







II. DÉVELOPPER L'ACCÈS DES PATIENTS À L'INNOVATION EN PRENANT RÉSOLUMENT LE VIRAGE DE LA MÉDECINE PERSONNALISÉE
A. PERMETTRE À LA FRANCE DE RETROUVER SON ATTRACTIVITÉ POUR ACCUEILLIR LA RECHERCHE CLINIQUE
1. Moderniser l'examen des demandes d'essais cliniques
a) Un bilan en demi-teinte du raccourcissement des délais d'autorisation
Le 8 e CSIS comportait un engagement sur la réduction des délais d'autorisation des essais cliniques respectivement à 60 jours au niveau des CPP , 45 jours pour les médicaments et les dispositifs médicaux (DM) et dispositifs médicaux de diagnostic in vitro (DMDIV) et 110 jours pour les médicaments de thérapie innovante (MTI) au niveau de l'ANSM.
Cet engagement a été globalement respecté par l'ANSM, puisqu'en 2019, le délai moyen de décision sur tous les essais 33 ( * ) s'est établi à 46,5 jours. Ce délai s'est légèrement dégradé en 2020, à 51 jours, en raison de la charge de travail consécutive à la crise sanitaire. Toutefois, grâce à une priorisation et à la mise en oeuvre de procédures accélérées, sur 105 essais cliniques portant sur la covid-19, la durée moyenne de décision a été de 26 jours.
Les actions d'amélioration des délais d'autorisation mises en oeuvre par l'ANSM
L'ANSM a mis en place deux dispositifs de traitement rapide (« fast-track ») qui ont permis de réduire drastiquement les délais d'autorisation des essais cliniques répondant à des critères de priorité de santé publique, avec des délais inférieurs aux exigences règlementaires :
- le dispositif « fast-track 1 » - accès à l'innovation - permet un accès rapide pour les patients aux traitements innovants dans les essais (autorisation en 40 jours au maximum). Les critères d'éligibilité à ce dispositif sont les suivants : essais précoces en oncopédiatrie et hémato-pédiatrie, maladie rare, design complexe, MTI, soutien à l'innovation (c'est-à-dire suite d'une recherche du programme hospitalier de recherche clinique - PHRC, d'un centre labellisé de phase précoce - CLIP...) ;
- le dispositif « fast-track 2 » - soutien au développement - permet d'accélérer la mise en place des essais cliniques pour les molécules ou les associations de molécules déjà évaluées par l'ANSM (autorisation en 20 jours maximum). Les critères d'éligibilité à ce dispositif sont les suivants : molécule ou association de molécules déjà évaluées en France et dans la même indication que l'essai concerné, MTI.
Source : Agence nationale de sécurité du médicament et des produits de santé
Les progrès dans l'instruction des demandes d'essais cliniques par les CPP restent plus mesurés. Pour mémoire, les délais règlementaires pour les avis des CPP à compter de la date de recevabilité du dossier s'établissent à 45 jours pour des dossiers ne suscitant pas de questions du CPP et à 60 jours pour les dossiers pour lesquels le CPP adresse des demandes d'informations complémentaires au promoteur. Selon les données de la direction générale de la santé, le délai médian global des essais cliniques de médicaments, tous domaines confondus, s'établit à 77 jours sur les trois premiers mois de 2021. Néanmoins ce délai est calculé à partir de la date du tirage au sort pour la désignation du CPP, et non pas à compter de la date de réception du dossier complet conditionnant sa recevabilité, et comprend également le délai de réponse et de mise en conformité applicable au promoteur.
La conférence nationale des CPP (CNCPP) estime que le temps médian d'évaluation des CPP pourrait s'établir à 53 jours en retranchant le temps de réponse des promoteurs. En décomptant également de ce délai une dizaine de jours pour rendre le dossier complet et recevable, la CNPP considère que le délai médian serait ramené à un niveau proche du délai règlementaire de 45 jours pour les dossiers ne faisant pas l'objet de questions. Néanmoins, concernant plus spécifiquement les recherches en cancérologie, la CNCPP constate une dégradation du délai médian global d'évaluation à 79 jours au premier trimestre 2021, alors que le délai règlementaire d'évaluation prévoit une décision du CPP en 45 jours. À cela s'ajoute une augmentation du délai de réponse des promoteurs qui aggrave le délai global d'autorisation des essais.
Les actions d'amélioration des délais d'instruction des CPP
Un décret du 19 mars 2021 34 ( * ) est venu simplifier le fonctionnement des CPP :
- il ouvre la possibilité d'organiser des réunions par conférence téléphonique ou audiovisuelle pour tout ou partie des membres ;
- il tire les conséquences de la loi de 2018 sur la modulation du tirage au sort des CPP : le système d'information mis en place par la DGS ne met en concurrence que les CPP disponibles et disposant de la compétence nécessaire à l'examen du projet, ces critères devant être précisés par un arrêté. Lorsque le CPP désigné estime qu'il n'est pas en capacité de traiter le dossier, il peut demander au ministre de la santé dans un délai de deux jours de le renvoyer vers un autre CPP ;
- il impose au promoteur un délai maximal de douze jours pour répondre aux questions du CPP. Avec l'entrée en vigueur de cette disposition, à ce jour, sur 348 dossiers, 232 demandes ont dépassé le délai de douze jours et ont donc été réputées caduques.
À la suite d'un audit, la direction générale de la santé a lancé un plan « ambition CPP 2019-2022 » qui contribue à la formation des membres des CPP à l'utilisation du nouveau système d'information SIRIPH2G et à l'application des règlements européens sur les dispositifs médicaux. 11 CPP volontaires vont également être formés spécifiquement pour être les pilotes dans l'application de la procédure européenne d'évaluation des demandes pour les essais cliniques de médicaments selon le règlement européen. L'objectif est, par ailleurs, de doter chaque CPP d'1,5 équivalent temps plein (ETP) permanent, soit 58,5 ETP au total. Les CPP ont obtenu 850 000 euros supplémentaires pour le financement de 17 ETP d'assistants de recherche clinique, mais seuls cinq ou six ETP ont jusqu'ici été effectivement recrutés compte tenu des procédures de recrutement complexes au sein des hôpitaux. En outre, la loi de financement de la sécurité sociale pour 2021 35 ( * ) a procédé à une augmentation de la contribution sur le chiffre d'affaires des industriels pharmaceutiques dont le rendement supplémentaire doit être affecté au financement des CPP.
Source : Conférence nationale des comités de protection des personnes
b) Des essais cliniques précoces insuffisamment accueillis en France
L'accueil d'essais cliniques est un enjeu concurrentiel majeur. La mise en oeuvre en France d'essais précoces est déterminante pour inciter un industriel à poursuivre le développement clinique et la production d'une thérapie innovante sur notre territoire.
Or, en dépit de progrès observés dans la période récente, la France peine encore à se positionner en première ligne pour attirer des essais de phase précoce. Selon les données du Leem 36 ( * ) , la France se situait, au premier semestre 2019, au 4 e rang européen en participant à 12,9 % des essais mondiaux, derrière l'Allemagne (16 %) et le Royaume-Uni et l'Espagne (14,5 % chacun), mais occupait le 5 e rang européen pour l'accueil d'essais de phase 1 en n'accueillant que 5 % de ces essais.
L'ANSM met toutefois en avant une augmentation du nombre d'essais précoces autorisés en France au cours des dernières années, même si notre pays reste sérieusement concurrencé par d'autres pays européens. De 2018 à 2020, la part de la France dans les autorisations d'essais précoces innovants en oncologie s'établit à 21 %, soit un niveau supérieur à l'Allemagne (18 %) et la Belgique (13 %), mais derrière l'Espagne (26 %) et le Royaume-Uni (22 %).
Demandes d'autorisation d'essai clinique enregistrées par l'ANSM
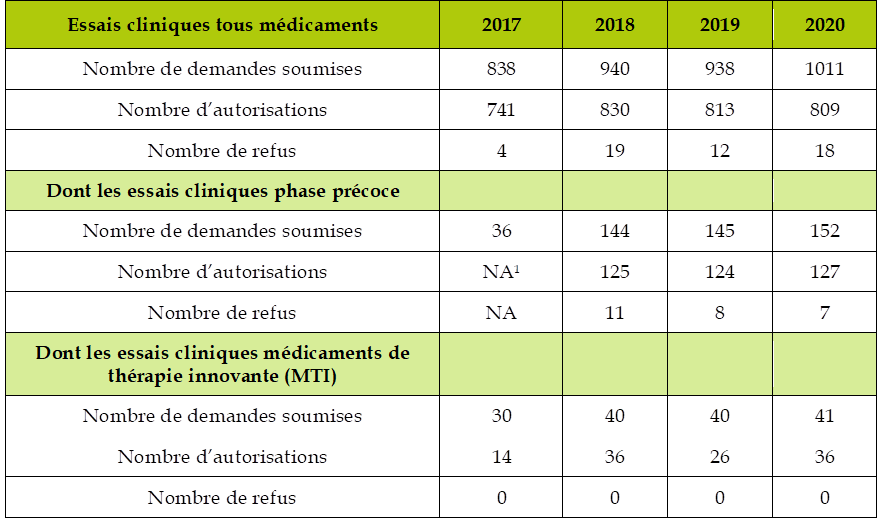
Source : Agence nationale de sécurité du médicament et des produits de santé
Certains groupes pharmaceutiques privilégient clairement d'autres pays que la France pour l'accueil d'essais cliniques. Aucun essai de phase 1 ou 2 n'a ainsi été lancé en France par la biotech française Valneva. Son PDG, M. Franck Grimaud, regrette une attitude très théorique et insuffisamment pragmatique des instances responsables de l'instruction des demandes d'essais cliniques dans l'évaluation des bénéfices-risques. Lors de son audition par la commission 37 ( * ) , il a pointé un problème culturel en France dans l'approche des demandes d'essais cliniques : « l'innovation dans le domaine des biotechnologies repose forcément sur de nouvelles approches, de nouveaux types de médicament, et emprunte souvent un nouveau mode de “ delivery ”. Par essence, la plupart des innovations en biotechnologie sortent des cases, de ce qui est déjà connu. Or, en France mais pas seulement, on privilégie une approche “ to the book ” : si on ne rentre pas dans les cases, dans des choses déjà décrites, on demande un niveau de protection maximal, alors que d'autres pays adoptent une approche plus pragmatique du rapport bénéfices-risques. »
c) Une expertise parfois critiquée
Nombreux sont en effet les promoteurs qui déplorent une certaine frilosité de la part des experts mobilisés par l'ANSM ou les CPP qui manqueraient de pragmatisme dans l'évaluation du rapport bénéfices-risques en suivant une approche essentiellement théorique et monodimensionnelle. Cette frilosité résulterait :
- d'une insuffisante connaissance des nouveaux mécanismes des innovations de rupture qui peut conduire les experts à faire preuve d'une aversion disproportionnée au risque ;
- des contraintes liées à la prévention des conflits d'intérêts qui disqualifieraient nombre d'experts disposant d'une expérience au sein de l'industrie. Cette conception rigide ne ferait qu'aggraver l'éloignement des experts « officiels » de l'ANSM et des CPP vis-à-vis de l'expertise développée dans les territoires qui se nourrit des interactions entre la recherche académique et l'industrie.
Dans un article de 2018 38 ( * ) , le professeur Didier Truchet souligne qu'« on a trop tendance en pratique à confondre liens d'intérêts et conflits d'intérêts, alors pourtant que les textes font la distinction », en rappelant qu'un lien d'intérêts, qui doit être déclaré, « peut conduire à écarter l'expert ou ce dernier à se déporter, mais sous cette réserve, ne le disqualifie normalement pas, en particulier en présence d'une pluralité d'experts ».
À cet égard, la commission plaide pour un renforcement des capacités de la commission nationale des recherches impliquant la personne humaine (CNRIPH) afin de constituer un annuaire des experts mobilisables dans différentes aires thérapeutiques à la disposition des CPP et de mener un benchmark de la gestion des liens d'intérêts dans les autres pays pour en tirer un guide des bonnes pratiques . Elle préconise également la mise en place d'un déontologue de l'expertise sanitaire au sein de la CNRIPH qui permettrait de conseiller les CPP dans leur recours aux experts, dans un souci de préservation des principes d'impartialité, de transparence, de pluralité et du contradictoire de l'expertise sanitaire 39 ( * ) .
d) Des CPP embolisés par l'examen des études observationnelles
Le nombre de recherches examinées par les CPP augmente en moyenne de 8 % par an. En dix ans, le nombre de demandes d'autorisation de recherches impliquant la personne humaine (RIPH) a doublé, pour atteindre 3 987 demandes initiales en 2020 . Cette augmentation est en grande partie alimentée par les demandes portant sur les RIPH de catégorie 3, correspondant aux études observationnelles. Globalement, plus de 35 % des dossiers examinés par les CPP en 2020 étaient des RIPH de catégorie 3, contre 26 % de recherches sur les médicaments et 12 % de recherches sur les dispositifs médicaux.
Répartition des demandes d'autorisation de RIPH par catégories
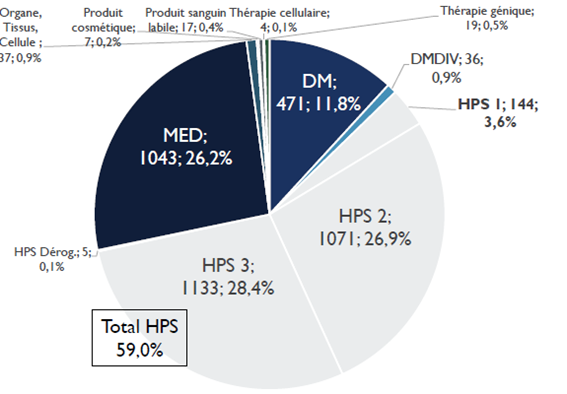
Source : Conférence nationale des comités de protection des personnes
Nombre de RIPH toutes catégories confondues
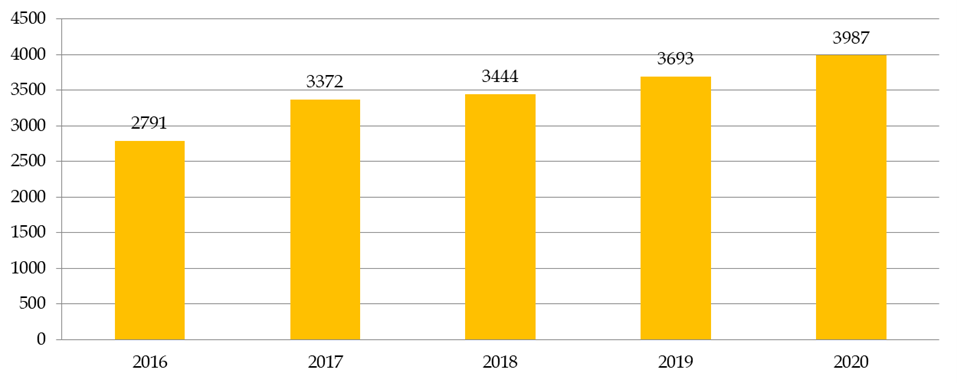
Source : Commission des affaires sociales du Sénat, d'après la direction générale de la santé
Les principales aires thérapeutiques concernées par les RIPH
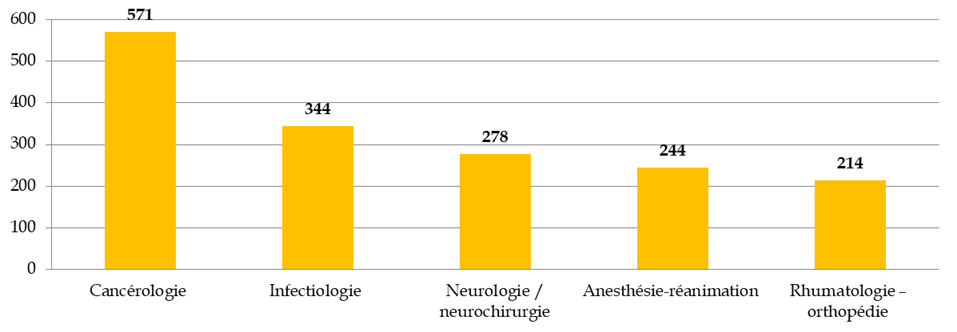
Source : Commission des affaires sociales du Sénat, d'après la direction générale de la santé
Les CPP, au nombre de 39 sur le territoire, ne disposent pas des moyens humains, financiers et techniques pour absorber une telle charge de travail. La loi d'accélération et de simplification de l'action publique 40 ( * ) a, certes, simplifié les procédures applicables aux recherches non interventionnelles ne portant pas sur des produits de santé (HPS) auprès des CPP 41 ( * ) . Cette mesure, encore insuffisamment connue des promoteurs, peine à produire ses effets. Elle ne comprend, du reste, pas d'automaticité pour l'obtention d'un avis favorable du CPP sur des protocoles dont la méthodologie a pourtant déjà été validée par la commission nationale de l'informatique et des libertés (CNIL).
La proportion des RIPH de catégorie 3 a, en outre, vocation à augmenter, les fabricants de dispositifs médicaux étant appelés à réaliser des recherches post-commercialisation. La commission estime donc indispensable d'alléger la charge de travail des CPP pour leur permettre de se concentrer sur l'examen des projets de recherches interventionnelles, en transférant l'examen des RIPH de catégorie 3 à un seul comité d'éthique qui serait spécialisé sur les recherches non interventionnelles et la protection des données de santé personnelles, ainsi que le préconise la présidente de la commission des affaires sociales, Mme Catherine Deroche, dans une proposition de loi déposée en 2019 42 ( * ) .
Outre cette mesure, la commission appelle également à moderniser le fonctionnement des CPP en mettant en oeuvre les mesures déclinées dans la proposition ci-dessous.
Proposition n° 11
: moderniser le fonctionnement des CPP par :
- le transfert de
l'examen des RIPH de catégorie 3 à un comité
d'éthique spécialisé sur les recherches non
interventionnelles et la protection des données de santé
personnelles ;
- le renforcement de la
formation de leurs membres aux mécanismes des innovations de
rupture ;
- la constitution, au plus tard avant la fin 2021,
sous l'égide de la CNRIPH, d'un annuaire d'experts selon
différentes aires thérapeutiques à la disposition des CPP,
avec publication de leurs déclarations d'intérêts ;
- l'assouplissement, sous le contrôle de la
CNRIPH qui serait dotée d'un déontologue de l'expertise
sanitaire, des conditions de mobilisation de l'expertise pertinente en
s'appuyant, à cet égard, sur un
benchmark
des pratiques
dans d'autres pays européens en matière de prévention des
conflits d'intérêts ;
- la rénovation
des modes d'indemnisation des membres et experts des CPP pour valoriser leur
engagement, notamment en reconnaissant l'investissement des présidents
et vice-présidents
43
(
*
)
;
- la mise en place d'un
audit indépendant et périodique des CPP conditionnant le
renouvellement de leur agrément.
La commission appelle également à clarifier la situation des études portant sur la personne humaine mais n'ayant pas de finalité biologique ou médicale , dites « études hors loi Jardé 44 ( * ) ». Ces recherches, qui nourrissent pour partie les sciences humaines et sociales, restent l' angle mort de l'encadrement de la recherche sur la personne humaine , alors que certaines d'entre elles peuvent présenter des risques éthiques pour les personnes 45 ( * ) . À l'heure actuelle, ces recherches sont examinées par des comités d'éthique de la recherche (CER) qui ne font l'objet d'aucun encadrement juridique et dont les pratiques restent très hétérogènes. En outre, par souci de sécurisation juridique, les études impliquant la collecte de nouvelles données et supposant le consentement des personnes à l'utilisation de ces données empruntent bien souvent la voie des RIPH de catégorie 3 et sont alors envoyées pour avis aux CPP, ce qui tend à augmenter d'autant la charge de travail de ces derniers.
Proposition n° 12 : inscrire dans la loi un statut des comités d'éthique de la recherche chargés d'examiner les protocoles de recherche n'ayant pas de finalité biologique et médicale et clarifier les méthodologies de référence applicables à ces recherches.
e) Prendre le virage des essais adaptatifs et des études post-commercialisation
La commission insiste sur l'importance du développement des essais adaptatifs et des études post-commercialisation , après obtention de l'autorisation de mise sur le marché (AMM). Les essais cliniques traditionnels, par définition, procèdent à une évaluation de l'efficacité d'un médicament ou d'une thérapie selon plusieurs biais, notamment les indications visées ou les caractéristiques des patients recrutés, et ne prévoient pas la possibilité de modifier le protocole en cours de mise en oeuvre.
Or, dans une approche de l' innovation au service de l'amélioration continue des soins , la France doit prendre le virage des essais adaptatifs et de l'évaluation en vie réelle. La crise sanitaire a été un facteur d'accélération dans la diffusion des méthodologies adaptatives : les essais « Recovery », au Royaume-Uni, et « Discovery », en France, ont prévu la possibilité d'abandonner en temps réel des traitements expérimentaux apparus inefficaces et de les remplacer, le cas échéant, par d'autres thérapies en fonction de l'évolution des connaissances.
La collecte de données en vie réelle , après la commercialisation d'un produit, permet, en outre, de disposer de données plus robustes pour objectiver l'apport d'un médicament dans la prise en charge des patients mais aussi pour l'efficience du système de santé, en contribuant à l'analyse médico-économique.
Proposition n° 13 : prendre le virage des essais adaptatifs, des études post-AMM et de la collecte de données en vie réelle dans un objectif d'amélioration continue de la qualité des soins.
Une partie significative des médicaments innovants ou des nouvelles indications thérapeutiques pour des médicaments existants résultera, à l'avenir, de la mise en corrélation d'une grande variété de données sur les effets d'une molécule. L'accès aux données du Health Data Hub , qui devrait permettre de disposer d'une multitude de points de mesure, représente une opportunité de réduction des coûts non négligeable pour la conception et l'organisation d'essais cliniques.
La méthodologie de référence MR004 établie par la CNIL, ne requérant qu'un simple engagement de conformité au référentiel, a d'ailleurs permis de faciliter la réutilisation de données issues d'une recherche antérieure ou du soin dans le cadre de recherches translationnelles ou du partage de données avec un chercheur académique ou un industriel. En revanche, la fédération nationale des centres de lutte contre le cancer Unicancer a relevé une lourdeur administrative dans l'appariement de ces données avec celles du Health Data Hub , dès lors que la procédure requiert une autorisation de la CNIL après avis favorable du comité éthique et scientifique pour les recherches, les études et les évaluations dans le domaine de la santé (Cesrees).
Pour mémoire, l' article 18 du projet de loi relatif à la bioéthique permet, par dérogation à l'exigence d'un consentement exprès et préalable, de réutiliser des échantillons humains prélevés initialement à d'autres fins afin de réaliser des examens génétiques à la condition que la personne concernée ait été dûment informée de l'utilisation potentielle de ses échantillons dans le cadre d'un programme de recherche 46 ( * ) , et qu'elle n'a pas exprimé son opposition. Ce mécanisme pourrait utilement être étendu à la réutilisation des données collectées à l'occasion de soins ou d'essais cliniques : le patient serait alors informé de la possibilité de s'opposer à la réutilisation de ses données dans le cadre de recherches s'inscrivant dans un programme de recherche dans le domaine thérapeutique concerné.
Proposition n° 14 : mettre en place une procédure accélérée ( fast-track ) pour permettre l'accès des chercheurs et des promoteurs et investigateurs d'essais cliniques aux données de santé du Health Data Hub pour évaluer l'ensemble des effets d'une thérapeutique.
Proposition n° 15 : étudier la faisabilité de la transposition du dispositif de passerelles soin/recherches de l'article 18 du projet de loi relatif à la bioéthique à la réutilisation de données de santé dans le cadre de programmes de recherche.
Enfin, la crise sanitaire a montré que l'accès aux données de santé constitue une véritable carte à jouer pour attirer des essais cliniques . En témoignent la mise en place au Royaume-Uni d'un registre national de volontaires pour participer à des essais cliniques sur des vaccins ou des traitements contre la covid-19 ou encore l'accès du laboratoire Pfizer aux données de santé des personnes vaccinées en Israël.
La commission appelle à saisir cette opportunité afin d'attirer des essais cliniques, notamment de phase précoce, en mettant en place des registres nationaux de volontaires sur de grands enjeux thérapeutiques (cancers à pronostic sombre, VIH, maladies infectieuses ne disposant pas de traitement ou de vaccin comme Zika, la dengue ou Lyme...).
Proposition n° 16 : créer des registres nationaux de candidats à des essais cliniques sur de grands enjeux thérapeutiques.
2. Rénover le modèle de financement de la recherche clinique
Les missions d'enseignement, de recherche, de référence et d'innovation 47 ( * ) (Merri), censées rétribuer l'investissement des hôpitaux dans la recherche, ne sont en réalité pas vues par ces derniers comme une dotation propre dédiée à leur activité de recherche mais plutôt comme une compensation de la perte de recettes liée au fait que leurs équipes se consacrent à des projets de recherche et non à des activités rémunérées dans le cadre de la tarification à l'activité (T2A).
À cela s'ajoute une approche du financement de la recherche à l'hôpital qui reste centrée sur le volume de publications scientifiques, et apparaît insuffisamment tournée vers la valeur ajoutée de la recherche clinique, qui devrait être appréciée à l'aune des applications industrielles potentielles ou des sauts thérapeutiques ou organisationnels poursuivis. Bien qu'il ait été rénové en avril 2021, le modèle de fixation de la dotation des Merri est ainsi articulé autour de trois enveloppes : l'enveloppe « publications » (61 % de la dotation), l'enveloppe « recherches-inclusions » (15 %) et l'enveloppe « enseignement » (24 %) 48 ( * ) . En conséquence, une part non négligeable de la recherche clinique hospitalière est uniquement accompagnée par les appels à projets et non par les crédits récurrents des Merri.
La commission recommande, dès lors, de revaloriser l'enveloppe « recherches-inclusions » au sein des Merri à hauteur de 25 % de la dotation socle - soit une augmentation de dix points. Cette enveloppe doit également permettre de soutenir les projets d'innovation organisationnelle, notamment en télémédecine et en télésurveillance.
Proposition n° 17 : augmenter l'enveloppe « recherches-inclusions » au sein des Merri à hauteur de 25 % de la dotation socle.
Lors de son audition par la commission, M. Franck Mouthon, président de France Biotech, s'est félicité des progrès réalisés dans la généralisation de la convention unique , mise en place à la suite du CSIS de 2013, qui permet de facturer à l'industriel, dans le cadre d'un seul contrat, les surcoûts engendrés pour les établissements de santé dans l'organisation d'un essai clinique. Il a néanmoins relevé une faiblesse dans l' établissement de l'annexe financière de la convention qui requiert bien souvent une longue négociation avec plusieurs établissements.
Selon des données du Leem, le délai médian pour la conclusion de la convention unique s'est établi à 76 jours en 2018 et 70 jours en 2019, un délai qui s'ajoute aux délais d'autorisation des essais par l'ANSM et les CPP : en moyenne, 204 jours sont ainsi nécessaires en France pour inclure le premier patient depuis le dépôt de la demande d'essai auprès de l'ANSM, contre un objectif cible de 120 jours . La part des essais ayant franchi l'étape de la convention unique dans des délais inférieurs à ce qui serait nécessaire pour répondre à l'objectif cible a stagné à 21 % en 2019, alors que la part des essais ayant franchi l'étape de l'ANSM et celle des CPP dans des délais infra-règlementaires a plus nettement progressé 49 ( * ) .
Proposition n° 18 : confier aux fédérations d'établissements de santé et aux organisations représentatives des promoteurs le soin d'établir des référentiels de coûts et de surcoûts et des méthodologies de calcul afin d'accélérer la négociation de l'annexe financière de la convention unique.
Enfin, la commission plaide pour une simplification du bénéfice du CIR pour couvrir l'ensemble des études et démarches associées au lancement d'un essai clinique . À l'heure actuelle, seules les étapes de conception et de méthodologie des essais cliniques, et d'analyse et de publications ouvrent droit au CIR. L'extension du CIR aux étapes de faisabilité et de mise en place d'un essai clinique pourrait ainsi constituer un facteur puissant d'attractivité .
Proposition n° 19 : étendre l'éligibilité au CIR à l'ensemble des étapes de faisabilité (démarches pour trouver des patients, évaluation de la disponibilité des centres, monitoring ...) et de mise en place des essais cliniques (choix du pays où se dérouleront les essais, démarches règlementaires et d'éthique, formation des personnels des centres...).
* 33 Qu'il s'agisse d'essais sur des médicaments, des MTI ou d'essais hors produits de santé.
* 34 Décret n° 2021-301 du 19 mars 2021 modifiant certains articles du titre II du Livre I er de la première partie du code de la santé publique (partie règlementaire) relatif aux recherches impliquant la personne humaine.
* 35 Loi n° 2020-1576 du 14 décembre 2020 de financement de la sécurité sociale pour 2021.
* 36 http://www.leem.org/recherche-et-developpement .
* 37 Table ronde sur l'innovation en santé du 2 juin 2021
* 38 Didier Truchet, « Déontologie des experts en santé, perspectives critiques », Revue de droit sanitaire et social , Sirey, Dalloz, 2018.
* 39 Principes énumérés à l'article L. 1452-1 du code de la santé publique, dans sa rédaction résultant de la loi n° 2011-2012 du 29 décembre 2011 relative au renforcement de la sécurité sanitaire du médicament et des produits de santé.
* 40 Article 31 de la loi n° 2020-1525 du 7 décembre 2020 d'accélération et de simplification de l'action publique.
* 41 En prévoyant la remise d'un dossier comprenant des attestations de conformité à la règlementation et aux référentiels de la commission nationale de l'informatique et des libertés (CNIL) sur les traitements de données.
* 42 Proposition de loi relative à l'évaluation éthique de la recherche impliquant la personne humaine, déposée au Sénat le 7 novembre 2019.
* 43 Qui jouent un rôle déterminant dans l'animation des CPP et la fixation de leur ordre du jour.
* 44 Loi n° 2012-300 du 5 mars 2012 relative aux recherches impliquant la personne humaine.
* 45 Notamment des recherches en neurologie ou sciences cognitives, comme les recherches relatives à l'« homme augmenté ».
* 46 Plus large qu'un protocole de recherche, il est défini comme un ensemble d'activités de recherche organisées en vue de faciliter et d'accélérer les découvertes dans un domaine scientifique déterminé.
* 47 Dont la dotation est incluse dans les missions d'intérêt général d'aide à la contractualisation (Migac).
* 48 Ministère des solidarités et de la santé, « Évolution du modèle de répartition de la dotation socle au titre des Merri », avril 2021.
* 49 87 % en 2019 contre 54 % en 2018 pour l'étape « ANSM » et 33 % en 2019 contre 25 % en 2018 pour l'étape « CPP ».