







B. PRÉSERVER L'ATTRACTIVITÉ ET L'EFFICACITÉ DU SYSTÈME DES ATU EN L'ADAPTANT AUX RÉALITÉS NOUVELLES DE L'INNOVATION
Les auditions conduites par vos rapporteurs ont fait apparaître plusieurs points de crispation dans le mode de fonctionnement actuel du dispositif des ATU , qui ne serait aujourd'hui plus entièrement adapté aux nouveaux modèles de développement et de mise sur le marché des innovations médicamenteuses.
Deux difficultés concentrent les critiques : la question des extensions d'indication de médicaments bénéficiant ou ayant déjà bénéficié d'une ATU, que le dispositif actuel ne permet pas de prendre en charge de manière satisfaisante, et celle de la régulation financière du dispositif ; cette dernière a fait l'objet d'ajustements récents qui se heurtent au modèle économique des entreprises de biotechnologies, devenues des acteurs importants du développement de molécules innovantes.
Au-delà de ces deux sujets, d'autres ajustements apparaissent aujourd'hui nécessaires afin de préserver la pérennité, l'excellence et l'attractivité du dispositif des ATU, et ainsi de garantir un accès rapide, systématique et universel des patients français aux innovations.
1. Améliorer le suivi du dispositif pour en accroître la réactivité
a) Avec le recul, une évaluation hétérogène des produits sous ATU, reflet d'une prise de risque inhérente au dispositif
• L'ensemble des médicaments entrant dans le cadre des ATU ne constituent pas nécessairement des ruptures d'innovation si l'on considère, a posteriori , les résultats de leur évaluation par la Haute Autorité de santé.
Le rapport de septembre 2017 à la commission des comptes de la sécurité sociale indique à cet égard que si « cette évaluation confirme le plus souvent l'efficacité globale des traitements », « leur performance par rapport aux alternatives thérapeutiques, lorsqu'elles existent, n'est pas toujours avérée » 26 ( * ) .
Ainsi, 77 % des molécules admises en ATU depuis 2013 présentent un service médical rendu (SMR) important, 7 % un SMR modéré, 2 % un SMR faible et 5 % un SMR insuffisant et donc ne justifiant pas leur inscription au remboursement.
S'agissant du critère de l'amélioration du service médical rendu (ASMR), seules un peu plus d'un quart des spécialités pharmaceutiques mises à disposition sous ATU ont obtenu un ASMR II ou III , qui signent une amélioration thérapeutique importante ou modérée (2 % une ASMR II et 26 % une ASMR III). La grande majorité des produits (58 %) ont obtenu une ASMR IV, soit une amélioration thérapeutique mineure , et 14 % d'entre eux une ASMR V, soit aucune amélioration thérapeutique par rapport aux comparateurs existants.
• Vos rapporteurs soulignent que ces résultats mitigés doivent être lus avec la nuance qui s'impose, en prenant en compte deux aspects.
En premier lieu, ils sont malgré tout meilleurs que ceux obtenus par l'ensemble des médicaments évalués (ou réévalués) par la commission de la transparence de la HAS 27 ( * ) . Par ailleurs, pour la plupart des médicaments ayant obtenu une ATU, il n'existe en réalité et par définition aucune alternative thérapeutique satisfaisante.
Ainsi, ces résultats sont interprétés de manière divergente par les différents interlocuteurs entendus par vos rapporteurs.
Des fédérations hospitalières ont pu voir, sur ce point comme sur l'augmentation du nombre de produits pris en charge sous ATU, le signe d'une « dérive » dans l'application des critères prévus par l'article L. 5121-12 du code de la santé publique. Autrefois réservées à la prise en charge de pathologies mortelles pour lesquelles il n'existait aucun traitement, les ATU seraient aujourd'hui accordées de manière très large avant de déboucher sur des évaluations médiocres et souvent décevantes.
Le Leem estime pour sa part que le fait que la majorité des produits ayant bénéficié d'une ATU finissent par obtenir une ASMR de niveau IV ou V témoigne de la faible lisibilité du système d'évaluation des médicaments, ainsi que de son absence de prévisibilité.
L'ANSM relève quant à elle que l'hétérogénéité relative qui apparaît dans l'évaluation a posteriori des produits sous ATU est le reflet d'une indispensable politique de prise de risque endossée par l'agence .
Un « verrouillage » en amont du dispositif n'est pas souhaitable ; ce serait contraire à sa vocation de permettre un accès précoce des patients à des traitements prometteurs. En revanche, ces constats plaident pour un meilleur suivi en aval des produits sous ATU.
b) Renforcer le suivi des produits bénéficiant d'une ATU
• Dans la mesure où l'on se trouve, au stade de l'octroi de l'ATU, dans un contexte de simple présomption d'un rapport bénéfices/risques favorable, le recueil et l'exploitation des données cliniques au cours de la mise en oeuvre de l'autorisation constituent des enjeux majeurs pour la bonne évaluation finale des produits supposés innovants.
Ainsi que l'a souligné la présidente de la HAS, le déploiement des ATU constitue en effet une première utilisation des médicaments en vie réelle , qui plus est à grande échelle. Alors que les dossiers d'AMM ne recensent que quelques dizaines ou centaines de situations d'utilisation des produits évalués, les ATU peuvent concerner des milliers de patients.
La phase d'ATU devrait dès lors être l'occasion de conforter l'efficacité et la sécurité des produits pressentis comme innovants, mais aussi d'analyser les pratiques afin de développer leur efficience .
Les représentants de l'INCa ont par ailleurs souligné que la phase d'ATU offre une occasion précieuse de recueil de données intermédiaires entre les informations obtenues au cours des essais cliniques, qui portent sur des patients très sélectionnés, et celles ressortant de l'utilisation en vie réelle, qui se caractérise par une prescription large et donc parfois non totalement pertinente.
• Or, la plupart des interlocuteurs entendus par vos rapporteurs se sont accordés à considérer comme insuffisant le suivi des données d'utilisation, de sécurité et d'efficacité au stade de l'ATU . En dépit des obligations 28 ( * ) qui pèsent sur les professionnels de santé et les laboratoires, le suivi des patients traités dans le cadre des ATU (de cohorte comme nominatives) reste lacunaire ou de médiocre qualité.
Le respect du protocole d'utilisation thérapeutique et de recueil d'informations (PUT) 29 ( * ) , transmis à l'ANSM et auquel la délivrance des ATU de cohorte et de certaines ATU nominatives est pourtant conditionné, n'est par ailleurs pas suffisamment sanctionné.
Les services ministériels ont ainsi estimé que « la visibilité sur les résultats des données d'efficacité et de tolérance, dont le recueil est pourtant intangible à l'octroi d'une ATU, peut faire défaut ».
La commission de la transparence de la HAS déplore quant à elle que les résultats du suivi des médicaments sous ATU ne soient pas de qualité suffisante pour lui permettre d'enrichir son évaluation , notamment lorsque les médicaments sont indiqués dans le traitement de maladies rares, pour lesquels de telles données sont particulièrement précieuses.
|
Fonctionnement et utilisation des protocoles
Dans le cadre des ATU de cohorte et pour certaines ATU nominatives, le recueil des données d'utilisation est obligatoire dans le cadre des protocoles d'utilisation thérapeutique et de recueil d'information. Le PUT est établi entre l'ANSM et le titulaire des droits d'exploitation du médicament. Il permet de fixer les modalités de suivi des patients traités, le recueil de données portant sur l'efficacité, les effets indésirables, les conditions réelles d'utilisation, les caractéristiques de la population bénéficiant du médicament autorisé. Ces données de suivi sont régulièrement soumises à l'autorité compétente (ANSM) tout au long de l'ATU et du post-ATU. Les données issues des PUT sont également soumises dans le dossier de demande déposé auprès de la commission de la transparence de la HAS. Source : ANSM |
• Selon un rapport récemment remis à la ministre en charge de la santé sur les données en vie réelle pour les médicaments 30 ( * ) , ces difficultés s'expliquent en partie par des obstacles techniques apparaissant, à première vue, faciles à résoudre.
Ce rapport relève ainsi que « les suivis d'efficacité et de sécurité [prévus dans le cadre des ATU] s'effectuent dans des conditions qui ne permettent pas d'optimiser l'exploitation des données recueillies ». Le formulaire rempli, dans le cadre des ATU nominatives, par le médecin prescripteur pour obtenir la délivrance initiale puis le renouvellement de l'autorisation, est en effet renseigné manuellement. Les données de suivi collectées dans ce cadre pourraient donc très facilement être mieux valorisées grâce à la simple informatisation de leur recueil .
Le rapport relève à cet égard que dans les exemples étrangers, « cette informatisation conditionne l'exploitation effective des registres, et permet aussi d'impliquer les prescripteurs grâce aux retours d'information qu'elle permet ».
Les représentants du Leem ont néanmoins relevé que le raccourcissement de la phase d'ATU ne permet pas toujours le traitement d'un nombre suffisant de patients pour pouvoir générer des données robustes entre l'octroi de l'ATU et la demande d'AMM.
• Pour autant, vos rapporteurs estiment qu'il n'est pas de bonne gestion de se priver de données aussi précieuses pour l'évaluation des produits de santé, qui plus est alors qu'elles sont déjà produites.
Alors que les accès précoces aux médicaments devraient se développer au cours des prochaines années, sous l'effet de la poursuite du mouvement d'innovation, ces informations apparaissent indispensables à la réforme et à la consolidation de notre système d'évaluation.
Le recueil et l'exploitation de ces informations doivent dès lors être rendus véritablement effectifs .
Une telle évolution pourrait notamment passer par le renforcement des obligations pesant sur les laboratoires s'agissant de la périodicité des transmissions , tout au long de la période d'ATU comme de la séquence de post-ATU. Ces données pourraient par ailleurs être directement transmises à la HAS, et non pas seulement à l'ANSM.
Cela pourrait conduire à rendre les ATU révisables à tout moment , dès lors que l'exploitation des informations transmises n'a pas permis de mettre en évidence un progrès thérapeutique compatible avec le cadre particulièrement favorable des ATU. En contrepartie, la délivrance des ATU pourrait être rendue plus rapide et plus souple , de manière à les faire intervenir davantage en amont de l'octroi de l'AMM.
Un tel système permettrait de mettre fin aux configurations dans lesquelles un produit dont l'évaluation par la HAS ne met finalement pas en avant un progrès thérapeutique a précédemment bénéficié du cadre généreux de l'ATU et du post-ATU pendant, parfois, plusieurs années. Cette situation n'est en effet satisfaisante ni pour les industriels, qui se voient alors contraints de rembourser des montants importants correspondant à la différence entre l'indemnité ATU et le prix finalement fixé par le CEPS, ni pour les pouvoirs publics, qui versent des indemnités élevées ne correspondant pas toujours au service thérapeutique rendu aux patients.
|
Proposition n° 1 : Rendre le dispositif des ATU plus rapide et plus souple, mais révisable à tout moment sur la base des données obligatoirement produites au cours des phases d'ATU et de post-ATU. |
2. Répondre aux situations de rupture d'équité entre patients : améliorer la continuité des prises en charge
Selon les industriels, si les ATU permettent aux patients français de bénéficier d'un accès aux médicaments innovants avant leurs voisins allemands ou anglais, cet accès précoce ne concerne cependant que 10 % de la population cible des produits , en raison de l'étroitesse des critères définis pour la délivrance de l'ATU.
Au-delà de ce constat général, les auditions conduites par vos rapporteurs ont permis de mettre en évidence deux situations problématiques pour l'accès aux soins dans le cadre des ATU , relayées par les professionnels de santé.
• En premier lieu un défaut de formation et d'information des professionnels de santé ainsi qu'un défaut d'information des patients , seraient à l'origine de ruptures d'égalité selon les territoires et les établissements de santé.
France Assos santé considère ainsi que « l'accès équitable aux ATU est rarement constaté dans la pratique ».
La Fehap a souligné par ailleurs que ce défaut d'information des équipes soignantes pouvait être à l'origine de défaillances dans le lien entre le centre de référence dans lequel est traitée la pathologie faisant l'objet de l'ATU et le centre hospitalier dans lequel est habituellement suivi le patient. Elle relève que « les mécanismes d'ATU mériteraient de faire l'objet d'une synthèse pédagogique adressée aux professionnels de santé ».
• Se pose en second lieu le problème de l'entrée dans le régime de post-ATU , dont les critères d'accès ne permettent pas de couvrir la totalité de la population potentiellement bénéficiaire du médicament, alors même que ce régime a tendance à s'étirer dans le temps du fait de l'allongement de la procédure d'accès au marché de droit commun.
Ce problème d'accès aux soins se pose notamment pour les médicaments ayant fait l'objet d'ATU nominatives , dont la prise en charge en post-ATU est limitée aux seules poursuites de traitements engagés sous le régime de l'ATU. Selon les services ministériels, cette disposition avait été initialement introduite dans le but d'inciter les laboratoires à solliciter des ATU de cohorte qui permettent un accès plus transparent aux médicaments et les engagent plus formellement à mettre en place un suivi des patients traités.
Or, vos rapporteurs regrettent que cette limitation de la prise en charge retarde parfois durablement l'accès de patients atteints de maladies graves ou rares à des traitements appropriés . Un aménagement du dispositif de post-ATU pour les produits ayant fait l'objet d'ATU nominatives, de manière à permettre les initiations de traitement pendant cette période, permettrait d'assurer une plus grande équité entre patients.
|
Proposition n° 2 : Aménager le dispositif de post-ATU nominative pour autoriser les initiations de traitement après la délivrance de l'AMM. |
3. Les extensions d'indication, « trou dans la raquette » du dispositif : remédier à un besoin thérapeutique urgent
La question qui fait l'objet des critiques les plus unanimes de la part des professionnels de santé comme des patients et des institutions de la santé est celle des extensions d'indication pour les produits bénéficiant ou ayant bénéficié d'une ATU - qui constitue, selon une expression familière, le « trou dans la raquette » majeur du dispositif.
a) Un cadre juridique inadapté aux nouveaux enjeux liés au mode d'action des innovations oncologiques
(1) Les ATU sont délivrées par produit et non par indication
Les ATU sont délivrées pour un médicament innovant - en tant qu'il constitue une entité moléculaire nouvelle -, et non pour une ou plusieurs indications de ce médicament - en tant qu'il constitue un mode d'action thérapeutique.
Elles ne sont par ailleurs délivrées qu'en amont de la première AMM pour le produit considéré. Durant cette période, selon les éléments transmis par les services ministériels, « lorsqu'un médicament bénéficie d'une ATU de cohorte dans une ou plusieurs indications thérapeutiques, le directeur général de l'ANSM peut modifier l'ATU, à la demande du laboratoire, pour l'élargir à une nouvelle indication ».
Passée la délivrance de l'AMM, le périmètre de l'ATU se fige et se trouve limité au produit employé dans les indications ayant fait ou faisant l'objet de la demande d'AMM . Toute extension d'indication devient en revanche impossible au-delà de la délivrance de la première AMM.
Les premières indications demandées par le laboratoire au stade de la demande d'AMM pour leur produit deviennent de ce fait les seules autorisées jusqu'à la sortie du régime dérogatoire d'ATU.
Aux termes employés par l'INCa, dès lors que la première AMM faisant suite à un régime d'ATU de cohorte a été attribuée, seuls deux types de patients peuvent recevoir le médicament concerné en post-ATU : les patients répondant strictement aux indications de l'AMM (parfois plus étroites que celles de l'ATU de cohorte antérieure), et ceux relevant d'extensions d'indications demandées par l'industriel et en cours d'évaluation par l'agence du médicament européenne.
(2) Ce régime rencontre aujourd'hui ses limites scientifiques, entraînant d'inacceptables pertes de chances pour les patients
Les progrès scientifique, notamment dans le champ de l'oncologie, viennent questionner la pertinence de ce cadre.
L'un des domaines majeurs de développement de nouvelles molécules pour le traitement du cancer est celui de l'immunothérapie. Or, le mode d'action de ces nouveaux produits est radicalement différent des générations précédentes de médicaments anticancéreux. Dans la mesure où ils visent à renforcer le système immunitaire du patient en agissant sur des récepteurs présents dans différents organes, et non à cibler les cellules cancéreuses, ils peuvent être efficaces, de manière transversale, contre plusieurs types de cancers différents , là où les chimiothérapies actuelles visent généralement un organe particulier.
Ce mode d'action a notamment permis le développement rapide d'indications parallèles ou successives pour les anticorps anti-PD1 et anti-PDL1 . Les premières AMM pour ces produits, délivrées depuis 2011 aux États-Unis et depuis 2013 en Europe, ont ainsi successivement concerné le mélanome de stade IV, le cancer du poumon, les lymphomes de Hodgkin, les cancers de la vessie, du rein, de la tête et du cou. Le Livre blanc du Crio précité indique que des programmes de développement clinique sont en cours sur plus de 30 indications de cancers , ce qui annonce de nouvelles AMM pour les mois et les années à venir.
• Les personnes entendues par vos rapporteurs ont unanimement considéré que l'impossibilité de mettre en oeuvre une extension d'indication pour les immunothérapies sous ATU est constitutive d'importantes pertes de chances pour les patients. Le régime actuel ne garantit pas en effet aux patients de recevoir le traitement le plus efficace disponible. Des situations de besoin thérapeutique restent ainsi non couvertes.
La situation est d'autant plus absurde que les molécules concernées sont déjà présentes dans les pharmacies hospitalières, et pourraient dès aujourd'hui permettre de soigner de nombreux patients.
A notamment été cité le cas du nivolumab , qui, après avoir bénéficié d'une ATU en 2014, est aujourd'hui autorisé et admis au remboursement contre les mélanomes et les cancers du poumon. Alors même que les essais cliniques sont positifs et qu'une AMM est déjà donnée aux États-Unis dans ces indications, il est cependant impossible de le prescrire à des patients atteints, notamment, de cancers de la vessie ou ORL.
Les indications successives des nouvelles
immunothérapies anticancéreuses
et l'état des AMM et
des inscriptions au remboursement
aux États-Unis, en Europe et en
France
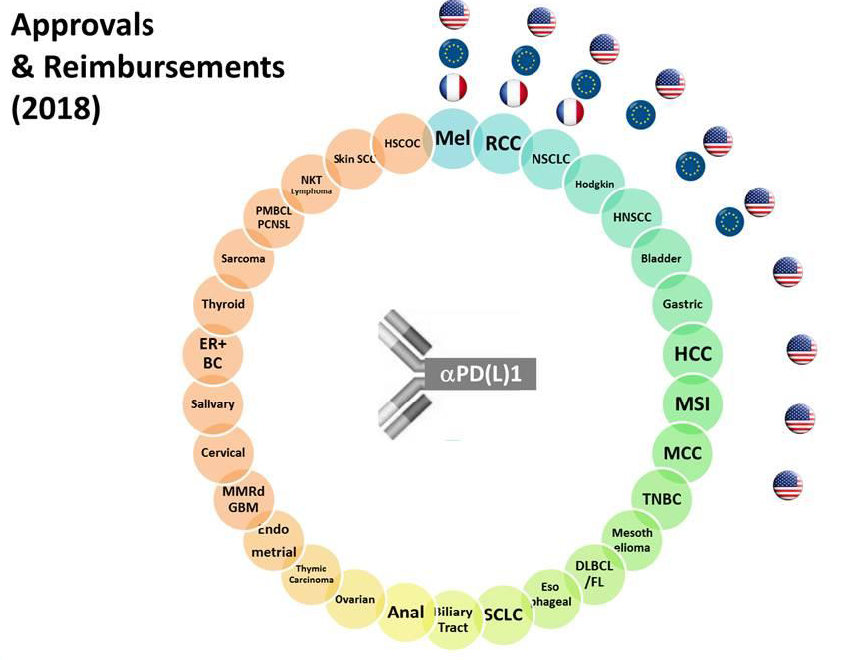
Source : Institut Gustave Roussy
Les patients subissent ainsi les conséquences des choix stratégiques des industriels, ou simplement de contraintes administratives et financières conduisant les laboratoires à ne pas pouvoir obtenir toutes les AMM simultanément.
Cette situation est également dommageable pour l'organisation des soins. Cette restriction est en effet susceptible de retarder l'adoption et l'appropriation par les professionnels et les établissements de santé des thérapies de demain.
L'INCa a ainsi relevé que, alors qu'elle était pionnière pour l'accès aux médicaments innovants en amont de leurs premières AMM, la France se retrouve dorénavant en « queue de peloton » pour les extensions d'indications.
Un exemple pratique illustrant cette situation a été cité à vos rapporteurs par le laboratoire MSD : si les patients français ont accédé, grâce au dispositif des ATU, au traitement d'immuno-oncologie du prembrolizumab pour le mélanome avancé 13 mois avant l'AMM européenne, ce même médicament, dans son extension d'indication pour le cancer bronchique (CBNPC), ne leur a été disponible que plus tardivement.
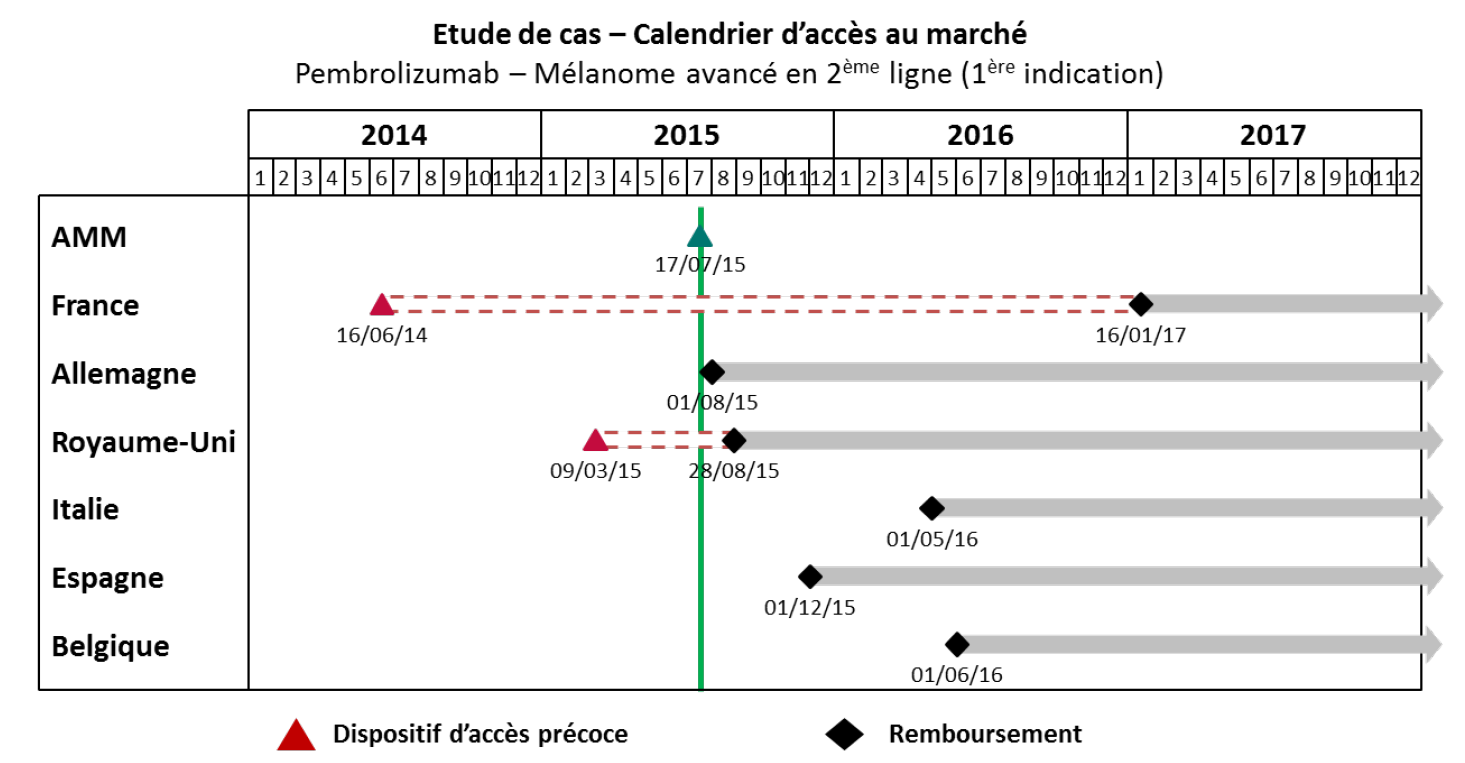
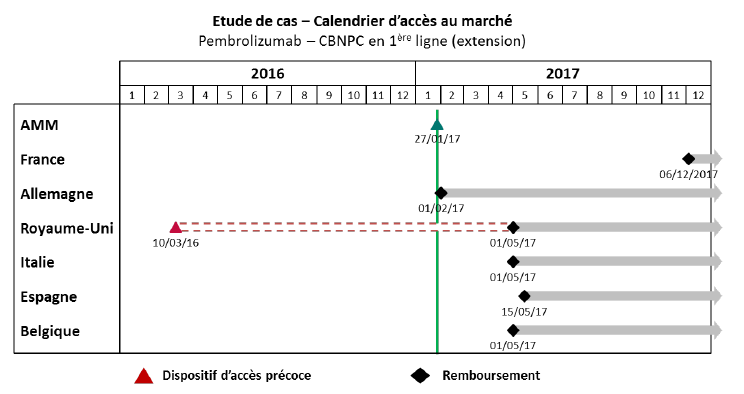
Source : MSD France
(3) Le dispositif des recommandations temporaires d'utilisation (RTU) n'apporte pas une réponse adaptée à ce besoin
• Introduites par la loi dite « Médicament » de 2011 31 ( * ) à la suite de l'affaire du Mediator®, les RTU autorisent l'utilisation d'un médicament en dehors des indications prévues par son AMM et dans le cadre d'un protocole dédié. Prévu à l'article L. 5121-12-1 du code de la santé publique, le dispositif est opérationnel depuis la parution de deux décrets d'application 32 ( * ) encadrant son régime ; il a ensuite été modifié par la LFSS pour 2014.
L'objet des RTU, accordées pour une durée de trois ans renouvelable, est d'encadrer et de sécuriser certaines pratiques de prescription constatées sur le terrain , sous condition que l'ANSM présume d'un rapport bénéfice/risque favorable dans l'indication considérée.
Une RTU ne peut être accordée qu'en l'absence d'une autre spécialité présentant le même principe actif, le même dosage et la même forme pharmaceutique, et disposant déjà d'une AMM ou d'une ATU dans l'indication considérée.
L'octroi d'une RTU emporte l'obligation pour le titulaire de l'AMM de mettre en place, à ses frais, un suivi des patients traités retraçant les données relatives à l'efficacité, à la sécurité et aux conditions d'utilisation du médicament dans l'indication considérée.
Octroyées par l'ANSM à la demande des acteurs institutionnels (autorités ministérielles, Cnam, INCa, HAS), les RTU permettent en somme d'encadrer le « bon hors AMM » . Le mécanisme vise à pallier le fait que dans de telles situations, l'industrie pharmaceutique ne demande pas toujours l'extension de l'AMM pour le produit concerné.
Dans la mesure où elles s'appliquent à des médicaments déjà autorisés et présents sur le marché, les RTU ne constituent pas à proprement parler un dispositif d'accès à des produits innovants. Dès lors cependant qu'elles pourraient permettre d'autoriser le recours à une nouvelle indication mise en évidence au cours de la vie de ces produits, elles peuvent être considérées comme un possible dispositif d'accès à l'innovation.
• Pour autant, la doctrine d'application de ce dispositif, telle qu'elle est envisagée par les différentes autorités compétentes rencontrées par vos rapporteurs, semble exclure son application au problème des extensions d'indication pour les produits ayant bénéficié d'une ATU.
Ainsi que les services ministériels l'ont rappelé, les RTU n'ont pas été conçues pour pallier cette difficulté ; du reste, en dépit de la proximité de leur dénomination, ces deux dispositifs n'ont aucun lien. Et, quand bien même on voudrait utiliser ce mécanisme à cette fin, il présenterait plusieurs faiblesses.
En premier lieu, le dispositif apparaît particulièrement lourd et rigide dans sa mise en place comme dans son fonctionnement. Il semblerait que les prescripteurs ne procèdent pas le plus souvent à l'inclusion des patients dans la RTU, du fait de contraintes administratives et procédurales jugées trop importantes.
Cette difficulté explique le faible nombre de RTU accordées depuis la mise en place du dispositif en 2012 : on en compte 47 dont 4 ont été arrêtées du fait de l'octroi d'une AMM dans l'indication considérée. Deux d'entre elles ont fait l'objet d'un traitement médiatique plus marqué : l'utilisation du Baclofène® dans le traitement de l'alcoolodépendance (mars 2014), ainsi que celle de l'Avastin® dans la dégénérescence maculaire liée à l'âge (DMLA) dans sa forme néovasculaire (juin 2015).
En outre, les RTU ne sont pas délivrées à la demande des laboratoires, ce qui exclut du circuit l'acteur majeur du développement des produits.
Enfin et surtout, un recours renforcé au mécanisme des RTU ainsi que son ouverture aux laboratoires pourraient conduire à déséquilibrer les négociations de prix conduites entre le CEPS et les industriels . La mise en oeuvre d'une RTU postérieurement à la conclusion de ces discussions aboutirait en effet à augmenter la population cible des médicaments concernés, et donc le volume de produits pris en charge par l'assurance maladie, sans que cette donnée ait été prise en compte dans la fixation préalable du prix. La demande d'une AMM dans une indication cible étroite puis d'une RTU visant une population plus large pourrait ainsi alimenter les stratégies industrielles, pour un impact financier certain sur les comptes de l'assurance maladie.
• Vos rapporteurs souscrivent cependant à l'observation des équipes hospitalières et de la HAS selon laquelle le dispositif des RTU est sous-exploité alors même qu'il pourrait répondre à d'importants besoins . Il peut par ailleurs constituer un levier d'incitation pour la mise en place d'essais cliniques par les laboratoires dans le but de parvenir à une extension d'indication de leur médicament.
Quoique cette question ne relève pas directement de l'objet du présent rapport, ils estiment dès lors indispensable de le rendre à la fois plus rapide, plus souple et plus simple , par exemple par l'introduction de délais maximum de procédure.
b) Adapter les ATU pour couvrir les besoins thérapeutiques liés aux extensions d'indication
Vos rapporteurs estiment en tout état de cause indispensable de mettre en place dans les meilleurs délais un élargissement du dispositif d'ATU aux nouvelles indications de produits disposant déjà d'une AMM et faisant l'objet de développements innovants dans de nouvelles indications , en cas de rapport bénéfices/risques satisfaisant et en l'absence d'alternative thérapeutique.
Suivant la piste avancée par la plupart des acteurs entendus, la mise en place d'un système d'ATU par indications apparaît comme la façon la plus simple et efficace de répondre à cet objectif. Cette solution reviendrait à repenser le principe du dispositif pour l'attribuer non plus une seule fois pour un produit dans quelques indications limitativement énumérées, mais, potentiellement, plusieurs fois successivement pour un même produit.
|
Proposition n° 3 : Délivrer les ATU par indication et non plus par produit, de manière à couvrir les situations d'extension d'indication survenant après la délivrance de la première AMM. |
4. Revenir sur un régime de régulation financière devenu excessivement complexe
a) Un encadrement financier renforcé dans le cadre des dernières LFSS
L'évolution du coût du dispositif des ATU a justifié un renforcement de son encadrement financier.
En particulier, l'article 97 de la loi de financement de la sécurité sociale pour 2017 33 ( * ) a été adopté dans l'objectif affiché par le précédent gouvernement de « préserver » les dispositifs d'ATU et de post-ATU en assurant leur soutenabilité financière pour l'assurance maladie. Il s'agissait de mettre en place un « accès précoce plus régulé », reposant sur « un partage des coûts entre l'industriel et la collectivité ».
Cet encadrement repose sur deux leviers.
• D'une part, un plafonnement de la dépense moyenne annuelle par patient au titre des produits sous ATU, selon un mécanisme à double détente : pour tout produit dont le chiffre d'affaires excède 30 millions d'euros par an, le coût annuel par patient est limité à 10 000 euros .
Le cas échéant, ce plafonnement se traduit par un remboursement rétroactif annuel à l'assurance maladie, par le laboratoire pharmaceutique, de la part de son chiffre d'affaires en dépassement de ces seuils, sous la forme d'une remise.
Selon les services ministériels, il s'agit d'un « garde-fou » visant à mettre fin au régime de totale liberté dont bénéficiaient les industriels dans la fixation de l'indemnité au titre des produits sous ATU , dans la mesure où il affaiblissait ensuite le pouvoir de négociation du CEPS dans la fixation du prix, en partant d'un niveau de référence élevé.
Applicable aux produits pour lesquels une ATU a été octroyée à compter du 1 er janvier 2017, ce mécanisme n'a, selon les services ministériels, encore jamais trouvé à s'appliquer.
Vos rapporteurs observent que l'absence de dépassement à ce jour de ce plafond signifie que les industriels ont probablement calibré le montant de l'indemnité demandée afin de l'inscrire dans la fourchette fixée par la loi, ce qui constitue une ébauche de régulation. Ils relèvent également que, selon les indications fournies par l'assurance maladie, ce système de plafonnement du coût des traitements onéreux existe dans la plupart des pays comparables à la France.
• D'autre part, un nouveau mode de calcul pour le montant du remboursement rétroactif du différentiel entre l'indemnité ATU fixée par l'industriel et le prix du médicament défini par le CEPS après son AMM.
L'article 97 a modifié le mode de calcul du montant rétroactivement reversé par le laboratoire à l'assurance maladie, sous forme de remise, dans tous les cas où le prix fixé par le CEPS est inférieur à l'indemnité déterminée par le laboratoire en ATU et post-ATU.
Ce montant est désormais calculé non plus par rapport au prix ou au tarif de remboursement (le « prix facial »), mais par rapport au « prix net de référence » (prix net des remises conventionnellement consenties par le laboratoire) pour le produit concerné.
Tableau comparatif des dispositions de l'article
L. 162-16-5-1
avant et après la réforme introduite par
l'article 97 de la LFSS pour 2017
|
[...] le laboratoire reverse aux organismes mentionnés à l'article l. 213-1 désignés par le directeur de l'agence centrale des organismes de sécurité sociale, sous forme de remise, |
|
|
avant la LFSS pour 2017 la différence entre le chiffre d'affaires facturé aux établissements de santé sur la base de l'indemnité et celui qui aurait résulté de la valorisation des unités vendues au prix ou au tarif de remboursement fixé par le comité . |
après la LFSS pour 2017 la différence entre le chiffre d'affaires facturé aux établissements de santé, au titre de la période s'étendant de l'obtention de l'autorisation mentionnée à l'article l. 5121-12 du code de la santé publique à la première date d'inscription au remboursement, minoré le cas échéant des remises mentionnées au ii du présent article au titre de cette même période, et celui qui aurait résulté de la valorisation des unités vendues au prix net de référence . |
Aux termes de l'article L. 162-18 du code de la sécurité sociale, ce prix net de référence est calculé à partir du niveau prévisionnel des prix nets de remises sur les trois années à venir . Il repose donc sur une prévision des volumes de vente pour les trois années suivant la sortie du dispositif ATU . Selon les indications fournies par le CEPS, il s'agit en somme de défalquer du prix facial le montant des remises conventionnelles qui pourraient être dues au titre de ces trois années - étant précisé qu'un prix net plus bas que celui qui résulterait de ce mode de calcul peut toujours être déterminé par voie conventionnelle.
Ce mécanisme, applicable aux chiffres d'affaires réalisés dès l'année 2016, viserait avant tout à inciter les laboratoires à conclure rapidement un accord de prix permettant la sortie du post-ATU .
La Cnam a qualifié le nouveau mode de calcul de « nécessité absolue » du fait de l'impact financier du régime des ATU. Selon la DSS, ce mécanisme se justifie par les prétentions très élevées des laboratoires en matière de prix . Ce mode de calcul a dès lors été conçu, comme le mécanisme de plafonnement, pour inciter les industriels à sortir rapidement du régime financier dérogatoire de l'ATU et du post-ATU.
Il ne semble pas, pour autant, que cet objectif soit atteint dans la mesure où il paraît contribuer, à l'opposé, à une crispation des positions.
b) Des dispositifs illisibles et contestés : des modalités à simplifier
Vos rapporteurs entendent les raisons qui ont justifié le renforcement de la régulation financière du dispositif des ATU, dans un contexte de croissance dynamique des coûts et de l'arrivée de nouvelles innovations dont les prix, réels ou fantasmés, sont très élevés.
Pour autant, les modalités prévues par l'article 97 de la LFSS pour 2017 soulèvent plusieurs difficultés.
• Le mécanisme de plafonnement aboutit, pour les industriels, à une dégradation de la valeur marchande de l'innovation .
On peut s'interroger en effet sur l'adaptation du seuil de 10 000 euros par patient à certains traitements innovants : les représentants d'Unicancer ont estimé qu'il n'était « absolument pas adapté aux nouvelles thérapies anticancéreuses », et donc susceptible de fragiliser le dispositif des ATU.
• Mais c'est avant tout le mode de calcul de l'indemnité rétroactive versée sous forme de remise par les laboratoires au sortir de la phase de post-ATU qui concentre les critiques.
Qualifié par les représentants du Leem de « mécanisme surréaliste », ce nouveau régime a, de l'avis unanime des industriels entendus par vos rapporteurs, fortement dégradé l'attractivité des ATU .
Outre que cette modification, prévue par voie d'amendement à un stade avancé de la procédure d'adoption de la LFSS pour 2017, pose la question générale de la sécurité juridique du système des ATU , la complexité de ce nouveau mode de calcul apparaît très dissuasive pour les laboratoires. Cette complexité est d'autant plus mal reçue qu'elle se justifie par des raisons strictement économiques, voire comptables - ce dont les autorités ministérielles ne se cachent d'ailleurs pas -, perçues comme non justifiées au regard des enjeux de promotion de l'innovation.
Surtout, ce mode de calcul est dénoncé comme remettant en cause la lisibilité et la prévisibilité du mécanisme de sortie des ATU , dans la mesure où le prix net qui sert de base à la détermination de la remise est calculé sur la base de volumes prévisionnels de vente . Les laboratoires estiment que cela revient à mettre en place un mécanisme de remise sur un chiffre d'affaires potentiel , qui est par nature incertain et soumis à de nombreux aléas - comme par exemple l'arrivée sur le marché d'un nouveau traitement concurrent.
Les industriels soulignent par ailleurs que le montant de la remise ainsi calculée est très difficile à absorber par les petits laboratoires de biotechnologies , qui constituent aujourd'hui des acteurs importants de l'innovation médicamenteuse, et d'autant plus lorsqu'ils sont monoproduit.
Ces difficultés sont renforcées par le caractère rétroactif du dispositif , qui, voté en 2017, s'applique aux chiffres d'affaires réalisés au titre des ATU en 2016. Les entreprises concernées n'ont dès lors pas toujours pu provisionner les sommes à reverser.
Les industriels avancent enfin que le raffinement technique de nouveau dispositif est très difficile à expliquer auprès des instances décisionnaires de celles des entreprises concernées dont la taille est mondiale et dont le siège se situe à l'étranger.
Vos rapporteurs notent que ces difficultés s'ajoutent à celles résultant du mode de calcul de la contribution sur le chiffre d'affaires des laboratoires , dite « clause de sauvegarde » ou « L » et prévue par l'article L. 138-10 du code de la sécurité sociale. Le montant de cette taxe, qui porte sur la progression du chiffre d'affaires des laboratoires d'une année sur l'autre, est en effet calculé sur la base d'un chiffre d'affaires net de remises pour l'année n-1, ce qui conduit à un gonflement artificiel du chiffre d'affaires pour l'année n et à une augmentation mécanique du montant de la contribution. Cette situation a été dénoncée au cours de l'examen des derniers PLFSS par votre commission des affaires sociales, qui a adopté à plusieurs reprises un amendement tendant à rendre comparables les assiettes prises en compte d'une année sur l'autre pour le calcul de la contribution due au titre de la clause de sauvegarde.
Cette contribution a été scindée, dans le cadre de la LFSS pour 2017, en une contribution Lh portant sur la progression du chiffre d'affaires réalisé à l'hôpital et une contribution Lv pour les médicaments de ville. Or, la plupart des médicaments innovants étant distribués à l'hôpital par un petit nombre de laboratoires, le montant de la contribution à leur charge s'en trouve mécaniquement augmenté.
• Vos rapporteurs estiment que, du fait de la particulière complexité du dispositif mis en place par l'article 97 de la LFSS pour 2017, il n'apparaît pas raisonnable de poursuivre son application .
L'objectif de maîtrise des dépenses de l'assurance maladie doit être concilié avec la nécessaire attractivité du dispositif d'ATU, afin de continuer à garantir aux patients cet accès essentiel à l'innovation médicamenteuse.
Compte tenu de l'allongement de la phase de post-ATU, qui expose l'assurance maladie au paiement d'une indemnité très élevées pendant une longue période, vos rapporteurs considèrent cependant indispensable de réfléchir à un nouveau mécanisme de régulation .
La proposition n° 1 formulée dans le cadre du présent rapport apporte un élément de réponse en garantissant l'adéquation du niveau de l'indemnité payée par l'assurance maladie en ATU avec les résultats thérapeutiques réellement observés. Des professionnels de santé entendus ont d'ailleurs souligné que, en toute logique, l'indemnité versée au titre des ATU devrait d'abord être faible, avant d'être révisée à la hausse en fonction des résultats obtenus.
|
Proposition n° 4 : Revenir sur le mode de calcul complexe de la remise rétroactivement versée par les laboratoires au titre de la récupération sur l'indemnité de la phase d'ATU, tel que prévu par l'article 97 de la LFSS pour 2017. |
*
Les difficultés relatives à la mise en oeuvre des ATU ne se comprennent que dans le cadre plus général des procédures de droit commun de mise sur le marché et de fixation des prix .
Les préconisations faites par les industriels traduisent au fond la volonté d'une valorisation commerciale rapide des innovations, aujourd'hui rendue difficile par l'allongement de la phase de post-ATU.
C'est finalement au moins autant le cadre général de l'évaluation des médicaments et de la fixation des prix, dont les délais apparaissent incompatibles avec l'accélération des innovations scientifiques, qui doit aujourd'hui évoluer.
* 26 Le British Medical Journal (BMJ), en conclusion d'un article d'octobre 2017 intitulé « Availability of evidence of benefits on overall survival and quality of life of cancer drugs approved by European Medicines Agency : retrospective cohort study of drug approvals 2009-2013 » et signé par Courtney Davis, Huseyin Naci, Evrim Gurpinar, Elita Poplavska, Ashlyn Pinto,Ajay Aggarwal, souligne par ailleurs que la plupart des médicaments anticancéreux approuvés par l'EMA de façon précoce, sur des critères intermédiaires, et mis sur le marché entre 2009 et 2013 n'ont pas apporté la preuve qu'ils amélioraient la survie globale ou la qualité de vie. Cet article a été porté à la connaissance de vos rapporteurs par les services ministériels.
* 27 Sur ce point, voir partie II. Le rapport d'activité de la HAS pour 2016 indique que sur 750 spécialités pharmaceutiques ayant fait l'objet d'un avis rendu au cours de l'année, seuls 25 médicaments (soit 3,3 % d'entre eux) ont été reconnus comme apportant une avancée thérapeutique par rapport aux traitements existants. Sur ces 25 produits, 6 seulement ont obtenu un ASMR de niveau II ou III, les 19 autres ayant reçu une ASMR IV.
* 28 En application de l'article l. 5121-12 du code de la santé publique.
* 29 Le régime des PUT est défini à l'article R. 5121-70 du code de la santé publique.
* 30 Bernard Bégaud, Dominique Polton et Franck von Lennep, « Les données de vie réelle, un enjeu majeur pour la qualité des soins et la régulation du système de santé. L'exemple du médicament », Rapport réalisé à la demande de la ministre de la santé Marisol Touraine, mai 2017.
* 31 Loi n° 2011-2012 du 29 décembre 2011 relative au renforcement de la sécurité sanitaire du médicament et des produits de santé.
* 32 Décret n° 2012-742 du 9 mai 2012 relatif aux recommandations temporaires d'utilisation des spécialités pharmaceutiques modifié et décret n° 2012-740 du 9 mai 2012 relatif à la prise en charge dérogatoire par l'assurance maladie des spécialités pharmaceutiques bénéficiant d'une recommandation temporaire d'utilisation ou de certains produits et prestations.
* 33 Loi n° 2016-1827 du 23 décembre 2016 de financement de la sécurité sociale pour 2017.