traitement par un laboratoire privé étranger à ce processus de découverte, de telle sorte que le traitement, dont le coût de production est très faible, est aujourd'hui facturé à hauteur de 40 000 euros à la sécurité sociale.
(2) La nécessité de promouvoir la recherche fondamentale
Selon le Professeur Hugues de Thé, les obstacles pesant sur le développement de nouvelles innovations thérapeutiques médicamenteuses - en France singulièrement, mais aussi de manière plus globale - sont aujourd'hui de plusieurs ordres.
En premier lieu, la recherche fondamentale ne serait pas suffisamment encouragée , que ce soit dans le secteur privé ou dans les laboratoires publics. Dans le secteur privé, cela résulterait de la stratégie de l'industrie pharmaceutique, qui tourne sa politique de recherche et de développement vers les secteurs les plus profitables financièrement, qui ne sont pas nécessairement les plus cruciaux pour la santé publique. Dans le secteur public, ce sont principalement les moyens alloués à ce type de recherche qui sont en cause.
Le Pr de Thé a pourtant souligné que de nombreuses révolutions thérapeutiques ont été le fait du hasard plus que d'une progression préprogrammée de recherches. Cette question de méthode se traduit dans le mode d'organisation des essais cliniques, qui ne permettraient plus de réaliser aujourd'hui les découvertes majeures qui ont révolutionné la pharmacopée dans le passé. Alors en effet que l'objectif des phases de recherche devrait être de rechercher la rupture, la conduite d'essais de grande ampleur sur des échantillons très importants vise au contraire à mettre en évidence de petits effets de manière irréfutable. La plupart des nouveaux médicaments sont ainsi conçus comme une amélioration de l'existant, et non comme un moyen de créer une rupture majeure dans le champ thérapeutique.
Il apparaît ainsi indispensable de conserver, au sein des établissements publics, une forme de recherche fondamentale libre qui ne soit ni spécifiquement ciblée, ni par trop limitée dans le temps, et la nécessité de préserver des budgets de recherche globaux à cet effet .
Il a également proposé devant vos rapporteurs de développer les cursus mixtes de médecine et de science , sur le modèle du programme créé par l'Inserm il y a une dizaine d'années.
Le développement de partenariats public-privé d'une part, et internationaux d'autre part , est bien évidemment à encourager par ailleurs. Le rapport du CSIS d'avril 2016 souligne les diverses initiatives déjà prises en matière de renforcement de la recherche partenariale : création d'Aviesan et d'Ariis, doublement par les industriels des montants de leurs partenariats entre 2009 et2012, soutien à la participation d'équipes française s à l'initiative européenne sur les médicaments innovants (IMI).
En second lieu, les travaux de recherche se trouveraient soumis à des contraintes réglementaires trop importantes , que ce soit en matière d'essais cliniques, de liens d'intérêt ou d'expérimentation animale. Selon le Pr de Thé, « l'hyperréglementation du champ de la recherche dans les pays développés sclérose le mode de recherche traditionnel dans le champ clinico-biologique ».
En matière d'expérimentation animale a notamment été regrettée l'inflation d'une réglementation devenue souvent irréaliste et inapplicable, en particulier sous l'influence de l'Union européenne.
S'agissant des essais cliniques, le dernier CSIS s'est saisi de la question en proposant, d'une part, de réduire à 45 jours les délais d'approbation, et, d'autre part, de créer une instance de concertation réunissant tous les acteurs de la recherche clinique afin d'identifier les points de blocage, sur le modèle du UK Clinical research council .
II. QUEL PÉRIMÈTRE DE PRISE EN CHARGE POUR QUEL USAGE ?
Environ 2 800 substances actives différentes, correspondant à plus de 11 000 spécialités, sont disponibles sur le marché français . Du point de vue de leur financement, elles se répartissent entre médicaments pris en charge et non pris en charge par l'assurance maladie.
En 2013, 85 % des médicaments vendus en France étaient remboursables . Cette proportion s'élevait à 97 % s'agissant des spécialités soumises à prescription médicale obligatoire (que l'on raisonne en valeur ou en volume). D'après l'ANSM, cette situation « résulte de ce que les pathologies graves, et prises en charge par les régimes d'assurance maladie, sont traitées par des médicaments qui impliquent obligatoirement une prescription médicale » 23 ( * ) .
Un médicament peut faire l'objet d'une prise en charge intégrale par la sécurité sociale soit en raison de l'intérêt qu'il présente 24 ( * ) , soit parce qu'il est prescrit à une personne pour traiter une pathologie correspondant aux critères de l'affection de longue durée (ALD) dite « exonérante » c'est-à-dire d'une affection « dont la gravité et/ou le caractère chronique nécessitent un traitement prolongé et une thérapeutique particulièrement coûteuse, et pour lesquelles le ticket modérateur est supprimé » 25 ( * ) .
Mais certains ne sont remboursés que partiellement, soit en taux, soit seulement pour certaines populations ou pour certaines indications.
La différenciation des taux de remboursement tient au service médical rendu par le médicament tel qu'il est évalué par la commission de la transparence de la Haute Autorité de santé (HAS) . Cette commission, qui existe depuis 1967, est chargée par le code de la sécurité sociale de rendre un avis sur :
- les demandes d'inscription et de renouvellement de l'inscription des médicaments sur la liste des médicaments remboursables ;
- la modification des conditions d'inscription d'un médicament.
La commission doit également réévaluer le service médical rendu des médicaments inscrits sur cette liste. La réévaluation a lieu par classe pharmaco-thérapeutique ou pour tous les médicaments à même visée thérapeutique.
Cette réévaluation a lieu notamment lorsque la commission propose l'inscription sur ces listes ou l'une de ces listes d'un médicament apportant une amélioration du service médical rendu (ASMR) majeure, susceptible de modifier substantiellement les stratégies thérapeutiques antérieures ou lorsque le contexte scientifique qui a fondé l'avis rendu précédemment a évolué de façon significative ou notoire.
Nombre d'avis rendus par la Commission de la transparence
|
Nombre d'avis rendus par type de demande |
2010 |
2011 |
2012 |
2013 |
2014 |
|
Primo-inscriptions |
296 |
219 |
216 |
169 |
190 |
|
Extensions d'indications |
29 |
22 |
32 |
31 |
45 |
|
Renouvellements et réévaluation
|
269 |
557 |
459 |
276 |
269 |
|
Autres avis rendus (radiations, modifications des conditions d'inscriptions...) |
201 |
194 |
163 |
144 |
126 |
|
Total des avis rendus |
795 |
992 |
870 |
620 |
630 |
Source : Rapport d'activité HAS
En 2011, d'après le syndicat des entreprises du médicament (Leem), les médicaments pris en charge à 100 % par l'assurance maladie représentent environ deux tiers des dépenses de remboursement par l'assurance maladie, le tiers restant se répartissant entre les médicaments remboursés aux autres taux 26 ( * ) .
Par ailleurs, les médicaments en libre accès, médicaments dits d'automédication, ne sont pas remboursés par la sécurité sociale 27 ( * ) .
Dans un contexte qui impose la meilleure allocation des financements sociaux aux traitements les plus efficaces, il convient de s'interroger sur la pertinence du périmètre actuel de prise en charge des médicaments. Ceci suppose d'étudier les raisons pour lesquelles certains médicaments ne sont pas remboursés, et de déterminer si les mécanismes d'ajustement du coût des médicaments remboursés garantissent la pérennité du système de prise en charge.
A. LE MÉDICAMENT NON PRIS EN CHARGE
Trois types de médicaments ne font pas l'objet d'une prise en charge par la sécurité sociale : les médicaments d'automédication, les médicaments prescrits en dehors de leur autorisation de mise sur le marché ou d'une indication spécifique, enfin les médicaments déremboursés.
1. L'automédication : liberté des prix et choix du patient
Le choix de faire figurer un médicament sur la liste des spécialités en automédication relève du titulaire de l'autorisation de mise sur le marché . L'ANSM contrôle cependant que le médicament ne relève pas du champ de la prescription médicale obligatoire et que le conseil pharmaceutique peut suffire au bon usage 28 ( * ) . Si les médicaments en automédication n'ont pas vocation à apporter des réponses thérapeutiques aux pathologies les plus graves, il n'y a pas de lien juridique entre leur service médical rendu et le fait qu'ils soient en accès libre.
Les industriels tendent le plus généralement à demander à ce qu'un de leur produit fasse l'objet d'un remboursement en raison de l'impact de la prise en charge sociale sur le chiffre d'affaires. Mais une autre stratégie industrielle peut consister à privilégier la liberté de fixation du prix, qui est le corollaire de l'accès direct, indépendamment du service médical rendu . Ainsi un laboratoire commercialisant un médicament dont la notoriété est établie peut faire le choix d'en assurer la vente en automédication, surtout si l'effet « marque » se trouve limité, en cas de remboursement, par la substitution au profit d'un générique, ou au moins le risque d'une telle substitution. Les débats récents autour du remboursement du Doliprane sont un exemple de l'évolution des stratégies industrielles par rapport au remboursement d'une spécialité, sans que le choix du laboratoire soit lié à l'efficacité du médicament.
L'automédication renvoie à plusieurs concepts dont les définitions reflètent les différences de point de vue des acteurs : producteurs, autorité sanitaire ou acteurs de santé. L'essentiel est cependant de déterminer la motivation des patients qui recourent à l'automédication et son impact en matière de santé publique.
L'association française de l'industrie pharmaceutique pour une automédication responsable (Afipa), qui représente 38 laboratoires et 80 % du marché de l'automédication, s'appuie sur une définition attribuée à l'OMS en 2000 pour caractériser l'automédication responsable : « L'automédication responsable consiste, pour les individus, à soigner leurs maladies grâce à des médicaments autorisés, accessibles sans ordonnance, sûrs et efficaces dans les conditions d'utilisation indiquées ». La notion de responsabilité est essentielle car l'OMS souligne également que « l'automédication qui peut aussi avoir des aspects positifs est inévitablement associée (...) à l'usage rationnel des médicaments ; ce dernier est mis en danger, dans le secteur public comme dans le secteur privé, par les ordonnances surchargées (parfois de cinq à six médicaments), l'abus des antibiotiques, des injections, les «conseils» donnés par les vendeurs à la sauvette, les médicaments périmés, etc., mais aussi par le manque de médicaments ou de personnel qualifié. Il existe également d'autres causes comme celles de la non-conformité au traitement qui relève non seulement de l'ignorance mais aussi du souhait de contrôler sa propre vie en refusant certains effets des médicaments ou en supprimant la prise lors de la disparition des symptômes. »
Les autorités sanitaires utilisent pour leur part le terme de médication officinale afin de marquer que les médicaments acquis sans ordonnance peuvent, voire doivent, faire l'objet d'un conseil par le pharmacien 29 ( * ) .
Les médicaments en accès direct constituent une partie des médicaments disponibles sans ordonnance en pharmacie. Ils sont disponibles directement, sans donc qu'il soit besoin d'en faire la demande, « s'ils répondent à certains critères (indication, dosage, notice, boîte spécialement étudiés) pour (...) permettre [à chacun] de les choisir et de les utiliser sans une prescription médicale » 30 ( * ) . D'autres médicaments situés derrière le comptoir ne sont accessibles que sur demande faite au pharmacien en raison de l'obligation que la vente soit accompagnée d'un conseil pharmaceutique.
L'attitude des pouvoirs publics face à l'automédication est ambivalente. La volonté d'assurer la plus grande sécurité du patient tend à ce que l'ANSM privilégie le niveau d'encadrement le plus élevé. L'automédication reste peu développée en France par rapport au reste de l'Europe et son chiffre d'affaires a même eu tendance à se contracter ces deux dernières années d'après les chiffres fournis à vos rapporteurs par l'Afipa.
L'économiste Claude Le Pen attribue cette situation au fait qu'il « existe une alliance anti-automédication en France ». « Cette alliance , affirme-t-il, rassemble les acteurs de santé qui craignent surtout de perdre des rentes de situation ». Pour autant les raisons que présente l'économiste tiennent autant à la santé publique qu'au souci de maintenir des situations acquises : « Les médecins craignent une perte de patientèle, le mésusage des médicaments, le mélange de produits incompatibles, les pertes de chance en cas d'erreur d'appréciation d'une pathologie existante. L'Etat se méfie des patients rendus plus autonomes avec les risques que cela peut générer (...) Les patients eux-mêmes sont parfois réticents car le remboursement des soins n'est plus au rendez-vous . » 31 ( * )
Pour autant, l'automédication est une part intégrante des modes d'accès au médicament. L'anthropologue Sylvie Fainzang, auteure de L'automédication ou les mirages de l'autonomie 32 ( * ) , considère que les pouvoirs publics et l'industrie promeuvent une autonomie limitée du patient au travers de l'automédication. Après avoir étudié les motivations des comportements d'automédication, elle a résumé pour la revue de l'Ordre des médecins l'un de ses principaux constats : « L'automédication et la volonté d'autonomie qui va avec peuvent résulter soit de la confiance, soit de la méfiance envers le médecin. Dans le premier cas, le patient considère son médecin comme compétent et cherche à reproduire les prescriptions médicales (ou croit reproduire les prescriptions médicales) antérieures. Dans le deuxième cas, le médecin est jugé incompétent pour résoudre un problème donné et le patient décide de gérer lui-même son mal en faisant ses propres choix, en fixant ses posologies, en développant ses stratégies de réduction des risques médicamenteux... Dans tous les cas, l'automédication traduit la mise en acte d'une autonomie avec parfois la volonté de s'affranchir de l'autorité médicale. »
En outre, « la pratique de l'automédication repose sur une évaluation des médicaments, faite tout à la fois de la connaissance [que les patients] ont acquise auprès des professionnels de santé, de celle qu'ils ont retirée de leur propre expérience, et de représentations des médicaments dont la logique symbolique court-circuite parfois les logiques pharmacologiques ». Ainsi le rapport à l'automédication apparaît complexe. En outre ses représentations varient selon l'origine culturelle et religieuse des personnes.
L'anthropologue Daniela Cerqui, interrogée par la revue de l'ordre des médecins, rappelle pour sa part que « l'accès à l'information n'est pas synonyme d'accès au savoir », ce qui renforce la nécessité d'un accompagnement par le conseil des professionnels et par les campagnes d'information publiques.
Il paraît donc nécessaire à vos rapporteurs de traiter la question de l'automédication non comme une source d'économie potentielle, mais d'abord comme un enjeu de santé publique . Il paraît dès lors souhaitable que le Gouvernement définisse des orientations claires relativement au développement de l'automédication.
2. L'usage hors AMM, un mal nécessaire ?
Un médicament déjà sur le marché, mais prescrit hors du champ de son autorisation de mise sur le marché, ne doit pas être pris en charge par la sécurité sociale sauf, après avis de la HAS, s'il fait l'objet d'une recommandation temporaire d'utilisation (RTU) par l'ANSM . Cette exception est fondée sur la nécessité de permettre l'usage de traitements dont l'efficacité n'avait pas été établie au moment de la demande d'AMM 33 ( * ) .
Le mécanisme de RTU a également été récemment utilisé pour réduire les coûts liés à un traitement sans préjudice pour le patient 34 ( * ) en permettant l'utilisation de l'Avastin dans le traitement de la dégénérescence maculaire néovasculaire liée à l'âge (DMLA).
La prescription hors AMM d'un médicament relève de la responsabilité du praticien et a été décrite comme nécessaire pour la prise en charge de certaines pathologies à la mission d'information sénatoriale sur le Mediator. Néanmoins, si, comme l'impose l'article 8 du code de déontologie médicale, la prescription hors AMM s'appuie sur l'état de la science, elle devrait aboutir à l'élaboration d'une RTU. De fait lorsqu'un produit est autorisé dans une indication hors AMM par les autorités sanitaires de plusieurs pays européens il devrait faire l'objet d'une RTU en France. Or le cas du Mediator montre que parfois le hors AMM peut relever du mésusage du médicament et que certains praticiens prescrivent comme remboursables des médicaments dont ils savent qu'ils les prescrivent hors AMM. En conséquence, la loi du 29 décembre 2011 relative au renforcement de la sécurité sanitaire du médicament et des produits de santé a inscrit dans le code de la santé publique 35 ( * ) les conditions de la prescription hors AMM et l'obligation de porter sur l'ordonnance la mention « prescription hors autorisation de mise sur le marché ».
Cependant, le rapport sur la surveillance et la promotion du bon usage des médicaments remis en septembre 2013 à la ministre de la santé par Dominique Costagliola et Bernard Bégaud relève « la difficulté à identifier ce qui est réellement du hors-AMM ou du « hors-AMM justifié »» mais insiste sur le fait que l'absence de données précises ne doit « cependant pas occulter, comme cela est trop souvent fait, que la France est l'un des pays dans lequel les prescriptions et l'usage irrationnels sont les plus prévalents ». S'agissant du coût social du mésusage, le rapport fait l'estimation suivante : « on ne dispose pas non plus d'une estimation crédible sur le coût supporté chaque année par l'assurance maladie (toutes branches confondues) du fait des conséquences de la haute prévalence des prescriptions et des utilisations non conformes. Ce coût est considérable (probablement supérieur à dix milliards d'euros par an) car, comme mentionné ci-dessus, il inclut non seulement les remboursements non justifiés de médicaments et des consultations et actes biologiques que ces prescriptions induisent (le dosage du « cholestérol » pour les statines) mais surtout les coûts induits par les maladies mal ou non prises en charge ou non prévenues et par la iatrogénie évitable (conséquences immédiates, hospitalisations, séquelles, examens complémentaires, etc.) ».
Ainsi le hors AMM est justifié sur le plan médical, encadré sur le plan législatif, fait l'objet d'une procédure spécifique pouvant aboutir à sa prise en charge par la sécurité sociale, mais reste source de dérives coûteuses et dangereuses pour les patients .
Les recommandations de l'ANSM en matière de prescription n'ont qu'un effet limité sur la prescription hors AMM. Des mesures contraignantes peuvent néanmoins être mises en place dans le cadre des relations conventionnelles du comité économique des produits de santé (Ceps) avec l'industrie du médicament. Ce comité a en effet compétence pour déterminer les obligations liées à la visite médicale et signe avec les laboratoires des engagements sur le bon usage des médicaments. La lettre d'orientation adressée au président du Ceps par ses ministres de tutelle le 2 avril 2013 précise ainsi : « en cas d'usage constaté d'un produit en dehors des indications de son AMM et des recommandations des autorités sanitaires compétentes, ainsi que de non-respect des engagements conventionnels souscrits par l'industriel pour contribuer à son bon usage, vous prononcerez les sanctions financières prévues.
Lorsque le chiffre d'affaires d'un produit ou d'une famille de produits excède manifestement la population cible de son AMM et des recommandations des autorités sanitaires compétentes, ou lorsque le montant remboursé excède manifestement la population cible de ses indications remboursables, vous veillerez à ce que les industriels concernés contribuent activement à la restauration du bon usage de leur produit et vous engagerez des baisses de prix en cas de refus ou d'implication insuffisante de leur part .
Vous proposerez, chaque fois que nécessaire, aux autorités compétentes, la mise en place de dispositifs de sensibilisation des prescripteurs au bon usage, et vous y contribuerez par le biais du dispositif conventionnel, notamment dans les cas évoqués précédemment ».
La Cnam s'est également saisie de la question du bon usage depuis plusieurs années au travers du suivi des volumes de remboursements. Au travers de son Hackathon organisé entre décembre 2015 et mai 2016 sur le thème de l'usage du médicament, elle a également primé les applications tendant à permettre une meilleure observance et une comparaison des pratiques de prescription.
Il paraît utile à vos rapporteurs, pour favoriser le bon usage du médicament, de développer des partenariats entre le Ceps et la Cnam pour mieux suivre l'utilisation des médicaments et agir sur les comportements de promotion, de prescription et d'usage.
3. Le médicament non pris en charge, excepté dans des indications thérapeutiques spécifiques
A l'initiative du laboratoire et après examen par l'ANSM, un médicament est commercialisé dans le cadre d'une autorisation de mise sur le marché pour des indications thérapeutiques spécifiques.
Le remboursement du médicament ne porte pas nécessairement sur l'ensemble de ces indications thérapeutiques puisque l'article L. 162-17 du code de la sécurité sociale, relatif aux conditions de remboursement des médicaments dispose que : « La liste précise les seules indications thérapeutiques ouvrant droit à la prise en charge ou au remboursement des médicaments ».
Ainsi, les antiviraux à action directe (AAD), nouveaux traitements de l'hépatite C, qui entraînent une disparition du virus dans le sang des malades, font l'objet d'une prise en charge spécifique par la sécurité sociale.
En effet, l'arrêté du 18 novembre 2014 inscrivant le Sovaldi sur la liste des spécialités remboursables dispose, dans son article 2, que : « Compte tenu du caractère irremplaçable et particulièrement coûteux du médicament relevant du présent arrêté (...) la participation de l'assuré aux frais d'acquisition de ce médicament est supprimée ». Il est donc pris en charge intégralement par l'assurance maladie.
Néanmoins l'article 1 er renvoie à une annexe la détermination des « seules indications thérapeutiques ouvrant droit à la prise en charge ou au remboursement du médicament ». Cette annexe fixe cinq indications thérapeutiques, ainsi, « au vu des exigences de qualité et de sécurité des soins », qu'une condition « relative à l'organisation des soins ». En l'occurrence, « l'initiation du traitement est subordonnée à la tenue, dans les pôles de référence hépatites (devenus services experts de lutte contre les hépatites virales), d'une réunion de concertation pluridisciplinaire ». L'arrêté du 12 juin 2015 prévoit le même dispositif pour l'Harvoni, médicament ayant la même autorisation de mise sur le marché que le Sovaldi.
La limitation de la prise en charge à certaines indications thérapeutiques et la condition relative à l'organisation des soins ont fait l'objet de critiques sévères . Du côté des soignants, l'association française pour l'étude du foie (Afef) a, pour sa part, préconisé en février 2016 le traitement universel. Les « recommandations Afef sur la prise en charge des hépatites virales C » comportent le paragraphe suivant : « Un traitement antiviral doit être proposé à tous les patients qui ont une hépatite chronique C, naïfs ou en échec d'un précédent traitement, avec une maladie hépatique compensée ou décompensée, à l'exception de ceux qui ont une comorbidité limitant leur espérance de vie à court terme. L'hépatite chronique virale C n'est pas une maladie uniquement hépatique mais une maladie générale. Les critères d'indication de traitement uniquement liés à la sévérité de la fibrose hépatique sont obsolètes. L'accès à un traitement universel est un objectif à court terme dans le but d'une disparition de l'épidémie d'hépatite C avant 2025. Il n'y aucun argument médical pour refuser à un patient un traitement efficace et sans effet indésirable majeur. Ceci nécessite une ouverture des indications au traitement pour tous dès 2016 ». Quelques semaines auparavant, les organisateurs de la 9th Paris Hepatitis Conference, qui s'est tenue les 11 et 12 janvier 2016 « ont critiqué les restrictions de remboursements des nouvelles molécules, qui rendent l'accès au traitement problématique pour les patients aux stades F2 » 36 ( * ) .
Les associations ont également fait part de leur opposition à l'avis de la commission de la transparence de la HAS du 14 mai 2014 sur lequel se fonde l'arrêté du 18 novembre 2014 37 ( * ) . Dans son rapport de 2015 VIH/Hépatites, la face cachée des discriminations , l'association Aides critique le caractère restrictif des recommandations de la HAS, estimant qu'elles ne s'inscrivent pas « dans une perspective de ralentissement de l'épidémie ». Aides oppose les recommandations de la HAS à celles « issues d'un consensus d'experts » proposées en mai 2014 dans un rapport de recommandations sur la prise en charge des personnes infectées par les virus de l'hépatite B ou de l'hépatite C. Ce rapport a été commandé par la ministre de la santé en janvier 2013 à l'agence nationale de recherche sur le Sida et les hépatites (ANRS) et l'association française pour l'étude du foie (Afef), il a été élaboré sous la direction du Pr Daniel Dhumeaux.
Face à la perspective portée par le rapport dit « Dhumeaux » d'une éradication de l'hépatite C, les restrictions préconisées par la HAS n'ont été vues que comme une conséquence du coût élevé du traitement, malgré les baisses de prix et remises obtenues par la négociation avec le comité économique des produits de santé. L'acceptation du prix élevé d'un médicament a donc été condamnée comme entraînant un rationnement des soins. Le journal Le Parisien du 5 avril 2016 rapporte ainsi les propos de Yann Mazens, responsable de l'association SOS Hépatites : « La France compte 250 000 cas d'hépatite C, dont 140 000 ont un besoin pressant de traitement. Pour raisons financières, seuls 26 000 cas ont été traités à ce jour ».
Le Formindep a pour sa part critiqué les prises de positions de professionnels tendant à une prise en charge plus large au nom des conflits d'intérêts dont seraient entachées les positions des auteurs de ces recommandations 38 ( * ) .
L'accès de tous les malades aux traitements est un des fondements de notre politique d'assurance maladie , tels qu'ils ont été rappelés par le Président de la République lors de son allocution aux côtés du Président Jacob Zuma le 23 mars 2016 39 ( * ) : « Dans une démocratie, il ne peut pas être dit à une personne, quels que soient son niveau de revenus, ses origines, son parcours : vous ne pourrez pas être soigné et guéri parce que c'est trop cher. »
L'impossibilité d'accéder aux soins remboursés entraînerait des comportements de contournement du circuit du médicament français. L'éditorial du Parisien du 5 avril 2016 s'interroge : « Combien sont-ils en France à devoir se résoudre, parfois sur les conseils de leur propre médecin, de se fournir en médicaments à l'étranger ? » puis constate : « Certains patients dont le médicament n'a pas encore d'autorisation ou dont le coût, prohibitif, oblige les hôpitaux à choisir sur [sic] les malades qui en bénéficieront, franchissent le pas. » Le quotidien relate ensuite le cas d'Audrey dont l'infection par l'hépatite C ne correspondait pas aux indications et à laquelle un hépatologue de l'AP-HP aurait conseillé d'aller en Egypte pour trouver un générique du Sovaldi.
S'agissant de la prise en charge des traitements relatifs à l'hépatite C, le Gouvernement a fait valoir que la prise en charge des patients français était plus large que celle des autres pays européens et que les indications fixées par la Haute Autorité de santé correspondent à la capacité de prise en charge des patients par les services hospitaliers spécialisés. Il n'aurait donc pas été possible de prendre en charge plus de patients, quand bien même le remboursement aurait prévu des indications plus larges.
La stratégie de prise en charge tendait donc à ouvrir d'abord l'accès aux traitements aux personnes aux stades les plus avancés de la maladie pour l'étendre progressivement, une fois les cas les plus graves traités. Cette approche en terme thérapeutiques s'appuie par ailleurs sur l'entrée sur le marché de nouveaux AAD concurrents du Sovaldi et entrainant donc des baisses de prix. Ainsi les malades les plus nombreux devraient être pris en charge dans des délais compatibles avec l'évolution de leur pathologie et pour un coût inférieur.
Evolution des volumes de
Sovaldi®
consommés en France et en Europe en 2016
C'est donc logiquement que la ministre de la santé a saisi le 19 mai 2016 la commission de la transparence sur les modalités de prise en charge des AAD pour les patients au stade F2 de fibrose hépatique. L'avis ayant été rendu le 25 mai 2016, la ministre a annoncé l'extension de la prise en charge des traitements d'AAD a ces patients et la prise en charge des traitements pour tous les patients atteint de l'hépatite C à partir de septembre, suite à un nouvel avis de la commission de la transparence.
4. Des déremboursements mal perçus par l'opinion publique et parfois difficiles à intégrer à une stratégie médicale.
a) Le réexamen régulier du service médical rendu a remplacé les vagues successives de déremboursement
Depuis 1999 et la mise en place du plan « Aubry », plusieurs vagues de déremboursement ont été conduites afin de concentrer la dépense sociale sur les molécules les plus efficaces .
|
Chronologie établie par
l'Irdes
Juin 2001 : la Commission de la transparence, aujourd'hui rattachée à la Haute Autorité de santé (HAS), publie une liste de 835 médicaments jugés insuffisants sur 4 890 produits évalués. Le gouvernement décide d'étudier ces produits au cas par cas. Août 2003 : la première vague de déremboursement concerne 72 spécialités (60 médicaments). Il s'agit essentiellement de "vieux" produits, dont certains peuvent être dangereux. Beaucoup d'entre eux sont retirés du marché. Février-mars 2006 : la deuxième vague de déremboursement concerne 282 spécialités pharmaceutiques (156 médicaments) : des expectorants, des fluidifiants bronchiques, des produits de phytothérapie, des oligoéléments et des médicaments contre les troubles digestifs. Pour 105 médicaments veinotoniques, les pouvoirs publics créent un taux de prise en charge provisoire de 15 % (au lieu de 35 %) destiné à préparer leur déremboursement total. Janvier 2007 : la troisième vague de déremboursement ne se fait pas. Contre l'avis de la HAS, le gouvernement ne dérembourse pas 89 médicaments jugés insuffisants, dont des vasodilatateurs. Le taux de remboursement de certains médicaments passe de 35 % à 15 %, avant leur déremboursement total prévu pour janvier 2008. C'est par exemple le cas d'antidiarrhéiques et d'antitussifs. 1 er janvier 2008 : déremboursement total des phlébotoniques et des veinotoniques. Arrêté du 30 septembre 2011 : il liste les 26 spécialités pharmaceutiques déremboursées à compter du 1 er décembre 2011. Les radiations concernent 26 molécules (80 radiations au total en tenant compte des présentations et des génériques). |
Ces mesures d'ampleur portées par le Gouvernement ont été arrêtées depuis 2012 au profit d'une gestion des taux de remboursement au travers des saisines de la commission de la transparence .
En effet, l'article R. 163-2 du code de la sécurité sociale dispose que l'inscription d'une spécialité sur la liste des médicaments remboursables est prononcée pour une durée de cinq ans renouvelable 40 ( * ) . La commission de la transparence est donc saisie chaque année de demandes de renouvellement de l'inscription de médicaments : « À cette occasion, la commission se prononce sur le maintien ou la modification du service médical rendu des spécialités concernées en prenant en compte à la fois des nouvelles données d'efficacité et de tolérance disponibles et des données acquises, des évolutions des pratiques concernant les pathologies et les stratégies thérapeutiques des indications concernées ».
En dehors des demandes émanant des industriels, la commission de la transparence peut être saisie par le ministère, le collège de la HAS, ou s'autosaisir de la réévaluation d'un médicament ou d'une classe de médicaments.
Dans la majorité des cas, le réexamen confirme le niveau de SMR déjà reconnu. Dans quelques rares cas, le réexamen peut conduire à la reconnaissance d'un service médical rendu plus élevé. Surtout, plusieurs dizaines de décisions tendent à la réduction du SMR ou à sa déclaration comme insuffisant. Ceci entraîne la recommandation d'une baisse du taux de remboursement, voire d'un déremboursement.
|
Décisions du rapport annuel 2014 de la HAS 79 avis ont été rendus en 2014, dont 1 qui concernait une saisine de réexamen de population cible et donc ni le SMR, ni l'ASMR. Parmi ces avis, 13 ont été rendus en réponse à la saisine conjointe de la Direction générale de la santé, de la Direction de la sécurité sociale et de la Direction générale de l'offre de soins qui fait suite aux travaux menés par le Conseil de l'hospitalisation pour mettre à jour la liste des médicaments facturables en sus des prestations hospitalières. La HAS donc a été saisie en octobre 2013 pour se prononcer sur l'ASMR de 13 des spécialités figurant sur cette liste. Compte tenu de l'ancienneté des évaluations antérieures de certains de ces produits, la commission a reconnu : 8 SMR important, 4 SMR faible, 1 SMR insuffisant et 8 ASMR V, 1 ASMR IV, 3 ASMR II. Parmi les 65 autres avis rendus suite à une réévaluation, la commission a maintenu ses conclusions précédentes à 36 reprises. Parmi les 29 avis ayant vu une modification des conclusions de la commission : - 6 ont conclu à une baisse du SMR le plus élevé entraînant une recommandation de prise en charge du médicament à un taux plus bas ; - 19 ont conclu à l'octroi d'un SMR insuffisant dans une seule indication ou une population, recommandant ainsi la restriction du périmètre de prise en charge du médicament concerné ; - 4 ont conduit la commission à conclure à un SMR insuffisant pour la totalité des indications thérapeutiques et, de ce fait, à une recommandation d'arrêt de la prise en charge du médicament. |
En 2015, seize arrêtés ont été pris par la ministre en charge de la sécurité sociale sur le fondement des réévaluations conduites par la commission de la transparence. Ils ont exclu du remboursement 351 spécialités, plusieurs spécialités pouvant correspondre aux différents conditionnements et dosages d'un même médicament .
Les vagues de déremboursement de 2011 et 2012 ont conduit, d'après l'Irdes, à une économie respectivement d'environ 30 et 50 millions d'euros. (Les économies liées aux déremboursements depuis cette date n'ont pas été chiffrées.) Cependant l'impact à moyen terme de ces mesures est contesté.
b) Des déremboursements contestés
La contestation des déremboursements s'exprime tant au sein de la presse et de l'opinion publique que parmi les professionnels de santé qui soulignent le risque d'un report de prescription des spécialités déremboursées vers les médicaments qui le sont.
A titre d'exemple, en novembre 2013, la commission de la transparence a considéré que les antiarthrosiques symptomatiques d'action lente, précédemment remboursés à hauteur de 15 % par la sécurité sociale, ont un effet minime « sur la douleur et la gêne fonctionnelle liées à l'arthrose (...) et sont de pertinence clinique discutable. Ils ne permettent pas de réduire la consommation d'anti-inflammatoires non stéroïdiens (AINS). De ce fait, le service médical rendu par ces médicaments est insuffisant pour justifier leur prise en charge par la solidarité nationale ».
En conséquence, un arrêté du 16 janvier 2015 a radié à compter du 1 er mars 2015 ces spécialités pharmaceutiques de la liste des médicaments remboursables.
L'Association française de lutte anti-rhumatismale (Aflar), qui indique, sur son site, regrouper des associations de patients et de professionnels de santé, a contesté cette décision. Elle a lancé une pétition en ligne destinée à dire « Non au déremboursement des traitements de l'arthrose » présentée comme envisagée par le Gouvernement. Cette pétition, ouverte le 25 juillet 2014, se prévaut de 162 789 signatures 41 ( * ) .
Les laboratoires ont également saisi la presse pour relayer les positions de l'Aflar. Ainsi, le laboratoire Expanscience a publié le 8 septembre 2015 un communiqué de presse intitulé « Six mois après le déremboursement des antiarthrosiques symptomatiques d'action lente : des patients en attente d'une forte mobilisation ». Il comporte notamment la citation suivante attribuée au Pr Pascal Richette, chef de service de rhumatologie à l'hôpital Lariboisière : « les patients qui prennent ces anti-arthrosiques, même s'ils ont une efficacité modeste, pourraient consommer moins d'anti-inflammatoires et moins d'antalgiques ».
L'origine de cette citation n'est pas clairement identifiable, mais un entretien accordé par le Pr Richette au quotidien Metronews le 3 mars 2015 42 ( * ) comporte les développements suivants : « Si la Haute Autorité de santé dit dérembourser les médicaments les moins performants pour prendre en charge les plus efficaces, dans le cas de l'arthrose, "la pharmacopée est réduite ".
Or, « quand vous déremboursez un médicament, le volume des prescriptions diminue de 50 % ". Le risque, donc, "c'est que les patients et les prescripteurs se tournent vers les antalgiques (tramadol, codéine) et les consommations d'antiinflammatoires non stéroïdiens (aspirine, ibuprofène)" alors que leurs effets indésirables sur le plan digestif et cardiovasculaires sont plus rares mais plus graves, surtout chez le sujet âgé.
(...) Il serait alors plus sûr que les médecins continuent à prescrire ces cinq médicaments malgré le déremboursement. D'autant que service médical rendu insuffisant signifie "insuffisant pour justifier une prise en charge financière par la solidarité nationale" et non médicament dangereux ou inefficace. "L'arsenal thérapeutique à notre disposition a des effets secondaires et une efficacité modeste mais celle-ci reste supérieure au placebo". »
La contestation porte donc sur la manière dont la mesure sera perçue par les patients (le médicament déremboursé n'est pas nécessairement inefficace ou dangereux mais risque d'être vu comme tel) et par les médecins (il faut montrer au patient qu'on lui donne le meilleur traitement possible). Elle n'est pas véritablement celle du remboursement ou non-remboursement, celui-ci étant déjà au taux le plus faible.
De fait, d'après l'étude conduite par l'Irdes en 2005 comparant la prise en charge des médicaments en Allemagne, Angleterre et France, « ce sont les comportements de prescription et non le nombre de produits pris en charge qui expliquent les écarts de dépenses entre les pays » 43 ( * ) .
Le déremboursement est donc une action sur la conséquence (le remboursement) et pas sur la cause (la prescription) .
Il s'expose ainsi au risque de n'avoir qu'une efficacité limitée. Le rapport sur la surveillance et la promotion du bon usage du médicament en France, remis en septembre 2013 par Bernard Bégaud et Dominique Costagliola à la ministre en charge de la santé, détaille les risques liés au report de prescription et propose une évolution des avis de la HAS.
« En l'absence de messages clairs sur l'attitude à adopter par les praticiens, le retrait du marché ou le déremboursement (partiel ou total) d'une spécialité ou d'une classe induit généralement son « remplacement ». Les « alternatives » étant souvent plus coûteuses et parfois plus mal tolérées », « l'économie » ayant motivé le déremboursement correspond de fait a posteriori à un surcoût parfois important. Avant toute décision, les conséquences sanitaires et économiques devraient être anticipées et prises en compte, et une surveillance mise en place. Pour les déremboursements partiels ou totaux, l'avis de la HAS devrait être accompagné d'une information aux prescripteurs indiquant l'attitude thérapeutique à privilégier et les alternatives de remplacement s'il y a lieu. Cet avis ne devrait pas se limiter à signaler que le médicament ou la classe « n'a pas sa place dans la stratégie thérapeutique » mais, justement, indiquer quelle est cette stratégie thérapeutique et ce qu'il convient de faire, et ce qu'il convient de ne pas faire. L'exemple du déremboursement de l'Euphytose® (anxiolytique et hypnotique « doux » à base d'extraits de plantes) ayant entraîné un report de prescription massif vers les benzodiazépines, avec les conséquences que l'on connaît, est emblématique de ce phénomène. Celui du déremboursement des « anti-arthrosiques » (pratiquement 900 000 utilisateurs en général âgés ou très âgés) poserait le même problème (iatrogénie prévisible dans cette classe d'âge) si un report massif se faisait vers les anti-inflammatoires non stéroïdiens . »
Le rapport propose donc de combiner les avis de la commission de la transparence en cas de déremboursement avec des recommandations permettant d'assurer la meilleure prise en charge du patient tout en limitant le report de prescription.
On peut relever que la HAS s'efforce déjà de faire le point sur les meilleures prises en charge lorsqu'elle procède à un déremboursement. Ainsi lors de celui des anti-arthrosiques symptomatiques d'action lente elle a publié un article expliquant les raisons du déremboursement et faisant le point sur les modalités de prise en charge des patients. Cet article indique notamment que « La prise en charge d'un patient atteint d'une arthrose symptomatique des membres inférieurs repose d'abord essentiellement sur des mesures hygiénodiététiques (alimentation, sport, kinésithérapie...) » et en cas de douleur sur le paracétamol en première intention 44 ( * ) .
|
Le problème de la polymédication L'enjeu du nombre de médicaments prescrits est non seulement financier, mais d'abord médical car la polymédication peut être source de iatrogénie néfaste. Les effets néfastes de la polymédication sont en effet établis. Le rapport de Chloé Le Cossec pour l'Irdes 45 ( * ) précise que : « la polymédication est associée à une augmentation du risque de placement en établissement médico-social et à une augmentation des hospitalisations. Les événements iatrogéniques sont responsables de 5 à 25 % des admissions hospitalières et de 10 % des admissions aux urgences. Enfin, des études de cohortes prospectives ont permis d'identifier une surmortalité associée à la polymédication ». Néanmoins, la nécessité d'agir pour une meilleure prescription a été particulièrement soulignée s'agissant des cas de polymédication des personnes âgées. Que choisir santé a ainsi publié en février 2015 une enquête concluant que sur près de 350 ordonnances de plus de cinq lignes concernant des personnes de plus de 75 ans, 39,5 % comportaient un médicament a priori inadapté. Le rapport publié par l'Irdes en décembre 2015 relève les problèmes de définition des seuils de polymédication de la manière suivante : « Il n'y a actuellement pas de consensus sur le seuil à adopter pour définir la polymédication. Cela vient probablement du fait qu'il n'y ait pas de norme médicale permettant de définir ce qui serait un nombre trop important de médicaments ». Dans un objectif de mise en place et d'évaluation de politiques publiques, il semble plus opportun d'agir sur les individus les plus touchés par la polymédication. Un seuil à 10 médicaments, ciblant aux alentours de 30 % des 75 ans ou plus en ambulatoire, semble plus opérationnel qu'un seuil à cinq médicaments pour lequel 80 % des mêmes individus sont considérés polymédiqués. Cela ne signifie en aucun cas une absence de risque d'interaction ou même une absence de charge pour le patient en dessous de dix médicaments pris de façon chronique ; il a d'ailleurs été montré une association significative entre polymédication et fragilité, dépendance, mortalité et chutes dès cinq médicaments ». La prévention de la iatrogénie médicamenteuse fait partie des objectifs de l'assurance maladie qui dispose d'un indicateur de suivi du nombre moyen de médicaments prescrits par ordonnance. Le programme de qualité et d'efficience « Maladie » rattaché au PLFSS pour 2016 note que ce nombre a connu une baisse rapide.
|
B. LES MOYENS D'ACTION SUR LE COÛT DU MÉDICAMENT
Pour agir sur le coût du médicament, les pouvoirs publics disposent de deux leviers : le taux de remboursement et la fixation du prix.
1. La limite de la différenciation des taux de remboursement
Il existe quatre taux de remboursement échelonnés de 15% à 100% . Leur impact est cependant limité par la prise en charge à 100 % des médicaments prescrits pour le traitement d'une affection de longue durée. Comme le souligne le rapport de Mme Dominique Polton 46 ( * ) , des médicaments à service médical rendu faible peuvent être utilisés dans le traitement de pathologies graves. Ainsi, des médicaments peu remboursés en théorie sont pris en charge à 100 % pour certaines affections.
Afin d'avoir une vision du niveau de prise en charge réel du médicament, il ne faut pas se limiter à prendre en compte le taux de remboursement tel qu'il est fixé par le ministre en charge de la sécurité sociale, mais connaître son taux de remboursement moyen effectif.
L'analyse conduite par Dominique Polton établit que pour les médicaments pris en charge à 15 % comme pour ceux pris en charge à 30 %, le taux effectif moyen de remboursement est de près de 40 % . Pour les médicaments pris en charge à 65 % à l'issue de leur évaluation par la HAS, ce taux s'élève à 81 %.
Taux moyen observé selon le taux théorique de remboursement
|
Taux théorique |
Taux observé |
Montants
|
% des dépenses prises en charge
|
|
15 % |
38 % |
287 |
27 % |
|
30 % |
40 % |
508 |
15 % |
|
65 % |
81 % |
11 065 |
46 % |
Source : Rapport de Dominique Polton, présidente de la commission des comptes de santé sur la réforme des modalités d'évaluation des médicaments, novembre 2015
Cette différence entre un taux de prise en charge théoriquement faible et un taux moyen plus élevé en raison de la prise en charge intégrale des affections de longue durée est légitime, dès lors qu'il existe des médicaments dont le service médical rendu est faible sauf pour une part restreinte de la population. Il s'agit, en pratique, d'une prise en charge différenciée selon les pathologies .
Le coût pour l'assurance maladie est cependant non négligeable et pose la question de la cohérence de l'évaluation . En effet, quand un médicament a plusieurs SMR ouvrant droit au remboursement, le taux de remboursement est unique et fixé au niveau le plus élevé pour toutes les indications. Cependant, 27 % des médicaments dont le SMR a été jugé le plus faible sont remboursés à 100 %.
L'une des solutions possibles est la prise en charge différenciée selon les indications . Un médicament serait donc, selon l'indication dans laquelle il est prescrit, remboursé à 15, 30, 65 ou 100 %. L'idée se heurte néanmoins à la difficulté d'encadrer les prescriptions afin que ce ne soit pas systématiquement le taux de prise en charge le plus élevé qui soit indiqué par les prescripteurs.
Le rapport de Dominique Polton se fonde sur l'exemple belge pour étudier les modalités d'encadrement des prescriptions avec prise en charge différenciée et indique que l'Allemagne s'est également engagée dans cette voie. Il souligne toutefois la nécessité d'une expertise technique du dispositif qui fait peser un fort niveau de contrainte sur le prescripteur dont il faut contrôler les prescriptions pour vérifier l'adéquation des indictions au taux de remboursement.
A l'inverse, le rapport envisage la possibilité de mise en place d'un taux unique de remboursement, tout en conservant une prise en charge intégrale pour les ALD, afin de renforcer la cohérence du système. Cette proposition a, semble-t-il, été écartée par le Gouvernement 47 ( * ) .
2. Les modalités de fixation du prix du médicament
L'Europe connaît trois systèmes de fixation du prix du médicament . La liberté, dont l'exemple le plus pur est le Royaume-Uni, la négociation, dont la France est un exemple, et la fixation par référence au prix pratiqué dans d'autres pays, qui est généralement pratiquée en Europe centrale et orientale.
La fixation du prix du médicament par référence à celui pratiqué dans d'autres pays est généralement le fait d'Etats de plus petite taille. Ils entendent ainsi bénéficier des prix consentis par les industriels à des pays dont le marché est plus important, pour lesquels les firmes ont un intérêt économique à baisser leurs tarifs ou ont une obligation de les négocier. Elle pousse les industriels à concentrer leur action sur maintien d'un prix facial des médicaments et à consentir aux Etats utilisés comme référence, des remises dont le montant est couvert par le secret des affaires. La négociation qui est conduite sur le prix facial du médicament intègre donc une dimension de report sur les autres Etats du bénéfice des laboratoires. Pour un prix donné, tout ce qui est consenti comme rabais, non public, à un Etat est, potentiellement, autant de chiffre d'affaires garanti auprès des Etats qui ne retiendront que le prix facial. Les laboratoires ont intérêt à jouer de la rivalité des Etats en matière de rabais, c'est-à-dire de prix réels.
Ces inconvénients ont été relevés par le directeur général de la Cnam 48 ( * ) : « Les professionnels défendent ce prix affiché en France, (...) quitte à consentir des prix nets intéressants. En effet, les remises négociées sont couvertes par le secret des affaires et ne sont pas connues des marchés financiers. La conséquence en est, d'une part, qu'il devient impossible d'effectuer des comparaisons internationales et, d'autre part, qu'il n'y a plus de transparence des prix, notamment vis à vis de la représentation nationale. Pour autant, il semble difficile de définir un cadre européen permettant de contourner les stratégies des laboratoires ».
Cette mise en concurrence des Etats par les laboratoires fonctionne car aucune tentative de négociation coordonnée des prix des médicaments au niveau européen n'existe. Sans même envisager un transfert de cette compétence au niveau de l'Union ou même une négociation menée par les Etats, la coordination entre entités en charge de la négociation, le comité économique des produits de santé en France, la fédération des caisses d'assurance maladie en Allemagne par exemple, est balbutiante 49 ( * ) . De même l'initiative prise par le président de la République d'aborder la question du prix des médicaments à l'occasion du G7 qui s'est réuni au Japon les 26 et 27 mai 2016 n'a pas aboutie à une prise de position commune.
Vos rapporteurs estiment néanmoins que la négociation au moins concertée entre la France et ces principaux partenaires européens est le seul moyen d'agir de manière efficace contre l'avantage dont jouissent les laboratoires pharmaceutiques quand ils négocient avec chaque Etat individuellement . Ils estiment donc nécessaire que le Gouvernement poursuivent ses efforts afin de définir un cadre commun de négociation du prix des médicaments les plus onéreux.
Il convient de relever que la négociation du prix proposé par les laboratoires est un modèle de plus en plus suivi. Depuis 2011, l'Allemagne a ainsi imposé une négociation des laboratoires avec les caisses d'assurance maladie pour le prix de remboursement des médicaments à l'issue d'une année de liberté des prix. Plus récemment, le ministre de la santé allemand a annoncé le dépôt d'un projet de loi réduisant la période de liberté des prix pour les médicaments particulièrement onéreux dès qu'ils auraient dépassé un chiffre d'affaire de 250 millions d'euros 50 ( * ) . Cette décision fait suite à la mise sur le marché des médicaments contre l'hépatite C. Elle a également suscité cette déclaration à l'issue du conseil des ministres franco-allemand de Metz le 7 avril 2016 : « La France et l'Allemagne reconnaissent la nécessité d'un dialogue international concernant les défis que posent les médicaments innovants ».
Dans ce contexte international qui tend à renforcer l'importance de la négociation des prix tout en privilégiant les solutions nationales, il convient d'examiner le mécanisme en oeuvre en France.
|
Comparaison internationale du prix du médicament Depuis 2013 le rapport annuel du Ceps comporte une annexe relative à la comparaison internationale des niveaux des prix des médicaments. Cette annexe pose les problèmes méthodologiques liés aux comparaisons de médicaments et propose des tableaux portant sur différentes classes de médicaments ou spécialités. Les résultats obtenus se fondent sur des études nationales mais les études internationales ou étrangères disponibles sont également présentées. L'annexe 8 du rapport du Ceps pour 2014 présente notamment les études relatives aux médicaments sous brevet à chiffres d'affaires élevés en France. Elle conclue que « Les prix Français sont dans 20 cas sur 40 (50% des produits) inférieurs au plus bas prix européen et pour 37 cas sur 40 (93% des produits) inférieurs à la moyenne des 5 pays. » |
3. La fixation et la révision des prix par le Ceps
Lors de son audition par la commission des affaires sociales le 8 avril 2015, le directeur général de la Cnam a fait l'analyse suivante : « Le système actuel de fixation du prix des médicament me paraît satisfaisant. Il est fondé sur une base conventionnelle : un accord-cadre complété par quelques dispositions législatives venues régler des situations individuelles. Il a permis d'atteindre l'objectif de baisse des prix au rythme d'un milliard d'euros par an, sans pour autant créer de retard dans la mise à disposition des innovations thérapeutiques. »
Le Ceps, qui détermine le prix du médicament, fait néanmoins l'objet de critiques récurrentes. Pour en mesurer l'intérêt, il faut voir quelle est l'action du comité et dans quel cadre juridique elle s'exerce.
a) Une baisse durable du prix moyen des médicaments
La manière dont le prix du médicament est fixé en France a été dénoncée comme trop favorable aux laboratoires. A l'inverse le Leem estime que le prix des médicaments en France est trop bas pour permettre de financer durablement les investissements du secteur pour trouver de nouvelles molécules.
Du côté de la dénonciation de prix trop élevés ont peut noter la comparaison conduite sur le prix de certains médicaments en France et en Italie. L'ouvrage de Mmes Rivasi, Bertella-Geffroy et de M. Radier, Le racket des laboratoires pharmaceutiques et comment s'en sortir , publié en 2015, reprend l'affirmation ancienne selon laquelle le coût des médicaments en Italie est inférieur à celui qui prévaut en France et documente cette affirmation par la comparaison du prix constaté de onze molécules en 2014.
Ces données doivent cependant être nuancées par l'analyse des données agrégées. En effet, la part des dépenses pharmaceutiques est à peu près équivalente dans les deux pays autour de 1,6 % du PIB. Surtout les dépenses pharmaceutiques par habitant s'élèvent en 2014 à 449 euros en France 51 ( * ) et à 438 euros en Italie 52 ( * ) .
Au-delà de différences de prix facial incontestables sur certaines molécules, le montant global des dépenses de médicament est donc très proche entre les deux pays.
L'un des éléments à prendre en compte pour expliquer ce phénomène est que l'Italie a, depuis 2004, attaché beaucoup d'importance au fait de limiter les prix faciaux des nouvelles molécules tandis que la France a plutôt privilégié les remises et les baisses de prix pour les médicaments plus anciens.
D'autres paradoxes en termes de prix apparaissent cependant . La grande majorité de ceux mis sur le marché avec plusieurs dosages ou plusieurs conditionnements ont des prix différents pour chacun. Or ces prix sont souvent non linéaires ce qui signifie pour les petits dosages sont proportionnellement beaucoup plus chers que les dosages supérieurs. Ainsi l'une des statines les plus prescrites en France, près de 5,7 millions de prescriptions en 2013, existe en trois dosages 5 mg, qui est la dose la plus prescrite (85% des montants remboursés), 10 mg et 20 mg. Pour un conditionnement identique de 30 comprimés la boite de 5 mg coute 16,8 euros, celle de 10 mg 24,9 euros et celle de 20 mg 32,39 euros. Il en résulte qu'une boite de 10 mg coûte 9 euros de moins que deux boites de 5 mg et qu'une boite de 20 mg coute moitié moins que quatre boites de 5 mg.
Ainsi, s'il était possible de diviser en quatre les comprimés de 20 mg pour les patients auxquels la dose de 5 mg a été prescrite l'économie pour l'assurance maladie aurait été d'environ 145 millions d'euros en 2013. De fait les comprimés n'étant pas sécables cette économie reste virtuelle mais pose une question de cohérence dans la fixation des prix et sur la stratégie du Ceps en la matière.
La non-linéarité des prix se retrouve aussi sur les conditionnements puisque les conditionnements les plus prescrits qui comportent le moins de gélules sont proportionnellement plus chers que les conditionnements plus importants.
Une meilleure sensibilisation des prescripteurs à ces enjeux paraît nécessaire.
Du côté de la dénonciation de prix trop bas, le Leem prend pour sa part l'exemple d'une autre statine dont le prix a diminué de 82 % depuis 1997.
Ici encore, afin d'avoir une plus juste appréciation de la régulation du prix du médicament il convient de se référer au prix moyen.
Les dépenses d'assurance maladie liées au médicament ont décru en France entre 2012 et 2014 avant de rebondir en 2015 du seul fait des médicaments contre l'hépatite C. Mais en moyenne, le prix des médicaments a baissé depuis plusieurs années et, à l'exception de 2015, de manière continue . Au cours des dix dernières années l'essentiel de la hausse du coût des médicaments pour la sécurité sociale est liées aux effets de structure « résultant d'une part de la déformation de la consommation de médicaments vers des produit innovants et donc coûteux et d'autre part de l'augmentation de la prise en charge par l'assurance maladie au titre notamment des affections de longue durée »53 ( * ).
Le comité économique des produits de santé (Ceps), qui est un « organisme interministériel et interinstitutionnel placé sous l'autorité conjointe des ministres chargés de la santé, de la sécurité sociale et de l'économie » et « est principalement chargé par la loi de fixer les prix des médicaments et les tarifs des dispositifs médicaux à usage individuel ainsi que des prestations pris en charge par l'assurance maladie obligatoire », a donc durablement atteint l'objectif qui lui a été fixé de contribuer au respect de l'Ondam. Le professeur Jean-Yves Fagon, alors vice-président du Ceps, a estimé fin 2014 54 ( * ) que les réductions de prix conduites par le Ceps avaient abouti à des réductions de dépenses pour l'assurance maladie de l'ordre de 1 milliard d'euros par an depuis quatre ans.
Il convient de regarder les mécanismes qu'il met en oeuvre avant de s'interroger sur leur capacité à faire face à l'arrivée de médicaments innovants et onéreux.
b) Un processus de fixation des prix particulièrement encadré par le droit national et international
Les missions du Ceps et ses méthodes de travail sont particulièrement encadrées . Son existence et ses missions sont définies par le code de la sécurité sociale 55 ( * ) . De plus, ses missions « sont exercées dans le cadre des orientations qu'il reçoit des ministres compétents, en application de la loi de financement de la sécurité sociale ». Elles « portent notamment sur les moyens propres à assurer le respect de l'objectif national de dépenses d'assurance maladie ».
En plus de ses missions de fixation des prix, le Ceps « assure un suivi [tous les quatre mois] des dépenses de médicaments ainsi que des produits et prestations mentionnés à l'article L. 165-1 en vue de constater si l'évolution de ces dépenses est compatible avec le respect de l'objectif national de dépenses d'assurance maladie ».
Le Ceps est placé ainsi clairement dans l'orbite de la sécurité sociale en tant qu'instrument de maîtrise du coût des médicaments. Il est de ce point de vue significatif que le comité soit également chargé de conclure avec les laboratoires la charte de « qualité des pratiques professionnelles » de la visite médicale 56 ( * ) .
Les critères déterminant la fixation du prix d'un médicament sont énumérés par l'article L. 162-16-4 du code de la sécurité sociale.
« Le prix de vente au public de chacun des médicaments mentionnés au premier alinéa de l'article L. 162-17 est fixé par convention entre l'entreprise exploitant le médicament et le Comité économique des produits de santé conformément à l'article L. 162-17-4 ou, à défaut, par décision du comité, sauf opposition conjointe des ministres concernés qui arrêtent dans ce cas le prix dans un délai de quinze jours après la décision du comité. La fixation de ce prix tient compte principalement de l'amélioration du service médical rendu apportée par le médicament, le cas échéant des résultats de l'évaluation médico-économique, des prix des médicaments à même visée thérapeutique, des volumes de vente prévus ou constatés ainsi que des conditions prévisibles et réelles d'utilisation du médicament. Lorsque la fixation du prix du médicament est fondée sur une appréciation de l'amélioration du service médical rendu différente de celle de la commission mentionnée à l'article L. 5123-3 du code de la santé publique, le Comité économique des produits de santé fait connaître à la commission les motifs de son appréciation.
Ce prix comprend les marges prévues par la décision mentionnée à l'article L. 162-38 ainsi que les taxes en vigueur . »
La dernière lettre d'orientation adressée par les ministres de tutelle au président du Ceps fixe pour sa part des conditions précises d'évolution du prix des médicaments lors de l'arrivée d'un générique .
Ainsi, le Ceps dispose incontestablement d'un pouvoir d'appréciation dans la conduite de ses négociations avec les industriels étant donné la multiplicité des produits dont il administre les prix mais les critères de fixation des prix qu'il utilise sont publics et sous le contrôle des ministres et du Parlement.
Les relations entre le Ceps et les industriels, représentés par le Leem, sont définies par un accord cadre triennal dont la dernière version a été signée le 31 décembre 2015. Cet accord prévoit parmi ses considérants la nécessité d'organiser « une régulation proportionnée à l'apport du médicament ». Cette régulation, fondée sur le constat que « les dépenses de médicament sont pour l'essentiel financées par la collectivité sur des ressources par nature limitées » s'exerce dans le cadre législatif et réglementaire précité mais aussi « dans des conditions équitables et transparentes ». Ces dernières découlent notamment de la directive 89/105/CEE du Conseil du 21 décembre 1988 qui fixe notamment un délai maximal de 90 jours pour l'instruction des demandes d'inscription sur la liste des médicaments remboursables formulées par les industriels.
L'accord-cadre prévoit également que la régulation doit respecter la propriété intellectuelle, les marques, la protection des données d'enregistrement et la confidentialité des affaires.
Ces différentes obligations, qui font souvent l'objet de critiques fondées sur l'absence de transparence des négociations conduites par le Ceps , trouvent un fondement en droit international dans l'accord sur les aspects des droits de propriété intellectuelle qui touchent au commerce (Adpic), texte annexé à l'accord créant l'organisation mondiale du commerce.
Spécifiquement, l'article 39 de l'accord fonde la protection des données d'enregistrement. Il dispose en effet (39.3) : « Lorsqu'ils subordonnent l'approbation de la commercialisation de produits pharmaceutiques ou de produits chimiques pour l'agriculture qui comportent des entités chimiques nouvelles à la communication de données non divulguées résultant d'essais ou d'autres données non divulguées, dont l'établissement demande un effort considérable, les membres protégeront ces données contre l'exploitation déloyale dans le commerce. En outre, les membres protégeront ces données contre la divulgation, sauf si cela est nécessaire pour protéger le public, ou à moins que des mesures ne soient prises pour s'assurer que les données sont protégées contre l'exploitation déloyale dans le commerce. »
Le point (39.2) de l'article fonde pour sa part la confidentialité des affaires. L'accord dispose que : « Les personnes physiques et morales auront la possibilité d'empêcher que des renseignements licitement sous leur contrôle ne soient divulgués à des tiers ou acquis ou utilisés par eux sans leur consentement et d'une manière contraire aux usages commerciaux honnêtes (...) » sous réserve de trois conditions cumulatives.
Ce fondement juridique est important car la notion de secret ou de confidentialité des affaires a pour l'essentiel en droit national un fondement jurisprudentiel 57 ( * ) . Le projet de directive sur la protection des savoir-faire et des informations commerciales non divulgués (secrets d'affaires) contre l'obtention, l'utilisation et la divulgation illicites 58 ( * ) qui a fait l'objet d'un accord entre le Parlement et le Conseil contient pour sa part une définition plus précise du secret des affaires qui s'appliquera en droit français.
Les considérants du projet de directive indiquent : « Les savoir-faire et les informations commerciales de valeur, qui ne sont pas divulgués et que l'on entend garder confidentiels, sont appelés secrets d'affaires . » Ils précisent que ces secrets couvrent « une large gamme d'informations, qui va des connaissances technologiques aux données commerciales telles que les informations relatives aux clients et aux fournisseurs, les plans d'affaires et les études et stratégies de marché ». Si la divulgation aux autorités publiques de certains éléments couverts par le secret des affaires est prévue par le droit de l'Union qui fait même obligation aux autorités de divulguer au public certaines données, le principe est celui de la protection du secret, garanti par le juge.
c) Des objectifs qui doivent être conciliés
Les missions confiées aux Ceps en matière de fixation du prix des médicaments ne concernent pas uniquement sa maîtrise dans le cadre du respect de l'Objectif national des dépenses d'assurance maladie, mais également l'accès des patients aux médicaments ainsi que des objectifs de valorisation de l'innovation et de développement industriel. Significativement, le respect de l'Ondam n'est que le cinquième des six objectifs fixés par les ministres de tutelle au Ceps dans la lettre de mission d'avril 2013.
|
Lettre d'orientation adressée au
président du Ceps
La politique économique des produits de santé du Gouvernement s'organise autour des objectifs suivants : - garantie d'un accès effectif pour tous à des soins de qualité ; - promotion du bon usage du médicament et efficience de la dépense ; - valorisation des innovations sources de progrès thérapeutique ; - transparence du processus de fixation des prix et cohérence des décisions ; - respect des objectifs annuels d'évolution des dépenses d'assurance maladie ; - soutien, conformément au Pacte national pour la compétitivité, la croissance et l'emploi, au dynamisme des industries de santé, qui sont un secteur d'avenir prioritaire, et au développement de l'emploi. Vous veillerez à fixer les prix et à animer la politique conventionnelle dans le respect de ces objectifs. |
Ceci explique que les critères de fixation du prix des médicaments, tels qu'ils sont énumérés à l'article L. 162-16-4 du code de la sécurité sociale, relèvent de l'analyse de l'apport du médicament plutôt que de son coût de production. Cet apport est mesuré par la commission de la transparence de la Haute Autorité de santé qui détermine l'amélioration du service médical rendu par un médicament et, de manière croissante, par l'évaluation médico-économique conduite par la commission évaluation économique et de santé publique créé en 2008 au sein de la Haute Autorité.
Cette évaluation, qui tend à garantir l'efficience de la dépense, découle de l'article 47 de la loi de financement de la sécurité sociale pour 2012, voulu par le Sénat. L'article R. 161-71-1 du code de la sécurité sociale a été modifié pour permettre sa mise en oeuvre. Il fixe deux conditions cumulatives pour qu'une telle évaluation soit conduite pour un médicament : « 1° La reconnaissance ou la confirmation d'une amélioration du service médical rendu ou du service attendu, majeure, importante ou modérée (...) est sollicitée par l'entreprise ;
« 2° Le produit ou la technologie a ou est susceptible d'avoir un impact significatif sur les dépenses de l'assurance maladie compte tenu de son incidence sur l'organisation des soins, les pratiques professionnelles ou les conditions de prise en charge des malades et, le cas échéant, de son prix. »
La logique médico-économique tend à se développer. Ainsi l'accord cadre Leem-Ceps de janvier 2016 rend obligatoire par son article 9 la réalisation d'un étude d'impact budgétaire pour tout produit innovant dont le chiffre d'affaires prévisionnel est supérieur à 50 millions d'euros à l'issue de la deuxième année de commercialisation . Mais elle n'a jamais été totalement absente des critères de fixation des prix. Ainsi, la diversification de l'offre de médicaments pour une même indication offre des alternatives thérapeutiques mais répond aussi à une logique économique.
L'autorisation de mise sur le marché de médicaments n'ayant pas d'apport thérapeutique par rapport à ceux déjà existant, mais se prévalant du même effet dans les mêmes indications est régulièrement contestée. Ces médicaments sont dits « me too ». Mais, du point de vue de la fixation du prix, l'arrivée de ces médicaments se justifie pour augmenter la concurrence et faire baisser les prix. Les baisses de prix obtenues du fait de la mise sur le marché des « me too », longtemps demandées par notre ancien collègue François Autain, figurent désormais dans le rapport annuel du Ceps.
A l'occasion de la contestation du prix du Sovaldi, plusieurs associations ont contesté le fondement de la fixation du prix sur l'apport du médicament. Pour établir une « juste rémunération » du laboratoire elles se sont fondées sur une étude du coût de production de la molécule qui devrait, selon elles, servir de base au calcul du prix du médicament. Or, il résulte clairement des missions assignées au Ceps que tant l'objectif de rémunération de l'innovation que celui de l'accès des patients aux médicaments lui imposent de rémunérer le travail intellectuel protégé par le brevet plus que le simple coût de production. Le risque, en effet, en fondant le prix de vente du médicament sur le coût de production, est de désinciter les entreprises à investir en matière de recherche et développement, mais aussi que les entreprises refusent de mettre à disposition leurs médicaments sur le marché français si elles considèrent que le prix proposé est trop bas.
Au-delà de la négociation, le moyen de faire baisser les prix des médicaments innovants de manière significative est une meilleure gestion de la déclinaison de la recherche publique française et des starts-ups qui en sont issues ainsi que la contestation des brevets qui paraissent abusifs. Cette compétence, qui ne figure pas parmi celles du Ceps, incombe au ministre en charge de la propriété industrielle sur le fondement de l'article L. 613-16 du code de la propriété intellectuelle.
|
Les règles relatives à la licence d'office Plusieurs associations ont évoqué la possibilité, voire la nécessité, de mettre en place un mécanisme de licence d'office pour fournir les AAD à tous les malades souffrant d'hépatite C. L'article L. 613-16 du code de la propriété intellectuelle ouvre la possibilité suivante : « Si l'intérêt de la santé publique l'exige et à défaut d'accord amiable avec le titulaire du brevet, le ministre chargé de la propriété industrielle peut, sur la demande du ministre chargé de la santé publique, soumettre par arrêté au régime de la licence d'office, dans les conditions prévues à l'article L. 613-17, tout brevet délivré pour : a) Un médicament, (...) Les brevets de ces produits, procédés ou méthodes de diagnostic ne peuvent être soumis au régime de la licence d'office dans l'intérêt de la santé publique que lorsque ces produits, ou des produits issus de ces procédés, ou ces méthodes sont mis à la disposition du public en quantité ou qualité insuffisantes ou à des prix anormalement élevés, ou lorsque le brevet est exploité dans des conditions contraires à l'intérêt de la santé publique ou constitutives de pratiques déclarées anticoncurrentielles à la suite d'une décision administrative ou juridictionnelle devenue définitive. Lorsque la licence a pour but de remédier à une pratique déclarée anticoncurrentielle ou en cas d'urgence, le ministre chargé de la propriété industrielle n'est pas tenu de rechercher un accord amiable. » L'accord sur les aspects des droits de propriété intellectuelle qui touchent au commerce (Adpic), auquel la France est partie, et qui est entré en vigueur en 1995, reconnaît la possibilité pour un Etat de prévoir une licence d'office, aussi appelée licence obligatoire, mais l'encadre. L'article 31 de l'accord prévoit ainsi qu'en cas de mise en place d'une licence d'office, « le détenteur du droit percevra une rémunération adéquate selon le cas d'espèce, compte tenu de la valeur économique de l'autorisation ». La référence à la valeur économique de l'autorisation fonde l'existence d'un niveau d'indemnisation proche du prix du médicament tel qu'il est établi par le laboratoire. Le risque en cas de recours à la licence d'office est donc que le juge national impose à l'Etat de payer le prix demandé par le laboratoire, lequel prix viendra, du point de vue des finances publiques, s'ajouter au coût d'achat ou au moins de production des médicaments produits sur le fondement de la licence d'office. |
4. La fiscalité du médicament
Sans représenter l'élément déterminant pour les entreprises dans les choix de localisation de leurs activités, la fiscalité est l'un des éléments constitutifs du degré d'attractivité du territoire français.
Outre les questions d'attractivité, la localisation des activités, en particulier des activités de recherche, peut avoir un impact en termes d'accès des patients aux molécules ou aux traitements les plus innovants dans le cadre des essais cliniques.
Plus que son niveau, difficile à apprécier, c'est le caractère évolutif et instable de la fiscalité applicable au secteur du médicament qui semble de nature à influer sur les choix des entreprises.
a) Une fiscalité générale peu lisible
Le secteur pharmaceutique est assujetti aux impositions de droit commun. Alors qu'elle se situait dans la moyenne européenne (33 % en 1999), elle a eu tendance à augmenter son taux d'imposition sur les sociétés, en imposant notamment une surtaxe à partir de 2011 alors que l'Allemagne ou le Royaume-Uni baissaient leur taux. Avec 34,4 % en 2015, contre 25 % en moyenne au sein de l'Union européenne, la France affichait le taux nominal d'impôt sur les sociétés le plus élevé de l'Union européenne.
Plusieurs dispositifs contribuent cependant à tempérer ce constat. Le régime d'intégration fiscale, qui permet d'exonérer en grande partie les remontées de dividendes au sein d'un groupe intégré, le mécanisme de suramortissement, qui conduit à subventionner à hauteur de 40 % des investissements et un régime fiscal favorable pour les PME innovantes permettent de réduire les montants d'imposition. Une étude 59 ( * ) situe la France dans la moyenne des quatorze pays européen étudiés.
Le crédit impôt recherche, puis le crédit d'impôt compétitivité emploi, et, plus récemment, la suppression de la surtaxe IS au 1 er janvier 2016 ont eu pour effet de réduire le produit de l'IS qui s'élevait à 34 milliards d'euros en 2015 contre 44 milliards en 2013.
En 2012, le secteur pharmacie-parfumerie-entretien était le deuxième secteur en termes de dépenses déclarées (13 %) et le troisième pour le crédit impôt recherche (11 % des dépenses) ; au sein de cet ensemble la place de la pharmacie est très importante (11 % des dépenses et 9 % du CIR). Sur un total de 5 milliards d'euros, l'industrie pharmaceutique bénéficie donc d'environ 600 millions d'euros de CIR.
Pour ce qui concerne le CICE, dont le seuil de sortie est de 2,5 Smic, l'effet du dispositif est plus marginal. D'après le rapport du comité de suivi du CICE, l'industrie pharmaceutique représente une masse salariale de 3,4 milliards d'euros dont 1,3 milliard d'euros, soit 38 %, entrent dans l'assiette éligible au CICE.
L'industrie pharmaceutique devrait en revanche bénéficier de la réduction de 1,6 point de la cotisation famille, applicable depuis le 1 er avril 2016 et dont le seuil de sortie est de 3,5 Smic.
b) Une fiscalité spécifique foisonnante aux objectifs brouillés
Notre pays se distingue par l'existence et l'ampleur d'une fiscalité spécifique applicable aux produits de santé. Sur le champ de l'industrie du médicament, sept taxes sont applicables, auxquelles s'ajoutent deux mécanismes de régulation, qui sont en fait des déclinaisons du mécanisme de fixation des prix.
Les dispositifs et leurs produits sont les suivants :
|
2012 |
2013 |
2014 |
2015 (p) |
2016 (p) |
|||
|
Contribution sur
|
Article L 245-6
|
Cnamts |
364 |
332 |
401 |
391 |
384 |
|
Contribution due
|
Articles L. 138-10
|
Cnamts RSI maladie, CCMSA - salariés
|
315 |
203 |
298 |
299 |
284 |
|
Contribution due
|
Article L. 245-1 du code de la sécurité sociale |
Cnamts, Haute autorité de la santé jusqu'en 2013 / Cnamts uniquement à partir de 2014 |
189 |
185 |
208 |
177 |
185 |
|
Droits perçus
|
Article 1635 bis AE du code général des impôts |
Cnamts |
164 |
67 |
70 |
71 |
73 |
|
Taxe sur les premières ventes de médicaments et produits de santé |
Article 1600-0 N du code général des impôts |
Cnamts |
39 |
45 |
_ |
_ |
|
|
Contribution
|
Article L. 245-5-1 A du code de la sécurité sociale |
Cnamts,
|
6 |
2 |
2 |
2 |
|
|
Taxe annuelle relative à l'enregistrement
|
Article L. 5211-5-1 du code de la santé publique,
|
Haute Autorité
|
4 |
0 |
0 |
0 |
|
|
Total |
1032 |
787 |
977 |
938 |
926 |
||
A la suite de la 5 ème réunion du Conseil stratégique des industries de santé (CSIS), tenue le 25 janvier 2012, les ministres ont commandé à l'inspection générale des affaires sociales et à l'inspection générale des finances un état des lieux de la fiscalité spécifique applicable sur le territoire français.
Cet état des lieux relève effectivement une situation singulière en France, du fait notamment du manque de lisibilité et de la complexité de la fiscalité spécifique.
Il conclut toutefois à un niveau voisin de pression fiscale sur le secteur en Allemagne, en Italie et en Espagne, qui ont également mis en place une fiscalité spécifique, une fois pris en compte les mécanismes de crédit d'impôt, dont le CIR dont il est noté qu'il représentait, en 2012, près des deux-tiers des produits de fiscalité spécifique .
Tout en constatant un niveau de pression fiscale moindre au Royaume-Uni et en Suisse, l'étude pointe globalement, à paramètres inchangés, le risque d'une dégradation de la position relative de la France en raison des mesures d'allègements fiscaux mis en place dans différents pays d'Europe.
Dans son état des lieux, l'étude relève par ailleurs, de « nombreuses imperfections » :
« - sa complexité et son manque de lisibilité ;
« - sa faible prévisibilité ;
« - l'éclatement de sa gouvernance et de son recouvrement, qui entraîne le risque d'une méconnaissance du secteur faute d'une vision globale, ainsi que des coûts d'administration sous-optimaux ;
« - son adaptation perfectible à des évolutions structurelles du secteur cohérence parfois incertaine avec l'ensemble des objectifs poursuivis par les politiques publiques
« - ses failles formelles, qui permettent à certains acteurs du secteur d'entretenir des contentieux d'autant plus nombreux et durables sur certaines taxes (taxes sur les dépenses de promotion des médicaments) qu'ils sont parfois fondés juridiquement ».
À la suite de ce rapport, différents correctifs ont été apportés, comme la suppression des taxes affectées aux agences, qui s'est traduite par un effort de rationalisation, mais le constat global de complexité et de variabilité reste valable.
Chaque année, une étude financée par le LEEM, dont le champ comprend l'ensemble des composantes de la fiscalité mais aussi les cotisations sociales, conclut à un désavantage comparatif pour la France en raison précisément de cette fiscalité spécifique. Le bilan économique du secteur comporte des développements sur ce handicap des entreprises présentes sur le sol français. Sur le fondement de calculs théoriques, cette étude fait apparaître un différentiel fiscal significatif avec le Royaume-Uni, l'Irlande ou encore la Suisse. Ce différentiel tient davantage aux cotisations sociales pour l'Allemagne ou l'Italie.
c) Une fiscalité palliative au mécanisme de fixation des prix
Vos rapporteurs ont souhaité distinguer deux mécanismes de régulation, respectivement institué et révisé par la loi de financement pour 2015, la contribution sur le chiffre d'affaires des médicaments visant à lutter contre le virus de l'hépatite C ou « mécanisme W » et le taux L, ou « clause de sauvegarde de l'Ondam ».
Ces mécanismes sont complémentaires de la fixation des prix du médicament, même si ils tendent à se rapprocher d'une taxation de rendement.
La clause de sauvegarde, prévue par l'article L 138-10 du code de la sécurité sociale créé par la LFSS pour 1999 du 23 décembre 1998 poursuivait un objectif de conventionnement des laboratoires avec le comité économique des produits de santé. Précédemment exonératoire, le conventionnement ne permet plus que de réduire le montant de la contribution à verser.
De manière comparable, le mécanisme W pallie les éventuelles difficultés rencontrées lors du processus de fixation du prix du médicament en assujettissant à une contribution la part du chiffre d'affaires dépassant un seuil cible. Le conventionnement ne permet là encore que de réduire le montant de la contribution qui prend alors la forme d'une remise. Au titre de 2014, 282 millions d'euros ont été notifiés pour cette contribution.
Ces mécanismes ont été fortement critiqués en ce qu'ils constituent des entorses au principe de négociation du prix des médicaments en ville en le CEPS et l'industrie. Leur existence acte effectivement le fait que le mécanisme de négociation des prix ne permet pas de parvenir à un résultat satisfaisant pour la puissance publique. On peut aussi craindre que l'industrie ne les intègre à l'avenir dans la négociation des prix.
Il convient toutefois de noter que la progression du chiffre d'affaires global de l'industrie pharmaceutique étant intégralement imputable aux médicaments de lutte contre le virus de l'hépatite C, dans un contexte de chiffre d'affaires en recul, l'absence de mécanisme spécifique aurait conduit à faire contribuer l'ensemble du secteur au titre du chiffre d'affaires réalisés pour ces seuls médicaments. Cela a été effectivement le cas, mais dans une moindre mesure.
Vos rapporteurs recommandent toutefois de ne pas multiplier à l'avenir ces mécanismes de régulation spécifique, qui risquent de porter atteinte à un mécanisme de négociation des prix qui a permis, jusqu'à présent, y compris pour les médicaments anti-VHC, de parvenir à des solutions soutenables pour l'assurance-maladie.
L'examen de la fiscalité applicable à l'industrie pharmaceutique conduit à un constat renouvelé de complexité et faible visibilité, parfois (taux L dans sa première version) de difficulté même de mise en oeuvre.
Certains dispositifs, conduisent à des conflits d'objectifs entre différentes politiques publiques : la taxe sur les dépenses de publicité porte majoritairement sur de la masse salariale dont les dispositifs généraux (CICE, allègements de cotisations) visent à diminuer le coût.
Un nouvel examen serait sans doute nécessaire à l'issue du pacte de responsabilité dans un souci de cohérence globale et avec un objectif de stabilité.
5. Le déploiement du générique et du biosimilaire
Le prix du médicament a vocation à baisser de manière substantielle dès l'échéance du brevet qui protège la propriété intellectuelle. Il convient de rappeler que ce brevet est plus long pour le médicament que pour les autres produits en raison des dépenses de recherche et développement qui sont nécessaires à son élaboration.
Deux types de médicaments peuvent, selon la nature du médicament dit princeps, être mis sur le marché à l'échéance d'un brevet : les génériques et les biosimilaires.
a) Les médicaments génériques
Le recours aux génériques reste plus faible en France que dans les autres pays européens. Néanmoins, leur développement a permis des économies substantielles qui peuvent être poursuivies.
Il apparaît d'abord nécessaire de préciser la définition du générique : « Dans tous les pays, un générique est un médicament qui possède les mêmes propriétés (même substance active) que le produit de référence (appelé « princeps ») et dont le brevet est tombé dans le domaine public. Cependant, le périmètre des génériques varie d'un pays à l'autre. En France, la définition des génériques est plus restrictive qu'ailleurs, ce qui explique en partie leur plus faible pénétration. Elle se réfère à la notion d'équivalent chimique (soit la même molécule), alors que dans d'autres pays, comme aux Pays-Bas et en Allemagne par exemple, elle s'appuie sur la notion d'équivalent thérapeutique qui autorise de plus larges possibilités de substitution et accroît leur pénétration. » 60 ( * ) .
Sur cette base, il est incontestable que non seulement le taux de recours aux génériques est plus faible en France qu'ailleurs, mais en outre leurs prix sont plus élevés. Les orientations données par les ministres de tutelle au Ceps en 2013 fixent ainsi des obligations très précises de baisse des prix du médicament princeps et des médicaments génériques. Par ailleurs, un plan triennal d'actions de promotion des médicaments génériques a été lancé le 24 mars 2015.
Le rapport « charges et produits » de la Cnam pour 2016 fait le constat suivant : « La croissance des dépenses de médicament est d'ores et déjà principalement tirée par l'arrivée des nouveaux médicaments de spécialité. Dans les années précédentes, cette dynamique a pu être financée par les économies associées à l'expiration progressive des brevets des blockbusters apparus dans les années 1990 (statines, médicaments antihypertenseurs, inhibiteurs de la pompe à protons...), combinées à des actions pour promouvoir la diffusion des génériques et à des politiques actives de baisse de prix.
Cet effet va cependant s'épuiser, car des classes entières sont aujourd'hui génériquées : les gains importants générés mécaniquement du fait des volumes élevés de prescriptions ont été réalisés, même si ces gains ont parfois été moins rapides que dans d'autres pays, du fait de la tendance française au report des prescriptions vers les médicaments récents non génériqués .»
Il ne faut donc pas surestimer les économies liées au déploiement des médicaments génériques. Ils demeurent cependant porteurs d'économies substantielles pour l'assurance maladie 61 ( * ) et peu développés en France en comparaison d'autres pays.
En effet, la Cnam, dans son rapport charges et produits pour 2015, indiquait elle-même : « Sur neuf classes représentant 70 % des prescriptions de médecine générale 50, un transfert de 1 point des prescriptions hors répertoire vers des produits génériques induirait une économie voisine de 14 millions d'euros, dont 3,3 millions d'euros sur les traitements de l'asthme et de la BPCO, ainsi que le paracétamol, qui ne sont pas à ce jour inscrits au répertoire même si une offre générique est disponible. Si l'on extrapole à l'ensemble du marché du médicament remboursable, on peut estimer à 25 millions d'euros l'économie globale par point de prescription supplémentaire dans le répertoire ».
b) Les biosimilaires
Le développement du médicament biosimilaire en France est pour sa part un chantier d'avenir. Le rapport charges et produits pour 2016, qui constatait que les économies liées aux génériques pourraient se réduire, indiquait en contrepoint : « Le développement des biosimilaires devrait cependant apporter des perspectives d'économies dans les prochaines années ». Un médicament biosimilaire est similaire à un médicament biologique (substance qui est produite à partir d'une cellule ou d'un organisme vivant ou dérivée de ceux-ci) de référence qui a déjà été autorisé en Europe et dont le brevet est tombé dans le domaine public.
L'article 47 de la loi de financement de la sécurité sociale pour 2014 a introduit dans le code de la santé publique un article L. 5125-23-3 prévoyant la possibilité de substitution par le pharmacien d'un médicament biologique prescrit en initiation de traitement par un médicament biosimilaire. « Les modalités d'application [de cet] article, et notamment les conditions de substitution du médicament biologique et d'information du prescripteur à l'occasion de cette substitution de nature à assurer la continuité du traitement avec le même médicament, sont précisées par décret en Conseil d'Etat. » Les dispositions relatives à la substitution ne sont à ce jour pas parues, en raison notamment de la volonté des industriels du secteur de prévoir des modalités de diffusion des médicaments biosimilaires distinctes de celles du médicament générique et reposant davantage sur les prescripteurs, ce qui semble un moyen de susciter chez eux les réticences qui entourent encore les génériques.
Le décret relatif au médicament biosimilaire est particulièrement attendu. Dans le rapport charges et produits pour 2015, la Cnam formulait la proposition suivante : « Favoriser l'usage des médicaments biosimilaires » et indiquait qu'elle menait une « action de sensibilisation et d'accompagnement en cours de réalisation par l'assurance maladie dans les établissements de santé sur le thème des EPO avec un message pour favoriser la prescription des biosimilaires ». En effet, il a été évalué que « L'introduction sur le marché Allemand du biosimilaire de l'EPO a permis de réaliser des économies annuelles de plus de 60 millions d'euros (- 17,3%) dans sa première année de commercialisation » 62 ( * ) .
Au-delà de cet exemple, l'ampleur des économies possibles grâce aux biosimilaires a été soulignée par la ministre Marisol Touraine qui a déclaré devant l'Opecst en janvier 2015 qu'« en France, sur les dix médicaments les plus couteux utilisés à l'hôpital, sept sont des médicaments biologiques ».
Le cadre permettant le déploiement du biosimilaire s'avère cependant complexe à mettre en oeuvre. En effet l'ANSM a publié sur son site internet, le 2 mai 2016, un état des lieux sur les médicaments biosimilaires, présentant une évolution au regard de ses précédentes recommandations datant de septembre 2013.
Afin que les textes en vigueur ne soient pas en décalage avec la pratique médicale, l'ANSM a procédé à des consultations auprès des professionnels de santé sur le sujet. Par ailleurs, à la suite d'une réflexion de l'Agence européenne du médicament (EMA) sur les médicaments biologiques, les recommandations des autorités sanitaires de certains Etats membres ont évolué et l'ANSM a également estimé nécessaire d'adapter son rapport initial de 2013 sur les biosimilaires.
L'ANSM a indiqué à vos rapporteurs qu'elle considère désormais, à l'instar de la position des autorités néerlandaises sur le sujet, que l'interchangeabilité des médicaments biologiques est envisageable sous certaines conditions strictes consistant à ce que le patient soit informé d'un éventuel changement de spécialité et qu'il ait manifesté son consentement, que le patient reçoive une surveillance clinique appropriée durant le traitement, et que la traçabilité des spécialités ayant été délivrées au patient soit assurée.
Dès lors, l'article L.5125-23-3 du Code de la santé publique issu de l'article 47 de la Loi de financement de la Sécurité Sociale pour 2014 devra faire l'objet d'une modification. Cet article prévoit actuellement un mécanisme permettant la substitution d'un médicament biologique par un biologique similaire du même groupe par le pharmacien, uniquement en initiation de traitement ou afin de poursuivre un traitement déjà initié avec un biosimilaire. Le mécanisme prévu par cet article prévoit en outre la nécessaire continuité de traitement avec le médicament initialement délivré au patient.
Des discussions devront donc être engagées dans le cadre de la préparation du PLFSS pour 2017.
En revanche, une première étape a d'ores et déjà été menée puisque le Ministère chargé de la santé a transmis au Conseil d'Etat un premier projet de décret, relatif uniquement à l'inscription des médicaments biologiques similaires sur la liste de référence des groupes biologiques similaires. Ce projet de décret a été examiné en section sociale au Conseil d'Etat le 21 juin.
III. QUEL MÉCANISME INSTITUTIONNEL POUR LA MEILLEURE PRISE EN CHARGE DU MÉDICAMENT ?
A. UN MÉCANISME DE MISE SUR LE MARCHÉ ACCUSÉ D'OPACITÉ
Les différentes étapes qui aboutissent à rendre un médicament disponible en officine avec un prix et un taux de remboursement relèvent d'organismes différents dont le nombre a été critiqué.
1. Des missions confiées à des organes distincts
S'agissant de l'arrivée sur le marché d'un médicament, on peut distinguer trois étapes différentes : l'autorisation de mise sur le marché, qui relève d'un processus encadré au niveau européen, l'évaluation du médicament pour la détermination de son taux de remboursement et de son prix et, dans le système français, la négociation du prix.
A ces trois étapes correspondent en France, par opposition, par exemple, avec l'Italie, trois instances : l'agence de sécurité du médicament et des produits de santé (ANSM) pour l'autorisation de mise sur le marché ; la Haute Autorité de santé, au travers de deux de ses commissions, la commission de la transparence et la commission de l'évaluation médico-économique, pour l'évaluation ; enfin, le comité économique des produits de santé (Ceps) pour la négociation du prix du médicament avec les industriels.
Le débat sur l'organisation du système actuel est double. Il porte sur le fait de savoir si les missions attribuées à différents organismes sont suffisamment distinctes et si elles ne pourraient être mieux exercées par d'autres instances.
La mission d'évaluation du médicament est conduite par l'ANSM d'une part, par la Haute Autorité de santé d'autre part. L'évaluation porte sur des points différents car l'ANSM évalue la sécurité du médicament . L'absence de nocivité d'un médicament n'est pas absolue mais résulte du rapport entre le bénéfice qu'il représente pour le patient et le risque qu'il comporte, dans les indications soumises par l'industriel 63 ( * ) .
L'évaluation conduite par la Haute Autorité de santé porte, pour sa part, sur l'apport du médicament afin de savoir s'il doit être pris en charge par la collectivité et dans quelle proportion.
Les deux démarches sont distinctes mais elles se fondent le plus souvent sur les mêmes études et sont parfois conduites devant les deux instances par les mêmes experts. Un rapprochement des deux évaluations pourrait donc être envisagé. Dans les suites de l'affaire du Mediator, les partisans d'un renforcement du contrôle exercé au moment de l'autorisation de mise sur le marché ont ainsi plaidé pour que ne soient admis que les médicaments présentant un réel apport pour les patients, ce qui rapprocherait le contrôle exercé aujourd'hui par l'ANSM de celui de la commission de la transparence de la HAS. Cette volonté de renforcer l'évaluation des bénéfices réels a conduit à généraliser les études présentant la supériorité du médicament contre comparateur actif plutôt que contre placebo uniquement 64 ( * ) .
L'article L. 5311-1 du code de la santé publique, dans sa version issue de la loi du 29 décembre 2011 dite « loi Bertrand », dispose que « l'ANSM peut demander que les essais cliniques portant sur des médicaments soient effectués sous forme d'essais contre comparateurs actifs et contre placebo. Si la personne produisant ou exploitant un médicament s'oppose aux essais contre comparateurs actifs, elle doit le justifier ».
Cependant, d'après le Pr Joseph Emmerich 65 ( * ) , alors directeur à la direction des médicaments (cardiologie, endocrinologie, gynécologie et urologie) de l'ANSM, il résulte de la directive 2004/27/CE du Parlement européen et du Conseil (EMA/119319/04), qu'« il n'est pas nécessaire, pour le profil bénéfice/ risque (...) d'un traitement expérimental, d'être aussi favorable qu'une autre ou qu'aux autres thérapeutique(s) établie(s) dans ce champ thérapeutique, pour obtenir une AMM ». Ainsi, même si l'on a recours à un comparateur actif plutôt qu'à un placebo, le bénéfice du médicament testé reste évalué en lui-même. Il n'est pas nécessaire qu'il soit supérieur à celui du médicament auquel il est comparé. Il suffit donc que le rapport entre le bénéfice et le risque du médicament testé soit positif, comme lorsqu'il y a comparaison avec un placebo, pour qu'il puisse être mis sur le marché . Il en résulte que la comparaison d'un nouveau médicament contre un médicament actif a essentiellement pour conséquence d'améliorer l'information des autorités sanitaires sur l'intérêt d'un médicament, mais n'a pas de conséquence sur l'octroi de l'AMM.
Dès lors que la directive européenne n'impose pas un renforcement des exigences lorsqu'un comparateur actif est utilisé, conditionner la mise sur le marché d'un médicament à sa supériorité en termes de bénéfices par rapport aux médicaments existants est impossible au plan national. Tout d'abord, la grande majorité des autorisations de mise sur le marché se font au niveau européen. Surtout, augmenter le niveau d'exigence en matière d'autorisation de mise sur le marché serait contraire au droit européen. Le niveau d'exigence ne peut donc être aussi important lorsqu'il s'agit de cette procédure, européenne, que lorsqu'il s'agit d'établir l'admission au remboursement qui relève de la seule compétence nationale.
Vos rapporteurs considèrent que dans un contexte où les projets d'AMM fractionnées porté notamment par l'EMA tendent à raccourcir les délais de mise sur le marché il est nécessaire de renforcer le niveau d'exigence concernant les médicaments autorisés sur le marché européen et de s'orienter vers une approche plus comparative des molécules et une prise en compte de leur efficacité . En conséquence ils souhaitent que la France prenne l'initiative au niveau européen d'une évolution de la directive 2004/27 afin de renforcer les exigences en matière de mise sur le marché.
Dans l'attente d'une telle évolution le pouvoir d'appréciation de la HAS pour évaluer l'apport d'un médicament comparé aux traitements existants est donc beaucoup plus important que celui de l'ANSM. Ceci d'autant plus que la HAS est une agence d'expertise qui, contrairement à l'ANSM, ne rend pas de décision, la décision d'inscrire ou non un médicament au remboursement et le taux de celui-ci relevant du ministre.
On ne peut donc juridiquement réserver l'autorisation de mise sur le marché aux seuls médicaments ayant le meilleur service médical rendu. A l'inverse, le ministre, à partir des évaluations des commissions compétentes de la HAS, peut choisir de ne rembourser que ceux-là.
S'agissant du rôle du Ceps, la question est régulièrement posée de son utilité ou de son fonctionnement. Pendant plusieurs années, une partie de l'industrie pharmaceutique a souhaité sa suppression et son remplacement par l'acceptation d'un prix européen du médicament atténué par des ristournes négociées directement avec le ministère. Pour des raisons inverses, une partie des associations d'usagers et plusieurs personnalités ou mouvements politiques ont considéré que les négociations dont le contenu est partiellement couvert par le secret des affaires sont trop favorables aux entreprises du médicament et qu'il convient de remplacer le Ceps par une détermination du prix du médicament par les pouvoirs publics, au plus près des coûts supportés effectivement par les entreprises.
Le pouvoir de négociation et de décision du Ceps est, on l'a vu, particulièrement encadré sur le plan législatif, réglementaire et dans le cadre des orientations données à son président par les ministres de tutelle . Le législateur, à l'initiative du Sénat, a souhaité renforcer la cohérence des positions du Ceps par rapport aux évaluations de la HAS. Le Ceps ne peut plus en effet définir de prix du médicament sans se fonder sur l'amélioration du service médical rendu tel qu'il a été évalué par la commission de la transparence de la HAS, à moins de justifier son choix.
L'article L. 162-17-3 du code de la sécurité sociale fixe les principes de la composition du Ceps : « Le comité comprend, outre son président et deux vice-présidents choisis par l'autorité compétente de l'Etat en raison de leur compétence dans le domaine de l'économie de la santé, quatre représentants de l'Etat, trois représentants des caisses nationales d'assurance maladie et un représentant de l'Union nationale des organismes d'assurance maladie complémentaire ».
Le détail de cette composition permet de constater qu'elle reflète la multiplicité de ses missions, mais qu'elle reste majoritairement dans l'orbite de la sécurité sociale. Outre le président et le vice-président du Ceps, ont voix délibérative la direction de la sécurité sociale, la direction générale de la santé, la Cnam, le RSI et la MSA qui partagent une voix, l'Unocam enfin. La DGCCRF et la direction générale des entreprises ont chacune voix délibérative. La direction générale de la recherche et la direction générale de l'offre de soins n'ont de voix que consultative. Les organismes de sécurité sociale (régime de base et complémentaire) disposent donc de trois des huit voix délibératives auxquelles s'ajoutent la DSS et la DGS.
Indépendamment du vote du président et du vice-président, on peut donc estimer que les objectifs de réduction du prix du médicament et de respect de l'Ondam sont dans une situation d'équilibre par rapport aux intérêts industriels.
Le poids des représentants de notre système social au sein du Ceps est essentiel. Pour le Leem, « le prix du médicament résulte de la confrontation de 3 perspectives : l'industriel, le régulateur, les patients ». Pour vos rapporteurs, il est important de préciser que la perspective du régulateur doit non seulement être celle de l'Etat stratège des industries de santé, mais aussi et peut-être surtout celle du payeur, c'est-à-dire de l'assurance maladie.
2. Quelle instance de négociation des prix ?
Reste donc à savoir si le Ceps est la meilleure instance de négociation du prix du médicament.
Les alternatives à la fixation du prix par le Ceps sont :
- la liberté de prix , qui est aujourd'hui remise en cause par l'Allemagne notamment, et qui occulte totalement le prix réel du médicament ;
- la négociation par une autre instance ou sous une autre forme.
La négociation, si elle n'est pas confiée au Ceps, pourrait relever des payeurs des médicaments, c'est-à-dire de l'Uncam et de l'Unocam.
Or, ces deux instances sont déjà représentées au sein du Ceps. Il semble toutefois que la Cnam pourrait souhaiter que la politique de prix du médicament soit conduite de manière encore plus rigoureuse. Interrogé par le rapporteur général de la commission des affaires sociales en octobre 2014, l'ancien directeur général de la Cnam a développé l'analyse suivante :
« Jean-Marie Vanlerenberghe : Faut-il renforcer le rôle de la Cnam dans la négociation du prix des médicaments ? 66 ( * ) (...)
Frédéric Van Roekeghem : Quant à la négociation du prix des médicaments, l'équilibre actuel résulte de la loi de 2004, qui a fixé la composition du Comité économique des produits de santé (CEPS) : sur ses dix membres, trois représentent l'assurance maladie, un l'assurance maladie complémentaire et les six autres les différents ministères et directions de l'Etat, lequel nomme le président et le vice-président. Toucher à cet équilibre serait délicat, car il s'agit du pilotage d'une activité économique qui représente tout de même en France une trentaine de milliards d'euros. En tant que directeur général de la Cnam, je devrais m'y déclarer favorable mais, connaissant la dimension politique des questions qui touchent le médicament, je considère qu'il est de la responsabilité du pouvoir politique de prononcer l'arbitrage. Je puis cependant vous dire, sans manquer à mon devoir de discrétion, qu'il n'est pas rare que les représentants de l'assurance maladie votent contre les décisions finalement adoptées par le CEPS -position d'autant plus facile, me direz-vous, que leurs voix ne sont pas décisives. Nous faisons régulièrement des propositions d'évolution touchant non la composition du CEPS, fixée par la loi, mais d'autres mesures de régulation. Nous avons ainsi recommandé l'année dernière l'adoption d'un système plus régulier et automatisé de révision des prix, pour tenir compte du développement des génériques. Nous avions également suggéré un droit de veto, ou un vote à la majorité des deux tiers. »
La solution préconisée par Frédéric Van Roekeghem est donc de préserver l'équilibre de la représentation des différents ministères au sein du Ceps, quitte à augmenter le pouvoir des représentants de la sécurité sociale. Vos rapporteurs considèrent qu'il serait nécessaire que les représentants de l'assurance maladie (base et complémentaire) se voient attribués la moitié des sièges au sein du Ceps.
Une solution complémentaire pourrait être de mettre en place des appels d'offres ou au moins d'un renforcement de la concurrence pour certaines catégories de médicaments, les médicaments en rétrocession hospitalière par exemple. Les médicaments génériques font déjà l'objet de telles mesures dans plusieurs pays européens.
Une étude de la Cnam 67 ( * ) note que : « Le système de fixation du prix des médicaments en France a permis de mener une politique de prix conduisant au cours des dernières années à des économies significatives pour le système de santé tout en préservant un partenariat conventionnel avec l'industrie du médicament.
Néanmoins, la France reste en retrait sur la politique du générique par rapport à ses voisins européens, aussi bien en termes de volumes que de prix.
En moyenne, les Anglais, les Allemands, les Hollandais consomment plus de génériques pour beaucoup moins cher. »
En effet, en 2011, le prix par unité standard de générique est deux fois plus élevé en France qu'au Royaume-Uni et aux Pays-Bas, et supérieur de 20 % à l'Allemagne.
« [En France], le long processus de négociations et la contrainte réglementaire d'étiquetage ne favorisent pas (...) une dynamique à court terme de modification des prix. Les procédures de mise en concurrence appliquées dans les pays voisins permettent quant à elles de révéler des prix de marché très rapidement.
Ces procédures reposent soit sur des appels d'offres par les assureurs comme en Allemagne et aux Pays-Bas, soit sur la mise en concurrence des industriels directement par les pharmaciens comme en Angleterre mais aussi aux Pays-Bas. »
Le point de repère de la Cnam relève toutefois que « si l'appel d'offre permet effectivement d'atteindre très rapidement un prix bas, les risques de rupture d'approvisionnement liés à la sélection d'un nombre restreint de fournisseurs et la fragilisation globale du secteur en limitent d'autant plus l'intérêt que les difficultés administratives associées sont significatives. Le système anglais, qui laisse la place aux mécanismes de marché, autorise en revanche une plus grande souplesse de gestion tout en permettant une régulation des impacts économiques sur les agents concernés : fabricants et acteurs de la distribution. »
Ce mécanisme d'appel d'offres impose en fait un fort niveau de contrainte aux usagers, dont les traitements évoluent en fonction des marchés passés, que ce soit par les pouvoirs publics au travers des appels d'offre ou par les pharmaciens. A ce titre, il paraît difficile à vos rapporteurs d'en envisager la mise en place en France.
Plus qu'un remplacement du Ceps par une autre instance de négociation, il peut donc paraître utile de mieux distinguer politique de l'innovation et politique du prix et de renforcer les mécanismes de concurrence, sans toutefois bouleverser les habitudes des patients qui suivent un traitement.
B. QUELLE PRISE EN CHARGE DE L'INNOVATION ?
L'accès des patients à l'innovation se fait sur des critères médicaux, le principe étant celui de l'accès le plus rapide aux traitements innovants au travers du mécanisme des autorisations temporaires d'utilisation (ATU) dont on a vu qu'elles sont considérées comme un vrai gain de chance par les autorités sanitaires et les patients mais qu'elles peuvent être, parfois, critiquées comme favorisant le recours à des médicaments mal évalués ou peu efficaces, sinon dangereux, en vie réelle.
La prise en charge des ATU est, par nature, spécifique puisqu'elle intervient avant tout le processus d'examen d'un médicament par les agences sanitaires. Le choix des pouvoirs publics a donc été de mettre en place une prise en charge à 100 % au prix demandé par le laboratoire. Cette dérogation aux règles de la prise en charge et de la fixation du prix est cependant temporaire. A l'issue de la négociation avec le Ceps, le laboratoire commercialisant le médicament est tenu de restituer, éventuellement sous la forme de ristournes, la différence entre le prix initial et le prix négocié pour l'ensemble des médicaments pris en charge dans le cadre de l'ATU.
S'agissant des médicaments particulièrement onéreux, comme le Sovaldi, la question peut toutefois se poser de l'acceptabilité du prix fixé par le laboratoire. Lors de son audition, Frédéric Van Roekeghem a formulé la proposition suivante : « Sur le fond, le mécanisme de l'ATU est très favorable au patient, puisqu'il favorise l'introduction rapide du médicament. L'expérience de cette année nous conduit toutefois à nous demander si ses modalités ne devraient pas évoluer, notamment par l'attribution au Gouvernement d'un droit d'opposition au prix proposé par le laboratoire . »
Vos rapporteurs estiment que cette proposition bien que fondée risque de retarder l'accès des patients aux médicaments les plus innovants alors que le coût pour l'assurance maladie doit être relativisé. En effet, une fois le prix du médicament fixé par le Ceps, l'industriel doit rembourser la différence entre le prix demandé pendant la phase d'ATU et de post ATU. Il ne paraît donc pas nécessaire de risquer de remettre en cause un mécanisme qui a fait ses preuves en matière de santé publique.
En dehors du mécanisme de l'ATU, la question de la rémunération de l'innovation est posée de manière récurrente par les laboratoires. La mise sur le marché des antiviraux à action directe (AAD) a mis en relief la nécessité de revoir les modalités d'évaluation du médicament par la HAS. Des solutions de remplacement du système français actuel ont également été proposées.
1. Une réforme de l'évaluation nécessaire, une gestation contrariée
Les critères de service médical rendu (SMR) permettant de fixer le taux de remboursement d'un médicament et d'amélioration du service médical rendu (ASMR) ont fait l'objet de critiques récurrentes fondées essentiellement sur la difficulté à distinguer véritablement les deux notions dès lors qu'il s'agit d'évaluer un médicament.
Afin de moderniser les techniques d'évaluation, la Haute Autorité de santé a donc construit en 2012 un projet d'indicateur unique , baptisé « Index Thérapeutique Relatif » (ITR). La HAS souhaitait l'inscription de ce dispositif dans la loi de financement de la sécurité sociale pour 2013.
Le ministère de la santé a jugé cette réforme prématurée et, « afin d'envisager l'opportunité et les conditions d'un éventuel changement de critère, la ministre des affaires sociales et de la santé a demandé au chef de l'Igas de mandater une courte mission d'appui à la Direction de la sécurité sociale ». Muriel Dahan, désignée pour cette mission, a conduit en 2013 un important travail de consultation et de débat. 68 ( * )
A l'issue de ses travaux, la mission de l'Igas a proposé une « trajectoire de réforme », selon un schéma largement inspiré de la méthode élaborée par la HAS, tout en y apportant les corrections suggérées par les tests réalisés pour mesurer l'impact de la réforme et les remarques des experts et membres des instances sanitaires et sociales concernées.
Au Sénat, deux rapporteurs généraux successifs ont souhaité la mise en place de l'ITR lors de la discussion du PLFSS pour 2014 et du PLFSS pour 2015. Le Gouvernement s'y est montré pour sa part défavorable.
« Mme Marisol Touraine, ministre69 ( * ). Je vais vous expliquer en détail pourquoi le Gouvernement émet un avis défavorable sur cet amendement.
La question du mode d'évaluation des produits de santé est, bien entendu, importante. Nous touchons là à l'évidence aux limites du système actuel.
C'est ce qui explique le regain d'attention en faveur du nouveau mode d'évaluation dit « intérêt thérapeutique relatif », ou ITR, pour la décision d'admission au remboursement des produits de santé.
(...)
D'aucuns disent parfois, et c'est ce que vous laissez entendre, monsieur le rapporteur général, qu'une telle solution serait « prête à l'emploi ».
Pourtant, il y a aujourd'hui un consensus, y compris au sein de la Haute Autorité de santé, pour considérer qu'un tel mécanisme n'est pas adapté.
D'abord, l'ITR ne s'applique pas à tous les produits de santé ; en particulier, il ne s'applique pas aux dispositifs médicaux. Or il ne paraît pas opportun de changer de mode d'évaluation seulement pour une partie des produits de santé, en utilisant un nouvel outil pour les médicaments tout en en gardant un autre pour les dispositifs médicaux.
En outre, l'ITR ne permet ni de fixer un taux de remboursement ni de prendre en compte l'évaluation médico-économique.
Ces problèmes expliquent que la phase de test menée par la Haute Autorité de santé ait été jugée non conclusive par cette dernière.
Toutefois, nous sommes convaincus de la nécessité de repenser nos critères d'admission au remboursement des produits de santé.
C'est pourquoi un rapport complémentaire a été commandé à l'Inspection générale des affaires sociales. Sa remise est prévue pour la fin de l'année. À cette date, je demanderai à toutes les institutions publiques compétentes sur le médicament - je pense à la Haute Autorité de santé, à l'ANSM, au CEPS, dont nous venons de parler, ainsi qu'à l'assurance maladie - de faire des propositions concrètes pour rénover les critères d'évaluation des produits de santé.
Une concertation approfondie doit par ailleurs avoir lieu avec l'ensemble des acteurs. Elle sera menée avec les représentants de l'industrie pharmaceutique, qui est concernée au premier chef par cette évolution. Nous devrons à nouveau mettre en place une phase de test, car nous ne pouvons pas nous permettre de ne pas avoir d'évaluation assurée à cet égard.
Les premières modifications qui pourraient en découler seront alors intégrées dans le futur projet de loi sur la santé, le cas échéant à l'Assemblée nationale ou au Sénat, selon le parcours législatif du texte.
Sur un sujet aussi important, qui entraînera des changements profonds, nous devons être certains des mesures que nous adoptons.
Or, aujourd'hui, compte tenu des évaluations qui ont été menées, il ne nous semble pas possible de considérer, indépendamment de toute considération quant à la date, que l'index thérapeutique relatif pourrait être mis en oeuvre. Je ne parle pas seulement de son entrée en vigueur anticipée en 2015. Nous avons abouti à la conclusion que, même en 2016, il ne faudrait pas s'engager sur cette voie.
Je vous prie donc de bien vouloir retirer votre amendement sous le bénéfice de ces explications, monsieur le rapporteur général.
J'indique également aux auteurs d'amendements allant dans le même sens que la réflexion n'est pas écartée ; elle est même accélérée et approfondie.
Je déplore d'ailleurs la situation ; nous placions beaucoup d'espoir dans l'ITR. Malheureusement, les tests menés ne nous permettent pas de franchir l'obstacle auquel nous nous heurtons. »
Le Sénat avait adopté l'amendement présenté par le rapporteur général et inscrit la mise en place de l'ITR dans le projet de loi de financement de la sécurité sociale pour 2015. Cette mesure n'a pas été retenue par l'Assemblée nationale, appelée à se prononcer en lecture définitive.
A l'issue de ces débats, le Gouvernement a demandé à Mme Dominique Polton de conduire une nouvelle réflexion sur l'évaluation du médicament. Ce rapport, remis à la ministre en novembre 2015 reprend l'ensemble des données relatives à l'évaluation et formule de nombreuses propositions. Cependant le Gouvernement n'a pas inclus de dispositions spécifiques relatives à l'évaluation du médicament dans la loi relative à la modernisation de notre système de santé et n'a à ce jour annoncé aucun calendrier de réforme.
|
PROPOSITIONS DE LA MISSION CONFIEE A MME DOMINIQUE
POLTON - RECAPITULATIF
OE Donner une place plus importante à l'évaluation comparative au travers d'une ASMR rénovée, la VTR (valeur thérapeutique relative) , utilisée en primo-inscription et en réévaluation, cotée selon un score à quatre niveaux et prenant en compte : ü la quantité d'effet par rapport au comparateur : efficacité, tolérance, ü la pertinence clinique de ces effets, ü la qualité de la démonstration (critères de jugement, utilisation d'un comparateur pertinent dans les essais), ü les avantages non cliniques (praticabilité), qu'il convient d'expliciter plus précisément, ü la couverture du besoin. Missionner la HAS pour élaborer dans un délai de six mois une grille explicitant la relation entre la VTR et l'évaluation du médicament sur ces différentes composantes. ç Clarifier et simplifier les critères du SMR, voire le supprimer , en fonction du scénario qui sera retenu pour les modalités de fixation des taux de remboursement : ü Si le SMR est maintenu, § ré-expliciter les critères qui le fondent : l'efficacité et la tolérance du produit : quantité d'effet (efficacité thérapeutique / effets indésirables), la pertinence clinique du résultat obtenu en termes d'efficacité, la qualité de la démonstration de ces résultats, la transposabilité attendue en vie réelle, ces éléments devant être appréciés en prenant en compte la couverture du besoin (autres thérapies disponibles) et la place du produit dans la stratégie thérapeutique, ainsi que la gravité de la maladie ; § ne plus inclure la visée du médicament (thérapeutique, symptomatique,...) qui n'a pas de caractère discriminant ; § documenter l'impact de santé publique uniquement pour un nombre limité de produits, sur demande de la Direction Générale de la santé ; ü Supprimer le SMR si un scénario d'évolution des modalités de remboursement est retenu : § soit l'évolution vers un taux unique de prise en charge, comme c'est le cas pour les dispositifs médicaux, mais cette option introduit une rupture radicale avec le système actuel et par conséquent un coût de transition élevé ; § soit la fixation des taux en fonction de la gravité de la pathologie ou en fonction du taux du comparateur, cette deuxième option apparaissant en première approche la plus praticable, mais devant être validée tant du point de vue de sa faisabilité que de ses impacts économiques et juridiques. é Consolider la place de l'évaluation médico-économique dans la décision de prix , avec les évolutions suivantes : ü aller au-delà de la seule critique méthodologique dans les avis de la CEESP pour émettre un véritable jugement (qualitatif) sur l'efficience ; ü systématiser l'analyse de l'impact budgétaire, qui est un complément d'information utile pour les négociations en CEPS ; ü publier les avis, dans un souci de transparence et de pédagogie ; ü mettre en oeuvre des évaluations médico-économiques également dans les cas des réinscriptions, de manière à éclairer le CEPS dans ses décisions de baisses de prix ; ü améliorer l'articulation entre la commission de la transparence (CT) et la CEESP. Suivi et évaluation en vie réelle / réévaluation è Aller vers des réévaluations de la valeur thérapeutique relative (VTR) par groupe de produits , plutôt que des réévaluations quinquennales produit par produit, afin que la HAS puisse redéfinir globalement, pour une situation clinique donnée, la place de chacun dans la stratégie thérapeutique. ê Pour des médicaments traitant des pathologies graves pour lesquelles il n'existe pas d'alternative disponible, mais dont l'effet est faible, mal démontré, et demande à être confirmé, envisager un mécanisme de remboursement temporaire conditionné à la mise en place d'études cliniques et médico-économiques en vie réelle . z Pour des médicaments aux multiples indications, dont certaines présentent un intérêt faible au regard d'alternatives existantes, mettre en oeuvre dès l'inscription au remboursement une procédure de suivi et d'encadrement de la prescription permettant de vérifier que la prescription respecte les restrictions (en termes d'indications ou de sous-populations) prévues lors de l'évaluation en primo-inscription (cette vérification pouvant intervenir a priori ou a posteriori). { Engager de manière prioritaire un projet de développement d'un outil de surveillance post-inscription pour un certain nombre de produits ou de domaines clés, en s'appuyant sur les données médico-administratives existantes (système national de données de santé, SNDS) et en les appariant avec des données cliniques complémentaires recueillies en continu par les prescripteurs. Les registres ainsi constitués pourront servir le double objectif de suivre le respect des recommandations et des indications d'une part, et de pouvoir compiler des données épidémiologiques utiles pour l'évaluation en vie réelle d'autre part. Ils permettront à moyen terme d'apporter une réponse plus structurelle aux deux difficultés évoquées ci-dessus (l'incertitude sur les effets de produits arrivant sur le marché et les indications multiples) qui représentent des situations de plus en plus fréquentes. Processus d'évaluation | Alléger les tâches de la HAS et renforcer le CEPS pour permettre une réduction des délais d'instruction des dossiers ; fixer des objectifs de raccourcissement de ces délais et mettre en place un dispositif de suivi } Mener dans les six prochains mois un travail commun CEPS - HAS pour améliorer encore l'utilisation des avis de la CT et de la CEESP, en élaborant conjointement des modèles d'avis présentant de manière standardisée l'ensemble des éléments techniques nécessaires au travail du CEPS. La réflexion est à poursuivre afin d'élaborer des propositions opérationnelles sur trois autres aspects concernant le processus de travail de la CT : - la valorisation de l'activité d'expertise, - l'utilisation des évaluations réalisées dans d'autres pays, - les modalités de vote. |
Au regard de travaux déjà conduits vos rapporteurs estiment nécessaire de mettre en oeuvre un indicateur unique d'évaluation comparative du médicament . Cet indicateur n'a pas nécessairement vocation à être étendu aux dispositifs médicaux si la HAS estime que leurs spécificités s'y opposent. Etant donnée l'étude approfondie des limites du la dualité des critères actuels de SMR et d'ASMR menés par la mission Polton il paraît aujourd'hui plus efficace de retenir sa proposition de mettre en place un indice de valeur thérapeutique relative (VTR). En conséquence, ainsi que le préconise le rapport Polton, il convient de donner mission à la HAS d'élaborer dans un délai de six mois une grille explicitant la relation entre la VTR et l'évaluation du médicament sur ces différentes composantes. Le prochain PLFSS pourrait être l'occasion d'inscrire cette mission dans la loi si le Gouvernement n'a pas d'ici là pris d'initiative en ce sens.
Par ailleurs vos rapporteurs estiment nécessaire de simplifier la multiplicité actuelle des taux de remboursement, qui est une spécificité française . En dehors du taux de remboursement à 100 % qui doit être maintenu et de la prise en charge intégrale des ALD, les taux de 65 %, 30 % et 15 % sont facteurs de confusion pour les patients voire d'inégalités. En effet, ainsi qu'on a pu le constater à l'occasion de l'analyse des déremboursements ou des stratégies des firmes en matière d'automédication, le lien entre l'efficacité du médicament et son taux de remboursement n'est pas facile à appréhender par la population. Ceci créé l'illusion dangereuse en terme de santé publique et couteuse pour l'assurance maladie que l'on est nécessairement mieux soigné par un médicament mieux remboursé.
Cette approche est renforcée par la pratique des assurances complémentaires qui tendent à dérembourser elles-mêmes les médicaments qui ne sont plus pris en charge par l'assurance de base. Il convient de rappeler qu'aucune obligation ne leur est faite en la matière et qu'une complémentaire peut prendre en charge intégralement ou partiellement des médicaments non pris en charge par l'assurance maladie. De fait de nombreuses couvertures complémentaires incluent de tels produits comme l'homéopathie par exemple.
En plus d'être difficile d'appréhension la multiplication des taux autres que le 100 % est d'application limitée du fait des ALD et source d'inégalités. Comme le démontre le rapport de la mission Polton, certains médicaments qui ont sont considérés comme supplémentaires se voient attribués un SMR et donc un taux de remboursement faible. Or ils peuvent être des traitements nécessaires pour les patients qui ne régissent pas aux traitements de première intention. Or « on ne voit pas pourquoi, pour les patients ayant une intolérance aux autres traitements, ce traitement supplémentaire, s'il est effectivement prescrit avec discernement, serait moins bien remboursé. Ceci revient à dire qu'un médicament de 2 eme intention aurait par construction un taux de remboursement plus bas qu'un médicament de 1ère intention (« double peine » pour le patient intolérant aux traitements jugés les plus efficaces) ».
Au regard de ces différents arguments vos rapporteurs considèrent que les taux de 15 %, 30 % et 65 % doivent être fusionnés en un taux unique après la mise en oeuvre de la VTR selon les étapes définies par le scénario 2 du rapport Polton. Le niveau de ce taux unique doit faire l'objet de concertations entre les différentes parties prenantes de la couverture maladie et les pouvoirs publics afin de limiter les restes à charge pour la population. Il ne pourrait être inférieur à 50 %.
Ces évolutions devraient notamment permettre de rémunérer de manière adéquate l'innovation en matière de médicament. D'autres propositions ont néanmoins été formulées.
2. Faut-il remplacer le système actuel de fixation des prix pour les médicaments innovants ?
Le rapport Polton comporte une analyse poussé des enjeux en matière de rémunération de l'innovation mais ne conclus pas à la mise en place de mesures spécifiques pour les médicaments innovants. Il est essentiellement préconisé de conduire de nouvelles études sur les différents propositions qui ont pu être formulée.
|
Préconisation du rapport Polton sur le prix et le financement de l'innovation La réflexion doit être approfondie par des travaux complémentaires, différentes pistes pouvant être explorées : les modalités pratiques et conditions de succès des accords de performance, et leur place dans la palette des « contrats d'accès au marché » ( managed entry agreements ) ; la faisabilité de modes de rémunération alternatifs (rémunération au parcours) pour certaines pathologies ; la prise en compte des coûts de recherche et développement ; les possibilités de rapprochement avec d'autres pays. |
A l'occasion de leurs auditions, plusieurs de ces propositions ont été soumises à vos rapporteurs, certaines ayant été proposée déjà depuis plusieurs années.
La contribution du Leem au thésaurus argumentaire sur le prix et le financement des médicaments innovants en recense plusieurs, susceptibles, aux yeux du syndicat d'être combinées. Les perspectives proposées sont notamment d'intégrer le coût de l'innovation à l'Ondam (ce qui revient à l'augmenter), d'ajuster dans le temps les remises consenties par les laboratoires pour faire baisser effectivement le prix payé lors de la montée en charge d'une nouvelle molécule, de créer un fonds d'amortissement et de financement de l'innovation thérapeutique et d'expérimentation d'un forfait parcours de soins fixant le prix de la prise en charge d'une pathologie de manière transversale entre la ville et l'hôpital.
Vos rapporteurs considèrent qu'il n'est pas réaliste de proposer une prise en charge segmentée du médicament selon qu'il soit ou non considéré comme une innovation de rupture, permettant la chronicisation d'une maladie mortelle ou améliorant le service médical.
Lors d'une conférence de presse organisée à Paris par le groupe suisse Roche le 23 octobre 2013, son directeur général, Severin Schwan, a plaidé pour un nouveau système de fixation des prix des médicaments qui prendrait en compte le bénéfice réel apporté au patient. La proposition de Roche a été maintes fois réitérée depuis.
Alors que dans la situation actuelle, le prix d'un produit est fixé, de façon indifférenciée, en fonction du volume (par flacon ou par milligramme de molécule), le laboratoire Roche estime qu'il est temps de s'orienter vers « des modèles de fixation de prix plus sophistiqués », pour créer un environnement tarifaire à la fois durable pour la société et incitatif pour les industriels engagés dans l'innovation.
Il s'agit de mettre en place un système de prix ou de remboursement correspondant à chaque indication du médicament, ce qui permettrait de valoriser exactement l'apport du médicament pour chaque patient.
Cette approche, outre une grande difficulté technique de mise en oeuvre, pose la question de l'aune à laquelle s'effectue l'évaluation en vie réelle. S'agit-il de mesurer les années de vie ajustées par leur qualité auxquelles serait attribuée une valeur sur la modèle britannique des Qaly (quality adjusted life years), ou d'évaluer l'économie réalisée par le système de santé du fait de l'amélioration de l'état du patient voir de sa guérison ?
Si c'est l'approche en termes de Qaly qui est retenue, le prix du médicament, ou en tout cas sa prise en charge, seront limités. Si c'est l'approche en termes d'économies générées pour le système de santé, le risque est plutôt celui d'une sur-rémunération.
a) La rémunération du médicament en fonction de l'apport en années de vie et en qualité de vie
Le conseil d'analyse économique a défini 70 ( * ) ainsi le Qaly : « L'approche coût-efficacité utilise le concept de Qaly pour fonder des arbitrages entre différents traitements permettant d'améliorer la santé. Les gains de santé sont quantifiés en termes d'années de vie gagnées, pondérées par leur qualité : chaque année de vie est pondérée par un coefficient compris entre 0 et 1 exprimant, depuis l'état de parfaite santé correspondant au coefficient 1, toutes les gradations de mal-être croissant jusqu'à la mort correspondant au 0 ».
La note préconise l'adoption en France d'une démarche de type britannique sur le fondement suivant : « de facto, définir un seuil par Qaly ou un budget total sont deux manières alternatives d'améliorer le plus efficacement possible la santé d'une population. Dans tous les cas, il faut définir les traitements à délivrer en priorité . » Constatant cependant que le débat sur la méthodologie du Qaly a empêché son développement en France, les auteurs de la note ajoutent : « Afin d'assurer sa légitimité, la définition d'un seuil acceptable en termes de coût par année de vie gagnée doit s'appuyer sur un processus transparent, associant de nombreux acteurs de la société ; enfin, il ne doit pas être appliqué mécaniquement, mais constituer une référence permettant d'indiquer les niveaux de coût acceptables, et les coûts manifestement déraisonnables ».
Par sa nature même, le mécanisme des Qaly tend à limiter le prix auquel un médicament innovant peut être pris en charge. En effet, seules les innovations susceptibles d'apporter des gains majeurs en termes de nombre d'années de vie et de qualité sont susceptibles d'obtenir un niveau de prise en charge élevé par l'assurance maladie.
Ainsi vos rapporteurs ont pu constater qu'en matière d'accès aux traitements innovants, l'approche britannique des Qaly s'est avérée plutôt source de restrictions pour les malades, que ce soit pour l'accès aux antiviraux à action directe (AAD) ou plus généralement aux traitements innovants en matière de cancer. Ecartés de la prise en charge par l'assurance maladie dans le cadre de l'application stricte de l'évaluation par Qaly, ces traitements font d'ailleurs l'objet d'un financement spécifique par les Cancer Drug Funds depuis avril 2011.
L'expérience des Qaly en termes d'accès aux traitements innovants va donc à l'encontre des pratiques et des débats actuels en France. Il ne faut cependant pas en conclure que l'approche médico-économique doive être proscrite. Incontestablement, le développement d'une telle compétence par la HAS doit servir d'appui aux négociations sur le prix du médicament et le Ceps est demandeur de telles études. Mais le critère médico-économique ne saurait devenir le critère unique ou déterminant de la prise en charge du médicament.
b) Rémunérer les médicaments en fonction des « économies » réalisées par l'assurance maladie
Dans le même but de mieux rémunérer les médicaments innovants, tant les laboratoires ayant mis sur le marché des AAD que le laboratoire Roche entendent, pour leur part, fonder leurs prix sur les sommes qui seraient économisées par l'assurance maladie en terme de fermeture de lits, de coûts de prise en charge, voire de transplantations.
Ainsi, une étude sur l'impact budgétaire et organisationnel des innovations thérapeutiques conduites par le cabinet Jalma pour le Leem, mais encore à paraître, tend à établir que « les économies attendues dans l'organisation des soins sur les dix prochaines années pour un seul cancer (celui de la prostate au stade métastatique) s'élèvent à 350 millions d'euros ».
Or, ces économies, qui supposeraient des adaptations rapide de notre système de soins et des structures hospitalières, sont généralement virtuelles à court ou moyen terme. Surtout, elles constituent une appropriation par les laboratoires de sommes qui ne sont disponibles que du fait de la socialisation des dépenses de santé. Poussée à son terme, la logique de fixation d'un prix en fonction des économies réalisées par un système de santé conduirait à faire payer plus cher les pays qui ont la meilleure prise en charge, et peu ou pas ce qui n'en ont aucune.
Auditionné par la commission des affaires sociales, le directeur général de la Cnam, Nicolas Revel 71 ( * ) a fait l'analyse suivante que partagent vos rapporteurs : « Il existe différentes propositions des laboratoires sur la différenciation du prix du médicament en fonction de l'efficacité évaluée en vie réelle. Nous la pratiquons déjà pour quelques médicaments ou en fonction des indications thérapeutiques, nous la pratiquons également pour un produit dont la prise varie dans une proportion de un à quinze. Pouvons-nous généraliser ces modèles ? La première piste est très exigeante : elle implique la fixation d'indicateurs d'efficacité et de suivi, partagés, sécurisés et incontestables. Cela n'est pas simple et nécessiterait de doter le Comité économique des produits de santé (CEPS) de plus de moyens. La seconde est envisageable uniquement si le service médical rendu (SMR) est très différent d'une indication à l'autre. Il serait difficile de justifier que l'on paie la même molécule à des prix différents selon les cancers. Cela ne signifie pas que le calcul des remises ne puisse intégrer la prise en compte des différentes indications thérapeutiques. Pour donner un exemple, des produits d'immuno-oncologie arrivent sur le marché, ils peuvent être utilisés pour soigner différents cancers. Nous devons fixer des indicateurs de résultats différents selon les pathologies. Pour autant, la fixation de prix faciaux différents ne s'impose pas.
Vous m'interrogez sur la prise en compte des économies réalisées par l'assurance maladie dans la fixation du prix des médicaments. L'idée a été soulevée à propos de traitements qui permettraient de diminuer les dépenses d'hospitalisation. Je ne suis pas certain que la question puisse être posée ainsi. Lorsqu'une innovation thérapeutique est « réelle », nous en tenons compte dans la détermination du prix afin d'encourager la recherche mais je suis sceptique sur l'idée d'introduire des clauses d'intéressement en fonction du nombre de lits ou de services fermés dans un horizon de temps donné, en lien avec l'utilisation d'un médicament.
M. Yves Daudigny. - Certains laboratoires revendiquent ce type de résultats...
M. Nicolas Revel.- Je le sais. C'est un argument de négociation... Jusqu'à présent, les prix consentis, même nets de remise, n'ont jamais freiné le déploiement de l'innovation. »
c) Adapter la rémunération à l'efficacité en vie réelle du médicament
Les contrats de performance qui soumettent le prix du médicament à son efficacité en vie réelle sont actuellement développés par le Ceps avec le soutien des industriels. Ils ont le mérite de mesurer les gains exacts d'une nouvelle molécule en matière de santé publique et d'ajuster sa rémunération en conséquence.
Elle n'est possible qu'à une condition particulièrement exigeante, que les pouvoirs publics mobilisent les données pharmacoépidémiologiques aujourd'hui trop éparses et insuffisamment développées pour suivre l'impact réel d'un médicament sur plusieurs années. A défaut d'une capacité d'expertise publique forte un tel système ne pourrait reposer en effet que sur les données fournies par les laboratoires eux-mêmes.
Sous réserve du développement de la capacité des agences sanitaires et du Ceps de développer l'évaluation en vie réelle du médicament, vos rapporteurs sont favorables au développement de ce type de rémunération des médicaments.
d) Quel bilan pour le système actuel de rémunération et d'accès des patients à l'innovation ?
Le système actuel de rémunération de l'innovation en France est complexe du fait de la mise en oeuvre d'un système d'évaluation et de fixation du prix lui-même divisé en plusieurs étapes d'une part, d'un mécanisme de fiscalité qui favorise l'innovation (le crédit impôt recherche) tout en plafonnant le chiffre d'affaire susceptible d'être réalisé par les laboratoires.
Force est néanmoins de constater, comme le directeur général de la Cnam, que le système actuel permet l'accès des patients au médicament, même si cela est fonction du stade de développement de leur pathologie, et que cette prise en charge a été possible sans dérapage majeur des dépenses de santé.
Il est intéressant de ce point de vue de comparer les dépenses liées aux AAD à celles réellement effectuées en 2014. Lors de son audition du 24 mars 2015 par la commission des affaires sociales, M. Christian Eckert, secrétaire d'Etat au budget, avait estimé le montant des dépenses liées à l'hépatite C à 1,2 milliard d'euros. Les données contenues dans le point de repère publié par la Cnam en février 2016 72 ( * ) tendent à établir un chiffre légèrement inférieur, entre 1 et 1,1 milliard. Ce montant est le montant net des remboursements, sans prise en compte des remises négociées par le Ceps, que le ministre a évaluées à 300 millions d'euros, et du mécanisme fiscal spécifique réduisant de moitié les dépenses au-delà de 450 millions d'euros. Le montant réellement dépensé pour les AAD a donc été 600 millions d'euros pendant la plus forte période de montée de prise en charge des patients. Parallèlement, le remboursement des traitements de l'hépatite C préexistants aux AAD a chuté, entraînant une économie de 90 millions d'euros par rapport à 2013. Le montant des dépenses supplémentaires est donc de l'ordre de 500 millions d'euros. Montant incontestablement significatif (un peu plus de 43 000 euros par patient traité) mais inférieur aux projections initiales.
Au total, les mécanismes français d'action sur les prix du médicament permettent aux patients d'accéder aux médicaments innovants dans des conditions plus favorables que dans les autres pays européens et d'une manière relativement soutenable pour l'équilibre des comptes de l'assurance maladie.
La vigilance est bien sûr nécessaire comme le rappel l'avis du 27 mai 2016 du comité d'alerte sur l'évolution des dépenses d'assurance maladie 73 ( * ) . Celui-ci relève que : « Les dépenses au titre des médicaments en ATU et en post-ATU (essentiellement les nouveaux médicaments anti-cancéreux) restent particulièrement dynamiques en 2016, et, dans l'attente de la conclusion des négociations de prix en cours, font peser un risque significatif sur le respect de l'ONDAM hospitalier en 2016 ».
Si le retour de l'innovation doit être salué et encouragé par des mécanismes en amont de la mise sur le marché, il convient de ne pas surestimer son importance et son coût. Face aux demandes des laboratoires, qui se nourrissent des attentes des patients et des soignants, une évaluation rénovée devrait permettre aux acteurs actuels de gérer l'arrivée des nouvelles molécules.
EXAMEN EN COMMISSION
_______
Réunie le mercredi 29 juin 2016, sous la présidence de M. Alain Milon, président, la commission entend une présentation de MM. Gilbert Barbier et Yves Daudigny de leur rapport d'information sur la politique du médicament.
M. Alain Milon, président . - Nous examinons maintenant le rapport de Gilbert Barbier et Yves Daudigny sur la politique du médicament.
M. Yves Daudigny, rapporteur . - Le médicament est un enjeu politique et un objet constant de polémique. Il est jugé tour à tour trop coûteux, trop dangereux, trop peu accessible, pas assez innovant, quand ce n'est pas tout simplement dépassé par les progrès dans les autres formes de prise en charge.
De trop nombreux dossiers, du Thalidomide au Mediator ou à la Dépakine jettent la suspicion sur les politiques d'encadrement, de contrôle et de suivi du médicament, peut-être plus dans notre pays que dans le reste de l'Europe !
Les polémiques se succèdent et se ressemblent sans paraître réellement permettre d'avancer. Industriels, ONG, médecins, pharmaciens, assurance maladie, lanceurs d'alerte expriment des points de vue tous légitimes, mais qui peinent à se croiser.
Dès lors, en quoi un nouveau rapport sur le médicament est-il nécessaire ? Comment peut-il faire autre chose qu'ajouter un point de vue, celui de la commission des affaires sociales du Sénat, à tous ceux qui s'expriment déjà largement dans la presse ?
Il nous est apparu que la diversité même des approches rend nécessaire la définition par les pouvoirs publics d'une position claire quant à la place du médicament dans notre système de santé - ceci d'autant plus que le cadre, déjà contraint, de nos finances sociales se trouve largement bousculé par le retour de l'innovation médicamenteuse.
Contrairement au Royaume-Uni par exemple, la France n'admet pas que certains ne puissent pas avoir accès à l'innovation ; elle a d'ailleurs mis en place des mécanismes d'accès précoce aux nouveaux traitements, comme les autorisations temporaires d'utilisation (ATU), largement saluées lors des auditions conduites. Le prix élevé des nouvelles molécules pose toutefois question. Après plusieurs années de baisse ayant permis une économie annuelle d'un milliard d'euros, l'enveloppe médicament a augmenté en 2014 du fait des seuls antiviraux d'action directe - traitement apparemment curatif contre l'hépatite C.
Le médicament ne peut donc plus uniquement apparaître comme un possible gisement d'économies pour l'assurance maladie. Il pose aussi de nouveaux problèmes de financement. Le retour de l'innovation thérapeutique, après un long plateau, pose ainsi une autre question essentielle pour l'avenir de notre système d'assurance maladie : celle de l'évaluation du médicament afin de déterminer son niveau de prise en charge et son prix.
Ces questions, qui sont au coeur de l'actualité, ont fait l'objet de propositions diverses, émanant de plusieurs acteurs, entre lesquelles le Gouvernement n'a pas tranché, voire même annoncé qu'il ne trancherait pas. Or, la continuité de la prise en charge du médicament par la sécurité sociale exige des décisions rapides pour mettre en place des réformes qui s'étaleront nécessairement sur plusieurs années.
Le rapport que nous vous présentons ne porte donc pas sur les enjeux de santé publique liés à l'usage du médicament, à la sécurité des produits ou à la pharmacovigilance, même s'il les mentionne nécessairement. La question qui nous a paru fondamentale est la suivante : quels sont les médicaments qui doivent être pris en charge par l'assurance maladie, et à quelles conditions ?
Cette question nous a tout d'abord conduits à tenter de distinguer entre les enjeux industriels et financiers, liés au processus productif du médicament, et les objectifs spécifiques à l'assurance maladie - payer au meilleur prix les médicaments les plus efficaces pour garantir l'accès de l'ensemble de la population aux meilleurs traitements. Nous avons ensuite étudié les différents types de médicament, selon qu'ils sont ou non pris en charge par l'assurance maladie. Nous avons enfin analysé le mécanisme de fixation du taux de remboursement et du prix du médicament afin d'apprécier son adaptation aux enjeux actuels.
M. Gilbert Barbier, rapporteur . - C'est donc tout d'abord dans sa dimension industrielle que nous avons étudié le médicament. Indéniablement, l'industrie du médicament constitue un secteur stratégique à la fois par la nature de sa production, qui constitue un outil indispensable au service de la santé des populations, mais aussi par son poids économique. Elle contribue en outre à la fois au rayonnement international de la recherche publique et privée et à celui de notre industrie.
En raison de la croissance continue de la demande et du caractère très innovant du secteur (que l'on pense au développement des biotechnologies, des thérapies ciblées, à l'immunothérapie ou encore à l'apparition de la médecine personnalisée), le médicament apparaît clairement comme un secteur économique d'avenir. Or, les entreprises françaises du secteur, qui constituent traditionnellement une part non négligeable de notre industrie nationale, affichent une érosion continue de leur compétitivité dans les dernières années.
Ainsi l'étude de Roland Berger cite : sur 130 molécules autorisées en Europe entre 2012 et 2014 : 8 étaient produites en France, 32 en Allemagne, 28 au Royaume Uni et 18 en Italie.
Dans ce contexte, le système d'administration des prix à la française est décrit comme illisible par de nombreux acteurs, qui y voient même une forme d'opacité, laissant place à toutes les suspicions. S'il s'avérait qu'il comprenne en effet traditionnellement une part de soutien à l'industrie nationale, cela sous-entendrait que les pouvoirs publics n'affirment pas suffisamment leurs exigences face aux laboratoires, en termes de prix comme en termes de santé publique. Nos travaux nous ont cependant permis de nuancer largement ce soupçon : l'enjeu majeur pour les administrations françaises de santé est aujourd'hui très clairement celui de la maîtrise du coût des médicaments. Cela n'interdit pas cependant la prise en compte des contraintes de l'industrie pharmaceutique, notamment dans le sens de la préservation des impératifs de santé publique : des hausses de prix peuvent ainsi être ponctuellement consenties pour des produits très anciens, dont le prix n'a pas été revalorisé depuis longtemps et dont l'exploitation devient de ce fait déficitaire, mais dont l'intérêt thérapeutique reste majeur.
Il est par ailleurs apparu que le modèle de production du médicament devra faire face, au cours des prochaines années, à de profonds bouleversements qui devront nécessairement être pris en compte par les pouvoirs publics dans leur dimension industrielle comme de santé publique.
On assiste en premier lieu à un phénomène de délocalisation et, dans une moindre mesure, de concentration des acteurs hors de France. Il pourrait s'intensifier au cours des prochaines années, dans la mesure notamment où les entreprises françaises n'auront pas pris le virage du médicament biologique. Cette évolution fait peser des risques importants sur le tissu productif français, mais aussi pour l'influence française dans le secteur des produits de santé. Dans ce contexte, il apparaît indispensable de préserver nos outils de compétitivité que constituent notamment notre exceptionnel environnement scientifique et médical, la qualité de nos structures industrielles, les avantages fiscaux tels que le crédit impôt-recherche (CIR) ou encore des mécanismes tels que celui des ATU.
En second lieu, on assiste à un phénomène de retour de l'innovation, qui remet en cause le modèle de fonctionnement des laboratoires fondé sur l'exploitation des blockbusters, pour lesquels de nombreux brevets sont désormais tombés. L'enjeu est ici d'accroître l'efficacité du système français, notamment en augmentant les moyens de la recherche publique fondamentale, en examinant les contraintes réglementaires qui pèsent sur elle, ou encore en encourageant le développement de partenariats de recherche publics-privés - ainsi que l'a d'ailleurs préconisé le conseil stratégique des industries de santé (CSIS) en avril dernier.
M. Yves Daudigny, rapporteur . - Revenons brièvement sur le parcours du médicament une fois la molécule élaborée par un laboratoire.
Contrairement à d'autres pays comme l'Italie, la France a réparti entre différents organismes les étapes permettant la distribution d'un médicament. Tout d'abord, la mise sur le marché d'un médicament, qui est régie par le droit européen, relève de l'Agence européenne du médicament (EMA, basée à Londres) et, en France, de l'Agence nationale de sécurité du médicament et des produits de santé (ANSM). Un médicament est mis sur le marché si son bénéfice est jugé supérieur aux risques qu'il présente pour l'indication de traitement présentée par l'industriel. A l'appui de leur demande, les laboratoires soumettent à l'agence des études cliniques dont le niveau d'exigence et de transparence fait l'objet de débat. Nous y reviendrons.
Le seul critère admis pour mettre un médicament sur le marché est celui du rapport bénéfice/risque et, en France, le refus de mise sur le marché d'un médicament en France ou son interdiction doivent être justifiés au regard de ce critère sous peine d'une condamnation par le juge. L'ANSM est aussi chargée du suivi de l'usage des médicaments et de la pharmacovigilance, donc des décisions de restriction ou d'interdiction d'une molécule en fonction des risques qui peuvent être associés à son usage en vie réelle. Cette mission est particulièrement importante, comme l'a illustré récemment le scandale du Mediator. Mais elle a aussi en charge les recommandations temporaires d'utilisation (RTU) qui permettent l'usage remboursé d'un produit en dehors de son autorisation de mise sur le marché (AMM). Cette possibilité est importante pour la santé publique quand il s'avère qu'un médicament utilisé dans une certaine indication a aussi des effets thérapeutiques pour une autre maladie.
La deuxième étape est l'évaluation du médicament pour déterminer s'il doit ou non être pris en charge par la sécurité sociale. Depuis 1967, cette évaluation a été confiée à une commission, dite de la transparence, qui fait aujourd'hui partie de la Haute Autorité de santé (HAS). La commission de la transparence détermine le service médical rendu (SMR) du médicament et propose un taux de remboursement en fonction de celui-ci. Elle détermine aussi l'amélioration du service médical rendu (ASMR) qui sert de base à la fixation du prix.
En 2013, 85 % des médicaments vendus en France étaient remboursables. Cette proportion s'élevait à 97 % s'agissant des spécialités soumises à prescription médicale obligatoire.
Un médicament peut faire l'objet d'une prise en charge intégrale par la sécurité sociale soit en raison de l'intérêt qu'il présente, soit parce qu'il est prescrit à une personne pour traiter une pathologie correspondant aux critères de l'affection de longue durée (ALD) dite « exonérante », c'est-à-dire d'une affection « dont la gravité et/ou le caractère chronique nécessitent un traitement prolongé et une thérapeutique particulièrement coûteuse, et pour laquelle le ticket modérateur est supprimé ».
En 2011, d'après le syndicat des entreprises du médicament (Leem), les médicaments pris en charge à 100 % par l'assurance maladie représentent environ deux tiers des dépenses de remboursement par l'assurance maladie, le tiers restant se répartissant entre les médicaments remboursés aux autres taux.
En effet, certains ne sont remboursés que partiellement, soit en taux (15 %, 30 % ou 65 %), soit seulement pour certaines populations ou pour certaines indications.
Par ailleurs, certains médicaments ne sont pas remboursables, soit que le laboratoire n'ait pas fait la demande d'inscription sur la liste des médicaments remboursables, soit que le médicament ait été déremboursé.
Les médicaments pour lesquels une prescription médicale est obligatoire sont presque tous remboursés. Ceux pour lesquels une prescription médicale n'est pas obligatoire sont dits d'automédication en langage courant. L'ANSM qui en assure la régulation préfère, pour sa part, la notion de médicament sur conseil pharmaceutique pour bien montrer que si le patient peut décider d'avoir recours au médicament pour une pathologie bénigne, il doit pouvoir bénéficier du conseil du pharmacien. Ces médicaments ne sont pas pris en charge par la sécurité sociale et leur prix est libre. Il peut d'ailleurs varier d'une officine à l'autre.
Le fait qu'un médicament soit en accès direct dépend largement du choix que fait le laboratoire de demander ou non le remboursement, et non de l'efficacité intrinsèque du médicament. Ainsi, le paracétamol est incontestablement efficace mais ses dosages les plus courants sont en accès libre. Après plusieurs années de contestation de l'éventualité de dérembourser le Doliprane, Sanofi envisage aujourd'hui de le mettre entièrement en automédication. Cet exemple montre bien que cette question relève essentiellement de la stratégie commerciale des firmes. Est-il plus rémunérateur d'avoir un médicament remboursé par la sécurité sociale ? Oui, dans la grande majorité des cas, mais plus nécessairement quand le nom de marque est particulièrement connu du grand public, que le médicament risque de devenir substituable sachant que le prix des médicaments en automédication est libre.
D'autres médicaments ne sont plus admis au remboursement du fait de la réévaluation de leur service médical rendu. Cette réévaluation est conduite par la commission de la transparence de la HAS de manière continue. Chaque molécule inscrite au remboursement doit être réévaluée au plus tard tous les cinq ans. A la demande des ministres successifs depuis 2002, différents plans de réévaluation des médicaments ont pu être conduits avec un périmètre élargi aboutissant au déremboursement de nombreuses spécialités. Depuis 2012, aucun nouveau plan d'ensemble n'a été mis en oeuvre mais la commission de la transparence continue ses réévaluations systématiques, molécule par molécule. Dans la majorité des cas, le réexamen confirme le niveau de SMR déjà reconnu. Dans quelques rares cas, le réexamen peut conduire à la reconnaissance d'un service médical rendu plus élevé. Surtout, plusieurs dizaines de décisions tendent à la réduction du SMR ou à sa déclaration comme insuffisant. Ceci entraîne la recommandation d'une baisse du taux de remboursement, voire d'un déremboursement.
En 2015, seize arrêtés ont été pris par la ministre en charge de la sécurité sociale sur le fondement des réévaluations conduites par la commission de la transparence. Ils ont exclu du remboursement 351 spécialités (plusieurs spécialités pouvant correspondre aux différents conditionnements et dosages d'un même médicament).
La réévaluation et le déremboursement sont un élément structurel de la gestion de la prise en charge des médicaments par l'assurance maladie.
Ces déremboursements ne vont pas sans contestation, que ce soit récemment pour les anti-arthrosiques symptomatiques d'action lente, ou actuellement pour les médicaments anti-Alzheimer. Néanmoins, la réévaluation elle-même par la commission de la transparence n'est que rarement remise en cause. C'est l'effet du déremboursement sur la prescription -c'est-à-dire notamment le risque de report vers des molécules remboursées mais peut-être inadaptées- qui est invoqué pour contester les mesures de déremboursement, dans l'idée que l'économie serait au mieux incertaine et le risque d'effets secondaires renforcé.
Incontestablement, sur cette question comme sur celle, complexe, de la prescription d'un médicament hors des conditions d'autorisation de sa mise sur le marché, les mesures de gestion de la prise en charge doivent s'accompagner de mesures d'information à destination des professionnels de santé et tout particulièrement des prescripteurs.
La question fondamentale est en fait celle des critères d'évaluation du médicament par la commission de la transparence. En effet, le service médical rendu (SMR) est le critère utilisé pour déterminer le taux de prise en charge, tandis que l'amélioration du SMR l'est pour déterminer le prix. Cette distinction, cohérente a priori, est en fait devenue, au fil du temps, de plus en plus floue et les deux critères tendent à se confondre, du fait notamment de la plus grande exigence de la commission de la transparence en matière de comparaison avec les traitements existants. La détermination du SMR est, dès lors, devenue plus difficile et potentiellement porteuse d'inégalités puisque, de deux médicaments traitant une même pathologie, le deuxième arrivant sur le marché est moins remboursé que le premier. Or, dans certains cas, ce médicament est le seul qui puisse traiter une partie des patients dont la pathologie ne répond pas au médicament mieux remboursé. Selon les termes de Dominique Polton, qui a remis le rapport le plus complet sur la question de l'évaluation du médicament, ces patients subissent une double peine puisqu'ils ne peuvent tirer bénéfice du premier traitement et qu'ils sont moins remboursés pour celui qui leur est nécessaire.
A la suite de la position portée par Jean-Luc Harousseau lorsqu'il était président de la HAS, et en cohérence avec la position du Sénat sous deux majorités différentes, il nous semble donc aujourd'hui nécessaire de fusionner les critères d'évaluation du SMR et de l'ASMR et de passer à un critère d'évaluation unique. Le rapport Polton propose la mise en place d'un « indice de valeur thérapeutique relative » qui intègre pleinement la dimension comparative de l'évaluation du médicament. Celle-ci nous paraît plus que jamais nécessaire. En effet, nos critères d'évaluation actuels sont marqués par le long plateau qu'a connu l'innovation en matière de médicament : dans un contexte de faible innovation, la moindre avancée était fortement valorisée. Dès lors qu'arrivent sur le marché des médicaments ayant un apport thérapeutique important, voire majeur, il faut des critères d'évaluation adaptés.
Le choix d'un indice unique pour l'évaluation du médicament s'accompagne pour nous d'un autre choix que le rapport Polton décrit, parmi d'autres, et que le Gouvernement n'a pas retenu. Il s'agit de la fusion de l'ensemble des taux de remboursement autre que le taux de 100 %. En effet les taux de 15 %, 30 % et 65 % établissent une hiérarchie complexe dans la prise en charge sociale du médicament, hiérarchie illisible pour les patients comme pour les professionnels de santé dans la mesure où un médicament pris en charge à 30 % n'est pas nécessairement plus efficace qu'un médicament pris en charge à 15 % ni moins efficace qu'un médicament pris en charge à 65 %.
Par ailleurs, le taux réel de prise en charge des médicaments est en fait très différent du taux affiché. Ceci résulte de la prise en charge à 100 % des molécules prescrites pour le traitement d'une affection de longue durée. Comme le montre le rapport Polton, les médicaments pris en charge en théorie à 15 % le sont en moyenne à 38 %, ceux pour lesquels le taux est fixé à 30 % le sont en réalité à 40 % et ceux relevant du taux de 65 % le sont à 81 %. Une réorganisation des taux de remboursement serait d'abord une mesure de clarification avant, éventuellement, d'être une mesure d'économie. Elle devrait limiter les écarts de prise en charge pour une même molécule entre personnes prises en charge en ALD et les autres malades.
Cette restructuration des taux de remboursement devra être accompagnée d'une révision de la liste des affections de longue durée ALD qui n'a cessé de s'allonger depuis les 6 premières maladies admises pour atteindre aujourd'hui plus de 30 pathologies.
A l'évidence, certaines d'entre elles nécessitent des traitements peu coûteux et supporteraient un remboursement moindre.
Les effets d'une telle mesure, qui ne pourrait être mise en place que progressivement, doivent bien sûr être étudiés, notamment l'impact financier du basculement d'une partie des molécules vers un remboursement à 100 %, mais elle apparaît nécessaire pour la cohérence du système. Le niveau du taux de remboursement autre que 100 % devrait dans la même logique être étudié et débattu, mais il apparaît clairement qu'il ne saurait être inférieur à 50 %.
M. Gilbert Barbier, rapporteur . - Un indice unique d'évaluation du médicament sera également un moyen d'agir de manière plus efficace sur la fixation du prix du médicament. En effet, dès lors qu'un médicament est admis au remboursement, son prix n'est plus libre. Il est fixé par le Comité économique des produits de santé, dont la composition et les compétences sont fixées par la loi : sur la base de l'évaluation de l'ASMR faite par la commission de la transparence et de l'évaluation médico-économique que conduit également, dans certains cas, la HAS, le CEPS négocie avec les industriels le prix de chaque molécule.
Les critères de fixation de ce prix, s'agissant des médicaments princeps et génériques, sont définis par le code de la sécurité sociale, et subsidiairement par la convention qui lie le CEPS aux entreprises du médicament et par la lettre d'orientation adressée au président du comité par les ministres en charge de la santé, de la sécurité sociale, des finances et de l'industrie.
La baisse des prix du médicament, en dehors de l'année 2014, est attribuable à la capacité de négociation du CEPS qui négocie et renégocie le prix des molécules en faisant jouer la concurrence entre médicaments ayant une même indication et en appliquant des baisses de prix dès lors qu'un médicament est génériqué.
Ce système a été critiqué, car il repose sur une négociation avec les industriels dont les clauses sont secrètes. En effet, les industriels proposent un prix qui fait l'objet d'une négociation, mais ils proposent aussi des remises : le prix réel n'est donc pas le prix affiché. Le montant total de ces remises versées à l'assurance maladie est connu : 700 millions d'euros en 2014.
Si le montant total des remises est publié chaque année par le CEPS, le montant des remises consenti par les industriels sur chaque médicament est protégé par le secret des affaires, qui résulte tant du droit international que de la jurisprudence de la commission d'accès aux documents administratifs. Une directive européenne est en cours d'élaboration sur cette question.
Quel que soit l'organisme chargé de négocier avec les industriels, il sera donc soumis au secret des affaires. La question n'est donc pas de savoir s'il peut être levé mais s'il existe un autre moyen d'agir sur le prix du médicament.
Certains pays, comme les Etats-Unis ou le Royaume-Uni, acceptent le prix proposé par les industriels. Cette solution n'est possible que dans des systèmes qui admettent la possibilité de ne pas prendre en charge socialement les médicaments les plus chers. Au Royaume-Uni, l'évaluation médico-économique en termes de qualité des années de vie gagnées du fait d'un médicament conduit à écarter sa prise en charge par le National Health Service (NHS) dès lors qu'il est trop cher. Ceci a par exemple été le cas pour les médicaments contre le virus de l'hépatite C (VHC).
En France, l'action sur les prix est nécessaire et, malgré ses défauts, la négociation paraît le seul moyen d'y parvenir. En effet, les mécanismes d'appel d'offres sur le modèle des Pays-Bas ou sur celui préconisé par la Commission européenne créent une instabilité pour les patients, qui deviennent dépendants des contrats passés par les autorités publiques pour la continuité d'une molécule.
Par ailleurs, l'idée d'une fixation unilatérale du prix des médicaments par les pouvoirs publics, voire de la mise en place d'une licence d'office, se heurte aux règles régissant le commerce international et spécifiquement à la nécessité d'une indemnité en cas d'expropriation.
Il n'en demeure pas moins que la différence entre le prix affiché et le prix remisé permet aux laboratoires de faire jouer la concurrence entre les Etats. Cette situation n'est pas acceptable au regard de l'arrivée de traitements particulièrement onéreux sur le marché. Mais elle ne peut trouver de solution que dans une concertation internationale ou au moins européenne. Il est donc particulièrement regrettable que les efforts de la ministre de la santé pour établir avec nos partenaires européens, ou au moins avec l'Allemagne, une position commune sur le Sovaldi aient échoué, tout comme le projet du Président de la République d'établir une position commune du G7.
La France doit mener une action résolue au niveau européen et international et doit renforcer sa capacité de négociation avec les laboratoires. Ceci signifie que l'information sur le prix du médicament à l'hôpital, dans les officines, à l'international doit être mieux suivie par le CEPS qui ne dispose que de neuf personnes, aussi qualifiées soient-elles dont son président, pour mener l'ensemble des négociations sur le médicament face aux grandes entreprises internationales du médicament.
Ce renforcement des moyens mis au service de la négociation doit permettre également le développement des contrats de performance signés avec les laboratoires afin de rémunérer un médicament sur ses effets réels et non sur les promesses issues des premiers essais, portés par les laboratoires eux-mêmes. Pour l'heure, ces contrats s'avèrent décevants pour les finances sociales en raison du manque de données publiques permettant de suivre l'usage et les effets des molécules. De tels instruments doivent être développés afin de rémunérer l'innovation de manière adéquate. En l'absence de tels contrats, les laboratoires proposent pour leur part de se voir attribuer la part des dépenses sociales qui serait économisée du fait de leurs médicaments. Il s'agirait simplement de fermer les services hospitaliers devenus inutiles... A supposer même que de telles réorganisations soient possibles, il n'est pas envisageable de considérer les dépenses d'assurance maladie comme une enveloppe acquise que les fournisseurs de produits de santé pourraient se partager.
Notre réflexion commune nous a également conduits à préconiser une évolution de la composition du CEPS afin que les représentants de l'assurance maladie y aient une place renforcée. C'est en effet l'assurance maladie qui est le payeur en dernier ressort du médicament et son point de vue doit être au moins égal à celui de l'Etat.
A l'issue de nos travaux, nous formulons donc les préconisations suivantes sur l'ensemble des aspects de la politique du médicament, que nous avons abordés :
- Mieux valoriser les innovations liées à la recherche publique française en renforçant les moyens de la recherche fondamentale et en menant une politique plus active en matière de brevets.
- Agir au niveau européen pour renforcer les exigences relatives à l'évaluation du médicament pour l'autorisation de mise sur le marché.
- Associer aux avis de la commission de la transparence en cas de déremboursement des recommandations permettant d'assurer la meilleure prise en charge du patient tout en limitant le report de prescription.
- Définir une politique de santé publique relative au développement de l'automédication.
- Développer des partenariats entre le CEPS et la Cnam afin de favoriser le bon usage du médicament et d'agir sur les comportements de promotion, de prescription et d'usage.
- Mener une action intergouvernementale avec nos principaux partenaires européens afin de définir un cadre commun de négociation du prix des médicaments les plus onéreux.
- Clarifier la notion de secret des affaires en transposant rapidement la directive européenne.
- Proscrire la mise en place de mesures fiscales ponctuelles.
- Prévoir une audition publique annuelle du CEPS devant les commissions des affaires sociales des Assemblées présentant les résultats de la négociation avec les industriels et la comparaison entre les prix du médicament en France et dans les pays voisins.
- Renforcer la place de l'assurance maladie au sein du CEPS et donner à celui-ci des moyens plus importants de contrôle et de comparaison.
- Mettre en oeuvre les préconisations du rapport Polton pour établir un critère unique d'évaluation comparative des médicaments, la valeur thérapeutique relative (VTR).
- Fusionner en un seul taux les trois taux de prise en charge à 15 %, 30 % et 65 % en s'appuyant sur les évaluations conduites par ce rapport.
M. Georges Labazée . - J'aurais souhaité que le rapport aborde davantage la question du générique et traite notamment du problème récurrent de la réticence des prescripteurs, voire de la population, vis-à-vis de ces médicaments.
M. René-Paul Savary . - J'adhère pour ma part aux propositions concrètes de ce rapport. Peut-être faudrait-il se pencher sur la question du rôle des complémentaires et sur le fait qu'elles ne remboursent pas les médicaments qui ne sont pas pris en charge par la sécurité sociale. Par ailleurs disposez-vous d'éléments sur le coût pour l'assurance maladie du remboursement du paracétamol ? Il m'apparaît également qu'il y a des molécules, que l'on pourrait qualifier de confort, qui sont nécessaires mais ne relèvent pas forcement d'un remboursement par la sécurité sociale.
Mme Laurence Cohen . - Je voudrai insister sur le fait que l'industrie du médicament a fait 856 milliards de chiffre d'affaires en 2012 et que la France était le deuxième marché après l'Allemagne. Le poids des laboratoires est donc très important. Certains Etats résistent, des expériences ont été menées au Brésil ou en Inde sur la licence. Ceci est particulièrement intéressant face notamment au monopole que détient un laboratoire sur le traitement contre l'hépatite C. Comment peut-on avancer ? Il faut également penser à un pôle public de la recherche pour faire pièce aux laboratoires. Nous partageons beaucoup des propositions du rapport et je pense qu'on ne peut plus rembourser des médicaments à 15 % soit ils sont efficaces et il faut mieux les prendre en charge, soit ils ne le sont pas.
Mme Corinne Imbert . - Je comprends que les rapporteurs aient choisi de se concentrer sur la question de la prise en charge car la question de la politique du médicament est particulièrement vaste. On aurait pu notamment parler de l'impact du tiers payant généralisé en officine qui a transformé le médicament en bien de consommation et qui a eu un impact réel sur les dépenses sociales.
Le médicament peut être un fleuron de notre industrie nationale. Certains laboratoires peuvent avoir un comportement répréhensible mais d'autres ont un comportement éthique, notamment en matière de recherche.
Il faut avoir conscience que depuis de nombreuses années le médicament a servi de variable d'ajustement à court terme pour faire des économies.
Sur le paracétamol, le problème est que l'on n'a pas désigné de groupe générique. Aujourd'hui, plutôt qu'une telle désignation, les laboratoires préfèrent aller vers le déremboursement.
J'ai lu le dernier rapport annuel du Ceps. Il indique que les efforts de maîtrise de prix ont constitué en 2014-2015 un quart des économies programmées dans l'Ondam. Les négociations menées par le Ceps ont également permis l'accès à tous les médicaments innovants ou orphelins et on constate que sur 23 médicaments utilisés pour faire des comparaisons européennes nous avons pour 20 d'entre eux le prix facial le plus bas ou le deuxième plus bas. Je souscris donc à l'idée d'auditionner le Ceps chaque année lors de la présentation de son rapport annuel, comme je partage l'idée d'encourager la recherche fondamentale, même si l'innovation ne se décrète pas. Par contre je ne suis pas sûr que la Cnam soit la mieux placée pour promouvoir le bon usage n'est-ce pas plutôt le rôle de la HAS ou de Santé publique France ?
Sur le taux de prise en charge, je pense qu'il faut veiller à l'articulation avec la complémentaire car même si elle est aujourd'hui devenue obligatoire, il ne faudrait pas réduire l'accès par l'intermédiaire du taux unique qui, par ailleurs, est une mesure intéressante d'économie.
Mme Michelle Meunier . - Je salue le caractère pédagogique de ce rapport et je suis pour ma part particulièrement intéressée par l'idée d'auditionner le Ceps qui a une responsabilité énorme avec peu de moyens.
M. Olivier Cigolotti . - Comment expliquez-vous la différence entre le taux théorique et le taux effectif de prise en charge ? Y-a-t-il un rapport avec les remises consenties par les laboratoires ?
M. Gérard Roche . - Je n'ai pas de question mais je pense que ce rapport doit nous servir de base pour approfondir nos réflexions et je partage les recommandations qu'il contient.
M. Yves Daudigny, rapporteur . - Pour répondre à M. Labazée j'indique que la Mecss a déjà publié un rapport sur le générique et que nous avons choisi parmi les thèmes possibles de notre nouveau travail, celui de la prise en charge. C'est sous cet angle que les génériques et les biosimilaires sont traités dans le rapport.
M. Gilbert Barbier, rapporteur . - M. Savary a abordé la question des complémentaires qui est un sujet en soi. Elles peuvent déjà prendre en charge les médicaments non remboursés. Sur le paracétamol, l'enjeu est de l'ordre de 500 millions d'euros pour la sécurité sociale. Il faudra veiller, s'il n'est plus remboursé, à ne pas mettre une partie de la population dans une situation difficile.
M. Yves Daudigny, rapporteur . -Mme Cohen, les grandes entreprises pharmaceutiques et l'assurance maladie se font face avec l'enjeu de permettre l'accès de tous nos concitoyens au médicament. La ministre peut-elle intervenir de manière plus brutale ? N'oublions pas que le laboratoire a toujours l'arme de ne pas mettre un médicament sur le marché. C'est rare mais cela peut arriver. La licence d'office est-elle la solution ? A nos yeux non car il faut indemniser le laboratoire ce qui peut en fait entrainer des dépenses supérieures. Nous préconisons donc le recours à la négociation.
Sur la recherche, il existe de beaux exemples de partenariat public-privé, notamment à l'hôpital Necker. Néanmoins notre première préconisation est de favoriser la recherche fondamentale car c'est d'elle que peuvent venir les ruptures en matière d'innovation.
M. Gilbert Barbier, rapporteur . - Sur la question de l'accès, on a constaté que le Levotirox était plus difficile à obtenir en France car il était mieux rémunéré à l'étranger. Aujourd'hui, les laboratoires ont une dimension mondiale et cela peut poser un problème.
S'agissant du traitement contre l'hépatite C, il n'y plus de monopole. Les prix baissent mais pas suffisamment. Je pense qu'il y a là un problème éthique.
Je rejoins Yves Daudigny pour dire que notre pôle public de recherche est devenu très faible et qu'il faut le renforcer notamment par les partenariats publics-privés.
J'indique à Mme Imbert que nous avons été très surpris par le manque de moyens du Ceps comparé aux effectifs de la commission de la transparence de la HAS et de l'ANSM. Cela m'interpelle.
M. Yves Daudigny, rapporteur . - J'indique à M. Cigolotti que la différence entre le taux théorique et le taux réel de remboursement est uniquement lié aux ALD. S'agissant des remises elles s'élèvent pour l'année dernière à 700 millions à comparer au montant total de remboursement du médicament de 30 milliards d'euros.
M. Alain Milon, président . - Je vous remercie.
La commission autorise la publication du rapport d'information.
LISTE DES PERSONNES AUDITIONNÉES
_______
Fédération des syndicats pharmaceutiques de France (FSPF)
Philippe Gaertner
,
président
Jocelyne Wittevrongel
,
vice-présidente
Conseil national de l'ordre des pharmaciens (CNOP)
Isabelle Adenot , présidente
Olivier Gross , directeur de l'exercice professionnel
Conseil national des assurances-maladies des travailleurs salariés (CNAM)
Dr Laurence Lévi , médecin conseil au cabinet du médecin conseil national
Dr Christelle Ratignier-Carbonneil , responsable du département des produits de santé
Haut Conseil de la santé publique
(HCSP)
Direction générale de la santé, ministère
des Affaires sociales, de la Santé et des droits des femmes
Dr Régine Lefait-Robin
, secrétaire
générale
Dr Daniel Floret
, président
du comité technique des vaccinations
Dr Corinne Le
Goaster,
Conseillère scientifique de la commission
spécialisée maladies transmissibles, du comité technique
des vaccinations et du comité des maladies transmissible liées
aux voyageurs et des maladies d'importations
Conseil National de l'Ordre des Médecins (CNOM)
Dr Jean Marc Brasseur , conseiller ordinal et membre de la section santé publique et démographie médicale, en charge du dossier sur les médicaments
Comité Economique des Produits de Santé (CEPS), ministère des Affaires sociales, de la Santé et des Droits des femmes
Dominique Giorgi , président
Agence nationale de sécurité du médicament (ANSM)
Dominique Martin , directeur général
Professeur Hugues de Thé, professeur au collège de France
Union des Syndicats de Pharmaciens d'Officine (USPO)
Gilles Bonnefond
,
président
Brigitte Bouzige
,
vice-présidente
Bénédicte Bertholom
,
responsable juridique
Association des laboratoires internationaux de recherche (LIR)
Agnès Soubrier
, directrice
générale
Corinne le Goff
,
présidente
Jean-François Brochard
,
vice-président
Association française pour l'industrie pharmaceutique pour une automédication responsable (Afipa)
Pascal Brossard
,
président
Daphné Lecomte
,
déléguée générale
Association E3M (Entraide aux malades de myofasciite à macrophages)
Didier Lambert,
président
Yves Ketterer
, administrateur
Celtipharm , pour une santé raisonnée
Jean-Yves Robin , directeur général
Les entreprises du médicament (Leem)
Patrick Errard,
président
Philippe Lamoureux,
directeur
général
Eric Baseilhac,
directeur des
affaires économiques
Muriel Carroll,
directeur des
affaires publiques
Clémentine Body,
responsable des
études économiques et statistiques
Sylvie Fainzang, directrice de recherche, Inserm, auteur de « l'automédication ou les mirages de l'autonomie »
Revue Prescrire
Pierre Chirac
Union Fédérale des Consommateurs - Que Choisir
Daniel Bideau
, administrateur
national
Mathieu Escot,
adjoint du responsable du
département des études,
en charge des dossiers de
santé
Pfizer
Catherine Raynaud,
directeur des affaires
institutionnelles
David Lepoittevin
, directeur Accès
au marché et Affaires publiques
Haute Autorité de santé (HAS)
Jean-Luc Harousseau,
président
Dominique Maigne,
directeur
Pr Loïc Guillevin,
membre du
collège et Président de la Commission de la
Transparence
Dr Jean-Patrick Sales,
directeur de
l'évaluation médicale, économique et de santé
publique
Gemme (générique, même médicament)
Pascal
Brière
, président
Frédéric
Girard
, vice-président
Catherine
Bourrienne-Bautista
, déléguée
générale
Alexandre Soufer, chargé
de
mission
Institut national du Cancer (Inca)
Professeur Agnès Buzyn
,
présidente
Chantal Bélorgey
, directrice des
recommandations et de la qualité de l'expertise
Collectif interassociatif sur la santé (CISS)
Christian Saout , secrétaire général délégué
Institut de recherche et de documentation en économie de la santé (Irdes)
Catherine Sermet
, responsable du pôle
médicament
Sylvain Pichetti
, maître de
recherche
SOS hépatites Fédération
Yann Mazens
, directeur
Jean
François Corty
, directeur des missions France de
médecins du monde
Théau Brigand
,
chargé de mission plaidoyer, Aides
Pierre-Yves Geoffard , économiste de la santé, directeur de recherche au CNRS
Organisation de coopération et de développement économiques (OCDE)
Valérie Paris, analyste
Médecins du Monde
Olivier Maguet
Céline
Grillon
Marie-Dominique Pauti
Direction générale des entreprises (DGE)
Benjamin Leperchey
, sous-directeur des
industries de santé et des biens de consommation
Alain-Yves
Bregent
, adjoint au chef de bureau des industries de santé et
de l'agroalimentaire
Autorité de la concurrence
Virginie Beaumeunier,
rapporteure
générale
Caroline Teyssié
, rapporteure
permanente
Roche
Yannick Plétan
, directeur
médical
Emmanuelle Garault
, directrice des affaires
publiques et gouvernementales
Claire Lhériteau
,
chargée de mission en affaires publiques
Bayer Healthcare
Philippe Mougin , directeur des affaires publiques
AbbVie
Valérie Hervé-Bannier
, directrice
des affaires institutionnelles et économiques
Aurélie
Andrieux-Bonneau
, responsable des affaires institutionnelles
•
Comité économique des
produits de santé
(Ceps)
Maurice-Pierre
Planel
, président
•
Caisse nationale de l'assurance maladie
des travailleurs salariés
(Cnamts)
Dominique
Polton
, conseillère auprès du directeur
général
•
Délégué
ministériel à l'innovation en
santé
Professeur Jean-Yves Fagon
* 1 Améliorer la qualité du système de santé et maîtriser les dépenses, Propositions de l'assurance maladie pour 2016.
* 2 Le débat sur les AMM « fractionnées » envisagées par l'agence européenne du médicament pour accélérer la mise sur le marché de certains médicaments en est l'une des dernières illustrations. Voir par exemple la Conférence-débat Pilule d'Or Prescrire 2016 d'Ancella Santos Quintano.
* 3 « Dépakine et Mediator?: repensons la pharmacovigilance », Le Monde, supplément science et médecine, 22 mars 2016.
* 4 « Industries de santé : état des lieux et principaux enjeux », Rexecode services, 23 février 2016.
* 5 Rapport du CSIS du 11 avril 2015.
* 6 Les produits de santé recouvrent les médicaments humains et vétérinaires, les dispositifs médicaux, les outils de diagnostic ainsi que les technologies médicales et les biotechnologies.
* 7 Données Eurostat.
* 8 Selon le rapport de l'ADEC des industries de santé de juin 2015.
* 9 Dont 28 000 pour la seule entreprise Sanofi, sur 37 000 emplois au total.
* 10 Selon une enquête sur les investissements productifs réalisée par l'observatoire Leem Polepharma en 2014.
* 11 L'indice Insee du prix des médicaments est calculé chaque année à qualité constante : les médicaments innovants n'intégrant l'échantillon suivi que l'année suivant leur mise sur le marché, l'indice ne retrace pas le fait qu'ils soient plus coûteux.
* 12 Selon le rapport économique du Leem pour 2015, « cette mesure phare a grandement contribué à la chute du chiffre d'affaires de l'industrie pharmaceutique : l'impact en chiffre d'affaires PFHT sur les princeps du répertoire en décembre 2013 s'élevait à - 1 milliard d'euros et à - 501 millions d'euros sur les molécules ciblées. Cette mesure a permis à la Sécurité sociale d'économiser 448 millions d'euros sur l'exercice 2013. Elle a également eu pour conséquence une forte augmentation des ventes de médicaments génériques : leur taux de pénétration dans le répertoire conventionnel a atteint 82,5 % à fin décembre 2013, le taux cible étant de 85 %. »
* 13 Etude publiée en juin 2012 par l'association des laboratoires de recherche internationaux (LIR) et la chaire santé de l'Essec, et portant sur la période 2000-2011.
* 14 L'étude relève cependant que la comparabilité de cet indicateur est « limitée ».
* 15 Allemagne, France, Finlande, Norvège, Espagne, Italie, Pays-Bas, Royaume-Uni.
* 16 Antibiotiques, anxiolytiques, antidépresseurs, antiulcéreux, hypolipémiants, antihypertenseurs, antidiabétiques et antiasthmatiques.
* 17 Le premier groupe mondial, Novartis, ne détient ainsi que 5,5 % du marché mondial.
* 18 Sur le changement de paradigme dans l'industrie pharmaceutique et le recul des médicaments chimiques traditionnels au profit des biotechnologies, cf. infra, p. ... .
* 19 Inspection générale des affaires sociales (Igas) : « Evaluation de la politique française des médicaments génériques », Dorothée Imbaud, Alain Morin, Sylvain Picard et François Toujas, septembre 2012.
* 20 Rapport d'information n° 864 (2012-2013) sur les médicaments génériques, fait par M. Yves Daudigny au nom de la commission des affaires sociales du Sénat.
* 21 80 % des matières premières utilisées viennent de Chine ou d'Inde.
* 22 Le laboratoire Pfizer, 3 ème groupe mondial en CA, commercialise un très large portefeuille de médicaments couvrant de nombreux publics.
* 23 ANSM, Analyse des ventes de médicament en France en 2013, juin 2014.
* 24 Ce sont les « médicaments irremplaçables pour affections graves et invalidantes ».
* 25 Définition du site ameli.fr de l'assurance maladie. Les ALD figurent sur une liste dont les critères sont fixés par un décret n° 2011-77 du 19 janvier 2011. Elles peuvent aussi être reconnues hors liste. Dans tous les cas, l'exonération du ticket modérateur a une durée limitée, en fonction de la pathologie.
* 26 62,15 % contre 37,85 %.
* 27 Décret n° 2008-641 du 30 juin 2008 relatif aux médicaments disponibles en accès direct dans les officines de pharmacie.
* 28 Sur son site, l'ANSM définit ainsi son rôle. « [L'agence établit] la liste des médicaments qui peuvent être présentés en accès direct dans les pharmacies selon des critères choisis pour garantir la sécurité sanitaire et la sécurité des patients : ces médicaments, du fait de leurs indications thérapeutiques, peuvent être utilisés sans intervention d'un médecin pour le diagnostic, l'initiation ou la surveillance d'un traitement ; de plus, ils présentent une posologie, une durée prévue de traitement et une notice adaptées. Le conditionnement correspond à la posologie et à la durée prévue de traitement. Pour des raisons de sécurité, certains médicaments ne sont pas éligibles, en particulier : les médicaments présentant des contre-indications majeures ou un risque important d'interactions médicamenteuses ; les médicaments destinés à la population pédiatrique, dont le niveau de sécurité ne serait pas suffisant pour une utilisation en automédication. »
* 29 « Le terme médication officinale désigne les médicaments destinés à traiter des symptômes courants et bénins, pour une durée limitée, sans l'intervention du médecin, avec l'aide du pharmacien ». Afssaps 2008.
* 30 Idem.
* 31 « Il faut un statut pour l'automédication », Espace social européen, 1090, du 20 mai au 2 juin 2016.
* 32 Presses universitaires de France, 2012.
* 33 L'ANSM note que deux conditions cumulatives doivent être réunies : « l'existence d'un besoin non couvert par une alternative médicamenteuse autorisée en France dans l'indication concernée et un rapport bénéfice/risque du médicament présumé favorable ».
* 34 Décision établissant la Recommandation Temporaire d'Utilisation d'Avastin (25/06/2015). Le Conseil d'Etat statuant en tant que juge des référés (ordonnance du 21 septembre 2015, société Roche SAS) a estimé qu'aucune des critiques formulées par la société Roche ne faisait sérieusement douter de la légalité de la décision.
* 35 Article L. 5121-12-1.
* 36 « Traitement de l'hépatite C : les patients pris en otage », Dr Chantal Guéniot, Egora, 18-24 janvier 2016.
* 37 Les considérants de l'arrêté comportent les mentions suivantes : « Considérant que, dans son avis du 14 mai 2014 susvisé, la commission de la transparence de la Haute Autorité de santé a précisé, au titre de ses recommandations, que, du fait de l'évolution lente de la pathologie, la mise sous traitement de certaines populations infectées par le virus de l'hépatite C pourrait être différée ;
Considérant que, dans sa recommandation de juin 2014 susvisée concernant la prise en charge de l'hépatite C par les médicaments antiviraux à action directe, le collège de la Haute Autorité de santé a précisé que la décision de mise sous traitement doit être adaptée au stade de fibrose hépatique concerné ainsi qu'à certaines caractéristiques des patients ;
Considérant que, s'agissant plus particulièrement des patients au stade de fibrose F2, le collège a estimé souhaitable une mise sous traitement, en recommandant toutefois de documenter au mieux le stade réel de fibrose avant de traiter et avec un degré de priorité moindre que pour les stades de fibrose plus avancés ; qu'ainsi il y a lieu de prendre en compte, dans les indications de prise en charge de ces médicaments, parmi ces patients, la sévérité constatée de la fibrose de stade 2 ;
Considérant que les ministres compétents, au regard notamment des appréciations concordantes susvisées de la Haute Autorité de santé et conformément à l'article L. 162-17, alinéas 2 et 3, du code de la sécurité sociale, estiment fondé de limiter la prise en charge du médicament relevant du présent arrêté aux indications thérapeutiques figurant en annexe et, au regard notamment de ces indications, de conditionner cette prise en charge au mode d'organisation des soins précisé dans cette même annexe.
* 38 « Quand l'hépatologie s'enhardit à propos de l'efficacité des nouveaux médicaments de l'hépatite C, mais reste muette sur les lourds liens d'intérêts qui l'unissent aux industriels », François Pesty, 10 juin 2015, www.formindep.org/Quand-l-hepatologie-s-enhardit-a.html#Propos-excessifs.
* 39 Déclaration du président de la République lors d'un déplacement à l'Hôpital Léon Bérard de Lyon.
* 40 Cette durée était de trois ans avant 1999.
* 41 www.stop-arthrose.org/petition
* 42 « Arthrose : 5 choses à savoir sur les médicaments déremboursés », www.metronews.fr/info/5-choses-a-savoir-sur-les-medicaments-contre-l-arthrose-qui-vont-etre-derembourses/mobA!MZjdDsIurtfk/ , consulté le 20 mai 2016.
* 43 « Les politiques de prise en charge des médicaments en Allemagne, Angleterre et France », Luc Nguyen-Kim, Zeynep Or, Valérie Paris, Catherine Sermet, Bulletin d'information en économie de la santé, n° 99 octobre 2005.
* 44 « Prise en charge de l'arthrose : le paracétamol en première intention lors des crises douloureuses »,
Actualités & Pratiques - N° 57 - Mars 2014.
* 45 La polymédication au regard de différents indicateurs de sa mesure : impact sur la prévalence, les classes thérapeutiques concernées et les facteurs associés, Chloé Le Cossec, Les rapports de l'Irdes, n° 562, décembre 2015.
* 46 Rapport sur la réforme des modalités d'évaluation des médicaments, remis à la ministre de la santé en novembre 2015.
* 47 « Médicaments : le taux unique de remboursement écarté », Solveig Godeluck, Les Echos, 9 septembre 2015.
* 48 Audition par la commission des affaires sociales du 8 avril 2015.
* 49 En témoigne la grande prudence de la lettre d'orientation adressée en avril 2013 au Président du comité économique des produits de santé par les ministres de tutelle : « Des contacts avec vos homologues européens seraient de nature à favoriser des échanges d'information et une coordination minimale dans la politique de fixation des prix de produits innovants. ».
* 50 Les Echos, Mercredi 13 avril 2016, « L'Allemagne veut encadrer davantage le prix des nouveaux médicaments ».
* 51 Drees, Comptes de la santé 2014.
* 52 National report on Medecines uses in Italy, 2014.
* 53 Programme de qualité et d'efficience Maladie annexé au PLFSS pour 2014.
* 54 « Nouveaux traitements de l'hépatite C et accès aux soins en France », Le Havre Forum de l'économie positive, 24 septembre 2014.
* 55 Articles L. 162-17-3 et suivants.
* 56 Article L. 162-17-8 du CSS.
* 57 La commission d'accès aux documents administratifs reconnaît trois éléments constitutifs du secret en matière industrielle et commerciale : le secret des procédés, le secret des informations économiques et financières et le secret des stratégies financières (conseil n° 20045291 du 6 janvier 2005).
* 58 COM (2013) 813 : Proposition de Directive du Parlement européen et du Conseil sur la protection des savoir-faire et des informations commerciales non divulgués (secrets d'affaires) contre l'obtention, l'utilisation et la divulgation illicites, dernière version adoptée par le Conseil le 26 avril 2016.
* 59 Cabinet EY, mai 2016, cité par les Echos 10 mai 2016.
* 60 Sylvie Delacroix-Lopes, Sakia van der Erf, Coût des génériques en Europe et mécanismes de régulation des prix en Allemagne, en Angleterre et aux Pays-Bas, CNAMTS, points de repère n° 39, novembre 2012.
* 61 Dans un communiqué inséré dans Le Quotidien du médecin du 6 juin 2016, le Gemme, qui représente les industriels du générique chiffre les économies supplémentaires possibles à 1,5 milliard chaque année.
* 62 Maxime Duffaud, Les enjeux des biosimilaires, du modèle économique à la valorisation, Sciences pharmaceutiques, 2015, <dumas-01172631>.
* 63 Vos rapporteurs ont été alertés lors des auditions de la commission des affaires sociales relatives à la sécurité des essais clinique sur la difficulté pour l'ANSM de mesurer la sincérité des dossiers d'étude pré-cliniques soumis par les laboratoires.
* 64 La méthodologie recommandée pour les essais cliniques destinés à étayer une demande d'autorisation de mise sur le marché dispose en effet que lorsque des essais sont conduits contre un comparateur actif ils le sont également contre placebo.
* 65 Bulletin de l'Association des Pharmaciens de l'Industrie et du Club de la Communication Santé, 4 décembre 2012.
* 66 Mardi 21 octobre 2014 - Loi de financement de la sécurité sociale pour 2015 - Audition de M. Frédéric Van Roekeghem, directeur général de la caisse nationale d'assurance maladie des travailleurs salariés.
* 67 « Coût des génériques en Europe et mécanismes de régulation des prix en Allemagne, en Angleterre et aux Pays-Bas », Sophie Delcroix-Lopes, Saskia van der Erf, Points de repères, Cnamts, novembre 2012 - n° 39.
* 68 Révision des critères d'évaluation des produits de santé en vue de leur prise en charge par l'assurance maladie Analyse de l'Index thérapeutique relatif (ITR) proposé par la HAS Mission d'appui à la Direction de la sécurité sociale, rapport établi par Muriel DAHAN Conseillère générale des établissements de santé, octobre 2013.
* 69 Séance du 14 novembre 2014.
* 70 Philippe Askenazy, Brigitte Dormont, Pierre-Yves Geoffard et Valérie Paris, « Pour un système de santé plus efficace », Les notes du conseil d'analyse économique, n° 8, juillet 2013.
* 71 Comptes rendus de la commission des affaires sociales mercredi 8 avril 2015.
* 72 Point de repère n° 44, Les antiviraux à action directe (AAD) dans le traitement de l'hépatite C : retour sur 18 mois de prise en charge par l'assurance maladie.
* 73 Avis du Comité d'alerte n° 2016-2 sur le respect de l'objectif national de dépenses d'assurance maladie.