







6. Revoir les modalités de l'évaluation et de la surveillance clinique des dispositifs médicaux implantables avant et après leur commercialisation
Alors que le marquage CE constitue avant tout une garantie de conformité des dispositifs médicaux avec des règles de sécurité et de fabrication, tous les acteurs du secteur s'accordent sur la nécessité de renforcer l'évaluation de ces produits de santé par la systématisation des études réalisées après leur mise en vente.
Cette préoccupation n'est pas nouvelle. Comme on l'a vu précédemment, la directive 2007/47/CE du 5 septembre 2007 a complété la directive de 1993 en rendant obligatoires les investigations cliniques pour les dispositifs médicaux implantables ainsi que pour ceux de classe III, « sauf si le recours aux données cliniques existantes peut être dûment justifié » (annexe X de la directive 93/42, §1.1 bis ; article R. 5211-36-2 du code de la santé publique). Malheureusement, le droit en vigueur est souvent méconnu et mal appliqué.
a) Corriger les ambiguïtés de la réglementation européenne
Jean-Luc Harousseau l'a déploré lors de son audition : « les essais cliniques, que la directive 2007/47/CE encourage pour les dispositifs de classe III, se mettent en place lentement, les produits étant souvent fabriqués par des toutes petites entreprises pour des publics très ciblés. [...] Il faudrait inciter, plus que ne le fait la directive, les industriels à lancer des études » . En l'absence de lignes directrices claires, leurs termes sont négociés entre le fabricant et l'organisme notifié qui examine son dossier. Les autorités nationales de régulation du secteur, comme l'ANSM en France, n'ont pas directement accès à ces données et ne peuvent pas s'assurer que les études sont réalisées selon un protocole scientifiquement irréprochable. Elles ne connaissent pas non plus le plan de surveillance établi par le fabricant afin d'obtenir sa certification CE.
Dans ces circonstances, il est regrettable de constater que les dispositions de l'annexe X de la directive 93/42, qui définit les modalités de l'évaluation clinique obligatoire pour les dispositifs médicaux implantables, soient de manière quasi systématique contournées par les fabricants et interprétées de telle sorte qu'ils soient exonérés de leurs obligations. En effet, ce texte prévoit que les investigations cliniques doivent être réalisées « sauf si le recours aux données cliniques existantes peut être dûment justifié » (§ 1.1 bis ). Malheureusement, aucun contrôle n'est effectué sur le recours à cette alternative qui aurait dû, dans l'esprit du texte, rester l'exception . La mauvaise application de cette réglementation lui ôte toute sa force. De plus, l'évaluation clinique a un caractère souvent biaisé lorsqu'elle est réalisée par des experts rémunérés par les industriels.
Il est donc indispensable que, dans le cadre de la refonte de la réglementation européenne qui s'annonce, les obligations en matière d'évaluation clinique soient précisées et rendues plus contraignantes. D'ores et déjà, les organismes notifiés pourraient se montrer plus exigeants au niveau de l'évaluation des données de conception des dispositifs médicaux implantables . La démarche d'évaluation de la performance clinique d'un dispositif médical par son fabricant devrait être soumise au régulateur. Afin de s'assurer de la qualité de l'expertise scientifique, il serait même souhaitable de mettre en place une double validation, par l'organisme notifié puis par l'autorité nationale compétente, des données cliniques avant la délivrance du marquage CE .
La procédure d'évaluation clinique par équivalence, basée sur la littérature scientifique existante, devrait être mieux encadrée afin qu'elle cesse d'être celle choisie dans près de 90 % des cas . L'organisme notifié et le régulateur devraient pouvoir demander plus de détails au fabricant sur la méthodologie utilisée et vérifier qu'elle est applicable à chaque cas d'espèce.
Surtout, il convient de mieux encadrer la notion même d'équivalence, afin d'éviter un « effet domino » qui permet aujourd'hui à des fabricants, par une suite d'équivalences successives, de considérer que la littérature scientifique relative à un dispositif médical existant est pertinente pour évaluer le comportement d'un nouveau produit sensiblement différent. Il faudrait donc définir clairement les critères permettant de l'apprécier et de la restreindre à une équivalence en lien direct entre deux dispositifs médicaux précis. Afin de garantir incontestablement le sérieux scientifique de cette voie d'évaluation et renforcer son bien-fondé, elle devrait être limitée aux cas où le dispositif médical auquel le fabricant prétend que son produit est équivalent a lui-même fait l'objet d'investigations cliniques avant sa commercialisation .
Proposition n° 20 :
Limiter
« l'effet domino » en définissant plus
précisément l'équivalence
pouvant être
acceptée entre deux dispositifs médicaux
pour satisfaire
à l'obligation d'évaluation clinique
Proposition n° 21 :
Imposer que le
dispositif médical auquel un nouveau produit est présenté
comme équivalent ait lui-même fait l'objet d'investigations
cliniques
Les mesures proposées par la mission d'information visent à mettre un terme à des pratiques et des comportements, de la part des fabricants, des organismes notifiés et des autorités nationales, qui ont eu pour conséquence de rendre quasi inopérantes, dans les faits, les dispositions de la réglementation européenne relatives à l'évaluation clinique des dispositifs médicaux implantables.
Profitant des imprécisions du texte et du peu d'empressement des certificateurs et des régulateurs à s'assurer de sa bonne application, les producteurs ont réussi jusqu'à présent à s'affranchir de leurs obligations en ayant massivement recours à la voie la moins contraignante qui, si elle peut être justifiée dans certaines situations spécifiques, devrait néanmoins rester exceptionnelle. C'est pourquoi, pour des raisons qui tiennent à la surveillance du marché, à la protection de la santé publique et au développement de l'expertise scientifique dans le domaine des dispositifs médicaux implantables, il est nécessaire de renforcer les contrôles et de clarifier les textes afin d'affirmer sans aucune ambiguïté que des essais cliniques scientifiquement crédibles et impartiaux doivent constituer une obligation préalable à la commercialisation des dispositifs médicaux implantables.
b) Assurer une meilleure évaluation post-inscription des dispositifs médicaux implantables remboursés en France
Pour l'instant, la principale forme d'évaluation conduite dans notre pays est celle faite par la CNEDiMTS, instance de la HAS, lorsque l'inscription d'un dispositif médical sur la LPP en vue d'obtenir sa prise en charge par l'assurance maladie est demandée. Elle mesure le rapport bénéfices-risques du dispositif en question par comparaison avec les stratégies thérapeutiques existantes, se fondant sur son service médical attendu (SMA). Toutefois, lorsque le dispositif médical doit être enregistré sous son nom de marque, en raison de son caractère innovant ou très spécifique, une étude clinique complète peut être menée à la demande de la CNEDiMTS et conditionner son remboursement.
Le récent accord-cadre entre le Comité économique des produits de santé (Ceps) et les organisations professionnelles des fabricants de dispositifs médicaux, signé le 16 décembre 2011, vient faciliter la réalisation de telles études. Comme l'a expliqué Jean-Luc Harousseau, président de la HAS, à la mission d'information, cet accord prévoit que, pour un dispositif médical, « quand un manque clinique sera constaté, une étude devra être mise en place dès son inscription. La CNEDiMTS devra ensuite réévaluer le produit » . Plus précisément, ses articles 10 à 15 donnent à la CNEDiMTS et au Ceps le droit de demander à un fabricant de mener une évaluation clinique complémentaire, sous l'égide d'un comité scientifique, dont les résultats doivent faire l'objet d'une publication. La même procédure est applicable lors du renouvellement de l'inscription d'un produit sur la LPP. Enfin, des études peuvent également être conduites sur des produits qui y sont inscrits sous description générique, sachant que la CNEDiMTS réévalue la cohérence et la pertinence thérapeutiques des lignes génériques tous les trois à cinq ans.
Entendu par la mission d'information, Jean-Michel Dubernard, président de la CNEDiMTS, a souligné la grande importance de cet accord-cadre, qui marque une nouvelle étape dans les relations entre les fabricants et les autorités chargées de déterminer si un dispositif médical est admissible au remboursement et quel doit être son prix. Ainsi, selon son article 19, « le choix entre l'inscription en nom de marque et l'inscription sous description générique relève exclusivement des ministres chargés de la santé et de la sécurité sociale sur proposition du Ceps après avis de la CNEDiMTS » . L'affirmation de ce principe général donne un pouvoir accru à ces deux organismes pour mettre au point une stratégie réfléchie d'évaluation des dispositifs médicaux implantables, qu'ils soient uniques et innovants ou fondus dans une ligne générique.
Il est évident qu'il ne sera pas possible de procéder, à brève échéance, à l'évaluation post-inscription de tous les dispositifs médicaux implantables présents sur le marché. Néanmoins, les dispositions de cet accord-cadre ainsi que celles de la loi « Médicament » 85 ( * ) sont des compléments bienvenus aux pouvoirs des autorités sanitaires afin qu'elles mettent en oeuvre une politique volontariste, sur le marché français, de suivi clinique des dispositifs médicaux, dans l'attente d'une modification de la réglementation européenne qui reste, sur ce sujet, trop imprécise.
c) Continuer à favoriser l'innovation
Les dispositifs médicaux évoluant au gré des changements technologiques et des avancées scientifiques, il est indispensable de soutenir l'innovation et de mettre en place des procédures spécifiques de financement de la recherche médicale et de prise en charge temporaire des produits les plus innovants. C'est la raison pour laquelle plusieurs programmes d'aide, intervenant aux différentes étapes de développement d'un dispositif médical, ont été mis en place par le législateur et le pouvoir réglementaire, sous l'égide du ministère de la santé.
Le programme de soutien aux techniques innovantes, coûteuses ou non (Pstic) , permet de tester les nouveaux produits de santé contre le standard existant et d'évaluer leur efficience, afin de valider leur utilité clinique et médico-économique. Cette procédure a donc lieu une fois que l'efficacité clinique a été démontrée. Un dispositif médical inscrit à ce programme doit disposer du marquage CE, et les résultats du Pstic ont vocation à faciliter l'évaluation qui doit ensuite être réalisée par la HAS pour permettre sa prise en charge par l'assurance maladie. En pratique, des appels à projet sont lancés annuellement afin de recueillir des candidatures, sélectionnées ensuite par le ministre chargé de la santé, après avis d'un comité d'experts. Les établissements de santé lauréats bénéficient ensuite d'un financement spécifique destiné à compenser le surcoût lié à l'innovation, sur une durée de vingt-quatre mois, dans le cadre des missions d'enseignement, de recherche, de référence et d'innovation (Merri).
A côté du Pstic, le « forfait innovation » prévu à l'article L. 165-1-1 du code de la sécurité sociale, conditionne la prise en charge temporaire, à titre dérogatoire, d'un produit innovant à la réalisation d'études cliniques. Dans le cas des dispositifs médicaux, il concerne ceux d'entre eux pour lesquels la HAS a jugé le service attendu insuffisant, en raison du manque de données disponibles. Les conditions générales de prise en charge par l'assurance maladie ne sont donc pas remplies. Néanmoins, si le dispositif innovant présente un intérêt potentiel, le forfait innovation constitue un moyen d'approfondir les recherches cliniques à son sujet tout en permettant son utilisation dans des établissements de santé prédéterminés. Contrairement au Pstic, il intervient à la suite de l'évaluation réalisée par la HAS. Le forfait innovation apporte un financement global prenant en charge à la fois l'acte, les frais d'hospitalisation et le produit en question. Son but est de démontrer que l'innovation concernée est efficace, sûre et présente une réelle utilité clinique.
Les conditions de recours au forfait innovation
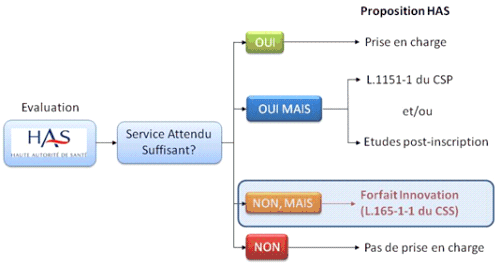 Source : Direction
générale de l'offre de soins
Source : Direction
générale de l'offre de soins
La France dispose donc d'outils de soutien à l'innovation de nature à couvrir les différents cas de figure qui peuvent se présenter lors du développement d'une technique nouvelle. Toutefois, la question des délais et de la mise à disposition des dispositifs médicaux les plus innovants aux praticiens et aux patients n'est pas encore résolue . Créé par la loi de financement de la sécurité sociale (LFSS) pour 2009 puis modifié par la loi « HPST », le forfait innovation n'a véritablement été opérationnel qu'à partir de 2011. Seulement deux dispositifs médicaux ainsi qu'un acte sont en phase d'examen. Il conviendrait donc d'accélérer sa mise en place et d'en élargir la portée.
* 85 Articles 35 et 37 de la loi n° 2011-2012 du 29 décembre 2011 relative au renforcement de la sécurité sanitaire du médicament et des produits de santé.