







3. Les responsabilités en matière d'évaluation, de contrôle et de surveillance des dispositifs médicaux
Les directives de 1990 et 1993 permettent d'identifier quatre acteurs concernés par la mise en oeuvre de la réglementation relative aux dispositifs médicaux :
- les fabricants ;
- les organismes notifiés ;
- les autorités compétentes , communautaires et nationales, chargées de veiller au respect de la réglementation et, le cas échéant, de son adaptation ;
- les utilisateurs (professionnels de santé, patients, tiers).
a) Les organismes notifiés évaluent les niveaux de performance et de sécurité des dispositifs médicaux
L'article 1 er de la directive du 14 juin 1993 définit, pour la première fois, la notion de fabricant d'un dispositif médical. Il s'agit de « la personne physique ou morale responsable de la conception, de la fabrication, du conditionnement et de l'étiquetage d'un dispositif en vue de sa mise sur le marché en son propre nom, que ces opérations soient effectuées par cette même personne ou pour son compte par une tierce personne » .
Cette définition privilégie la responsabilité du fabricant sur l'ensemble du cycle de vie du produit, au-delà de sa seule activité de production.
Il doit veiller à « réduire [...] le risque d'une erreur d'utilisation due aux caractéristiques ergonomiques du dispositif et à l'environnement dans lequel le dispositif doit être utilisé (conception pour la sécurité du patient) » et à « prendre en compte les connaissances techniques, l'expérience, l'éducation et la formation et [...] l'état de santé et la condition physique des utilisateurs auxquels les dispositifs sont destinés (conception pour les utilisateurs profanes, professionnels, handicapés ou autres) » . 13 ( * )
Le fabricant ou son mandataire doit, ainsi, « notifier aux autorités compétentes de l'Etat membre dans lequel il a son siège social, l'adresse de son siège social ainsi que la désignation des dispositifs concernés » 14 ( * ) .
Les Etats membres notifient auprès de la Commission européenne les organismes chargés d'évaluer les performances et le respect des caractéristiques de sécurité par les dispositifs médicaux.
Les organismes notifiés ont pour mission d'évaluer la conformité du dispositif médical sur la base d'un dossier technique établi par le fabricant. Ils sont conduits à mettre en oeuvre, dans le cadre de vérifications sur pièces, des procédures de certification et des audits des systèmes de gestion de la qualité.
Contrairement aux Etats-Unis où la responsabilité de l'ensemble de la régulation du secteur des dispositifs médicaux incombe à un seul organe, la Food and Drug Administration (FDA), l'octroi du marquage CE, condition de la commercialisation sur le marché européen, est réservé en Europe aux organismes notifiés, en règle générale sociétés de droit privé à but lucratif, seuls responsables de l'évaluation des données techniques et cliniques communiquées par le fabricant .
Les Etats membres de l'Union européenne, ainsi que les Etats de l'Espace économique européen et la Turquie, disposent en pratique d'un haut degré de liberté dans la désignation des organismes notifiés. Il existe donc potentiellement plus de vingt-sept systèmes nationaux de désignation et de surveillance de ces organismes. Dans ces conditions, un niveau uniforme d'évaluation et de sécurité des dispositifs médicaux n'est pas garanti au sein de l'Union européenne.
La liste des organismes notifiés comprend également des établissements situés en Suisse et en Turquie, dans la mesure où la réglementation européenne concerne des pays associés au marché commun européen, et en Australie, en application d'accords de reconnaissance mutuelle.
Certains critères communs dans la désignation par les Etats membres des organismes notifiés ont été élaborés à partir des directives relatives aux dispositifs médicaux et de la décision n° 768/2008/CE 15 ( * ) relative à un cadre commun pour la commercialisation des produits. Les critères principaux retenus par les directives sont :
- l'indépendance ;
- l'impartialité ;
- une expertise dans le domaine des dispositifs médicaux ;
- un personnel scientifique en nombre suffisant et doté d'une expérience et de connaissances suffisantes pour évaluer, sur le plan médical, le caractère fonctionnel et les performances des dispositifs ;
- du matériel et des locaux adaptés ;
- une bonne formation professionnelle portant sur l'ensemble des opérations d'évaluation et de vérification pour lesquelles l'organisme est désigné ;
- une connaissance satisfaisante des prescriptions relatives aux contrôles qu'il effectue ;
- une pratique suffisante des contrôles, l'aptitude requise pour rédiger les attestations, procès-verbaux et rapports qui constituent la matérialisation des contrôles effectués et une assurance de responsabilité civile.
La Commission européenne a également développé des lignes directrices (« Guidelines ») concernant la désignation et la surveillance des organismes notifiés qui sont plus complètes que les directives. Elles n'ont pas de caractère contraignant pour les Etats membres . Ces derniers doivent également s'assurer que les organismes notifiés maintiennent un niveau élevé de compétences. Toutefois, il n'existe pas de procédure de contrôle harmonisée au niveau communautaire : il revient aux Etats membres seuls de choisir les moyens et les méthodes des contrôles à mettre en oeuvre .
Lorsqu'il a connaissance d'un problème concernant un organisme notifié qui cesse de remplir les critères précités, l'Etat membre concerné doit retirer ou suspendre la notification de l'organisme après l'en avoir averti. L'information doit alors être communiquée à la Commission européenne ainsi qu'aux autres Etats membres. Le retrait de la notification n'affecte pas les certificats délivrés par l'organisme notifié tant que la preuve n'est pas apportée qu'ils doivent également faire l'objet d'un retrait.
Il n'est pas possible d'établir une carte européenne de la qualité des organismes notifiés : les prothèses mammaires PIP ont été certifiées par l'organisme allemand TÜV Rheinland et les prothèses de hanche métal-métal fabriquées par la société américaine DePuy, aujourd'hui retirées du marché, par l'organisme britannique BSI. Ils jouissent tous deux d'une excellente réputation auprès des régulateurs européens et des fabricants. De plus, ils font partie des huit organismes notifiés ayant obtenu l'homologation du « Canadian Medical Devices Conformity Assessment System » (CMDCAS) permettant d'autoriser l'entrée de dispositifs sur le marché canadien 16 ( * ) .
En France, le seul organisme habilité par l'Agence nationale de sécurité du médicament et des produits de santé (ANSM, ex-Afssaps) et notifié auprès de la Commission européenne est la structure G-Med , créée en 1994 et intégrée au laboratoire national de métrologie et d'essais (LNE). Le LNE, dont l'origine remonte à 1901, dispose du statut d'établissement public à caractère industriel et commercial (Epic).
b) Les autorités compétentes des Etats membres sont chargées de surveiller le marché national et les organismes notifiés
Il appartient aux autorités compétentes des Etats membres de veiller à ce que le marché national soit conforme aux exigences des directives. A ce titre, l'ANSM est chargée, en France :
- d'assurer la mise en oeuvre de la procédure de vigilance visée à l'article 8 de la directive du 20 juin 1990 et à l'article 10 de la directive du 14 juin 1993 prévoyant l'enregistrement dans un système centralisé des informations concernant les incidents liés à un dispositif ;
- de procéder à des enquêtes périodiques ou inopinées , en particulier lorsqu'elle reçoit d'utilisateurs - professionnels de santé ou patients - des informations concernant d'éventuels dysfonctionnements liés à un dispositif médical.
Dans le cadre de la surveillance du marché des dispositifs médicaux, l'action de l'ANSM est :
- soit réactive, en mettant en oeuvre les décisions administratives ou de police sanitaire requises en réponse à un signal d'alerte ;
- soit proactive, en sélectionnant chaque année des types de dispositifs médicaux qu'elle souhaite examiner, sur la base de critères précis. Elle tient compte de l'évolution très rapide des marchés, notamment en cas de saut technologique substantiel nécessitant de vérifier de manière globale la qualité des nouveaux dispositifs. Dans la période récente, l'ANSM s'est ainsi, par exemple, intéressée aux prothèses dentaires dont l'importation s'est considérablement développée.
Dans l'exercice de son contrôle, l'ANSM déploie trois modalités d'intervention susceptibles de se combiner : soit en examinant sur dossier , c'est-à-dire en demandant au fabricant de lui soumettre son dossier de marquage CE ; soit en inspectant l'entreprise sur site ; soit en effectuant un contrôle en laboratoire . Cette dernière possibilité est la plus compliquée à mettre en oeuvre, certains dispositifs (tel le gel de silicone) se prêtant mieux que d'autres (tel le stimulateur cardiaque) à des tests en laboratoire.
Principales étapes du processus de certification des dispositifs médicaux en Europe
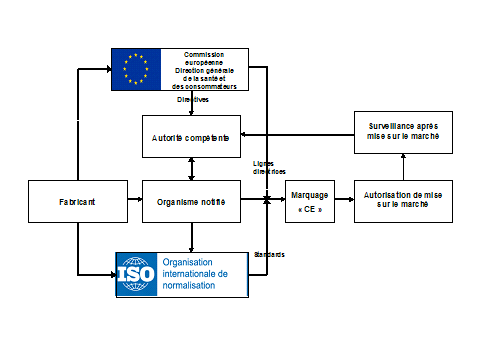
Les flèches pleines représentent les exigences formelles, les flèches en pointillés représentent les lignes directrices ou les conseils prodigués.
Source : « Clinical evaluation of
cardiovascular devices:
principles, problems, and proposals for European
regulatory reform »
in European Heart Journal Advance Access,
14 mai 2011.
* 13 Annexe 1 de la directive 1993/42/CEE du Conseil du 14 juin 1993.
* 14 L'article 14 de cette même directive prévoit l'« enregistrement des personnes responsables pour la mise des dispositifs sur le marché ».
* 15 Décision n° 768/2008/CE du Parlement européen et du Conseil du 9 juillet 2008 relative à un cadre commun pour la commercialisation des produits et abrogeant la décision 93/465/CEE du Conseil.
* 16 Ces huit organismes sont : BSI (Royaume-Uni), Kema (Pays-Bas), LGA Intercert (Allemagne), LNE/G-Med (France), NSAI (Irlande), TÜV Rheinland (Allemagne), TÜV Süd (Allemagne), et TÜV Nord (Allemagne).