







II. FLUIDIFIER L'ACCÈS AUX MÉDICAMENTS INNOVANTS APRÈS LEUR AUTORISATION DE MISE SUR LE MARCHÉ
Si le dispositif des ATU constitue, quelles qu'en soient les adaptations nécessaires, une force indéniable du modèle français pour l'accès précoce des patients aux innovations, dans les autres cas la situation est perçue comme moins satisfaisante.
Dans un contexte d'innovation dynamique, un grand nombre d'intervenants ont pointé les lenteurs et lourdeurs affectant l'accès « de droit commun » au marché français des médicaments innovants :
- d'une part, les délais entre l'autorisation de mise sur le marché et la mise à disposition effective aux patients sont plus longs que dans d'autres pays, même si ces inquiétudes sont à relativiser du fait des autres canaux d'accès précoce aux innovations ( via les ATU ou les essais cliniques) ;
- d'autre part, les procédures administratives séquencées précédant cette mise à disposition, indispensables pour définir les conditions de prise en charge solidaire des traitements innovants, soulèvent des interrogations, parfois sources d'incompréhensions entre les acteurs voire révélatrices d'un rapport de force autour des enjeux de régulation des prix.
Il ressort, plus globalement, le sentiment d'un système de santé qui subit l'innovation , et son coût, plutôt que de l'anticiper et de l'accompagner, en repensant l'équilibre délicat entre l'ambition d'un accès de plus en plus précoce à certains innovations prometteuses et les exigences tout aussi essentielles de sécurité des patients et de maîtrise budgétaire.
Si des évolutions se dessinent déjà, des mesures de fond seraient à envisager pour favoriser l' « accueil » des innovations, en renforçant la lisibilité ou la souplesse des procédures et la visibilité pour les acteurs.
A. AU-DELÀ DES LOURDEURS RÉGLEMENTAIRES, UN CLIMAT DE DÉFIANCE ET D'INCERTITUDES
1. L'accès au marché de droit commun des médicaments : une procédure séquencée et à forts enjeux
a) L'autorisation de mise sur le marché : un examen centralisé au niveau européen pour les médicaments innovants
En dehors de la procédure d'ATU ou de dispositifs d' early access équivalents, la commercialisation des médicaments et leur mise à disposition auprès des patients ont comme point de départ l'autorisation de mise sur le marché (AMM), accordée après évaluation de l'efficacité, de la sécurité et du bénéfice/risque dans une indication donnée ; elle est requise pour les « médicaments à usage humain destinés à être mis sur le marché dans les États membres et préparés industriellement ou fabriqués selon une méthode dans laquelle intervient un processus industriel. » 34 ( * )
Pour les médicaments présumés innovants, la procédure est centralisée au niveau de l'agence européenne du médicament ( European Medicines Agency - EMA) : l'AMM délivrée par la commission européenne après avis de l'EMA est unique pour l'ensemble du territoire de l'Union européenne.
|
Les procédures d'autorisation de mise sur le marché des médicaments en Europe Il existe quatre procédures d'autorisation de mise sur le marché des médicaments : une procédure nationale et trois procédures européennes. • La procédure centralisée au niveau européen, qui relève de l'EMA, est obligatoire pour les médicaments de thérapie innovante, ceux issus des biotechnologies, les médicaments contenant une nouvelle substance active et dont l'indication thérapeutique est le traitement de certaines affections (SIDA, cancer, maladie neurodégénérative, diabète, maladies auto-immunes et maladies virales) ainsi que les médicaments orphelins indiqués dans le traitement des maladies rares. Pour les autres pathologies, elle reste optionnelle. Cette procédure peut également être envisagée si le médicament présente un intérêt majeur pour les patients. En 2016, 114 AMM ont été délivrées en procédure centralisée par l'EMA dont 14 concernant des médicaments orphelins. L'agence française, l'ANSM, a été rapporteur ou co-rapporteur de 14 dossiers sur 114 (soit 12 %). • La procédure décentralisée s'applique pour les médicaments qui ne sont pas encore autorisés dans l'Union européenne et sont destinés à au moins deux États membres. L'industriel demande à un État membre d'agir en tant qu'état de référence parmi les États dans lesquels il souhaite commercialiser son médicament. • La procédure de reconnaissance mutuelle est fondée sur la reconnaissance d'une AMM déjà accordée dans un des États membres de l'Union européenne, appelé « État de référence », par d'autres États membres désignés par le laboratoire pharmaceutique titulaire de l'AMM. Dans ces deux cas (procédure décentralisée et de reconnaissance mutuelle), ce sont les autorités nationales compétentes (en l'occurrence l'ANSM en France) qui délivrent les AMM dont les annexes sont harmonisées. • La procédure nationale concerne des médicaments autorisés uniquement dans le pays concerné, ce qui est notamment le cas de génériques. En 2016, le nombre d'AMM délivrées par l'ANSM s'établit à 565 au total (502 en 2015) dont 245 AMM en procédure nationale, 295 en procédure décentralisée et 25 en procédure de reconnaissance mutuelle ; 72 % (soit 406 AMM) concernant des médicaments génériques. Source : ANSM - rapport d'activité 2016 (20 septembre 2017) |
Comme cela a été relevé à vos rapporteurs, les États-Unis sont aujourd'hui le premier marché pour la commercialisation d'un grand nombre de médicaments innovants 35 ( * ) . Toutefois, les enjeux de l'accès précoce ont conduit l'EMA, à l'image de son homologue américaine (la Food and Drug Administration - FDA), à mettre en place des outils pour accélérer le traitement des demandes d'AMM relevant de la procédure centralisée et plus généralement pour encourager l'accès précoce.
|
L'accès précoce aux médicaments en Europe Plusieurs outils ont été mis en place au niveau de l'Agence européenne du médicament pour favoriser l'accès précoce des patients aux nouveaux traitements éligibles à la procédure centralisée : - l'évaluation accélérée, qui peut être sollicitée par l'industriel pour des médicaments présentant un intérêt majeur du point de vue de la santé publique : cette procédure permet de réduire le délai maximum à 150 jours au lieu de 210 ; - l'octroi d'AMM conditionnelle ; cette procédure peut concerner des médicaments pour le traitement de maladies graves ou des situations d'urgence en réponse à des menaces sur la santé publique ou pour des médicaments dits orphelins ; elle requiert des données moins complètes que celles exigées dans la procédure de droit commun, en contrepartie de certaines obligations ; - l'usage compassionnel (programmes d' early access ) permettant aux États membres de mettre en place des dispositifs tels que les ATU en France ; - le programme PRIME ( Priority Medicines ) de soutien au développement de médicaments visant un besoin médical non satisfait ; - le programme Adaptative pathways issu d'un projet pilote de 2014, permettant l'autorisation précoce pour une population très réduite et une indication étroite, suivie de nouvelles phases itératives de développement cliniques en vue d'élargir progressivement l'indication (AMM dites « fractionnées »). |
Ces évolutions traduisent une volonté de gagner en réactivité dans un contexte dynamique d'innovation et donc une large prise de conscience des enjeux de l'accès précoce. Elles ne vont pas toutefois sans soulever des critiques et réinterrogent les procédures « classiques » d'évaluation qui suivent au niveau national pour déterminer l'accès au remboursement et le prix, pays par pays.
b) Un cadre national rigoureux en vue de la prise en charge des médicaments par la solidarité nationale
(1) La phase d'évaluation par la Haute Autorité de Santé (HAS) : une procédure lourde mais essentielle
L'évaluation scientifique précédant l'AMM est doublée, lorsque le laboratoire qui commercialise un médicament souhaite obtenir son inscription sur la liste des produits remboursables, d'une autre procédure d'évaluation thérapeutique et, dans certains cas, médico-économique. Elle est assurée en France par une autorité administrative indépendante, la Haute Autorité de santé (HAS).
• L'évaluation thérapeutique des médicaments relève plus spécifiquement de la commission de la transparence de la HAS, instance scientifique indépendante composée de vingt-et-un experts avec voix délibérative (médecins, pharmaciens, cliniciens, spécialistes en méthodologie et épidémiologie, membres des associations de patients et d'usagers) 36 ( * ) .
Cette commission est chargée de donner un avis aux ministres chargés de la santé et de la sécurité sociale en vue de la prise en charge des médicaments et de la fixation de leur prix , en appréciant :
- d'une part, le service médical rendu (SMR) qui sert à déterminer si un médicament doit être remboursé et à quel taux ;
- d'autre part, l' amélioration du service médical rendu (ASMR) pris en compte pour la fixation du prix.
|
SMR, ASMR : de quoi parle-t-on ? • Le service médical rendu (SMR) : Le médicament a-t-il suffisamment d'intérêt clinique pour être pris en charge par la solidarité nationale ? L'appréciation de ce critère prend en compte plusieurs aspects : - la gravité de la pathologie pour laquelle le médicament est indiqué, - l'efficacité et les effets indésirables du médicament, - sa place dans la stratégie thérapeutique notamment au regard des autres thérapies disponibles, - le caractère préventif, curatif ou symptomatique du médicament, - son intérêt pour la santé publique (gravité de la maladie et prévalence, besoin médical, impact sur la qualité de vie, impact sur l'organisation des soins...). Il existe plusieurs niveaux de SMR qui apportent un éclairage scientifique et clinique à l'Uncam et au ministre en charge de la santé pour déterminer si un médicament doit être remboursé et à quel taux : - SMR majeur ou important taux de remboursement 65 % - SMR modéré taux de remboursement 30 % - SMR faible taux de remboursement 15 % - SMR insuffisant pour justifier une prise en charge par la collectivité. Le SMR d'un médicament est mesuré à un moment donné. Il peut évoluer dans le temps et son évaluation se modifier. • L'amélioration du service médical rendu (ASMR) : Le médicament apporte-t-il un progrès par rapport aux traitements disponibles ? L'ASMR correspond au progrès thérapeutique que le médicament est susceptible d'apporter par rapport aux traitements déjà disponibles, au regard : - des données comparatives disponibles en termes d'efficacité et de tolérance (niveau de preuve, quantité d'effet, extrapolation en pratique clinique), - du besoin thérapeutique et de sa couverture, - de l'impact sur la qualité de vie. Les différents niveaux d'ASMR apportent un éclairage scientifique et clinique au CEPS et au ministre pour la fixation du prix des médicaments : - ASMR I, majeure, - ASMR II, importante, - ASMR III, modérée, - ASMR IV, mineure, - ASMR V, inexistante, signifie « absence de progrès thérapeutique » : dans ce cas, le médicament ne peut être inscrit au remboursement que s'il apporte une économie dans les coûts de traitement. Source : HAS |
La commission de la transparence de la HAS évalue (en première inscription ou pour des extensions d'indication) ou réévalue près de 800 produits chaque année. Entre 2014 et 2017, elle a eu à évaluer 261 nouveaux médicaments en première indication.
• L'évaluation médicale peut être complétée, dans certains cas encore rares, d'une évaluation médico-économique , qui met en regard le coût du médicament et ses conséquences sur l'organisation des soins.
Cette mission relève depuis 2008 de la commission d'évaluation économique et de santé publique (CEESP) de la HAS, dont la composition est différente de celle de la commission de la transparence 37 ( * ) . Celle-ci formule un avis sur le rapport coût/efficacité des actes, médicaments ou dispositifs médicaux susceptibles d'avoir un impact significatif sur les dépenses de l'assurance maladie. Elle est prise en compte comme critère de fixation du prix du médicament depuis 2012.
Pour les médicaments, cette procédure s'applique à ceux réputés les plus innovants (ASMR I à III) et pouvant présenter un impact « significatif » sur les dépenses de l'assurance maladie, évalué à plus de 20 millions d'euros les deux premières années de commercialisation.
Le nombre de produits concernés reste en pratique limité : vingt avis d'efficience de produits de santé ont été rendus en 2016 et autant en 2017 38 ( * ) .
(2) La phase de fixation du prix : une démarche conventionnelle qui s'inscrit dans un cadre budgétaire contraint
Sur la base des résultats de l'évaluation et notamment de l'ASMR, s'engage ensuite la négociation ou la fixation du prix du médicament devant le comité économique des produits de santé (CEPS) .
Le CEPS est un organisme interministériel placé sous l'autorité conjointe des ministres chargés de la santé, de la sécurité sociale et de l'économie. Sa formation délibérante en matière de médicament est composée, outre son président et son vice-président, de cinq représentants de l'administration 39 ( * ) , de trois représentants de l'assurance maladie obligatoire et d'un représentant des complémentaires.
Le mode de fixation des prix se fonde sur les dispositions prévues par le code de la sécurité sociale, mais également sur un accord-cadre signé le 31 décembre 2015 entre le CEPS et le Leem pour trois ans, une lettre d'orientation ministérielle en date du 17 août 2016 et une « doctrine » développée par le CEPS et formalisée dans son rapport annuel d'activité.
Le prix du médicament est fixé par voie de conventions bilatérales entre le CEPS et les entreprises pharmaceutiques.
Le présent rapport n'a pas vocation à examiner en détail ce processus complexe, qui a déjà fait l'objet d'un rapport de votre commission des affaires sociales publié en juin 2016 40 ( * ) et dont la Cour des comptes s'est encore récemment saisie 41 ( * ) .
Il est toutefois utile d'en rappeler quelques principes généraux.
|
La fixation du prix du médicament : principes généraux • Des dispositions distinctes du code de la sécurité sociale s'appliquent aux modalités de fixation de prix des médicaments, selon que le celui-ci est dispensé en officine ou à l'hôpital, et selon les différentes modalités de prise en charge (liste en sus 42 ( * ) , médicaments rétrocédables). Selon les articles L. 162-16-4 (médicaments vendus en officine) et L. 162-16-6 (liste en sus) du code de la sécurité sociale, la fixation du prix « tient compte principalement de l'amélioration du service médical rendu par le médicament, le cas échéant des résultats de l'évaluation médico-économique , des prix des médicaments à même visée thérapeutique , des volumes de vente prévus ou constatés ainsi que des conditions prévisibles et réelles d'utilisation du médicament. » • L'économie générale de la fixation des prix est articulée autour de « prix faciaux » sur lesquels s'appliquent des « remises conventionnelles » qui abaissent le « prix net » des médicaments. Le prix net n'est pas public, le montant des remises étant couvert par le secret des affaires. Les remises conventionnelles (article L. 162-18 du code de la sécurité sociale) sont négociées entre le CEPS et les entreprises pharmaceutiques et peuvent être conclues pour tous les médicaments quelle que soit leur ASMR. Leur montant brut, sur la base des ventes réalisées en 2016, s'établit d'après le CEPS à 1 milliard d'euros. 44 % de ces remises portent sur une dizaine de produits. Les engagements de type prix/volume représentent 41 % des remises dues. Ces remises sont recouvrées par les Urssaf pour le compte de l'assurance maladie. Celles liées à plusieurs catégories de médicaments pris en charge par l'assurance maladie (liste en sus, médicament sous et post-ATU, rétrocessions hospitalières) viennent alimenter en recette le fonds de financement de l'innovation pharmaceutique (FFIP) créé par l'article 95 de la loi de financement de la sécurité sociale pour 2017. Géré par la Cnam, ce fonds vise à lisser les dépenses de médicaments innovants et coûteux mais conduit à sortir ces dépenses du périmètre de l'Ondam. • Pour les médicaments innovants, une garantie de prix européen est prévue depuis 2003. Elle consiste à accorder à certains médicaments, pour une durée de cinq ans, un prix hors taxe qui ne peut être inférieur au plus bas prix pratiqué par un panel de quatre pays européens (Allemagne, Espagne, Italie, Royaume-Uni). Cette garantie ne concerne que le prix facial . Celle-ci s'applique, sauf exception justifiée, à l'ensemble des médicaments avec une ASMR I à III et, sous certaines conditions, à des produits avec une ASMR IV. Pour être éligible à cette garantie de prix le médicament doit avoir recueilli un avis de la commission d'évaluation économique de la HAS permettant au CEPS de recueillir les conditions de son efficience médico-économique. • Pour les médicaments hospitaliers , achetés par les hôpitaux, ceux de la liste en sus ont un prix (tarif de responsabilité) fixé par convention avec le CEPS (tarif déclaré par l'entreprise au CEPS qui a la possibilité de s'y opposer et de fixer son propre tarif) mais laissant la possibilité aux établissements de négocier un prix inférieur. Pour les spécialités rétrocédées, le prix de vente aux établissements de santé est déclaré par l'entreprise au CEPS qui peut s'y opposer et fixer ce prix. Sources : Rapport d'activité du CEPS pour 2016 et Cour des comptes (Ralfss 2017) |
(3) Des choix d'organisation différents entre pays
Ces étapes successives en vue de la commercialisation des médicaments et de leur admission au remboursement - AMM, évaluation, fixation du prix - visent à garantir la pleine sécurité des patients et à déterminer les conditions de leur prise en charge par la solidarité nationale.
Toutefois, comme l'ont relevé les représentants du Leem, cette procédure est comparable, en France, à « une succession de haies ».
D'autre pays ont opté pour un processus plus fluide d'accès au marché. C'est notamment le cas de l' Allemagne , fréquemment cité en exemple à vos rapporteurs : les médicaments sont disponibles dès l'obtention de l'AMM , sur la base d'un prix librement fixé par les industriels. La procédure d'évaluation en vue de la détermination du prix et de l'admission au remboursement intervient dans un délai d'un an suivant cette mise sur le marché et se déroule parallèlement à celle-ci.
Si ce modèle apparaît attractif, il faut noter qu'il fait peser une pression forte sur la procédure d'évaluation et de fixation du prix. Son attractivité au regard du modèle français est à tempérer en outre par l'existence en France des ATU.
D'une manière générale, si les modèles étrangers sont évidemment utiles à la réflexion, il est pour vos rapporteurs délicat voire non pertinent d'envisager toute transposition, tant les organisations des systèmes de santé et leur mode de régulation diffèrent par ailleurs.
|
Les procédures d'accès au marché des médicaments innovants en Europe : l'exemple de l'Allemagne, du Royaume-Uni et de l'Italie Allemagne La procédure de fixation du prix des médicaments innovants a été réformée par une loi entrée en vigueur en 2011. Le prix est fixé librement par le laboratoire pour la seule première année de mise sur le marché suivant l'AMM . Au-delà, le prix est négocié avec l'assurance maladie, sur la base de l'efficacité. Le processus dure douze mois : - dans les trois mois suivant l'accès au marché, l'institut pour la qualité et l'efficience des services de santé (IQWiG), organisme scientifique indépendant, doit avoir procédé à l'évaluation du médicament ; - dans les six mois suivant l'accès au marché, le Comité fédéral conjoint (G-BA), collège associant les fédérations de médecins, d'hôpitaux et des caisses d'assurance maladie, statue sur l'amélioration du service médical rendu : - si le médicament n'apporte pas de valeur ajoutée, l'assurance maladie peut fixer le montant remboursé de ce médicament en fonction d'un groupe de médicaments comparable. Les patients doivent payer l'éventuelle différence entre ce prix de référence et le prix de vente lorsque ce dernier est supérieur ; - si le médicament démontre une valeur ajoutée clinique, son niveau de remboursement fait l'objet de négociations entre le GKV-SV (représentant des fonds d'assurance maladie au niveau national) et les laboratoires pharmaceutiques, en liaison avec les représentants des assurances privées. L'Allemagne privilégie la négociation d'un montant maximal de remboursement, basée sur les prix européens, le prix des médicaments comparables, la valeur ajoutée et la population cible. Si aucun accord n'est trouvé à l'issu du délai de 12 mois à compter de la mise sur le marché, une commission d'arbitrage fixe le montant de remboursement, dans un délai de maximum trois mois, qui s'appliquera rétroactivement dès le treizième mois après la mise sur le marché du produit. Dès l'AMM, les hôpitaux peuvent acheter et utiliser les médicaments, pris en charge sur la base d'un montant forfaitaire, avec des mécanismes de financements additionnels pour des médicaments particulièrement onéreux. • Royaume-Uni Les prix sont fixés librement par les laboratoires mais une autre forme de régulation existe. Le National Institute for Health and Care Excellence (NICE), organisme indépendant, ne procède pas à l'évaluation de tous les médicaments ; l'évaluation est établie à l'initiative du Department of Health , dans un délai de 20 semaines. Elle se fonde notamment sur le rapport coût-efficacité du médicament (sur la base de l' Incremental Cost Effectiveness Ratio ou ICER, qui doit être situé entre 20 000 et 30 000 £) ; elle conduit à une recommandation de prise en charge par le NHS, le cas échéant avec restrictions, ou un refus , le cas échéant temporaire, de prise en charge (dans ce dernier cas le NHS n'a aucune obligation de mettre à disposition le produit pour les patients). Afin de pallier cette difficulté, le Cancer Drug Fund créé en 2011 permet de financer des médicaments anticancéreux qui n'ont pas été recommandés par le NICE. Ce dispositif a rencontré des limites en termes de financement, avec un budget initial de 200 millions de livres passé à 340 millions de livres en 2015 ; de nombreuses molécules et/ou indications ont été radiées. Le dispositif a été réformé en 2016 et réintégré dans le processus d'évaluation par le NICE, qui peut recommander le produit pour son utilisation via le Cancer drugs fund . L'évaluation initiale se fait de manière anticipée, avant l'AMM, avec un financement et un accès temporaires pour les patients dans l'attente de l'évaluation finale. • Italie Pour les médicaments classés comme remboursables par l' Agenzia Italiana del Farmaco (AIFA), la fixation du prix est déterminée, depuis 2004, après négociation avec le laboratoire. L'évaluation du taux de remboursement et du prix est réalisée sur la base du ratio coût-efficacité du produit, du bénéfice-risque, de l'impact financier prévisible, des volumes prévisionnels de vente, des prix et des consommations dans les autres pays européens. Les prix sont habituellement fixés pour deux ans. Si aucun accord sur le prix n'est trouvé, le médicament est reclassifié en non-remboursable. Les médicaments décrits comme des innovations thérapeutiques sont inscrits automatiquement sur les formulaires régionaux (cette étape supplémentaire est liée au co-paiement assuré par les autorités régionales). Depuis 2011, l'AIFA a fixé un prix de remboursement maximum pour les médicaments remboursables (classe A). Ce « cap » est basé sur une enquête des prix en cours dans les pays de l'Union Européenne. Le recours à des contrats de performance ou de partage de risques est particulièrement développé pour les médicaments onéreux, avec un accent sur le suivi en vie réelle via des registres nationaux. D'après l`INCa, en 2015 environ 200 millions d'euros soit 1 % des dépenses de médicaments ont été reversées au système de santé par des laboratoires pharmaceutiques pour des traitements anticancéreux dont les résultats n`ont pas été jugés concluants au regard des accords. Plus récemment, un mécanisme de paiement à la performance a été testé, le système de santé italien payant le traitement à l'industriel ex post pour les seuls patients répondeurs. Sources : « Accélération de l'accès à l'innovation pharmaceutique : état des lieux et perspectives », Sophie Joubert, Université d'Angers, 2015 ; « Innovation médicamenteuse en cancérologie, étude internationale sur la définition et l'accès à l'innovation », INCa, janvier 2018. |
2. La France à la traîne de l'Europe ?
Les choix d'organisation différents d'un État membre à l'autre conduisent à observer des variations, parfois importantes, dans les délais de commercialisation de certains médicaments innovants après l'obtention de l'AMM. Vos rapporteurs ont constaté que notre pays souffre à certains égards de cette comparaison.
L'analyse des données disponibles sur les délais de commercialisation des médicaments, entre celles fournies par les acteurs institutionnels et celles émanant des laboratoires est toutefois complexe car elles ne sont pas établies sur la base des mêmes périmètres. Cela plaiderait pour l'élaboration et le suivi dans le temps d'un indicateur commun à l'ensemble des acteurs, établi selon une méthodologie transparente et distinguant éventuellement selon les catégories de médicaments (par exemple en fonction de leur ASMR ou le bénéfice préalable d'une ATU).
On constate dans tous les cas que le délai de 180 jours fixé au niveau européen par la directive « transparence » 43 ( * ) pour les procédures d'inscription sur la liste des médicaments pris en charge par les systèmes d'assurance maladie n'est pas respecté .
• Au niveau institutionnel , ce délai, il faut le regretter, ne fait pas l'objet d'un suivi particulier par le ministère en charge de la santé .
La HAS et le CEPS communiquent des données qui ne permettent pas d'avoir une vision complète et consolidée de l'ensemble du processus.
- D'après la HAS, le temps d'examen des dossiers par la commission de transparence s'établit en moyenne à 105 jours en 2016 et 88 jours en 2017 pour l'évaluation des médicaments en première inscription , soit dans un délai désormais conforme au maximum réglementaire pour la part qui incombe à la phase d'évaluation. Il faut saluer les efforts déployés par la commission de transparence pour réduire ce temps d'évaluation .
- Le rapport d'activité 2016 du CEPS retrace quant à lui le temps moyen de traitement des demandes de première inscription au remboursement des médicaments en ville 44 ( * ) , qui s'établit à 166 jours en 2016 45 ( * ) , dont 284 jours pour les seuls médicaments non génériques ; il oscille, pour ces médicaments, entre 275 jours pour les dossiers ayant abouti à un accord ( 41 jours de plus qu'en 2015 ) et 326 jours pour ceux n'ayant pas abouti à un accord (par abandon, retrait ou rejet).
Pour les dossiers d'inscription de médicaments à l'hôpital , la seule procédure d'examen par le CEPS, après leur évaluation et le cas échéant leur inscription sur la liste en sus pour lesquelles vos rapporteurs n'ont pas eu communication de données spécifiques, est en moyenne de 100 jours en 2016 46 ( * ) (contre 97 jours en 2015), dont 114 jours pour ceux de la liste en sus des prestations d'hospitalisation et 55 jours pour les médicaments rétrocédables.
• Les seules données permettant d'établir une comparaison avec la situation dans les autres pays européens émanent de la fédération européenne des laboratoires pharmaceutiques (EFPIA) 47 ( * ) .
Si certains peuvent en critiquer les biais, force est de constater que ces données sont plus lisibles et davantage appropriées par les acteurs.
Ces données ne correspondent pas, pour la France, aux chiffres des acteurs institutionnels, car elles ne portent ni sur le même périmètre de médicaments ni sur la même période de référence (sont concernés des médicaments ayant obtenu une AMM sur une période considérée, et non les dossiers traités dans l'année par la HAS et le CEPS). Elles ne sont pas superposables avec le délai de 180 jours, en partant notamment de l'octroi de l'AMM et non du dépôt du dossier par l'industriel à la HAS et au CEPS, ce qui peut inclure des délais imputables à l'entreprise.
Ces données apportent néanmoins un éclairage intéressant même si elles appellent quelques nuances : d'une part, elles ne prennent pas en compte les produits ayant fait l'objet en France d'une procédure d'ATU , ce qui contribue à « dégrader » le classement relatif de notre pays ; d'autre part, elles n'éclairent pas pleinement sur l'équité d'accès aux médicaments dans les différents pays, notamment en termes de modalités de prise en charge financière.
D'après cet indicateur 48 ( * ) , la France se positionne relativement loin derrière des pays comme l'Allemagne, le Royaume-Uni ou encore l'Italie, avec un délai médian entre l'AMM et la commercialisation de nouveaux médicaments de 405 jours en France pour ceux ayant obtenu une première AMM entre 2013 et 2015.
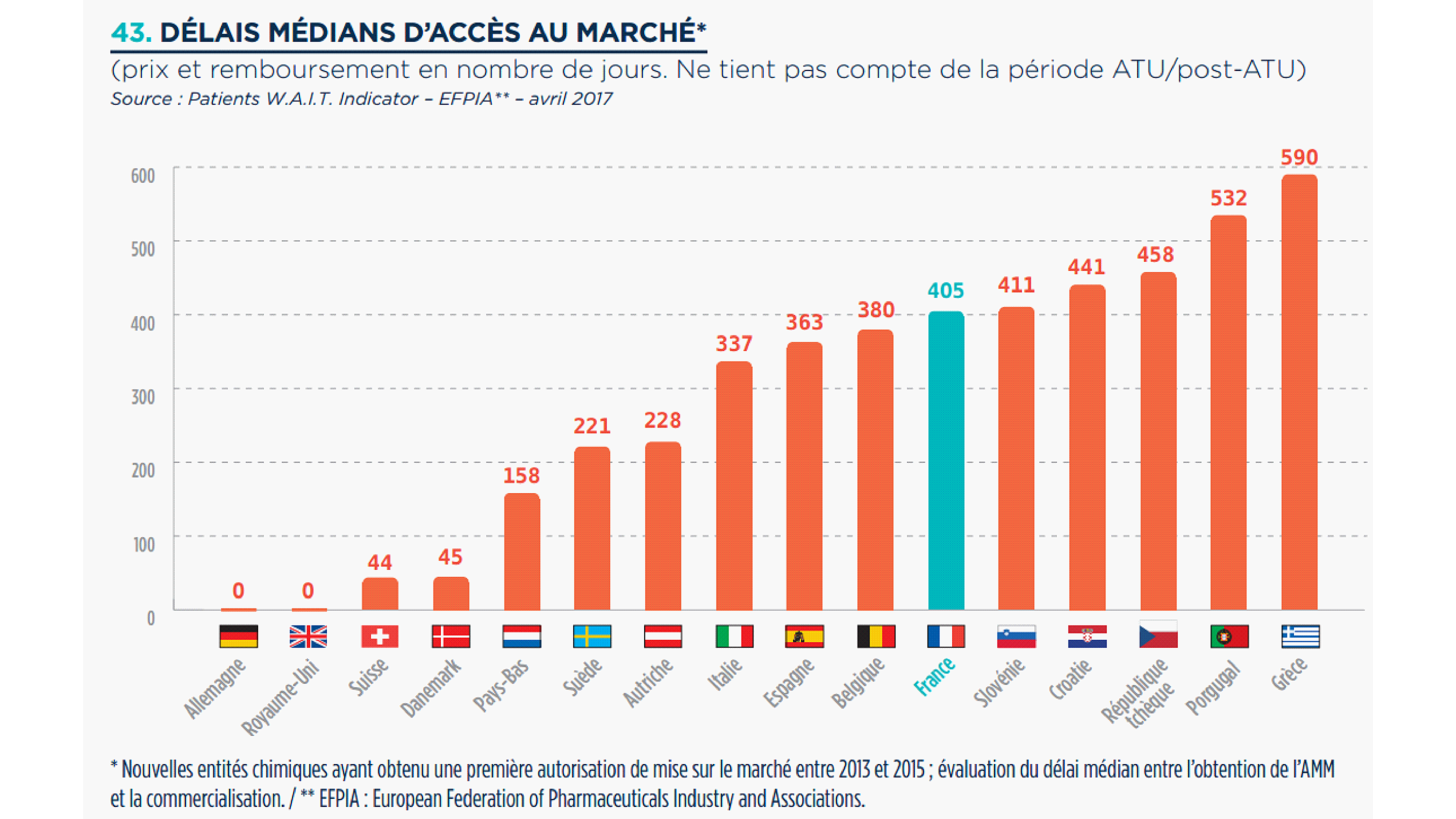
Ce délai médian pour l'accès au marché français est en augmentation puisqu'il était de 363 jours sur la période 2011-2014 et s'établirait à 460 jours sur la période 2014-2016 (d'après des données provisoires).
Le délai moyen passe dans le même temps de 408 jours (sur la base de 139 médicaments ayant obtenu une AMM sur la période 2011-2014) à 530 jours (sur la base de 36 médicaments ayant obtenu une AMM sur la période 2014-2016).
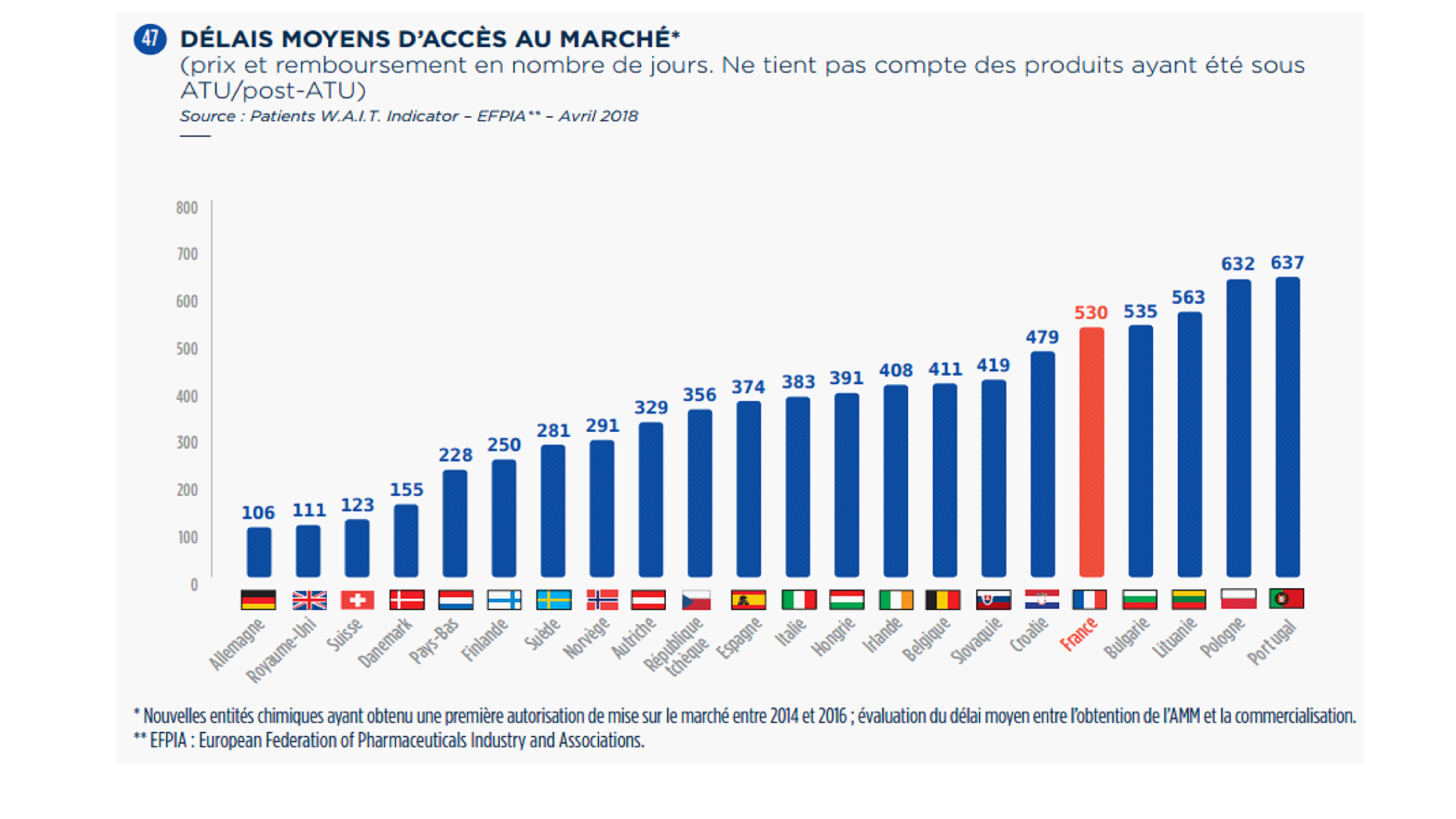
Selon ce même indicateur, le taux de disponibilité des nouveaux médicaments ayant reçu une AMM entre 2011 et 2014 est de 56,6 % en France quand il dépasse les 80 % en Allemagne, en Suisse ou encore en Suède.
• La procédure n'est guère plus rapide pour les médicaments ayant fait au préalable l'objet d'une ATU : une récente étude du Leem ciblée sur 36 produits montre des délais moyens entre l'AMM et la publication du prix au Journal officiel de 389 jours en moyenne, et de 361 jours à partir du dépôt du dossier par l'industriel, soit bien au-delà des 180 jours réglementaires.
Délais moyens et médians de la
procédure d'évaluation
et de fixation du prix de
médicaments ayant fait l'objet d'une ATU
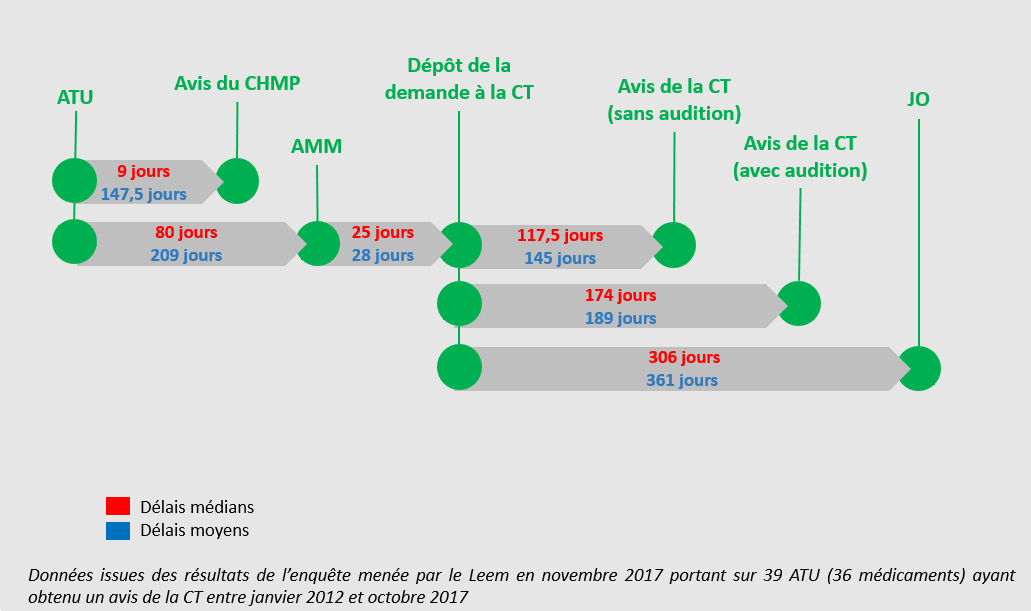
Source : Leem
3. Quels sont les limites et les freins identifiés ?
a) L'évaluation par la HAS : une rigueur reconnue, des modalités réinterrogées dans un contexte d'accélération de l'innovation
Nombre d'interlocuteurs ont souligné les efforts déployés par la HAS pour accélérer ses procédures, accompagner en amont les industriels et permettre un accès plus précoce des patients aux médicaments innovants.
Il faut ainsi souligner que d'après une étude internationale de l'INCa précitée sur l'accès aux innovations médicamenteuses en cancérologie, les agences d'évaluation allemandes et françaises se positionnent en Europe plutôt rapidement par rapport aux autres pays étudiés et sont plus enclines à se positionner positivement.
Toutefois, si la rigueur scientifique de cette procédure et son caractère stratégique sont indéniables, force est de constater que l'accélération des innovations et les enjeux de leur accès précoce conduisent à interroger les modalités actuelles de cette évaluation.
Si cette phase n'est pas perçue comme le frein principal à l'accès précoce des patients français aux molécules innovantes, elle ne va pas sans soulever des interrogations, parfois sources d'incompréhensions : est-elle encore adaptée dans le contexte actuel de l'innovation ?
• Du point de vue de la HAS, le manque de robustesse des données dont la commission de la transparence dispose pour apprécier l'efficacité et la valeur-ajoutée des médicaments présentés comme innovants rend l'évaluation plus complexe .
Cela tient au fait que les AMM sont délivrées de manière de plus en plus précoce par l'EMA, voire à titre conditionnel, sur la base de données jugées immatures (préliminaires, c'est-à-dire en phase 2 des essais cliniques).
Comme l'a relevé sa présidente, ce niveau de preuve conduit la HAS à faire un « double pari » , à la fois sur la réalité de l'efficacité du produit et sa tolérance, incertaines à long terme, mais aussi sur l'impact sur les finances publiques compte tenu des prix revendiqués par les industriels.
• Pour certains industriels, cela conduirait la commission de la transparence à la prudence voire à un excès de prudence dans l'évaluation de l'ASMR de médicaments qu'ils considèrent comme innovants.
Les données disponibles sur l'évolution des ASMR attribuées depuis 2008 ne permettent pas, toutefois, de confirmer une tendance à l'affaiblissement des ASMR I à III correspondant à des innovations importantes ou de rupture qui demeurent extrêmement rares. Le graphe suivant traduit une certaine stabilité, même si le nombre d'ASMR IV est depuis 2015 d'un niveau supérieur aux années précédentes.
Évolution de la répartition des ASMR
attribuées par la HAS
(demandes de première inscription ou
d'inscription
dans une nouvelle indication)
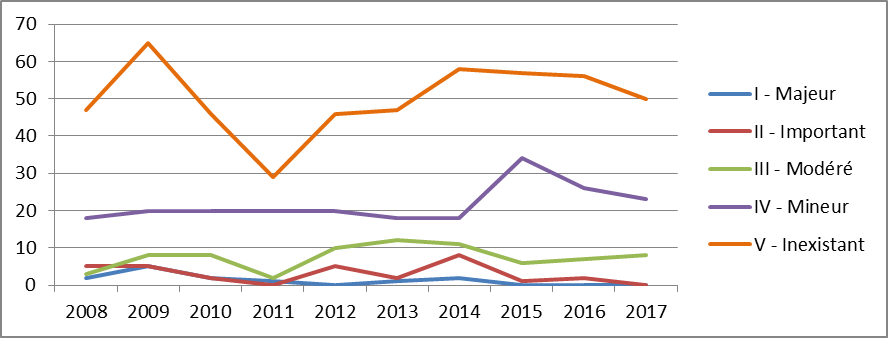
Source : HAS. Ces données ne portent pas sur les demandes d'avis ayant fait l'objet d'une procédure simplifiée.
• D'autres interrogations portent sur l'adaptation de cette procédure aux besoins et à la réalité des thérapies innovantes .
Vos rapporteurs ont ainsi pu constater les incompréhensions que cette procédure peut susciter en particulier chez certaines entreprises de biotechnologies parfois spécialisées sur un produit ou une pathologie :
- l'évaluation ne tiendrait pas suffisamment compte des spécificités des médicaments orphelins liés aux maladies rares : les données portent sur des populations forcément très restreintes et les maladies sont méconnues des experts de la commission de transparence qui peuvent remettre en cause certains critères ou analyses pourtant validés au stade de l'AMM. La prévention des conflits d'intérêt, quelle que soit la légitimité de ces règles et garanties, conduit à ce que cette évaluation ne soit pas réalisée par des spécialistes des pathologies qui seraient jugés plus capables de mesurer l'enjeu de certains nouveaux traitements, a fortiori dans le cas de maladies rares pour lesquelles il n'existe parfois qu'un seul spécialiste en France. En outre, au stade de l'évaluation médico-économique, la méthodologie « standard » de la CEESP ne serait pas jugée adaptée à des études portant sur des populations forcément restreintes ;
- certains comparateurs pris en compte pour l'évaluation de l'ASMR sont jugés contestables , créant là aussi des situations d'incompréhension, par exemple lorsqu'un médicament qui a fait l'objet d'une ATU - et donc, par définition, répond à un besoin médical non couvert - obtient par la suite une ASMR nulle en état comparé à un traitement jugé non pertinent.
Des praticiens hospitaliers ont confirmé le caractère paradoxal, à leur sens, des résultats de l'évaluation de certains traitements qu'ils perçoivent comme efficaces , pouvant parfois conduire à remettre en question leur prise en charge.
• Pour autant, vos rapporteurs saluent, dans ce contexte, les efforts déployés par la commission de la transparence pour réduire les délais d'examen des dossiers, mais aussi pour développer le dialogue en amont avec les industriels : plusieurs outils mis en place par la HAS visent à leur permettre de mieux appréhender ses exigences et donc à accélérer l'évaluation des médicaments présumés innovants.
Si, d'une façon générale, les industriels se saisissent encore peu de ces outils qui manquent selon le Leem d'agilité et de souplesse, certains ont reconnu leur intérêt et la nécessité de les renforcer.
|
Les outils mis en place par la HAS pour
accélérer l'évaluation
• Les « rencontres précoces » peuvent se tenir au niveau national ou européen en amont des demandes d'inscription au remboursement, c'est-à-dire avant les phases III des essais cliniques. L'objectif est d'identifier, dans une phase préliminaire de développement du produit, les points utiles pour l'évaluation future du médicament par la HAS ; il ne s'agit pas toutefois d'une pré-évaluation. Cette mission a été renforcée par la loi de modernisation du système de santé de 2016 pour les produits innovants. 19 rencontres précoces ont été réalisées en 2017. • Un processus d'évaluation anticipée , permettant à l'industriel, pour un médicament présumé innovant, de déposer son dossier auprès de la HAS dès le dépôt de la demande d'AMM , ce qui permet de démarrer de manière anticipée l'instruction du dossier. Il est également possible de déposer un pré-dossier dès l'avis du comité se prononçant sur l'AMM au niveau européen 49 ( * ) . Ce dernier processus n'a été utilisé, d'après les données de la HAS, que pour 31 des 261 nouveaux médicaments évalués entre 2014 et 2017, et pour seulement 22 % des produits en ATU. |
b) Des tensions qui se focalisent sur la phase de négociation du prix
Vos rapporteurs ont constaté, en lien direct avec la question des délais d'accès au coeur de leur réflexion, que c'est sur l'étape de fixation des prix, phase cruciale pour les industriels, que se concentrent les difficultés.
• Si la lettre d'orientation ministérielle de 2016 affiche clairement comme objectif l'accès des patients aux innovations thérapeutiques « dans les meilleurs délais chaque fois que leur état de santé le justifie » , en invitant le président du CEPS à rechercher « le juste prix de ces innovations » à la fois « compatible avec la soutenabilité de la diffusion de ces traitements et un retour sur investissement pour les industriels » , la plupart des acteurs ont relevé un allongement de ces délais en raison d'un durcissement des positions de part et d'autre :
- d'un côté, les exigences financières des industriels sont jugées trop élevées, parfois même extravagantes , notamment au regard de résultats de l'évaluation. Ainsi, pour la Cnam, les promesses des industriels ne sont pas toujours tenues : une analyse du prix des médicaments contre le cancer a ainsi mis en avant « une augmentation régulière et significative des prix par année de vie gagnée » , de 11 % chaque année entre 1995 et 2016 50 ( * ) . Plusieurs campagnes publiques, à l'initiative de Médecins du monde ou de cancérologues ont alerté récemment sur les prix des médicaments et la pression financière exercée par les industriels sur les systèmes de santé.
Certains dénoncent par ailleurs les stratégies des entreprises tendant à se recentrer sur des aires thérapeutiques ciblées à plus fort potentiel économique et à commercialiser des produits d'abord dans une indication visant une population restreinte, pour obtenir un prix plus élevé, avant d'élargir à des indictions visant des populations plus larges ;
- de l'autre, les industriels estiment que l'innovation n'est pas toujours reconnue, valorisée et rémunérée à sa juste valeur . Ce constat émane notamment des entreprises de biotechnologies spécialisées parfois sur un petit nombre de produits. Certains ont estimé que la pression sur les prix était plus forte que dans d'autres pays européens, comme l'Allemagne, avec des prix nets (après remises) bien inférieurs à ceux négociés outre-Rhin, ou encore que le recours à des accords prix-volume conduisait à plafonner rapidement les gains. Les prix nets négociés pays par pays n'étant pas transparents au niveau européen, il n'est toutefois pas possible pour vos rapporteurs d'apprécier la réalité et l'ampleur de ce différentiel 51 ( * ) .
• Pour le Leem, la lettre d'orientation ministérielle de 2016 contribue, plus généralement, à tendre les négociations devant le CEPS et devrait être revue. Dans l'étude précitée de septembre 2017 sur la fixation du prix des médicaments, la Cour des comptes relève en effet que ces lettres d'orientation « sont inspirées de manière croissante par des objectifs de maîtrise du coût des médicaments » et que les orientations de 2016 « marquent un tournant en assignant au président du CEPS des objectifs plus volontaristes et des principes plus rigoureux dans la fixation du prix des médicaments. »
La lettre d'orientation ministérielle de 2016 fixe notamment les principes suivants :
- pour les médicaments sans ASMR (soit ASMR V), le prix doit être « inférieur au prix du comparateur le moins cher » afin de permettre de réaliser une économie dans le coût de traitement ; le CEPS reconnaît que certains produits « font état d'améliorations qualifiées d'incrémentales par l'industriel, qui touchent la forme galénique ou les modalités d'administration dont les impacts hypothétiques en termes d'observance, de qualité de vie ou de coûts d'administration n'ont pas donné lieu à une valorisation au travers de l'ASMR » 52 ( * ) ;
- pour les médicaments avec une ASMR mineure (soit ASMR IV), qui couvrent un champ très large, l'indication concernée « ne doit pas entraîner d'augmentation dans les dépenses de coût de traitement défini par rapport au coût net du comparateur le moins cher pour cette indication », ce qui constitue un durcissement de l'appréciation 53 ( * ) .
Pour le CEPS, cela s'inscrit dans une doctrine récurrente selon laquelle « le bénéfice de l'innovation pour l'entreprise consistera dans l'accroissement de ses parts de marché, sans qu'il y ait lieu d'y ajouter un avantage de prix ».
• Si vos rapporteurs souscrivent aux objectifs de soutenabilité et de nécessaire maîtrise de la dépenses qui sous-tendent ces principes, force est de constater que, dans ce contexte, la phase de négociation suscite des tensions voire des incompréhensions. Comme le reconnaît le CEPS, « le différentiel entre les attentes du laboratoire et les prix résultant de cette approche n'ont pas toujours permis de parvenir à un accord » 54 ( * ) . Vos rapporteurs n'ont pas eu communication toutefois de données globales permettant d'étayer ce constat.
Parmi les critiques exposées par les laboratoires, la question des « comparateurs économiquement pertinents » 55 ( * ) pris en compte, parfois anciens, est à noter : comme pour les comparateurs cliniques intervenant lors de la phase d'évaluation, certains sont contestés dans leur pertinence, a fortiori quand ils s'appliquent à des médicaments ayant fait l'objet préalablement d'une ATU et qui répondent, par définition, à un besoin non couvert.
Au-delà, un laboratoire a récemment manifesté publiquement les tensions dans les négociations de prix devant le CEPS : le laboratoire Vertex, spécialisé dans le traitement de la mucoviscidose, a ainsi fait état, en début d'année 2018, du blocage des discussions pour son produit Orkambi® ayant reçu une ASMR IV alors que le laboratoire s'attendait à une évaluation plus favorable. Cette entreprise a évoqué, dans cette situation d'incertitude, son intention de ne pas débuter des essais cliniques en France pour un nouveau produit. Tout en entendant les arguments des uns et des autres et les rigidités des procédures, vos rapporteurs ne peuvent toutefois adhérer à une position qui conduirait à prendre en otage des patients.
* 34 Code communautaire relatif aux médicaments à usage humain, transposé à l'article L. 5121-8 du code de la santé publique.
* 35 D'après une étude internationale de l'INCa publiée en janvier 2018 sur l'accès aux innovations médicamenteuses en cancérologie, les États-Unis se positionnent en effet souvent en premier dans les décisions d'autorisation de mise sur le marché, devant l'agence européenne.
* 36 Ces membres titulaires ayant voix délibérative sont nommés par décision du collège de la HAS pour une durée de trois ans renouvelable deux fois ; par ailleurs, sept membres suppléants assistent aux séances avec voix consultative ; des membres ont enfin une voix consultative (le directeur de la sécurité sociale, le directeur général de la santé, le directeur général de l'offre de soins, le directeur général de l'ANSM, les directeurs de la Cnam et de la MSA, ou leurs représentants).
* 37 Cette commission est composée de 33 membres titulaires (professionnels de santé, économistes, épidémiologistes, sociologues et autres disciplines des sciences humaines et sociales, membre d'une association de patients et d'usagers) ayant voix délibérative.
* 38 Rapport de mandature de la Commission évaluation économique et santé publique (2015-2017), HAS, février 2018.
* 39 Direction de la sécurité sociale, direction générale de la santé, direction générale de la concurrence, de la consommation et de la répression des fraudes, direction générale des entreprises et, avec voix consultative, direction générale de l'offre de soins.
* 40 « Le médicament : à quel prix ? », rapport d'information n° 739 (2015-2016), fait par MM. Gilbert Barbier et Yves Daudigny, au nom de la commission des affaires sociales, Sénat, 29 juin 2016.
* 41 Rapport sur l'application des lois de financement de la sécurité sociale (Ralfss), Chapitre VIII - « La fixation du prix des médicaments : des résultats significatifs, des enjeux toujours majeurs d'efficience et de soutenabilité, un cadre d'action à fortement rééquilibrer », Cour des comptes, septembre 2017.
* 42 Cf. partie III ci-après.
* 43 Directive 89/105/CEE ou Directive Transparence, qui harmonise les délais réglementaires d'accès au marché en Europe. Ce délai s'applique entre la demande d'inscription sur les listes des spécialités prises en charge et les décisions de prise en charge ou de prix.
* 44 Ce délai est apprécié du dépôt du dossier par les industriels (parallèlement au dépôt de ce dossier à la commission de transparence) à sa conclusion (par la clôture du dossier ou la parution du prix au Journal Officiel), incluant donc une phase d'évaluation par la HAS dont l'évaluation est toutefois déconnectée des données de la HAS et bien inférieure (soit 80 jours pour les médicaments non génériques et 29 jours en moyenne).
* 45 Sur un échantillon de 935 demandes de première inscription auxquelles le CEPS a répondu en 2016, dont 65 % relatives à des médicaments génériques.
* 46 Sur 64 dossiers de médicaments à l'hôpital traités par le CEPS en 2016, dont 39 concernaient des médicaments rétrocédables et 25 des médicaments facturés en sus des prestations d'hospitalisation.
* 47 European Federation of Pharmaceutical Industries Associations (EFPIA).
* 48 L'EFPIA a mis en place le « Patients W.A.I.T. indicator », montrant le taux de disponibilité des nouveaux médicaments pour les patients à travers l'Europe ainsi que le délai moyen ou médian entre l'AMM et l'accès au patient.
* 49 Committee for Medicinal Products for Human Use (CHMP).
* 50 Rapport sur les charges et produits de l'Assurance maladie pour 2018, Cnamts, juillet 2017.
* 51 On peut toutefois noter que la dépense de médicaments remboursés connaît en Allemagne sur la période 2012-2016 une croissance de + 4,7 % en moyenne par an (d'après la Cour des comptes, Ralfss 2017). D'après le rapport d'activité 2016 du CEPS, le marché des médicaments remboursables progresse en France de + 2,6 % en 2016 contre + 0,6 % en 2015, la croissance étant principalement tirée par les produits en ATU.
* 52 CEPS, rapport d'activité 2016, annexe 4 (les méthodes de fixation du prix du médicament).
* 53 La précédente lettre d'orientation ministérielle de 2013 prévoyait que le prix d'un médicament avec une ASMR IV ne devait « pas entraîner de surcoût pour l'assurance maladie » ; depuis 2016, l'absence de surcoût n'est plus considérée sur la base d'une moyenne pondérée des comparateurs mais par rapport au coût net du comparateur le moins cher.
* 54 CEPS, rapport d'activité pour l'année 2016.
* 55 La lettre d'orientation ministérielle du 17 août 2016 au président du CEPS définit comme suit le recours à ces comparateurs : « Vous prendrez en considération l'ensemble des comparateurs cités par la HAS mais en l'absence de tels comparateurs, ou si cela peut se justifier de manière appropriée, vous utiliserez des comparateurs économiquement pertinents au regard des connaissances médicales avérées qu'il s'agisse de produits de santé, d'actes ou de prestations. »