







Rapport n° 237 (2019-2020) de Mmes Corinne IMBERT , Muriel JOURDA , MM. Olivier HENNO et Bernard JOMIER , fait au nom de la commission spéciale, déposé le 8 janvier 2020
Disponible au format PDF (6,1 Moctets)
Tableau comparatif au format PDF (2 Moctets)
Synthèse du rapport (250 Koctets)
-
L'ESSENTIEL
-
A. RÉPONDRE À DES ATTENTES
SOCIÉTALES EN MATIÈRE DE PROCRÉATION DANS LE RESPECT DES
PRINCIPES FONDAMENTAUX DE NOTRE DROIT
-
1. Ouvrir l'accès à l'assistance
médicale à la procréation aux couples de femmes et
aux femmes non mariées sans revenir sur le cadre existant pour les
couples infertiles
-
2. Autoriser, sans la décourager,
l'autoconservation de gamètes à des fins de
prévention
-
3. Permettre l'accès aux données
personnelles du donneur par les personnes nées de dons, quelle que
soit leur date de naissance et dans le respect de la vie
privée
-
1. Ouvrir l'accès à l'assistance
médicale à la procréation aux couples de femmes et
aux femmes non mariées sans revenir sur le cadre existant pour les
couples infertiles
-
B. FAVORISER UNE POLITIQUE AMBITIEUSE DE DON
D'ORGANES, DE TISSUS OU DE CELLULES
-
C. POUR UNE GÉNÉTIQUE ÉTHIQUE
ET RESPONSABLE : TROUVER LE POINT D'ÉQUILIBRE ENTRE
LIBERTÉ INDIVIDUELLE, ACCOMPAGNEMENT MÉDICAL ET
SOLIDARITÉ
-
D. ACCOMPAGNER UNE RECHERCHE LIBRE ET RESPONSABLE
AU SERVICE DE LA SANTÉ
-
E. GARANTIR LA SÉCURITÉ ET LE
CARACTÈRE ÉTHIQUE DE CERTAINES PRATIQUES ET TECHNOLOGIES
MÉDICALES
-
1. Rénover le cadre juridique du diagnostic
prénatal
-
2. Sécuriser les pratiques d'interruption de
grossesse pour motif médical et de réduction embryonnaire ou
foetale
-
3. Autoriser, en l'encadrant strictement, le
recours au diagnostic préimplantatoire
-
4. Mieux encadrer le recours à
l'intelligence artificielle et aux neurosciences dans le domaine de la
santé
-
1. Rénover le cadre juridique du diagnostic
prénatal
-
F. ADAPTER LA GOUVERNANCE BIOÉTHIQUE AU
RYTHME DES PROGRÈS SCIENTIFIQUES ET TECHNOLOGIQUES
-
A. RÉPONDRE À DES ATTENTES
SOCIÉTALES EN MATIÈRE DE PROCRÉATION DANS LE RESPECT DES
PRINCIPES FONDAMENTAUX DE NOTRE DROIT
-
EXAMEN DES ARTICLES
-
TITRE 1ER
ÉLARGIR L'ACCÈS AUX TECHNOLOGIES
DISPONIBLES SANS S'AFFRANCHIR
DE NOS PRINCIPES ÉTHIQUES
-
CHAPITRE IER
Permettre aux personnes d'exercer un choix éclairé
en matière de procréation dans un cadre maîtrisé
-
Article premier
Ouverture de l'accès à l'assistance médicale
à la procréation aux couples de femmes et aux femmes non mariées
-
Article 1er bis
Rapport au Parlement sur la structuration
des centres d'assistance médicale à la procréation
-
Article 2
Assouplissement du don de gamètes
et autorisation de leur autoconservation
-
Article 2 bis
Définition, par arrêté ministériel,
de mesures de lutte contre les causes d'infertilité
-
Article premier
-
CHAPITRE II
Reconnaître et sécuriser les droits des enfants
nés d'assistance médicale à la procréation
-
Article 3
Droit des personnes nées d'une assistance médicale
à la procréation avec tiers donneur d'accéder à certaines données
non identifiantes et à l'identité du donneur à leur majorité
-
Article 4
Établissement de la filiation des enfants nés du recours
à une assistance médicale à la procréation
avec tiers donneur par un couple de femmes
-
Article 4 bis (nouveau)
Interdiction de la transcription totale d'un acte de naissance
ou d'un jugement étranger établissant la filiation d'un enfant
né d'une gestation pour autrui lorsqu'il mentionne le parent d'intention
-
Article 3
-
TITRE II
PROMOUVOIR LA SOLIDARITÉ
DANS LE RESPECT DE L'AUTONOMIE DE CHACUN
-
CHAPITRE IER
Conforter la solidarité dans le cadre
du don d'organes, de tissus et de cellules
-
Article 5 A (nouveau)
Statut de donneur d'organes
-
Article 5
Extension du don croisé d'organes
-
Article 5 bis
Extension de l'information sur le don d'organes
prévue par le code de la santé publique aux patients de 16 ans et plus
-
Article 6
Possibilité de prélever des cellules souches hématopoïétiques
sur un mineur ou un majeur protégé au bénéfice de ses parents
-
Article 7
Levée partielle de l'interdiction des dons d'organes,
de tissus et de cellules applicable aux majeurs protégés
-
Article 5 A (nouveau)
-
CHAPITRE IER BIS
Conforter la solidarité dans le cadre du don de sang
-
CHAPITRE II
Permettre la solidarité dans le cadre
de la transmission d'une information génétique
-
Article 8
Réalisation d'examens des caractéristiques génétiques sur une personne décédée ou hors d'état d'exprimer sa volonté au profit des membres de sa famille
-
Article 9
Transmission d'une information génétique au profit de la parentèle
ou dans les situations de rupture du lien de filiation biologique
-
Article 8
-
TITRE III
APPUYER LA DIFFUSION DES PROGRÈS SCIENTIFIQUES
ET TECHNOLOGIQUES DANS LE RESPECT
DES PRINCIPES ÉTHIQUES
-
Article 10
Consentement à l'examen des caractéristiques génétiques
-
Article 10 bis (nouveau)
Encadrement de l'accès aux tests génétiques à visée généalogique
-
Article 10 ter
(nouveau)
Expérimentation de l'accès en population générale
aux examens des caractéristiques génétiques
-
Article 11
Encadrement du recours
à un traitement algorithmique à des fins médicales
-
Article 12
Encadrement du recours aux techniques
d'imagerie cérébrale et interdiction des discriminations
fondées sur les résultats de ces techniques en matière d'assurance
-
Article 13
Interdiction par décret des dispositifs ayant pour effet de modifier l'activité cérébrale en cas de danger pour la santé humaine
-
Article 10
-
TITRE IV
SOUTENIR UNE RECHERCHE LIBRE ET RESPONSABLE
AU SERVICE DE LA SANTÉ HUMAINE
-
CHAPITRE 1ER
Aménager le régime actuel de recherches sur l'embryon
et les cellules souches embryonnaires
-
Article 14
Différenciation des régimes juridiques d'autorisation
s'appliquant à l'embryon et aux cellules souches embryonnaires
-
Article 15
Régulation, en recherche fondamentale,
de certaines utilisations des cellules souches pluripotentes induites
-
Article 16
Limite de conservation des embryons proposés à la recherche
-
Article 14
-
CHAPITRE II
Favoriser une recherche responsable
en lien avec la médecine génomique
-
TITRE V
POURSUIVRE L'AMÉLIORATION
DE LA QUALITÉ ET DE LA SÉCURITÉ
DES PRATIQUES DU DOMAINE BIOÉTHIQUE
-
CHAPITRE IER
Renforcer la qualité et la sécurité des pratiques
-
Article 19
Actualisation du régime du diagnostic prénatal
-
Article 19 bis A
Abrogation du double diagnostic préimplantatoire (DPI-HLA)
et demande de rapport sur le sang placentaire
-
Article 19 bis
Etat des lieux du diagnostic prénatal
et du diagnostic préimplantatoire par l'Agence de biomédecine
-
Article 19 ter
(nouveau)
Expérimentation du diagnostic préimplantatoire
pour la recherche d'aneuploïdies
-
Article 19 quater (nouveau)
Réalisation en première intention d'un examen
des caractéristiques génétiques chez le nouveau-né
dans le cadre du dépistage néonatal pour la recherche d'anomalies
génétiques ciblées susceptibles de mesures de prévention ou de soins
-
Article 20
Suppression du délai de réflexion
dans l'interruption de grossesse pour raison médicale
et encadrement de la réduction embryonnaire ou foetale
-
Article 21
Clarification des conditions d'interruption de grossesse
pour raison médicale pour les mineures non émancipées
-
Article 21 bis
Prise en charge des enfants présentant une variation
du développement génital
-
Article 22
Autorisation de greffe de tissu germinal
pour rétablir une fonction hormonale et clarification
du devenir des gamètes et tissus germinaux conservés
-
Article 19
-
CHAPITRE II
Optimiser l'organisation des soins
-
Article 23
Élargissement des missions des conseillers en génétique
-
Article 24
Garantie d'une transmission sécurisée
des résultats de génétique entre laboratoires
-
Article 25
Aménagement, pour les patients concernés,
d'une passerelle entre la génétique somatique
et la génétique constitutionnelle
-
Article 26
Sécurisation de l'utilisation du microbiote fécal
-
Article 27
Réalisation d'un médicament de thérapie innovante
préparé ponctuellement dans le cadre d'une seule intervention médicale
-
Article 28
Diverses mises en cohérence
-
Article 23
-
TITRE VI
ASSURER UNE GOUVERNANCE BIOÉTHIQUE
ADAPTÉE AU RYTHME DES AVANCÉES RAPIDES
DES SCIENCES ET DES TECHNIQUES
-
Article 29 A
Création, dans chacune des deux assemblées du Parlement,
d'une délégation parlementaire à la bioéthique
-
Article 29
Élargissement des missions du comité consultatif national d'éthique
des sciences de la vie et de la santé
-
Article 30
Évolution des missions et des instances
de l'agence de la biomédecine
-
Article 29 A
-
TITRE VII
DISPOSITIONS FINALES
-
Article 31
Habilitation à légiférer par voie d'ordonnance
-
Article 32
Clause de révision et évaluation de la loi de bioéthique
-
Article 33
Rapport au Parlement présentant l'état des stocks
des gamètes en France et les conditions de recours à ces derniers
-
Article 34
Rapport au Parlement sur l'application des dispositions
encadrant l'entretien avec les proches en matière
de prélèvement d'organes et de tissus
-
Article 31
-
TRAVAUX DE LA COMMISSION
-
I. AUDITIONS
-
M. François Clavairoly,
président de la Fédération protestante de France,
M. Haïm Korsia, Grand rabbin de France,
Mgr Pierre d'Ornellas, archevêque de Rennes,
et M. Olivier Wang-Genh, président
de l'Union des bouddhistes de France
-
M. Jean-François Delfraissy,
président,
et Mme Karine Lefeuvre, vice-présidente
du Comité consultatif national d'éthique (CCNE)
-
Mmes Marie-Thérèse Besson,
présidente,
et Joëlle Mounier, membre de la Commission nationale
éthique-bioéthique de la Grande Loge Féminine de France
M. Edouard Habrant, Grand Maître,
et Mme Christiane Vienne, Grand Maître Adjoint
aux affaires extérieures de la Grande Loge Mixte de France
-
Pr Nathalie Rives, CECOS de Rouen, CHU de
Rouen
Pr Catherine Guillemain, CECOS de Marseille,
Assistance publique-hôpitaux de Marseille,
et Dr Sophie Mirallie, CECOS de Nantes, CHU de Nantes
Pr Rachel Lévy, Pr Nelly Achour-Frydman
et Dr Patrice Clément, pour la Fédération des BLEFCO
(Biologistes des laboratoires d'étude
de la fécondation de l'oeuf)
-
Mme Cécile Martinat, présidente
de la Société française de recherche sur les cellules souches,
et M. Marc Peschanski, directeur de l'Institut des cellules souches
pour le traitement et l'étude des malades monogénétiques (I-Stem)
-
Pr Israël Nisand, président du
Collège national
des gynécologues et obstétriciens français (CNGOF),
Pr Jean-François Mattei, vice-président de l'Académie nationale de médecine,
et Pr René Frydman, professeur émérite des universités, gynécologue obstétricien
-
M. Hugues Fulchiron, professeur de droit
privé
à l'Université Jean Moulin Lyon 3, directeur du centre de droit de la famille, Mme Marie Mesnil, maîtresse de conférences en droit privé
à l'Université de Rennes 1 et M. Jean-René Binet,
professeur de droit privé à l'Université de Rennes
-
Mme Alexandra Benachi,
présidente de la Fédération française
de centres pluridisciplinaires de diagnostic prénatal
-
Mme Huguette Mauss, présidente,
et de M. Jean-Pierre Bourély, secrétaire général,
du Conseil national pour l'accès aux origines personnelles (CNAOP)
-
M. Jean-Marie Le Méné,
président de la Fondation Jérôme Lejeune
-
Mme Emmanuelle Cortot-Boucher,
directrice générale de l'Agence de la biomédecine
-
Mmes Nicole Belloubet, garde des sceaux, ministre
de la justice,
et Frédérique Vidal, ministre de l'enseignement supérieur,
de la recherche et de l'innovation, et M. Adrien Taquet,
secrétaire d'État auprès de la ministre des solidarités et de la santé
-
MM. Alexandre Urwicz, président,
et Fabien Joly, porte-parole de l'Association des familles homoparentales (ADFH), Mme Marie-Claude Picardat
et M. Dominique Boren, porte-parole de l'Association des parents
et futurs parents gays et lesbiens (APGL), Mmes Catherine Michaud, présidente de l'association GayLib, Laurène Chesnel,
déléguée Familles de l'Inter-LGBT et Véronique Cerasoli,
administratrice et porte-parole de l'association SOS homophobie
-
M. François Clavairoly,
-
II. EXAMEN DU RAPPORT
-
I. AUDITIONS
-
LISTE DES PERSONNES ENTENDUES
PAR LES RAPPORTEURS ET CONTRIBUTIONS ÉCRITES
N° 237
SÉNAT
SESSION ORDINAIRE DE 2019-2020
Enregistré à la Présidence du Sénat le 8 janvier 2020
RAPPORT
FAIT
au nom de la commission spéciale (1) sur le projet
de loi, adopté
par l'Assemblée nationale, relatif
à la
bioéthique
,
Par Mmes Corinne IMBERT, Muriel JOURDA,
MM. Olivier HENNO et
Bernard JOMIER,
Sénateurs
(1) Cette commission est composée de : M. Alain Milon, président ; M. Olivier Henno, Mme Corinne Imbert, M. Bernard Jomier et Mme Muriel Jourda, rapporteurs ; MM. Philippe Bas, Jacques Bigot, Mme Catherine Deroche, M. Gérard Longuet, Mmes Michelle Meunier, Élisabeth Doineau, M. Michel Amiel, Mmes Véronique Guillotin, Laurence Cohen et M. Daniel Chasseing, vice-présidents ; M. Bernard Bonne, Mme Marie-Pierre de la Gontrie et M. Jean-Marie Mizzon, secrétaires ; Mmes Éliane Assassi, Martine Berthet, Maryvonne Blondin, MM. Guillaume Chevrollier, Jean-Pierre Corbisez, Yves Daudigny, Mmes Jacky Deromedi, Chantal Deseyne, Catherine Di Folco, M. Loïc Hervé, Mme Christine Herzog, MM. Xavier Iacovelli, Roger Karoutchi, Dominique de Legge, Hervé Marseille, Mme Marie Mercier, M. Thani Mohamed Soilihi, Mmes Laurence Rossignol, Patricia Schillinger et M. Yannick Vaugrenard.
Voir les numéros :
|
Assemblée nationale ( 15 ème législ.) : |
2187 , 2243 et T.A. 343 |
|
Sénat : |
63 et 238 (2019-2020) |
L'ESSENTIEL
_______
Le projet de loi de bioéthique soumis à l'examen du Sénat engage, dans le prolongement des lois de 2004 et de 2011, la troisième « grande » révision des lois de bioéthique adoptées par le Parlement en 1994.
Conformément à ce qu'avait souhaité le législateur en 2011, l'élaboration de ce texte a été précédée d'une large concertation.
Le Comité consultatif national d'éthique (CCNE) a ouvert le 18 janvier 2018 la consultation citoyenne dans le cadre des États généraux de la bioéthique , qui ont révélé ou ravivé l'intérêt de nos concitoyens pour ces questions touchant directement, comme l'a relevé le CCNE, « le “noyau dur” de l'humain » . Ce comité a rendu son avis le 25 septembre 2018.
De nombreuses instances ont également contribué à nourrir la réflexion du Parlement en vue du réexamen de la loi : l'Agence de la biomédecine et le Conseil d'Etat ont rendu publiques des études et propositions tant sur les aspects scientifiques que juridiques des enjeux bioéthiques posés à la société d'aujourd'hui. En outre, l'Office parlementaire d'évaluation des choix scientifiques et technologiques a procédé à des travaux approfondis sur l'évaluation de la précédente loi 1 ( * ) .
Chaque loi de bioéthique s'inscrit dans un équilibre délicat , produit de la volonté d'accueillir des avancées médicales, technologiques ou scientifiques dans le respect des principes fondamentaux formant « l'éthique à la française ». Dans ce contexte, la commission spéciale a abordé ses travaux dans un esprit de responsabilité, d'écoute et de respect mutuels, sur des sujets impliquant fortement les convictions personnelles. Elle a tenu à entendre, à travers ses auditions plénières et celles conduites par ses rapporteurs, l'expression d'une grande diversité de points de vue.
Les évolutions sociétales engagées par le projet de loi en matière d'assistance médicale ont soulevé d'importants questionnements en ce qu'elles impliquent des choix collectifs et des attentes individuelles. La commission spéciale, au terme d'un large débat, a ouvert la voie à ces évolutions tout en réaffirmant des principes essentiels, que ce soit sur la finalité de notre système de sécurité sociale ou sur le respect de la vie privée pour ce qui est de la levée de l'anonymat des donneurs de gamètes.
Sur d'autres sujets comme la génétique ou la recherche, qui connaissent d'importants bouleversements ou sont exposés à des enjeux renouvelés, la commission spéciale a souhaité accompagner des évolutions qui lui sont apparues porteuses de progrès en matière de santé, tout en les encadrant de garde-fous, plutôt que de freiner a priori des développements qui posent inévitablement de nouveaux questionnements éthiques.
La commission spéciale a adopté le projet de loi en y apportant, ainsi, des modifications substantielles. Elle l'a enrichi de 137 amendements, dont 124 à l'initiative de ses rapporteurs.
A. RÉPONDRE À DES ATTENTES SOCIÉTALES EN MATIÈRE DE PROCRÉATION DANS LE RESPECT DES PRINCIPES FONDAMENTAUX DE NOTRE DROIT
1. Ouvrir l'accès à l'assistance médicale à la procréation aux couples de femmes et aux femmes non mariées sans revenir sur le cadre existant pour les couples infertiles
• La commission spéciale, au terme d'un large débat, a ouvert l'assistance médicale à la procréation aux couples de femmes et aux femmes non mariées tout en modifiant substantiellement les modalités prévues par l' article 1 er du projet de loi.
Elle a ainsi maintenu les conditions médicales actuelles de recours à l'AMP pour les couples hétérosexuels , sur la base d'une infertilité pathologique ou afin d'éviter la transmission à l'enfant ou à l'autre membre du couple d'une maladie d'une particulière gravité.
Seul le recours à l'assistance médicale à la procréation pour ces raisons médicales ou pathologiques sera pris en charge par l'assurance maladie , conformément à la vocation de la sécurité sociale d'assurer la « protection contre le risque et les conséquences de la maladie ».
La commission spéciale a également assoupli les conditions d'âge pour accéder à l'assistance médicale à la procréation, en les renvoyant à une recommandation de bonnes pratiques plutôt qu'à un décret en Conseil d'Etat, afin de ménager plus de souplesse dans l'appréciation par les équipes médicales des situations individuelles.
Elle a prévu la participation, au sein de l'équipe médicale pluridisciplinaire des centres d'AMP, d'un pédopsychiatre et a introduit le principe d'une évaluation psychologique et, en tant que de besoin, sociale des demandeurs. Elle a clarifié la portée, en outre, de l'appréciation de la « motivation » des demandeurs.
La commission spéciale a supprimé des dispositions introduites par l'Assemblée nationale pour interdire toute discrimination en matière d'accès à l'AMP, jugées redondantes au regard des dispositions du code de déontologie médicale, et stigmatisantes pour les équipes médicales.
Elle a enfin étendu à tous les établissements de santé l'activité d'accueil d'embryons , qui ne peut actuellement être exercée qu'au sein du secteur public ou privé non lucratif.
• La commission spéciale a adopté l' article 4 , tirant les conséquences de l'ouverture de l'assistance médicale à la procréation aux couples de femmes en termes de filiation , dans sa rédaction issue des travaux de l'Assemblée nationale.
Afin de donner une portée pleine et entière à l'interdiction de la gestation pour autrui (GPA) en France, elle a introduit un article 4 bis interdisant la transcription totale de l'acte de naissance étranger d'un enfant né d'une GPA lorsqu'il mentionne comme mère une autre femme que celle qui a accouché ou deux pères.
2. Autoriser, sans la décourager, l'autoconservation de gamètes à des fins de prévention
• La commission spéciale a approuvé, à l' article 2 , l'autoconservation de gamètes dans une démarche de prévention de l'infertilité, que le Gouvernement entend autoriser « sans l'encourager ».
Afin cependant de rendre cette possibilité effective, elle a assoupli les critères d'âges pour en bénéficier en renvoyant cette appréciation aux équipes médicales , sur la base d'une recommandation de bonnes pratiques, plutôt qu'à un décret en Conseil d'Etat, en soulignant le caractère restrictif des bornes d'âge envisagées.
Elle a également étendu cette pratique à l'ensemble des établissements de santé , alors que le projet de loi prévoyait d'en exclure les établissements de santé à but lucratif qui assurent à l'heure actuelle plus de la moitié de l'activité d'assistance médicale à la procréation et constituent la seule offre médicale disponible sur de nombreux territoires.
Elle a précisé, en parallèle, les informations apportées aux personnes en amont de cette démarche, notamment sur les risques liés aux grossesses tardives, afin que cette possibilité ne soit pas perçue comme une « solution miracle » contre l'infertilité.
La commission spéciale a par ailleurs maintenu le consentement du conjoint ou partenaire au don de gamètes , d'autant plus utile compte tenu de l'accès possible à l'identité du donneur par les enfants issus du don.
Elle a également mieux encadré les conditions d'importation et d'exportation de gamètes afin d'éviter toute dérive mercantile.
Parallèlement aux évolutions proposées concernant l'activité d'accueil d'embryons, elle a autorisé, à titre dérogatoire, des établissements de santé privés lucratifs à pratiquer l'activité de don de gamètes , en l'absence d'une autre offre médicale disponible dans un département.
• S'agissant de l'autoconservation de gamètes ou tissus germinaux pour motif pathologique, dont l' article 22 du projet de loi étend la finalité à la restauration d'une fonction hormonale afin d'améliorer la qualité de vie des patientes, la commission spéciale a encadré ces dispositions notamment afin de protéger la situation des personnes mineures au moment du recueil ou du prélèvement et de renforcer leur information ainsi que le suivi des personnes concernées. Elle a également autorisé expressément ce recueil en cas de changement de sexe à l'état civil.
3. Permettre l'accès aux données personnelles du donneur par les personnes nées de dons, quelle que soit leur date de naissance et dans le respect de la vie privée
La commission spéciale a approuvé, à l' article 3 , la création d'un droit reconnu à toute personne née d'un don de gamètes ou d'embryons d' accéder aux données non identifiantes de son donneur.
Elle a toutefois souhaité permettre au donneur d'accepter ou de refuser l'accès à son identité au moment de la demande exprimée par la personne issue de son don, considérant que cette solution, qui avait également la préférence du Conseil d'État, présentait une temporalité plus respectueuse de la vie privée du donneur .
Elle a fait le choix de confier les missions relatives à l'accès aux données personnelles du donneur au Conseil national pour l'accès aux origines personnelles (CNAOP) qui existe depuis près de 18 ans et a acquis une expérience forte en matière d'accès aux origines et d'accompagnement, plutôt que de créer une commission ad hoc distincte.
Pour faciliter le travail du CNAOP, elle l'a habilité à interroger le répertoire national d'identification des personnes physiques avec le numéro d'inscription (code NIR) du donneur.
Constatant que la situation des enfants déjà nés avait été ignorée par le Gouvernement, la commission spéciale a confié au CNAOP la mission de recontacter en toute discrétion les anciens donneurs en cas de demandes d'accès provenant de personnes nées de dons sous l'ancien régime d'anonymat, et de les interroger sur leur volonté ou non de communiquer leurs données personnelles , sans attendre qu'ils se manifestent spontanément.
Pour améliorer l'échange de données médicales entre donneurs et personnes nées de dons de gamètes via leurs médecins, elle a introduit à l'article L. 1244-6 du code de la santé publique la possibilité de mettre à jour ces données auprès des CECOS .
B. FAVORISER UNE POLITIQUE AMBITIEUSE DE DON D'ORGANES, DE TISSUS OU DE CELLULES
1. Reconnaître un « statut de donneur »
En parallèle du plan greffes pour la période 2017-2021, qui fixe des objectifs ambitieux en matière de développement du don du vivant, la commission spéciale a inséré un article 5 A posant les bases d'un statut de donneur d'organes afin de favoriser une meilleure reconnaissance de ce geste altruiste et d'en faire une priorité de la promotion du don.
Ce statut ouvre droit, d'une part, à une forme de reconnaissance symbolique sous la forme de l'accès à une décoration honorifique, et conduit, d'autre part, à reconnaître explicitement le principe de neutralité financière du don pour le donneur.
2. Préciser le recours au don croisé d'organes
Alors que cette possibilité ouverte par le législateur en 2011 a permis à ce jour la réalisation de seulement 12 greffes rénales, l' article 5 assouplit de façon bienvenue les conditions de recours à un don croisé d'organes en cas d'incompatibilité entre donneur et receveur en portant à quatre au lieu de deux le nombre de paires impliquées et en autorisant le recours dans une chaîne à un organe prélevé sur une personne décédée.
La commission spéciale a réintroduit dans la loi le nombre maximal de paires impliquées dans un don croisé, que l'Assemblée nationale avait renvoyé à un décret tout en prévoyant l'information du Parlement en cas de modification. Elle a porté ce nombre à six afin de ménager une souplesse dans la mise en oeuvre de cette procédure, tout en restant compatible avec le délai de 24 heures fixé pour la réalisation des opérations de prélèvement.
3. Mieux articuler consentement et vulnérabilité des mineurs et majeurs protégés
• La commission spéciale a approuvé la possibilité, ouverte à l'article 6, de recourir aux cellules souches hématopoïétiques d'un enfant mineur ou d'un majeur protégé au bénéfice de l'un de ses parents, tout en encadrant la procédure afin de renforcer la protection du donneur pressenti.
Elle a toutefois choisi, s'agissant du mineur, d'abaisser l'âge du consentement à 16 ans afin qu'il puisse exprimer lui-même son consentement devant le juge, sans recourir à la nomination d'un administrateur ad hoc.
• Favorable au renforcement de l'autonomie des majeurs protégés, la commission spéciale a adopté l' article 7 qui vise à faire entrer dans le droit commun du don d'organes, de tissus et de cellules les majeurs qui font l'objet des mesures de protection juridique les plus légères, soit l'assistance ou la représentation aux biens.
Elle a toutefois refusé d'appliquer le régime du consentement présumé en matière de prélèvement d'organe post mortem à tous les majeurs protégés, considérant que le consentement éclairé des personnes faisant l'objet d'une mesure de protection avec représentation à la personne ne peut être présumé.
• Afin de permettre aux majeurs protégés et aux mineurs de participer à la solidarité nationale par un geste citoyen, la commission spéciale a souhaité, en introduisant un article 7 bis , ouvrir le don du sang aux majeurs faisant l'objet d'une mesure de protection juridique avec représentation aux biens et assistance ainsi qu'aux mineurs de 17 ans.
C. POUR UNE GÉNÉTIQUE ÉTHIQUE ET RESPONSABLE : TROUVER LE POINT D'ÉQUILIBRE ENTRE LIBERTÉ INDIVIDUELLE, ACCOMPAGNEMENT MÉDICAL ET SOLIDARITÉ
La génétique connaît aujourd'hui une véritable révolution. Elle est source d'espoir pour le diagnostic et la prise en charge thérapeutique, notamment dans les domaines de la médecine prédictive et de la médecine de précision, en permettant, par exemple, la mise au point de thérapies géniques ciblées dans le traitement de maladies rares ou de cancers. Le développement des techniques de séquençage haut-débit et la démocratisation, sur le plan économique, de l'accès aux tests génétiques font néanmoins craindre une mauvaise appréhension par la société de données génétiques de plus en plus massives dont la signification diagnostique et les conséquences médicales restent encore en grande partie indéterminées.
1. Pour une génétique au service de la prévention et du soin
Dans le prolongement d'une proposition de loi adoptée par le Sénat en juin 2018 2 ( * ) , l' article 8 du projet de loi autorise la réalisation, dans l'intérêt de la parentèle, d'un examen des caractéristiques génétiques sur une personne décédée ou hors d'état d'exprimer sa volonté , sauf opposition de cette personne manifestée de son vivant, lorsqu'est suspectée une anomalie génétique pouvant être responsable d'une affection grave justifiant des mesures de prévention ou de soins qui pourraient bénéficier aux membres de la famille.
Ces examens génétiques post mortem seront effectués à partir d'échantillons déjà conservés ou prélevés dans le cadre d'une autopsie. Les pratiques de conservation des échantillons biologiques prélevés par les laboratoires de biologie médicale pouvant varier d'un laboratoire à un autre, votre commission a adopté un amendement visant à harmoniser ces pratiques par la publication de règles de bonnes pratiques en matière de conservation et de traçabilité de ces échantillons .
Dans un souci de prévention et de maximisation de l'efficacité des soins, votre commission a également introduit, dans le projet de loi, un article 19 quater autorisant, dans le cadre du dépistage néonatal , les examens génétiques ciblés sur la recherche chez le nouveau-né de quelques anomalies génétiques associées à des pathologies graves pour lesquelles des traitements et thérapies géniques administrés suffisamment tôt permettent d'améliorer significativement l'espérance et la qualité de vie.
2. Renforcer la prise en compte de l'intérêt des apparentés biologiques dans le traitement d'informations à caractère génétique
À l' article 9 du projet de loi, votre commission a souhaité renforcer la transmission d'une information génétique dans les situations de rupture du lien de filiation biologique , c'est-à-dire entre tiers donneurs et personnes nées d'un don, et entre parents de naissance et personnes nées dans le secret, dans un souci d' égalité d'accès de ces personnes , qui de fait n'ont pas connaissance des antécédents médicaux de leurs apparentés biologiques, aux mesures de prévention ou de soins , tout en préservant rigoureusement l'anonymat des personnes concernées.
Comme l'Assemblée nationale l'a fait pour les tiers donneurs et les personnes nées d'un don, votre commission a ainsi adopté un amendement rendant automatique l'alerte, par l'intermédiaire du médecin prescripteur de l'examen et du Cnaop, de la personne née dans le secret ou du parent de naissance sur l'existence d'une information génétique potentiellement majeure, voire vitale. En aucun cas, ce mécanisme d'alerte ne donnera lieu à la révélation de l'identité de la personne initialement concernée par l'examen génétique, ni à la révélation de l'anomalie génétique en cause ou des risques associés, dans le respect du droit de toute personne d'être tenue dans l'ignorance d'éventuelles prédispositions génétiques.
3. Prévenir les dérives associées aux tests génétiques en accès libre
L' article 10 du projet de loi maintient le principe selon lequel un examen des caractéristiques génétiques ne peut être envisagé qu'en cas d'antécédent familial connu ou de symptôme d'une maladie d'origine potentiellement génétique.
Rien n'est en revanche prévu pour prémunir les personnes ayant recours à des tests génétiques en accès libre, notamment sur Internet, contre les risques qu'emporte la délivrance d'une information génétique en dehors de toute consultation médicale, l'interdiction de ces tests restant aujourd'hui purement virtuelle .
En cohérence avec la position du CCNE sur ce sujet, votre commission a par conséquent adopté un amendement permettant, à titre expérimental, d' ouvrir l'accès en population générale aux examens génétiques , notamment dans le cadre d'un dépistage préconceptionnel pour un couple s'engageant dans un projet parental, en l'absence d'antécédent familial connu ou de symptôme, pour la recherche de mutations génétiques dont la signification diagnostique est connue. Le Gouvernement pourra ainsi limiter les anomalies recherchées à une liste de mutations génétiques établie en concertation avec l'agence de la biomédecine et la Haute Autorité de santé.
En outre, face à l'ineffectivité de l'interdiction du recours aux tests génétiques en accès libre, votre commission a adopté un dispositif tendant à autoriser l'accès des examens génétiques à visée généalogique sous réserve du respect de conditions de nature à préserver les droits des personnes dans le traitement de données aussi sensibles. Conformes à un référentiel de qualité établi par l'agence de la biomédecine, ces tests ne pourront avoir pour objet de délivrer une information génétique d'ordre médical , ni ne pourront servir de fondement à des actions visant à faire valoir des droits patrimoniaux ou extrapatrimoniaux, notamment dans le cadre d'une démarche d'établissement d'un lien de filiation.
D. ACCOMPAGNER UNE RECHERCHE LIBRE ET RESPONSABLE AU SERVICE DE LA SANTÉ
La mise en place, par l' article 14 du projet de loi, d'un régime de déclaration préalable pour les recherches sur les cellules souches embryonnaires humaines permet d'acter la différence de nature de ces recherches avec celles sur l'embryon, qui continueront de faire l'objet d'un régime d'autorisation. Les cellules souches embryonnaires humaines n'ont en effet pas la capacité de former spontanément un embryon : les recherches portant sur ces cellules ne soulèvent donc pas les mêmes enjeux éthiques qu'une intervention sur l'embryon.
1. Consacrer la spécificité des recherches sur l'embryon
Afin de sécuriser sur le plan juridique les recherches menées sur les embryons surnuméraires , votre commission a précisé leurs prérequis dont certains sont inadaptés au contexte scientifique actuel et sont aujourd'hui exploités dans le cadre de contentieux quasi -systématiques. Elle a ainsi rappelé que les recherches sur l'embryon peuvent non seulement s'inscrire dans une finalité médicale déterminée mais également poursuivre un objectif d'amélioration de la connaissance de la biologie humaine dans le cadre de travaux de recherche fondamentale qui ne peuvent par définition anticiper avec précision les résultats de la recherche et les applications médicales qui pourraient en être tirées.
De même, votre commission a clarifié le critère de l'absence de méthodologie alternative en précisant que ce dernier devra être apprécié au regard de la pertinence scientifique des modèles alternatifs à la recherche sur l'embryon par rapport aux objectifs de la recherche considérée. Même si les cellules souches embryonnaires et les cellules souches pluripotentes induites permettent aujourd'hui d'envisager la constitution de « modèles embryonnaires à usage scientifique » susceptibles de mimer certaines étapes du développement embryonnaire, ces modèles ne permettent pas d'égaler les propriétés de l'embryon humain et donc de faire l'économie d'une recherche sur ce dernier.
Par ailleurs, dans le souci de permettre des avancées dans la compréhension du développement embryonnaire dans le respect des principes éthiques, votre commission a adopté un amendement tendant à autoriser, à titre dérogatoire, le développement in vitro d'embryons jusqu'au 21 e jour suivant leur constitution dans le cadre de protocoles de recherche spécifiquement dédiés à l'étude des mécanismes de développement embryonnaire au stade de la gastrulation. La limite de culture de l'embryon à 14 jours est en effet aujourd'hui réinterrogée par plusieurs pays qui envisagent de la repousser pour permettre des recherches indispensables à une meilleure connaissance du processus de différenciation des cellules souches embryonnaires.
2. Mieux encadrer certaines recherches sur les cellules souches embryonnaires ou pluripotentes induites
Certaines recherches conduites sur les cellules souches embryonnaires ou les cellules souches pluripotentes induites requièrent une vigilance particulière. Il s'agit notamment de la possibilité de différencier ces cellules en gamètes, de les insérer dans des tissus extra-embryonnaires afin de constituer des modèles mimant l'embryon ou de les insérer dans des embryons provenant d'autres espèces.
Ce dernier type de recherche n'est pas sans soulever d'importantes questions éthiques quant aux limites à poser au franchissement de la barrière des espèces . Par conséquent, votre commission a adopté un amendement visant à supprimer la possibilité, à l' article 14 du projet de loi, de créer des embryons chimériques résultant de l' insertion de cellules souches embryonnaires humaines dans un embryon animal .
Elle a également posé deux « verrous » à la création d'embryons chimériques par l'adjonction à un embryon animal de cellules pluripotentes induites d'origine humaine à l' article 15 du projet de loi :
- ces embryons ne pourront donner lieu à parturition , si bien qu'en cas de transfert chez la femelle, la gestation sera obligatoirement interrompue dans un délai approuvé par l'agence de la biomédecine ;
- la contribution des cellules d'origine humaine au développement de l'embryon chimérique ne saurait dépasser un seuil approuvé par l'agence de la biomédecine.
Par coordination, votre commission a adopté un amendement, à l' article 17 du projet de loi, visant à maintenir dans le champ de l'interdiction de création des embryons chimériques l'insertion de cellules souches embryonnaires humaines dans un embryon animal.
E. GARANTIR LA SÉCURITÉ ET LE CARACTÈRE ÉTHIQUE DE CERTAINES PRATIQUES ET TECHNOLOGIES MÉDICALES
1. Rénover le cadre juridique du diagnostic prénatal
L' article 19 vise à actualiser la définition du diagnostic prénatal qui s'entend, à l'heure actuelle, des « pratiques médicales, y compris l'échographie obstétricale et foetale, ayant pour but de détecter in utero chez l'embryon ou le foetus une affection d'une particulière gravité » 3 ( * ) . Il prend également en compte les développements importants qui ont concerné cette discipline et permettent à la fois de limiter son caractère invasif comme d'améliorer la précision du diagnostic posé. Les démarches d'accompagnement des couples en cas de révélation de données génétiques incidentes pouvant justifier des investigations complémentaires sont notamment précisées.
La commission spéciale a ajusté la définition adoptée par l'Assemblée nationale regroupant le diagnostic prénatal sous le terme plus général de médecine foetale, encore trop restrictive au regard de la réalité des pratiques consistant à prendre en charge in utero des pathologies présentant des degrés de sévérité divers.
2. Sécuriser les pratiques d'interruption de grossesse pour motif médical et de réduction embryonnaire ou foetale
La suppression , par l' article 20 , de la proposition systématique d'un délai de réflexion en cas d'interruption médicale de grossesse (IMG) pour motif foetal et la clarification des dispositions applicables en matière d'IMG chez la femme mineure sont bienvenues et permettent de garantir aux femmes enceintes les mêmes droits, en termes de consentement et d'autonomie, qu'en cas d'interruption volontaire de grossesse (IVG). De même, l'introduction dans la loi d'un encadrement des pratiques de réduction embryonnaire ou foetale en cas de grossesse multiple participe d'une sécurisation et d'une plus grande transparence de ces interventions réalisées pour prévenir des complications pour la mère, les embryons ou les foetus.
En revanche, rien ne justifie d'introduire , comme cela est prévu à l' article 21 , dans le code de la santé publique, une clause de conscience spécifique des professionnels de santé en matière d'IMG , dès lors qu'une clause de conscience générale, permettant de ne pas accomplir un acte contraire à ses convictions, bénéficie déjà aux professionnels de santé intervenant dans les procédures d'IVG ou d'IMG. Ceux qui souhaitent l'exercer n'ont pas attendu le projet de loi pour le faire.
Pour mémoire, l'existence dans la loi d'une clause de conscience spécifique à l'IVG découle des débats parlementaires qui avaient entouré l'adoption de la loi sur l'IVG du 17 janvier 1975 : le Gouvernement avait alors consenti à cette disposition dans un souci d'apaisement.
En conséquence, votre commission a supprimé la mention dans le projet de loi d'une clause de conscience spécifique des professionnels de santé en matière d'IMG .
3. Autoriser, en l'encadrant strictement, le recours au diagnostic préimplantatoire
• La commission spéciale a inséré un article 19 ter complétant les dispositions relatives au diagnostic préimplantatoire qui consiste en l'examen génétique de cellules prélevées sur des embryons après une fécondation in vitro et avant leur transfert in utero . Au-delà des conditions de recours actuelles limitées à la non transmission à l'enfant d'une maladie grave dont est porteur l'un des parents, cet article autorise, à titre expérimental, le diagnostic préimplantatoire pour la recherche d'aneuploïdies (DPI-A) en vue d'améliorer l'efficience de l'assistance médicale à la procréation dans certaines indications médicales ciblées.
Cette évolution strictement encadrée , soutenue par les sociétés savantes en médecine de la reproduction et les cliniciens de l'AMP et dont le CCNE avait préconisé la mise en oeuvre, vise à améliorer la prise en charge de femmes ayant des antécédents d'échec de fécondation in vitro ou de fausses-couches à répétition.
• En outre, la commission spéciale a supprimé l'article 19 bis A inséré par l'Assemblée nationale, rétablissant ainsi le recours possible à la technique du double diagnostic préimplantatoire (DPI-HLA) . Celle-ci permet que l'enfant à naître, en plus d'être indemne de l'anomalie génétique grave affectant un frère ou une soeur, présente des caractéristiques compatibles avec l'aîné malade pour envisager une greffe.
Elle a considéré que cette technique introduite en 2004 à titre expérimental et pérennisée par le législateur en 2011 pouvait, dans certaines situations certes exceptionnelles mais strictement encadrées sur le plan éthique, apporter une solution à des familles et sauver la vie d'enfants atteints de maladies rares.
4. Mieux encadrer le recours à l'intelligence artificielle et aux neurosciences dans le domaine de la santé
• L' article 11 institue un cadre juridique pour l'utilisation d'un traitement algorithmique de données massives lors de la réalisation d'un acte médical. Le développement de l' intelligence artificielle (IA) est très important en médecine et peut avoir des effets positifs pour le patient, en améliorant l'efficacité des diagnostics ou en aidant à la prise de décision thérapeutique. Mais l'usage de ces technologies n'est pas sans risque, notamment car il peut présenter des biais. C'est pourquoi les professionnels de santé doivent demeurer décisionnaires en matière de soins apportés aux patients.
La commission spéciale a donc renforcé les garanties applicables à l'utilisation de ces technologies, en prévoyant notamment que le patient soit informé en amont de l'utilisation d'un traitement algorithmique et qu'aucune décision médicale ne puisse exclusivement se fonder sur un tel traitement.
• S'agissant des neurosciences, l' article 12 propose notamment de modifier l'article 16-14 du code civil qui régit le recours aux techniques d'imagerie cérébrale , créé par la loi du 7 juillet 2011 relative à la bioéthique. Ces techniques ne sont aujourd'hui autorisées qu'à des fins médicales, de recherche scientifique ou dans le cadre d'expertises judiciaires, avec le consentement exprès de la personne concernée.
La commission spéciale a maintenu le droit en vigueur qui paraît satisfaisant sur ce point, ce qu'avait également conclu le Conseil d'État dans son étude en 2018.
• Enfin, l' article 13 confère au ministre de la santé le pouvoir d'interdire, en cas de danger grave ou de suspicion de danger grave, les actes, procédés, techniques ou équipements qui ont pour objet de modifier l'activité cérébrale.
La commission spéciale a exclu les équipements qui sont des dispositifs médicaux, car ils relèvent déjà des pouvoirs de police de l'Agence nationale de sécurité du médicament et des produits de santé (ANSM).
F. ADAPTER LA GOUVERNANCE BIOÉTHIQUE AU RYTHME DES PROGRÈS SCIENTIFIQUES ET TECHNOLOGIQUES
1. Supprimer la mise en place non justifiée de délégations parlementaires à la bioéthique
La commission spéciale a supprimé l' article 29 A , qui visait à la création de délégations parlementaires à la bioéthique afin d'informer les assemblées de la politique suivie par le Gouvernement au regard de ses conséquences sur la bioéthique et de suivre l'application des lois.
Elle a considéré qu'il existait déjà une structure permanente spécialisée bicamérale, l'Office parlementaire d'évaluation des choix scientifiques et technologiques (Opecst), et que des commissions permanentes, en particulier la commission des affaires sociales et, dans une moindre mesure, la commission des lois, remplissaient déjà pleinement cette mission.
2. Consacrer le rôle de l'agence de la biomédecine dans la proposition d'adaptations de la législation aux évolutions scientifiques et techniques
À l' article 30 du projet de loi, votre commission a rétabli la mission de l'agence de la biomédecine dans l'élaboration d'un référentiel permettant d'évaluer la qualité des tests génétiques en accès libre, en cohérence avec la mise en place, à l'article 10 bis , d'un encadrement législatif des examens génétiques à visée généalogique. En outre, elle a confié à l'agence le soin d'établir un bilan annuel des évolutions législatives et réglementaires qui pourraient être justifiées par l' évolution des connaissances et des techniques dans les domaines relevant de sa compétence mais aussi par des situations qui ne seraient pas couvertes par le droit en vigueur et nécessiteraient des autorisations de dérogation.
Répartition des articles

EXAMEN DES ARTICLES
TITRE
1ER
ÉLARGIR L'ACCÈS AUX TECHNOLOGIES
DISPONIBLES SANS
S'AFFRANCHIR
DE NOS PRINCIPES ÉTHIQUES
CHAPITRE IER
Permettre aux personnes d'exercer un choix
éclairé
en matière de procréation dans un cadre
maîtrisé
Article premier
Ouverture de
l'accès à l'assistance médicale
à la
procréation aux couples de femmes et aux femmes non mariées
Cet article ouvre aux couples de femmes ainsi qu'aux femmes non mariées l'accès aux techniques d'assistance médicale à la procréation, en supprimant pour l'ensemble des demandeurs la référence à toute indication médicale, étend à ces nouveaux publics les conditions actuelles de prise en charge de ces actes par l'assurance maladie, lève l'interdiction de recours à un double don de gamètes et procède à diverses modifications ou coordinations.
La commission spéciale a conservé le critère notamment d'infertilité d'accès à cette technique pour les couples hétérosexuels et a réservé la prise en charge par l'assurance maladie de l'assistance médicale à la procréation aux seules demandes fondées sur ces indications médicales. Elle a également prévu une évaluation psychologique et, en tant que de besoin, sociale des demandeurs. Elle a par ailleurs ouvert l'activité d'accueil d'embryons aux établissements privés à but lucratif.
I - Le dispositif proposé : un accès à l'assistance médicale à la procréation étendu aux couples de femmes et aux femmes seules, déconnecté de tout critère médical
1. L'assistance médicale à la procréation aujourd'hui : « une réponse médicale à l'infertilité naturelle » 4 ( * )
• La première loi « bioéthique » de 1994 5 ( * ) autorisant l'assistance médicale à la procréation a réservé l'accès à ces techniques à deux finalités strictement thérapeutiques :
- d'une part, remédier à l'infertilité dont le caractère pathologique a été médicalement diagnostiqué ;
- d'autre part, éviter la transmission à l'enfant d'une maladie d'une particulière gravité , étendue ensuite à la prévention de la transmission « à un membre du couple » principalement afin d'éviter la transmission virale, pour le VIH notamment.
Par voie de conséquence, le législateur a réservé cette pratique aux couples formés d'un homme et d'une femme « en âge de procréer ».
• Les lois de 2004 et de 2011 n'ont modifié qu'à la marge ces critères .
La loi de juillet 2011 a supprimé, pour mettre en avant la finalité thérapeutique de la démarche, la mention selon laquelle l'AMP est « destinée à répondre à la demande parentale d'un couple ».
Cette même loi a supprimé toute référence au statut juridique des couples ainsi que la mention d'une durée de vie commune d'au moins deux ans pour les couples non mariés. Néanmoins, elle a conservé la condition selon laquelle l'insémination ou le transfert d'embryons ne peuvent intervenir en cas non seulement de décès d'un des membres du couple ou cas de divorce, séparation ou cessation de la communauté de vie.
Comme l'a rappelé l'Agence de la biomédecine, la situation de « couple » est déclarative, l'équipe du centre d'AMP n'ayant pas accès à des documents que le couple ne lui fournirait pas. Les actes de mariage et de PACS sont fournis le cas échéant par les couples. En cas de concubinage, une attestation de vie commune est demandée.
L'assistance médicale à la procréation : quelques chiffres clés
• L'assistance médicale à la procréation recouvre différentes activités : elle « s'entend des pratiques cliniques et biologiques permettant la conception in vitro, la conservation des gamètes, des tissus germinaux et des embryons, le transfert d'embryons et l'insémination artificielle » (article L. 2141-1 du code de la santé publique).
• A l'exception de l'insémination artificielle et de la stimulation ovarienne, ces activités cliniques et biologiques ne peuvent être pratiquées que dans des établissements de santé et des laboratoires spécifiquement autorisés (l'autorisation pouvant porter sur une ou plusieurs activités d'AMP).
En 2017, d'après l'Agence de la biomédecine, 104 centres clinico-biologiques ont assuré les activités d'AMP (dont 1 a eu pour seule activité, le prélèvement, le recueil et la mise à disposition de gamètes dans le cadre du don) et 183 laboratoires ont assuré les préparations de sperme en vue d'insémination artificielle.
• L'Agence de la biomédecine recense pour l'année 2017 un total de 151 611 tentatives d'AMP (3,8 % de plus qu'en 2014), dont :
- 49 367 inséminations artificielles (-13 % par rapport à 2014) ;
- 102 244 fécondations in vitro (+14 % par rapport à 2014) pour lesquelles différentes techniques sont utilisées : la fécondation naturelle des gamètes mises en contact (FIV classique) ou la fécondation par micro-injection des spermatozoïdes dans l'ovocyte (FIV-ICSI 6 ( * ) ) ; des embryons peuvent également être conservés pour être transférés ultérieurement (on parle de TEC pour transfert d'embryon congelé).
• 96% de ces tentatives d'AMP sont réalisées en intraconjugal c'est à dire avec les gamètes des deux membres du couple.
Dans 4% des cas, il est fait recours à un tiers donneur pour des gamètes (un don de sperme dans 70 % des cas ou d'ovocyte) ou, plus marginalement, pour un accueil d'embryons (147 cas en 2017).
• 25 614 naissances sont recensées par l'Agence de la biomédecine pour la même année 2017 (soit 1 naissance pour 6 tentatives d'AMP environ), ce qui représente 3,3 % des enfants nés de la population générale (contre 2,6 % en 2009).
2. Le projet de loi : vers une AMP pour toutes les femmes
• La nouvelle rédaction proposée pour l'article L. 2124-2 du code de la santé publique (1° du I) modifie les conditions de recours à l'assistance médicale à la procréation :
- en ouvrant l'accès à l'AMP non seulement à tout couple formé d'un homme et d'une femme mais également à tout couple formé de deux femmes , quel que soit leur statut - marital ou non - comme à l'heure actuelle, et à toute femme non mariée , sous réserve de leur consentement préalable ;
- en supprimant, parallèlement, toute référence aux conditions médicales jusqu'alors fixées pour y recourir .
Par coordination avec ce dernier point, le projet de loi supprime l'article L. 2141-7 du code de la santé publique réservant l'accès à l'AMP avec tiers donneur à ces situations pathologiques.
• Le projet de loi retient la notion juridique de « femme non mariée » plutôt que les termes de « femme seule » ou « femme célibataire » qui ne correspondent à aucune notion juridique. En effet, la femme mariée peut par son seul statut imposer à son mari de devenir père de l'enfant dont elle accoucherait, par le jeu de la présomption de paternité. Ce choix sémantique avait été jugé nécessaire par le Conseil d'Etat dans son avis sur le projet de loi, afin d'éviter tout effet de ce projet sur un conjoint qui n'y aurait pas pris part. En théorie, cette formulation n'interdit pas l'accès à l'AMP à une femme seule qui serait par ailleurs en concubinage ou aurait conclu un pacte civil de solidarité. L'homme non marié ne serait pas alors contraint d'établir sa filiation avec l'enfant.
• Quant à la suppression de tout critère médical , il s'agit pour le Gouvernement d'éviter de « créer une nouvelle inégalité, source potentielle de contentieux » alors que, comme la ministre en charge de la santé l'a avancé lors des débats à l'Assemblée nationale ou comme l'indique l'étude d'impact, le critère pathologique d'infertilité n'est pas identifié dans 10 à 15 % des cas, sans que cela fasse aujourd'hui obstacle à la prise en charge de ces couples : le diagnostic d'hypofertilité est alors basé sur un constat, celui d'absence de grossesse. Pour des professionnels entendus, il s'agit bien néanmoins dans ces situations d'un diagnostic d'infertilité, même si son origine médicale précise ne peut pas toujours être identifiée en l'état des connaissances.
Un autre argument à la levée de tout critère médical dans l'accès à l'AMP, avancé dans l'étude d'impact en écho à des observations du Haut Conseil à l'égalité entre les femmes et les hommes ou de la Commission nationale consultative des droits de l'homme, est que le principe de l'AMP avec tiers donneur ne serait pas un « traitement » stricto sensu de l'infertilité mais un moyen de la pallier. L'ouverture de l'AMP à des cas d'infertilité « situationnelle », pour reprendre une expression du CCNE dans son compte rendu des Etats généraux de la bioéthique, serait donc un prolongement logique et naturel de cette technique, mise à disposition d'autres publics.
• Comme à l'heure actuelle, plusieurs situations, s'agissant d'un couple, continueraient à faire obstacle à la poursuite de la démarche , à savoir à l'insémination artificielle ou au transfert des embryons :
- le décès d'un des membres du couple ;
- le dépôt d'une demande en divorce, la signature d'une convention de divorce par consentement mutuel ou la cessation de la communauté de vie. Ces situations, pour des couples mariés, emportent en effet des conséquences sur la filiation dans la mesure où l'article 313 du code civil permet dans certaines situations d'écarter la présomption de paternité en cas de demande en divorce ou en séparation de corps ;
- la révocation, par l'un des membres du couple, du consentement écrit auprès du médecin chargé de mettre en oeuvre l'AMP.
Le Gouvernement fait ainsi le choix d' écarter l'ouverture de l'AMP post mortem (c'est-à-dire le transfert d'embryons ou de gamètes conservés dans le cadre d'une AMP après le décès du conjoint) dont le CCNE avait préconisé l'autorisation sous conditions 7 ( * ) , et que plusieurs associations ou sociétés savantes appellent de leurs voeux dès lors que la femme concernée aurait désormais accès, en tant que femme seule, à un accueil d'embryon d'un autre couple ou à un don de sperme d'un donneur anonyme.
Il faut rappeler à cet égard que dans une décision du 31 mai 2016 , le Conseil d'Etat a estimé que, en dépit de l'interdiction générale posée par la loi, l'autorisation d'exporter des gamètes aux fins de procéder à une AMP post mortem devait être accordée dans certaines circonstances exceptionnelles où un refus aurait pour effet de porter une atteinte excessive au droit du demandeur au respect de sa vie privée (article 8 de la convention européenne de sauvegarde des droits de l'homme et des libertés fondamentales). En l'occurrence, l'homme avait expressément consenti avant son décès à ce que son épouse puisse bénéficier d'une insémination artificielle avec ses gamètes à titre posthume en Espagne, pays d'origine où celle-ci est retournée vivre, qui autorise contrairement à la France l'insémination post mortem .
3. Le renvoi au décret des conditions d'âge pour recourir à l'AMP
• Depuis 1994, la loi réserve l'accès à l'AMP aux couples « en âge de procréer » (article L. 2141-1 du code de la santé publique).
Cette limite a répondu à des considérations tant sociales - l'intérêt de l'enfant - que biologiques, en prenant en compte les risques pour la santé de la femme et la limite « naturelle » de reproduction.
L'Agence de la biomédecine, dans son rapport de janvier 2018 d'évaluation de la précédente loi de bioéthique, a relevé que « cette disposition, en ce qu'elle ne comporte pas de limite chiffrée, pose des difficultés d'application aux praticiens , auxquels il revient d'apprécier si les membres du couple qui les consultent sont en âge de procréer et de leur imposer des limites. Les différences éventuelles d'appréciation d'une équipe médicale à l'autre sont en outre susceptibles de créer des inégalités d'accès à l'AMP. »
Pour les femmes, les conditions de prise en charge par l'assurance maladie - à savoir jusqu'à leur 43 ème anniversaire - constituent le plus souvent une limite de fait. Pour les hommes, les équipes médicales sont, comme le note l'Agence, plus en difficulté.
A l'occasion de contentieux contre des décisions de l'Agence de la biomédecine de refus d'exportations de gamètes, par deux récentes décisions du 17 avril 2019, le Conseil d'Etat a estimé que l'âge de 60 ans pouvait être admis pour l'homme comme la limite supérieure de l'« âge de procréer » 8 ( * ) .
• Le 1° du I (dernier alinéa de la rédaction proposée pour l'article L. 2141-2 du code de la santé publique) remplace la notion d'âge de procréer, à l'appréciation des équipes médicales, par le renvoi à un décret en Conseil d'Etat pris après avis de l'Agence de la biomédecine, des conditions d'âge requises pour bénéficier d'une AMP .
Il est précisé que ces conditions prennent en compte :
- d'une part, les risques médicaux de la procréation liés à l'âge ;
- d'autre part, l'intérêt de l'enfant à naître, ce qui implique notamment de considérer la place de l'enfant dans les générations familiales.
Dans un avis du 8 juin 2017 , le conseil d'orientation de l'Agence de la biomédecine a préconisé une limite d'âge à 43 ans pour les femmes, avec une appréciation au cas par cas possible entre 43 et 45 ans en cas d'ovocytes préalablement conservés ou de don d'ovocyte, et à 60 ans pour les hommes.
4. L'évaluation médicale et psychologique des demandeurs
• Depuis 1994 et en application de l'article L. 2141-10 du code de la santé publique, la mise en oeuvre d'une procédure d'AMP doit être précédée d' entretiens particuliers des demandeurs avec les membres de l'équipe médicale pluridisciplinaire du centre, laquelle comporte des médecins cliniciens (gynécologues obstétriciens, endocrinologues, gynécologues médicaux) et des biologistes.
Ces entretiens, qui peuvent associer en tant que de besoin le service social, ont aux termes de cet article un objectif essentiellement informatif :
- vérifier la motivation de chaque membre du couple, en leur rappelant les possibilités ouvertes par la loi en matière d'adoption ;
- informer sur les techniques d'AMP (possibilités de réussite et d'échec, notamment en fonction de l'âge ou de la situation médicale, effets secondaires et risques à court et long terme, contraintes...) ;
- informer de l'impossibilité de réaliser un transfert des embryons conservés en cas de rupture du couple ou de décès d'un de ses membres ;
- remettre un « dossier-guide » rappelant les dispositions applicables et décrivant les différentes techniques.
Ce même article fixe un délai de réflexion d'un mois entre le dernier entretien et la confirmation de la demande d'AMP. Ce délai peut être prolongé si cela est jugé nécessaire « dans l'intérêt de l'enfant à naître ».
L'arrêté du 30 juin 2017 relatif aux règles de bonnes pratiques cliniques et biologiques d'AMP précise qu'il s'agit de délivrer, à travers ces entretiens, « une information loyale, claire et appropriée sur toutes les étapes de la prise en charge » . Il rappelle également que « l'équipe pluridisciplinaire peut à tout moment différer ou refuser la prise en charge dans les limites fixées par la loi et le code de déontologie , dans la mesure où tout médecin doit tenir compte des avantages et inconvénients des différentes investigations et thérapeutiques possibles » . En cas de refus de prise en charge, les voies de recours de droit commun s'appliquent et la responsabilité des professionnels de santé peut être recherchée. Le rapporteur n'a pas obtenu de données à ce sujet, en dehors de contentieux mettant en cause des établissements de santé, fondés sur des motifs tirés de l'âge des demandeurs 9 ( * ) , le refus de procéder à une AMP pour un couple homosexuel 10 ( * ) ou d'exporter des gamètes d'une personne décédée 11 ( * ) .
• Le projet de loi complète les dispositions de l'article L. 2141-10 pour ajouter une « évaluation médicale et psychologique » des demandeurs , qu'il s'agisse d'un couple ou d'une femme seule, comme préalable à l'accès à l'AMP ( 1° du I , article L. 2141-2 du code de la santé publique et 4° du I , article L. 2141-10 du code de la santé publique), en plus de la vérification de leur « motivation ».
L'arrêté du 30 juin 2017 précité prévoit à l'heure actuelle que l'équipe du centre d'AMP peut faire appel, seulement « si nécessaire » , à un médecin qualifié en psychiatrie ou un psychologue. Un entretien avec un psychiatre ou psychologue est en revanche systématiquement proposé pour un couple candidat à un accueil d'embryon.
5. La levée de l'interdiction du double don de gamètes
• La rédaction globale proposée par le 1° du I pour l'article L. 2141-3 du code de la santé publique procède à des coordinations et supprime la précision selon laquelle un embryon conçu in vitro « ne peut être conçu avec des gamètes ne provenant pas d'un au moins des membres du couple » .
Outre la prise en compte des remarques générales de l'Agence de la biomédecine ( cf . ci-après), cette évolution vise à tirer les conséquences de l'ouverture de l'AMP aux femmes seules, qui pourraient, si elles étaient dans l'incapacité de concevoir avec leurs propres ovocytes, uniquement avoir recours à un don d'embryon. Or, d'après l'étude d'impact, le Gouvernement souhaite que les nouveaux publics éligibles puissent avoir accès « à toutes les techniques disponibles ».
• Plus généralement, dans son rapport sur l'application de la loi de bioéthique, l'Agence de biomédecine relevait que si le double don de gamètes (ovocytes et spermatozoïdes) est exclu du champ de l'AMP, « la loi permet par ailleurs l'accueil d'embryons provenant de couples qui n'ont plus de projet parental » dont la conception n'a fait intervenir les gamètes d'aucun des deux membres du couple . Elle notait ainsi que, dans ces conditions, « l'interdiction du double don de gamètes peut soulever des incompréhensions , à la fois pour les professionnels qui sont amenés à traiter des cas de double infertilité masculine et féminine, et pour les couples potentiellement concernés ». Or, « l'activité d'accueil d'embryons peine à se développer et ne permet pas, aujourd'hui, de répondre à la demande des couples. »
En effet, l' accueil d'embryon reste une activité confidentielle : d'après les données de l'Agence de la biomédecine, 138 couples ont bénéficié d'un accueil d'embryon en 2017 et 18 enfants sont nés vivants. La même année, 582 couples ont consenti à l'abandon de leur projet parental et ont proposé les embryons conservés (1 654 soit 13 % des embryons ayant fait l'objet d'un consentement à l'abandon du projet parental) à l'accueil par un autre couple.
Au-delà de la complexité de la procédure, qui implique une collaboration formalisée avec l'un des 19 centres autorisés, les professionnels de santé entendus par le rapporteur et la commission spéciale ont fait état des barrières psychologiques pour le don, comme l'accueil, d'un embryon résultant d'un projet parental d'un couple et s'inscrivant dans une fratrie. En ce sens, le recours à un double don présenterait selon eux une charge symbolique moindre.
Les conditions d'accueil d'embryon ont néanmoins été récemment simplifiées par la loi du 23 mars 2019 12 ( * ) : le régime d'autorisation judiciaire - pouvant donner lieu à « toutes investigations permettant d'apprécier les conditions d'accueil que ce couple est susceptible d'offrir à l'enfant à naître sur les plans familial, éducatif et psychologique » - a été supprimé au profit d'un régime de consentement simple devant notaire. Le projet de loi ne revient pas sur cette évolution dont le bilan ne peut encore être tiré.
6. Une prise en charge intégrale par la sécurité sociale de l'AMP étendue à tous les demandeurs
• Le II fixe les conditions de prise en charge par l'assurance maladie de l'assistance médicale à la procréation, dans les conditions d'accès étendues telles que proposées par cet article 1 er .
A l'heure actuelle, le 12° de l'article L. 160-14 du code de la sécurité sociale 13 ( * ) prévoit l'exonération du ticket modérateur, c'est-à-dire la prise en charge intégrale par l'assurance maladie, des frais de santé concernant « les investigations nécessaires au diagnostic de la stérilité et pour le traitement de celle-ci, y compris au moyen de l'insémination artificielle » .
Afin de tenir compte de la suppression du critère d'infertilité comme condition d'accès à l'AMP, ces dispositions sont scindées en deux, afin de mentionner l'exonération de ticket modérateur, selon les conditions prévues au même article L. 160-14 susmentionné :
- d'une part, des investigations nécessaires au diagnostic de l'infertilité ( 1° du II ) ;
- d'autre part, de l'assistance médicale à la procréation dans les conditions prévues par le code ( 2° du II ).
Comme le note l'étude d'impact, afin d'assurer une équité dans l'accès à l'AMP, l'intention du Gouvernement est d'étendre la prise en charge de l'AMP selon les règles en vigueur pour les couples hétérosexuels à toutes les femmes , à savoir une prise en charge à 100 % par la sécurité sociale après entente préalable, jusqu'au 43 ème anniversaire de la femme, dans la limite de six inséminations artificielles (à raison d'une par cycle) et de quatre tentatives de fécondation in vitro pour obtenir une grossesse 14 ( * ) .
• L'étude d'impact évalue le coût de cette mesure pour l'assurance maladie entre 10 et 15 millions d'euros soit moins de 5 % du coût total de l'assistance médicale à la procréation évalué à environ 300 millions d'euros 15 ( * ) .
Cette évaluation repose sur des estimations par définition fragiles, comme le rapporteur le souligne ci-après concernant l'impact de cette ouverture sur les gamètes disponibles pour le don.
7. Les coordinations diverses rendues nécessaires
D'autres articles du code de la santé publique, dont le projet de loi propose la réécriture globale, sont modifiés par coordination afin d'ouvrir l'ensemble des techniques d'AMP aux nouveaux publics demandeurs.
C'est ainsi le cas des articles L. 2141-5 et L. 2141-6 relatifs au don et à l'accueil d'embryon .
Les modifications visent à étendre la possibilité à une femme non mariée , et non seulement à un couple, de consentir à l'accueil des embryons conservés dans le cadre d'une procédure d'AMP, dès lors que le projet parental n'est plus poursuivi, ou de bénéficier d'un tel don.
Parallèlement, les conditions fixées pour accueillir un embryon , c'est-à-dire lorsque les techniques d'AMP au sein du couple « ne peuvent aboutir » ou que celui-ci y renonce, sont supprimées par coordination avec la suppression du critère médical d'accès à l'AMP.
Est également prévue l' information des personnes consentant à l'accueil d'un embryon sur les dispositions de l'article 3 du projet de loi relatives à l'accès des enfants nés d'un don, à leur majorité, aux données non identifiantes ou à l'identité des tiers donneurs.
II - Les modifications adoptées par l'Assemblée nationale
• La commission spéciale a introduit, outre des amendements rédactionnels ou de coordination de son rapporteur Jean-Louis Touraine, plusieurs modifications de fond au texte, visant à :
- remplacer le principe d'une « évaluation médicale et psychologique » préalable à l'AMP par des « entretiens particuliers » des demandeurs avec les membres de l'équipe pluridisciplinaire du centre, selon les termes du droit en vigueur (amendement présenté par Martine Wonner et les membres du groupe La République en marche, adopté avec l'avis favorable du rapporteur et du Gouvernement), considérant, d'après l'exposé sommaire, que l'évaluation psychologique fait partie intégrante de la prise en charge par l'équipe pluridisciplinaire. Pour les auteurs de l'amendement, le fait d'introduire une évaluation psychologique au moment où l'on ouvre l'AMP aux couples de femmes et aux femmes seules était perçu comme « de nature à semer la confusion ». La commission spéciale a parallèlement précisé, à l'initiative des mêmes auteurs, que cette équipe pluridisciplinaire sera « composée notamment d'un psychiatre, d'un psychologue, ou d'un infirmier ayant une compétence en psychiatrie , le cas échéant extérieur au centre » . Par cohérence, elle a maintenu comme finalité à ces entretiens l'évaluation médicale des demandeurs, sans mentionner expressément le principe d'une évaluation psychologique (cinq amendements identiques de Anne-France Brunet et Martine Wonner (La République en marche), Bruno Fuchs (Modem), Hervé Saulignac (Socialistes et apparentés), Sylvia Pinel (Libertés et Territoires)) ;
- affirmer le principe de non-discrimination dans l'examen des demandes , en précisant, d'une part, que l'accès à l'AMP « ne peut faire l'objet d'aucune différence de traitement notamment au regard du statut matrimonial ou de l'orientation sexuelle des personnes » (amendement présenté par Guillaume Chiche et les membres du groupe La République en marche, adopté avec l'avis favorable du Gouvernement) et, d'autre part, que l'évaluation médicale ne peut conduire à « débouter le couple ou la femme non mariée en raison de son orientation sexuelle, de son statut marital ou de son identité de genre » (amendement présenté par Hervé Saulignac et des membres du groupe socialistes et apparentés, adopté avec l'avis favorable du rapporteur) ;
- proposer une étude de suivi au couple receveur ou à la femme receveuse, sous réserve de leur consentement (amendement de Jean-Louis Touraine, rapporteur), afin de disposer de données scientifiques sur le devenir des receveurs de gamètes ou d'embryons issus de tiers donneurs et des enfants issus de ce don ;
- refuser, au-delà de tout paiement, toute « contrepartie » au don d'embryon (amendement présenté par Laurence Vanceunebrock-Mialon et les membres du groupe La République en marche) ;
- prévoir l' information des couples demandeurs sur les dispositions prévues en cas de décès de l'un d'eux (amendement de Jean-Louis Touraine, rapporteur) ;
- inclure dans le dossier-guide remis aux demandeurs des éléments d'information sur l'accès aux données non identifiantes et à l'identité du tiers donneur, ainsi que la liste des associations et organismes susceptibles de compléter leur information sur ce sujet, par coordination avec les dispositions prévues à l'article 3 (amendement de Jean-Louis Touraine, rapporteur) ;
- préciser que le médecin prenant la décision in fine d'accepter ou non la prise en charge doit avoir participé aux entretiens préalables avec les demandeurs (amendement de Jean-Louis Touraine, rapporteur) ;
- remettre au Parlement en 2025 un rapport d'évaluation sur les dispositions du présent article (amendement de Marie Tamarelle-Verhaeghe et de membres du groupe La République en marche).
• En séance publique , outre des précisions rédactionnelles ou coordinations, l'Assemblée nationale a adopté les modifications suivantes :
- elle a précisé que l'AMP « est destinée à répondre à un projet parental » (amendement présenté par Raphaël Gérard et des membres du groupe La République en marche, adopté avec l'avis favorable de la commission spéciale et l'avis de sagesse du Gouvernement). Pour mémoire, une formule proche - selon laquelle l'AMP « est destinée à répondre à la demande parentale d'un couple » - était employée dans le code de la santé publique jusqu'à la loi de juillet 2011 qui l'a supprimée, d'après son exposé des motifs, « afin de mieux mettre en exergue la condition médicale d'infertilité permettant à un couple d'accéder à ces techniques » ;
- elle a prévu une consultation préalable du couple sur le devenir des embryons conservés en cas de décès d'un de ses membres (amendement présenté par Elsa Faucillon et des membres du groupe de la Gauche démocrate et républicaine, adopté contre l'avis défavorable de la commission spéciale 16 ( * ) mais avec l'avis favorable du Gouvernement). Une modification similaire a été introduite à l'article 16 du projet de loi ;
- elle a remplacé le terme de renoncement aux embryons par le terme de consentement à leur accueil (amendements présentés par Annie Genevard et des membres du groupe Les Républicains, adoptés avec l'avis favorable de la commission et du Gouvernement) ;
- elle a précisé que les entretiens avec l'équipe pluridisciplinaire pouvaient être effectués par d' autres professionnels de santé que des médecins, comme des sages-femmes ou psychologues (amendement de Marie Tamarelle-Verhaeghe et de membres du groupe La République en marche) ;
- elle a renvoyé à un décret en Conseil d'Etat la composition de l'équipe clinicobiologique pluridisciplinaire (amendement de Martine Wonner et de membres du groupe La République en marche). Cette composition n'est aujourd'hui pas précisée, en dehors des indications figurant dans l'arrêté de bonnes pratiques du 30 juin 2017 ;
- elle a précisé que l'information donnée aux demandeurs sur les possibilités de réussite ou d'échec, ainsi que les risques et contraintes de l'AMP se fait « complètement et au regard de l'état des connaissances scientifiques » (amendement de Jean-Louis Touraine, rapporteur) afin, d'après l'exposé sommaire, de « favoriser un consentement éclairé » ;
- elle a précisé que lors des entretiens particuliers préalables à la mise en oeuvre de l'AMP, « les membres du couple sont incités à anticiper et créer les conditions qui leur permettront d'informer l'enfant, avant sa majorité, de ce qu'il est issu d'un don » (amendement présenté par Martine Wonner et des membres du groupe La République en marche, adopté contre l'avis défavorable de la commission spéciale 17 ( * ) mais avec l'avis favorable du Gouvernement), « au nom de l'intérêt supérieur de l'enfant à savoir qu'il est né d'un don » d'après l'exposé sommaire ;
- elle a enfin précisé, afin de lever toute ambiguïté, que sont pris en charge par la sécurité sociale sans ticket modérateur les investigations nécessaires non seulement au diagnostic de l'infertilité mais également à son traitement comme c'est le cas à l'heure actuelle (amendement de Thibault Bazin, Les Républicains, adopté avec l'avis favorable de la commission spéciale et du Gouvernement).
III - La position de la commission : préciser les conditions d'accès et de prise en charge
1. Une évolution sociétale guidée par un choix politique et non pas fondée sur un principe juridique d'égalité
Cet article 1 er du projet de loi traduit, au-delà d'un engagement présidentiel en faveur d'une « PMA pour toutes », une réponse à des demandes exprimées par des femmes en couple avec une autre femme ou vivant seules de voir reconnu leur désir légitime à vouloir devenir mères.
La satisfaction de ce désir, le législateur l'a déjà rendue possible par le biais de l'adoption, ouverte à toutes les personnes seules - femme ou homme - et à tous les couples mariés.
Autoriser l'accès à un geste médical relativement simple, l'insémination artificielle ou la fécondation in vitro , a été perçu comme un prolongement naturel dans la reconnaissance par la société de la diversité des moyens de « faire famille » et de l'égale valeur des différents modèles familiaux. Au-delà, le fait de ne pas avoir accès en France à des techniques autorisées dans un grand nombre de pays voisins a suscité un sentiment d'injustice voire de discrimination.
Cette évolution est largement soutenue par la communauté médicale, au nom d'un « principe de réalité » : les femmes souhaitant avoir un enfant - à tout le moins pour celles qui en ont les moyens - se rendent aujourd'hui au-delà de nos frontières, principalement en Espagne ou en Belgique, pour réaliser un projet qui leur est refusé en France.
Cette question éminemment sociétale a été au coeur des Etats généraux de la bioéthique, au cours desquels se sont exprimées des attentes d'égalité mais aussi des dissensions dont le compte rendu du CCNE fait état.
Avant de relever les interrogations que ce texte soulève pour le rapporteur et des membres de la commission spéciale, il faut d'abord rappeler que les travaux préparatoires au projet de loi ont confirmé que si un sentiment de discrimination s'exprime, l'évolution engagée par le projet de loi n'est pas motivée par un motif juridique : dans son étude du 28 juin 2018 sur la révision de la loi de bioéthique, le Conseil d'État a clairement rappelé qu' « aucun argument juridique n'impose en effet l'extension de l'AMP. Ni le fait que l'adoption soit ouverte aux couples de femmes et aux personnes seules, ni le principe d'égalité, ni le droit au respect de la vie privée, ni la liberté de procréer, pas plus que l'interdiction des discriminations ne rendent nécessaire l'ouverture d'accès à l'AMP. »
C'est d'ailleurs ce que le Conseil constitutionnel avait acté en 2013 en considérant que l'ouverture du mariage et de l'adoption aux couples de même sexe n'imposait pas, au nom du principe d'égalité, de modifier la législation en matière d'assistance médicale à la procréation, dès lors que « les couples formés d'un homme et d'une femme sont, au regard de la procréation, dans une situation différente de celle des couples de personnes de même sexe » 18 ( * ) .
Le « sentiment » de discrimination mis en avant ne recouvre donc pas la notion juridique de discrimination qui consiste à ne pas accorder les mêmes droits à des personnes placées dans une situation identique.
Par ailleurs, l'argument selon lequel il serait « réaliste » d'ouvrir un certain nombre de droits au motif qu'ils peuvent être exercés dans d'autres pays ne peut être suivi. Il suffirait alors au législateur français d'observer les systèmes juridiques les moins contraignants pour les traduire en droit national. Prôner cela aurait pour conséquence de faire systématiquement le choix du « moins-disant » éthique.
L'extension de l'AMP relève donc d'un choix politique dont le principe et les conséquences ne suscitent pas un large consensus tant ils soulèvent d'interrogations.
2. Une évolution qui soulève des interrogations de différentes natures
• Une première interrogation, dès lors que l'on ouvre un nouveau droit, porte sur les conditions permettant de s'assurer de son effectivité .
En l'occurrence, les spécialistes de la médecine de reproduction alertent sur les besoins supplémentaires en don de gamètes , principalement mais pas exclusivement de sperme : l'ouverture de l'accès à l'AMP à de nouveaux bénéficiaires supposera d'après la fédération des CECOS de multiplier par 2 voire 3 environ les gamètes disponibles pour le don, sur la base des évaluations - forcément fragiles - du Gouvernement de 2 000 à 4 000 demandes supplémentaires attendues. Comme l'ont relevé des sociétés savantes, 2 000 patientes supplémentaires impliqueraient de l'ordre de 6 000 tentatives en plus dès lors que plusieurs inséminations pourraient être nécessaires afin d'aboutir à une grossesse.
Si le don de sperme permet aujourd'hui de couvrir les besoins (avec aujourd'hui de l'ordre de 2 000 couples recourant chaque année à une AMP avec don de sperme), tel n'est pas le cas du don d'ovocyte pour lequel jusqu'à trois ans d'attente sont requis selon les centres (avec 2 726 couples en attente d'un don fin 2017, en augmentation continue depuis 2014) ce qui conduit déjà de nombreux couples à se rendre à l'étranger.
Or, les autres évolutions engagées par le projet de loi aux articles 2 et 3 (dissociation entre don et autoconservation, levée de l'anonymat des donneurs) comme la levée de l'interdiction du double don de gamètes ouvrent à cet égard une période d'incertitude et donc de probable tension.
Pour votre rapporteur, la tentation d'une marchandisation dans un contexte de pénurie qui aurait été mal anticipé ne serait pas acceptable ; elle propose en ce sens à l'article 2 des évolutions pour mieux encadrer les importations de gamètes.
Plus généralement, le projet de loi pourrait conduire indirectement à remettre en cause un des principes fondamentaux de la bioéthique qui est la gratuité du don. C'est d'ailleurs la question posée dans une récente tribune 19 ( * ) par le Professeur Israël Nisand, invitant à débattre « sans tabou » de la gratuité du don de gamètes qui serait une source de sa pénurie.
• Au-delà de ce sujet, l'ouverture de l'AMP à toutes les femmes soulève pour le rapporteur deux principales interrogations de fond.
- D'une part, telle que la propose le projet de loi, elle induit un changement de nature de la finalité de l'assistance médicale à la procréation en la dissociant de tout critère médical .
La finalité « sociale » de contribuer à un projet parental prendrait ainsi le pas sur la finalité « médicale » de remédier à une situation d'infertilité d'un couple dont le caractère pathologique a été diagnostiqué ou d'éviter la transmission d'une maladie génétique grave et incurable.
Dans 96 % des cas à l'heure actuelle, cette technique est réalisée en intraconjugal, c'est-à-dire sans intervention d'un tiers donneur, l'acte médical en lui-même permettant de remédier à une infertilité de fait, que la cause précise en soit identifiée ou non en l'état des connaissances scientifiques.
La déconnexion de cette technique médicale de tout critère pathologique recentre sa finalité sur la satisfaction d'une demande ou d'un désir, celui d'avoir un enfant. Quelle que soit la légitimité de cette demande ou de ce désir, cette évolution appelle à s'interroger, comme le relève le CCNE dans son compte rendu des États généraux de la bioéthique : « Un désir - même compréhensible - peut-il aboutir à un droit ? ».
Cette question de principe sur la finalité du recours à un acte médical se pose d'autant plus que celui-ci serait intégralement pris en charge par la solidarité nationale . La volonté d'assurer l'équité dans l'accès à ce droit conduit à endosser une logique nouvelle de recours à notre système d'assurance maladie aujourd'hui fondé sur « la protection contre le risque et les conséquences de la maladie » 20 ( * ) .
- D'autre part, l'évolution proposée par cet article conduit le législateur à banaliser l'absence de filiation paternelle, ce qui pose la question de l'intérêt de l'enfant à naître.
Si le Conseil d'État a estimé que « la notion juridique d'intérêt supérieur de l'enfant ne saurait faire obstacle à l'ouverture de l'AMP aux couples de femmes et aux femmes seules » , il est toutefois permis de s'interroger sur les conséquences d'un tel projet de loi sur l'enfant.
A l'heure actuelle, la technique proposée par l'AMP « mime » une procréation charnelle qui serait théoriquement possible et conduit à l'établissement d'une filiation sexuée vraisemblable. Dans le cas d'un couple de femmes ou d'une femme seule, l'accès à l'AMP conduirait à accepter le fait qu'un enfant soit privé dès le départ de sa lignée paternelle , conséquence, selon les propos du rapporteur de la mission d'information de l'Assemblée nationale sur l'évaluation de la loi de bioéthique, d'une « procréation sans sexe pour tous ».
Si le Gouvernement considère dans l'étude d'impact, sur la base d'études la plupart étrangères, que le projet de loi est conforme à la priorité qu'il attache au respect des droits de l'enfant et à la protection de l'enfance, des pédopsychiatres entendus par le rapporteur ont attiré l'attention sur les conséquences de ces conditions de venue au monde sur les fondations psychiques de l'enfant , comme celles de l'absence de diversité des sexes dans son développement. Dans le même sens, l' Académie de médecine , comme l'a mis en avant Jean-François Mattéi lors de son audition par la commission spéciale, a émis, en se plaçant du point de vue de l'intérêt de l'enfant, des réserves sur « de potentielles conséquences d'ordre médical » : l'Académie a ainsi conclu à la nécessité de poursuivre les études sur ce sujet, au vu des résultats discordants, discutables ou incertains au plan scientifique des études disponibles. Elle a également estimé que la figure du père restait fondatrice pour la personnalité de l'enfant.
Bien évidemment, il existe de facto une diversité des situations familiales ; des couples de femmes et des femmes seules élèvent des enfants auxquels elles sont liées soit par un lien de procréation charnelle, soit par un lien adoptif.
Ouvrir l'AMP aux femmes seules et aux couples de femmes revêt une portée différente de celle consistant à prendre acte de situations existantes ou à permettre l'adoption d'enfants déjà nés. Cela consiste en effet à institutionnaliser l'absence de filiation paternelle.
Or, dans l'intérêt de l'enfant, et à rebours des évolutions ici engagées, le législateur a tenu, au cours de récentes évolutions concernant l'exercice conjoint de l'autorité parentale et la résidence alternée 21 ( * ) , le congé de paternité ou encore le partage plus équitable du congé parental 22 ( * ) , à renforcer la place des pères dans l'éducation de leurs enfants.
Si l'appréciation de l'intérêt de l'enfant à travers ces textes peut être discutable, il a toujours été associé à une plus grande présence du père auprès des enfants. L'exposé des motifs du projet de loi relatif à l'égalité réelle entre les femmes et les hommes présentait ainsi la réforme du congé parental comme visant à « favoriser un meilleur partage des responsabilités parentales et permettre aux pères qui souhaitent s'investir auprès de leurs enfants de ne plus être confrontés au poids des résistances culturelles » . Dans l'avis 23 ( * ) de la commission des affaires sociales sur ce même texte, notre collègue Michelle Meunier, rapporteure, rappelle en effet que « de nombreuses études soulignent les retombées extrêmement positives d'une participation précoce des pères à l'éducation de leurs enfants, en particulier en termes de socialisation. »
L'accès aux origines du tiers donneur ouvert par l'article 3 ne saurait tenir lieu de succédané de père.
L'ouverture de l'AMP aux couples de femmes conduit enfin le Gouvernement à opérer à l'article 4 une « révolution » dans le droit civil en établissant une filiation maternelle d'intention, détachée de toute référence aux conditions naturelles de la procréation fondées sur la vraisemblance biologique. Cette ouverture ne va pas sans susciter des craintes d'un glissement vers une autorisation de la gestation pour autrui, fondée sur une parentalité d'intention, au nom du même principe d'égalité - cette fois à l'égard des couples d'hommes notamment - face au désir d'enfant.
- L'intérêt de l'enfant à naître conduit enfin à faire preuve d'une vigilance toute particulière à l'égard de l'ouverture de l'AMP aux femmes seules .
Certains professionnels de la médecine de reproduction ont formulé plus de réserves sur ce point, à l'instar du CCNE qui envisageait dans son avis n° 126 de réfléchir à des « dispositions d'accompagnement des demandes de femmes seules (...) qui pourraient s'inspirer de celles qui s'appliquent au cadre de l'adoption plénière, ou prendre d'autres formes plus spécifiques à ce type de situations nouvelles » 24 ( * ) . Les représentantes associatives entendues font valoir une monoparentalité choisie et non subie, qui emporte en effet une grande différence. Le rapporteur s'interroge toutefois, même dans ce contexte, sur la plus grande vulnérabilité inévitable de ces situations, qui se traduisent notamment en Belgique à un plus fort taux de refus des demandes après enquête sociale (de l'ordre de 30 %). Cela a été confirmé par les associations familiales ainsi que les pédopsychiatres entendus, quelles que soient d'ailleurs leurs divergences d'appréciation sur le principe même de l'ouverture de l'AMP ; ceux-ci ont attiré l'attention sur l'importance de la présence d'une altérité dans la relation mère-enfant.
• Ces différentes considérations ont conduit le rapporteur à présenter un amendement de suppression de l'article.
Au terme d'un large débat, la commission spéciale a cependant approuvé l'ouverture aux couples de femmes et aux femmes non mariées de l'assistance médicale à la procréation tout apportant de nombreux ajustements à cet article.
3. Les modifications adoptées par la commission spéciale : le maintien d'un critère médical d'accès à l'AMP pour les couples hétérosexuels et la prise en charge des seules demandes fondées sur ces indications médicales
• A l'initiative de son rapporteur (amendement COM-171), la commission spéciale a maintenu les conditions médicales d'accès à l'assistance médicale à la procréation pour les couples hétérosexuels , fondées sur une infertilité pathologique ou la non transmission à l'enfant ou à l'autre membre du couple d'une pathologie d'une particulière gravité. Elle a inscrit au sein d'un nouvel article du code de la santé publique l'accès à cette procédure des couples de femmes et des femmes non mariées.
Pour l'ensemble des demandeurs, la commission spéciale a assoupli les conditions d'âge pour accéder à l'AMP, en les renvoyant à une recommandation de bonnes pratiques plutôt qu'à un décret en Conseil d'Etat, afin de ménager plus de souplesse dans l'appréciation par les équipes médicales des situations individuelles (même amendement COM-171).
Elle a adopté, en outre, plusieurs amendements de coordination ou de précision rédactionnelle ( amendements COM-172, COM-173, COM-174, COM-175 et COM-180 du rapporteur et COM-102 de Thani Mohamed Soilihi ). Par cohérence, elle a également rétabli l'article L. 2141-7 du code de la santé publique sur les conditions de recours à une assistance médicale à la procréation avec tiers donneur tout en élargissant ces conditions aux nouveaux publics demandeurs ( amendement COM-182 du rapporteur ).
• La commission spéciale a adopté parallèlement l'amendement COM-181 de son rapporteur réservant la prise en charge par l'assurance maladie, selon les conditions actuellement en vigueur, de l'assistance médicale à la procréation aux seules demandes fondées sur ces indications médicales , dès lors que le système de prise en charge solidaire des soins a vocation à assurer la « protection contre le risque et les conséquences de la maladie » aux termes du code de la sécurité sociale (article L. 111-2-1).
• Concernant les conditions de l'accès à la procédure d'AMP, la commission spéciale a par ailleurs maintenu le caractère médical de l'équipe pluridisciplinaire des centres d'AMP en y associant un pédopsychiatre ou pédopsychologue , plutôt qu'un « infirmier ayant une compétence en psychiatrie » comme prévu par l'Assemblée nationale, dès lors qu'il est opportun de placer les investigations sous l'angle de l'intérêt de l'enfant à naître ( amendement COM-176 du rapporteur ).
Elle a substitué à la formulation de la vérification de la « motivation » des demandeurs, ambigüe dès lors qu'il ne s'agit pas de juger de la légitimité d'un projet parental, celle de leur volonté de poursuivre la démarche après avoir reçu les informations utiles, en rétablissant le rappel des possibilités ouvertes par la loi en matière d'adoption, supprimé par le projet de loi ( amendement COM-177 du rapporteur ).
Elle a également rétabli explicitement le principe d'une évaluation non seulement médicale mais également psychologique des demandeurs en amont de la démarche, en y ajoutant, en tant que de besoin, une évaluation sociale ( amendement COM-132 rectifié de Roger Karoutchi ).
• Parallèlement, la commission spéciale a supprimé des dispositions ajoutées par l'Assemblée nationale ne présentant pas de portée suffisante :
- d'abord, la précision selon laquelle l'évaluation médicale ne peut conduire à débouter un demandeur « en raison de son orientation sexuelle, de son statut marital ou de son identité de genre », qui semblait jeter le doute sur la capacité des équipes médicales à examiner de manière loyale et impartiale l'ensemble des demandes. Cette précision est en outre inutile dès lors que le code de déontologie médicale comporte déjà un principe général de non-discrimination ( amendement COM-179 du rapporteur ) ;
- ensuite, l'invitation des couples à « anticiper et créer les conditions qui leur permettront d'informer l'enfant, avant sa majorité, de ce qu'il est issu d'un don », dont la formulation est ambigüe et la portée normative discutable. Cette disposition est en outre satisfaite sur le fond par l'information sur ce sujet qui sera délivrée aux demandeurs dans le dossier-guide ( amendement COM-178 du rapporteur ) ;
- enfin, la demande de rapport d'évaluation de cet article, qui n'est pas nécessaire dès lors que les travaux préparatoires au réexamen général de la loi fourniront de ce point de vue les éléments d'évaluation utiles ( amendement COM-163 du rapporteur ).
• Enfin, la commission spéciale a élargi à l'ensemble des établissements de santé privés l'activité de mise en oeuvre de la procédure d'accueil d'embryons , actuellement réservée, comme celle de don de gamètes, aux seuls établissements publics et privés à but non lucratif ( amendement COM-63 de Jacques Bigot et des membres du groupe socialiste et républicain). Cette évolution vise à réduire les délais importants constatés dans l'accès à cette procédure.
La commission a adopté cet article ainsi modifié.
Article
1er bis
Rapport au Parlement sur la structuration
des centres
d'assistance médicale à la procréation
Cet article, inséré par l'Assemblée nationale, demande la remise d'un rapport au Parlement évaluant la structuration des centres d'assistance médicale à la procréation et leurs taux de réussite. La commission spéciale a supprimé cette demande qu'elle a considérée satisfaite notamment par la définition des missions de l'Agence de la biomédecine.
I - Le dispositif adopté par l'Assemblée nationale
Cet article a été introduit par la commission spéciale à l'initiative de Philippe Berta et de plusieurs députés du groupe du Modem, avec un avis de sagesse du rapporteur Jean-Louis Touraine. Il a fait l'objet de modifications rédactionnelles lors de l'examen en séance publique.
Il demande au Gouvernement la remise d'un rapport au Parlement, dans un délai d'un an, portant sur la structuration des centres d'assistance médicale à la procréation, leurs taux de réussite respectifs et l'opportunité d'une évolution structurelle, dans l'objectif notamment, mis en avant par l'auteur de l'amendement, d'assurer une égalité des chances pour les personnes qui recourent à cette technique. Le texte adopté précise, en outre, que ce rapport peut faire l'objet d'un débat dans les conditions prévues par les règlements des deux assemblées.
II - La position de la commission
La structuration et l'évaluation des centres d'assistance médicale à la procréation sont d'ores et déjà encadrées par l'Agence de la biomédecine .
D'une part, les activités cliniques et biologiques d'AMP sont soumises à autorisation des agences régionales de santé (ARS) après avis de l'Agence de la biomédecine 25 ( * ) , laquelle est chargée d'évaluer ces activités.
D'autre part, la loi de juillet 2011 a confié à l'Agence de biomédecine la mission de publier régulièrement les résultats de chaque centre d'AMP « selon une méthodologie prenant en compte notamment les caractéristiques de leur patientèle et en particulier l'âge des femmes » afin de parvenir à des résultats comparables. Au vu de ces données, l'Agence peut diligenter des missions d'appui et de conseil - ce qui a été le cas pour deux centres jusqu'à présent d'après les indications transmises au rapporteur - voire proposer des recommandations d'indicateurs chiffrés (4° de l'article L. 1418-1 du code de la santé publique).
Certes, des progrès sont toujours possibles en termes de transparence des résultats mis à disposition des patients, comme l'a récemment préconisé la Cour des comptes 26 ( * ) .
Cependant, la commission spéciale a estimé qu'un nouveau rapport sur le sujet n'aurait qu'une portée limitée au regard des missions qui incombent déjà à l'Agence de la biomédecine y compris pour formuler des recommandations au Parlement ou répondre aux sollicitations de ses commissions permanentes. En outre, la loi n'a pas à prévoir un débat dans les assemblées parlementaires sur un tel rapport du Gouvernement.
Pour ces raisons, la commission spéciale a adopté, à l'initiative de son rapporteur, l'amendement COM-157 de suppression de l'article .
La commission a supprimé cet article.
Article
2
Assouplissement du don de gamètes
et autorisation de leur
autoconservation
Cet article autorise l'autoconservation de gamètes indépendamment d'une démarche de don en mettant fin à la possibilité ouverte en 2011 pour les donneurs n'ayant pas procréé de conserver des gamètes à des fins autologues, et supprime le consentement du conjoint au don de gamètes.
La commission spéciale a rétabli le consentement du conjoint au don de gamète et a assoupli les conditions d'âge pour bénéficier de l'autoconservation tout en renforçant l'information des demandeurs. Elle a également autorisé les établissements de santé privés à pratiquer cette activité, de même, qu'à titre dérogatoire, l'activité de don de gamètes. Elle a encadré les dispositions, enfin, relatives à l'importation de gamètes.
I - Le dispositif proposé : « autoriser sans encourager » l'autoconservation des gamètes
1. La clarification et l'assouplissement des conditions du don de gamètes, notamment par la suppression du consentement du conjoint
• La loi du 6 août 2004 a levé la condition pour le donneur de gamètes de faire partie d'un couple ayant procréé mais a maintenu la double condition d'avoir procréé d'une part et, pour le donneur en couple, de consentement de l'autre membre du couple d'autre part.
L'article 29 de la loi du 7 juillet 2011, tout en levant l'obligation de maternité ou paternité antérieure ( cf. ci-après), a confirmé le principe du consentement du conjoint, lequel, comme celui du donneur, est recueilli par écrit et peut être révoqué à tout moment jusqu'à l'utilisation des gamètes.
• Le I clarifie la rédaction de l'article L. 1244-2 du code de la santé publique afin de lever la condition de principe d'avoir procréé pour être donneur, puisque celle-ci acceptait depuis 2011 une dérogation générale.
Dans le même temps, cela répond à une limite de rédaction qu'avait soulignée l'Agence de la biomédecine, puisque le consentement au don et sa révocabilité ne sont pas traités de façon explicite, à l'heure actuelle, pour le donneur n'ayant pas procréé.
Les autres modifications proposées répondent à plusieurs objectifs :
- réaffirmer la condition pour le donneur d' être majeur , en excluant explicitement cette possibilité pour le mineur émancipé ;
- prévoir, par coordination avec les dispositions de l'article 3 du projet de loi, l' information préalable du donneur sur les dispositions relatives à la communication des données non identifiantes le concernant ou à son identité par les personnes nées d'un don ;
- supprimer les dispositions relatives au consentement des membres du couple « receveur » et à sa révocabilité, qui figurent déjà à l'article 311-20 du code civil, dont les dispositions sont reprises à l'article 342-10 dans la rédaction prévue par l'article 4 du projet de loi ;
- supprimer, enfin, le consentement du conjoint au don .
Sur ce dernier point, l'étude d'impact relève que la France est le seul pays en Europe à solliciter le consentement du conjoint pour le don de gamètes : or, la possibilité de sa révocation « prive le donneur de la liberté de maintenir son don » , autant qu'elle pose question, d'après le Gouvernement, avec la possibilité, ouverte par le projet de loi, d'accès à l'identité du tiers donneur. L'Agence de la biomédecine avait quant à elle considéré, dans son rapport de janvier 2018 sur l'application de la loi de bioéthique, que cette condition ne paraissait plus d'actualité .
2. L'autorisation de l'autoconservation de gamètes pour des raisons non médicales et de manière déconnectée d'un don
a) L'autoconservation conditionnée à une démarche de don ouverte par la loi de 2011 : une possibilité faisant l'objet de critiques
- Une possibilité réservée aux donneurs n'ayant pas procréé et limitée à une partie résiduelle des gamètes recueillis en vue d'un don
• L'autoconservation de gamètes ou de tissus germinaux, en vue notamment de la réalisation ultérieure d'une assistance médicale à la procréation, n'est autorisée en France qu' en cas de pathologie ou de traitement affectant la fertilité 28 ( * ) .
Plus ponctuellement, elle peut également être décidée par l'équipe médicale, dans certaines circonstances, dans le cadre d'un parcours d'AMP, en vue d'une mise en fécondation différée 29 ( * ) .
La loi de bioéthique du 7 juillet 2011 a ouvert une nouvelle possibilité : en étendant le don de gamètes aux personnes majeures n'ayant pas procréé , le législateur a prévu, dans cette situation, que ces donneurs puissent se voir proposer le recueil et la conservation d'une partie des gamètes ou tissus germinaux prélevés en vue de la réalisation ultérieure, à leur bénéfice, d'une assistance médicale à la procréation 30 ( * ) .
Ces dispositions ont été précisées par le décret du 13 octobre 2015 31 ( * ) et l'arrêté du 24 décembre 2015. Leur mise en oeuvre n'a ainsi été effective qu'à compter de 2016.
Ces textes réglementaires ont notamment fixé les règles de répartition entre les gamètes orientées vers le don et celles conservées au bénéfice du donneur , l'autoconservation étant impossible en cas d'obtention d'une quantité insuffisante de gamètes :
- pour les donneuses, jusqu'à 5 ovocytes matures obtenus, tous les ovocytes sont destinés au don ; de 6 à 10 ovocytes, au moins 5 sont destinés au don ; au-delà de 10, au moins la moitié est dirigée vers le don ;
- pour les donneurs, au-delà de 3 recueils de sperme, un recueil peut être proposé en vue d'une autoconservation.
• Le décret introduit une consultation annuelle sur le maintien ou non de la conservation ; si la personne ne souhaite plus maintenir la conservation à son bénéfice, celle-ci se poursuit exclusivement en vue de don . Il en est de même en l'absence de réponse du donneur, consulté à plusieurs reprises, lorsque la durée de conservation a dépassé dix ans, ainsi qu'en cas de décès du donneur ou si celui-ci n'est plus en âge de procréer (article R. 1244-7 du code de la santé publique).
- Un bilan mitigé pour une demande néanmoins émergente
L'application récente de cette mesure, depuis 2016, ne permet pas de bénéficier d'un recul suffisant pour évaluer, quantitativement, les effets de cette mesure sur le nombre de donneurs.
On constate toutefois, entre 2016 et 2017, années pour lesquelles des données de l'Agence de la biomédecine sont disponibles, une hausse significative du nombre de donneurs ou donneuses n'ayant pas procréé , qui dépasse en 2017, s'agissant des hommes, le nombre de ceux ayant procréé.
Évolution du nombre de donneurs, de donneurs
n'ayant pas procréé
et de l'autoconservation dans le cadre
d'un don
|
2015 |
2016 |
2017 |
|
|
Nombre de donneurs de spermatozoïdes dans l'année |
255 |
399 |
404 |
|
dont hommes n'ayant pas procréé |
- |
148 |
205 |
|
(en % du nombre total de donneurs) |
- |
37 % |
51 % |
|
Part des donneurs n'ayant pas procréé ayant bénéficié d'une autoconservation |
- |
47 % |
43 % |
|
Nombre de donneuses d'ovocytes dans l'année |
540 |
741 |
756 |
|
dont femmes n'ayant pas procréé |
- |
144 |
245 |
|
(en % du nombre total de donneurs) |
- |
19 % |
32 % |
|
Part des donneuses n'ayant pas procréé ayant bénéficié d'une autoconservation |
- |
31 % |
45 % |
Source : Agence de la biomédecine, rapport médical et scientifique. Pour 2016, les données concernant les hommes n'ayant pas procréé sont issues d'un premier bilan incomplet des CECOS, cité dans l'étude d'impact du projet de loi
D'après l'Agence de la biomédecine, l'âge moyen des donneuses ayant bénéficié d'une autoconservation dans le cadre d'un don était de 30,5 ans en 2016. Les données des CECOS citées dans l'étude d'impact mettent en évidence un âge moyen de 33,4 ans des donneurs de sperme ayant réalisé une autoconservation la même année.
- Un dispositif critiqué comme une forme de leurre et de contrepartie au don
Les dispositions introduites en 2011 ont soulevé, dans le cadre des travaux préparatoires au projet de loi, des appréciations critiques.
• Le Conseil d'Etat, dans son étude du 28 juin 2018 sur la révision de la loi bioéthique, note ainsi qu' « un consensus se dégage pour considérer que le dispositif actuel d'autoconservation contre don est contraire au principe de gratuité » en ce qu'il « consiste à inciter à donner ses ovocytes en créant une forme de contrepartie au don ».
• Le CCNE, dans son avis n° 129, partage ce constant en relevant « une certaine contradiction avec l'article L. 1211-4 du code de la santé publique » , aux termes duquel « aucun paiement, quelle qu'en soit la forme, ne peut être alloué à celui qui se prête au prélèvement d'éléments de son corps ou à la collecte de ses produits » . Il préconisait de séparer clairement les deux démarches : un don gratuit d'une part, et la prise en charge d'une autoconservation de précaution d'autre part, sous certaines conditions. Le CCNE envisageait ainsi « pour une femme qui n'aurait pas eu l'opportunité de réaliser son désir d'enfant plus tôt, l'autoconservation de ses ovocytes à un âge (30-35 ans) où sa fertilité est encore optimale, associée à une limite de conservation et un âge maximal de conservation ».
• Dans un rapport de juin 2017 32 ( * ) , l' Académie de médecine dresse un bilan également très critique de cette possibilité, pouvant être perçue comme « un chantage » ou « un leurre au détriment des donneuses motivées prioritairement par le projet de conserver des ovocytes pour elles-mêmes » , et « médicalement et éthiquement inacceptable » en donnant des chances extrêmement minces aux donneuses de pouvoir obtenir une grossesse avec les ovocytes conservés. S'appuyant sur des données internationales, l'Académie estime, avec un taux de grossesses pour un ovocyte dévitrifié entre 4,5 et 12 %, qu'il faudrait en effet au moins 15 à 20 ovocytes, avec plusieurs cycles de recueil, pour raisonnablement espérer obtenir une naissance. Or, le nombre moyen d'ovocytes recueillis étant de 8 à 13 par cycle, seuls 3 à 6 ovocytes peuvent être autoconservés avec les règles existantes. Ce rapport se prononçait également pour découpler le don de la conservation pour soi-même, en prévention de l'infertilité liée à l'âge.
b) Le texte proposé : l'ouverture, hors démarche de don, de la possibilité de conserver des gamètes pour prévenir l'infertilité liée à l'âge
- La suppression de l'autoconservation liée au don
Le I propose une nouvelle rédaction globale de l'article L. 1244-2 du code de la santé publique, conduisant à supprimer la faculté ouverte en 2011, pour les donneurs n'ayant pas procréé, de conserver des gamètes pour soi à l'occasion d'une démarche de don.
- L'autorisation, sous conditions, d'une autoconservation « de précaution »
Le II insère un article L. 2141-12 au sein du code de la santé publique qui autorise parallèlement, pour les femmes comme pour les hommes , le recueil de gamètes en vue de leur conservation pour la réalisation ultérieure, à leur bénéfice, d'une assistance médicale à la procréation, et en fixe les conditions. Cette possibilité ne serait plus réservée, comme à l'heure actuelle dans le cadre d'un don, aux personnes n'ayant pas procréé, à l'instar de l'autoconservation réalisée dans un contexte pathologique.
• Cette possibilité est soumise, en premier lieu, à des conditions d'âge , renvoyées à un décret en Conseil d'Etat après avis de l'Agence de la biomédecine.
Comme le note l'étude d'impact, les bornes d'âge envisagées par le Gouvernement traduisent la préoccupation de « ne pas encourager » cette pratique en l'encadrant strictement. Celle-ci ne serait ainsi autorisée, pour les femmes comme les hommes, qu' à partir de 32 ans . En effet, en deçà de cet âge, la femme aurait « toutes les chances de procréer naturellement sans recourir aux ovocytes qu'elle aurait conservés » , dès lors que 78 % des femmes ont leur premier enfant avant 35 ans. L' âge supérieur serait, quant à lui, celui retenu pour les donneurs de gamètes , afin de garantir la qualité des ovocytes ou spermatozoïdes recueillis et donc les chances de grossesse ultérieure 33 ( * ) , à savoir moins de 38 ans pour les femmes et de 45 ans pour les hommes .
• L'autoconservation autorisée par cet article s'inscrit , en outre, dans une prise en charge médicale par l'équipe clinicobiologique pluridisciplinaire des centres autorisés. Celle-ci sera notamment chargée de délivrer une information préalable sur « les conditions, les risques et les limites de la démarche et de ses suites » .
• La conservation des gamètes est subordonnée au consentement écrit de l'intéressé, qui sera par ailleurs consulté chaque année sur le point de savoir s'il souhaite maintenir cette conservation (II de l'article L. 2141-12).
Au cas où la personne ne souhaiterait plus la poursuivre, trois possibilités lui seront offertes :
- consentir à ce que ses gamètes fassent l'objet d'un don ;
- consentir à ce que ses gamètes fassent l'objet de recherche ;
- consentir enfin à leur destruction.
L'absence de réponse de la personne aux sollicitations pendant dix années consécutives ou son décès conduiront à mettre fin à la conservation.
- Une activité réservée aux secteurs public et privé non lucratif
Le projet de loi réserve l'activité d'autoconservation, pour ce qui concerne le recueil, le prélèvement comme la conservation des gamètes, aux seuls établissements de santé publics et privés à but non lucratifs habilités à exercer le service public hospitalier , de manière, selon le Gouvernement, à garantir l'absence de facturation de dépassements de tarifs et d'honoraires 34 ( * ) . En outre, les praticiens hospitaliers ne peuvent, par voie de conséquence, pratiquer cette activité dans le cadre de leur activité libérale dans laquelle ils peuvent déroger à ces dispositions 35 ( * ) . Les établissements de santé du secteur privé lucratif en sont enfin exclus.
Cette disposition est justifiée, d'après l'étude d'impact, par le fait que le don de gamètes, vers lequel les gamètes autoconservés pourront être orientés, ne peut se pratiquer que dans des centres du secteur public ou privé non lucratif. Elle reprend en outre une préconisation de l'Académie de médecine dans son avis sur le sujet de juin 2017, destinée notamment à éviter, sur ce champ, toutes « démarches mercantiles » .
Pour autant, elle conduit à exclure de cette pratique les 42 centres du secteur privé lucratif autorisés à pratiquer les activités d'assistance médicale à la procréation , qui représentent plus de 41 % de l'offre médicale et assurent 51 % des tentatives d'AMP. Pour 14 des 52 départements disposant d'une telle offre médicale, celle-ci a exclusivement un caractère privé lucratif.
- Une prise en charge partielle par l'assurance maladie
• Le III complète l'article L. 160-8 de la sécurité sociale afin de préciser les modalités de prise en charge par l'assurance maladie de base des actes liés à l'autoconservation de précaution.
Cet article introduit à cet article du code la prise en charge de l'assistance médicale à la procréation, qui n'était pas distinguée jusqu'à présent des actes de soins de manière générale, du fait de son indication strictement médicale.
Il inclut en outre dans la prise en charge les « frais relatifs aux actes et traitements liés à la préservation de la fertilité », c'est-à-dire l'acte de ponction ovocytaire et les traitements afférents et celui de recueil de sperme.
En revanche, resteraient à la charge des personnes intéressées les frais afférents à la conservation annuelle des gamètes , sauf lorsque celle-ci est réalisée pour des motifs pathologiques en application de L. 2141-11 du code de la santé publique. Leur montant est de 40,50 euros par an d'après la nomenclature des actes de biologie médicale.
Comme le note l'étude d'impact, cette prise en charge partielle est justifiée par le fait que l'autoconservation instaurée par cet article constitue une technique de médecine préventive , pour la prévention de l'infertilité liée à l'âge, et qu'elle assurera une meilleure efficience de l'AMP. Il s'agit en outre de permettre, conformément aux préconisations formulées par le CCNE comme le Conseil d'Etat dans les travaux préparatoires, une équité d'accès à cette technique, déjà accessible à l'étranger pour les femmes les plus fortunées.
II - Les modifications adoptées par l'Assemblée nationale
• La commission spéciale a introduit, outre des amendements rédactionnels, plusieurs modifications de fond au texte :
- pour prévoir une étude de suivi des donneurs de gamètes, sous réserve de leur consentement, afin, comme le précise l'auteur de l'amendement, de disposer de données sur la motivation du don et son évolution dans le temps (amendement du rapporteur Jean-Louis Touraine) ;
- pour informer les intéressés de la possibilité de consentir à tout moment à ce qu'une partie des gamètes recueillis pour autoconservation fasse l'objet d'un don (deux amendements identiques de Laurianne Rossi, députée LREM, et d'Aurore Bergé et des membres du groupe LREM, adoptés avec l'avis favorable du rapporteur) ;
- pour réaffirmer l' interdiction d'importation de gamètes à titre commercial (amendement de Laurianne Rossi et des membres du groupe LREM, adopté contre l'avis défavorable du rapporteur) ;
- pour interdire toute prise en charge par l'employeur de l'autoconservation des gamètes, en introduisant des dispositions en ce sens dans le code de la sécurité sociale (amendement d'Aurore Bergé et des membres du groupe LREM, adopté avec l'avis favorable du rapporteur) et dans un nouveau paragraphe interdisant en outre à une entreprise toute compensation « de manière directe ou indirecte » (amendement de Thibault Bazin, député Les Républicains, adopté contre l'avis défavorable du rapporteur) ;
- pour élargir aux établissements de santé privés lucratifs l'activité de prélèvement, de recueil et de conservation des gamètes (amendement de Cyrille Isaac-Sibille, député du Modem, adopté avec l'avis favorable du rapporteur et l'avis défavorable du Gouvernement).
• En séance publique , outre des précisions rédactionnelles ou coordinations, l'Assemblée nationale est revenue sur certaines dispositions introduites par la commission spéciale :
- elle a circonscrit aux seuls spermatozoïdes recueillis pour autoconservation la possibilité de consentir à leur don partiel (amendement d'Aurore Bergé et de membres du groupe LREM, adopté avec l'avis favorable de la commission et du Gouvernement) ;
- elle a supprimé l'élargissement, par la commission spéciale, de l'activité de prélèvement, de recueil et de conservation des gamètes aux établissements de santé privés lucratifs , en revenant au texte initial (cinq amendements identiques présentés par Agnès Firmin-Le Bodo et des membres du groupe UDI, Agir et Indépendants, Thomas Mesnier et des membres du groupe LREM, Xavier Breton et des membres du groupe Les Républicains, Patrick Hetzel et Thibault Bazin, députés Les Républicains, adoptés avec l'avis défavorable de la commission, compte tenu de sa position antérieure, mais un avis de sagesse du rapporteur à titre personnel, et un avis favorable du Gouvernement). Comme l'a relevé Agnès Firmin-Le Bodo, présidente de la commission spéciale, à l'appui de cet amendement, « tout profit doit être exclu de l'activité afin de ne pas inciter à cette autoconservation et éviter les risques de démarchage mercantiles » ;
- elle a précisé que l'interdiction de prendre en charge ou compenser l'autoconservation des gamètes s'applique aux entreprises publiques et privées ainsi qu'aux personnes morales de droit privé (amendement de Laurianne Rossi et de députés du groupe LREM, sous-amendé par le rapporteur, avec l'avis favorable de la commission et du Gouvernement).
L'Assemblée nationale a adopté, enfin, avec l'avis favorable du Gouvernement, un amendement de précision de son rapporteur Jean-Louis Touraine alignant le régime applicable en cas d'absence de réponse d'une personne pendant dix ans sur celui d'une personne décédée pour décider de mettre fin à la conservation des gamètes.
III - La position de la commission : « autoriser sans décourager »
• La possibilité d'autoconservation ouverte par cet article suscite des observations et réactions contrastées.
- D'un côté, la mesure est largement soutenue par les associations de patientes infertiles ainsi que par la profession médicale et les spécialistes de l'assistance médicale à la procréation, qui y voient un acte de prévention de l'infertilité liée à l'âge .
Le recours à des gamètes, et singulièrement des ovocytes, conservés à un âge plus précoce permettra d'améliorer les chances de succès d'une procédure ultérieure d'assistance médicale à la procréation , alors que, comme l'ont rappelé les professionnels de santé, il n'existe pas à ce jour de traitement du vieillissement ovocytaire, ce qui rend l'AMP peu efficace au-delà d'un certain âge 36 ( * ) . Cela permettra à des couples d'avoir un enfant « biologique » en évitant le recours à un tiers donneur qui peut ne pas correspondre à leur projet parental et apportera une réponse à la pénurie de gamètes destinés au don, alors que le délai d'attente pour un don d'ovocyte peut atteindre trois ans, ce qui exclut de facto les demandeurs les plus âgés.
Comme cela a été relevé au rapporteur lors des auditions, cette évolution fait écho à un contexte social qui conduit les femmes à reporter de plus en plus l'âge de la première grossesse : si les causes en sont forcément multiples, le fait de rencontrer plus tardivement le conjoint avec lequel les femmes souhaitent construire une famille, et la tendance de certains hommes à reporter l'échéance de ce projet parental, ont été davantage mises en exergue que la volonté des femmes de « faire carrière ».
- D'un autre côté, la possibilité d'autoconservation ouverte par cet article ne va pas sans susciter certaines réserves , exprimées d'ailleurs par le CCNE dans son avis n° 126 du 15 juin 2017 sur les demandes sociétales de recours à l'assistance médicale à la procréation : celui-ci avait souligné la nécessité d'accompagner une telle mesure d'une information claire sur les risques liés aux grossesses tardives et le succès incertain de la démarche pour ne pas en faire un leurre en la présentant, à tort, comme une « solution magique » au décalage de l'âge de la grossesse.
Une sensibilisation des femmes et des hommes au déclin de la fertilité lié notamment à l'âge et un accompagnement adéquat de la société pour leur permettre de concilier, au moment souhaité, projet parental et vie professionnelle paraissent en effet indispensables.
Cette mesure ne peut en outre remplacer la mise en oeuvre d'une véritable politique familiale.
- Ensuite, la mesure ne saurait être présentée comme une réponse opérante au déficit de gamètes en vue de don. Comme cela a été confirmé lors des auditions, les examens requis lors d'un don 37 ( * ) ne pourraient être généralisés ex ante pour des raisons de coût et de faisabilité, si bien que les personnes devront accepter de les réaliser ultérieurement, au moment où elles consentiront, le cas échéant, à dédier les gamètes conservés au don.
Si le fait de dissocier cette démarche d'autoconservation d'un contexte de don apparaît comme une évolution bienvenue, elle ne va pas sans poser toutefois des interrogations sur l'évolution du nombre de donneurs , dans le contexte général posé par le projet de loi (extension de l'accès à l'assistance médicale à la procréation conjuguée à la levée de l'anonymat des donneurs). Les chiffres cités plus haut mettent en effet en évidence une part importante de donneurs également motivés par la possibilité d'autoconservation, sans qu'il soit possible de savoir quelle a été la motivation première de leur démarche. Il faut souhaiter que les personnes qui souhaiteront effectuer une conservation à des fins autologues soient sensibilisées, à cette occasion, à l'intérêt et à l'importance du don.
• Pour autant, la commission spéciale a considéré que l'ouverture de la possibilité d'autoconservation entrait bien dans une démarche de prévention de l'infertilité et qu'elle offrait, à la condition d'être bien expliquée et accompagnée, une liberté aux femmes et aux hommes quant à la réalisation de leur projet de maternité ou de paternité.
Comme l'a relevé l'Académie de médecine dans son étude de 2017 sur le sujet, l'expérience des pays étrangers autorisant cette pratique montre que moins de 10 % des femmes y ayant recours utilisent leurs ovocytes autoconservés, sans contribuer en soi à un recul de l'âge des grossesses.
Dès lors, la commission spéciale s'est interrogée sur l'approche du Gouvernement consistant à autoriser cette conservation « sans l'encourager » selon les termes utilisés à plusieurs reprises par la ministre de la santé lors des débats à l'Assemblée nationale.
D'après les spécialistes entendus, le recours à cette possibilité ne devrait pas être massif, notamment en raison de la lourdeur de la démarche et de son caractère contraignant pour les femmes (stimulation ovarienne suivie d'une ponction ovocytaire, acte réalisé sous anesthésie locale ou générale).
En l'état, la possibilité ouverte par la loi pourrait faire ainsi de nombreux déçus , ce qui a conduit certains spécialistes de l'assistance médicale à la procréation entendus par le rapporteur à souhaiter que cette conservation puisse être autorisée « sans la décourager » .
En ce sens, la commission spéciale a adopté un amendement COM-162 de son rapporteur renvoyant les conditions d'âge pour accéder à cette possibilité de conservation à l'appréciation des équipes médicales , sur la base d'une recommandation de bonnes pratiques qui pourra être définie par l'Agence de la biomédecine en liaison avec les sociétés savantes 38 ( * ) . Les professionnels entendus ont considéré restrictives les bornes d'âges envisagées par le Gouvernement. Le fait de les encadrer dans un décret en Conseil d'Etat n'offrira pas la souplesse nécessaire pour apprécier la diversité des situations individuelles . La balance bénéfices-risques, comme la probabilité plus faible d'y avoir ensuite recours, ne devront pas conduire, bien entendu, à ce que des femmes trop jeunes soient autorisées à conserver leurs ovocytes, en dehors de toute justification médicale.
Parallèlement, la commission a précisé la nature des informations dispensées aux demandeurs concernant notamment la baisse de la fertilité liée à l'âge et les risques liés aux grossesses tardives ( amendement COM-133 de Roger Karoutchi ).
La commission spéciale a par ailleurs décidé d' ouvrir cette activité à l'ensemble des catégories d'établissements de santé, afin de ne pas exclure les établissements de santé privés lucratifs qui constituent, comme cela a été souligné, la seule offre médicale disponible dans un grand nombre de territoires ( amendement COM-87 de Thani Mohamed Soilihi ) 39 ( * ) . A l'instar des autres activités d'assistance médicale à la procréation, comme l'autoconservation pour motif pathologique, cette activité sera soumise aux autorisations prévues par l'article L. 2142-1 du code de la santé publique.
Concernant l'autoconservation, la commission spéciale a adopté, en outre, des amendements visant à :
- supprimer la référence à une étude de suivi des donneurs de gamètes, déjà partiellement satisfaite par l'article L. 1418-1 du code de la santé publique qui confie à l'Agence de la biomédecine la mission de mettre en oeuvre un suivi de l'état de santé des donneurs d'organes ou d'ovocytes, afin d'évaluer les conséquences du prélèvement sur leur santé ( amendement COM-159 du rapporteur ) ;
- confirmer l'absence de pression de l'employeur dans l'objet de différer un projet de maternité par le biais du financement de l'autoconservation, en clarifiant la rédaction découlant de deux alinéas insérés par l'Assemblée nationale ( amendement COM-140 du rapporteur ) ;
- mieux encadrer les conditions d'importation ou d'exportation de gamètes , en supprimant des dispositions inutiles au regard de la rédaction actuelle du code de la santé publique mais en précisant le champ des autorisations délivrées par l'Agence de la biomédecine, à savoir la poursuite d'un projet parental ou la restauration de la fertilité pour le demandeur suivant la pratique actuelle ( amendement COM-160 du rapporteur ) ;
- prévoir une disposition transitoire afin de soumettre au régime mis en place par l'article 2 les gamètes autoconservés dans le cadre des dispositions actuellement en vigueur, c'est à dire lors d'une démarche de don ( amendement COM-164 du rapporteur ).
Elle a par ailleurs ajusté le régime du consentement à la conservation ou à l'arrêt de la conservation des gamètes :
- en prévoyant que, lors de la consultation annuelle, la personne est invitée à se prononcer sur les conditions de conservation des gamètes en cas de décès ( amendement COM-135 de Roger Karoutchi ) ;
- en prévoyant que cette même intention est également recueillie dès le consentement initial au prélèvement et à la conservation des gamètes ( amendement COM-168 du rapporteur ). La personne pourra ensuite modifier son choix à l'occasion de chaque consultation annuelle ;
- en allégeant la procédure de confirmation à trois mois du consentement, jugée lourde à mettre en place par les professionnels concernés, par coordination avec des modifications similaires proposées aux articles 16 et 22 ( amendement COM-161 du rapporteur ).
• En outre, la commission spéciale a rétabli le consentement du conjoint au don de gamètes (amendement COM-183 de son rapporteur 40 ( * ) ) , en considérant que celui-ci demeure pertinent alors, précisément, que le projet de loi ouvre l'accès des enfants issus de ce don à l'identité du donneur. Elle a étendu à ce conjoint, par coordination, l'information sur les dispositions relatives au don ( amendement COM-109 d'André Reichardt ).
• La commission a enfin autorisé, à titre dérogatoire, des établissements de santé privés lucratifs à pratiquer l'activité de don de gamètes , sur décision du directeur général de l'agence régionale de santé, en l'absence d'offre disponible au sein du secteur public ou privé à but non lucratif ( amendement COM-67 de Catherine Conconne et des membres du groupe socialiste et républicain). Cette disposition vise, pour les auteurs de l'amendement, à éviter une « rupture d'égalité dans l'accès à la procréation médicalement assistée avec tiers donneur » dans certains territoires notamment ultra-marins où il n'existe aucun Cecos 41 ( * ) .
D'après les données de l'Agence de la biomédecine, 32 centres (dont 25 centres hospitaliers universitaires) sont autorisés pour l'activité de don d'ovocytes et 30 pour l'activité de don de sperme. Un seul centre est situé dans les outre-mer, à La Réunion.
La commission a adopté cet article ainsi modifié .
Article
2 bis
Définition, par arrêté ministériel,
de
mesures de lutte contre les causes d'infertilité
Cet article, inséré par l'Assemblée nationale, renvoie à un arrêté interministériel la définition d'un plan national de lutte contre l'infertilité. En relativisant sa portée normative, la commission spéciale a décidé de supprimer cet article.
I - Le dispositif adopté par l'Assemblée nationale
Cet article a été adopté en séance publique à l'initiative de la présidente de la commission spéciale, Agnès Firmin Le Bodo, avec l'avis favorable de la commission et du Gouvernement.
L'amendement, cosigné par l'ensemble des rapporteurs et issu d'une démarche transpartisane, vise d'après son exposé sommaire à « alerter le Gouvernement sur l'urgence de mesures fortes, soutenues et coordonnées de lutte contre l'infertilité » et à « favoriser la mise en place d'un plan national de lutte contre l'infertilité » à l'image du plan cancer. Il a réuni au total la signature de 423 députés issus de différents groupes politiques.
L'article ainsi inséré dans le projet de loi renvoie à un arrêté interministériel - pris par les ministres en charge de l'éducation nationale, de la santé, de la recherche et de l'écologie - la définition de « mesures nationales et pluriannuelles d'organisation » concernant :
- la prévention et l'éducation du public,
- l'information sur la fertilité féminine et masculine,
- la formation des professionnels de santé,
- la coordination en matière de recherche et de protocolisation, dans l'objectif de « lutter contre toutes les causes d'infertilité, notamment comportementales et environnementales ».
II - La position de la commission
Cet article répond à une intention politique louable. L'ensemble des spécialistes de l'infertilité entendus par la commission spéciale ou le rapporteur, ainsi que les associations actives sur ce sujet, ont souligné la nécessité de davantage sensibiliser le grand public aux enjeux de la baisse de la fertilité liée à l'âge ou à d'autres facteurs, que l'amélioration des techniques médicales ne parvient pas totalement à compenser.
Pour autant, la portée normative de cet article « d'affichage » est discutable puisqu'il se contente de renvoyer la définition de ce « plan d'actions » au pouvoir réglementaire.
Or, rien n'empêche le pouvoir réglementaire de prendre des mesures en ce sens, comme rien ne garantit que la présence de cette disposition dans la loi ne soit une condition de l'effectivité de ce plan.
Pour ces raisons, la commission spéciale a décidé de supprimer cet article (amendement COM-158 de son rapporteur).
La commission a supprimé cet article.
CHAPITRE II
Reconnaître et sécuriser les droits des enfants
nés d'assistance médicale à la procréation
Article 3
Droit des personnes nées d'une assistance médicale
à la procréation avec tiers donneur d'accéder à
certaines données
non identifiantes et à l'identité du
donneur à leur majorité
Cet article propose de créer au bénéfice des personnes nées d'une AMP avec don de gamètes ou d'embryons un droit d'accéder, à leur majorité, à certaines données dites non identifiantes et à l'identité de leur donneur, le consentement de celui-ci étant recueilli préalablement au don et de manière irrévocable. Soucieuse du respect de la vie privée, la commission a souhaité que le donneur soit consulté au moment de la demande de communication de son identité et puisse s'y opposer. Par ailleurs, elle a préféré confier les missions relatives à l'exercice de ce droit d'accès au Conseil national de l'accès aux origines personnelles (CNAOP), plutôt que de créer une nouvelle structure. Elle a par ailleurs ouvert la possibilité aux personnes déjà nées d'AMP avec tiers donneur de saisir le CNAOP pour qu'il contacte et sollicite le consentement des donneurs concernés.
I - Le dispositif proposé : créer un droit d'accès aux données non identifiantes et à l'identité du donneur égal pour tous
1. L'état du droit : un principe d'anonymat opposable aux enfants nés d'un don de gamètes ou d'embryons
Depuis l'encadrement de l'assistance médicale à la procréation (AMP) par la loi du 29 juillet 1994, les dons de gamètes et d'embryons sont soumis au même principe d'anonymat que les autres dons d'éléments ou de produits du corps humain, la loi consacrant ainsi la pratique des centres d'étude et de conservation des oeufs et du sperme humains (CECOS).
Dans ce cadre - et c'est une particularité par rapport aux autres dons -, l'anonymat est opposable non seulement au couple receveur du don qui bénéficie de l'AMP, mais également à l'enfant conçu grâce à ce don .
Le principe d'anonymat est inscrit dans le code civil, à l'article 16-8, et dans le code de la santé publique, à l'article L. 1211-5 42 ( * ) . Ces dispositions législatives ont été validées par le Conseil constitutionnel qui, dans sa décision du 27 juillet 1994, a jugé que « l'interdiction de donner les moyens aux enfants ainsi conçus de connaître l'identité des donneurs ne saurait être regardée comme portant atteinte à la protection de la santé telle qu'elle est garantie par ce Préambule » 43 ( * ) . Le principe d'anonymat a été conservé lors de la révision de la loi relative à la bioéthique en 2011 , malgré la proposition initiale du Gouvernement d'intégrer un titre V intitulé « Accès à des données non identifiantes et à l'identité du donneur de gamètes » dans le projet de loi.
Par décision du 28 décembre 2017 44 ( * ) , le Conseil d'État a précisé que l'anonymat était opposable à toutes les demandes de communication d'informations présentées postérieurement à l'entrée en vigueur de la loi du 29 juillet 1994 , y compris à celles qui se rapportaient à un don effectué antérieurement.
Dans cette même décision, il a considéré que le refus d'un CECOS de communiquer des documents relatifs au donneur de gamètes à l'origine de la conception du demandeur ne portait pas une atteinte excessive aux droits et libertés protégés par la Convention européenne de sauvegarde des droits de l'homme et des libertés fondamentales, rappelant que « plusieurs considérations d'intérêt général ont conduit le législateur à interdire la divulgation de toute information sur les données personnelles d'un donneur de gamètes puis à écarter toute modification de cette règle de l'anonymat, notamment la sauvegarde de l'équilibre des familles et le risque majeur de remettre en cause le caractère social et affectif de la filiation , le risque d'une baisse substantielle des dons de gamètes , ainsi que celui d'une remise en cause de l'éthique qui s'attache à toute démarche de don d'éléments ou de produits du corps ».
Le principe d'anonymat connaît à ce jour une seule exception instituée dès 1994 afin de permettre l'accès aux informations médicales .
L'accès aux informations médicales non identifiantes
Les donneurs sont sélectionnés selon des critères médicaux d'acceptabilité après avoir passé une consultation médicale et des examens biologiques. Les candidats au don doivent ainsi présenter un bon état de santé général, un examen clinique et des résultats de marqueurs infectieux satisfaisants et n'avoir aucun signe connu de transmission de pathologies liés à des antécédents personnels ou familiaux graves 45 ( * ) . Près d' une personne sur deux est écartée du don pour motif médical . Compte tenu de cette sélection, les enfants issus d'un don ont moins de risques de développer des pathologies héréditaires que les autres enfants.
Nonobstant ces précautions préalables au don, le législateur a souhaité ménager un accès aux informations médicales du donneur dans l'intérêt de l'enfant.
En application de l'article L. 1244-6 du code de la santé publique, un médecin peut ainsi accéder aux informations médicales non identifiantes du donneur en cas de nécessité thérapeutique concernant un enfant conçu à partir de gamètes issus de son don. Le même dispositif est prévu à l'article L. 2141-6 en cas de don d'embryons. Ces informations sont transmises de médecin à médecin et l'anonymat est respecté, tant vis-à-vis de l'enfant issu du don que de ses parents.
La jurisprudence a apprécié la nécessité thérapeutique comme comprenant les actions à des fins de prévention, ce qui permet à un couple de personnes issues l'une et l'autre de dons de gamètes de prévenir tout éventuel risque de consanguinité en faisant interroger leurs centres d'AMP par leur médecin 46 ( * ) .
Par ailleurs, depuis la deuxième loi relative à la bioéthique du 7 juillet 2011, un donneur peut autoriser son médecin prescripteur à communiquer au CECOS où il a procédé à ses dons les résultats d'un test génétique « lorsqu'est diagnostiquée une anomalie génétique grave dont les conséquences sont susceptibles de mesures de prévention ». Un médecin du centre porte alors à la connaissance des personnes issues de son don qu'une information médicale est susceptible de les concerner et les invite à se rendre à une consultation de génétique.
Le mécanisme d'accès aux informations médicales serait renforcé dans le cadre du présent projet de loi : la nécessité « thérapeutique » serait remplacée par la notion plus large de nécessité « médicale » (alinéa 2 de l'article 3) et la possibilité pour un donneur d'informer les personnes conçues grâce à son don des résultats d'un test génétique détectant une anomalie grave est transformée en obligation (article 9).
2. Les demandes exprimées par certaines personnes issues d'un don pour connaître leur donneur
En 1994, lorsque la règle de l'anonymat du don de gamètes et d'embryons a été inscrite dans la loi, la possibilité que les enfants issus de ces dons expriment le besoin de connaître l'identité de leurs donneurs avait déjà été évoquée. Ainsi Jean Chérioux, rapporteur au Sénat, écrivait-il : « si l'on perçoit bien en effet les fondements du principe d'anonymat, qui évite des contestations en matière de filiation et des problèmes découlant pour l'enfant de la présence de deux « pères », et si l'on comprend qu'en l'absence d'anonymat, le don de gamètes serait probablement découragé, il n'est pas possible de méconnaître l'importance chez l'enfant des quêtes identitaires , fréquentes en particulier au moment de l'adolescence et de la post-adolescence, et l'impérieux besoin qui est le sien de se situer dans l'histoire familiale » 47 ( * ) .
Aujourd'hui les demandes d'accès aux origines, anticipées en 1994, existent bel et bien et sont largement relayées auprès des médias par les associations Origines, PMAnonymes, Dons de gamètes solidaires ou encore le Collectif pour le droit aux origines dont les représentants ont été entendus par le rapporteur. Certains enfants nés grâce à des dons de gamètes portent leur combat pour « humaniser leur mode de conception » devant la justice 48 ( * ) ou utilisent les tests génétiques à visée généalogique, malgré leur interdiction actuelle en France 49 ( * ) , pour retrouver leur géniteur.
La médiatisation de ces initiatives ne doit pas masquer le fait que la grande majorité des 70 000 personnes issues de don ne s'expriment pas , tandis que d'autres ne sont pas favorables à la levée de l'anonymat.
Le rapporteur a ainsi entendu des représentants de l'Association des enfants du don (ADEDD) qui se sont déclarés opposés à titre personnel à un accès aux origines qui pourrait changer la nature altruiste du don et conduire à une démarche plus commerciale des donneurs. À leurs yeux, l'anonymat total est structurant pour l'enfant et ses parents, qui n'ont pas à faire de place au donneur sur le plan symbolique. Ils mettent également en garde contre le « faux Graal » que constitue la quête du donneur qui apparaît à certains comme le moyen d'apaiser leurs souffrances, alors qu'un travail sur soi pourrait être plus bénéfique.
Un consensus existe désormais sur la nécessité de révéler à l'enfant son mode de conception dès que possible, pour éviter une découverte tardive qui peut se révéler traumatisante car révélatrice d'un mensonge. Les CECOS eux-mêmes plaident pour une annonce concertée par les parents à un jeune âge 50 ( * ) .
Mais la question de « l'accès aux origines » reste plus discutée , le terme « origines » étant lui-même contesté car « les origines d'un enfant ne se trouvent pas dans un matériel génétique, mais bien plutôt dans son histoire, qui lui est racontée par ses parents. Même si sa vie découle de ce capital génétique, celui-ci ne résume ni son histoire ni ses origines » 51 ( * ) .
Ce débat est éminemment complexe et conduit à s'interroger sur les places respectives que la société veut reconnaître à la parentalité sociale et à la paternité biologique, et sur l'équilibre à instituer entre l'aspiration de l'enfant à connaître l'identité de son géniteur et le respect de la vie privée du donneur et de sa famille. L'ouverture envisagée de l'AMP aux couples de femmes ou aux femmes non mariées ajoute une interrogation supplémentaire sur le lien que pourrait souhaiter nouer avec le géniteur un enfant qui n'a pas de filiation paternelle établie 52 ( * ) .
3. Le mécanisme choisi par le Gouvernement : un accès garanti, à partir de 18 ans, aux données non identifiantes et à l'identité des donneurs
a) Les deux options envisagées par le Gouvernement
Aucune norme constitutionnelle ou conventionnelle n'impose à ce jour la mise en place d'un dispositif d'accès à des informations sur le donneur ou la donneuse de gamètes ou à son identité.
La Cour européenne des droits de l'homme, dans son arrêt Odièvre 53 ( * ) , rendu en matière d'accouchement sous X, a précisé que l'article 8 de la Convention protège un droit à l'identité et à l'épanouissement personnel de l'individu au titre duquel figurent « l'établissement des détails de son identité d'être humain et l'intérêt vital, protégé par la convention, à obtenir des informations nécessaires à la découverte de la vérité concernant un aspect important de son identité personnelle, soit par exemple l'identité de ses géniteurs ». Toutefois, la Cour n'a pas, en l'état de sa jurisprudence 54 ( * ) , consacré le droit d'une personne conçue grâce à un don de gamètes d'accéder à des informations sur le donneur, mais reconnu une marge d'appréciation aux États.
En matière de droit aux origines des personnes conçues grâce à un don de gamètes, les pays européens ont adopté diverses solutions selon l'équilibre qu'ils ont choisi d'établir entre les droits des parents, des donneurs et des enfants. Un certain nombre d'entre eux ont souhaité permettre l'accès d'un enfant aux données identifiantes de son donneur, comme la Suède - dès 1985 -, la Suisse, l'Autriche, la Norvège ou les Pays-Bas. D'autres ont fait le choix d'autoriser un accès partiel - Islande, Belgique ou Danemark - et d'autres encore, de maintenir l'anonymat comme l'Espagne, la Pologne ou la Grèce.
Le Gouvernement a soumis au Conseil d'État deux versions alternatives de l'article 3 de son projet de loi. L'une prévoyait que tout donneur consent, avant même de procéder au don, à ce que l'enfant accède, à sa majorité, s'il le demande, à des données non identifiantes comme à son identité. L'autre, proche des dispositions du projet de loi déposé en 2010, conditionnait l'accès de l'enfant à l'identité du donneur au consentement de ce dernier au moment de la demande de l'enfant devenu majeur. C'est la première alternative qui a été présentée par le Gouvernement à l'Assemblée nationale.
b) Le choix d'un consentement irrévocable du tiers donneur à communiquer ses données non identifiantes et son identité dès le don
Le Gouvernement a souhaité conditionner le don de gamètes et d'embryons à l'acceptation préalable du tiers donneur 55 ( * ) à la communication de ses données non identifiantes, ainsi que de son identité , aux personnes issues de ses dons qui en exprimeraient la volonté à partir de leur majorité. Les enfants issus de dons pourraient ainsi tous avoir accès au même degré d'informations , sans dépendre de la volonté de leur donneur.
L'anonymat du don serait maintenu à l'égard des parents, qui ne pourraient choisir leur donneur « sur catalogue » comme certaines sociétés privées étrangères le proposent 56 ( * ) . Cet anonymat deviendrait toutefois provisoire , d'une durée minimum de 18 ans, et pourrait être levé à la majorité de l'enfant.
Deux types d'informations deviendraient alors accessibles s'il le souhaite 57 ( * ) :
- l'identité du donneur : celle-ci, non définie par le projet de loi, consisterait a priori en ses nom, prénoms, date et lieu de naissance 58 ( * ) ;
- ses données dites non identifiantes , qui seraient définies par l'article L. 2143-3 du code de la santé publique et précisées par décret en Conseil d'État. Il s'agirait de : l'âge ; l'état général tel que décrit au moment du don ; les caractéristiques physiques ; la situation familiale et professionnelle ; le pays de naissance ; les motivations du don. Le choix du qualificatif « non identifiantes » - qui se comprend par opposition à l'identité du donneur - a été critiqué par la Commission nationale de l'informatique et des libertés (CNIL) dans son avis du 11 juillet 2019 59 ( * ) . La Commission a relevé que la transmission de ces données pouvait engendrer des risques possibles de ré-identification des donneurs. Il conviendrait donc qu'ils en soient informés.
Toutes ces données seraient recueillies par le médecin du CECOS au moment du consentement au don et il reviendrait à ce dernier d'apprécier le caractère identifiant ou non d'une information. En cas de doute, il pourrait solliciter l'avis de la commission ad hoc mise en place pour traiter les demandes d'accès.
c) La création d'un registre national des donneurs de gamètes et d'embryons et d'une commission ad hoc
À ce jour, les données relatives aux donneurs sont conservées localement par les vingt-neuf centres CECOS et il n'y a aucune centralisation au niveau national, ce qui ne permet pas un suivi satisfaisant du respect de l'article L. 1244-4 du code de la santé publique, qui limite le recours aux gamètes d'un même donneur à la naissance de dix enfants.
Le projet de loi prévoit de créer une base unique de données relatives aux tiers donneurs, à leurs dons et aux personnes nées à la suite de ces dons, dont le traitement serait confié à l'Agence de la biomédecine qui possède déjà une expérience en la matière puisque qu'elle gère d'autres fichiers 60 ( * ) . Dans ce cadre, elle assurerait la mise en réseau des CECOS qui sont ses interlocuteurs traditionnels et élaborerait un système d'information garantissant la qualité et la sécurité des données recueillies.
Une commission ad hoc , placée auprès du ministre de la santé, serait constituée pour servir d'interface entre l'Agence de la biomédecine et les personnes conçues par AMP avec donneur qui voudraient exercer leur droit d'accès aux origines.
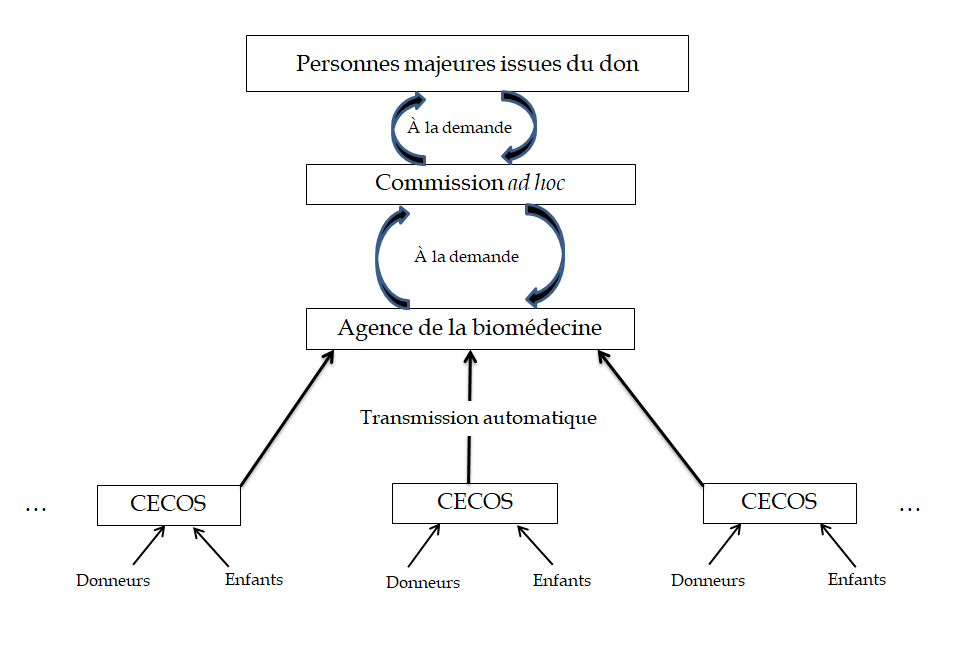
Source : Commission spéciale du Sénat
Cette commission aurait une composition assez proche de celle du Conseil national d'accès aux origines personnelles (CNAOP) régi par l'article L. 147-1 du code de l'action sociale et des familles 61 ( * ) : magistrat de l'ordre administratif, membre de la juridiction administrative, représentants des ministères de la justice et des ministères de l'action sociale et de la santé, personnalités qualifiées et représentants d'associations.
d) Un dispositif transitoire qui suscite de nombreuses inquiétudes
La mise en place du nouveau système serait progressive et le Gouvernement a prévu trois phases :
- une première phase d'environ un an 62 ( * ) pendant laquelle seraient créées la base de données auprès de l'Agence de la biomédecine et la commission ad hoc ;
- une deuxième phase, dont la durée serait déterminée par décret , pendant laquelle seraient recrutés des donneurs autorisant l'accès à leurs données personnelles qui constitueraient de nouveaux stocks de gamètes et d'embryons , les AMP avec donneurs continuant à être opérées avec les stocks collectés antérieurement sous le régime de l'anonymat ;
- une troisième phase, pendant laquelle ne seraient plus utilisés que les gamètes et embryons de donneurs ayant accepté de donner accès à leur identité et leurs données non identifiantes, les éventuels stocks constitués sous l'ancien régime de l'anonymat restants étant alors détruits .
Cette période transitoire, qui pourrait aboutir à la destruction de stocks, crée des inquiétudes parmi les professionnels et les associations qui craignent une éventuelle pénurie de gamètes en raison des nombreuses incertitudes qui existent quant au comportement des donneurs face à la levée de l'anonymat 63 ( * ) et aux conséquences de la concomitance de cette mesure avec l'ouverture de l'AMP aux couples de femmes et aux femmes non mariées 64 ( * ) .
Selon le Planning familial, les conséquences pourraient être, au-delà d'une pénurie de gamètes si ces mesures ne s'accompagnent pas de campagnes efficaces sur le don, « un risque de "suppression du don altruiste mais protégé" et à long terme une dérive vers l'indemnisation des donneurs/euses comme dans d'autres pays d'Europe » 65 ( * ) . La question financière a été directement évoquée par le professeur Israël Nisand dans une tribune intitulée « Indemniser le don de gamètes: et si on en débattait sans tabou? » 66 ( * ) .
La Fédération française des CECOS a indiqué : « à l'heure actuelle, il n'est pas possible d'apprécier de manière fiable la durée de la période de transition qui sera nécessaire pour recevoir les nouveaux candidats au don, en nombre suffisant pour répondre à l'ensemble des demandes, couples infertiles, couples de femmes et femmes non mariées, dans des délais comparables aux délais actuellement proposés pour les couples infertiles, à savoir un délai moyen national de 12 mois » 67 ( * ) .
Sa présidente a par ailleurs évoqué la situation des pays ayant modifié les conditions du don et leur recours aux importations pour pallier la baisse des dons : « (...) le nombre de dons n'est pas plus important qu'avant la modification de la loi. En effet, il est très compliqué de recruter les donneurs et ces pays importent des paillettes . Nous ne souhaitons pas rencontrer cette situation. Il serait regrettable d'aboutir à un système faisant appel à des structures qui ne respectent pas les mêmes règles éthiques » 68 ( * ) .
M. Adrien Taquet, secrétaire d'État, auditionné par la commission, a reconnu qu'il n'y avait pas « d'éléments de comparaison avec d'autres pays » compte tenu de la concomitance de l'ouverture de l'AMP à toutes les femmes et de la possibilité d'accéder à ses origines. Pour autant, il a écarté tout choix du Gouvernement d'importer des gamètes 69 ( * ) .
II - Le dispositif adopté par l'Assemblée nationale : un dispositif retouché à la marge
L'Assemblée nationale n'a pas modifié l'économie globale de l'article 3 du projet de loi.
En commission , les députés ont adopté divers amendements apportant des précisions rédactionnelles, notamment :
- pour prévenir tout risque de « double guichet » en inscrivant dans la loi que la personne qui ne consent pas à la communication de ses données non identifiantes et de son identité est exclue du don 70 ( * ) ;
- pour conforter l'absence de contrôle d'opportunité de la commission ad hoc sur les demandes d'accès qui ne « statuerait » plus sur les demandes d'accès, mais serait désormais « chargée d'y faire droit » 71 ( * ) .
Un amendement prévoyant la remise par le Gouvernement d'un rapport d'évaluation au Parlement en 2025 72 ( * ) a également été adopté.
En séance , les députés ont ajouté :
• la possibilité d'une réactualisation des données non identifiantes par le donneur 73 ( * ) ;
• la possibilité pour le donneur d'interroger la commission ad hoc pour prendre connaissance du nombre d'enfants nés grâce à son don avec la précision de leur sexe et de leur année de naissance 74 ( * ) .
Ils ont également précisé que les anciens donneurs pourront consentir à ce que leurs gamètes ou embryons en cours de conservation continuent à être utilisés dans le cadre du nouveau régime, dès lors qu'ils se manifestent auprès des CECOS pour exprimer leur consentement à se soumettre aux nouvelles règles 75 ( * ) . Ce consentement pourrait être donné avant le jour fixé par décret de « basculement » dans le nouveau régime, ce qui éviterait la destruction des gamètes ou embryons donnés.
III - La position de la commission : solliciter l'accord exprès du donneur au moment de la demande d'accès à l'identité, confier au CNAOP les missions relatives au nouveau droit d'accès et traiter les cas des personnes déjà nées d'AMP avec tiers donneur
1. Subordonner l'accès à l'identité du donneur à un accord de celui-ci au moment où l'enfant en fait la demande
Le rapporteur a considéré que l'article 3 tel que proposé par le Gouvernement ne respectait pas suffisamment les droits des donneurs . Tout d'abord, le temps écoulé entre le consentement, qui serait exprimé au moment du don, et la communication des données, qui aurait lieu au minimum 18 ans après, semble problématique. Le CCNE lui-même l'a relevé 76 ( * ) , de même que la présidente de la Fédération française des CECOS lors de son audition du 20 novembre 2019. Par ailleurs, même si le droit d'accès à l'identité du tiers donneur n'est pas un « droit de rencontre », selon le Gouvernement, ses conséquences sur la vie du donneur et son entourage demeurent incertaines.
Le Conseil d'État, dans son avis du 18 juillet 2019, a considéré qu'un système à deux vitesses, distinguant la communication des données non identifiantes de celle de l'identité du donneur, ménagerait un plus juste équilibre des intérêts en présence : ceux de l'enfant qui pourrait en tout état de cause avoir accès à des informations non identifiantes sur le donneur et à son identité si celui-ci y consent ; ceux du donneur « en lui permettant d'exprimer son consentement ou son refus dans un contexte plus propice à une décision éclairée , celui né de sa vie privée et familiale telle qu'elle est constituée au moment où se fait la demande d'accès aux origines » ; enfin l'intérêt général en prévenant une éventuelle baisse des dons liés à la levée de l'anonymat.
À l'initiative du rapporteur, la commission a souhaité permettre au donneur d'accepter ou de refuser l'accès à son identité au moment de la demande exprimée par la personne issue de son don, comme cela avait été envisagé par le Gouvernement dans l'article 3 bis de son avant-projet.
La commission a adopté l'amendement COM-264 à cette fin.
2. S'appuyer sur le Conseil National pour l'accès aux origines personnelles
Le Conseil d'État dans son étude de 2018 77 ( * ) envisageait de faire reposer l'accès aux origines des personnes issues de dons de gamètes sur le Conseil national pour l'accès aux origines personnelles (CNAOP) qui existe depuis près de 18 ans et a acquis une expérience forte en matière d'accès aux origines et d'accompagnement. De nombreux juristes se sont également déclarés en faveur de cette solution 78 ( * ) . Elle a été écartée par le Gouvernement semble-t-il devant la réticence du CNAOP lui-même qui, par la voix de sa présidente, a mis en avant le fait que le CNAOP ne travaillait pas avec des partenaires médicaux, mais avec le réseau des services départementaux de l'aide sociale à l'enfance, et que les publics concernés et leurs histoires étaient très différents.
À l'initiative de son rapporteur, la commission a fait le choix de passer outre ces réticences et de confier les missions relatives à l'accès aux données non identifiantes et à l'identité du donneur au CNAOP plutôt que de créer une commission ad hoc distincte. Les moyens supplémentaires prévus pour cette commission seraient affectés au CNAOP pour l'aider à développer ses nouvelles compétences 79 ( * ) , tout en utilisant son expérience développée auprès des personnes adoptées et des pupilles de l'État. Une formation distincte adaptée aux nouvelles missions serait constituée au sein du CNAOP.
Afin de faciliter l'accomplissement de ces nouvelles missions, la commission a conféré une habilitation législative au CNAOP pour qu'il puisse utiliser le numéro d'inscription au répertoire national d'identification des personnes physiques (code NIR) du donneur. Le décret n° 2019-341 du 19 avril 2019 relatif à la mise en oeuvre de traitements comportant l'usage du numéro d'inscription au répertoire national d'identification des personnes physiques ou nécessitant la consultation de ce répertoire pourrait ainsi être mis à jour, dans le respect du Règlement général sur la protection des données (RGPD).
La commission a adopté l'amendement COM-239 en conséquence.
3. Mieux régler la situation passée et la situation transitoire
La commission a constaté que le projet de loi ne règle en rien la situation des enfants déjà nés et les demandes portées par les associations telles Origines et PMAnonyme.
Pour y remédier, elle a adopté deux amendements identiques COM-265 et COM-252 de son rapporteur et de son président, afin de confier au CNAOP la mission de recontacter les anciens donneurs en cas de demandes d'accès provenant de personnes nées de dons sous l'ancien régime d'anonymat, et de les interroger sur leur volonté ou non de communiquer leurs informations personnelles, sans attendre qu'ils se manifestent spontanément. Le CNAOP pourrait contacter les CECOS qui seraient tenus de lui communiquer les données nécessaires en application du dispositif déjà prévu au E du VII de l'article 3 du projet de loi.
À l'initiative de son rapporteur, la commission a adopté un amendement COM-242 afin de respecter l'accord donné par le conjoint 80 ( * ) du donneur au moment du don, qui suppose que son accord soit également recueilli au moment de la levée de l'anonymat 81 ( * ) .
La commission a également adopté sur proposition de son rapporteur :
- l'amendement COM-234 pour permettre aux donneurs de gamètes ou aux personnes issues d'un don de mettre à jour les données médicales non identifiantes accessibles dans le cadre de l'article L. 1244-6 du code de la santé publique ;
- l'amendement COM-236 pour supprimer les données relatives à l'état général du donneur parmi les données non identifiantes - l'état général crée une confusion avec les données médicales du donneur - et préciser que la rédaction des motivations se ferait en concertation avec le médecin du CECOS 82 ( * ) ;
- l'amendement COM-237 pour préciser la durée maximale de conservation des données et prévoir que la CNIL serait consultée sur différents projets de décret en Conseil d'État dont celui fixant la date de conservation des données du fichier tenu par l'Agence de la biomédecine ;
- l'amendement COM-238 pour supprimer toute possibilité pour le donneur d' obtenir des informations sur les enfants issus de ses dons afin de ne pas porter atteinte à la nature purement altruiste de celui-ci ;
- l'amendement COM-241 supprimant la demande de rapport au Gouvernement, les rapports de l'Agence de la biomédecine ou du CNAOP étant suffisants à l'information du Parlement ;
- ainsi que cinq amendements rédactionnels et de coordination COM-232, COM-233, COM-235, COM-240 et COM-243.
La commission a adopté cet article ainsi modifié.
Article 4
Établissement de la filiation des enfants nés du
recours
à une assistance médicale à la
procréation
avec tiers donneur par un couple de femmes
Cet article vise, en conséquence de l'ouverture aux couples de femmes de l'assistance médicale à la procréation (AMP) avec tiers donneur, à établir la filiation des enfants qui en seraient issus par des modalités spécifiques. Malgré la proposition du rapporteur d'établir la filiation de la mère d'intention par la voie d'une procédure d'adoption rénovée, la commission a adopté cet article sans modification.
I. La situation actuelle : les modalités d'établissement de la filiation des enfants nés d'une AMP sont calquées sur le modèle de la procréation charnelle, qui ne permettent pas d'établir de double filiation maternelle
La filiation désigne traditionnellement le lien de parenté unissant l'enfant à son père (filiation paternelle) ou à sa mère (filiation maternelle) 83 ( * ) . Ce modèle, fondé depuis 1804 sur la procréation charnelle, gouverne tant le titre VII « De la filiation » que le titre VIII « De la filiation adoptive » du code civil.
Il a évolué depuis 2013 avec l'ouverture du mariage aux personnes de même sexe et de leur accès à la filiation adoptive conjointe. Il est désormais remis en question par l'ouverture de l'assistance médicale à la procréation (AMP) aux couples de femmes prévue à l'article 1 er du projet de loi.
1. Le régime actuel d'établissement de la filiation pour les couples ayant recours à une AMP est calqué sur le modèle de la procréation charnelle
Lorsqu'un couple a recours une AMP, qu'il y ait tiers donneur ou non, l'établissement de la filiation de l'enfant qui en est issu se fait depuis 1994 84 ( * ) selon les modes classiques d'établissement de la filiation, en s'inscrivant dans le modèle de la « vraisemblance biologique ».
En l'état du droit, la filiation d'un enfant s'établit légalement, hors actions contentieuses, « par l'effet de la loi, par la reconnaissance volontaire ou par la possession d'état constatée par un acte de notoriété », en vertu de l'article 310-1 du code civil.
Lorsque la filiation est établie par l'effet de la loi, il correspond, à l'égard de la mère, à la règle de l'article 311-25 du code civil qui dispose que la filiation découle de « la désignation de celle-ci dans l'acte de naissance de l'enfant » et, à l'égard du père, à la présomption de paternité de l'article 312 du même code, selon laquelle l'enfant conçu ou né pendant le mariage a pour père le mari 85 ( * ) .
S'il n'est pas marié avec la mère, le père doit reconnaître l'enfant avant ou après la naissance, par acte reçu par l'officier de l'état civil ou par tout autre acte authentique (article 316 dudit code) 86 ( * ) .
Enfin, l'établissement de la filiation peut aussi résulter d'un acte de notoriété constant les règles de la possession d'état 87 ( * ) , lorsque l'assistance médicale à la procréation se fait sans recours à un tiers donneur et que l'enfant n'a pas été reconnu.
En cas de conflit 88 ( * ) , l'article 332 du code civil précise que la contestation de la maternité n'est possible qu' « en rapportant la preuve que la mère n'a pas accouché de l'enfant ». L'article 325 du même code permet également à l'enfant de faire établir sa filiation par une action en recherche de maternité, par laquelle il « est tenu de prouver qu'il est celui dont la mère prétendue a accouché ».
Ainsi, la mère de l'enfant est toujours la femme qui accouche, conformément à l'adage latin mater semper certa est, selon lequel la mère est toujours certaine. Comme l'indiquaient notre collègue Yves Détraigne et notre ancienne collègue Catherine Tasca dans leur rapport d'information sur le recours à l'assistance médicale à la procréation et à la gestation pour autrui : « l'utilisation de techniques d'assistance médicale à la procréation n'a, dès lors, jamais suscité de difficultés juridiques particulières concernant l'établissement du lien de filiation de l'enfant à l'égard de sa mère, y compris dans l'hypothèse d'un don d'ovocyte » 89 ( * ) .
Toutefois, en application de l'article 332, « la paternité peut être contestée en rapportant la preuve que le mari ou l'auteur de la reconnaissance n'est pas le père ». Or, en vertu de l'article 310-3 du code civil, la filiation « se prouve et se conteste par tous moyens » et essentiellement par la preuve biologique, grâce au recours à des tests génétiques, autorisés dans le cadre d'une procédure judiciaire.
Dès lors, lorsque le couple qui a recours à une AMP fait appel à un tiers donneur, une action en contestation de la filiation paternelle aboutirait nécessairement. Pour que l'absence de caractère biologique ne fragilise le lien de filiation ainsi institué, le législateur de 1994 l'a soumis à des règles spécifiques aux articles 311-19 et 311-20 du code civil 90 ( * ) .
L'article 311-20 du code civil dispose ainsi que les couples « doivent préalablement donner, dans des conditions garantissant le secret, leur consentement au notaire 91 ( * ) , qui les informe des conséquences de leur acte au regard de la filiation ». Il résulte de ce consentement que la filiation établie à la suite d'une AMP avec tiers donneur, contrairement au droit commun de la filiation, « interdit toute action aux fins d'établissement ou de contestation de la filiation, à moins qu'il ne soit soutenu que l'enfant n'est pas issu de la procréation médicalement assistée ou que le consentement a été privé d'effet ».
Le consentement peut être privé d'effet dans plusieurs hypothèses, lorsqu'elles surviennent avant la réalisation de l'AMP :
- le décès de l'un des membres du couple ;
- et l'introduction d'une demande de divorce ou en séparation de corps ou la cessation de la communauté de vie.
Le consentement est également privé d'effet dans une autre hypothèse, celle dans laquelle « l'homme ou la femme le révoque, par écrit et avant la réalisation de la procréation médicalement assistée, auprès du médecin chargé de mettre en oeuvre cette assistance ».
À l'exception de ces hypothèses, celui qui, après avoir consenti à l'AMP, ne reconnaît pas l'enfant qui en est issu, engage sa responsabilité envers la mère et envers l'enfant. En outre, sa paternité est judiciairement déclarée 92 ( * ) . La parenté est donc imposée au couple demandeur qui, une fois l'AMP réalisée, ne peut plus revenir en arrière à l'exception éventuelle, pour la mère, de l'accouchement sous le secret.
Contrairement à certaines affirmations, comme l'a précisé la garde des sceaux lors du débat à l'Assemblée nationale 93 ( * ) , ce n'est pas le consentement donné chez le notaire qui fonde aujourd'hui la filiation des enfants nés d'une AMP avec tiers donneur, mais bien les modalités de droit commun précédemment exposées (désignation de la mère dans la déclaration de naissance, présomption de paternité ou reconnaissance). Ce consentement n'a pour effet, comme le souligne l'étude d'impact sur le projet de loi, que de « sceller » la filiation paternelle ultérieure 94 ( * ) .
Ces dispositions n'ont donné lieu à aucune difficulté de mise en oeuvre dont la Cour de cassation aurait été saisie, comme l'ont indiqué Rachel Le Cotty et Domitille Duval-Arnould, respectivement conseillère référendaire et conseillère à la première chambre civile, lorsqu'elles ont été entendues par le rapporteur.
Enfin, afin d'éviter toute confusion entre l'établissement de la filiation à l'égard du couple qui a eu recours à une AMP et la filiation biologique de l'enfant, aucun lien de filiation ne peut être établi entre l'auteur du don et l'enfant issu de la procréation et aucune action en responsabilité ne peut être exercée à l'encontre du donneur (article 311-19 du code civil). Cette interdiction a été déclarée conforme à la Constitution en 1994 95 ( * ) . La Cour européenne des droits de l'homme n'a, quant à elle, jamais eu à juger cette question. Elle a plusieurs fois jugé que le droit de connaître ses origines, ainsi que le droit de voir établir sa filiation sont des éléments constitutifs du droit au respect de la vie privée et familiale, mais elle ne confond pas les deux 96 ( * ) .
D'après les magistrates de la Cour de cassation précitées, l'interdiction d'établir un lien de filiation avec l'auteur du don ne semble pas, en l'état du droit, inconventionnelle. Sa levée exclurait, en pratique, toute possibilité d'un don de gamètes.
2. L'établissement d'une double filiation maternelle est aujourd'hui impossible sauf en matière d'adoption
• La double filiation de même sexe n'est aujourd'hui possible que par la voie de l'adoption
Les couples de même sexe sont exclus de l'application des dispositions du titre VII du code civil par les articles 6-1 et 320.
L'ouverture de l'adoption, en la forme simple et plénière, aux époux de même sexe par la loi n° 2013-404 du 17 mai 2013 ouvrant le mariage aux couples de personnes de même sexe, a fait entrer pour la première fois dans notre droit la possibilité d'une double filiation monosexuée maternelle ou paternelle.
Saisi du projet de loi, le Conseil constitutionnel a écarté la consécration d'un droit pour l'enfant à une filiation hétérosexuée. Il a considéré qu' « en tout état de cause, doit être écarté le grief tiré de la méconnaissance d'un principe fondamental reconnu par les lois de la République en matière de "caractère bilinéaire de la filiation fondé sur l'altérité sexuelle" ; qu'il en va de même du grief tiré de la méconnaissance d'un principe constitutionnel garantissant le droit de tout enfant de voir sa filiation concurremment établie à l'égard d'un père et d'une mère » 97 ( * ) .
Le législateur a toutefois explicitement réservé cette double filiation maternelle ou paternelle à la filiation adoptive. L'article 6-1 du code civil créé par la loi de 2013 à l'initiative du Sénat dispose en effet que « le mariage et la filiation adoptive emportent les mêmes effets, droits et obligations reconnus par les lois, à l'exclusion de ceux prévus au titre VII du livre I er du présent code, que les époux ou les parents soient de sexe différent ou de même sexe. »
Ce faisant, comme l'a rappelé la Cour de cassation dans un avis rendu par sa première chambre civile le 7 mars 2018, « les modes d'établissement du lien de filiation prévus au titre VII du livre I er du code civil, tels que la reconnaissance ou la présomption de paternité, ou encore la possession d'état, n'ont donc pas été ouverts aux époux de même sexe, a fortiori aux concubins de même sexe » 98 ( * ) .
Or, l'article 320 du code civil qui dispose que, « tant qu'elle n'a pas été contestée en justice, la filiation légalement établie fait obstacle à l'établissement d'une autre filiation qui la contredirait », s'oppose à ce que deux filiations maternelles ou deux filiations paternelles soient établies à l'égard d'un même enfant.
Deux personnes de même sexe ne pourraient demander à bénéficier cumulativement de l'une des règles de la présomption de maternité ou de paternité, qui distribuent la filiation selon le sexe de l'auteur. De même, la filiation d'un enfant ne pourrait être établie sur le fondement de la reconnaissance volontaire, à l'égard de deux personnes de même sexe, car il y aurait contradiction au sens de l'article 320 du code civil : la première reconnaissance effectué par un homme ou une femme interdit à toute personne du même sexe d'en effectuer une à son tour.
La possession d'état n'est pas non plus possible car elle a bien un fondement de vraisemblance biologique. Elle fait foi jusqu'à la preuve du contraire (article 317 du code civil), qui est, soit la preuve de l'absence d'accouchement s'agissant de la mère, soit la preuve ADN pour le père. Elle doit en outre être « continue, paisible, publique et non équivoque » (article 311-2 du code civil). Or, la double filiation de même sexe est équivoque dans la mesure où il est impossible de savoir si la seconde filiation de même sexe ne vient pas en contradiction des droits d'une autre personne.
• En l'absence d'ouverture de l'AMP aux couples de femmes, il n'existe pas de règle de droit en France organisant spécifiquement la filiation des enfants dans ces situations
Deux situations doivent cependant être distinguées, selon que les enfants éventuellement conçus par une AMP à l'étranger par des françaises y naissent également ou naissent en France.
S'ils naissent en France, comme l'indiquaient nos collègues Yves Détraigne et Catherine Tasca dans leur rapport précité, l'adoption par les couples de même sexe a permis, dans les faits, « à des enfants issus d'une AMP avec donneur, pratiquée à l'étranger (en Belgique et en Espagne notamment), de faire l'objet d'une demande d'adoption de la part de l'épouse de leur mère » 99 ( * ) .
Après que plusieurs tribunaux se sont placés sur le terrain de la fraude à la loi 100 ( * ) pour faire obstacle à l'adoption de l'enfant par l'épouse de la mère, la Cour de cassation a, dans deux avis du 22 septembre 2014 rendus en assemblée plénière, estimé que « le recours à l'assistance médicale à la procréation, sous la forme d'une insémination artificielle avec donneur anonyme à l'étranger, ne fait pas obstacle au prononcé de l'adoption par l'épouse de la mère de l'enfant né de cette procréation dès lors que les conditions légales de l'adoption sont réunies et qu'elle est conforme à l'intérêt de l'enfant » 101 ( * ) . Toutefois, comme le relevaient nos collègues sénateurs, « à moins de le révéler », ce qui semble être le cas des affaires ayant donné lieu à ces jurisprudences, « le recours à une telle pratique est indécelable » 102 ( * ) .
Cette interprétation de la Cour de cassation a, sans conteste, eu pour effet de faciliter l'adoption de l'enfant de l'épouse de la mère. Entre les mois de septembre 2014 et de mai 2019, sur 2 964 requêtes en adoption de l'enfant de la conjointe au sein des couples de même sexe, seuls 35 refus de prononcer l'adoption ont été enregistrés 103 ( * ) .
Si l'enfant issu de l'AMP avec donneur naît à l'étranger, l'enfant dispose d'un acte de naissance établi dans le pays de naissance. La filiation est établie à l'égard de la mère qui accouche ; la seconde filiation maternelle est établie ou non en fonction de la loi du pays de naissance de l'enfant.
Dans tous les cas, un acte de naissance établi à l'étranger peut être transcrit 104 ( * ) sur les registres de l'état civil français si l'un des parents est français, dans les conditions prévues par l'article 47 du code civil 105 ( * ) . D'une part, le fait d'avoir eu recours à une technique interdite en France n'y fait pas obstacle. D'autre part, la transcription n'est possible que si l'acte a été rédigé dans les formes requises par l'État de naissance, s'il est exempt de fraude et si les mentions qu'il énonce correspondent à la réalité. À cet égard, la Cour de cassation a jugé à plusieurs reprises et, notamment, dans un arrêt du 5 juillet 2017 106 ( * ) que, « concernant la désignation de la mère dans les actes de naissance, la réalité, au sens de ce texte, est la réalité de l'accouchement », ce qui fait obstacle à la transcription d'une seconde filiation maternelle.
Toutefois, elle vient de rendre une décision le 18 décembre 2019 dans laquelle elle juge que « ni la circonstance que l'enfant soit né d'une assistance médicale à la procréation ni celle que [l'acte de naissance] désigne la mère ayant accouché et une autre femme en qualité de mère ou de parent ne constituent un obstacle à sa transcription sur les registres français de l'état civil » , dès lors l'acte est probant au sens de l'article 47 du code civil, c'est-à-dire régulier, exempt de fraude et conforme au droit de l'État dans lequel il a été établi 107 ( * ) .
Un acte de naissance étranger désignant la mère ayant accouché et une autre femme comme « mère » ou « parent » pourrait donc être transcrit sur les registres de l'état civil français 108 ( * ) .
La décision rappelle toutefois qu'une action aux fins de transcription de l'acte de naissance étranger d'un enfant n'est pas une action en reconnaissance ou en établissement de la filiation même si, dans les faits, il est peu probable que la filiation soit contestée.
II. Le dispositif proposé : la création d'un mode d'établissement de la filiation spécifique aux couples de femmes ayant recours à une AMP avec donneur
Le Gouvernement estime que l'ouverture de l'AMP aux femmes non mariées n'implique aucun aménagement particulier du droit de la filiation, les modes classiques d'établissement de la filiation permettant de répondre à l'ensemble des situations envisageables. Il semble en effet logique de laisser vacante la seconde filiation pour permettre son éventuel établissement ultérieur.
L'autorisation de l'AMP aux couples de femmes conduit en revanche à s'interroger sur le droit de la filiation applicable à l'enfant qui en est issu, lequel pourrait évoluer selon plusieurs scénarii .
En effet, le principe d'égalité en matière de filiation exige que tous les enfants et tous les parents disposent des mêmes droits et obligations les uns envers les autres quel que soit le mode d'établissement de la filiation, mais il n'exige pas qu'il y ait une similitude dans la façon d'établir la filiation.
1. Les deux options écartées par le Gouvernement
• Le refus de l'extension aux couples de femmes de l'établissement de la filiation sur le modèle de la procréation charnelle
Plusieurs juristes - notamment Lisa Carayon, maîtresse de conférences à l'Université Paris 13 - Paris Nord, Marie Mesnil, maîtresse de conférences en droit privé à l'Université de Rennes et Victor Deschamps, maître de conférences en droit privé, Université Paris 2 Panthéon-Assas -ainsi que certaines associations de parents homoparentaux entendus par la commission, ont suggéré l'extension aux couples de femmes des règles applicables aux couples de même sexe mariés ou non mariés : la femme qui accouche serait mère par l'accouchement et son épouse par la présomption de co-maternité. Hors mariage, la compagne de la mère pourrait reconnaître l'enfant, avant ou après la naissance de celui-ci.
Or, cela apparaît en contradiction avec les fondements des modes d'établissement classiques de la filiation qui reposent sur la vraisemblance, le sens de la présomption et de la reconnaissance étant de refléter une vérité biologique. Par la reconnaissance de l'article 316 du code civil, le père déclare que l'enfant est issu de sa relation charnelle avec la mère et par le jeu de la présomption de l'article 312 du même code, la filiation se déduit de la preuve du mariage, cadre dans lequel s'inscrit la procréation charnelle. Comme l'indiquait le Conseil d'État en 2018 dans son étude préalable à la révision de la loi de bioéthique, ce système « conduirait à une remise en cause des principes fondateurs du droit de la filiation fixés par le titre VII du livre 1 er du code civil qui régit l'ensemble des situations » 109 ( * ) .
Cela conduirait inévitablement à fonder l'ensemble du système de filiation sur la responsabilité et l'engagement dans le projet parental plutôt que sur la seule vraisemblance procréative. Or, dans les modes classiques d'établissement de la filiation, la volonté est toujours encadrée par la vraisemblance biologique voire la vérité biologique en cas de contestation.
Le Gouvernement n'a donc pas fait ce choix, reprenant à son compte les arguments du Conseil d'État, comme l'a rappelé la garde des Sceaux lors des débats à l'Assemblée nationale 110 ( * ) .
• Le refus de modifier le droit en vigueur pour les couples de sexe différent ayant recours à une AMP avec donneur
L'article 4 du projet de loi ne modifie pas les conditions actuelles d'établissement de la filiation pour les couples de sexe différent qui ont recours à une AMP avec donneur 111 ( * ) . Seul l'article 311-20 du code civil serait modifié pour étendre les modalités de recueil chez le notaire du consentement à une AMP avec donneur à la femme non mariée 112 ( * ) .
Créer un mode d'établissement de la filiation sur le fondement de la volonté identique pour tous les couples ayant recours à une AMP avec donneur aurait des répercussions importantes pour les couples hétérosexuels qui ont recours à un tiers donneur, dont l'enfant bénéficie aujourd'hui d'une filiation fondée sur la vraisemblance biologique.
En effet, le mode d'établissement de la filiation apparaît sur l'acte de naissance. Or, l'admission d'une filiation d'intention vient nécessairement signaler qu'il y a eu recours à un tiers donneur, ce qui a une incidence différente qu'il s'agisse d'un couple de sexe différent ou d'un couple de femmes.
Si l'intervention du tiers donneur relève de l'évidence s'agissant d'un couple de femmes, elle ne l'est pas s'agissant de la femme seule ; quant au couple hétérosexuel, l'intervention d'un tiers donneur est l'indice d'une infertilité, voire d'une pathologie.
Cette solution présente donc un risque d'atteinte à la vie privée voire d'atteinte au secret médical, puisqu'elle viendrait signaler le caractère infertile du couple.
Or, rien ne vient juridiquement justifier ce changement pour ces couples, dès lors que les couples de femmes ne sont pas placés dans la même situation que les couples hétérosexuels au regard de la procréation.
Cette solution préserve ainsi la liberté des parents de choisir ou non de révéler le mode de conception de l'enfant même si cela est fortement conseillé 113 ( * ) et évite également la révélation à des tiers, v ia une mention dans l'acte de naissance, des conditions de sa conception et d'une infertilité du couple. Elle permet enfin de ne pas distinguer, au sein des couples hétérosexuels, les modalités d'établissement de la filiation.
Défendue par une partie de la doctrine et notamment les professeurs Anne-Marie Leroyer, professeur à l'Université de Paris 1 et Hugues Fulchiron, professeur de droit privé à l'Université Jean Moulin Lyon 3, auteurs du rapport publié en 2014 avec la sociologue Irène Théry qui le préconisait, au nom de l'égalité d'accès à leurs origines pour tous les enfants 114 ( * ) , cette option n'a pas été retenue par le Gouvernement qui a préféré ne pas troubler la paix des familles.
2. Le choix du Gouvernement : l'établissement d'une double filiation maternelle ab initio et l'admission d'une filiation d'intention
Dans son projet de loi initial, le Gouvernement fait le choix d'autoriser l'établissement d'une double filiation maternelle ab initio en créant un titre ad hoc dans le code civil (titre VII bis ) et qui n'est ni la filiation adoptive, ni la filiation charnelle.
L'article 4 instituerait un nouveau mode d'établissement de la filiation par « déclaration anticipée de volonté » pour les seuls couples de femmes ayant recours à la procréation médicalement assistée. Ces dispositions constitueraient un nouveau titre VII bis composé de quatre nouveaux articles (articles 342-9 à 342-12 du code civil) et intitulé « De la filiation par déclaration anticipée de volonté », au sein du livre I er du code civil.
La création d'un régime spécifique d'établissement de la filiation pour les couples de femmes ne pose pas de problème constitutionnel d'égalité ou de difficulté d'ordre conventionnel, les couples de femmes n'étant pas dans la même situation que les couples hétérosexuels au regard de la procréation.
Les modalités d'établissement de la
filiation
des enfants issus d'une AMP avec donneur dans les pays
étrangers
Si les dix États membres de l'Union européenne qui ont ouvert l'assistance médicale à la procréation aux couples de femmes ont retenu comme principes communs que la femme qui accouche est la mère de l'enfant et que le nombre de filiations pour un même enfant est limité à deux, ils privilégient des modes d'établissement de la filiation différents pour la mère d'intention.
Dans certains pays, comme les Pays-Bas, la Suède et la Finlande, le lien de filiation avec la conjointe, la partenaire ou la concubine de la mère est établi par adoption.
En Belgique et en Autriche, la filiation est établie par extension aux couples de femmes du régime de droit commun prévu pour les couples de sexes différents.
Au Danemark et à Malte, l'établissement de la filiation est en revanche fondé sur le consentement exprès à l'assistance médicale à la procréation.
Dans d'autres pays, des systèmes mixtes ont été mis en place. En Espagne, l'épouse de la mère doit faire une déclaration de consentement à devenir parent et, pour les couples de femmes non mariées, seule la voie de l'adoption permet à la compagne de la mère d'établir sa filiation. Au Portugal, la filiation est établie par présomption si le couple est marié ou si le concubin présente le consentement conjoint à l'assistance médicale à la procréation qui a été donné avant le début de la procédure de procréation. Au Royaume-Uni, la présomption de parenté est étendue aux femmes unies par un partenariat enregistré pour devenir les parents légaux de l'enfant ; s'agissant de deux concubines, le lien de filiation est établi avec la compagne de la mère aux termes d'une convention passée entre elles, à défaut, cette dernière peut solliciter auprès du juge l'exercice de l'autorité parentale ou engager une procédure d'adoption.
Source : étude d'impact du projet de loi, p. 185
En premier lieu, les couples de femmes devraient, comme les autres couples ou la femme non mariée, consentir à l'AMP avec donneur devant notaire et, en même temps, déclarer conjointement leur volonté de devenir les parents de l'enfant issu de l'AMP, déclaration irrévocable à compter de la réalisation de l'AMP.
La filiation serait « établie à l'égard de la femme qui accouche et de l'autre femme » de manière indivisible, par leur désignation dans la déclaration anticipée de volonté, remise à l'officier de l'état civil par l'une de ses auteurs lors de la déclaration de naissance de l'enfant 115 ( * ) .
L'officier de l'état civil en porterait mention sur l'acte de naissance. La déclaration de volonté apparaîtrait donc sur l'acte de naissance de l'enfant et sur la copie intégrale, à l'instar des autres modes d'établissement de la filiation mais non sur l'extrait d'acte de naissance.
La filiation ainsi établie serait assortie du principe d'interdiction d'en établir une avec le donneur et d'exercer une action en responsabilité à son égard, par renvoi de l'article 311-19 du code civil.
Le consentement donné par le couple et la déclaration anticipée de volonté de devenir parent, qui sont des actes établis en la forme authentique par le notaire, auraient les mêmes effets que ceux prévus à l'actuel article 311-20 : ils interdiraient toute action aux fins d'établissement ou de contestation de la filiation, à moins qu'il ne soit soutenu que l'enfant n'est pas issu de la procréation médicalement assistée ou que le consentement ou la déclaration ont été privés d'effet. Les effets de la déclaration anticipée de volonté cesseraient également concomitamment à ceux du consentement. À cet égard, les représentants du Conseil supérieur du notariat (CSN) ont regretté que le projet de loi autorise la rétractation du consentement devant le médecin ou le notaire, alors que le premier est le mieux à même de faire cesser la démarche de l'AMP.
Comme dans le droit en vigueur, le consentement au don devant le notaire serait donc une condition sine qua non d'accès à l'AMP avec tiers donneur. S'agissant des couples de femmes, le consentement serait, en outre, la condition pour effectuer une déclaration anticipée de volonté par laquelle les deux femmes établissent leur filiation par anticipation, celle-ci ne devenant effective qu'au moment de la naissance de l'enfant.
L'innovation majeure du texte est donc de permettre l'établissement d'une double filiation maternelle ab initio et d'admettre une filiation d'intention.
Un dispositif similaire à celui de la paternité judiciairement déclarée serait également prévu. Si la déclaration anticipée de volonté n'a pas été remise et que la filiation n'est par conséquent établie qu'à l'égard de la femme qui a accouché, la déclaration anticipée de volonté pourrait alors être « communiquée au procureur de la République à la demande de l'enfant majeur, de son représentant légal s'il est mineur ou de toute personne ayant intérêt à agir en justice ». La mention de cette déclaration serait portée en marge de l'acte de naissance.
Lors des débats à l'Assemblée nationale, la garde des sceaux relevait qu'il s'agit de « cas très particuliers et relativement rares », vraisemblablement des « cas de fraude ou de séparation d'un couple » 116 ( * ) .
Dans cette hypothèse, une filiation paternelle aura même pu être mentionnée sur l'acte de naissance de l'enfant. Dès lors, la mère d'intention ne pourrait faire reconnaître sa maternité qu'après avoir contesté la filiation paternelle, en établissant que l'enfant est bien issu de l'AMP.
De plus, celle des deux femmes qui après avoir consenti à l'assistance médicale à la procréation, ferait obstacle à la remise de la déclaration anticipée de volonté à l'officier de l'état civil engagerait sa responsabilité, comme le père, aujourd'hui, qui ne reconnaît pas l'enfant issu de l'AMP.
L'article 4 prévoit également des règles de choix et de dévolution du nom, selon des modalités comparables à ce qui est prévu au titre VII ainsi que pour la filiation adoptive au titre VIII du livre I er du code civil : choix du nom de famille au moment de la déclaration de naissance, par les deux parents désignés dans la déclaration anticipée de volonté qui opteraient soit pour le port du nom de l'un d'entre eux, soit pour celui de leurs deux noms accolés dans l'ordre qu'ils souhaitent, mais dans la limite d'un nom par parent. A défaut d'accord, l'officier de l'état civil retiendrait le nom de chacun des parents dans l'ordre alphabétique.
Enfin, s'agissant des effets de la filiation, ils seraient les mêmes pour tous les enfants, comme le prévoit déjà le droit en vigueur aux articles 310 117 ( * ) et 358 118 ( * ) du code civil. Le projet de loi prévoit toutefois de substituer à ces dispositions un nouvel article 6-2 placé en tête du code civil dans le titre préliminaire, selon lequel tous les enfants dont la filiation est légalement établie ont, dans leurs rapports avec leurs parents, les mêmes droits et les mêmes devoirs, sous réserve des dispositions relatives à l'adoption simple et qu'ils entrent dans la famille de leurs parents.
Tous les modes d'établissement de la filiation produiraient donc les mêmes effets, comme aujourd'hui.
S'agissant du champ d'application territorial de l'article 4 du projet de loi, elles s'appliqueraient de plein droit en Guadeloupe, en Guyane, en Martinique, à Mayotte, à La Réunion, ainsi qu'à Saint-Barthélemy, à Saint-Martin, et à Saint-Pierre et Miquelon. Les dispositions relatives à l'état des personnes sont applicables de plein droit à Wallis-et-Futuna mais nécessiteraient une adaptation eu égard à l'absence de notaires. Elles s'appliqueraient également de plein droit en Polynésie Française sans qu'il soit besoin d'une mention expresse.
En revanche ces dispositions ne seraient pas applicables en Nouvelle-Calédonie 119 ( * ) .
L'article 4 du projet de loi n'exclut pas, non plus, son application aux enfants nés d'une AMP réalisée à l'étranger, dès lors que le couple aurait préalablement fait un consentement à l'AMP et une reconnaissance conjointe devant notaire en France. En l'absence de reconnaissance conjointe, la femme qui n'a pas accouché devra, comme aujourd'hui, adopter l'enfant de sa conjointe - ce qui suppose qu'elles soient mariées.
Enfin, il faut préciser que les conséquences du recours à une AMP en violation des conditions légales - cas de l'AMP dite « artisanale » ou « amicale » sont complètement différentes s'agissant de la filiation de l'enfant qui en est issu. Les éventuels engagements pris par la femme de ne jamais agir pour faire établir le lien de filiation à l'égard du donneur complaisant ou de ce dernier de ne jamais reconnaître l'enfant ou faire établir sa filiation, sont dépourvus de toute valeur juridique car l'article 323 du code civil dispose, en vertu du principe de l'indisponibilité de l'état des personnes, que « les actions relatives à la filiation ne peuvent faire l'objet de renonciation ».
L'ouverture de l'AMP aux couples de femmes et aux femmes seules ne changerait rien à ces hypothèses dans lesquelles la filiation demeurerait établie et contestable selon les règles classiques du titre VII du livre I er du code civil.
3. Le choix de l'Assemblée nationale : approuver l'esprit du texte initial et opérer des modifications symboliques pour répondre aux revendications d'égalité
Le texte de l'article 4 résultant des travaux de l'Assemblée nationale est issu de deux amendements identiques du Gouvernement et de la rapporteur, notre collègue députée Coralie Dubost, adoptés en commission, dont l'économie générale n'a pas été modifié en séance publique.
Tout en maintenant l'établissement de la filiation par des modalités spécifiques pour les couples de femmes ayant recours à une AMP avec donneur, l'article 4 adopté par l'Assemblée nationale présente deux modifications substantielles par rapport au texte initial, justifiées selon les auteurs des amendements afin que « les distinctions opérées entre les différents modes d'établissement de la filiation n'apparaissent pas comme enfermant dans une catégorie juridique à part les couples de femmes qui auraient recours à l'AMP » 120 ( * ) .
En premier lieu, là où le texte initial créait un nouveau titre VII bis au sein du livre I er du code civil, propre à l'établissement de la filiation des enfants nés de couples de femmes ayant eu recours à une AMP avec tiers donneur, l'article 4 complèterait le titre VII qui établit la filiation sur la base de la vraisemblance biologique d'un nouveau chapitre consacré au recours à l'AMP avec tiers donneur pour tous les couples, de femmes ou de sexe différent.
En second lieu, la « déclaration anticipée de volonté » a été supprimée du dispositif ; la mention de « la femme qui accouche » aussi.
Pour les couples de femmes, la filiation serait établie, « à l'égard de chacune d'elles, par la reconnaissance qu'elles ont faite conjointement devant le notaire lors du recueil du consentement ». Cette « reconnaissance », terme emprunté à la reconnaissance volontaire de l'article 316 du code civil, serait remise à l'officier de l'état civil lors de la déclaration de naissance de l'enfant. L'acte de naissance de l'enfant porterait alors la mention selon laquelle il a été reconnu par ses deux mères.
En l'état du texte adopté par l'Assemblée nationale, les incidences de l'intégration des nouvelles dispositions au sein du titre VII en lieu et place de la création d'un titre VII bis sont plus symboliques que juridiques. Le droit resterait inchangé pour les couples de sexe différent. Les effets juridiques de la « déclaration anticipée de volonté » et de la « reconnaissance conjointe » devant le notaire sont exactement les mêmes.
D'ailleurs, dans les deux cas, la filiation de la femme qui accouche - même mentionnée - serait établie de manière indivisible, comme à l'égard de l'autre femme, par sa désignation l'acte authentique, déclaration anticipée de volonté ou reconnaissance conjointe, remis à l'officier de l'état civil lors de la déclaration de naissance de l'enfant. Interrogée sur ce point par le rapporteur, les services de la Chancellerie ont expliqué que : « l'établissement simultané de la filiation pour les deux femmes était une demande de la part des associations que nous avions entendues tout au long des travaux préparatoires. Il était alors souhaité une parfaite égalité entre les deux femmes et que l'une ne soit pas mère avant ou "davantage" ou "plus sûrement" que l'autre. Ce nouveau mode d'établissement de la filiation permet de consacrer le projet parental du couple en fondant la filiation sur l'intention déclarée conjointement et formalisée par un acte authentique. »
La reconnaissance conjointe apparaîtrait donc sur l'acte de naissance et sur la copie intégrale.
Interrogés sur le risque de confusion relevé par plusieurs juristes entendus par la commission entre la reconnaissance de l'article 316 du code civil - aveu de filiation biologique - et la reconnaissance conjointe créée par le projet de loi, la Chancellerie a indiqué à la rapporteur que « la ressemblance dans les termes était voulue et assumée pour réduire au maximum la différence entre les couples hétérosexuels et les couples de femmes dans l'établissement de la filiation ».
Si les deux termes n'auront pas le même objet juridique, le législateur pourrait cependant, s'agissant d'actes différents n'ayant pas la même portée, recourir à des termes distincts.
III. La position de la commission : approuver le texte issu des travaux de l'Assemblée nationale, malgré la proposition du rapporteur d'établir la filiation de la mère d'intention par la voie d'une procédure d'adoption rénovée
Aux termes des travaux de la commission, il est clair que l'article 4 du projet de loi tend à établir la filiation d'un enfant sur le fondement de la volonté à l'égard d'un couple de femmes ayant recours à une assistance médicale à la procréation (AMP) avec donneur.
Or, selon l'analyse du rapporteur, l'introduction d'un critère de volonté pure risque, à terme, de rendre le critère de la vraisemblance biologique caduque et de fragiliser tout le système français de filiation. Le projet de loi vise donc à prendre en compte le désir d'enfant des couples de femmes, alors qu'elles ne sont pas, au regard de la procréation, dans la même situation que les couples de sexe différent.
Or, le rôle du législateur est de garantir la protection de l'intérêt supérieur de l'enfant, récemment érigé au rang d'exigence constitutionnelle 121 ( * ) .
Considérant que cette réforme ne le permettrait pas, le rapporteur a proposé à la commission un amendement COM-253 de suppression de l'article 4 du projet de loi, en cohérence avec l' amendement COM-167 de suppression de l'article 1 er .
Pour le rapporteur, les écueils de cette réforme de la filiation sont de deux ordres.
En premier lieu, l'établissement obligatoire d'une double filiation maternelle aurait pour effet de priver délibérément un enfant de filiation paternelle, ce qui n'est pas, selon certains pédopsychiatres, sans conséquences pour le développement et l'équilibre de l'enfant 122 ( * ) . Certes, les avis sont divergents sur ce point 123 ( * ) , les études françaises manquent et le caractère scientifique des études étrangères est contesté 124 ( * ) . Toutefois, dans le même temps, le projet de loi tend à permettre aux enfants nés d'un don de connaître leurs origines biologiques : preuve en est donc que le père de l'enfant ne peut pas être éludé.
Cette réalité est d'ailleurs flagrante en droit de la famille où le législateur n'a cessé de renforcer le rôle du père, que ce soit à l'issu de la grossesse de la mère (création du congé de paternité et du congé parental) ou pour l'éducation de l'enfant, que les parents soient en couple ou se séparent (partage de l'autorité parentale et résidence alternée notamment).
Outre l'intérêt de l'enfant, insuffisamment pris en compte, cette réforme imposerait en second lieu une remise en cause de l'ensemble du système français de filiation : il s'agirait d'assumer de fonder la filiation sur la seule volonté.
Tel qu'il a été adopté par l'Assemblée nationale, l'article 4 établirait la filiation de l'enfant sur ce fondement pour les deux femmes, même celle qui accouche, au mépris du principe selon lequel la mère est toujours certaine en raison de l'accouchement ( mater semper certa est ). Mentionnée dans le projet de loi initial, la « femme qui accouche » n'apparaît plus dans la version adoptée par l'Assemblée nationale.
Ce mode indivisible d'établissement de la filiation a été unanimement critiqué lors des auditions.
Comme l'a relevé Astrid Marais, professeur de droit privé à l'Université de Paris 8 entendu par la rapporteur, cette solution « nie l'accouchement en faisant reposer la filiation de l'enfant sur la seule volonté du couple » 125 ( * ) .
La femme qui accoucherait au sein d'un couple de femmes serait ainsi la seule pour laquelle la filiation ne serait pas établie selon le principe mater semper certa est , alors qu'il n'est nullement nécessaire que le mode d'établissement de la filiation soit identique et simultané pour les deux femmes. En 2018, le Conseil d'État indiquait d'ailleurs que, « s'agissant de la femme qui accouche, il conviendrait de maintenir la règle selon laquelle la mère est toujours certaine (mater certa est) en n'imposant pas à cette dernière de présenter cette déclaration à l'officier de l'état civil pour obtenir l'établissement de son lien de filiation à l'égard de l'enfant, le simple fait qu'elle ait donné naissance à l'enfant devant demeurer suffisant pour l'établir » 126 ( * ) .
Ce choix aurait en outre, de l'avis notamment de Jean-René Binet, professeur de droit privé à l'Université de Rennes et des représentantes de la première chambre civile de la Cour de cassation, comme conséquence que la femme ne pourrait plus accoucher dans le secret, en l'absence d'accord de la femme qui n'accouche pas, en raison de l'indivisibilité des filiations et de l'engagement pris de remettre la reconnaissance conjointe à l'officier de l'état civil 127 ( * ) .
Au final, le rapporteur estime que la reconnaissance d'une filiation d'intention sans aucune vraisemblance biologique reviendrait à admettre une forme de filiation contractuelle qui pourrait, à terme, supplanter le mode de filiation actuel et remettre en cause les principes de la parentalité.
En effet, s'il est possible d'établir la filiation sur le fondement de la seule volonté, la limitation à deux du nombre de filiations possibles - hors adoption - à l'égard d'un enfant pourrait être réinterrogée, puisqu'elle découle directement des possibilités de la procréation charnelle.
La consécration d'une parentalité d'intention ne manque pas, non plus, de renvoyer à la question de la gestation pour autrui (GPA) qui fait, dans les pays où elle est permise, l'objet d'un contrat et dans laquelle l'un des parents, voire les deux, le sont par la seule volonté. La reconnaissance légale d'une parentalité d'intention pour les enfants nés d'une GPA s'inscrit dans la continuité de celle des enfants nés d'une AMP demandée par un couple de femmes. Or, il ne peut exister, selon le rapporteur, de « GPA éthique ». Il s'agit toujours et avant tout d'une forme de réification de l'enfant que la France doit continuer à prohiber.
La commission spéciale étant majoritairement favorable à l'ouverture de l'AMP aux couples de femmes 128 ( * ) , le rapporteur a toutefois jugé plus cohérent de retirer son amendement COM-253 de suppression de l'article 4, pour soumettre uniquement à la commission son amendement COM-254 rect bis proposant d'établir la filiation de la mère d'intention par la voie d'une procédure d'adoption rénovée.
Selon le rapporteur, l'adoption est en effet la seule possibilité de notre droit d'établir un lien de filiation par la seule volonté. Elle est régie par le titre VIII du livre I er du code civil. L'adoption étant une filiation élective, elle est nécessairement établie par jugement, afin de vérifier que ses conditions légales sont remplies et qu'elle est conforme à l'intérêt de l'enfant.
Cette conception rejoint le sens des observations de plusieurs juristes entendus lors des auditions, notamment Aline Cheynet de Beaupré, professeur de droit privé à l'Université d'Orléans, Aude Mirkovic, maître de conférences en droit privé et sciences criminelles à l'Université d'Évry et Claire Neirinck, professeur émérite de droit privé et de sciences criminelles de l'Université de Toulouse 1 - Capitole.
Afin d'atteindre cet objectif, le dispositif proposé par le rapporteur avait trois objets.
En premier lieu, il insérait un nouvel article au sein du titre VII du livre I er du code civil pour interdire explicitement l'établissement de deux filiations maternelles ou paternelles à l'égard d'un même enfant.
En second lieu, il créait un nouveau titre VII bis au sein du même livre I er du code civil, regroupant les dispositions applicables à la filiation en cas de recours à une AMP avec donneur, sans rien modifier ni pour les couples de sexe différent, ni pour la femme qui accouche quelle que soit sa situation conjugale.
Pour sécuriser l'établissement de la filiation de l'enfant issu d'une AMP lorsque cette technique a été demandée par un couple de femmes, l'amendement proposait que le consentement à une AMP avec donneur vaille, pour la mère qui accouche, consentement à l'adoption de l'enfant issu de l'AMP par l'autre membre du couple. Le second membre du couple se serait engagé à faire une demande d'adoption de l'enfant, sans quoi sa responsabilité aurait pu être engagée, ainsi que l'adoption prononcée à la requête de la mère de l'enfant.
En troisième lieu, l'amendement modifiait les conditions requises pour l'adoption - qu'elle soit demandée en la forme simple ou plénière - afin de permettre l'adoption de l'enfant issu d'une AMP par la mère d'intention. L'article 1 er du projet de loi propose en effet que tous les couples de femmes puissent recourir à une AMP quelle que soit leur situation conjugale.
L'amendement rendait donc l'adoption possible pour les couples liés par un pacte civil de solidarité (PACS) ou en concubinage, alors qu'elle est aujourd'hui réservée aux époux 129 ( * ) . Il permettait aussi, pour l'adoption individuelle, l'adoption de l'enfant du partenaire de PACS ou du concubin, sur le même modèle que l'adoption de l'enfant du conjoint. Dans cette situation, qui correspondrait à celle de la mère d'intention au sein d'un couple de femmes recourant à une AMP, l'adoptant bénéficie d'une procédure simplifiée : aucune condition d'âge ni agrément ne sont exigés.
À cet égard, l'amendement proposait de simplifier encore davantage la procédure lorsque l'enfant était issu d'une AMP avec donneur. La condition d'accueil au foyer de l'adoptant de six mois n'était pas exigée et le tribunal de grande instance aurait eu un mois pour rendre son jugement, contre six pour les autres procédures d'adoption.
Compte tenu de la diligence de l'adoptant, la filiation adoptive aurait pu être établie, lorsque l'enfant est issu d'une procédure d'AMP avec donneur, le jour même de sa naissance, puisque l'adoption produit ses effets à compter du jour du dépôt de la requête.
La solution proposée avait le mérite de ne pas modifier les fondements de la filiation et d'utiliser les règles existantes conformément à la réalité. Au terme d'un débat nourri entre ses membres, la commission n'a pas retenu le dispositif proposé et a préféré en rester à la rédaction issue des travaux de l'Assemblée nationale.
La commission a adopté cet article sans modification.
Article 4 bis
(nouveau)
Interdiction de la transcription totale d'un acte de naissance
ou d'un jugement étranger établissant la filiation d'un
enfant
né d'une gestation pour autrui lorsqu'il mentionne le parent
d'intention
Cet article introduit en commission propose d'interdire la transcription totale de l'acte de naissance ou du jugement étranger établissant la filiation d'un enfant né d'une gestation pour le compte d'autrui (GPA) lorsqu'il mentionne comme mère une autre femme que celle qui a accouché ou deux pères.
Introduit en commission par l'adoption d'un amendement COM-99 rect. ter à l'initiative de notre collègue Bruno Retailleau et avec l'avis favorable du rapporteur, l'article 4 bis tend à interdire la transcription totale de l'acte de naissance ou du jugement étranger établissant la filiation d'un enfant né d'une gestation pour le compte d'autrui (GPA) sur les registres de l'état civil français concernant le parent d'intention.
Il précise que ces dispositions ne font pas obstacle à la transcription partielle de l'acte ou du jugement qui établit la filiation, c'est-à-dire la transcription de l'acte pour le père biologique, ni à l'établissement ultérieur du lien de filiation avec le parent d'intention par la voie de l'adoption.
Cet article vise, afin de donner une portée pleine et entière à l'interdiction de la GPA, prohibée en France par l'article 16-7 du code civil 130 ( * ) depuis la loi n° 94-653 du 29 juillet 1994 relative au respect du corps humain et sanctionnée par les articles 227-12 et 227-13 du code pénal, à faire obstacle aux dernières jurisprudences de la Cour de cassation rendues en la matière.
Alors qu'en 2013, la Cour de cassation jugeait que la fraude à la loi faisait obstacle à la transcription, même partielle, d'un acte de naissance étranger d'un enfant conçu par GPA 131 ( * ) , elle a progressivement infléchi sa jurisprudence sous l'influence de la Cour européenne des droits de l'homme (CEDH) et autorisé la transcription partielle de l'acte de naissance en ce qui concerne le père biologique de l'enfant.
La Cour de cassation a également jugé, dans quatre arrêts rendus le 5 juillet 2017 132 ( * ) , que le refus de la transcription totale d'un acte de naissance étranger en ce qui concerne le parent d'intention ne portait pas une atteinte disproportionnée au droit à la vie privée de l'enfant, dès lors que la voie de l'adoption était ouverte au parent concerné et permettait de créer, si les conditions légales en sont réunies et si elle est conforme à l'intérêt de l'enfant, un lien de filiation entre l'enfant et l'épouse ou l'époux de son père.
Consultée par la Cour de cassation sur un cas de recours à la GPA par deux époux de sexe différent, la Cour européenne des droits de l'homme a, dans un avis publié le 10 avril 2019 133 ( * ) , jugé que cette solution était conforme à la Convention européenne des droits d'homme et des libertés fondamentales.
Elle a en effet rappelé que, si le droit au respect de la vie privée de l'enfant, au sens de l'article 8 la Convention précitée, requiert que le droit interne offre une possibilité de reconnaissance d'un lien de filiation entre l'enfant et la mère d'intention, désignée dans l'acte de naissance légalement établi à l'étranger comme étant « la mère légale », il ne requiert pas que cette reconnaissance se fasse par la transcription sur les registres de l'état civil de l'acte de naissance. Elle peut donc se faire par une autre voie, telle l'adoption de l'enfant par la mère d'intention.
Elle exige toutefois que, selon l'appréciation des circonstances de chaque cas, le mécanisme effectif permettant la reconnaissance de la filiation existe au moment où le lien entre l'enfant et la mère d'intention s'est concrétisé, ce à quoi peut répondre la procédure d'adoption, dès lors que ses modalités permettent une décision rapide, afin d'éviter que l'enfant soit maintenu longtemps dans l'incertitude juridique quant à ce lien.
En droite ligne de cet avis, la CEDH a jugé le 19 novembre 2019 134 ( * ) que la procédure française de l'adoption de l'enfant du conjoint garantissait l'effectivité et la célérité nécessaires. Dans les cas d'espèce, les enfants nés du recours à une GPA à l'étranger par des époux hétérosexuels avaient respectivement sept et trois ans au moment où ils auraient pu être adoptés 135 ( * ) , soit bien après la concrétisation du lien entre eux et leur mère d'intention.
Pourtant, la CEDH juge que « dans les circonstances de la cause, ce n'est pas imposer aux enfants concernés un fardeau excessif que d'attendre des requérants qu'ils engagent maintenant une procédure d'adoption » 136 ( * ) afin de concrétiser le lien de filiation, constatant qu'une décision judiciaire d'adoption de l'enfant du conjoint est obtenue, en moyenne, en un peu plus de 4 mois.
La Cour de cassation, dans deux arrêts du 18 décembre 2019 137 ( * ) , semble être allée plus loin en jugeant que le recours à une GPA légalement réalisée à l'étranger ne faisait pas obstacle à la transcription totale de l'acte de naissance désignant le père biologique et son époux, dès lors que l'acte est probant au sens de l'article 47 du code civil 138 ( * ) c'est-à-dire régulier, exempt de fraude et conforme au droit de l'État dans lequel il a été établi. La Cour de cassation a considéré que sa jurisprudence du 5 juillet 2017 ne pouvait trouver application « lorsque l'introduction d'une procédure d'adoption s'avère impossible ou inadaptée à la situation des intéressés », alors que dans l'un des deux cas d'espèce, les requérants sont mariés et que le parent d'intention peut recourir à l'adoption de l'enfant du conjoint.
Une telle transcription avait également été admise, dans les mêmes conditions s'agissant du caractère probant de l'acte, dans le cas d'un couple hétérosexuel marié 139 ( * ) mais il semblait à l'époque que le jugement était d'espèce puisque cette solution était présentée comme la seule possible, « s'agissant d'un contentieux qui perdure depuis plus de quinze ans, en l'absence d'autre voie permettant de reconnaître la filiation dans des conditions qui ne porteraient pas une atteinte disproportionnée au droit au respect de la vie privée » des enfants alors âgés de plus de 18 ans.
Cela revient toutefois à faire perdre toute consistance à la prohibition de la GPA en France. Dans ces conditions, la commission a donc estimé qu'il serait justifié de cantonner par la loi la transcription d'un acte de naissance d'un enfant issu d'une GPA à l'étranger à la transcription de la filiation biologique.
Le rapporteur observe toutefois que, pour garantir les conditions exigées par la CEDH d'effectivité et de célérité de l'établissement du lien de filiation avec le parent d'intention, le législateur devrait au moins probablement modifier les conditions requises de l'adoption pour permettre à tous les couples d'adopter, quelle que soit leur situation conjugale (époux, partenaires d'un pacte civil de solidarité ou concubins).
La commission a adopté cet article additionnel ainsi rédigé.
TITRE II
PROMOUVOIR
LA SOLIDARITÉ
DANS LE RESPECT DE L'AUTONOMIE DE CHACUN
CHAPITRE IER
Conforter la solidarité dans le cadre
du don
d'organes, de tissus et de cellules
Article 5 A (nouveau)
Statut de donneur d'organes
Cet article additionnel, inséré par la commission spéciale, pose les bases d'un statut de donneur d'organes en France.
Alors que la loi de bioéthique de 2004 a érigé à l'article L. 1231-1 A du code de la santé publique le prélèvement et la greffe d'organes au rang de « priorité nationale », le CCNE, dans son avis n° 129 préparatoire au réexamen de la loi de bioéthique, a dressé un bilan plutôt nuancé : « malgré les efforts des professionnels de santé et des pouvoirs publics, de trop nombreuses personnes décèdent encore chaque année faute d'avoir pu bénéficier à temps d'une greffe d'organes. Plus de 6000 greffes d'organe ont été réalisées en 2017, mais le nombre de malades en attente d'un organe est près de quatre fois supérieur aux greffes réalisées et, dans le même temps, en moyenne 550 d'entre eux décèdent chaque année, depuis plusieurs années, même si la situation française est plutôt bonne au niveau européen. »
Si les greffes à partir de donneur vivant représentent 15 % des greffes rénales contre 10 % en 2011, la France reste encore derrière d'autres pays comme les Pays-Bas et les pays scandinaves, se situant au 9 ème rang en Europe en 2015. Le plan greffe 2017-2021 a fixé l'objectif, ambitieux, d'atteindre 1 000 greffes rénales à partir de donneur vivant à l'horizon 2021, soit 14,7 par million d'habitants (pmh) contre 8 pmh en 2015.
Dans ce contexte, une meilleure reconnaissance des donneurs d'organes paraît susceptible de contribuer au développement du don . C'est ce qui avait conduit le CCNE, suivant des préconisations portées par des associations, comme Renaloo entendue par le rapporteur, à envisager la création d'un statut de donneur.
Le présent article additionnel, issu de l' amendement COM-141 du rapporteur adopté par la commission spéciale, complète en ce sens l'article L. 1231-1 A du code de la santé publique, afin :
- d'une part, d'ouvrir droit à une forme de reconnaissance symbolique 140 ( * ) sous la forme de l'accès à une décoration honorifique,
- et, d'autre part, de reconnaître explicitement le principe de neutralité financière du don pour le donneur d'organes.
Ce principe, symétrique à celui de gratuité du don posé par l'article 16-1 du code civil, résulte, à l'heure actuelle, de plusieurs dispositions législatives ou réglementaires éparses 141 ( * ) .
Pour autant, ces dispositions trop peu connues, y compris des établissements de santé, induisent des démarches souvent complexes pour les donneurs . Cela avait ainsi conduit le CCNE dans son avis n° 129 à préconiser la clarification d'un « statut » du donneur impliquant « la reconnaissance effective de ses droits (ne pas subir les conséquences financières, professionnelles ou assurantielles de son don, bénéficier tout au long de sa vie d'un suivi médical, pas nécessairement hospitalier, et de la gratuité des soins éventuellement prodigués en cas de complications, même tardives...) », afin notamment que le donneur vivant n'ait pas à supporter les conséquences financières de son geste généreux.
Si l'Agence de la biomédecine a publié un « Guide de prise en charge financière des donneurs vivants d'éléments du corps humain » visant à permettre « une amélioration des pratiques de prise en charge financière des donneurs vivants » , il est important d'ériger le principe de neutralité financière du don au plan législatif pour lui donner toute la visibilité nécessaire et en faire une priorité dans la politique de promotion du don.
La commission a adopté cet article additionnel ainsi rédigé.
Article 5
Extension du don croisé d'organes
Cet article propose d'étendre le don croisé d'organes à quatre paires de donneurs et receveurs tout en autorisant le recours, dans une chaîne de don croisé, à un organe prélevé sur une personne décédée, et autorise l'Agence de la biomédecine à recourir à des experts inscrits sur une liste nationale afin de compléter la composition des comités d'experts pour donneurs vivants, en dehors des seuls cas d'urgence vitale.
La commission spéciale est revenue sur une disposition adoptée à l'Assemblée nationale renvoyant à un décret la fixation du nombre maximal de paires impliquées dans un don croisé, en fixant ce nombre à six afin de ménager une souplesse dans la mise en oeuvre de cette procédure.
I - Le dispositif proposé
1. L'assouplissement du don croisé d'organes afin d'en renforcer l'effectivité
a) Le « don croisé », une extension du don du vivant introduite par la loi bioéthique de 2011
• Le don du vivant est depuis la première loi de bioéthique de 1994 une source complémentaire de greffons pour les personnes en attente d'une greffe rénale et, plus marginalement, d'une greffe hépatique , dont le nombre ne cesse d'augmenter, en France comme dans les autres pays : pour le rein, l'Agence de la biomédecine recensait ainsi 6 889 patients en attente de greffe en 2009 et 15 190 au 1 er janvier 2019 (soit + 120 %), avec un rythme de plus de 5 000 nouveaux inscrits par an pour environ 3 500 greffes réalisées.
Afin de répondre à la pénurie de greffons prélevés sur donneurs décédés, le législateur, au fil de l'examen des lois de bioéthique successives, a élargi les possibilités de prélèvements sur donneur vivant.
Dans le prolongement de la loi de 1994 qui a autorisé le don du vivant en le limitant au cercle familial restreint (père ou mère, fils ou filles, frères ou soeurs du receveur) ou en cas d'urgence au conjoint, la loi du 6 août 2004 l'a étendu, par dérogation et après autorisation, aux autres membres de la famille 142 ( * ) (conjoint même sans cas d'urgence, grands-parents, oncles ou tantes, cousins germains, conjoint du père ou de la mère du receveur) ainsi qu'à « toute personne apportant la preuve d'une vie commune d'au moins deux ans avec le receveur » .
La loi du 7 juillet 2011 a poursuivi cette évolution :
- d'une part, en élargissant le don du vivant à toute personne ne faisant pas partie de la famille du receveur ou sans lien légal avec lui mais pouvant apporter la preuve d'un « lien affectif étroit et stable » depuis au moins deux ans avec celui-ci. Depuis 2014, ce don représente de 7 à 8 % des greffes rénales à partir de donneur vivant, soit 40 à 50 greffes par an 143 ( * ) ;
- d'autre part, en autorisant, si le don ne peut se faire directement entre parents ou amis, le principe d'un don croisé d'organes entre deux paires de donneurs-receveurs. Cette possibilité vise à répondre aux cas d' incompatibilité biologique (du fait des groupes sanguins ou antigènes des leucocytes humains - HLA) entre un receveur et un proche prêt à lui faire don d'un organe, principalement d'un rein, rendant impossible la greffe.
Cette loi a conservé ainsi l'exigence d'un lien d'affection ou de parenté entre donneur et receveur qui constitue une dérogation à la règle de l'anonymat posée par l'article 16-8 du code civil, en excluant le recours à un donneur altruiste ou « bon samaritain » que d'autres pays autorisent 144 ( * ) . Comme l'a souligné le Conseil d'Etat dans son étude du 28 juin 2018 sur la révision de la loi de bioéthique, « le législateur part du principe que le désintéressement requis pour effectuer un tel geste sans aucune contrepartie se rencontre principalement dans le cadre de relations amicales ou familiales proches » .
• Le don croisé d'organes s'inscrit en cohérence avec cette conception mais vise à offrir une alternative en cas de paires constituées d'un patient et d'un donneur incompatibles (situation qui, d'après l'étude d'impact, se présenterait dans « plus de la moitié des cas ») : il s'agit de les « rassembler » afin de permettre aux patients d'échanger leurs donneurs et donc de bénéficier d'une greffe compatible. Cette procédure est définie par l'article L. 1231-1 du code de la santé publique : elle « consiste pour le receveur potentiel à bénéficier du don d'une autre personne ayant exprimé l'intention de don et également placée dans une situation d'incompatibilité à l'égard de la personne dans l'intérêt de laquelle le prélèvement peut être opéré (...), tandis que cette dernière bénéficie du don du premier donneur. »
Le don croisé est assuré dans le respect de l'anonymat entre donneur et receveur de la même « chaîne ».
Afin de prévenir le risque de rétractation d'un donneur après la greffe de son proche, les actes de prélèvement et de greffe sur les deux donneurs et les deux receveurs doivent se dérouler de façon simultanée .
Le cadre applicable aux donneurs vivants (autre que le père ou la mère), lequel comporte plusieurs étapes, s'applique au don croisé :
- d'abord, l'information préalable du donneur par un comité d'experts, qui vérifie la bonne compréhension de l'information reçue, s'assure que celui-ci a bien mesuré tous les risques qu'il encourt et les conséquences éventuelles du prélèvement et répond à ses questions ;
- ensuite, l'expression par celui-ci de son consentement « libre et éclairé » au don croisé devant le président du tribunal judiciaire ;
- enfin, le comité d'expert délivre ou non l' autorisation de prélèvement, sur la demande du receveur.
b) Un dispositif au cadre restrictif et au succès modeste
• Le programme de don croisé a débuté en octobre 2013. D'après les données de l'Agence de la biomédecine, il n'a donné lieu qu'à la réalisation de 12 greffes rénales (soit 6 échanges croisés) entre 2014 et février 2018.
Ce bilan est extrêmement modeste au regard du nombre déjà réduit de greffes réalisées à partir d'un donneur vivant : celles-ci représentent de l'ordre de 15 % des greffes rénales en France contre 57 % aux Pays-Bas, 41 % au Danemark ou 32 % en Suède 145 ( * ) . Si elles ont connu une progression depuis la dernière loi de bioéthique (l'Agence de la biomédecine recensait 357 greffes de rein à partir de donneur vivant en 2012), dans le cadre du plan greffe 2012-2016 qui en a fait une priorité, elles marquent depuis un certain ralentissement, comme le montre le tableau ci-après.
Parallèlement, le nombre de décès de personnes en attente de greffe n'a cessé de progresser, passant, s'agissant du rein, de 304 en 2014 à 396 en 2018 d'après les données de l'Agence de la biomédecine.
Nombre de greffes réalisées par organe entre 2014 et 2018
|
2014 |
2015 |
2016 |
2017 |
2018 |
|
|
Greffes hépatiques |
1 280 |
1 355 |
1 322 |
1 374 |
1 323 |
|
dont à partir de donneurs vivants |
12 |
15 |
5 |
18 |
14 |
|
Greffes rénales |
3 232 |
3 486 |
3 615 |
3 782 |
3 543 |
|
dont à partir de donneurs vivants |
514 |
547 |
576 |
611 |
537 |
|
Total greffes d'organes |
5 357 |
5 746 |
5 891 |
6 105 |
5 781 |
|
dont à partir de donneurs vivants |
526 |
562 |
581 |
626 |
551 |
Source : Agence de la biomédecine
• Comme l'ont reconnu les représentants de l'Agence entendus par votre rapporteur, la France fait partie des pays d'Europe dont le programme de dons croisés est le moins performant .
La limitation à deux de la chaîne de donneurs-receveurs fait que les possibilités d'appariement sont réduites . Dans son rapport de janvier 2018, l'Agence rappelle ainsi que ce programme a porté sur des cycles d'appariement trimestriels incluant chacun 10 à 20 paires de donneurs-receveurs, alors que la modélisation mathématique comme l'expérience des autres pays montrent que l'appariement est efficace lorsqu'au moins 50 paires donneur-receveur sont inscrites et que l'on « chaîne » plus de deux paires.
Un autre argument avancé par les représentants de l'Agence pour expliquer ce faible taux de recours tient au fait que le don croisé est concurrencé par le développement des greffes dites « incompatibles », comportant un risque immunologique ; celles-ci peuvent être réalisées lorsque le receveur reçoit un traitement immunosuppresseur spécial.
Enfin, la condition de simultanéité des actes de prélèvement et de greffe sur les deux donneurs et les deux receveurs s'avère contraignante du point de vue logistique : cette exigence est interprétée de manière stricte, avec des opérations de prélèvement qui débutent le même jour et à la même heure, rendant nécessaire la synchronisation de quatre salles opératoires.
c) Les travaux préparatoires ont mis en avant l'opportunité d'une évolution de la législation relative au don croisé
L'Agence de la biomédecine 146 ( * ) identifiait ainsi, au vu des contraintes actuelles du dispositif, plusieurs axes d'évolution : introduire la notion de « chaîne », en envisageant alors « au minimum trois paires donneur-receveur » ; autoriser l'introduction d'un donneur décédé (ou d'un donneur altruiste si ce don devait être autorisé) pour « amorcer » la chaîne ; enfin, étendre l'ouverture du don croisé à l'international pour accroître le nombre de paires disponibles à chaque lancement d'appariement, selon un axe du plan greffe 2017-2021. Une coopération est déjà en place avec la Suisse et des discussions sont engagées avec la Belgique.
Dans son avis n° 129, le CCNE jugeait de même souhaitable la mise en place d'une « chaîne » de donneurs successifs, éventuellement initiée avec le rein d'un donneur décédé, tout en s'assurant du respect du consentement éclairé des donneurs comme des patients à greffer.
Les rapporteurs de l'OPECST s'étaient également prononcés en faveur de l'extension de la chaîne de dons croisés, mais à titre expérimental, permettant notamment d'évaluer les résultats en fonction de l'ajout ou non, en début de chaîne, d'un don post mortem . Ils s'étaient montrés opposés à l'intervention, en début de chaîne, d'un donneur vivant ou « bon samaritain » sans lien de parenté avec un receveur de la chaîne, suivant l'avis « réservé » exprimé sur ce point par le Conseil d'Etat dans son étude de juin 2018.
Le Conseil d'Etat avait en effet considéré que « le régime du prélèvement d'organes sur donneurs vivant doit rester précautionneux pour ne pas heurter les principes d'inviolabilité et de non patrimonialité du corps humain » . Il avait en revanche envisagé un assouplissement possible de l'obligation de simultanéité des prélèvements et greffes ainsi que l'initiation d'une chaîne de don par un don post mortem , de nature à soulever moins d'objections de principe que l'introduction d'un donneur vivant solidaire.
d) Les dispositions du projet de loi : lever des verrous pour rendre plus efficace le don croisé
Le 1° modifie l'article L. 1231-1 du code de la santé publique en apportant trois changements notables visant à améliorer les possibilités de recours au don croisé d'organes.
• En premier lieu, il porte à quatre maximum le nombre de paires de donneurs et de receveurs consécutifs , alors que le texte ne prévoit aujourd'hui le don croisé qu'entre deux paires de donneurs et receveurs.
Pour le Gouvernement, cette proposition constitue « un choix rationnel » retenu au vu des expériences internationales et en tenant compte des capacités opérationnelles des équipes hospitalières françaises. Aux Pays-Bas, la limite est fixée de même à quatre paires ; aux États-Unis comme au Royaume Uni, le cadre n'est pas limité mais l'expérience américaine - où le don croisé se pratique depuis 2007 - permet de constater, d'après l'étude d'impact, un nombre moyen de 4,6 paires impliquées dans une chaîne.
• En outre, le projet de loi autorise l'introduction dans une procédure de don croisé d'un organe prélevé sur un donneur décédé , « pour augmenter les possibilités d'appariement entre donneurs et receveurs engagés dans un don croisé et en substitution au prélèvement de l'un des donneurs vivants ».
Cela permettra de pallier une « rupture de chaîne » due à un problème médical ou à un désistement, ou d'initier une chaîne de don en offrant une solution à une situation d'impasse immunologique pour l'un des receveurs, lorsque l'appariement ne sera trouvé que pour trois paires sur quatre.
En cas d'échec de prélèvement ou de greffe, l'Agence de la biomédecine devra procéder, en outre, à l'attribution d'un greffon selon les règles d'équité de répartition de l'article L. 1231-1 B du code de la santé publique « les plus favorables » au receveur. Ce dernier sera en pratique inscrit dans une catégorie prioritaire.
• Enfin, le projet de loi prévoit de desserrer la condition de simultanéité des opérations de prélèvement et de greffe d'organe : les opérations de prélèvement devront se dérouler dans un délai de 24 heures et les opérations de greffes être réalisées, quant à elles, « consécutivement », sans condition de délai. La greffe devra toutefois se faire dans un délai contraint par la durée maximale de conservation du greffon, qui est aujourd'hui d'environ 21 heures pour le rein une fois prélevé.
Comme l'ont indiqué les représentants de l'Agence de la biomédecine, ce délai maximal de 24 heures constitue un compromis entre l'efficacité opérationnelle et le risque théorique de désistement d'un donneur ou d'arrêt de procédure , en raison par exemple d'une difficulté opératoire. Le risque de « rupture de chaîne » reste cependant très faible : aux Etats-Unis, où le nombre de paires est potentiellement illimité, 20 chaînes ont connu un problème de ce type sur 1 748 greffes réalisées. Cela corrobore les conclusions d'une étude scientifique de 2017 citée dans l'étude d'impact, mettant en évidence un « taux de remords » évalué à 1,7 %.
2. Les ajustements visant à faciliter la réunion des comités d'experts
• En contrepartie de l'élargissement du cercle des donneurs vivants, la loi de 2004 a encadré la protection des donneurs d'un point de vue éthique en confiant à des comités indépendants, les comités d'experts ou « comités donneurs vivants » la double mission d'informer le donneur et de délivrer, in fine , l'autorisation de prélèvement par une décision collégiale , le consentement étant recueilli entre temps par un magistrat.
L'autorisation de ce comité est obligatoire pour les donneurs admis à titre dérogatoire (cercle familial élargi ou liens affectifs) et facultative, sur décision du magistrat qui recueille le consentement, pour les donneurs admis par principe (le père ou la mère du receveur). La décision n'a pas à être motivée et ne peut faire l'objet d'un recours. 38 refus ont été prononcés entre juin 2005 et fin 2016, pour 2 989 autorisations.
Il existe aujourd'hui neuf comités d'experts répartis sur le territoire. L'Agence de la biomédecine en assure le secrétariat centralisé et organise, afin de les harmoniser, les échanges de pratiques entre eux.
Comme le souligne l'Agence dans son rapport d'évaluation de la loi de bioéthique de juin 2018, ces comités « offrent aux donneurs potentiels un indispensable lieu d'expression libre et indépendant , échappant à toute pression, qu'elle émane de la famille du malade ou de l'équipe médicale. » Elle constate cependant que l'augmentation de l'activité de ces comités les confronte à une difficulté à trouver des experts disponibles pour y siéger. En outre, le déroulement de la procédure apparaît « trop long et contraignant » pour les équipes médicales au regard de leurs besoins de planification des interventions chirurgicales, suscitant en outre l'incompréhension de certains donneurs engagés dans un parcours médical déjà long.
• Le 2° modifie ponctuellement l'article L. 1231-3 du code de la santé publique précisant la composition de ces comités.
Les règles de composition des comités d'experts
Les comités d'experts siègent en deux formations de cinq membres désignés pour trois ans par arrêté du ministre chargé de la santé, à raison de quatre suppléants pour un titulaire.
Ils comportent dans tous les cas deux médecins et une personne qualifiée dans le domaine des sciences humaines et sociales , communs aux deux formations. Le comité comporte en outre :
- un psychologue et un médecin lorsqu'il se prononce sur les prélèvements sur personne majeure (don du vivant, don croisé, prélèvement de cellules hématopoïétiques) ;
- une personne qualifiée dans le domaine de la psychologie de l'enfant et un pédiatre lorsqu'il se prononce sur les prélèvements de cellules hématopoïétiques sur personne mineure.
A l'heure actuelle, en cas d'urgence vitale, les membres d'un comité d'experts peuvent être désignés par l'Agence de la biomédecine parmi les membres disponibles figurant sur la liste établie par arrêté ministériel.
La modification proposée (a) du 2°) tend à généraliser, au-delà des seuls cas d'urgence vitale, le recours à des experts figurant sur la liste arrêtée au niveau national pour compléter la composition d'un comité lorsqu'un membre titulaire et ses quatre suppléants sont empêchés.
D'après l'étude d'impact, il s'agit de faciliter la réunion des comités pour accompagner le développement du don du vivant, alors que le principe actuel de liste d'experts par ressort territorial restreint la faculté de les mobiliser dans des délais rapides . Les représentants de l'Agence de la biomédecine auditionnés par votre rapporteur ont confirmé qu'une liste nationale, leur permettant de combiner par voie informatique les disponibilités des membres, a vocation à alléger les contraintes des réunions tout en optimisant les déplacements, notamment dans des régions où les experts s'avèrent peu disponibles.
La composition de ces comités n'est pas modifiée, en revanche, afin de conserver leur caractère pluridisciplinaire.
• Le 3° modifie quant à lui l'article L. 1231-4 du code de la santé publique qui fixe les conditions d'application du chapitre relatif au don du vivant par décret en Conseil d'Etat.
Sont visés :
- comme à l'heure actuelle, les dispositions applicables aux dons croisés d'organes, en précisant que cela comprendra « les modalités d'information des donneurs et receveurs engagés dans celui-ci » ;
- de manière plus succincte qu'à l'heure actuelle, les conditions de fonctionnement du comité d'experts, en ne faisant plus référence explicitement à leur nombre, compétence territoriale ou conditions de désignation et de rémunération.
II - Les modifications adoptées par l'Assemblée nationale
• Outre l'adoption d'amendements rédactionnels, la commission spéciale a décidé, à l'initiative de Jean-Louis Touraine (La République en Marche), de renvoyer à un décret en Conseil d'État après avis de l'Agence de la biomédecine la définition du nombre maximum de paires de donneurs et receveurs impliquées dans un don croisé plutôt que le fixer dans la loi à quatre.
Cet amendement, visant à permettre une adaptation plus souple aux progrès de la médecine, a été adopté avec l'avis favorable du rapporteur Hervé Saulignac ainsi que de la ministre des solidarités et de la santé. Cette dernière a rappelé que « l'Agence de la biomédecine repose sur des comités d'experts dont le conseil d'orientation scientifique et médical valide toutes les décisions » et ajouté que « procéder par décret permettrait d'aller progressivement à trois paires, et peut-être à quatre paires » .
• En séance publique, l'Assemblée nationale a complété ces dispositions, avec l'avis favorable du Gouvernement, afin de prévoir que le Parlement est tenu informé lorsque ce nombre maximal fait l'objet d'une modification (amendement présenté par Hervé Saulignac, rapporteur).
III - La position de la commission : préciser le nombre de paires de donneurs dans la loi
• Cet article, le seul du projet de loi initial concernant la greffe d'organes, répond à une intention louable.
Il vise à contribuer, selon les priorités du plan greffe 2017-2021, au développement du don du vivant. En ce sens, les assouplissements proposés concernant le don croisé d'organes, par l'extension du nombre de paires impliquées dans la chaîne et le recours possible à un donneur décédé, doivent être accueillis positivement : ils font écho à des propositions convergentes formulées dans les travaux préparatoires pour lever des contraintes pesant sur le développement de cette pratique.
Il est cependant fort peu probable que cette disposition, qui ne porte que sur un aspect ponctuel, suffise en elle-même à répondre aux enjeux majeurs du plan greffe, qui renvoient également à d'autres défis, comme les associations entendues par le rapporteur l'ont souligné, en termes notamment d'information et de sensibilisation ou encore d'équité territoriale dans l'accès à la greffe, en particulier pour nos concitoyens ultra-marins.
• Si le projet de loi traduit, en matière de don croisé, une position d'équilibre, le texte issu des travaux de l'Assemblée nationale n'est pas pleinement satisfaisant : tout en renvoyant au décret en Conseil d'Etat la fixation du nombre maximal de paires, les députés ont tenu à prévoir une information du Parlement en cas de modification de ce décret.
La commission spéciale a jugé préférable de réintroduire au niveau de la loi le nombre maximal de paires de donneurs-receveurs pouvant être impliqué dans un don croisé, en portant ce nombre à six au lieu de quatre dans le projet de loi initial afin de ménager une certaine souplesse. Elle a adopté en ce sens l'amendement COM-142 de son rapporteur.
Cette évolution, conforme aux indications de l'étude d'impact sur la « taille souhaitable » des chaînes au vu des expériences internationales, reste compatible, au plan logistique, avec le délai de 24 heures prévu pour la réalisation des opérations de prélèvement. D'après les débats à l'Assemblée nationale, le Gouvernement n'aurait pas envisagé d'aller au-delà de ce nombre de six paires.
La commission spéciale a adopté, en outre, un amendement COM-184 de coordination de son rapporteur.
La commission a adopté cet article ainsi modifié.
Article 5 bis
Extension de l'information sur le don d'organes
prévue par le code de la santé publique aux patients de 16
ans et plus
Cet article, inséré par l'Assemblée nationale, propose d'étendre à toutes les personnes de plus de 16 ans l'information sur le don d'organes réalisée par les médecins, actuellement ciblée sur les jeunes de 16 à 25 ans. Tout en relativisant sa portée, la commission spéciale l'a adopté sans modification.
I - Le dispositif proposé
Cet article résulte de l'adoption, par la commission spéciale, d'un amendement présenté par Laurianne Rossi, députée La République en Marche, contre l'avis « plutôt défavorable » du rapporteur Hervé Saulignac mais avec l'approbation de la ministre des solidarités et de la santé.
Il modifie l'article L. 1211-3 du code de la santé publique afin d' élargir à l'ensemble des personnes de plus de 16 ans l'information aujourd'hui ciblée sur les jeunes de 16 à 25 ans délivrée par les médecins destinée à s'assurer que leurs patients « sont informés des modalités de consentement au don d'organes à fins de greffe » . A défaut, les médecins doivent leur délivrer individuellement cette information « dès que possible » .
Cette disposition avait été introduite par la loi du 6 août 2004 relative à la bioéthique, dans l'objectif de sensibiliser en priorité un public jeune. Toutefois, l'auteure de l'amendement comme la ministre ont considéré que cette borne d'âge, assez théorique, était inutilement restrictive.
II - La position de la commission
L'enjeu d'une plus large sensibilisation des Français à la législation relative au don d'organes est évidemment majeur.
Au-delà de la mission générale confiée à l'Agence de la biomédecine en matière de promotion du don d'organes, le législateur a cherché, au fil de l'examen des lois de bioéthique, à introduire de multiples canaux d'information. La loi de 2011 a ainsi prévu de mentionner sur la carte vitale 147 ( * ) et le dossier médical partagé 148 ( * ) la mention selon laquelle la personne a été informée de la législation en matière de don d'organe. Cette loi a également prévu une information ad hoc dispensée dans les lycées et les établissements d'enseignement supérieur 149 ( * ) .
Il est cependant permis de s'interroger sur la portée concrète de ces dispositions et, singulièrement, de l'obligation législative d'information de leurs jeunes patients par les médecins posée par la loi de bioéthique de 2004.
Tout en relativisant la portée et donc l'intérêt de la modification introduite par cet article, la commission spéciale n'a toutefois pas trouvé de raisons de s'y opposer.
La commission a adopté cet article sans modification.
Article 6
Possibilité de prélever des cellules souches
hématopoïétiques
sur un mineur ou un majeur
protégé au bénéfice de ses parents
Cet article propose de permettre le prélèvement de cellules souches hématopoïétiques sur un mineur ou un majeur protégé au bénéfice de l'un de ses parents en prévoyant un mécanisme particulier de représentation par un administrateur ad hoc pour assurer la représentation de l'enfant mineur ou du majeur protégé. La commission a souhaité permettre au mineur de 16 ans d'exprimer lui-même son consentement, considérant qu'il était en capacité de le faire sans nécessité de s'en remettre à un administrateur ad hoc .
I - Le dispositif proposé : permettre le prélèvement de cellules souches hématopoïétiques sur un mineur ou un majeur protégé au bénéfice de l'un de ses parents
La greffe de cellules souches hématopoïétiques permet de traiter certains cancers et maladies du sang ou du système immunitaire en faisant « une remise à zéro » du système sanguin ou immunitaire. L'opération consiste à remplacer les cellules malades par des cellules saines recueillies dans la moelle osseuse ou le sang périphérique 150 ( * ) d'un donneur HLA compatible 151 ( * ) , hors cas de greffe à soi-même (ou autologue).
Le prélèvement des cellules souches hématopoïétiques
Les cellules souches hématopoïétiques (CSH) sont fabriquées par la moelle osseuse et sont à l'origine des différentes cellules du sang : les globules rouges, les globules blancs et les plaquettes.
Les CSH sont prélevées :
- soit dans le sang depuis une veine du bras, comme pour le don de sang 152 ( * ) , après une stimulation préalable de la moelle osseuse par voie médicamenteuse ; cette méthode, « par aphérèse », représente 75 % des prélèvements 153 ( * ) ;
- soit directement dans la moelle osseuse, par ponction dans les os iliaques, au niveau du bassin ; ce prélèvement est réalisé à l'hôpital sous anesthésie générale.
Depuis quelques années, la recherche médicale a permis d'élargir considérablement le champ de donneurs potentiels de cellules souches hématopoïétiques en rendant possible la réalisation de greffes à partir de donneurs haplo-identiques , c'est-à-dire semi-compatibles seulement.

Source : Étude d'impact
Ainsi, lorsqu'il est à la recherche d'un greffon, un praticien peut désormais recourir à trois types de donneurs, avec par ordre de préférence : les donneurs géno-identiques (les frères et soeurs du patient) ; les donneurs phéno-identiques, inscrits dans les fichiers des banques de donneurs non apparentés ; et en dernier ressort, les donneurs haplo-identiques (père et mère ou enfants du patient).
C'est le développement de cette technique de greffe haplo-identique qui pose aujourd'hui la question des prélèvements de cellules souches hématopoïétiques sur un enfant mineur ou un majeur protégé au profit de l'un de ses parents.
1. La situation actuelle : un prélèvement interdit
L'article L. 1241-2 du code de la santé publique interdit tout prélèvement de tissus ou de cellules ou toute collecte de produits du corps humain en vue de don « sur une personne vivante mineure ou sur une personne vivante majeure faisant l'objet d'une mesure de protection légale ». Une interdiction similaire existe en matière de don d'organes 154 ( * ) . Ce principe connaît depuis son origine des exceptions au profit de membres de la famille, hors père et mère .
a) Les exceptions prévues pour les mineurs à l'article L. 1241-3
L'exception à l'interdiction de prélèvement d'organes, de tissus ou de cellules chez un mineur existe depuis la loi Cavaillet de 1976 155 ( * ) au profit d'un frère ou d'une soeur . Comme aujourd'hui, cette exception supposait le respect d'une procédure particulière destinée à protéger le mineur, comprenant le consentement du représentant légal du mineur et une autorisation donnée par un comité composé de trois experts. Par ailleurs, le refus du mineur d'accepter le prélèvement devait « être toujours respecté ».
La loi de 1994 156 ( * ) a restreint cette exception au seul prélèvement de moelle osseuse - au caractère régénérable - au profit du frère ou de la soeur du donneur et a ajouté comme garantie l'intervention du président du tribunal de grande instance devant lequel le consentement doit être exprimé 157 ( * ) . La loi de 2004 158 ( * ) a ensuite élargi le cercle des receveurs familiaux possibles en intégrant, à titre exceptionnel , en cas d'absence d'alternative, les cousins germains ou cousines germaines, les oncles ou tantes, neveux ou nièces.
En 2011 159 ( * ) , la loi relative à la bioéthique est venue soumettre au même régime, le prélèvement - moins invasif - de cellules souches hématopoïétiques dans le sang périphérique par aphérèse (cf. encadré supra ).
b) Les exceptions prévues pour les majeurs protégés à l'article L. 1241-4
La loi du 6 août 2004 relative à la bioéthique a introduit une exception similaire à celle qui existe pour les mineurs pour les majeurs protégés, en prévoyant qu'en l'absence d'autre solution thérapeutique , un prélèvement de cellules hématopoïétiques issues de la moelle osseuse pouvait être fait sur une personne vivante majeure faisant l'objet d'une mesure de protection légale au bénéfice de son frère ou de sa soeur .
Là encore, une procédure particulière a été mise en place, sous le contrôle du juge des tutelles qui :
- autorise le prélèvement lorsque le donneur est sous tutelle ;
- apprécie si la personne a la faculté de consentir lorsqu'elle est sous curatelle ou fait l'objet d'une sauvegarde de justice. Dans l'affirmative, le prélèvement doit encore être autorisé par le comité d'experts.
Lorsque les personnes font l'objet d'une mesure de curatelle ou de sauvegarde de justice et ont été reconnues comme ayant la faculté de consentir au prélèvement par le juge des tutelles compétent après avoir été entendues par celui-ci, le cercle des receveurs intrafamiliaux peut être étendu , à titre exceptionnel, aux collatéraux : cousins germains ou cousines germaines, oncles ou tantes, neveux ou nièces.
Ces exceptions respectent l'article 14 du Protocole additionnel à la Convention sur les droits de l'homme et la biomédecine relatif à la transplantation d'organes et de tissus d'origine humaine du Conseil de l'Europe, dite « convention d'Oviedo » du 4 avril 1997.
En 2011, comme pour les mineurs, le dispositif a été étendu au prélèvement de cellules souches hématopoïétiques dans le sang périphérique.
2. La proposition du Gouvernement : permettre tout en encadrant
Le Gouvernement propose d'autoriser les prélèvements de cellules souches hématopoïétiques d'un mineur ou d'un majeur protégé lorsque le receveur est un parent ou, s'agissant de la personne protégée, la personne exerçant la mesure de protection ou un descendant ou un collatéral de la personne chargée de la mesure de protection, ce qui pourrait concerner environ une cinquantaine de greffes par an.
Dans ces deux cas, le refus exprimé par le donneur pressenti continuerait à faire obstacle au prélèvement et les dérogations seraient entourées de précautions particulières compte tenu du fait que le receveur est le représentant légal du donneur ou un proche de ce représentant légal :
- pour le mineur , un administrateur ad hoc serait désigné par le président du tribunal judiciaire 160 ( * ) en dehors du cercle familial - ni un ascendant, ni un collatéral - pour représenter le mineur en lieu et place de ses parents . L'administrateur ad hoc serait informé par le praticien ayant posé l'indication de greffe des risques encourus par le mineur et des conséquences éventuelles du prélèvement.
Le président du tribunal judiciaire autoriserait le prélèvement après avoir entendu le mineur - s'il est capable de discernement -, ses parents, ainsi que l'administrateur ad hoc , et après avoir recueilli l'avis du comité d'experts donneur vivant . Cette procédure serait ainsi renforcée par rapport au don intrafamilial au profit d'une autre personne que le père ou la mère. Dans ce cas, le président du tribunal judiciaire se contente en effet de recueillir le consentement et de s'assurer qu'il est libre et éclairé, le prélèvement étant autorisé par le comité d'experts donneur vivant ;
- pour le majeur faisant l'objet d'une mesure de protection juridique avec représentation à la personne 161 ( * ) , un administrateur ad hoc serait nommé par le juge des tutelles pour recevoir les informations médicales sur les risques encourus par le majeur protégé et les conséquences éventuelles du prélèvement.
Le juge des tutelles agirait ensuite selon deux modalités :
- soit il estime, après l'avoir entendue, que la personne protégée a la faculté de consentir au prélèvement , et le droit commun s'applique : il reçoit ce consentement au prélèvement, qui ne peut être réalisé qu'après avoir été autorisé par le comité d'experts donneur vivant
- soit il estime après l'avoir entendue, que la personne protégée n'a pas la faculté de consentir au prélèvement , et il peut autoriser le prélèvement après avoir recueilli l'avis de la personne concernée - lorsque cela est possible -, de la personne chargée de la mesure de protection, lorsque celle-ci n'est ni le receveur, ni un descendant, ni un collatéral du receveur, du comité d'experts et, le cas échéant, de l'administrateur ad hoc .
Pour un majeur faisant l'objet d'une mesure de protection juridique avec représentation aux biens, aucune procédure dérogatoire ne s'appliquerait plus en cohérence avec l'article 7 du projet de loi.
Le régime choisi met ainsi en avant l'autonomie et le respect des choix de la personne protégée conformément à la loi du 5 mars 2007 portant réforme de la protection juridique des majeurs 162 ( * ) , en réservant l'autorisation par le juge au seul cas où le majeur ne peut consentir.
Le projet de loi prévoit également la mise en place d'un suivi de l'état de santé de tous les donneurs de cellules souches hématopoïétiques, non apparentés comme apparentés, par l'Agence de la biomédecine 163 ( * ) . Jusqu'à présent, seuls les donneurs non apparentés faisaient l'objet d'un suivi par l'Agence de la biomédecine en vertu de sa mission de gestion du fichier des donneurs volontaires de cellules souches hématopoïétiques 164 ( * ) .
Il met enfin en cohérence les infractions pénales prévues à l'article L. 1271-4 du code de la santé publique et l'article L. 511-5 du code pénal réprimant le non-respect des conditions de prélèvement chez un mineur ou chez un majeur protégé.
II - Les modifications adoptées par l'Assemblée nationale
L'Assemblée nationale n'a pas apporté de modifications substantielles à l'article 6.
Seuls trois amendements rédactionnels du rapporteur ont été adoptés en commission 165 ( * ) .
III - La position de la commission : permettre aux mineurs de 16 ans d'exprimer eux-mêmes leur consentement devant le juge
À l'initiative de son rapporteur, la commission a souhaité abaisser l'âge du consentement afin qu'un mineur de 16 ans puisse lui-même consentir au prélèvement de cellules souches hématopoïétiques (CSH) au bénéfice de l'un de ses parents.
Cette proposition est née d'un double constat :
- comme l'a souligné la présidente de la Fédération nationale des administrateurs ad hoc (FENAAH), à partir du moment où les deux parents sont en accord pour ce don, ainsi que l'enfant donneur lui-même, il n'y a pas réellement de conflit d'intérêts 166 ( * ) et la désignation d'un administrateur ad hoc ne se justifie nullement ; un enfant aura naturellement envie de « sauver » un parent en danger de mort et ce n'est pas la nomination d'un mandataire ad hoc qui pourrait écarter la pression qui pèse de toute manière sur l'enfant ; un accompagnement psychologique adapté serait plus propice à résoudre les difficultés que la désignation d'un administrateur ad hoc peu formé à ce genre de situation ;
- à ce jour, selon les informations obtenues de l'Agence de la biomédecine, seules deux demandes de dérogation relatives à des dons de cellule souches hématopoïétiques d'un mineur vers l'un de ses parents ont été formulées et il s'agissait dans les deux cas de mineurs de plus de 17 ans .
Cet âge s'explique par la nécessaire adéquation qui doit exister entre le poids du donneur pressenti et celui du patient. En effet, la quantité de cellules souches hématopoïétiques prélevée est fonction du poids du donneur tandis que la quantité de cellules souches nécessaires au malade pour assurer une bonne prise de greffe est calculée en fonction du poids de celui-ci. Le prélèvement de cellule souches hématopoïétiques au bénéfice d'un parent ne concernera pas les jeunes enfants .
Compte tenu de ce constat, il est apparu souhaitable au rapporteur d'abaisser l'âge du consentement, afin que, dès 16 ans, un adolescent, qui a alors la « faculté de consentir » 167 ( * ) , puisse lui-même assumer la décision et l'exprimer directement. Par cohérence, cet abaissement de l'âge du consentement jouerait également dans les autres hypothèses de dons intrafamiliaux de cellules souches hématopoïétiques.
Le consentement libre et éclairé du mineur de 16 ans pourrait être vérifié par le président du tribunal judiciaire auprès duquel il comparaitrait personnellement, éventuellement accompagné d'un avocat, et par le comité d'experts donneur vivant par lequel il serait reçu. Ce comité d'experts, lorsqu'il se prononce sur les prélèvements sur personne mineure, comporte une personne qualifiée dans le domaine de la psychologie de l'enfant et un pédiatre 168 ( * ) .
La fixation de la condition d'âge à 16 ans apparaît par ailleurs cohérente avec d'autres dispositions légales qui prévoient la capacité d'exercice sans autorisation parentale telles que l'émancipation, l'acquisition de la nationalité française ou plus récemment, l'âge minimum requis pour s'inscrire sur les réseaux sociaux (règlement général sur la protection des données).
Pour garder une certaine souplesse et permettre de faire face aux situations - a priori rares - où les donneurs pressentis seraient des mineurs de moins de 16 ans, le rapporteur a souhaité ne pas interdire les prélèvements en deçà de cet âge et maintenir l'intervention d'un administrateur ad hoc dans cette hypothèse. La FENAAH et l'Union des associations familiales de France (UNAF) ont souligné avec raison la nécessité qui existerait alors de prévoir une formation adaptée de l'administrateur ad hoc , un cadre référentiel, ainsi qu'une rémunération adéquate, sachant que l'offre est déjà insuffisante pour couvrir certains territoires.
Dans ces conditions, la commission a adopté l'amendement COM-248 abaissant l'âge du consentement à 16 ans pour le don de cellules souches hématopoïétiques intrafamilial.
La commission a par ailleurs adopté les amendements de coordination COM-139 et COM-247 présentés par le rapporteur.
La commission a adopté cet article ainsi modifié.
Article 7
Levée partielle de l'interdiction des dons d'organes,
de tissus et de cellules applicable aux majeurs protégés
Cet article propose d'autoriser les majeurs bénéficiant d'une protection juridique avec représentation aux biens ou assistance à faire des dons d'organes, de tissus et de cellules. Il vise également à soumettre tous les majeurs protégés au régime de droit commun du prélèvement d'organes post mortem . La commission a considéré qu'il n'était pas possible de présumer le consentement éclairé des personnes majeures faisant l'objet d'une protection juridique avec représentation à la personne et les a sorties du dispositif.
I - Le dispositif proposé : permettre aux majeurs protégés de donner leurs organes, tissus et cellules selon le régime de droit commun
1. Les mesures de protection des majeurs
Les majeurs protégés sont des personnes qui sont dans l'impossibilité de pourvoir seules à leurs intérêts en raison d'une altération de leurs facultés mentales ou de leurs facultés corporelles de nature à empêcher l'expression de leur volonté 169 ( * ) . Ils bénéficient de ce fait de mesures de protection juridique résultant d'un contrat - le mandat de protection future -, ou d'une décision de justice.
La personne chargée de leur protection :
- soit, les assiste et les conseille dans tous les actes importants de leur vie 170 ( * ) , les personnes protégées restant autonomes pour la gestion des affaires courantes ;
- soit, les représente dans tous les actes de la vie civile , c'est-à-dire prend les décisions à leur place 171 ( * ) , tout en veillant à les informer et à respecter au mieux leur volonté.
Les différentes mesures de protection juridique des majeurs
Le mandat de protection future (articles 477 et suivants du code civil)
La personne anticipe le moment où elle pourrait ne plus être en mesure de pourvoir seule à ses intérêts pour une des causes justifiant une mesure de protection et désigne une ou plusieurs personnes chargées de l'assister ou la représenter . Le mandat prend effet lorsqu'il est établi médicalement que le mandant ne peut plus pourvoir seul à ses intérêts.
Les mesures de protection judiciaire (articles 428 et suivants du code civil)
Ces mesures ne sont mises en place qu'à titre subsidiaire , c'est-à-dire si un mandat de protection future, le droit commun de la représentation, les droits et devoirs entre époux, le régime matrimonial ou encore une habilitation familiale ne peuvent suffire à pourvoir aux intérêts du majeur. La mesure prononcée doit être proportionnée et individualisée en fonction du degré d'altération des facultés personnelles de l'intéressé.
- l' habilitation familiale permet au juge des tutelles d'habiliter un proche du majeur protégé à l'assister ou le représenter pour des actes ponctuels ;
- la sauvegarde de justice peut être prononcée pour les personnes qui ont besoin d'une protection temporaire ou d'être représentées pour l'accomplissement de certains actes déterminés ;
- la curatelle est destinée aux personnes ayant besoin d'être contrôlées ou assistées d'une manière continue pour les actes importants de la vie civile ;
- la tutelle , qui est la mesure la plus protectrice, concerne les personnes ayant besoin d'être représentées de manière continue dans les actes de la vie civile.
Une mesure de protection juridique vise à protéger tant la personne du majeur protégé que ses biens . Elle peut toutefois être expressément limitée à l'une ou l'autre de ces missions - ou modulée selon les intérêts à protéger - par le juge ou dans le mandat de protection future. Un majeur protégé peut ainsi par exemple faire l'objet d'une représentation pour la protection de ses biens et d'une simple assistance pour la protection de sa personne.
Une mesure de représentation pour la protection de la personne (« représentation à la personne ») peut être mise en place dans le cadre d'une tutelle , d'une habilitation familiale ou d'un mandat de protection future . Il y a actuellement environ 400 000 personnes concernées par ces mesures 172 ( * ) , principalement des majeurs sous tutelle.
2. La situation actuelle : les majeurs protégés ne peuvent pas être donneurs vivants d'organes, de tissus et de cellules
En matière de don d'organes, de tissus et de cellules de son vivant , le code de la santé publique a édicté une interdiction pour protéger les majeurs en situation de vulnérabilité 173 ( * ) :
- l'article L. 1241-2 interdit tout prélèvement de tissus ou de cellules et toute collecte de produits du corps humain en vue de don sur une personne vivante majeure faisant l'objet d'une protection légale 174 ( * ) ;
- l'article L. 1231-2 exclut tout prélèvement d'organes sur une telle personne.
Cette dernière interdiction connaît toutefois une exception lorsqu'un organe fonctionnel est prélevé lors d'une intervention chirurgicale effectuée dans l'intérêt de la personne opérée et peut être transplanté au bénéfice d'un tiers (« greffe en domino ») ou utilisé à des fins scientifiques 175 ( * ) . Le don d'un organe prélevé dans l'intérêt thérapeutique du majeur protégé, et non en vue d'un don, peut être donné selon le droit commun - après information et sous condition de non opposition - sauf s'il est sous tutelle. Dans ce cas, l'article L. 1235-2 dispose qu'une telle utilisation est permise à condition que le majeur protégé ne la refuse pas et si son tuteur, également dûment informé, ne s'y oppose pas.
S'agissant du prélèvement d'organes post mortem , le cas des majeurs sous tutelle a également fait l'objet d'une disposition spécifique : l'article L. 1232-2 ne permet un prélèvement sur une personne sous tutelle qu'à la condition que son tuteur y consente par écrit . Ce mécanisme déroge au régime commun qui repose sur le consentement présumé des personnes, auxquelles il revient de manifester leur refus de leur vivant , principalement en s'inscrivant sur le registre national des refus tenu par l'Agence de la biomédecine 176 ( * ) . Cet article n'est toutefois plus applicable car l'article 418 du code civil, issu de la réforme de 2007 de la protection juridique des majeurs 177 ( * ) , prévoit que le décès du majeur protégé met fin à la mission du tuteur.
Le cas particulier des dons de cellules souches hématopoïétiques a été évoqué supra 178 ( * ) .
3. Lever l'interdiction pour les majeurs faisant l'objet d'une protection juridique avec représentation aux biens ou assistance
L'article 7 du projet de loi vise à mettre en cohérence le régime de consentement au don d'organes, de tissus et de cellules pour les majeurs protégés du code de la santé publique avec les dispositions actuelles du code civil relatives au consentement des majeurs protégés en matière de santé. Il tend également à favoriser l'autonomie des personnes protégées en leur permettant d'exprimer leur consentement.
Le consentement des majeurs protégés en matière de santé
L'article 458 du code civil précise la liste des actes strictement personnels pour lesquels aucune assistance ou représentation n'est possible ; il s'agit de manière non exhaustive de la déclaration de naissance d'un enfant, sa reconnaissance, les actes de l'autorité parentale relatifs à la personne d'un enfant, la déclaration du choix ou du changement du nom d'un enfant et le consentement donné à sa propre adoption ou à celle de son enfant.
L'article 459 prévoit quant à lui que pour les autres actes de nature personnelle , y compris en matière de santé , la personne prend seule les décisions, « dans la mesure où son état le permet » . Ce n'est que lorsque son état ne lui permet pas de prendre une décision personnelle éclairée, que le juge des tutelles peut prévoir la mise en place d'une assistance ou, lorsque l'assistance est inadaptée à la situation, autoriser la représentation du majeur protégé.
Depuis 2019 179 ( * ) , cette représentation peut jouer pour toute décision médicale « y compris pour les actes ayant pour effet de porter gravement atteinte à son intégrité corporelle » sans autorisation préalable du juge. Le juge n'intervient pas, sauf urgence et en cas de désaccord entre l'intéressé et la personne en charge de sa représentation, pour autoriser l'un ou l'autre à prendre la décision.
En cohérence avec l'article 459 du code civil, l'article 7 prévoit de soumettre les personnes bénéficiant d'une assistance ou d'une représentation mais uniquement pour la gestion de leurs biens 180 ( * ) , au droit commun applicable en matière de don d'organes, de tissus et de cellules de son vivant considérant qu'ils sont aptes à exprimer un consentement éclairé en matière médicale.
Le majeur protégé bénéficierait alors des mêmes protections que tout donneur vivant , et en particulier, en cas de don d'organes, d'une procédure en trois temps : information par le comité d'experts donneur vivant, vérification du caractère libre et éclairé de son consentement par un juge, puis autorisation donnée par ce même comité d'experts.
Seules les personnes faisant l'objet d'une mesure de protection juridique avec représentation à la personne resteraient soumises à la restriction des articles L. 1231-2, L. 1235-2 et L. 1241-2 du code de la santé publique. Les infractions pénales incriminées par les articles L. 1272-2 du code de la santé publique et 511-3 du code pénal réprimant le non-respect des conditions de prélèvement chez un mineur ou chez un majeur protégé seraient mises en cohérence.
L'article 7 propose également de soumettre l'ensemble des majeurs protégés, quelles que soient les mesures de protection dont ils bénéficient, au droit commun en matière de prélèvement post mortem , c'est-à-dire de considérer qu'ils sont présumés donneurs 181 ( * ) .
II - La position de la commission : ne pas permettre le prélèvement post mortem sur les majeurs faisant l'objet d'une protection juridique avec représentation à la personne
À l'initiative de son rapporteur, la commission a souhaité ne pas appliquer le droit commun du prélèvement post mortem aux majeurs faisant l'objet d'une protection juridique avec représentation à la personne , considérant que leur consentement éclairé ne pouvait pas être présumé et qu'effectuer des prélèvements dans ces conditions ne serait pas respectueux de leur personne.
En effet, il est peu probable qu'un majeur dont, par définition, les facultés mentales ou corporelles sont altérées et l'empêchent de pourvoir seul à ses intérêts, ait la capacité d'autonomie , voire de discernement , pour être informé du système du consentement présumé, en comprendre les enjeux et s'inscrire sur le registre national des refus - un dispositif par ailleurs peu connu de la population en général malgré les campagnes d'information de l'Agence de la biomédecine - ou exprimer un refus à son entourage.
D'autre part, ce choix de s'inscrire ou non de son vivant sur le registre national des refus et de laisser ou non prélever ses organes après sa mort, est un choix éminemment personnel . Il n'est pas pris dans l'intérêt de la personne, mais dans un but purement altruiste . Il ne semble pas, de ce fait, relever de la mission d'un représentant légal .
Dans ces conditions, la commission a considéré qu'il n'y avait pas lieu de laisser pratiquer des prélèvements d'organes après la mort des majeurs faisant l'objet d'une protection juridique avec représentation à la personne , ce d'autant plus que le contrôle a minima prévu dans le droit actuel - le consentement écrit du tuteur - ne peut être maintenu compte tenu de la cessation de sa mission au décès du majeur protégé en application de l'article 418 du code civil.
Cette position est cohérente avec le régime choisi par le Gouvernement en matière de dons d'organes, de tissus et de cellules de son vivant. Ils restent interdits pour les majeurs faisant l'objet d'une protection juridique avec représentation à la personne, sauf en cas de greffe en domino ou de don de cellules souches hématopoïétiques, deux exceptions justifiées : soit l'organe est déjà prélevé dans l'intérêt thérapeutique du majeur protégé, soit son don bénéficie directement à un membre de sa famille.
La commission a en conséquence adopté l'amendement COM-249 afin de ne permettre aucun prélèvement post mortem sur une personne faisant l'objet d'une protection juridique avec représentation à la personne .
Elle a également adopté , sur proposition du rapporteur, un amendement de coordination COM-266 .
La commission a adopté cet article ainsi modifié.
CHAPITRE IER BIS
Conforter la solidarité dans le cadre du don de
sang
Article 7 bis (nouveau)
Levée partielle de l'interdiction du don du
sang applicable aux majeurs protégés et abaissement de
l'âge du don du sang pour les mineurs
Cet article, inséré par la commission, propose de permettre aux majeurs bénéficiant d'une protection juridique avec représentation aux biens ou assistance à faire des dons du sang. Il vise également à autoriser les mineurs de 17 ans à donner leur sang, à condition qu'un parent ou un tuteur y consente par écrit.
Le don du sang soigne chaque année un million de malades. C'est un acte citoyen et civique, à portée de tous, qui permet de participer à la solidarité nationale comme en témoigne la hausse spectaculaire de la collecte de sang au lendemain des attentats du 13 novembre 2015 182 ( * ) .
L'article 7 bis, introduit à l'initiative du rapporteur par l'adoption en commission de son amendement COM-250 , vise à ouvrir le don du sang :
- aux majeurs faisant l'objet d'une mesure de protection juridique avec représentation aux biens et assistance, alignant ainsi le don du sang sur le régime des dons d'organes, de tissus et de cellules par donneur vivant proposé dans le cadre du projet de loi à l'article 7 ;
- aux mineurs de 17 ans , reprenant une disposition de la proposition de loi visant à la consolidation du modèle français du don du sang déposée par le député Damien Abad 183 ( * ) et votée à l'unanimité le 11 octobre 2018 à l'Assemblée nationale.
S'agissant des mineurs, le rapporteur a relevé que le don du sang est promu lors de la Journée défense et citoyenneté , à laquelle doivent participer les jeunes entre 16 et 18 ans. Il est donc logique de leur permettre de pratiquer ce don, conforme aux valeurs républicaines que l'on souhaite leur transmettre, sans attendre leur majorité.
L'article adopté prend en compte le cadre européen posé par la directive du 22 mars 2004 184 ( * ) qui fixe les critères d'admissibilité pour les donneurs de sang et en application de laquelle l'âge du don ne peut être inférieur à 17 ans et le consentement écrit de l'un des parents ou du tuteur légal doit être donné pour un mineur.
La commission a adopté cet article additionnel ainsi rédigé.
CHAPITRE II
Permettre la solidarité dans le cadre
de la
transmission d'une information génétique
Article 8
Réalisation d'examens des caractéristiques
génétiques sur une personne décédée ou hors
d'état d'exprimer sa volonté au profit des membres de sa
famille
Cet article autorise la réalisation d'un examen des caractéristiques génétiques d'une personne qui ne peut exprimer son consentement, notamment lorsqu'elle est décédée ou hors d'état d'exprimer sa volonté, lorsqu'un tel examen peut permettre de conseiller et prendre en charge les apparentés vivants en matière de surveillance, de prévention ou de soins en cas d'anomalie génétique prédisposante.
Afin de faciliter la réalisation des tests génétiques post mortem , la commission spéciale a adopté un amendement prévoyant la publication, par arrêté du ministre chargé de la santé, de règles de bonnes pratiques en matière de conservation et de traçabilité d'échantillons biologiques humains prélevés à des fins diagnostiques ou thérapeutiques ou à l'occasion d'une autopsie réalisée à des fins médicales.
I - Le dispositif proposé : la possibilité de réaliser des examens dans l'intérêt médical de la parentèle
1. Le droit en vigueur : le conditionnement des examens des caractéristiques génétiques au consentement préalable et exprès des personnes concernées
a) L'obligation du recueil préalable du consentement écrit
L'examen des caractéristiques génétiques d'une personne ou son identification par ses empreintes génétiques sont aujourd'hui encadrés par les articles 16-10 à 16-13 du code civil et les articles L. 1131-1 à L. 1131-7 du code de la santé publique.
Aux termes de l'article 16-10 du code civil, « l'examen des caractéristiques génétiques d'une personne ne peut être entrepris qu'à des fins médicales ou de recherche scientifique. » Cet examen ne peut être réalisé qu'avec « le consentement exprès de la personne [qui] doit être recueilli par écrit préalablement à la réalisation de l'examen, après qu'elle a été dûment informée de sa nature et de sa finalité. Le consentement mentionne la finalité de l'examen. Il est révocable sans forme et à tout moment. » Sans préjudice des règles applicables en matière d'examens de génétique menés dans le cadre de recherches impliquant la personne humaine, l'article L. 1131-1 du code de la santé publique opère un renvoi, à son premier alinéa, aux dispositions du code civil et du code de la santé publique 185 ( * ) encadrant l'examen des caractéristiques génétiques.
Outre l'exigence de consentement exprès de la personne concernée inscrite à l'article 16-10 du code civil, deux autres principes posés par le même code conditionnent la réalisation d'un examen des caractéristiques génétiques en impliquant un consentement préalable de l'intéressé :
- l'article 16-1 du même code fixe le principe fondamental d'inviolabilité du corps humain, étant rappelé que « le respect dû au corps humain ne cesse pas avec la mort » aux termes de l'article 16-1-1 dudit code ;
- l'article 16-3 du code civil interdit de porter atteinte à l'intégrité du corps humain, sauf « en cas de nécessité médicale pour la personne ou à titre exceptionnel dans l'intérêt thérapeutique d'autrui ». Il est toutefois précisé au même article que « le consentement de l'intéressé doit être recueilli préalablement hors le cas où son état rend nécessaire une intervention thérapeutique à laquelle il n'est pas à même de consentir. »
b) Des examens qui ne peuvent être entrepris que dans l'intérêt propre de la personne
Le second alinéa de l'article L. 1131-1 du code de la santé publique dispose qu'il peut être dérogé au principe de recueil du consentement exprès de la personne « lorsqu'il est impossible de recueillir le consentement de cette personne ou, le cas échéant, de consulter la personne de confiance 186 ( * ) , la famille ou, à défaut, un de ses proches ». Dans ce cas précis, l'examen des caractéristiques génétiques ne peut être entrepris qu'« à des fins médicales, dans l'intérêt de la personne. »
Une autre dérogation permet d'envisager l'examen des caractéristiques génétiques d'une personne sans son consentement exprès, mais uniquement dans le cadre circonscrit des autopsies médicales.
L'article L. 1211-2 du code de la santé publique prévoit en effet que les autopsies, pratiquées « dans le but d'obtenir un diagnostic sur les causes du décès », peuvent, à titre exceptionnel, « être réalisées malgré l'opposition de la personne décédée, en cas de nécessité impérieuse pour la santé publique et en l'absence d'autres procédés permettant d'obtenir une certitude diagnostique sur les causes de la mort. »
Les dérogations au principe du consentement exprès prévues aujourd'hui par la législation ne permettent donc d'envisager des examens des caractéristiques génétiques chez des personnes hors d'état d'exprimer leur volonté ou décédées que dans l'intérêt propre de ces personnes ou pour des motifs impérieux de santé publique dans le cadre d'autopsies pour la recherche des causes d'un décès. La réalisation d'examens des caractéristiques génétiques d'une personne hors d'état d'exprimer sa volonté, qu'elle soit vivante ou décédée, n'est donc pas possible lorsque ces examens auraient pour finalité d'informer la parentèle de la personne d'anomalies génétiques.
2. Les évolutions proposées par le projet de loi en matière d'examens des caractéristiques génétiques chez des personnes hors d'état d'exprimer leur volonté ou des personnes décédées
a) La clarification des conditions dans lesquelles un examen des caractéristiques génétiques d'une personne hors d'état d'exprimer sa volonté peut être réalisé dans l'intérêt médical de cette personne
Dans un avis de janvier 2016 187 ( * ) , le CCNE a estimé que le renvoi opéré par le premier alinéa de l'article L. 1131-1 du code de la santé publique aux principes posés par le code civil et le code de la santé publique en matière d'examens des caractéristiques génétiques « rend difficile la compréhension de l'alinéa 2 qui suit 188 ( * ) et qui prévoit des exceptions à cette règle de principe, faute qu'elle soit explicitée. » Dans le même ordre d'idées, selon l'étude d'impact annexée au projet de loi, le second alinéa de l'article L. 1131-1 du code de la santé publique n'est pas conforme aux modalités requises pour le recueil du consentement d'une personne hors d'état de l'exprimer : ni la personne de confiance, ni la famille ou les proches se sont en effet en capacité de consentir à ces examens en lieu et place de l'intéressé.
En conséquence, le III de l'article 8 du projet de loi prévoit l'introduction, dans le code de la santé publique, d'un nouvel article L. 1130-3 afin de clarifier les conditions dans lesquelles un examen des caractéristiques génétiques peut être entrepris chez une personne hors d'état d'exprimer sa volonté. Il est ainsi rappelé que, par dérogation au principe du recueil du consentement exprès inscrit aux articles 16-10 et 16-11 189 ( * ) du code civil, l'examen ou l'identification par les empreintes génétiques peuvent être entrepris à des fins médicales dans l'intérêt de la personne hors d'état d'exprimer sa volonté (premier alinéa du nouvel article L. 1130-3 du code de la santé publique).
Dans ce cas, il reviendra au médecin de s'assurer que la personne ne s'y est pas opposée antérieurement auprès de sa personne de confiance, de sa famille, de ses proches ou de la personne chargée d'une mesure de protection juridique avec représentation à la personne. Il est ainsi substitué à l'obligation de consentement exprès (écrit) l'absence d'opposition de la personne hors d'état d'exprimer sa volonté et ce sera donc au médecin de se substituer à la personne, et non à l'entourage de cette dernière, pour décider de la réalisation d'un examen des caractéristiques génétiques (second alinéa du nouvel article L. 1130-3 du code de la santé publique).
b) La possibilité de réaliser des examens génétiques chez des personnes hors d'état d'exprimer leur volonté ou décédées, dans l'intérêt médical de la parentèle
• En outre, le même III de l'article 8 du projet de loi crée un nouvel article L. 1130-4 au sein du code de la santé publique prévoyant la possibilité de réaliser un examen des caractéristiques génétiques d'une personne hors d'état d'exprimer sa volonté ou d'une personne décédée, à des fins médicales dans l'intérêt des membres de sa famille potentiellement concernés, dès lors qu'un médecin suspecte une anomalie génétique pouvant être responsable d'une affection grave justifiant des mesures de prévention, y compris de conseil génétique, ou de soins. Il est précisé que, dans le cas d'une personne décédée, l'examen sera réalisé à partir d'échantillons déjà conservés ou prélevés à l'occasion d'une autopsie médicale (I du nouvel article L. 1130-4 du code de la santé publique).
Un examen des caractéristiques génétiques d'une personne hors d'état d'exprimer sa volonté ou d'une personne décédée ne pourra intervenir que si la personne n'a pas fait antérieurement connaître son refus. La recherche de l'absence d'opposition incombera au médecin dans les mêmes conditions que celles prévues par le nouvel article L. 1130-3 du code de la santé publique dans le cas d'un examen pratiqué dans l'intérêt médical de la personne (II du nouvel article L. 1130-4 du code de la santé publique). Selon l'étude d'impact annexée au projet de loi, le médecin prescripteur pourra être « celui qui prend en charge la personne hors d'exprimer son consentement ou celui qui intervient dans le cadre de l'autopsie d'une personne décédée ou encore celui qui est sollicité par des membres de la famille potentiellement concernés (onco-généticien par exemple qui aura besoin d'accéder à des échantillons biologiques conservés de la personne décédée). »
• Le nouvel article L. 1130-4 du code de la santé publique précise également, en ses II et III, les modalités d'information de la parentèle en cas de réalisation d'un test génétique chez une personne hors d'état d'exprimer sa volonté ou une personne décédée :
- le médecin informera les membres de la famille potentiellement concernés, dont il possède les coordonnées 190 ( * ) , qu'il estime plausible l'existence d'une telle anomalie génétique. Il leur précisera qu'elles auront la possibilité d'accepter ou de refuser par écrit la réalisation de l'examen, en leur rappelant qu'il suffit que l'un des membres ait donné son accord pour que cet examen soit réalisé (II du nouvel article L. 1130-4 du code de la santé publique) ;
- une fois l'examen réalisé, le médecin informera tous les membres de la famille potentiellement concernés, y compris ceux qui ont refusé que l'examen soit pratiqué, que l'information sur la présence ou l'absence d'une anomalie génétique leur est accessible. En cas de confirmation de l'anomalie génétique, les personnes ayant souhaité recevoir l'information se verront invitées par le médecin à se rendre à une consultation chez un médecin qualifié en génétique, sans que leur soient dévoilés l'anomalie en question et les risques associés. Le cas échéant, les membres de la famille qui le souhaitent pourront alors bénéficier d'un examen de leurs caractéristiques génétiques dans les conditions prévues par l'article L. 1131-1 du code de la santé publique (III du nouvel article L. 1130-4 du code de la santé publique).
• En conséquence, l'article 8 du projet de loi procède à des coordinations au sein des dispositions encadrant l'accès au dossier médical d'une personne décédée :
- à l'article L. 1110-4 du code de la santé publique, il est précisé que le secret médical ne fait pas obstacle à ce que les informations concernant une personne décédée soient délivrées au médecin prenant en charge une personne susceptible de faire l'objet d'un test génétique, à la condition que la personne décédée ne s'y soit pas opposée de son vivant ( I de l'article 8 du projet de loi). En l'état actuel du droit en vigueur, l'accès au dossier médical d'une personne décédée n'est possible, sauf opposition de sa part avant son décès, que pour connaître les causes de la mort, défendre sa mémoire ou faire valoir ses droits ;
- à l'article L. 1111-7 du code de la santé publique, le champ des personnes susceptibles de demander l'accès au dossier médical d'une personne décédée, aujourd'hui restreint à ses ayants droit, son concubin ou son partenaire lié par un pacte civil de solidarité, est élargi au médecin prenant en charge un membre de la parentèle susceptible de faire l'objet d'un test génétique ( II de l'article 8 du projet de loi).
• Enfin, l'article 8 du projet de loi insère dans le code de la santé publique un nouvel article L. 1130-6 renvoyant à un décret en Conseil d'État la définition des modalités d'application de ces nouvelles dispositions.
Il reviendra, en outre, au ministre chargé de la santé d'arrêter, sur proposition de l'agence de la biomédecine, les critères déterminant les situations médicales justifiant la réalisation d'un test génétique chez une personne hors d'état d'exprimer sa volonté ou une personne décédée dans l'intérêt médical de la parentèle. Ces critères, élaborés par l'agence de la biomédecine en concertation avec les professionnels de santé et les représentants de sociétés savantes, doivent permettre d'identifier des situations pour lesquelles des mesures de prévention ou de soins pourront être mises en oeuvre chez les membres de la parentèle potentiellement concernés, notamment en cas de décès subits sans antécédent pour lesquels une pathologie d'origine génétique est suspectée ou de nécessité d'identification d'une mutation génétique transmissible responsable de cancers héréditaires en l'absence de cas index en vie.
II - Les modifications adoptées par l'Assemblée nationale
L'Assemblée nationale a apporté des modifications d'ordre rédactionnel et visant à opérer des coordinations à l'article 8 du projet de loi.
III - La position de la commission : une avancée qui s'inscrit dans le droit fil d'une proposition de loi adoptée par le Sénat
• En juin 2018, le Sénat a adopté à l'unanimité une proposition de loi relative à l'autorisation d'analyses génétiques sur personnes décédées, déposée par le président de la commission des affaires sociales, Alain Milon. Ce texte visait à revenir sur l'impossibilité juridique de procéder à l'examen des caractéristiques génétiques d'une personne décédée dans l'intérêt médical de ses proches, quand bien même celle-ci y aurait consenti de son vivant. Le diagnostic d'une anomalie génétique liée à une pathologie grave susceptible de mesures préventives ou thérapeutiques au sein des membres d'une famille permet en effet de prévenir les pertes de chances qui résulteraient de l'absence d'informations, pourtant disponibles, quant au fait d'être potentiellement porteur de l'anomalie prédisposante.
Le texte adopté par le Sénat autorise ainsi les examens génétiques post mortem dans l'intérêt de la parentèle d'une personne décédée, sauf opposition manifestée par la personne de son vivant, par analogie avec le principe de consentement présumé applicable en matière de don d'organe. Il est prévu que l'examen ne puisse être prescrit que par un médecin généticien ou par un membre d'une équipe pluridisciplinaire comprenant un médecin qualifié en génétique. Comme prévu par l'article 8 du projet de loi, la proposition de loi n'autorise la réalisation de l'examen qu'à partir d'éléments du corps de la personne prélevés soit préalablement à son décès, soit dans le cadre d'une autopsie médicale.
• L'autorisation des examens des caractéristiques génétiques réalisés sur des personnes décédées dans l'intérêt des membres de leur famille est cohérente avec les recommandations tant de l'agence de la biomédecine 191 ( * ) que du CCNE 192 ( * ) . Comme il l'a été indiqué à votre commission par la professeure Dominique Stoppa-Lyonnet, l'article 8 du projet de loi, qui s'inscrit dans le droit fil de la proposition de loi adoptée par le Sénat, opère une dissociation entre la personne à partir de laquelle est réalisé l'examen génétique, qu'elle soit décédée ou hors d'état d'exprimer sa volonté, et la personne qui consent à l'examen, à savoir le membre de la famille qui aura donné son accord pour que l'examen soit effectué et à partir de laquelle l'examen sera facturé pour une prise en charge par l'assurance maladie. Deux conditions seront nécessaires pour la réalisation de l'examen : l'absence d'opposition manifestée par la personne antérieurement à son décès et le consentement d'au moins un membre de la famille de la personne décédée.
L'information sur la présence chez une personne décédée d'une anomalie génétique pouvant être responsable d'une pathologie grave peut se révéler particulièrement utile en cas de mort subite, par exemple pour les cardiomyopathies génétiques susceptibles d'affecter des sujets jeunes.
La nécessité d'harmoniser les pratiques en matière de conservation des échantillons biologiques humains
• Les conditions de conservation des tissus, cellules et liquides biologiques prélevés à l'occasion d'actes diagnostiques ou de soins varient selon les établissements. Or le bon état de conservation et la traçabilité de ces échantillons seront déterminants pour donner un caractère pleinement opérationnel à la possibilité de réaliser des examens génétiques post mortem . Seuls certains types de prélèvements font aujourd'hui l'objet d'une réglementation de leur durée de conservation, notamment dans le cadre de l'assistance médicale à la procréation, de la recherche sur l'embryon et les cellules souches embryonnaires humaines ou du diagnostic prénatal.
Les conditions de conservation de certains examens de biologie médicale, relevant par exemple de l'anatomocytopathologie, sont également précisées par un arrêté du 26 novembre 1999 193 ( * ) qui a officialisé le guide de bonne exécution des analyses de biologie médicale (GBEA) et ses annexes. Ce guide précise que « la durée de conservation pour chaque cas particulier doit, si elle n'est pas réglementée, être fixée par le biologiste et inscrite sur les procédures opératoires. »
Dans un souci d'harmonisation des pratiques entre laboratoires de biologie médicale, qu'il s'agisse d'un laboratoire privé ou d'un laboratoire situé au sein d'un établissement de santé public ou privé, votre commission a adopté un amendement renvoyant à un arrêté du ministre chargé de la santé, pris après avis de l'agence de la biomédecine, de la Haute Autorité de santé et des représentants des établissements de santé et des laboratoires de biologie médicale, le soin de préciser les règles de bonnes pratiques en matière de conservation et de traçabilité d'échantillons biologiques humains prélevés à des fins diagnostiques ou thérapeutiques ou à l'occasion d'une autopsie réalisée à des fins médicales (amendement COM-185 du rapporteur).
• Votre commission a adopté enfin un amendement procédant à des corrections et harmonisations rédactionnelles à l'article 8 du projet de loi (amendement COM- 223 du rapporteur).
La commission a adopté cet article ainsi modifié.
Article 9
Transmission d'une information génétique au profit
de la parentèle
ou dans les situations de rupture du lien de
filiation biologique
Cet article actualise les dispositions relatives à l'information de la parentèle du patient faisant l'objet d'un examen des caractéristiques génétiques en cas de découverte d'une anomalie génétique pouvant être responsable d'une affection justifiant des mesures de prévention ou de soins afin de tenir compte notamment des situations de rupture du lien de filiation biologique pour des personnes nées d'un don ou dans le cadre d'un accouchement sous le secret.
La commission spéciale a souhaité renforcer l'effectivité de la transmission d'une information génétique dans les situations de rupture du lien de filiation biologique, c'est-à-dire entre tiers donneurs et personnes nées d'un don, et entre parents de naissance et personnes nées dans le secret, dans un souci d'égalité d'accès de ces personnes, qui de fait n'ont pas connaissance des antécédents médicaux de leurs apparentés biologiques, aux mesures de prévention ou de soins, tout en préservant rigoureusement l'anonymat des personnes concernées.
I - Le dispositif proposé : organiser la transmission de l'information en cas de découverte d'une anomalie génétique
1. Le droit en vigueur : un dispositif d'information des apparentés biologiques incomplet s'agissant des personnes en situation de rupture du lien de filiation biologique
Le dispositif d'information de la parentèle en cas d'anomalie génétique grave découverte chez un patient ayant fait l'objet d'un examen des caractéristiques génétiques a été introduit dans le code de la santé publique par la loi relative à la bioéthique de 2004 194 ( * ) . Il s'appuie sur le principe d'information des membres de la famille en cas d'anomalie génétique détectée chez l'un d'eux reconnu, sur le plan conventionnel, par l'article 18 du protocole additionnel à la convention sur les droits de l'homme et la biomédecine, dite convention d'Oviedo, du 4 avril 1997 195 ( * ) , qui a été signé par la France le 13 décembre 2011. Cet article 18 stipule ainsi que « lorsque les résultats d'un test génétique réalisé sur une personne peuvent être pertinents pour la santé d'autres membres de sa famille, la personne ayant fait l'objet du test doit en être informée. »
L'article L. 1131-1-2 du code de la santé publique détermine les conditions d'information tant de la personne que des membres de sa famille éventuellement concernés sur le diagnostic résultant d'un examen des caractéristiques génétiques. Il prévoit ainsi que, préalablement à la réalisation de l'examen, le médecin doit informer la personne concernée « des risques qu'un silence ferait courir aux membres de sa famille potentiellement concernés si une anomalie génétique grave dont les conséquences sont susceptibles de mesures de prévention, y compris de conseil génétique, ou de soins était diagnostiquée. » Le médecin est alors tenu de définir avec la personne, dans un document écrit éventuellement complété après le diagnostic, les modalités de transmission de l'information à la parentèle.
a) Le droit de « ne pas savoir » et les modalités d'information de la personne à partir de laquelle l'examen des caractéristiques génétiques a été effectué
L'article L. 1131-1-2 précité consacre le droit de la personne à « ne pas savoir », c'est-à-dire d'être tenue dans l'ignorance du diagnostic. Le refus de connaître les résultats de l'examen doit être exprimé par écrit.
S'agissant des modalités de communication des résultats à la personne, cet article dispose que la découverte d'une anomalie génétique grave doit donner lieu à un document rédigé de manière loyale, claire et appropriée, signé et remis par le médecin au patient, résumant l'information médicale relative à cette anomalie. La remise de ce document à la personne est attestée par cette dernière et le médecin l'informe, lors de l'annonce du diagnostic, de l'existence éventuelle d'associations de malades susceptibles de l'accompagner et de lui donner des renseignements complémentaires.
b) Le dispositif d'information des apparentés : trois modalités possibles
- La responsabilité de la personne en matière d'information de ses apparentés
Aux termes du troisième alinéa de l'article L. 1131-1-2 du code de la santé publique, la personne a l'obligation d'informer les membres de sa famille potentiellement concernés dont elle possède ou peut obtenir les coordonnées, dès lors que des mesures de prévention ou de soins peuvent leur être proposées. Ce caractère obligatoire de l'information de la parentèle résulte de la loi relative à la bioéthique de 2011 qui permet désormais que le manquement de la personne à cette obligation puisse servir de fondement à une action en responsabilité à son encontre.
- La possibilité de délégation au médecin prescripteur de l'acte de transmission de l'information à la parentèle
Néanmoins, il existe deux situations dans lesquelles la personne peut déléguer au médecin prescripteur le soin d'informer la parentèle : dans le cas où la personne ne souhaite pas informer elle-même les membres de sa famille ou dans le cas où elle a souhaité être tenue dans l'ignorance du diagnostic. Dans ces deux hypothèses, la délégation au médecin de l'acte de transmission de l'information reste facultative pour la personne concernée par l'examen des caractéristiques génétiques. Cette demande de délégation est faite à partir d'un document écrit, le médecin devant attester de cette demande. La personne est alors tenue de lui communiquer les coordonnées des intéressés dont elle dispose.
Le médecin chargé par la personne d'informer ses apparentés doit alors se limiter à porter à leur connaissance l'existence d'une information médicale à caractère familial susceptible de les concerner, et les inviter à se rendre à une consultation de génétique. Le médecin ne doit leur dévoiler ni le nom de la personne ayant fait l'objet du test, ni l'anomalie génétique, ni les risques qui lui sont associés.
- L'information des enfants issus d'un don en cas d'anomalie génétique grave détectée chez le donneur ou chez l'un des membres d'un couple ayant consenti à donner un embryon
La loi relative à la bioéthique de 2011 a complété le dispositif d'information des apparentés biologiques en prévoyant qu'en cas de détection d'une anomalie génétique grave chez une personne dont le don de gamètes aurait abouti à la conception d'un ou plusieurs enfants ou chez l'un des membres d'un couple qui aurait consenti à effectuer un don d'embryon à la suite d'une procédure d'AMP, la personne concernée peut autoriser le médecin prescripteur à saisir le responsable du centre d'AMP afin qu'il procède à l'information des enfants issus du don.
2. Le dispositif proposé par le projet de loi : l'organisation de la transmission de l'information dans les situations de rupture du lien de filiation biologique
L'article 9 du projet de loi procède à une réorganisation de l'architecture des dispositions du code de la santé publique relatives aux examens des caractéristiques génétiques. Au sein d'un chapitre intitulé « Modalités de mise en oeuvre des examens des caractéristiques génétiques et des identifications par empreintes génétiques et information de la parentèle » ( 1° du I de l'article 9 du projet de loi), les articles L. 1131-1 à L. 1131-1-2 seront spécifiquement consacrés aux modalités d'information des apparentés en cas de détection d'une anomalie génétique grave chez une personne.
L'encadrement des conditions dans lesquelles il peut être procédé à l'examen des caractéristiques génétiques d'une personne sera, lui, précisé à l'article 16-10 du code civil dans sa version proposée par l'article 10 du projet de loi. Quant aux règles applicables aux examens des caractéristiques génétiques réalisés dans des situations particulières, elles seront développées aux articles L. 1130-3, L. 1130-4 et L. 1130-6 du code de la santé publique créés par l'article 8 du projet de loi pour les examens effectués sur des personnes hors d'état d'exprimer leur volonté ou décédées, et à l'article L. 1130-5 du même code créé par l'article 18 du projet de loi pour les examens entrepris à des fins de recherche scientifique.
a) Les modalités de transmission de l'information aux apparentés lorsque l'examen a été réalisé sur un majeur protégé ou une personne hors d'état d'exprimer sa volonté ou lorsque la personne est décédée avant l'annonce des résultats
Dans sa rédaction proposée par le projet de loi ( 2° du I de l'article 9 du projet de loi), l'article L. 1131-1 du code de la santé publique reprend, dans ses I et II, les dispositions de l'actuel article L. 1131-1-2 relatives au dispositif d'information de la parentèle, moyennant quelques ajustements rédactionnels, à l'exception des dispositions relatives à la transmission d'une information de nature génétique entre un donneur et une personne née du don de ce dernier qui feront l'objet de l'article L. 1131-1-1 proposé par l'article 9 du projet de loi.
Une clarification rédactionnelle conduit ainsi à centrer l'information sur une anomalie génétique « pouvant être responsable d'une affection grave justifiant de mesures de prévention, y compris de conseil génétique, ou de soins ». Contrairement à ce que laissait jusqu'ici entendre l'article L. 1131-1-2 du code de la santé publique, ce n'est en effet pas l'anomalie génétique qui est grave en soi, mais la pathologie dont elle peut être à l'origine.
Le III de l'article L. 1131-1 proposé par l'article 9 du projet de loi permet de préciser les modalités d'information de la parentèle lorsque l'examen des caractéristiques génétiques a été effectué à partir d'une personne faisant l'objet d'une mesure de protection juridique avec représentation à la personne ou d'une personne hors d'état d'exprimer sa volonté et que l'examen a été réalisé dans son intérêt. L'information de la parentèle sera alors garantie dans les mêmes conditions que lorsqu'une personne refuse d'informer elle-même les membres de sa famille : le médecin prescripteur informera ces derniers de l'existence d'une information médicale à caractère familial et les invitera à se rendre chez un médecin qualifié en génétique, sans dévoiler le nom de la personne, l'anomalie et les risques associés.
Le IV de l'article L. 1131-1 tient compte d'une autre hypothèse non encore envisagée par le droit en vigueur : les modalités d'information de la parentèle lorsque la personne décède avant l'annonce du résultat ou avant d'avoir pu informer les membres de sa famille. Dans ces situations, il est prévu que le médecin procède à l'information des apparentés dont il possède les coordonnées dans les mêmes conditions que lorsqu'une personne refuse d'informer elle-même les membres de sa famille, sauf si la personne s'y était opposée antérieurement.
Enfin, le V de l'article L. 1131-1 précise que le médecin qualifié en génétique qui sera consulté par la personne apparentée devra être informée par le médecin prescripteur de l'anomalie génétique en cause.
b) Les modalités de transmission de l'information aux apparentés dans les situations de rupture du lien de filiation biologique
- Entre tiers donneur et personne née d'un don
L'article L. 1131-1-1 du code de la santé publique proposé par l'article 9 du projet de loi ( 3° du I ) organise la transmission d'une information génétique entre le tiers donneur et la personne née du don de ce dernier :
- lorsque l'anomalie génétique est détectée chez le tiers donneur 196 ( * ) , ce dernier peut autoriser le médecin prescripteur à saisir le responsable du centre d'AMP afin que celui-ci procède à l'information des personnes issues du don, dans les mêmes conditions que lorsqu'une personne refuse d'informer elle-même les membres de sa famille (sans divulgation du nom du tiers donneur, de l'anomalie et des risques associés), ou à l'information des parents ou du tuteur si la personne née du don est mineure ;
- lorsque l'anomalie est détectée chez la personne issue d'un don (de gamètes ou d'embryon), cette dernière ou ses représentants légaux peuvent autoriser le médecin prescripteur à saisir le responsable du centre d'AMP afin qu'il procède à l'information du tiers donneur, dans les mêmes conditions que lorsqu'une personne refuse d'informer elle-même les membres de sa famille (sans divulgation du nom de la personne issue du don, de l'anomalie et des risques associés).
Dans les deux cas, la saisine du responsable du centre d'AMP par le médecin prescripteur reste soumise à l'accord de la personne ayant fait l'objet de l'examen génétique, qui peut refuser que l'information soit transmise.
- Entre les parents de naissance et les enfants nés sous le secret
L'article L. 1131-1-2 du code de la santé publique proposé par l'article 9 du projet de loi ( 4° du I ) organise la transmission d'une information génétique entre une personne née sous le secret et les parents de naissance. Selon l'étude d'impact annexée au projet de loi, le Cnaop a été saisi, depuis la loi relative à la bioéthique de 2011, de plusieurs situations dans lesquelles se posait la question de la transmission d'une information médicale d'ordre génétique entre une personne née lors d'un accouchement sous le secret et un parent de naissance. Or le troisième alinéa de l'article L. 1131-1-2 du code de la santé publique, en application duquel l'obligation d'information de la parentèle ne s'impose à une personne ayant reçu une information génétique que pour les apparentés dont il détient ou peut obtenir les coordonnées, ne permet pas la transmission d'une information médicale d'ordre génétique entre des personnes dont le lien de filiation biologique a été rompu dans le cadre d'un accouchement sous X.
Par conséquent, il est prévu par le 4° du I de l'article 9 du projet de loi que soit le parent de naissance d'un enfant né sous le secret 197 ( * ) , soit l'enfant né sous le secret (ou, le cas échéant, des représentants légaux, son tuteur ou ses descendants directs majeurs en cas de décès) 198 ( * ) pourra autoriser le médecin prescripteur à saisir le Cnaop afin d'identifier l'enfant né dans le secret ou le parent de naissance susceptible de bénéficier d'une telle information. Dans chaque cas, ni l'anomalie génétique en cause, ni les risques associés ne seraient mentionnés dans la saisine.
Il reviendra alors au Cnaop de procéder à l'information de la personne qu'il aura identifiée « dans des conditions de nature à préserver le secret de cette transmission définies par décret », sans dévoiler le nom de la personne ayant fait l'objet de l'examen, ni aucune autre information permettant d'identifier cette personne. La personne identifiée par le Cnaop sera dès lors invitée à se rendre à une consultation de génétique. Il appartiendra en outre au Cnaop de transmettre au médecin consulté par la personne ainsi informée les coordonnées du médecin prescripteur pour la communication de l'anomalie génétique en cause.
Dans tous les cas, la saisine du Cnaop par le médecin prescripteur reste soumise à l'accord de la personne ayant fait l'objet de l'examen génétique, qui peut refuser que l'information soit transmise.
Par coordination, le II de l'article 9 du projet de loi complète l'article L. 147-1 du code de l'action sociale et des familles relatif aux missions du Cnaop afin de préciser que ce dernier est chargé de porter à la connaissance des personnes nées dans le secret ou des parents de naissance d'une personne née dans le secret une information médicale d'ordre génétique. Il complète également l'article L. 147-2 du même code, relatif aux saisines du Cnaop, afin de tenir compte de la possibilité que lui soit transmise une saisine du médecin prescripteur d'un examen génétique concernant une personne née sous le secret ou un parent de naissance d'une personne née sous le secret.
II - Les modifications adoptées par l'Assemblée nationale
Les seuls amendements adoptés par l'Assemblée nationale à l'article 9 du projet de loi ont été examinés en commission.
À l'initiative de notre collègue Philippe Berta, la commission des affaires sociales de l'Assemblée nationale a adopté, avec un avis de sagesse de son rapporteur, Hervé Saulignac, un amendement tendant à rendre obligatoire la saisine du responsable du centre d'AMP par le médecin prescripteur en cas d'anomalie génétique détectée chez un tiers donneur ou une personne née d'un don. La suppression du caractère facultatif de la transmission d'une information médicale d'ordre génétique entre des personnes dont le lien de filiation biologique a été rompu permet en effet d'aligner les droits de ces personnes dans l'accès à des mesures de prévention ou de soins en cas d'anomalie génétique potentiellement à l'origine d'une affection grave sur ceux dont disposent les membres d'une famille dont les liens de parenté biologique sont connus.
Par ailleurs, outre plusieurs amendements rédactionnels, la commission a adopté un amendement de son rapporteur précisant que la personne née dans le secret ou le parent de naissance d'une personne née dans le secret informés par le Cnaop de l'existence d'une information médicale les concernant seront invités à se rendre chez un médecin qualifié en génétique, par parallélisme avec les dispositions relatives à l'information des apparentés inscrites à l'article L. 1131-1 du code de la santé publique.
III - La position de la commission : renforcer l'information des personnes
• Le V de l'article L. 1131-1 du code de la santé publique, dans sa rédaction prévue par l'article 9 du projet de loi, prévoit que, « dans tous les cas, le médecin qualifié en génétique consulté par la personne apparentée est informé par le médecin prescripteur de l'anomalie génétique en cause. »
Dans les cas prévus aux II 199 ( * ) , III 200 ( * ) et IV 201 ( * ) de l'article L. 1131-1 précité, l'information de la parentèle sur l'existence d'une information médicale d'ordre génétique sera assurée par le médecin prescripteur. La transmission des coordonnées de ce dernier aux personnes contactées se fera donc logiquement sans difficulté.
En revanche, dans le cas où l'information de la parentèle est assurée par la personne ayant fait l'objet de l'examen génétique, il convient de s'assurer que cette dernière communique aux apparentés qu'elle contacte les coordonnées de son médecin prescripteur afin que celui-ci puisse être sollicité sur l'anomalie génétique en cause par le médecin qualifié en génétique qui sera consulté par un apparenté. Votre commission a ainsi adopté un amendement apportant cette précision (amendement COM-186 du rapporteur).
• Lorsque le responsable du centre d'AMP doit informer le tiers donneur ou la personne née d'un don de l'existence d'une information génétique susceptible de les concerner, il n'est renvoyé, pour la transmission de cette information prévue au nouvel article L. 1131-1-1 du code de la santé publique, qu'au II de l'article L. 1131-1 du même code qui permet d'assurer l'information de la parentèle tout en préservant l'anonymat de la personne qui a fait l'objet de l'examen, de même que le droit de tout apparenté biologique de ne pas connaître l'anomalie en cause ou les risques associés.
Rien n'est précisé, en revanche, s'agissant de la transmission des coordonnées du médecin prescripteur initial qui, seul, détient l'information sur l'anomalie génétique à rechercher. Votre commission a donc adopté un amendement visant à préciser que le responsable du centre d'AMP transmettra à la personne qui a choisi d'aller en consultation de génétique médicale les coordonnées du médecin prescripteur afin de permettre à son médecin de connaître l'anomalie qu'il convient de rechercher (amendement COM-227 du rapporteur).
• L'article 9 du projet de loi maintient le caractère facultatif de la délégation par la personne ayant effectué un examen de ses caractéristiques génétiques au médecin prescripteur de l'information de la parentèle en cas de détection d'une anomalie génétique grave dans les deux cas prévus aujourd'hui par la loi : lorsque la personne a souhaité être tenue dans l'ignorance du diagnostic ou lorsqu'elle ne souhaite pas informer elle-même les membres de sa famille. Dans l'un et l'autre de ces cas, le choix de la personne de ne pas autoriser le médecin prescripteur à informer ses apparentés peut conduire à une perte de chance potentiellement grave pour ces derniers qui ne pourraient alors pas avoir accès à des mesures de prévention ou de soins. En cas d'opposition de la personne à ce que l'information puisse être transmise à ses apparentés, sa responsabilité pourra être engagée.
Dans ses réponses au questionnaire de votre commission, le ministère des solidarités et de la santé explique que le maintien de la possibilité pour la personne de s'opposer à une information de sa parentèle, quand bien même la loi lui impose de procéder à cette information, s'inscrit dans la volonté de préserver la relation de confiance entre le médecin et le patient, de respecter le secret médical comme le principe du consentement des personnes. Le choix effectué par le projet de loi est donc de ne procéder à aucune levée du secret médical contre le gré du patient concerné qui reste, quant à lui, responsabilisé. L'article R. 1131-20-1 du code de la santé publique prévoit, à l'heure actuelle, que le médecin informe la personne « des conséquences d'un éventuel refus de sa part de transmettre, soit elle-même, soit par son intermédiaire, l'information aux membres de sa famille potentiellement concernés ». Parmi ces conséquences figure la possibilité qu'elle soit poursuivie en justice au titre de sa responsabilité pour ne pas avoir permis à ses apparentés de bénéficier de mesures de prévention ou de soins.
• L'article 9 du projet de loi prévoit qu'en cas de décès de la personne avant l'annonce du résultat ou avant qu'elle ait pu informer les membres de sa famille, le médecin ne procèdera à l'information de ces derniers que si la personne ne s'y était pas opposée antérieurement.
Or on saisit mal l'objet de cette opposition, notamment s'il s'agit du refus de la personne d'informer sa parentèle ou de son souhait d'être tenue dans l'ignorance du diagnostic. En conséquence, votre commission a adopté un amendement de clarification tendant à préciser que le médecin ne peut procéder à l'information de la parentèle que dans les cas où la personne s'était auparavant opposée à être informée du résultat ou s'était opposée à ce que les membres de sa famille potentiellement concernés bénéficient de cette information (amendement COM-188 du rapporteur).
• Alors que l'Assemblée nationale a rendu obligatoire la transmission par le médecin prescripteur d'une information médicale d'ordre génétique entre un tiers donneur et une personne née d'un don, la transmission de ce type d'information à des personnes concernées par une rupture du lien de filiation biologique dans le cadre d'une naissance dans le secret reste subordonnée à la volonté de la personne ayant fait l'objet de l'examen.
Or, dans tous les cas de figure, la transmission d'une telle information reste entourée de toutes les garanties de nature à préserver l'anonymat de la personne ayant fait l'objet de l'examen des caractéristiques génétiques, sans risque que son identité soit dévoilée à son apparenté biologique. La non-transmission de l'information pourrait, en revanche, entraîner une perte de chance particulièrement préjudiciable à la personne entretenant un lien biologique avec la personne chez laquelle l'anomalie aurait été découverte, puisqu'elle ne pourrait bénéficier de mesures de prévention ou de soins.
Votre commission a, par conséquent, adopté un amendement tendant à rendre obligatoire, en cas de détection d'une anomalie génétique susceptible d'être à l'origine d'une pathologie grave chez une personne née sous le secret ou le parent de naissance d'une personne née sous le secret, la saisine par le médecin prescripteur du Cnaop afin que celui-ci procède à l'information de la personne entretenant un lien biologique avec la personne chez qui l'anomalie a été découverte sur l'existence d'une information médicale la concernant (amendement COM-189 du rapporteur).
Il convient de rappeler que la nature de l'anomalie et ses risques associés ne sont jamais communiqués par le Cnaop et que la personne contactée par le Cnaop conserve le droit « de ne pas savoir », en refusant de se rendre à une consultation de génétique, en application des règles de droit commun en matière d'examen des caractéristiques génétiques.
• Afin de rendre pleinement effective l'information des personnes entretenant un lien biologique avec une personne chez laquelle une anomalie génétique a été détectée, que ce lien biologique découle d'une procédure d'AMP ou d'une naissance dans le secret, il convient de s'assurer que les personnes qui se soumettent à un examen de leurs caractéristiques génétiques informent le médecin prescripteur du fait qu'elles sont un tiers donneur, une personne issue d'un don, une personne née dans le secret ou une personne entretenant un lien biologique avec une personne née dans le secret.
L'article R. 1131-20-3 du code de la santé publique en vigueur prévoit que « le médecin prescripteur interroge la personne sur l'existence éventuelle de sa part d'un don de gamètes ou, le cas échéant, d'un consentement du couple dont elle est membre à l'accueil de ses embryons par un autre couple. » Il conviendra de compléter, après publication de la loi, ces dispositions réglementaires afin de prévoir que les personnes nées d'un don ou nées dans le secret ou les personnes parents de naissance d'une personne nées dans le secret soient amenées à informer le médecin prescripteur de l'existence d'un tel lien de filiation biologique qui pourrait justifier une information d'éventuels apparentés biologiques.
• Dans ses réponses au questionnaire de votre commission, le Cnaop indique qu'il lui est nécessaire de pouvoir utiliser le numéro d'inscription des personnes au répertoire national d'identification des personnes physiques (RNIPP) et consulter ce répertoire, afin de pouvoir accomplir ses missions en matière d'information des personnes nées dans le secret ou des parents de naissance d'enfants nés sous le secret sur l'existence d'une information médicale d'ordre génétique pouvant les concerner. Votre commission a ainsi adopté un amendement garantissant l'accès du Cnaop à ces données, dans des conditions fixées par décret en Conseil d'État, pris après avis de la CNIL (amendement COM-224 du rapporteur).
• Enfin, votre commission a adopté un amendement rédactionnel COM-187 du rapporteur.
La commission a adopté cet article ainsi modifié.
TITRE
III
APPUYER LA DIFFUSION DES PROGRÈS SCIENTIFIQUES
ET
TECHNOLOGIQUES DANS LE RESPECT
DES PRINCIPES ÉTHIQUES
Article 10
Consentement à l'examen des caractéristiques
génétiques
Cet article ouvre la possibilité d'informer la personne, sous réserve de son consentement, de découvertes de caractéristiques génétiques sans relation avec l'indication initiale de l'examen dès lors que cette information peut permettre à cette personne ou ses apparentés de bénéficier de mesures de prévention ou de soins.
La commission spéciale a supprimé la précision, introduite en première lecture par l'Assemblée nationale, de l'interdiction de la publicité en faveur des examens de génétique constitutionnelle, ce type de publicité relevant de pratiques commerciales trompeuses déjà sanctionnées par le droit en vigueur.
I - Le dispositif proposé
1. Le droit en vigueur : des informations tirées d'un examen génétique circonscrites au champ de l'indication initiale de l'examen
Dans sa rédaction en vigueur, l'article 16-10 du code civil prévoit qu'un examen des caractéristiques génétiques ne peut être entrepris qu'à des fins médicales ou de recherche scientifique et est soumis au consentement exprès de la personne, qui doit être consigné par écrit préalablement à la réalisation de l'examen, après que la personne a été dûment informée de la nature et de la finalité de l'examen. Le consentement doit mentionner la finalité de l'examen et reste révocable sans forme et à tout moment.
L'article R. 1131-1 du code de la santé publique détaille les finalités médicales que peut poursuivre un examen des caractéristiques génétiques. Celui-ci peut ainsi avoir pour objet :
- de poser, de confirmer ou d'infirmer le diagnostic d'une maladie à caractère génétique chez une personne ;
- de rechercher les caractéristiques d'un ou plusieurs gènes susceptibles d'être à l'origine du développement d'une maladie chez une personne ou les membres de sa famille potentiellement concernés ;
- d'adapter la prise en charge médicale d'une personne selon ses caractéristiques génétiques.
L'article R. 1131-4 du même code précise, en outre, le contenu de l'information qui doit être délivrée à la personne à l'occasion du recueillement de son consentement : « la personne est informée des caractéristiques de la maladie recherchée, des moyens de la détecter, du degré de fiabilité des analyses ainsi que des possibilités de prévention et de traitement. En outre, elle est informée des modalités de transmission génétique de la maladie recherchée et de leurs possibles conséquences chez d'autres membres de sa famille. » Dans ces conditions, les informations tirées de l'examen des caractéristiques génétiques doivent être strictement circonscrites à la maladie recherchée.
Lorsque l'examen des caractéristiques génétiques est entrepris à des fins de recherche, les dispositions relatives aux recherches impliquant la personne humaine sont applicables. Trois modalités de consentement sont alors possibles :
- lorsqu'il entre dans le cadre d'une recherche interventionnelle, que celle-ci comporte une intervention sur la personne non justifiée par sa prise en charge habituelle (1° de l'article L. 1121-1 du code de la santé publique) ou seulement des risques et contraintes minimes (2° de l'article L. 1121-1 du code de la santé publique), l'examen est conditionné au consentement exprès, libre et éclairé de la personne (premier et deuxième alinéas de l'article L. 1122-1-1 du code de la santé publique) ;
- lorsqu'il entre dans le cadre d'une recherche non interventionnelle (3° de l'article L. 1121-1 du code de la santé publique), l'examen est conditionné à l'absence d'opposition de la personne après information de cette dernière sur les finalités du projet de recherche (avant-dernier alinéa de l'article L. 1122-1-1 du code de la santé publique) ;
- lorsqu'il est réalisé à partir d'éléments du corps de la personne prélevés à d'autres fins, l'examen est conditionné à l'absence d'opposition de la personne après information de cette dernière sur les finalités du projet de recherche (article L. 1131-1-1 du code de la santé publique).
Le non-respect du principe du consentement exprès et écrit posé par l'article 16-10 du code civil est sanctionné pénalement : aux termes de l'article 226-25 du code pénal, « le fait de procéder à l'examen des caractéristiques génétiques d'une personne à des fins autres que médicales ou de recherche scientifique, ou à des fins médicales ou de recherche scientifique, sans avoir recueilli préalablement son consentement dans les conditions prévues par l'article 16-10 du code civil, est puni d'un an d'emprisonnement et de 15 000 euros d'amende. »
2. Le développement du séquençage haut-débit et la collecte de données additionnelles
L'utilisation de plus en plus courante de tests pangénomiques 202 ( * ) grâce aux techniques de séquençage haut-débit, permettant l'analyse de l'ensemble du génome ou de l'exome 203 ( * ) , conduit à la découverte de données additionnelles sans lien avec l'indication initiale de l'examen, mais révélant la présence de variations génétiques susceptibles d'être en lien avec une pathologie. Ces données sont généralement classées en deux catégories :
- les données incidentes : elles sont découvertes de manière fortuite ;
- les données secondaires : elles sont recherchées activement à partir d'une liste de gènes préétablie.
L'interprétation et, le cas échéant, la prise en charge des variations génétiques découvertes de façon incidente peuvent s'avérer problématiques dès lors que leurs pertinence et utilité cliniques n'ont pas, pour certaines d'entre elles, encore pu être clairement établies par les données scientifiques disponibles. Certaines variations génétiques peuvent être considérées comme « actionnables » au regard de l'état des connaissances médicales et ainsi donner lieu à des mesures de prévention ou de soins. Pour d'autres, leur signification pathologique et la pénétrance 204 ( * ) de la maladie associée à la mutation génétique sont encore mal appréhendées.
Dans son avis 205 ( * ) de juin 2018 sur la révision de la loi de bioéthique, le Conseil d'État, observant que « l'information donnée dans ce cadre reste souvent probabiliste, exprimée sous la forme d'un risque accru », redoute ainsi que la communication de ces anomalies génétiques découvertes fortuitement ne conduise à la révélation d'un « statut génétique » qui consistera essentiellement pour la personne concernée en « un passage d'une incertitude à une autre. » Néanmoins, saisi en 2013 d'un projet de décret relatif aux conditions de mise en oeuvre de l'information de la parentèle dans le cadre d'un examen des caractéristiques génétiques à finalité médicale, le Conseil d'État avait alerté le Gouvernement « sur la nécessité d'adapter les termes de l'article 16-10 du code civil et de l'article R. 1131-4 du code de la santé publique pris pour son application qui apparaissent, compte tenu des évolutions scientifiques, partiellement obsolètes et empêchent de prévoir de façon explicite une information de la parentèle conforme aux objectifs légitimes de prévention et de soins. »
Selon l'étude d'impact annexée au projet de loi, le recours au séquençage haut-débit et aux tests pangénomiques s'est intensifié au cours de la période récente : le nombre de laboratoires réalisant des examens à partir de séquenceurs de nouvelle génération a augmenté de près de 71 % sur la période 2013-2017 et les examens résultant de l'utilisation de ces séquenceurs a représenté 15,8 % de l'ensemble des examens de génétique moléculaire en 2017, contre 9,3 % en 2015. Par ailleurs, le plan « France médecine génomique 2025 », qui prévoit la création de 12 plateformes de séquençage génomique haut-débit, soulève la question de la communication sur des variations génétiques découvertes de manière aléatoire dans le cadre de projets de recherche.
3. Le dispositif proposé par le projet de loi : la possibilité de consentir à la révélation de données génétiques incidentes
L'article 10 du projet de loi procède à une réécriture de l'article 16-10 du code civil. Il précise le champ d'application du régime de consentement exprès et écrit en le limitant aux seuls examens des caractéristiques génétiques constitutionnelles, c'est-à-dire héritées ou acquises à un stade précoce du développement prénatal. Sont ainsi exclus de ce régime :
- les examens des caractéristiques génétiques somatiques, acquises ultérieurement et non transmissibles, visées par le nouvel article L. 1130-2 du code de la santé publique proposé par l'article 25 du projet de loi ;
- les analyses génétiques effectuées à partir d'échantillons biologiques prélevés et conservés à d'autres fins qui feront l'objet d'un régime de consentement tacite (en l'absence d'opposition), prévu par le nouvel article L. 1130-5 du code de la santé publique proposé par l'article 18 du projet de loi.
Si le nouvel article 16-10 du code civil proposé par le projet de loi maintient le principe du consentement exprès et écrit préalablement à la réalisation d'un examen des caractéristiques génétiques constitutionnelles et ses garanties 206 ( * ) , il précise, dans son II, le contenu de l'information que la personne doit recevoir avant de donner son consentement. Cette information devra ainsi indiquer :
- la nature de l'examen ;
- l'indication de l'examen, s'il est entrepris à des fins médicales, ou son objectif, s'il est entrepris à des fins de recherche scientifique ;
- le cas échéant, la possibilité que l'examen révèle incidemment des caractéristiques génétiques sans relation avec l'indication initiale ou avec son objectif initial mais dont la connaissance permettrait à la personne ou aux membres de sa famille de bénéficier de mesures de prévention, y compris de conseil génétique, ou de soins ;
- la possibilité de refuser la révélation de données génétiques incidentes. Ce droit d'opposition est néanmoins contrebalancé par une information sur les risques qu'un refus ferait courir aux membres de la famille potentiellement concernés dans le cas où l'anomalie diagnostiquée pourrait être responsable d'une affection grave justifiant de mesures de prévention ou de soins.
Il est, en outre, précisé que la communication des résultats révélés incidemment sera assurée dans les mêmes conditions que la communication des résultats correspondant à l'indication initiale.
En cas d'examen des caractéristiques génétiques réalisé dans le cadre d'un projet de recherche à partir d'échantillons prélevés à d'autres fins, notamment à l'occasion de soins ou d'essais cliniques, il est renvoyé, au III de l'article 16-10 du code civil dans sa rédaction envisagée par le projet de loi, aux dispositions du nouvel article L. 1130-5 du code de la santé publique proposé par l'article 18 du projet de loi, visant à faciliter l'utilisation secondaire de tels échantillons par des recherches nécessitant le recours à des examens génétiques, en conditionnant la réalisation de ces examens à l'absence d'opposition de la personne dûment informée.
II - Les modifications adoptées par l'Assemblée nationale
Outre un amendement rédactionnel adopté en commission à l'initiative de son rapporteur, l'Assemblée nationale a adopté en séance, en dépit d'un avis défavorable de la commission et du Gouvernement, un amendement de nos collègues députés Pascal Brindeau et Jean-Christophe Lagarde (UDI) visant à interdire tout démarchage publicitaire portant sur l'examen des caractéristiques génétiques constitutionnelles d'une personne.
Cet amendement intervient en réaction à la diffusion sur plusieurs chaînes de télévision (BFM-TV, CNews et LCI), à l'été 2018, d'une publicité en faveur de tests ADN émanant d'un annonceur étranger. Dans la mesure où le recours à des tests génétiques en dehors du cadre prévu par la loi est puni pénalement 207 ( * ) , ce message publicitaire pouvait inciter à des comportements délinquants, ce qui a amené le conseil supérieur de l'audiovisuel (CSA) à adresser une mise en garde aux chaînes concernées.
III - La position de la commission
L'interdiction de toute publicité en faveur de tests génétiques adoptée par l'Assemblée nationale n'est pas assortie de sanctions pénales. Votre commission s'interroge donc sur l'opportunité et l'effectivité d'une telle disposition.
Il convient, en effet, de préciser que la réalisation d'examens des caractéristiques génétiques en dehors du cadre légal, notamment les tests génétiques disponibles sur Internet et effectués en dehors d'une consultation de génétique médicale, fait déjà l'objet d'un dispositif de sanctions pénales :
- l'article 226-25 du code pénal punit d'un an d'emprisonnement et de 15 000 euros d'amende le fait de procéder à des tests génétiques à d'autres fins que médicales ou de recherche scientifique ou sans le consentement préalable de la personne ;
- l'article 226-28-1 du même code punit de 3 750 euros d'amende le fait pour une personne de solliciter l'examen de ses caractéristiques génétiques, de celles d'un tiers ou l'identification d'une personne par ses empreintes génétiques en dehors des conditions prévues par la loi.
La publicité en faveur d'actes médicaux pris en charge par l'assurance maladie est déjà prohibée. Aux termes de l'article R. 4127-19 du code de la santé publique, repris à l'article 19 du code de déontologie médicale, « la médecine ne doit pas être pratiquée comme un commerce » et « sont interdits tous procédés directs ou indirects de publicité et notamment tout aménagement ou signalisation donnant aux locaux une apparence commerciale. »
Enfin, la publicité en faveur de tests génétiques commerciaux disponibles en accès libre relève de pratiques commerciales trompeuses qui font déjà l'objet de sanctions pénales. En application du 9° de l'article L. 121-4 du code de la consommation, est réputée trompeuse toute pratique commerciale qui a pour objet de déclarer ou de donner l'impression que la vente d'un produit ou la fourniture d'un service est licite alors qu'elle ne l'est pas. L'article L. 132-2 du même code punit les pratiques commerciales trompeuses d'un emprisonnement de deux ans et d'une amende de 300 000 euros.
Dans ces conditions, votre commission a adopté un amendement supprimant l'alinéa introduit en première lecture par l'Assemblée nationale et précisant, à l'article 10 du projet de loi, que la publicité en faveur des tests génétiques est interdite (amendement COM-83 de Michel Amiel).
La commission a adopté cet article ainsi modifié.
Article 10 bis (nouveau)
Encadrement de l'accès aux tests
génétiques à visée généalogique
Cet article, inséré par la commission, autorise le recours aux tests génétiques à visée généalogique sous réserve du respect de certains critères, dont l'interdiction de délivrer des informations à caractère médical.
I - Le droit en vigueur : une interdiction devenue virtuelle
Le recours aux tests génétiques en accès libre, c'est-à-dire en dehors d'une consultation de génétique médicale ou ne s'inscrivant pas dans le cadre d'un projet de recherche en santé, est aujourd'hui interdit :
- l'article 226-25 du code pénal punit d'un an d'emprisonnement et de 15 000 euros d'amende le fait de procéder à des tests génétiques à d'autres fins que médicales ou de recherche scientifique ou sans le consentement préalable de la personne ;
- l'article 226-28-1 du même code punit de 3 750 euros d'amende le fait pour une personne de solliciter l'examen de ses caractéristiques génétiques, de celles d'un tiers ou l'identification d'une personne par ses empreintes génétiques en dehors des conditions prévues par la loi.
La France et la Pologne sont aujourd'hui les seuls pays, en Europe, à interdire le recours aux tests génétiques commerciaux en accès libre.
En dépit de cette interdiction, nombreux sont les Français qui ont recours aux tests génétiques disponibles sur Internet, commercialisés par des sociétés étrangères telles que « Ancestry.com », « 23andme » ou « MyHeritage ». Selon les estimations communiquées à votre commission, plus de 100 000 Français par an se procureraient des kits de prélèvement en vue de réaliser un examen de leurs caractéristiques génétiques à des prix de plus en plus abordables compte tenu des progrès techniques réalisés dans ce domaine. Une association spécialisée en génétique généalogique estime ainsi à près d'1,5 million le nombre de personnes en France ayant potentiellement déjà utilisé des tests génétiques à visée généalogique et ont alimenté ainsi les bases de donnée des sociétés commercialisant ces tests. Au niveau mondial, on estime que plus de 15 millions de personnes ont déjà acheté des tests génétiques sur Internet.
Dans ses réponses au questionnaire de votre commission, François Molins, procureur général près la Cour de cassation, a confirmé l'ineffectivité des sanctions pénales en matière de tests génétiques effectués en dehors du cadre légal, ces sanctions ne permettant pas d'enrayer ce phénomène croissant. Il rappelle ainsi que les tests génétiques commerciaux constituent aujourd'hui un marché particulièrement attractif que certains observateurs valorisent à 22 milliards de dollars à l'horizon 2024, avec un prix des tests toujours plus accessible, compris entre 59 et 99 dollars.
Les buts poursuivis par ces tests sont variés : il peut s'agir, selon les sociétés les proposant, de tests généalogiques, de tests de paternité 208 ( * ) mais aussi de tests proposant de détecter des variants génétiques susceptibles d'être liés au développement d'une pathologie. Selon les cas, plusieurs de ces services peuvent être proposés à partir d'un seul auto-prélèvement réalisé par la personne demandeuse.
L'interdiction des tests génétiques en accès libre reste aujourd'hui essentiellement virtuelle. La direction des affaires criminelles et des grâces du ministère de la justice indique qu'aucune procédure en la matière n'a été portée à sa connaissance et aucun jugement n'est recensé à ce jour sur les infractions prévues par l'article 226-28-1 du code pénal. Devant l'ampleur de ce phénomène, l'agence de la biomédecine a été chargée par la loi relative à la bioéthique de 2011 de « mettre à disposition du public une information sur l'utilisation des tests génétiques en accès libre et d'élaborer un référentiel permettant d'en évaluer la qualité » 209 ( * ) .
Si elle n'a pas élaboré de référentiel des examens génétiques vendus sur Internet, l'agence a déployé, en revanche, un site Internet à destination du grand public visant notamment à alerter sur les risques liés à l'utilisation de ces tests en accès libre et valorisant l'encadrement médical proposé en France dans l'accompagnement des personnes lors de la communication des résultats ( www.genetique-medicale.fr ).
II - Le dispositif adopté par la commission : autoriser les tests à visée généalogique tout en les encadrant
L'enthousiasme des Français pour les tests génétiques généalogiques ne semble pas prêt d'être entamé par leur interdiction légale qui ne s'est jusqu'ici jamais matérialisée par une quelconque condamnation ou amende. Nombreux sont ceux de nos compatriotes qui ont déjà consenti ou envisagent de consentir à la conservation de leurs données génétiques personnelles dans des bases de données valorisées par des sociétés commerciales étrangères qui pourraient s'affranchir des règles applicables en France et dans l'Union européenne en matière de protection des données personnelles, pour ensuite céder ces données à d'autres opérateurs privés ou publics, commerciaux, pharmaceutiques, académiques ou institutionnels 210 ( * ) .
Une partie du recours à ces tests est motivée par la recherche d'origines généalogiques, historiques ou géographiques, notamment par des personnes nées d'un don ou nées dans le secret qui cherchent à identifier des apparentés biologiques qui auraient eux-mêmes eu recours à ces tests. D'autres tests proposent néanmoins de révéler la présence chez une personne de variants génétiques identifiés comme prédisposants à une pathologie, sans aucun accompagnement médical.
Découle de l'interdiction absolue des tests génétiques commerciaux une situation particulièrement préjudiciable : la cession de données génétiques personnelles à des sociétés étrangères en dehors de tout contrôle et la délivrance d'informations génétiques d'ordre médical sans conseil délivré par des professionnels qualifiés.
Dans ces conditions, votre commission a adopté un amendement COM-190 du rapporteur tendant à autoriser, par dérogation à l'article 16-10 du code civil, les tests génétiques exclusivement à visée généalogique, sous réserve que ces tests remplissent plusieurs conditions :
- leur conformité au référentiel de qualité élaboré par l'agence de la biomédecine devra être attestée dans le cadre d'une procédure d'évaluation définie par voie réglementaire. L'attestation de cette conformité devra être déclarée à l'agence de la biomédecine qui disposera ainsi d'une visibilité complète des tests génétiques s'engageant à respecter son référentiel ;
- ces tests ne peuvent avoir pour objet de délivrer une information génétique d'ordre médical et ne peuvent, en conséquence, faire l'objet d'une prise en charge par la solidarité nationale ;
- les informations tirées de ces tests ne peuvent servir de fondement à des actions visant à faire valoir des droits patrimoniaux ou extrapatrimoniaux, notamment dans le cadre d'une démarche d'établissement d'un lien de filiation ;
- les données collectées à l'occasion de la réalisation de ces tests ne pourront être utilisées lors de la conclusion de tout contrat, notamment d'un contrat relatif à une protection complémentaire d'assurance maladie ou d'un contrat d'assurance.
La commission a adopté cet article additionnel ainsi rédigé.
Article 10 ter (nouveau)
Expérimentation de l'accès en
population générale
aux examens des caractéristiques
génétiques
Cet article permet l'expérimentation de l'accès aux examens des caractéristiques génétiques en population générale sans nécessité de présenter un antécédent familial de pathologie d'origine génétique ou de contexte clinique justifiant la réalisation d'un tel examen.
I - Le droit en vigueur : la nécessité d'antécédents familiaux ou de signes cliniques
En application de l'article 16-10 du code civil, la prescription par un médecin d'un examen des caractéristiques génétiques est subordonnée à l'identification d'une maladie d'origine génétique prédéterminée à partir d'un examen clinique ou de l'historique médical familial du patient. Aussi, en application de l'article R. 1131-5 du code de la santé publique, la personne éligible à un tel examen doit-elle faire état soit d'un antécédent familial d'une pathologie dont l'un des facteurs serait une mutation génétique, soit de symptômes ou d'indices cliniques d'une telle pathologie, notamment en oncologie.
Or une personne peut hériter d'une mutation génétique pouvant être responsable d'une affection grave, sans pour autant que cette affection se soit manifestée dans son contexte familial ou sans que cette personne ne dispose d'informations complètes sur l'historique médical de ses apparentés biologiques. À titre d'exemple, dans le cadre de la prescription d'un médicament indiqué dans le traitement du cancer du sein lié aux mutations BRCA 1 et 2, il apparaît que les patientes auxquelles ce traitement a été administré n'auraient pu, en l'absence d'indices familiaux, se voir prescrire un examen préalable des caractéristiques génétiques dans près d'un cas sur deux. De même, dans certaines formes de l'amyotrophie spinale infantile, dont la forme la plus sévère conduit au décès à deux ans de 50 % des enfants concernés, il s'avère dans près de neuf cas sur dix qu'aucun antécédent familial n'est reporté.
II - Le dispositif adopté par la commission : un accès facilité mais encadré aux examens génétiques
L'intérêt grandissant des Français pour les tests génétiques en accès libre et la très grande sensibilité des informations d'ordre médical sollicitées à cette occasion plaident pour un accès facilité mais encadré aux examens génétiques effectués dans un parcours médical. Il est en effet préférable que les personnes souhaitant savoir si elles présentent une mutation génétique en soient informées dans le cadre d'une consultation de génétique médicale.
Dans cette logique, votre commission a adopté un amendement COM-191 du rapporteur tendant à autoriser, à titre expérimental, la prescription d'examens des caractéristiques génétiques en l'absence d'antécédent familial ou de contexte clinique justifiant la recherche d'une anomalie génétique prédéterminée, sous réserve que les conditions suivantes soient remplies :
- le consentement de la personne doit être obtenu dans les mêmes conditions que celles prévues par l'article 16-10 du code civil, après que la personne a reçu les informations listées par le même article, à l'exception de l'indication de l'examen ;
- l'examen réalisé dans le cadre de cette expérimentation ne peut faire l'objet d'une prise en charge par la solidarité nationale. Il devra être pris en charge directement par la personne mais pourra faire l'objet, le cas échéant, d'une prise en charge, partielle ou totale, par son organisme complémentaire d'assurance maladie.
Cet article ménage la possibilité pour le pouvoir réglementaire de définir une liste des anomalies génétiques susceptibles d'être recherchées dans le cadre de cette expérimentation, dans le cas où des consultations menées auprès de l'agence de la biomédecine et des sociétés savantes en génétique médicale concluraient à la pertinence d'une telle liste. À l'heure actuelle, quatre pathologies pourraient justifier, selon certaines de ces sociétés savantes, la conduite d'études pilotes permettant d'évaluer la prévalence de certaines mutations génétiques : la mucoviscidose, l'amyotrophie spinale infantile et les deux hémoglobinopathies que sont la drépanocytose et la bêta-thalassémie.
Cette expérimentation reprend une proposition formulée par le CCNE dans son avis 129 211 ( * ) dans lequel il « se propose d'examiner, de façon plus approfondie, les possibilités d'extension du dépistage génétique à la population générale ». Il y plaide pour la « mise en place [d'] une étude pilote de recherche opérationnelle portant sur plusieurs régions ou sur des tranches d'âge différentes et que soient évaluées les conséquences de cette extension en termes de santé publique, de retentissement psychologique et de coût. » Lors de son audition par votre commission, le président du CCNE s'est déclaré favorable à l'assouplissement de l'accès aux examens génétiques, en dehors d'un projet parental et sans antécédent familial, sous réserve de sa réalisation dans le cadre d'une consultation de génétique avec, le cas échéant, une prise en charge des frais associés par la personne demandeuse.
La commission a adopté cet article additionnel ainsi rédigé.
Article 11
Encadrement du recours
à un traitement algorithmique
à des fins médicales
Cet article propose de créer un cadre juridique pour l'utilisation d'un traitement algorithmique de données massives lors de la réalisation d'un acte médical. La commission a adopté plusieurs amendements afin de renforcer les garanties assurées au patient lors de l'utilisation de ces technologies dites d' « intelligence artificielle ».
I - Le dispositif proposé : la création d'un cadre juridique dédié à l'utilisation d'un traitement algorithmique de données massives en médecine pour préserver une forme de garantie humaine
Alors que l'intelligence artificielle est en constant développement dans le domaine de la santé, l'article 11 du projet de loi tend à prévoir un encadrement spécifique pour l'utilisation d'un traitement algorithmique de données massives en vue de la réalisation d'un acte médical à visée préventive, diagnostique ou thérapeutique.
Sont ici visés les actes médicaux prodigués au patient à titre individuel et non l'utilisation de l'intelligence artificielle à des fins de recherche ou de gestion sanitaire.
• Le développement de l'intelligence artificielle en santé constitue un progrès au service du patient mais soulève des enjeux éthiques et juridiques
L'intelligence artificielle se définit comme « la science qui consiste à faire faire aux machines ce que l'homme ferait moyennant une certaine intelligence » 212 ( * ) . Un algorithme, qui répond à cette définition, se définit au sens strict selon la Commission nationale de l'informatique et des libertés (CNIL), entendue par le rapporteur, comme « la description d'une suite finie et non ambigüe d'étapes ou d'instructions permettant d'obtenir un résultat à partir d'éléments fournis en entrée » 213 ( * ) . Le traitement algorithmique visé par le projet de loi correspond aux algorithmes informatiques « permettant de combiner des informations les plus diverses pour produire une grande variété de résultats » 214 ( * ) .
Ces outils peuvent être très positifs pour le patient en améliorant l'efficacité et la précocité des diagnostics, en fournissant une aide à la décision thérapeutique, en améliorant le suivi de l'évolution d'une maladie ou encore en prévenant la survenance de pathologies. Le rapport sur les enjeux du numérique en santé commandé par le Comité consultatif national d'éthique (CCNE) 215 ( * ) , en propose diverses illustrations dont l'aide au diagnostic de cancers grâce au traitement par intelligence artificielle d'une très grande quantité de clichés d'imagerie médicale, qui permet de repérer de façon précoce les zones à risque tumoral.
Marc Cuggia, professeur d'informatique médicale et praticien hospitalier au centre hospitalier universitaire (CHU) de Rennes, également entendu par le rapporteur, citait comme autre exemple les logiciels de prescription de médicaments dotés de modules d'intelligence artificielle et permettant l'émission d'alertes visant à éviter les erreurs de doses ou les interactions médicamenteuses.
Comme l'a indiqué la CNIL au rapporteur, le développement et l'usage d'algorithmes va très souvent nécessiter le recours à des données personnelles, l'inverse étant possible mais plus rare. Les données de santé recueillies dans ce cadre seront donc soumises au règlement général sur la protection des données (RGPD) s'il s'agit de données à caractère personnel 216 ( * ) . Les données du patient saisies dans un traitement algorithmique sont en premier lieu utilisées en vue d'aider le professionnel de santé à statuer sur la situation personnelle dudit patient. Mais elles peuvent aussi être ultérieurement utilisées à d'autres fins et, notamment, pour alimenter un ou plusieurs algorithmes ou pour un projet de recherche par exemple 217 ( * ) .
Il faut toutefois préciser qu'aux termes de l'article L. 1111-8 du code de la santé publique, les hébergeurs de données de santé à caractère personnel recueillies à l'occasion d'activités de prévention, de diagnostic, de soins ou de suivi social et médico-social ne peuvent utiliser les données qui leur sont confiées à d'autres fins que l'exécution que la prestation d'hébergement et que tout acte de cession à titre onéreux de telles données est sanctionné des peines prévues à l'article 226-21 du code pénal 218 ( * ) .
Outre la question de la gestion des données, l'utilisation de l'intelligence artificielle soulève la question de la responsabilité du médecin lors de l'utilisation d'un traitement algorithmique.
Dans son étude publiée en 2018, le Conseil d'État estimait que « les règles actuelles de la responsabilité médicale sont susceptibles de s'adapter aux évolutions issues du développement des systèmes d'intelligence artificielle » 219 ( * ) .
L'intelligence artificielle relève du droit des biens et les dommages qu'elle est susceptible de causer peuvent être appréhendés par les règles de la responsabilité du fait des choses 220 ( * ) . De ce fait, la responsabilité du professionnel reconnu comme le gardien de la chose peut être engagée sans faute sauf en cas de force majeure. Si le bien est défectueux, la responsabilité du producteur peut aussi être engagée sur le fondement de la responsabilité du fait des produits défectueux 221 ( * ) .
La Cour de justice de l'Union européenne (CJUE) a récemment confirmé qu'un logiciel d'aide à la prescription constituait un dispositif médical, même si un tel logiciel n'agissait pas directement dans ou sur le corps humain222 ( * ). Il en résulte que les dispositifs d'intelligence artificielle qui exploitent les données d'un patient pour assister une décision médicale doivent semble-t-il être regardés au sens du droit européen comme des dispositifs médicaux.
Les dispositifs médicaux
Ils sont régis par le règlement (UE) 2017/745 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux, modifiant la directive 2001/83/CE, le règlement (CE) n° 178/2002 et le règlement (CE) n° 1223/2009 et abrogeant les directives du Conseil 90/385/CEE et 93/42/CEE.
Ce règlement impose notamment une évaluation clinique du dispositif médical avant sa mise sur le marché, pour démontrer sa conformité avec les exigences générales en matière de sécurité et de performance. Les fabricants de dispositifs médicaux sont également tenus d'établir et de garantir la fiabilité et la robustesse du traitement tout au long de sa durée de vie.
Pour être qualifié de dispositif médical (DM) ou de dispositif médical de diagnostic in vitro (DM DIV), le dispositif doit présenter les critères cumulatifs suivants :
- être destiné à une utilisation à des fins médicales au sens de la définition du DM ou du DM DIV. Il doit permettre, par exemple, un diagnostic, une aide au diagnostic, un traitement ou une aide au traitement ;
- donner un résultat propre au bénéfice d'un seul patient ;
- effectuer une action sur les données entrantes, telle qu'une analyse afin de fournir une information médicale nouvelle. Cette action doit être différente d'un stockage, d'une communication, ou d'une simple recherche via une base de données ou une bibliothèque numérique intégrant des données dans un but exclusif d'archivage sans les exploiter.
En conséquence, le traitement algorithmique qui réunit ces critères cumulatifs est un dispositif médical.
Le champ des traitements algorithmiques ne pouvant être limitativement défini, la CNIL estime que certains de ces traitements pourraient être exclus de la catégorie des dispositifs médicaux, d'où l'intérêt de prévoir un cadre juridique ad hoc .
• La création d'un cadre juridique applicable à l'utilisation d'un traitement algorithmique de données massives vise à préserver une forme de garantie humaine
Compte tenu de ces enjeux, l'article 11 du projet de loi tend à instituer une forme de « garantie humaine » afin d'éviter une déshumanisation de la relation de soin entre le patient et le médecin et la dépossession de ce dernier de son autonomie décisionnelle.
Ce concept de garantie humaine dans l'utilisation de l'intelligence artificielle, développé notamment dans les travaux du professeur David Gruson, déjà cité, serait consacré au sein d'un nouvel article L. 4001-3 du code de la santé publique et se déclinerait en trois points.
Premièrement, il instituerait un principe d'information du patient lorsqu'un professionnel de santé 223 ( * ) a recours à un traitement algorithmique de données massives pour des actes à visée préventive, diagnostique ou thérapeutique, au moment de la communication des résultats de ces actes. Le professionnel de santé en question devrait également informer la personne concernée des « modalités d'action de ce traitement ».
Deuxièmement, le nouvel article L. 4001-3 du code de la santé publique préciserait que l'adaptation des paramètres du traitement algorithmique utilisé par un professionnel de santé ne pourrait être réalisée sans son intervention, formulation qui a été précisée en commission à l'Assemblée nationale à l'initiative de notre collègue député Philippe Berta, rapporteur.
Troisièmement, ce même article instituerait une obligation de traçabilité des actions du traitement algorithmique ainsi que des données qui sont utilisées dans ce cadre. Les informations qui en résultent devraient être accessibles aux professionnels de santé qui l'utilisent.
II - La position de la commission : renforcer les garanties offertes au patient lors de l'utilisation d'un traitement algorithmique à des fins médicales
La commission a approuvé le principe proposé à l'article 11 du projet de loi. Elle a adopté plusieurs amendements sur proposition du rapporteur afin de renforcer les garanties applicables à l'utilisation de ces technologies en matière médicale.
• Encadrer le recours aux traitements algorithmiques sans exclure ceux qui ne font pas intervenir de données massives au stade de leur utilisation
La commission a adopté un amendement COM-256 du rapporteur qui supprime les termes de « données massives » pour encadrer plus largement le recours aux traitements algorithmiques en matière médicale. Cette modification semble nécessaire pour deux raisons.
Les auditions ont tout d'abord mis en évidence que la définition des « données massives » n'était pas consensuelle, comme l'ont indiqué au rapporteur Bénédicte Bévière-Boyer, maître de conférences en droit privé et sciences criminelles à l'Université de Paris 8 et Sonia Desmoulin-Canselier, docteur en droit privé et chargée de recherche au Centre national de la recherche scientifique (CNRS). À cet égard, si le droit français ne connaît pas la notion de « traitement algorithmique de données massives », il connaît déjà celle de « traitement algorithmique », introduite par la loi n° 2018-493 du 20 juin 2018 relative à la protection des données personnelles 224 ( * ) pour les décisions administratives individuelles et reprise par la loi n° 2019-222 du 23 mars 2019 de programmation 2018-2022 et de réforme pour la justice 225 ( * ) pour les services en ligne de conciliation et de médiation.
Ensuite, la référence aux données massives au stade de l'utilisation d'un traitement algorithmique réduit inutilement le champ d'application de l'article car certains traitements algorithmiques peuvent être « entraînés » sur des données massives mais ensuite utilisés sur un nombre bien plus faible de données. Certains traitements algorithmiques peuvent même ne pas s'appuyer sur des données fournies en très grande quantité : par exemple, lorsqu'il s'agit de cohortes de patients spécifiques ou souffrant d'une maladie rare.
• Prévoir l'information du patient en amont du recours à un traitement algorithmique
Par l'adoption du même amendement COM-256 du rapporteur, la commission a souhaité que le recours à un traitement algorithmique fasse l'objet d'une information préalable à son utilisation par le professionnel de santé, alors que le projet de loi ne prévoit qu'une information au moment de la communication des résultats.
Ce principe a fait l'unanimité lors des auditions. Préconisé par la Commission nationale de l'informatique et des libertés (CNIL) 226 ( * ) et le Comité consultatif national d'éthique (CCNE) 227 ( * ) , il a en outre recueilli l'avis favorable du conseil national de l'ordre des médecins. L'information préalable du patient sur le recours à un traitement algorithmique est nécessaire à deux égards : en vertu du traitement de ses données personnelles et de leur archivage, ainsi qu'au titre des principes généraux définis par la loi n° 2002-303 du 4 mars 2002 relative aux droits des malades et à la qualité du système de santé, impliquant que le patient doit accepter la prescription du médecin.
Comme l'indique notre collègue député Philippe Berta dans son rapport sur le projet de loi, il s'agit de « préserver la portée des principes posés par les articles L. 1111-2 et L. 1111-4 du code de la santé publique selon lesquels le patient a le droit d'être informé sur son état de santé et les traitements qui lui sont proposés et qu'il prend, avec le professionnel de santé et compte tenu des informations et des préconisations qu'il lui fournit, concernant sa santé » 228 ( * ) . À cet égard, l'exigence d'information préalable prévue à l'article L. 1111-2 du code de la santé publique sur laquelle se fonde le consentement libre et éclairé du patient à un acte ou traitement médical porte sur les « différentes investigations, traitements ou actions de prévention qui sont proposés (...) ». Il est donc logique, s'agissant de dispositions spéciales applicables à l'utilisation de traitements algorithmiques, de prévoir un principe similaire d'information préalable du patient.
Il y serait toutefois fait exception en cas d'urgence ou d'impossibilité d'informer, pour ne pas faire peser sur le professionnel de santé une obligation disproportionnée.
Prolongeant le principe de l'information préalable, la commission a également précisé que le professionnel de santé devrait expliquer au patient sous une forme intelligible la manière dont le traitement serait mis en oeuvre à son égard. Cette rédaction paraît plus claire que celle adoptée par l'Assemblée nationale qui imposait au médecin d'expliquer au patient « les modalités d'action de ce traitement ».
• Garantir la supervision du traitement algorithmique par le professionnel de santé
La commission a ensuite garanti la supervision du traitement algorithmique par le professionnel de santé.
Par l'adoption d'un amendement COM-257 du rapporteur, la commission a indiqué que la saisie d'informations relatives au patient dans le traitement algorithmique se faisait sous le contrôle du professionnel de santé qui y a recourt.
La rédaction adoptée par l'Assemblée nationale qui prévoyait que l'adaptation des « paramètres » d'un traitement algorithmique ne pouvait être réalisée sans un professionnel de santé ne paraissait pas adaptée car elle relevait plutôt du domaine technique.
Les termes utilisés étaient d'ailleurs ambigus, ce qu'ont confirmé les auditions : s'agissait-il pour le professionnel de santé de modifier lui-même les « paramètres » d'un traitement, notion qui renvoie à la conception même du dispositif, ou plutôt de saisir lui-même les informations relatives à son patient dans l'outil pour obtenir le résultat final ?
D'après les éléments portés à la connaissance du rapporteur par les services du ministère de la santé, il semblerait que la seconde interprétation soit la bonne. En effet, le professionnel de santé est rarement en mesure de modifier directement le traitement algorithmique. C'est la raison pour laquelle la commission a levé la confusion pour atteindre le même objectif : la supervision du traitement algorithmique par le professionnel de santé.
La marge d'appréciation du médecin, mis en concurrence avec un algorithme dont la puissance de calcul et de diagnostic est supérieure à l'homme, peut en effet se réduire. Dans son étude publiée en 2018 sur la révision de la loi de bioéthique, le Conseil d'État estimait qu'il convenait « d'écarter le risque que les progrès de l'intelligence artificielle dans le domaine de la santé aient pour effet de marginaliser l'autonomie du médecin dans l'acte médical » 229 ( * ) .
Or, si ces outils peuvent être très positifs pour le patient, ils n'en comportent pas moins des biais qui peuvent être liés, comme l'a indiqué la Commission nationale de l'informatique et des libertés (CNIL), à l'âge, au sexe, à l'origine, ou encore à l'environnement.
Compte tenu de ces risques, il importe que le médecin continue d'assumer les décisions médicales qu'il prend, quitte à s'écarter des solutions proposées par une intelligence artificielle.
Si le Conseil d'État relève que les dispositions en vigueur en matière de déontologie médicale permettent d'ores et déjà de prévenir une aliénation du médecin à la machine 230 ( * ) et de fonder l'interdiction d'un diagnostic établi uniquement par un système d'intelligence artificielle 231 ( * ) , il juge que « cette interdiction mériterait d'être rappelée de manière plus nette, à l'instar de ce que prévoit l'article [47] de la loi du 6 janvier 1978 qui prévoit qu'aucune décision produisant des effets juridiques à l'égard d'une personne ne peut être prise sur le seul fondement d'un traitement automatisé de données. 232 ( * ) »
Dans cet esprit, la commission a donc consacré le principe selon lequel « aucune décision médicale ne peut être prise sur le seul fondement d'un traitement algorithmique » par l'adoption d'un amendement COM-258 du rapporteur.
Par l'adoption d'un amendement COM-259 du rapporteur, la commission a fixé une obligation de transparence du fonctionnement d'un traitement algorithmique. Cette rédaction se substituerait à celle adoptée par l'Assemblée nationale dont les termes ont été jugés imprécis. Il était ainsi fait référence à une obligation de « traçabilité » sans préciser sa définition ni par qui elle était assurée.
Il s'agirait de garder trace des opérations effectuées par la machine, afin que les professionnels de santé, ayant accès aux informations, puissent a posteriori en reconstituer le raisonnement.
Dès lors, sans exiger que les professionnels de santé aient accès à la « traçabilité » de toutes les étapes, qui renvoie plutôt au domaine technique, la commission a souhaité que le traitement algorithmique ne fonctionne pas comme une « boîte noire », c'est-à-dire que le professionnel de santé ne puisse pas expliquer pour quelles raisons le traitement est arrivé à un résultat donné.
Enfin, par l'adoption d'un amendement COM-260 du rapporteur, la commission a renvoyé au pouvoir réglementaire, après avis de la Commission nationale de l'informatique et des libertés (CNIL), le soin de préciser les conditions d'application de ce nouveau cadre juridique.
La commission a adopté cet article ainsi modifié.
Article 12
Encadrement du recours aux techniques
d'imagerie
cérébrale et interdiction des discriminations
fondées
sur les résultats de ces techniques en matière d'assurance
Cet article propose d'étendre le champ d'application de l'article 16-14 du code civil qui régit le recours aux techniques d'imagerie cérébrale, d'exclure le recours à l'imagerie fonctionnelle en justice et d'interdire toute discrimination fondée sur les résultats de ces techniques. La commission a préféré en rester au droit en vigueur pour ce qui concerne le recours à l'imagerie fonctionnelle en justice, considérant que sa modification n'était pas nécessaire.
I - Le cadre actuel du recours aux techniques d'imagerie cérébrale
Dans son avis n° 129 préalable à la révision de la loi de bioéthique, le Comité consultatif national d'éthique (CCNE) définit les neurosciences comme ayant « pour objet l'étude du fonctionnement du système nerveux depuis ses aspects les plus fondamentaux (moléculaires, cellulaires, synaptiques) jusqu'à ceux, plus fonctionnels, qui concernent les comportements ou les processus mentaux » 233 ( * ) .
Parmi les différentes disciplines qui les composent, l'imagerie cérébrale est utilisée pour étudier la structure du cerveau et son activité. Comme le précise Luc Buée, président de la société des neurosciences, consulté par le rapporteur, elle permet de caractériser l'évolution normale ou pathologique du cerveau à tous les stades de la vie et de tenter de comprendre les règles de son fonctionnement. Elle comprend différentes techniques parmi lesquelles on trouve le scanner cérébral 234 ( * ) , l'imagerie par résonnance magnétique (IRM) 235 ( * ) , la tomographie par émissions de positons 236 ( * ) (TEP) ou l'électroencéphalographie 237 ( * ) .
Dans son avis n° 129, le CCNE attire l'attention sur l'IRM qui, en particulier, permet l'étude du cerveau, non seulement sur le plan morphologique ou biochimique (imagerie anatomique), mais aussi sur le plan fonctionnel (imagerie fonctionnelle) en valorisant l'activation du cerveau lors de tâches diverses (motrices, sensorielles, cognitives) ou encore lors d'états psychologiques variés (peur, angoisse, plaisir, satisfaction). Le CCNE précise que ces deux potentialités de l'IRM n'ont pas la même finalité : « l'imagerie anatomique peut permettre de déceler des anomalies éventuellement susceptibles de contribuer à expliquer un comportement particulier », alors que l'imagerie fonctionnelle « vise à observer l'activité cérébrale afin d'en déduire des conséquences sur le psychisme » 238 ( * ) .
Le recours aux techniques d'imagerie cérébrale, régi par l'article 16-14 du code civil, n'est aujourd'hui autorisé qu'à des fins médicales, de recherche scientifique ou dans le cadre d'expertises judiciaires. Il est soumis au consentement exprès et écrit de la personne concernée, informée de la finalité de l'examen, celle-ci pouvant révoquer ledit consentement sans forme et à tout moment.
Issues de la loi n° 2011-814 du 7 juillet 2011 relative à la bioéthique, ces dispositions visaient à apporter des garanties à l'utilisation de techniques ne faisant jusqu'alors l'objet d'aucun encadrement. Comme l'indiquait à l'époque notre collègue François-Noël Buffet, rapporteur pour avis au nom de la commission des lois, la crainte sous-jacente était que l'on puisse considérer que « les configurations neuronales du cerveau, siège de la pensée, portent la vérité de la personne ou que l'on utilise ces savoirs pour prédire son comportement ou ce qui relève de son for intérieur » 239 ( * ) .
À l'origine, Jean Leonetti - rapporteur de la loi en 2011 à l'Assemblée nationale - entendait cantonner l'usage des techniques en justice à l'objectivation d'un préjudice au niveau du cerveau ou à l'évaluation de la responsabilité d'un prévenu sur le fondement de l'article 122-1 du code pénal 240 ( * ) , dans l'objectif d'éviter que l'imagerie cérébrale puisse être utilisée contre celui sur lequel elle serait pratiquée pour prouver sa culpabilité.
La crainte est avant tout celle de l'utilisation de cette technologie comme un détecteur de mensonge, grâce à l'utilisation de l'imagerie cérébrale fonctionnelle. Si la rédaction finalement retenue à l'article 16-14 du code civil, plus large, n'exclut pas formellement cette possibilité, elle réserve l'utilisation de cette technique à un magistrat du siège, seul compétent pour ordonner une expertise judiciaire, tant en matière civile 241 ( * ) que pénale 242 ( * ) .
Cette précaution n'a pas permis d'éviter les critiques, notamment celles du CCNE, qui dans son avis n° 129 précité, considère que l'imagerie fonctionnelle pourrait être utilisée comme détecteur de mensonge et pour cette raison, est « très défavorable à l'utilisation de l'IRMf 243 ( * ) dans le domaine judiciaire », d'autant plus que cette technique n'est « pas reconnue comme présentant une fiabilité suffisante ». Dans le même esprit, l'Office parlementaire d'évaluation des choix scientifiques et technologies (OPECST), relevait à l'occasion de son rapport d'évaluation de la loi du 7 juillet 2011 relative à la bioéthique, « qu'il demeure un risque d'emploi abusif, quant à leur valeur prédictive réelle ou étayée, des techniques d'imagerie cérébrale fonctionnelle dans le cadre des expertises judiciaires » 244 ( * ) .
II - Le dispositif proposé : l'exclusion du recours à l'imagerie fonctionnelle en justice (IMRf), l'extension du champ des techniques visées à l'article 16-14 du code civil et la sanction des discriminations
• Exclure le recours à l'IMRf en justice et étendre le champ des techniques visées à l'article 16-14 du code civil
Tenant compte de ces critiques, l'article 12 du projet de loi tend à exclure l'utilisation de l'imagerie cérébrale fonctionnelle pour les expertises judiciaires. L'Assemblée nationale, approuvant ce principe, a toutefois renvoyé au pouvoir réglementaire le soin de déterminer la liste des techniques interdites, en adoptant en séance publique un amendement de notre collègue député Jean-François Éliaou, rapporteur, avec les avis favorables de la commission et du Gouvernement.
L'article 12 du projet de loi propose également de faire évoluer le champ des techniques visées par l'article 16-14 du code civil aujourd'hui relatif à la seule imagerie cérébrale. Le texte adopté par l'Assemblée nationale propose, à l'initiative d'amendements du rapporteur adoptés en séance publique avec les mêmes avis favorables précités, de faire référence à l'imagerie et à « l'exploration » de l'activité cérébrale. Le projet de loi initial lui, ne visait plus que « l'enregistrement » de l'activité cérébrale.
• Sanctionner les discriminations fondées sur les résultats de techniques mentionnées à l'article 16-14 du code civil
L'article 225-1 du code pénal définit les différents critères de discrimination, parmi lesquels figure notamment l'état de santé d'une personne. La commission de telles infractions est punie de trois ans d'emprisonnement et de 45 000 euros d'amende lorsqu'elles consistent par exemple à refuser la fourniture d'un bien ou d'un service ou à refuser d'embaucher une personne (article 225-2 du code pénal).
L'article 225-3 du code pénal déroge à ces principes en permettant aux personnes physiques ou morales de procéder à certaines distinctions fondées sur les critères énumérés à l'article 225-1, lorsqu'elles apparaissent légitimes. Il précise à cet égard que les discriminations fondées sur l'état de santé d'une personne peuvent être légitimes lorsqu'elles consistent en des opérations ayant pour objet la prévention et la couverture du risque décès, des risques portant atteinte à l'intégrité physique de la personne ou des risques d'incapacité de travail ou d'invalidité.
Ces discriminations ne peuvent toutefois se fonder :
- ni sur la prise en compte de résultats de tests génétiques prédictifs ayant pour objet une maladie qui n'est pas encore déclarée ;
- ni sur la prise en compte des conséquences sur l'état de santé d'un prélèvement d'organe.
L'article 12 du projet de loi propose d'ajouter à ces deux exclusions la prise en compte de données issues des techniques mentionnées à l'article 16-14 du code civil. Dans sa version initiale, il excluait les données issues de techniques « d'enregistrement » de l'activité cérébrale. Dans la version finalement adoptée par l'Assemblée nationale, il vise désormais les techniques « d'imagerie et d'exploration » de l'activité cérébrale, par coordination avec la modification du champ des techniques visées à l'article 16-14 du code civil.
III - La position de la commission : maintenir le droit en vigueur à l'article 16-14 du code civil et approuver l'interdiction de l'usage des résultats d'examens du cerveau à des fins de discrimination dans le domaine assurantiel
La commission n'a pas été convaincue par les propositions d'évolution législative de l'article 16-14 du code civil et a préféré, par l'adoption d'un amendement COM-261 du rapporteur, en rester au droit existant pour plusieurs raisons.
S'agissant de l'usage de l'imagerie fonctionnelle en justice, le Conseil d'État considérait dans son étude de 2018 245 ( * ) que la modification des dispositions en vigueur ne s'avérait pas nécessaire. Les services de la Chancellerie, interrogés par le rapporteur, ont confirmé ne pas avoir connaissance de recours abusifs aux techniques d'imagerie cérébrale par les juridictions pénales, qui justifieraient d'encadrer plus strictement leur usage.
Le Conseil d'État relevait en outre à la lumière des travaux parlementaires de 2011 « qu'en aucun cas, le législateur n'a entendu permettre le recours à ces techniques aux fins de détecter le mensonge ».
Il semble d'ailleurs douteux de déduire de l'article 16-14 du code civil qu'un tel usage serait autorisé. L'utilisation des techniques d'imagerie cérébrale aux fins de détecter le mensonge d'un prévenu pourrait, au demeurant, être contraire au principe selon lequel « nul n'est tenu de s'accuser », qui découle de l'article 9 de la Déclaration des droits de l'homme et du citoyen de 1789 relatif à la présomption d'innocence, selon une jurisprudence établie du Conseil constitutionnel depuis 2004 246 ( * ) . Comme l'indique le commentaire aux cahiers de cette décision : « aucun autre principe ou règle de valeur constitutionnelle ne fait obstacle à ce qu'une personne reconnaisse sa culpabilité si elle le fait volontairement, consciemment et librement, c'est-à-dire en dehors de tout « chantage » , de tout « marchandage », de tout malentendu et de toute contrainte » 247 ( * ) . Dans ces conditions, il est fort probable que l'obtention d'aveux sous la contrainte au moyen d'une technique faisant office de détecteur de mensonge relèverait d'une procédure contraire à la Constitution.
Comme l'indique Sonia Desmoulin-Canselier, docteur en droit privé et chargée de recherche au Centre national de la recherche scientifique (CNRS), dans son étude sur le sujet, le droit en vigueur permet de ne « pas fermer la porte à un emploi utile des différentes techniques d'imagerie, au service des droits de la défense, de la découverte de la vérité et de la protection des victimes » 248 ( * ) . En pratique, comme elle l'a indiqué au rapporteur lors de son audition, l'analyse cérébrale fonctionnelle n'aurait été utilisée qu'une seule fois depuis 2011 devant la justice, pour déterminer le caractère grave et irréversible des lésions cérébrales d'un patient.
Une telle expertise peut donc être utile au juge, sans toutefois jamais se substituer à sa libre appréciation. Le Conseil d'État relevait à cet égard « qu'une telle expertise ne peut être imposée à l'intéressé, qu'elle ne constitue qu'un élément parmi d'autres dans le cadre du procès, soumis au débat contradictoire y compris quant à la méthode employée, et qu'elle ne saurait, comme l'a rappelé la Cour de cassation, se substituer à l'appréciation du juge sur les questions qui relèvent de son office » 249 ( * ) , comme c'est le cas pour toutes les expertises 250 ( * ) .
S'agissant de l'extension des techniques entrant dans le champ d'application de l'article 16-14 du code civil, le Gouvernement s'est inspiré d'une préconisation de l'étude du Conseil d'État suggérant de faire référence à l'enregistrement cérébral en lieu et place de l'imagerie cérébrale, pour prendre en compte l'évolution des technologies intervenue depuis 2011.
Il semble toutefois que ni la notion d' « enregistrement » cérébral ni celle d' « exploration » ne soient pertinentes. Il est en effet ressorti des auditions des représentants de l'Institut national de la santé et de la recherche (INSERM), Étienne Hirsch et Bernard Poulain, ainsi que de celle de Stanislas Dehaene, professeur au collège de France, que cela pourrait conduire à interdire des dispositifs de neuro-modulation non invasifs utilisés à des fins paramédicales. Or, ces dispositifs sont aujourd'hui autorisés et pour ceux qui présenteraient des risques, un encadrement spécifique est proposé à l'article 13 du présent projet de loi 251 ( * ) .
Les échanges avec les services du ministère de la santé ont également mis en évidence que cette extension du champ d'application de l'article 16-14 du code civil pourrait être contraire au droit de l'Union européenne, en ce qu'elle constituerait un obstacle à la libre circulation d'un produit sans justification de santé publique.
Pour ce qui concerne la sanction des discriminations fondées sur des données issues de l'imagerie cérébrale, en l'état du droit, la prise en compte de données issues de techniques d'imagerie cérébrale ne fait pas partie des exclusions prévues à l'article 225-3 du code pénal.
Il est donc souhaitable de renforcer les garanties applicables aux personnes subissant de tels examens, afin que les résultats ne puissent être utilisés en leur défaveur pour majorer le coût d'une assurance décès ou invalidité en raison de risques déduits de l'examen du cerveau.
La commission a adopté cet article ainsi modifié.
Article 13
Interdiction
par décret des dispositifs ayant pour effet de modifier
l'activité cérébrale en cas de danger pour la santé
humaine
Cet article propose d'encadrer les dispositifs de neuro-modulation en permettant leur interdiction par décret, pris après avis de la Haute autorité de santé, en cas de danger grave ou de suspicion de danger grave pour la santé humaine. La commission a exclu les équipements qui sont des dispositifs médicaux, relevant déjà des pouvoirs de police de l'Agence nationale de sécurité du médicament et des produits de santé (ANSM).
I - Le dispositif proposé : encadrer le développement de la « neuro-modulation » en permettant l'interdiction de ces dispositifs par décret lorsqu'ils représentent un risque pour la santé humaine
Constatant le développement de dispositifs dits de « neuro-modulation », c'est-à-dire ayant pour objet de modifier le fonctionnement du cerveau, l'article 13 du projet de loi tend à permettre au pouvoir réglementaire, après avis de la Haute autorité de santé (HAS), d'interdire tout acte, procédé, technique ou équipement qui aurait un tel objet, dès lors qu'il présenterait « un danger grave ou une suspicion de danger grave pour la santé humaine ». Toute décision de levée de l'interdiction serait prise, par parallélisme des formes, également par décret. Un nouvel article L. 1151-4 serait inséré dans le code de la santé publique à cet effet.
Les techniques concernées relèvent de deux catégories : le « neuro-traitement » à des fins thérapeutiques ou la « neuro-amélioration », destinée à l'amélioration des capacités cérébrales de personnes non malades. Consultés par le rapporteur, les spécialistes des neurosciences Olivier Oullier, professeur à l'Université d'Aix-Marseille et président d'une société de neuroinformatique et Laura Pignatel, docteur en droit de l'Université d'Aix-Marseille, précisent que ces termes sont avant tout issus du secteur commercial et doivent donc être employés avec prudence. Ils permettent toutefois d'identifier les familles de dispositifs concernés.
Elles comprennent ainsi, pour la première, des médicaments (comme les psychostimulants ou les anxiolytiques) ou des dispositifs médicaux invasifs destinés à la stimulation transcrânienne 252 ( * ) ou à la stimulation cérébrale profonde 253 ( * ) pour lutter contre des maladies neurodégénératives, comme les maladies de Parkinson ou d'Alzheimer, les troubles obsessionnels compulsifs ou les états dépressifs majeurs.
Ces dispositifs sont encadrés par les règles applicables à la mise sur le marché et au contrôle des médicaments 254 ( * ) ou des dispositifs médicaux 255 ( * ) . L'article L. 5312-1 du code de la santé publique dispose à cet égard que l'Agence nationale de sécurité du médicament et des produits de santé (ANSM) peut interdire la mise en service ou l'utilisation de dispositifs médicaux, lorsqu'il existe un danger grave ou une suspicion de danger grave pour la santé humaine.
En revanche, certains dispositifs de « neuro-amélioration » sans finalité médicale vendus dans le commerce ne font pas l'objet d'un tel encadrement, ne relevant ni de la catégorie des médicaments ni forcément de celle des dispositifs médicaux - seuls ceux destinés à la stimulation cérébrale transcrânienne le sont. Ils sont toutefois soumis à l'obligation de sécurité générale des produits. Comme le précisent Olivier Oullier et Laura Pignatel, déjà cités, la neuro-amélioration constitue le plus souvent un acte de détournement de techniques initialement développées pour traiter des cas pathologiques afin d'améliorer les capacités cognitives chez des personnes non malades. Peut être citée, à titre d'illustration, la technique du « neurofeedback », processus d'apprentissage destiné à modifier et à réguler son activité cérébrale grâce à la stimulation transcrânienne.
Le Comité consultatif national d'éthique (CCNE), qui avait déjà consacré un rapport au sujet en 2012 256 ( * ) , relève dans son avis n° 129 préalable à la révision de la loi de bioéthique que « les effets réels (efficaces et délétères) de ces techniques chez le sujet non malade demeurent très mal connus » 257 ( * ) .
Outre les risques de l'ambition transhumaniste, qui consiste à vouloir dépasser la vulnérabilité inhérente à l'espèce humaine, les services du ministère de la santé ont indiqué au rapporteur que les effets à court terme ou à long terme de ces équipements sans finalité médicale sont aujourd'hui encore insuffisamment évalués. Ils sont susceptibles de présenter des risques pour la santé des consommateurs. La mesure proposée par l'article 13 donnerait à l'administration les moyens d'intervenir.
Elle s'inspire du dispositif existant à l'article L. 1151-3 du code de la santé publique, introduit dans notre droit par la loi n° 2009-879 du 21 juillet 2009 portant réforme de l'hôpital et relative aux patients, à la santé et aux territoires, qui permet au pouvoir réglementaire d'interdire, dans le domaine des actes à visée esthétique, ceux qui présenterait un danger grave ou une suspicion de danger grave pour la santé humaine.
La HAS, en charge des avis préalables aux décrets, ne tire pas un bilan très positif de l'application de ces dispositions en vigueur, comme elle a pu l'indiquer lors de son audition du rapporteur. Elle considère que l'absence de définition des notions de « danger grave » ou de « suspicion de danger grave » obère l'efficacité du dispositif et, surtout, qu'elle manque de données pour étayer les avis destinés à justifier une interdiction. Elle craint que le dispositif proposé par l'article 13 du projet de loi rencontre les mêmes écueils. Les services du ministère de la santé estiment, quant à eux, que les notions relatives au danger auquel le projet de loi fait référence sont familières du juge administratif et permettent une juste appréciation du risque.
L'Assemblée nationale n'a pas modifié, lors de l'examen du texte, son économie générale, outre l'adoption d'amendements rédactionnels du rapporteur, Philippe Berta, en commission et en séance publique avec l'avis favorable du Gouvernement.
II - La position de la commission : approuver le principe de l'article 13 tout en excluant les dispositifs médicaux qui relèvent déjà des pouvoirs de police de l'ANSM
La commission a approuvé le principe de l'article 13 du projet de loi, qui tend à pallier un vide juridique pour les dispositifs de « neuro-amélioration » en permettant d'intervenir en cas de danger pour la santé humaine.
Il n'est pas sans présenter des limites : comme l'a relevé la HAS, la rareté des données disponibles ne facilitera pas la mise en oeuvre du dispositif. Est-ce pour cela qu'il faut y renoncer ? La commission, suivant l'analyse du rapporteur, juge au contraire qu'il s'agit d'une mesure utile de prévention des risques, qu'il n'est pas interdit de compléter à l'avenir.
La commission a également relevé que la rédaction des notions de « danger grave » ou de « suspicion de danger grave » était volontairement large pour permettre une appréciation in concreto par les services et, en cas de litige, par le juge. Comme l'indique le Conseil d'État dans son avis sur le projet de loi « l'extrême difficulté, relevée par l'étude du Conseil d'Etat de 2018, à distinguer la neuro-amélioration à des fins de performance chez la personne non malade et les neuro-traitements à des fins thérapeutiques, conduit à ce que les dispositions ne différencient pas l'utilisation des techniques de neuro-modulation selon leurs finalités mais selon leur dangerosité ».
La commission a également repris à son compte les observations du Conseil d'État selon lesquelles les limitations à la liberté d'entreprendre qui pourraient résulter de l'application du nouvel article L. 1151-4 du code de la santé publique pouvaient être admises au regard des principes constitutionnels de protection de la santé et de sauvegarde de la dignité de la personne humaine.
Par l'adoption d'un amendement COM-262 du rapporteur, la commission spéciale a en revanche exclu les équipements qui sont des dispositifs médicaux, car ils relèvent déjà des pouvoirs de police de l'Agence nationale de sécurité du médicament et des produits de santé (ANSM), en application des articles L. 5311-1 et 5312-1 du code de la santé publique. Par le même amendement, elle a également procédé à une modification rédactionnelle.
La commission a adopté cet article ainsi modifié.
TITRE
IV
SOUTENIR UNE RECHERCHE LIBRE ET RESPONSABLE
AU SERVICE DE LA
SANTÉ HUMAINE
CHAPITRE 1ER
Aménager le régime actuel de recherches
sur l'embryon
et les cellules souches embryonnaires
Article 14
Différenciation des régimes juridiques
d'autorisation
s'appliquant à l'embryon et aux cellules souches
embryonnaires
Cet article procède à une différenciation des régimes juridiques s'appliquant aux recherches sur l'embryon et aux recherches sur les cellules souches embryonnaires, en maintenant pour les premières le régime d'autorisation préalable en vigueur et en instituant pour les secondes un régime de déclaration préalable auprès de l'agence de la biomédecine.
La commission spéciale a adopté plusieurs amendements tendant à sécuriser sur le plan juridique les recherches menées sur les embryons surnuméraires, en précisant notamment les prérequis applicables à ces recherches, tout en autorisant, à titre dérogatoire, le développement in vitro d'embryons jusqu'au 21 e jour afin de permettre des avancées dans la compréhension des mécanismes du développement embryonnaire. Elle a en outre renforcé la vigilance sur certaines recherches sur les cellules souches embryonnaires humaines susceptibles de soulever des questions éthiques majeures, notamment en supprimant la possibilité de constituer des embryons chimériques par l'adjonction de ces cellules à un embryon animal, estimant que ce type d'expérimentation est de nature à transgresser une « ligne rouge » en termes de risque de franchissement de la barrière des espèces.
I - Le dispositif proposé
1. Le droit en vigueur : un régime d'autorisation relativement récent, qui explique le lent développement de la recherche française dans ce domaine
a) Un cadre légal évolutif
Initialement interdite 258 ( * ) par la loi relative à la bioéthique de 1994 259 ( * ) , la recherche sur l'embryon a connu une évolution progressive de son cadre juridique au cours des quinze dernières années :
- bien que maintenant le principe d'interdiction de la recherche sur l'embryon, la loi relative à la bioéthique de 2004 260 ( * ) avait institué un régime dérogatoire d'autorisation des recherches sur l'embryon et les cellules souches embryonnaires pour une durée limitée à cinq ans ;
- étendant le principe d'interdiction de la recherche sur l'embryon aux recherches sur les cellules souches embryonnaires et les lignées de cellules souches, la loi relative à la bioéthique de 2011 261 ( * ) a pérennisé l'autorisation de dérogation de ces recherches sous réserve du respect de plusieurs conditions 262 ( * ) ;
- la loi du 6 août 2013 263 ( * ) , d'initiative parlementaire 264 ( * ) , a mis fin à l'interdiction de principe des recherches sur l'embryon et les cellules souches embryonnaires qu'elle soumet, désormais, à un régime d'autorisation ;
- la loi « Santé » 265 ( * ) du 26 janvier 2016 a inséré, dans l'article L. 2151-5 du code de la santé publique, des dispositions autorisant des recherches biomédicales dans le cadre d'une procédure d'assistance médicale à la procréation sur des gamètes destinés à constituer un embryon ou sur un embryon avant ou après son transfert à des fins de gestation, sous réserve du consentement du couple.
En application de l'article L. 2151-5 du code de la santé publique, seuls peuvent aujourd'hui être autorisés par l'agence de la biomédecine les protocoles de recherche qui remplissent les critères suivants :
« 1° La pertinence scientifique de la recherche est établie ;
2° La recherche, fondamentale ou appliquée, s'inscrit dans une finalité médicale ;
3° En l'état des connaissances scientifiques, cette recherche ne peut être menée sans recourir à ces embryons ou ces cellules souches embryonnaires ;
4° Le projet et les conditions de mise en oeuvre du protocole respectent les principes éthiques relatifs à la recherche sur l'embryon et les cellules souches embryonnaires. »
En particulier, le respect de deux principes fondamentaux conditionne la possibilité de conduire des recherches sur l'embryon ou les cellules souches embryonnaires :
- la recherche ne peut être menée qu'à partir d'embryons dits « surnuméraires », conçus in vitro dans le cadre d'une assistance médicale à la procréation et qui ne font plus l'objet d'un projet parental et ont été cédés à la recherche par le couple dont le consentement exprès écrit a été préalablement recueilli. En conséquence, il est précisé à l'article L. 2151-5 précité que « les embryons sur lesquels une recherche a été conduite ne peuvent être transférés à des fins de gestation » ;
- la conception in vitro d'embryon à des fins de recherche est interdite, sur le fondement de l'article L. 2151-2 du code de la santé publique.
Sur le plan de la procédure, l'autorisation est accordée par l'agence de la biomédecine dont la décision est assortie d'un avis du conseil d'orientation. Cette décision est communiquée aux ministres chargés de la santé et de la recherche qui disposent d'un délai d'un mois pour demander, conjointement, un nouvel examen du dossier dans les deux cas suivants :
- en cas de doute sur le respect des principes éthiques ou sur la pertinence scientifique du protocole. L'agence procède à un nouvel examen dans un délai de trente jours et, en cas de confirmation de sa décision, la validation du protocole est réputée acquise ;
- dans l'intérêt de la santé publique ou de la recherche scientifique, lorsque le protocole a été refusé. L'agence procède à un nouvel examen dans un délai de trente jours et, en cas de confirmation de sa décision, le refus du protocole est réputé acquis.
b) Un encadrement qui n'est pas parvenu pleinement à relancer la recherche française sur l'embryon et les cellules souches
Selon l'étude d'impact annexée au projet de loi, 92 protocoles de recherche sur l'embryon et les cellules souches embryonnaires ont été autorisés depuis 2005 par l'agence de la biomédecine et huit ont été refusés.
Sur les 19 protocoles sur l'embryon humain autorisés depuis 2005, 11 ont été autorisés entre 2006 et 2008, dont la majorité visait à dériver des lignées de cellules souches embryonnaires. Un seul protocole sur l'embryon a été autorisé entre 2009 et 2013. 7 protocoles sur l'embryon ont par la suite été autorisés entre 2014 et 2017. 79 protocoles de recherche sur les cellules souches embryonnaires ont été autorisés depuis 2005.
Des éléments d'information actualisés communiqués par l'agence de la biomédecine à votre commission font état de 97 protocoles de recherche autorisés par l'agence depuis 2005, dont 23 portent spécifiquement sur l'embryon.
Il ressort des auditions conduites par votre commission que l'environnement réglementaire et le risque contentieux associé ont contribué à décourager la recherche sur l'embryon et les cellules souches embryonnaires en France. Selon Cécile Martinat, présidente de la société française de recherche sur les cellules souches (« French Society for Stem Cell Research » - FSSCR), environ 50 % des programmes de recherche font encore l'objet d'un processus juridique très largement à l'initiative de la fondation Jérôme Lejeune.
2. Le cadre proposé par le projet de loi : un dispositif qui acte la différence de nature entre l'embryon et les cellules souches embryonnaires
Les différents chercheurs auditionnés par votre commission ont insisté sur la différence de nature entre les recherches conduites sur l'embryon et celles conduites sur des cellules souches embryonnaires humaines (CSEh), qui soulève des interrogations éthiques différentes.
Obtenues à partir d'embryons au stade blastocyste, c'est-à-dire cinq à sept jours après la fécondation in vitro , les cellules souches embryonnaires humaines présentent deux principales propriétés. D'une part, par leur caractère pluripotent, elles ont la capacité de se différencier pour se spécialiser et former n'importe quel tissu du corps humain. D'autre part, elles peuvent se multiplier indéfiniment en laboratoire et former ainsi des lignées « immortelles ».
Comme le rappelle la société française de recherche sur les cellules souches dans sa contribution aux états généraux de la bioéthique du CCNE de 2018, la loi relative à la bioéthique de 2004 avait inclus les cellules souches embryonnaires dans le périmètre du régime applicable aux recherches sur l'embryon au motif que le processus de dérivation de ces cellules entraînait nécessairement la destruction de l'embryon. Elle rappelle, néanmoins, que les cellules souches embryonnaires « ne représentent qu'une fraction de l'embryon et il est clairement admis qu'elles n'ont pas le potentiel d'un embryon humain entier. En particulier, après avoir été extraites de l'embryon originel puis cultivées in vitro afin de les immortaliser elles sont devenues incapables de former spontanément un nouvel embryon. À l'évidence, les cellules CSEh ne constituent donc plus une personne humaine potentielle. »
Dans son avis 129, le CCNE estime pour sa part que les cellules souches embryonnaires humaines « n'ont rien du caractère symbolique de « personne potentielle » attribué à l'embryon ». Si le processus de dérivation des cellules souches embryonnaires doit faire l'objet d'une autorisation dès lors qu'il aboutit à la destruction de l'embryon, le maintien dans un régime d'autorisation des recherches menées à partir de cellules souches embryonnaires humaines déjà dérivées ne semble désormais plus justifié à nombre de chercheurs spécialisés dans ce type de recherche.
Les recherches menées sur ces cellules n'impliquent plus en effet la destruction d'un embryon. En France, 28 lignées de cellules souches embryonnaires ont été dérivées, dont 25 sont issues du diagnostic préimplantatoire et présentent des cellules porteuses d'une mutation ou d'un déséquilibre chromosomique. La dernière lignée a été dérivée en 2008. Seules 11 de ces lignées sont encore utilisées dans des protocoles autorisés actuellement.
Depuis l'entrée en vigueur des dispositions introduites par la loi relative à la bioéthique de 2004 autorisant l'importation de cellules souches embryonnaires humaines à des fins de recherche, 70 lignées de cellules souches embryonnaires humaines ont été importées et utilisées par des équipes de recherche. Certaines lignées sont très largement utilisées par les équipes de recherche, notamment les lignées H1, H9 et RC09. Toutes les lignées de cellules souches embryonnaires humaines disponibles en Europe sont répertoriées dans le registre hPSCReg 266 ( * ) qui comptabilisait, au 1 er novembre 2019, 742 lignées. Selon ce registre, le Royaume-Uni a déclaré 88 lignées de cellules souches embryonnaires quand la France n'en a déclaré que 22.
Compte tenu de ces considérations, l'article 14 du projet de loi opère une différenciation des régimes juridiques applicables aux recherches sur l'embryon et aux recherches sur les cellules souches embryonnaires.
a) Les recherches sur l'embryon dans le cadre d'une assistance médicale à la procréation
La loi « Santé » de 2016 a introduit, à l'article L. 2151-5 du code de la santé publique relatif au régime juridique des recherches conduites sur l'embryon et les cellules souches embryonnaires, un régime spécifique aux recherches biomédicales menées dans le cadre de l'assistance médicale à la procréation sur les gamètes destinés à constituer un embryon ou sur un embryon conçu in vitro destiné à être transféré à des fins de gestation. Le renvoi aux dispositions du titre II du livre I er de la première partie du code de la santé publique permet de soumettre ces recherches aux mêmes conditions d'autorisation que celles applicables aux recherches impliquant la personne humaine, notamment les essais cliniques de médicament, qui nécessitent l'avis favorable d'un comité de protection des personnes (CPP) et une autorisation de l'agence nationale de sécurité du médicament et des produits de santé (ANSM).
Dans le souci de différencier ce type de recherche de celles menées sur des embryons ne faisant plus l'objet d'un projet parental, le I de l'article 14 du projet de loi transfère ces dispositions au sein d'un nouvel article L. 2141-3-1 du code de la santé publique, au sein des dispositions générales relatives à l'assistance médicale à la procréation. L'expression « recherches biomédicales », choisie en 2016 pour désigner ces recherches sur l'embryon destiné à être transféré, est remplacée par le mot « recherches », dès lors que les recherches biomédicales impliquent la personne humaine et qu'un embryon n'a pas le statut de personne.
Les termes « in vitro » sont également supprimés afin de tenir compte du fait que des recherches sur l'embryon après son transfert in utero sont possibles. Enfin, par coordination avec les nouvelles modalités de recours à l'AMP envisagées par le projet de loi, il est précisé que le consentement à ces recherches est exprimé soit par chaque membre du couple, soit par la femme non mariée.
Le II procède, en conséquence, à une coordination de référence à l'article L. 1125-3 du code de la santé publique relatif à l'autorisation par l'ANSM des recherches impliquant la personne humaine menées dans le cadre de l'AMP.
b) Le maintien d'un régime d'autorisation pour les recherches sur l'embryon
Le 1° du III de l'article 14 du projet de loi procède à une réécriture de l'article L. 2151-5 du code de la santé publique qui sera spécifiquement dédié au régime d'autorisation des recherches sur l'embryon, l'encadrement des recherches sur les cellules souches embryonnaires humaines faisant l'objet d'un nouvel article L. 2151-6 du même code.
Les critères conditionnant l'autorisation d'un protocole de recherche sur l'embryon ne sont pas sensiblement modifiés par le projet de loi et continuent d'inclure :
- la pertinence scientifique de la recherche ;
- la finalité médicale poursuivie par la recherche, que celle-ci soit fondamentale ou appliquée ;
- la démonstration qu'en l'état des connaissances scientifiques, la recherche ne peut être menée sans recourir à des embryons humains ;
- le respect de principes éthiques fondamentaux.
C'est ce dernier critère qui est précisé par l'article 14 du projet de loi. À l'heure actuelle, il est fait référence aux « principes éthiques relatifs à la recherche sur l'embryon et les cellules souches embryonnaires. »
Les principes éthiques auxquels renvoie le 4° du I de l'article L. 2151-5 du code de la santé publique tel que modifié par l'article 14 du projet de loi recouvrent, en premier lieu, les principes posés par le titre V « Recherche sur l'embryon et les cellules souches embryonnaires » du livre I er de la deuxième partie du code. Il s'agit de :
- l'interdiction de la conception in vitro d'embryons et de la constitution par clonage d'embryons humains à des fins de recherche 267 ( * ) ;
- l'interdiction de l'adjonction de cellule d'une autre espèce dans un embryon humain 268 ( * ) ;
- l'interdiction de la conception et du clonage d'embryons et de leur utilisation à des fins commerciales ou industrielles 269 ( * ) ;
- l'interdiction de constituer par clonage un embryon humain à des fins thérapeutiques 270 ( * ) .
Sont également visés, en deuxième lieu, les principes fondamentaux prévus par les articles 16 à 16-8 du code civil :
- l'interdiction de toute atteinte à la dignité de la personne 271 ( * ) ;
- le droit au respect de l'être humain dès le commencement de la vie 272 ( * ) ;
- le droit au respect du corps humain, l'inviolabilité et l'impossibilité, pour celui-ci, ainsi que pour les éléments et les produits qui en sont issus, de faire l'objet d'un droit patrimonial 273 ( * ) ;
- la possibilité de ne porter atteinte à l'intégrité du corps humain qu'en cas de nécessité thérapeutique, et après avoir recueilli le consentement de l'intéressé 274 ( * ) ;
- l'interdiction de porter atteinte à l'intégrité de l'espèce humaine 275 ( * ) ;
- la prohibition des pratiques eugéniques ayant pour objet la sélection des personnes 276 ( * ) ;
- l'interdiction des modifications des caractères génétiques transmissibles à la descendance 277 ( * ) ;
- l'interdiction de rémunération de celui qui se prête à une expérimentation, au prélèvement d'éléments de son corps ou à la collecte de produits de celui-ci 278 ( * ) ;
- le principe de l'anonymat du don portant sur un produit ou un élément du corps humain 279 ( * ) .
En troisième lieu, les principes éthiques applicables à la recherche sur l'embryon et les cellules souches embryonnaires humaines incluent les principes mentionnés au titre I er du livre II de la première partie du code de la santé publique applicables au don et à l'utilisation des éléments et produits du corps humain. Parmi ceux-ci figurent notamment :
- les principes des articles 16 à 16-8 du code civil rappelés ci-dessus ;
- le principe du consentement au don ;
- le principe de la gratuité du don ;
- le principe de l'anonymat du don.
Dans une décision du 1 er août 2013 280 ( * ) , le Conseil constitutionnel a jugé conformes à la Constitution les dispositions encadrant l'attribution des autorisations de recherche sur l'embryon sous réserve du respect, notamment, de l'ensemble des principes précités.
Le II de l'article L. 2151-5 du code de la santé publique dans sa version proposée par le projet de loi dispose que les recherches ne peuvent porter que sur des embryons conçus in vitro dans le cadre d'une assistance médicale à la procréation qui ne font plus l'objet d'un projet parental et sont cédés à la recherche.
En l'état du droit en vigueur, la recherche est subordonnée au consentement écrit préalable du couple ou du membre survivant du couple, après que ceux-ci ont été informés des possibilités d'accueil par un autre couple ou d'arrêt de la conservation des embryons. Il est, en outre, rappelé que le consentement au don à la recherche doit faire l'objet d'une confirmation à l'expiration d'un délai de trois mois, sauf dans deux cas : lorsque le couple consent au don à l'issue d'un diagnostic préimplantatoire concluant au diagnostic d'une anomalie génétique responsable d'une maladie d'une particulière gravité 281 ( * ) ou lorsque le couple consent au don des embryons non susceptibles d'être transférés ou conservés à l'issue d'une fécondation in vitro 282 ( * ) .
L'article 14 du projet de loi procède à plusieurs coordinations audit II de l'article L. 2151-5 du code de la santé publique :
- il est tenu compte du fait que l'embryon peut résulter d'une procédure d'AMP engagée par une femme non mariée ;
- par souci de simplification, les modalités de consentement au don de l'embryon à la recherche sont renvoyées aux dispositions du 2° du II de l'article L. 2141-4 du code de la santé publique dont l'article 16 du projet de loi propose une nouvelle rédaction, du dernier alinéa de l'article L. 2131-4 du même code et du troisième alinéa de l'article L. 2141-3 du même code.
L'autorisation par l'agence de la biomédecine est maintenue, étant précisé que l'agence se prononce non seulement sur le respect des critères prévus par le I de l'article L. 2151-5 du code de la santé publique mais également sur le respect des conditions prévues par son II (embryons surnuméraires cédés à la recherche). Compte tenu de la sensibilité de ces décisions, elles continueront d'être assorties d'un avis de son conseil d'orientation, instance chargée de veiller au respect par l'agence des patients, des donneurs et des principes éthiques dans l'exercice de ses missions. Dans les mêmes conditions qu'à l'heure actuelle, les décisions d'autorisation ou de refus de l'agence devront être communiquées aux ministres chargés de la santé et de la recherche qui pourront, le cas échéant, demander un réexamen du dossier.
L'agence de la biomédecine conserve, en outre, son pouvoir de suspension ou de retrait de l'autorisation à l'issue d'inspections indépendantes.
c) L'inscription dans la loi d'une durée limite de culture in vitro des embryons destinés à la recherche
Le 1° du III de l'article 14 du projet de loi maintient, au IV de l'article L. 2151-5 du code de la santé publique, la disposition selon laquelle les embryons sur lesquels une recherche a été conduite ne peuvent être transférés à des fins de gestation. Il la complète par une disposition tendant à limiter la durée de culture in vitro des embryons à 14 jours suivant leur constitution.
À l'heure actuelle, aucune disposition en droit français ne fixe de limite à la durée de développement in vitro des embryons. Jusqu'à récemment, les protocoles de recherche autorisés sur l'embryon ne prévoyaient pas de culture au-delà de sept jours, date qui correspond généralement à la limite marquant le moment de l'implantation. Toutefois, compte tenu des progrès réalisés dans la mise au point de milieux de culture propices au développement embryonnaire, la question du développement in vitro d'embryons au-delà de sept jours se pose désormais.
Dans ses réponses au questionnaire de votre commission, l'agence de la biomédecine rappelle que la limite de 14 jours pour la culture d'un embryon dans le cadre d'un protocole de recherche a été définie en 1984 par la commission d'enquête sur la fertilisation humaine et l'embryologie mise en place au Royaume-Uni et présidée par la philosophe Mary Warnock, dite « commission Warnock ». Cette limite est fondée sur :
- un argument philosophique : le 15 e ou le 16 e jour de développement marque, pour certains, le moment où l'embryon humain perd sa capacité à se scinder en deux embryons indépendants. Les tenants de cet argument considèrent alors que l'embryon acquiert son indivisibilité et y voient donc l'acquisition d'une nature humaine de valeur supérieure ;
-un argument scientifique : les premières cellules embryonnaires différenciées 283 ( * ) apparaissent à partir du 15 e ou du 16 e jour de développement de l'embryon. Parmi ces cellules différenciées, on trouve les cellules du neurectoderme, à l'origine du futur cerveau. Il avait semblé utile, aux membres de la commission Warnock, d'empêcher la formation du neurectoderme dans les embryons in vitro dans l'hypothèse, qui reste non vérifiée, où leur émergence marquerait la naissance d'une conscience humaine.
L'agence de la biomédecine souligne le fait que cette limite de développement in vitro de 14 jours fait aujourd'hui l'objet d'un consensus international et a été intégrée par 17 pays dans leur législation ou dans des recommandations. Elle n'a pas été remise en cause jusqu'à récemment car il existait une limite technique : les conditions de culture ne permettaient pas la survie d'un embryon au-delà du stade correspondant à son implantation dans l'utérus, c'est-à-dire à sept jours de développement.
En 2016, deux études ont rapporté la mise au point de « plateformes d'implantation » permettant le développement de l'embryon in vitro au-delà de cette limite. Les auteurs ont volontairement arrêté la culture des embryons après 13 jours, mais il semble aujourd'hui que la limite des 14 jours puisse être franchie. De fait, deux études récentes ont montré qu'il était possible de procéder à la culture d'embryons de macaques jusqu'à 20 jours en utilisant des plateformes d'implantation similaires.
Par ailleurs, le 2° du III de l'article 14 du projet de loi procède à une renumérotation d'articles du code de la santé publique afin de tenir compte des nouvelles dispositions prévues par le projet de loi en matière d'encadrement des recherches sur les cellules souches embryonnaires humaines.
d) L'introduction d'un régime de déclaration préalable des recherches menées sur les cellules souches embryonnaires humaines
Le 3° du III de l'article 14 du projet de loi vise à introduire, à l'article L. 2151-6 du code de la santé publique, un régime de déclaration préalable auprès de l'agence de la biomédecine. Il est précisé que ces recherches ne peuvent être menées qu'à partir de lignées dérivées autorisées dans les conditions suivantes :
- de cellules souches embryonnaires dérivées d'embryons humains dans le cadre d'un protocole de recherche sur l'embryon autorisé en application de l'article L. 2151-5 du code de la santé publique ;
- de cellules souches embryonnaires ayant fait l'objet d'une autorisation d'importation accordée par l'agence de la biomédecine.
Le directeur général de l'agence de la biomédecine est appelé à s'opposer à la réalisation du protocole si celui-ci n'est pas réalisé à partir de lignées dérivées autorisées dans les conditions évoquées précédemment et s'il ne respecte pas les principes suivants :
- le protocole doit s'inscrire dans une finalité médicale ;
- la pertinence scientifique de la recherche doit être établie ;
- le protocole et ses conditions de mise en oeuvre doivent respecter les mêmes principes éthiques fondamentaux auxquels sont astreints les protocoles de recherche sur l'embryon.
Le délai dans lequel le directeur général de l'agence pourra s'opposer aux protocoles de recherche sera fixé par voie réglementaire. Il pourrait être de deux mois, selon les informations communiquées par le ministère des solidarités et de la santé à votre commission.
Il est à noter que la condition de démonstration de l'absence d'une méthodologie alternative, applicable aux recherches sur l'embryon, ne sera plus applicable aux recherches sur les cellules souches embryonnaires humaines.
En outre, il est prévu que certaines recherches considérées comme particulièrement sensibles au regard de leurs enjeux éthiques devront faire l'objet d'une vigilance renforcée. Il s'agit, dans la version initiale du projet de loi, de deux types de recherche :
- la différenciation de cellules souches embryonnaires humaines en gamètes ;
- l'agrégation de cellules souches embryonnaires humaines avec des cellules précurseurs de tissus extra-embryonnaires, c'est-à-dire la constitution de modèles embryonnaires à usage scientifique susceptibles de mimer la phase de gastrulation et parfois dénommés « gastruloïdes ».
Pour ces recherches, une décision d'opposition du directeur général de l'agence de la biomédecine devra être précédée d'un avis public du conseil d'orientation scientifique de l'agence. Dans ce cas, l'agence estime que le délai d'opposition de l'agence pourrait être porté à trois mois.
Enfin, toute décision de suspension ou d'interdiction des recherches sur les cellules souches embryonnaires humaines qui ne respecteraient plus les conditions prévues par le code de la santé publique devra être précédée d'un avis public du conseil d'orientation de l'agence de la biomédecine.
À titre transitoire, le VI de l'article 14 du projet de loi prévoit que les protocoles de recherche sur des cellules souches embryonnaires humaines en cours d'instruction à la date de la publication de la loi par l'agence de la biomédecine en vue, initialement, de leur autorisation seront réputés, si leur dossier est complet, satisfaire à l'obligation de déclaration préalable. Le délai d'opposition de l'agence est fixé à quatre mois pour ces protocoles à compter de la réception du dossier complet de la demande d'autorisation.
Le 4° du III de l'article 14 du projet de loi tend à abroger l'article L. 2151-7 du code de la santé publique dont les dispositions relatives aux autorisations de conservation d'embryons ou de cellules souches embryonnaires à des fins de recherche ont vocation à figurer dans un nouvel article L. 2151-9 du même code prévu par le projet de loi.
Le 5° du III de l'article 14 du projet de loi procède à une clarification rédactionnelle à l'article L. 2151-8 284 ( * ) du code de la santé publique relatif à l'autorisation par l'agence de la biomédecine de l'importation de cellules souches embryonnaires humaines à des fins de recherche.
e) Le maintien du régime d'autorisation de la conservation d'embryons et de cellules souches embryonnaires humaines
En application de l'article L. 2151-7 du code de la santé publique en vigueur, la conservation d'embryons ou de cellules souches embryonnaires humaines à des fins de recherche est soumise à une autorisation délivrée par l'agence de la biomédecine, qui se prononce au regard du respect des principes éthiques inscrits au code de la santé publique, des règles en matière de sécurité des personnes employées sur le site de conservation et des dispositions applicables en matière de protection de l'environnement et de sécurité sanitaire.
L'ANSM est informée de ces activités de conservation lorsque le lieu de conservation est situé sur le même site que des activités de préparation et de conservation des tissus et de leurs dérivés et des préparations de thérapie cellulaire autorisées par l'ANSM.
Le 6° du III de l'article 14 du projet de loi crée un nouvel article L. 2151-9 au sein du code de la santé publique, reprenant les dispositions de l'article L. 2151-7 précité, moyennant les modifications suivantes :
- dans l'attribution des autorisations de conservation, l'agence de la biomédecine veillera au respect, outre des critères déjà prévus par le droit en vigueur, des principes éthiques fondamentaux inscrits aux articles 16 à 16-8 du code civil ;
- les laboratoires de biologie médicale accrédités 285 ( * ) pour exercer les activités biologiques d'assistance médicale à la procréation pourront conserver des embryons proposés à la recherche 286 ( * ) sans être titulaires d'une autorisation spécifique en ce sens par l'agence de la biomédecine ;
- par coordination, la cession d'embryons ne pourra se faire qu'au profit soit d'un organisme lui-même autorisé pour la conservation d'embryons, soit d'un organisme autorisé, en application de l'article L. 2151-5 du code de la santé publique, à pratiquer une recherche sur l'embryon. De même, des cellules souches embryonnaires humaines ne pourront être cédées qu'à un organisme lui-même autorisé à conserver des cellules souches embryonnaires ou à un organisme ayant déclaré un protocole de recherche sur ces cellules en application de l'article L. 2151-6 du même code, à la condition que l'agence de biomédecine ne se soit pas opposée à ce protocole.
Le 7° du III de l'article 14 du projet de loi procède à une coordination à l'article L. 2151-10 du code de la santé publique qui, tel qu'il résulte de l'article 14 du projet de loi, reprend les dispositions de l'actuel article L. 2151-7-1 du code de la santé publique relatif à la clause de conscience en vertu de laquelle un chercheur, un ingénieur, un technicien ou un auxiliaire de recherche, de même qu'un médecin ou un auxiliaire médical peuvent refuser de participer à quelque titre que ce soit à une recherche sur des embryons humains ou des cellules souches embryonnaires humaines. Cette coordination vise à tenir compte du fait que les recherches sur les cellules souches seront déclarées et non plus autorisées. Cette clause de conscience avait été introduite dans le code de la santé publique par l'Assemblée nationale en première lecture lors de l'examen de la loi relative à la bioéthique de 2011.
f) Les sanctions pénales applicables en matière de conservation et de cession d'embryons et de cellules souches embryonnaires humaines
Le IV de l'article 14 du projet de loi tire les conséquences de la réécriture, à l'article L. 2151-9 du code de la santé publique, des dispositions relatives à la conservation et à la cession des embryons et des cellules souches embryonnaires humaines, en réécrivant l'article 511-19-2 du code pénal qui punit les activités ne se conformant pas à ces dispositions de deux ans d'emprisonnement et de 30 000 euros d'amende. De même, les dispositions « miroir » de l'article L. 2163-7 du code de la santé publique, qui reproduit les dispositions de l'article 511-19-2 du code pénal, sont actualisées, au V de l'article 14 du projet de loi, pour tenir compte de ces modifications.
II - Les modifications adoptées par l'Assemblée nationale
• En commission , outre plusieurs amendements rédactionnels, la commission des affaires sociales de l'Assemblée nationale a adopté des amendements de son rapporteur, Philippe Berta, tendant à :
- maintenir la précision selon laquelle l'embryon, sur lequel des recherches cliniques peuvent être effectuées en dehors ou sein de l'utérus dans le cadre d'une assistance médicale à la procréation, est « conçu in vitro ». Cette précision concerne le mode de conception de l'embryon et non les modalités des recherches cliniques concernées ;
- préciser que la recherche non-clinique sur l'embryon humain peut porter sur les causes de l'infertilité, au motif que les pays développés à économie de marché sont concernés par une hausse conséquente des situations d'infertilité ;
- compléter les recherches sur les cellules souches embryonnaires humaines devant faire l'objet d'une vigilance particulière 287 ( * ) de la part de l'agence de la biomédecine par celles consistant à créer des embryons chimériques par l'insertion dans un embryon animal de cellules souches embryonnaires humaines en vue de son transfert chez la femelle. Ces recherches pourraient, dans le futur, représenter un intérêt pour explorer la possibilité d'obtenir des organes humains développés chez l'animal. En posant la question du franchissement de la barrière des espèces, ces recherches sont néanmoins susceptibles de soulever des interrogations éthiques majeures ;
- tirer les conséquences de l'institution de régimes distincts d'autorisation et de déclaration préalable, applicables respectivement aux recherches sur l'embryon et aux recherches sur les cellules souches embryonnaires humaines, pour la conservation de ces éléments :
- la conservation d'embryons continuera de faire l'objet d'une autorisation par l'agence de la biomédecine ;
- la conservation de cellules souches embryonnaires humaines devra faire l'objet d'une déclaration préalable auprès de l'agence de la biomédecine qui gardera la possibilité de suspendre ou interdire à tout moment cette conservation en cas de non-respect des mêmes critères que ceux conditionnant l'autorisation de la conservation des embryons ;
- l'organisme auquel des cellules souches embryonnaires humaines auront été cédées devra lui aussi procéder à une déclaration de conservation de ces cellules auprès de l'agence ;
- afin de tenir compte de l'institution de régimes distincts en matière de conservation, les coordinations nécessaires au sein des dispositions relatives aux sanctions pénales applicables en cas de non-respect de ces régimes d'autorisation ou de déclaration sont apportées.
• En séance , outre plusieurs amendements rédactionnels, l'Assemblée nationale a adopté en séance un amendement tendant à interdire, dans le cadre des recherches menées sur un gamète ou un embryon dans le cadre de l'assistance médicale à la procréation, toute intervention ayant pour objet de modifier le génome des gamètes ou de l'embryon. Cet amendement intervient en réaction aux expérimentations annoncées en novembre 2018 ayant conduit, selon le responsable de ces expérimentations, à la conception de deux bébés dont le génome avait été modifié par la technique « CRISPR-Cas9 » 288 ( * ) afin qu'une mutation génétique leur confère une immunité contre l'infection au VIH. Ces expérimentations avaient suscité l'émoi de la communauté scientifique internationale.
L'article 16-4 du code civil prévoit déjà que « sans préjudice des recherches tendant à la prévention et au traitement des maladies génétiques, aucune transformation ne peut être apportée aux caractères génétiques dans le but de modifier la descendance de la personne. »
III - La position de la commission
• Tenir compte de la spécificité des recherches menées sur un embryon destiné à être transféré dans le cadre de l'assistance médicale à la procréation
Depuis l'adoption, dans la loi « Santé » de 2016, d'une disposition soumettant les recherches menées sur un embryon destiné à être transféré dans le cadre de l'AMP au régime applicable aux recherches impliquant la personne humaine, plusieurs recherches de ce type ont autorisées par l'ANSM et un comité de protection des personnes (CPP).
L'évolution du cadre juridique applicable
aux recherches
menées sur l'embryon dans le cadre de l'assistance
médicale à la procréation
La loi n° 2011-814 du 7 juillet 2011 relative à la bioéthique avait modifié l'article L. 2151-5 du code de la santé publique afin de prévoir, en son VI, qu'« à titre exceptionnel, des études sur les embryons visant notamment à développer les soins au bénéfice de l'embryon et à améliorer les techniques d'assistance médicale à la procréation ne portant pas atteinte à l'embryon peuvent être conduites avant et après leur transfert à des fins de gestation si le couple y consent [...] », ces études étant alors autorisées par l'agence de la biomédecine en application du IV de ce même article.
La loi n° 2013-715 du 6 août 2013 tendant à modifier la loi n° 2011-814 du 7 juillet 2011 précitée, en autorisant sous certaines conditions la recherche sur l'embryon et les cellules souches embryonnaires, a à son tour modifié l'article L. 2151-5 du code de la santé publique en supprimant notamment son alinéa VI et de ce fait l'autorisation par l'agence de la biomédecine de ce type d'études. La suppression de ce VI de l'article L. 2151-5 du code de la santé publique avait été motivée par le souhait de soumettre les études sur l'embryon à la réglementation sur les recherches biomédicales, pour lesquelles l'ANSM est l'unique régulateur en France avec les CPP pour les aspects éthiques.
Toutefois, lors de l'examen du décret, pris en application de la loi du 6 août 2013 susmentionnée, le Conseil d'État a estimé qu'en l'état du droit les recherches biomédicales interventionnelles portant sur les gamètes destinés à constituer un embryon et sur un embryon avant son transfert in utero n'étaient pas permises. En effet, le Conseil d'État a considéré qu'il n'appartenait pas au pouvoir réglementaire de rattacher les études sur l'embryon au droit des recherches biomédicales au vu du peu d'éléments à disposition dans les débats parlementaires et du silence de la loi de 2013 sur ce point.
Le législateur est donc à nouveau intervenu par la loi n° 2016-41 du 26 janvier 2016 (article 155, III) pour ajouter un V à l'article L. 2151-5 du code de la santé publique ainsi rédigé : « Sans préjudice du titre IV du présent livre I er , des recherches biomédicales menées dans le cadre de l'assistance médicale à la procréation peuvent être réalisées sur des gamètes destinés à constituer un embryon ou sur l'embryon in vitro avant ou après son transfert à des fins de gestation, si chaque membre du couple y consent. Ces recherches sont conduites dans les conditions fixées au titre II du livre I er de la première partie. »
Le législateur a donc confié à l'ANSM la compétence sur les recherches impliquant la personne humaine (RIPH) (anciennement appelées « recherches biomédicales ») menées dans le cadre de l'AMP. En effet, l'ANSM est l'autorité compétente pour toutes les RIPH qu'elles portent sur des médicaments, des dispositifs médicaux, des cosmétiques, des organes, des tissus, des cellules humains et du sang, ainsi que sur les RIPH ne portant pas sur un produit de santé.
En pratique, l'ANSM a déjà autorisé des RIPH dans le cadre de l'AMP, dont, à titre d'exemple :
- une RIPH promue par l'AP-HP intitulée « NATural Ovarian Stimulation (NATOS) : un protocole de stimulation ovarienne innovant associant croissance folliculaire multiple et environnement estrogénique physiologique destiné aux patientes de bon pronostic prises en charge en fécondation in vitro (FIV) » a été autorisée par l'ANSM le 26 août 2016 avec pour objectif de comparer l'efficacité de la FIV en utilisant NATOS ou un protocole de stimulation ovarienne antagoniste classique ;
- une RIPH intitulée « Comparaison du nombre cumulé d'ovocytes recueillis avec 2 stimulations ovariennes sur le même cycle par Fertistartkit® (DUOSTIM) versus 2 stimulations conventionnelles chez les patientes avec réserve ovarienne altérée en FIV. Étude BISTIM », promue par le centre hospitalier intercommunal de Créteil, a été autorisée le 14 février 2018. Elle a pour objectif de montrer que 2 stimulations par Fertistartkit® sur un même cycle (phase folliculaire puis phase lutéale : DUOSTIM) permettent de recueillir 1,5 ovocyte de plus que le cumul de 2 stimulations conventionnelles (phase folliculaire) sur 2 cycles différents consécutifs ou non chez les patientes avec un CFA<5 et/ou AMH<1,2 ng/ml (groupe 3/4 des mauvaises répondeuses selon les critères de Poséïdon).
Source : Agence nationale de sécurité du médicament et des produits de santé
De l'avis de certains professionnels de l'assistance médicale à la procréation, le régime d'autorisation des essais cliniques par l'ANSM apparaît néanmoins peu pertinent, en pratique, pour l'examen des projets de recherche portant sur des embryons destinés à être transférés dans le cadre d'une procédure d'AMP. Les enjeux de l'AMP et de la recherche sur l'embryon ne constituent pas en effet le coeur de l'expertise de l'ANSM dont l'examen de projets d'essais cliniques porte généralement sur les risques potentiels que présentent pour une personne humaine l'administration d'un médicament ou la mise en oeuvre d'une thérapie.
L'article R. 1125-18 du code de la santé publique prévoit que toute demande d'autorisation de recherche menée sur un gamète ou un embryon dans le cadre de l'assistance médicale à la procréation est transmise, pour avis, par l'ANSM à l'agence de la biomédecine. Votre commission considère essentiel que l'agence de la biomédecine reste impliquée dans cette procédure d'autorisation, étant la mieux indiquée pour se prononcer sur la pertinence scientifique de ce type de projet de recherche.
Votre commission a adopté un amendement COM-192 de la rapporteure supprimant, à l'article L. 1125-3 du code de la santé publique, les termes « impliquant la personne humaine » utilisés pour qualifier les recherches menées sur les gamètes ou l'embryon dans le cadre de l'AMP. Il convient en effet de mettre un terme à toute confusion sur le statut du gamète ou de l'embryon concernés qui, au stade de la recherche, n'ont pas le statut de « personne humaine ».
• Sécuriser sur le plan juridique les recherches menées sur l'embryon
De nombreux représentants de la communauté scientifique ont insisté sur le risque contentieux qui pèse sur les protocoles de recherche concernant l'embryon et les cellules souches embryonnaires humaines. Les décisions d'autorisation prononcées par l'agence de la biomédecine, rendues publiques, sont quasi -systématiquement contestées devant les juridictions administratives par des associations opposées à ce type de recherche, dont principalement la fondation Jérôme Lejeune.
Depuis 2008, 48 requêtes au fond ont été déposées contre des décisions de l'agence de la biomédecine autorisant soit des protocoles de recherche sur l'embryon ou les cellules souches embryonnaires humaines, soit l'importation de lignées de cellules souches embryonnaires humaines. Sur ces 48 requêtes, quatre concernaient des décisions d'autorisation de recherche sur l'embryon. À cela s'ajoutent dix requêtes en référé contre des décisions d'autorisation de recherche, déposées en parallèle des requêtes au fond.
Pour l'agence de la biomédecine, le coût de ces contentieux avoisine les 800 000 euros, consistant essentiellement en des honoraires d'avocat. Ce coût ne tient pas compte de la charge de travail résultant de la gestion de ces contentieux par les membres du personnel de l'agence.
Les recours déposés contre les décisions d'autorisation de l'agence s'appuient sur différents motifs ayant trait au respect des critères d'autorisation : la pertinence scientifique, la finalité médicale, l'absence d'alternative au recours aux embryons, de même que la traçabilité des embryons ou cellules souches embryonnaires humaines utilisés. Lors de son audition par votre commission, Cécile Martinat a rappelé qu'en mars 2019, deux autorisations de recherche ont été annulées par le tribunal administratif de Versailles, sur le fondement de la pertinence scientifique et de la traçabilité.
En particulier, les chercheurs insistent sur la difficulté opérationnelle à démontrer le respect de deux critères dans leur libellé actuel, maintenus par le projet de loi :
- toute recherche participe potentiellement de l'ambition de réaliser des progrès médicaux sans qu'il puisse être démontré avec précision et ab initio l'intérêt d'une recherche fondamentale en termes thérapeutiques. Les projets de recherche fondamentale posent, par nature, des questions dont les réponses et les conséquences dans le domaine médical ne peuvent en l'état des connaissances scientifiques être anticipées. Dans ces conditions, les équipes de recherche défendant des protocoles en recherche fondamentale peuvent être amenées à formuler une finalité médicale « artificielle » puisqu'il est bien souvent impossible d'anticiper l'applicabilité thérapeutique de résultats qui restent eux-mêmes hypothétiques voire tout simplement inattendus ;
- la démonstration de l'absence de méthodologie alternative au recours aux embryons humains reste également un exercice difficile. Si de nouveaux paradigmes expérimentaux sont en train d'émerger, tels que la constitution de modèles embryonnaires à usage scientifique par l'agréation de cellules souches embryonnaires à des tissus extra-embryonnaires, en vue de mimer différentes phases du développement embryonnaire, comme les « blastoïdes » et les « gastruloïdes », ils sont encore bien loin de reproduire fidèlement les propriétés d'un embryon. A fortiori , il reste indispensable de pouvoir comparer ces modèles expérimentaux aux embryons humains et d'en évaluer les éventuelles limites afin, le cas échéant, de les améliorer.
Afin de sécuriser, sur le plan juridique, les décisions d'autorisation des protocoles de recherche sur l'embryon, votre commission a donc adopté deux amendements tendant à en préciser les prérequis :
- à la finalité médicale pourra être substitué l'objectif d'amélioration de la connaissance de la biologie humaine, plus pertinent en matière de recherche fondamentale (amendement COM-193 de la rapporteure) ;
- toute méthode alternative au recours aux embryons ne sera recevable que s'il est démontré qu'elle présente une pertinence scientifique comparable avec l'embryon humain (amendement COM-194 de la rapporteure).
Par coordination, votre commission a adopté un amendement COM-197 de la rapporteure permettant, dans les exigences auxquelles doivent répondre les protocoles de recherche sur les cellules souches embryonnaires humaines, de tenir compte du fait que ces protocoles doivent poursuivre soit une finalité médicale, soit un objectif d'amélioration de la connaissance de la biologie humaine.
Enfin, votre commission a adopté un amendement COM-226 de la rapporteure tendant à rappeler que, conformément à l'article 18 289 ( * ) de la convention d'Oviedo, l'interdiction de la constitution d'embryons à des fins de recherche s'entend de la conception d'un embryon humain par fusion de gamètes. En effet, par définition, un embryon humain est le résultat de la fusion de gamètes humains. À l'heure actuelle, des modèles embryonnaires à usage scientifique peuvent être créés par l'agréation de cellules souches embryonnaires humaines avec des cellules précurseurs de tissus extra-embryonnaires.
Ces modèles, qui sont déjà utilisés en recherche, ne constituent pas des embryons humains, dès lors qu'ils ne résultent pas de la fusion de deux gamètes. Il convient de lever toute ambiguïté à l'égard de ces modèles embryonnaires à usage scientifique.
• Permettre des avancées dans la compréhension du développement embryonnaire dans le respect des principes éthiques
À l'heure où la France s'apprête pour la première fois à inscrire dans sa législation une durée limite de culture d'embryons in vitro , plusieurs pays, dont le Royaume-Uni, s'interrogent sur la pertinence d'une durée fixée à 14 jours et pourraient prochainement la réviser à la hausse.
Une meilleure compréhension des étapes du développement embryonnaire entre le 14 e et le 21 e jour, qui concernent la période dite de gastrulation, présente un intérêt scientifique majeur. Des études réalisées chez le primate non humain, le singe macaque, publiées en novembre 2019, ont montré des mécanismes de développement propres aux primates différents de ceux observés chez les rongeurs. Plusieurs scientifiques insistent dès lors sur l'importance d'étudier ces mécanismes de développement chez l'embryon humain afin de mieux appréhender le contrôle de la différenciation des cellules souches embryonnaires humaines et pluripotentes induites et les retombées médicales potentiellement associées.
L'argument scientifique soutenant une limite de culture in vitro à 14 jours est désormais contesté, dans la mesure où il apparaît difficile de soutenir qu'une conscience humaine puisse émerger à partir d'une structure embryonnaire aussi primitive que le neurectoderme. La société française de recherche sur les cellules souches, dans une contribution transmise à votre commission, estime ainsi qu'« il ne fait aucun doute que les pays anglo-saxons [...] autoriseront [le fait de repousser la durée limite de culture d'un embryon humain à 21 jours] dans les prochaines années. »
Dans le souci de permettre à la France de prendre une position de leadership dans la compréhension des étapes du développement embryonnaire, votre commission a adopté un amendement COM-195 de la rapporteure tendant à autoriser, à titre dérogatoire, le développement in vitro d'embryons jusqu'au 21 e jour suivant leur constitution dans le cadre de protocoles de recherche spécifiquement dédiés à l'étude des mécanismes de développement embryonnaire au stade de la gastrulation. Cette durée limite apparaît d'autant plus pertinente que le projet de loi prévoit d'ores et déjà la possibilité de constituer de modèles embryonnaires à usage scientifique dits « gastruloïdes », mimant le stade de la gastrulation, par l'agrégation de cellules souches embryonnaires humaines avec des cellules précurseurs de tissus extra-embryonnaires.
• Clarifier la distinction entre les recherches menées sur l'embryon dans le cadre de l'assistance médicale à la procréation et les recherches sur les embryons surnuméraires n'ayant pas vocation à être transférés à des fins de gestation
Votre commission a adopté un amendement COM-225 de la rapporteure visant à clarifier la distinction entre les recherches menées sur l'embryon dans le cadre de l'assistance médicale à la procréation et les recherches sur les embryons surnuméraires n'ayant pas vocation à être transférés à des fins de gestation, en précisant que les embryons qui ne peuvent être transférés sont ceux sur lesquels une recherche a été autorisée et conduite en application de l'article L. 2151-5 du code de la santé publique.
• Supprimer une disposition déjà satisfaite
À l'initiative de l'Assemblée nationale, l'article 14 du projet de loi prévoit désormais que la recherche sur un embryon non destiné à être implanté peut porter sur les causes de l'infertilité. Or, en l'état actuel de la législation, rien n'interdit de mener, sur les embryons, des recherches portant sur les causes de l'infertilité. À ce jour, sur les 23 protocoles de recherche sur l'embryon qui ont été autorisés par l'agence de la biomédecine, neuf concernent l'étude du développement embryonnaire préimplantatoire avec un lien direct sur sa qualité ou la capacité d'implantation de l'embryon. Il s'agit de causes reconnues d'infertilité encore mal documentées et nécessitant une amélioration des connaissances.
Sans ouvrir de nouvelles voies de recherche par rapport au droit existant, cette disposition participe essentiellement d'une volonté d'affichage. Les recherches en matière d'infertilité n'ont d'ailleurs pas vocation à se limiter à l'identification de leurs causes, mais à en améliorer le traitement, ce qui supposerait de compléter cette disposition, si elle était maintenue, pour préciser l'ensemble du champ possible de la recherche dans ce domaine. Compte tenu de l'absence de réelle valeur ajoutée de cette précision par rapport au droit en vigueur, votre commission a donc adopté un amendement COM-196 de la rapporteure tendant à la supprimer.
• Renforcer la vigilance sur des recherches soulevant des questions éthiques majeures
À l'heure actuelle, les seules recherches possibles impliquant la constitution d'embryons chimériques consistent en l'adjonction de cellules souches pluripotentes induites (iPS) d'origine humaine à un embryon animal, les iPS n'étant pas aujourd'hui encadrées par le code de la santé publique. En limitant l'interdiction de création d'embryons chimériques à la seule adjonction de cellules animales à un embryon humain, l'article 17 du projet de loi ouvre la possibilité de constitution d'embryons chimériques résultant de l'insertion de cellules souches embryonnaires humaines à un embryon animal. Cette nouvelle possibilité soulève d'importantes questions éthiques quant aux limites à poser face au risque de franchissement de la barrière des espèces.
Par conséquent, votre commission a adopté un amendement visant à écarter la possibilité de constitution d'embryons chimériques résultant de l'insertion de cellules souches embryonnaires humaines dans un embryon animal, procédure qui franchit une « ligne rouge » difficilement acceptable au regard du risque de franchissement de la barrière des espèces (amendement COM-201 de la rapporteure).
• Enfin, votre commission a adopté un amendement COM-267 de la rapporteure procédant à diverses coordinations au sein du code pénal et du code de la santé publique.
La commission a adopté cet article ainsi modifié.
Article 15
Régulation, en recherche fondamentale,
de
certaines utilisations des cellules souches pluripotentes induites
Cet article vise à encadrer certaines finalités, sensibles sur le plan éthique, des recherches menées sur les cellules souches pluripotentes induites.
La commission spéciale a souhaité encadrer les expérimentations impliquant la création d'embryons chimériques par l'adjonction de cellules souches pluripotentes induites humaines à un embryon animal, en interdisant la possibilité que le transfert de ces embryons chez la femelle donne lieu à parturition et en posant le principe d'un seuil à ne pas dépasser en termes de contribution des cellules d'origine humaine au développement de l'embryon chimérique.
I - Le dispositif proposé
1. Les cellules pluripotentes induites : un outil à fortes potentialités médicales, non dénué d'enjeux éthiques
En 2007, le professeur japonais Shinya Yamanaka a mis au point la technique permettant de transformer des cellules adultes spécialisées en cellules pluripotentes induites, plus communément désignées sous le sigle « iPS » issu de l'anglais « induced Pluripotent Stems Cells ». Cette découverte, pleine de promesses pour la médecine notamment génique et régénérative, lui a valu l'attribution du prix Nobel de médecine en 2012. Les iPS sont obtenues en reprogrammant une cellule somatique différenciée 290 ( * ) vers un état de pluripotence. Ces lignées cellulaires ont en commun, avec les cellules souches embryonnaires humaines, d'être pluripotentes : elles sont capables de se multiplier indéfiniment et de se différencier en tout type de cellule qui compose l'organisme.
Toutefois, comme le rappelle le professeur Pierre Savatier dans ses réponses au questionnaire de votre commission, si les cellules souches embryonnaires humaines et les cellules souches pluripotentes induites présentent, par définition, les mêmes propriétés, c'est-à-dire l'autorenouvèlement et la pluripotence, elles se distinguent par les éléments structurels et fonctionnels suivants :
- les cellules iPS gardent parfois en mémoire des traces épigénétiques de leur origine somatique. En effet, pour un même individu, l'épigénome peut varier d'une cellule à l'autre. La régulation de l'expression des gènes des iPS étant altérée, leurs potentialités de différenciation peuvent s'en trouver modifiées ;
- des différences dans la qualité de l'ADN mitochondrial des iPS ont été récemment décrites. Les cellules iPS présenteraient des altérations de ce génome qui auraient pour conséquences l'apparition d'une réponse immunitaire dirigée contre les cellules iPS et leurs dérivés différenciés ;
- les cellules iPS peuvent contenir des variations génomiques provenant soit de la cellule somatique d'origine, soit induite par la reprogrammation elle-même et le stress cellulaire qu'elle entraîne, ce qui peut résulter dans des altérations basales au sein des lignées d'iPS dérivées. Pour les applications cliniques, il est nécessaire de réaliser un contrôle de la qualité des cellules iPS en séquençant leur génome pour s'assurer qu'il ne contient pas de mutations délétères.
Selon le professeur Pierre Savatier, ces différences structurelles entre les cellules souches embryonnaires humaines et les cellules pluripotentes induites font que les premières constituent l'original et les secondes n'en sont que la copie imparfaite.
La pluripotence des iPS ouvre des perspectives de recherche susceptibles de soulever des interrogations éthiques, dans la mesure où ces cellules résultent d'un processus de reprogrammation (donc de modification) génétique et dès lors qu'il pourrait, dans le futur, être envisagé de les différencier en gamètes, même si cette possibilité n'est pas encore d'actualité.
2. Le dispositif proposé par le projet de loi : un régime déclaratif ciblé sur les recherches « sensibles » sur le plan éthique
Le I de l'article 15 du projet de loi modifie l'intitulé du titre V du livre I er de la deuxième partie du code de la santé publique, qui devient : « Recherche sur l'embryon humain, les cellules souches embryonnaires humaines et les cellules souches pluripotentes induites ». Ce nouvel intitulé permet :
- de tenir compte de l'introduction dans le code de la santé publique de dispositions encadrant certaines recherches sur les cellules pluripotentes induites ;
- de préciser l'origine humaine des embryons et des cellules souches embryonnaires faisant l'objet des recherches régies par les dispositions du titre V précité. La place de cette précision dans l'intitulé dispense de la décliner dans l'ensemble des articles de ce titre et permet de lever toute ambiguïté sur leur non-applicabilité aux recherches sur les embryons et cellules souches embryonnaires d'origine animale.
Le II de l'article 15 du projet de loi rétablit, au sein du code de la santé publique, un article L. 2151-7 définissant, en son I, les cellules souches pluripotentes induites comme les « cellules qui ne proviennent pas d'un embryon et sont capables de se multiplier indéfiniment ainsi que de se différencier en tous les types de cellules qui composent l'organisme. » Le II du même article L. 2151-7 prévoit l'obligation pour deux types de recherche considérés comme sensibles sur le plan éthique de faire l'objet d'une déclaration préalable auprès de l'agence de la biomédecine. Les protocoles de recherche visés devant être déclarés ont pour objet :
- la différenciation de cellules souches pluripotentes induites en gamètes, afin de permettre par exemple l'étude de la gamétogenèse et de la fécondance des gamètes ;
- l'agrégation de cellules souches pluripotentes induites avec des cellules précurseurs de tissus extra-embryonnaires, cette technique étant susceptible de donner lieu à la constitution de modèles embryonnaires à usage scientifique mimant différents stades du développement embryonnaire comme des blastoïdes ou des gastruloïdes.
L'utilisation des iPS restera soumise, en outre, au régime commun applicable à l'utilisation des éléments et produits du corps humain à des fins scientifiques, notamment à l'article 1243-3 du code de la santé publique relatif à la déclaration auprès du ministre chargé de la recherche des activités de conservation et de préparation à des fins scientifiques de tissus et de cellules issus du corps humain et à la constitution et à l'utilisation de collections d'échantillons biologiques humains. Elle relèvera également de la recherche impliquant la personne humaine en application de l'article L. 1121-1 du code de la santé publique dans le cas où elle nécessiterait le prélèvement de tissus ou cellules à partir de patients ou volontaires sains.
Le III du nouvel article L. 2151-7 du code de la santé publique prévoit que le directeur général de l'agence de la biomédecine pourra s'opposer, dans un délai fixé par voie réglementaire, à la réalisation du protocole de recherche déclaré qui ne respecterait pas les principes éthiques fondamentaux de notre législation. Cette décision d'opposition devra être précédée d'un avis public du conseil d'orientation de l'agence. Le directeur général de l'agence conserve également la possibilité de suspendre ou d'interdire à tout moment, après avis public du conseil d'orientation, les recherches qui ne se conformeraient plus à ces exigences (IV du nouvel article L. 2151-7 du code de la santé publique).
Afin d'assurer le respect des nouveaux régimes de déclaration introduits en matière de recherche sur les cellules souches embryonnaires humaines et sur les cellules souches pluripotentes induites, le III de l'article 15 du projet de loi procède aux coordinations nécessaires au sein des articles du code de la santé publique et du code pénal prévoyant les sanctions applicables en la matière.
Le chapitre III du titre VI « Dispositions pénales » du livre I er de la deuxième partie du code de la santé publique est ainsi intitulé « Recherche sur l'embryon humain, les cellules souches embryonnaires humaines et les cellules souches pluripotentes induites ». Les manquements constatés dans la réalisation des recherches sur les cellules souches, embryonnaires humaines ou pluripotentes induites, seront ainsi punis de deux ans d'emprisonnement et de 30 000 euros d'amende.
II - Les modifications adoptées par l'Assemblée nationale
Outre plusieurs amendements rédactionnels, la commission des affaires sociales de l'Assemblée nationale a adopté, à l'initiative de son rapporteur Philippe Berta un amendement tendant à compléter la liste des recherches sur les iPS susceptibles de faire l'objet d'une décision d'opposition de l'agence de la biomédecine sur le fondement du respect des principes éthiques fondamentaux, après avis public de son conseil d'orientation : est ainsi ajoutée l'insertion d'iPS dans un embryon animal en vue de son transfert chez la femelle, dans le cadre de la constitution d'un embryon chimérique.
III - La position de la commission : encadrer la création d'embryons chimériques aux iPS
Votre commission approuve l'introduction d'un régime juridique spécifique encadrant les recherches sur les cellules souches pluripotentes induites qui repose sur un équilibre entre, d'une part, la reconnaissance d'une liberté d'initiative scientifique dans ce domaine, sous réserve du respect des dispositions applicables en matière de conservation de tissus et cellules issus du corps humain et de recherches impliquant la personne humaine, et, d'autre part, le souci d'une vigilance renforcée lorsque la recherche, en envisageant l'expérimentation de procédés de recréation du vivant in vitro , soulève des questions éthiques sensibles, notamment en termes de manipulation du vivant.
Néanmoins les expérimentations impliquant la création d'embryons chimériques par l'adjonction de cellules souches pluripotentes humaines à un embryon animal, bien que déjà possibles aujourd'hui en l'absence de cadre législatif ou réglementaire, ne peuvent manquer de susciter des interrogations quant au risque de franchissement de la barrière des espèces.
Dans son avis 129, le CCNE rappelle que « certains développements, dont l'obtention de gamètes à partir d'une cellule adulte reprogrammée, ou encore le développement in vivo de cellules différenciées humaines de type germinal ou neuronal dans des chimères chez le gros animal, représentent cependant un risque de transgression. » Le CCNE insiste alors sur la nécessité d'un encadrement des recherches qui consisteraient à « d'intégrer des cellules pluripotentes humaines dans des embryons d'un plus gros animal, dans l'idée de produire des organes humains chez l'animal - source potentielle de greffons. » Dans ces conditions, il estime que « si ces approches ne relèvent pas directement de la recherche sur l'embryon humain, un encadrement semble néanmoins nécessaire, en particulier si les embryons chimériques sont transférés chez des femelles et donnent naissance à des animaux chimères avec le risque, chez le gros animal, que les cellules humaines se développent et induisent certaines caractéristiques humaines (morphologiques, neurologiques). »
Il convient donc de s'assurer que les cellules d'origine humaine introduites dans un embryon animal ne participent que de façon minoritaire au développement de cet embryon, qui doit rester un embryon animal. Par conséquent, votre commission a adopté un amendement COM-199 de la rapporteure visant à renforcer l'encadrement de la création d'embryons chimériques recourant aux iPS en posant deux « verrous » :
- ces embryons ne peuvent donner lieu à parturition (mise-bas), si bien qu'en cas de transfert chez la femelle, la gestation sera obligatoirement interrompue dans un délai approuvé par l'agence de la biomédecine au regard des délais gestationnels propres à l'animal concerné ;
- la contribution des cellules d'origine humaine au développement de l'embryon chimérique (taux de chimérisme) ne saurait dépasser un seuil approuvé par l'agence de la biomédecine. En tout état de cause, aucun embryon chimérique ne peut présenter une proportion de cellules d'origine humaine supérieure à 50 %.
La commission a adopté cet article ainsi modifié.
Article 16
Limite de conservation des embryons proposés à
la recherche
Cet article fixe une durée de conservation de cinq ans pour les embryons cédés à la recherche qui n'auraient pas été inclus dans un protocole de recherche à l'expiration de ce délai.
La commission spéciale a assoupli le principe de confirmation dans un délai de trois mois du premier consentement au devenir des embryons ne faisant plus l'objet d'un projet parental. Elle a, en outre, porté de cinq à dix ans le délai de conservation des embryons cédés à la recherche qui n'auraient toujours pas été inclus dans un protocole de recherche.
I - Le dispositif proposé
• À l'heure actuelle, seule la conservation des embryons congelés dans le cadre d'un projet parental et la conservation des embryons cédés pour l'accueil par un autre couple que celui dont ils sont issus sont soumises à un délai à l'expiration duquel il peut être procédé à la destruction des embryons. L'article L. 2141-4 du code de la santé publique prévoit en effet que :
- les embryons initialement congelés dans le cadre d'un projet parental mais pour lesquels l'un des deux membres du couple, consultés à plusieurs reprises, ne répond pas à la question de savoir s'il maintient ou non son projet parental sont détruits si leur durée de conservation est au moins égale à cinq ans. Il en va de même en cas de désaccord au sein du couple sur le maintien du projet parental ou le devenir des embryons ;
- les embryons destinés à être accueillis par un autre couple que celui dont ils sont issus sont également appelés à être détruits s'ils n'ont pas été accueillis dans un délai de cinq ans à compter du jour où le consentement écrit à l'accueil a été donné.
Aucune disposition ne prévoit, en revanche, qu'il soit mis fin à la conservation des embryons cédés à la recherche qui n'auraient pas été inclus dans un protocole de recherche au bout d'un certain temps.
Les embryons sont conservés par les centres d'assistance médicale à la procréation où ils ont été congelés.
• Selon les données du rapport d'activité médicale et scientifique de l'agence de la biomédecine, on dénombrait, au 31 décembre 2017, 246 263 embryons en cours de conservation pour 82 485 couples. Parmi ces embryons, 32 878 embryons (13 %) ne faisaient plus l'objet d'un projet parental dont :
- 21 727 291 ( * ) étaient cédés à la recherche ;
- 11 151 292 ( * ) pouvaient faire l'objet d'un accueil par un autre couple.
Selon l'étude d'impact annexée au projet de loi, ce sont 2 855 embryons qui ont été proposés à la recherche en 2016 et qui sont donc venus alimenter le stock d'embryons conservés et proposés à la recherche, un nombre relativement constant au cours de la période récente. Étant précisé que le stock d'embryons cédés à la recherche reste lui aussi plus ou moins constant au cours des dernières années, l'étude d'impact en déduit que le nombre d'embryons donnés chaque année à la recherche par les couples qui n'ont plus de projet parental est à peu près équivalent à celui des embryons effectivement inclus dans un protocole de recherche la même année.
Le nombre d'embryons inclus dans un protocole de recherche compris entre 2 500 et 3 000 chaque année représenterait entre 12,5 % et 15 %. Dans son rapport d'évaluation de la loi relative à la bioéthique de 2011, l'office parlementaire d'évaluation des choix scientifiques et éthiques (Opecst) estime, néanmoins, à moins de 10 % des 20 000 embryons conservés et proposés à la recherche la proportion des embryons effectivement utilisés dans des projets de recherche.
• Le I de l'article 16 du projet de loi procède à une réécriture de l'article L. 2141-4 du code de la santé publique.
Est maintenu le principe selon lequel les membres du couple ou la femme non mariée sont consultés chaque année sur leur souhait de maintenir leur projet parental : en cas de confirmation par écrit de ce maintien, la conservation de leurs embryons sera poursuivie.
En cas d'abandon du projet parental, il est toujours prévu que les deux membres du couple ou la femme mariée ou, en cas de décès de l'un des membres du couple, le membre survivant du couple consentent par écrit à l'une des trois possibilités suivantes pour le devenir de leurs embryons :
- l'accueil de ces embryons par un autre couple ;
- l'utilisation de ces embryons dans le cadre d'un protocole de recherche, dans les conditions fixées par l'article L. 2151-5 du code de la santé publique relatif aux autorisations de recherches sur l'embryon, ou l'utilisation, dans les conditions applicables en matière de recherches biomédicales, de cellules dérivées à partir de ces embryons dans une préparation de thérapie cellulaire ou un médicament de thérapie innovante à des fins exclusivement thérapeutiques ;
- la destruction de ces embryons.
Comme c'est déjà le cas aujourd'hui, le consentement à l'une de ces trois possibilités doit faire l'objet d'une confirmation écrite après un délai de réflexion de trois mois à compter du premier consentement. Il est en outre précisé que, lorsque les embryons sont cédés à la recherche, le consentement du couple est révocable tant qu'ils n'ont pas été inclus dans un protocole de recherche.
Les III, IV et V de l'article L. 2141-4 du code de la santé publique envisagent les différents cas de figure pour lesquels il doit être mis fin à la conservation des embryons ne faisant plus l'objet d'un projet parental, en retenant une durée limite de conservation de cinq ans. Les embryons seront ainsi détruits :
- en cas de silence du couple ou de la femme non mariée, consultés annuellement à au moins deux reprises, sur le maintien de leur projet parental, si les embryons ont été conservés pour une durée égale ou supérieure à cinq ans. Il en va de même en cas de désaccord au sein du couple sur le maintien du projet parental ou en l'absence de confirmation du consentement dans un délai de trois mois ;
- lorsque les embryons proposés pour un accueil par un autre couple n'ont pas été accueillis dans un délai de cinq ans à compter de la confirmation du consentement ;
- lorsque les embryons cédés à la recherche n'ont pas été inclus dans un protocole de recherche dans un délai de cinq ans à compter de la confirmation du consentement.
Le VI de l'article L. 2141-4 du code de la santé publique prévoit, en outre, une destruction des embryons en cas de décès des deux membres du couple en l'absence de consentement préalable à un accueil par un autre couple ou à une utilisation dans le cadre d'un protocole de recherche.
Le II de l'article 16 du projet de loi tient compte de la situation des embryons actuellement donnés à la recherche et conservés depuis plus de cinq ans à la date de la promulgation de la loi : ces embryons seront détruits, à l'exception de ceux qui présentent un intérêt particulier pour la recherche en raison de leur conservation à un stade précoce de leur développement.
Aujourd'hui congelés et vitrifiés cinq jours après la fécondation, ce stade de développement étant jugé plus compatible avec une implantation dans le cadre de l'assistance médicale à la procréation, les embryons pouvaient être congelés auparavant à des stades bien plus précoces du développement embryonnaire, un à trois jours après la fécondation. Aussi existe-t-il, dans le stock des embryons conservés depuis une durée supérieure à cinq ans, des zygotes ne présentant qu'une seule cellule ou des embryons formés de deux cellules au premier jour de la fécondation, mais aussi des embryons formés de quatre cellules, à J+2, ou de huit cellules, à J+3.
Ces embryons présentent un intérêt particulier pour la recherche dès lors qu'ils permettent d'étudier les phases précoces du développement embryonnaire. Le projet de loi prévoit donc que les établissements autorisés pour les pratiques cliniques et biologiques de l'assistance médicale à la procréation déclareront les embryons susceptibles de présenter un intérêt particulier pour la recherche à l'agence de la biomédecine qui se prononcera sur la poursuite de leur conservation, dans des conditions fixées par décret en Conseil d'État.
II - Les modifications adoptées par l'Assemblée nationale
Outre des amendements rédactionnels adoptés par la commission, l'Assemblée nationale a adopté un amendement de notre collègue député Pierre Dharréville et d'autres membres du groupe « Gauche démocrate et républicaine » ouvrant la possibilité pour le couple, à l'occasion de la consultation annuelle sur le point de savoir s'il maintient son projet parental, de formuler des directives anticipées sur le devenir des embryons en cas de décès de l'un des membres du couple.
De cette façon, les deux membres du couple peuvent manifester un consentement conjoint à ce que, en cas de décès de l'un d'eux, les embryons soit puissent être accueillis par un autre couple, soit fassent l'objet d'une recherche. Le membre survivant du couple sera interrogé sur la confirmation de ce consentement à l'expiration d'un délai d'un an à compter du décès, sauf initiative anticipée de sa part. Il conserve ainsi la possibilité de révoquer son consentement, auquel cas il sera mis fin à la conservation des embryons.
III - La position de la commission
• La pertinence d'une confirmation écrite dans un délai de trois mois à compter de la date du premier consentement sur le devenir d'embryons ne faisant plus l'objet d'un projet parental est aujourd'hui fortement remise en cause, d'autant que très peu de couples reviennent sur leur premier consentement, comme l'ont confirmé devant votre commission la fédération des Cécos et l'association des biologistes des laboratoires d'étude de la fécondation et de la conservation de l'oeuf (Blefco).
Au-delà du caractère lourd et chronophage de cette procédure pour les centres d'AMP et les Cécos 293 ( * ) , il apparaît délicat d'interroger à nouveau sur le devenir de leurs embryons un couple qui a déjà entamé le « deuil » de son projet parental. Le silence gardé par les membres du couple sur cette confirmation est bien souvent moins le signe d'une remise en cause de leur consentement initial que le souhait de ne plus avoir à réexaminer la question du devenir de leurs embryons.
Or l'absence de cette confirmation, après deux relances, est supposée conduire à la destruction des embryons, alors même que le couple a pu consentir initialement à ce qu'ils soient accueillis par un autre couple ou donnés à la recherche.
Par conséquent, votre commission a adopté un amendement tendant à supprimer le principe d'une confirmation par le couple du consentement sur le devenir des embryons ne faisant plus l'objet d'un projet parental (amendement COM-202 de la rapporteure).
• Les embryons cédés à la recherche par les couples pris en charge dans le cadre d'une assistance médicale à la procréation sont aujourd'hui dispersés entre les 103 centres de fécondation in vitro autorisés à conserver ces embryons en application de l'article L. 2151-7 294 ( * ) du code de la santé publique. Tant l'institut national de la santé et de la recherche médicale (Inserm) que les sociétés savantes et les représentants des organismes assurant la conservation de ces embryons (centres d'AMP, Cécos) déplorent l'absence d'organisation structurée de cette activité de conservation, de même que l'absence de visibilité et de traçabilité des stocks d'embryons conservés et donnés à la recherche.
Au vu du nombre d'embryons conservés, que ceux-ci continuent de s'inscrire dans un projet parental ou soient destinés à la recherche ou à un accueil par un autre couple, les capacités de stockage des centres concernés tendent à diminuer. L'absence de cartographie des embryons conservés, précisant les caractéristiques des cohortes embryonnaires, est en outre préjudiciable à la recherche, les scientifiques rencontrant des difficultés à identifier de façon optimale les embryons les plus pertinents en fonction de l'objet de la recherche.
Par une décision du 25 juin 2015, l'agence de la biomédecine a autorisé le centre de ressources biologiques (CRB) multi-sites « Germethèque » à exercer des activités de conservation des embryons. Fondé en 2007 et constitué par onze centres hospitaliers universitaires (CHU), ce CRB est coordonné par le CHU de Toulouse et « a pour vocation d'organiser la collecte, le stockage, la gestion et l'exploitation d'échantillons biologiques humains (gamètes, tissu germinal testiculaire et ovarien, ADN, liquide séminal) afin de permettre l'étude des causes, mécanismes et conséquences des altérations de la reproduction et du développement humain ». Véritable interface entre les laboratoires ayant accompagné la conception des embryons et les équipes de recherche, le CRB met à la disposition des centres associés dans le cadre de ce partenariat plusieurs outils permettant de valoriser les collections d'embryons : logiciel et système d'information, processus de certification ISO, procédures de qualité...
Bien qu'il permette de valoriser, sur le plan scientifique, les collections d'échantillons biologiques conservés par les centres d'AMP et les Cécos, ce CRB pâtit de l'insuffisance de ses moyens financiers, le CHU coordonnateur de Toulouse rencontrant des difficultés pour obtenir des financements publics, notamment dans le cadre du programme d'investissements d'avenir (PIA).
L'article 42 de la loi relative à la bioéthique de 2011 avait prévu la remise par le Gouvernement d'un rapport au Parlement, avant le 1 er juillet 2012, sur les conditions de mise en place de centres de ressources biologiques sous la forme d'un système centralisé de collecte, de stockage et de distribution des embryons surnuméraires dont il a été fait don à la science. Ce rapport n'a jamais été établi et aucun effort n'a été déployé par les pouvoirs publics en faveur de la mise en place d'une cartographie précise de l'ensemble des stocks d'embryons cédés à la recherche, assortie de données relatives aux caractéristiques de ces embryons (stade de développement embryonnaire au moment de la congélation, anomalies géniques ou chromosomiques identifiées par DPI, date de la congélation...).
• Selon l'étude d'impact annexée au projet de loi, « il n'existe aucun consensus sur la période de conservation des embryons mais la plupart des pays ont choisi cinq ans. » La durée de conservation varie en effet d'un pays à l'autre, pouvant ainsi aller jusqu'à dix ans en Israël et en Australie, voire jusqu'à quinze ans au Royaume-Uni (moyennant dans ce pays un renouvellement de l'autorisation de conservation tous les cinq ans).
Une durée de conservation de cinq ans ne permet pas néanmoins de tenir compte des contraintes qui caractérisent aujourd'hui le montage et la mise en oeuvre d'un protocole de recherche, opérations qui peuvent nécessiter bien plus que cinq ans, en particulier lorsqu'une autorisation de recherche fait l'objet d'une contestation en justice. Le stock d'embryons donnés à la recherche accumulé jusqu'ici résulte d'ailleurs, selon les personnes auditionnées par votre commission, du lent décollage de la recherche sur l'embryon en France.
Deux types de considérations plaident alors pour un délai de conservation des embryons destinés à la recherche supérieur à cinq ans :
- compte tenu du tempo de la recherche sur l'embryon en France, qui reste lent comparativement à d'autres pays, un délai de conservation de cinq ans pourrait conduire à terme à une diminution substantielle du stock d'embryons disponibles. Une partie significative du stock existant pourrait ainsi faire l'objet d'une destruction (tous les embryons conservés depuis plus de cinq ans, à l'exception de ceux présentant un intérêt particulier pour la recherche). Au bout d'un certain temps, ne seront alors plus disponibles pour la recherche que les embryons conservés depuis moins de cinq ans, le stock n'étant renouvelé que par l'introduction de nouveaux embryons donnés à la recherche, étant rappelé que le nombre d'embryons nouvellement cédés à la recherche chaque année correspond peu ou prou au nombre d'embryons nouvellement inclus dans un protocole de recherche. En outre, il convient de garder à l'esprit que rien ne garantit que les embryons attribués par l'agence de la biomédecine à une équipe de recherche résisteront à la décongélation, l'échec de la décongélation pouvant alors nécessiter de recourir à d'autres embryons ;
- comme l'indique la fédération des Cécos dans ses réponses au questionnaire de votre commission, certaines pathologies à révélation tardive ou bien rares nécessitent que ce délai de conservation soit rallongé afin de laisser aux chercheurs la possibilité de mettre en place des recherches sur des embryons présentant des anomalies géniques ou chromosomiques à l'origine de ces pathologies. De plus, la disponibilité des embryons précoces diminue et leur utilisation dans le cadre de différents protocoles de recherche augmente afin de caractériser les différentes étapes du développement embryonnaire précoce.
Considérant que le délai de cinq ans n'est pas adapté et apparaît trop court au regard des exigences de la recherche sur l'embryon, la fédération des Cécos n'estime pas nécessaire d'imposer une limite à la conservation des embryons destinés à la recherche.
Soucieuse de respecter le tempo de la recherche sur l'embryon, votre commission a adopté un amendement visant à porter le délai de conservation des embryons destinés à la recherche à dix ans, qui reste intermédiaire par à d'autres pays prévoyant une durée de conservation plus longue, comme le Royaume-Uni (amendement COM-203 de la rapporteure). La révision de la législation relative à la bioéthique devrait permettre, avec un recul d'au moins cinq ans, d'évaluer les premiers effets d'un délai de dix ans sur le stock actuel d'embryons cédés à la recherche et de déterminer si celui-ci peut être ramené à cinq ans, sans préjudice pour la recherche sur l'embryon.
• Votre commission a également adopté un amendement de précision rédactionnelle, permettant d'inclure, dans les possibilités de devenir des embryons conservés examinées par le couple à l'occasion de la consultation annuelle pour la détermination de directives anticipées en cas de décès de l'un des membres du couple, la possibilité de mettre fin à la conservation des embryons (amendement COM-84 de Michel Amiel).
La commission a adopté cet article ainsi modifié.
CHAPITRE II
Favoriser une recherche responsable
en lien avec la
médecine génomique
Article 17
Utilisation des outils de modification ciblée
du
génome en recherche fondamentale
Cet article supprime l'interdiction de création d'embryons transgéniques, limite l'interdiction de création d'embryons chimériques aux seuls embryons résultant de l'adjonction à un embryon humain de cellules provenant d'autres espèces et étend les possibilités de recours aux techniques d'édition génomique dans la recherche en santé, sous réserve du respect du principe d'interdiction de transformation des caractéristiques génétiques transmissibles à la descendance.
La commission spéciale a clarifié l'interdiction relative à la constitution d'embryons chimériques, en maintenant l'impossibilité de constituer de tels embryons non seulement par l'insertion dans un embryon humain de cellules provenant d'autres espèces mais aussi par l'insertion dans un embryon animal de cellules souches embryonnaires humaines. Elle a, en outre, rappelé que la constitution d'embryons chimériques par l'insertion de cellules souches pluripotentes induites humaines était soumise aux exigences éthiques posées par l'article 15 du projet de loi dans sa version adoptée par la commission spéciale.
I - Le dispositif proposé : La suppression de l'interdiction de la création d'embryons transgéniques et la limitation de l'interdiction de création d'embryons chimériques
L'article 16-4 du code civil pose le principe selon lequel il est interdit d'apporter au génome humain, tant chez une personne déjà née au niveau de ses cellules germinales que chez l'embryon, des modifications qui pourraient se transmettre à la descendance. Cette interdiction, qui résulte de l'article 13 de la convention d'Oviedo, est néanmoins assortie d'exceptions : des interventions sur le génome humain peuvent en effet être autorisées dans le cadre de recherches à des fins de prévention ou de traitement des maladies génétiques.
Le II de l'article 17 du projet de loi étend le champ des recherches pour lesquelles une modification du génome humain peut être envisagée : il pourra s'agir également de recherches menées à des fins diagnostiques, et ces recherches ne seront pas tenues de se limiter aux seules maladies génétiques mais pourront concerner potentiellement toutes les maladies.
La possibilité d'édition génomique d'embryons restera cantonnée aux recherches conduites sur des embryons autorisées en application de l'article L. 2151-5 du code de la santé publique. En effet, l'article L. 2141-3-1 du même code, créé par l'article 14 du projet de loi, prévoit, dans sa version issue des travaux de l'Assemblée nationale en première lecture, que les recherches menées sur des embryons dans le cadre de l'assistance médicale à la procréation, et donc destinés à être implantés, ne peuvent avoir « pour objet de modifier le génome des gamètes ou de l'embryon ».
En parallèle, le I de l'article 17 du projet de loi supprime les dispositions inscrites à l'article L. 2151-2 du code de la santé publique aux termes desquelles la création d'embryons transgéniques est interdite. La levée de cette interdiction doit permettre d'ouvrir la voie à des expérimentations sur des embryons surnuméraires recourant aux techniques d'édition génomique telles que l'outil « CRISPR-Cas9 », afin d'en évaluer l'innocuité, notamment en identifiant les potentiels effets non-ciblés d'une modification génomique.
À l'heure actuelle, la portée de l'interdiction de création d'embryons transgéniques se limite au seul ajout dans le génome d'un embryon d'une ou plusieurs séquences d'ADN exogène, c'est-à-dire l'introduction de fragments d'ADN étrangers. Comme le souligne le Conseil d'État dans son étude de juin 2018 sur la révision de la loi de bioéthique 295 ( * ) , cette interdiction ne fait pas aujourd'hui obstacle aux recherches ayant pour seule finalité de supprimer ou d'inactiver un fragment du génome de l'embryon concerné, seul son remplacement par un fragment d'ADN étranger étant prohibé. L'étude recommandait ainsi « soit d'interdire la technique dans son ensemble, soit de l'autoriser, mais de ne pas maintenir cette situation asymétrique qui ne repose sur aucune logique cohérente. »
• La limitation du champ de l'interdiction de création d'embryons chimériques
L'article L. 2151-2 du code de la santé publique interdit également, à l'heure actuelle, la création d'embryons chimériques. Toutefois, la place de cette interdiction au sein d'un chapitre consacré aux recherches sur l'embryon humain et les cellules souches embryonnaires humaines est à l'origine d'un flou juridique concernant la création d'embryons chimériques par l'adjonction de cellules d'origine humaine à un embryon animal. Dans son étude précitée de juin 2018, le Conseil d'État estime en effet que la localisation de l'interdit « dans le code de la santé publique au sein de la partie consacrée à la « Santé sexuelle et reproductive, droits de la femme et protection de la santé de l'enfant, de l'adolescent et du jeune adulte » ainsi que les travaux préparatoires [parlementaires] laissent à penser qu'il n'a pas vocation à couvrir la recherche réalisée sur l'embryon animal. »
Suivant la même logique, le Gouvernement soutient également que l'interdiction actuelle ne porte que sur l'utilisation d'embryons humains ayant pour objectif la formation de chimère. L'article L. 2151-2 du code de la santé publique ne serait pas applicable à l'introduction de cellules d'origine humaine dans un embryon animal dès lors qu'il s'agirait d'une expérimentation conduite sur un embryon animal. Par conséquent, dans un souci de clarification, le I de l'article 17 du projet de loi prévoit que seule « la modification d'un embryon humain par adjonction de cellules provenant d'autres espèces est interdite. »
L'agence de la biomédecine a indiqué à votre commission qu'aucun protocole de recherche impliquant la création d'embryons chimériques résultant de l'adjonction de cellules embryonnaires humaines à un embryon animal n'a été autorisé à ce jour en France. Les seules recherches menées en France sur des embryons chimériques ont porté sur :
- l'adjonction, au sein d'une même espèce animale, notamment chez le lapin, de cellules d'un individu animal à un embryon d'un autre individu animal de la même espèce, avec transfert de l'embryon chez la femelle et interruption de la gestation avant la naissance, afin d'étudier la mise en place et les différents états de la pluripotence chez l'animal et de comparer les résultats avec les données disponibles chez l'homme ;
- l'adjonction de cellules souches pluripotentes induites humaines à un embryon animal. La création de ce type d'embryons chimériques est en effet juridiquement possible, dès lors que les cellules souches pluripotentes induites humaines ne rentrent pas, aujourd'hui, dans le périmètre d'action de l'agence de la biomédecine. De tels embryons ont pu être conçus in vitro à partir d'embryons de lapin ou de singes macaques, mais la durée de culture de ces embryons a été limitée à trois jours et ces derniers n'ont jamais été transférés chez la femelle.
Plus généralement, l'agence de la biomédecine explique que l'utilisation d'embryons chimériques présente deux intérêts majeurs pour la recherche :
- d'une part, elle permet l'étude du développement embryonnaire précoce ;
- d'autre part, l'utilisation d'embryons chimériques rend possible, sur le plan théorique, la formation d'un organe humain chez l'animal. Une telle approche est présentée comme constituant l'une des solutions possibles pour combler la pénurie d'organes à des fins de transplantation. Actuellement, les recherches internationales concernent essentiellement le pancréas et se limitent à la souris et le rat. Chez le mouton et le porc, les tentatives de création de chimère par adjonction de cellules humaines ont montré un très faible taux de chimérisme (proportion de cellules humaines capables de se développer par rapport au nombre de cellules animales) : une cellule humaine sur 10 000 chez le mouton, une sur 100 000 chez le porc. Dans les deux cas, la gestation a été interrompue au 28 e jour 296 ( * ) .
Ce faible taux de chimérisme ne permet pas d'envisager un développement satisfaisant à court terme. À ce jour aucune équipe de recherche française n'a d'ailleurs mené de travaux dans ce domaine, et aucune équipe de recherche dans le monde n'a donné naissance à un animal chimérique contenant des cellules humaines, bien qu'une dérogation ait été accordée cet été à une équipe japonaise.
Législation internationale sur les embryons chimériques
|
Pays |
Loi, recommandations
|
Définitions embryons |
Embryon chimérique :
|
|
Australie |
Le « Prohibition of Human Cloning for Reproduction Act » interdit, expressément, la création d'embryons chimériques par introduction de cellules animales dans un embryon humain mais pas par introduction de cellules humaines dans un embryon animal 1 . |
Embryon chimérique (a) un embryon humain dans lequel a été introduit une cellule ou tout autre composant cellulaire provenant d'un animal (b) toute chose déclarée par la loi comme étant un embryon chimérique. Embryon hybride : (a) un embryon créé par fécondation d'un ovocyte humain par du sperme animal (b) un embryon créé par fécondation d'un ovocyte animal avec du sperme humain (c) un ovocyte humain dans lequel a été introduit le noyau d'une cellule animale (d) un ovocyte animal dans lequel a été introduit le noyau d'une cellule humaine (e) toute chose déclarée par la loi comme étant un embryon hybride. |
Embryons chimériques : Restrictif : embryon humain + cellules animales Permissif : embryon animal + cellules humaines. Embryons hybrides : permissif avec autorisation si culture inférieure à 14 jours Restrictif si culture au-delà de 14 jours. |
|
Canada |
La loi sur la Procréation Assistée (L.C. 2004, ch. 2) interdit la création de chimères, définies comme des embryons humains dans lesquels ont été introduites des cellules d'autres animaux ou humains. La loi n'interdit pas la création d'embryons animaux rendus chimériques par injection de cellules humaines. Cependant la principale agence de financement au Canada interdit expressément la création des deux types d'embryons chimériques2. |
Embryon chimérique : a) embryon humain dans lequel a été introduite au moins une cellule provenant d'une autre forme de vie b) embryon consistant en cellules provenant de plusieurs embryons, foetus ou êtres humains (chimera). |
Embryons chimériques : Restrictif pour les embryons humains auxquels on ajoute des cellules animales. Pas de restriction pour les embryons animaux auxquels on ajoute des cellules humaines mais pas autorisé par l'agence de financement de la recherche. |
|
États-Unis |
Aucune loi fédérale n'interdit la création de chimères en partie humaine. Les recherches sur l'embryon sont régulées au niveau des différents États, avec des lois locales plus ou moins permissives. Néanmoins, le NIH (« the National Institutes of Health ») a instauré, en 2015, un moratoire sur le financement fédéral des recherches impliquant des chimères humain-animal, le temps d'examiner les questions éthiques soulevées par l'introduction de cellules souches embryonnaires humaines dans un embryon animal 3 . À l'heure actuelle, le NIH mène une enquête publique sur la possibilité de lever ce moratoire 4 . |
Pas de définition dans la loi ? |
Embryons chimériques Actuellement, pas de restriction fédérale mais interdiction de financer les projets utilisant des embryons chimériques avec les fonds publics. Le NIH envisage de modifier le moratoire pour permettre le financement de projets faisant appels à des chimères créées par l'injection de cellules souches embryonnaires humaines dans un embryon animal. Les recommandations actuelles du NIH interdisent l'introduction de cellules pluripotentes humaines dans un embryon de primate et la reproduction d'animaux chimériques dont les cellules pluripotentes humaines ont pu modifier les lignées germinales 5 . Pour la levée du moratoire, le NIH prévoit de mettre en place un comité de pilotage interne qui évaluera en détail, les recherches dans lesquelles : • Des cellules pluripotentes humaines sont introduites dans des embryons de primates non-humain jusqu'à la fin de la phase de gastrulation, • Des cellules humaines sont introduites, après la phase de gastrulation dans des mammifères non-humains (excluant les rongeurs) dans lesquels les cellules humaines pourraient contribuer significativement ou induire une modification fonctionnelle substantielle du cerveau de l'animal. |
|
Royaume-Uni |
Le « Human Fertilisation and Embryology Act 2008 » interdit, entre autres, la conservation d'un embryon humain mélangé (« admixed ») pour plus de 14 jours de culture ou au-delà de l'apparition des feuillets primitifs, ainsi que de placer un embryon humain modifié dans un animal en vue de son développement. Les embryons humains « mélangés » incluent les embryons humains altérés par l'introduction d'une ou plusieurs cellules animales, ainsi que les embryons contenant à la fois de l'ADN humain et animal dans lequel l'ADN animal n'est pas prédominant 6 . L'académie des Sciences médicales a recommandé que certaines catégories de recherche impliquant des chimères soient soumises à l'avis d'un comité d'experts (incluant les recherches qui rendrait plus « humains » les fonctions cérébrales, le comportement ou l'apparence physique) tandis que l'académie a rejeté en bloc certaines catégories de recherche (incluant les recherches impliquant la création de chimères humains-primate non-humains avec des fonctions cérébrales apparentées à l'Homme ou la reproduction d'animaux contenant des cellules germinales humaines 7 . |
Embryon « mélangé » : a) Tout embryon créé par remplacement du noyau d'un ovocyte animal ou d'une cellule animale ou de deux pronuclei animaux avec : - deux pronuclei humains - le nucleus d'un gamète humain ou de tout autre cellule humaine ou - un gamète humain ou toute autre cellule humaine. b) Tout autre embryon créé en utilisant : - des gamètes humains et des gamètes animaux ou - un pronucleus humain et un pronucleus animal c) Un embryon humain qui a été modifié par l'introduction de toute séquence d'ADN nucléaire ou mitochondrial d'un animal dans une ou plusieurs cellules de l'embryon d) Un embryon qui a été altéré par l'introduction d'une ou plusieurs cellules animales o e) Tout embryon ne rentrant pas dans les définitions (a) à (d) qui contiennent, à la fois de l'ADN nucléaire ou mitochondrial humain et de l'ADN nucléaire ou mitochondrial animal mais dans lequel l'ADN animal n'est pas prédominant. |
Embryons chimériques : Restrictif : embryon humain + cellules animales Permissif : embryon animal + cellules humaines soumis à autorisation par le Secretary of State for the Home Office en application du Animals (Scientific Procedures) Act 1986. L'académie des sciences médicales recommande des restrictions pour certaines recherches. |
|
Japon |
Selon la loi « Act on Regulation of Human Cloning Techniques »), les embryons chimériques, humain-animal, peuvent uniquement être cultivés jusqu'à l'apparence des feuillets primitifs. Ils ne peuvent pas être transférés dans un utérus animal ou humain 8 . Le comité japonais d'expert en bioéthique a recommandé l'allègement de ces restrictions et de limiter les pratiques interdites à la création d'un cerveau humain chez l'animal et de chimères humain-primate non-humain 9 . |
Embryon chimérique humain-humain : Tous les embryons suivants (incluant chaque embryon produit successivement par séparation simple ou multiple du dit embryon) : a) Un embryon résultant de l'agrégation de deux ou plusieurs embryons humains fécondés, d'embryons fragmentés, d'un transfert nucléaire d'embryon humain ou d'une cellule somatique humaine. b) Un embryon humain produit par l'agrégation d'un embryon humain fécondé, d'un embryon humain fragmenté, d'un transfert nucléaire d'un embryon humain ou d'une cellule somatique humaine. Embryon hybride humain-Animal : Tous les embryons suivants (incluant chaque embryon produit successivement par séparation simple ou multiple du dit embryon) : a) Un embryon produit par une fécondation entre une cellule germinale humaine et une cellule germinale animale b) Un embryon produit par la fusion d'un ovocyte énucléé humain ou animal avec un embryon au stade une cellule décrit en (a) ou avec une cellule embryonnaire ayant le noyau d'un embryon décrit en (a). Embryon clone humain-animal : Tous les embryons suivants (incluant chaque embryon produit successivement par séparation simple ou multiple du dit embryon) : a) Un embryon produit par la fusion d'un ovocyte animal énucléé avec une cellule somatique humaine ou un embryon humain fécondé, ou un embryon humain fragmenté, ou un transfert nucléaire d'un embryon humain ou d'un embryon humain au stade une cellule provenant d'un transfert nucléaire d'une cellule somatique humaine, ou d'un embryon chimérique humain-humain. b) Un embryon produit par la fusion d'un ovocyte humain énucléé avec un embryon au stade une cellule décrit en (a) ou une cellule embryonnaire avec un noyau d'un embryon décrit en (a). Embryon chimérique humain-animal : Tous les embryons ci-dessous qui ne sont pas des embryons chimériques humain-humain, des embryons animaux ou des embryons chimériques animal-humain (incluant chaque embryon produit successivement par séparation simple ou multiple du dit embryon) : a) Un embryon produit par l'agrégation de deux ou plus embryons (incluant un embryon produit par agrégation d'un embryon et d'une cellule somatique ou embryonnaire). b) Un embryon produit par agrégation d'un embryon et d'une cellule somatique ou embryonnaire. c) Un embryon produit par la fusion d'une cellule embryonnaire contenant le noyau d'une cellule d'un embryon listé en (a) et (b) et un ovocyte énucléé humain ou animal. |
Embryons issus d'un transfert nucléaire d'une cellule somatique humaine, embryons hybrides humain-animal, embryon clone humain-animal, embryon chimérique humain-animal : Permissif : embryon animal + cellules humaines in vitro jusqu'à l'apparition des feuillets primitifs 8 . Interdiction de transfert dans un utérus humain ou animal. Une seule dérogation pour les recherches concernant la production d'organes dérivant de cellules humaines transplantables chez l'homme 10 . Cette restriction devrait être allégée si le législateur suit les recommandations du comité japonais d'expert en bioéthique 9 . |
|
Allemagne |
La loi allemande sur la protection de l'embryon de 1990 interdit la création d'embryons hybrides ou chimériques 11 . Le conseil allemand d'éthique recommande en plus d'interdire dans la loi de protection de l'embryon d'autres recherches sur les chimères (incluant l'interdiction de transfert d'embryons animaux chez l'humain, l'interdiction d'insérer du matériel animal dans les cellules germinales humaines et l'interdiction de création de chimères capables de former des gamètes humaines) 12 . Le conseil d'éthique recommande aussi de modifier la loi sur le bien-être animal pour interdire l'ajouter de cellules nerveuses humaines dans le cerveau des grands singes 12 . |
Embryon chimérique ou hybride : Embryon créé en : 1) fusionnant des embryons ayant un matériel génétique différent avec au moins une cellule d'un embryon humain 2) fusionnant un embryon humain avec une cellule contenant un matériel génétique différent du sien 3) fécondant un ovocyte humain avec le sperme d'un animal ou en fécondant un ovocyte animal avec le sperme d'un homme dans le but de générer un embryon capable de se développer. |
Embryons chimériques ou hybrides : Restrictif : interdiction de créer des embryons chimériques N.B : L'Allemagne interdit les recherches sur l'embryon humain. Permission uniquement d'importer des cellules souches embryonnaires humaines dérivées avant mai 2007. |
|
France |
Loi de bioéthique 2011. La création d'embryons chimériques et transgéniques est interdite. |
Embryons chimériques Restrictif : interdit. |
|
|
Suisse |
L'article 36 du « Reproductive MedicineAct » du 18 décembre 1998 interdit la création d'embryons chimériques à partir de cellules embryonnaires 13 . Cependant, le texte de loi n'interdit pas la création d'embryons chimériques à partir de cellules souches pluripotentes induites humaines injectées dans un embryon animal. |
Embryon : Représente la descendance depuis la fusion des noyaux cellulaires (caryogamie) jusqu'à la fin du développement des organes. Embryon chimérique : La formation d'une chimère signifie la fusion de cellules totipotentes provenant de deux ou plus embryons génétiquement différents. Les cellules embryonnaires sont totipotentes si elles sont capables de se différencier en n'importe quel type cellulaire du corps. |
Embryons chimériques : Restrictif à partir de cellules souches embryonnaires humaines. Possible à partir de cellules souches pluripotentes induites. |
|
Recommanda-tions internationales |
La société internationale de recherche sur les cellules souches « The International Society for Stem Cell Research (ISSCR) » recommande d'interdire les recherches concernant des chimères en parties humaines si elles impliquent la reproduction de chimères humain-animal ayant la capacité de former des gamètes humains 14 . L'ISSCR recommande aussi que les recherche impliquant un chimérisme aussi bien au niveau du système nerveux central que des lignées germinales soient soumises au contrôle d'un comité spécialisé qui évaluera les questions de bien-être animal. |
1 Health. Prohibition of Human Cloning for Reproduction Act 2002 [Internet]. Disponible sur: http://www.legislation.gov.au/Details/C2017C00306
2 Branch LS. Consolidated federal laws of canada, Assisted Human Reproduction Act [Internet]. 2019. Disponible sur: https://laws-lois.justice.gc.ca/eng/acts/a-13.4/
3 NOT-OD-15-158: NIH Research Involving Introduction of Human Pluripotent Cells into Non-Human Vertebrate Animal Pre-Gastrulation Embryos. Disponible sur: https://grants.nih.gov/grants/guide/notice-files/NOT-OD-15-158.html
4 NOT-OD-16-128: Request for Public Comment on the Proposed Changes to the NIH Guidelines for Human Stem Cell Research and the Proposed Scope of an NIH Steering Committees Consideration of Certain Human-Animal Chimera Research. Disponible sur: https://grants.nih.gov/grants/guide/notice-files/NOT-OD-16-128.html
5 Pullen LC. NIH to Lift Moratorium on Chimera Research Funding. Am J Transplant. 2016;16(12):3311?2.
6 Participation E. Human Fertilisation and Embryology Act 2008 [Internet]. Disponible sur: https://www.legislation.gov.uk/ukpga/2008/22/contents
7 Ethics of animal-human chimera the Academy of Medical Sciences (UK).
8 Japanese Law Translation - [Law text] - Act on Regulation of Human Cloning Techniques [Internet]. Disponible sur: http://www.japaneselawtranslation.go.jp/law/detail_header/?id=112&vm=2&re=02
9 Mizuno H, Akutsu H, Kato K. Ethical acceptability of research on human-animal chimeric embryos: summary of opinions by the Japanese Expert Panel on Bioethics. Life Sci Soc Policy. 22 nd December 2015;11(1):15.
10 Japanese Guidelines for Handling of Specified Embryos. 2001.
11 German Embryo Protection Act (October 24 th , 1990): Gesetz zum Schutz von Embryonen (Embryonenschutzgesetz-ESchG). Hum Reprod. 1 avr 1991;6(4):605?6.
12 German ethics council, Human-animal mixtures in research, Opinion, September 27 th , 2011.
13 Suisse CC 810.11 Federal Act of 18 December 1998 on Medically Assisted Reproduction (Reproductive Medicine Act, RMA) [Internet]. Disponible sur: https://www.admin.ch/opc/en/classified-compilation/20001938/index.html
14 ISSCR: Guidelines for Stem Cell Research [Internet]. Disponible sur: http://www.isscr.org/membership/policy/2016-guidelines/guidelines-for-stem-cell-research-and-clinical-translation
Source : Agence de la biomédecine
II - Les modifications adoptées par l'Assemblée nationale
Si aucun amendement n'a été formellement adopté par l'Assemblée nationale à l'article 17 du projet de loi, une modification légistique opérée à son dernier alinéa a pour conséquence de modifier la portée de cet alinéa par rapport au texte initial du projet de loi.
III - La position de la commission : encadrer la création d'embryons chimériques d'origine animale
• La correction d'une erreur matérielle
Si l'Assemblée nationale n'a pas adopté d'amendement à l'article 17 du projet de loi, il a néanmoins été procédé à une réécriture du dernier alinéa de l'article pour des raisons vraisemblablement légistiques. Il est désormais prévu qu'au dernier alinéa de l'article 16-4 du code civil le mot « génétiques » soit supprimé. Or le dernier alinéa de l'article 16-4 du code civil comprend deux occurrences du mot « génétiques », l'une après le mot « maladies », l'autre après le mot « caractères ».
Le projet de loi initial ne visait que la suppression du mot « génétiques » après le mot « maladies » afin de permettre l'extension des recherches recourant aux techniques d'édition génomique à des fins de prévention, de diagnostic et de traitement pour toutes les maladies, et pas seulement les maladies génétiques. En revanche, il n'était pas prévu de modifier les dispositions selon lesquelles, en dehors de ces recherches, « aucune transformation ne peut être apportée aux caractères génétiques dans le but de modifier la descendance de la personne. »
En conséquence, votre commission a adopté un amendement tendant à corriger cette erreur matérielle et à maintenir le principe selon lequel les caractères génétiques ne peuvent être modifiés lorsqu'ils sont transmissibles aux générations suivantes (amendement COM-204 du rapporteur).
• L'indispensable encadrement des recherches conduisant à la création d'embryons chimériques d'origine animale
Si le projet de loi opère une clarification de l'interdit en matière de création d'embryons chimériques, en prohibant l'insertion de cellules provenant d'autres espèces dans un embryon humain, il ouvre en revanche pleinement la voie aux recherches conduisant à la création d'embryons chimériques à partir d'embryons animaux auxquels seraient ajoutées des cellules d'origine humaine. Dans son avis sur le projet de loi, le Conseil d'État relève ainsi que le texte proposé « ne traite pas des enjeux que peut soulever l'adjonction, à l'inverse, de cellules d'origine humaine, en particulier de cellules pluripotentes, au sein d'un embryon animal, susceptibles pourtant de soulever des questionnements éthiques soulignés notamment par le CCNE dans son avis n° 129 préalable à la présente révision de la loi de bioéthique. »
Dans son avis n° 129, le CCNE estime en effet qu'« un encadrement semble néanmoins nécessaire, en particulier si les embryons chimériques sont transférés chez des femelles et donnent naissance à des animaux chimères avec le risque, chez le gros animal, que les cellules humaines se développent et induisent certaines caractéristiques humaines (morphologiques, neurologiques). » Deux paramètres doivent ainsi faire l'objet d'une attention particulière afin de prévenir la conception d'organismes qui, au cours de leur développement, présenteraient des caractéristiques propres à l'espèce humaine et remettraient en question la « ligne rouge » du franchissement de la barrière des espèces :
- le taux de « chimérisme » doit être maintenu à un niveau inférieur à un seuil au-delà duquel la contribution des cellules d'origine humaine au développement embryonnaire serait jugée excessive, sauf à courir le risque d'une « humanisation » de l'organisme ainsi créé ;
- la gestation doit être interrompue avant que le développement du cerveau n'atteigne un stade pour lequel il existerait un risque de propagation des cellules d'origine humaine dans le corps de l'animal et, le cas échéant, dans le cerveau de l'animal (par exemple, au niveau des neurones). De même, l'interruption de cette gestation apparaît incontournable pour prévenir l'émergence chez l'animal de caractéristiques phénotypiques humaines.
Votre commission a adopté un amendement COM-200 du rapporteur visant à rétablir l'interdiction de la création d'embryons chimériques. L'amendement prévoit néanmoins une dérogation pour les recherches conduites à partir de cellules souches pluripotentes induites humaines (iPS) : comme ces cellules font désormais l'objet d'un encadrement inséré dans le chapitre consacré aux recherches sur l'embryon et les cellules souches embryonnaires humaines, l'interdiction de la création d'embryons chimériques s'appliqueraient aux embryons chimériques constitués par l'adjonction d'iPS humaines à des embryons animaux, alors que des embryons chimériques de ce type sont aujourd'hui possibles au regard du droit en vigueur.
La commission ayant encadré la création de tels embryons chimériques en modifiant l'article 15 du projet de loi, il est opéré, à l'article L. 2151-2 du code de la santé publique, un renvoi aux nouvelles dispositions de l'article L. 2151-7 du même code consacré aux recherches sur les cellules iPS qui posent les conditions suivantes pour la création d'embryons chimériques par l'adjonction de cellules iPS à un embryon animal :
- l'interruption obligatoire de la gestation au terme d'un délai approuvé par l'agence de la biomédecine en fonction de la durée gestationnelle propre à l'espèce animale, mais aussi au regard du stade de développement embryonnaire ou foetal afin de prévenir les risques d'« humanisation » (liés à la propagation de cellules d'origine humaine dans le reste de l'organisme animal) ;
- la proportion de cellules d'origine humaine introduites dans l'embryon animal ne doit pas dépasser un seuil approuvé par l'agence de la biomédecine, qui ne pourra en tout état de cause être supérieure à 50 %. Cette dernière condition doit permettre de s'assurer que les cellules d'origine humaine ne contribuent pas au développement embryonnaire ou foetal au point que l'embryon ne puisse plus être considéré comme un embryon animal.
La commission a adopté cet article ainsi modifié.
Article 18
Développement des « passerelles
soin/recherches »
par l'utilisation facilitée
d'échantillons conservés à d'autres fins
Cet article définit les conditions dans lesquelles il peut être procédé à un examen des caractéristiques génétiques d'une personne à partir d'échantillons biologiques prélevés sur cette personne initialement à d'autres fins, dans un souci de conciliation du respect des droits des personnes dont sont issues ces échantillons et du développement de la recherche, notamment dans le domaine génomique.
La commission spéciale a adopté un amendement tendant à sécuriser les droits que les personnes, dont les échantillons peuvent faire l'objet d'examens génétiques dans le cadre d'une recherche scientifique, tiennent du RGPD en matière de protection de leurs données à caractère personnel.
I - Le dispositif proposé
Par dérogation au principe du consentement préalable et exprès pour tout examen des caractéristiques génétiques d'une personne 297 ( * ) , l'article L. 1131-1-1 du code de la santé publique prévoit aujourd'hui la possibilité qu'un tel examen soit réalisé à des fins de recherche scientifique à partir d'éléments du corps de la personne prélevés à d'autres fins 298 ( * ) , à la condition que la personne, dûment informée du projet de recherche, n'ait pas exprimé son opposition. L'obligation d'information n'est néanmoins pas applicable en cas d'impossibilité de retrouver la personne : dans ce cas, un comité de protection des personnes s'assure que la personne ne s'était pas opposée à l'examen de ses caractéristiques génétiques et émet un avis sur l'intérêt scientifique de la recherche.
Dans un contexte de multiplication des projets de recherche requérant l'utilisation de données massives génomiques, le CCNE préconise, dans son avis n° 129, « la rédaction de consentements éclairés élargis qui mentionnent de façon explicite les objectifs de la collecte des données en ne ciblant plus une maladie donnée (celle pour laquelle la personne participe initialement) mais un ensemble de projets de recherche qui recevront des accords de comités d'éthique. »
Dans son étude de juin 2018299 ( * ), le Conseil d'État évoque, pour sa part, différentes pistes, dont celle envisageant la mise en place d'un consentement par délégation dans le cadre duquel, « une fois le consentement initial obtenu pour une finalité précise, la possibilité de réutiliser les données de l'individu serait subordonnée à l'avis d'un « courtier honnête » (à l'instar du CPP consulté pour l'application de l'article L. 1131-1-1 du code de la santé publique lorsque la personne concernée ne peut être retrouvée) et au respect par le protocole de recherche d'exigences renforcées (qualité scientifique, rigueur éthique, etc.). » Le Conseil d'État ne tranche pas, toutefois, entre un consentement réinterrogé à chaque réutilisation pour un nouveau projet de recherche ou un consentement par délégation.
S'il ne va pas jusqu'à mettre en place un mécanisme de consentement élargi ou de consentement par délégation, l'article 18 du projet de loi assouplit les conditions de mise en oeuvre de l'obligation d'information de la personne par le responsable de la recherche.
Le I de l'article 18 du projet de loi procède ainsi à une réécriture des dispositions de l'actuel article L. 1131-1-1 300 ( * ) du code de la santé publique qui devient un nouvel article L. 1130-5 du même code. Ce nouvel article précise les modalités dérogatoires de consentement applicables à la réalisation d'examen des caractéristiques génétiques entrepris à des fins de recherche à partir d'échantillons biologiques prélevés à d'autres fins et permet de renforcer l'information de la personne en cas de découverte, à l'occasion de cet examen, d'anomalies génétiques susceptibles d'être à l'origine d'une affection justifiant des mesures de prévention ou de soins.
Est maintenu, au I dudit article L. 1130-5, le principe selon lequel, par dérogation à l'exigence d'un consentement préalable et exprès, l'examen des caractéristiques génétiques d'une personne peut être réalisé dans le cadre d'une recherche scientifique à partir d'éléments du corps de cette personne prélevés initialement à d'autres fins à la condition que :
- la personne ait été dûment informée du « programme de recherche » au sens de l'article L. 1243-3 du code de la santé publique. La notion de programme de recherche recouvre un champ d'activités de recherche plus large qu'un projet de recherche qui correspond, en règle générale, à un protocole de recherche bien déterminé. Le II de l'article 18 du projet de loi modifie en effet l'article L. 1243-3 précité afin de définir un programme de recherche comme « un ensemble d'activités de recherche organisées en vue de faciliter et d'accélérer les découvertes dans un domaine scientifique déterminé, défini par un organisme exerçant des activités de recherche ou en assurant la promotion » ;
- la personne n'ait pas exprimé son opposition.
Le périmètre du consentement, par absence d'opposition, de la personne à la réalisation d'examens de ses caractéristiques génétiques est ainsi élargi au champ du programme de recherche qui peut englober plusieurs projets ou protocoles de recherche. Le IV du nouvel article L. 1130-5 du code de la santé publique précise que cette dérogation au régime du consentement préalable et exprès en matière d'examens génétiques n'est pas applicable aux recherches dont la publication des résultats est susceptible de permettre la levée de l'anonymat des personnes concernées.
La CNIL note, dans son avis sur le projet de loi, que cette disposition tend à assouplir la réglementation actuelle qui exclut du régime dérogatoire au consentement les recherches dont les résultats, et pas seulement leur publication, pourraient être ré-identifiants 301 ( * ) .
Le II de l'article L. 1130-5 du code de la santé publique introduit, en outre, un dispositif d'information de la personne en cas de découverte incidente, dans le cadre de la recherche, d'une anomalie génétique susceptible de mesures de prévention ou de soins. Dans ce cas de figure, il reviendra au responsable de la recherche ou au médecin détenteur de l'identité de la personne d'informer la personne de l'existence d'une information médicale la concernant et de l'inviter à se rendre à une consultation chez un médecin qualifié en génétique, à la condition que la personne ne se soit pas préalablement opposée à la transmission de cette information.
Le III de l'article L. 1130-5 du code de la santé publique détermine les situations dans lesquelles l'opposition ne peut être manifestée par la personne ou dans lesquelles la personne ne peut être informée du programme de recherche :
- lorsque la personne est mineure, l'opposition sera exprimée par les détenteurs de l'autorité parentale, c'est-à-dire les parents ou le tuteur ;
- lorsque la personne fait l'objet d'une mesure de protection juridique avec représentation à la personne, elle exprimera seule son opposition, le cas échéant assistée de la personne chargée de la mesure de protection ;
- lorsqu'il est impossible au responsable de la recherche de procéder à l'information de la personne, soit que celle-ci ne peut être retrouvée par le responsable de la recherche, soit qu'elle est décédée ou qu'elle est hors d'état d'exprimer sa volonté, il appartiendra à un comité de protection des personnes de confirmer l'impossibilité d'informer la personne et déterminera dans ce cas s'il peut être procédé à l'examen des caractéristiques génétiques de la personne, au regard de la pertinence éthique et scientifique de la recherche.
Un décret simple précisera les modalités d'information des personnes concernées et les modalités d'expression de leur opposition.
Outre la définition de la notion de « programme de recherche », le II de l'article 18 du projet de loi précise, à l'article L. 1243-3 du code de la santé publique relatif aux conditions d'utilisation en recherche des collections d'échantillons biologiques, que le ministre chargé de la recherche et le directeur général de l'agence régionale de santé ayant autorité sur l'organisme de recherche pourront solliciter de ce dernier la transmission de toute information permettant de garantir que les activités sont bien poursuivies dans le respect de la législation, notamment en matière d'obligation d'information de la personne et de vérification de l'absence d'opposition de celle-ci.
Par coordination, le III de l'article 18 du projet de loi met en cohérence les dispositions de l'article 75 de la loi du 6 janvier 1978 302 ( * ) , dite « Informatique et libertés », avec les nouvelles dispositions applicables en matière d'examen des caractéristiques génétiques dans le cadre d'une recherche scientifique.
II - Les modifications adoptées par l'Assemblée nationale
Outre des amendements rédactionnels et de coordination, l'Assemblée nationale a modifié l'article 18 du projet de loi afin de préciser que seul le médecin détenteur de l'identité de la personne sera tenu de porter à la connaissance de celle-ci l'existence d'une information médicale la concernant en cas de découverte d'une anomalie génétique pouvant être responsable d'une affection justifiant des mesures de prévention ou de soins. En effet, l'alerte de la personne sur l'existence d'une information de ce type ne peut également être confiée au responsable de la recherche qui ne détient pas l'identité de la personne.
III - La position de la commission
• Sécuriser les droits que les personnes tiennent du RGPD en matière de protection de leurs données à caractère personnel
Dans son avis 303 ( * ) du 11 juillet 2019 sur le projet de loi, la CNIL relève que l'opposition de la personne à l'examen de ses caractéristiques génétiques dans le cadre d'une recherche pourra s'exprimer, aux termes du nouvel article L. 1130-5 du code de la santé publique, sans forme tant qu'il n'y a pas eu d'intervention sur l'élément concerné dans le cadre de la recherche. Interrogé sur la portée de cette disposition, le Gouvernement a confirmé que le droit d'opposition ne s'exercerait que sur la réalisation de l'examen des caractéristiques génétiques et non sur le traitement des données associées au prélèvement ou produites ultérieurement après son analyse.
Or la CNIL souligne que « le droit d'opposition prévu à l'article 21 du RGPD ainsi que le droit à l'effacement prévu par l'article 17 du RGPD sont applicables dans les conditions prévues par le RGPD, et ce, même après manipulation. Elle demande que des précisions soient apportées en ce sens dans le projet. » L'article 17 du RGPD reconnaît en effet à toute personne un droit à l'effacement de ses données à caractère personnel (« droit à l'oubli »), notamment lorsqu'elle entend retirer son consentement au traitement desdites données ou lorsqu'elle souhaite exercer son droit d'opposition en application de l'article 21 du RGPD, lequel droit d'opposition peut s'exercer « à tout moment, pour des raisons tenant à sa situation particulière ».
Des situations permettant au responsable de la recherche de poursuivre le traitement des données à caractère personnel en dépit de l'exercice d'un droit d'effacement ou d'un droit d'opposition postérieur à la réalisation de l'examen génétique, sont néanmoins envisagées par le RGPD : il pourrait s'agir notamment de traitements nécessaires aux fins de médecine préventive ou de médecine du travail, ou encore de traitements répondant à des motifs d'intérêt public en matière de santé publique (comme la prévention des menaces sanitaires transfrontalières ou la sécurité des produits de santé).
Dans un souci de clarification, votre commission a adopté un amendement tendant à préciser que le droit d'opposition dont dispose la personne pour la réalisation de l'examen de ses caractéristiques génétiques dans le cadre d'une recherche, et qui peut s'exercer tant que l'examen n'a pas été réalisé, est distinct des droits qu'elle tient des articles 17 et 21 du RGPD en matière d'utilisation des données à caractère personnel qui pourraient découler de cet examen (amendement COM-206 du rapporteur).
Dans la mesure où le recours aux tests génomiques complets, grâce au séquençage à haut débit, s'intensifie, notamment dans le cadre de la recherche scientifique, et dès lors que ces tests présentent un risque accru de ré-identification, il semble nécessaire de s'assurer que les personnes pourront exercer, postérieurement à la réalisation de l'examen des caractéristiques génétiques, leur droit à l'effacement de données potentiellement ré-identifiantes ou leur droit d'opposition à l'utilisation de ces données tant que les résultats de la recherche n'ont pas été publiés.
• Garantir les droits des personnes hors d'état d'exprimer seules leur volonté
Dans son avis précité sur le projet de loi, la CNIL s'est également interrogée sur le fait que, parmi les situations dans lesquelles l'impossibilité d'information de la personne sur le projet de recherche peut être établie a priori , sont envisagés par le projet de loi non seulement les cas où la personne a été perdue de vue ou est décédée mais aussi le cas où la personne est hors d'état d'exprimer sa volonté. Or ce dernier cas recouvre des situations pour lesquelles une information des personnes susceptibles de défendre les intérêts de la personne reste possible : dans ce cas, le droit d'opposition pourrait être exercé par délégation par la personne de confiance ou, à défaut, la famille ou l'entourage de la personne, comme cela est prévu par le droit en vigueur.
En effet, l'article L. 1131-1- du code de la santé publique dispose, à l'heure actuelle, que « lorsque la personne est un majeur hors d'état d'exprimer son consentement et ne faisant pas l'objet d'une tutelle, l'opposition est exprimée par la personne de confiance prévue à l'article L. 1111-6, à défaut de celle-ci, par la famille ou, à défaut, par une personne entretenant avec l'intéressé des liens étroits et stables. »
Le Gouvernement indique que le recours à la personne de confiance n'a pas été retenu parce qu'il n'est pertinent que pour des situations que la personne a pu anticiper et vis-à-vis desquelles elle a pu exprimer une volonté, la personne de confiance n'étant censée que rendre compte de cette volonté déjà exprimée 304 ( * ) . Or, le consentement recherché à l'article 18 du projet de loi n'est pas un consentement général, in abstracto , mais un consentement à l'examen des caractéristiques génétiques dans le cadre d'un programme de recherche donné dont les tenants et aboutissants seront exposés à la personne concernée par le consentement. Par définition, ce type de consentement ne peut être couvert par une volonté ou un refus général précédemment exprimé. Le Gouvernement estime donc, dans le cas des personnes hors d'état d'exprimer leur volonté, que le renvoi au CPP est plus adapté.
En revanche, il est prévu par le projet de loi que la personne faisant l'objet d'une mesure de protection juridique, avec représentation à la personne, exprime seule son opposition, moyennant le cas échéant l'assistance de la personne chargée de la mesure de protection. Toutefois, peuvent se présenter des situations dans lesquelles la personne faisant l'objet d'une mesure de protection juridique n'est pas en capacité de prendre seule des décisions relatives à sa personne, auquel cas il sera considéré qu'elle est hors d'état d'exprimer seule sa volonté et que la recherche sera alors soumise à l'avis du comité de protection des personnes.
Par conséquent, votre commission a adopté un amendement COM-207 du rapporteur de clarification rédactionnelle tendant à préciser que la personne faisant l'objet d'une mesure de protection juridique exprime seule son opposition « dans la mesure où son état le permet », en cohérence avec les dispositions de l'article 459 du code civil 305 ( * ) .
• Votre commission a également adopté un amendement permettant de mentionner qu'une découverte incidente d'anomalie génétique à l'occasion d'un examen génétique réalisé dans le cadre d'une recherche scientifique peut bénéficier aux membres de la famille de la personne et non uniquement à la personne concernée (amendement COM-55 de Jacques Bigot).
• Enfin, votre commission a adopté un amendement d'harmonisation rédactionnelle et de coordination (amendement COM-205 du rapporteur).
La commission a adopté cet article ainsi modifié.
TITRE
V
POURSUIVRE L'AMÉLIORATION
DE LA QUALITÉ ET DE LA
SÉCURITÉ
DES PRATIQUES DU DOMAINE BIOÉTHIQUE
CHAPITRE IER
Renforcer la qualité et la sécurité
des pratiques
Article 19
Actualisation du régime du diagnostic prénatal
Cet article actualise la définition du diagnostic prénatal et modifie les modalités d'information de la femme enceinte tout au long du processus de prise en charge, afin de prévoir d'une part l'information de l'autre membre du couple lorsqu'elle vit en couple, et de préciser d'autre part les démarches en cas de révélation de données génétiques incidentes pouvant justifier des investigations complémentaires.
La commission spéciale a ajusté la définition de la médecine foetale résultant des travaux de l'Assemblée nationale pour la mettre en cohérence avec la réalité des pratiques.
I - Le dispositif proposé
1. L'actualisation de la définition du diagnostic prénatal
a) Une discipline connaissant d'importantes évolutions de pratique
• D'après le I de l'article L. 2131-1 du code de la santé publique, « le diagnostic prénatal s'entend des pratiques médicales, y compris l'échographie obstétricale et foetale, ayant pour but de détecter in utero chez l'embryon ou le foetus une affection d'une particulière gravité. »
Si cette définition a été introduite par la première loi de bioéthique en 1994, la mention expresse de l'échographie obstétricale et foetale résulte de la loi du 7 juillet 2011. Cette dernière loi a par ailleurs précisé les différentes étapes de la démarche de diagnostic prénatal, au cours desquelles est assurée l'information de la femme enceinte ( cf . ci-après).
Depuis 2011, cette discipline a connu des développements importants qui permettent à la fois de limiter son caractère invasif ( cf. pour le dépistage de la trisomie 21 comme retracé dans l'encadré) et d'améliorer la précision du diagnostic posé. Comme l'a relevé l'Agence de la biomédecine dans son rapport d'information au Gouvernement et au Parlement de décembre 2017, « les performances des appareils d'échographie multiplient les actions de dépistages et rendent les actions diagnostiques plus fiables et les pronostics plus précis. De plus, les nouvelles techniques d'imagerie (imagerie par résonnance magnétique, tomodensitométrie par exemple) s'appliquent désormais aussi au foetus in utero. Dans des indications précises, elles apportent des précisions diagnostiques importantes pour le pronostic. »
Les chiffres clés du diagnostic prénatal (DPN)
• L'expertise en matière de diagnostic prénatal est réunie au sein de 48 centres pluridisciplinaires de diagnostic prénatal (CPDPN) autorisés par l'Agence de la biomédecine, répartis sur l'ensemble du territoire.
Ces centres réunissent des équipes pluridisciplinaires dont la mission est d'aider les équipes médicales et la femme enceinte dans l'analyse, la prise de décision et le suivi de la grossesse lorsqu'une malformation ou une anomalie foetale est détectée ou suspectée et lorsque le risque de transmission d'une maladie génétique amène à envisager un diagnostic préimplantatoire ou prénatal.
Ces centres ont reçu en 2016 plus de 46 000 dossiers et ont vu en consultation plus de 33 150 femmes , dont 96% en cours de grossesse, soit 24% de plus qu'en 2013 alors que le nombre de naissances a diminué en parallèle de 5%.
Pendant une grossesse, dans 22% des cas (7 366) une attestation de particulière gravité en vue d'une interruption médicale de grossesse (IMG) a été délivrée, soit pour motif foetal soit, plus rarement, pour motif maternel. Dans plus de 53% des cas (17 039), le diagnostic posé est celui d'une pathologie considérée comme curable ou ne comportant pas une particulière gravité, permettant la poursuite de la grossesse.
• Les actes proposés par les CPDPN sont principalement des actes d'imagerie (avec 95 997 échographies diagnostiques ou 8 840 échographies cardiaques foetales en 2017), des prélèvements invasifs à visée diagnostique (15 554 en 2017, dont principalement des amniocentèses, en diminution de 18 % depuis 2013), des gestes à visée thérapeutique (1 625 en 2017, principalement des drainages amniotiques ou le traitement par laser sur le placenta du syndrome « transfuseur-transfusé ») et des gestes d'arrêt de vie in utero (6 529 en 2017).
Les prélèvements soit sur le foetus et ses annexes (liquide amniotique, villosité choriale, sang foetal) soit sur le sang de la mère se rapportent à des techniques comme la cytogénétique (étude du nombre et de la forme des chromosomes foetaux), la génétique moléculaire (étude de l'ADN foetal circulant dans le sang maternel) et les autres disciplines biologiques (hématologie, immunologie, bactériologie, virologie, biochimie foetale).
• L'examen de dépistage concernant le plus grand nombre de femmes est celui de la trisomie 21 par les marqueurs sériques maternels : proposé systématiquement depuis 2010, cet examen a concerné 637 547 femmes enceintes en 2017 soit 83% d'entre elles. Depuis 2017 (sur la base d'une recommandation de la Haute Autorité de santé), un diagnostic non invasif des principales trisomies (13, 18, 21), portant sur l'ADN libre circulant dans le sang maternel (ADNlc) est proposé en deuxième ligne (et remboursé) en fonction d'un calcul de risque (seuil de 1/1000).
Suite au diagnostic d'une anomalie chromosomique (4 203 cas dont 1 967 trisomies 21), le taux de recours à une interruption médicale de grossesse s'établit en 2017 à près de 63% (dont 73,7% pour la trisomie 21) ; près de 12% des enfants sont nés vivants (dont 4,9% pour la trisomie 21).
Source : Agence de la biomédecine - rapport d'activité du diagnostic prénatal
b) Une définition élargie à la médecine foetale
Le a) du 1° modifie , pour la mettre en cohérence avec l'évolution des connaissances et des pratiques, la définition du diagnostic prénatal donnée par le I de l'article L. 2131-1 du code de la santé publique :
- d'une part, est mentionnée au titre des pratiques médicales mises en oeuvre dans ce cadre l' imagerie obstétricale et foetale au-delà de la seule « échographie », puisque des technologies comme l'IRM ou le scanner sont rapportées dans l'activité des centres de DPN ;
- d'autre part, le but du diagnostic prénatal est étendu à la médecine foetale c'est-à-dire la prise en charge in utero d'une affection d'une particulière gravité chez l'embryon ou le foetus. La médecine foetale est définie comme « les soins médicaux et chirurgicaux » apportés à l'embryon et au foetus. Cette évolution permet d'intégrer les évolutions technologiques ayant notamment conduit au développement de la thérapie foetale in utero 306 ( * ) .
La médecine foetale ne fait jusqu'à présent l'objet que d'une définition au niveau réglementaire, à la suite d'un décret du 14 janvier 2014 307 ( * ) qui a listé, à l'article R. 2131-1 du code de la santé publique les différents examens permettant d'évaluer le risque que l'embryon ou le foetus présente une affection susceptible de modifier le déroulement ou le suivi de la grossesse : pour le V de cet article, « La médecine foetale s'entend de la prise en charge adaptée ou des traitements apportés au foetus en cas de pathologie. »
La possibilité de soin ou de « prise en charge adaptée du foetus ou de l'enfant né » était cependant évoquée dans l'article L. 2131-1 dès la loi de juillet 2011, lorsqu'est mentionnée l'information délivrée à la femme enceinte par le prescripteur sur les résultats des examens, en cas de risque avéré.
Le maintien toutefois, dans la définition du diagnostic prénatal, de la référence à une affection de particulière gravité est justifiée, d'après l'étude d'impact, par la volonté du Gouvernement de prévenir un risque de judiciarisation en évitant des demandes de diagnostic non fondées.
Cela rejoint d'ailleurs une recommandation du Conseil d'Etat dans son étude de juin 2018 sur l'évaluation de la loi de bioéthique qui estimait, alors que de nouveaux tests permettant de séquencer le génome du foetus apparaissent, que la finalité du DPN tendant à détecter in utero une affection d'une particulière gravité gagnerait à être préservée.
2. Le renforcement de l'information de la femme enceinte
• La loi du 7 juillet 2011 a précisé les différentes étapes du diagnostic prénatal en assurant l'information et l'accompagnement de la femme enceinte tout au long de ce processus :
- en amont, pour l'informer de la possibilité de solliciter des examens,
- lors de la communication des résultats,
- avant la réalisation de nouveaux examens en cas de risque avéré,
- préalablement au recueil de son consentement à la réalisation de certains examens, pour les informer sur les objectifs, les modalités, les risques, les limites et le caractère non obligatoire de ces examens.
• Le b) du 1° modifie à la marge les modalités de communication des premiers résultats par le prescripteur de l'examen prévues au III de l'article L. 2131-1, afin de prévoir que cette information est également communiquée à l'autre membre du couple lorsque la femme enceinte vit en couple. De même, la prise en charge éventuelle par un centre pluridisciplinaire de diagnostic prénatal est proposée systématiquement à l'autre membre du couple, et non plus seulement si la femme enceinte le souhaite.
Le d) du 1° renvoie par ailleurs à un décret en Conseil d'Etat les modalités d'information de l'autre membre du couple dans les différents cas prévus. Comme le note l'étude d'impact, « si le diagnostic prénatal a la particularité de concerner une femme enceinte et son foetus la plaçant au centre du dispositif (c'est elle qui prend toutes les décisions relatives à sa grossesse), il est toutefois recommandé d'impliquer le plus souvent possible le couple, en particulier lors du rendu des résultats. »
• Le c) du 1° complète les dispositions sur les informations transmises préalablement du recueil du consentement de la femme enceinte à la réalisation de certains examens prévues au VI de l'article L. 2131-1, pour tenir compte de la possibilité que ces examens révèlent des données incidentes, c'est-à-dire des caractéristiques génétiques foetales sans relation certaine avec l'indication initiale de l'examen .
Là aussi, le texte prévoit que les résultats de ces examens sont communiqués à la femme enceinte et le cas échéant à son conjoint. Ils le sont toutefois « sauf opposition de leur part ». Des investigations supplémentaires pourraient être réalisées, notamment des examens des caractéristiques génétiques de chaque parent, ou l'orientation vers un médecin qualifié en génétique.
Ces dispositions font écho à celles prévues par l'article 9 du projet de loi. Elles prennent également en compte la montée en charge de nouvelles techniques utilisées dans l'activité de diagnostic prénatal .
Comme le soulignent l'étude d'impact et les données de l'Agence de la biomédecine 308 ( * ) , en matière d'activité de DPN en génétique moléculaire (qui a concerné 2 734 foetus en 2017 pour 322 maladies génétiques différentes 309 ( * ) ), les nouvelles techniques d'étude de l'ADN (séquençage de nouvelle génération) permettent d'élargir le diagnostic des maladies rares. En outre, de nouvelles techniques utilisées permettent de révéler des anomalies chromosomiques de plus petite taille non décelables par le caryotype : c'est le cas de l'ACPA (analyse chromosomique par puce à ADN) qui connaît une montée en charge continue avec 8 580 examens en 2017 versus 1 756 en 2013 310 ( * ) .
3. Une base légale aux arrêtés de bonnes pratiques
Le 2° donne enfin une base légale à divers arrêtés de bonnes pratiques relatives aux activités du diagnostic prénatal, pris aujourd'hui sur une base réglementaire, afin d'en sécuriser la portée 311 ( * ) .
Il complète ces dispositions en prévoyant spécifiquement la définition de recommandations de bonnes pratiques, sur proposition de l'Agence de la biomédecine, sur « les critères médicaux justifiant la communication à la femme enceinte des caractéristiques génétiques foetales sans relation certaine avec l'indication initiale de l'examen » .
II - Les modifications adoptées par l'Assemblée nationale
• La commission spéciale , outre des amendements rédactionnels, a adopté, avec l'avis favorable de la ministre des solidarités et de la santé, des amendements, à l'initiative de Thibault Bazin (Les Républicains) et de son rapporteur Jean-François Eliaou, visant à laisser la femme enceinte choisir si son partenaire, lorsqu'elle vit en couple, doit également être informé et, le cas échéant, pris en charge par l'équipe médicale, suivant ce que prévoit le droit en vigueur.
Elle a également adopté, avec l'avis de sagesse de la ministre, un amendement de son rapporteur visant à prévoir un dispositif d' information du tiers donneur si les examens révèlent, en données incidentes, une anomalie génétique pouvant être responsable d'une affection grave.
• En séance publique , l'Assemblée nationale a adopté un amendement de cohérence de son rapporteur, visant à tirer les conséquences du choix laissé à la femme enceinte d'informer son partenaire en matière d'annonce des premiers résultats.
Elle a également adopté, à l'initiative de son rapporteur Jean-François Eliaou et avec l'avis favorable du Gouvernement, une nouvelle définition de la médecine foetale visant à clarifier « une ambiguïté quant à l'articulation entre médecine foetale et diagnostic prénatal. » Plutôt que de distinguer, d'un côté, le diagnostic prénatal, étendu à la prise en charge in utero, et, de l'autre, la médecine foetale, la rédaction adoptée regroupe sous le terme de « médecine foetale » l'ensemble des actes visant à poser un diagnostic, procéder à l'évaluation pronostique et, le cas échéant, traiter l'affection d'une particulière gravité chez l'embryon ou le foetus.
III - La position de la commission
• Les évolutions adoptées par l'Assemblée nationale afin de laisser à la femme enceinte la décision d'informer son conjoint, comme c'est le cas à l'heure actuelle, sont conformes à l'expérience des professionnels concernés ainsi que l'a indiqué la présidente de la fédération des centres pluridisciplinaires de diagnostic prénatal. Imposer la présence du conjoint aurait pu poser des difficultés en pratique, dès lors que la présence seule de la femme enceinte est la situation la plus fréquemment rencontrée.
• Quant à la définition de la médecine foetale, qui englobe le diagnostic prénatal, le principe de son actualisation répond à une préconisation du CCNE notamment dans son avis n° 129 : celui-ci relevait alors que « cette spécialité a beaucoup évolué en 24 ans et [que] la définition qui lui est attribuée paraît restrictive en suggérant que peu ou pas de traitements sont disponibles et que l'interruption médicale de grossesse est la seule issue » . Il proposait de faire référence, plus généralement, au diagnostic et au traitement, dès lors que cela est possible, des « pathologies ou malformations pendant la grossesse ».
A cet égard, comme l'ont regretté les professionnels concernés, la définition retenue, tant dans le projet de loi initial que dans le texte adopté par l'Assemblée nationale, demeure restrictive.
Tout en visant à mettre en cohérence la définition du diagnostic prénatal à la réalité des pratiques, le projet de loi fait le choix de maintenir la référence à la prise en charge d'une affection d'une particulière gravité, selon la terminologie employée pour le recours à une interruption médicale de grossesse, alors que l'étude d'impact souligne explicitement que cet acte « n'est pas l'objectif premier du diagnostic prénatal ». Les professionnels de santé assurent la prise en charge de pathologies présentant des degrés divers de gravité.
La commission spéciale a ainsi adopté l'amendement COM-165 de sa rapporteure tendant à élargir le champ de la définition proposée , suivant une préconisation du CCNE et des professionnels concernés, en visant plus généralement la prise en charge in utero d'affections susceptibles d'avoir un impact sur le devenir du foetus ou de l'enfant à naître.
Elle a également adopté les amendements COM-143 et COM-169 de la rapporteure de clarification rédactionnelle. Ce dernier tend à lever une ambiguïté, en ouvrant aux deux parents la possibilité de s'opposer à la communication de résultats pouvant, en matière d'examen des caractéristiques génétiques, avoir des implications pour chacun.
La commission a adopté cet article ainsi modifié.
Article 19 bis A
Abrogation du double diagnostic préimplantatoire
(DPI-HLA)
et demande de rapport sur le sang placentaire
Cet article, inséré par l'Assemblée nationale, supprime le recours possible à la technique du double diagnostic préimplantatoire (DPI-HLA) et demande au Gouvernement de rendre compte dans un rapport des progrès réalisés dans la collecte et le stockage d'unités de sang placentaires.
La commission spéciale a décidé de supprimer cet article en considérant que cette technique introduite en 2004 pouvait dans certaines situations certes exceptionnelles mais strictement encadrées sur le plan éthique apporter une solution à des familles et sauver la vie d'enfants atteints de maladies rares.
I - Le dispositif adopté par l'Assemblée nationale
Cet article a été introduit par l'Assemblée nationale lors de l'examen du texte en séance publique, à l'initiative d'Annie Genevard et de plusieurs députés du groupe Les Républicains, contre l'avis défavorable de la commission spéciale et avec un avis de sagesse du Gouvernement.
Il abroge l'article L. 2131-4-1 du code de la santé publique autorisant la technique du double diagnostic préimplantatoire (DPI-HLA) 312 ( * ) et demande au Gouvernement, en parallèle, de rendre compte dans un rapport des progrès réalisés dans la collecte et le stockage d'unités de sang placentaires .
1. Le DPI-HLA : un dispositif strictement encadré, introduit en 2004 et pérennisé en 2011
• La loi du 6 août 2004 313 ( * ) relative à la bioéthique a autorisé, à titre expérimental et sous des conditions strictes, le recours au diagnostic préimplantatoire (DPI) couplé au typage HLA 314 ( * ) des embryons . Cette technique dite DPI-HLA vise à ce que l'enfant à naître, en plus d'être indemne de l'anomalie génétique grave affectant un frère ou une soeur, présente des caractéristiques d'HLA2 compatibles avec l'aîné malade pour que les cellules souches du sang de cordon ombilical soient susceptibles de lui être greffées.
• Le caractère expérimental de ce dispositif a été supprimé par la loi « bioéthique » du 7 juillet 2011 , qui a ainsi pérennisé la possibilité de recours à cette « double sélection », selon les conditions suivantes définies par l'article L. 2131-4-1 du code de la santé publique :
- le couple demandeur doit avoir donné naissance à un enfant atteint d'une maladie génétique entraînant la mort dès les premières années de la vie et reconnue comme incurable au moment du diagnostic ;
- le pronostic vital de cet enfant peut être amélioré, de façon décisive, par l'application d'une thérapeutique ne portant pas atteinte à l'intégrité du corps de l'enfant né du transfert de l'embryon in utero ;
- le diagnostic préimplantatoire a pour seuls objets de rechercher la maladie génétique ainsi que les moyens de la prévenir et de la traiter, et de permettre l'application de la thérapeutique susmentionnée ;
- toutes les autres possibilités thérapeutiques offertes par les articles L. 1241-1 à L. 1241-7 du code de la santé publique - à savoir les greffes de cellules hématopoïétiques, de sang de cordon ou de sang placentaire à partir d'un donneur intrafamilial ou volontaire - doivent avoir été épuisées. Cette condition a été introduite en 2011 par un amendement issu du Sénat ;
- enfin, s'appliquent les dispositions de l'article L. 2141-3 du code de la santé publique sur l'interdiction du cumul embryonnaire applicables à toutes les démarches d'assistance médicale à la procréation, selon lesquelles un couple dont des embryons ont été conservés ne peut bénéficier d'une nouvelle tentative de fécondation in vitro avant le transfert de ceux-ci, sauf si un problème de qualité affecte ces embryons.
La réalisation du diagnostic est soumise à la délivrance d'une autorisation par l'Agence de la biomédecine.
2. Une technique lourde qui n'est plus pratiquée depuis 2014
• Dans son rapport au Parlement sur l'application de la loi de bioéthique de janvier 2018, l'Agence de la biomédecine rappelle que l'hôpital Antoine Béclère, le seul à avoir pratiqué le DPI associé au typage HLA en France, a cessé depuis 2014 cette activité .
Jugée « longue et lourde, tant pour les couples que pour l'équipe médicale » , cette technique ne présente en outre qu'une faible probabilité (3 chances sur 16 en théorie, mais seulement 11% en pratique pour des raisons notamment de recombinaison génétique 315 ( * ) ) d'identifier un embryon à la fois indemne de la maladie génétique recherchée et HLA-compatible avec un aîné malade. Comme l'a rappelé la ministre lors des débats à l'Assemblée nationale, cette probabilité est encore plus faible si l'on prend en compte les chances de succès d'une fécondation in vitro, qui sont de 20%.
En outre, comme le souligne l'Agence de la biomédecine, « les couples ont tendance à refuser l'implantation d'embryons seulement indemnes de la maladie, et cela leur interdit d'accéder à une nouvelle fécondation in vitro » .
• Entre 2006 et 2014, 38 demandes d'autorisations de DPI-HLA ont été accordées . La démarche a été entreprise par 25 couples, donnant lieu à 59 stimulations ovariennes et à l'analyse de 135 embryons.
Au final, les naissances obtenues dans ce cadre ont permis d'envisager la greffe de 3 enfants . Dans le même temps, 8 couples ont tenté une grossesse spontanée : ils ont donné naissance à 7 enfants indemnes, dont 3 compatibles permettant d'envisager également 3 greffes 316 ( * ) .
II - La position de la commission
• La commission spéciale mesure les questions éthiques attachées à la technique du DPI-HLA , dont la dénomination courante - cependant ambigüe et contestable - de « bébé-médicament » ou, de manière plus élégante, « bébé du double espoir », révèle les enjeux.
Ces questionnements éthiques, conjugués aux difficultés techniques ayant conduit à l'abandon de cette pratique en France depuis 2014, avaient ainsi conduit nos collègues Annie Delmont-Koropoulis et Jean-François Eliaou, rapporteurs au nom de l'OPECST, à en préconiser l'abrogation 317 ( * ) .
Il est vrai que la complexité technique et administrative de la procédure pour les équipes de soins ainsi que son coût, ainsi que ses délais de mise en oeuvre (plus de 24 mois en 2014 à l'instar des délais d'attente du DPI « classique »), la rendent peu compatible avec la situation médicale et le pronostic de l'enfant malade en attente de greffe.
• Pour autant, les raisons ayant conduit le Parlement à autoriser cette procédure exceptionnelle en 2004, en la confirmant en 2011 selon des modalités strictement définies, appellent à mettre en doute les motifs invoqués pour supprimer aujourd'hui la possibilité de sa mise en oeuvre .
Comme l'ont confirmé les experts comme les associations entendus par la rapporteure, le recours à une greffe de moelle intrafamiliale demeure une option thérapeutique pertinente pour certaines maladies du sang telles que des hémoglobinopathies sévères (drépanocytose ou thalassémie) ou l'anémie de Fanconi, et même la plus efficace. La ministre en charge de la santé l'a confirmé lors des débats à l'Assemblée nationale : les greffes, lorsqu'elles sont réalisées avec des donneurs volontaires compatibles, donnent de moins bons résultats que des greffes réalisées à partir de frères ou de soeurs compatibles.
Ainsi, l'évolution des thérapeutiques disponibles ou des connaissances scientifiques depuis 2011 n'a pas rendu caduque le recours, en solution de dernier recours comme le prévoit expressément la loi, au DPI-HLA, au point de justifier son abrogation .
Comme l'a confirmé la direction générale de la santé, la collecte et le stockage d'unités de sang placentaire augmentent théoriquement les chances de trouver un greffon compatible mais, en pratique, la probabilité reste extrêmement faible.
Dans ce contexte, il ne faut pas sous-estimer la violence du message renvoyé aux 38 familles ayant sollicité le recours à cette pratique pendant les neuf années d'activité du centre de Clamart-Necker ainsi qu'aux enfants nés. L'abrogation du DPI-HLA a ainsi suscité l'incompréhension chez les associations entendues. Les questionnements éthiques qui entourent cette pratique supposent de l'encadrer, mais ne sauraient conduire à dévaluer le désir d'enfant d'un couple qui se conjuguerait avec un espoir de soigner un ainé malade . Le caractère exceptionnel de la procédure, du fait des maladies rares concernées et de la complexité de la démarche, ne suffit pas à en caractériser, en outre, l'inutilité.
L'abrogation du dispositif priverait des couples français de la possibilité de prise en charge par la sécurité sociale des démarches engagées à l'étranger 318 ( * ) , dans des pays comme la Belgique où le DPI-HLA est autorisé 319 ( * ) . D'après les informations données à la rapporteure, une famille aurait sollicité à cette fin l'assurance maladie en 2018.
Enfin, le fait qu'aucun centre ne propose cette pratique en France depuis 2014 ne doit pas être un motif suffisant pour en écarter, définitivement, la possibilité . Elle interroge plutôt sur les difficultés que rencontrent aujourd'hui les centres de DPI, en raison notamment d'un manque de moyens et de problèmes d'organisation structurels. D'après les indications de l'Agence de la biomédecine, il n'est pas exclu qu'un autre centre souhaite mettre en oeuvre cette procédure. Or, il serait regrettable de perdre une compétence technique en interdisant définitivement cette pratique.
Pour l'ensemble de ces raisons, la commission spéciale a décidé de maintenir l'autorisation du DPI-HLA et a ainsi adopté, à l'initiative de sa rapporteure, l'amendement COM-145 de suppression de l'article.
La commission a supprimé cet article.
Article 19 bis
Etat des lieux du diagnostic prénatal
et du
diagnostic préimplantatoire par l'Agence de biomédecine
Cet article, inséré par l'Assemblée nationale, sollicite un état des lieux du diagnostic préimplantatoire et du diagnostic prénatal. La commission spéciale l'a supprimé en considérant que cette mission incombe déjà à l'Agence de la biomédecine.
I - Le dispositif adopté par l'Assemblée nationale
Cet article a été adopté par la commission spéciale à l'initiative de Jean-François Eliaou, rapporteur, avec un avis de sagesse du Gouvernement.
Il sollicite la réalisation, par l'Agence de la biomédecine, d'un état des lieux du diagnostic préimplantatoire et du diagnostic prénatal , avant le réexamen de la loi prévu dans un délai de cinq ans par l'article 32.
II - La position de la commission
L'Agence de la biomédecine réalise, dans son rapport médical et scientifique, des rapports annuels d'activité du diagnostic préimplantatoire et du diagnostic prénatal : ce bilan portant sur la mise en oeuvre des diagnostics préimplantatoire et prénatal est expressément prévu par l'article L. 1418-1-1 du code de la santé publique introduit par la loi de bioéthique de 2011.
L'Agence a par ailleurs, au titre de ses missions, celle « d'assurer une information permanente du Parlement et du Gouvernement sur le développement des connaissances et des techniques pour les activités relevant de sa compétence et de leur proposer les orientations et mesures qu'elles appellent » 320 ( * ) . Elle apportera ainsi sa contribution à la préparation du réexamen de la loi, comme elle l'a fait dans son rapport de janvier 2018, sans qu'il soit besoin de solliciter un rapport spécifique.
La demande d'état des lieux formulée par cet article apparaît donc superfétatoire . Pour ces raisons, la commission spéciale a adopté l'amendement COM-146 de sa rapporteure de suppression de l'article.
La commission a supprimé cet article.
Article 19 ter (nouveau)
Expérimentation du diagnostic
préimplantatoire
pour la recherche d'aneuploïdies
Cet article additionnel, inséré par la commission spéciale, autorise à titre expérimental le diagnostic préimplantatoire pour la recherche d'aneuploïdies (DPI-A) en vue d'améliorer l'efficience de l'assistance médicale à la procréation dans certaines indications ciblées.
• Le diagnostic préimplantatoire (DPI) est un examen génétique d'une à deux cellules prélevées sur des embryons entre le troisième et le cinquième jour après une fécondation in vitro.
Autorisé en France par la première loi de bioéthique de 1994 321 ( * ) , il ne peut être mis en oeuvre qu' « à titre exceptionnel » et dans des conditions strictement encadrées par l'article L. 2131-4 du code de la santé publique :
- d'une part, un médecin exerçant dans un centre pluridisciplinaire de diagnostic prénatal doit attester que le couple, du fait de sa situation familiale, a une forte probabilité de donner naissance à un enfant atteint d'une maladie génétique d'une particulière gravité reconnue comme incurable au moment du diagnostic ;
- d'autre part, le diagnostic ne peut être effectué que lorsqu'a été préalablement et précisément identifiée, chez l'un des parents ou l'un de ses ascendants immédiats dans le cas d'une maladie gravement invalidante, à révélation tardive et mettant prématurément en jeu le pronostic vital, l'anomalie ou les anomalies responsables d'une telle maladie.
Le diagnostic est ainsi ciblé sur l'anomalie identifiée, responsable d'une affection dont l'un des parents ou ascendants est lui-même porteur.
D'après les données de l'Agence de la biomédecine, 797 demandes de DPI ont été acceptées en 2017 sur 1 018 demandes examinées par les cinq centres autorisés par cette même Agence à pratiquer cette activité 322 ( * ) . 270 enfants sont nés vivants à l'issue d'un DPI la même année.
• Les travaux préparatoires au réexamen de la loi de bioéthique ont introduit un débat sur l'opportunité d'élargir le DPI à la recherche d'anomalies chromosomiques (ou aneuploïdies), au-delà des seules anomalies préalablement identifiées dans la famille.
En effet, le développement des technologies de séquençage à haut débit du génome rendent plus simple et moins coûteuse l'analyse du génome entier à partir d'une cellule embryonnaire, posant alors l'enjeu, comme le relevait l'Agence de la biomédecine dans son rapport d'information sur les évolutions scientifiques et technologiques, de devoir « « dégrader » l'information obtenue par la technique pour ne connaître le résultat que sur la maladie d'une particulière gravité qui fait l'objet du DPI. »
En outre, les sociétés savantes en médecine de la reproduction 323 ( * ) et cliniciens de l'assistance médicale à la procréation soutiennent le recours à cette technique dans des situations spécifiques, afin d'améliorer la prise en charge de leurs patientes ayant des antécédents d'échec d'implantation embryonnaire ou fausses-couches à répétition ou dont l'âge les prédispose à des anomalies responsables d'échec d'implantation ou d'échec de développement embryonnaire . Cette position est également soutenue par l'association des cytogénéticiens de langue française.
C'est ce qui a conduit le CCNE, dans son avis n° 129, à préconiser l'autorisation du DPI indépendamment d'un contexte de maladie familiale, à savoir pour la recherche d'anomalies chromosomiques prédictives d'échecs de FIV, associée ou non au diagnostic préimplantatoire « classique ».
Comme l'a rappelé le Professeur Michaël Grynberg, chef du service d'AMP de l'hôpital Antoine-Béclère, lors de son audition par la rapporteure, le recours au DPI-A ne permettrait pas d'augmenter les taux de naissance en AMP, mais d'arriver plus rapidement à une grossesse en implantant en premier l'embryon le plus susceptible de se développer, afin d'éviter plusieurs échecs douloureux pour la femme et pour le couple. C'est également ce qu'a mis en avant le Professeur René Frydman lors de son audition par la commission spéciale : « Allons-nous augmenter les taux de succès ? Paradoxalement non, mais nous allons améliorer le management. Pourquoi transférer un embryon alors qu'il mènera à un échec ou à une fausse couche ? (...) Nous cherchons à éviter des échecs répétés. À une époque sensible aux féminicides et atteintes faites aux femmes, je le considère comme une violence psychique, physique et économique. » 324 ( * )
Certes, comme l'ont rappelé l'Agence de la biomédecine comme le Professeur Julie Steffann, chef du service de génétique moléculaire à l'hôpital Necker, les données de la littérature scientifique sont contrastées en termes d'efficacité et de population cible. Le risque identifié serait notamment d'aboutir à des « faux négatifs », c'est-à-dire des embryons sur lesquels une anomalie chromosomique non viable aurait été détectée mais donnant naissance à un enfant indemne, en raison d'un phénomène de « mosaïque » 325 ( * ) .
La poursuite de la recherche, notamment dans le cadre du programme hospitalier de recherche clinique (PHRC) évoqué par la ministre lors des débats à l'Assemblée nationale, permettra ainsi d'affiner les indications médicales les plus pertinentes.
Il serait toutefois regrettable de reporter plus encore le recours en France à cette technique pratiquée « en routine » dans d'autres pays européens, comme l'Espagne.
• Le présent article additionnel, issu de l'adoption de l'amendement COM-166 de la rapporteure, vise à autoriser, à titre expérimental et dans des conditions strictement encadrées, le diagnostic préimplantatoire pour la recherche d'aneuploïdies (DPI-A) non compatibles avec le développement embryonnaire .
Ce DPI-A n'aurait pas vocation à concerner l'ensemble des femmes ayant recours à l'AMP , mais seulement celles répondant à des critères médicaux - en raison de leur âge 326 ( * ) et/ou d'antécédents de fausses-couches ou d'échecs de FIV - qui auront été préalablement définis après avis de l'Agence de la biomédecine et en concertation avec les sociétés savantes.
Ce diagnostic ne pourrait avoir pour seul objectif d'améliorer l'efficience de la procédure d'assistance médicale à la procréation, à l'exclusion de la recherche du sexe de l'enfant à naître.
Il serait bien entendu réalisé sous réserve du consentement des intéressés, après que ceux-ci ont reçu une information complète sur les conditions, les risques et les limites de la démarche.
Pour des conditions de recevabilité financière au regard de l'article 40 de la Constitution, il est précisé que ce DPI-A ne puisse donner lieu à prise en charge par l'assurance maladie. Si des résultats probants étaient confirmés dans certaines indications, cette prise en charge serait toutefois pleinement justifiée et indispensable afin d'assurer l'équité dans l'accès à cette technique.
Cet article additionnel ouvre ainsi la voie à des avancées au service de la recherche et de la qualité de la prise en charge des patientes en AMP, alors que moins d'une tentative sur cinq aboutit.
La commission a adopté cet article additionnel ainsi rédigé.
Article 19 quater (nouveau)
Réalisation en première intention
d'un examen
des caractéristiques génétiques chez le
nouveau-né
dans le cadre du dépistage néonatal pour
la recherche d'anomalies
génétiques ciblées
susceptibles de mesures de prévention ou de soins
Cet article, inséré par la commission, vise à permettre de proposer aux parents, dans le cadre du dépistage néonatal, la recherche en première intention par le biais d'un examen des caractéristiques génétiques d'anomalies génétiques ciblées pouvant être responsables d'une affection d'une particulière gravité susceptible de mesures de prévention ou de soins.
Certaines maladies génétiques d'une particulière gravité font aujourd'hui l'objet de thérapies géniques prometteuses qui représentent un véritable gain de chances pour les personnes concernées lorsque ces thérapies sont administrées à un stade précoce chez le jeune enfant, idéalement avant l'apparition des premiers symptômes. C'est le cas notamment pour certaines formes de l'amyotrophie spinale infantile.
Le dépistage de ces maladies génétiques ne peut néanmoins être réalisé par le biais des tests enzymatiques biochimiques réalisés en première intention dans le cadre du dépistage néonatal à partir d'une goutte de sang prélevé sur le nouveau-né (test de Guthrie ou test néonatal du buvard).
Le programme national de dépistage néonatal
L'arrêté du 22 février 2018 relatif à l'organisation du programme national de dépistage néonatal recourant à des examens de biologie médicale prévoit la recherche des maladies suivantes :
* pour l'ensemble des nouveaux-nés :
- la phénylcétonurie ;
- l'hypothyroïdie congénitale ;
- la mucoviscidose ;
* pour les nouveaux-nés à partir de 32 semaines d'aménorrhée ;
- l'hyperplasie congénitale des surrénales ;
* pour les nouveaux-nés présentant un risque particulier de développer la maladie :
- la drépanocytose.
Généralement réalisé 72 heures après la naissance, au plus tôt à 48 heures de vie du nouveau-né, le test de Guthrie, gratuit, est obligatoirement proposé aux parents. En cas de résultat positif, des examens complémentaires, notamment génétiques, seront requis pour confirmer le diagnostic. Il s'agit de détecter des maladies rares susceptibles de mesures de prévention ou de soins. À titre d'exemple, la détection très tôt de la phénylcétonurie permet d'adapter le régime alimentaire du nourrisson en diminuant le taux de phénylalanine et de prévenir ainsi l'apparition de problèmes de développement cognitif.
Source : Commission spéciale
Le dépistage de ces maladies nécessite en effet un examen ciblé de génétique moléculaire qui permet d'identifier la présence d'une mutation génétique bien précise. Ce test, peu coûteux (de l'ordre de quatre euros), permettrait de faire bénéficier les nouveaux-nés concernés par cette mutation de traitements qui amélioreraient significativement leurs espérance et qualité de vie. Dans la mesure où les examens génétiques ne peuvent être pratiqués qu'en cas de symptôme déjà présent de la maladie ou d'antécédent familial, il est nécessaire de déroger aux articles 16-10 du code civil et L. 1131-1 du code de la santé publique pour les pratiquer en première intention.
Les thérapies disponibles dans le traitement de l'amyotrophie spinale
• L'oligonucléotide nusinersen (Spinraza®)
Ce produit, développé par les laboratoires Ionis Pharmaceuticals et Biogen, cible le gène SMN2 pour lui faire fabriquer la protéine SMN, qui est manquante dans l'amyotrophie spinale. Après avoir obtenu l'autorisation européenne de mise sur le marché en juin 2017, le nusinersen (Spinraza®) peut être prescrit en France, au sein des consultations spécialisées Maladies neuromusculaires, et remboursé pour les personnes atteintes d'amyotrophie spinale proximale de type I, II et III.
Si les résultats de l'essai « ENDEAR » et de l'essai « CHERISH » (sur lesquels est basée l'autorisation de mise sur le marché) ont montré une efficacité du Spinraza® sur les capacités motrices des nourrissons et enfants atteints de SMA 327 ( * ) (âgés de moins de 9 ans), d'autres essais cliniques sont encore en cours pour évaluer à long terme l'efficacité et la sécurité du nusinersen dans l'amyotrophie spinale proximale liée à SMN1 et de nombreuses équipes de cliniciens publient leur expérience en pratique clinique avec le nusinersen.
• La thérapie génique Zolgensma® (AVXS-101)
La thérapie génique consiste à remplacer le gène SMN1 défectueux par un gène SMN1 thérapeutique. Développé par le laboratoire AveXis et maintenant par le laboratoire Novartis, le Zolgensma® est né de recherches menées à Généthon par l'équipe de Martine Barkats.
Les résultats de différents essais du Zolgensma® montrent que le produit de thérapie génique est bien toléré et qu'il améliore la survie et les performances motrices dans la SMA. En mai 2019, le Zolgensma® a reçu l'autorisation de mise sur le marché pour les enfants atteints d'amyotrophie spinale infantile proximale liée à SMN1 de moins de deux ans, aux États-Unis. Une demande d'autorisation de mise sur le marché auprès des autorités de santé européenne et japonaise est en cours d'examen.
En France, depuis mars 2019, des nourrissons atteints de SMA de type I ont pu bénéficier du traitement pour la première fois, dans le cadre de l'essai « STR1VE-EU » et d'autorisations temporaires d'utilisation (ATU) nominatives. Des essais cliniques sont en cours aux États-Unis afin d'évaluer le produit chez des patients SMA type II et des bébés asymptomatiques.
• Le composé RO7034067 (risdiplam ou RG7916)
Le RO7034067 (RG7916 ou risdiplam), développé par le laboratoire Roche en partenariat avec PTC Therapeutics, augmente la quantité de protéine SMN fabriquée à partir du gène SMN2.
Plusieurs essais internationaux sont actuellement en cours pour évaluer la sécurité d'utilisation, la tolérance, le devenir dans l'organisme (pharmacocinétique) et l'efficacité du risdiplam chez des personnes atteintes de SMA de type II ou III (essai « SUNFISH » et « JEWELFISH ») et chez des nourrissons atteints de SMA de type I (essai « FIREFISH »). Des résultats préliminaires de l'essai « SUNFISH » et de l'essai « FIREFISH », annoncés en juin 2017 et juin 2018 (lors des conférences « Cure SMA ») ont confirmé que le candidat-médicament est bien toléré. Les essais continuent afin d'évaluer son efficacité à plus long terme.
En novembre 2019, les laboratoires Roche et PTC Therapeutics annoncent avoir déposé une demande d'autorisation de mise sur le marché auprès des autorités règlementaires américaines.
Source : AFM-Téléthon ( http://www.afm-telethon.fr/amyotrophie-spinale-proximale-liee-gene-smn1-1019 )
Votre commission a donc adopté un amendement COM-198 de la rapporteure tendant à autoriser la réalisation d'examens génétiques en première intention, dans le cadre du dépistage néonatal, pour la recherche chez le nouveau-né d'anomalies génétiques pouvant être responsables d'une affection grave justifiant de mesures de prévention ou de soins. Une liste des anomalies génétiques susceptibles d'être recherchées dans le cadre d'un examen des caractéristiques génétiques réalisé en première intention chez le nouveau-né sera ainsi fixée par arrêté du ministre chargé de la santé, après avis de l'agence de la biomédecine et de la Haute Autorité de santé.
La commission a adopté cet article additionnel ainsi rédigé.
Article 20
Suppression du délai de réflexion
dans
l'interruption de grossesse pour raison médicale
et encadrement de la
réduction embryonnaire ou foetale
Cet article supprime la proposition systématique à la femme d'un délai de réflexion d'au moins une semaine en cas d'interruption médicale de grossesse pour motif foetal et encadre les pratiques de réduction embryonnaire ou foetale en cas de grossesse multiple.
La commission spéciale a ouvert la possibilité pour les femmes susceptibles de recourir à une interruption de grossesse pour motif médical ou à une réduction embryonnaire ou foetale de désigner soit un médecin soit une sage-femme pour participer à la concertation pluridisciplinaire sur ces types d'intervention.
I - Le dispositif proposé
1. La suppression de la proposition systématique d'un délai de réflexion en cas d'interruption médicale de grossesse pour motif foetal
L'article L. 2213-1 du code de la santé publique prévoit aujourd'hui qu'un délai de réflexion d'au moins une semaine doit être proposé à la femme qui envisage une interruption de grossesse au motif qu'il existe une forte probabilité que l'enfant à naître soit atteint d'une affection d'une particulière gravité reconnue comme incurable au moment du diagnostic, une fois que cette probabilité a été attestée par deux médecins membres d'une équipe pluridisciplinaire. Ce délai, introduit à l'occasion de l'adoption de la loi relative à la bioéthique de 2011, n'est cependant proposé qu'« hors urgence médicale ».
Or des incertitudes pèsent sur le point de départ d'un tel délai : il n'est en effet pas précisé s'il doit être calculé à compter de l'annonce du diagnostic d'une anomalie chez le foetus susceptible d'être responsable d'une affection d'une particulière gravité, ou à compter du souhait exprimé par la femme de recourir à une interruption médicale de grossesse (IMG). En outre, la mise en oeuvre de ce délai emporte le recueil d'un consentement supplémentaire pour l'équipe médicale, permettant d'établir si la femme a accepté ou a refusé de bénéficier de ce délai de réflexion.
Par ailleurs, la décision par la femme de procéder à une IMG intervient nécessairement à l'issue d'un processus comprenant des examens complémentaires évaluant le pronostic d'une pathologie particulièrement grave chez un foetus présentant une anomalie. À l'annonce du diagnostic succèdent généralement un temps d'explications délivrées par l'équipe médicale à la femme ou aux membres du couple, un temps d'échanges entre l'équipe médicale et la patiente et un temps de réflexion qui peuvent s'étaler sur plusieurs jours. Enfin, une fois actée la décision de la femme, l'organisation de la prise en charge exige un temps de préparation médicale incompressible qui peut prendre deux à trois jours. Dans les faits, une IMG est donc, en règle générale, pratiquée au moins une semaine après l'annonce du diagnostic de l'anomalie foetale.
Dès lors, considérant que l'observance d'un délai de réflexion d'au moins une semaine avant la réalisation d'une IMG ne tient pas compte de la temporalité individuelle de chaque décision d'IMG et semble peu pertinente au regard des conditions pratiques de mise en oeuvre d'une telle intervention, l'article 20 du projet de loi propose de supprimer l'obligation pour l'équipe médicale de proposer systématiquement ce délai.
2. L'encadrement des réductions embryonnaires ou foetales en cas de grossesse multiple
Les interruptions volontaires partielles en cas de grossesse multiple ne font, à l'heure actuelle, l'objet d'aucun encadrement législatif ou réglementaire.
Une grossesse multiple peut en effet présenter un risque tant pour la santé de la mère que pour celle des embryons ou des foetus. Selon les informations transmises à votre commission, une réduction embryonnaire est généralement envisagée en cas de grossesses triples ou de rang supérieur, résultant en majorité d'une assistance médicale à la procréation (de 70 % à 80 % des cas).
En ce qui concerne la santé des embryons ou des foetus, le risque principal tient à la probabilité d'une grande prématurité et de ses conséquences à court terme (risque accru de mortalité périnatale) et long terme (risques de séquelles neuro-développementales). Les spécialistes de médecine foetale ont indiqué que la réduction embryonnaire ne permet pas de prévenir le risque de fausse-couche, qui est plus élevé en cas de grossesse multiple qu'en cas de grossesse « singleton ». En revanche, la réduction embryonnaire permet une réduction substantielle du risque d'accouchement prématuré : elle permet de réduire par deux le risque d'accouchement survenant entre 29 et 32 semaines.
Il a été indiqué à votre commission que la réduction embryonnaire peut être envisagée en cas de grossesse gémellaire dans des contextes maternels particuliers : malformation utérine en elle-même à l'origine d'un haut risque d'accouchement prématuré, pathologie maternelle antérieure à la grossesse exposant la mère à un sur-risque significatif en cas de grossesse gémellaire...
L'article 20 du projet de loi propose ainsi d'encadrer, en introduisant un II au sein de l'article L. 2213-1 du code de la santé publique, les interruptions volontaires partielles de grossesse multiple lorsqu'elles permettent de réduire les risques pour la santé de la femme, des embryons ou des foetus. Celles-ci ne pourront être réalisées que dans les conditions suivantes :
- la réduction embryonnaire ou foetale ne peut intervenir qu'avant la fin de la douzième semaine de grossesse, soit jusqu'à la quatorzième semaine d'aménorrhée. Les risques associés aux grossesses multiples pour la santé maternelle, embryonnaire ou foetale se situent, en effet, entre la 10 e et la 12 e semaine d'aménorrhée. Il convient de rappeler qu'une interruption partielle de grossesse est d'autant plus dangereuse pour la poursuite de la grossesse que cette interruption intervient tardivement. Les interruptions de grossesse qui seraient réalisées au-delà de la douzième semaine de grossesse répondraient à des problématiques différentes tenant à la mise en péril grave de la santé de la mère ou à une anomalie foetale d'une particulière gravité pouvant justifier une IMG à tout moment de la grossesse. Il n'est pas à exclure qu'une interruption partielle soit nécessaire après la douzième semaine de grossesse, notamment lorsque le caractère multiple de la grossesse menace la grossesse dans son ensemble et peut occasionner chez la mère des risques pour sa santé physique et mentale d'une particulière gravité : dans ces cas précis, l'interruption partielle interviendrait dans le cadre d'une IMG pour motif maternel ;
- deux médecins membres d'une équipe pluridisciplinaire doivent attester que les conditions médicales, notamment obstétricales et psychologiques, sont réunies pour une telle intervention, après l'avis d'une équipe pluridisciplinaire comprenant au moins un médecin qualifié en gynécologie-obstétrique, membre d'un centre pluridisciplinaire de diagnostic prénatal, un médecin choisi par la femme, un médecin qualifié en psychiatrie ou, en l'absence de médecin psychiatre, un psychologue ;
- une réduction embryonnaire ou foetale ne peut être entreprise sur la base d'aucun critère relatif aux caractéristiques des embryons ou des foetus, y compris leur sexe.
II - Les modifications adoptées par l'Assemblée nationale
Le motif de mise en péril de la santé des embryons ou des foetus pour justifier une réduction embryonnaire ou foetale avait été supprimé, dans un premier temps, par la commission, à l'initiative de son rapporteur, Jean-François Eliaou. L'Assemblée nationale a néanmoins rétabli ce motif en séance, par la voie d'un amendement du Gouvernement. Il est en effet établi que le caractère multiple d'une grossesse peut présenter des risques pour le développement des embryons ou foetus et justifier une interruption volontaire partielle d'une grossesse multiple.
En outre, l'Assemblée nationale a adopté un amendement de notre collègue députée Anne-France Brunet tendant à simplifier la composition de l'équipe pluridisciplinaire chargée d'examiner la demande de réduction embryonnaire ou foetale. Dans un souci d'uniformisation des procédures d'examen des demandes d'IMG ou de réduction embryonnaire ou foetale, cette équipe serait celle d'un centre pluridisciplinaire de diagnostic prénatal qui se sera adjoint, lorsqu'il l'estime nécessaire, le concours d'un psychiatre ou, à défaut d'un psychologue.
L'article R. 2131-12 du code de la santé publique prévoit en effet déjà que chaque centre pluridisciplinaire de diagnostic prénatal est constitué d'une équipe composé d'au moins :
- un médecin spécialisé en gynécologie-obstétrique ;
- un praticien spécialisé en échographie du foetus ;
- un pédiatre ;
- un médecin qualifié en génétique ;
- un psychiatre ou un psychologue ;
- un médecin spécialisé en foetopathologie ;
- un conseiller en génétique.
III - La position de la commission
• L'examen des demandes d'IMG ou de réduction embryonnaire ou foetale donne lieu à une décision collégiale prise par deux médecins membres d'une équipe pluridisciplinaire afin d'attester que la poursuite de la grossesse met en péril grave la santé de la femme dans le cadre d'une IMG pour motif maternel, ou qu'il existe une forte probabilité que l'enfant à naître soit atteint d'une maladie grave incurable dans le cadre d'une IMG pour motif foetal, ou que les conditions obstétricales et psychologiques sont réunies pour une réduction embryonnaire ou foetale. Cette décision est systématiquement précédée de l'avis d'une équipe pluridisciplinaire dont la configuration varie en fonction de l'intervention envisagée :
- dans le cadre d'une IMG pour motif maternel, elle comprend un médecin qualifié en gynécologie-obstétrique, membre d'un centre pluridisciplinaire de diagnostic prénatal, un praticien spécialiste de l'affection dont la femme est atteinte, un médecin choisi par la femme et une personne qualifiée tenue au secret professionnel qui peut être un assistant social ou un psychologue ;
- dans le cadre d'une IMG pour motif foetal ou d'une réduction embryonnaire ou foetale, l'équipe pluridisciplinaire est celle d'un centre pluridisciplinaire de diagnostic prénatal, prévue à l'article R. 2131-12 précité du code de la santé publique. Dans le cas d'une demande d'IMG pour motif foetal, il est précisé qu'un médecin choisi par la femme peut, à la demande de celle-ci, être associé à la concertation.
Bien souvent, le suivi de la grossesse peut être assuré par une sage-femme qui a connaissance du contexte clinique dans lequel s'inscrit la grossesse et de la situation de la femme, et qui bénéficie donc de la confiance de cette dernière. En conséquence, votre commission a adopté un amendement visant à préciser que la femme peut désigner soit un médecin, soit une sage-femme pour faire partie de l'équipe pluridisciplinaire chargée d'examiner une demande d'IMG pour motif maternel (amendement COM-208 du rapporteur). De même, l'amendement précise que la personne désignée par la femme pour être associée à la concertation au sein de l'équipe pluridisciplinaire chargée d'examiner une demande d'IMG pour motif foetal peut être soit un médecin, soit une sage-femme.
• Votre commission a également adopté un amendement permettant de préciser qu'une réduction embryonnaire ou foetale peut être entreprise lorsque le caractère multiple met en péril le développement des embryons ou des foetus (amendement COM-85 de Patricia Schillinger). En effet, une grossesse multiple peut présenter un danger pour la santé de la femme mais également poser un risque pour le développement d'embryons ou de foetus ne présentant initialement pas de problème de santé. Ce risque peut par exemple entraîner des problèmes de développement morphologique ou des séquelles neuro-développementales pour les embryons ou les foetus et donc les enfants à naître.
• En outre, votre commission a adopté un amendement tendant à permettre à la femme de désigner un médecin ou une sage-femme pouvant être associé, si la femme le souhaite, à la concertation prenant place au sein de l'équipe pluridisciplinaire chargée d'examiner une demande de réduction embryonnaire ou foetale (amendement COM-210 du rapporteur). La femme dispose, en effet, de la possibilité de désigner un professionnel de santé « de confiance » participant à la concertation au sein de l'équipe pluridisciplinaire examinant les demandes d'IMG pour motif maternel ou foetal. Il paraît pertinent qu'elle dispose de cette faculté également dans le cadre d'une réduction embryonnaire ou foetale.
• Enfin, votre commission a adopté un amendement de précision rédactionnelle rappelant que l'avis de l'équipe pluridisciplinaire chargée d'examiner la demande de réduction embryonnaire ou foetale est consultatif, comme c'est le cas pour les demandes d'IMG pour motif maternel ou foetal (amendement COM-209 du rapporteur).
La commission a adopté cet article ainsi modifié.
Article 21
Clarification des conditions d'interruption de
grossesse
pour raison médicale pour les mineures non
émancipées
Cet article vise à clarifier les dispositions applicables en matière d'interruption de grossesse pour raison médicale pratiquée chez une femme mineure et à préciser les conditions dans lesquelles la clause de conscience des professionnels de santé s'exerce en cas d'interruption médicale de grossesse.
La commission spéciale a supprimé l'introduction dans le code de la santé publique d'une clause de conscience spécifique des professionnels de santé pour l'interruption de grossesse pour motif médical.
I - Le dispositif proposé : la clarification des dispositions applicables en matière d'interruption de grossesse pour raison médicale pratiquée chez une femme mineure
En l'état du droit, l'article L. 2213-2 du code de la santé publique se contente de renvoyer aux articles L. 2212-2 et L. 2212-8 à L. 2212-10 du même code, applicables en matière d'interruption volontaire de grossesse (IVG) pratiquée avant la douzième semaine de grossesse, afin d'encadrer les conditions de réalisation des interruptions de grossesse pratiquées pour motif médical. L'article L. 2212-7 dudit code, qui encadre les modalités de consentement de la femme mineure à une IVG, se trouve néanmoins exclu de ce renvoi. En découle une incertitude sur les dispositions applicables en matière d'interruption médicale de grossesse (IMG) chez une femme mineure qui désire garder le secret à l'égard de ses parents.
Dans un souci de clarification de la réglementation encadrant la pratique des IMG, l'article 21 du projet de loi supprime, au sein du chapitre dédié à l'IMG, le mécanisme du renvoi aux dispositions applicables en matière d'IVG avant la douzième semaine de grossesse.
Son 1° consacre ainsi l'article L. 2213-2 du code de la santé publique aux modalités de consentement de la femme mineure non émancipée à une IMG. Comme pour une IVG envisagée avant la douzième semaine de grossesse, il est prévu que le consentement de l'un des titulaires de l'autorité parentale sera recherché par le médecin. Dans le cas où la femme mineure souhaiterait garder le secret, le médecin devra s'efforcer, dans l'intérêt de celle-ci, d'obtenir son consentement pour que les parents soient consultés ou de vérifier que la femme mineure a elle-même informé ses parents. Si la femme mineure refuse d'effectuer cette démarche ou de donner son consentement au médecin pour consulter ses parents, il pourra être procédé, à la demande de l'intéressée, à l'IMG. Comme en matière d'IVG, la femme mineure désirant garder le secret à l'égard de ses parents devra se faire accompagner dans sa démarche par la personne majeure de son choix.
Le 2° de l'article 21 du projet de loi inscrit, au sein d'un nouvel article L. 2213-2-1 du code de la santé publique, les dispositions selon lesquelles :
- l'IMG ne peut être pratiquée que par un médecin, disposition déjà prévue à la seconde phrase de l'article L. 2213-2 du code de la santé publique ;
- l'IMG ne peut être pratiquée que dans un établissement de santé, public ou privé. En l'état du droit en vigueur, le renvoi à l'article L. 2212-2 du code de la santé publique rendait applicable aux IMG l'exigence d'une intervention au sein d'un établissement de santé public ou privé. L'organisation des IMG est en effet placée aujourd'hui sous la responsabilité des centres pluridisciplinaires de diagnostic prénatal (CPDPN), au nombre de 48 dont seulement deux sont des établissements privés habilités à participer au service public hospitalier.
• Les conditions d'exercice de la clause de conscience des professionnels de santé en cas d'interruption médicale de grossesse
Le 3° de l'article 21 du projet de loi introduit, dans le code de la santé publique, un article L. 2213-2-2 instituant une clause de conscience spécifique pour les professionnels de santé en matière d'IMG. Pour mémoire, le renvoi actuel à l'article L. 2212-8 rendait déjà applicable aux IMG la clause de conscience spécifique que peuvent exercer les professionnels de santé (médecins, sages-femmes, infirmiers et auxiliaires médicaux) en matière d'IVG.
II - Les modifications adoptées par l'Assemblée nationale
L'Assemblée nationale a adopté, à l'article 21 du projet de loi, des amendements de précision rédactionnelle et a procédé à une renumérotation des dispositions introduites par cet article dans le chapitre du code de la santé publique dédié à l'IMG. Elle a également adopté un amendement maintenant, dans le chapitre du code de la santé publique consacré à l'IMG, une disposition renvoyant à un décret en Conseil d'État la définition des conditions d'application du chapitre.
III - La position de la commission : l'absence de nécessité d'une clause de conscience spécifique
Votre commission considère qu'il n'y a pas lieu d'introduire, dans le code de la santé publique, une clause de conscience spécifique des professionnels de santé en matière d'IMG.
Une clause de conscience générale, permettant de ne pas accomplir un acte contraire à ses convictions, bénéficie déjà aux professionnels de santé intervenant dans les procédures d'IVG ou d'IMG :
- l'article R. 4127-47 du code de la santé publique, qui reprend l'article 47 du code de déontologie des médecins, prévoit que, « hors le cas d'urgence et celui où il manquerait à ses devoirs d'humanité, un médecin a le droit de refuser ses soins pour des raisons professionnelles ou personnelles » ;
- les sages-femmes bénéficient de la même clause de conscience générale en application de l'article R. 4127-328 du code de la santé publique. Il en va de même pour les infirmiers en application de l'article R. 4312-12 du même code.
En revanche, l'inscription dans la loi de l'obligation pour le médecin, qui exercerait sa clause de conscience en matière d'IMG, d'informer immédiatement l'intéressée de son refus et de lui communiquer le nom de praticiens susceptibles de réaliser l'IMG, permet d'en garantir l'effectivité. En conséquence, votre commission a adopté un amendement tendant à supprimer, au sein du nouvel article L. 2213-4 du code de la santé publique prévu par l'article 21 du projet de loi, la mention selon laquelle « un médecin n'est jamais tenu de pratiquer une interruption de grossesse pour motif médical ».
Afin de ne maintenir dans la loi que le principe selon lequel tout refus de pratiquer une IMG s'accompagne d'une obligation de référer la patiente à un praticien susceptible de réaliser l'intervention, est ainsi préférée la formulation suivante : « un médecin qui refuse de pratiquer une interruption de grossesse pour motif médical doit informer, sans délai, l'intéressée de son refus et lui communiquer immédiatement le nom de praticiens susceptibles de réaliser cette intervention » (amendement COM-211 du rapporteur).
Par coordination, l'amendement supprime toute mention d'une clause de conscience spécifique à l'IMG pour les autres professionnels de santé, les sages-femmes et les infirmiers bénéficiant déjà, comme les médecins, d'une clause de conscience générale.
La commission a adopté cet article ainsi modifié.
Article 21 bis
Prise en charge des enfants présentant une variation
du développement génital
Cet article propose d'inscrire dans la loi l'orientation systématique des enfants présentant une variation du développement génital vers les centres de référence des maladies rares du développement génital, afin qu'ils puissent être pris en charge après concertation d'équipes pluridisciplinaires spécialisées. La commission a intégré dans le dispositif tous les centres de référence des maladies rares compétents et précisé que le diagnostic et la prise en charge donneraient lieu à l'édiction de recommandations de bonnes pratiques à des fins d'harmonisation.
I - Le dispositif adopté par l'Assemblée nationale : une orientation systématique des enfants vers les centres de références dédiés
1. Un dispositif destiné aux enfants présentant une « variation du développement génital »
La notion de variations du développement génital renvoie à des situations médicales congénitales caractérisées par un développement atypique du sexe chromosomique (ou génétique), gonadique (c'est-à-dire des glandes sexuelles, testicules ou ovaires) ou anatomique (le sexe morphologique visible). Ces situations correspondent soit à un arrêt avant son terme d'une des voies de différentiations, masculine ou féminine, sans stimulation de l'autre, soit à la stimulation simultanée des deux voies, l'une prédominant sur l'autre sans qu'aucune des deux ne soit aboutie 328 ( * ) .
L'appellation de « variations du développement génital » a été utilisée par le Conseil d'État dans son étude du 28 juin 2018 329 ( * ) . Pour le Défenseur des droits, elle serait trop réductrice, puisqu'elle limiterait le sexe d'un individu aux seuls organes génitaux extérieurs 330 ( * ) . Le Comité consultatif national d'éthique pour les sciences de la vie et de la santé (CCNE) a, quant à lui, choisi de retenir les termes de « variations du développement sexuel », adoptés en Suisse. C'est cette terminologie que la délégation aux droits des femmes a retenue dans son rapport remarqué de 2017 intitulé « Variations du développement sexuel : lever un tabou, lutter contre la stigmatisation et les exclusions » 331 ( * ) .
Plutôt que « variations » du développement génital, les médecins préfèrent pour leur part l'emploi du terme « anomalies » ou « différences ». En tout état de cause, ces situations doivent être distinguées des cas de dysphorie de genre 332 ( * ) que les qualificatifs d'« intersexes » ou « intersexuées » pourraient comprendre.
De même qu'il n'existe pas de consensus sur les termes à employer, il n'existe pas, à ce jour, de consensus au sein du monde médical sur le type de traitements à effectuer ainsi que sur le moment auquel ils doivent intervenir , comme l'a rappelé le Conseil d'État dans son étude de 2018. Si les traitements visant à éviter des complications susceptibles d'engager le pronostic vital de l'enfant ne posent pas de difficulté (par exemple, les traitements hormonaux pour traiter les cas d'hyperplasie congénitale des surrénales avec perte de sel), les chirurgies effectuées sur des enfants en bas âge et visant à « normaliser » l'apparence de leurs organes génitaux, sont controversées et certaines associations et organismes internationaux de défense des droits de l'homme, tels que l'Assemblée parlementaire 333 ( * ) ou le Commissaire aux droits de l'homme du Conseil de l'Europe 334 ( * ) , appellent à les interdire.
2. Une orientation systématique des enfants vers les centres de références dédiés
L'article additionnel 21 bis a été introduit en séance publique à l'Assemblée nationale par l'adoption, avec l'avis favorable du Gouvernement, d'un amendement du groupe La République en Marche 335 ( * ) . Le lien avec le texte initial du projet de loi peut interroger 336 ( * ) .
Cet article vise à améliorer la prise en charge des enfants présentant une variation du développement génital, en les orientant systématiquement vers les centres de référence des maladies rares du développement génital afin que le diagnostic et des propositions thérapeutiques - incluant l'abstention - puissent être établis en réunions de concertation pluridisciplinaire , dans le respect du principe de proportionnalité 337 ( * ) mentionné à l'article L. 1110-5 du code de la santé publique 338 ( * ) .
Ce dispositif permettrait une prise en charge par des équipes pluridisciplinaires spécialisées et expérimentées qui auraient à connaître de l'ensemble des cas, ce qui faciliterait une réflexion globale au niveau national.
Le centre de référence des maladies rares du développement génital
Un centre de référence rassemble une équipe hospitalière hautement spécialisée ayant une expertise avérée pour une maladie rare ou un groupe de maladies rares, et qui développe son activité dans les domaines de la prévention , des soins , de l'enseignement et de la recherche . Cette équipe est médicale, mais intègre également des compétences paramédicales, psychologiques, médico-sociales, éducatives, sociales et des partenariats avec les associations de personnes malades 339 ( * ) .
Le centre de référence du développement génital : du foetus à l'adulte (DEV-GEN) a été labellisé en septembre 2006. Depuis 2017 340 ( * ) , il est composé d'un site coordonnateur (Lyon) et de trois sites constitutifs : Paris-Bicêtre, Lille et Montpellier. Ces quatre sites - parfois aussi appelés « centres de référence » - s'appuient sur un réseau de centres de compétence qui couvrent l'ensemble du territoire.
Source : https://www.developpement-genital.org/
Tous les centres de référence et de compétence ont l'obligation de renseigner BaMaRa, l'application de la Banque nationale des données maladies rares (BNDMR).
Le centre DEV-GEN fait partie de la filière maladies rares FIRENDO qui regroupe sept centres de référence s'occupant de pathologies endocriniennes rares, dont le centre de référence des maladies rares endocriniennes de la croissance et du développement (CERMERCD) avec lequel il collabore.
3. Une demande de rapport afin d'avoir une connaissance chiffrée des cas
Les données statistiques ne permettent pas de connaître précisément le nombre de naissances concernées : la Banque nationale des données maladies rares (BNDMR) renseignée par les centres de référence des maladies rares ne donne pas à ce jour des chiffres complets dans la mesure où tous les cas de variations du développement génital ne sont pas traités par ces centres.
Certaines associations évoquent le chiffre de 1,7 % des naissances concernées par ces variations. Les données les plus précises concernent les cas d'hyperplasie congénitale des surrénales (HCS) qui concerneraient environ quarante cas dépistés par an 341 ( * ) . Le centre de référence des maladies rares du développement génital évalue le nombre d'anomalies du développement génital à 1 naissance sur 4 500 à 5 000 . Il estime que parmi tous ces cas, seuls 25 ou 30 cas par an posent un problème d'assignation de genre .
Afin de remédier à cette absence de données chiffrées fiables, l'article 21 bis adopté par l'Assemblée nationale prévoit la remise au Parlement d'un rapport présentant des éléments chiffrés dans un délai de douze mois à compter de la publication de l'arrêté réservant aux centres de référence des maladies rares du développement génital la prise en charge des enfants présentant une variation du développement génital. Ce rapport permettrait un suivi statistique de l'ensemble des cas dont les centres auraient à traiter.
II - La position de la commission : soumettre le diagnostic et la prise en charge de ces enfants à des recommandations de bonnes pratiques élaborées par la HAS en concertation avec les associations de patients
Le dispositif adopté par l'Assemblée nationale met en oeuvre l'une des recommandations du Conseil d'Etat dans son étude du 28 juin 2018, ainsi qu'une recommandation du Comité consultatif national d'éthique pour les sciences de la vie et de la santé figurant dans son avis n° 132 du 19 septembre 2019 342 ( * ) qui a relevé : « la nécessité d'une centralisation des cas est essentielle, en raison de leur très grande spécificité et du faible nombre d'occurrences critiques. Le savoir-faire, l'approche pluridisciplinaire, un nombre suffisant de cas nécessaire pour favoriser le discernement dans la prise de décision , sont autant de facteurs qui justifient que toutes les situations, examinées au cas par cas, soient centralisées sur le Centre de référence, qui peut faire aussi appel aux vingt centres de compétences qui ont développé une expertise particulière dans ces domaines » . La commission y est favorable .
Elle a souhaité, sur la proposition de son rapporteur, inscrire dans la loi une autre recommandation du CCNE 343 ( * ) qui a notamment préconisé que :
- les quatre sites du centre de référence DEV GEN rapprochent leurs pratiques, pour « arriver à une position commune respectueuse de bonnes pratiques, sans exclure des questionnements inhérents à toute pratique médicale » ;
- que les échographistes comme les personnels des maternités, en néonatologie ou en pédiatrie, soient formés pour annoncer aux parents l'existence d'une variation du développement génital de la façon la plus pertinente avant que les familles ne soient orientées vers les centres de référence.
Par adoption de l'amendement COM-263 déposé par son rapporteur, la commission a souhaité inciter à une harmonisation des pratiques sur le territoire national. Elle a décidé de soumettre à la fois le diagnostic et la prise en charge des enfants présentant une variation du développement génital à des recommandations de bonnes pratiques arrêtées par la Haute Autorité de santé , dans le cadre de sa mission définie au 2° l'article L. 161-37 du code de la sécurité sociale. Conformément à la méthode de « Recommandations par consensus formalisé » mise en oeuvre par la HAS, les associations de patients seraient invitées à participer à leur élaboration , ce qui permettrait l'instauration d'un dialogue.
À l'initiative de son rapporteur, la commission a également adopté un amendement COM-246 afin :
- d'intégrer dans le dispositif tous les centres de référence des maladies rares compétents pour la prise en charge des enfants présentant une variation du développement génital, et en particulier, le centre de référence des maladies rares endocriniennes de la croissance et du développement (CERMERCD) qui s'occupe des cas d'hyperplasie congénitale des surrénales ;
- d' étendre à dix-huit mois le délai de remise du rapport au Parlement afin de permettre aux centres de référence d'avoir un regard sur une année complète d'activité.
La commission a adopté cet article ainsi modifié.
Article 22
Autorisation de greffe de tissu germinal
pour rétablir
une fonction hormonale et clarification
du devenir des gamètes et
tissus germinaux conservés
Cet article étend la finalité de conservation de gamètes ou tissus germinaux pour motif pathologique à la restauration d'une fonction endocrine au-delà de la seule finalité procréative et clarifie les dispositions relatives au devenir des gamètes ou tissus conservés dans ce cadre.
La commission spéciale a apporté plusieurs précisions visant notamment à protéger la situation des personnes mineures au moment du recueil ou du prélèvement et renforcer leur information ainsi que le suivi des personnes concernées. Elle a également autorisé expressément ce recueil en cas de changement de sexe à l'état civil.
I - Le dispositif proposé
1. Une possibilité offerte depuis 2004 dans un objectif de préservation de la fertilité de personnes malades
• La loi du 6 août 2004 relative à la bioéthique a autorisé le recueil et la conservation de gamètes ou de tissu germinal en vue de la réalisation ultérieure d'une assistance médicale à la procréation , au bénéfice d'une personne atteinte d'une pathologie ou exposée à un traitement risquant d'altérer prématurément sa fertilité.
Cette possibilité ouverte aux personnes majeures comme mineures a été étendue en 2008 344 ( * ) , au-delà de la seule finalité de réalisation d'une AMP, à la préservation et à la restauration de la fertilité, ce que permet notamment une greffe des tissus ovariens cryoconservés.
• D'après l'Agence de la biomédecine 345 ( * ) , 78 290 patients disposaient de gamètes et/ou de tissus germinaux 346 ( * ) conservés dans le cadre de ces dispositions au 31 décembre 2017, près de 30 % de plus qu'en 2016 ; il s'agit de spermatozoïdes dans 89 % des cas.
Cette activité est pratiquée dans 50 centres autorisés pour l'activité d'assistance médicale à la procréation parmi lesquels 26 centres hospitaliers universitaires, 4 sites de l'AP-HP, 5 établissements publics de santé et 17 laboratoires privés.
La conservation de gamètes et de tissus
germinaux
pour préservation de la fertilité en
2017

Source : Agence de la biomédecine, rapport médical et scientifique
Le nombre de patients concernés traduit une forte augmentation , en raison d'une meilleure information des patients mais également de l'évolution des techniques et notamment de la mise en oeuvre de la congélation ovocytaire qui a permis d'élargir progressivement les indications d'autoconservation.
Évolution du nombre de patients ayant
réalisé une autoconservation
dans le cadre de la
préservation de la fertilité dans l'année
|
2008 |
2009 |
2010 |
2011 |
2012 |
2013 |
2014 |
2015 |
2016 |
2017 |
|
|
Ovocytes |
||||||||||
|
Patientes |
19 |
17 |
41 |
132 |
202 |
324 |
493 |
799 |
1382 |
1666 |
|
Tissus ovariens |
||||||||||
|
Patientes |
160 |
224 |
235 |
320 |
264 |
277 |
338 |
323 |
301 |
346 |
|
Spermatozoïdes |
||||||||||
|
Patients |
4343 |
4434 |
4644 |
3934 |
4091 |
4190 |
4397 |
4707 |
4909 |
5339 |
|
Tissus testiculaires |
||||||||||
|
Patients |
50 |
99 |
38 |
50 |
81 |
86 |
87 |
82 |
126 |
123 |
Source : Direction générale de la santé et Agence de la biomédecine
Comme indiqué dans le bilan d'activité du Grecot 347 ( * ) , les patients concernés sont principalement atteints de cancer et appelés à subir des traitements gonadotoxiques (46% ont des maladies hématologiques malignes et 41% des tumeurs solides) ; dans 13 % des cas, il s'agit de pathologies non malignes (drépanocytose, maladies génétiques ou auto-immunes, syndrome de Turner, endométriose ou kystes de l'ovaire, etc.). Environ 27 % ont moins de 12 ans et près de la moitié (47 %) moins de 18 ans .
La même année 2017, plus de 920 tentatives d'AMP avec les gamètes conservés ont été réalisées, donnant lieu à 185 naissances. Une vingtaine de greffes de fragments d'ovaire ont lieu chaque année et un enfant est né en 2017 d'une patiente greffée après une procédure d'AMP 348 ( * ) . La réutilisation des tissus germinaux reste encore limitée, notamment car certains patients sont encore très jeunes (le prélèvement de tissu testiculaire est effectué chez des patients dont l'âge moyen est de 7 ans d'après la fédération des Cecos et peut concerner des nourrissons) ou en raison de techniques pas encore stabilisées. La greffe de tissu ovarien peut être enfin contrindiquée lorsqu'il existe un risque de réintroduire la maladie.
Le recueil et la conservation sont pris en charge pour les patients, avec un financement aux centres d'AMP autorisés dans le cadre des MIG (missions d'intérêt général).
2. Des dispositions complétées par le projet de loi dans un double objectif
a) Un élargissement de la finalité de la conservation
• La nouvelle rédaction proposée ( I ) pour le I de l'article L. 2141-11 du code de la santé publique conduit à étendre la finalité du recueil et de la conservation de gamètes ou tissus germinaux au « rétablissement d'une fonction hormonale » , au-delà du seul objectif de procréation, pour des patients n'ayant pas ou plus de désir d'enfant.
Il s'agit, comme le note l'étude d'impact, de restaurer, par la greffe de tissu ovarien, la fonction endocrine « naturelle » de l'ovaire (à savoir la sécrétion d'hormones), ce qui permet d'éviter aux patientes concernées la prise de traitements hormonaux substitutifs. La présidente du Grecot a confirmé que cette technique permettait, selon le témoignage de patientes greffées, d' améliorer leur qualité de vie .
Fin 2017, 3 244 patientes disposent de tissus ovariens conservés et seraient ainsi potentiellement concernées.
Cette faculté est également ouverte aux tissus testiculaires (concernant 708 patients fin 2017) même si, dans ce cas, l'état des connaissances scientifiques est à ce jour moins avancé.
• Comme à l'heure actuelle, le recueil et la conservation sont subordonnés au consentement de l'intéressé ou de ses représentants légaux pour un mineur. En revanche, le projet de loi apporte une précision s'agissant des majeurs protégés en matière personnelle , en requérant soit le consentement du mandataire soit celui de la personne exerçant l'habilitation familiale ou de la personne chargée de le représenter en matière personnelle.
b) Sécuriser le devenir des gamètes et tissus germinaux conservés
La nouvelle rédaction proposée pour les II, III et IV de l'article L. 2141-11 vise en outre à compléter et sécuriser le cadre juridique relatif au devenir des gamètes ou tissus germinaux conservés .
• En l'absence de dispositions législatives, ces questions sont aujourd'hui encadrées au niveau réglementaire , par les articles R. 2141-18 et R. 2141-19 du code de la santé publique 349 ( * ) .
Or, comme le souligne l'étude d'impact, « les professionnels hésitent à détruire les gamètes et tissus germinaux de crainte que leur responsabilité ne puisse être engagée » . Notamment, les dispositions réglementaires précitées n'ont pas prévu les cas dans lesquels une personne est « perdue de vue », c'est-à-dire ne répond pas, pendant une certaine durée, aux sollicitations annuelles sur le devenir des produits conservés 350 ( * ) .
Or, comme l'ont rappelé les représentants de la fédération des Blefco 351 ( * ) , les professionnels sont confrontés à un manque de place pour le stockage en azote liquide de prélèvements de plus en plus nombreux, et conservés pour certains sur un temps très long, selon des conditions strictement réglementées.
• La rédaction proposée pour l'article L. 2141-11 fixe, pour la poursuite de la conservation, deux régimes distincts selon que la personne est mineure (II) ou majeure (III et IV), qui reprennent pour l'essentiel les dispositions actuellement prévues au niveau réglementaire.
- Dans le cas d'une personne mineure, même émancipée, seul le décès peut conduire à mettre fin à la conservation . Les parents sont contactés chaque année par écrit pour recueillir les informations nécessaires à la poursuite de la conservation.
- Dans le cas d'une personne majeure, le consentement à la poursuite de la conservation doit être renouvelé chaque année par écrit. Si elle ne souhaite pas poursuivre cette conservation, trois options s'offrent à elle : proposer ses gamètes au don , permettre que ses gamètes ou tissus germinaux fassent l'objet de recherche ou mettre fin à leur conservation.
Les conditions sont analogues à celles prévues par l'article 2 du projet de loi pour les cas d'autoconservation de gamètes hors situations pathologiques : le consentement doit être confirmé dans un délai de trois mois et il est révocable jusqu'à l'utilisation ou la fin de la conservation des gamètes ou tissus germinaux.
De même, il est mis fin à la conservation en l'absence de réponse de la personne durant dix années consécutives - ce délai courant à partir de la majorité si le recueil a été fait sur une personne mineure - ou en cas de décès sans que la personne ait exprimé un consentement au don ou à la recherche (IV de la rédaction proposée pour l'article L. 2141-11).
Comme l'indique l'étude d'impact, le délai d'absence de réponse est fixé à dix ans, contre cinq ans dans le cas de la conservation d'un embryon par un couple ayant eu recours à une assistance médicale à la procréation (article L. 2141-4 du code de la santé publique), « au regard des risques de perte définitive des chances de procréation avec ses propres gamètes ou tissus germinaux en cas de destruction. »
On ne dispose pas aujourd'hui de données, cependant, quant au devenir des gamètes et tissus germinaux destinés à la recherche et au don. Les professionnels des Cecos et Blefco ont indiqué appliquer un « principe de précaution » en ne destinant pas au don des gamètes prélevés sur des personnes malades ; cette destination serait selon le Grecot néanmoins possible pour certaines pathologies non malignes.
• Le II fixe une disposition transitoire pour les gamètes et tissus germinaux conservés à la date de publication de la loi : il sera mis fin à leur conservation en cas de décès de la personne. C'est ce que prévoyait expressément le III de l'article R. 2141-18 du code de la santé publique.
La nécessité juridique de cette disposition est discutable mais vise, selon la direction générale de la santé, à conforter les équipes qui hésitent toujours à mettre fin à la conservation de ces gamètes ou tissus germinaux.
II - Les modifications adoptées par l'Assemblée nationale
• Outre des amendements rédactionnels, la commission spéciale a apporté les modifications suivantes :
- à l'initiative de Raphaël Gérard et de membres du groupe La République en marche, elle a prévu l'information systématique de la personne, lors de l'annonce de la proposition médicale, sur cette possibilité ainsi que les risques et les limites de la démarche ;
- à l'initiative de son rapporteur Jean-François Eliaou, elle a précisé, au sein des dispositions transitoires (II), que les gamètes ou tissus germinaux conservés à la date de publication de la loi ne seront détruits, en cas de décès d'une personne majeure, que si celle-ci n'a pas préalablement consenti à ce qu'ils fassent l'objet d'un don ou d'une recherche.
• En séance publique, l'Assemblée nationale a adopté, en outre :
- deux amendements identiques de Danielle Brulebois (La République en marche) et Emmanuelle Anthoine (Les Républicains), avec l'avis défavorable de la commission 352 ( * ) et favorable du Gouvernement, renvoyant à l'article 458 du code civil les conditions d'expression du consentement des majeurs protégés afin d'élargir leur sphère d'autonomie. Cette modification, portée notamment par l'Union nationale des associations familiales (UNAF), conduit à assimiler le recueil et la conservation des gamètes ou tissus germinaux aux actes « dont la nature implique un consentement strictement personnel [qui] ne peut jamais donner lieu à assistance ou représentation de la personne protégée » 353 ( * ) , alors que le projet de loi soumettait cette démarche au consentement du mandataire ou de la personne chargée de représenter le majeur protégé en matière personnelle ;
- un amendement de son rapporteur Jean-François Eliaou, avec l'avis favorable du Gouvernement, visant à permettre que les gamètes ou tissus germinaux d'une personne mineure venant à décéder puissent faire l'objet d'une recherche , avec le consentement de ses parents, comme cela est possible en cas de décès d'une personne majeure. Comme l'a souligné l'auteur de l'amendement, « cette recherche rendrait possible l'étude de la spermatogenèse, ainsi que la vérification de la non-contamination des gamètes ou tissus germinaux conservés par la maladie résiduelle - c'est-à-dire la maladie cancéreuse - et souvent prélevés dans une phase aiguë de la maladie. »
III - La position de la commission
• Les évolutions proposées par cet article consolident le cadre instauré par la loi de bioéthique de 2004 : elles répondent à des attentes des professionnels de santé concernés et contribuent à préserver l'intégrité physique des personnes atteintes de pathologies graves .
L'augmentation continue du nombre de patients pris en charge est à cet égard une évolution positive, alors qu'un rapport conjoint de l'Agence de la biomédecine et de l'Institut national du cancer avait relevé en 2013 un accès inégal à l'information et une prise en compte « insuffisante » des risques d'infertilité auxquels sont exposés ces patients, « alors qu'elle devrait être systématique » : « la révélation de l'infertilité se fait encore trop souvent au moment où précisément ils souhaitent concevoir. » 354 ( * )
Comme l'ont souligné les représentants des Cecos, l'enjeu de l'information par les médecins prescripteurs reste important. De même, l'accès équilibré sur l'ensemble du territoire à ces soins de préservation de la fertilité, relevé par ce rapport, reste un enjeu majeur pour une meilleure prise en charge des patients comme l'a relevé la présidente du Grecot, compte tenu du faible nombre de centres d'AMP autorisés.
• Tout en approuvant le sens des évolutions proposées par cet article, la commission spéciale a apporté, outre des précisions rédactionnelles (amendement COM-151 du rapporteur), plusieurs compléments à la rédaction proposée .
Dans la mesure où ces dispositions les concernent quasiment pour moitié, elle a ainsi tenu à renforcer l'implication des personnes mineures dans ces actes touchant à leur intimité et à leur intégrité physique :
- en instituant une information ad hoc par les équipes médicales du centre responsable de la conservation dans l'année où ces personnes atteignent l'âge de la majorité afin de parler de « l'après » ( amendement COM-148 du rapporteur clarifiant en outre la rédaction de la disposition introduite par l'Assemblée nationale relative à l'information délivrée aux patients - majeurs - sur la démarche). Une autre évolution souhaitable à cet égard, mais qui ne ressort pas du niveau législatif, serait de l'avis des professionnels la création d'un fichier centralisé avec un numéro unique pour renseigner facilement les personnes sur le lieu de conservation de leurs gamètes ou tissus ;
- en sollicitant leur consentement à chaque fois que leur discernement permet de l'exprimer ( amendement COM-150 du rapporteur ) ;
- en rétablissant la précision selon laquelle il ne peut être mis fin à la conservation des gamètes ou tissus d'une personne mineure qu'en cas de décès ( amendement COM-152 du rapporteur ).
Dans le même objectif, la commission spéciale a décidé de porter à vingt ans, au lieu de dix, la durée de conservation des gamètes en l'absence de réponse de la personne aux sollicitations annuelles à compter de sa majorité ( amendement COM-154 du rapporteur ). Cette proposition, qui fait écho à une recommandation du Grecot, vise à prendre en compte la situation de jeunes patients qui n'auraient pas encore formé, à 28 ans, de projet parental mais qui pourraient nourrir des regrets plus tard.
En parallèle, elle a précisé (même amendement) que le consentement au don des gamètes conservés ou à leur orientation vers la recherche en cas de décès peut être exprimé dès le consentement à la prise en charge initial, afin de favoriser l'orientation vers le don ou la recherche sans pour autant instituer, comme à l'article 16 concernant les embryons conservés dans le cadre d'une AMP, une consultation annuelle sur ce point qui pourrait être délicate s'agissant de personnes malades parfois encore en cours de traitement. Elle a également renvoyé à des recommandations de bonnes pratiques la fixation d'un âge limite à la conservation 355 ( * ) .
La commission spéciale a prévu, en parallèle, d'alléger la procédure de confirmation à trois mois du consentement, source de lourdeurs administratives, par cohérence avec des modifications similaires introduites aux articles 2 et 16 du projet de loi ( amendement COM-155 du rapporteur ).
Elle a également prévu un suivi des patients bénéficiant d'une telle prise en charge ( amendement COM-147 du rapporteur ), afin de contribuer à améliorer la connaissance scientifique sur le réel impact des traitements reçus ou de la maladie sur le fonctionnement des gonades pour mieux cibler les indications de recours à cette technique de préservation de la fertilité. Les données de l'Agence de la biomédecine ne sont que partielles à cet égard puisque ne sont suivies que les personnes ayant ensuite recours à une démarche d'AMP.
Elle a enfin introduit la précision explicite selon laquelle la modification de la mention du sexe à l'état civil ne fait pas obstacle à l'application de ces dispositions et donc au bénéfice de l'autoconservation de gamètes ou tissus germinaux pour motif pathologique ( amendement COM-17 de Laurence Cohen et des membres du groupe communiste, républicain, citoyen et écologiste).
La commission a adopté cet article ainsi modifié.
CHAPITRE II
Optimiser l'organisation des soins
Article 23
Élargissement des missions des conseillers en
génétique
Cet article ouvre la possibilité pour les conseillers en génétique, dans un contexte d'augmentation de l'activité de génétique moléculaire en France, de prescrire certains examens de génétique.
La commission spéciale a ouvert la possibilité pour les conseillers en génétique de communiquer les résultats d'un examen génétique, qu'ils comportent ou pas la découverte d'une anomalie génétique, avec l'accord et sous la supervision du médecin généticien.
I - Le dispositif proposé
1. Le conseiller en génétique : une compétence précieuse dans un contexte d'augmentation de l'activité de génétique moléculaire
• La profession de conseiller en génétique a été créée par la loi « Santé » de 2004 356 ( * ) . Dans un contexte de développement de la génétique médicale et des techniques de séquençage du génome, il s'agissait de permettre aux médecins généticiens de faire face à une augmentation des besoins des couples et familles faisant l'objet d'un examen de leurs caractéristiques génétiques et qui nécessitent d'être informés et accompagnés, en s'appuyant sur une équipe pluridisciplinaire au sein de laquelle le conseiller en génétique occuperait une place déterminante.
Aux termes de l'article L. 1132-1 du code de la santé publique, le conseiller en génétique est ainsi chargé de participer à la délivrance des informations et des conseils aux personnes et familles susceptibles de faire l'objet d'un examen de leurs caractéristiques génétiques et à la prise en charge médico-sociale, psychologique et au suivi de ces personnes.
En application de l'article L. 1132-2 du code de la santé publique, seuls peuvent exercer cette profession les titulaires d'un titre de formation figurant sur une liste arrêtée par le ministre chargé de la santé. En vertu d'un arrêté du 10 avril 2008 357 ( * ) , seuls les titulaires du master « Sciences de la santé », mention « Pathologie humaine », spécialité « Conseil en génétique et médecine prédictive », délivré par l'université Aix-Marseille, peuvent se voir délivrer, par le directeur de l'agence régionale de santé, une autorisation d'exercice de la profession de conseiller en génétique 358 ( * ) .
• Selon l'étude d'impact annexée au projet de loi, 155 conseillers en génétique exercent aujourd'hui au sein d'une équipe pluridisciplinaire, généralement en milieu hospitalier, notamment au sein des centres de référence de maladies rares, des centres pluridisciplinaires de diagnostic prénatal et des centres de lutte contre le cancer. Le nombre de généticiens médicaux était, lui, de 251 en 2017, en progression de 12,5 % par rapport à 2013. Dans le même temps, le nombre d'individus ayant bénéficié d'un examen de génétique, qui s'est établi en 2017 à 410 801, a augmenté de près de 15 % sur la période 2013-2017.
2. La nécessité de reconnaître la profession de conseiller en génétique par un élargissement de ses missions
Afin de permettre aux médecins généticiens de répondre à une demande de conseil génétique en augmentation, l'article 23 du projet de loi étend aux conseillers en génétique la possibilité de prescrire des examens de génétique pré- et postnatals :
- le I supprime ainsi, à l'article L. 1132-1 du code de la santé publique, l'obligation pour le conseiller en génétique d'exercer toujours dans le cadre d'une prescription médicale. Est maintenue, en revanche, l'exigence selon laquelle il exerce sous la responsabilité d'un médecin qualifié en génétique. Un nouvel alinéa est, en outre, inséré audit article afin de préciser que le conseiller en génétique peut prescrire certains examens de biologie médicale dans des conditions fixées par décret en Conseil d'État pris après avis de l'académie nationale de médecine ;
- par coordination, le II modifie l'article L. 4161-1 du code de la santé publique afin d'exclure les conseillers en génétique qui prescriraient des examens de génétique des personnes susceptibles d'être poursuivies pour exercice illégal de la médecine.
Selon l'étude d'impact annexée au projet de loi, la prescription d'un examen de génétique par un conseiller en génétique devrait permettre, dans certains cas, aux personnes d'obtenir la communication de leurs résultats dès leur première consultation avec le médecin généticien. En résulterait une réduction des délais d'attente.
II - Les modifications adoptées par l'Assemblée nationale
Outre un amendement rédactionnel adopté par sa commission des affaires sociales, l'Assemblée nationale a adopté, en séance, avec un avis défavorable de la commission mais un avis de sagesse du Gouvernement, un amendement de notre collègue Sereine Mauborgne 359 ( * ) (LaREM) visant à permettre au conseiller en génétique de communiquer les résultats d'un examen de génétique aux personnes concernées, sous réserve que ces résultats ne révèlent pas d'anomalie génétique et que cette communication soit réalisée en accord avec le médecin sous la responsabilité duquel il intervient. Les situations dans lesquelles le conseiller en génétique pourra communiquer les résultats seront précisées par le décret en Conseil d'État censé fixer les conditions de prescription d'examens de génétique par les conseillers en génétique.
III - La position de la commission
Votre commission souscrit à un élargissement des missions des conseillers en génétique, dans un souci d'allègement de la charge de travail pesant sur les médecins généticiens et de réduction des délais d'attente pour les personnes concernées. Les médecins généticiens et les conseillers en génétique définissent déjà ensemble, au sein de l'équipe pluridisciplinaire, les indications pour lesquelles certaines personnes peuvent être prises en charge par le conseiller en génétique de la première consultation jusqu'au rendu du résultat, la consultation par le médecin généticien pour la prescription de l'examen restant néanmoins aujourd'hui obligatoire.
Les conseillers en génétique exercent toujours sous la responsabilité d'un médecin qualifié en génétique et sollicitent régulièrement l'avis de ce dernier dans le cadre des prises en charge qu'ils assurent. Rien ne s'oppose donc à ce que les conseillers en génétique puissent non seulement prescrire des examens de génétique mais également en communiquer les résultats, pour autant que cette communication soit réalisée avec l'accord du médecin généticien.
En outre, le fait de cantonner les conseillers en génétique à l'annonce de résultats ne révélant pas d'anomalie génétique aurait pour effet de créer une asymétrie entre les patients appelés pour le rendu de leurs résultats entre, d'une part, ceux convoqués pour une consultation avec un conseiller en génétique qui anticiperont un diagnostic rassurant en l'absence d'anomalie génétique, et, d'autre part, ceux convoqués pour une consultation avec un médecin généticien, qui s'attendront d'emblée, avant même l'annonce du résultat, à un diagnostic problématique.
Par conséquent, votre commission a adopté un amendement ouvrant la possibilité aux conseillers en génétique d'annoncer le résultat d'un examen de génétique, que ce résultat comporte ou pas l'annonce d'une anomalie génétique, sous réserve que cette communication soit réalisée avec l'accord et sous la supervision du médecin généticien (amendement COM-212 de la rapporteure).
La commission a adopté cet article ainsi modifié.
Article 24
Garantie d'une transmission sécurisée
des
résultats de génétique entre laboratoires
Cet article permet au laboratoire qui a réalisé un examen de génétique à partir d'un échantillon collecté par un laboratoire intermédiaire de transmettre directement le résultat de l'examen au prescripteur, et précise que seul le médecin prescripteur d'un examen de génétique prénatal peut communiquer les résultats à la personne concernée.
La commission spéciale a adopté un amendement rédactionnel à l'article 24 du projet de loi.
I - Le dispositif proposé
1. La transmission sécurisée des résultats de génétique entre laboratoires et prescripteurs
• Aux termes de l'article L. 1131-1-3 du code de la santé publique, seul le médecin prescripteur de l'examen des caractéristiques génétiques est habilité à communiquer les résultats de cet examen à la personne concernée ou, le cas échéant, à la personne de confiance qu'elle a désignée, à sa famille ou ses proches. Cette disposition répond à l'exigence de transmission d'informations médicales sensibles, auxquels s'attachent de forts enjeux de confidentialité et nécessitant une interprétation par un spécialiste qualifié en génétique, dans le cadre d'une consultation médicale individuelle.
Le I de l'article 24 du projet de loi maintient ce principe, tout en supprimant, dans les personnes susceptibles de se voir communiquer les résultats de l'examen, la référence aux personnes mentionnées au second alinéa de l'article L. 1131-1 du code de la santé publique, à savoir la personne de confiance, la famille ou, à défaut, un des proches. Il complète en revanche ledit article L.1131-1 afin de prévoir que les résultats sont communiqués, s'agissant d'un majeur faisant l'objet d'une mesure de protection juridique avec représentation à la personne, à la personne chargée de la mesure de protection.
• Par ailleurs, plusieurs laboratoires peuvent intervenir dans la réalisation de l'examen de génétique. L'échantillon peut en effet être prélevé par un premier laboratoire qui peut, ensuite, déléguer le soin à un autre laboratoire d'effectuer tout ou partie de l'examen à partir de l'échantillon qu'il lui transmet, notamment lorsque le laboratoire auquel s'est adressé la personne ne dispose pas des moyens ou de l'habilitation pour rechercher certaines caractéristiques génétiques.
Dans ce cas, ce sont les dispositions de droit commun, inscrites à l'article L. 6211-11 du code de la santé publique, qui trouvent à s'appliquer : lorsque l'une des phases de l'examen est réalisée par un autre laboratoire de biologie médicale que celui auquel le patient s'est adressé, les résultats correspondants sont transmis au biologiste-responsable du premier laboratoire à qui il revient de transmettre l'intégralité des résultats de l'examen au médecin prescripteur. Toutefois, l'intermédiation du premier laboratoire auquel s'est adressé le patient dans la transmission des résultats au médecin prescripteur peut représenter un risque pour la préservation de la confidentialité de ces données.
En conséquence, le I de l'article 24 du projet de loi complète l'article L. 1131-1-3 du code de la santé publique par un paragraphe prévoyant, dans le cas de la transmission des résultats d'un examen de génétique, des dispositions dérogatoires du droit commun. Il reviendra au laboratoire autorisé à réaliser l'examen de transmettre directement les résultats au prescripteur. Dans le cas où l'examen aurait été réalisé à partir d'un échantillon prélevé initialement par un autre laboratoire, celui-ci sera informé par le laboratoire ayant effectué l'examen de la transmission des résultats sans pour autant que les résultats lui soient révélés, dans un souci de préservation de la confidentialité.
2. La transmission sécurisée des résultats d'un examen de génétique prénatal entre le médecin prescripteur et la patiente
Aux termes du III de l'article L. 2131-1 du code de la santé publique, seul le prescripteur, médecin ou sage-femme, est habilité à communiquer à la femme enceinte les résultats d'un examen de biologie médicale ou d'imagerie réalisé dans le cadre d'un diagnostic prénatal.
Le IV de ce même article prévoit, en cas de risque avéré, que des examens complémentaires à visée diagnostique peuvent être prescrits par un médecin au cours d'une consultation adaptée à l'affection recherchée. Toutefois, il n'existe pas de disposition encadrant le rendu des résultats de ces examens complémentaires. Or il s'agit généralement, selon l'étude d'impact annexée au projet de loi, d'examens de génétique (cytogénétique et génétique moléculaire), réalisés après prélèvement foetal in utero , notamment par amniocentèse ou choriocentèse 360 ( * ) .
Afin d'encadrer le rendu des résultats issus de ces examens complémentaires, le II de l'article 24 du projet de loi complète le VII de l'article L. 2131-1 du code de la santé publique en précisant que les résultats de l'examen sont transmis directement au prescripteur par le laboratoire autorisé à réaliser l'examen. Dans le cas où l'examen aurait été réalisé à partir d'un échantillon transmis par un autre laboratoire, ce laboratoire sera informé par le laboratoire ayant réalisé l'examen de la transmission au prescripteur des résultats.
II - Les modifications adoptées par l'Assemblée nationale
L'Assemblée nationale a adopté cet article sans modification.
III - La position de la commission
Votre commission a adopté un amendement rédactionnel COM-213 de la rapporteure.
La commission a adopté cet article ainsi modifié.
Article 25
Aménagement, pour les patients
concernés,
d'une passerelle entre la génétique
somatique
et la génétique constitutionnelle
Cet article étend aux examens des caractéristiques génétiques résultant d'altérations somatiques une partie des garanties prévues par la loi en matière de diagnostic génétique constitutionnel, notamment en termes d'information préalable et de prise en charge dans le cadre d'une consultation de génétique constitutionnelle en cas de découverte d'une anomalie génétique constitutionnelle.
La commission spéciale a adapté la définition des examens de génétique somatique en rappelant la possibilité que des altérations génétiques somatiques soient recherchées à partir de cellules germinales.
I - Le dispositif proposé
Le 1° de l'article 25 du projet de loi introduit, dans le code de la santé publique, un nouvel article L. 1130-1 définissant un examen des caractéristiques génétiques constitutionnelles comme un examen consistant à « analyser les caractéristiques génétiques d'une personne héritées ou acquises à un stade précoce du développement prénatal. » Compte tenu de l'impact potentiel de ces caractéristiques génétiques héréditaires ou transmissibles pour la personne et ses apparentés biologiques, le diagnostic de génétique constitutionnelle est assorti de garanties protectrices des droits des personnes concernées, notamment en termes de consentement, d'information de la parentèle et de prise en charge dans le cadre d'une consultation de génétique médicale, conformément aux articles 16-10 361 ( * ) à 16-13 du code civil.
Par ailleurs, le développement de l'oncogénétique et des thérapies ciblées a donné lieu à la multiplication des examens des caractéristiques génétiques réalisés à partir de cellules issues de tissus tumoraux permettant d'identifier des altérations génétiques dites « somatiques » : ces variations génétiques sont acquises au cours de la vie, présentes dans quelques cellules du corps (le « soma » en grec ancien) et absentes des gamètes, donc non susceptibles de transmission à la descendance.
Comme le souligne l'étude d'impact annexée au projet de loi, l'analyse des altérations génétiques somatiques est particulièrement utile pour déterminer la « signature » génétique d'une tumeur et définir une prise en charge thérapeutique ciblée sur le profil génétique de cette tumeur.
Or les examens des caractéristiques génétiques acquises ultérieurement ne font pas l'objet d'un encadrement législatif, les dispositions du code de la santé publique relatives aux examens des caractéristiques génétiques à des fins médicales ne s'appliquant qu'aux examens de génétique constitutionnelle. Les examens de génétique somatique peuvent pourtant donner lieu à la découverte de caractéristiques génétiques constitutionnelles, liées ou pas à l'indication initiale de l'examen.
Dans ces conditions, le 1° de l'article 25 du projet de loi introduit également dans le code de la santé publique un nouvel article L. 1130-2 prévoyant que :
- lorsque les résultats des examens des caractéristiques génétiques acquises ultérieurement sont susceptibles de révéler des caractéristiques constitutionnelles, la personne est invitée à se rendre chez un médecin qualifié en génétique pour une prise en charge dans le cadre d'une consultation de génétique constitutionnelle. Dans ce cadre, la révélation de caractéristiques génétiques constitutionnelles sera entourée des garanties législatives en matière de consentement et d'information de la parentèle ;
- la réalisation d'un examen de génétique somatique sera nécessairement précédée d'une information de la personne sur la possibilité que les résultats justifient une orientation vers une consultation de génétique constitutionnelle.
Par coordination, le 2° de l'article 25 du projet de loi complète le 1° de l'article L. 1131-6 du code de la santé publique afin de prévoir que le décret en Conseil d'État fixant les conditions de prescription et de réalisation des examens des caractéristiques génétiques constitutionnelles d'une personne déterminera également les conditions de prescription des examens des caractéristiques génétiques acquises ultérieurement.
II - Les modifications adoptées par l'Assemblée nationale
L'article L. 1130-2 du code de la santé publique envisagé initialement par l'article 25 du projet de loi fait référence aux « examens des caractéristiques génétiques acquises ultérieurement », sans que soit précisé le point de départ d'acquisition de ces caractéristiques génétiques, même si l'on devine qu'il s'agit des caractéristiques génétiques somatiques acquises ultérieurement à un stade précoce du développement prénatal et non transmissibles. En conséquence, outre un amendement tendant à corriger une erreur de référence, l'Assemblée nationale a adopté, avec l'avis favorable du Gouvernement, un amendement du rapporteur, Jean-François Eliaou, visant à introduire dans le code de la santé publique une définition, au nouvel article L. 1130-2, des examens des caractéristiques somatiques consistant à « analyser les caractéristiques génétiques qui ne sont ni héritées ni transmissibles, à partir de cellules autres que les cellules germinales. »
L'Assemblée nationale a adopté, en outre, un amendement du Gouvernement visant à préciser que le dispositif d'encadrement des examens des caractéristiques génétiques s'envisage dans l'intérêt non seulement des patients mais aussi de leur parentèle.
III - La position de la commission
Si elle est bienvenue, la définition des examens des caractéristiques génétiques somatiques introduite par l'Assemblée nationale pose deux problèmes :
- en définissant l'examen de génétique somatique comme l'analyse exclusive de caractéristiques génétiques qui ne sont ni héritées ni transmissibles, elle tend à poser une frontière en apparence imperméable entre génétique somatique et génétique constitutionnelle, peu compatible avec la réalité. Les tests génétiques tumoraux, réalisés notamment en oncologie, visent en effet à établir la signature génétique d'une tumeur qui est, en théorie, somatique, mais peut également se révéler constitutionnelle. Il apparaît ainsi que, dans le cas des cancers de l'ovaire, les altérations des gènes BRCA1 et BRCA2, présentes dans 20 % de ces cancers, sont constitutionnelles dans 70 % des cas. Par conséquent, si un examen de génétique somatique vise, en première intention, à identifier des altérations non constitutionnelles, il emporte bien souvent, compte tenu des facteurs responsables d'apparition de tumeurs, la découverte incidente de caractéristiques génétiques constitutionnelles. Votre commission a donc adopté un amendement visant à préciser que les examens de génétique somatique consistent à « rechercher en première intention » des caractéristiques génétiques ni héritées ni transmissibles (amendement COM-214 de la rapporteure) ;
- la présence d'altérations génétiques somatiques dans des cellules germinales ne peut être, dans l'absolu, exclue. En effet, certaines tumeurs germinales peuvent résulter d'altérations de ces cellules à partir desquelles un examen des caractéristiques génétiques somatiques pourra, à titre exceptionnel, être envisagé. En conséquence, votre commission a adopté un amendement tendant à supprimer la précision selon laquelle les examens de génétique somatique ne sont réalisés qu'à partir de cellules autres que les cellules germinales (amendement COM-215 de la rapporteure).
En outre, votre commission a adopté un amendement d'harmonisation rédactionnelle (amendement COM-216 de la rapporteure).
La commission a adopté cet article ainsi modifié.
Article 26
Sécurisation de l'utilisation du microbiote
fécal
Cet article vise à encadrer le recueil de selles d'origine humaine destinées à une utilisation thérapeutique dans le cadre de la préparation du microbiote fécal.
La commission spéciale a adopté un amendement soumettant la transplantation de microbiote fécal aux principes éthiques du bénévolat et de l'anonymat du don, tout en tenant compte de la spécificité des dons intrafamiliaux dans ce domaine.
I - Le dispositif proposé
Dans ses réponses au questionnaire de votre commission, l'ANSM indique avoir été régulièrement sollicitée depuis 2014 par certains opérateurs souhaitant développer et mettre à disposition des patients des préparations de microbiote fécal dans certaines indications thérapeutiques telles que l'infection récidivante à Clostridium difficile , les maladies inflammatoires chroniques de l'intestin (MICI), les complications liées aux traitements antibiotiques, cytotoxiques ou de greffe prescrits dans le cadre de pathologies oncohématologiques (leucémie, GVHD 362 ( * ) )... À ce jour, seule l'indication infection récidivante à Clostridium difficile est validée par des études cliniques de phase III.
L'ANSM rappelle que sept essais cliniques de phases I ou II utilisant la transplantation de microbiote fécal en tant que médicament expérimental fabriqué dans une pharmacie à usage intérieur ou un établissement pharmaceutique sont en cours en France dans les indications suivantes :
- maladie de Crohn ;
- rectocolite hémorragique ;
- décolonisation de bactéries hautement résistantes émergentes, d'entérobactéries productrices de bêta-lactamases à spectre élargi (E-BLSE) ou productrices de carbapénèmases (EPC), chez des sujets par ailleurs sains ou chez des patients hospitalisés ;
- prévention des complications de la dysbiose par transplantation de microbiote fécal autologue chez des patients atteints de leucémie aiguë myéloïde sous traitement intensif ;
- traitement de la maladie du greffon contre l'hôte réfractaire aux corticoïdes.
La transplantation de microbiote fécal consiste en l'introduction de préparation de selles d'un ou de plusieurs donneurs sains dans le tube digestif d'un patient receveur afin de rééquilibrer la flore intestinale altérée de l'hôte. Cette approche thérapeutique suscite un intérêt grandissant et a fait l'objet de plusieurs essais cliniques montrant des résultats encourageants.
Afin d'accompagner cette innovation thérapeutique, l'ANSM a réuni un comité scientifique spécialisé temporaire (CSST) dédié à la transplantation de microbiote fécal à l'issu duquel elle a élaboré des recommandations intitulées La transplantation de microbiote fécal et son encadrement dans les essais cliniques en mars 2014, réactualisées deux fois depuis.
L'ANSM rappelle que le microbiote fécal étant utilisé à visée curative à l'égard de maladies humaines, il doit être considéré comme un médicament conformément à l'article L. 5111-1 du code de la santé publique 363 ( * ) . À ce stade du développement du microbiote fécal et en l'absence d'autorisation de mise sur le marché, celui-ci peut être utilisé dans le cadre législatif et réglementaire applicable :
- soit aux préparations magistrales et hospitalières 364 ( * ) . Dans ce cadre, les préparations de microbiote fécal sont soumises aux bonnes pratiques de préparation fixées par décision du directeur général de l'ANSM 365 ( * ) ;
- soit aux médicaments expérimentaux destinés à un essai clinique 366 ( * ) ;
- soit via des autorisations temporaires d'utilisation (ATU) 367 ( * ) , notamment pour des patients nominativement identifiés. À ce jour, l'ANSM a délivré 16 ATU nominatives.
Selon l'ANSM, les selles, qui servent à la fabrication de ce médicament, ne sont pas considérées comme un élément constitutif du corps humain en tant que tel mais sont le résultat du processus de digestion et sont donc issues d'une transformation effectuée par le corps humain en vue de leur élimination par celui-ci. Elles sont entre autre constituées de cellules mortes, de bactéries et de virus. Non assimilables à un produit du corps humain, elles sont identifiées comme la matière première de départ pour la fabrication d'un médicament, le microbiote fécal. En conséquence, le I de l'article 26 du projet de loi exclut explicitement, à l'article L. 1211-8 du code de la santé publique, les selles collectées pour la fabrication du microbiote fécal de l'application des dispositions de droit commun relatives aux éléments et produits du corps humain.
Le II de l'article 26 du projet de loi introduit, au titre III du livre I er de la cinquième partie du code de la santé publique, un chapitre consacré au « Recueil de selles d'origine humaine destinées à une utilisation thérapeutique ». Composé de quatre articles, ce nouveau chapitre prévoit :
- la déclaration à l'ANSM de toute activité de collecte de selles destinées à la préparation de microbiote fécal à des fins thérapeutiques, à l'exception de la collecte réalisée dans le cadre d'une recherche clinique (nouvel article L. 513-11-1 du code de la santé publique) ;
- la définition par décision du directeur général de l'ANSM de règles de bonnes pratiques auxquelles seront soumises les opérations de collecte, de contrôle, de conservation, de traçabilité et de transport des selles effectuées par les organismes déclarant des activités de collecte de selles (nouvel article L. 513-11-2 du code de la santé publique) ;
- la suspension ou l'interdiction des activités de collecte de selles menées par des organismes qui méconnaîtraient leurs obligations légales (nouvel article L. 513-11-3 du code de la santé publique) ;
- le renvoi à un décret de la définition des modalités d'application du chapitre (nouvel article L. 513-11-4 du code de la santé publique).
Enfin, par coordination, le III de l'article 26 du projet de loi modifie, à l'article L. 5311-1 du code de la santé publique, le champ des compétences de l'ANSM afin d'y inclure le contrôle des activités de collecte de selles d'origine humaine.
II - Les modifications adoptées par l'Assemblée nationale
Outre des amendements rédactionnels, la commission des affaires sociales de l'Assemblée nationale a adopté un amendement tendant à substituer à la déclaration à l'ANSM des activités de collecte de selles une autorisation expresse de ces activités par l'ANSM, au regard des risques que peuvent présenter les traitements recourant au microbiote fécal pour les patients.
En séance, l'Assemblée nationale a adopté, avec des avis favorables du Gouvernement, deux amendements du rapporteur visant à préciser que :
- les règles de bonnes pratiques relatives à la collecte, au contrôle, à la conservation, à la traçabilité et au transport des selles destinées à la fabrication du microbiote fécal sont également applicables dans le cadre des recherches impliquant la personne humaine ;
- par coordination avec l'introduction d'un mécanisme d'autorisation des activités de collecte de selles à des fins thérapeutiques, sera également soumise à autorisation par l'ANSM l'importation de selles destinées à la préparation de microbiote fécal.
III - La position de la commission
L'article 26 du projet de loi a trait à la sécurité sanitaire par l'encadrement d'une technique thérapeutique ne soulevant aucune question éthique particulière : on peut raisonnablement s'interroger sur l'opportunité de son examen dans un texte relatif à la bioéthique.
En cas de transplantation de microbiote fécal, l'article 26 du projet de loi ne mentionne pas de règle liée à l'anonymat du don. Il s'agit aujourd'hui, en effet, souvent de dons dirigés ou intrafamiliaux. Pour autant, d'autres types de dons pourraient émerger. Dans cette perspective, votre commission a adopté un amendement rappelant que la transplantation de microbiote fécal s'effectue dans l'intérêt du receveur et est soumise aux principes éthiques du bénévolat et de l'anonymat du don (amendement COM-217 du rapporteur). Il est précisé que les règles d'anonymat du don ne sont pas applicables lorsque le don est intrafamilial.
La commission a adopté cet article ainsi modifié.
Article 27
Réalisation d'un
médicament de thérapie innovante
préparé
ponctuellement dans le cadre d'une seule intervention médicale
Cet article vise à autoriser et à encadrer la réalisation de médicaments de thérapie innovante préparés ponctuellement (MTI-PP) par les établissements de santé dans le cadre d'une seule intervention médicale. La commission spéciale l'a adopté sans modification.
I - Le dispositif proposé
1. Les médicaments de thérapie innovante, une catégorie récente qui traduit l'émergence de la médecine biologique
• La loi du 22 mars 2011 368 ( * ) a transposé dans le droit français la catégorie des « médicaments de thérapie innovante préparés ponctuellement », définie par le 17° de l'article L. 5121-1 du code de la santé publique comme « tout médicament tel que défini dans le règlement (CE) n° 1394/2007 du Parlement européen et du Conseil, du 13 novembre 2007, concernant les médicaments de thérapie innovante [...], fabriqué en France selon des normes de qualité spécifiques et utilisé dans un hôpital en France, sous la responsabilité d'un médecin, pour exécuter une prescription médicale déterminée pour un produit spécialement conçu à l'intention d'un malade déterminé ».
Ces médicaments sont régulés au niveau national et ont été exclus du champ d'application de la directive européenne de 2001 instituant un code communautaire relatif aux médicaments à usage humain 369 ( * ) . Leur délivrance ne passe pas, ainsi, par l'autorisation de mise sur le marché. Ils doivent toutefois faire l'objet d'une autorisation de l'ANSM , pouvant être assortie de conditions particulières ou de restrictions d'utilisation, et sont soumis aux exigences du droit européen en matière de qualité et de sécurité.
Les médicaments de thérapie innovante
La notion de « médicament de thérapie innovante » recouvre trois catégories :
• Les médicaments de thérapie innovante (MTI) stricto sensu sont :
- les médicaments de thérapie génique
- les médicaments de thérapie cellulaire somatique
- les médicaments issus de l'ingénierie tissulaire et cellulaire
- les médicaments combinés de thérapie innovante
Ils sont régulés au niveau national pour les essais cliniques et au niveau européen pour leur mise sur le marché et les procédures de suivi post-autorisation.
• Les médicaments de thérapie innovante préparés ponctuellement (MTI-PP) sont des MTI fabriqués et utilisés au sein d'un unique Etat membre.
Un MTI-PP est ainsi un MTI qui, en raison de ses caractéristiques et de sa destination, est préparé de façon ponctuelle à l'attention d'un malade déterminé , que les produits soient ou non d'origine autologue.
Le MTI-PP est régulé au niveau national : il ne peut être utilisé que dans l'état membre ou il est fabriqué et autorisé. Exemptés de la clause de l'AMM centralisée, les MTI-PP suivent un cadre réglementaire national qui doit être équivalent aux règles communautaires applicables en matière de qualité et de sécurité. Les étapes du don, de l'obtention et du contrôle des tissus et cellules sont ainsi encadrées par la directive « tissus et cellules humains » (directive 2004/23/CE du Parlement européen et du Conseil du 31 mars 2004).
Ce nouveau type de produits créé par le règlement européen 1394/2007 (article 28) est ainsi appelé « exemption hospitalière ».
• Les préparations cellulaires ou tissulaires
Ce sont des produits cellulaires ou tissulaires à finalité thérapeutique qui, contrairement aux MTI et MTI-PP, ne sont pas des médicaments au sens du code de la santé publique mais des produits de santé placés sous la compétence de l'ANSM.
A contrario , un produit est un MTI ou un MTI-PP (et non une préparation) si deux critères sont vérifiés :
- des modifications « substantielles » des cellules ou tissus sont réalisées au cours de la production des cellules, c'est-à-dire entraînant une modification des propriétés biologiques initiale de ces cellules ou tissus ;
- les cellules ou les tissus ne sont pas destinés à être utilisés pour la (les) même(s) fonction(s) essentielle(s) chez le receveur et chez le donneur.
Source : ANSM
2. L'objectif du projet de loi : autoriser, dans l'intérêt du patient, la réalisation d'un MTI-PP dans le cadre d'une seule intervention médicale
• L'article L. 4211-9-1 du code de la santé publique réserve la préparation, la conservation ou encore la distribution des médicaments de thérapie innovante préparés ponctuellement (MTI-PP) aux établissements bénéficiant d'une autorisation - accordée pour une durée de cinq ans - par l'ANSM après avis de l'Agence de la biomédecine.
Dix établissements ont reçu une autorisation sur le fondement de cet article 370 ( * ) , en plus des autorisations délivrées pour cette activité aux structures de l'Etablissement français du sang et à certains établissements pharmaceutiques.
Or, comme le relève l'étude d'impact, « les médecins investigateurs engagés dans les essais cliniques souhaitent, dans l'intérêt du patient, que l'ensemble des opérations de prélèvement des cellules/tissus, de préparation et d'administration du produit fini soit fait dans le cadre d'une seule et même intervention médicale réalisée au bloc opératoire, sans passer par un établissement ou un organisme dédié à la préparation et ce, afin d'éviter par exemple que le patient fasse l'objet de deux anesthésies (l'une pour le prélèvement et l'autre pour l'administration). »
• Le 3° du II complète ainsi l'article L. 4211-9-1 afin de permettre que la préparation, la distribution et l'administration des MTI-PP soit réalisée dans le cadre de la même intervention médicale que celle du prélèvement des tissus ou cellules autologues dans un établissement de santé ou un hôpital des armées ne disposant pas d'une autorisation à ce titre.
Plusieurs conditions sont toutefois fixées par le texte pour garantir la sécurité des patients :
- d'une part, la préparation « au lit du patient » et la distribution - c'est-à-dire la mise à disposition en vue de son administration au patient - du MTI-PP sera réalisée sous la responsabilité d'un établissement ou organisme autorisé à ce titre par l'ANSM. Cette dernière s'assurera du respect des bonnes pratiques applicables et de l'exercice de ses responsabilités par la personne qui représentera l'établissement autorisé ;
- d'autre part, elle se déroulera en conformité avec les bonnes pratiques fixées par l'Agence et « dans le cadre d'un contrat écrit » entre l'établissement de santé assurant la prise en charge du patient et l'établissement autorisé à préparer des MTI-PP. Ce contrat aura vocation à préciser les responsabilités de chacune des parties ;
- enfin, un décret en Conseil d'Etat précisera les conditions applicables à la préparation ainsi que le type de médicaments concernés, étant par ailleurs précisé que l'ANSM vérifiera la nécessité de l'administration des MTI-PP dans le cadre d'une même intervention lorsqu'elle évaluera les demandes d'autorisation de MTI-PP ou la demande d'autorisation d'essai clinique. Comme l'a indiqué l'ANSM, le décret pourra écarter les préparations nécessitant des manipulations substantielles, comme les produits de thérapie génique, pour ne viser que les médicaments pour lesquels un bénéfice à ne pas passer par un établissement autorisé peut être fourni.
D'après les indications de l'ANSM, la disposition prévue au présent article pourrait notamment concerner l'utilisation, en chirurgie orthopédique, de concentrés de moelle autologue, ou encore l'association de la moelle osseuse autologue concentrée à un substitut osseux pour améliorer la consolidation des fractures des os longs.
• Les autres alinéas de l'article prévoient pour l'essentiel des coordinations , concernant l'actualisation de la dénomination de l'ANSM ( 2° du II et III ) ou afin de tenir compte des évolutions proposées par l'article ( I ).
II - Les modifications adoptées par l'Assemblée nationale
• Outre un amendement rédactionnel, la commission spéciale a dans un premier temps adopté, à l'initiative de son rapporteur Jean-François Eliaou, une précision selon laquelle la vérification par l'ANSM de la nécessité de l'administration d'un MTI-PP au cours d'une même intervention, est opérée le cas échéant « en coordination avec l'Agence régionale de santé ».
• Lors de l'examen du texte en séance publique , l'Assemblée nationale a cependant supprimé cette dernière disposition, sur la proposition du même rapporteur, considérant qu'il ne relève pas de la compétence de l'ARS d'intervenir dans le cadre de l'évaluation du produit.
En effet, comme l'a indiqué l'ANSM, les établissements ou organismes autorisés par l'ANSM à préparer des MTI-PP sont inspectés par l'ANSM. Dans ce cadre, l'ANSM interviendra pour s'assurer du respect des bonnes pratiques dans la préparation « au lit du patient » du MTI-PP et de l'exercice de ses responsabilités par l'établissement autorisé. Quant aux ARS, elles demeurent compétentes pour ce qui concerne les établissements de santé (bloc opératoire) ce qui nécessitera des interactions entre ANSM et ARS en tant que de besoin.
Au final, l'Assemblée nationale n'a donc adopté qu'un amendement de clarification rédactionnelle de son rapporteur.
III - La position de la commission
Si l'on peut s'interroger sur la présence de cette disposition dans un projet de loi de bioéthique, cette mesure de simplification va dans le sens d'une amélioration des conditions de prise en charge des patients et d'une plus large diffusion de certaines thérapies innovantes.
Cette forme de sous-traitance, encadrée de strictes garanties en termes de sécurité sanitaire, répond à une préconisation formulée par l'ANSM, comme ses représentantes l'ont confirmé au rapporteur lors de leur audition, et n'appelle pas en l'état d'observation particulière.
La commission a adopté cet article sans modification.
Article 28
Diverses mises en cohérence
Cet article procède à diverses mises en cohérence au sein du code de la santé publique, du code pénal et du code des douanes, concernant les examens des caractéristiques génétiques, l'assistance médicale à la procréation, le diagnostic prénatal ainsi que le contrôle de tissus et cellules. La commission spéciale l'a adopté sans modification.
I - Le dispositif proposé
1. Une base légale aux sanctions administratives en cas de manquements dans le domaine des examens de caractéristiques génétiques
Le I insère au sein du code de la santé publique un nouvel article L. 1131-2-2 qui prévoit la suspension ou le retrait de l'autorisation pour un établissement de santé ou un laboratoire de biologie médicale de procéder à l'examen des caractéristiques génétiques d'une personne à des fins médicales en cas notamment de violation des prescriptions applicables à ces examens.
Cette sanction s'applique dans les conditions prévues à l'article L. 6122-13 du code de la santé publique, c'est-à-dire après notification par le directeur général de l'Agence régionale de santé et procédure contradictoire.
• Ces dispositions sont similaires à celles prévues, dans le même code, en cas de manquements constatés par un établissement ou laboratoire aux prescriptions applicables en matière d'assistance médicale à la procréation (article L. 2142-3) et de diagnostic prénatal (article L. 2131-3). D'après les indications transmises par la direction générale de la santé, ces dispositions n'auraient pas trouvé à s'appliquer au cours des dernières années.
2. Diverses modifications ou actualisations des dispositions relatives au diagnostic prénatal et à l'assistance médicale à la procréation
a) L'actualisation des dispositions relatives aux techniques d'AMP
Le III modifie l'article L. 2141-1 du code de la santé publique :
- pour y supprimer la référence, devenue sans objet, à un rapport sollicité par la loi de juillet 2011 dans les trois mois suivant sa promulgation , sur la liste des procédés biologiques utilisés en assistance médicale à la procréation ainsi que les critères d'inscription sur cette liste ; celui-ci a été remis par l'Agence de la biomédecine et a servi de base au décret n° 2012-360 du 14 mars 2012 et à l'arrêté du 18 juin 2012 371 ( * ) ;
- pour y supprimer l'autorisation explicite de la « technique de congélation ultra-rapide des ovocytes » introduite par la loi de juillet 2011 ; comme le note l'étude d'impact, la vitrification des gamètes comme des embryons ou zygotes est devenue une technique régulièrement utilisée et visée par l'Agence de la biomédecine dans la liste des techniques visant à améliorer l'efficacité ou la sécurité des procédés biologiques autorisés ;
- pour ne plus cibler l'arrêté ministériel définissant les bonnes pratiques de l'assistance médicale à la procréation sur les seules pratiques d'AMP « avec tiers donneur » . En effet, l'arrêté du 30 juin 2017 pris sur cette base (modifiant l'arrêté du 11 avril 2008) encadre les activités d'AMP avec ou sans tiers donneur : il s'agit donc de lui donner une base légale claire.
b) Diverses mises en cohérence et harmonisation
• Les II et IV visent à harmoniser la référence aux conditions d'implantation des établissements en matière de diagnostic prénatal et d'AMP en modifiant les articles L. 2131-5 et L. 2142-4 du code de la santé publique qui renvoient ces modalités à un décret en Conseil d'Etat (les conditions d'implantation étant à l'heure actuelle fixés aux articles R. 2131-5-5 et suivants pour le DPN et R. 2142-1 et suivants pour l'AMP).
• Le V et VI procèdent à une mise en cohérence avec les évolutions en matière d' accueil d'embryon introduites par la loi du 23 mars 2019 de réforme pour la justice. Ils modifient l'article L. 2162-6 du code de la santé publique et l'article 511-25 du code pénal, dont les dispositions sont en miroir, afin de les mettre en cohérence avec l'article L. 2141-6 du code de la santé publique : la loi précitée a supprimé le consentement du couple receveur devant le juge ainsi que la faculté de ce dernier de diligenter une enquête sociale, en y substituant un consentement devant notaire. Est donc supprimée, par coordination , la mention au 1° de ces articles des peines applicables au fait de réaliser les activités nécessaires à l'accueil d'un embryon sans s'être préalablement assuré de l'obtention de l'autorisation judiciaire.
3. Une base légale au contrôle douanier des tissus et cellules d'origine humaine
• Le VII vise à pallier l'absence de base légale aux mesures de police sanitaire relatives aux tissus et cellules issus du corps humain.
Il modifie en ce sens l'article L. 1245-1 du code de la santé publique afin d'étendre la possibilité de retrait des autorisations données aux établissements ou organismes pour la pratique de prélèvements, greffes ou préparation d'organes ou de tissus en cas de violation des prescriptions en matière d'importation et d'exportation des tissus ou préparations de thérapie cellulaire (1°) .
Le 2° modifie par coordination les articles L. 1245-5 et L. 1245-5-1 du code de la santé publique qui ont été réécrits (pour le premier) ou créés (pour le second) par la loi du 23 février 2017 372 ( * ) . Ils encadrent la possibilité d'import et d'export de produits issus du corps humain (tissus, dérivés et cellules ainsi que préparations de thérapie cellulaire) en provenance d'un Etat non membre de l'Union européenne ou n'étant pas partie à l'accord sur l'Espace économique européen en la soumettant à une autorisation délivrée par l'Agence nationale de sécurité du médicament et des produits de santé (ANSM), après avis de l'Agence de la biomédecine.
L'évolution proposée permet ainsi à l'ANSM d'asseoir ses pouvoirs de suspension ou de retrait d'autorisation en matière d'import et d'export, compte tenu des enjeux de sécurité sanitaire qui s'y rattachent .
Comme l'a indiqué l'ANSM, le nombre d'unités de thérapies cellulaires autorisés à exporter est en augmentation, compte tenu des demandes faites pour les cellules destinées à la fabrication de Car-T-cells.
• Dans le même sens, le VIII modifie l'article 38 du code des douanes afin de donner, d'une part, une base légale au contrôle par les services des douanes des tissus et cellules importés à des fins de fabrication de médicaments ou dispositifs médicaux, en application de l'article L. 1245-5-1 du code de la santé publique ( 1° ).
Il procède, d'autre part, à une harmonisation avec les dispositions de l'article L. 2151-6 du code de la santé publique qui ne portent que sur l'autorisation d'importer ou d'exporter des cellules souches embryonnaires à des fins de recherche et non des tissus ou cellules foetaux ( 2° ).
II - Les modifications adoptées par l'Assemblée nationale
• La commission spéciale a adopté cinq amendements de son rapporteur, dont deux d'ordre rédactionnel.
Les autres modifications tendent à :
- supprimer par cohérence le dernier alinéa de l'article L. 1131-2-1 du code de la santé publique, dont les dispositions, relatives au retrait d'autorisation en cas de manquement aux prescriptions législatives et réglementaires applicables à l'examen des caractéristiques génétiques d'une personne, sont reprises dans un nouvel article L. 1131-2-2 ;
- prévoir, comme en dispose le dernier alinéa de l'article L. 1131-2-1, que ce retrait peut intervenir en cas de manquement aux prescriptions applicables à l'identification d'une personne par empreintes génétiques ;
- renvoyer à des critères énoncés par décret en Conseil d'Etat, après avis de l'Agence de la biomédecine les conditions selon lesquelles l'autorisation des pratiquer des examens génétiques peut être retirée dans les cas où « le volume d'activité ou la qualité des résultats sont insuffisants », afin d'éviter tout arbitraire en ce domaine.
• En séance publique , l'Assemblée nationale a adopté, à l'initiative de son rapporteur Jean-François Eliaou, un amendement visant à donner une base légale au contrôle, par les services des douanes, des selles destinées à la préparation de microbiote fécal et des préparations de microbiote fécal à des fins thérapeutiques, par coordination avec l'article 26 du projet de loi.
Elle a également adopté un amendement de coordination formelle.
III - La position de la commission
Les coordinations, actualisations ou mises en cohérence diverses apportées par cet article n'appellent pas d'observation particulière.
La commission a adopté cet article sans modification.
TITRE
VI
ASSURER UNE GOUVERNANCE BIOÉTHIQUE
ADAPTÉE AU RYTHME
DES AVANCÉES RAPIDES
DES SCIENCES ET DES TECHNIQUES
Article 29 A
Création, dans chacune des deux assemblées du
Parlement,
d'une délégation parlementaire à la
bioéthique
Cet article tend à la création de délégations parlementaires à la bioéthique afin d'informer les assemblées de la politique suivie par le Gouvernement au regard de ses conséquences sur la bioéthique et de suivre l'application des lois. La commission a supprimé cet article, considérant que l'Office parlementaire d'évaluation des choix scientifiques et technologiques et des commissions permanentes remplissaient déjà pleinement cette mission.
I - Le dispositif adopté par l'Assemblée nationale : créer des délégations parlementaires à la bioéthique
Cet article additionnel résulte de l'adoption en séance par l'Assemblée nationale d'un amendement présenté par la rapporteure, Mme Romeiro Dias pour mettre en oeuvre la proposition n° 60 de la mission d'information de l'Assemblée nationale sur la révision de la loi relative à la bioéthique 373 ( * ) de créer une délégation parlementaire permanente chargée des sujets relatifs à la bioéthique , afin « de réduire le risque d'emballement médiatique et de crispation sociétale qui pourrait résulter de chaque nouvel examen de la loi relative à la bioéthique ».
Ces délégations seraient constituées de trente-six membres à la représentation proportionnelle des groupes parlementaires et équilibrée des hommes et des femmes, ainsi que des commissions permanentes.
Leur rôle consisterait à informer les assemblées de la politique suivie par le Gouvernement au regard de ses conséquences sur la bioéthique et à assurer dans ce domaine le suivi de l'application des lois .
II - La position de la commission : maintenir la compétence de l'Opecst et des commissions permanentes
L'initiative des députés a surpris le rapporteur car il existe déjà une structure permanente spécialisée bicamérale dont la mission est « d'informer le Parlement des conséquences des choix de caractère scientifique et technologique afin, notamment, d'éclairer ses décisions » 374 ( * ) et à qui la loi a confié des compétences spécifiques en matière de bioéthique . Il s'agit de l'Office parlementaire d'évaluation des choix scientifiques et technologiques (Opecst).
Depuis 1994 375 ( * ) , la loi lui confie l'évaluation de l'application des lois relatives à la bioéthique . C'est dans ce cadre que M. Jean-François Eliaou, député, et Mme Annie Delmont-Koropoulis, sénatrice, ont déposé un rapport le 25 octobre 2018 376 ( * ) . Le présent projet de loi se propose de reconduire ce dispositif d'évaluation par l'Office dans son article 32.
Par ailleurs, en application de l'article L. 1412-1-1 du code de la santé publique - introduit par la loi du 7 juillet 2011 et modifié par la loi du 2 février 2016 377 ( * ) - l'Opecst est associé aux États généraux de la bioéthique qui doivent être organisés, à l'initiative du Comité consultatif national d'éthique pour les sciences de la vie et de la santé (CCNE), « avant tout projet de réforme sur les problèmes éthiques et les questions de société soulevés par les progrès de la connaissance dans les domaines de la biologie, de la médecine et de la santé » .
L'Office est tout d'abord consulté sous forme d'avis en amont de l'organisation des États généraux ; puis, il procède ensuite à l'évaluation du rapport que lui présente le CCNE à la suite du débat public, en faisant ressortir les éléments scientifiques indispensables à la bonne compréhension des enjeux de la réforme envisagée.
C'est dans ce cadre que l'Office a rendu un rapport le 9 juillet 2019 378 ( * ) préalablement à la révision de la loi de bioéthique.
Enfin, le rapporteur relève que certaines commissions permanentes, - en particulier, la commission des affaires sociales au premier chef et la commission des lois - sont déjà en charge de sujets bioéthiques dans le cadre de leurs compétences sectorielles. Il est de ce point de vue très surprenant d'envisager de constituer des délégations à la bioéthique à la représentation équilibrée de toutes les commissions permanentes.
Travaux des commissions permanentes en matière de bioéthique
Commission des affaires sociales :
- Proposition de loi relative à l'autorisation d'analyses génétiques sur personnes décédées, rapport n° 523 (2017-2018) du 30 mai 2018 de Mme Catherine Deroche ;
- Proposition de loi créant de nouveaux droits en faveur des malades et des personnes en fin de vie, rapport n° 103 (2015-2016) du 21 octobre 2015 de MM. Michel Amiel et Gérard Dériot ;
- Proposition de loi tendant à modifier la loi n° 2011-814 du 7 juillet 2011 relative à la bioéthique en autorisant sous certaines conditions la recherche sur l'embryon et les cellules souches embryonnaires, rapport n° 10 (2012-2013) du 3 octobre 2012 de M. Gilbert Barbier ;
Commission des lois :
- Communication sur la jurisprudence rendue pour l'application des dispositions relatives aux droits des malades et à la fin de vie du 19 juin 2019, de Mme Muriel Jourda ;
- « Défendre les principes, veiller à l'intérêt des enfants - Quelle réponse apporter au contournement du droit français par le recours à l'AMP et à la GPA à l'étranger ? », rapport d'information n° 409 (2015-2016) du 17 février 2016 de M. Yves Détraigne et Mme Catherine Tasca ;
- Proposition de loi créant de nouveaux droits en faveur des malades et des personnes en fin de vie, avis n° 506 (2014-2015) du 10 juin 2015 de M. François Pillet.
Dans ces conditions, la création de délégations parlementaires à la bioéthique ne semble pas justifiée, ce d'autant plus, que le Sénat fait l'effort depuis 2009 de rationaliser ses différentes structures de contrôle et d'évaluation, dans une volonté d'assurer l'efficacité et la cohérence du travail parlementaire 379 ( * ) . Il faut selon les termes des rapporteurs du groupe de réflexion sur les méthodes de travail du Sénat, MM. Karoutchi et Richard, dont les propositions ont été adoptées par le Bureau du Sénat en 2015 380 ( * ) « éviter la dispersion des sénateurs et donc la multiplication, la polysynodie des structures ».
En conséquence, la commission a adopté les deux amendements de suppression COM-18 du groupe communiste républicain citoyen et écologiste et COM-244 de son rapporteur.
La création d'une délégation relevant du règlement de chaque assemblée, il est tout à fait loisible à l'Assemblée nationale de créer sa propre délégation à la bioéthique sans qu'une disposition spécifique soit votée dans le présent projet de loi, qui aurait pour seul effet d'imposer cette création au Sénat.
La commission a supprimé cet article.
Article 29
Élargissement des missions du comité
consultatif national d'éthique
des sciences de la vie et de la
santé
Cet article élargit le champ de compétences du CCNE aux questions et problèmes de santé résultant de progrès scientifiques et technologiques dans d'autres domaines que ceux de la biologie, de la médecine et de la santé, lui confie l'organisation de débats publics annuels et simplifie sa gouvernance.
La commission spéciale a supprimé la précision selon laquelle, s'agissant du député et du sénateur nommés membres du CCNE, l'un est issu de la majorité et l'autre de l'opposition, compte tenu du caractère inapplicable en l'espèce de la notion de majorité/opposition.
I - Le dispositif proposé
1. L'élargissement des missions du CCNE
Aux termes de l'article L. 1412-1 du code de la santé publique, le CCNE « a pour mission de donner des avis sur les problèmes éthiques et les questions de société soulevés par les progrès de la connaissance dans les domaines de la biologie, de la médecine et de la santé. »
Toutefois, les questions éthiques qui peuvent surgir dans le débat public dépassent désormais le strict champ de la biologie, de la médecine et de la santé. Afin de tenir compte de l'évolution des techniques et des progrès des connaissances scientifiques dans des domaines tels que l'intelligence artificielle, les neurosciences, le numérique en santé ou encore l'environnement et les nanobiotechnologies, le 1° du I de l'article 29 du projet de loi précise, à l'article L. 1412-1 précité, que les avis du CCNE peuvent également porter des problèmes éthiques et des questions de société soulevés « par les conséquences sur la santé des progrès de la connaissance dans tout autre domaine. »
Afin de tenir compte de cet élargissement, une coordination est opérée à l'article L. 1412-1-1 du code de la santé publique ( a) du 2° du I de l'article 29 du projet de loi).
2. L'organisation de débats publics annuels sur des questions éthiques
Le CCNE se voit, par ailleurs, confier le soin d'animer, chaque année, des débats publics sur un ou plusieurs des problèmes éthiques et des questions de société relevant de son champ de compétences, en lien avec les espaces de réflexion éthique régionaux ou interrégionaux prévus à l'article L. 1412-6 du code de la santé publique ( b) du 2° du I de l'article 29 du projet de loi).
3. La simplification de la gouvernance du CCNE
Le 3° du I de l'article 29 du projet de loi procède à la réécriture de l'article L. 1412-2 du code de la santé publique, relatif à la composition du CCNE. Dans un souci de simplification, il est renvoyé au décret la liste des autorités chargées de désigner une partie des 40 membres du comité, dont le président et cinq personnalités qualifiées représentatives des principales familles philosophiques et spirituelles continueront d'être nommés par le Président de la République.
Un décret fixera ainsi la liste des ministres auxquels il reviendra de désigner quinze personnalités qualifiées choisies en raison de leur compétence et de leur intérêt pour les problèmes d'éthique, ainsi que la liste des organismes chargés de désigner quinze personnalités appartenant aux secteurs de la recherche et de la santé. Outre les six membres nommés par le Président de la République, les quatre membres restants du CCNE seront toujours un député et un sénateur, un membre du Conseil d'État désigné par le vice-président du Conseil d'État et un membre de la Cour de cassation désigné par le premier président de la Cour de cassation.
La durée du mandat du président du comité, aujourd'hui nommé pour une durée de deux ans renouvelable, et la durée du mandat des autres membres, aujourd'hui de quatre ans renouvelable, sont alignées sur une durée de trois ans renouvelable une fois.
Le 4° du I de l'article 29 du projet de loi renvoie à un décret en Conseil d'État, aujourd'hui prévu à l'article L. 1412-5 du code de la santé publique, la détermination des modalités de désignation des membres du CCNE, notamment celles permettant d'assurer le respect de la parité. Il est ajouté que ce décret fixera également les modalités selon lesquelles est organisé un renouvellement par moitié de l'instance, le principe de ce renouvellement étant aujourd'hui de niveau réglementaire 381 ( * ) .
Le II de l'article 29 du projet loi prévoit une application des nouvelles règles de nomination et de renouvellement des membres du CCNE à compter du 26 décembre 2021. À titre transitoire, il est précisé que les mandats des membres nommés selon les règles actuelles entre la promulgation de la loi et le 26 décembre 2021 prennent fin le 25 décembre 2021 ( III de l'article 29 du projet de loi) et que les mandats de ces personnes ne seront pas comptabilisés comme un mandat pour l'application du principe de limitation des mandats à deux ( IV de l'article 29 du projet de loi).
II - Les modifications adoptées par l'Assemblée nationale
En séance, l'Assemblée nationale a adopté, outre des amendements rédactionnels, plusieurs amendements tendant à modifier l'article 29 du projet de loi :
- à l'initiative de la rapporteure Laëtitia Romeiro Dias, des amendements sont venus substituer 382 ( * ) aux commissions parlementaires permanentes compétentes et à l'Opecst les délégations parlementaires à la bioéthique dont la création est prévue par l'article 29 A du projet de loi adopté par l'Assemblée nationale ;
- un amendement de notre collègue Alexandre Freschi (LaREM), adopté contre l'avis de la commission mais avec un avis de sagesse du Gouvernement, tend à préciser que, s'agissant du député et du sénateur membres du CCNE, l'un doit être issu de la majorité, l'autre de l'opposition ;
- un amendement de la rapporteure Laëtitia Romeiro Dias précise, par cohérence, que les modalités de désignation fixées par décret en Conseil d'État ne concernent pas les parlementaires membres du CCNE. En effet, les règles générales de désignation des parlementaires membres d'organismes extra-parlementaires sont déjà fixées par la loi n° 2018-699 du 3 août 2018 383 ( * ) , dont l'article 1 er prévoit notamment que « lorsque l'Assemblée nationale et le Sénat sont appelés, en application d'une loi, à nommer, respectivement, un député et un sénateur pour siéger, en cette qualité, au sein d'un organisme extérieur au Parlement, ils désignent alternativement, chacun en ce qui le concerne, une femme et un homme. »
III - La position de la commission
• Votre commission a adopté un amendement de coordination afin de tirer les conséquences de la suppression de l'article 29 A, initialement introduit par l'Assemblée nationale et créant dans chaque assemblée une délégation parlementaire à la bioéthique (amendement COM-229 de la rapporteure).
• Votre commission a également adopté deux amendements identiques tendant à supprimer la précision selon laquelle, s'agissant du député et du sénateur nommés membres du CCNE, l'un est issu de la majorité et l'autre de l'opposition (amendements COM-218 de la rapporteure et COM-61 de Jacques Bigot). La notion de majorité/opposition est en effet inapplicable en l'espèce puisqu'elle recouvre des réalités différentes selon la configuration propre à chaque assemblée parlementaire.
Les groupes politiques déclarés d'opposition à l'Assemblée nationale peuvent appartenir à une famille politique disposant pourtant de sénateurs appartenant à la majorité du Sénat. Par ailleurs, certains groupes peuvent, au Sénat, se déclarer minoritaires, sans pour autant se placer dans l'opposition à la majorité du Sénat. Chaque assemblée parlementaire doit donc conserver son autonomie dans la définition de ses pratiques en matière de désignation de parlementaires membres d'organismes extra-parlementaires et de respect de l'équilibre interne entre groupes politiques.
La commission a adopté cet article ainsi modifié.
Article 30
Évolution des missions et des instances
de
l'agence de la biomédecine
Cet article vise à supprimer trois domaines du champ de compétences de l'agence de la biomédecine et à faire évoluer la composition de ses organes de gouvernance.
La commission spéciale a rétabli la mission de l'agence de la biomédecine dans l'élaboration d'un référentiel permettant d'évaluer la qualité des tests génétiques en accès libre, en cohérence avec la mise en place, à l'article 10 bis , d'un encadrement législatif des examens génétiques à visée généalogique. En outre, elle a confié à l'agence le soin d'établir un bilan annuel des évolutions législatives et réglementaires qui pourraient être justifiées par l'évolution des connaissances et des techniques dans les domaines relevant de sa compétence mais aussi par des situations qui ne seraient pas couvertes par le droit en vigueur et nécessiteraient des autorisations de dérogation.
I - Le dispositif proposé
1. La suppression de trois compétences de l'agence de la biomédecine
Le 1° du I de l'article 30 du projet de loi modifie l'article L. 1418-1 du code de la santé publique afin de supprimer trois compétences que détient aujourd'hui l'agence de la biomédecine :
- les nanobiotechnologies : l'étude d'impact annexée au projet de loi soutient que l'agence n'est pas en capacité de s'entourer de l'expertise nécessaire dans ce domaine que l'étude juge trop éloigné de son coeur de métier ;
- l'élaboration d'un référentiel permettant d'évaluer la qualité des tests génétiques en accès libre : selon l'étude d'impact, l'élaboration de ce référentiel s'avère trop complexe à mettre en oeuvre et présente le risque de légitimer ces tests dont l'utilisation est, en l'état du droit en vigueur, punie par la loi ;
- l'information du Parlement et du Gouvernement sur le développement des connaissances et des techniques dans le domaine des neurosciences : là encore, l'étude d'impact précise que l'agence n'est pas en mesure de s'adjoindre le concours de l'expertise nécessaire.
2. L'actualisation des missions de l'agence de la biomédecine pour tenir compte des changements apportés par le projet de loi
• Par coordination avec d'autres dispositions du projet de loi, le 1° du I de l'article 30 du projet de loi prévoit également d'actualiser les missions de l'agence de la biomédecine qui est nouvellement chargée :
- de mettre en oeuvre un suivi de l'état de santé des donneurs de cellules souches hématopoïétiques, conformément à l'article 6 du projet de loi ;
- de gérer les traitements de données relatifs aux tiers donneurs, à leurs dons et aux enfants nés de ces dons, à l'exclusion des données médicales recueillies ultérieurement au don, conformément à l'article 3 du projet de loi ;
- d'être destinataire des déclarations de protocoles de recherche sur les cellules souches embryonnaires humaines, conformément à l'article 14 du projet de loi, et sur les cellules souches pluripotentes induites humaines, conformément à l'article 15 du projet de loi.
• Par coordination, le 2° du I de l'article 30 du projet de loi actualise, à l'article L. 1418-2 du code de la santé publique, le champ d'intervention des inspecteurs de l'agence de la biomédecine chargés du contrôle et des investigations relatifs au suivi des activités médicales et biologiques.
3. La modification de la composition du conseil d'administration et du conseil d'orientation de l'agence de la biomédecine
• Le 3° du I de l'article 30 du projet de loi modifie l'article L. 1418-3 du code de la santé publique afin de mettre fin au principe de parité selon lequel le collège des représentants de l'État et institutionnels et le collège des personnalités qualifiées, des représentants d'associations et des représentants du personnel de l'agence sont de même taille. L'étude d'impact annexée au projet de loi soutient que cette parité rend difficile toute modification de la composition du conseil d'administration de l'agence.
Par ailleurs, est supprimée la mention selon laquelle les ministres chargés de la santé et de la recherche peuvent interdire ou suspendre la réalisation d'un protocole de recherche autorisé par l'agence, compte tenu de l'incohérence de cette disposition avec les dispositions de l'article L. 2151-5 du code de la santé publique qui prévoient que ces ministres ne peuvent que demander à l'agence un nouvel examen du protocole de recherche.
• L'étude d'impact mentionne qu'en raison d'une erreur aux deuxième et troisième alinéas de l'article L. 1418-4 du code de la santé publique, le conseil d'orientation de l'agence de la biomédecine comprend aujourd'hui quatre députés et quatre sénateurs. En effet, le deuxième alinéa de cet article prévoit que, « outre son président, trois députés et trois sénateurs », le conseil d'orientation comprend à parts égales d'autres membres listés par les 1° à 4° dudit article. Or le 1° de l'article L. 1418-4 précité mentionne également « des représentants du Parlement », aux côtés de représentants du Conseil d'État, de la Cour de cassation, du CCNE et de la commission nationale consultative des droits de l'homme.
En conséquence, le 4° du I de l'article 30 du projet de loi modifie l'article L. 1418-4 précité afin de préciser que le conseil d'orientation de l'agence comprend trois députés et trois sénateurs. Il opère également une clarification rédactionnelle dans la désignation des représentants d'associations de malades et d'usagers du système de santé.
Enfin, le II de l'article 30 du projet de loi prévoit une entrée en vigueur de la nouvelle composition du conseil d'orientation de l'agence de la biomédecine à compter du 22 juin 2021, avec prorogation jusqu'à cette date des mandats des membres arrivant à expiration dans l'intervalle.
II - Les modifications adoptées par l'Assemblée nationale
Outre des amendements rédactionnels, l'article 30 du projet de loi a été modifié par l'Assemblée nationale par l'adoption :
- en commission, d'un amendement de la rapporteure Laëtitia Romeiro Dias tendant à rétablir la compétence de l'agence de la biomédecine dans le domaine des nanobiotechnologies ;
- en séance, d'un amendement de la rapporteure Laëtitia Romeiro Dias, ayant reçu un avis favorable du Gouvernement, visant à préciser la mission de l'agence dans la mise en oeuvre des dispositifs d'assistance médicale à la procréation, en indiquant qu'elle élabore des règles d'attribution des gamètes et des embryons.
III - La position de la commission
• Votre commission a confirmé le maintien, souhaité par l'Assemblée nationale, de la compétence de l'agence de la biomédecine dans le domaine des nanobiotechnologies. L'utilisation croissante des nanobiotechnologies comme vecteurs d'administration de traitements médicamenteux et la possibilité, bien qu'encore théorique, pour certaines nanobiotechnologies d'augmenter les capacités physiques et mentales humaines peuvent soulever de vraies questions éthiques. L'intervention de l'agence dans ce domaine est pleinement justifiée, aux côtés du CCNE et du commissariat à l'énergie atomique et aux énergies alternatives (CEA).
Compte tenu de son souhait d'encadrer le recours aux tests génétiques à visée généalogique en accès libre, votre commission a adopté deux amendements identiques tendant à rétablir la mission de l'agence de la biomédecine dans l'élaboration d'un référentiel permettant d'évaluer la qualité des tests en accès libre (amendements COM-219 de la rapporteure et COM-78 de Bernard Jomier).
• Votre commission a également adopté un amendement procédant à diverses coordinations et corrections rédactionnelles (amendement COM-220 de la rapporteure) et un amendement de coordination visant à modifier l'article L. 1418-1-1 du code de la santé publique afin de préciser que le rapport annuel de l'agence de la biomédecine comporte une analyse des décisions d'opposition à certains protocoles de recherche sur les cellules souches embryonnaires humaines et les cellules souches pluripotentes induites (amendement COM-221 de la rapporteure).
• Afin de faire la transparence sur la pratique des « dérogations » accordées par l'agence de la biomédecine dans le silence de la loi, votre commission a en outre adopté un amendement, à l'initiative de notre collègue Bernard Jomier, précisant que le rapport annuel de l'agence de la biomédecine comporte un bilan des autorisations qu'elle délivre en matière de greffes, de dons et de recherches en l'absence de dispositions législatives ou réglementaires applicables aux cas d'espèce (amendement COM-81 de Bernard Jomier).
• Votre commission a, enfin, adopté un amendement rétablissant le principe de parité, au sein du conseil d'administration de l'agence de la biomédecine, entre le collège de représentants de l'État et institutionnels et le collège des personnalités qualifiées, des représentants d'associations et des représentants du personnel de l'agence (amendement COM-222 de la rapporteure).
La commission a adopté cet article ainsi modifié.
TITRE
VII
DISPOSITIONS FINALES
Article 31
Habilitation à légiférer par voie
d'ordonnance
Cet article vise à habiliter le Gouvernement à légiférer par voie d'ordonnance, en application de l'article 38 de la Constitution, afin de prendre les mesures nécessaires :
- à l'application de la loi, sous réserve des adaptations nécessaires, dans certaines collectivités ultra-marines (I) et pour les diverses mises en cohérence qui apparaîtraient utiles (IV) ;
- à l'adaptation du droit national dans le domaine des dispositifs médicaux, des recherches impliquant la personne humaine (II) et des médicaments de thérapie innovante (III).
La commission spéciale a restreint le champ de cette habilitation en ce qui concerne dispositions relatives aux investigations cliniques.
I - Le dispositif proposé
1. Les habilitations directement justifiées par l'application du projet de loi
• L'application dans certaines collectivités d'outre-mer (I)
Le I habilite le Gouvernement à légiférer par voie d'ordonnances, dans un délai de 18 mois suivant la promulgation de la loi, afin d'appliquer ses dispositions, sous réserve des adaptations nécessaires :
- à Saint-Pierre-et-Miquelon et Mayotte, qui relèvent du principe d'identité législative mais disposent d'un régime particulier de sécurité sociale ;
- en Nouvelle-Calédonie, en Polynésie Française et à Wallis-et-Futuna, collectivités régies par le principe de spécialité législative où certaines dispositions du projet de loi doivent être explicitement étendues pour trouver à s'appliquer 384 ( * ) .
• Les diverses mises en cohérence (IV)
Le IV autorise quant à lui le Gouvernement à prendre par ordonnances, dans le même délai, les mesures nécessaires pour procéder à diverses mises en cohérence à droit constant avec les dispositions du projet de loi ou des ordonnances (cohérence rédactionnelle, harmonisations, correction d'erreurs matérielles...).
Ces mises en cohérence pourront inclure, comme l'a relevé le Conseil d'Etat dans son avis sur le projet de loi dans un objectif de clarté et d'intelligibilité de la loi, des adaptations du code civil si la reconnaissance d'une double filiation maternelle était actée, en particulier des articles rédigés sur le modèle de l'altérité sexuelle des parents, comme ceux relatifs à l'établissement de l'acte de naissance ou l'exercice de l'autorité parentale.
2. Les habilitations visant à adapter le droit national au droit européen
• Dans le domaine des dispositifs médicaux (II)
Le II propose d'adapter par ordonnance, dans un délai d'un an, le code de la santé publique en vue de l'entrée en application de deux règlements n° 2017/745 et n° 2017/746 du Parlement européen et du Conseil du 5 avril 2017 relatifs, respectivement, aux dispositifs médicaux et aux dispositifs médicaux de diagnostic in vitro 385 ( * ) .
Ces règlements ont abrogé les directives de 1990 et 1993 386 ( * ) formant le cadre réglementaire européen des dispositifs médicaux et une directive de 1998 387 ( * ) relative aux dispositifs médicaux de diagnostic in vitro.
Ils poursuivent deux objectifs présentés comme indissociables : garantir le bon fonctionnement du marché intérieu r des dispositifs médicaux, sur lequel sont actifs un grand nombre de petites et moyennes entreprises, et fixer des normes élevées de qualité et de sécurité pour les patients et usagers, afin de faire face aux enjeux communs de sécurité relatifs à ces produits.
Le règlement 2017/745 sera d' application directe le 26 mai 2020 dans tous les Etats membres après une période transitoire de trois ans qui a débuté le 26 mai 2017. Son champ intègre des dispositifs sans finalité médicale, par exemple à finalité esthétique. Pour l'ANSM, ces dispositions devraient permettre d'harmoniser les règles applicables en favorisant la mise en place d'un réseau organisé des autorités compétentes à l'échelle européenne. En matière de matériovigilance, ce règlement a notamment décidé de la création d'Eudamed, une base de données européennes, dont l'un des modules concerne spécifiquement les incidents. Il impose en outre des obligations aux fabricants en matière de surveillance de ses produits après commercialisation, dans le cadre notamment d'un rapport périodique actualisé de sécurité permettant d'apprécier en continu le rapport bénéfices-risques.
Pour les dispositifs de diagnostic in vitro, les modifications les plus importantes portées par le règlement 2017/746 portent sur une nouvelle classification en quatre classes et de nouvelles règles limitant l'autocertification (dans l'objectif de la réduire de 80 % à 20 %).
Les ordonnances concerneront :
- d'une part, en application de ces nouvelles exigences européennes, le renforcement du rôle de l'ANSM, la mise en cohérence du système de matério ou réactovigilance et les modalités de traçabilité des dispositifs médicaux notamment au sein des établissements de santé ( 1° du II ) ;
- d'autre part, les modalités de réalisation des investigations cliniques qui devront être réalisées en application de ces règlements ( 2° du II ).
Sur ce dernier point, les règlements précités font de l'investigation clinique la règle pour l'évaluation des dispositifs implantables et des dispositifs relevant de la classe III 388 ( * ) . Les fabricants devront résumer les résultats de l'évaluation clinique dans un document destiné à être rendu public. Si l'investigation clinique pourra être réalisée de manière coordonnée dans plusieurs Etats membres, l'examen par un comité éthique - les comités de protection de la personne en France - restera du niveau national. A l'issue de la procédure d'évaluation, chaque Etat membre rend sa décision.
Comme l'a indiqué l'ANSM, cette procédure est facultative jusqu'au 25 mai 2027 et devient obligatoire à compter du 26 mai 2027 pour les dispositifs médicaux, et de 2029 pour les dispositifs de diagnostic in vitro .
• Dans le domaine des médicaments de thérapie innovante (III)
Le III vise à autoriser le Gouvernement à prendre une autre ordonnance dans un délai d'un an afin de mettre en cohérence la législation avec le règlement n° 1394/2007 sur les médicaments de thérapie innovante (MTI) 389 ( * ) sur deux points ponctuels :
- d'une part, pour abroger les références aux préparations de thérapie cellulaire xénogénique et de thérapie génique (définies aux 12° et 13° de l'article L. 5121-1 du code de la santé publique), couvertes par la catégorie de MTI ;
- d'autre part, pour supprimer de la catégorie de produits cellulaires à finalité thérapeutiques les cellules servant à transférer du matériel génétique et donc génétiquement modifiées, qui doivent être considérées comme des MTI et non des préparations de thérapie cellulaire du fait de cette modification substantielle.
Compte tenu de ce champ très circonscrit, on peut se demander pour quelles raisons ces ajustements n'ont pas été apportés directement au texte de l'article 27 du projet de loi relatif aux médicaments de thérapie innovante préparés ponctuellement.
II - Les modifications adoptées par l'Assemblée nationale
L'Assemblée nationale a adopté un amendement rédactionnel de sa rapporteure.
III - La position de la commission
Si des habilitations à légiférer par ordonnance ouvertes par cet article sont justifiées par l'objet même du projet de loi, d'autres portent sur des sujets larges ne présentant qu'un lien ténu avec le texte. On peut ainsi s'interroger sur la justification, dans un projet de loi de bioéthique, d'une telle mesure concernant le régime général applicable aux dispositifs médicaux.
A cet égard, du fait des enjeux attachés à ce sujet sur le plan de la sécurité sanitaire, comme l'ont montré de récents évènements, la commission spéciale a tenu à exclure du champ des habilitations les dispositions relatives aux investigations cliniques dans ce domaine ( amendement COM-231 de son rapporteur).
Alors qu'une proposition de loi transpartisane a été récemment déposée au Sénat sur le thème plus général de l'évaluation éthique de la recherche impliquant la personne humaine 390 ( * ) , ce sujet qui nécessitera une modification de la loi Jardé 391 ( * ) , mérite d'être débattu au sein du Parlement. Lors de l'examen de la loi de modernisation de notre système de santé de 2016, le Sénat et sa commission des affaires sociales s'étaient de même opposés au principe de légiférer par voie d'ordonnance pour adapter la législation française au droit européen en matière de recherches biomédicales 392 ( * ) .
La commission a adopté cet article ainsi modifié.
Article 32
Clause de révision et évaluation de la loi de
bioéthique
Cet article propose de fixer à cinq ans le délai dans lequel la loi de bioéthique devrait faire l'objet d'un nouvel examen par le Parlement, après évaluation par l'OPECST. La commission en a approuvé le principe et y a apporté des modifications rédactionnelles.
L'article 32 du projet de loi reprend la clause de révision présente dans la loi n° 2011-814 du 7 juillet 2011 relative à la bioéthique. Elle figurait également dans l'une des lois fondatrices de 1994 (loi n° 94-654 du 29 juillet 1994 relative au don et à l'utilisation des éléments et produits du corps humain, à l'assistance médicale à la procréation et au diagnostic prénatal) 393 ( * ) .
Fixée à cinq ans en 1994 puis à sept en 2011, le projet de loi propose de revenir à cinq ans. Comme précédemment, la révision législative serait précédée d'une évaluation de la loi par l'Office parlementaire d'évaluation des choix scientifiques et technologiques (OPECST), dans les quatre ans de sa promulgation.
Introduite en 2011 dans la loi à l'initiative du Sénat, la commission a de nouveau approuvé, en adoptant l'amendement COM-255 du rapporteur, le principe de cette révision périodique, réintroduite dans la loi de 2011 à l'initiative du Sénat.
La commission a adopté cet article ainsi modifié.
Article 33
Rapport au
Parlement présentant l'état des stocks
des gamètes en
France et les conditions de recours à ces derniers
Cet article, inséré par l'Assemblée nationale, demande la remise d'un rapport au Parlement sur l'état des stocks de gamètes en France et les conditions de recours à ces gamètes. La commission spéciale l'a supprimé en considérant que cette mission incombe déjà à l'Agence de la biomédecine.
I - Le dispositif adopté par l'Assemblée nationale
Cet article est issu d'un amendement présenté par Maxime Minot et d'autres députés du groupe Les Républicains, adopté en séance publique avec l'avis favorable de la commission spéciale et un avis de sagesse du Gouvernement.
Il sollicite la remise, dans un délai de deux ans, d'un rapport au Parlement « présentant l'état des stocks des gamètes et France et les conditions des recours à ces dernières » . Il s'agit notamment d'évaluer l'impact de l'ouverture, par l'article 1 er , de l'assistance médicale à la procréation aux couples de femmes et aux femmes non mariées sur les « stocks » de gamètes.
II - La position de la commission
L'Agence de la biomédecine met d'ores et déjà à disposition du Parlement et du public, chaque année, dans son rapport médical et scientifique , les données relatives à l'assistance médicale à la procréation et au don de gamètes, qui relèvent de son champ de compétences.
Il lui appartiendra bien évidemment d'évaluer l'impact du projet de loi - notamment de la conjugaison de ses articles 1 er , 2 et 3 concernant l'ouverture de l'accès à l'AMP, la dissociation entre les démarches de don et d'autoconservation et la levée de l'anonymat des donneurs - sur le don de gamètes et les délais d'accès pour les demandeurs.
Si ces enjeux sont essentiels, solliciter un rapport supplémentaire sur le sujet paraît superfétatoire et sans réelle portée. C'est la raison pour laquelle la commission spéciale a adopté l'amendement COM-156 de sa rapporteure de suppression de l'article.
La commission a supprimé cet article.
Article 34
Rapport au Parlement sur l'application des dispositions
encadrant l'entretien avec les proches en matière
de
prélèvement d'organes et de tissus
Cet article, inséré par l'Assemblée nationale, demande la remise d'un rapport au Parlement sur l'application des dispositions encadrant l'entretien avec les proches en matière de prélèvement d'organes et de tissus, comportant l'évaluation de l'organisation de ces prélèvements au sein des établissements. Constatant sa portée trop relative quelle que soit l'importance du sujet abordé, la commission spéciale a supprimé cet article.
I - Le dispositif adopté par l'Assemblée nationale
Cet article est issu d'un amendement de Jean-Louis Touraine et de membres du groupe La République en Marche, adopté en séance publique contre l'avis de la commission 394 ( * ) mais avec l'avis favorable du Gouvernement.
Il sollicite, dans un délai d'un an, la remise d'un rapport au Parlement sur l'application des dispositions de l'arrêté du 16 août 2016 portant homologation des règles de bonnes pratiques relatives à l'entretien avec les proches en matière de prélèvement d'organes et de tissus, pris en application de la loi de modernisation de notre système de santé de janvier 2016 ( cf. encadré ci-après). Ce rapport devra notamment évaluer l'organisation de ces prélèvements au sein des établissements.
D'après l'auteur de l'amendement, les dispositions de l'arrêté mentionné n'auraient contribué qu'à modifier « à la marge » des pratiques qui « demeurent trop hétérogènes selon les territoires et selon les équipes » .
Les évolutions apportées par la loi de modernisation de notre système de santé du 26 janvier 2016 en matière de prélèvement d'organes
La loi du 26 janvier 2016 a réaffirmé et clarifié le principe du consentement présumé au don.
Jusqu'alors, si le médecin n'avait pas directement connaissance de la volonté du défunt, il devait « s'efforcer de recueillir auprès des proches l'opposition au don d'organes éventuellement exprimée de son vivant par le défunt, par tout moyen » .
La loi de 2016 a supprimé cette disposition : l'article L. 1232-1 du code de la santé publique prévoit désormais que le médecin informe les proches du défunt, préalablement au prélèvement envisagé, de sa nature et de sa finalité , « conformément aux bonnes pratiques arrêtées par le ministre chargé de la santé sur proposition de l'Agence de la biomédecine. »
L'arrêté du 16 août 2016 a ainsi précisé les différentes étapes et finalités de cet entretien qui intervient dans un contexte singulier après l'annonce du décès. Il poursuit plusieurs objectifs : recueillir l'éventuelle expression d'un refus de prélèvement exprimé par le défunt de son vivant, recueillir des informations sur des antécédents médicaux ou sur les circonstances du décès pour réduire les risques de transmission de pathologie, ou encore informer sur la nature et les modalités du prélèvement dès lors qu'ils sont possibles et que la personne ne s'y est pas opposée.
Comme l'a noté la Cour des comptes dans une récente étude 395 ( * ) , cet entretien permet également d'identifier les cas dans lesquels « en raison du contexte, le prélèvement n'a pas été possible », qui permet aux équipes hospitalières de ne pas pratiquer le prélèvement en cas d'opposition des familles, sachant que l'accord des familles reste dans les faits déterminants.
Parallèlement, le décret n° 2016-1118 du 11 août 2016 a précisé les modalités d'expression de l'opposition au prélèvement d'organes et de tissus, dont le principal moyen est l'inscription au registre national des refus géré par l'Agence de la biomédecine. Cette inscription peut se faire en ligne depuis janvier 2017.
II - La position de la commission
• Les règles de bonnes pratiques définies par l'arrêté du 16 août 2016 relatives à l'abord des proches ont fait l'objet, comme l'a indiqué l'Agence de la biomédecine, d'une large concertation.
Elles imposent une préparation de l'entretien, sa formalisation et l'accompagnement des proches, qu'il y ait ou non don d'organes. Le président de la Société française de médecine des prélèvements d'organes et de tissus a souligné le caractère vertueux de ce référentiel pour renforcer la professionnalisation de l'entretien. D'après l'Agence, cela aurait déjà permis d'harmoniser les pratiques des professionnels des coordinations de prélèvement, même si cela nécessite, en raison du renouvellement des personnels, un effort de formation important.
• Il est de bon sens de vouloir faire un bilan de l'application par les équipes de terrain de cet arrêté. Cependant, cela relève pleinement de la mission de l'Agence de la biomédecine , à travers l'audit des coordinations de prélèvements auquel elle procède.
Il appartiendra à celle-ci d'en rendre compte dans ses différentes publications, comme sur tout sujet relevant de son champ de compétences, notamment dans la perspective d'un nouveau « plan greffes » à compter de 2022.
Quelle que soit l'importance du sujet et la réalité des enjeux, il apparaît dès lors inutile de solliciter un rapport spécifique . C'est la raison pour laquelle la commission spéciale a adopté l'amendement COM-230 de son rapporteur de suppression de l'article.
La commission a supprimé cet article.
TRAVAUX DE LA COMMISSION
___________
I. AUDITIONS
M.
François Clavairoly,
président de la Fédération
protestante de France,
M. Haïm Korsia, Grand rabbin de France,
Mgr
Pierre d'Ornellas, archevêque de Rennes,
et M. Olivier Wang-Genh,
président
de l'Union des bouddhistes de France
M. Alain Milon , président . - Nous entamons nos travaux sur le projet de loi relatif à la bioéthique avec l'audition des représentants des cultes. Je souhaite la bienvenue à M. François Clavairoly, président de la Fédération protestante de France, à M. Haïm Korsia, Grand rabbin de France, à Mgr Pierre d'Ornellas, archevêque de Rennes, et à M. Olivier Wang-Genh, président de l'Union des bouddhistes de France. Le Conseil français du culte musulman (CFCM) n'a pas pu envoyer de représentant et transmettra à la commission une contribution écrite. Je crois, messieurs, que vous êtes coutumiers de cet exercice auquel vous vous êtes déjà livrés, à plusieurs reprises, à l'Assemblée nationale...
M. François Clavairoly, président de la Fédération protestante de France . - Je vous remercie de nous accueillir pour évoquer le projet de loi relatif à la bioéthique qui comprend notamment l'extension de l'assistance médicale à la procréation (AMP) aux couples de femmes et aux femmes seules. Nous avons été également reçus par l'Assemblée nationale. Il est très bon que les cultes puissent être entendus, mais nous souhaiterions débattre aussi avec d'autres courants philosophiques, et non seulement dans cette seule configuration. Il n'existe évidemment pas de « front des religions », générique, sur ce genre de sujets.
Les trois principes de notre réflexion, pour les
protestants, sont la liberté
- d'interprétation,
citoyenne, d'exercice du culte et de conscience -, la
responsabilité - rendre compte de cette liberté devant les
autres - et la solidarité ou fraternité - nous ne sommes pas
seulement des consciences individuelles, mais liées en
société.
Pour les protestants, l'éthique est un ensemble d'interrogations et non pas un dogme donnant des réponses. C'est une inquiétude pour se frayer un chemin, pour atteindre l'expression apaisée d'un vivre ensemble, sous-tendue par l'injonction du Christ « tu aimeras le prochain comme toi-même »...
M. Haïm Korsia, Grand rabbin de France . - Vous exagérez !
M. François Clavairoly . - ... largement portée par le judaïsme, et issue du Lévitique.
M. Haïm Korsia . - Oui, tout de même !
M. François Clavairoly . - La réponse de l'homme, c'est parfois de se défausser de sa responsabilité : suis-je le gardien des actes de mon frère ? Nous devons être ensemble coresponsables de nos actes.
Les protestants sont partagés sur l'extension de l'AMP, comme la plupart de l'opinion publique et des confessions. Cette façon de vivre le débat en partage est un signe de bonne santé spirituelle.
Nous ne sommes pas opposés à cette extension, mais nous émettons des réserves et des interrogations : nous récusons une éthique purement naturelle, « ce qui est doit devenir ce qui doit être », tout comme une vision purement technicienne, « ce qui est faisable doit être fait ». Entre ces deux visions, l'une conservatrice et l'autre progressiste, se trouve une place pour le débat.
La liberté ne se confond ni avec l'individualisme, ni avec la loi de l'offre et de la demande, ni avec la loi du plus fort. Nous défendons l'amour du prochain, au nom d'une transcendance.
Plusieurs techniques répondent à l'infertilité des couples hétérosexuels. Nous saluons ces avancées. Mais la médecine de la fertilité devient de plus en plus réparatrice d'un désir manqué ou d'un projet parental impossible pour des femmes seules ou des couples de femmes. L'évolution de la médecine reproductive répond à des attentes sociétales et nécessite notre vigilance. Il faut accompagner ces nouvelles formes de parentalité, de plus en plus présentes. Au départ purement réparatrice, la médecine reproductive devient une médecine sociétale qui s'inscrit dans le processus civilisationnel occidental, accompagné par des décisions de justice. La loi du 17 mai 2013 qui a ouvert le mariage aux couples de même sexe et les avis de la Cour de cassation de septembre 2014 qui ont validé l'adoption des enfants issus d'AMP pratiquée à l'étranger par la conjointe de la mère ont contribué à reconnaître ces nouvelles formes de familles. L'insémination artificielle par des paillettes de sperme d'un donneur n'est plus seulement utilisée par des couples hétérosexuels mariés ou des partenaires de fait souffrant d'infécondité d'origine masculine, mais par des femmes célibataires et des couples de femmes, sans problème d'infertilité, dans d'autres pays d'Europe. Cette évolution s'est traduite dans les législations, selon des modalités différentes, en Grèce, en Estonie, en Autriche, en Suède, en Norvège, au Danemark, aux Pays-Bas, en Espagne, et au Royaume-Uni.
Le contexte institutionnel traduit des évolutions civilisationnelles. Nous ne pouvons pas rester sans cadre juridique, sinon nous aboutirons à des parentalités clandestines, à des injustices, à des situations de fragilité, notamment pour les femmes seules avec enfant.
Quelle compréhension avons-nous de la médecine reproductive, sur fond de déni de la finitude humaine, au risque de la marchandisation des corps humains ? Comment accompagner les nouvelles formes de parentalité et de filiation en veillant à protéger les plus vulnérables - notamment l'enfant à naître et les femmes seules - tout en refusant les discriminations, et en faisant preuve de solidarité et en assurant l'égalité des droits ?
Des études existent, certaines sont interprétables, mais toutes encouragent les questionnements. De fait, il existe une nouvelle réalité à accompagner.
Les confessions, notamment le protestantisme, doivent veiller à ce que ces principes de liberté, responsabilité et fraternité soient préservés contre la tentation de l'exploitation du corps, contre des désirs injustifiés et contre le risque de dérive eugéniste qui nous guette, si notre société se laisse uniquement guider par la technique.
M. Alain Milon , président . - Pour répondre à votre première observation, la loi du 7 juillet 2011 a mis en place les États généraux de la bioéthique, organisés par le Comité consultatif national d'éthique (CCNE) sur l'ensemble du territoire. L'hémicycle du Sénat ne suffirait pas à regrouper l'ensemble des acteurs concernés par les lois de bioéthique, et il aurait été impossible de les rassembler le même jour à la même heure, alors que le projet de loi sera débattu en séance publique durant la seconde quinzaine de janvier...
M. Haïm Korsia . - Je remercie le pasteur Clavairoly d'avoir énoncé, en hébreu, la phrase de Caïn après avoir tué Abel. Les Juifs ont une certaine antériorité sur les évangiles pour dire : « Tu aimeras ton prochain comme toi-même »...
Réviser régulièrement la loi, et ainsi la rendre temporaire, n'est-ce pas en amoindrir la portée ? Je comprends l'objectif, mais cette fois-ci, on risque de fragiliser en permanence la vocation de la loi à dépasser les situations particulières. Elle ne doit pas juste constater et entériner ce qu'une majorité des Français font à un moment donné. Le bien doit nous élever.
Il serait plus juste qu'une commission permanente du Parlement examine régulièrement toutes les incidences éthiques ou bioéthiques des projets de loi. Cela permettrait une mise à jour régulière des sujets éthiques ou bioéthiques dans la loi, et éviterait l'écueil que chaque loi éthique ou bioéthique soit considérée comme datée au terme d'un certain délai.
Autre interrogation, qu'en est-il du désir individuel face au désir collectif ? Le désir individuel est au coeur de la volonté de chaque citoyen. Mais lorsque le désir individuel percute la volonté commune ou le sens collectif, il faut trouver un équilibre. Oui à la liberté, mais faites-le en mettant le manteau de Noé : ne nous demandez pas de valider. Le règlement fixerait une limite d'âge pour le recours à l'AMP pour les femmes. Celles qui veulent le faire peuvent se rendre en Espagne ; à leur retour, la Sécurité sociale ne va pas refuser de les prendre en charge, mais va prendre en compte les soins liés à leur grossesse - fort heureusement. Mais il ne faut pas faire comme si cet état de fait était un idéal. La loi doit gérer l'idéal, même si elle doit traiter aussi du concret.
Transgresser les règles tout en respectant leur bien-fondé n'est pas la même chose que de leur dénier tout bon sens. Ainsi, malgré la limitation de vitesse à 130 kilomètres par heure, je peux rouler à 150 kilomètres par heure, en raison d'une urgence. Si je me fais verbaliser, je comprends la raison d'être de la règle, mais à ce moment-là, j'avais arbitré en faveur de mon intérêt individuel et non de l'intérêt collectif - c'est mon choix. Ce n'est pas la même chose que quelqu'un qui transgresse la loi sans respecter son bien-fondé. On ne va pas changer la règle parce que certains, voire beaucoup, la transgressent ! On ne peut pas, du moins au sein du Gouvernement ou du Parlement, pour justifier une loi, dire que le père ne sert à rien, ou qu'une grand-mère peut jouer le rôle d'un père. Il faut un peu de bon sens. Certains veulent créer une réalité abstraite, théorique, qui n'existe pas. Dites aux Français : « faites comme vous voulez, mais pas en notre nom ». Certains ont des pulsions contradictoires : ce sont les mêmes qui voudraient donner accès aux origines et en même temps dire à un enfant qu'il n'a pas de père ! Ceux qui veulent légiférer sont un peu schizophrènes : ils veulent reconnaître certains pères, et organiser la disparition d'autres. Ce n'est pas une démarche sereine.
Comme le disait justement le pasteur Clavairoly, nos fragilités, nos impossibilités, nos imperfections et nos manques sont notre humanité. La médecine se conçoit pour réparer l'impossibilité, pour un homme et une femme, d'avoir un enfant, par la procréation médicalement assistée. C'est sa vocation de pallier un manque, mais si elle pallie un désir, jusqu'où ira-t-elle, et pour quel type de désir ? En sociologie des organisations, la priorité d'une organisation est d'assurer sa survie, et non de répondre aux désirs de chacun ou de chacune. Je ne dis pas qu'il ne faut pas le faire. La matriarche Rachel hurlait sa douleur de ne pas avoir d'enfant, et a donné sa servante à son mari. Je ne dis pas que c'est bien, ni que c'est l'idéal, mais elle l'a fait. Entendons cette douleur, mais ne légiférons pas seulement pour répondre à ce cas particulier.
Est-ce que tout ce qui est possible est légitime ? Je fais confiance à votre sagacité pour trouver les réponses.
M. Roger Karoutchi . - Avis de sagesse !
Mgr Pierre d'Ornellas, archevêque de Rennes . - Dans le premier testament, que je citerai en grec, le äïêéìÜæåéí est le discernement, le mot de l'éthique, qui dit quelque chose de la sagesse pratique pour discerner les voies justes afin que l'institution soit juste. La révolution intervient lorsque l'institution représentant le peuple est injuste. Discerner l'institution juste est le point majeur de la réflexion actuelle.
Plusieurs points animent notre réflexion.
Avec ce projet de loi, certains enfants auront le droit d'avoir un père mais pas d'autres. On peut y remédier en permettant à un enfant né de deux femmes de pouvoir dire « père ».
Le projet de loi tel qu'issu des travaux de l'Assemblée nationale postule que tout repose sur le « projet parental ». L'enfant est donc soumis à un projet d'adulte qui lui est imposé. Ce concept a été critiqué par les juristes, car il aboutit au retour d'un droit des puissants - les adultes - sur un enfant. Peut-on considérer qu'il s'agit d'une institution juste ?
L'égalité dans les modes de filiation n'est-elle pas en outre une pétition de principe ? Les modes d'établissement de la filiation ne sont pas identiques : l'enfant issu d'une femme seule n'aura jamais pas la même filiation que l'enfant d'un homme et d'une femme, mais on voudrait faire croire le contraire.
Qu'est-ce qui est vraisemblable ou invraisemblable ? Qu'une filiation soit vraisemblable a toujours existé, mais instaurer l'invraisemblable pose question. La loi propose « d'inciter les parents au récit le plus exact possible ». La loi s'immiscerait alors dans ce que doit faire la famille ; n'est-ce pas dangereux, au vu de la politique menée par certains États en ce sens ? Selon le doyen Carbonnier, la famille est le domaine dans lequel on doit le moins entrer par la loi... Il est nécessaire que le récit dise ce qu'il est advenu en raison d'un accident de la vie ; cela aidera considérablement la conscience psychologique et psychique de l'enfant. Mais il est différent d'accepter le récit d'une volonté qui s'impose. Permettra-t-il au psychisme de se construire paisiblement ?
La logique du marché de la procréation est considérable. Voyez les publicités qui s'affichaient sur nos écrans d'ordinateur une demi-heure après le vote de l'article 1 er en première lecture à l'Assemblée nationale : la marchandisation de gamètes existe dans différents pays. Cela nous interroge sur le principe de non patrimonialité du corps humain et de gratuité du don : est-il possible de le violer sans violer le principe de dignité humaine ? Une institution juste peut-elle le permettre ?
Nous assistons à un grand débat sur la réalité du principe de dignité. Le Conseil d'État a clairement mis en évidence ce sujet dans le débat. Pouvons-nous dire que c'est faire justice de considérer qu'il y a un bien qui nous précède tous, un bien de la planète et de l'humanité ? Nous sommes tous inscrits, par notre généalogie, sur une planète qui nous précède. Ce bien nous est donné, il est constitué et rationnel. Nos aînés ont pu dire quelque chose d'intelligible, cela nous invite à la responsabilité. Le concept de dignité irrigue tout le modèle français de bioéthique, et relativise le concept d'autonomie, qui est toujours relationnelle - comme le soulignait très bien la loi de 2002 sur les soins donnés au patient. C'est toujours avec le personnel soignant que le patient prend ses décisions. La liberté n'est jamais celle d'un sujet autonome, absolu, sans aucun lien avec ce qui le précède, mais c'est une liberté responsable, car un bien le précède. C'est dans ce lien que s'imagine le principe de dignité.
L'article 2 bis du projet de loi a introduit une étude sur l'infertilité. Ce sujet mériterait d'être approfondi, car c'est un vrai mal de notre temps. Ce serait une étude capitale pour la santé publique mais aussi pour l'environnement.
Promouvoir l'adoption serait d'une grande utilité sociale. Auparavant, cette solution était obligatoirement proposée verbalement aux personnes souhaitant s'engager dans une AMP. Le projet propose que cela soit mentionné uniquement dans le dossier sans échange avec les personnels soignants. Or, avoir un échange oral est important. Si notre société est fraternelle, elle doit mettre en lumière l'adoption comme réparation d'un accident de la vie et comme moyen de donner des parents à un enfant qui en manque. Cela mériterait une plus grande attention dans le projet de loi.
M. Olivier Wang-Genh, président de l'Union des bouddhistes de France. - Il est toujours difficile de parler au nom des bouddhistes sur des sujets aussi complexes. Il existe une tradition bouddhiste avec des spécificités et des cultures propres. Il n'y a pas d'opinion faisant l'unanimité. Mais nous avons quelques principes, partagés par tous, touchant à l'intention précédant la décision ou l'action. Si l'intention est bienveillante, elle a toute sa valeur. Si elle est malveillante, elle est malvenue. Mais une intention initialement bienveillante peut être utilisée à des fins malsaines, égoïstes, commerciales.
Les bouddhistes insistent sur l'éthique individuelle. Ces grands principes éthiques se rapportent à la prise de conscience de chacun. Cette conscience est infiniment complexe, et nous n'en saisissons qu'une petite partie. Avec l'évolution de l'intelligence artificielle (IA), on développe une partie infime du fonctionnement de la conscience, celle qui a trait à la rationalité, aux probabilités, à l'intelligence et au calcul. Cette IA, de plus en plus, prend le pas sur notre société, dans le cadre d'un développement technologique. Nous commençons à observer les effets d'un abandon excessif aux algorithmes ou aux prévisions fondées sur des empilements d'expérience. La conscience de l'être humain est infiniment plus vaste que cela. Lorsque l'être humain pense, c'est toute son humanité qui interagit. Cette richesse risque d'être mise à mal par ces évolutions technologiques. Tout sens de la nuance risque d'être proscrit, face à des machines qui assèneraient des vérités évidentes, impossibles à contredire. On aura alors tendance à faire davantage confiance à l'IA qu'à notre propre jugement...
Mme Corinne Imbert , rapporteure . - Je vous remercie de vos interventions. Comment analysez-vous les demandes sociétales en faveur de l'élargissement aux couples de femmes ou aux femmes non mariées ? Votre analyse diffère-t-elle selon qu'il s'agit d'une femme non mariée ou d'un couple ? Quels enseignements tirez-vous des exemples étrangers autorisant déjà cette extension de l'AMP ?
Mme Muriel Jourda , rapporteur . - Considérez-vous que l'accès aux données non identifiantes ou à l'identité d'un tiers donneur, prévu à l'article 3 du projet de loi, soit un progrès pour le droit de l'enfant à connaître ses origines, en écho aux demandes des personnes nées d'un don de gamète ?
M. Bernard Jomier , rapporteur . - Vous êtes les premières personnes auditionnées...
M. Alain Milon , président . - On me l'a reproché !
M. Bernard Jomier . - Je suis très intéressé par vos propos sur les valeurs. Mais les premiers articles de ce projet de loi n'ont pas grand-chose à voir avec la bioéthique : ce sont des droits sociétaux. Les lois de bioéthique doivent prendre en compte les évolutions des biotechnologies et apporter des réponses collectives aux questions soulevées.
Vous avez mis en évidence les dérives que vous craigniez : eugénisme, marchandisation, risque de perte de dignité humaine. Que trouvez-vous de positif dans les avancées de la médecine ? Si nous légiférons, c'est parce que les biotechnologies allègent des souffrances, et apportent des réponses plus humaines. Elles permettent, par exemple, d'éviter que des enfants souffrent de maladies incurables. Nous cherchons ainsi à promouvoir une éthique de vulnérabilité, une autonomie de l'être humain. La revendication de plus d'égalité doit être entendue par le législateur. Quelles sont les dispositions du projet de loi bioéthique que vous considérez comme porteuses de progrès et de valeurs et que vous soutenez ?
Je vous citerai un exemple : les greffes d'organes se sont développées, et ont permis de sauver des vies grâce à l'évolution des biotechnologies. Autrefois autorisées au seul sein de la famille nucléaire, elles sont désormais possibles au sein à la famille élargie - cousins, cousines. La réflexion du projet de loi porte sur le nombre de paires de donneurs. Qu'en pensez-vous ?
M. Alain Milon , président . - En effet, ce projet de loi comporte 41 articles, et nous parlons des quatre premiers uniquement, ceux du titre I er . Nous considérons pourtant que l'éthique médicale, c'est l'autorisation, ou la non-autorisation, de techniques qui permettent ou bien de donner la vie, ou bien de l'améliorer. Certaines dérives ne relèvent pas de l'éthique médicale, mais de la morale sociétale.
Il s'agit d'un autre problème. J'aurais préféré que l'extension de l'AMP ne soit pas dans le projet de loi relatif à la bioéthique, mais dans une loi spécifique, sociétale.
M. François Clavairoly . - L'esprit critique des religions fait que, devant un texte, l'exégèse se met à l'oeuvre. En amont, la fabrication de la loi par les élus est une responsabilité éminente, qui étend les droits et permettre d'accroître l'égalité. Personnellement, je regarde donc ce texte positivement, ce qui ne m'empêche pas d'émettre un certain nombre d'alertes, sans mauvaise humeur, mais avec un sens des responsabilités : la gratuité, il faudra qu'on y revienne - et j'ai évoqué la tentation de l'eugénisme, tout comme la ministre Agnès Buzyn, d'ailleurs.
Oui, il y a une avancée. Depuis 1994, la France peut s'enorgueillir d'avoir travaillé sur les questions de bioéthique. Un chemin a été parcouru, ce qui n'a pas été le cas dans tous les pays d'Europe.
Pour les femmes seules, la Fédération protestante a alerté sur un point : la corrélation entre famille monoparentale et vulnérabilité, fragilité sociale, pauvreté, précarité. La loi va peut-être autoriser la fabrication de familles monoparentales. Il faudra donc être d'autant plus attentif à ne pas reproduire à l'identique de la précarité ou de la vulnérabilité potentielle. Cette corrélation est documentée en France, en Europe et dans le monde : les familles monoparentales sont plus fragiles et précaires.
Pour ce qui concerne l'accès aux origines, je me réjouis qu'on ait changé de logiciel, en levant l'interdit : avec l'ouverture d'un accès à l'identité, je pense que la loi va dans le bon sens.
M. Haïm Korsia . - Chacun peut trouver un chemin de crête entre ses aspirations et la loi. Cela a toujours été permis. La technique nous donne à présent d'autres possibilités, et je ne jette la pierre à personne. Voyez comment les gens sont questionnés de manière indigne pour monter des dossiers d'adoption : on leur demande de se justifier, ils sont mis en situation d'accusés. Je veux bien qu'on trouve des gens stables, mais de là à cautionner un système presque inquisitorial, il y a un pas ! À vouloir protéger des enfants, on a parfois cassé les volontés d'adoption, poussant les gens à des extrêmes dont ils pourraient se passer si on leur ouvrait cette possibilité d'adoption.
Les données non identifiantes sont déjà collectées pour des questions médicales. Il faut aussi protéger la possibilité, pour quelqu'un, d'avoir le droit de ne pas savoir, et de rester avec cette idée magnifique de se dire : « Mes parents sont mes parents. » Si le donneur ne souhaite pas que son identité soit dévoilée, il faut respecter sa capacité à ne pas être un jour sommé de devoir assumer la subsistance d'un fils qui n'est son fils que par la génétique.
Cette notion de transparence absolue est donc assez ambiguë, et vient percuter la liberté du don. Comment améliorer les choses ? S'agit-il de répondre au désir d'un enfant de connaître son origine, ou de répondre à une pulsion de judiciarisation ? On répond trop souvent à une souffrance de l'opinion publique en légiférant. Une ministre de l'intérieur a ainsi déposé cinq projets de loi contre les chiens qui mordent les enfants...
M. Roger Karoutchi . - Deux !
M. Haïm Korsia . - Je suis séfarade, j'ai le droit d'exagérer un peu... La seule façon d'améliorer les choses, est-ce de légiférer ? Parfois, cela relève plus de la main tendue, d'une prise en compte collective de la souffrance des personnes.
On a longtemps dit que les religions étaient contre les greffes. C'est faux et caricatural ! Je sais qu'on manque de greffons, et qu'il faut trouver à répondre à ces besoins. Mais la greffe doit demeurer rare et précieuse, pour qu'on ne banalise pas le fait de donner une partie de soi. Je me méfie beaucoup du don intrafamilial. Il y a deux principes pour les greffes : gratuité et anonymat. Avec le don intrafamilial, vous entrez dans une culpabilisation. Difficile de refuser à son frère ou à sa soeur ! En organisant cette catégorie de don, on créée une pression qui n'est pas l'expression de la liberté : on empêche de dire non.
Mgr Pierre d'Ornellas . - Monsieur le président, je suis bien d'accord avec vous : je ne sais pas si le titre I er relève d'une loi de bioéthique. Je ne crois pas qu'il y ait une avancée scientifique au sujet de l'extension de l'AMP. On parle d'un choix politique clairement énoncé. Le mettre dans une loi de bioéthique fragilise le magnifique édifice bioéthique français qui a été construit depuis 1994. Comment la France pourra-t-elle parler au reste du monde sur cette question-là ? Nous allons toucher au principe de gratuité. Nous ne pourrons plus dire que tous les hommes naissent égaux en dignité et en droits puisque certains n'auront pas le droit d'avoir un père.
On ne distingue plus entre la pathologie et la non-pathologie. Ce n'est pas tant qu'on étende la PMA d'un couple hétérosexuel à un couple homosexuel. Le problème vient de ce qu'on supprime le critère de pathologie. Du point de vue de l'organisation de notre société, ne plus distinguer ce qui relève de la pathologie de ce qui n'en relève pas crée un flou. On ne sait plus très bien quel sera notre modèle social de solidarité, ni à quoi notre sécurité sociale sera consacrée. Cela mérite réflexion. Il y a eu des débats pour savoir ce qui doit être remboursé par la sécurité sociale, ce qui témoigne de la difficulté qu'il y a à ne plus faire de distinction entre ce qui est pathologique et ce qui ne l'est pas.
Je suis inquiet de voir que toutes les femmes seraient mises sur un pied d'égalité en matière d'AMP. Une femme mariée avec un homme sans aucun problème d'infertilité aura accès à une AMP, par exemple via une fécondation in vitro, ce qui ouvre clairement une voie vers l'eugénisme. On avance vers une plus grande fragilité dans notre société. Sur les femmes seules, certains exemples étrangers sont parlants. Le Royaume-Uni a décidé de ne plus accorder d'aides financières aux femmes qui choisissent, et non qui subissent, le fait d'être seules avec leur enfant. D'ailleurs, 91 % des Français pensent qu'il ne faut pas ouvrir l'AMP aux femmes seules. Outre le simple bon sens, cela pose la question de l'altérité, dont l'enfant a besoin pour ne pas être enfermé dans une relation exclusive avec sa mère, ce qui peut être dangereux.
L'accès à l'identité du donneur me paraît être une bonne chose, mais elle pose deux questions. Si on permet la connaissance de l'identité du donneur, c'est qu'il s'agit de réparer un mal. On reconnaît donc qu'il y a un mal. C'est très bien de réparer un mal. Mais, d'un autre côté, on promeut l'AMP avec tiers donneur, c'est-à-dire qu'on organise le mal ! Il y a là une contradiction interne. Or, s'agissant de l'AMP, il faudrait mettre en oeuvre le premier principe de la médecine qui est : primum non nocere.
Si on ouvre l'accès à l'identité du donneur, c'est qu'on reconnaît que cela nuit à l'enfant de ne pas connaître sa source biologique.
Par ailleurs, on peut aujourd'hui contester la paternité en s'appuyant sur une preuve biologique. Un enfant qui naîtrait sur la base d'un projet parental pour un couple de femmes ne pourrait rien contester du tout. Cela lui serait imposé pour toute sa vie. Or, avec l'une des deux femmes, il n'aura aucun lien biologique. Elle aura simplement érigé sa volonté comme étant ce qui lui permet d'être désignée comme mère. Imaginons qu'à l'âge de dix-huit ans, il connaisse le donneur de sperme dont il est issu, et que cet homme, dans le fond, ne soit pas si mal que ça - même si c'est un homme - et que cet enfant se prenne d'affection pour lui et demande une reconnaissance en paternité. Aurait-il le droit de contester la maternité de la mère qui ne l'est que par volonté ? Ce droit me paraîtrait important pour l'enfant, que nous reconnaîtrions comme une personne à part entière. Je trouverais cela plus juste que de priver pour toujours l'enfant d'avoir un père.
Pour le don d'organes, se pose une question très difficile : plus on augmentera les paires, plus on accroîtra la distance, y compris affective. On risque d'en arriver à demander une contrepartie au don, qui ne serait plus gratuit. Plus on s'éloigne du cercle où il y a un authentique lien affectif, plus on va vers un besoin de reconnaissance. Je ne suis pas convaincu par le registre du don, selon lequel on est un donneur potentiel sauf si on en a exprimé le refus. Ce n'est pas respecter l'être humain, qui est capable de faire un acte de don. En Ille-et-Vilaine, il y a 357 établissements scolaires privés catholiques. Je m'y rends pour faire une éducation au don d'organes. Je suis frappé de voir que les collégiens et les lycéens sont prêts à faire un don, librement. C'est la grandeur de l'être humain d'être capable de faire un don.
Enfin, vous parlez des progrès qui permettraient cette loi. Mais comment réfléchir à l'embryon humain ? Si c'est une loi de bioéthique, quand allons-nous réfléchir sereinement, paisiblement, en écoutant les scientifiques, en écoutant les philosophes, en écoutant les sages, pour penser ? L'embryon humain n'est pas pensé : il n'est pas une personne, il n'est pas une chose. Résultat : la recherche sur l'embryon humain est encore accrue. Pourtant, détruire un embryon humain ne devrait jamais nous paraître banal. On le voit d'ailleurs dans les réponses aux questionnaires que doivent compléter les couples dans les centres d'AMP, s'agissant des embryons qui ne font plus l'objet d'un projet parental. Le plus souvent il n'y a pas de réponse... Difficile de répondre ! Or le projet de loi accentue la recherche sur l'embryon humain, en augmentant à quatorze jours le temps pendant lequel on peut le garder en culture, ce qui correspond à la première différenciation dans le tissu nerveux. Pourquoi pas ? Mais pourquoi pas plus tard ? Jusqu'où aller ? Je ne suis pas sûr qu'on aille vers le progrès, si on ne réfléchit pas de manière plus approfondie. Nous nous laissons guider par la fascination de la technique, sans réfléchir à ce qu'est la technique. Tous ceux qui ont réfléchi sur la technique lui demandent un supplément d'âme, comme dit magnifiquement Bergson. Et je ne vois pas comment la technique serait un progrès si elle n'engage pas un supplément éthique consubstantiel à notre fraternité.
Il n'y a pas de progrès en bioéthique si l'on ne considère pas l'écologie. Si on ne respecte pas la planète, ce n'est pas un progrès. Nous avons pris conscience qu'il y a des techniques qu'il ne faut pas utiliser pour la planète.
Il y a un écosystème à protéger, je pense que l'être humain est aussi un écosystème à protéger et qu'il y a des techniques qu'il ne faut pas utiliser, parce qu'elles sont malfaisantes pour l'être humain, tout comme des techniques ne sont pas utilisées parce qu'elles sont malfaisantes pour la planète et l'environnement. On ne peut plus séparer la bioéthique de l'écologie. Peut-être, avec la sagesse qui lui est reconnue, le Sénat pourrait-il l'inscrire dans ce texte.
M. Olivier Wang-Genh . - Tous les cultes sont sensibles aux effets négatifs potentiels. D'un point de vue bouddhiste, tout ce qui peut amener à soulager les souffrances d'un être humain est bienvenu. L'attention de départ, autant que l'intention, est fondamentale. Le rôle des lois est de faire ce qu'on fait avant un mariage : même si on s'aime à la folie, on rédige tout de même un contrat de mariage, dans lequel on évoque des situations pas toujours agréables ! En tant qu'éveilleurs de conscience, nous sommes là pour attirer l'attention sur ces dangers potentiels, plus que pour certifier que cela va effectivement amener beaucoup de bienfaits dans les années à venir. Jusqu'à quand l'être humain restera-t-il maître de ces évolutions technologiques ? Jusqu'à quand restera-t-il dans cette conscience d'une globalité ? Il ne devrait jamais se séparer d'une réflexion beaucoup plus globale sur le monde dans lequel il est et avec lequel il est en unité, en totale interdépendance.
M. Michel Amiel . - J'ai grand plaisir à vous retrouver, puisque nous nous étions rencontrés lors de la préparation du projet de loi sur la fin de vie, dont j'ai été rapporteur.
Le débat a été un peu confisqué par les quatre premiers articles du texte : c'est dommage ! Nous aurions préféré une dissociation entre la réflexion sur l'extension de l'AMP et d'autres questions de bioéthique comme les recherches sur les cellules souches embryonnaires, ou les cellules pluripotentes... Pour autant, les quelques questions que j'ai à vous poser vont concerner aussi l'extension de l'AMP ! Un député a dit : « il n'existe pas de vérité biologique, il n'existe de vérité que sociale et politique. » Qu'en pensez-vous ?
Vous avez évoqué l'idée de se passer d'un père. Ne s'agit-il pas plutôt d'une question d'effacement de la masculinité, dans un processus néo-féministe ? Déjà, en 1985, Robert Badinter parlait de déclin de la masculinité.
Vous avez parlé d'un chemin de crête. On a l'impression que le débat se situe entre le naturel et le culturel, mais aussi entre le naturel et le contractuel. La procréation est un phénomène naturel. Vouloir l'ouvrir à une volonté affirmée préalablement, pour des questions juridiques complexes liées au droit de la filiation, n'a-t-il pas pour résultat que le contractuel se substitue à un processus naturel ?
Le manque de gamètes dont il a été question, qui est déjà une réalité et qui pourrait être accentué par la suppression de l'anonymat du donneur, ne risque-t-il pas d'ouvrir la voie à un grand marché de la procréation ?
Enfin, dans le bouddhisme, il y a les quatre vérités saintes : tout est souffrance, la souffrance naît du désir et, pour supprimer la souffrance, supprimons le désir !
M. Roger Karoutchi . - Merci à tous les représentants des religions de se prêter à cet exercice, qui n'est pas facile.
J'ai été interpellé par les propos tenus, notamment par le grand rabbin et par Mgr l'archevêque. Nous n'avons pas tout à fait la même conception de la loi. M. Korsia dit qu'il faut laisser les choses en l'état et que, s'il y a transgression, il y a transgression, mais qu'il ne faut pas en faire une règle. Mgr d'Ornellas, lui, dit que nous devons nous souvenir que nous sommes des héritiers, et qu'il y a un bien qui nous précède. Sur le fond, je suis assez d'accord, parce que je suis un conservateur, de par mon éducation. Mais quand on fait la loi, on ne vote pas pour ses conceptions morales, on établit un cadre, sans avoir nécessairement envie de le suivre.
Ainsi, depuis 1976, nous avons eu plusieurs textes, sur la contraception, l'avortement, le Pacs, le mariage... Je n'en remets aucun en cause, mais je ne m'en appliquerais aucun à moi-même. Quand je vote la loi, je ne le fais pas pour moi ou pour ma conception, je le fais pour créer un cadre pour éviter des souffrances, des poursuites judiciaires, des départs à l'étranger... Ne faut-il pas dissocier loi et morale individuelle ?
Sur ce texte, je suis bien d'accord avec tous, on aurait dû dissocier l'extension de l'AMP d'un texte bioéthique. Pour autant, quelle que soit ma conception personnelle, est-ce qu'une conception publique peut intégrer ceux qui n'adhèrent pas à la même conception que moi de la morale ? La loi, ce n'est pas un texte général fondé sur ma conception morale personnelle.
M. Dominique de Legge . - Vous avez été plusieurs à souligner une certaine forme de contradiction entre créer une famille sans père mais avec deux mères et autoriser l'accès aux origines. Peut-on traiter de la même façon un don d'organe, gratuit et anonyme, et un don de gamètes qui, par définition, a vocation à donner la vie ? Le texte permet l'accès aux origines selon certaines modalités. Faut-il l'approuver ? Qui cherche-t-on à protéger ? L'enfant ? Le donneur ? La mère ? L'ouvrir, n'est-ce pas recréer un équilibre ? Ou n'est-ce pas fragiliser ?
M. Jean-Pierre Corbisez . - Pour ma part, j'ai été greffé quatre fois. En tant que receveur, je peux vous dire qu'on n'est jamais à la recherche des origines du donneur. Aujourd'hui, si on ne s'inscrit pas sur un registre des refus, on est par principe donneur. Auparavant, il fallait l'inscrire sur une pièce d'identité ou une carte de donneur sanguin. Certes, on bénit le donneur tous les jours, car il a eu un geste de supra-citoyenneté, estimant pouvoir être encore citoyen après sa mort. En revanche, les familles des donneurs décédés ont tendance à essayer de projeter le parent défunt dans celui qui a reçu l'organe. Le manque de donneurs décédés en France ou en Europe et l'importance des critères de cross-match amènent à poser le problème des dons intrafamiliaux. La gêne et la honte sont surtout du côté du futur receveur, qui ne souhaite demander l'identité du donneur. Une épouse peut même donner un rein à son mari, vu l'avancée de la recherche.
Le problème de l'extension de l'AMP relève de la même logique : il ne faut surtout pas qu'on puisse remonter aux donneurs. Le droit du mariage entre parfois en contradiction avec l'AMP. Certains des articles du code civil qui l'encadrent datent du code napoléonien. Ainsi de l'article 215, qui prévoit que « les époux s'obligent mutuellement à une communauté de vie » : les jeunes d'aujourd'hui vivent sous le même toit avant de se marier ! En cas de divorce, si vous avez un nouveau projet de vie et que, pour des raisons de santé, vous voulez avoir recours à l'AMP, mais que vous n'êtes pas divorcé, les services hospitaliers vous refusent l'accès à cette technique. Il va falloir trouver dans ce projet de loi de bioéthique un moyen de répondre à la problématique de la durée de la procédure de divorce.
M. François Clavairoly . - J'aimerais revenir sur les remarques qui viennent d'être faites sur les apparentes contradictions entre le naturel et le contractuel, le biologique et le culturel, etc. C'est justement dans ces ambivalences, dans ces tensions, que s'inscrit le travail du législateur. La société des hommes n'est pas exactement celle des fourmis ou des abeilles, elle ne reproduit pas à l'identique, de génération en génération, la même sociabilité ; les choses évoluent.
Nous sommes des héritiers - je rejoins les propos de Mgr d'Ornellas -, nous ne commençons pas le monde, mais nous sommes aussi des pionniers. La loi de la République nous déplace et elle contient finalement une forme de déracinement - j'ai entendu ce mot hier soir dans la bouche du Président de la République à l'occasion de l'inauguration du centre européen du judaïsme -, car elle est toujours en train de se chercher. En l'espèce, nous construisons par la loi le cadre de nouvelles formes de parentalité qui sont effectivement décalées par rapport à celles du code Napoléon... Alors, adaptons-nous ! La vie des hommes et des femmes est ainsi faite qu'elle ne reproduit jamais les choses à l'identique.
En ce qui concerne le don d'organes, le cadre proposé par le projet de loi est plus large, plus efficient. Et si la question se pose, c'est qu'il existe aujourd'hui une carence. J'entends les propos de Mgr d'Ornellas, il faut apprendre à donner, le don est une belle responsabilité, mais la loi doit l'encadrer. Je rejoins ce que disait M. Karoutchi, on peut ne pas être d'accord avec les termes de la loi, mais le cadre qu'elle fixe est général et s'applique à tous. Lors des débats, anciens maintenant, sur la loi dépénalisant l'avortement, beaucoup de femmes protestantes - je pense à ma mère par exemple - disaient en même temps qu'elles n'avorteraient pas elles-mêmes, mais qu'il fallait mettre un terme aux situations insupportables que nous connaissions alors, que ce soit la clandestinité ou la nécessité d'aller à l'étranger.
Les débats éthiques sont toujours compliqués, mais ce sont des sujets qui concernent l'ensemble de la société. Je crois avoir lu que 3 % des enfants naissent aujourd'hui grâce à à l'AMP et l'élargissement qui est prévu dans le projet de loi ne changera pas fondamentalement cet ordre de grandeur. L'enjeu sociétal est réel, mais limité. En revanche, pour les femmes qui seront concernées, c'est très important. L'attention à l'humain doit être au coeur de nos préoccupations.
M. Haïm Korsia . - Chacun voit les choses de son point de vue, comme vient de le dire M. le sénateur Corbisez dans son émouvante intervention sur le don d'organe. La question se pose naturellement pour celui qui reçoit - il peut être gêné de demander cela à quelqu'un de sa famille -, mais elle se pose aussi pour les membres de la famille, sur lesquels se posent tous les regards. Ce n'est pas la seule question, mais elle se pose aussi. D'ailleurs, l'exemple que vous avez donné, monsieur le sénateur, est surprenant : trouver une convergence génétique entre un mari et sa femme est douteux me semble-t-il. Il nous faut prendre des précautions et organiser les choses sans imposer un choix.
La question de la loi se retrouve aussi dans celle des origines : qui protège-t-on ? Selon les études, 15 % des gens n'ont pas pour père celui qu'ils croient et 35 % pour ceux qui demandent un test génétique. Freud expliquait, avant l'existence des tests ADN, que c'est la mère qui désigne le père ; la construction paternelle est donc une question culturelle. Les aspects naturels et culturels sont donc bien tous deux nécessaires, ce qui me paraît essentiel.
Autre question, la loi est-elle pour moi ou pour les autres ? J'adhère parfaitement aux propos de M. le pasteur. Monsieur Karoutchi, vous savez fort bien qu'on ne se sent jamais soi-même soumis aux lois. Il existe tant de domaines dans lesquels nous ne sommes pas compétents ; je pourrais disserter des heures sur la vache rousse dans la Bible, mais je n'ai jamais vu de vache rousse moi-même ! Je vais prendre un autre exemple de la difficulté de légiférer pour les autres : il existe une grande différence entre celui qui viendrait à la synagogue le jour de Kippour, en disant : « j'ai mangé du jambon, mais je souhaite participer à l'office », et celui qui viendrait à la synagogue en mangeant du jambon et qui me demanderait de dire qu'il a le droit de le faire et de participer à l'office. Cette différence s'appelle le bon sens !
La loi est faite pour tous, mais chacun peut librement se positionner par rapport à elle. Pour reprendre les statistiques qui viennent d'être citées, il faut que les 97 % aient l'intelligence de dire que les 3 % peuvent exister et vivre selon leurs choix, mais on ne doit pas être sommé de dire que ces 3 % représentent un idéal. La loi est quelque chose qui doit nous élever ; c'est une aspiration, un horizon, un projet de société, et pas simplement le constat de ce que nous faisons - je réponds en cela à la question sur les origines.
Par ailleurs, je voudrais rassurer ceux qui s'inquiètent de l'existence d'un front des religions... En effet, nous ne sommes pas d'accord sur tout, grâce à Dieu, pourrais-je dire. C'est par exemple le cas sur la question de la recherche sur les embryons. À ce sujet, je crois qu'il faut distinguer les embryons potentiels et les embryons théoriques potentiels. Si vous prenez deux embryons au même stade de développement, que l'un est placé dans une éprouvette et que l'autre est implanté dans le ventre d'une femme, la différence est évidente neuf mois après !
M. Alain Milon , président . - Vous avez quand même intérêt à congeler l'embryon qui est en éprouvette, si vous voulez faire des expériences de ce type !
M. Haïm Korsia . - Effectivement ! Mais il y a bien une différence entre un potentiel théorique et un potentiel réel. Cette différence rend plus simples les questions du développement des cellules pluripotentes et de la gestion du « trop-plein » - je crois qu'il existe aujourd'hui environ 150 000 embryons congelés.
À mon sens, la notion de don est toujours liée à celle de gratuité - c'est d'ailleurs ce qui me gêne profondément dans la gestation pour autrui. Pour reprendre la phrase de M. le pasteur, avec le don, je suis le gardien de mon frère, je suis responsable, je ne suis pas indifférent à la souffrance de l'autre. En donnant quelque chose, je permets à un espoir de perdurer - c'est quelque chose d'éminemment républicain.
M. Alain Milon , président . - Une remarque sur embryon potentiel et théorique : le projet de loi précise qu'il s'agit d'embryons théoriques, sur lesquels il n'y a plus de projet parental.
M. Haïm Korsia . - En effet, mais qu'est-ce que cela veut dire exactement ? C'est évidemment une question d'appréciation. Les données peuvent changer, de manière dramatique parfois. Pouvons-nous vraiment faire une différence entre la souffrance d'une femme seule - est-ce un choix ? En est-elle responsable ? - et une autre souffrance ? Je ne veux pas faire de hiérarchie entre les souffrances. Le projet parental peut évoluer. On pourrait comparer cette décision à celle que l'on doit prendre en fin de vie, partir de directives anticipées, préparées longtemps à l'avance lorsque la personne était en bonne santé. Un choix fait à un moment donné peut évoluer en fonction des circonstances.
M. Alain Milon , président . - D'un point de vue médical, la souffrance est une pathologie dont il faut s'occuper !
M. Haïm Korsia . - C'est une évolution récente...
Mgr Pierre d'Ornellas . - Premièrement, en ce qui me concerne, je pense qu'il y a une vérité dans le biologique ; sinon, l'écologie n'aurait plus de sens. Il y a une biologie dans la nature qu'on voudrait d'ailleurs respecter de plus en plus. Les agriculteurs le reconnaissent tous. Je me souviens d'une directrice de recherche de l'Institut national de recherche pour l'agriculture (INRA) qui m'expliquait son travail : au milieu de sa présentation apparaissait sur une diapositive un enfant noir ballonné extrêmement maigre et j'ai demandé à cette directrice de recherche pourquoi elle avait intégré cette photo à sa présentation ; elle m'a répondu avec une grande émotion que tout son travail sur la biologie servait justement à nourrir ces enfants. Il est donc indéniable qu'il y a une vérité dans le biologique.
Ensuite, pour l'être humain, on ne peut pas distinguer entre l'esprit d'un côté, qui serait l'amour, la volonté, etc., et le biologique de l'autre. Il me semble que nous sommes une unité indissociable d'esprit et de corps ; nous sommes un être d'esprit de condition corporelle dont la réalité la plus spirituelle s'exprime de façon corporelle. Il n'est pas possible d'exprimer du spirituel, c'est-à-dire les valeurs les plus nobles de l'esprit, sans que le biologique y participe - ce n'est pas possible. Et ces valeurs les plus nobles de l'esprit sont parfois suscitées par des réactions du biologique, parce que le biologique est touché, voire blessé. Je ne crois donc pas que l'on puisse formuler, en particulier sous les ors de la République, l'affirmation péremptoire, selon laquelle il n'y a pas de vérité dans le biologique. En tout cas, cette affirmation que je viens d'entendre de la bouche d'un sénateur doit faire tressaillir beaucoup de philosophes. Je pense que c'est une vue de l'esprit de vouloir opérer cette distinction.
Deuxièmement, au sujet de la loi, je comprends l'idée d'une morale individuelle qui ne pourrait pas s'imposer à tous, mais je ferai deux distinctions.
J'ai parlé de sagesse pratique. Quand on lit Cicéron, on voit bien que ceux qui ont le plus besoin de cette sagesse pratique, donc des philosophes et des sages, ce sont précisément les gouvernants. Je crois qu'il ne s'agit pas de partir de sa morale individuelle, mais plutôt de ce besoin d'une sagesse pratique pour tous. C'est plus que l'intérêt général, on peut appeler cela le bien commun, c'est-à-dire toutes les conditions justes qui permettent précisément la croissance et le respect de tout être humain, quel qu'il soit. Il me semble que l'élaboration de la loi ne se fait pas à partir d'une morale individuelle, mais à partir d'une recherche collective, d'une sagesse pratique pour tous. Dans le fond, le lien entre le philosophe et le politique est intrinsèque à la bonne marche de la gouvernance d'une société et le philosophe ne pense pas une morale pour lui. Je pourrais aussi citer Hans Jonas chez qui le concept du « collectif » est très important. C'est aussi l'universel que pense Emmanuel Kant : il y a quelque chose dans la personne humaine, dans l'individu humain, qui est de l'ordre de l'universel et c'est cela qui est à découvrir, pas à imposer.
Troisièmement, de nombreuses lois ont été citées, mais il existe aujourd'hui un grand débat - Sylviane Agacinski le met en lumière de façon très forte, mais elle n'est pas la seule - sur la question du vraisemblable et de l'invraisemblable. Ce vocabulaire simple n'est pas aussi simple qu'il n'y paraît, il acquiert une charge éthique et philosophique considérable. Il me semble que nous avons passé un cap : nous imaginons mettre dans la loi ce qui ne correspond pas au réel, c'est-à-dire l'invraisemblable. Comment justifier que la loi promeuve ce qui est irréel, invraisemblable ? Il me semble que nous passons un seuil, puisque la loi va dire que l'invraisemblable est le réel.
Nous rejoignons ici le grand débat philosophique qui a eu lieu entre Guillaume d'Ockham et Thomas d'Aquin aux XIII e et XIV e siècles : Thomas d'Aquin insistait toujours sur le réel, tandis que, pour Guillaume d'Ockham, c'était la volonté qui comptait. Ce débat reste d'actualité et nous sommes en train de passer à un ockhamisme, parfois appelé nominalisme : il suffit de vouloir quelque chose pour que cette chose soit réelle, alors qu'elle n'est pas vraisemblable. Nous touchons là un point fondamental. Par exemple, la non-distinction entre la femme qui accouche et celle qui n'accouche pas est, me semble-t-il, problématique, dans la mesure où la loi fait de l'invraisemblable une réalité. Il en est de même pour l'embryon chimérique : est-ce que nous n'abolissons pas toute différence entre l'animal et l'humain, quelque chose qui contredit l'article 16 du code civil sur le respect du réel, c'est-à-dire l'intégrité de l'espèce humaine ? On aboutirait à quelque chose d'invraisemblable : un animal destiné à produire des organes humains ou susceptibles d'être greffés sur l'être humain. Nous toucherions ainsi des limites que bien des penseurs n'ont jamais pu imaginer : la condition humaine devient invraisemblable !
Quelque chose de nouveau - penser l'invraisemblable - apparaît dans notre société. Certains vont par exemple qualifier la gestation pour autrui d'éthique. Mais comment qualifier une transgression d'éthique ? Nous atteignons vraiment une ligne rouge, et je crois que nous devons mettre en place un moratoire, comme le demandait Jacques Testart lorsqu'il est arrivé dans le laboratoire où l'embryon était fécondé dans l'éprouvette - il raconte cet épisode dans son livre L'OEuf transparent.
Une réflexion doit aujourd'hui être menée sur cette loi pour faire en sorte qu'elle soit une loi civilisatrice, qui nous tire vers le haut et qui représente véritablement la République des droits fondamentaux de l'être humain.
M. Olivier Wang-Genh . - Je voudrais répondre à la question sur l'origine de la souffrance qui concernait spécifiquement le bouddhisme. Il y a 2 600 ans, le bouddhisme identifiait déjà la souffrance comme étant la soif insatiable de désir et de satisfaction. Tout l'enseignement du Bouddha consiste justement à prendre conscience de cette soif qui nous consume sous des aspects extrêmement divers et à faire en sorte de l'apaiser à travers la méditation, l'éthique, le comportement, la réflexion, la sagesse, le discernement, etc.
Aujourd'hui, nous assistons à une forme d'emballement comme un cheval lancé au galop qu'on ne tente même pas d'arrêter, mais qu'on essaye simplement d'accompagner. Tout cela ne peut que créer davantage de désir, d'attachement, donc de sources de souffrances. Ces lois et technologies cherchent paradoxalement à apaiser certaines formes de souffrance, en en créant de nouvelles, et la complexité ainsi créée ne permet plus de comprendre le pourquoi de tout cela. Soyons conscients qu'en ce moment même des recherches touchent, dans divers endroits du monde, à des domaines totalement nouveaux qui transformeront finalement l'invraisemblable en vraisemblable. Nous sommes aujourd'hui dans le vraisemblable de l'invraisemblable... Nous devons prendre cette réalité en compte.
M. Alain Milon , président . - Le rôle du politique et de la loi est justement d'encadrer ce qui peut devenir vraisemblable pour éviter l'inhumain.
Ce point de l'ordre du jour a fait l'objet d'une captation vidéo qui est disponible en ligne sur le site du Sénat .
M.
Jean-François Delfraissy, président,
et Mme Karine Lefeuvre,
vice-présidente
du Comité consultatif national
d'éthique (CCNE)
M. Alain Milon , président . - Mes chers collègues, nous poursuivons nos travaux sur le projet de loi relatif à la bioéthique avec l'audition de M. Jean-François Delfraissy, président, et de Mme Karine Lefeuvre, vice-présidente du Comité consultatif national d'éthique (CCNE).
J'indique que cette audition fait l'objet d'une captation vidéo retransmise en direct sur le site du Sénat et consultable à la demande.
Aux termes de la loi, « tout projet de réforme sur les problèmes éthiques et les questions de société soulevés par les progrès de la connaissance dans les domaines de la biologie, de la médecine et de la santé doit être précédé d'un débat public sous forme d'états généraux, organisés à l'initiative du CCNE ».
Je vous rappelle que ni le Gouvernement ni l'Assemblée nationale ne souhaitaient, à l'époque, que la loi de bioéthique soit révisable; le Sénat l'a imposé. C'est donc grâce à sa volonté que des états généraux, dont un rapport de synthèse a été publié en juin 2018, se sont tenus au premier semestre de la même année. Cette association des citoyens à la réflexion est la marque du processus de révision de la loi et nous permet d'entamer le processus parlementaire avec cet apport.
Dans un second temps, en septembre 2018, le CCNE a publié l'avis n° 129 qui détaille ses prises de position sur les différents sujets soumis à la consultation adoptées sinon sur un mode consensuel, ce qui, compte tenu des sujets, semble un art difficile, mais selon le principe de l'assentiment majoritaire.
Le Conseil réfute la loi de Gabor, du nom d'un ancien prix Nobel de physique selon lequel tout ce qui est techniquement possible sera fait tôt ou tard. Cette réfutation laisse un espace pour la réflexion éthique qui consiste précisément à définir, au sein de ce que la technique biomédicale permet, ce qui est souhaitable pour le patient et, plus largement, pour une société dotée de règles communes.
En conclusion, le CCNE appelle de ses voeux une loi de « confiance » qui réponde aux défis posés par les questions de bioéthique en perpétuelle évolution et aux enjeux sociétaux qui s'y rattachent. Vous nous direz dans quelle mesure le texte adopté par l'Assemblée nationale répond à ce souhait exprimé d'une loi de confiance.
Je vous laisse la parole pour un propos introductif avant de la passer à nos rapporteurs, puis à ceux de nos collègues qui souhaiteront vous interroger.
M. Jean-François Delfraissy, président du Comité consultatif national d'éthique (CCNE) . - Je tiens tout d'abord à vous remercier de cette invitation. Comme vous l'avez souligné, monsieur le président, c'est grâce à la volonté du Sénat que la « démocratie sanitaire » a pu prendre une dimension nettement plus importante que d'habitude.
L'organisation des états généraux était une tâche nouvelle pour le CCNE dont il s'est acquitté pour la première fois en 2018. Dans ma vie professionnelle, j'ai eu à accomplir des missions importantes, notamment dans la lutte contre le sida ou contre le virus Ébola. Toutefois, je peux dire qu'animer la démocratie sanitaire et la discussion autour des sujets de bioéthique, face auxquels il faut faire preuve de beaucoup d'humilité, est la plus difficile tâche que l'on m'ait jamais confiée. Il s'agit d'un choix de société relativement important. Un des thèmes des états généraux de la bioéthique s'intitulait d'ailleurs : « Quel monde voulons-nous pour demain ? » Il s'agit d'un choix difficile qui oscille entre une vision individuelle, et donc très diverse, et une vision plus collective en ce qui concerne non seulement la bioéthique, mais aussi la santé.
Aux États-Unis, par exemple, le choix individuel l'emporte en ce moment ; en Asie, ce sont les choix d'État qui dominent. En Europe, notamment en France, nous avons une culture de la réflexion bioéthique. À l'occasion des états généraux, nous avons retrouvé un socle de valeurs partagées sur lesquelles nous pourrons revenir dans le cours de notre discussion.
La construction collective n'est pas seulement faite d'apports individuels. Quelque chose de plus vaste se construit. On dit des Français qu'ils deviennent de plus en plus égoïstes. Après les états généraux de la bioéthique, je n'en suis pas persuadé. Nous avons tous fait, à un moment de notre vie, des choix individuels. Mais tous, nous sommes aussi capables d'avoir une vision plus collective. Et c'est là qu'est tout l'enjeu, qu'il s'agisse des questions de procréation ou d'accès à certains tests génomiques. Nous sommes dans cette oscillation entre vision individuelle et construction collective. Cette dernière, pour généreuse qu'elle soit, ne doit pas non plus écraser un choix qui touche à l'intime sur un certain nombre de sujets délicats.
Nous avons tenté d'aborder des questions difficiles, sinon conflictuelles, au travers des états généraux, bien sûr, mais aussi de la réflexion que nous avons menée au sein du CCNE et qui se poursuit dans le pays, de manière plus globale, autour de la construction de cette loi. Nous avons dû le faire dans un délai contraint. Toutefois, entre les travaux du CCNE, ceux de l'Office parlementaire d'évaluation des choix scientifiques et technologiques (Opecst) et ceux des deux commissions spéciales de l'Assemblée nationale et du Sénat, environ 1 200 heures auront été consacrées à la réflexion sur la bioéthique, avant même que le débat parlementaire ne soit engagé - c'est bien.
En France, on a souvent tendance à se tirer une balle dans le pied et à dénigrer notre démocratie. Je me rends régulièrement à l'étranger pour exposer ce que nous faisons. Je peux vous assurer que beaucoup de grands pays regardent avec intérêt ce qui se passe chez nous. Le Japon, par exemple, va probablement organiser des états généraux sur des questions aussi difficiles que la génomique ou la recherche sur l'embryon avec une participation citoyenne.
On pourrait s'interroger sur la place respective du législateur, des experts et des sachants - médecins, scientifiques, philosophes, etc. - et du citoyen de base. Il est difficile de toucher ce dernier sur des sujets aussi complexes. Nous vivons dans une démocratie, c'est donc vous qui allez trancher en votant. Mais le débat qui aura eu lieu avant votre vote se poursuivra encore après. La réflexion sur les sujets de bioéthique ne s'arrêtera pas à ce texte. C'est la raison pour laquelle il est essentiel de faire participer nos concitoyens à cette réflexion.
Nous ne sommes qu'au début de la construction d'une démocratie sanitaire. La discussion sur les enjeux majeurs qui sont devant nous doit pouvoir s'appuyer sur ce triangle que j'évoquais entre parlementaires, experts et citoyens. Il s'agit d'une construction commune. La santé se prête bien à ce type de discussion.
Vous m'avez très gentiment fait parvenir une quarantaine de questions, jeudi soir. J'ai bien compris votre clin d'oeil et j'y répondrai dans les délais proposés, à savoir d'ici au 20 novembre prochain. Toutefois, je peux déjà dire que le CCNE et son président se retrouvent globalement dans cette loi qui est bien de confiance et d'ouverture, comme nous l'avions appelée de nos voeux.
Le texte comporte ainsi des titres et des sous-titres explicatifs qui permettent d'aborder un certain nombre de points importants. Ces derniers ne sont donc pas noyés dans des articles auxquels nos concitoyens - moi le premier - ne comprennent pas grand-chose, faute d'être des spécialistes.
Par ailleurs, alors que la société change profondément, la science n'est pas forcément source de progrès. C'est d'ailleurs la raison pour laquelle des choix bioéthiques se posent et qu'un équilibre doit être trouvé entre évolution profonde de la société et nouvelles possibilités offertes par la science. Cette loi, qui peut encore être améliorée sur certains points, se situe sur cette ligne de crête.
Avant d'évoquer le sujet de l'accès à l'assistance médicale à la procréation (AMP) aux femmes seules et aux couples de femmes, que nous ne pourrons éviter, je souhaite parler d'abord de la génomique. De nouveaux tests de séquençage du génome à haut débit sont aujourd'hui disponibles. Avec un peu de votre salive, je peux avoir, demain, en milieu d'après-midi, le séquençage de votre génome, pour 300 euros. Dans un an, il ne faudra plus que 50 euros ! La loi des coûts et l'innovation technologique rendent possible une génomique du quotidien.
À l'heure actuelle, la loi interdit l'utilisation des tests génomiques récréatifs ou de recherche des origines. Pourtant, au cours des douze derniers mois, des publicités vantaient ces tests sur BFMTV. C'est la raison pour laquelle le CCNE avait proposé l'utilisation de tests génomiques en population générale. Par crainte d'une certaine forme d'eugénisme, les ministres de la recherche et de la santé ont préféré les interdire, tout en fermant les yeux sur ce qui se passe réellement. Le CCNE avait préconisé de rester dans un modèle à la française, soit un modèle permettant aux personnes qui l'auraient souhaité d'avoir accès à un conseil génétique. On le sait, dans la majorité des cas, les personnes intéressées auraient renoncé à leur projet de test. Cette solution a l'avantage de dépister un certain nombre de mutations concernant des pathologies parfois létales. Par ailleurs, elle permet de repérer des mutations non classiques, la personne concernée bénéficiant ainsi d'une surveillance particulière, sans avoir à attendre un événement clinique.
Le CCNE continue de préconiser un dépistage préconceptionnel pour la population qui le souhaite, dans un contexte médicalisé et de conseil génétique. En effet, une interdiction pure et simple de ces tests risque d'entraîner une utilisation « à la sauvage », par le biais d'Internet.
Par ailleurs, la loi, dans sa forme actuelle, clarifie la différence entre recherche sur embryons et recherche sur cellules souches. La recherche sur embryon est un sujet difficile. Moi-même, il m'a fallu du temps pour comprendre en quoi elle ne pouvait avoir d'alternative.
Les taux de réussite en matière de fécondation in vitro (FIV) sont relativement faibles. Ainsi, sur les embryons implantables, seulement 16 % ou 17 % d'entre eux vont finalement « prendre ». Un tel taux d'échec, considérable, n'aurait jamais été admis dans le cadre d'une autorisation de mise sur le marché d'un médicament ou d'un vaccin. Nous avons donc besoin de comprendre ce qui se passe durant les tout premiers jours de l'embryon qui se trouve en contact avec son milieu naturel.
Quant aux cellules souches, elles sont soit d'origine embryonnaire, soit d'origine adulte (les IPS). On a longtemps cru que les cellules souches d'origine adulte avaient les mêmes qualités de plasticité et de durée de vie que les cellules souches embryonnaires, ce qui n'est pas tout à fait le cas. Somme toute, l'important, ce n'est pas d'où viennent les cellules souches, mais ce que l'on va en faire. C'est la communauté scientifique elle-même qui réclame une régulation sur ce point.
Les cellules souches permettent d'aboutir à deux choses. Premièrement, elles peuvent devenir un cartilage d'épaule ou de hanche, et elles représentent la médecine du futur, qui doit se développer. Deuxièmement, elles peuvent se différencier en spermatozoïdes ou en ovocytes, avec lesquels on pourra créer des embryons nouveaux, sans acte sexuel. C'est le domaine de la crête, de la ligne rouge qu'il ne faut pas franchir. La loi installe un phénomène de régulation, qui était nécessaire et n'existait pas jusqu'à maintenant.
J'en viens au diagnostic préimplantatoire : parmi les embryons créés par FIV pourrait-on mieux cerner ceux qui seront viables ? Il existe des tests génétiques permettant de repérer les modifications chromosomiques. Même s'il ne s'agit pas de tests définitifs, ils permettent toutefois de disposer d'une base solide de différentiation, en cas d'anomalies importantes au niveau chromosomique. Au demeurant, nous faisons déjà ce type de choix dans le cadre du diagnostic de la trisomie 21. Pourtant, la loi ne va pas jusqu'à les autoriser, considérant qu'il y a là quelque chose de très sensible. Quant à la communauté médicale, elle souhaite mettre en oeuvre ces tests, car, dans ce domaine, la souffrance est immense. Le CCNE regrette que ce point n'ait pas été plus approfondi. Mais vous avez encore la possibilité d'écouter ce que disent les spécialistes de médecine foetale. Pour eux, ce sujet constitue un vrai point d'interrogation. Ne les accusons pas de vouloir construire l'eugénisme !
Dans le cadre des états généraux, deux sujets nouveaux ont été mis sur la table : « Intelligence artificielle et santé » et « Santé et environnement ». Ce dernier n'a pas aussi bien fonctionné que le premier. Il n'apparaît pas dans la loi, sauf dans les préconisations concernant le périmètre du CCNE.
S'agissant du groupe de travail « Intelligence artificielle et santé », il aurait été « inéthique » de ne pas se pencher sur les nouveaux outils de l'innovation technologique. Il s'agit de ne pas laisser passer certaines chances, tout en conservant un modèle dans un domaine où la France a encore un rôle majeur à jouer, en raison, notamment, de l'importance des bases de données de la Caisse nationale de l'assurance maladie (CNAM). Comment faire pour que l'homme garde la main ? Toute la question tourne autour de la notion de consentement.
Enfin, la loi n'aborde absolument pas le sujet des coûts de l'accès à l'innovation, qui soulève des questions éthiques. Pour ma part, je n'ai jamais autant ressenti la présence du business dans le domaine de la santé. Je pense non seulement aux cliniques gérées par des fonds d'investissement, mais aussi au coût des médicaments. Ainsi, pour traiter certains cancers, il existe aujourd'hui des traitements dont le coût s'élève à plus de 500 000 euros par patient et par an.
Quelle relation avec une loi de bioéthique ? À la fois aucune et beaucoup ! Soit on laisse s'installer une période de non-choix et on privilégie l'innovation - c'est ce que l'on a toujours fait en France, et c'est ce que j'ai toujours préconisé de faire -, soit on procède à des choix. Car si l'on finance l'accès à ces nouveaux traitements, on ne financera pas du personnel aux urgences ou dans les établissements d'hébergement pour personnes âgées dépendantes (Ehpad). Ce choix éthique ne peut pas être du seul ressort des médecins ou des politiques. Il doit être fait par les citoyens. Cette question n'est absolument pas abordée dans la loi.
M. Olivier Henno , rapporteur . - J'ai apprécié votre propos sur la notion d'équilibre et vos précisions sur le génome. Nous avons le sentiment que nous n'avons pas encore arbitré entre, d'une part, l'amélioration et la prolongation de la vie et, d'autre part, la crainte de bouleverser la vie et la peur du vide. Il faut aborder ce sujet de manière plus frontale.
Sur l'innovation, le débat est absolument fondamental. Nous n'avons pas décidé s'il fallait trancher dans le tout-remboursement, même si certains médicaments sont déremboursés. Nous continuons à croire qu'on peut tout financer. Sans aller jusqu'au modèle anglais, il faut avoir le courage de dire que ce qui relève du banal ne sera plus remboursé pour que la France puisse rester au top en matière d'innovation et éviter tout risque de décrochage.
M. Jean-François Delfraissy. - Il existe plusieurs définitions de la bioéthique. En tant que médecin, la mienne est simple : c'est l'équilibre à trouver entre les avancées scientifiques et les modifications de la société. Pour éviter d'être dans un monde virtuel, il faut tenir compte de la capacité de la société à réclamer et à utiliser les avancées scientifiques, qui ne sont pas toujours source de progrès. Cette loi de bioéthique est la première à sortir du domaine des sachants, en abordant ces questions complexes avec une approche plus sociétale. Mais cela n'est pas suffisant, et le CCNE poursuivra dès 2020 un débat continu et approfondi sur ces sujets, sans attendre la prochaine loi.
Nous menons actuellement une réflexion sur l'accès à l'innovation. Dans le modèle américain, il faut vendre sa maison pour payer les 500 000 euros du coût de l'immunothérapie après un cancer... Ce modèle individuel est inacceptable en France. Mais à force de faire des non-choix, nous allons devoir en arriver à des choix drastiques.
Il faut mettre un frein à l'industrie pharmaceutique. Même si celle-ci est importante et nécessaire, son budget atteint aujourd'hui un niveau inacceptable, notamment d'un point de vue éthique.
Le CCNE rendra un avis sur l'accès à l'innovation thérapeutique au printemps 2020.
Mme Corinne Imbert , rapporteure . - Merci pour votre présentation, qui était très claire. Dans son avis, le CCNE explique que le possible n'est pas toujours souhaitable. Puisque vous nous avez dit que vous vous retrouviez dans ce projet de loi, j'en déduis qu'il n'y a rien qui ne soit pas souhaitable dans ce texte.
S'agissant de la recherche sur l'embryon et les cellules souches embryonnaires, le CCNE souhaite, dans son avis, que « le nouvel encadrement législatif afférent à la recherche sur l'embryon soit précisé, clarifié sur les points suivants : la création d'un d'embryon transgénique, la création d'embryons chimériques et la limite temporelle au temps de culture sur l'embryon ».
Je vous remercie de ne pas avoir commencé votre propos par la question de l'assistance médicale à procréation, qui n'est pas le seul sujet du texte. Les questions relatives à l'embryon et aux cellules souches sont passionnantes : le CCNE est-il satisfait du texte eu égard aux propositions qu'il a formulées sur ces sujets ?
Ma seconde question, qui peut paraître anecdotique, concerne la gouvernance de la bioéthique. Êtes-vous favorable à l'extension du champ de compétences du CCNE ?
M. Jean-François Delfraissy . - En ce qui concerne la recherche sur l'embryon, nous avons essayé, après la synthèse des états généraux, d'être aussi neutres que possible. Nous avons eu un débat avant de donner notre avis, lequel peut servir de table d'orientation pour les décideurs politiques sur un certain nombre de grands sujets. Une large majorité du CCNE a soutenu la production de l'avis n° 129, mais certains membres y étaient opposés en soutenant que nous dépassions notre rôle d'observateurs.
Le CCNE a été profondément changé par les états généraux. Constitué d'un tiers de médecins et de chercheurs, mais aussi de philosophes, d'économistes et de grands juristes, il représente un monde d'intellectuels, parisiens dans leur majorité, composé à parité d'hommes et de femmes. En tant qu'élus, vous côtoyez vos concitoyens chaque week-end, mais tel n'est pas le cas des membres du CCNE. Les états généraux leur ont fait le plus grand bien, en leur permettant de prendre connaissance de la vision qu'avait la société, ce qui nous a fait évoluer sur certains sujets. Sur cette base, nous avons émis un certain nombre de recommandations.
S'agissant des embryons, l'idée est de ne pas faire d'embryons à visée de recherche. C'est ce que prévoit la loi et c'est ce que nous avions également recommandé, dans le respect de la convention d'Oviedo. Mais un certain nombre d'embryons surnuméraires peuvent, en l'absence de tout projet parental, être utilisés. Cette situation soulève une série de questions scientifiques, notamment sur les conséquences d'une modification du génome à J 8.
Nous avons évoqué un allongement du délai de J 8 à J 10 ou J 12, pour nous aligner sur la communauté internationale. Pourquoi les scientifiques formulent-ils cette demande ? Parce que les embryons ne deviennent surnuméraires qu'à partir de J 4 ou J 5 : il existe donc une période « grise », entre J 0 et J 5, pendant laquelle aucune étude ne peut être menée. Je suis plutôt favorable à un prolongement de la date d'utilisation, en précisant qu'il ne s'agit pas d'embryons de recherche, mais d'embryons surnuméraires sur lesquels on fait de la recherche.
Sur le périmètre du CCNE, je veux soulever deux points.
D'abord, le mécanisme de nomination des membres du CCNE, qui était jusqu'à présent défini dans la loi, relèverait désormais d'un décret en Conseil d'État : ce mécanisme permettra de procéder plus facilement à des modifications. J'ai demandé leur avis à des conseillers d'État, lesquels estimaient qu'il s'agissait plutôt d'une mesure de simplification bienvenue.
Ensuite, sur l'extension du périmètre du CCNE au numérique, d'une part, et à l'environnement et à la santé, d'autre part, je vous donne rendez-vous dans deux ans ! Il s'agit d'une question démocratique importante. Après la première FIV, le CCNE a été conçu par François Mitterrand et ses conseillers, qui ont compris que la biologie-santé serait l'un des enjeux majeurs du début du XXI e siècle. Mais il en existe d'autres : l'intelligence artificielle, le numérique... Lors du dernier renouvellement, nous avons fait entrer au sein du CCNE trois membres issus du milieu du numérique.
Si l'on met en place un comité d'éthique du numérique, qui ne s'intéresserait pas seulement aux questions du numérique et de la santé, mais aussi, par exemple, à la voiture autonome et à la reconnaissance faciale, doit-il faire partie du CCNE ou être une entité à part entière ?
Il a été décidé de créer un comité pilote du numérique, sous l'égide du CCNE : il devrait permettre de diffuser un certain nombre de savoir-faire, comme la multidisciplinarité, le partage des valeurs et la construction commune, auprès des intervenants du monde du numérique, mais aussi de nous ouvrir à des idées nouvelles. La recherche dans le numérique se fait pour moitié dans des start-up : nous avons besoin de jeunes dans ce comité ! Leur vision est très différente de celle du CCNE.
J'ai reçu une lettre de mission du Premier ministre fin juillet pour mettre en place ce comité pilote, qui couvrira l'ensemble des questions du numérique et pas seulement celles qui sont relatives à la santé. La première réunion se tiendra le 4 décembre prochain sous l'égide du CCNE : ce comité n'est pas une nouvelle entité administrative. Nous nous donnons un délai de deux ans - c'est la raison pour laquelle je vous ai donné ce rendez-vous - pour réfléchir à la suite. Soit on en fait une entité autonome - on peut imaginer faire la même démarche pour l'environnement -, ce qui conduit à multiplier le nombre d'organismes ; soit on garde le CCNE, en mettant en place des piliers - sciences de la vie, numérique, environnement - et en organisant des réunions communes, mais il faut alors veiller à ne pas créer un « machin ».
Nous ne voulons pas être une agence : nous souhaitons garder notre autonomie par rapport aux élus et au Gouvernement.
M. Bernard Jomier , rapporteur . - Pour élaborer une bonne loi bioéthique, il faut suivre l'évolution des biotechnologies, d'un côté, et confronter cette évolution à nos valeurs éthiques fondamentales, mais aussi à ce que sont les nouvelles demandes de la société, les valeurs « montantes » que celle-ci exprime, de l'autre. Quelles sont, selon vous, ces valeurs nouvelles ?
On observe parfois un décalage entre l'avis du CCNE et la version du projet de loi adopté par l'Assemblée nationale, notamment sur les questions de médecine génomique. Vous venez d'ailleurs vous-même d'indiquer que vous ne vous y retrouviez pas tout à fait sur le diagnostic préimplantatoire. Comment expliquez-vous un tel décalage ?
Ma dernière question est beaucoup plus précise : dans le projet de loi, les modalités d'application de l'une des dispositions relatives aux greffes d'organes sont renvoyées à un décret en Conseil d'État. Cela vous paraît-il une bonne chose que le Conseil d'État soit juge en la matière ?
M. Jean-François Delfraissy . - S'agissant des valeurs, on peut tirer trois enseignements des états généraux de la bioéthique.
Le premier est qu'il existe incontestablement un certain nombre de valeurs de bioéthique, dites « à la française », qui sont partagées. Je pense notamment à l'attention portée aux plus faibles, à la gratuité du don et à la nécessaire bienveillance.
Une fois de plus, les états généraux de la bioéthique ne sont pas un sondage et ne reflètent pas ce que pense la population française. Néanmoins, certains débats en région, notamment ceux qui se sont déroulés en présence de jeunes étudiants, ont révélé l'existence d'un déséquilibre entre des aspirations individuelles, qui s'accroissent fortement en France, et une vision collective. Pour autant, je précise que ces deux visions, l'une individuelle et anglo-saxonne, l'autre plus collective, ne sont pas incompatibles.
Le deuxième enseignement, c'est l'interrogation d'une partie de la société vis-à-vis des médecins et des scientifiques, une forme de remise en cause des sachants. J'alerte moi-même régulièrement les experts à propos de la fragilité actuelle de la confiance dont témoignent les Français à l'égard de la communauté scientifique dans le domaine biomédical, le risque étant d'entrer dans un modèle à l'anglo-saxonne.
Le troisième enseignement, assez inattendu et qui est ressorti de manière très forte, c'est l'importance que prend la question de la place du citoyen dans le système de soins et celle de la gouvernance du futur système, dans un monde où tout s'accélère à la vitesse grand V.
Mme Karine Lefeuvre, vice-présidente du Comité consultatif national d'éthique . - La question des valeurs est absolument indispensable et anime tous les débats du CCNE.
Les notions de consentement et d'information ont représenté un fil rouge lors des états généraux, alors même que celles-ci ne sont pas du tout nouvelles et que la loi en fait déjà des droits fondamentaux.
Ayant très attentivement relu le texte de l'Assemblée nationale, je note que ces questions ont été traitées avec beaucoup de prudence. On recherche le consentement, qui peut être refusé ou révoqué ; on prévoit un délai de réflexion, voire plusieurs. Quant à l'information, elle doit être claire, loyale et détaillée. Tous ces termes visent à encadrer le plus possible les démarches biomédicales et à accompagner le plus possible les patients. De mon point de vue, cette démarche est très positive. Ainsi, le projet de loi donne toute son effectivité au principe de « consentement éclairé », déjà tant employé en droit.
Deuxième point positif, le thème de l'opinion citoyenne ne cesse de prendre de l'ampleur. Nous sommes nous-mêmes en train de réfléchir sur la place que peut prendre l'expression citoyenne, et pas uniquement les représentants des usagers, dans la réflexion et l'élaboration des avis du CCNE. J'observe que le texte en cours d'examen confie au Comité l'organisation de débats publics annuels sur la bioéthique.
Dans cette perspective, nous avons déjà commencé à travailler sur la mise en place d'un maillage territorial beaucoup plus serré, avec le soutien des espaces de réflexion éthique régionaux, mais aussi d'autres instances. Tout cela devrait contribuer à nourrir une culture de la réflexion éthique en France.
Le défi que nous avons à relever consiste à mieux associer l'ensemble des citoyens à ces questions, en particulier les plus jeunes, ce qui permettra de construire une véritable culture de l'éthique.
M. Jean-François Delfraissy . - Pour répondre à votre question sur la génomique, monsieur Jomier, je reprendrai à mon compte les arguments formulés par la ministre de la santé, à savoir que nous assistons à la mise en place d'une démarche commerciale avec des tests proposés qui ne sont pas totalement fiables. Aujourd'hui, on fait dire à la génétique des choses qu'elle ne peut pas dire. Je suis d'accord avec la ministre lorsqu'elle affirme que notre avenir n'est pas dicté par nos gènes. Je partage en outre son interrogation : n'est-on pas au début d'une certaine forme d'eugénisme ?
Je comprends tout à fait que le législateur ait la main qui tremble sur ces sujets. Toutefois, je vous mets en garde sur un point : en interdisant sans être capable d'interdire, ne laisse-t-on pas la voie libre à une forme sauvage d'utilisation des tests génétiques ? On sait très bien que le prix de ces tests va baisser et qu'il sera bientôt possible d'en offrir à des occasions aussi diverses que Noël ou Halloween, sans pour autant que les usagers puissent bénéficier du conseil génétique « à la française ». Je regrette profondément cette prudence. Le CCNE, pour sa part, est plutôt favorable à une ouverture prudente, encadrée, médicalisée, à la française.
S'agissant des greffes d'organes, je ne répondrai pas à votre question sur le rôle du Conseil d'État. Nous regrettons seulement que le sujet n'ait pas été mis davantage en avant, alors même que l'on observe une baisse du don d'organes en France. Pour faire écho aux demandes des associations, nous souhaiterions que la loi instaure un véritable statut du donneur vivant, ce qui permettrait de faciliter les choses et d'accroître le nombre de donneurs.
Mme Muriel Jourda , rapporteur . - J'ai deux questions à poser à Mme Lefeuvre. Tout d'abord, en quoi l'ouverture de l'assistance médicale à la procréation aux couples de femmes ou aux femmes seules est-elle un sujet de bioéthique ? Ensuite, l'ouverture de l'AMP doit-elle systématiquement engendrer la suppression de toute référence à une pathologie, notamment aux maladies transmissibles et à l'infertilité, dans le texte ?
Mme Karine Lefeuvre . - Pour répondre à votre première question, l'ouverture de l'AMP est un sujet de bioéthique, tout simplement parce que ces femmes ou ces couples de femmes demandent à avoir accès au même processus que les couples hétérosexuels. Très concrètement, ce texte défend une vision beaucoup plus large, qui est celle d'une AMP sociale.
Faire référence aux textes antérieurs ajouterait de la confusion. Le texte dans sa version actuelle permet de respecter le principe de non-discrimination et d'égal accès de tous les citoyens à cette procédure, qu'il s'agisse d'un couple homme-femme, d'un couple de femmes ou d'une femme seule.
Proposer une telle ouverture n'empêche pas d'encadrer la démarche, ce que fait d'ailleurs très précisément le texte dans sa version actuelle : il y est ainsi fait référence à la recherche du consentement, au fait notamment que la mise en oeuvre d'une AMP nécessitant un tiers donneur devra donner lieu à un consentement préalable devant notaire.
Lors de notre l'audition à l'Assemblée nationale, j'ai déjà indiqué que le texte faisait à chaque fois référence soit aux couples de femmes soit à « toute femme non mariée », ce qui renvoie à la présomption de paternité dans le cadre du mariage. Il me semble que l'on devrait assumer jusqu'au bout cette distinction en ouvrant la PMA soit à un couple, soit à une femme seule non mariée. Ce serait beaucoup plus clair ainsi.
Mme Catherine Deroche . - Vous avez évoqué, s'agissant de l'innovation, son coût et sa soutenabilité pour l'Assurance maladie. Nous avons travaillé sur le sujet, notamment sur l'accès à l'innovation pour l'ensemble des territoires. Je ne crois pas qu'il faille opposer l'innovation aux autres dépenses sanitaires, à l'instar du budget consacré à l'hôpital ou du remboursement des traitements plus classiques par exemple. Certes, des choix s'imposent, mais il convient de favoriser le développement de l'innovation et son accessibilité, notamment en termes de coût.
M. Jean-François Delfraissy . - L'innovation constitue un sujet de débat depuis deux ans environ, mais elle n'apparaît pas dans le projet de loi. J'ai travaillé à de nombreuses reprises avec l'industrie pharmaceutique sur le sida et diverses maladies infectieuses : il ne s'agit nullement de la condamner, mais d'interroger la viabilité de son modèle au regard des calculs réalisés par plusieurs experts. Il faut désormais compter plus de 500 000 euros pour certains traitements anticancéreux comme les checkpoint blockers - peut-être davantage pour les bithérapies - auxquels un tiers des patients répondent favorablement. Le coût du médicament devient un sujet majeur, tandis que les dividendes versés aux actionnaires des grands laboratoires pharmaceutiques apparaissent, rapportés au chiffre d'affaires, supérieurs à ceux qui sont versés par l'industrie du luxe. Le secteur est devenu financier - il ne crée plus, mais rachète des start-up - et il n'est pas illégitime de questionner son modèle s'agissant de l'accès à l'innovation, car l'argent que nous y consacrons ne bénéficie pas à d'autres politiques. À mon sens, le temps humain d'une aide-soignante dans un service des urgences ou dans un Ehpad n'est pas moins précieux pour nos concitoyens les plus fragiles.
M. Michel Amiel . - N'aurait-il pas été opportun de séparer la PMA du reste du projet de loi afin que le sujet ne confisque pas le débat ? Je ne lis pas de proposition en ce sens dans votre avis n o 129. Vous n'avez, par ailleurs, pas évoqué le sujet des neurosciences et ses corollaires : la neuro-éthique et la neuro-loi.
M. Jean-François Delfraissy . - Fallait-il externaliser le débat sur la PMA ? Je vous rappelle que le CCNE n'est responsable que de l'organisation des états généraux de la bioéthique, pas de la définition du périmètre du projet de loi. Les états généraux ont abordé le sujet de la PMA, comme celui de la fin de vie, qui n'est pas traité par le texte, mais représente un débat de société majeur.
Le CCNE n'a effectivement pas pris position sur l'intégration de la PMA au projet de loi, mais son avis n o 129 traite des nouvelles techniques de procréation. Il ne semble pas illogique que le Gouvernement ait choisi d'intégrer la PMA au texte ; l'inverse se serait d'ailleurs avéré délicat. En raison des évolutions technologiques - utérus et gamètes artificiels, différenciation des cellules souches par exemple -, il s'agit plus que des faits sociétaux ; nous nous trouvons à l'aube de questions bouleversantes sur la procréation.
Je vous remercie de votre question sur les neurosciences. Je ne suis pas neuroscientifique moi-même, mais le CCNE compte trois experts de haut niveau. Le plafond de verre de la connaissance est sur le point d'être brisé, notamment dans les domaines de la psychiatrie et de la génomique, grâce au big data. Le CCNE a peiné à établir des propositions constructives dans le cadre du présent texte - l'Agence de la biomédecine n'y a elle-même pas consacré plus de cinq lignes -, mais le sujet va devenir central dans les prochaines années.
Mme Élisabeth Doineau . - Le CCNE publie des rapports de synthèse à la portée de tous, y compris sur des sujets scientifiques complexes, et je salue son travail de vulgarisation.
Lors des débats sur la loi du 24 juillet 2019 relative à l'organisation et à la transformation du système de santé, nous avons évoqué le sujet des données de santé dont d'aucuns s'inquiètent de l'usage. Comment ces données, qui peuvent conduire à des avancées réelles, seront-elles utilisées ? Quels remparts seront érigés contre d'éventuels abus, notamment dans le domaine de la santé mentale ?
S'agissant des liens entre la santé, tout particulièrement la fertilité, et l'environnement, nous avons entendu, dans le cadre de la préparation de la loi précitée, le professeur Jean-Marc Ayoubi, chef de service de gynécologie-obstétrique à l'hôpital Foch, professeur à la faculté de médecine de l'Université de Versailles-Saint-Quentin-en-Yvelines et membre de l'Institut santé. Nous sommes aux prémices de l'utilisation des données de santé, mais il faudrait aller plus loin compte tenu des implications avec la PMA.
M. Jean-François Delfraissy . - Le sujet du big data en santé apparaît majeur. Après les états généraux de la bioéthique, le CCNE a lancé une réflexion sur le sujet avec des personnalités extérieures, en particulier de la haute administration, et a publié un avis. La France affiche une position originale, étant l'un des rares pays à disposer, avec la CNAM, d'une prodigieuse base de données. Le Danemark ou la Hollande, plus avancés en matière de big data, ne possèdent pas, en l'absence de masse critique, une telle base.
L'utilisation des données de santé inquiète, mais elle peut s'avérer utile. Ainsi des chercheurs de l'Institut national de la santé et de la recherche médicale (Inserm) ont-ils montré que l'étude des données de la CNAM aurait pu permettre de donner l'alerte sur le Mediator cinq ans avant. Le big data comprend cependant des limites, notamment parce que 18 % de la population française ne dispose pas d'un accès aisé à Internet. À titre d'illustration, la prise de rendez-vous en ligne dans les hôpitaux parisiens, pour pratique qu'elle semble, prive certaines personnes âgées, isolées ou handicapées d'une réservation facile d'un créneau de consultation. Le CCNE est engagé sur le sujet et a plaidé pour que le texte le traite.
Je partage votre perception des liens entre fertilité et environnement. Vous entendrez, lors des auditions, des pontes de la gynécologie aborder le sujet. L'environnement porte effectivement en partie la responsabilité de la réduction de la fécondité et l'enjeu apparaît crucial. Les trois groupes de travail établis par le CCNE concernent d'ailleurs respectivement les liens entre l'environnement et la santé, les nouvelles techniques de procréation et les neurosciences. Nous souhaitons également intégrer des experts de l'environnement à nos travaux, mais n'avons guère rencontré de succès dans notre entreprise lors des états généraux de la bioéthique : les secteurs de la santé et de l'environnement se connaissent mal et travaillent encore peu ensemble.
Mme Michelle Meunier . - Je vous suis reconnaissante d'avoir abordé le projet de loi sans focaliser votre propos sur la seule PMA.
Nos débats, comme l'évolution accélérée de la société, plaident pour une révision plus fréquente, quinquennale par exemple, de la législation relative à la bioéthique, voire pour un rendez-vous annuel entre le CCNE et le Parlement, au-delà des seules auditions proposées par la commission des affaires sociales.
Comme ma collègue Élisabeth Doineau, je m'interroge sur les règles régissant l'utilisation des données de santé.
Mme Karine Lefeuvre . - Le projet de loi rend obligatoires le consentement et l'information des assurés avant toute utilisation de leurs données de santé. Il me semble toutefois que l'article 11 mériterait de préciser davantage, compte tenu du risque d'abus, la nature de l'information fournie.
M. Jean-François Delfraissy . - La bioéthique est-elle une matière figée ? Elle s'appuie sur un socle de valeurs, mais son contenu peut évoluer, comme le montre le nombre de nouveaux sujets abordés par le texte - big data, séquençage génomique, imagerie médicale notamment. L'évolution des techniques interroge logiquement le corpus de la bioéthique. Le CCNE est favorable à une révision quinquennale de la loi, comme le projet de loi le prévoit.
D'aucuns souhaitent élargir le périmètre du CCNE : nous n'y sommes pas opposés, mais il conviendrait alors de nous allouer des moyens supplémentaires. Nos collègues anglais, canadiens et allemands disposent, à titre d'exemple, d'un budget de deux tiers supérieur au nôtre.
M. Alain Milon , président . - Je vous remercie.
Ce point de l'ordre du jour a fait l'objet d'une captation vidéo qui est disponible en ligne sur le site du Sénat .
Mmes
Marie-Thérèse Besson, présidente,
et Joëlle
Mounier, membre de la Commission nationale
éthique-bioéthique
de la Grande Loge Féminine de France
M. Edouard Habrant, Grand
Maître,
et Mme Christiane Vienne, Grand Maître Adjoint
aux
affaires extérieures de la Grande Loge Mixte de France
Mme Élisabeth Doineau , présidente . - Nous poursuivons nos travaux sur le projet de loi relatif à la bioéthique avec l'audition de représentants des courants de pensée. Je vous prie de bien vouloir excuser l'absence de M. Alain Milon, qui préside notre commission. Cette audition fait l'objet d'une captation vidéo retransmise en direct sur le site Internet du Sénat et consultable à la demande.
Nous recevons, dans l'ordre des réponses reçues à notre invitation, pour la Grande Loge féminine de France, Mmes Marie-Thérèse Besson, présidente de la Commission nationale « éthique-bioéthique », et Joëlle Mounier, membre de cette même commission ; pour la Grande Loge mixte de France, M. Édouard Habrant, Grand Maître, et Mme Christiane Vienne, Grand Maître adjoint aux affaires extérieures ; pour la Grande Loge de France, M. Alain-Noël Dubart, ancien Grand Maître, et M. Jean-Jacques Zambrowski, ancien Grand Chancelier ; et pour le Grand Orient de France, M. Pascal Neveu, président de la commission de santé publique et de bioéthique, et M. Thierry Lagrange, conseiller de l'ordre.
Mme Marie-Thérèse Besson, présidente de la Commission nationale « éthique-bioéthique » de la Grande Loge féminine de France . - Il était important que notre obédience, la Grande Loge féminine de France, strictement féminine donc directement concernée, puisse s'exprimer sur l'ensemble des sujets qui concernent la bioéthique. Notre qualité de franc-maçonne et le travail que nous effectuons dans nos loges nous conduisent à faire preuve de discernement, ce qui est indissociable, pour nous, des notions de justice et d'équité. Les quelque 14 000 femmes de la Grande Loge féminine de France sont représentées dans trois commissions : la commission d'éthique, la commission des droits des femmes et la commission de la laïcité. Elles ne pratiquent pas la pensée unique. Pourtant, nous nous retrouvons toutes sur les valeurs issues du siècle des Lumières, à savoir la liberté, la fraternité, l'égalité, la dignité absolue de tous les êtres humains, et l'usage de la raison, sur laquelle s'est fondée l'autonomie de la conscience.
Nous sommes satisfaites des progrès de cette loi dans son ensemble, malgré quelques réserves et quelques interrogations.
Nous sommes bien entendu favorables à l'ouverture de la procréation médicalement assistée (PMA) à toutes les femmes. Nous souhaitons toutefois attirer votre attention sur le fait qu'il faudra être extrêmement vigilant et apporter une aide soutenue et réelle aux femmes seules, dont la précarité psychique et financière a été très bien documentée, en particulier chez les veuves et les femmes divorcées.
La conservation des gamètes est un réel progrès pour la liberté et l'égalité entre les hommes et les femmes. Jusque-là, on ne pouvait conserver que le sperme, dans le cadre de la convenance personnelle avec un projet parental, la congélation ovocytaire n'étant pas possible sauf pour raisons médicales. Or, pour les femmes, la fertilité naturelle baisse dès 35 ans. Et l'âge de la procréation, eu égard à l'engagement professionnel des femmes, a reculé. Il apparaît toutefois que seules les structures publiques pourront procéder, après autorisation, au prélèvement, au recueil et à la conservation des gamètes. Pourquoi ? Cela risque d'allonger encore les délais, qui sont déjà souvent de l'ordre d'une année, voire davantage. Et si les couples vont en Espagne, c'est à cause des délais. N'allons-nous pas organiser la pénurie, en rendant encore plus difficile l'accès au sperme ? Peut-on faire une loi qui serait, dans certains domaines, peut-être inapplicable ? Où est la solidarité, la justice pour tous, dans la prise en charge ? Il suffirait d'encadrer les pratiques, en limitant par exemple le nombre de tentatives remboursées par la sécurité sociale. Il faudrait élargir le diagnostic préimplantatoire à toutes les femmes de plus de 38 ans, en raison des risques causés par les anomalies génétiques liées à l'âge, avec des embryons de moins bonne qualité.
Concernant la filiation pour les couples de femmes, est-il vraiment nécessaire d'indiquer la reconnaissance conjointe dans l'acte de naissance ? Ne risque-t-on pas de créer une stigmatisation de certains enfants ? Interdire à l'enfant, en ce cas précis, de pouvoir établir un jour un lien avec son géniteur, n'est-ce pas le spolier du légitime désir de vouloir se replacer dans une filière que l'on pourrait qualifier de naturelle ? Naître, c'est aussi être mis en face d'une liberté d'être et d'advenir, qui pourrait disparaître pour certains. La loi va modifier complètement la définition de la famille. Il nous semble donc important que le législateur soit attentif aux conséquences des lois sur la filiation et à l'égalité de traitement des femmes et des enfants.
Concernant le don d'organes, à propos du don croisé à quatre paires, nous ne pouvons qu'apprécier cette fraternité au-delà de la famille, véritable valeur maçonnique de solidarité, qui augmentera bien évidemment les chances de réussite des greffes. Mais nous nous interrogeons sur la notion de liberté à propos du don intrafamilial et de l'inévitable pression psychologique qui risque d'exister.
Mme Joëlle Mounier, membre de la Commission nationale « éthique-bioéthique » de la Grande Loge féminine de France . - En ce qui concerne les manipulations génétiques, il y a un enjeu éthique dans la modification des cellules germinales et de ce qui toucherait au patrimoine de la descendance. On ne connaît pas les conséquences exactes, mais il pourrait y avoir des mutations inattendues. Jusqu'où peut-on aller dans la modification du génome humain ? Sommes-nous au seuil de l'invraisemblable et du moralement non souhaitable ? Il nous paraît essentiel de ne pas autoriser ces modifications et, par conséquent, d'interdire les manipulations sur les cellules germinales humaines, car la tentation sera de dériver vers l'eugénisme.
À la suite de tests prescrits lors d'un conseil génétique, des maladies non recherchées, dites incidentes, peuvent être découvertes. La loi permet l'information de la personne concernée et de sa famille. Faut-il révéler ces anomalies génétiques découvertes fortuitement, ou bien les cacher ? Ce serait vraiment une avancée que de donner cette liberté de choix et de connaissance médicale aux membres de la famille.
En ce qui concerne les tests génétiques dits récréatifs, les résultats sont vraiment difficiles à interpréter pour des personnes qui n'ont aucune connaissance scientifique et médicale, ce qui peut apporter confusion et inquiétudes. Ces tests donnent l'illusion de comprendre son profil génétique. La loi les interdit, mais comment la faire respecter ? Le débat porte aussi sur la confidentialité des informations récoltées : en donnant son ADN à une société commerciale, on communique aussi des informations génétiques qui concernent celui de ses parents et de sa famille, qui n'ont pas forcément donné leur accord pour communiquer ces renseignements. La réflexion doit se poursuivre sur l'impact de toutes ces technologies, dont l'utilisation de l'intelligence artificielle, qui va bouleverser nos vies.
La recherche sur les cellules souches embryonnaires pourrait être soumise uniquement à une déclaration obligatoire auprès de l'Agence de biomédecine. Cela faciliterait certainement beaucoup les conditions de manipulations et d'échanges dans les collaborations internationales. Il faudrait poursuivre les recherches sur l'embryon, mais uniquement dans le cas d'embryons venant d'un abandon de projet parental, avec le consentement éclairé du couple - comme c'est déjà la procédure actuelle -, mais non pour des embryons qui seraient conçus à cet effet, ce qui serait de la manipulation génétique sur l'embryon. Il nous paraît totalement justifié de poursuivre la recherche sur l'embryon, en la conditionnant à une autorisation encadrée - très encadrée - de l'Agence de la biomédecine. Le régime juridique serait donc distinct pour les deux types de recherche, les enjeux éthiques étant différents. Il faudrait sans doute inscrire dans la loi les deux prérequis à la recherche sur l'embryon que sont la finalité médicale et l'absence d'alternative.
Mme Marie-Thérèse Besson . - Cette première présentation est incomplète, mais nous répondrons bien évidemment au questionnaire que vous nous avez adressé dans les délais impartis.
Il nous semblerait plus judicieux de mettre en place une commission permanente, qui pourrait assurer le suivi de cette loi dans son application, plutôt que de faire une évaluation à quatre ans - sans l'exclure pour autant. Ces projets de loi ne devront pas enfermer nos libertés individuelles, au risque de déshumaniser nos existences.
M. Édouard Habrant, Grand Maître à la Grande Loge mixte de France . - Nous nous exprimons au nom de la Grande Loge mixte de France et de sa commission de bioéthique, mais pas au nom de chacun et chacune des 6 000 membres de la Grande Loge mixte de France, qui conservent naturellement leurs opinions propres, avec des nuances et des subtilités.
La commission de bioéthique de la Grande Loge mixte de France estime que le rôle d'un débat bioéthique est d'informer, d'expliquer et de clarifier les enjeux, et non pas d'imposer un mode de pensée. Nous avons été invités au titre des courants de pensée, ce qui sonne comme un oxymore ! La pensée, c'est justement de ne pas suivre le courant, et parfois d'aller contre le courant.
La bioéthique est l'affaire de toutes et de tous. Ce n'est pas l'ordre public, ce ne sont pas les experts officiels ou les traditions religieuses qui doivent définir le contenu de la bioéthique. Il n'y a pas de nature humaine gravée dans le marbre : nous devons faire face de façon pragmatique aux phénomènes nouveaux soulevés par la science et les nouvelles pratiques sociales.
Notre valeur ajoutée n'est pas celle d'experts ou de médecins. C'est celle de maçons. Ce sont les valeurs et principes de la Grande Loge mixte de France qui peuvent donner une orientation à cette réflexion.
Le premier principe est la recherche du bonheur individuel et collectif, en veillant à ne pas faire souffrir les tiers. L'épanouissement individuel et l'émancipation doivent être la finalité de toute organisation sociale dans une démocratie. Autres principes : la faculté de se déterminer librement - l'autonomie ; l'abolition des rapports de domination - et en premier lieu dans les rapports entre hommes et femmes - ; le refus de toute assignation à un genre, à une classe sociale ou à une communauté ; la primauté de la raison, la défiance à l'égard de tout ordre naturel, où l'individu ne s'appartiendrait pas, mais appartiendrait à une condition qui le dépasse ; la foi dans le progrès, dans le projet des Lumières, dans le développement du savoir ; la tradition humaniste, qui conçoit l'idée que la condition humaine n'est pas frappée du sceau de lois immuables et extérieures à l'humain ; une approche laïque : ce ne sont pas les églises, ni les convictions religieuses, qui doivent structurer la cité. Enfin, la fraternité et la solidarité, car notre recherche est de renforcer sans cesse notre commune humanité.
Quelles sont les conséquences de ces principes sur une loi de bioéthique ? D'abord, ne pas privilégier des principes immuables, qui proviennent le plus souvent de religions. Ne pas obéir toujours à des principes transcendants, tels que celui d'une nature humaine qui serait inscrite dans les cieux, où l'idée que la maternité impliquerait un lien profond et interne avec le foetus. Puis, le refus du catastrophisme, des fantasmes, des discours apocalyptiques : nous ne croyons pas en matière de bioéthique à la pente fatale, où la libre disposition de soi mènerait inexorablement à la marchandisation du corps et à l'esclavage. Plutôt qu'une éthique des convictions, nous croyons en une éthique de la responsabilité. Les personnes sont responsables de leurs choix, et la loi ne doit intervenir que quand des comportements causent un préjudice à un tiers.
Le principe de responsabilité et le principe d'émancipation excluent de faire de la bioéthique une réflexion qui serait seulement sous le contrôle de l'État, s'agissant de questions qui concernent la relation à notre corps, à notre personnalité et à notre reproduction. Or, le droit français reste marqué par un ordre public du corps et de la vie - et je ne parle pas de la confiscation de la mort. Le corps est encore sous l'emprise de Léviathan, des pouvoirs publics, de l'État. Nous devons sortir de ce paternalisme d'État, tout en veillant à défendre les plus fragiles. Il faut donc passer d'un ordre public de direction à un ordre public de protection.
Nous relevons certaines avancées, et en particulier la reconnaissance que la filiation est désormais la conséquence d'une intention, d'un projet. Nous sommes déçus, par contre, par le rejet de l'assistance médicale à la procréation (AMP) post mortem, car cela implique que l'on force la femme dont le conjoint est décédé à donner ou détruire ses embryons, tout en ayant la faculté de procéder seule à une insémination avec les gamètes d'un donneur. Il s'agit, selon nous, d'une injonction arbitraire, qui est d'autant plus injuste au niveau de l'enfant qu'il pourrait sans doute, dans ce cas, avoir un accès plus facile à ses origines, et à l'histoire de sa famille. Il en va de même du refus de reconnaître la filiation d'enfants conçus par grossesse pour autrui (GPA) dans un pays étranger où la pratique est autorisée. L'arrêt de la Cour de cassation du 4 octobre 2019 valide pourtant l'idée de transcription automatique, même si c'est dans un cas d'espèce, où elle a reconnu que l'adoption n'était pas appropriée.
Benjamin Constant disait : « Que l'autorité se borne à être juste, nous nous chargerons d'être heureux ! »
Mme Christiane Vienne, Grand Maître adjoint aux affaires extérieures de la Grande Loge mixte de France . - À la lumière des travaux préparatoires, et notamment du rapport Breton-Touraine, nous sommes déçus que ce texte soit essentiellement technique et manque d'une vision globale de la bioéthique, qui est, par nature, une matière transversale. Ce texte manque un peu d'âme, même pour des laïcs ! On ne voit pas quelle vision de la société est portée, puisque les mesures sont très techniques et n'ont pas nécessairement de lien entre elles. J'aimerais toutefois souligner quelques évolutions de la société que l'on retrouve en partie dans le texte.
Toutes les études le montrent, et la pratique le confirme, le désir de nos contemporains est d'avoir des enfants issus de leur capital génétique. Deuxième évolution sociétale, plutôt sympathique : l'investissement des hommes dans la paternité. Ils ont le désir d'être père, même lorsqu'ils sont homosexuels, et celui d'élever des enfants, d'avoir une garde alternée, bref de participer à l'éducation. Troisième élément : les carrières féminines, les études font que beaucoup de femmes repoussent leur désir de maternité à un âge où leur fécondité est en baisse.
Le texte comporte un certain nombre d'évolutions que nous considérons comme positives, notamment en matière d'AMP pour toutes les femmes, qu'elles soient homosexuelles ou non : après tout, en observant l'appareil génital d'une femme, je ne vois pas comment on peut mesurer si elle est hétérosexuelle ou homosexuelle. Nous sommes satisfaits de la reconnaissance du droit de l'enfant à connaître ses origines. Il s'agit d'un débat de fond qui, au sein de l'Union européenne, a trouvé une solution depuis quelques années déjà. Nous approuvons aussi le don d'organes croisé et la lisibilité en matière de recherche sur l'embryon et les cellules embryonnaires. Nous regrettons cependant qu'il n'y ait pas un mot dans le texte sur l'accompagnement des adultes et des enfants transgenres. Certains adolescents préfèrent acheter des hormones sur Internet plutôt que d'avoir un véritable accompagnement médical. Pour nous, c'est une lacune.
Sur la reconnaissance des enfants nés de GPA réalisées à l'étranger dans les pays où cela est autorisé, il y a un rendez-vous manqué. Quant à la question de la fin de vie, c'est un sujet qui nous tient beaucoup à coeur, et qui est resté hors du périmètre.
Nous ne faisons pas de différence dans le désir d'enfant d'une femme selon son orientation sexuelle et la nature du couple qu'elle forme - comme l'Union européenne depuis de nombreuses années. Toutes les études montrent qu'il n'y a pas d'impact sur l'évolution et le développement de l'enfant.
La question de l'autoconservation des gamètes est un sujet important pour les femmes. C'est un immense progrès pour les femmes occidentales que de pouvoir choisir le moment de la maternité. L'autoconservation des gamètes permettrait à de nombreuses femmes de devenir mères quand elles le souhaiteront réellement.
L'information du public et la formation des professionnels sont-elles suffisantes ? Elles ne le seront jamais, puisque les évolutions scientifiques se développent à un rythme très rapide. Il faut constamment y revenir et avoir une attitude ouverte. Le texte parle très peu du médecin généraliste, alors qu'il a un rôle fondamental à jouer.
La thérapie génique du patrimoine génétique de cellules somatiques ne nous pose pas de problèmes, puisque ces cellules ne transmettent jamais à leur descendance les mutations qu'elles ont pu subir. Il ne faut pas voir de l'eugénisme partout ! C'est le rôle de l'État d'empêcher de telles dérives, et il peut le faire. Il ne faut pas brandir des monstres du Loch Ness bioéthiques... Les nanotechnologies aussi suscitent de nombreux fantasmes. Or, grâce à une puce implantée dans le cerveau, une personne hémiplégique peut remarcher. On ne peut que s'en réjouir ! Il faut donc poursuivre le travail.
M. Jean-Jacques Zambrowski, ancien Grand Chancelier de la Grande Loge de France . - Merci d'avoir prévu l'audition de représentants des courants de pensée, et particulièrement des obédiences maçonniques, qui regroupent environ 200 000 hommes et femmes à travers la France, de métropole et d'outre-mer.
L'histoire de la Grande Loge de France remonte aux premières décennies du XVIII e siècle. Elle compte à ce jour près de 34 000 membres. La devise de la Grande Loge de France a été « Liberté, Égalité, Fraternité », cinquante ans avant que la République ne l'adopte ! Nous y ajoutons aujourd'hui la spiritualité, avec, pour corollaire, la liberté absolue de penser, la liberté absolue de conscience, et l'humanisme, au sens où l'homme et son épanouissement sont au coeur de notre projet.
Nous n'avons pas demandé l'avis de chacun des 34 000 frères de la Grande Loge de France sur les différents éléments techniques de cette loi. Notre processus ne consiste pas à nous poser des questions sur l'actualité ou les questions de société, mais nous défendons de manière très ferme des valeurs et des principes liés à l'éthique, à la morale et à des aspirations spirituelles ou philosophiques, sans être nécessairement religieuses.
Il ne peut donc pas y avoir un point de vue de la Grande Loge de France sur les questions extrêmement pertinentes que vous vous posez légitimement, et auxquelles vous devez apporter des réponses. Notre propos sera de vous rappeler, s'il en était besoin, à votre mission, qui est de respecter des valeurs et des principes fondamentaux et de ne pas les oublier au motif que la réflexion est très largement technique, pour ne pas dire scientifique. Si elle est aussi médicale, elle est surtout profondément humaine, profondément spirituelle, au sens le plus large de ce terme.
Nous avons toutefois des commissions qui travaillent techniquement sur ces sujets.
M. Alain-Noël Dubart, ancien Grand Maître de la Grande Loge de France . - Merci de nous donner la parole.
Premier regret : ce projet de loi est essentiellement technique, alors qu'il va apporter des modifications substantielles au fonctionnement même de la société. Il nous aurait semblé nécessaire de privilégier une réflexion fondée sur ce qu'est la société, ce qu'est la finalité de la société, la vie bonne en société, et sur les problèmes éthiques. Répondre à des questions de société par des modifications purement techniques, sans préalable de considérations éthiques, ne me semble pas la bonne solution.
Ce projet existe, et a été adopté en première lecture par l'Assemblée nationale. Il va dans la bonne direction concernant la possibilité pour les couples homosexuels féminins d'utiliser la PMA dans toute son amplitude. C'est une réponse logique au progrès de la société. Seule réserve : il est absolument nécessaire pour l'enfant d'avoir un accès complet à ses origines, y compris génétiques. C'est un droit fondamental. Il y a une petite lacune dans le projet de loi sur la levée de l'anonymat pour les dons de sperme effectués avant ce texte.
En ce qui concerne la PMA pour les femmes célibataires, nous avons une certaine réserve, car il paraît différent sur le plan éthique de faire naître un orphelin, ou de devenir orphelin par accident. Le principe d'égalité nous renvoie au couple homosexuel féminin. Il me semble que le principe d'égalité n'est pas le principe fondamental, cela dit. Il y a aussi le principe de l'organisation de la société, qui peut aller parfois contre le principe d'égalité. Il faut y réfléchir.
Il n'y a pas de droit à l'enfant, il y a des droits de l'enfant. L'enfant à naître possède les mêmes droits que les personnes adultes, tout simplement parce qu'il est un sujet de droit. Nous avons hérité cela de la philosophie des Lumières : l'autonomie de la personne humaine s'entend pour l'autonomie de l'enfant. J'ai même le sentiment que les droits de l'enfant sont supérieurs au droit des parents, car ils se projettent plus loin dans l'avenir.
La conservation des gamètes est un progrès technique et un progrès pour les femmes qui veulent choisir le moment de leur maternité. La procréation post-mortem peut raisonnablement s'envisager, mais avec des conditions médicales et techniques bien précises. Ce n'est pas un droit absolu.
La commission de l'Assemblée nationale nous avait dit que la GPA ne serait pas à l'ordre du jour. Pourtant, un rapporteur a introduit un amendement, qui a été voté par l'Assemblée nationale et retiré par la suite. Pour nous, la GPA pose un énorme problème : celui de l'indisponibilité du corps humain et celui de sa non-marchandisation. Sur ce sujet, il faut être clair : nous ne sommes pas favorables à la GPA, dans quelque circonstance que ce soit, sauf circonstances médicales très particulières. Nous sommes issus du siècle des Lumières. À ce titre, nous sommes fondamentalement attachés à la philosophie de John Locke et d'Emmanuel Kant. Or, l'impératif catégorique d'Emmanuel Kant s'énonce de la manière suivante : tu considéreras la maxime de son action de telle sorte que, pour autrui comme pour toi-même, tu ne prendras jamais de personne humaine comme un moyen, mais toujours comme une fin. Utiliser le ventre d'une femme comme moyen est inacceptable pour un franc-maçon.
Tout est techniquement possible, et la recherche doit être absolument libre. Mais ce n'est pas parce que c'est techniquement possible que c'est éthiquement souhaitable. L'éthique est là pour mettre des barrières à l'utilisation du progrès technique.
M. Jean-Jacques Zambrowski . - Même si le Comité consultatif national d'éthique (CCNE) avait fait le constat que le don d'organes était à la fois fiable et nécessaire, il s'avère que le don croisé d'organes, légalisé par la loi de bioéthique de 2011, ne va peut-être pas assez loin, parce que les donneurs sont trop peu nombreux, et que les dispositions à prendre sont insuffisamment connues. Le principe de non-opposition au prélèvement post mortem, qui vient d'être évoqué s'agissant des gamètes, réaffirmé par la loi de janvier 2016 de modernisation du système de santé, ne va pas non plus assez loin. Il faut adopter une véritable politique si l'on veut favoriser le don d'organes, qui est une nécessité pour traiter un certain nombre de situations pathologiques, acquises ou innées.
L'article qui permet un examen génétique d'une personne hors d'état d'y consentir devrait à cet égard être adopté, dans l'intérêt des membres de sa famille qui sont potentiellement concernés. Les réponses aux questions de la gratuité et de l'anonymat du don sont encore imprécises. Il faudrait les clarifier, expliquer et élargir le cadre actuel, tout en préservant la notion d'équité, qui vient d'être évoquée au travers de cet héritage philosophique dont nous nous inspirons.
Il faut bien voir la différence de nature qui existe entre le don d'organes ou de tissus, d'une part, et la création d'un embryon humain qui possède les mêmes droits naturels et imprescriptibles que tout individu d'autre part. Favoriser les progrès scientifiques ou technologiques dans les domaines de l'intelligence artificielle ou des neurosciences - sur lequel certains d'entre nous passent l'essentiel de leur temps - est évidemment primordial : un franc-maçon, par définition, est un cherchant, quand il n'est pas un chercheur. Nous sommes donc favorables aux dispositions soutenant la recherche à condition qu'elle soit libre, responsable, et non gouvernée par les seuls intérêts matériels du petit nombre au détriment de la collectivité. C'est le sens même du progrès. Idem pour tout ce qui concerne l'imagerie cérébrale. Les textes doivent simplement être attentifs à ce que les valeurs éthiques et le respect de l'individu qui servira de sujet d'expérimentation soient parfaitement respectés. La création d'embryons chimériques doit rester formellement interdite. L'article 14 du projet de loi qui confirme le distinguo entre embryons et cellules souches embryonnaires doit être bien entendu regardé avec précision puisque le régime juridique de ces deux entités est parfaitement différent.
Il faut donner un cadre éthique et juridique - c'est au fond le leitmotiv de notre intervention - à tout ce qui concerne la génétique ou le microbiote, sur lesquels certains d'entre nous travaillent à longueur d'année, 75 heures par semaine. Ce cadre éthique et juridique est au moins aussi indispensable que la promotion et le développement de la technique elle-même : si celle-ci se développe en dehors de tout cadre relevant de l'intérêt collectif, de l'intérêt moral et de la préservation de notre société qui, dans sa diversité, partage certains fondamentaux, nous allons à la catastrophe et nous laisserons les intérêts d'un tout petit nombre prendre le pas sur l'intérêt collectif dont vous êtes les garants.
M. Thierry Lagrange, Conseiller de l'Ordre du Grand Orient de France . - Permettez-moi de vous adresser d'abord les chaleureuses salutations du grand maître du Grand Orient de France, Jean-Philippe Hubsch, retenu ce jour.
La bioéthique suppose de se poser des questions fondamentales sur la vie, même sur la façon dont nous, êtres humains, tentons d'intervenir sur nos formes d'existence. Y a-t-il un seul instant où les questions de bioéthique ne se posent pas ? Les esprits religieux l'ont bien compris, et c'est pourquoi nous devons, nous, maçons, formuler des réponses sur ces questions. Car, en matière de bioéthique, il ne saurait y avoir de dogme, d'obscurantisme, de réponse unique ou figée. La réflexion bioéthique appelle des réponses, celles que nous élaborons en nous écoutant, en débattant, en émettant des avis. Il n'y a pas d'autres méthodes pour en comprendre les enjeux.
En 2018, nous avons assisté à une première dans l'histoire : en amont de la révision des lois relatives à la bioéthique, des états généraux de la bioéthique ont été organisés, pensés par leurs animateurs comme un temps de démocratie sanitaire et de santé démocratique. Le Comité consultatif national d'éthique s'est donc proposé de construire un avis sur l'ensemble des huit thèmes débattus : recherche sur l'embryon et cellules souches embryonnaires, examens génétiques et médecine génomique, transplantations d'organes, neurosciences, numérique et santé, santé et environnement, procréation et accompagnement de la fin de vie - même si ce dernier thème est sorti de nos réflexions, nous aurions des propositions à vous faire sur ce sujet.
L'éthique ne peut se penser hors sol. Elle ne peut pas non plus être déléguée à quelques experts, car elle concerne chacun d'entre nous. Nous, maçons, devons étudier en amont à travers le filtre de nos valeurs les questions d'avenir, construire notre réflexion éthique et établir une morale provisoire. C'est ce que notre commission nationale de santé publique et de bioéthique nous propose et je laisse la parole à son président.
M. Pascal Neveu, président de la Commission nationale de santé publique et de bioéthique du Grand Orient de France . - Merci de recevoir le Grand Orient de France, dont la Commission nationale de santé publique et de bioéthique travaille depuis presque trente ans sur les thèmes que nous allons aborder. En vue de la révision de la loi bioéthique, le CCNE a pris en compte, de manière inédite, l'avis des citoyens, et non pas seulement celui des experts. De nouveaux espaces de réflexion en matière de santé se sont ouverts, s'appuyant non pas sur des avancées scientifiques et techniques, mais sur des demandes émanant d'une partie de la société. Nous souscrivons aux avis du CCNE et souhaitons les soutenir.
Concernant la recherche sur l'embryon et les cellules souches embryonnaires, le CCNE est favorable à la recherche sur les embryons surnuméraires issus des fécondations in vitro qui n'ont pas été conduites jusqu'au bout par les parents. Il est en revanche opposé à la création d'embryons par la culture de lignées de cellules souches embryonnaires. L'idée est d'utiliser les matériaux biologiques existants, de recycler ceux qui ont été créés pour un projet parental, mais non - pour éviter tout eugénisme - d'autoriser une nouvelle forme de création de la vie en laboratoire. La difficulté de définir une législation dans ce domaine tient à l'évolution relativement rapide des biotechnologies à base de cellules souches. Il faut donc être vigilant sur la finalité médicale, l'absence d'alternative, la robustesse du protocole sont à questionner.
Dans le domaine des examens génétiques et de la médecine génomique, la finalité médicale rend les décisions plus faciles à trancher sur le plan éthique. Le diagnostic génétique préconceptionnel, c'est-à-dire le dépistage en amont d'anomalies qui peuvent être engendrées par des géniteurs porteurs sains, telles que des monosomies ou des déficits immunitaires par exemple, est ainsi légitime. C'est un acte médical de prévention qui devrait être pris en charge par l'assurance maladie ; le consentement éclairé devrait être recueilli à tous les stades. Diagnostic préimplantatoire ou diagnostic prénatal doivent pouvoir être étendus à la population générale et une étude doit être menée sur vingt-quatre mois afin d'en établir les bénéfices éventuels.
Concernant les dons et transplantations d'organes, le prélèvement d'organes chez des patients décédés souffre aujourd'hui de fortes disparités régionales. L'offre de greffons arrive trop tôt ou trop tard, ce qui compromet le principe d'égalité lorsqu'il s'agit de bénéficier d'une greffe. En accord avec le CCNE, nous sommes favorables au don d'organes, qu'il faut amplifier, d'abord en formant mieux les personnels médicaux, ensuite en réalisant une large campagne d'information auprès du public. L'inscription au registre national des refus est accessible partout sur Internet depuis quelques années. Le plus important est de faire prévaloir le consentement éclairé et le choix de la personne sur les arrangements médicaux ou les pressions. Il en va de même pour les greffes d'organes à partir de donneurs vivants. Un statut de donneur dans le respect du principe d'équité entre tous les patients inscrits en liste d'attente reste fondamental ; il serait souhaitable de raccourcir les délais de remboursement des frais avancés par le donneur vivant, afin qu'il ne soit pas amené à supporter les conséquences financières de ce geste généreux.
Concernant les neurosciences, ce domaine de la recherche scientifique s'appuie principalement sur l'énergie cérébrale pour approfondir la connaissance du fonctionnement du corps humain, ainsi que les possibilités du diagnostic d'un certain nombre de pathologies. L'étude de ces mécanismes cérébraux soulève néanmoins un certain nombre de questions éthiques relatives à la frontière entre le normal et le pathologique. Une imagerie cérébrale anormale chez un criminel peut-elle expliquer ses actes criminels ? Quelles sont les limites du neuro-marketing, voire d'une neuro-politique ? Un employeur potentiel pourrait-il passer votre cerveau dans une machine afin de savoir s'il va vous embaucher ? Si la neuro-éthique est très développée dans les pays anglo-saxons depuis une quinzaine d'années, elle l'est encore assez peu en France. Les concepts de dignité humaine, d'autonomie, de non-malfaisance et d'équité restent fondamentaux. Les techniques de neuro-amélioration concernant des dispositifs médicaux et surtout non médicaux doivent être encadrées par la loi et une information doit être diffusée.
L'insuffisante utilisation du numérique dans le domaine de la santé, qu'il s'agisse de prise en charge des patients, de recherche ou de pilotage par les données, induit sur une large échelle des situations non éthiques au sein de notre système de santé. La résorption de ces problèmes est un enjeu prioritaire et qui ne peut passer que par la loi. La diffusion du numérique en santé semble inévitable, mais face au développement de ces technologies, le recours au droit opposable doit être circonscrit au maximum, comme le précise le CCNE : « Compte tenu des marges de gain de qualité et d'efficience permises par un recours élargi au numérique dans le nouveau système de santé, mettre en oeuvre des réglementations restrictives est contraire à l'éthique. » Le contact humain, pour nous, reste essentiel, car lui seul est en mesure de transmettre l'ensemble des informations concernant le patient dans le cadre de nos parcours de soins. Comme dans d'autres domaines, le consentement libre et éclairé du patient est indispensable et fondamental pour le recours aux techniques d'intelligence artificielle.
La procréation est un des points les plus clivants. Depuis la fin des années 1960, une forte pression sociale s'est exercée en faveur de la liberté de la procréation humaine. En libérant la sexualité d'une finalité procréatrice, la possibilité pour un couple de faire un enfant quand il le veut et s'il le veut est devenu un droit revendiqué. Il implique, lorsque la procréation spontanée se heurte à une difficulté, d'utiliser une technique d'assistance médicale à la procréation. En apportant une réponse médicale à un problème d'infertilité, l'AMP recouvre un ensemble de techniques conçu par le corps médical, puis organisé par le législateur pour répondre à des infertilités dues à des dysfonctionnements de l'organisme. Mais elle soulève des problèmes éthiques d'ordre général, qui sont depuis le début au centre des travaux du CCNE et même à l'origine de sa création, avec le premier bébé-éprouvette.
Les demandes sociétales d'accès à l'AMP, c'est-à-dire à d'autres fins que de pallier l'infertilité pathologique chez les couples hétérosexuels, augmentent, alors qu'elles étaient autrefois très marginales. Elles sont portées à la fois par les évolutions de la société, de la loi française et de certains pays étrangers, et de la technique. Le CCNE est favorable à l'ouverture de l'assistance médicale à la procréation pour les couples de femmes et les femmes seules. Il demeure favorable au maintien de l'interdiction de la gestation pour autrui. Il souhaite par ailleurs que soit rendue possible la levée de l'anonymat des futurs donneurs de sperme pour les enfants issus de ces dons. Le CCNE est favorable à l'ouverture de l'AMP post mortem, c'est-à-dire au transfert in utero d'un embryon cryogéné après le décès de l'homme sous réserve d'un accompagnement médical et psychologique de la conjointe. Le Grand Orient de France précisait dans un communiqué officiel du 29 septembre 2017 : « Le Grand Orient de France souhaite que cette évolution vers plus d'égalité et de justice sociale se réalise rapidement. Il suffit pour cela que le législateur prenne toutes ses responsabilités, conformément aux principes de notre République laïque. Il serait contre-productif de relancer à cette occasion d'éternels débats de société qui font la part belle aux lobbies politico-religieux, voire provoquent des déferlements d'homophobie, comme en 2013. Le droit de toutes les femmes à la PMA, leur égalité quels que soient leurs préférences sexuelles et leurs modes de vie, ne doivent pas plus être otages des campagnes politiciennes que des anathèmes religieux. [...] Le vrai débat, qui revient au Parlement, doit porter sur la faisabilité technique et financière - notamment les conditions de remboursement - de cette ouverture de la PMA. Le Grand Orient de France met en garde contre tout amalgame avec l'indispensable réflexion sur la GPA, sujet de nature différente, qui pose d'autres types de questions que l'on ne peut considérer tranchées à ce jour. »
M. Thierry Lagrange . - Le Grand Orient de France a été auditionné par diverses institutions lors de la révision des lois de bioéthique. Nous avons mis en avant nos principes de liberté, d'égalité, de fraternité, mais bien évidemment, de laïcité et de solidarité. Il nous importe - et il vous importe en tant que sages - de réfléchir en avance au travers des filtres de nos valeurs, aux questions d'avenir, de construire notre réflexion éthique, d'établir un cadre sociétal provisoire sans doute, sans jamais cesser de penser un avenir qui sera celui de nos enfants et petits-enfants - mais le penser comme un advenir sur certains sujets inéluctables et face auquel le cadre légal doit se positionner, car la loi se positionne trop souvent a posteriori, n'étant pas suffisamment en lien avec ce qui se pratique déjà en coulisses. La loi doit ainsi s'inscrire dans un champ des possibles et surtout la prospective d'un meilleur vivre-ensemble. Si la bioéthique doit être l'établissement d'une morale provisoire avec l'incertitude, le doute et l'imperfection, il est indispensable de rappeler que l'éthique emporte avec elle la solidarité et la responsabilité. La réflexion du Grand Orient de France a pour objet l'amélioration à la fois de l'humain et de la société, éclairée par sa devise qui se confond avec celle de notre République : liberté, égalité, fraternité. Il ne s'agira pas de répondre à la question « pour ou contre ? », mais d'examiner comment et pourquoi l'amélioration simultanée de l'humain et de la société peut et doit être pensée.
Au-delà du cadre législatif qui, n'en doutons pas, sera tôt ou tard transgressé, la question majeure est celle de la solidarité et de la responsabilité. L'exercice de la responsabilité multiple doit engager solidairement le politique et le scientifique ; un choix doit être fait, considéré comme provisoire et donc révisable ; un suivi rigoureux doit être mis en place avec la promotion d'études à fort niveau de preuves pour évaluer l'effet de ces choix ; être responsable consistera ensuite à partager avec les citoyens le résultat de ses choix ; enfin, il conviendra d'assumer face au débat citoyen les conséquences des choix proposés et assumer, si nécessaire, leur révision. Notre avenir et le bonheur d'une vie à penser et à construire est entre vos mains.
Mme Élisabeth Doineau , présidente . - Quelle responsabilité vous nous donnez là !
Mme Muriel Jourda , rapporteur . - Madame Besson, s'agissant de la filiation, vous avez indiqué deux choses : la première, que la reconnaissance conjointe dans l'acte de naissance pourrait être vécue comme une stigmatisation pour l'enfant, mais je n'ai pas compris la seconde. Pourriez-vous me la réexpliquer ?
Mme Marie-Thérèse Besson . - Nous avons compris que, dans le texte, un enfant qui serait mis au monde à partir d'un couple de femmes ne pourrait pas avoir accès à l'identité du donneur, contrairement à ce qui se passerait pour les autres enfants. Si c'était le cas, cela constituerait une inégalité de traitement entre les enfants ; il serait quand même problématique pour un enfant que de ne pas pouvoir inscrire sa vie dans quelque chose qui serait de l'ordre d'une lignée, d'avoir accès à ses origines. C'est important dans la constitution d'un être humain.
Mme Muriel Jourda , rapporteur . - Vous parliez donc en termes d'accès aux origines, et non en termes de reconnaissance d'une filiation paternelle. (Mme Marie-Thérèse Besson le confirme)
M. Bernard Jomier , rapporteur . - Merci à toutes et à tous de nous avoir bien exposé les valeurs qui sous-tendaient votre approche du projet de loi. Madame Besson, vous avez parlé justement de la notion de discernement. J'imagine que vous ne faisiez pas référence à la notion juridique, mais plutôt à l'intelligence ou à la sagesse. Pourriez-vous préciser ? Monsieur Dubart, vous avez dit : « On doit pouvoir parfois faire passer les questions d'organisation de la société avant les questions d'égalité. » Cette dernière est pourtant une valeur essentielle et fondamentale de notre République ! Madame Vienne, vous avez dit que le risque d'eugénisme était très faible dans ce projet de loi. Pouvez-vous préciser ?
Mme Marie-Thérèse Besson . - Nous avons des méthodes de travail communes dans nos loges, quelles que soient nos obédiences. Celles-ci nous amènent avant toute chose à travailler sur nous-mêmes avant de travailler sur l'altérité et de réfléchir à tous les grands axes auxquels nous sommes confrontés à travers notre vie quotidienne. Faire preuve de discernement, c'est à la fois savoir prendre suffisamment de recul par rapport à un texte et c'est aussi savoir prendre en compte tout ce qui peut être afférent, tout ce qui pourrait être mis en jeu, en termes d'évolution de la société et des rapports humains, car le monde de demain ne sera pas notre monde d'aujourd'hui, en termes de filiation, par exemple. C'est pour cela que je l'ai associé à la notion de justice et d'équité.
M. Alain-Noël Dubart . - Sur la notion d'égalité, je comprends que vous ayez été un peu surpris. L'égalité, comme tous les principes de la République, n'est pas seule ; elle est encadrée par la liberté, la fraternité - c'est-à-dire l'humanisme au sens large. On ne peut donc pas mettre en avant isolément un seul des trois principes qui collaborent ensemble à l'organisation de la société. Celle-ci, depuis la nuit des temps - en fait depuis Aristote et Platon -, c'est l'organisation de la vie bonne pour tous les hommes et les femmes qui composent la société, en fonction d'un certain nombre de principes éthiques qui dépassent la revendication d'un droit isolé.
M. Jean-Jacques Zambrowski . - Une dimension qui ne doit pas non plus nous échapper, c'est celle de l'anticipation. Légiférer, c'est déterminer un cadre pour le temps présent, en se fondant sur l'expérience du passé ou sur l'immédiate actualité ; mais évidemment, tout texte crée des conséquences qui se développeront dans l'avenir. Il faut donc anticiper, le temps qu'une nouvelle législation vienne éventuellement corriger des dérives. Il faut se demander quels dominos seront renversés, de quels effets le battement de l'aile de papillon agitée ici affectera l'autre bout de l'univers, quelles pourront être les conséquences sociales, sociétales profondes qu'une législation rapidement examinée sur des aspects purement techniques pourra engager. C'est le sens de ce qu'ont dit tout à l'heure M. Dubart, mais aussi les autres frères. Vous voyez qu'au-delà de nos différences nos préoccupations sont les mêmes.
Vous ne faites pas une législation et un cadre pour les trois prochaines années ; or dans trois ans, la technologie, qu'il s'agisse de manipulation génétique ou d'intelligence artificielle - ou plutôt de prétendue intelligence, car il s'agit de bien autre chose - aura évolué. Cette notion d'anticipation doit nous garder des dérives de l'intuition, de ce qu'on pourrait imaginer dans l'instant et par effet de mode, ou pour répondre à l'urgence. La sagesse, qui est peut-être la dimension particulière du travail législatif des sénateurs, doit prévaloir et tenir compte de cette dimension dans la durée au moins à l'échelle de ce que nous pouvons anticiper comme étant la durée d'un cadre législatif.
Mme Christiane Vienne . - Ce qui se trouve dans le texte n'ouvre pas la porte à l'eugénisme. Les cellules humaines somatiques qui seraient manipulées ne seraient pas transmissibles dans le capital génétique ; c'est donc de la recherche qui vise à soigner. Dans votre questionnaire, vous revenez sur la question des cellules souches pluripotentes induites et vous les mettez en parallèle avec les cellules souches embryonnaires ; rien n'empêche de poursuivre la recherche dans les deux domaines. Votre question donne l'impression qu'il faut choisir entre les deux, mais la possibilité de transformer une cellule adulte spécialisée en cellule immature, capable de donner n'importe quelle cellule, donc en une cellule souche pluripotente, c'est un peu un joker en la matière : cela ouvre vraiment des portes intéressantes sur, notamment, la modélisation d'un certain nombre de pathologies et en médecine régénérative. Tant que l'on aborde ces questions de travail sur la cellule, on n'est pas dans l'eugénisme.
J'ai entendu parler tout à l'heure de ce génome, mais le texte ne parle pas de manipulation du génome humain. Nous avons tous signé la convention européenne qui date des années 1960 sur l'interdiction du travail sur le génome humain. Il n'empêche que l'année dernière ou il y a quelques mois, un chercheur chinois, en utilisant ce qu'on a appelé d'une manière un peu simple des ciseaux génétiques, a mené une expérience qui a permis de manipuler le génome humain et d'enlever avec l'accord de ceux qui ont souhaité participer à l'expérience, la transmission du virus HIV. Or cela est transmissible et nous entrons là dans des domaines extrêmement dangereux. D'ailleurs, la communauté internationale a réagi, la condamnation a été unanime, même en Chine. Lorsque l'on manipule le génome, la manipulation est transmissible, héréditaire, et donc on ne parle pas tout à fait de la même chose.
La crainte d'eugénisme est plus légitime dans le domaine de la GPA : la situation d'une femme née sans utérus, victime d'un syndrome de Rokitansky, dont la mère, la soeur, la cousine porte l'enfant, dans une logique de générosité, est différente de celle d'un couple, qu'ils soit d'hommes ou de femmes ou hétérosexuel, car la GPA ne concerne pas que les couples homosexuels, qui choisirait un géniteur ou une mère porteuse parce qu'il veut avoir à tout prix un bel enfant blond aux yeux bleus. On peut toujours imaginer des dérives, mais rien dans le texte ne porte à croire qu'il y a un risque d'eugénisme.
Mme Joëlle Mounier . - Les cellules qui sont reprogrammées ne peuvent pas être travaillées comme des cellules souches d'origine, puisqu'elles gardent une mémoire épigénétique sur ce qu'elles ont vécu. Il faut donc être très prudent. Par ailleurs, elles peuvent avoir acquis des mutations qui provoqueraient la formation de cellules cancérigènes. Il ne s'agit pas d'eugénisme, mais quand on dit qu'on peut les mettre en parallèle avec des cellules souches embryonnaires classiques, ce n'est pas tout à fait exact ; il faut vraiment vérifier en fonction des critères de travail de ces manipulations. Désolé si c'est un peu technique...
Mme Corinne Imbert , rapporteure . - Mmes Besson et Mounier se sont exprimées sur les tests génétiques récréatifs...
Mme Joëlle Mounier . - ... dits récréatifs.
Mme Corinne Imbert , rapporteure . - En effet. Les représentants des autres obédiences maçonniques pourraient-ils rapidement nous donner leur position ? Puisque la semaine prochaine, le Sénat va discuter du projet de loi de financement de la sécurité sociale, une absence de prise en charge par l'assurance maladie de l'assistance médicale à la procréation - contrairement au texte de l'Assemblée - vous semblerait-elle contraire à vos principes d'égalité, de solidarité, tels que vous les avez tous exprimés ?
M. Alain-Noël Dubart . - Il en va des tests génétiques récréatifs comme du cannabis récréatif : des gens expédient un échantillon de telle partie de leur muqueuse buccale à l'étranger pour qu'on leur renvoie une analyse de leur ADN et une présomption d'origine géographique. Quelles garanties de qualité, quelles garanties sur l'effet que va produire la révélation du résultat sur l'individu, quelles garanties a-t-on ensuite sur le fait qu'on trouvera à l'individu des parentés géographiques avec telle ou telle région à l'insu de son plein gré - si vous me permettez l'expression ? Tout cela manque terriblement d'anticipation. Il y a certainement des entreprises qui pourraient proposer en France des tests auxquels la sécurité sociale n'aurait aucune raison de participer et qui pourraient avoir de l'intérêt.
Autant les tests génétiques chez des individus dans la famille desquels existent des pathologies susceptibles d'avoir une transmission héréditaire comme le cancer ou le diabète ou bien d'autres pathologies, ont une raison médicale, scientifique, de prévention, permettant de proposer à la personne concernée une information qui lui servira à gouverner sa liberté et ses choix ou lui déconseiller formellement telle ou telle direction, autant les tests récréatifs faits par des opérateurs non contrôlés relèvent du jeu d'argent : c'est un trafic comme un autre, sur la base d'une découverte scientifique. Le rôle du législateur est de protéger les individus contre les dérives, qu'elles soient attachées à un passé révolu ou à un futur sans contrôle. Il est de votre responsabilité d'établir des barrières qui éviteront que les plus fragiles, les moins capables de discernement puissent se prémunir contre les risques. Les barrières, c'est contraignant ; c'est vrai que distinguer le bon grain de l'ivraie est parfois un exercice difficile. Dans le cas des tests génétiques récréatifs, la pratique présente infiniment plus de risques et de dangers que de bénéfices.
Cela n'a rien à voir avec un test prescrit par un conseiller en génétique, car beaucoup plus de pathologies que ce que nous croyions encore il y a encore une dizaine d'années ont un substratum génétique inné ou acquis, transmissible ou non, qui fait peser un danger que non seulement l'individu et sa famille assumeront, mais qu'assumera la collectivité tout entière au travers de l'assurance maladie et des institutions d'aide aux personnes en situation de handicap. Bien sûr, il faut favoriser ces nouveautés.
Pour le reste, la collectivité doit être au moins aussi attentive à la prévention des risques liés à ces tests génétiques récréatifs qui n'ont pas de raison d'être qu'à l'addiction aux jeux de hasard.
Mme Christiane Vienne . - Sur cette question-là vous abordiez dans votre questionnaire la question d'Internet, par lequel passent ces tests récréatifs. Derrière cette demande, il y a un peu de curiosité, la demande n'est pas nécessairement extrêmement scientifique, mais je ne vois pas par quels mécanismes législatifs, on pourrait interdire l'accès à Internet dans ce domaine-là. Dès lors, pourquoi se fatiguer ?
À l'inverse, il me semble essentiel d'informer et de mettre en garde. Toute la question qui est posée à travers ce texte est aussi celle de l'information, de la communication, de l'éducation : la médecine évolue à une telle vitesse que le fait d'être capable de maîtriser, de comprendre les enjeux devient en soi un défi. Un jeune de dix-huit ou vingt ans qui veut savoir quelles sont ses origines ne voit pas nécessairement quoi que ce soit de dangereux dans ces tests. Le texte est peut-être un peu faible sur ce sujet-là.
Mme Marie-Thérèse Besson . - Sur le remboursement par l'assurance maladie de l'AMP, il ne faut pas poser la question d'une manière binaire - notre approche à nous, en maçonnerie, c'est d'éviter d'être binaire. Si je ne m'abuse, actuellement, l'assurance maladie prend en charge quatre essais, pendant lesquels on récupère à chaque fois 3 à 5 ovocytes. Cela fait donc une récolte de 15 à 20 ovocytes. S'il faut prendre en compte le côté économique de la chose, on pourrait penser que peu de femmes utilisent 15 à 20 ovocytes dans leurs démarches, et que l'on pourrait ne rembourser que trois essais. Cela représenterait une économie conséquente, ce qui permettrait à chacun d'avoir accès à la prise en charge de cet acte par l'assurance maladie.
M. Thierry Lagrange . - Vous avez pu voir tout au long de notre propos qu'au triptyque « liberté, égalité, fraternité » nous associons très souvent laïcité et solidarité. Au titre de la solidarité et de l'égalité, comment ouvrir un nouveau droit et en priver certaines femmes ? Il est donc évident que nous sommes favorables au remboursement. C'est peut-être un peu binaire, mais cela a le mérite d'être clair.
M. Olivier Henno , rapporteur . - J'ai été très marqué par une audition, celle de la présidente de l'Agence de la biomédecine qui disait ceci : ce qui, pour nous, relevait de la science-fiction il y a seulement cinq ans, est devenu possible.
Compte tenu de cette vitesse qui s'accroît de manière presque exponentielle, veiller au principe humain est absolument indispensable. J'ai cru comprendre que vous n'avez pas vu dans ce texte de question qui remettait en cause l'humanisme, les valeurs de notre pays ou la bioéthique à la française - même s'il en aurait été autrement s'il avait autorisé la GPA.
En examinant les travaux du CCNE, je me suis dit que le statut de donneur pouvait être intéressant ; mais cela ne remet-il pas quelque peu en cause les principes d'anonymat et de gratuité ? N'y a-t-il pas une forme de contradiction ? Vous avez évoqué à plusieurs reprises la question du consentement éclairé. Dans le cas des examens génétiques sur personnes décédées, ce consentement est transféré de la personne à la famille. Qu'en pensez-vous ?
M. Alain-Noël Dubart . - Le principe de gratuité nous semble fondamental. Il implique la non-marchandisation du corps humain, des greffes d'organes, des dons de toute nature qui ont trait à la personne humaine. Nous nous situons en cela très clairement dans un monde différent du monde anglo-saxon, nous avons d'autres valeurs. Quant au principe de l'anonymat, bien sûr, les dons d'organes sont anonymes, comme les dons de sperme. Mais il existe une différence entre donner un organe pour assurer la vie de quelqu'un et transmettre des ovocytes ou des spermatozoïdes pour « fabriquer » un autre être humain, avec la possibilité de transmettre des maladies héréditaires et des prédispositions. Il est fondamental que l'enfant à naître, qui possède les mêmes droits que les personnes ayant fait un projet parental, puisse accéder à son origine génétique, sans, bien entendu, qu'il y ait de filiation. On ne naît pas de rien, on naît d'un père et d'une mère. Dans une procréation médicalement assistée, on naît toujours de l'union d'un spermatozoïde et d'un ovocyte. Peut-être que dans quelques générations, ce sera différent, que se posera le problème du clonage, qui est interdit... Il nous semble donc légitime que tout enfant qui naît puisse avoir accès à ses origines. Cela posera d'autres problèmes, comme celui du nombre de donneurs. Mais avoir accès à ses origines nous semble consubstantiel à l'identité de la personne humaine.
M. Jean-Jacques Zambrowski . - Sur le prélèvement post mortem, on a vu récemment un militaire congeler quelques-uns de ses spermatozoïdes avant de partir en opération extérieure, et son épouse, alors qu'il était décédé en opération, demander à porter un enfant de lui. On est là dans une situation particulière qu'il faut naturellement savoir reconnaître légitimement et autoriser. Il serait inhumain, non éthique et non conforme à notre tradition, à nos valeurs culturelles de l'interdire. Il en est autrement de tout ce qui serait de l'ordre du commercial.
Sur le prélèvement des organes post mortem, nous faisons partie du petit nombre de pays qui, pour l'instant, se contente de la non-opposition. Malgré les campagnes, il nous semble qu'il serait opportun d'aller plus loin, d'informer largement les gens sur la possibilité qu'a chacun de refuser à ce qu'un prélèvement soit opéré, sur la carte vitale par exemple. Les accidentés de la route fournissent tragiquement un large contingent d'individus jeunes dont les organes feraient la survie d'un nombre très important d'individus, mais il faut que la population - donneurs et receveurs potentiels - en soit informée et que cela fasse partie d'un consensus culturel. Or pour l'instant, l'Agence de la biomédecine a été extrêmement discrète dans sa communication, et c'est dommageable à l'expansion nécessaire du don d'organes, qu'il s'agisse de coeurs, de reins, de poumons ou de foies. Le prélèvement post mortem est donc une opportunité formidable, à condition que chacun soit éclairé sur le besoin et sur le fait que l'absence d'opposition vaut implicitement consentement.
Nous avons, en matière de greffes d'organes, un dispositif remarquable, une circulation des organes à travers la France qui est merveilleusement organisée, une gratuité des organes que beaucoup de pays nous envient, des équipes chirurgicales prêtes dans chaque établissement, des infirmières de greffes qui sont prêtes à gérer la chose pour les équipes chirurgicales en permanence, un financement par l'assurance maladie qui ne pose aucun problème. Or mieux vaut être greffé du rein que d'être à la dialyse trois fois par semaine pour le restant de ses jours.
M. Thierry Lagrange. - Sur le don d'organes post mortem, je partage totalement les propos du professeur Zambrowski. Par ailleurs, le texte traite de manière équilibrée la question de l'anonymat : la levée de celui-ci sur la base du volontariat semble une très bonne chose. Enfin, comme vous vous en doutez, nous sommes très attachés à la gratuité du don.
Mme Joëlle Mounier. - Pour en revenir au sujet de l'anonymat, je considère qu'un adulte a besoin et a le droit de connaître ses racines. Cette information est nécessaire à la construction de son individualité biologique et génétique, le risque étant qu'il soit perturbé toute sa vie par la zone d'ombre qui entoure celles-ci. Cela étant, il faut respecter la liberté de choix : toutes les personnes ne souhaitent pas forcément connaître leurs origines.
Mme Christiane Vienne. - Le projet de loi ouvre la possibilité à celui ou celle qui s'interrogerait sur une pathologie pouvant être liée au patrimoine génétique d'une personne décédée de pratiquer des tests génétiques sur ce mort. Le dispositif repose sur la notion de consentement éclairé qui, dans ce cas précis, est un peu biaisée. D'une certaine façon, on peut considérer que le droit des vivants prévaut sur celui des personnes déjà mortes.
Mme Michelle Meunier . - Merci à tous pour la clarté de vos propos. Plusieurs d'entre vous ont regretté le caractère un peu trop technique du texte. Auriez-vous adressé les mêmes reproches au précédent projet de loi relatif à la bioéthique ?
Certains d'entre vous ont replacé ce texte dans le contexte géopolitique actuel et évoqué la nécessité de tenir des débats annuels. Pour faire vivre ce projet de loi, jugez-vous utile de prévoir un délai de réexamen du texte plus resserré ?
Mme Vienne a évoqué la problématique des personnes transgenres, hélas absente du texte. Toutefois, un amendement visant à améliorer la prise en charge des enfants intersexes a été adopté à l'Assemblée nationale. L'idée d'un consentement des familles aux traitements proposés à ces enfants figure désormais dans le projet de loi. Qu'en pensez-vous ?
Mme Christiane Vienne. - Le sujet des enfants et des adultes transgenres est délicat. Nous regrettons que cette thématique ait été occultée, car les personnes concernées, l'enfant lui-même, les parents, et parfois même les médecins, sont en détresse.
Dès que l'enfant identifie clairement le genre qui lui correspond le mieux, il faut déterminer le meilleur moment à partir duquel on peut lui administrer un traitement hormonal. En France, on considère généralement qu'il faut le faire avant la fin de sa maturité sexuelle. À l'inverse, en agissant trop vite, on risque de perturber le développement de l'enfant.
Mme Marie-Thérèse Besson. - Le texte est en effet extrêmement technique, ce qu'illustre parfaitement le questionnaire que vous nous avez adressé, puisqu'il comporte des questions auxquelles il est impossible de répondre si l'on n'est pas le spécialiste du sujet !
La formation des médecins est un enjeu essentiel. Certains médecins, qui y sont confrontés au quotidien, sont sûrement très au clair sur les sujets de bioéthique, mais je ne suis pas certaine que ce soit le cas de l'ensemble des médecins généralistes. Il faudrait peut-être réfléchir à mieux former ces médecins, car ce sont à eux que s'adressent les patients la plupart du temps quand ils ont une question en la matière.
Mme Joëlle Mounier. - Le texte comporte beaucoup d'aspects techniques, mais c'est le propre d'un projet de loi relatif à la bioéthique que de répondre à des questions complexes, comme la recherche sur les cellules souches ou l'embryon. Ce texte doit certes promouvoir des principes éthiques généraux, mais il doit aussi encadrer de manière précise tous les usages et les procédures dans leur complexité.
M. Pascal Neveu. - La technicité du texte n'est pas gênante. Il est bien construit, même si des aménagements pourraient être envisagés, notamment pour tenir compte des suggestions que nous pourrions faire au travers des réponses à votre questionnaire.
En tout cas, le texte doit encadrer les usages existants. Idéalement, il faudrait essayer d'anticiper les évolutions, car la loi arrive souvent trop tardivement. En réalité, il s'agit d'un débat de prospective : il faut déjà penser le monde de demain et s'interroger sur ce qui se passera dans trente ou cinquante ans. Aujourd'hui, nous estimons qu'il faut légaliser les usages, tout en les encadrant pour éviter les débordements.
Prenons l'anonymat du don : plusieurs études montrent que le nombre de donneurs a chuté dans les pays nordiques après sa levée. Je pense pour ma part qu'il faut maintenir un droit à l'anonymat, tout en inventant un dispositif qui permettrait de dévoiler le nom du donneur au bout d'un certain temps, dix-huit ou vingt ans par exemple. Après tout, l'identité d'un individu ne se construit pas uniquement à partir de ses origines. Cela étant, certaines problématiques génétiques et médicales soulèvent de grandes interrogations, qui légitiment la création d'un fichier centralisé des données relatives aux donneurs.
M. Thierry Lagrange. - Il nous semble que les choix que reflétera la loi une fois promulguée devront être évalués et réajustés avant même la révision prévue dans la prochaine loi relative à la bioéthique.
Mme Élisabeth Doineau , présidente . - Notre questionnaire sert à ouvrir la discussion. Nous avons besoin de vos lumières, car il est normal que nous doutions sur des sujets qui sont très techniques, mais qui font aussi écho à notre histoire personnelle.
M. Jean-Jacques Zambrowski. - Vous n'allez pas manquer d'auditionner des techniciens capables d'apporter l'expertise la plus juste sur les multiples questions que vous vous posez. Beaucoup d'entre nous ont aussi une connaissance technique de ces dossiers. Seulement, nous nous exprimons en tant que représentants des courants de pensée et souhaitons apporter un simple témoignage sur ces enjeux éthiques.
De notre point de vue, le texte ne pose pas de problème sur le plan technique. Tel qu'il est, il nous paraît approprié. Notre rôle est de faire en sorte que vous ne perdiez pas de vue le contexte éthique témoignant de l'ensemble des valeurs qui fondent notre identité nationale depuis plusieurs siècles. La mode ou la technique ne doit pas balayer les fondements de notre réflexion sur ces sujets.
La notion d'anticipation est essentielle. Aujourd'hui, par exemple, nous parlons du réchauffement climatique et des mesures à prendre en urgence pour en limiter les effets. Mais qu'aurait-on pu ou dû faire pour l'éviter ? Les décisions que vous prendrez pourront paraître parfaitement licites et vraisemblables dans le contexte actuel, mais il faudra veiller à ne pas mettre en cause nos valeurs fondamentales, sauf à décider qu'il faut impérativement en changer. Nous ne sommes pas conservateurs par principe : nous accueillons le progrès, mais pas à n'importe quel prix !
M. Alain-Noël Dubart. - Je voudrais simplement revenir sur la notion de suivi. Jusqu'à présent, les lois relatives à la bioéthique étaient révisées à intervalles réguliers sur des sujets qui ne touchent pas fondamentalement l'organisation de la société.
Avec cette loi, il n'en est plus de même, car de nouveaux problèmes apparaissent. En nous contentant d'une révision tous les cinq ans, nous nous exposons à un certain nombre de dérives. En effet, la loi a un caractère général et ne peut pas traiter certains cas particuliers qui nécessiteraient d'être abordés dans un délai restreint. Nous proposons, comme nous l'avons fait lors de notre audition à l'Assemblée nationale, que le Parlement crée une commission de suivi permanente à côté du Comité consultatif national d'éthique, de sorte à pouvoir suivre l'évolution des techniques, l'application de la loi et l'évolution des situations nécessitant des réponses circonstanciées. Cette commission de suivi se composerait des représentants des différents courants de pensée et aurait pour mission d'examiner des sujets très pratiques et concrets.
M. Jean-Jacques Zambrowski. - Il ne faudrait pas qu'une instance administrative et, a fortiori, une décision individuelle viennent finalement clore un débat qui nécessite un consensus sociétal. Le rôle de l'administration ne consiste pas à décider à la place du législateur sur des sujets sur lesquels celui-ci n'a pas voulu trancher.
Mme Marie-Thérèse Besson. - Nous sommes favorables à l'idée d'une commission de suivi, ce qui n'exclut pas une révision de la loi tous les cinq ans pour appréhender les sujets dans leur globalité.
Mme Muriel Jourda , rapporteur . - Monsieur Zambrowski, vous avez estimé que le rôle du législateur n'était pas de chercher à conserver un passé révolu, mais pas non plus de promouvoir un futur sans contrôle. Dans la mesure où le lien de filiation serait créé ab initio, non plus sur le fondement de la vérité biologique ni même de la vraisemblance, mais sur le fondement de la volonté individuelle, comment exercer ce contrôle ?
M. Jean-Jacques Zambrowski. - Le contrôle s'inscrit dans la démarche de prudence dont j'ai déjà parlé. Nous n'admettons pas de vérité que nous n'ayons pas dûment scrutée, ce qui suppose de bien peser le pour et le contre avant d'ouvrir le champ des expérimentations. Ce contrôle pourrait s'exercer, si vous le jugez utile, au travers d'une instance qui regrouperait des personnes intéressées par ces sujets, qui suivraient l'évolution de la loi dans l'intervalle de deux révisions pour vérifier que la loi ne comporte aucun vice caché.
Aujourd'hui, on est conscient que le laxisme mène à l'imprudence et à de possibles catastrophes. On ne peut pas faire n'importe quoi au nom de la liberté individuelle.
Mme Élisabeth Doineau , présidente . - Justement, nos concitoyens nous disent parfois que l'ouverture de la PMA revient à faire n'importe quoi et qu'elle mène à la GPA. Il est parfois difficile de trouver le ton juste pour bien faire comprendre les choses.
M. Jean-Jacques Zambrowski. - Sauf à imaginer que la PMA entraîne automatiquement la GPA à moyen terme, l'idée qui paraît faire consensus aujourd'hui, c'est que le fait d'aider un couple, notamment de femmes, à aller jusqu'au bout du désir de maternité, qui est différent du désir de grossesse en ce qu'il implique un désir de transmission et en ce qu'il semble correspondre à la biologie même de notre espèce, est compréhensible.
En revanche, le désir d'une femme célibataire d'avoir un enfant doit être examiné très soigneusement, sauf à fabriquer une espèce d'orphelin. Le droit à l'enfant doit-il prendre le pas sur le droit de l'enfant ? Il vous appartient de porter un regard éthique sur cette question, regard que nous pouvons éclairer sans pour autant gouverner à votre place.
Les couples d'homosexuels masculins ayant un désir semblable nous paraissent poser un autre problème. En conséquence, on introduit ipso facto une inégalité de traitement entre les couples homosexuels masculins et féminins. Il faut là encore s'interroger sur les conséquences d'une telle mesure, non pas seulement sur les enfants de ces couples, en raison de potentiels risques de nature psychologique, mais sur la société qui en résultera.
Mme Christiane Vienne. - Je souhaite attirer votre attention sur le fait que, dans le domaine de la parentalité, on parle généralement d'engendrement et pas seulement de filiation. Les modèles familiaux sont multiples et évoluent au cours de la vie : une femme seule ne restera peut-être pas seule, un couple marié ne le reste pas toujours très longtemps. On doit travailler sur des modèles en évolution permanente pour aborder ce genre de thématiques. C'est la raison pour laquelle la question de l'égalité est devenue tellement importante, beaucoup plus qu'elle ne l'était à l'époque où ces modèles étaient figés. Aujourd'hui, ce qui est vrai pour l'un doit l'être pour l'autre, quelles que soient les circonstances de sa vie, ce qui rend le travail du législateur passionnant.
Mme Marie-Thérèse Besson. - Notre société est en constante évolution. C'est pourquoi notre méthode de travail repose sur un questionnement permanent.
Sur ces sujets de bioéthique, en particulier la famille et la filiation, il est difficile de dissocier raison et sentiments. Il faut donc veiller à les aborder dans leur globalité.
Mme Catherine Di Folco . - Madame Besson, sauf erreur de ma part, vous avez parlé tout à l'heure d'un enfant né d'un couple de femmes. Même si le monde est en pleine mutation, cela me paraît tout à fait impossible.
Mme Marie-Thérèse Besson. - Vous avez raison, madame la sénatrice. J'aurais dû parler d'un enfant né du désir de deux femmes vivant en couple.
Mme Élisabeth Doineau , présidente . - Merci à tous pour votre contribution à la réflexion sur l'évolution de notre société.
Ce point de l'ordre du jour a fait l'objet d'une captation vidéo qui est disponible en ligne sur le site du Sénat .
Pr Nathalie Rives, CECOS de
Rouen, CHU de Rouen
Pr Catherine Guillemain, CECOS de
Marseille,
Assistance publique-hôpitaux de Marseille,
et Dr Sophie
Mirallie, CECOS de Nantes, CHU de Nantes
Pr Rachel Lévy, Pr Nelly
Achour-Frydman
et Dr Patrice Clément, pour la
Fédération des BLEFCO
(Biologistes des laboratoires
d'étude
de la fécondation de l'oeuf)
M. Alain Milon , président . - Nous poursuivons nos travaux sur le projet de loi relatif à la bioéthique par l'audition commune de professionnels directement impactés par les dispositions relatives à l'assistance médicale à la procréation. Cette audition fait l'objet d'une captation vidéo, retransmise en direct sur le site du Sénat. Elle sera ensuite consultable à la demande.
Pour la Fédération nationale des Centres d'Études et de Conservation des OEufs et du Sperme (CECOS), nous recevons le professeur Nathalie Rives, présidente, le professeur Catherine Guillemain, vice-présidente, et le docteur Sophie Mirallie, secrétaire générale. Pour la Fédération des biologistes des laboratoires d'étude de la fécondation et de la conservation de l'oeuf (BLEFCO), nous accueillons le Professeur Rachel Levy, vice-présidente, le Professeur Nelly Achour-Frydman, coordinatrice, ainsi que le docteur Patrice Clément, membre de la Fédération.
Pr Rachel Levy, vice-présidente de la Fédération nationale des BLEFCO . - La Fédération des BLEFCO est la société savante réunissant l'ensemble des praticiens des laboratoires publics et privés de l'assistance médicale à la procréation en France. Nous représentons à ce jour 290 membres adhérents actifs et l'ensemble des centres d'AMP français. Nous saluons le projet de révision de la loi de bioéthique, en particulier l'accès à l'AMP pour les femmes en couple ou célibataires et la possibilité d'autoconservation des ovocytes en dehors d'une pathologie.
Nous souhaitons aborder la mise en place d'un plan national sur la fertilité et l'AMP. Le premier versant porterait sur la prévention, la pédagogie et l'éducation. L'objectif serait l'information aux adolescents, jeunes adultes et adultes, femmes et hommes, au sujet de la physiologie de la reproduction, des effets de l'âge, mais également des modes de vie, sur la fertilité. Le deuxième versant serait consacré à la recherche, notamment sur les causes d'infertilité. Nous plaidons pour un soutien fort à la recherche clinique et fondamentale dans le domaine de la fertilité par le biais d'appels d'offres dédiés. Enfin et surtout, nous souhaitons l'engagement d'une réflexion médico-économique sur la situation actuelle des activités biologiques des activités d'AMP et l'impact des modifications prévues par la loi de bioéthique. De notre point de vue, elles devraient aboutir à une juste valorisation de ces activités.
Notre discipline relève d'une biologie hautement spécialisée et interventionnelle. Il n'est pas possible de déplacer des gamètes pour centraliser cette activité sur un seul site, d'automatiser cette activité ou de travailler en série. Aucune économie d'échelle n'est envisageable en AMP. Par ailleurs, nous devons faire face à des évolutions technologiques rapides et à des contraintes réglementaires coûteuses et chronophages. À côté de la diminution récente de la cotation de l'acte de micro-injection, nous avons salué la cotation en B des activités de vitrification ovocytaires et embryonnaires. Cependant, nous restons vigilants à l'égard des négociations à venir en 2020 et 2022. Pour que le texte de loi soit applicable dans des conditions satisfaisantes, il est indispensable qu'une cartographie précise soit réalisée région par région au sujet de l'adéquation entre l'offre de soins actuelle des centres, leurs moyens et les futures demandes afin que chaque centre d'AMP puisse disposer des moyens adéquats en temps médical et paramédical et en équipements. Enfin, nous souhaitons être interrogés sur la question de l'AMP post mortem.
Dr Patrice Clément, membre de la Fédération nationale des BLEFCO . - Je vous remercie de nous faire participer à vos travaux. Je souhaite intervenir au sujet de la problématique de l'autoconservation telle qu'elle est décrite dans l'article L. 2141-12 proposé par l'article 2 du projet de loi, qui limite aux centres d'AMP publics et privés à but non lucratif la possibilité de réaliser l'autoconservation des gamètes. Cet article est discriminatoire vis-à-vis du secteur privé à but lucratif, puisqu'il prive les patients de la possibilité de choisir librement leur centre d'AMP. Interdire aux praticiens libéraux de pratiquer l'autoconservation des gamètes signifiera une orientation de la patientèle vers un nombre nécessairement limité de structures d'accueil publiques et privées à but non lucratif. Cette mesure aura pour effet direct et immédiat une entrave à l'accès aux soins des patients ainsi qu'un allongement du délai de prise en charge. Elle aura des conséquences contraires à l'intérêt supérieur des patients qui attendaient une ouverture vers une AMP accessible à tous, dans des délais compatibles avec la capacité réelle des centres.
Les professionnels médicaux libéraux ont les mêmes compétences que leurs collègues du secteur public. Ils réalisent 47 000 tentatives annuelles d'AMP parmi les 92 000 recensées par l'Agence de la biomédecine. Ils sont soumis aux mêmes inspections de l'Agence régionale de santé et de l'Agence de la biomédecine. Ces seuls chiffres sur l'activité prouvent l'indiscutable attachement et la confiance des patients vis-à-vis de ce mode d'exercice. La demande de modification de l'article L. 2141-12 est soutenue par la grande majorité des acteurs de l'assistance médicale à la procréation en France, qu'ils relèvent du secteur public ou du secteur libéral, ainsi que par la grande majorité des sociétés savantes de notre discipline.
Pr Nelly Achour-Frydman, coordinatrice de la Fédération nationale des CECOS . - Je vous propose d'aborder l'évolution technologique en AMP et le diagnostic préimplantatoire, qui est ma spécialité. La suppression du diagnostic préimplantatoire associé au typage HLA soulève des questions éthiques évidentes et paraît surprenante. Cette pratique concernait un faible nombre de couples, ayant un enfant atteint d'une maladie génétique rare, souvent issus d'un niveau socioéconomique peu favorisé et, surtout, sans représentation associative. Ils représentaient donc une cible sans défense. De ce point de vue, la suppression de cet accès aux soins est condamnable.
Par ailleurs, les débats à l'Assemblée nationale montrent que la majorité des députés n'a pas souhaité l'autorisation en pratique courante de la recherche d'anomalies chromosomiques sur l'embryon. Cette technique est appelée « diagnostic préimplantatoire des anomalies chromosomiques ». Le risque de dérives eugénistes, évoqué dans les débats, a heurté la communauté médicale et mérite d'être discuté. Le gouvernement a renvoyé cette question à l'article 14 du projet de loi qui rendrait possible cette pratique sous couvert d'une recherche clinique. Par conséquent, un financement dans le cadre du plan relatif à la fertilité, abordé à l'origine, doit être consacré à ce type de protocole de recherche.
Pr Nathalie Rives, Présidente de la Fédération nationale des CECOS . - La Fédération nationale des centres d'étude et de conservation des oeufs et du sperme humain comprend vingt-neuf centres répartis en France métropolitaine et dans les départements d'outre-mer, associés au sein des centres hospitaliers universitaires et des praticiens qui, pour la majorité d'entre eux, exercent une activité biologique et clinique d'enseignement et de recherche.
Nous saluons un certain nombre d'avancées proposées dans le cadre du projet de loi adopté par l'Assemblée nationale. Il s'agit de l'ouverture de l'AMP avec tiers donneur aux couples de femmes et aux femmes non mariées, de l'autorisation sans l'encourager de la conservation des gamètes en dehors des indications médicales, des conditions d'âge qui seront définies pour cette conservation des gamètes, mais également des conditions d'âge pour le recours à l'AMP.
Le projet de loi prévoit la possibilité d'arrêter la conservation des gamètes et des tissus germinaux, ce qui nous paraît extrêmement important pour gérer les difficultés rencontrées en matière de stockage des échantillons pour les patients qui ne s'inquiètent plus des échantillons conservés. Il nous permet également de mettre en place une évaluation de la loi à l'issue de cinq années. Cela nous semble important afin d'évaluer l'impact de l'AMP avec tiers donneur sur le fonctionnement des activités et l'accessibilité aux soins. Le projet de loi maintient un principe fondamental et fondateur pour tout type de don d'éléments et produits du corps humain, à savoir la gratuité du don en accord avec la non-marchandisation des éléments et produits du corps humain. Nous sommes particulièrement attachés à ce principe. Il met également en avant un certain nombre de points vis-à-vis desquels les professionnels des CECOS émettent un avis favorable, réservé, voire défavorable.
Notre avis réservé concerne un ajout apporté au texte de loi, qui met en doute la capacité de l'équipe médicale à prendre en charge sans discrimination les différentes demandes. Cet accès ne peut faire l'objet d'aucune différence de traitement, notamment au regard du statut matrimonial et de l'orientation sexuelle des demandeurs, car nous ne pouvons pas exercer notre pratique médicale en mettant en avant une discrimination.
Ce projet de loi autorise la transmission des données non identifiantes, à laquelle nous sommes favorables, ainsi que l'identité du donneur aux personnes majeures qui le souhaiteraient. À ce sujet, nous avons un avis plus réservé. Nous pensons que l'accompagnement doit être amplifié et que l'information de l'identité du donneur ne répondra pas à la plupart des interrogations des adultes conçus par don.
Par ailleurs, nous avons défendu depuis de nombreuses années la remise en place d'un registre national des donneurs. Nous souhaitons également la création d'une commission pour la transmission des données non identifiantes et identifiantes ainsi qu'une adaptation du mode de filiation aux couples de femmes qui ont recours à l'AMP avec tiers donneur, ceci en évitant toute discrimination vis-à-vis des couples infertiles et des femmes non mariées.
Le premier élément mis en avant dans ce texte est l'intérêt suprême de l'enfant, mais nous regrettons que les couples, voire les femmes seules soient mis de côté. Le consentement exprimé à l'origine par le donneur vis-à-vis de la transmission de l'identité au jeune adulte qui en exprimerait la demande peut évoluer au cours du temps. Or le donneur n'aura aucune possibilité de revenir sur sa position. En outre, le projet de loi a peu mis l'accent sur un certain nombre de sujets majeurs liés à l'AMP. Il n'identifie plus spécifiquement l'AMP comme une procédure permettant de répondre à l'infertilité pathologique de certains couples et d'éviter la transmission d'une maladie d'une particulière gravité.
La notion d'infertilité mériterait d'être réintroduite dans le texte de loi sans discrimination entre les différentes demandes. Les infertilités pathologiques existent. Leur remise en question fréquente au cours des discussions, y compris au sein de l'Assemblée nationale, va à l'encontre d'un ajout extrêmement positif à la loi, qui concerne la mise en place d'un plan sur la fertilité ou l'infertilité, depuis la prévention jusqu'à la recherche. La notion de l'infertilité doit donc réapparaître dans le texte de manière différente qu'au travers du plan sur la fertilité.
Le projet de loi associe un ensemble de propositions relevant de la réflexion éthique et scientifique et de la décision politique. Cependant, elles ne peuvent être dissociées des conditions optimales de leur mise en oeuvre grâce à des moyens humains et matériels permettant de répondre aux attentes des demandeurs. Le projet de loi doit donc être accompagné de moyens à la hauteur de ces ambitions. Il s'agit de maintenir une prise en charge équivalente à celle que nous proposons actuellement aux couples infertiles.
Ce projet de loi implique également la constitution de nouvelles banques de gamètes, et plus spécifiquement celle de spermatozoïdes conservés en vue de dons, et doit également favoriser le recrutement de nouvelles donneuses. Il doit prévoir une période de transition permettant l'évolution des dispositions réglementaires vers les nouvelles conditions fixées par la loi. Ce sera une période d'instabilité, qui doit durer le moins longtemps possible.
Mme Corinne Imbert , rapporteure . - Madame le Professeur Nelly Achour-Frydman, vous avez évoqué la décision de l'Assemblée Nationale de supprimer le diagnostic préimplantatoire associé au typage HLA, que vous estimez condamnable. Vous soulignez que le risque d'eugénisme évoqué a heurté la communauté médicale. Au-delà de l'éthique qui guide votre exercice au quotidien, comment rassurer les autorités et les concitoyens vis-à-vis de ce risque ?
Pr Nelly Achour-Frydman . - J'estime condamnable que l'accès au DPI avec typage HLA soit ôté de la loi. Le DPI des aneuploïdies est un autre problème. Le DPI avec typage HLA a concerné vingt-cinq couples en dix ans. Cette activité est vraiment confidentielle. Sa mise en place s'est avérée complexe. Ce sont essentiellement les équipes des hôpitaux Antoine-Béclère et Necker qui ont effectué ce type de diagnostic. En effet, il n'était pas possible de congeler les embryons sains et non compatibles afin de recommencer des tentatives. Il est nécessaire que nous puissions congeler les embryons non compatibles pour recommencer et tenter de disposer d'embryons compatibles. Les couples n'abandonneront pas ces embryons. L'objectif est d'essayer d'avoir un enfant sain et compatible pour sauver l'enfant précédent, gravement malade.
L'argument éthique selon lequel il n'est pas envisageable de concevoir un enfant au bénéfice d'un tiers et non plus pour lui-même, peut s'entendre. Toutefois, ces couples ont déjà essayé spontanément d'avoir ces enfants. La recommandation des médecins de retenter une grossesse pour essayer d'avoir un enfant compatible spontanément ne gêne personne. Par conséquent, cette discussion n'a pas de sens.
Concernant la recherche des aneuploïdies, il faut tenir compte d'un double problème. Le fait d'avoir refusé l'autorisation en pratique courante sous couvert de l'existence d'un risque d'eugénisme a beaucoup heurté la communauté médicale. Le DPI est mis en place depuis une vingtaine d'années dans le cadre d'anomalies génétiques. Jamais un couple ne répondant pas aux indications n'a été pris en charge. Les médecins respectent la loi à la lettre.
La deuxième raison du refus tient à la discussion sur la possibilité de dépister une trisomie 21. Le corps médical y voit une forme d'incohérence, puisque la trisomie 21 est systématiquement recherchée au stade foetal. Il paraît donc contradictoire de l'autoriser au stade prénatal, et non au stade préimplantatoire. Selon l'expérience internationale, le dépistage de la trisomie 21 sur l'embryon implantatoire concerne moins de 3 % des embryons. Il ne s'agit pas de l'anomalie majoritaire susceptible d'être dépistée, ni même celle que l'on souhaite forcément rechercher. L'objectif est d'identifier des anomalies qui empêchent la grossesse ou provoquent des fausses couches. Nous sommes favorables au fait de ne pas rechercher la trisomie 21. Le deuxième argument que j'estime compréhensible consiste à rappeler que l'efficacité de cette technique n'est pas prouvée à 100 %. Certaines études montrent qu'elle présente un intérêt. D'autres études se permettent de poser des questions. La recherche clinique proposée dans le cadre de l'article 14 semble une bonne solution, mais il faut avoir les moyens de mener cette recherche. En termes de financement, le taux de succès du PHRC est un peu bas.
Mme Muriel Jourda , rapporteur . - Ma première question, à laquelle vous m'avez invitée, concerne l'AMP post mortem. Ma deuxième question porte sur la réintroduction de la notion d'infertilité dans la loi. Pouvez-vous y revenir ? Par ailleurs, quelle est votre opinion sur les conditions de levée de l'anonymat et sur la possibilité de levée de l'anonymat des donneurs de l'ancien régime ?
Pr Rachel Levy . - La Fédération des BLEFCO a renvoyé le questionnaire complété, qui comprend les réponses aux questions que vous soulevez.
Concernant l'AMP post mortem, je rappelle qu'en cas de décès d'un membre du couple, le projet de loi prévoit de consulter le membre survivant au sujet du devenir des embryons obtenus en AMP et conservés.
En cas de décès de son conjoint, la partenaire d'un couple disposant d'embryons conservés se verrait contrainte de choisir entre leur destruction, leur utilisation en recherche ou l'accueil de ces embryons par un couple ou par une femme célibataire. Devenue veuve, elle pourrait solliciter pour elle-même l'accueil d'un embryon d'un autre couple, mais n'aurait pas la possibilité de disposer des embryons conçus dans le cadre de son projet parental. De même, en cas de décès du conjoint ayant autoconservé ses spermatozoïdes dans le cadre d'un projet parental, la veuve serait autorisée à requérir un don de spermatozoïdes, mais ne pourrait pas avoir accès aux gamètes autoconservés de son conjoint, que la loi contraint aujourd'hui à détruire. Dans les deux cas, la partenaire survivante du couple subirait la perte de son conjoint et l'impossibilité d'avoir accès aux gamètes conservés et aux embryons conçus dans le cadre d'un projet parental. Enfin, si son âge est avancé, elle aurait très peu de chances de pouvoir mener un nouveau projet parental avec un nouveau partenaire. Dans ce cadre, l'ouverture de l'AMP aux femmes célibataires rend inique le maintien de l'interdiction d'une AMP post mortem. Nous demandons que le couple puisse donner son accord ou non pour l'utilisation des embryons en cas de décès du conjoint. De même, l'utilisation post mortem de gamètes conservés lorsque le conjoint y a consenti de son vivant et dans le contexte d'un désir d'enfant paraît légitime.
Dans les deux cas, il est indispensable de faire preuve de vigilance quant aux conditions de délais de réalisation et au contexte de ces demandes et de s'assurer du bien-être de l'enfant. Nous proposons donc la mise en place d'une commission multidisciplinaire.
Pr Catherine Guillemain, vice-présidente de la Fédération des CECOS . - Vous avez également posé une question sur la disparition de la notion d'infertilité dans le texte. Nous pensons qu'il est important de maintenir cette notion de prise en charge potentielle des couples infertiles pour des raisons médicales, mais également pour éviter de transmettre une maladie grave. Il s'agit également d'un moyen de justifier la surveillance de nos pratiques destinées à améliorer les résultats des tentatives d'assistance médicale à la procréation. Il est important de maintenir le fait que la prise en charge concerne les couples confrontés à une infertilité constatée médicalement et de pouvoir prendre en charge des couples en cas de risque de transmission d'une maladie grave. Cela permet de justifier les actions de communication visant à rappeler à la population générale que la fertilité est potentiellement limitée dans le temps et peut être altérée par un certain nombre de facteurs environnementaux. Il est important de disposer de protocoles et de moyens d'études sur les facteurs, la gamétogenèse et la capacité à nous reproduire.
Mme Muriel Jourda , rapporteur . - Cela signifie-t-il que vous limiteriez le recours à l'AMP aux couples hétérosexuels infertiles ?
Pr Catherine Guillemain . - Non. La prise en charge des nouvelles demandes de couples de femmes et de femmes non mariées ne pose pas de problème. En revanche, le fait de ne pas mentionner les couples médicalement infertiles nous dérange. En outre, cela représente un danger à moyen terme et à long terme, car ces situations sont prises en charge par l'Assurance Maladie.
Mme Muriel Jourda , rapporteur . - Je me suis sans doute mal exprimée. Je n'évoquais pas le cas des femmes seules et des couples homosexuels, pour lesquels j'ai compris que vous êtes favorables à leur possibilité de recours à l'AMP. Ma question porte sur les couples hétérosexuels et sur la limitation de l'accès à l'AMP à ceux qui ont des problèmes d'infertilité plutôt que l'ouverture à des personnes qui n'ont a priori pas besoin de recourir à ces techniques.
Pr Catherine Guillemain . - Ces situations sont marginales. La prise en charge dans le cas de couples hétérosexuels pour lesquels aucune anomalie n'a pas été diagnostiquée représente moins de 10 % des cas. Il s'agit d'une infertilité du fait du temps sans conception.
Pr Nathalie Rives . - La médecine identifie des symptômes qui seront considérés comme idiopathiques dans la mesure où nous ne disposons pas de moyens diagnostiques pour mettre en évidence l'origine de la pathologie. Concernant les couples infertiles, les moyens actuels ne permettent pas dans un certain nombre de cas d'identifier l'origine de l'infertilité. Par ailleurs, le fait de ne pas être reconnus comme infertiles est très mal vécu par ces couples. À titre d'exemple, il n'est pas possible de dire à un garçon né avec une absence de testicules qu'il n'a pas de pathologie. Une femme ayant reçu des traitements anti-cancer extrêmement toxiques et rendue infertile peut difficilement entendre qu'elle n'est pas infertile et qu'il ne s'agit pas d'une pathologie. La reconnaissance de l'infertilité de couples doit être maintenue dans la loi tout en acceptant les autres demandes.
S'agissant des couples de femmes, notre avis est favorable. Concernant les femmes seules, notre avis est plus réservé. Nous ne remettons pas en question la capacité d'une femme seule à élever un enfant, mais nous avons des craintes d'ordre médical. Qu'advient-il si une femme enceinte dont la grossesse aura été induite au travers de notre action rencontre des complications liées à sa grossesse et décède à l'accouchement ?
La prise en charge des couples infertiles n'est pas systématique. Certaines prises en charge sont refusées pour diverses raisons, notamment médicales ou sociales. Comment pourrons-nous faire accepter nos refus ? Nous devons également nous assurer du bien-être de l'enfant à venir, conformément aux textes. S'agissant des couples de femmes et des femmes seules, le taux de refus de prise en charge à l'étranger est supérieur à 50 %, notamment en Belgique. En cas de refus, nous risquons d'entendre dire que nous pratiquons une discrimination comme le laisse entendre l'ajout à l'article 1 er . Il faut maintenir la reconnaissance que l'infertilité existe et qu'elle n'est pas relative.
Par ailleurs, le texte de loi ne stipule pas que nous devons recontacter les donneurs. Il indique que les donneurs de gamètes ou d'embryons exprimant ce souhait pourront faire part d'un consentement différent auprès de la commission et accepter la transmission de l'identité au jeune adulte conçu par don. S'il s'agit d'une démarche spontanée du donneur, cela ne nous gêne pas. En revanche, le fait de recontacter les donneurs ayant fait un don sous les conditions actuelles de la loi met en jeu des scénarios extrêmement différents selon le contexte du donneur.
Si le donneur a eu des enfants conçus par don au travers de différents couples infertiles alors qu'il reste encore des paillettes en cours de distribution, le fait de le recontacter pour lui demander d'exprimer un nouveau consentement et lever son identité lors de futures conceptions rompt le consentement établi et le contrat avec les couples infertiles. Lorsqu'un donneur leur a été attribué, ce donneur avait consenti de donner de manière anonyme. Cette garantie a été apportée au couple. Si nous modifions cette situation en recontactant le donneur, le consentement établi avec le couple se trouve rompu.
S'il s'agit d'un donneur dont les paillettes sont en cours de distribution et s'il n'y a pas eu d'enfants conçus à partir de ce don, rien n'empêche de le recontacter. En revanche, celui-ci peut nous demander d'obtenir des informations sur l'aboutissement de son don. La loi ne permet pas la transmission d'informations de ce type. Si le don n'a pas donné lieu à une naissance, le donneur peut s'interroger sur l'utilisation qui en a été faite. Il est souhaitable de recontacter les donneurs dans le seul but de leur demander un consentement pour une utilisation du don auprès des couples de femmes ou des femmes seules. Si nous modifions la finalité de l'utilisation des spermatozoïdes de donneurs déjà stockés et qui sont utilisés auprès de couples infertiles, ou pour éviter la transmission d'une maladie d'une particulière gravité, il faut en informer le donneur, qui doit émettre son consentement.
Si ce type de procédure est mis en oeuvre, chaque scénario devra être pris en compte afin d'éviter les ruptures de consentement émis et de contrats établis avec des couples infertiles qui pensent avoir conçu un enfant à partir d'un donneur anonyme. Je pense qu'un consentement doit être respecté.
Concernant les conditions de levée de l'anonymat, la question porte sur la possibilité pour les jeunes adultes majeurs conçus par don qui en exprimeraient la demande d'accéder aux données non identifiantes ou au prénom et au nom du donneur. Parmi les jeunes adultes que nous avons pu rencontrer, certains d'entre eux se manifestent, mais beaucoup ne s'expriment pas du tout. Le fait de transmettre uniquement un nom et un prénom ne suffira probablement pas. Il faudra certainement répondre à toutes les interrogations. Nous souhaitons amplifier l'accompagnement des demandes déjà en cours et des nouvelles demandes dans le cadre de la conception au travers de l'AMP avec tiers donneur. Actuellement, nous raisonnons au sujet de plaintes issues de jeunes adultes conçus il y a au moins une vingtaine d'années, mais ce contexte sociétal était différent de la société dans laquelle évolueront les nouveaux enfants conçus, une fois atteint l'âge adulte.
Concernant notre proposition relative aux données non identifiantes, elle se fonde sur l'enquête que nous avons menée auprès des donneurs et des donneuses, des couples infertiles et d'associations de jeunes adultes conçus. Ces dernières ont rarement répondu. Les donneurs et les donneuses sont favorables à la transmission de données non identifiantes. Les couples infertiles, pour leur part, sont extrêmement réticents, mais ne sont pas très demandeurs de transmission d'autres données, contrairement aux donneurs. La seule position commune entre les donneurs, les couples infertiles et les professionnels concerne la transmission des données médicales.
Nous accueillons des donneurs âgés de 18 à 45 ans. Le fait de pouvoir accueillir des donneurs et des donneuses qui n'ont pas d'enfant réduit l'âge des candidats au don. La manière d'aborder les difficultés de la société à cet âge n'est pas comparable à celle des personnes ayant plus de quarante ans, surtout celles qui ont l'expérience de la parentalité. Pour autant, nous n'avons pas de réelles solutions. Faut-il permettre au donneur ou à la donneuse de changer d'avis ? Le texte de loi l'autorise pour les anciens donneurs. En revanche, les nouveaux candidats se voient imposer une seule possibilité.
M. Olivier Henno , rapporteur . - L'un des effets du projet de loi est de modifier le contexte éthique et juridique du don. Cette évolution aura-t-elle un impact sur le nombre et les types de dons ? Quel est le nombre d'embryons surnuméraires actuellement congelés en France ? Qu'en est-il de la lignée des cellules-souches ? Enfin, quelle est la part des gamètes conservés et respectivement affectés aux dons et à la recherche et qui sera finalement détruite ? Pensez-vous que ce projet de loi sécurise la question de la destruction pour les professionnels ?
Pr Rachel Levy . - Selon le dernier rapport médical scientifique de l'Agence de la biomédecine, 177 968 embryons surnuméraires étaient conservés en France au 31 décembre 2017 pour un projet parental. Parmi les 32 878 embryons qui n'étaient plus investis d'un projet parental, 21 727 ont été donnés à la recherche. Dans de nombreux cas, la confirmation à trois mois n'a pas été obtenue. Il est difficile de répondre de façon robuste.
Pr Nelly Achour-Frydman . - La plupart des demandes de travaux de recherche sur les cellules-souches embryonnaires concernent des lignées importées ou déjà dérivées en France. Le rapport médico-scientifique de l'Agence de la Biomédecine montre que moins de dix lignées ont été obtenues en France. Globalement, l'activité de dérivation c'est-à-dire le fait d'obtenir des lignées de cellules-souches à partir d'embryons données à la recherche est quasi arrêtée. Les chercheurs travaillent sur des lignées importées ou des lignées déjà établies.
Dr Sophie Mirallie, secrétaire générale de la Fédération nationale des CECOS . - Nous nous efforçons d'interroger les personnes qui se présentent dans notre centre pour faire des dons sur leur positionnement dans l'hypothèse où la levée de l'anonymat serait possible. La plupart affirment qu'elles poursuivraient leur démarche de don et accepteraient la transmission de leurs données identifiantes, car ces demandes émanent d'enfants en souffrance. L'évolution du profil des donneurs est probable, mais elle est déjà entamée. Les personnes ouvertes à la levée de l'anonymat sont de plus en plus nombreuses.
Parmi les couples receveurs que nous recevons en consultation, certains réfléchissent à la possibilité d'attendre la promulgation de la loi pour obtenir un don de sperme afin que leur enfant, une fois majeur, puisse faire un choix. Je ne pense pas qu'une grave pénurie de dons soit à craindre à l'avenir.
Pr Nathalie Rives . - L'équipe du Professeur Louis Bujan, spécialiste de la reproduction, à Toulouse, a mené durant deux ans une étude achevée début 2018, dont les résultats ne sont pas encore publiés. Il y a une dizaine d'années, 80 % des candidats au don étaient favorables au maintien de l'anonymat du don. 20 % auraient potentiellement accepté de donner si le don n'était pas anonyme. Selon cette étude, 46 % accepteraient potentiellement de donner dans un contexte où la transmission de l'identité du donneur serait possible. Toutefois, ces personnes ont accepté de répondre à l'enquête tout en sachant que le don est actuellement effectué dans les conditions garantissant l'anonymat. De plus, le contexte varie suivant le profil des donneurs entre Paris et la province.
Lors du changement de la loi, il faudra ne pas ignorer la situation constatée dans tous les pays ayant modifié les conditions du don. Les propositions sont moins nombreuses durant une certaine période, puis une hausse est ensuite observée, mais le nombre de dons n'est pas plus important qu'avant la modification de la loi. En effet, il est très compliqué de recruter les donneurs et ces pays importent des paillettes. Nous ne souhaitons pas rencontrer cette situation. Il serait regrettable d'aboutir à un système faisant appel à des structures qui ne respectent pas les mêmes règles éthiques.
Par ailleurs, vous avez soulevé la question de la conservation des échantillons des patients pour la préservation de la fertilité. Nous transmettons à l'Agence de la biomédecine les informations relatives au nombre d'embryons conservés et à leur devenir, soit le maintien du projet parental, le don pour la recherche, la destruction ou le maintien de la conservation.
En revanche, nous ne transmettons pas de données relatives à la conservation des gamètes et des tissus germinaux. La mise en oeuvre du dispositif relatif aux tissus germinaux date du début des années 2000. Nous allons peut-être pouvoir les conserver durant au moins quarante ans, en fonction de la date d'ouverture du centre. Pour l'instant, nous n'avons pas la possibilité de détruire les échantillons lorsque le patient ne répond plus. Le fait de limiter la durée de conservation et de pouvoir les détruire si les patients ne répondent plus pendant dix ans est une bonne chose. Nous verrons si des réajustements sont nécessaires.
En fonction de nos activités, environ 10 % des échantillons sont destinés à la recherche, contre 20 % d'échantillons pour lesquels nous pouvons envisager la destruction. La question de la requalification vers le don pourra se poser pour les conservations hors indications médicales.
Pr Catherine Guillemain . - La très grande majorité des échantillons actuellement conservés concerne des patients atteints de pathologies graves qui reçoivent un traitement gonadotoxique et ont autoconservé leurs gamètes pour en prévenir les risques. La plupart de ces pathologies sont des pathologies malignes. Certains patients ne souhaitant plus utiliser à l'issue de quelques années les paillettes conservées souhaiteraient les donner à des couples infertiles. Toutefois, en vertu du principe de précaution, nous ne sommes généralement pas en mesure de les accepter au titre du don, car ces patients sont porteurs d'une pathologie maligne induisant un risque potentiel de susceptibilité génétique.
La question peut se poser si nous évoluons vers une conservation pour des indications non médicales. Un argument consiste à affirmer qu'au cas où certaines personnes ne souhaitant plus utiliser les gamètes conservés, cela bénéficiera au don de gamètes. Nous serions alors dans une situation de don de seconde intention. Actuellement, lorsqu'une personne souhaite une conservation de gamètes pour elle-même, un certain nombre d'examens sont nécessaires à la constitution du dossier.
Les candidats au don, pour leur part, doivent réaliser des examens supplémentaires, notamment un caryotype. Lorsqu'une personne souhaite après de nombreuses années que ses gamètes conservés servent dans le cadre du don, elle peut signer un consentement en ce sens, mais cela ne peut suffire. Cette personne doit accepter d'effectuer des tests supplémentaires. En pratique, lorsque les embryons sont conservés dans le cadre d'une AMP intraconjugale et qu'un souhait de don est exprimé de nombreuses années plus tard, certaines personnes y renoncent, car elles n'ont pas envie d'effectuer ces examens supplémentaires. Nous pensons donc que le don de seconde intention peut être un leurre.
De plus, certaines sérologies ne font pas partie du bilan d'autoconservation, mais sont obligatoires pour le don. Prenons le cas d'une personne ayant conservé des ovocytes décide ultérieurement de les donner et si les examens de sérologie montrent qu'elle a été en contact avec le CMV. Nous ne pourrons pas connaître la date de sa mise en contact avec le virus. Il est parfois possible de tester les gamètes conservés, mais cela implique de détruire les paillettes. La réalisation immédiate d'un bilan biologique plus large pour l'ensemble des conservations apportait une réponse à cette problématique. Toutefois, la mise en oeuvre systématique d'un caryotype ne serait pas forcément simple.
Dr Patrice Clément . - L'argument selon lequel les centres privés ne pourront pas procéder à l'autoconservation dans le cadre de l'article L. 2141-12 en raison de cette finalité de don a été avancé lors des discussions à l'Assemblée nationale. En pratique, cet argument est erroné. Nous réalisons déjà ce type d'activité et menons des échanges avec les centres publics pour l'accueil d'embryons. La problématique serait donc gérable. Cependant, le don de seconde intention serait, comme cela vient d'être souligné, compliqué.
Pr Nelly Achour-Frydman . - Je pense que la sensibilisation des femmes pourrait intervenir lors de l'autoconservation. Pour autant, cette gestion serait complexe, particulièrement si les patientes sont perdues de vue et ne peuvent pas revenir pour la réalisation des examens. La prescription des examens à toutes les femmes semble peu probable. Je soutiens d'ailleurs les propos du Docteur Clément. Il est possible de conclure des accords avec les laboratoires privés qui conservent des gamètes pour réorienter les éventuelles donneuses d'ovocytes vers les centres agréés pour le don.
Professeur Rachel Levy . - Vous nous avez interrogés sur les dispositions relatives à la recherche du consentement pour la conservation des embryons et leur utilisation à d'autres fins qu'un projet parental, et notamment sur la confirmation à trois mois. Celle-ci est extrêmement lourde pour les couples et chronophage pour les équipes des centres, compte tenu du manque de réponses et de la nécessité d'envoyer des lettres recommandées aux personnes concernées.
Lorsque nous obtenons cette confirmation à trois mois, nous constatons une concordance quasiment totale des réponses avec le choix initial. En cas de non-réponse à la confirmation à trois mois alors que nous avions obtenu une réponse lors de la première relance, les embryons sont considérés comme perdus de vue et seront détruits dans un délai de cinq ans. Cela soulève une question éthique puisque, dans certains cas, la volonté initiale du couple n'est pas respectée ou reportée à cinq ans. Cette relance à trois mois nous paraît donc inutile. Nous proposons sa suppression.
Le deuxième point concerne les capacités de stockage des centres d'AMP, qui est déjà mise à mal. En termes de surface disponible, de nombreux centres n'ont pas la capacité d'assurer à la fois la conservation des prélèvements déjà confiés et ceux qui vont l'être dans le cadre de la loi de bioéthique. Malgré des investissements conséquents, nous arrivons à un seuil de saturation. Il est envisageable de lever l'obligation de conservation sur le site où a été effectuée la congélation. La conservation serait placée sous la responsabilité de l'équipe, mais à distance.
M. Alain Milon , président . - Les chercheurs spécialisés dans les cellules-souches embryonnaires et les embryons, notamment le Professeur Marc Peschanski, nous demandent de préciser la distinction entre l'embryon qui part à la recherche et l'embryon qui part en AMP et de considérer la notion d'embryon et de préembryon, qui existe dans d'autres pays. Quel est votre point de vue ?
Pr Nelly Achour-Frydman . - Vous abordez une question sémantique. L'embryon issu d'une fécondation in vitro est conservé ou cultivé jusqu'au cinquième jour dans le cadre du soin. Sur le plan technique, nous ne savons pas aller plus loin. Nous l'appelons « embryon préimplantatoire ». Je ne pense pas que le changement de nom de l'embryon peut permettre à certains de mieux accepter la recherche ou le diagnostic préimplantatoire.
M. Alain Milon , président . - À combien de cellules ces cinq jours correspondent-ils ?
Pr Nelly Achour-Frydman . - À cinq jours, le blastocyste comprend une centaine de cellules. Les cellules qui formeront le placenta, le trophectoderme et la masse cellulaire interne sont déjà différenciées.
Dr Patrice Clément . - 21 000 embryons sont donnés à la recherche. Le nombre d'utilisations est bien moindre.
Pr Nelly Achour-Frydman . - Parmi ces 21 000 embryons, nous ignorons quel sera le nombre de confirmations à trois mois. Il faut donc prendre ce chiffre avec beaucoup de précautions.
Pr Nathalie Rives . - Cette confirmation à trois mois est imposée depuis deux ans pour la conservation des gamètes et tissus germinaux. Le nombre de patients et patientes à recontacter est presque dix fois supérieur au nombre de couples à recontacter dans le cas de la conservation d'embryons en vue d'un projet parental ou d'un autre type de projet. L'évolution n'apporterait rien, mais les professionnels de santé devraient effectuer des tâches administratives supplémentaires.
- Présidence de Mme Catherine Deroche -
Mme Michelle Meunier . - Nous vous remercions d'avoir abordé ces nombreux sujets. Je souhaite notamment remercier le Professeur Achour-Frydman pour la clarté de ses réponses sur l'AMP post mortem ainsi que le Docteur Clément pour son intervention relative aux relations entre le secteur public et le secteur privé. Je souhaite poser une question au CECOS au sujet de l'allongement de la durée de conservation des gamètes. J'ai entendu parler d'une conservation pendant quarante ans. Par ailleurs, le don personnalisé est autorisé en Belgique, mais cela n'est pas le cas en France. Il semble que le gouvernement ait maintenu son positionnement à l'Assemblée Nationale. Quel est votre avis ? Chez les couples de femmes, il me semble intéressant de proposer un don d'ovocytes lorsqu'une femme ne veut pas porter l'enfant.
Pr Nathalie Rives . - Les délais de conservation des tissus germinaux par rapport au moment de l'utilisation peuvent être beaucoup plus longs que ceux des gamètes, des ovocytes ou des spermatozoïdes. Cela pose la question de la pérennité de cette conservation au regard de la transmission des données d'une génération à une autre et des conditions de stockage. La multiplication des centres de stockage nous interroge sur la nécessité d'une centralisation permettant de renforcer les garanties de sécurité à très long terme, car nous ne pourrons pas augmenter indéfiniment le volume des salles. S'agissant des spermatozoïdes et des ovocytes, la mise en place de règles de limites de conservation si les hommes ou les femmes ne répondent pas à dix ans permettra une meilleure gestion de la situation.
Dans le cas de la conservation hors indications médicales, ce délai de dix ans me paraît beaucoup trop long. L'âge à partir duquel ce dispositif sera autorisé serait 32 ans. Dans le cas d'une démarche volontaire de conservation des gamètes hors indications médicales, je pense qu'il faudrait arrêter la conservation en l'absence de réponse pendant cinq ans. Je pense qu'il est nécessaire de différencier la préservation de la fertilité de la conservation hors indications médicales.
Par ailleurs, je ne pense pas qu'il faille aborder la question du don dirigé au sein de couples de femmes. Lorsqu'un couple hétérosexuel conçoit seul ses enfants, les spermatozoïdes du conjoint donnés à la conjointe ne constituent pas un don intraconjugal. Chez un couple de femmes, imaginons que la femme souhaitant porter l'enfant présente une insuffisance ovarienne et soit infertile et que sa compagne ne souhaite pas porter l'enfant. L'utilisation des ovocytes pour que l'autre conjointe porte l'enfant ne serait pas un don, mais une conception en contexte intraconjugal, grâce à des gamètes qui ne sont pas les spermatozoïdes et l'ovocyte du couple. Le texte de loi actuel ne l'empêche pas, puisqu'il ne s'agit pas d'un don.
Pr Catherine Guillemain . - Le don dirigé concernant un couple qui souhaiterait recevoir les gamètes d'une personne qu'il connaît a déjà eu lieu en France, même avant la promulgation de la première loi de bioéthique. Les professionnels impliqués dans ces pratiques n'étaient pas favorables à ce type de prise en charge, plus ou moins bien perçue au sein des familles. De plus, l'implication des personnes concernées sur le moyen et long terme est difficilement prévisible au moment de la prise en charge.
Pr Nathalie Rives . - Une collègue de la Fédération des CECOS souligne qu'il serait créé une différence entre les couples recourant à un don anonyme et les couples qui auront recours au don dirigé. Cette notion ne serait pas respectée.
Pr Nelly Achour-Frydman . - Ce cas soulève la question de la levée d'anonymat, qui n'est pas prévue dans l'esprit de la loi. Il convient également de rappeler la notion de dette entre la donneuse et le couple receveur. Cette situation est souvent comparée avec le don d'organe, mais il n'y a pas d'enfant issu du don d'organe. L'enfant issu du don sera élevé par un couple et la troisième personne peut peser sur la vie de cette famille.
Ce point de l'ordre du jour a fait l'objet d'une captation vidéo qui est disponible en ligne sur le site du Sénat .
Mme Cécile Martinat,
présidente
de la Société française de recherche
sur les cellules souches,
et M. Marc Peschanski, directeur de l'Institut
des cellules souches
pour le traitement et l'étude des malades
monogénétiques (I-Stem)
M. Alain Milon , président . - Mes chers collègues, nous poursuivons nos travaux sur le projet de loi relatif à la bioéthique avec une audition consacrée à la recherche sur l'embryon, les cellules souches embryonnaires et les cellules souches pluripotentes induites.
J'indique que cette audition fait l'objet d'une captation vidéo retransmise en direct sur le site du Sénat et consultable à la demande.
Nous recevons ce matin Mme Cécile Martinat, présidente de la Société française de recherche sur les cellules souches et M. Marc Peschanski, directeur scientifique de l'Institut des cellules souches pour le traitement et l'étude des malades monogénétiques.
Je souhaite par ailleurs la bienvenue aux étudiants de M. Pierre Calvel d'AgroParisTech que j'ai convié, il y a déjà de nombreuses semaines, à assister à nos travaux.
Je laisse la parole à nos invités pour un bref propos liminaire avant que nos rapporteurs n'interviennent, puis les commissaires qui le souhaiteront.
Mme Cécile Martinat, présidente de la Société française de recherche sur les cellules souches (FSSCR) . - Madame et Messieurs les Sénateurs, Mesdames et Messieurs, je vous remercie beaucoup pour cette invitation.
Je vais revenir en guise d'introduction sur les cellules souches embryonnaires humaines. Elles sont issues d'embryons obtenus après cinq à sept jours de fécondation in vitro. Elles sont caractérisées par leurs capacités d'autorenouvellement et de pluripotence. Elles ont été dérivées pour la première fois il y a plus d'une vingtaine d'années. Depuis, un cap a été franchi quant à leur utilisation, en particulier dans les domaines biomédicaux. Il existe à ce jour une vingtaine d'essais cliniques internationaux portant sur les dérivés de cellules souches embryonnaires, principalement pour des programmes de médecine régénératrice.
Depuis 2007, le professeur Yamanaka a identifié un procédé permettant de convertir des cellules adultes dans d'autres cellules présentant des caractéristiques identiques aux cellules souches embryonnaires humaines.
Il est nécessaire de distinguer le fait que ces cellules souches induites à la pluripotence sont différentes des cellules souches embryonnaires humaines, du fait d'un phénomène de reprogrammation. La régulation d'expression des gènes est modifiée.
En revenant sur les processus de révision de la loi de bioéthique, nous pouvons, en tant que citoyens, nous féliciter que la loi prenne en compte les évolutions scientifiques et technologiques. Nous pouvons tout de même regretter que le texte de loi de 2013, qui aurait dû nous laisser une certaine flexibilité quant à l'utilisation des cellules, n'ait été que source de problèmes. Nos programmes de recherche se sont vu attaquer en justice auprès de l'Agence de la biomédecine.
A ce stade, environ 50 % de programmes de recherche font encore l'objet d'un processus juridique. C'est ce constat qui a donné lieu à la création de la Société française de recherche sur les cellules souches en 2017. Elle compte plus de 450 membres actifs, scientifiques et cliniciens, couvrant la recherche fondamentale et appliquée. Les domaines traités sont vastes : cellules souches embryonnaires humaines, mésenchymateuses, mais aussi cancéreuses. Notre objectif était de nous faire entendre dans le cadre de la révision sur la loi de bioéthique. Nos revendications à ce sujet étaient triples : une distinction doit être opérée entre un embryon et des cellules souches embryonnaires ; les textes de loi doivent être adaptés, car ils sont trop limitants ; le troisième point porte sur la recherche sur l'embryon. Je ne l'aborderai pas aujourd'hui.
En tant que scientifiques, le fait de passer d'un système d'autorisation à un système déclaratif nous réjouit. Nous espérons vraiment qu'il nous protégera contre ces attaques juridiques.
Nous soulignons l'existence de prérequis, et en particulier le terme de « finalité médicale ». Il est limitant, restrictif et ne prend pas en compte le continuum de la recherche. Un « agrandissement des connaissances de la biologie chez l'homme et de l'amélioration de sa santé » serait plus adapté.
M. Marc Peschanski, directeur scientifique de l'Institut cellules souches pour le traitement et l'étude des malades monogénétiques (I-Stem) . - Bonjour à tous. Je vais aujourd'hui vous parler d'avenir. A ce titre, je dois d'abord mentionner la façon dont les scientifiques français ont abordé l'exploitation des cellules souches embryonnaires. L'angle abordé était à l'époque thérapeutique.
L'I-Stem s'est orienté de façon déterminée vers l'utilisation thérapeutique. Nous avons promis que nous allions travailler sur les possibilités d'exploitation des cellules souches embryonnaires à des fins thérapeutiques. De nombreux opposants ont discuté ces axes. Ils se sont trompés. Nous avons non seulement des programmes de recherche sur de nouvelles thérapies, mais surtout des résultats.
Nous avons exploré deux axes de recherche en particulier. Nous avons tout d'abord travaillé à partir de cellules souches dérivées d'embryons rejetés lors d'un diagnostic préimplantatoire. Les médecins donnent la possibilité à des parents ayant eu un enfant atteint d'une maladie génétique extrêmement grave d'avoir un bébé ne portant pas le gène muté responsable de cette maladie. Les embryons porteurs de la mutation sont donc éliminés. Nous nous sommes associés à des centres permettant d'obtenir des cellules souches embryonnaires porteuses des mutations. Nous les avons étudiées et y avons identifié les anomalies liées à ces mutations afin de les utiliser comme marqueur. Nous avons ensuite testé des composés pour étudier leurs éventuels effets sur les cellules. Après 15 ans de recherche, nous avons publié l'année dernière les résultats de l'essai clinique sur la maladie de Steinert. Il a démontré que la metformine, médicament antidiabétique, permettait aux malades de gagner 10 à 15 % de périmètre de marche. D'autres molécules ont montré des effets positifs sur deux patientes atteintes d'un autisme sévère. Nous ouvrons aujourd'hui la voie pour des maladies génétiques sur lesquelles nous n'avions précédemment aucun impact.
Ce travail donne lieu à des contacts de plus en plus étroits avec des industriels, intéressés par notre approche. Il ne s'agit pas pour eux d'un repositionnement, mais d'un travail vers de nouvelles entités chimiques. L'usage dépasse la recherche et devient du développement.
La thérapie cellulaire est un sujet de recherche plus connu, puisqu'elle bénéficie des faveurs de la médiatisation. Il s'agit d'une greffe de cellules dans un organe qui en a perdu. C'est d'ailleurs l'objectif le plus courant des scientifiques travaillant sur les cellules souches embryonnaires. Les premiers travaux cliniques ont débuté dès 2010 aux Etats-Unis et en Angleterre. A partir de 2014, des travaux ont été menés en France par Philippe Menasché. Ils portaient sur le traitement de pathologies liées à l'infarctus du myocarde. Les résultats lui ont permis de développer et améliorer son approche.
Je vous annonce aujourd'hui que nous avons lancé un essai clinique auprès de patients atteints de rétinites pigmentaires. Il s'agit d'une dégénérescence de cellules au fond de la rétine, que nous remplaçons par des cellules entièrement construites en laboratoire. Nous avons d'ores et déjà implanté deux patients, dix autres sont en attente pour l'année à venir. L'essai vise à retrouver de la vision chez des patients en train d'évoluer vers la cécité.
Quarante-deux essais cliniques sont dénombrés. Nous ne sommes plus dans les frémissements et dans les balbutiements, mais sommes entrés dans la phase d'application. Nous sommes à présent en lien avec des industriels qui y consacrent des moyens de plus en plus importants.
J'avais interrogé il y a quelques mois votre commission sur la façon dont les parlementaires pouvaient anticiper ce qu'il se passera dans les cinq à sept années à venir. En effet, nous changeons de registre puisque nos résultats le permettent et que des industriels s'en saisissent. Nous allons donc passer de la recherche à la médecine durant cette période. Les industriels préparent cette médecine pour des centaines de milliers de malades, et plus pour quelques dizaines de patients atteints de maladies rares. Nous allons vers une industrialisation, ce qui demande des investissements considérables.
Ces investissements ne doivent pas être handicapés par des lois les remettant en cause. La loi, concernant aujourd'hui la recherche, doit anticiper l'avenir du domaine médical. La France est en retard en termes d'infrastructures pour la bioproduction. Nous ne devons pas rater le virage industriel. Nous devons à ce titre trouver des modalités juridiques leur permettant de travailler.
Mme Corinne Imbert , rapporteure . - Merci pour votre exposé intéressant.
J'ai cru comprendre que dans le texte présenté aujourd'hui au Sénat, le régime de déclaration préalable pour les recherches sur les cellules souches embryonnaires vous satisfaisait. Vous considérez qu'il freinera les attaques potentielles sur les prochains projets. Pouvez-vous nous rappeler sur quels arguments se basent ces attaques dont font l'objet les programmes de recherche ? Nous les connaissons déjà, mais il serait intéressant de les repréciser à nos auditeurs.
Vous avez évoqué le fait que le pays a quelques années de retard sur certains sujets. Quelle place la France occupe-t-elle aujourd'hui en matière de recherche sur les cellules souches embryonnaires ? Comment exprimer cette nécessaire anticipation au travers du texte de loi ? Pouvez-vous nous apporter, au travers de votre expertise, des éléments permettant d'améliorer la loi dans ce sens ?
M. Bernard Jomier , rapporteur . - Je vous remercie de nous avoir rappelé que vos recherches produisaient du mieux-être, de la lutte contre des pathologies rares et incurables pour la plupart. L'encadrement en la matière est consubstantiel à vos recherches.
Vous estimez que les cellules souches reprogrammées en cellules pluripotentes ne sont pas réellement une alternative à la recherche sur les cellules souches embryonnaires. Pouvez-vous développer ce point ?
Ma deuxième question porte sur l'encadrement auquel nous devons procéder. Doit-il selon vous porter sur l'origine des cellules ou sur leur capacité ? Je pense notamment aux cellules pluripotentes qu'il est possible de faire évoluer et différencier vers des cellules de type germinal.
Pourriez-vous nous éclairer sur les perspectives de la recherche sur la transformation en tissus et en organes, donc sur la thérapie cellulaire ? La problématique la plus dense à ce sujet porte actuellement sur les reins.
Enfin, le texte transmis de l'Assemblée nationale constitue-t-il à vos yeux un cadrage suffisant pour la poursuite de vos recherches ?
M. Alain Milon , président . - Vers qui doit-on orienter un patient qui souffre d'une rétinite pigmentaire avancée ?
M. Marc Peschanski . - Ces patients sont à orienter vers le docteur Mohand-Saïd, ophtalmologue au CIC des Quinze-Vingts. Les rétinites pigmentaires auxquelles nous nous attaquons aujourd'hui touchent principalement l'épithélium pigmentaire rétinien. Certaines mutations sont spécifiques, d'autres s'étendent à d'autres populations cellulaires. Elles peuvent toucher les photorécepteurs. Notre capacité de thérapie cellulaire n'est à ce jour pas adaptée à leur remplacement. Nous travaillons assidûment sur le sujet afin de renforcer nos approches.
Aujourd'hui, trois grandes mutations nous concernent : RPE65, MRTK et LRAT qui sont toutes trois spécifiques à l'épithélium pigmentaire rétinien.
Mme Cécile Martinat . - Je vais rappeler les motifs des attaques dont nous faisons l'objet pour notre auditoire plus jeune, que je remercie d'ailleurs d'être présent.
Avant les lois de 2011 et 2013, d'interdiction puis d'autorisation, trois prérequis étaient indispensables à l'obtention d'une autorisation : la finalité médicale, la pertinence scientifique et le fait qu'on ne puisse pas effectuer les travaux de recherche avec une autre source de matériel. La situation s'est complexifiée lorsque les cellules souches induites à la pluripotence sont apparues.
Les motifs des attaques ont été multiples. En mars, deux autorisations de recherche portant sur la pertinence scientifique et sur la traçabilité ont été annulées par le tribunal de Versailles. Plus de la moitié des autorisations étant à l'heure actuelle en cours de processus juridique portent sur ces sujets. J'aimerais comprendre comment un juge peut déterminer la pertinence scientifique d'un projet. Avec le temps, les attaques nous visant ont sans cesse trouvé de nouveaux motifs. Elles concernaient au départ les sources alternatives, puis la traçabilité, et enfin les consentements. Les cellules utilisées sont « immortelles ». La majorité des laboratoires utilisent aujourd'hui des cellules embryonnaires humaines dérivées à la fin des années 1990, souvent dans les pays de l'Est. Le consentement à l'époque ne correspondait pas forcément aux prérequis actuels en la matière.
Pour répondre sur la place de la France dans le domaine de la recherche sur les cellules souches embryonnaires humaines, il est évident que nous avons pris du retard. Il s'agit d'ailleurs d'un des motifs de création de notre société savante. Nous l'avons lancée suite à une certaine lassitude de voir nos programmes attaqués. De plus, un congrès international regroupe tous les experts internationaux sur les cellules souches, en particulier pluripotentes. Nous avons remarqué que la France n'apparaissait même pas dans le top 20 des pays les plus représentés lors de celui-ci en 2017. Nous avons donc décidé de fédérer notre communauté scientifique, ce qui a fonctionné puisqu'en 2018, nous apparaissions enfin dans le top 15.
Nous n'avons pas abordé le problème de la lourdeur administrative et des délais des autorisations qui conduisent beaucoup d'équipes à se limiter dans l'utilisation des cellules souches embryonnaires humaines. Nous ne pouvons qu'espérer pouvoir aller beaucoup plus vite et franchir d'autres barrières à l'avenir. Malgré notre retard, notamment par rapport aux Etats-Unis et à l'Angleterre, nous avons tout de même montré une certaine excellence scientifique, et nous nous en réjouissons.
Je considère que les IPS ne sont pas une source alternative de matériel aux cellules souches embryonnaires humaines. Les cellules souches induites à la pluripotence sont obtenues par modification génétique de cellules adultes. Nous devons pouvoir les comparer aux cellules souches physiologiques. Des exemples montrent que l'utilisation de ces cellules dans le cadre de l'étude de certaines maladies pourrait présenter des défauts liés à la reprogrammation. Elles ne sont donc pas forcément l'outil le plus pertinent. Des disparités existent toujours et il est nécessaire de continuer à creuser cette question d'un point de vue scientifique.
En ce qui concerne le texte de l'Assemblée nationale, je crois pouvoir dire que celui-ci est plus intelligent et plus raisonnable que le premier. Nous pouvons nous réjouir en France du système permettant de revoir régulièrement cette loi. De nouvelles évolutions apparaîtront dans les années à venir, telles que la technologie CRISPR ou les embryons chimériques et synthétiques. Nous devons permettre aux scientifiques de travailler sur ces notions de sorte à ne pas prendre de retard.
M. Alain Milon , président . - Je voudrais adresser aux jeunes présents dans l'auditoire un message publicitaire sur l'utilité du Sénat. Je vous rappelle que l'autorisation de recherche sur les cellules souches embryonnaires vient du Sénat. C'est en outre le Sénat qui a imposé la révision de la loi bioéthique tous les 7 ans. Il semblerait que l'Assemblée nationale soit cette année plus encline à accepter une révision tous les cinq ans, ce que nous demandions au départ.
- Présidence de Mme Elisabeth Doineau, vice-présidente -
M. Marc Peschanski . - Je salue le travail que vous avez fait à l'époque, qui a largement bénéficié aux scientifiques, bien que les résultats n'aient pas été aussi bénéfiques qu'espérés. Il était très important de montrer que l'avis n'était pas unanime dans la représentation nationale.
Je souhaite compléter les propos de Mme Martinat au sujet de la place de la France dans les cellules souches. Lorsque nous avons créé l'I-Stem, lorsque les chercheurs ont pu commencer à travailler sur le sujet, personne en France ne savait manipuler ces cellules puisque c'était jusqu'alors interdit. Cette manipulation demande une pratique et une compétence sophistiquées. J'ai donc cherché à l'étranger des scientifiques acceptant de venir construire cet institut avec moi. Nous avons, grâce à l'appui du directeur général de l'INSERM, pu accueillir des post-doctorants au sein de son établissement ou de l'université d'Evry. Ce retard de 7 ans a été quelque peu compensé par l'utilisation des compétences acquises à l'étranger. Nous avons toutefois été limités par le manque de moyens. Le développement de la recherche a été freiné par l'exclusion des cellules souches embryonnaires de certains appels d'offres. L'Agence de la biomédecine les exclut par exemple de ses financements.
L'I-Stem a eu la chance d'être soutenu par le Téléthon. Cependant, un déploiement d'équipes sur le territoire n'a pas été permis, contrairement à la Grande-Bretagne qui a compté une quarantaine d'équipes dès 2006. La place de la France a été la conséquence de ce retard et de l'absence de financement spécifique.
Nous progressons dans les classements grâce à notre choix de cette orientation thérapeutique, appuyée sur des travaux fondamentaux réalisés à l'étranger. Nous les avons adaptés dans une recherche translationnelle aboutissant à des essais cliniques et des connexions industrielles. En regardant la carte des essais cliniques fondés sur les cellules souches embryonnaires, l'Europe n'est pas en pointe sur ces sujets. Elle compte une demi-douzaine d'essais en Grande-Bretagne, en Israël et en France. La structuration des centres d'investigation clinique opérée à la fin des années 1990 par l'INSERM en commun avec les hôpitaux joue un rôle majeur dans le cadre de la recherche translationnelle. Cette structuration n'existe pas de la même façon dans les autres pays européens. Elle nous apporte un énorme bénéfice pour faire passer les résultats d'une recherche de laboratoire dans la recherche clinique.
Concernant les changements de la loi, demander à des chercheurs en biologie d'avoir une finalité médicale revient à leur demander d'abandonner des pans entiers de la recherche fondamentale, ou de mentir.
Les chercheurs ayant découvert la technique CRISPR, des ciseaux qui coupent l'ADN, travaillaient sur la fermentation des bactéries. Son utilisation a commencé dans des laboratoires de recherche sur des cellules humaines avant de passer à la thérapeutique. Il s'agit du parcours permanent de notre recherche biologique, qui n'est pas unidirectionnel.
Depuis des années, cette finalité médicale oblige les chercheurs à écrire des choses sur lesquelles ils n'ont aucune certitude. Nos opposants nous attaquent sur des présentations ne pouvant être solides puisque les résultats de nos recherches sont toujours des paris faits par les scientifiques.
Ensuite, dans le cadre du changement de la loi, le fait de passer d'un système d'autorisation préalable à un système de déclaration correspond à la façon dont nous travaillons dans nos laboratoires. Lorsque nous travaillons sur des agents radioactifs, nous le faisons dans un cadre réglementaire et pas géré par une loi bioéthique de la radioactivité. Cette réglementation passe par une déclaration et une formation des scientifiques, ainsi que la restriction de l'utilisation de ces agents au domaine de la recherche.
Nous connaissons le cadre réglementaire et le système déclaratif et savons qu'il ne va pas nous ralentir. La demande d'autorisation préalable, au contraire, nous freine en nécessitant quatre mois de gestion par l'Agence de la biomédecine, qui n'ouvre cette possibilité que deux ou trois fois par an. Elle peut aboutir à des échecs par compétition d'autres équipes.
En ce qui concerne les cellules IPS par rapport aux cellules souches embryonnaires humaines, je voudrais juste dire quelque chose qui paraît banal. Lorsque vous avez le choix entre un maïs transgénique et un maïs bio, si le prix est le même, vous aurez tendance à privilégier le second. Nous considérons tous spontanément que ce qu'a produit la nature est meilleur. Quand on modifie le vivant, on ne sait jamais exactement ce que cela va donner. C'est la même chose avec les IPS. En 2007, nous les avons qualifiées de cellules repoussées à la case départ. Dès les mois qui ont suivi, les équipes ont modéré cette affirmation. Nous connaissons de mieux en mieux les éléments qui les différencient. De même que Monsanto observe la disparition d'abeilles après utilisation de son maïs transgénique, nous observons des dégâts collatéraux imprévus par les manipulations génétiques.
Aujourd'hui, les perspectives de la recherche tissulaire et en organes sont l'avenir. La construction d'organe est une des grandes pistes sur laquelle les équipes sont toutes engagées. J'ai abordé précédemment la modification de l'épithélium pigmentaire rétinien. L'équipe de Christelle Monville est en train de travailler sur un moyen de créer deux couches entre l'épithélium et les photorécepteurs. Cette technique permettra la création d'un tissu plus accessible pour les patients ne possédant pas les mutations spécifiques. Nous avons mis au point le traitement du syndrome de Phelan-McDermid à partir de cellules corticales cultivées en deux dimensions. Nous laissons aujourd'hui se développer les différentes couches du cortex pour regarder les interactions cellulaires entre celles-ci. Nous pouvons alors remarquer les altérations apparaissant au cours du développement du cerveau de certains autistes. Cette technique permet de voir comment ces désorganisations d'interactions cellulaires peuvent éventuellement être compensées par certaines approches thérapeutiques.
A I-Stem, nous ne travaillons pas sur le rein, mais collaborons avec une équipe hollandaise spécialisée sur cette question. Il est spectaculaire de voir se développer des glomérules et des tubules. Elles donnent accès à des désordres que nous ne pouvions pas observer jusqu'alors. Personne n'a à ce jour trouvé la façon de construire l'architecture rénale manquante, mais nous en approchons. L'étape suivante consistera à combiner nos cellules avec des moules en biomatériaux pour recréer un organe.
Les pistes d'ingénierie et de chirurgie du génome sont les prochaines pistes à suivre, grâce à CRISPR. Cette technique est très facile à introduire dans les laboratoires et permet de plus en plus d'avancées. Il était auparavant possible d'effectuer des coupures très précises dans le génome. Nous avons ensuite pu y insérer des morceaux d'ADN très précis. Par la suite, nous avons pu le faire au niveau d'une seule base sur les 6 milliards de bases. Les derniers travaux, publiés il y a quelques semaines à peine, ont montré que des chercheurs ont réussi à modifier le système CRISPR. Il peut ne plus couper l'ADN, mais simplement permettre la modification d'une base. C'est un élément extrêmement important pour la sécurité, empêchant des coupures imprévues.
Je répète que la loi de bioéthique concerne aujourd'hui la recherche sur l'embryon et les cellules embryonnaires. Nous espérons qu'elle aboutira à une réglementation à ce sujet. Nous devons commencer à réfléchir à l'application, à l'industrialisation et au développement des traitements. La présence d'industriels dans une salle comme celle-ci est indispensable pour qu'ils puissent exprimer leurs besoins, leurs perspectives, et leur optimisme sur ces différentes pistes. Les chercheurs peuvent apporter un certain nombre d'éléments, mais ils ne sont pas les seuls.
Mme Élisabeth Doineau , vice-présidente . - Je me tourne vers l'ensemble des sénateurs présents.
Mme Laurence Cohen . - Merci pour ce riche exposé.
Il me semble important de distinguer la recherche sur l'embryon de celle sur les cellules souches embryonnaires. J'ai l'impression que les questions éthiques ne se posent pas de la même manière sur ces deux cas.
La limite de la culture des embryons à des fins de recherche était auparavant de sept jours. Elle est passée à quatorze jours dans la proposition de loi, en prenant exemple sur les pays voisins. Pensez-vous que ce chiffre est positif et suffisant ?
De plus, vous parlez du virage industriel à ne pas rater et des moyens à investir. J'ai fait un parallèle avec les moyens insuffisants accordés à la recherche. Je plaide personnellement pour la création d'un pôle public du médicament et de la recherche. Je me suis interrogée sur les garde-fous à mettre en place. Ne serait-il pas nécessaire de mettre en place une charte éthique pour éviter les dérives ?
Mme Michelle Meunier . - J'aimerais demander à Mme Martinat : qui sont vos détracteurs ? Vous avez abordé les motifs des attaques, mais pas leur source. Sont-ils des professionnels, des collègues, des confrères ?
Je souhaite également obtenir votre éclairage sur le « bébé-médicament », enlevé du texte de l'Assemblée nationale.
Mme Cécile Martinat . - Nous nous félicitons que la proposition du texte de loi prenne en compte la distinction entre l'embryon et les cellules souches embryonnaires. Il s'agissait, au niveau de la FSSCR, du point d'ancrage pour commencer à travailler un texte de loi sur les cellules souches embryonnaires humaines.
Je ne travaille pas sur l'embryon, et considère donc que je n'ai pas les connaissances scientifiques nécessaires pour en parler. Pierre Sabatier a dû aborder cette problématique. Si je suis les propos de mes collègues de la société savante, nous regrettons cette limitation à quatorze jours. Deux arguments ont été utilisés pour placer cette limite. La première raison est purement philosophique. La seconde est scientifique : à partir de quatorze jours, nous commençons à voir apparaître des structures neuroectodermiques qui sont les prémices de l'élaboration du cerveau. Il a donc été considéré que l'apparition de la conscience commençait à ce stade, ce qui n'a jamais été démontré. Étendre au-delà de ce délai nous permettrait de mieux comprendre les mécanismes expliquant et contrôler la mise en place de ces structures. Cette compréhension serait extrêmement importante, en particulier dans le cadre de la procréation médicalement assistée et l'infertilité. Beaucoup de femmes consomment ou ont consommé du Gynéfam puisqu'il contient de l'acide rétinoïque, contribuant à la mise en place de ces premières structures.
Continuer la recherche sur ces structures devrait nous permettre de mieux appréhender les mécanismes.
M. Marc Peschanski . - La loi de bioéthique de 1994, interdisant tout travail de recherche sur l'embryon, a été suivie d'un arrêt de tout travail pour améliorer la fécondation in vitro. La France, qui était un pays en pointe en termes de résultats de FIV, est alors tombée très bas dans les classements. Nos collègues demandent donc qu'il soit possible d'étendre le travail sur les embryons jusqu'à quatorze jours, voire au-delà. Dans le cadre de FIV et de grossesses en général surviennent malheureusement régulièrement des défauts de développement, de nidation ou d'interactions avec les structures utérines. Ils doivent pouvoir observer les cellules responsables de la nidation, qui leur échappent aujourd'hui.
Une loi de bioéthique doit éviter que les résultats obtenus par les chercheurs se traduisent par des dysfonctionnements dans leur application à la société, à l'espèce humaine et à toutes les espèces en général d'ailleurs. Les chercheurs ne peuvent travailler qu'à condition d'aller dans des terrains inconnus, qui ne peuvent donc pas être réglementés. Les connaissances apportées seront donc, ou non, appliquées à la société. C'est là que se situe la barrière à mettre en place.
Il ne faudrait pas que les révisions de lois de bioéthique périodiques continuent de nous freiner. La recherche doit être libre, réglementée et surtout qu'elle permette aux scientifiques de s'intéresser à des terrains inconnus, sans quoi ils ne pourront apporter de connaissances nouvelles.
Je partage l'avis de madame la Sénatrice sur le déficit financier de nos recherches, mais rappelle toutefois l'accord de Lisbonne de l'an 2000. Le gouvernement français s'était engagé à aboutir à 3 % de PIB dans la recherche en 10 ans. Nous sommes à 2,2 % en 2019, ce qui se traduit donc par un déclin qui continuera jusqu'à ce que les moyens nécessaires soient mis en oeuvre. L'Allemagne a démarré en 2000 avec 2,15 % du PIB consacré à la recherche, comme nous. Elle a dépassé les 3 % depuis 2010. J'en profite pour vous adresser ce message au nom de la communauté scientifique.
Au sujet du virage industriel, il est aujourd'hui indispensable que vous écoutiez les industriels. Vous travaillez aujourd'hui sur le passé de nos recherches. La loi de bioéthique est régulièrement révisée sur la base des connaissances que nous vous apportons. Il est maintenant utile de regarder vers l'avant et d'anticiper. Le Sénat était très en pointe en 2011 sur sa réflexion bioéthique dans ses choix et ses anticipations. Nous avons la possibilité d'être en avance sur la bioproduction cellulaire, et devons donc l'être. Nous nous dirigeons vers une nouvelle médecine régénératrice, recherchant des traitements sur la base de cellules développées en laboratoire.
Je pense qu'aujourd'hui la question pour les grands acteurs pharmaceutiques est de savoir s'ils vont se développer ici ou de l'autre côté de la frontière. En 2009, nous avons été contactés par Roche, un industriel souhaitant développer ses approches de médicaments, ce qu'il a fait avec succès. Ils avaient compris qu'ils pourraient trouver chez nous des développements introuvables ailleurs. Nous avons donc travaillé deux ans et demi ensemble, en formant leurs équipes et construisant des laboratoires. Nous leur avons proposé d'en construire un à Evry, ce qu'ils ont refusé du fait de leur incertitude par rapport à la loi de 2009. Ils ont eu peur de perdre leurs investissements dans les années suivantes. Nous l'avons donc construit à Bâle.
Mme Laurence Cohen . - N'y a-t-il pas besoin d'une charte éthique ?
M. Marc Peschanski . - Il est nécessaire de leur parler. Je ne peux pas m'exprimer à leur place. Je serais partisan d'une charte, ou en tout cas d'un engagement commun autour de ces développements. J'estime qu'ils seraient heureux de pouvoir le faire maintenant, et surtout d'avoir des certitudes sur l'environnement réglementaire et légal dans lequel ils veulent construire. Certains veulent construire en France.
Le bébé-médicament n'est pas mon domaine de recherche. La position que Cécile Martinat et moi pouvons vous donner est une position de citoyen. La possibilité d'accès à des cellules provenant du frère ou de la soeur d'un bébé atteint d'une leucémie, passant par un tri d'embryons, n'est pas un domaine de recherche. Nous ne sommes donc plus dans une loi de bioéthique s'appliquant à la recherche.
Mme Cécile Martinat . - Pour compléter ces propos, en tant que citoyenne et mère, je défends le point de vue de Marc Peschanski. J'ajoute que nous avons assisté dernièrement à une modification de génome avec les « bébés-CRISPR ». Nous sommes contre toute manipulation par modification génétique.
Ce n'est pas un secret, toutes les attaques émanent de la Fondation Jérôme Lejeune. L'Agence de la biomédecine a dû totalement reconvertir son activité sur la réponse à ces attaques juridiques. J'aimerais connaître le montant dépensé à ce titre par celle-ci, et donc par l'Etat. Nous avions cosigné en 2017 une tribune dans Le Monde dénonçant le fait que cette fondation était d'intérêt public.
Dans la mesure où cette possibilité est utile, je ne vois pas de raison de l'empêcher. La position pragmatique est de voir qu'aujourd'hui, le bébé-médicament n'a pas servi. Nous ne savons pas si ça sera le cas à l'avenir. Ce n'est pas du domaine dans lequel nous pouvons intervenir en tant que scientifiques.
Mme Marie Mercier . - En tant que médecin et mère, je comprends totalement vos propos sur ces avancées thérapeutiques ouvrant des horizons vertigineux et très positifs. Cependant, nous sommes des législateurs. C'est toute la rigueur du droit que nous devons trouver. La loi est pensée pour rendre la société meilleure. Il y a quinze ans, nous nous posions déjà la question suivante : la dignité humaine est-elle dans les gènes ? Mais les gènes de qui ? La réponse est bien entendu négative. Cette position d'équilibre que nous recherchons tous est quelque peu chimérique. Si nous ouvrons toutes les portes, nous ignorons comment certains pourraient les exploiter. Je partage l'avis de ma collègue Laurence Cohen sur la nécessité d'une charte éthique. Il est navrant de constater que ce laboratoire va s'installer de l'autre côté de la frontière. Ne pourrions-nous pas trouver une position « à la française » ? Nous avons en effet des lois qui sont peut-être trop contraignantes et qui freinent ces avancées et le développement de la recherche ? Ne pourrions-nous pas travailler avec les industriels de façon à promouvoir une recherche éthique, via la signature d'une charte, sur les cellules souches embryonnaires tout en nous garantissant d'usages que nous ne pourrions absolument pas partager ?
M. Marc Peschanski . - Il est nécessaire de différencier les pratiques dans les laboratoires de recherche et l'utilisation de leurs résultats. Je tiens beaucoup à la liberté de la recherche, car elle a pour finalité de rendre le monde meilleur en apportant des connaissances, des perspectives et des moyens nouveaux. Il appartient ensuite à la société de décider s'il convient d'exploiter ces connaissances, ces perspectives et ces moyens dans la perspective d'un monde meilleur. Je pense vraiment qu'il faut anticiper ce passage. La recherche est un monde inconnu dans lequel nous entrons progressivement. Nous avons aujourd'hui apporté des résultats, y compris en essais cliniques, permettant de voir que la médecine régénératrice ne relève plus de la science-fiction. De nouveaux acteurs entrent en jeu, entraînant de nouvelles discussions. Nous y assumerons notre rôle de scientifiques en vous apportant les éléments qui vous permettront de comprendre ce que les industriels veulent faire de nos résultats. Il faudra cependant tenir compte de la parole de ces industriels. En effet, lorsque nous réalisons un essai clinique chez douze patients atteints de rétinite pigmentaire, cela épuise à la fois nos ressources humaines et financières. Un tel essai peut amputer nos budgets d'un million d'euros. Nous pouvons fort heureusement compter sur le Téléthon, car l'INSERM ne pourrait pas assumer de tels coûts. Il est donc inenvisageable de se projeter sur 12 000, 120 000 ou 1,2 million de patients. Pour atteindre de telles masses, il faut changer de cadre et évoluer vers un modèle industriel qui implique des développements robotiques, la construction d'usines, la formation de centaines de nouveaux techniciens et ingénieurs. Nous jouerons notre rôle d'experts dans ce cadre, mais pas plus. Les industriels doivent être assurés qu'ils pourront travailler dans un cadre législatif et réglementaire qui leur garantira que leurs investissements et leurs efforts ne seront pas freinés après quelques années.
Mme Cécile Martinat . - J'ajoute que je me méfie toujours un peu des chartes éthiques. Au regard des résultats de la convention d'Oviedo, leur pertinence mérite d'être remise en question. Ce texte, s'il a été pertinent à une époque, ne l'est plus et devrait être renouvelé. Un certain nombre de pays, comme les Pays-Bas, ont d'ailleurs décidé de retirer leur signature, ce texte ne reflétant plus du tout les travaux qu'ils mènent. Il faut être vigilant à la mise en place de chartes, qui peuvent se transformer indirectement en des formes de verrous.
Il ne faut pas oublier que la recherche française est dépendante de tutelles. Ainsi l'INSERM est supervisé par un Comité d'éthique qui surveille les avancées de travaux, évalue leur pertinence et qui est en mesure de rappeler que certaines recherches ne sont pas autorisées. Ces comités institutionnels contribuent donc à la définition d'un cadre.
Ce point de l'ordre du jour a fait l'objet d'une captation vidéo qui est disponible en ligne sur le site du Sénat .
Pr
Israël Nisand, président du Collège national
des
gynécologues et obstétriciens français (CNGOF),
Pr
Jean-François Mattei, vice-président de l'Académie
nationale de médecine,
et Pr René Frydman, professeur
émérite des universités, gynécologue
obstétricien
M. Alain Milon , président . - Nous poursuivons nos travaux sur le projet de loi sur la bioéthique. Cette audition est consacrée à l'assistance médicale à la procréation. Nous examinerons jeudi les conséquences sur la filiation des évolutions proposées dans ce domaine par le projet de loi en auditionnant des juristes. Je vous informe par ailleurs que cette audition fait l'objet d'une captation retransmise sur internet.
Je laisse la parole aux professeurs Jean-François Mattei, Israël Nisand et René Frydman.
M. Jean-François Mattei, vice-président de l'Académie nationale de médecine . - Je m'exprime en premier, n'y voyez aucune question de préséance mais simplement la conséquence que je devrai vous quitter avant la fin de la réunion. La position de l'Académie de médecine a engendré des polémiques. Je parle ici en son nom. Son rapport a été préparé par un comité d'éthique constitué de quatorze personnes. Les sujets ont été répartis par tandems, puis discutés. Un premier projet a été rédigé, amendé puis adopté par le comité. Il a ensuite été retravaillé par le conseil d'administration. Les académiciens l'ont reçu une semaine avant le vote, afin de déposer d'éventuels amendements.
La procréation médicalement assistée et son extension sont un sujet à double entrée. Nous n'avons rien à redire sur la question du désir de maternité, que chaque femme peut légitimement exprimer. La question de l'enfant n'a en revanche pratiquement pas été évoquée au cours des discussions préalables. Notre but est d'informer et non de polémiquer. Nous avons donc arrêté de communiquer à ce sujet. L'Académie estime qu'il n'est pas de sa compétence de s'exprimer en faveur ou contre un sujet d'essence sociétale. Elle s'est simplement intéressée aux potentiels aspects médicaux. Nous avons d'ailleurs émis des réserves sur de potentielles conséquences d'ordre médical pour l'enfant. Cette question a bien évidemment donné lieu à un débat.
Nous avons travaillé avec les moyens et les compétences de l'Académie de médecine. Nous avons sollicité le professeur Bruno Falissard, psychiatre et biostatisticien. Celui-ci a revu toute la littérature, fortement polluée par les a priori des auteurs. Ils s'accordent tous pour éliminer la possibilité d'une forte augmentation de mal-être et de psychopathologies chez les enfants de couples de même sexe. Cependant, pour une augmentation modérée à faible, le phénomène à étudier et les variables à prendre en compte sont tellement complexes que la littérature n'est pas stabilisée. Devant cet état de fait, les médecins ont décidé d'approfondir la recherche. J'ai mentionné les publications concernées dans le questionnaire que je vous ai envoyé.
Bruno Falissard a analysé quatre-vingt-dix-neuf références datant de 2005. Elles se révèlent totalement ininterprétables, et non recevables méthodologiquement parlant : échantillons volontaires et non aléatoires, absence de contrôle, nombres trop réduits ou encore durées trop restreintes.
En 2012, deux études de puissance statistique très satisfaisante ont été menées. La première ne montre aucune différence entre les enfants issus de couples homosexuels et hétérosexuels. La seconde répertorie en revanche plus de tendances suicidaires et dépressives chez les enfants de couples homoparentaux. Aucune conclusion formelle ne peut en être tirée. L'ensemble des études réalisées recommande de plus amples recherches.
J'ai pris connaissance hier d'une publication refusée par le New England journal of medicine. Elle porte sur un groupe de soixante-dix-sept personnes âgées de 0 à 25 ans. Elle ne relève aucune différence entre les enfants de couples hétérosexuels ou de même sexe. Les chercheurs ajoutent que leur étude est limitée par la taille de l'échantillon et la signification statistique pas interprétable. Les résultats sont donc à examiner avec la plus grande précaution.
Devant des résultats discordants, nous avons émis des réserves sur de potentielles conséquences d'ordre médical impactant les enfants. Nous ne pouvions pas prendre une autre position.
J'ajoute que l'extension de la PMA relève de l'insémination artificielle, avec sperme de donneur anonyme. Se pose donc la question de la quête des origines.
Dans un cas de couple hétérosexuel infertile ayant subi une insémination artificielle, l'enfant connaît une vraisemblance parentale. Lorsqu'il s'interroge sur ses origines, il recherche alors dans la majorité des cas son géniteur, mais pas son père, qu'il connaît déjà. Un enfant ayant deux mères et pas de père se posera des questions différentes. S'il retrouve son donneur, nul ne connaît le lien qu'il pourra établir avec celui-ci.
Deux faux arguments ont été utilisés. Il a d'abord été dit que puisque les femmes seules ou homosexuelles étaient déjà en mesure d'adopter, elles devaient aussi pouvoir procréer médicalement. En réalité, dans l'adoption, un enfant n'a pas du tout de parent. Il est préférable de lui en donner un ou deux, que de le laisser seul. Dans le cadre de la PMA pour une femme seule ou en couple avec une autre femme, nous assistons en revanche à une privation volontaire de père. De plus, le nombre croissant de familles monoparentales a également été évoqué. Celles qui existent aujourd'hui sont cependant plus souvent une conséquence des aléas de la vie que de réels choix.
Nous considérons qu'il est nécessaire de séparer l'extension de la PMA aux couples de femmes, et aux femmes seules. Dans ces couples, il existe une altérité au regard de l'enfant. Il n'y a pas de fusion entre la mère et l'enfant qui ne nous semble pas souhaitable. Les familles monoparentales s'inscrivent en outre dans un contexte social et économique plus difficile. Je ne suis donc pas certain qu'il soit souhaitable d'en créer de nouvelles. Mon avis n'est cependant pas formel.
M. Israël Nisand, président du Collège national des gynécologues et obstétriciens français (CNGOF) . - Je m'exprime au nom du CNGOF, que nous avons interrogé à plusieurs reprises. Nous avons une idée assez précise de l'opinion des professionnels réalisant la PMA en France. Pourquoi les médecins interviennent-ils dans un débat sociétal et éthique ? Ils émettent certes un avis en tant que citoyens, mais ils sont également témoins directs de cette avancée.
Le projet de loi tel qu'il a été voté à l'Assemblée nationale en octobre entraîne une haute satisfaction au sein de la profession. Il permet enfin aux femmes homosexuelles ou seules de profiter des services de la médecine. Les spécialistes n'y sont pas réticents.
J'évoquerai le côté court de cette loi, car certains points écrits ne nous conviennent pas et ne sont pas dans l'intérêt des patientes. Je n'évoquerai donc pas le DPI pour les aneuploïdies, qu'abordera René Frydman, mais deux autres sujets me paraissent importants.
J'aimerais citer les femmes perdant leur compagnon en cours de processus de FIV. Ce cas m'est arrivé au moins une fois. J'ai dû répondre à une femme qu'elle avait le choix entre donner ses embryons à une autre femme ou les détruire. Elle m'a répondu que cette réponse était obscène. Nous n'avons pas la même posture vis-à-vis des gamètes. Ceux-ci disparaissent ou sont jetés sans poser de problèmes. Nous ne pouvons en revanche pas dire des embryons qu'ils ont d'une part une certaine dignité, et vouloir d'autre part les détruire alors qu'une femme souhaite se les faire réimplanter. Il est bien évidemment nécessaire d'encadrer ce cas. Nous pensions que vos collègues de l'Assemblée nationale partageaient cette posture, c'est pourquoi nous ne comprenons pas la mesure prise. Elle concerne certes très peu de femmes, mais est terrible pour les personnes concernées. En l'état, nous ne pouvons même pas leur donner leurs embryons pour qu'elles se les fassent implanter à l'étranger. La profession entière vous demande donc de faire en sorte que nous n'ayons plus à leur fournir cette réponse à l'avenir.
Le point le plus important concerne l'autoconservation des ovocytes. Les hommes peuvent conserver leurs spermatozoïdes comme ils le souhaitent. Une quarantaine de centres publics autorisés se voient réserver la pratique d'autoconservation ovocytaire. Cette mesure sera donc inaccessible pour des zones entières du pays. De même, les quelques centres autorisés verront leur liste d'attente s'allonger considérablement. Il a été reproché aux centres privés de pousser à la consommation dans ce domaine. Je ne vois pas en quoi cette critique est valide. Ces centres ont les mêmes règles du jeu. Nous faisons de plus confiance à l'ABM qui contrôle remarquablement l'activité des centres d'aide médicale à la procréation.
Le don d'ovocytes ne fonctionne pas dans notre pays. Nous dépensons énormément d'argent pour les femmes de plus 40 ans pour des résultats très modestes. Mon équipe me demande d'ailleurs d'arrêter de les prendre en charge. Si une partie de ces femmes pouvaient disposer de leurs ovocytes, nous n'aurions pas à les envoyer dans des pays étrangers pour qu'elles s'en procurent d'autres, achetés à des étudiantes. Nous ne l'acceptons pas, mais le finançons pourtant avec notre sécurité sociale. J'affirme qu'il est préférable pour les femmes d'autoconserver ses propres cellules que d'avoir un don d'ovocytes, entraînant un réel deuil. Ne retirez pas d'une main ce que vous avez donné de l'autre.
Les centres publics devront pousser vers les centres privés certaines activités qu'ils ne seront plus capables d'accomplir. Laissons la totalité des gestes d'AMP dans les centres possédant un agrément pour le faire, et contrôlons en aval. Si l'objectif de la limitation de l'offre est de limiter les dépenses, d'autres moyens existent. La Hollande l'a prouvé. Le don d'ovocytes comporte deux phases. Lors de la première, l'autoconservation, la femme n'est pas malade et est en capacité de procréer. Je souhaite préciser que ce ne sont pas les femmes qui procrastinent, mais leur conjoint. Les hommes sont en effet largement responsables de l'arrivée tardive de femmes en demande de grossesse. La maltraitance des femmes est souvent évoquée, c'en est une. Le fait d'autoriser les femmes à autoconserver leurs ovocytes durant la première phase représente un coût d'environ 2 000 euros. Lorsqu'elles viennent demander de récupérer leurs ovocytes autoconservés, ou les donner à une autre femme, elles pourraient être remboursées de ce qu'elles ont dépensé dans cette phase. Cette méthode permettrait un équilibre des dons. Je comprends que le gouvernement souhaite limiter les dépenses, mais je n'accepte pas qu'il réduise à ce titre l'offre. L'offre de soin est énorme et gratuite en France, pourtant nous avons à gérer d'interminables listes d'attentes de personnes ne trouvant pas satisfaction.
Je suis heureux que nous revenions en arrière sur la question de l'anonymat. Nous avons le droit de vouloir à tout prix un enfant, à condition de lui expliquer ensuite ce que nous avons fait pour le concevoir. Nous avons une obligation de transparence totale vis-à-vis des actes que nous réalisons. Nous ne comprenons donc pas pourquoi cadenasser les listes de données relatives aux gamètes et embryons. Je préférerais que moins de dons soient réalisés, mais que nous fassions face à moins de reproches. Nous avons l'impression de faire plus de mal que de bien. Je signale toutefois que se débarrasser des dons effectués avant le passage de cette loi serait un gigantesque gâchis. Nous jetterons volontiers les dons de donneurs et donneuses en faisant la requête, mais nous aimerions être autorisés à les contacter. Ils pourraient alors autoriser la conservation, ou au contraire demander l'abandon de leurs dons. Nous savons que nous rencontrerons des difficultés.
La loi me semble suffisante concernant la filiation. Chaque personne ayant recours à un tiers donneur rédige une déclaration à un notaire ou magistrat pour accepter le don. Nous sommes attachés à une symétrie totale entre les couples hétérosexuels et homosexuels ayant recours à un tiers donneur. Des modalités d'instauration de la filiation dépendant de la structure familiale seraient dommageables.
M. René Frydman, professeur émérite des universités, gynécologue obstétricien . - Merci de votre invitation. Je souhaiterais réagir à un certain nombre de points avant d'intervenir au sujet de l'analyse de l'embryon.
Il est vrai que certaines interrogations n'emportent pas la conviction de tout le monde. Nous devrions renforcer l'accueil, l'écoute et l'accompagnement des couples hétérosexuels, homosexuels et des femmes seules. L'indication même de la FIV devrait être faite dans toutes les circonstances, en cas de problème médical bien sûr, mais également social, économique ou encore d'équilibre psychologique... Un point de vue pluridisciplinaire dans la loi serait intéressant et permettrait de résoudre certaines questions qui nous posent aujourd'hui problème.
Concernant le post-mortem, chacun s'accorde pour dire qu'un temps de réflexion et un délai final sont nécessaires, mais nous ne pouvons pas laisser les concernées dans cette souffrance.
L'autoconservation est également une nouvelle proposition à prendre en charge différemment, avec un réel accompagnement et une réelle présence. Ne devraient être agréés que les centres, publics ou privés, faisant preuve d'une concentration efficace et d'un accompagnement suffisant. Je pense, ici aussi, au pluridisciplinaire pour répondre aux questionnements pouvant se poser vis-à-vis de l'enfant.
Le don ne fonctionne pas en France, effectivement, à notre grand dam. Beaucoup de femmes se rendent à l'étranger. Il s'agit également d'une source de commercialisation redoutable à mettre de côté. Comment répondre à la demande qui ne va pas diminuer ? Des femmes de plus en plus âgées nous sollicitent. Je vous demande de nous laisser faire campagne et donner nos arguments. Il est nécessaire de passer par un plan national. Si je demande aux citoyens la date de la dernière campagne de don de gamète, personne n'aura la réponse. Nous devons pouvoir diffuser nos arguments pour convaincre. Les Françaises sont aussi généreuses que les autres, mais il faut les informer. Nous devons changer notre façon de faire.
J'ajouterai mon point de vue personnel concernant la possible levée de l'anonymat. Je suis pour une grande liberté des gens dans un cadre établi. Jusqu'à preuve du contraire, les personnes élevant l'enfant pourront décider de ce qu'ils lui diront. C'est leur responsabilité. Si l'enfant est mis au courant, il a la possibilité de s'adresser à une commission pour connaître, pourquoi pas, l'identité du donneur. Que ce dernier accepte d'être contacté si l'enfant le souhaite me semble positif. Il est maintenant nécessaire de s'interroger sur une possibilité de réflexion pour répondre à ce contact.
Un enfant né sous X peut maintenant s'adresser au CNAOP s'il cherche des réponses au sujet de sa filiation. Ce conseil mènera l'enquête et contactera sa mère. Nous nous sommes engagés il y a des années auprès de ces femmes à conserver leur anonymat. Elles ont maintenant la possibilité d'accepter ou non de répondre à leur enfant s'il souhaite découvrir ses origines. Les parents sont libres d'informer leur enfant, ce dernier est libre de chercher des réponses.
Sur le point médical, il me semble qu'il existe une confusion concernant la recherche sur l'embryon dans la proposition de loi. Il est normal que ce débat ait lieu. Les articles L. 2141-3-1 et L. 2151-5 sont contradictoires. La recherche sur des gamètes ou embryons avant ou après transfert à des fins de gestation est autorisée. Pourtant, aucune recherche ne peut être entreprise sans autorisation, mais surtout les embryons ayant fait l'objet d'une recherche ne peuvent être transférés à des fins de gestation. La confusion vient du fait que nous n'avons pas ramené suffisamment la notion de recherche sur deux situations totalement différentes. La première fait obligatoirement l'objet d'un projet parental. La seconde non. Nous sommes dans deux systèmes différents, ce qui n'est pas précisé dans la loi.
En 2011, quand la loi a autorisé la technique de vitrification, dont nous ne parlons plus aujourd'hui, nous avons dû nous battre. Elle était comprise comme de la recherche sur l'embryon. Si nous ne précisons pas ce que nous voulons, nous arrivons à cette confusion et à ces blocages. Nous devons différencier la recherche d'innovation de la recherche sans projet parental.
60 % des embryons, et 80 % à l'approche de la quarantaine, ne s'implanteront pas. Nous sommes confrontés, en fonction de l'âge, à des taux de succès bas. Il nous semble important d'essayer de comprendre la potentialité d'un embryon de s'implanter, et non de connaître la couleur de cheveux, d'yeux ou encore le QI du potentiel futur enfant.
Allons-nous augmenter les taux de succès ? Paradoxalement non, mais nous allons améliorer le management. Pourquoi transférer un embryon alors qu'il mènera à un échec ou à une fausse couche ? Toute cette gestion devrait être proposée, mais pas à toutes les patientes. Certaines populations sont à risque. L'âge est un de ces risques. Une publication datant de septembre portant sur 700 embryons montre qu'une analyse chromosomique avant 35 ans ne sert à rien. Elle est en revanche utile après 35 ans. Nous cherchons à éviter des échecs répétés. À une époque sensible aux féminicides et atteintes faites aux femmes, je le considère comme une violence psychique, physique et économique.
Dans cette approche de compréhension de l'embryon, l'eugénisme est abordé. Qui, parmi les 200 médecins et biologistes en faveur de cette proposition, serait pour l'eugénisme ? Cette notion essaie d'isoler volontairement une population par ses caractéristiques. Elle vise à améliorer génétiquement le patrimoine humain en limitant la reproduction d'individus porteurs de caractéristiques défavorables. Pratiquer cette analyse embryonnaire ne me semble pas acceptable.
Concernant le diagnostic anténatal, nous pourrions tendre vers cet eugénisme. Nous n'avons pas décidé d'arrêter les prises de sang, mais de les encadrer. 48 centres de médecine anténatale existent en France. Sur 700 000 femmes pouvant bénéficier d'une connaissance de leur statut embryonnaire, un certain nombre le refuse. 7 000 demandes ont été faites en 2016. Les médecins en ont refusé 120. Il est surtout à noter que 1 200 avis ont été jugés acceptables par les équipes médicales, mais refusées par les patientes. Cet espace de liberté est à valoriser. Bien entendu, il y a toujours des zones grises. Un diagnostic chromosomique permet de connaître le sexe, ce qui ne nous paraît pas utile en tant que médecin. Les biologistes n'ont donc pas le droit de communiquer cette information, n'entrant pas en ligne de compte. Dans le cas d'une anomalie chromosomique, nous ne modifions rien. La femme aura un choix à faire trois mois plus tard. Si, informé de cette situation, le couple ne veut pas procéder à cette implantation, je respecte leur choix. La liberté de décision est celle qui m'importe le plus.
M. Jean-François Mattei . - Je vois que l'ensemble des points a été abordé par mes collègues.
Je suis du même avis que mes collègues concernant l'insémination artificielle ou le transfert d'embryons post-mortem. La situation était différente en 1994. À l'époque, un enfant né dans un délai supérieur à neuf fois après la mort de son père ne pouvait être reconnu comme son fils.
Le ministère de la Justice s'y opposait. Il n'était de plus pas question d'inséminer une femme seule, la pratique étant réservée aux couples souffrant d'une infertilité pathologique. Ce sujet a toujours été traité avec une émotion lourde dans les hémicycles. Je considère qu'il serait incompréhensible de refuser à une femme veuve une réimplantation.
L'académie est évidemment favorable au principe d'autoconservation des ovocytes. Nous sommes choqués qu'elle soit présentée comme une garantie de grossesse au moment venu. C'est en réalité une fécondation in vitro, dont nous connaissons les risques et les échecs. Nous voudrions informer les femmes sur le fait qu'elle constitue une possibilité, mais qu'il est toujours mieux d'utiliser les voies naturelles.
Je rejoins monsieur Nisand sur la question des centres publics et privés. Les premiers pourraient être retenus pour établir des protocoles dans un premier temps, mais nous auront à l'étendre aux seconds.
Nous avons peu évoqué la pénurie de gamètes. Nous recevons environ 363 dons de sperme par an. Déjà 1 000 demandes ont été déposées par couples hétérosexuels, qui patientent en moyenne un à deux ans. Je doute que les dons augmentent parallèlement aux demandes. Je ne suis pas sûr que nous arrivions à établir l'offre et la demande de manière équitable.
J'insiste sur le fait qu'il y aurait une seule liste de candidats à une insémination artificielle, confondant les indications médicales et les demandes sociétales. Pour un couple souffrant d'une infertilité pathologique, le délai sera allongé, ce qui pourrait paraître incompréhensible. Nous considérons à ce titre qu'il est nécessaire d'établir deux listes distinctes.
Ensuite, sur le point de l'anonymat, je rejoins René Frydman. Le CNAOP a maintenant 17 ans d'existence et fonctionne très bien. Je ne comprends pas l'apparition d'une nouvelle commission. Créer une structure en plus de celle existante me paraît absurde.
Vous n'avez pas parlé du double don, auquel je ne vois pas d'obstacle. Il serait pertinent de favoriser l'accueil d'embryons. 221 538 embryons étaient congelés au 31 décembre 2015. Un embryon congelé n'est ni plus ni moins qu'un double don. Le modèle avait à l'époque été construit sur l'« adoption la plus précoce qui soit ». Je pense qu'il est nécessaire de faire table rase et d'adopter une posture facilitatrice.
Les esprits ont évolué depuis 1994 sur la recherche sur l'embryon. La refuser au prétexte qu'un embryon est une personne est paradoxal puisque cela revient alors à la priver d'un accès aux soins. Nous ne pouvons faire de thérapie sur l'embryon sans effectuer de recherches.
Concernant le CRISPR Cas9, nous devons être vigilants et ne devons pas aller trop vite. Personne ne connaît sa fiabilité et ses effets collatéraux. Nous devons autoriser les recherches grâce à cette technique et ses dérivés, mais pas dans le cas d'une implantation.
Nous savons qu'une fausse couche sur deux est liée à une anomalie chromosomique. Le diagnostic préimplantatoire avec recherche d'aneuploïdie chromosomique est utile. Cependant, s'il est trop précis, vous ouvrez la porte à la critique de l'eugénisme.
Enfin, je voudrais faire une remarque d'ordre général. Cette loi en dit beaucoup trop peu sur le CRISPR. Vous n'attendrez pas cinq ans avant de devoir légiférer à ce sujet. Nous parlons également trop peu d'intelligence artificielle en bioéthique.
Mme Muriel Jourda . - Merci Monsieur le président. Ma question s'adresse à tous les intervenants. Est-il selon vous nécessaire de supprimer la mention de l'infertilité comme cause de recours à l'AMP ?
M. Jean-François Mattei . - J'estime qu'il faut préciser « infertilité pathologique et maladies génétiquement transmissibles ». Nous ne pouvons pas faire l'économie d'indications médicales.
M. René Frydman . - Je ne comprends pas pourquoi nous ne définissons pas l'indication, même si elle est sociétale. Nous avons besoin de pouvoir encadrer, accueillir et définir la raison de ces tentatives. Le suivi des couples serait également nécessaire. Nous faisons tout de même face à beaucoup de grossesses spontanées chez les couples en projet de fécondation in vitro.
M. Israël Nisand . - De nombreuses personnes m'ont fait remarquer que nous devions être vigilants aux discriminations. Des couples de femmes venant nous consulter par le passé se considéraient comme des couples hétérosexuels faisant face à une infertilité masculine. Elles ne comprenaient donc pas pourquoi elles se voyaient refuser les services de l'AMP. Elles y percevaient une stigmatisation. Il n'est bien entendu pas question de retirer les indications, cependant il est nécessaire de veiller à ne pas prioriser certaines lignes.
Mme Muriel Jourda . - Pour rebondir sur les propos de monsieur Mattei, pensez-vous donc qu'il faille établir deux listes différentes ?
M. Jean-François Mattei . - Certaines personnes sont malades, d'autres bien portantes, handicapées ou valides. Certains ont droit à des avantages, qu'ils méritent amplement, d'autres non. Je pense que les personnes n'étant pas en mesure de procréer, ou étant susceptibles de transmettre des maladies à leurs enfants, doivent être considérées à part.
Nous n'avons pas abordé la question de l'assurance maladie. Il est clair que l'intégralité des demandes médicales doit être prise en charge. En réalité, l'ensemble des grossesses doit être pris en charge, il n'est pas question de revenir sur ce point. Il me semble cependant que les couples de femmes et femmes seules pourraient prendre en charge la question du don de gamètes et de l'insémination. L'autoconservation est déjà à la charge des patientes. L'Assurance maladie présente un déficit de 5 milliards d'euros. Elle n'est pas en capacité de financer des médicaments innovants pour le cancer. Elle ne peut donner aux Ehpad les moyens nécessaires pour les personnes dépendantes ou souffrant d'Alzheimer. Il faut donc, à mon sens, raison garder et dissocier les malades des personnes souhaitant bénéficier des techniques médicales. Il ne s'agit pas d'une discrimination.
M. Israël Nisand . - Le risque de ces listes est en effet de mener à une discrimination. À titre d'exemple, les indications d'IVG sont extrêmement variables. Jamais personne ne s'est permis de dire, en pénurie de moyens, que certaines d'entre elles pouvaient être prises en charge ou non selon la raison évoquée. Nous ne le faisons d'ailleurs dans aucun domaine de la médecine.
Il faut en effet gérer la pénurie de moyens. Nous pouvons revenir sur la prise en charge à 100 %, qui choque d'ailleurs toutes nos équipes. Elle nous coûte énormément d'argent, mais ne vaut rien aux yeux des patients. Ils la considèrent comme acquise, même en cas de probabilité de réussite extrêmement faible. Nous pouvons donc revenir en arrière sur ce sujet, mais pour tous les patients, et pas seulement une partie.
M. René Frydman . - Nous devons être vigilants sur deux points : ne pas être discriminants, et offrir une médecine actuelle à tout le monde.
Nous devons mettre en oeuvre les moyens nécessaires pour informer les Français sur le don. Nous ne pourrons pas combler la pénurie si nous ne le faisons pas. Nous en discutons depuis 40 ans, mais n'avons rien mis en place pour diffuser l'idée même du don.
La France et Israël sont les seuls pays au monde à rembourser intégralement 4 essais de fécondation in vitro. Nous sommes de plus en plus confrontés à des patientes souhaitant faire ces tentatives, puisqu'elles y ont droit, même après leur avoir expliqué qu'elles n'avaient aucune chance d'aboutir. Je suis donc en faveur d'une participation financière de principe, dans certaines situations, ayant pour but de responsabiliser ces femmes.
Je ne suis pas favorable aux doubles listes. Nous pouvons en revanche signaler les indications. Nous pouvons discuter de l'accompagnement de ce genre de situations. En Belgique, les femmes seules ou homosexuelles ont accès depuis longtemps à l'insémination artificielle. Il est pourtant important de noter que 15 % des demandes sont refusées. Les équipes médicales pluridisciplinaires se réservent donc la possibilité de contester. Il me semble important de l'accepter. Il n'est pas question d'accepter toutes les demandes, même hétérosexuelles. N'oublions pas que certaines personnes ne sont pas prêtes, que ce soit psychologiquement ou matériellement parlant.
Mme Corinne Imbert , rapporteure . - Merci à tous pour vos interventions passionnantes.
Professeur Mattei, vous avez rapidement évoqué le bébé-médicament. Que pensez-vous de la suppression de cette possibilité ?
Le CCNE souhaitait une nouvelle définition du diagnostic prénatal. Vous satisfait-elle ?
Le diagnostic prénatal était possible pour cinq maladies avant le projet de loi. Le CCNE souhaite l'élargir aux déficits immunitaires héréditaires. Quelle est votre position à ce sujet ?
M. Jean-François Mattei . - Nous connaissons tous trois le diagnostic prénatal. Nous y avons tous beaucoup réfléchi, mais n'y avons jamais trouvé de solution réellement satisfaisante, puisque le sujet est éminemment évolutif. Il était au départ chromosomique, puis est rapidement devenu échographique. Il est ensuite passé par les protéines du liquide amniotique, l'alpha-foetoprotéine. C'est pour cette raison que le double diagnostic me gêne. Il nous est arrivé fréquemment de réaliser une amniosynthèse pour une trisomie 21, non trouvée, mais entraînant la découverte d'une alpha-foetoprotéine élevée signalant une spina-bifida.
Une discussion portant sur le diagnostic prénatal non invasif a eu lieu au Parlement allemand. Il est réalisé sur les cellules foetales triées du sang maternel. Il permet une étude génétique suffisamment poussée, à la recherche d'anomalies pouvant entraîner une interruption de grossesse. Je suis moins inquiet d'une DPI assortie d'une aneuploïdie si elle est globalisée. Elle doit être une indication pour que le médecin choisisse un embryon plutôt qu'un autre.
Je suis en revanche épouvanté concernant le diagnostic préimplantatoire non invasif. Plus les séquençages à haut débit de l'ADN se feront sur des plateformes spécialisées, plus nous aurons accès à un inventaire génétique de ce qui nous paraîtra normal ou non. Nous avons identifié près de la totalité de nos gènes, mais n'avons pas, selon le généticien américain Craig Venter, compris 1 à 2 % du fonctionnement du génome. En effet, nous connaissons le gène, mais nous ne savons pas comment il s'exprimera, en raison des séquences d'ADN non codantes de part et d'autre de celui-ci par exemple.
J'ai assisté la semaine dernière à un congrès portant sur le cancer du sein, où nous avons bien évidemment abordé la génétique. La plupart des généticiens impliqués expliquent être ennuyés. En effet, s'ils trouvent un BRCA1 ou BRCA2, ils sont capables d'agir. S'ils sont face à deux variants associés qu'ils ne savent pas traduire, ils sont démunis. Nous prêtons aujourd'hui beaucoup plus à la génétique que ce qu'elle est réellement capable d'apporter.
M. Israël Nisand . - La définition du diagnostic prénatal doit être modifiée. Un grand nombre de chapitres de la loi antérieure indiquaient que celui-ci n'était que génétique. Il est depuis devenu imagerie, puis échographie ou encore IRM.
Je pense que certaines règles de l'OMS s'imposent à nous pour définir ce qu'est le dépistage prénatal. La maladie doit être relativement fréquente et poser un problème de santé publique. Elle doit également pouvoir être découverte à un stade présymptomatique, afin de pouvoir traiter l'enfant avant qu'il ne devienne malade. C'est le cas de certains déficits immunitaires.
Je suis favorable à ce que des maladies viennent se rajouter au buvard placé sur le talon des bébés. Nous pourrions ainsi dépister d'autres maladies ne s'exprimant pas à la naissance, mais étant longues à traiter après leur diagnostic.
Sous couvert de l'analyse épidémiologique et de santé publique, et dans le cas où il existe un traitement pour soigner la maladie après son dépistage, je suis donc favorable à l'extension du diagnostic.
M. Jean-François Mattei . - Un diagnostic prénatal d'une maladie que nous ne sommes pas en mesure de traiter équivaudrait à plonger la famille dans la maladie avant même qu'elle ne s'exprime.
M. Jean-François Mattei quitte la séance.
M. René Frydman . - Je n'ai pas du tout abordé CRISPR, qui est une intervention sur l'embryon. Nous n'avons parlé que de diagnostic. L'intervention fait partie d'un tout autre registre n'ayant pas pour objectif d'améliorer des résultats de la FIV.
Madame Imbert, vous avez posé la question du double espoir, et donc du bébé-médicament. Vous savez que j'ai initié cette technique en France. Onze cas ont été réalisés en une vingtaine, mais plus aucun depuis cinq ans. Les banques étaient à l'époque en pénurie de cellules de sang du cordon, que nous devions même acheter à l'étranger. Dans la plupart des cas où un enfant malade aurait besoin de transfusion de cellules adaptées, nous pouvons maintenant les trouver dans les banques nationales et internationales. J'accepte la création d'une loi pour interdire les bébés-médicaments, mais m'interroge sur la nécessité de légiférer sur tout.
Sur le problème du diagnostic, un point me semble important. La réticence peut parfois venir du caractère invasif du diagnostic, consistant à prélever une cellule même si celle-ci ne constitue pas l'embryon en lui-même. Nous disposons les embryons dans un milieu de culture pendant cinq à six jours. L'analyse de milieu peut nous donner des informations sur leur capacité à s'implanter, que j'ai abordée plus tôt. Sommes-nous réticents au principe même de choisir, ou simplement aux modalités ? Choisir des techniques de diagnostic non invasives est-il plus tolérable que des techniques invasives, ayant pourtant prouvé qu'elles n'intervenaient pas dans le développement embryonnaire ?
Les équipes médicales souhaitent pouvoir procéder à ces analyses sur la capacité à l'embryon de s'implanter, à l'instar des autres pays européens.
M. Israël Nisand . - La différence fondamentale entre le double don et l'accueil d'embryon réside dans le fait que dans le deuxième cas, l'embryon a des petits frères et des petites soeurs. Ce n'est pas le cas avec les doubles dons. Nous avons vu un grand nombre de couples partir à l'étranger, car ils ne le savaient pas. Ils n'avaient pas compris que derrière le don d'embryon se cachait toute une famille présente dans les fantasmes et l'imaginaire des receveurs. Les couples où la femme et l'homme sont tous deux stériles se voyant proposer le choix privilégieront le double don. Je me réjouis totalement de son acceptation nouvelle. Je suis bien évidemment préoccupé par la quantité d'embryons congelés, qu'aucun de nous ne méprise. Pourtant, interdire le double don pour obliger les familles à accueillir un embryon reviendrait à oublier ces frères et soeurs que j'évoquais.
M. René Frydman . - Le don d'embryon n'est pas simple pour les donneurs. Nous ne pouvons pas considérer l'embryon comme une personne potentielle, et dans le même temps le donner comme s'il s'agissait d'un simple gâteau au chocolat.
Si le double don est accepté, nous serons confrontés à des femmes seules d'un certain âge ou présentant des réserves ovariennes déficientes. Elles ont bien besoin de sperme, mais n'ont pas elles-mêmes les ovocytes adéquats. Nous devons donc aller au bout de cette satisfaction, sans oublier de les accompagner durant tout le processus.
M. Bernard Jomier , rapporteur . - J'aimerais transmettre une question à monsieur Mattei. Il nous a fait part du fait que la construction de l'altérité d'un enfant dans un couple de femmes ne pose pas de problème. Il a cependant mentionné qu'elle poserait problème dans le cas d'un enfant élevé par une femme seule. J'aimerais savoir sur quelles études et données il se base pour l'affirmer.
La rédaction actuelle des conditions à la recherche sur l'embryon vous satisfait-elle ? Je m'interroge en particulier sur la question de la finalité médicale.
Enfin, le taux de succès de l'AMP n'est pas très élevé en France. Monsieur Frydman, vous assuriez que la pratique du diagnostic préimplantatoire avec recherche d'aneuploïdies ne l'améliorerait pas significativement. Quelle autre disposition faudrait-il alors prendre ?
M. René Frydman . - Il existe selon moi une confusion entre les deux articles. La recherche conçue comme une innovation dans le but de transférer l'embryon doit être distinguée, identifiée et cadrée. Les textes sont contradictoires, indiquant d'un côté que nous pouvons le transférer, interdisant cette pratique d'un autre côté. Ce point clé est à préciser. Les recherches conçues comme une innovation dans le cadre d'un projet parental sont à distinguer des recherches n'ayant aucune visée de parentalité.
En ce qui concerne les taux de succès, il y a une sorte d'omerta sur les résultats en France. Je vous invite à vous intéresser aux résultats de l'AMP quant aux résultats français. Si vous cherchez les résultats américains, leur affichage est totalement compréhensible, ce qui n'est absolument pas le cas dans notre pays. Pourtant, en décryptant les résultats, nous voyons clairement se distinguer un top 10 et un top 100.
Bien entendu, il est complexe d'analyser ce qui est comparable. Les conclusions seront évidemment différentes entre un centre dont la moyenne d'âge des patients atteint plus de 40 ans en comparaison à un centre n'accueillant que des femmes d'une trentaine d'années.
Pourquoi est-ce important ? Certains centres sont isolés et ont peu de matériel et de personnel. Analysons les raisons de la queue de peloton afin de l'améliorer. J'estime qu'il est nécessaire de se référer à ce qui se fait de mieux pour aider les centres en difficulté. Les problèmes peuvent être multiples : défaut de formation, perte d'équipement, de savoir-faire ou de matériel.
M. Israël Nisand . - Je partage l'avis de monsieur Frydman. Nous ne pouvons pas comparer les résultats de centres payants et gratuits. Lorsqu'un médecin vous indique que votre taux de chance de réussite est infinitésimal, et que vous devez payer, vous ne tentez pas la FIV. Une sélection s'opère donc par le prix.
Pour améliorer les taux de succès des centres, il suffit de prendre des couples jeunes avec des indications extrêmement précises d'infertilité. La question est de savoir si nous soignons les patients ou les statistiques. En soignant les patients, nous devons accepter les couples n'ayant que 2 ou 3 % de chances de concevoir. En soignant nos statistiques, nous réfutons ces couples.
Je ne pense pas que le niveau des taux de succès soit uniquement dû à des problèmes techniques et de qualité des laboratoires, ce qui serait inquiétant. C'est pour cette raison que je suis favorable à l'élaboration d'un fichier « bas risque » par centre. Il pourrait par exemple regrouper tous les couples de moins de 35 ans présentant une infertilité tubaire. Sur ce cluster, nous pourrons alors nous intéresser aux taux de réussite dans des conditions optimales. Nous pourrions alors réaliser que la France est plutôt efficiente. Le fait que nous acceptons les couples ayant très peu de chances de réussite impacte forcément négativement les taux de succès français.
M. Olivier Henno , rapporteur . - Je vous remercie de la qualité de nos échanges. Je partage votre avis sur l'insémination post mortem qui est selon moi une question de dignité humaine.
Le gouvernement s'est quelque peu opposé à l'autoconservation des gamètes, s'inquiétant d'un risque potentiel d'utilisation massive. Je n'y vois pas le problème. Elle correspond pour moi à une évolution sociétale. Qu'en pensez-vous ?
Sur le CRISPR cas9, pensez-vous qu'il y ait aujourd'hui une possibilité d'un encadrement éthique dans un but thérapeutique ?
Enfin, nous voyons le choc des biotechnologies et de l'intelligence artificielle émerger en auditions. La question du consentement éclairé se pose, de même que l'encadrement essentiel et nécessaire. Disposons-nous de suffisamment d'éléments pour avancer sur ce sujet ?
M. René Frydman . - Je reviens rapidement sur la question antérieure. Nous pourrions peut-être revisiter la carte des centres aujourd'hui. Plusieurs centres tournant six ou sept jours sur sept et réalisant 700 tentatives regroupés permettraient bien plus de possibilités médicales et de recherche.
Vous évoquez CRISPR. Nous sommes dans une démarche d'expérimentation qui doit être faite sur l'animal au préalable. Nous ne savons pas ce que nous modifions en coupant l'ADN. À l'hôpital Necker il y a quelques années, nous avons connu le cas des enfants bulle présentant une déficience immunologique. Nous avons vu qu'en intervenant dans le cadre d'une thérapie génique, nous pouvions bloquer cette immunité à leur environnement. Nous avons dû arrêter les essais suite au développement de leucémies chez deux de ces patients. En coupant une zone d'ADN, nous ne savions pas ce qui se passerait dans le reste des gènes. Nous sommes ici dans un cadre de recherche très éloigné du projet parental.
Concernant une potentielle utilisation massive de l'autoconservation des ovocytes, je ne pense pas que nous ferons face à un tsunami de demandes. Je ne pense pas que le problème soit quantitatif. Les femmes seules parties à l'étranger dans ce but ne représentent pas à ce jour des sommes faramineuses. De plus, la mise en place d'un accompagnement et d'une écoute permettra à certaines femmes de faire marche arrière.
M. Israël Nisand . - Il s'agit d'une liberté que nous accordons aux femmes. Je ne pense pas que nous puissions inciter à cette demande. Il serait en revanche incompréhensible que les centres agréés AMP, prélevant et congelant des ovocytes, ne puissent pas réaliser ce travail.
M. René Frydman . - Il est indispensable d'avoir pris les dispositions nécessaires en termes d'accompagnement et de temps. L'agrément et le mode de fonctionnement doivent être identiques.
M. Alain Milon , président . - Y a-t-il d'autres questions ?
J'aimerais simplement ajouter que j'ai réalisé une étude sur la sécurité sociale en Espagne avec des collègues. L'équivalent espagnol du CCNE nous y a appris que chaque année, 7 000 femmes françaises se rendent en Espagne dans le cadre d'une PMA.
M. René Frydman . - Avec l'argent de la sécurité sociale française ?
M. Alain Milon , président . - Ce point serait à préciser. Merci à tous.
Ce point de l'ordre du jour a fait l'objet d'une captation vidéo qui est disponible en ligne sur le site du Sénat .
M. Hugues Fulchiron,
professeur de droit privé
à l'Université Jean Moulin
Lyon 3, directeur du centre de droit de la famille, Mme Marie Mesnil,
maîtresse de conférences en droit privé
à
l'Université de Rennes 1 et M. Jean-René Binet,
professeur de droit privé à l'Université de Rennes
Mme Élisabeth Doineau , présidente . - Mes chers collègues, je salue la présence de M. Hugues Fulchiron, professeur de droit privé à l'Université Jean Moulin Lyon 3, directeur du centre de droit de la famille, de Mme Marie Mesnil, maîtresse de conférences en droit privé à l'Université de Rennes 1 et de M. Jean-René Binet, professeur de droit privé à l'Université de Rennes 1.
Nous poursuivons nos travaux sur le projet de loi relatif à la bioéthique avec une audition consacrée aux évolutions proposées par le texte dans le domaine du droit de la famille. J'indique que cette audition fait l'objet d'une captation vidéo retransmise en direct sur le site du Sénat et consultable à la demande. Notre souhait d'entendre une pluralité d'analyses et de points de vue nous a conduits à solliciter de nombreux intervenants. Tous n'ont pas été en mesure de répondre à notre invitation, mais nous accueillons avec plaisir ce matin ceux qui ont pu se rendre disponibles.
Je laisse la parole à nos invités pour un bref propos liminaire avant que nos rapporteurs n'interviennent, puis les sénateurs qui le souhaiteront.
M. Jean-René Binet, professeur de droit privé à l'Université de Rennes 1 . - Je vous remercie, madame la Présidente, pour votre invitation à m'exprimer devant vous dans le cadre de la révision de la loi de bioéthique.
Avant d'en venir aux questions consacrées à la filiation, j'aimerais exprimer mon sentiment par rapport au processus en cours. Ce processus inscrit dans la logique des précédentes révisions semble marqué par une continuité historique depuis les lois de 1994. Cependant, cette continuité n'est qu'apparente. Le processus législatif me semble avoir été marqué par une accélération du temps que l'on constate aisément si l'on envisage chacune des étapes qui l'ont jalonné. Les états généraux de la bioéthique qui se sont déroulés l'année dernière n'ont duré que cent jours, soit beaucoup moins que la fois précédente. La mission parlementaire d'information à l'Assemblée nationale, constituée en juin dernier, a duré peu de temps et les débats n'ont pas été approfondis. Enfin, l'examen à l'Assemblée nationale n'a pas permis le soutien de très nombreux amendements et a empêché l'approfondissement de très nombreuses questions. Le processus actuellement en cours est donc bien différent de ce qui s'était passé précédemment.
Ensuite, le projet de loi a été présenté par le Gouvernement comme un projet révolutionnaire. Il ne s'agit donc pas, comme l'avaient fait les lois du 6 août 2004 et du 7 juillet 2011, de faire évoluer la loi de bioéthique de 1994, mais bien de révolutionner la matière. Comme l'avait indiqué le Conseil d'État, le risque est très fort de voir des principes structurants profondément remis en cause par la révision.
Avant d'en venir à la filiation, je souhaiterais évoquer certains principes fondamentaux de ce droit de la bioéthique. L'alinéa 1 er de l'article 16-4 du code civil affirme la protection de l'intégrité de l'espèce humaine. Que restera-t-il de ce principe si l'on autorise demain la création d'embryons transgéniques ou chimériques ? C'est ce que fait le projet de loi à l'article 17-I en modifiant les dispositions de l'alinéa 2 de l'article L 2151-2 du code de la santé publique. Que restera-t-il du respect dû à cette personne humaine potentielle qu'est l'embryon si, demain, l'essentiel des recherches conduites sur l'embryon et ses cellules est sorti du cadre contraignant de l'article L. 2151-5 du code de la santé publique? Ce principe de respect est affirmé à l'article 16 du code civil, mais serait remis en question par les futurs articles L. 2141-3 et L. 2151-7 du code de la santé publique.
J'en viens au principe de primauté de l'intérêt de l'enfant, qui est au coeur des conditions d'accès à l'assistance médicale à la procréation. Les conditions sont aujourd'hui fixées à l'article L. 2151-2 du code de la santé publique. Le texte prévoit que l'assistance médicale à la procréation est réservée à un couple formé d'un homme et d'une femme vivants et en âge de procréer. Ces conditions s'expliquent par le souci du législateur, en 1994, de conférer à l'enfant à naître des techniques d'assistance médicale à la procréation, une filiation crédible. Il doit pouvoir se représenter comme étant effectivement issus des parents que la loi lui désigne. Le législateur s'est conformé à l'obligation qui lui est imposée par la convention internationale des droits de l'enfant : assurer une considération primordiale à l'intérêt supérieur de l'enfant.
Ces conditions sont radicalement modifiées par l'article 1 er du projet de loi qui, en ouvrant l'accès à l'assistance médicale à procréation (AMP) aux couples de femmes et aux femmes seules et en supprimant les indications médicales du recours à l'assistance médicale à la procréation, déverrouille l'accès à ces techniques. Ces conditions orientent, comme y invite le rapport Touraine, vers la consécration d'un droit à la procréation sans sexe pour tous.
Cette révolution dans les conditions d'accès n'est nullement la conséquence d'une obligation qui pèserait sur le législateur français. Le Conseil d'État, la Cour européenne des droits de l'homme et le Conseil constitutionnel ont rappelé qu'aucune obligation ne pesait sur le législateur.
Si le législateur entend y procéder, il doit nécessairement tenir compte de cet intérêt supérieur de l'enfant, qui doit être sa considération primordiale. Il doit alors se poser cette question : est-il en mesure de garantir la prise en compte de l'intérêt de l'enfant en le privant de toute possibilité d'avoir une filiation paternelle ? L'enfant ne risque-t-il pas de souffrir de cette privation délibérée de père ? À cette question, les réponses ne peuvent venir que des études entreprises. Or, comme le rappelle le Comité consultatif national d'éthique (CCNE), celles-ci ne sont pas fiables. Elles sont affectées par de nombreux biais méthodologiques, souvent menées par des militants, et font l'objet de contestations ; la prudence commande de ne pas s'y fier. Par conséquent, le législateur peut adopter trois attitudes différentes. Une attitude de risque : accepter l'article 1 er tel qu'il est écrit. Une attitude de refus du risque : rejeter cet article 1 er .
Une attitude de prudence : modifier les dispositions pour prévoir qu'elles n'entreront en vigueur que lorsque des études méthodologiquement incontestables auront garanti que l'enfant ne risque pas de souffrir en raison de la privation délibérée de père et de toute possibilité d'établissement d'une filiation paternelle.
Si vous deviez accepter cette extension de l'assistance médicale à la procréation aux couples de femmes et aux femmes seules, il vous serait toutefois toujours possible de maintenir les indications médicales pour les couples formés d'un homme et d'une femme qui figurent actuellement dans le code de la santé publique. Cela aurait le mérite d'éviter un déverrouillage complet dans l'accès à l'assistance médicale à la procréation.
Vous pourriez également prévoir, comme cela avait été suggéré par le Conseil de l'ordre des médecins, une clause de conscience au profit des professionnels de santé.
Vous pourriez également maintenir le caractère subsidiaire du recours au don de gamètes que le projet de loi entend supprimer.
Il vous faudrait également élaborer l'établissement du lien de filiation de l'enfant né de cette technique dans un couple de femmes. Tel est l'objet de l'article 4 du projet de loi.
Actuellement, la filiation établie en cas de recours à l'assistance médicale à la procréation l'est conformément aux dispositions du titre VII du livre I er du code civil. Elle l'est conformément au droit commun en cas de procréation endogène, c'est-à-dire intraconjugale sans recours à un don de gamètes. En cas de don de gamètes, de procréation exogène, les conditions de droit commun s'appliquent pour l'essentiel, à quelques détails près. La filiation doit être obligatoirement établie et sa contestation est essentiellement impossible.
Les dispositions du titre VII du livre I er , qui sont fondées sur la vraisemblance ou la vérité biologique, ne permettent toutefois pas d'établir le lien de filiation à l'égard de deux personnes de même sexe, de deux femmes. C'est la raison pour laquelle le Conseil d'État avait suggéré quatre options en écartant d'emblée celles qui consistaient à modifier le titre VII. Il estimait dans son étude de 2018 que les modifications du titre VII seraient en contradiction avec la philosophie des modes d'établissement classiques de la filiation, qui reposent sur la vraisemblance. Il a attiré spécifiquement l'attention sur le fait qu'elles conduiraient à une remise en cause des principes fondateurs du droit de la filiation fixés par ce titre.
C'est la raison pour laquelle il avait préconisé la création d'un titre VII bis dans lequel seraient inscrites les règles conférant un double lien de filiation monosexuée à l'égard de cet enfant.
L'Assemblée nationale a été saisie d'un amendement gouvernemental inscrivant dans ce titre les modifications visant à établir le lien de filiation monosexuée. Si cette solution devait également être celle du Sénat, je me permets d'attirer votre attention sur deux points qui, techniquement, posent problème. Le premier point est l'utilisation du mot « reconnaissance » pour le couple de femmes. La reconnaissance conjointe, telle qu'elle est prévue par le projet de loi, risquerait de créer une confusion avec la reconnaissance prévue à l'article 316 du code civil. Ces deux reconnaissances figureraient en effet dans le même titre, mais avec des sens très différents. La reconnaissance de l'article 316 est un aveu de paternité, mais la reconnaissance conjointe ne peut être qu'un acte de volonté destiné à fonder la filiation. Cette confusion terminologique pourrait fragiliser la véritable reconnaissance. Il conviendrait de remplacer ce mot.
Autre problème, la filiation, telle qu'elle est prévue dans le projet de loi, ne distingue pas la femme qui accouche de l'autre femme. Ce faisant, elle méconnaît l'application de la règle mater semper certa est. Il faudrait impérativement opérer une dissociation entre les deux femmes : l'accouchement ferait la mère pour l'une tandis que la déclaration ferait la maternité de l'autre.
Enfin, le projet de loi envisage de créer un droit à l'accès aux origines personnelles et il s'inscrit dans une démarche tendant à une meilleure prise en compte de l'intérêt de l'enfant. Ce projet aurait dû arriver à son terme dès 2011. Au regard de l'affirmation par la Cour européenne des droits de l'homme d'un véritable droit à la connaissance de la filiation réelle, il me semble qu'il est désormais temps d'y procéder.
En 2011, le législateur avait cédé devant les craintes d'un tarissement des dons de gamètes exprimées par les professionnels du secteur. L'intérêt des professionnels avait donc prévalu sur l'intérêt de l'enfant. Fort heureusement, les enfants, dont l'intérêt supérieur a été méconnu, grandissent et demandent des comptes. C'est une leçon qu'il faut savoir méditer.
M. Hugues Fulchiron, professeur de droit privé à l'Université Jean Moulin Lyon 3 . - C'est un honneur d'être appelé à m'exprimer aujourd'hui devant vous dans le cadre de la révision des lois bioéthiques. L'ouverture de l'assistance médicale à la procréation aux femmes et aux femmes seules pose de délicates questions d'ordre éthique. Elle pose aussi des questions d'ordre juridique. Comment construire la filiation entre l'enfant et les deux femmes ayant porté le projet parental ? Comment construire juridiquement cette filiation dans le respect des droits de l'enfant de connaître ses origines ?
Avant d'aborder ces questions, je ferai une remarque préalable. Il ne m'appartient pas en tant qu'expert juridique de prendre parti sur les questions dont les enjeux sociétaux sont bien connus. Les choix à faire vous appartiennent. En tant qu'expert, il me revient seulement d'essayer d'éclairer ces choix et d'en souligner les enjeux d'un triple point de vue : tout d'abord, au regard de la pertinence technique des règles qui traduisent juridiquement les choix qui vous appartiennent ; ensuite, au regard de la cohérence du système dans lequel ces normes s'inscrivent, et, enfin, au regard des droits et libertés de la personne.
Le droit de la filiation procréative - terme plus exact que filiation charnelle - que je distingue de l'adoption, a pour objet de rattacher l'enfant à ceux qui lui ont donné la vie.
Comme tout système juridique régissant la filiation, le droit français tente de trouver un équilibre entre les données naturelles - autrefois le sang et aujourd'hui la biologie -, la volonté des individus, le vécu, les valeurs et les principes qui structurent la société.
Notre système français repose sur trois piliers : l'hétérosexualité de la parenté pour la filiation procréative, l'unicité de la parenté et la recherche de la vérité des filiations. Cette vérité est avant tout conçue comme une vérité biologique, autrefois le sang, même si une large place est faite à d'autres aspects comme le vécu en particulier, la volonté des intéressés, le souci de ne pas troubler l'ordre social, la paix des familles. Lorsque la filiation, à défaut d'être vraie, est vraisemblable, il y a des hypothèses dans lesquelles le droit s'en contentera.
L'ouverture de l'assistance médicale à la procréation aux couples hétérosexuels n'a pas bouleversé le droit de la filiation ainsi conçu, car on avait fait le choix, à l'époque, de transposer les règles conçues pour la filiation charnelle. De même, l'ouverture du mariage aux couples de même sexe n'a pas bouleversé le droit de la filiation procréative puisqu'on a choisi de prendre un autre modèle de filiation pour établir le lien : l'adoption.
En revanche, l'ouverture de l'assistance médicale à la procréation constitue un bouleversement d'une tout autre ampleur. C'est une rupture avec le modèle traditionnel de l'assistance médicale à la procréation, comme l'a souligné le professeur Binet. C'est aussi une rupture avec le modèle traditionnel de la filiation procréative.
La question est de savoir comment construire la filiation de l'enfant. Trois orientations étaient possibles : passer par l'adoption, étendre aux couples de femmes les règles construites traditionnellement pour la filiation charnelle, construire un nouveau système qui tienne compte des données particulières à cette nouvelle forme d'engendrement par deux femmes qui recourent aux gamètes d'un tiers. C'est donc cette troisième voie qui a été choisie.
Elle pouvait toutefois se subdiviser en deux chemins. Le premier chemin consistait à repenser dans sa globalité le modèle de construction de la filiation lorsque deux personnes recourent aux gamètes d'un tiers, que le couple soit homosexuel ou hétérosexuel. Le second chemin était la construction d'un système spécifique pour les couples de femmes puisque, dans ce cas-là, il n'est évidemment plus possible de rattacher la filiation à un modèle procréatif traditionnel.
Les techniques de mise en oeuvre peuvent être diverses sur ce point, mais c'est le choix de la reconnaissance conjointe qui a été fait par l'Assemblée nationale. Ce point, et je rejoins le professeur Binet, me semble, du point de vue juridique, poser trois problèmes. Dans la version retenue, il est question de reconnaissance anticipée. Le terme « reconnaissance » est déjà utilisé ailleurs dans la filiation procréative. Cela ne peut que créer de la confusion. Ensuite, cette reconnaissance entraîne l'établissement de la filiation pour les deux mères, celle qui a accouché et l'autre femme. Certes, les deux femmes ont porté ensemble le projet parental, mais ce système rompt avec un principe qui gouverne l'ensemble du droit de la filiation, à savoir que la mère est celle qui accouche. Certes, ce principe est aujourd'hui beaucoup moins évident qu'il n'y paraît, car la maternité elle-même est devenue, dans certaines hypothèses, divisible, une femme pouvant porter un embryon conçu avec les gamètes d'une autre femme. Toutefois, si l'on souhaite reconstruire la maternité, il faut, à mon avis, une construction d'ensemble et non pas une reconstruction partielle, comme celle qui est proposée et qui ne peut qu'être source d'incohérences et d'inégalités.
La troisième critique tient au fait que ce système semble nier la réalité de la grossesse et de l'accouchement puisque la filiation repose sur la seule volonté des parents. Dans cette hypothèse, l'équilibre entre réalité biologique, volonté et vécu est rompu au profit de la seule volonté des parents qui expriment le projet parental.
On change complètement le modèle sur lequel était construit le droit de la filiation. Ce modèle, qui est possible, mais pas souhaitable, appelle deux remarques. Il faut penser ce modèle dans sa globalité, en prenant en compte ses conséquences sur l'ensemble du droit de la filiation. La pire chose, de mon point de vue, est de l'introduire dans un cas particulier, sans en mesurer les conséquences, comme une technique permettant d'arbitrer entre les intérêts contradictoires. Par ailleurs, ce système pense la filiation par rapport au choix et aux intérêts des parents, ce qui me semble contraire à une des évolutions des plus importantes du droit contemporain de la famille, à savoir la reconnaissance des droits de l'enfant. Je dis bien : des droits de l'enfant, pas de l'intérêt de l'enfant. C'est à partir des droits et libertés de l'enfant qu'il faudrait reconstruire le droit de la filiation pour l'adapter aux nouvelles formes d'engendrement. Pour dire les choses autrement, il faudrait abandonner une fois pour toutes la perspective archaïque ou post-moderne qui pense la filiation à partir des parents.
Si l'on pense le système à partir de l'enfant, si l'on place le droit de l'enfant, notamment le droit de l'enfant au respect de son identité, au centre du nouveau système de filiation, il conviendrait de distinguer deux hypothèses.
La première hypothèse est celle de l'enfant issu des gamètes de ses parents, par un acte charnel ou par l'assistance médicale à la procréation. Je rappelle que l'acte charnel demeure le mode de conception le plus courant. Il me semble bon de penser la filiation et de construire un droit de la filiation à partir de l'hypothèse la plus générale même si l'on doit penser en même temps aux exceptions.
La seconde hypothèse regrouperait tous les cas dans lesquels l'enfant est né avec les gamètes d'une autre personne que celles du couple ayant porté le projet parental. À cette hypothèse, il faudrait ajouter celle d'une femme seule qui recourt à l'assistance médicale à la procréation. Dans toutes ces hypothèses où l'enfant est conçu avec les gamètes d'un tiers, que le couple soit hétérosexuel ou homosexuel, il faudrait placer au centre de cette seconde reconstruction le droit de l'enfant à connaître ses origines. Nous rejoignons là un autre aspect du projet de loi, à savoir l'inégalité qu'il crée au regard du droit de l'enfant à ses origines. Selon que l'enfant est né dans un couple de femmes ou dans un couple hétérosexuel, son droit ne sera pas le même, mais nous aurons certainement l'occasion d'en discuter.
Mme Marie Mesnil, maîtresse de conférences en droit privé à l'Université de Rennes 1 . - Je vous remercie de m'avoir invitée à discuter des enjeux liés à l'extension de l'assistance médicale à la procréation à toutes les femmes et aux questions de filiation qui en découlent.
Je souhaiterais faire deux remarques sur l'article 1 er qui étend l'accès à l'AMP à toutes les femmes. Cet article renforce deux mouvements déjà existants en droit et il ne s'agit en aucun cas d'un texte révolutionnaire au sens où M. Binet pourrait l'entendre. Tout d'abord, il s'agit de renforcer l'affirmation du rôle social de la médecine de la reproduction. Dans tous les cas, qu'il s'agisse d'un couple hétérosexuel, d'une femme seule ou d'un couple de femmes, la médecine de la reproduction ne remplit pas exclusivement une mission médicale au sens strict. Ensuite, l'article 1 er réaffirme la reconnaissance d'un pluralisme familial. Je pense que le terme de pluralisme familial est plus intéressant pour penser justement ces droits familiaux.
Ce pluralisme familial existe déjà en droit puisque, depuis 1966, une personne seule peut avoir recours à l'adoption pour fonder une famille. Depuis la loi du 17 mai 2013, les couples de personnes de même sexe peuvent avoir reconnaissance légale de leur famille.
J'en viens à la question de l'établissement de la filiation pour les couples de femmes et en particulier pour la seconde femme du couple lesbien, celle qui n'accouche pas. Au sens du droit, la filiation est un lien juridique. Il s'agit donc toujours d'une construction sociale et ce n'est jamais la reconnaissance par le droit d'une situation pré-existante de l'existence d'un lien biologique.
Certes, la mère, c'est la femme qui accouche. Pour le père, c'est différent, car il n'accouche pas. L'établissement de la filiation paternelle non contentieuse, c'est-à-dire dans la très grande majorité des cas, se fait donc grâce à la présomption de paternité, si l'homme est le mari de la femme qui accouche, ou par reconnaissance. Ces modes d'établissement de la filiation ne reposent pas sur l'existence d'un fondement biologique au moment de l'établissement non contentieux de la filiation. Cela signifie qu'un homme peut, par le biais de ces mécanismes juridiques, choisir d'être le père d'un enfant dont il sait qu'il n'est pas le géniteur. C'est uniquement dans le cadre d'un contentieux que la preuve génétique viendra départager les hommes qui revendiqueraient la paternité de l'enfant et uniquement si le délai de prescription le permet.
Il faut également souligner qu'il existe aujourd'hui deux grands types de filiation. Le titre VII « De la filiation » n'a pas d'adjectif qualificatif et constitue le droit commun de la filiation. Ce titre a parfois été qualifié de filiation par procréation charnelle, mais cette qualification est inexacte. Il ne s'agit en effet pas toujours de filiation biologique. Cela a conduit d'ailleurs à un glissement sémantique puisqu'aujourd'hui on parle plus volontiers de filiation par vraisemblance biologique.
Le titre VIII est celui de la filiation adoptive. Il consiste à donner une famille à un enfant qui n'en a pas, à laisser la trace de cette histoire dans l'acte de naissance de l'enfant et surtout à permettre un contrôle par le juge, dans l'intérêt de l'enfant. Ces deux types de filiation emportent aujourd'hui les mêmes effets en termes de droits et de devoirs à l'égard des enfants. Un mouvement, qui a commencé dans les années 70 et s'est achevé au début des années 2000, a effectivement mis fin aux différences existant entre les filiations légitimes et naturelles. Pourquoi vouloir de nouveau, comme c'était le cas à l'époque, distinguer les filiations, si ce n'est pour marquer une désapprobation sociale par rapport aux conditions de la naissance de l'enfant ?
J'en viens à présent aux quatre options envisageables.
La première option consisterait à ne pas changer l'état du droit et à continuer de demander à la seconde mère d'adopter l'enfant de sa conjointe. Elle a été jugée bien trop problématique par rapport à l'intérêt de l'enfant, qui est de voir sa filiation établie dans tous les cas.
Les deuxième et troisième options consistent à créer un nouveau mode d'établissement de la filiation spécifique notamment pour les couples lesbiens. C'est l'option actuellement retenue à la suite de l'avis du Conseil d'État. Il aurait aussi pu être envisagé de repenser plus généralement le système de filiation pour tous les enfants conçus par don de gamètes.
Dans ce cas-là, les couples hétérosexuels, les couples lesbiens, voire les femmes seules ayant recours au don de sperme, auraient été concernés.
Ces deux options soulèvent des questions au regard de ce qu'est actuellement le droit de la filiation. Tout d'abord, on crée une confusion problématique entre la filiation d'un côté et le mode de conception de l'enfant de l'autre. En effet, le mode d'établissement de la filiation, en apparaissant sur l'acte de naissance de l'enfant, aurait signifié le recours à un don de gamètes. On aurait donc une confusion à l'état civil entre la filiation - le lien de droit qui existe entre un enfant et ses parents - et les origines génétiques de l'enfant. Par ailleurs, pour les couples hétérosexuels, cette information constitue une atteinte au secret médical puisque le recours au don de gamètes traduit pour ces couples l'existence d'une stérilité. Ce nouveau mode d'établissement de la filiation constitue une stigmatisation relative aux conditions de la naissance, qu'elle soit liée à un recours de gamètes ou à l'orientation sexuelle de leurs parents.
La solution actuellement retenue, celle d'une reconnaissance conjointe prénatale pose des problèmes techniques. La difficulté ne repose pas sur l'emploi du terme « reconnaissance ». Je ne partage pas l'avis de mes collègues sur ce point. À mon sens, cette reconnaissance n'est pas un aveu de paternité ou de maternité, c'est-à-dire de l'existence d'un lien biologique, mais un engagement à assumer un rôle parental auprès de l'enfant.
La réelle difficulté technique a déjà été soulevée par M. Fulchiron : il s'agit de l'indivisibilité des filiations maternelles. La reconnaissance conjointe sécurise la filiation puisqu'elle est établie dès la naissance de l'enfant, de la même manière pour les deux femmes, mais l'une ne peut être établie sans l'autre. L'accouchement n'est alors plus le fondement de la filiation maternelle pour la femme qui accouche de l'enfant. Cela me semble particulièrement problématique. Certes, nous devons penser l'établissement de la filiation à l'égard de la seconde femme, celle qui n'accouche pas de l'enfant, mais pourquoi nier l'accouchement de la mère alors que c'est un élément de l'histoire narrative de l'enfant ?
Cela veut-il dire que l'accouchement comme fondement de la maternité serait remis en cause, y compris au stade de contentieux, pour les couples lesbiens ayant eu recours à un don de sperme ?
Par ailleurs, l'autre difficulté technique importante qui découle de l'indivisibilité des filiations est que la seconde mère est obligée pour pouvoir établir sa filiation de communiquer à l'officier d'état civil la reconnaissance conjointe prénatale qui établira également la filiation à l'égard de la femme qui accouche, quand bien même celle-ci aurait demandé le secret, c'est-à-dire aurait souhaité accoucher sous X. Cette situation soulève une vraie question quant au recul des droits des femmes à pouvoir recourir à l'accouchement sous X - protecteur à la fois de la santé des femmes et des enfants.
Une question se pose également concernant l'accès au juge. La filiation de la femme qui accouche est établie automatiquement sur présentation du document, et non à l'issue d'une procédure devant le juge qui offre de nombreuses garanties importantes, notamment au regard de l'article 6 de la Convention européenne des droits de l'homme.
Au vu de ces difficultés techniques, il me semble qu'il serait plus simple de garder le système actuel mis en place en 1994 pour les couples hétérosexuels ayant recours à un don de sperme, qui a d'ailleurs fait ses preuves, comme nous pouvons le constater au regard du très faible nombre, voire de l'absence de contentieux.
Cela nous conduit à la quatrième option, l'extension du dispositif actuel dont bénéficient les couples hétérosexuels. Celui-ci consiste à recueillir, préalablement à la mise en oeuvre de l'AMP avec don de gamètes, le consentement du couple devant notaire. Auparavant, les couples concernés avaient le choix entre le notaire et le juge, ce qui offrait la possibilité d'un accès gratuit au recueil du consentement.
Ce consentement exerce une fonction importante d'information des parents quant aux conséquences du recours au don de gamètes en matière de filiation. Ces conséquences sont de deux natures : d'une part, la possibilité d'établir la filiation à l'égard de l'homme uniquement sur la base de son consentement au don - quand bien même il ne serait pas le géniteur, il serait le père - et, réciproquement, l'impossibilité d'établir la filiation à l'égard du géniteur, c'est-à-dire du donneur de sperme.
Ce dispositif pourrait être étendu aux couples de femmes qui pourraient, de la même manière, consentir devant notaire, voire devant le juge, au recours au don de gamètes, et qui, sur présentation de ce consentement à l'officier d'état civil, pourraient établir la filiation de la seconde femme par reconnaissance.
Il est important que le consentement puisse être présenté à l'officier d'état civil. En effet, il ne s'agit pas de permettre à tous les couples de femmes d'établir leur filiation à l'égard d'un enfant, mais bien de rendre possible, uniquement dans le cas d'un recours au don de sperme, un double établissement de la filiation maternelle dès la naissance : pour la femme qui accouche, par la mention de son nom dans l'acte de naissance, et pour l'autre femme par reconnaissance - y compris devant l'officier d'état civil qui pourrait annexer le consentement au don à l'acte de naissance pour en prouver la légalité.
Cette solution permet de préserver l'intérêt de l'enfant, sa filiation étant établie dès la naissance à l'égard des deux femmes. De plus, elle assure une certaine souplesse du droit, la filiation étant divisible entre les deux mères et elle rend possible le recours aux mécanismes de droit commun mis en oeuvre sans difficulté depuis 1994 et qui ne créent aucun effet de stigmatisation à l'égard des enfants qui ne sont pas responsables de l'orientation sexuelle de leurs parents.
Par ailleurs, d'un point de vue légistique il s'agit de la solution la plus simple, qui demande le moins de modifications, la plus rapide et la plus efficace. Or la rapidité et l'efficacité sont les deux critères mis en avant par la Cour européenne des droits de l'homme (CEDH) dans son avis du 10 avril 2019 concernant la filiation.
Mme Élisabeth Doineau , présidente . - Je vous remercie.
Mme Muriel Jourda , rapporteur . - Si le droit de la filiation est basé en partie sur la volonté - nous avons beaucoup parlé de reconnaissance -, celle-ci est néanmoins bridée par la vraisemblance, voire la vérité. Or, dans le projet qui nous est présenté, une filiation semble uniquement basée sur la volonté, à savoir la filiation de la mère qui n'accouche pas.
Aucune vraisemblance ne s'exerce sur ce point, deux femmes ne pouvant pas avoir un enfant. Cette femme est donc mère par un pur acte de volonté, et nous ne pouvons pas prétendre que ce couple a pu avoir cet enfant.
Comment pourrions-nous empêcher des évolutions ultérieures, à l'image des demandes de projet parental à trois - avec une mère qui accouche, et deux autres personnes reconnaissant l'enfant par un pur acte de volonté - exprimées dans certains pays ?
M. Jean-René Binet . - La filiation du titre VII est effectivement fondée sur la vérité ou la vraisemblance. La présomption de paternité, comme la reconnaissance, est soutenue par la vraisemblance : il est vraisemblable que le mari de la mère soit le père de l'enfant, et il est vraisemblable que l'homme qui reconnaît un enfant en soit le père.
Si l'on brise le paradigme actuel du titre VII fondé sur la vraisemblance, nous ne voyons pas quelles limites conceptuelles pourraient s'opposer à des hypothèses de pluriparentalité. Lors des débats à l'Assemblée nationale, certains députés ont d'ailleurs suggéré d'aller plus loin dans cette logique en permettant l'établissement d'un lien de filiation pour plus de deux personnes. Si la volonté sert de fondement, rien ne l'empêcherait. Comment nous assurer que nous n'irons jamais vers cela ? Je l'ignore. C'est tout le risque d'un changement paradigmatique, qui nous fait basculer par définition dans un monde inconnu.
Mme Marie Mesnil . - Le changement de paradigme ne me semble qu'apparent, en l'espèce, puisque nous gardons les mêmes conditions de réalisation de l'AMP, que ce soit pour un couple de femmes ou un couple hétérosexuel.
La possibilité pour la femme qui ne porte pas l'enfant de fournir ses ovocytes à l'autre dans l'hypothèse où celle-ci n'en aurait pas d'assez bonne qualité a été écartée tant par le Gouvernement que lors des débats parlementaires. C'est ce que l'on appelle la technique de réception des ovocytes de la partenaire (ROPA). Or cette possibilité fournirait un fondement biologique y compris à l'égard de la seconde mère. Mais ce système n'emporte pas l'adhésion des députés. Je ne sais pas ce qu'il en sera pour vous.
La filiation repose effectivement, pour la seconde femme, uniquement sur la volonté, aussi parce qu'on la prive de la possibilité d'utiliser les gamètes présents au sein du couple, même lorsque ce serait médicalement possible, et alors que cela procurerait un fondement biologique à cette filiation.
Quant aux limites, ce sont vous qui les fixez. Elles sont fixées par la loi. Or, actuellement, le texte ne prévoit aucunement la possibilité de recourir à l'AMP et d'établir un lien de filiation au-delà de deux personnes.
M. Hugues Fulchiron . - Le droit de la filiation se construit à partir de la biologie, de la volonté, du vécu et du cadre retenu pour structurer la société. La place essentielle accordée à la volonté dans le projet de loi, sur la base de l'idée de projet parental qui ferait la filiation de l'enfant, m'inquiète.
Nous pouvons effectivement imaginer d'autres hypothèses, par exemple que notre droit s'ouvre à des possibilités de pluriparenté, sur lesquelles je ne me prononcerai pas. Cela existe déjà dans certains pays, notamment pour le cas où un couple de femmes a recours aux gamètes d'un ami pour réaliser son projet parental.
Se pose alors la question de la place de cet homme qui souhaiterait établir sa filiation. Ce genre d'hypothèse est donc envisageable. Certains systèmes juridiques la reconnaissent. Et si l'on reconstruit le droit uniquement sur la volonté, il est en effet facile de l'englober techniquement.
Mme Corinne Imbert , rapporteure . - Que pensez-vous de l'AMP post mortem, c'est-à-dire du fait d'autoriser une femme dont le couple poursuivait un projet parental par voie d'AMP et dont le mari est décédé avant l'implantation de l'embryon à mener à bien ce projet, sachant que cette AMP se ferait dans des conditions particulièrement douloureuses pour la femme concernée ?
M. Hugues Fulchiron . - C'est peut-être un regret que j'éprouve devant ce projet de loi : qu'ait été écartée l'AMP post mortem dans des conditions strictement définies. Les affaires que nous avons pu connaître par le passé, notamment celles qui ont été jugées par le Conseil d'État, mais aussi d'autres affaires plus anciennes, montrent que, dans certaines circonstances, alors que le processus médical du projet parental a déjà commencé, il est d'une cruauté particulière de ne pas permettre à la femme de le poursuivre, notamment lorsque des embryons ont été conçus.
Je serais assez favorable à l'idée d'étudier les systèmes existants à l'étranger qui autorisent, dans un cadre très strict et dans des délais limités, l'accomplissement de ce projet dès lors que l'on est certain qu'il était parfaitement voulu, mûri, construit et que seul le décès de l'homme l'a interrompu alors qu'il était en cours de réalisation.
Mme Marie Mesnil . - Je partage en très grande partie ces propos. La question de l'AMP post mortem se pose avec d'autant plus de force que le projet de loi prévoit l'ouverture de l'AMP aux femmes seules. Ces femmes pourraient donc recommencer un projet parental seules, ce qui impliquerait de reprendre la chose du début. Mais elles ne pourraient pas utiliser les ressources biologiques déjà disponibles, notamment le sperme de leur conjoint décédé. Cela soulève la question du consentement de celui-ci à son utilisation post mortem, mais nous pourrions envisager un système où soit ce consentement est prévu explicitement soit l'homme en question est informé explicitement qu'en cas de décès son sperme pourrait être utilisé par sa compagne.
De plus, la qualité et la quantité des ovocytes diminuent fortement avec le temps. La possibilité d'utiliser ces ovocytes se trouverait donc dans ce cas reportée dans le temps, à raison de plusieurs mois, voire de plusieurs années, selon la capacité des centres d'études et de conservation des oeufs et du sperme (CECOS) à prendre en charge cette femme. Le seul matériel génétique disponible restant pourrait être ses embryons auxquels on lui empêcherait d'accéder.
Par ailleurs, cette femme pourrait consentir à ce que ces embryons soient accueillis par un autre couple. L'enfant devenu majeur pourrait alors la retrouver dans le cadre de la levée de l'anonymat, et se présenter chez elle avec le matériel génétique de son conjoint décédé. Elle pourrait même le souhaiter pour pouvoir rencontrer l'enfant qu'elle aurait pu avoir.
M. Jean-René Binet . - Je me permettrai d'émettre sur ce point une voix discordante. Si l'on envisage les choses sous l'angle de la compassion que l'on doit à la femme durement éprouvée par le décès de son mari ou de son compagnon, nous ne pourrions qu'être enclins à lui apporter ce secours. Mais il faut avoir conscience de plusieurs réalités.
Tout d'abord, il existerait un risque de confusion de sentiments. Le deuil est une épreuve. Ajouter au deuil la possibilité ou non du transfert des embryons est une question qui peut difficilement s'appréhender dans un tel moment. Cela supposerait nécessairement de retarder dans le temps l'implantation des embryons. Cette démarche réglerait une question et en susciterait une autre. L'enfant viendrait au monde plusieurs mois, voire une année ou deux après le décès de son père. Et il arrivera un moment où cet enfant prendra conscience du fait qu'il est né bien trop longtemps après le décès de son père pour que les choses aient été tout à fait ordinaires. Comment un enfant pourra-t-il se construire en s'imaginant avoir été procréé par un mort ? En effet, au moment où il a commencé sa vie utérine, son père était mort depuis déjà six mois au moins. C'est une question terriblement compliquée.
Lors de la précédente révision de la loi bioéthique, l'idée de l'AMP post mortem avait été avancée dans le projet de loi. Or les citoyens réunis en états généraux de la bioéthique avaient estimé qu'il n'existait pas de bonne solution en la matière. S'appuyer sur le fait que des enfants naissent orphelins pour favoriser la naissance d'enfants orphelins ne paraissait pas un bon argument méritant d'être suivi par le législateur.
Je crois que nous faisons face à une question éthique très compliquée, qui résulte de l'existence d'embryons congelés. Peut-être pourrions-nous nous interroger sur la pertinence de la poursuite de cette congélation, sachant que l'évolution des techniques permettrait de s'en passer.
C'est l'interdiction de cette technique en Italie qui a rendu possible la réussite de la cryoconservation des ovocytes. Aujourd'hui, avec la vitrification ovocytaire, est-il vraiment pertinent de continuer à congeler des embryons, d'autant que les recommandations actuelles tendent vers le transfert d'un ou de deux embryons seulement ? En avoir en stock ne semble donc pas indispensable. Et en l'absence de stock la question du transfert d'embryons post mortem ne se posera plus.
Mme Muriel Jourda , rapporteur . - La question délicate de l'implantation post mortem de l'embryon n'entre-t-elle pas en contradiction avec le droit pour un enfant à être élevé par ses deux parents ?
M. Hugues Fulchiron . - Je ne pense pas que l'on puisse invoquer un tel droit, qui n'existe que si les deux parents sont vivants. C'est en tout cas dans cette perspective que la CEDH l'a posé dans sa jurisprudence. Je ne crois donc pas que l'on puisse en inférer une interdiction de donner la vie, dans cette hypothèse particulière, à un enfant dont l'un des parents serait décédé.
Mme Muriel Jourda , rapporteur . - Pourriez-vous développer le point que vous avez évoqué plus haut concernant l'inégalité de droit qui se présenterait entre les enfants nés d'une AMP réalisée au sein d'un couple de femmes et les enfants nés d'une AMP au sein d'un couple hétérosexuel ?
M. Hugues Fulchiron . - Dans le système actuel, une inégalité se présenterait. En effet, l'enfant né d'une AMP au sein d'un couple de femmes aurait la possibilité d'accéder à la connaissance de ses origines, alors que, dans un couple hétérosexuel, le secret serait maintenu. Tout dépendrait donc de ce que les parents lui diraient, car ils resteraient maîtres du secret. Ce n'est qu'une fois ce secret levé sur les circonstances de sa conception que l'enfant pourrait bénéficier du système mis en place par la loi lui donnant accès à ses origines.
Si je comprends très bien cette inégalité sur le plan humain, elle me paraît difficile à justifier sur le plan juridique. Les parents ont-ils un droit au secret, que l'on opposerait au droit de l'enfant à connaître ses origines ? Je ne le crois pas. Et si nous faisons la balance entre les droits et intérêts en présence, il me semble que le droit de l'enfant doit l'emporter.
Cela est très difficile à entendre pour certains couples hétérosexuels ayant eu recours à l'AMP et qui souhaitent conserver la maîtrise du moment de la révélation du secret, s'ils le révèlent. Mais personne ne la leur enlèverait. En revanche, il me semble qu'il serait préférable, au regard de l'égalité entre les enfants, que, dans toute hypothèse, l'enfant ait un égal accès à la connaissance de ses origines.
Mme Muriel Jourda , rapporteur . - Une modification du titre VII serait-elle alors nécessaire ?
M. Hugues Fulchiron . - Dans ma perspective, cette disposition s'inscrit dans une refonte des règles relatives à la filiation des enfants nés, de façon générale, par AMP - dans les couples hétérosexuels comme dans les couples homosexuels.
Mme Marie Mesnil . - Il faut avoir en tête que la solution proposée aujourd'hui est une solution amoindrie, qui concerne uniquement les couples de femmes. Mais le rapport intitulé Filiation, origines, parentalité - Le droit face aux nouvelles valeurs de responsabilité générationnelle, du groupe de travail « Filiation, origines, parentalité » présenté par Irène Théry et Anne-Marie Leroyer en 2014, et précédemment le rapport intitulé Accès à la parenté : assistance médicale à la procréation et adoption publié par Terra Nova en 2010, proposaient initialement une refonte de la filiation sur le modèle de l'adoption pour tous les enfants conçus par don de gamètes. L'idée était de faire en sorte que leur filiation traduise le recours à ce don, pour remettre en cause le secret.
Or nous nous trouvons face à un mode d'établissement de la filiation qui a été pensé pour révéler le recours au don de sperme ou d'ovocytes, mais qui n'est appliqué qu'aux couples de femmes - seuls à même de ne pas pouvoir cacher le recours au don de gamètes. Leurs enfants pourront donc plus facilement s'ils le souhaitent solliciter la commission ad hoc pour obtenir des informations sur le donneur.
Toutefois, l'accès à la connaissance des origines est ouvert dans le projet de loi à toute personne ayant été conçue par don de gamètes, ce qui suppose de savoir préalablement que l'on a été conçu ainsi.
Peut-être faudrait-il éclaircir ce point dans le texte de loi, pour que toute personne qui le souhaite ait la possibilité de solliciter la commission afin de savoir si elle a été conçue par don de gamètes avant même de demander des informations sur l'identité d'un éventuel donneur. Cette commission pourrait alors être utilisée comme voie d'accès à un registre.
En revanche, la solution consistant à étendre le mode d'établissement de la filiation à tous les enfants conçus par don de gamètes, qui révélerait leurs conditions de conception, soulève la question importante du respect du secret médical. Ce n'est pas la place du recours au don de gamètes de figurer à l'état civil. Ce n'est pas le rôle de la filiation que de traduire l'existence ou non d'un don de gamètes. Il s'agit d'une confusion contre laquelle il faut se battre.
Certes, une inégalité se présente, mais elle résulte simplement d'une différence de situations. Les enfants ne sont pas placés dans la même situation dans l'un et l'autre cas.
M. Jean-René Binet . - Dès l'origine, l'anonymat du don de gamètes pose problème. Si la solution de l'anonymat a été retenue dès l'origine de l'AMP, c'est en raison d'une fausse analogie avec le don de sang. Le don de sang étant anonyme, il a été considéré que le don de gamètes devait l'être également. Pourtant, les conséquences ne sont pas les mêmes. La naissance d'un enfant, qui pourra un jour vouloir savoir d'où il vient, mérite amplement des solutions différentes.
Il ne faudrait donc pas que le don de gamètes soit anonyme, ou qu'il le soit le moins possible. Il faudrait favoriser la levée de cet anonymat, de la manière la plus générale et la plus simple qui soit.
M. Olivier Henno , rapporteur . - Merci pour la qualité de vos exposés. C'est la matière et la richesse du droit que d'avoir des interprétations différentes.
J'ai bien aimé la formule employée par le professeur Nisand le 27 novembre : nous pouvons faire beaucoup de choses pour faire naître un enfant, à condition qu'on lui fasse connaître tout ce qu'on a fait pour qu'il puisse naître.
Les pays européens voisins où l'AMP existe déjà ont-ils fait évoluer leur droit de la filiation, ou ce droit reposait-il sur d'autres principes que le nôtre et appelait donc des évolutions moins nombreuses et moins importantes ?
M. Hugues Fulchiron . - Les solutions sont très différentes selon les pays. Une grande diversité de choix a été effectuée, généralement entre trois options. La première est celle de l'adoption. Nous restons là dans un cadre très classique. La deuxième a trait à l'extension des règles de filiation par procréation aux hypothèses de procréation avec AMP. Et de nouveaux systèmes ont pu aussi être imaginés.
De nombreuses solutions existent donc, mais une grande prudence est de mise en matière de droit comparé. En effet, il faut prendre en compte l'ensemble du système de filiation dans lequel s'inscrivent ces solutions. Le droit comparé est très éclairant, mais il convient de le mettre en regard des principes du système français de la filiation. À titre d'exemple, l'attachement que nous avons pour la filiation procréative à ce que nous appelons la possession d'état - le vécu - est invraisemblable dans d'autres systèmes juridiques.
Si le droit comparé est utile, il faut toujours le resituer dans notre système français, a fortiori dans ces matières où s'expriment des particularismes très forts au-delà de la lettre de la loi.
Mme Marie Mesnil . - Pour donner quelques exemples, la Belgique et l'Autriche ont étendu sans difficulté le système de droit commun qui s'appliquait aux couples hétérosexuels aux couples de femmes. Il en va de même, outre-Atlantique, pour le Québec. D'autres États mentionnent le recours aux dons, notamment l'Irlande ou encore l'État de Victoria en Australie.
Le droit comparé peut certes fournir des éléments supplémentaires, mais il en apporte moins que l'expérience que nous pouvons avoir de systèmes ayant fait leur preuve en France.
Mme Laurence Rossignol . - Je considère, pour ma part, qu'il n'existe pas de totale similarité possible entre des enfants nés d'un couple hétérosexuel et des enfants nés d'un couple homosexuel, pour une raison de vraisemblance. Ce n'est peut-être pas là qu'il convient de chercher la résorption de ce qui est moins une inégalité qu'une différence. On ne peut pas toujours confondre différence et inégalité.
Le système prévu dans le projet de loi crée-t-il, comme je le crois, une différence entre les enfants nés par AMP dans un couple lesbien et ceux nés par ce que les Québécois appellent « l'assistance amicale à la procréation » (AAP) ? N'est-ce pas là la différence qu'il conviendrait de résorber ?
Par ailleurs, je suis très sensible aux propos de Mme Mesnil sur le risque de l'effacement de la filiation maternelle par accouchement dans une confusion des deux filiations maternelles. Cela me semble très important.
Dès lors que la filiation maternelle est une filiation par accouchement et la filiation paternelle une construction sociale par présomption de paternité, serait-il possible de dupliquer la construction employée dans le cadre de la distinction entre filiation issue du mariage et filiation hors mariage pour l'appliquer aux couples lesbiens ? Nous mettrions alors en place une présomption de co-maternité pour l'épouse de la mère et un acte de reconnaissance pour la compagne de la mère lorsqu'il s'agit d'une filiation hors mariage.
Si la question de savoir s'il existe un droit au secret pour les parents semble pertinente, une question parallèle se pose : existe-t-il un droit, pour l'État, d'interdire le secret ? Comment qualifierions-nous un État qui interdirait le secret des parents ? La première question relève du droit de la famille, la seconde du droit politique.
Enfin, ne pensez-vous pas que la question de la multiparentalité - au-delà de deux personnes -, que l'on aborde toujours du point de vue de l'homoparentalité, ne devrait pas plutôt nous guider dans une réforme de l'adoption ?
M. Jean-René Binet . - Le projet de loi n'offre pas de place particulière à la question de l'assistance amicale à la procréation. Car cette question est hors du droit. Soit les conditions posées par la loi sont remplies et les conséquences peuvent en être tirées, soit les individus se placent hors du droit et ne peuvent alors espérer tirer les conséquences prévues pour d'autres situations.
Le projet pourrait envisager néanmoins cette question, mais cela impliquerait la mise en place d'une sorte de système optionnel. En ce cas, soit les couples auraient recours au don de gamètes dans un centre, soit ils trouveraient dans leur cercle amical une personne susceptible de les aider.
Le risque serait alors de voir jouer l'article 323 du code civil, qui interdit toute renonciation aux actions en filiation par anticipation. Rien n'empêcherait celui qui a donné amicalement ses gamètes de revendiquer sa paternité sur l'enfant. Pour l'instant, ces questions sont telles qu'elles existent, c'est-à-dire hors du droit.
Serait-il possible d'étendre aux couples de femmes la présomption de paternité prévue à l'article 312 et la reconnaissance de paternité prévue à l'article 316 ? Non, car en l'état toutes deux sont fondées sur la vérité - une vérité qui n'a pas besoin d'être démontrée, mais qui servira d'arbitre en cas de contentieux. En cas de contentieux - reconnaissance mensongère, contestation de la filiation à l'égard du mari de la mère, etc. -, c'est la preuve biologique qui servira à arbitrer. Nous ne pouvons donc pas étendre simplement ces deux règles aux situations dont nous parlons. Car cela impliquerait de revoir toutes les actions relatives à la filiation. C'est la cohérence du titre VII qui est ici en jeu.
S'agissant du droit au secret des parents ou du droit de l'État à interdire le secret, j'avoue ne pas avoir de réponse pertinente à donner.
Par ailleurs, penser la multiparenté dans le cadre de l'adoption reviendrait à poser la même question que le fait de la penser dans le cadre de la filiation du titre VII. Nous sommes toujours dans le paradigme d'un double lien de filiation. Et penser la multiparenté conduirait à sortir de ce cadre. Je crois donc qu'il faut penser les choses globalement, et ne pas imaginer que le fait de penser cette question dans le cadre de l'adoption pourrait tout régler - sauf à l'envisager au sens de l'adoption simple, où l'on maintient les liens de l'enfant à l'égard de sa famille d'origine tout en lui offrant des liens à l'égard de sa famille adoptive.
Mme Marie Mesnil . - La question de l'assistance amicale à la procréation se trouve plutôt à mon sens en dehors du cadre médical. Le projet de loi pourrait tout à fait appréhender cette hypothèse.
La difficulté est qu'en pareil cas l'ami qui fournirait son sperme pourrait établir sa filiation et faire obstacle à l'établissement de la filiation de la seconde femme. En effet, s'il est démontré que l'enfant n'est pas issu du tiers donneur il serait possible pour le géniteur de remettre en cause la filiation à l'égard de la seconde mère et d'établir sa propre filiation.
Dans l'hypothèse où un couple de femmes irait devant notaire faire une reconnaissance prénatale conjointe, et finalement préférerait avoir recours à un ami plutôt que se rendre dans un centre de PMA, ces femmes pourraient tout à fait établir leur filiation par le biais de la reconnaissance prénatale conjointe - puisque l'on ne vérifierait pas que l'enfant est bien issu du don. Mais si l'homme se manifeste, en tant que géniteur il aurait le droit d'établir sa paternité, ce qui remettrait en cause la filiation des deux femmes - si la filiation est indivisible au stade de son établissement. La femme qui accouche devrait donc établir sa filiation à la suite de cette remise en cause, à titre contentieux, sur le fondement de l'accouchement.
La difficulté est que le nouveau système ne pense pas toutes les situations potentiellement problématiques, notamment les conflits possibles entre les femmes. Nous ne pouvons pas idéaliser la conjugalité lesbienne. Les cas de recours à l'adoption de l'enfant d'une conjointe le montrent bien : il existe des situations de rupture dans lesquelles la filiation n'est pas établie. Et toutes ces situations conflictuelles soulèveraient des difficultés au regard du mécanisme d'indivisibilité des filiations maternelles.
S'agissant de la question de l'extension du droit commun, il ne s'agit pas d'ouvrir tout le titre VII aux couples lesbiens, ni de permettre à toutes les femmes d'établir un lien de filiation par présomption de co-maternité ou par reconnaissance, mais d'étendre le droit commun uniquement à celles qui peuvent justifier d'un recours à un don de gamètes en présentant l'acte notarié.
Il n'est donc pas question de remettre intégralement en cause le système du titre VII qui serait fondé sur la vraisemblance biologique, mais uniquement d'étendre le dispositif existant déjà pour les couples hétérosexuels ayant recours à un don de sperme, pour lesquels nous savons qu'il n'existe pas de vérité biologique. Dans ce cas, le fondement de la filiation paternelle est le consentement au don. Le même fondement s'appliquerait pour la seconde femme au sein d'un couple lesbien.
Par ailleurs, la question du secret est importante pour les couples hétérosexuels ayant eu recours à un don de gamètes. Une proposition faite devant l'Assemblée nationale visait à permettre aux parents qui le souhaitaient de refuser que les centres de PMA procèdent à l'appariement. Dès lors, les centres ne choisissaient pas le donneur de sperme ou la donneuse d'ovocytes en fonction du phénotype du parent stérile, donnant ainsi aux parents la possibilité d'échapper au mimétisme biologique visant à créer un enfant le plus ressemblant possible pour maintenir le secret. L'idée était de laisser aux parents non le choix du donneur, mais le choix de l'aléa - soit le choix le plus proche de la biologie.
S'agissant de la multiparenté, il est déjà possible d'avoir trois parents, dans le cadre de la procédure d'adoption de l'enfant du conjoint. En revanche, en cas de recomposition familiale, si chacun des parents a refait sa vie, un seul des deux parents peut établir un lien de filiation. Des évolutions pourraient s'avérer nécessaires sur ce point.
M. Hugues Fulchiron . - Il ne faut effectivement pas confondre différence et inégalité. Toute différence de traitement n'est pas forcément discriminatoire.
Cependant, au regard des problèmes en cause, il me semble qu'il existe vraiment une inégalité. L'existence de modes différents d'établissement de la filiation pour un enfant né dans un couple de femmes ou pour un enfant né de façon générale au moyen d'une AMP relève en effet de la différence puisqu'il s'agit de situations différentes.
Mais il en va tout autrement si l'on se place sous l'angle du droit de l'enfant à la connaissance de ses origines en cas de recours à un tiers donneur. Sous cet angle, je ne vois pas en quoi les situations d'enfants nés dans un couple de femmes et d'enfants nés dans un couple hétérosexuel sont différentes. Une différence s'applique en revanche entre la conception effectuée grâce à l'assistance d'un tiers donneur et la conception issue de l'acte procréatif d'un père et d'une mère.
S'agissant de la procréation amicalement assistée, le recours à l'AMP est strictement encadré en France pour de bonnes raisons. Est-il nécessaire de faire entrer indirectement dans la loi des hypothèses se situant en dehors du cadre légal ? Cela me paraîtrait un peu étonnant.
Mme Laurence Rossignol . - Un enfant né par procréation amicalement assistée aura-t-il le même statut et bénéficiera-t-il du même établissement de filiation à l'issue de la loi bioéthique qu'un enfant né d'une AMP ? La filiation à l'égard de la mère « non-accouchante » sera-t-elle établie de la même façon ? Et pouvons-nous nous accommoder de l'idée selon laquelle la technique médicale change le mode d'établissement de la filiation ?
M. Hugues Fulchiron . - La technique médicale ne changerait pas le mode d'établissement de la filiation. Mais nous pouvons imaginer deux femmes ayant eu recours à l'assistance amicale à la procréation et faisant une déclaration devant notaire en prétendant avoir eu recours à l'AMP dans le cadre légal.
En ce cas, la filiation serait toutefois fragilisée, car elle n'entrerait pas dans le cadre légal. Et il s'agit tout de même, de la part de ces femmes, d'une façon de contourner la loi. En effet, elles utiliseraient le procédé mis en place dans le cadre de l'AMP tel que prévu par la loi pour créer de la filiation dans une hypothèse située en dehors du cadre légal. Sans porter aucun jugement de valeur, je trouve que le fait de faire produire ainsi des effets à une déclaration prévue dans un système particulier pour des hypothèses s'étant délibérément placées en dehors de ce système pose problème.
Mme Marie Mesnil . - Faisons le parallèle avec ce que l'on permet aux hommes qui ont recours à l'assistance médicale à la procréation, dans le cas d'un couple hétérosexuel : le consentement devant notaire peut servir à établir de force la filiation de l'homme. Mais si les deux parents, l'homme et la mère, sont d'accord pour qu'il n'établisse pas sa filiation parce qu'ils sont séparés, il n'y a aucune difficulté. Le droit permet donc, en matière de filiation, un peu de souplesse lorsque toutes les parties sont d'accord. Dans l'hypothèse d'une assistance médicale à la procréation concernant deux femmes, le consentement devant notaire pourrait permettre d'établir la filiation, notamment à l'égard de la seconde femme, car c'est là qu'est l'enjeu. Mais pour sécuriser leur filiation, elles devront avoir recours à la procédure de l'adoption de l'enfant de la conjointe : la filiation sera alors inattaquable.
M. Hugues Fulchiron . - Il faut différencier les cas où la loi permet quelque chose et ceux où les personnes se placent dans les interstices de la loi. Ce que vous évoquiez, c'est plus une utilisation des interstices de la loi.
Je considère qu'il est tout à fait néfaste, pour la cohérence et la sécurité juridique de notre système ainsi que la compréhension des problèmes, d'étendre des règles qui ont été pensées, conçues et appliquées pour la filiation charnelle à d'autres types de filiations radicalement différents. Toutes ces règles reposent en effet sur une forme de vraisemblance. Les étendre à des hypothèses dans lesquelles il n'y a aucune vraisemblance serait inutile et dangereux. Il me semblerait beaucoup plus simple et cohérent de construire un système adapté à ces nouvelles configurations de filiation.
Sur la question du droit des parents au secret, je ne vois pas sur quoi fonder ce droit : la convention européenne des droits de l'homme ? La charte des droits fondamentaux ? Les pactes de l'ONU ? Socialement et humainement, les parents ont peut-être cette liberté. Mais juridiquement, ce droit n'existe pas. En revanche, il existe un droit de l'enfant à connaître ses origines. Quant au droit de l'État à imposer le secret, il n'a aucun fondement juridique. L'enfant reste libre d'accéder à ses origines ou non, donc l'État n'impose pas la révélation du secret, il la permet seulement.
La multiparenté existe en adoption, dans le cas par exemple d'une recomposition familiale au sein d'un couple, homosexuel ou hétérosexuel, avec plusieurs personnes qui assument la charge de parents. Le cas est différent lorsqu'il s'agit de la naissance d'un enfant dont la filiation serait plurielle. L'engendrement à plusieurs, à statuts différents, d'un enfant, ce n'est plus la même logique que dans le cas de l'adoption.
M. Dominique de Legge . - Je remercie nos intervenants. Je dois reconnaître que, lorsque l'on constate que le champ des possibles ne recouvre pas le champ de la vraisemblance, nous sommes pris de tournis.
Je voudrais revenir sur la question de la filiation fondée sur la volonté. La volonté peut évoluer. Un projet parental, est-ce élever ensemble un enfant ou reconnaître l'un des parents comme deuxième parent ? Dans la société traditionnelle, même si le projet parental évolue vers une séparation, les parents restent parents. Comment conjuguer la question de la volonté, autour de l'adhésion à un projet parental susceptible d'évoluer, et celle de la filiation, qui doit être permanente ? Ne serait-on pas en train d'inventer l'idée d'une volonté irréversible ?
M. Jean-René Binet . - La volonté peut sembler plus fragile que les liens du sang consacrés par la loi dans le cadre de la filiation du titre VII. Mais on pourrait imaginer que cette volonté devienne définitive et qu'il ne soit plus jamais possible d'y revenir. Si l'on reconstruisait le titre VII autour de l'idée que c'est d'abord la volonté qui fait la filiation avant toute autre considération, il faudrait envisager des conséquences en cascade. Par exemple, aujourd'hui, si une femme tombe enceinte à la suite d'une relation épisodique ou furtive avec un homme, et que cet homme ne souhaite pas faire établir sa paternité à l'égard de l'enfant, la femme peut parfaitement agir au nom de l'enfant aux fins d'établir la filiation paternelle : l'homme ne pourra pas s'abriter derrière le fait qu'il n'a jamais voulu être père de l'enfant. C'est la vérité biologique qui, ici, sert d'arbitre. Demain, dans un système fondé sur la volonté, l'homme pourrait refuser cette filiation. Faire de la volonté l'assise de la filiation aurait donc un certain nombre de conséquences qu'il faut envisager avant de s'engager dans une réforme par petits bouts.
Mme Marie Mesnil . - Pour le moment, personne n'a proposé de faire de la volonté le fondement principal de la filiation du titre VII. Quelles places respectives donne-t-on à la volonté et à la biologie ? On ne peut pas nier que, dans un certain nombre de cas, les enfants sont procréés par leurs parents. Mais quid des autres ? Depuis 1994, les lois bioéthiques encadrent la situation dans laquelle un couple a recours à un don de sperme : jusqu'au moment où les embryons sont implantés, le père est libre de retirer son consentement ; mais une fois que le processus biologique est enclenché et s'il aboutit, son acte de volonté devient irréversible. Le consentement au recours au don de gamètes, donné devant notaire, rend sa paternité inattaquable - sauf à démontrer que l'enfant n'est pas issu de l'assistance médicale à la procréation. Ce couple hétérosexuel dans lequel l'homme est stérile est dans la même situation, au regard de l'assistance médicale à la procréation, qu'un couple lesbien qui a également besoin de recourir à un don de sperme. On pourrait donc établir la maternité de la seconde femme dans les mêmes conditions, sans qu'elle puisse remettre en cause cette filiation qui repose uniquement sur un fondement volontariste.
Le titre VII fait reposer la filiation à titre principal sur la vraisemblance biologique - ce qui ne signifie pas vérité biologique. Mais sortir les enfants conçus par don de gamètes du titre VII, c'est faire comme si la filiation de ce titre reposait uniquement sur la vérité biologique, ce qui est strictement faux. Le système se veut équilibré et permet d'établir des filiations qui existent socialement ; ce n'est que dans les cas contentieux que l'on ira chercher le géniteur. Dans l'hypothèse exposée par Monsieur Binet, l'homme verra sa paternité établie, mais la femme n'est pas obligée de dire qui est le géniteur ; elle peut très bien choisir de laisser le lien de filiation libre, pour éventuellement qu'il soit comblé par une personne avec qui elle fera sa vie et qui jouera le rôle de père ou de mère auprès de l'enfant.
M. Hugues Fulchiron . - L'hypothèse d'un enfant qui naît dans un couple de femmes est la seule hypothèse où il y aurait une filiation fondée sur la volonté. Le rapprochement avec l'hypothèse d'un enfant qui naît dans un couple hétérosexuel grâce à l'assistance médicale à la procréation est quand même très relatif puisque, certes, il y a l'expression d'une volonté, mais les instruments juridiques qui sont mis en oeuvre sont des instruments classiques de la filiation procréative : la présomption de paternité, la reconnaissance, etc. Ce qui fait cette filiation, ce sont les procédés classiques de la filiation procréative, ce n'est pas la volonté du père. Dans le cas d'une procréation médicalement assistée avec deux femmes, la volonté fait la filiation pour le parent biologique et le parent non biologique.
Dans les modes de filiation classiques, la volonté joue un rôle important, mais elle vient au secours de la vérité et fait cette vraisemblance de la filiation qui fonde actuellement la filiation du titre VII.
Pour éviter les confusions, plutôt que de parler de volonté, nous devrions parler d'engagement. Lorsque l'on reconnaît l'enfant, on fait un aveu, au sens juridique du terme - on reconnaît que cet enfant est le sien parce qu'on lui a donné la vie -, et en même temps on s'engage à le reconnaître. Cette notion d'engagement devrait être mise au centre du droit de la filiation, plus que l'idée de volonté qui renvoie à l'autonomie du sujet. Or l'engagement à l'égard de l'enfant auquel on a donné la vie justifie que cet engagement soit irréversible. Une volonté peut être variable, l'engagement est irréversible.
Mme Marie Mesnil . - J'entends « société traditionnelle », « procédés classiques » : les familles formées par des couples de personnes de même sexe font partie de la société au même titre que les autres et les procédés classiques ne devraient pas exclure ce pluralisme familial !
Si le titre VII est réservé aux enfants des couples hétérosexuels, ça veut dire qu'on crée un droit dérogatoire de la filiation. L'intérêt de l'enfant doit primer. Mais est-il de l'intérêt de l'enfant d'avoir un type de filiation différent lié à l'orientation sexuelle de ses parents ? C'est stigmatisant pour les enfants qui, effectivement, sont dans une situation différente, mais qui n'en sont absolument pas responsables. Ils porteront, sur leur acte de naissance, la trace déjà qu'ils ont deux mères, mais en outre, que l'établissement de leur filiation est différent. Les cours de biologie permettent déjà de prendre la mesure de l'impossibilité pour deux femmes de procréer ; je ne pense pas que le droit ait besoin de le signifier à nouveau.
M. Jean-René Binet . - Je n'ai pas proposé de refonder le système de la filiation sur la volonté, mais d'autres l'ont proposé ; c'est la raison pour laquelle je m'en faisais l'écho. Un de nos collègues a écrit un livre sur la famille contractuelle, entièrement fondée sur la volonté, où il envisage justement toutes ces situations à partir de l'assistance médicale à la procréation.
M. Hugues Fulchiron . - Il n'est, bien sûr, pas question de réserver le titre VII à la famille hétérosexuelle, puisque la distinction ne passe pas entre homosexuels et hétérosexuels, mais entre enfants nés d'un acte procréation charnelle et enfants nés grâce à l'assistance médicale à la procréation, qui concerne donc des couples hétérosexuels comme des couples homosexuels : je n'oppose absolument pas les uns aux autres, je pars de l'enfant.
Je crois qu'il est toujours extrêmement périlleux d'invoquer l'intérêt de l'enfant qui n'est pas encore né. Je me place sur le plan des droits de l'enfant. L'intérêt de l'enfant ne peut être apprécié que dans une situation particulière, même si l'enfant n'est pas encore né. Tant qu'il n'est pas né, dire que l'intérêt de l'enfant c'est ceci ou cela est hasardeux.
Je suis toujours un peu perplexe quand on parle de la surstigmatisation de l'enfant : l'enfant risque-t-il de sentir sur stigmatisé ? Je n'en sais rien. Il n'est pas question de marquer l'enfant au fer rouge, mais de dire qu'il est né dans telle ou telle circonstance ; la société porte sur ces questions un regard qui me semble relativement apaisé et qui le sera encore plus lorsque l'enfant aura grandi.
M. Jean-René Binet . - L'intérêt de l'enfant est une notion vague, c'est même la notion vague par excellence et il est toujours périlleux de l'envisager. Cependant, je ne crois pas que l'on doive réserver la question de l'intérêt de l'enfant à l'enfant déjà né ou à l'enfant dans sa situation particulière. Il y a plusieurs niveaux d'appréhension de cet intérêt. Il y a tout d'abord l'intérêt général des enfants dont le législateur est le garant. La question de la gestation pour autrui en offre un parfait exemple : le législateur français estime qu'il n'est pas de l'intérêt de l'enfant d'être l'objet d'une convention qui conduit une femme à le porter, puis à l'abandonner pour qu'il soit accueilli par une autre ; l'article 16-7 interdit ces pratiques. Mais puisque ces pratiques ont cours ailleurs, le juge est parfois confronté à la question particulière d'un enfant ; la Cour européenne des droits de l'homme estime qu'il est de l'intérêt particulier de cet enfant déjà né de voir sa filiation reconnue dans le territoire où il se trouve. On voit bien qu'il y a deux niveaux d'appréciation : l'un appartient au législateur, l'autre au juge ; l'un est général, l'autre particulier. Mais, dans tous les cas, c'est bien de l'intérêt de l'enfant qu'il s'agit.
Mme Élisabeth Doineau , présidente . - Permettez-moi de vous interroger sur l'article 3 relatif à l'accès aux origines. Nous avons entendu hier les professeurs Mattéi, Frydman et Nisand, qui étaient assez unanimement d'accord pour élargir les missions du Conseil national pour l'accès aux origines personnelles (CNAOP) plutôt que de faire appel à une commission spéciale. Qu'en pensez-vous ?
M. Jean-René Binet . - Je partage leur sentiment. Créer une commission simplement chargée d'enregistrer les demandes et de les satisfaire ne me semble pas indispensable. Le CNAOP a démontré sa parfaite maîtrise de ces sujets délicats. Sauf à ce que ce qu'il ne croule déjà sous les demandes et ne puisse en satisfaire de nouvelles, il est le mieux indiqué pour répondre à ce type de besoin.
M. Hugues Fulchiron . - C'est également mon sentiment. Le CNAOP fonctionne bien, il a une certaine expérience et pourrait très bien se voir confier cette mission, en lien avec l'agence de biomédecine qui conservera les données.
Mme Marie Mesnil . - J'ai tendance à plaider pour l'extension des dispositifs qui fonctionnent. Au regard de son expertise, le CNAOP me semble à même de remplir cette nouvelle mission, avec peut-être plus de moyens pour prendre en charge ces nouvelles demandes.
M. Hugues Fulchiron . - Permettez-moi d'évoquer un sujet qui m'inquiète beaucoup et qui ne semble pas susciter beaucoup de réactions dans la société : il s'agit du développement d'agences qui recueillent les données personnelles des individus au travers des tests génétiques. Ces tests sont certes interdits en France, mais ils se développent et des publicités fleurissent sur Internet et même à la télévision française, dans une indifférence quasi générale. Or ces sociétés privées sont en train de recueillir les données de millions de personnes sans aucune garantie réelle pour les intéressés. Elles se constituent des trésors monnayables, mais surtout un pouvoir extrêmement inquiétant par la connaissance des personnes, de leurs ancêtres et de leurs descendants. Peut-être faudrait-il profiter de ce projet de loi pour agir et rendre effective l'interdiction de ces pratiques en France.
Mme Élisabeth Doineau , présidente . - Je vous remercie d'avoir mis l'accent sur cette question. Nous l'avons déjà en partie abordée, mais nous aurons prochainement des auditions qui y seront spécifiquement consacrées.
Je vous remercie de toutes vos réponses qui vont alimenter notre réflexion et je vous invite à répondre par écrit au questionnaire qui vous a été adressé.
Ce point de l'ordre du jour a fait l'objet d'une captation vidéo qui est disponible en ligne sur le site du Sénat .
Mme
Alexandra Benachi,
présidente de la Fédération
française
de centres pluridisciplinaires de diagnostic
prénatal
M. Alain Milon , président . - Nous poursuivons nos travaux sur le projet de loi relatif à la bioéthique avec l'audition de Mme Alexandra Benachi, présidente de la Fédération française de centres pluridisciplinaires de diagnostic prénatal. Cette audition fait l'objet d'une captation vidéo.
Mme Alexandra Benachi, présidente de la Fédération française de centres pluridisciplinaires de diagnostic prénatal . - Je suis présidente de la Fédération des centres pluridisciplinaires de diagnostic prénatal et je commence une quatrième année de mandat. La Fédération française de centres pluridisciplinaires de diagnostic prénatal a été créée en 2007. Les centres pluridisciplinaires de diagnostic prénatal ont, quant à eux, été créés en 1999, à la suite de la loi de bioéthique de 1994. Notre rôle est d'accueillir et de gérer toutes les grossesses pour lesquelles un foetus présente une pathologie.
Depuis 1994, notre métier a beaucoup évolué. À l'époque, le diagnostic prénatal consistait à dépister et diagnostiquer des pathologies d'une particulière gravité, avec l'idée que certaines grossesses pouvaient donner lieu à une interruption.
Aujourd'hui, nous sommes capables de proposer des traitements au foetus afin d'améliorer le pronostic. Cela peut être des traitements simples, tels que des transfusions ou des poses de petits drains, ou des chirurgies plus lourdes. C'est la raison pour laquelle nous avons demandé une modification de la définition de notre spécialité, afin qu'elle ne soit plus exclusivement associée aux interruptions de grossesse. Nous sommes en effet capables de faire beaucoup d'autres choses.
Nous avions également demandé d'autres modifications. Le nouveau texte de loi prévoyait que la patiente devait informer son conjoint avant de prendre une décision. Cela représentait une véritable entrave à l'autonomie des femmes mais aussi un problème de fonctionnement, pour nous, au quotidien. En effet, de nombreuses femmes consultent seules. Devoir demander l'avis du conjoint, qui parfois ne peut pas, ne veut pas venir ou n'existe pas, rend la prise en charge compliquée.
Le diagnostic préimplantatoire (DPI-A) n'entre pas exactement dans notre champ d'intervention mais nous concerne puisque, dans les centres de diagnostic prénatal, les DPI sont également pris en charge. Il n'existe que cinq centres en France, mais le mien, Antoine Béclère, travaille en étroite collaboration avec Necker. Ce sujet n'a pas été retenu par l'Assemblée nationale mais il est très important pour nous.
Un article du texte est relatif aux réductions embryonnaires. Il s'agit de la possibilité d'interrompre le développement d'un foetus dans le cas de grossesses multiples ou hypermultiples afin de réduire les risques liés à la grossesse. Cette technique n'était jusqu'à présent pas encadrée et était réalisée sans surveillance par certains professionnels. Pour nous, il s'agit donc d'une bonne chose.
Vous m'avez aussi posé des questions sur le DPI-HLA. Le DPI-HLA est la possibilité, lorsque l'on fait un diagnostic pré-implantatoire, de sélectionner soit un embryon indemne de la pathologie génétique que transmettent les parents, soit un embryon HLA qui serait compatible avec un enfant atteint en attente d'une greffe. Cela concerne notamment la drépanocytose ou la bêta-thalassémie. Dans le nouveau texte, l'interdiction de réaliser des DPI-HLA est apparue. Nous ne les pratiquions plus en France depuis quelques années mais cette volonté émane des centres, et non pas des professionnels.
Mme Corinne Imbert , rapporteure . - Merci pour vos propos introductifs. Vous avez demandé la modification de votre activité professionnelle. Êtes-vous satisfaite de la définition qui apparaît aujourd'hui dans le projet de loi ? Vous venez d'évoquer le DPI-HLA. Pouvez-vous nous expliquer l'intérêt de cette technique ? Que pensez-vous de la position de l'Assemblée nationale ? Je souhaiterais revenir sur le diagnostic préimplantatoire. Quel intérêt existe-t-il à le proposer ?
Mme Alexandra Benachi . - Le texte adopté est meilleur que celui déposé à l'Assemblée nationale. Toutefois, le terme « dépistage » a disparu. Le « diagnostic prénatal » a été remplacé par « médecine foetale ». C'est une bonne chose, parce que c'est beaucoup plus large, la médecine foetale incluant le diagnostic et le dépistage. Cependant, dans notre spécialité, le spectre de l'eugénisme revient systématiquement. Je le comprends mais le dépistage est autorisé dans notre pays. C'est le cas du dépistage de la trisomie 21. Des échographies non obligatoires, mais recommandées, permettent de déceler des anomalies au cours de la grossesse. Le but de ces échographies est de dépister des anomalies ou des retards de croissance chez le foetus. Nous aurions souhaité que le terme « dépistage » soit également inclus. Nous avions fait une proposition et demandé que la notion de particulière gravité soit retirée afin que les dépistages puissent être élargis. Une anémie chez un foetus ne représente pas une particulière gravité mais peut s'aggraver si l'on ne fait rien. Dans ce cas, une transfusion par le cordon ombilical est nécessaire, sans entraîner une interruption de grossesse.
La dernière partie de la phrase de cet article est identique au texte sur l'interruption de grossesse. C'est un problème. Lorsque l'on propose un traitement chirurgical à une patiente pour son bébé atteint d'une pathologie d'une particulière gravité, elle peut demander l'interruption de grossesse. En laissant ce terme dans le texte de loi, nous pourrions nous retrouver en difficulté. C'est la raison pour laquelle nous avions demandé que le terme de « particulière gravité » soit retiré de la définition.
Concernant le DPI-HLA, seule l'équipe de Necker-Béclère le pratiquait. Nous avons cessé de la faire car c'était extrêmement lourd. La pratique est très régulée. Pour chaque demande d'une patiente, il fallait demander l'autorisation à l'Agence de la biomédecine et la loi française ne nous autorise pas à refaire une tentative s'il existe des embryons sains pour la pathologie. C'est-à-dire que l'on part du principe qu'un couple va demander un DPI-HLA parce qu'il souhaite un nouvel enfant indemne de la pathologie. Si l'enfant peut être compatible avec l'enfant malade qui attend sa greffe, tant mieux. Mais le but du DPI, c'est d'avoir un enfant sain. Quand on faisait le DPI, s'il n'y avait que des embryons sains de la pathologie, mais incompatibles HLA, les patientes refusaient de faire l'implantation de l'embryon.
Au départ, leur souhait était d'avoir un enfant sain, mais nous savons très bien que c'était aussi pour avoir un enfant compatible avec le premier enfant. La loi française ne nous autorise pas à refaire une tentative tant qu'il y a des embryons sains au congélateur. Pour faire simple, nous avions des embryons sains au congélateur et des couples qui refusaient le transfert. Cela n'avait donc pas de sens de monter un projet aussi lourd.
Nous nous sommes aussi rendu compte que, faute de moyens, il y a presque deux ans d'attente pour ce qui relève de la biologie moléculaire. Face aux coûts et à la lourdeur du processus, les patients faisaient plus simple et sans nous. Nous n'avions toutefois pas demandé le retrait de cette possibilité, afin que les patientes puissent avoir accès à cette technique remboursée en Europe, notamment en Belgique ou en Espagne.
Le DPI-A intervient lors d'une procédure d'assistance médicale à la procréation. Aujourd'hui, on réimplante les embryons sans regarder s'il existe des anomalies chromosomiques. Or, plus une patiente vieillit, plus ses embryons seront porteurs d'anomalies chromosomiques. Généralement, quand un embryon est porteur d'anomalies, il ne s'implante pas ou conduit à une fausse couche.
Le DPI-A permet d'améliorer les performances de la fécondation in vitro. C'est efficace chez les patientes entre 35 et 42 ans et cela permet d'obtenir un enfant plus vite. Cela limite ainsi le nombre de tentatives de stimulation. Pour certains c'est une technique qui sélectionne des embryons indemnes d'anomalies chromosomiques. Pour moi, cela me permet de ne pas stimuler plusieurs fois une patiente déjà âgée, et d'éviter de l'exposer à des risques de thrombose ou de cancer à long terme.
Il y a donc deux éthiques qui s'affrontent : la crainte de la sélection des embryons et le souhait d'éviter aux patientes de subir des stimulations à répétition, des fausses couches qui peuvent également être extrêmement lourdes à porter. La plupart de ces embryons porteurs d'anomalies se termineront en fausses couches ou ne s'implanteront pas. C'est donc une fausse idée de dire que l'on va sélectionner des embryons.
La trisomie 21 représente 3 % des anomalies chromosomiques. En France, le dépistage de la trisomie 21 est autorisé pour les patientes qui souhaitent en bénéficier. Si l'enfant est porteur de l'anomalie et si la patiente le souhaite, elle pourra faire une interruption de grossesse. C'est le choix de nombreuses patientes en France. Pourquoi faire supporter tout cela aux patientes alors que nous disposons de cette technique, qui serait accessible ? La littérature nous démontre qu'elle est efficace, économiquement souhaitable et qu'elle va dans le sens de l'intérêt des patientes âgées de 35 à 42 ans.
Une demande de projet de recherche a été déposée par un de nos collègues. Il s'agit d'essayer de convaincre tout le monde afin d'évaluer la technique en France puis de l'autoriser dans deux ans si ce projet fonctionne. Cela permettrait d'évaluer la situation. Si le projet n'est pas adopté en 2020, il faudra attendre cinq ans avant la prochaine loi. En termes de recherche, nous allons perdre énormément. Je me rends régulièrement dans des congrès et je constate à quel point, n'ayant pas accès au DPI-A, nos résultats d'assistance médicale à la procréation (AMP) sont moins bons. Même les spécialistes français ne pourront plus publier. Je pense vraiment que ce n'est ni un problème d'eugénisme ni de sélection d'embryons puisque la plupart d'entre eux ne donneront pas naissance à un enfant.
M. Olivier Henno , rapporteur . - Madame, vous êtes convaincante. Certains défendent des alternatives thérapeutiques au DPI-HLA. Qu'en pensez-vous ?
Pourriez-vous développer la question des centres de diagnostic prénatal, leur répartition sur le territoire, et leur activité ?
Mme Alexandra Benachi . - Il n'existe pas d'alternative thérapeutique identique. Il est possible d'avoir recours à un donneur extérieur mais, avec un patrimoine génétique différent, les chances sont moindres pour l'enfant.
Concernant la répartition des centres de diagnostic prénatal, le document que je vous ai remis synthétise les informations. Il existe une prépondérance des centres dans la région parisienne et quelques régions, comme Bordeaux, sont moins bien dotées.
L'Agence de la biomédecine est notre instance de tutelle et nous lui présentons chaque année les résultats de notre activité. Son rapport annuel détaille les actes, le nombre d'interruptions de grossesse, de patients pris en charge, de gestes réalisés. Dans l'ensemble, les actes sont bien répartis sur le territoire. La Fédération a également un rôle de coordination et offre la possibilité aux responsables de se retrouver deux fois par an pour discuter des protocoles, échanger. Nous essayons d'homogénéiser la prise en charge et les protocoles de soins.
Les centres de diagnostic prénatal qui font de la chirurgie in utero sont en revanche mal répartis sur le territoire. C'est toutefois une activité de niche, qui représente moins de 800 chirurgies en incluant les transfusions, les lasers pour les jumeaux, les poses de drains. C'est peu par rapport aux 750 000 naissances annuelles.
Les centres maladies rares, créé il y a quelques années, nous permettent de regrouper des compétences. Il ne serait pas opportun de les répartir sur le territoire car il s'agit de pathologies qui demandent une réelle expertise. Depuis l'apparition de ces centres, la prise en charge des patients atteints de pathologies rares a été largement améliorée.
Mme Véronique Guillotin . - Il existe en effet un double enjeu éthique. Je suis convaincue par votre exposé et par votre vision éthique vis-à-vis des femmes, qui permet d'éviter de multiples implantations infructueuses.
M. Daniel Chasseing . - S'il n'y a pas de diagnostic pré-implantatoire, les femmes continueront à aller dans d'autres pays.
Mme Alexandra Benachi . - Il existe un tourisme procréatif évident, mais pas encore pour le DPI-A. Seules les patientes qui ont fait beaucoup de fausses couches - on voit des patientes qui ont fait plus de cinq fausses couches et qui approchent de l'âge fatidique de 42 ans - s'orientent de plus en plus vers l'Espagne ou la Belgique, car ces pays font du DPI-A. Cela concerne à mon avis peu de patientes, car beaucoup ignorent qu'elles auraient plus de chances en faisant un DPI-A. Le bénéficie de cette technique est réel pour les patientes de plus de 35 ans.
Mme Patricia Schillinger . - J'adhère aussi totalement à vos propos. Y-a-t-il beaucoup de chercheurs qui travaillent sur ce domaine-là ? Les femmes sont orientées par leur gynécologue. Comment sont-ils formés sur la suspicion ?
Mme Alexandra Benachi . - Des personnes qui font de l'assistance médicale à la procréation dans les centres universitaires travaillent sur ces sujets-là. Ils sont accompagnés par des équipes plus spécialisées sur les anomalies des chromosomes. En France, nous avons de bonnes équipes. Nous avons été cependant limités pour élargir les recherches sur l'embryon et nous contentons du nouveau texte.
Les gynécologues n'ont pas le droit de réorienter les patientes à l'étranger. Toutefois, certaines patientes, comme une femme de 39 ans qui a fait deux interruptions de grossesse pour Trisomie 21 et trois fausses couches, sont de très bonnes candidates pour un DPI-A. En l'espèce, cette patiente a été orientée vers un centre à l'étranger qui le pratique car il y a clairement quelque chose dans ses ovocytes ou dans les spermatozoïdes de son conjoint qui ne fonctionne pas. Pour elle, le DPI-A, c'est la solution.
Mme Michelle Meunier . - Y a-t-il dans ce texte des éléments qui mériteraient un développement ou une amélioration ?
Mme Alexandra Benachi . - Je fais ce métier depuis vingt ans et me dire que l'on ne pourrait pas faire de diagnostic d'anomalies génétiques et éviter des interruptions de grossesse à ces couples me semble inenvisageable. Je comprends très bien que ce soit compliqué pour certains, mais il y a tout de même des pathologies fréquentes, avec un pronostic très sévère, comme l'amyotrophie spinale, la mucoviscidose. Certes il existe des traitements mais pour certains cela fait vingt ans que j'en entends parler. Si nous pouvions, comme dans de nombreux pays, proposer aux jeunes couples de rechercher les principales anomalies génétiques pour leur éviter l'interruption de grossesse, soit en proposant un diagnostic préimplantatoire soit pour envisager une grossesse spontanée, cela éviterait de nombreuses souffrances. Aujourd'hui, il faut avoir souffert pour avoir droit au diagnostic prénatal. Cela soulève des questions. Quelle pathologie est considérée comme étant très grave ? Faut-il que l'enfant décède à la naissance, à un mois, à quatre mois, à six mois, à deux ans ? Où se situe le seuil ? Il existe des pathologies pour lesquelles nous connaissons le pronostic et où l'espérance de vie ne dépasse pas l'âge de six mois, avec de réelles souffrances comme l'amyotrophie spinale. À titre personnel, je pense que ce serait une bonne chose que l'on autorise le pré-conceptionnel pour certaines pathologies.
Nous n'aimons pas le concept de listes dans le domaine du diagnostic prénatal et ne pouvons établir des listes de pathologies de particulière gravité. Cela ne serait pas envisageable à l'égard des patients atteints de la pathologie. Également, la particulière gravité est quelque chose d'extrêmement subjectif pour les couples. En revanche, pour des raisons médico-économiques, cela serait souhaitable pour des pathologies relativement fréquentes comme la mucoviscidose. Une personne sur 25 est porteuse du gène.
M. Michel Amiel . - Sur ce sujet, il me semble qu'en droit, à l'heure actuelle, les tests sont possibles pour les couples lorsqu'il y a déjà un enfant né atteint de la maladie. Cela signifie qu'il faut d'abord avoir un enfant gravement malade pour pouvoir vérifier que les suivants ne le soient pas. C'est tout de même un vrai problème. On voit bien que vous avez raison. Il faut faire évoluer notre droit.
Mme Alexandra Benachi . - C'est exactement cela ! Pour avoir droit au diagnostic, il faut avoir souffert et avoir eu un enfant porteur de la maladie. Je comprends aussi que certains disent que l'on ne s'arrêtera jamais. Des dérives peuvent en effet exister mais notre système français est bien encadré, notamment par l'Agence de la biomédecine.
Mme Corinne Imbert , rapporteure . - Le comité consultatif national d'éthique (CCNE) était favorable au diagnostic pré-conceptionnel après une consultation spécialisée. Pourrions-nous l'organiser pour quelques maladies et proposer son remboursement par l'assurance maladie ? Qu'en pensez-vous ?
Mme Alexandra Benachi . - Tout test génétique doit être encadré. C'est inscrit dans la loi. Le patient doit bénéficier de l'information la plus correcte possible. Dans notre spécialité c'est un élément important. Ces tests doivent être précédés d'une consultation génétique. À l'étranger, peu de couples demandent une analyse génétique. Je pense qu'en France peu le demanderaient également mais cela pourrait tout à fait être organisé dans notre pays.
M. Yves Daudigny . - Pourriez-vous nous expliquer le lien entre le diagnostic prénatal et la DPI ?
Mme Alexandra Benachi . - Le DPI fait partie du diagnostic prénatal également. C'est le même texte de loi. Le DPI, c'est le diagnostic pré-implantatoire. On fait les analyses sur l'embryon, avant de le retransférer dans l'utérus. C'est une analyse très limitée. On sait faire des analyses pour la recherche des maladies génétiques - mais en général cela concerne une pathologie - et des analyses pour évaluer des anomalies des chromosomes. En France, nous n'avons pas le droit de faire les deux. Le diagnostic prénatal correspond aux analyses faites sur l'embryon et sur le foetus une fois qu'il est dans l'utérus. Pour être plus précise, le DPI fait partie du diagnostic prénatal.
M. Alain Milon , président . - Vous avez dit regretter les textes sur la recherche sur l'embryon. Jusqu'où souhaiteriez-vous que nous allions ?
Mme Alexandra Benachi . - C'est déjà beaucoup mieux ! Travailler sur les cellules-souches embryonnaires sans avoir à formuler une demande d'autorisation mais simplement un acte l'information, c'est très important. La régulation des études sur les cellules-souches pluripotentes, les IPS, vont concerner la réalisation de gamètes et c'est une vraie demande des professionnels. Nous pouvons également débattre pour savoir s'il faut autoriser la création d'embryons humains pour la recherche, mais, je suis également membre du CCNE, certaines limites s'imposent. Pouvoir faire l'étude de l'embryon jusqu'au 14 ème jour est aussi une très bonne chose.
Mme Corinne Imbert , rapporteure . - Que pensez-vous des recherches sur l'embryon au-delà du 14ème jour, notamment en Grande-Bretagne ?
Mme Alexandra Benachi . - À partir du moment où l'embryon n'est pas réimplanté, je crois que personne n'est capable de dire à partir de quel stade il représente un être humain. Or, dès lors que l'on ne peut pas réimplanter l'embryon, on ne peut rien en faire.
Il faudrait, je pense, poser la question à ceux qui travaillent vraiment sur l'embryon. Selon plusieurs sociétés scientifiques, 14 jours, c'est une durée qui permet d'en savoir beaucoup sur le moment de l'implantation.
M. Alain Milon , président . - Merci.
Ce point de l'ordre du jour a fait l'objet d'une captation vidéo qui est disponible en ligne sur le site du Sénat .
&&Audition commune d'associations&&
M. Alain Milon , président . - Nous poursuivons nos travaux sur le projet de loi relatif à la bioéthique avec une audition commune d'associations. Nous accueillons, en cette fin d'après-midi, Mmes Caroline Roux, déléguée générale adjointe et coordinatrice des services d'écoute, et Blanche Streb, directrice de la formation et de la recherche de l'Alliance Vita ; Mme Pascale Morinière, présidente, et M. Bertrand Lionel-Marie, secrétaire général de la Confédération nationale des associations familiales catholiques (CNAFC) ; Mmes Ludovine de La Rochère, présidente, et Anaïs Doisneau, déléguée jeunes de la Manif pour tous ; et MM. Bertrand du Marais, animateur, et Michel Simonnet, trésorier de l'association Les Poissons roses.
J'indique que cette audition fait l'objet d'une captation vidéo retransmise en direct sur le site du Sénat et consultable à la demande.
Avant de donner la parole à nos invités, je voudrais tout d'abord expliquer cet horaire un peu atypique : j'ai souhaité avancer à ce soir une audition précédemment prévue jeudi 5 décembre prochain, jour de grève et je remercie nos invités de s'être montrés disponibles et de permettre ainsi à nos collègues de les entendre et de pouvoir regagner leur département.
J'indique en second lieu que les demandes d'auditions sont très nombreuses et qu'il nous appartient par conséquent de faire des choix. Je veille au pluralisme des expressions, mais il est évident que nous ne pourrons satisfaire toutes les demandes.
Mme Caroline Roux, déléguée générale adjointe et coordinatrice des services d'écoute d'Alliance VITA . - Alliance VITA a été fondée au moment des premières lois de bioéthique, fin 1993. Nous sommes très sensibles aux évolutions de la loi pour que soit prise en compte la protection de la vie des plus fragiles, spécialement dans les domaines de la procréation, du handicap et du traitement réservé aux embryons humains. Nous accompagnons plus de 2 500 femmes et couples chaque année avec notre service d'écoute SOS Bébés sur les questions de maternité, d'infertilité, du handicap, etc. Nous considérons que nous devrions évaluer la façon dont le législateur élabore les lois de bioéthique. Le législateur s'affranchit toujours plus de l'éthique, en essayant d'ériger des digues qui n'arrêtent pas de s'effondrer et, surtout, sans évaluer les conséquences sociales et humanitaires de ces pratiques.
L'assistance médicale à procréation (AMP) constitue un parcours du combattant, avec des traitements qui affectent physiquement et psychologiquement les femmes, mais aussi les hommes. Nous ne pouvons que partager, bien sûr, la joie des parents qui donnent naissance à un enfant tant espéré en ayant recours à ces techniques, mais celles-ci ne sont qu'un palliatif et ne guérissent pas l'infertilité. Je rappelle qu'un couple sur deux n'aura pas d'enfant, à l'issue de cette procédure. On ne parle pas beaucoup, non plus, de la souffrance de certains couples, face à la question du devenir de leurs embryons surnuméraires. Nous sommes aussi témoins de leur désir profond d'être « guéris », comme certains le disent, de pouvoir procréer de manière autonome. Des femmes réussissent à avoir des enfants naturellement, après avoir suivi un parcours d'AMP sans succès. Cette perspective doit nous mobiliser.
Notre deuxième observation concerne la pratique du diagnostic prénatal. La multiplication des tests et des diagnostics de plus en plus précis rend les grossesses plus anxiogènes. L'annonce d'un risque de handicap, qu'il soit avéré ou non, provoque souvent un véritable tsunami émotionnel et relationnel pour le couple. Le soutien des équipes médicales est nécessaire pour envisager les prises en charge adaptées, mais la perspective, en toile de fond, des propositions d'interruption médicale de grossesse (IMG) peut détourner de véritables thérapies ou d'une prise en charge adaptée. Ce projet de loi, justement, ne va pas dans le sens d'une amélioration de l'accompagnement. Il est en rupture avec les précédentes lois en ce qui concerne l'assistance médicale à la procréation, car il entend supprimer le critère d'infertilité pour y avoir recours : c'est une préoccupation majeure pour nous et les couples infertiles sont les grands oubliés du texte. Ce changement de paradigme conduit à des injustices et des reculs en cascade. Il faut craindre, tout d'abord, l'extension de l'AMP avec tiers donneur, alors qu'il s'agit d'une pratique très minoritaire aujourd'hui - qui concerne moins de 5 % des enfants nés chaque année avec l'AMP. Cette pratique n'est pas anodine. Ce sont d'ailleurs les enfants eux-mêmes qui sont demandeurs de la levée de l'anonymat, même si celle-ci ne supprimera pas l'injustice d'une filiation éclatée et peut aussi se révéler une bombe à retardement. Nous devrions plutôt développer la recherche, notamment sur les causes d'infertilité masculine, afin de limiter le plus possible le recours à ce type d'AMP.
Avec le double don de gamètes et l'autorisation de l'AMP avec tiers donneur pour des femmes sans partenaire masculin et sans infertilité médicale, on peut craindre que de nombreux enfants se retrouvent victimes de l'injustice que constitue le fait d'être privé délibérément de toute origine ou de père.
La congélation ovocytaire est une fausse promesse. Les femmes à qui on la proposera se retrouveront sous une emprise médicale croissante. Elle ne constitue nullement une assurance maternité : selon certains experts, trois femmes sur quatre n'auraient pas d'enfant avec cette technique.
Le texte présente aussi le risque de constituer un détournement de la mission de la médecine et de l'assurance maladie. Sur la base de quels critères les médecins pourront-ils refuser l'AMP ? La loi reste muette sur ce point. Finalement, ce projet de loi constitue une atteinte sans précédent à la déontologie médicale.
Comment prétendre aussi, avec ces glissements, interdire ensuite la gestation pour autrui (GPA) et les mères porteuses ? Le législateur doit prendre la mesure de sa responsabilité, car la souffrance et le désir de certains ne peuvent être le seul critère de discernement quand il s'agit de l'intérêt des enfants. Le législateur n'a pas à céder à des modes ou à des pressions de quelques-uns, mais doit servir l'intérêt de tous.
Notre association regrette l'impasse sur trois enjeux fondamentaux. S'agissant de la lutte contre l'infertilité, si nous sommes favorables à l'amendement qui a été voté à l'Assemblée nationale, comment mettre réellement en oeuvre, d'une manière volontariste, une politique de lutte contre l'infertilité si elle n'est plus mentionnée parmi les critères de recours à l'AMP ? Nous avions proposé que l'Agence de la biomédecine fasse un recensement systématique des causes des demandes d'AMP pour mieux orienter les recherches sur l'infertilité.
Le deuxième enjeu est la lutte contre l'eugénisme et la prise en charge du handicap. Alors que nous célébrons la Journée internationale des personnes handicapées, comment peut-on concilier les appels de Sophie Cluzel pour une société toujours plus inclusive et, en même temps, donner des signes pouvant laisser à penser que certaines personnes handicapées auraient mieux fait de ne pas naître ? Il faut s'inquiéter de la pression exercée en faveur de la réalisation de tests génétiques de plus en plus précis, comme le dépistage prénatal non invasif notamment, dont l'encadrement serait renvoyé à des décrets et échapperait au législateur. On peut se demander d'ailleurs qui, parmi nous, aurait échappé à ces tests génétiques... La France devrait rééquilibrer ses politiques de dépistage, d'annonce et de prise en charge du handicap.
Enfin, la dernière cause qui devrait préoccuper le législateur est celle de l'intégrité de l'espèce humaine, et je laisse à Blanche Streb le soin de présenter nos positions sur ce sujet.
Mme Blanche Streb, directrice de la formation et de la recherche de l'Alliance VITA . - J'évoquerai le sujet de l'embryon humain dans ce projet de loi. On ne peut raisonner comme si l'embryon évoluait dans deux domaines étanches, celui de l'AMP, d'un côté, et celui de la recherche, d'un autre côté, dans lequel les cellules sont instrumentalisées, entrent dans des protocoles scientifiques ou sont mises en culture. Ces deux domaines s'interpénètrent de plus en plus à cause de la dynamique internationale de surenchère technicienne autour de la procréation assistée et des biotechnologies. Derrière la question de l'infertilité, il y a des marchés à prendre !
Dans trois pays, à nos portes, les premiers bébés génétiquement modifiés sont déjà nés, grâce à la méthode CRISPR ou grâce à la technique de la fécondation in vitro. L'article 17 du projet de loi ouvre la voie à ces techniques, dans le cadre de la recherche fondamentale. Mais il importe de penser dès maintenant à l'étape suivante, à l'utilisation de la technique dans la thérapie embryonnaire. Le texte ouvre aussi la voie à la recherche sur les gamètes artificiels.
Ces techniques s'inscrivent dans la logique de l'enfant à tout prix, à n'importe quel prix, quand bien même ce serait l'enfant lui-même qui aurait à en payer le prix. Immense paradoxe ! Le diagnostic préimplantatoire se développe, y compris pour des pathologies qui ne sont pas héréditaires. C'est révélateur d'une dynamique de contrôle qualité de l'embryon, de passage au crible des enfants à naître. La perspective sous-jacente est bien celle décrite par Jean-Louis Touraine d'un droit à la « procréation sans sexe pour tous ». Deux projets de société s'affrontent : celui de l'enfant accueilli sans condition et celui de l'enfant programmé et bricolé.
En ce qui concerne la modification du régime qui encadre l'embryon humain, le passage de l'autorisation à la déclaration constitue une nouvelle façon de banaliser un petit peu plus l'instrumentalisation de l'embryon humain, en supprimant la nécessité de prouver qu'il n'y a pas d'alternative à ces cellules. Au fond, cela pousse indirectement à la consommation de l'embryon humain. On réduit l'encadrement de l'utilisation de l'embryon humain et des cellules souches embryonnaires et, parallèlement, on ouvre la voie à de nouvelles perspectives, de nouvelles transgressions : l'utilisation de ces cellules pour créer des gamètes artificiels ou des chimères, comme le permet l'article 17. Ce changement de régime n'a qu'un seul objectif : faciliter le travail administratif de quelques chercheurs. On aimerait pourtant entendre d'autres scientifiques qui travaillent sur les perspectives prometteuses de la thérapie génique et de la médecine régénératrice avec des ressources biologiques qui ne posent pas les problèmes éthiques importants que soulève l'utilisation de l'embryon humain.
La culture des embryons est autorisée jusqu'à 14 jours in vitro. À ce stade de développement, l'embryon commence à développer son système nerveux. Il peut être implanté depuis le septième jour, ce qui oblige à modifier la manière de le mettre en culture. Le texte ouvre la voie à ces modifications parce que des équipes commencent à procéder de la sorte à l'étranger. C'est bien la logique du projet de loi : ouvrir, tester, et voir ce qu'il adviendra... En somme, après la loi, le déluge ! Si la bioéthique est conditionnée à la faisabilité technique, alors la perspective utilitariste n'aura plus de limite. Toutefois, l'argument de la compétition internationale n'est pas recevable en bioéthique.
Deux principes fondateurs de la bioéthique sont en jeu : la protection de l'intégrité de l'espèce humaine, d'abord, fragilisée par l'article 17 avec les embryons transgéniques et les chimères ; la primauté de la personne, ensuite. L'article 16 du code civil interdit toute atteinte à la dignité de la personne et garantit le respect de l'être humain dès le commencement de sa vie. Nul ne conteste plus qu'un embryon humain est un être humain et que la fécondation, in vitro ou in utero, constitue le commencement d'une vie humaine.
L'embryon nous crie son humanité. Il est déjà l'être humain qu'il deviendra. Qu'il fasse, ou non, partie d'un projet parental, ne modifie nullement sa nature. Nous vous appelons à réfléchir ensemble à une science qui ne se laisserait pas happer dans une dynamique toujours plus technicienne, mais qui se mettrait pleinement au service de l'homme, sans jamais le réduire à un objet.
Mme Ludovine de La Rochère, présidente de La Manif pour tous . - Un très grand nombre d'élus se déclarent expressément opposés à la gestation pour autrui, mais certains semblent plutôt favorables à la dérégulation de l'assistance médicale à la procréation. Or, si l'on veut vraiment éviter la légalisation de la GPA lors de la prochaine révision de la loi bioéthique, vers 2025, le maintien de l'encadrement actuel de l'AMP est incontournable.
En effet, ouvrir l'AMP, comme le fait le projet de loi, revient à accepter de priver des enfants de père pour toujours, comme si le père n'était qu'un simple géniteur, dépourvu d'importance pour l'enfant. Si l'on dit aujourd'hui qu'un enfant peut bien se passer de père, parce que telle est la volonté de certains adultes, on dira évidemment, demain, que l'enfant peut bien se passer de mère, si telle est la volonté de certains adultes.
Légitimer l'accès à l'AMP pour des personnes fécondes au motif de l'existence d'un projet parental prépare aussi le terrain pour la GPA, car des hommes seuls ou des couples d'hommes peuvent avoir eux aussi un projet parental.
Inscrire deux mères sur l'acte de naissance d'un enfant, c'est considérer qu'il n'y aurait aucune différence entre les deux femmes, entre celle qui l'a attendu et mis au monde, et sa conjointe, la belle-mère de l'enfant. C'est faire comme si la maternité, l'accouchement, n'étaient rien dans la vie de la femme. C'est nier une réalité que toute mère ressent puissamment dans son corps, son psychisme, et bien au-delà lors de la grossesse et de la naissance.
Si la grossesse et l'accouchement ne sont rien, alors, inversement, une femme qui a été enceinte et a accouché pourrait aussi ne pas être reconnue comme mère. C'est le principe de la pratique des mères porteuses, que les militants pro-GPA, justement, nomment « femmes porteuses » et non « mères porteuses » : à leurs yeux, elles ne sont pas mères !
Si l'on considère que la conjointe d'un enfant né par AMP est une « mère sociale », alors on ne pourra que reconnaître la mère sociale d'un enfant né par GPA. En effet, dans les deux cas, le projet parental existe ; dans les deux cas, la femme est la conjointe d'un parent biologique et élève l'enfant ; dans les deux cas, on dira qu'il faut « sécuriser la filiation dans l'intérêt de l'enfant »... Cela aboutit à construire une filiation invraisemblable, c'est-à-dire à démolir la filiation, tout en prétendant la sécuriser. Un comble ! En fait, ce mécanisme est déjà à l'oeuvre, pour la GPA, devant les tribunaux. La filiation sociale, introduite avec l'AMP pour les couples de femmes, prépare ainsi les conditions de la GPA.
Les femmes seules et les couples de femmes sont dans une situation différente des couples hétérosexuels au regard de la procréation. Mais des militants et des députés ne cessent de dire qu'il faut étendre l'accès à l'AMP aux femmes seules et aux couples de femmes par souci d'égalité. C'est faux, comme l'ont souligné le Conseil d'État, le Conseil constitutionnel et la Cour européenne des droits de l'Homme. Malgré les différences de situations, on s'achemine néanmoins vers l'AMP pour les femmes sans partenaire masculin. En même temps, Agnès Buzyn et Nicole Belloubet déclarent qu'on n'ouvrira pas la GPA parce que la situation des hommes est différente de celle des femmes au regard de la procréation. Aujourd'hui, on met de côté les différences entre les couples de femmes, les femmes seules et les couples constitués d'un homme et d'une femme. Demain, on mettra de côté cette différence entre les hommes et les femmes que l'on évoque aujourd'hui pour affirmer que la GPA ne sera pas légalisée par la suite. Bientôt, médiatiquement et politiquement, apparaîtront des campagnes pour l'égalité entre les hommes et les femmes.
Ouvrir l'AMP, sans motif médical, c'est détourner la médecine de sa finalité. Une fois celle-ci devenue prestation de service pour réaliser le projet parental de femmes seules et de couples de femmes, que dira-t-on aux femmes qui ont un problème d'utérus que l'AMP ne peut résoudre ? que dira-t-on aux couples ayant dépassé l'âge de la fécondité, mais qui ont un projet parental, ou aux veuves qui ont pu faire congeler le sperme de leur conjoint décédé ? Et que dira-t-on aux hommes ? Si vous entérinez le principe du détournement de la médecine, si vous acceptez de priver volontairement des enfants de père, quel argument évoquerez-vous face à la pression en faveur de la GPA ?
Ouvrir l'AMP aux femmes seules et aux couples de femmes, c'est ouvrir la porte à la marchandisation humaine. Tous les pays qui ont étendu l'AMP au-delà des cas médicalement justifiés rémunèrent les hommes qui fournissent leurs cellules sexuelles, ou alors achètent les gamètes dans les pays qui rémunèrent les hommes. Le Danemark vend ainsi des cellules sexuelles à la Grande-Bretagne et à la Belgique. Les campagnes d'appels à dons ne suffisent pas à couvrir les besoins, comme l'explique le professeur Israël Nisand, dans une tribune publiée hier dans Le Figaro. L'AMP, dite pour toutes, va de pair avec le commerce des gamètes. Étendre l'AMP revient à ouvrir la marchandisation humaine.
L'AMP sans motif médical, de même que l'autoconservation des gamètes sans condition médicale, constitue une technicisation excessive dès lors que les personnes concernées ne souffrent pas d'infertilité. Cette dérégulation constitue une incitation qui risque de démultiplier considérablement les recours à l'AMP. « La procréation sans sexe pour tous », en effet !
Business international, technicisation.... tout cela est aussi contraire à l'écologie dont se réclament le Président de la République, le Gouvernement, et La République En Marche. Nous avons fait n'importe quoi avec la planète, la faune et la flore et nous ne parvenons pas à mettre un terme à nos dérives. Ne faisons pas de même avec la procréation humaine !
Un mot enfin sur la location de la femme, qui concerne la GPA, mais non l'AMP. Si l'AMP sans père est légalisée, tout sera prêt pour que la GPA soit légalisée lors de la prochaine révision des lois de bioéthique. Il ne restera qu'à s'habituer à utiliser des femmes comme incubatrices pour tel ou tel qui veut un enfant. Il suffira de dire et de répéter, pendant quelques années, que c'est « altruiste », « généreux »... On l'entend déjà largement. On en est déjà presque parvenus à croire que l'on peut priver l'enfant de son père : 2 000 enfants naissent déjà sans père selon l'estimation d'Agnès Buzyn.
En réalité, la GPA est déjà sur la scène ; elle est déjà présente dans les séries télévisées et au cinéma ; elle a déjà fait l'objet de propositions de lois et d'amendements. Elle est, en outre, déjà reconnue par la France dès lors que nos concitoyens recourent à une mère porteuse à l'étranger, comme si la dignité des femmes ne devait être respectée que sur notre territoire... La Cour de cassation et la cour d'appel de Rennes, en novembre, ont déjà reconnu la filiation sociale dans le cas d'enfants nés par GPA à l'étranger.
La déréglementation de l'AMP jette les bases de la légalisation de la GPA : droits de l'enfant piétinés, déni de la paternité et de la maternité, création de la filiation sociale, détournement de la médecine, technicisation à outrance de la procréation, marchandisation humaine et contexte idéologique. Certes, le Premier ministre a dit que la GPA ne serait pas légalisée. Mais sa phrase se terminait par « sous ce quinquennat » !
Il faut du courage pour résister à ce tsunami de pressions, de désirs. De grâce, pour les générations à venir, ne franchissez pas la ligne rouge de l'AMP sans père ! Anaïs Doisneau va vous parler de la vie sans père.
Mme Anaïs Doisneau, déléguée jeunes de La Manif pour tous . - J'ai vingt ans et, depuis l'âge de trois mois, je n'ai plus de père. Autant dire que je n'en ai pas eu parce que je n'ai même pas eu à souffrir de la séparation ou à faire l'expérience du deuil ; je n'ai connu que cet état. Il est habituel pour moi de ne pas avoir de père. Et pourtant, je peux vous dire que l'absence d'un père représente un manque profond dans la construction de la personnalité et l'existence d'une jeune fille. Que diraient de moi les études anglo-saxonnes sur lesquelles se fonde le débat ? J'ai fait de brillantes études, j'ai des amis, je chante, je ris, j'écris, je ne suis ni hystérique ni névrosée, je n'ai pas fait dépression ni de tentative de suicide... Alors, tout va bien ? Oui et non ! Oui, parce que je fais avec. Je supporte tant bien que mal cette béance intime, ce vide affectif qui aurait dû être comblé par un père. J'accepte aussi, tant bien que mal, le fait de ne pas avoir connu cette altérité unique entre un père et sa fille.
Et pourtant non ! Car je porte ma croix habituelle, si habituelle que je pourrais même être tentée de l'ignorer. Je n'ai jamais connu mon père, et pourtant je ne pourrai jamais le remplacer, alors même que tout mon être ne cesse de le chercher dans les bras, les mots, les yeux et l'affection de tous les hommes. J'ai grandi seule avec ma mère, dans une famille soudée et équilibrée ; j'ai été choyée, bien éduquée, mais tout cela ne remplace pas un père... La souffrance indicible persiste. Dans mon coeur de femme, il manque cette ouverture décisive à l'altérité rassurante, apaisée. Il manque cette place capitale qu'aurait comblée un père et qui reste néanmoins vide. Je passerai ma vie à devoir accepter de ne pas avoir le droit d'avoir un père. La faute à la vie, c'est ce que je me dis.
Maintenant, je me mets dans la peau d'un enfant issu d'une AMP sans père et je me demande, comme tous les enfants sans père : « pourquoi n'ai-je pas le droit d'avoir un père ? » On me dira que c'est parce que ma mère l'a voulu. Mesurez-vous les conséquences d'une telle situation sur le coeur d'un enfant, qui déjà est privé de père, mais qui est aussi privé du droit de s'en désoler, car il est tenaillé entre son amour et sa culpabilité face à sa mère, qui est la responsable de son inconfort affectif.
Madame, monsieur, au nom de tous les sans-père, vous êtes responsables de ces futurs orphelins que la République envisage de créer. J'espère que, courageusement, vous vous y opposerez.
Mme Pascale Morinière, présidente de la Confédération nationale des associations familiales catholiques (CNAFC) . - Membre de l'Union nationale des associations familiales, la Confédération nationale des associations familiales catholiques porte la parole de 30 000 familles adhérentes et de 300 associations locales issues de tous les territoires de la République. Mouvement familial, de laïcs, fondé sur l'enseignement social de l'Église catholique, nous portons une parole qui n'est pas celle d'un lobby, mais de familles attachées au bien commun. Nous ne revendiquons aucun nouveau droit ou intérêt particulier pour nous-mêmes, mais nous efforçons de promouvoir une certaine vision de la personne humaine, de sa dignité intangible et de la manière dont s'organise la société autour de sa cellule de base, la famille. La famille est le meilleur écosystème qui soit pour le petit d'homme. Toutefois, comme tous les écosystèmes, il est fragile. Nous avons la responsabilité collective d'en prendre soin.
Quelle est l'unité de ce texte ? La bioéthique est invoquée pour justifier des réformes sociétales en matière de procréation. Si la loi de bioéthique est remise en cause au cours de chaque quinquennat, pour promouvoir les « avancées » que la précédente révision n'a pas obtenues, comment la loi pourra-t-elle faire consensus, être respectée dans son rôle normatif ?
L'essentiel de ce texte organise le cadre légal de la procréation artificielle et marchande avec l'installation d'une chaîne de production humaine.
Décrivons la situation. Un couple ou une personne désire un enfant. Des gamètes sont donnés, des embryons sont créés et conservés. Des enfants naissent. Les gamètes et les embryons sont gratuits, pour le moment, en France, mais tout ce processus ne l'est pas : il coûterait 300 millions d'euros en France, 20 milliards d'euros dans le monde. Ainsi, à travers le monde, il existe un gigantesque marché de la procréation. Les embryons servent à la recherche. La recherche améliore la productivité de la chaîne qui reste faible : 25 % de réussite à chaque cycle de fécondation in vitro (FIV), 12 % pour les inséminations artificielles.
Le caryotype systématiquement pratiqué sur le don de gamètes permet de vérifier les normes de qualité ; le diagnostic préimplantatoire pourrait permettre de faire la même chose pour l'embryon conçu. Dans cette chaîne de production, la recherche sur l'embryon fait le travail de recherche et développement, comme pour tout produit dans une entreprise. Elle n'a pourtant pas permis d'avancée thérapeutique majeure depuis 20 ans. Peu à peu, une philosophie « qualitariste » du produit se met en place, rendue possible par le double don de gamètes, et donc le double caryotype. L'article 17 autorise aussi les embryons transgéniques, et donc renforce, potentiellement, la qualité du produit final. La perspective de développement est la GPA, que l'on commence déjà à reconnaître insidieusement lorsqu'elle est pratiquée à l'étranger. Puis, pour rétablir l'égalité avec les couples qui n'ont pas les moyens d'aller à l'étranger, on la légalisera en France !
Qui tire profit de cette chaîne de production artificielle ? Tout d'abord, les adeptes de l'idéologie moderniste qui cherchent à améliorer l'humain, à l'affranchir et l'émanciper toujours plus, acceptant, mais seulement pour autrui, le risque prométhéen de créer des personnes désaffiliées de leur ancrage biologique, charnel ou généalogique.
Ensuite, le business technique et marchand de la bioéconomie qui est en plein développement et auquel la France, jusqu'alors, avait su résister. Pourtant entre 2016 et 2017, le nombre de procréations médicalement assistées (PMA) a augmenté de 5 %. Le marché des technologies de la reproduction englobe de nombreuses industries à travers le monde : laboratoires pharmaceutiques et biomédicaux, personnels de santé mentale, cliniques spécialisées en procréation artificielle, laboratoires de séquençage génomique, banques de gamètes, agences et cliniques spécialisées en GPA, experts juridiques, réseaux commerciaux... Le chiffre d'affaires est de 20 milliards de dollars.
Enfin, les hommes et les femmes, quelle que soit leur orientation sexuelle, qui ont recours à ces techniques ou dont les corps servent à ces techniques. Sont-ils utilisateurs ou matières premières de la bioéconomie ? « L'enjeu de ces débats, c'est la confusion entre les personnes et les biens », disait avec justesse Sylviane Agacinski, dans le Point, le 27 septembre 2019.
Nous, associations familiales catholiques, savons que la vie bonne est une vie reliée, insérée dans des générations cohérentes, accueillie quelles que soient ses faiblesses et aimée pour elle-même, et non parce qu'elle viendrait combler un manque. La dignité de la personne humaine ne tient pas aux normes de qualité auxquelles elle satisferait, ni à son autonomie ou à sa valeur économique. Le respect de cette dignité est ce qui fait le degré de développement d'une civilisation. Rappelons-nous qu'il n'est rien resté de Sparte qui pratiquait le tri des enfants à la naissance !
Nul doute que dans vos territoires ruraux ou plus citadins on ne vous réclame pas ces évolutions à cors et à cris. En fait, ça n'intéresse pas beaucoup de monde. La loi ne peut répondre à toutes les souffrances ou toutes les douleurs, c'est malheureux, mais c'est ainsi. Son rôle est d'empêcher les abus, comme celui qui vise à créer un droit à l'enfant et à l'enfant parfait. Le législateur doit résister à la poussée de la bioéconomie. Le rôle de la loi est de mettre des limites aux désirs de toute-puissance afin que la vie en société reste possible.
Merci pour votre courage et votre clairvoyance. Votre courage, car, comme le disait Péguy, « il faut toujours dire ce que l'on voit » et votre clairvoyance, car « il faut toujours, ce qui est plus difficile, voir ce que l'on voit. »
M. Bertrand Lionel-Marie, secrétaire général de la CNAFC . - Ce projet de loi, dont nous pourrions croire, en cédant à l'air du temps, qu'il participe de l'octroi de nouveaux droits et d'un progrès prétendument inéluctable, doit être questionné et il est de votre noble et lourde responsabilité, en tant qu'élus de la Nation, de prendre le temps de le faire, avec courage. Nous vous proposons un éclairage à la lumière des valeurs de la République auxquelles nous sommes attachées.
La liberté, d'abord. Le débat qui a animé notre société, depuis les États généraux de la bioéthique jusqu'au processus législatif en cours, appelle des observations sur la liberté d'expression, la liberté de réunion ou la liberté de manifestation, constitutionnellement garanties. Il soulève, aussi, une interrogation, profonde, sur la capacité de la démocratie, qui délègue son pouvoir à des sachants et à des experts, - dont certains ne « [savent] pas même ce que sont le bien et le mal » -, à régir « les techniques devenues folles » comme disait Camus dans son discours de réception du prix Nobel. La question de la condition de l'homme dans l'univers technique est aussi posée. Comme l'écrivait, Jacques Ellul, cité ces temps-ci par des personnalités aussi variées qu'Alain Juppé, Denis Tillinac ou José Bové : « l'homme idéal inventé, produit par le génie génétique, n'aurait aucune liberté, puisqu'il serait le modèle qu'il était programmé d'être. Et quand même la liberté absente, c'est un gros morceau » !!
La convocation de la médecine et du droit est, par ailleurs, de nature à heurter la libre conscience de médecins, de notaires ou d'officiers d'état civil.
L'égalité, ensuite. Comme l'a rappelé le Conseil d'État, dans une décision du 28 septembre 2018, « les couples formés d'un homme et d'une femme sont, au regard de la procréation, dans une situation différente de celle des couples de personnes de même sexe. [...] La différence de traitement, résultant des dispositions [de l'article L. 2141-2 du code de la santé publique], entre les couples formés d'un homme et d'une femme et les couples de personnes de même sexe, est en lien direct avec l'objet de la loi qui l'établit et n'est, ainsi, pas contraire au principe d'égalité. » En appeler au principe d'égalité participe, donc, d'une confusion.
Il est, en revanche, inégalitaire de créer différentes catégories d'enfants : si un enfant à naître est, en droit, fondé à obtenir réparation du préjudice d'affection d'avoir été, accidentellement, privé d'un père, celui amputé de père, par la loi, le pourra-t-il ? À défaut, il y aurait bien discrimination. Et si tel est le cas, quelle sera la cohérence d'une action publique qui créerait, par la loi, les conditions d'un tel droit à réparation.
Comment, enfin, soutenir sérieusement et garantir juridiquement que cette invocation du principe d'égalité ne débouchera, jamais, comme cela est affirmé aujourd'hui, sur la légalisation par la République, des mères porteuses, à la demande de couples ou d'hommes célibataires ? Le Premier ministre déclarait ainsi, dans le Huffington Post, en février 2013 - avant son virement de bord : « Nous nous opposerons résolument à la PMA pour les couples homosexuels féminins, et à la GPA qui, au nom de l'égalité, ne manquera pas d'être réclamée par la suite. »
Enfin, dernier élément de notre devise, la fraternité. Celle-ci est la grande oubliée de ce projet. Quand l'enfant sera né d'« une procréation sans sexe pour tous », telle que prônée par le rapport Touraine, qu'il sera possiblement issu de la rencontre programmée de deux gamètes de tiers donneurs, voire, après-demain, de deux gamètes artificiels, sélectionnés après screening et tri de multiples embryons, dont certains cryoconservés pendant des années, puis porté, un jour peut-être, dans un utérus artificiel, sera-t-il encore notre frère en humanité ? Quel regard porterons-nous sur lui ? Devra-t-on parler de naissance ou de livraison ?
Alors que nous prenons, trop lentement, conscience des méfaits du productivisme sur notre environnement, sur notre « maison commune », selon l'expression du Pape François, n'est-il pas paradoxal, au mépris des générations futures, de céder, en matière de procréation humaine, à l'artificialisation à tous crins et à un prométhéisme, drapé dans un progressisme de bon aloi ? Un authentique humanisme ne devrait-il pas prendre soin, au premier chef, des générations futures ?
Si une transition écologique est requise, la bioéthique ne peut rester en dehors de son champ. Demain, d'autres Greta Thunberg se lèveront pour agir contre la République française sur le fondement des articles 3 et 7 de la Convention internationale des droits de l'enfant.
« Agis de façon que les effets de ton action soient compatibles avec la permanence d'une vie authentiquement humaine sur la terre », lançait Hans Jonas, en 1979, dans Le Principe Responsabilité. Son interpellation, à l'origine du principe de précaution, demeure d'actualité !
M. Bertrand du Marais, administrateur de l'association Les Poissons roses . - Monsieur le président, mesdames, messieurs les sénateurs, je vous remercie de nous recevoir, mais aussi d'être plus accueillants que vos collègues de l'Assemblée nationale.
L'association Les Poissons roses est une plateforme de chrétiens de gauche, inspirés par la pensée d'Emmanuel Mounier, fondateur de la revue Esprit et à l'origine du courant personnaliste en France.
Créée en 2010, l'association représente une gauche humaniste et privilégie une approche de la politique par le bas, à partir des réalités sociales qui fondent la construction de la société et de la politique. Pour nous, le social, l'économie, l'environnemental et le spirituel sont intimement liés, selon une démarche d'écologie intégrale.
Nous disposons de plusieurs associations locales en province, notamment à Nantes, Bordeaux et Dijon. En 2016, la publication de notre livre À contre-courant visait à interpeller la gauche face à sa dérive sociétale. Depuis, nous avons publié une dizaine d'analyses, disponibles sur notre site www.poissonsroses.org : un rapport sur la famille durable, qui va au-delà des fascinations biotechniques, un rapport sur la laïcité ou encore un projet pour l'Europe. Notre dernier rapport intitulé Enquête sur les invisibles de la République, sera publié aux éditions du Cerf en mars 2020, et notre prochain groupe de travail aura pour thème « démocratie et écologie ».
Nous fonctionnons principalement par groupes de travail, en auditionnant les acteurs et experts d'une question, dans une démarche que nous voulons ouverte et respectueuse des différences. Pour l'élaboration de notre rapport sur la famille durable, nous avons, notamment, auditionné la présidente de La Manif pour tous et l'association David et Jonathan, la première association française d'homosexuels - hommes et femmes -, datant de 1972. Nous avons également reçu une très intéressante contribution écrite d'un mouvement de lesbiennes, gays, bisexuels et transgenres (LGBT), qui a préféré garder l'anonymat.
S'agissant de l'extension de l'assistance médicale à la procréation (AMP), sur laquelle je vous propose de concentrer mon intervention, notre position est le maintien du statu quo.
Nous sommes venus défendre ici la loi de la République, défendre votre rôle de parlementaires, et non pas le projet de loi qui nous est proposé.
C'est la première fois, dans notre République, qu'une loi vous est présentée officiellement sans se référer, soit à un motif d'intérêt général, soit à une méconnaissance d'une norme supérieure. En effet, le motif retenu par le Gouvernement est le droit au désir d'enfant, inscrit à la page 40 de l'étude d'impact, et l'égalité du désir. Ce désir, tout à fait légitime, anime les couples hétérosexuels aussi bien que les couples homosexuels et les célibataires. Nous reconnaissons l'ampleur de la douleur de ceux qui ne peuvent satisfaire à ce désir. Cependant, nous souhaitons mettre en garde la représentation nationale contre un glissement très sensible et inédit auquel procèdera cette loi.
Le recours à la loi républicaine, à l'imperium du législateur, est ici légitimé par l'égalité de désir et non pas, comme l'ont indiqué le Conseil d'État et le Conseil constitutionnel, par l'égalité des conditions, que protège l'article 6 de la Déclaration des droits de l'homme et du citoyen.
Nous ne sommes pas naïfs, les propositions de textes législatifs ne sont pas neutres. Les niches fiscales, par exemple, permettent à tel ou tel groupe d'intérêt d'obtenir un dispositif législatif. Mais ces demandes sont, à chaque fois, fondées sur un intérêt général. Or ce projet de loi met en jeu, non pas l'intérêt collectif, mais l'intérêt d'un collectif - aussi sympathique soit-il. Il crée un droit spécifique, dérogatoire à de nombreux autres principes, qui est attribué du seul fait de l'égalité de désir, en raison de la seule appartenance à une orientation sexuelle qui, heureusement, n'est plus un délit et n'est pas un handicap.
Ce faisant, le projet de loi porte atteinte au principe de la souveraineté de la loi dans notre démocratie. La loi est l'expression de la souveraineté nationale et donc de l'expression de l'intérêt général, précise l'article 6 susmentionné. Le principe de l'universalité de la souveraineté nationale figure aux articles 1 et 3 de la Constitution, ce dernier reprenant ledit article 6.
Or le Conseil constitutionnel a déjà consacré toute sa valeur constitutionnelle à ce principe de souveraineté nationale, dans sa décision du 30 décembre 1976, relative à l'élection au suffrage universel direct de l'Assemblée de communautés. Plus précisément, dans sa décision n o 99-412-DC, du 15 juin 1999, sur la Charte des langues régionales, le Conseil constitutionnel indique : « Le principe d'unicité du peuple français, dont aucune section ne peut s'attribuer l'exercice de la souveraineté nationale, a également valeur constitutionnelle. Considérant que ces principes fondamentaux s'opposent à ce que soient reconnus des droits collectifs à quelque groupe que ce soit, défini par une communauté d'origine, de culture, de langue ou de croyances. » Nous pourrions ajouter « ou d'option personnelle, d'option sexuelle », par exemple.
Le projet de loi nous entraîne officiellement dans ce que le grand juriste et philosophe italien Natalino Irti appelle le « nihilisme juridique », dans lequel la loi ne tire plus sa légitimité que de principes procéduraux. Je tirerai trois conséquences à ce projet de loi, qui crée un dangereux précédent pour la loi de la République, et je ferai un constat.
D'abord, nous ne disposons plus d'arguments pour empêcher n'importe quel collectif d'intérêt, n'importe quel groupe d'expression - et donc les plus puissants d'entre eux, évidemment - d'avoir accès, au nom de l'égalité de désir, à la légitimité de la loi républicaine.
La deuxième conséquence est l'extension de ce désir à la GPA, qui deviendra alors légitime et conduira, inéluctablement, à une nouvelle forme d'esclavagisme.
Enfin, le Parlement n'a plus toute sa raison d'être, puisqu'il vous appartient, dans une démocratie qui n'est pas le césarisme, de maintenir l'équilibre entre les intérêts collectifs qui forment la société.
J'en viens au constat : l'extension de l'AMP qui nous est aujourd'hui proposée est finalement un épiphénomène. Je suis désolé du caractère un peu brutal de ce terme qui peut choquer ceux qui, légitimement, cherchent à satisfaire le désir d'enfant, mais il s'agit bien d'un phénomène minoritaire. Le vrai sujet de politique et d'intérêt général est le suivant : quelles familles la société française souhaite-t-elle pour aujourd'hui et les générations futures ?
Nous estimons entre 10 et 15 millions le nombre de Français qui se débattent aujourd'hui avec des problèmes compliqués, graves, de familles éclatées, de familles monoparentales, de définition de la distinction entre l'autorité et la responsabilité parentale... C'est la raison pour laquelle, nous demandons que soient organisés des états généraux de la famille durable.
Par ailleurs, la marchandisation du corps humain est une conséquence inéluctable de cette loi. Ce projet de loi ne pourra être appliqué, compte tenu de la pénurie de gamètes, qui ne peut que s'aggraver dans le futur.
La présentation de l'équilibre entre l'offre et la demande de gamètes, par l'étude d'impact, est au mieux irénique, au pire fallacieuse, ce qui pose un problème s'agissant de la relation entre les pouvoirs législatif et exécutif. Selon le calcul le plus favorable à l'effectivité de la réforme, les chiffres donnés aux pages 23 et 25 de l'étude d'impact conduisent à une insuffisance, dans un facteur de un à cinq. En effet, le nombre de donneurs est aujourd'hui de 363, alors que, selon l'estimation la plus favorable, il en faudrait environ 1 600. Non seulement la production actuelle ne peut satisfaire à toutes les demandes, mais le stock est épuisé en une année.
Outre les conséquences de la levée de l'anonymat, dont nous ne savons pas trop quel effet elle aura, il y aura nécessairement des phénomènes d'allongement des files d'attente, des risques de marché noir... La mission d'enquête de l'Assemblée nationale s'en est d'ailleurs inquiétée à la page 61 de son rapport. Les représentants des centres d'études et de conservation des oeufs et du sperme (CECOS), au cours des auditions de l'Assemblée nationale, ont avancé le prix de 500 euros, un chiffre qui est en ligne avec ceux des États-Unis : 1 500 dollars pour un homme qui donnerait trois fois par mois.
Nous nous opposons donc à la marchandisation du corps humain, malheureusement inéluctable, car nous connaissons la force destructrice du marché. Le marché est nécessaire, mais il s'agit d'un rapport de force.
Le Gouvernement considère qu'il s'agit d'un faux problème et que l'offre s'ajustera spontanément. Notre association a donc réalisé une expérience, que va vous présenter Michel Simonnet.
M. Michel Simonnet, trésorier de l'association Les Poissons roses . - J'aborderai la politique par le bas. Ce projet de loi nous paraît inapplicable étant donné la pénurie de gamètes. Sans gamètes tout ce qui est évoqué ici n'aura aucun effet.
Je rappellerai quelques chiffres. En 2016, nous comptions 363 donneurs et 3 500 femmes traitées pour infertilité pathologique. Ce projet de loi prévoit 2 000 à 3 000 nouvelles candidates à une AMP. Ce constat de pénurie a conduit l'association Les Poissons roses à élaborer un kit de donneur - une burette et un certificat de traçabilité signé par le donneur.
Le 9 juillet dernier, nous avons présenté ce kit à l'Assemblée nationale pour recueillir la semence de 95 députés de moins de 45 ans. Nous n'avons pas oublié les sénateurs, de moins de 45 ans. Nous leur avons préparé un kit personnalisé, avec leurs noms.
La liste des 110 ministres et parlementaires est consultable sur le site de l'Établissement français de semence des ministres et parlementaires.
Cette proposition peut vous paraître un peu potache, mais comment souligner autrement l'incohérence du projet de loi qui oublie que le don de gamètes est essentiel ? Le problème serait résolu, selon M. Touraine, si 0,01 % des hommes français acceptaient de donner leur sperme. Or, nous devons le constater, les hommes, trop conscients de l'importance de ce geste, refusent d'effectuer ce don. Aussi, il conviendra d'envisager - pourquoi pas ? - une procédure similaire à celle du droit au logement opposable (DALO) gérée par le préfet.
Par ailleurs, cette pénurie créera, d'une part, un marché gris des gamètes, où régneront l'inégalité et l'injustice et, d'autre part, une marchandisation des gamètes. Nous refusons d'entrer dans cette logique marchande.
En conclusion, ce projet de loi bute sur une réalité humaine oubliée : la réticence masculine à donner ses gamètes. L'aveuglement à ignorer cette réalité est surprenant. Aussi, nous ne croirons à l'AMP pour tous lorsque plus de 50 parlementaires auront donné leurs gamètes, montrant ainsi l'exemple à la nation tout entière.
Pouvez-vous, messieurs les sénateurs, nous donner des garanties à ce sujet ?
M. Alain Milon , président . - Je vous remercie pour vos interventions. Avant de laisser la parole aux rapporteurs, je souhaiterais revenir sur les propos de M. du Marais, relatifs à l'intérêt général. Je ne partage pas votre observation dans la mesure où vous avez essentiellement évoqué l'AMP, qui ne représente qu'un article de la loi. Par ailleurs, lors de l'élaboration de la dernière loi bioéthique, nous avions décidé qu'elle était révisable tous les sept ans - le Sénat avait proposé cinq ans. Je pourrais suivre votre argumentaire si le projet de loi concernait uniquement l'AMP.
Mme Muriel Jourda , rapporteur . - Je vous remercie pour vos présentations très complètes. Seule une question n'a pas été évoquée, celle du diagnostic préimplantatoire (DPI). Cependant, ce point étant du domaine de compétence de Mme Imbert, je lui laisse la parole.
Mme Corinne Imbert , rapporteure . - Je vous remercie également pour vos interventions.
Plusieurs d'entre vous ont évoqué la question de la marchandisation, inéluctable, du corps humain. Je comprends votre inquiétude, mais alors expliquez-vous le fait que le don du sang est toujours gratuit, alors que nous manquons parfois de donneurs ?
S'agissant du diagnostic préimplantatoire, de la même façon que vous avez rappelé le courage dont nous faisons preuve pour aborder ce projet de loi, ne pensez-vous pas que les couples qui engagent une démarche de procréation médicalement assistée font, eux aussi, montre d'un grand courage ? L'AMP est une expérience douloureuse, notamment quand les femmes sont confrontées à plusieurs échecs. Il leur faut beaucoup de courage, et une attente réelle, pour bénéficier d'un DPI avec recherche d'aneuploïdie (DPIA).
Par ailleurs, si vous avez rappelé la considération que vous avez pour les femmes, vous avez également indiqué que ce texte était une défaite pour ces dernières, avec une emprise médicale. Ne pensez-vous pas que ce DPIA pourrait améliorer les chances de réussite d'une fécondation in vitro, notamment pour les femmes de 35 à 42 ans, et éviter des fausses couches qui sont, elles aussi, toujours très douloureuses ?
Mme Streb Blanche . - Nous ne pouvons pas comparer le don de sang au don de gamètes. Le don du sang sauve des vies et fait appel à la fraternité, à la solidarité. En revanche, le don de sperme ne sauve pas de vies, il en crée. Il me semble d'ailleurs que si nous comptons si peu de donneurs de gamètes, c'est bien parce que les hommes ont conscience que ce don n'est pas anodin. Il s'agit d'une paternité : ils sont les pères biologiques d'un ou de plusieurs enfants et, avec l'avancée de cette loi, les enfants nés de ces dons, auront la possibilité, dès leurs 18 ans, de le rencontrer.
Ce don est si peu anodin qu'un amendement a été adopté, aux termes duquel les donneurs de gamètes pourraient connaître le nombre et le sexe des enfants nés de leur don, ce qui explique pourquoi ces cellules, porteuses de vie, sont si peu données en France.
Concernant le DPI, le risque d'emballement que permet, et que permettra, la technique, nous inquiète. C'est un risque que nous observons à l'étranger, l'embryon étant rendu disponible par la fécondation in vitro.
Et pourquoi ne pas ouvrir le DPI pour toutes ? Pour celles qui bénéficient de fécondations in vitro pour raison d'infertilité, comme pour celles qui voudraient y avoir recours uniquement pour faire analyser leurs embryons, un risque identifié par Mme Buzyn.
À l'étranger, le DPI est très limité. Il n'est autorisé que pour des maladies très graves, incurables, et il est ouvert aux familles chez qui il existe un risque héréditaire. Ouvrir le DPI à des problèmes génétiques ou chromosomiques qui ne sont pas héréditaires reviendrait à permettre le contrôle qualité de l'embryon. Les aneuploïdies ne sont pas des problèmes chromosomiques héréditaires, ce sont des accidents de fécondation.
Le DPI est une bombe à retardement. L'enjeu est énorme : un eugénisme technologique à grande échelle. La Grande-Bretagne a établi une liste de plus de 2 000 maladies qui donnent aux familles l'accès au DPI. Elles vont des maladies extrêmement graves jusqu'à des prédispositions - et même au strabisme. Par ailleurs, le DPI est aujourd'hui commercialisé par des cliniques privées dans différents pays, notamment à Chypre et aux États-Unis, afin de permettre aux couples qui le souhaitent de choisir, par exemple, les embryons en fonction de leur sexe.
Mme Ludovine de La Rochère . - Madame Jourda, contrairement à ce que vous avez indiqué, nous n'avons pas développé tous nos arguments. En dix minutes, il nous a été difficile de hiérarchiser l'essentiel, tant ces sujets sont vastes, et les conséquences et implications, sur un plan éthique, mais également humain, stupéfiantes.
S'agissant du DPI, le projet de loi, dans son article 1 er , ouvrirait l'AMP aux couples homme-femme féconds. Pourquoi ces couples souhaitent-ils avoir recours à l'AMP ? Pour bénéficier d'un diagnostic prénatal, bien entendu. Nous pouvons alors deviner l'enchaînement qui, forcément, s'ensuivra.
Concernant l'achat des gamètes, le constat de tous les pays, sans exception, qui ont ouvert l'AMP au-delà des cas liés à une pathologie et sont rentrés dans la bourse des gamètes, si je puis m'exprimer ainsi, en suivant la loi de l'offre et de la demande. La banque de sperme danoise Cryos International en est un exemple. D'ailleurs, le projet de loi évoque l'importation des gamètes, en posant des conditions. Par exemple, le don de gamètes ne doit pas se faire au profit d'une entreprise commerciale. Mais en réalité, il existe bien des façons d'entrer dans une forme de trafic international de gamètes.
Le 31 août 2018, la Grande-Bretagne a exposé publiquement le fait que, dans le cadre du Brexit, elle aurait besoin de passer des accords sur l'AMP, sinon elle ne pourrait plus importer de sperme, d'ovocytes et d'embryons pour continuer à pratiquer des PMA, comme elle le souhaite.
Mme Pascale Morinière . - Vous avez rappelé, madame la rapporteure, que le don de sang était gratuit ; heureusement ! Il fait appel à la solidarité et à la fraternité. Quand les hôpitaux sont en manque de sang, ils lancent un appel à la population, qui y répond.
L'appel aux dons de gamètes n'a pas la même efficacité, ou alors cela m'a échappé. Depuis cinq ans, l'Agence de la biomédecine lance, chaque année, des campagnes pour le don de gamètes, avec de charmants slogans tels que « donneur de bonheur », « le plus petit des cadeaux ». Nous sommes passés de 363 donneurs en 2016 à 404 en 2017. Ces 404 donneurs ont permis la naissance de 1 027 enfants. L'étude d'impact avance le chiffre de 2 000 à 3 000 femmes intéressées par le don de gamètes. Il s'agit, en fait, d'une estimation, car personne ne peut prévoir combien de femmes seront intéressées. Mais nous pouvons imaginer aisément le nombre de donneurs qui manque.
La seule solution, pour pallier la pénurie de gamètes, serait de rétribuer les donneurs. Mais le risque est que les donneurs de sang, les donneurs de rein, et autres, réclament, eux aussi, une rétribution, de sorte que notre bioéthique à la française, fondée sur la fraternité - une valeur forte -, pourrait s'effondrer. Voilà quelques semaines, un journaliste américain s'étonnait de la gratuité du don de gamètes. La gratuité est vraiment une spécificité française qui nous honore.
S'agissant des PMA, elles ont augmenté de 5 % entre 2016 et 2017, alors que, dans le même temps, l'infertilité des femmes n'a certainement pas augmenté de 5 %.
Lors de mes études de médecine, le principe appliqué était le suivant : un couple devait avoir entretenu deux ans de relation régulière avant de procéder à des recherches sur l'infertilité puis éventuellement, à pratiquer une PMA. Aujourd'hui, dès qu'un couple consulte, le médecin propose une restauration de la fertilité, sans mener au préalable un travail de compréhension, de recherche des causes. Le marché est là : il encourage et incite les médecins à pratiquer des PMA. Pourquoi ? Parce qu'une PMA coûte 4 100 euros, pour une tentative de FIV - quatre tentatives sont remboursées par la sécurité sociale - et 1 000 euros pour chaque tentative d'insémination. La PMA n'est donc pas gratuite pour tout le monde. Il s'agit, je le répète, d'un énorme marché, l'idée de la gratuité n'étant qu'un leurre.
Je vous recommande de vous intéresser aux transferts d'argent, aux coûts et à ce marché.
Mme Ludovine de La Rochère . - Un coût qui serait remboursé par la sécurité sociale, ce qui poserait un autre problème.
M. Bertrand Lionel-Marie . - Je souhaite rappeler une donnée factuelle : selon l'INSEE, en 2018, 266 000 personnes vivaient en couple avec un conjoint de même sexe : 44 % de ces couples sont des couples de femmes, soit 58 520 couples - je laisse volontairement de côté la question des femmes célibataires.
Par ailleurs, cet institut indique que 14 630 couples de femmes vivent avec des enfants, ce qui signifie que, potentiellement, 43 890 couples de femmes sont concernés par l'ouverture à l'AMP. Si nous supprimons les couples dont les femmes ne sont plus en âge de procréer, 2 000 couples pourraient être potentiellement intéressés.
Le rapport de l'Agence de biomédecine de 2017 indique que « 1 961 couples ont effectué au moins une AMP avec les spermatozoïdes d'un donneur dans l'année », sachant que nous sommes déjà en tension, que l'attente pour obtenir des spermatozoïdes est d'environ un an. C'est la raison pour laquelle le risque de marchandisation est fort, d'autant que la Belgique a ouvert la voie en achetant du sperme au Danemark.
Par ailleurs, nous sommes bien obligés de tenir compte de l'état actuel du marché. Je vous invite à visiter le site de Cryos International, la plus grande banque de sperme en Europe, mais également à visionner le documentaire intitulé « Bébés sur mesure ». Dans ce documentaire, le médecin, qui a créé cette banque de sperme à Aarhus, ville étudiante, se compare à un fournisseur de lait, et nous apprenons qu'il envoie tous les jours des doses de sperme en France. Or, depuis deux ans, seules deux naissances ont été déclarées liées à du sperme fourni par Cryos International.
Mme Marie Mercier . - Vous avez le mérite de réfléchir. Nous aussi, au Parlement, nous pratiquons le doute cartésien, et organisons de très nombreuses auditions, sur un très large spectre. Plus on avance, plus on réfléchit - plus on recule, parfois, aussi ! En tous cas, nous n'avons pas d'idées préconçues.
Mademoiselle, je pense comprendre votre souffrance et la nécessité de mettre un nom sur un mal-être ; sans être psychiatre, je ne suis pas sûre que ce soit la seule réponse.
Il faut comparer des choses comparables. Se comparer aux autres pays ne me satisfait pas : la France est la France, avec sa culture, la façon dont elle s'est construite, en avançant, en reculant... Ce qui est bien ailleurs n'est peut-être pas bien pour nous, et vice-versa. Il y a de très bonnes choses chez nous. Se comparer n'est pas forcément toujours une bonne chose, pas plus qu'imiter ce qui se fait ailleurs. Le Sénat, vous savez, est une chambre de refroidissement...
M. Alain Milon , président . - Pas de congélation !
Mme Marie Mercier . - Nous avons le mérite de réfléchir, je crois, avec sérénité, et nous sommes aussi dans l'accueil et dans une forme de don - ce qui ne nous enlève pas le droit d'être quelquefois à contre-courant ! Pour nous, la vertu est juste au milieu de la rivière.
M. Olivier Henno , rapporteur . - Je n'aurais pas dit mieux ! La volonté d'améliorer la vie, de la prolonger, est une quête humaine qui ne date pas de 2019. Et l'usage de la science est une question légitime de l'éthique, posée par les avancées scientifiques. Par nature, en démocratie, en République, les convictions sont diverses. Que pensez-vous de l'autoconservation des ovocytes, et même de l'autoconservation des gamètes ? Trouvez-vous que c'est une aspiration féminine légitime, qui relève d'une recherche d'égalité ? Selon le professeur Nisand, cela répond à des aspirations personnelles de femmes qui veulent prolonger leur vie professionnelle sans renoncer à leur droit à la maternité et à l'enfant.
Mme Caroline Roux . - Pour moi, c'est une défaite des femmes, une fausse promesse. On fait miroiter aux femmes qu'elles auront des enfants, plus tard, par PMA. Nous sommes intervenus déjà pour la loi de 2011. Il y a un vrai décalage, dans notre société, qui doit tous nous interroger. C'est au cours de la meilleure période de fertilité, c'est-à-dire entre 20 et 30 ans, qu'on observe les plus forts taux d'avortement. Nous recevons dans nos services d'écoute des jeunes filles de 25 ans, enceintes dans un couple stable, qui hésitent à poursuivre leur grossesse, car les femmes enceintes qu'elles voient dans la rue sont plus vieilles. Ne devrions-nous pas changer de regard sur la maternité ? Nous demandons depuis longtemps qu'on aménage les conditions sociales pour que les femmes jeunes qui poursuivent leurs études ou entrent sur le marché du travail puissent concilier leur activité avec une vie maternelle si elles le souhaitent.
Nous travaillons aussi beaucoup, dans nos services d'écoute, sur le deuil. La technique appelant la technique, on fait miroiter aux femmes la possibilité d'avoir des enfants plus tard. Mais quid si elles ne trouvent pas de compagnon ? Auront-elles un enfant toutes seules, grâce à la PMA ? Cela suggère une quête insatiable, alors que normalement, à partir d'un certain âge, les femmes doivent faire le deuil de la maternité et s'ouvrir à d'autres fécondités, selon leurs talents. Nous accompagnons des femmes qui se demandent, à 40 ou 45 ans, comment avoir des enfants. Elles sont souvent dans une autre vie, recomposée, avec un autre homme, et ces réflexions sur les autres fécondités qu'elles pourraient avoir - autres, puisque la loi, en France, interdit encore - leur ouvrent aussi des chemins. En interdisant, la loi est protectrice, au fond.
Par rapport aux aspirations beaucoup plus écologiques des jeunes femmes d'aujourd'hui, ce n'est pas non plus une réponse adéquate : il s'agit de traitements lourds, pour des personnes qui n'ont pas de problèmes de santé... Cela semble tout à fait disproportionné. Notre société doit vraiment réfléchir à une manière d'intégrer la maternité non pas comme un handicap pour les femmes quand il s'agit de travailler, mais de manière à ce que celle-ci fasse pleinement partie de l'organisation de notre société.
Mme Blanche Streb . - En tant que pharmacien, cette question me paraît assez symbolique de ce projet de loi bioéthique, qui prend tout de même quelque peu la médecine à l'envers ! Il s'agit de proposer à des femmes plutôt jeunes, si l'on veut que cela réussisse, des traitements vraiment lourds - stimulation ovarienne, ponction ovarienne - qui ne sont pas dénués d'effets secondaires. L'imprégnation hormonale crée des risques, qui sont estimés à 1,5 %, de développer plus tard des cancers. Et cela peut abîmer les ovaires. C'est donc prendre la médecine à l'envers. Des sociétés privées vont entrer en concurrence pour réaliser le stockage... On a l'impression qu'il s'agit d'une sorte de guet-apens sur le ventre des femmes pour créer un nouveau secteur économique. Le rapport du CCNE sur cette question montrait d'ailleurs qu'il ne s'agissait aucunement d'une demande des femmes, mais bien plutôt des sociétés savantes, et des centres d'assistance à la procréation. On entend parfois que ce serait une nouvelle liberté, une nouvelle autonomie. N'est-ce pas plutôt s'enchaîner à la technique ? A quoi rime de mettre en banque des ovocytes qui peut-être ne serviront même pas ? Si une femme rencontre ensuite l'homme avec qui construire sa vie et faire des enfants, elle se retrouvera avec des ovocytes dont elle ne saura pas quoi faire. Faudra-t-il qu'elle les donne ? Bref, c'est une manière étonnante de considérer la médecine.
Mme Ludivine de la Rochère . - Et cela va instaurer une forme de pression nouvelle sur la femme. Imaginez une jeune femme qui attend un enfant naturellement à 25 ans, à 28 ans, à 30 ans. Son employeur va la regarder de travers, car elle aurait pu faire conserver ses ovocytes, avoir un enfant plus tard ! Je ne dis pas que l'employeur lui fera un reproche : tout restera implicite. Et l'on connaît déjà, dans l'entreprise, la culpabilité de la jeune femme qui attend un bébé. Souvent, elle n'ose pas le dire, et met longtemps à le faire - et elle démissionne d'elle-même si elle était en période d'essai. Il y a déjà une forte pression ; si l'on sait, en plus, que n'importe quelle jeune femme peut faire conserver ses ovocytes pour bénéficier ultérieurement d'une AMP... Il s'agit d'une technicisation inutile de la procréation, à un âge où la femme ne sera plus féconde, où son utérus aura vieilli.
Au lieu que la société, longtemps organisée par une majorité d'hommes, s'adapte à la femme à mesure que celle-ci prend davantage de responsabilités, on oblige la femme à s'adapter à un monde qui a été fait, pour une grande part, par les hommes. En fait, c'est un mépris stupéfiant de la femme que révèle cette idée, dont les conséquences vont être lourdes sur toutes les jeunes femmes, pendant de longues années.
Évidemment qu'à 25 ans, ce n'est pas le bon moment ; qu'à 30 ans, ce n'est pas le bon moment ; qu'à 40 ans, quand on devient responsable, directrice, directrice adjointe, ce n'est pas non plus le moment... Un bébé, en clair, sur le plan professionnel, ce n'est jamais le moment ! Pensez aussi aux conjoints : « Ma chérie, tu pourrais peut-être le faire plus tard, nous ne sommes pas pressés, nous n'avons pas encore assez d'argent : congèle tes ovocytes, et on verra plus tard ! ». Mais, plus tard, il y aura d'autres charges : ce n'est jamais le moment. Il faut au contraire que l'entreprise s'adapte à la femme et à la maternité.
M. Bertrand du Marais . - La congélation des ovocytes va dans le sens de la création et du développement d'un marché de la procréation. Cette question est liée à la vie professionnelle des femmes, et à la façon dont elles font famille. Ce sont des questions de nature à la fois scientifique et bioéthique. Pourtant, elles sont isolées de la réflexion plus générale. Quelle famille voulons-nous ? Comment aménager le développement démographique de la trajectoire des familles et de la trajectoire des individus ? Aujourd'hui, à 50 ans, on est pour ainsi dire à mi-vie, et il reste une longue vie en couple à mener - alors que, dans les années 1950, l'homme décédait vers 60 ou 65 ans. Il y a un lien entre le débat sur le type de cellule familiale qu'on veut, et celui sur les dispositifs techniques qu'on autorise. Nous pensons que ces deux réflexions doivent être connectées. Sinon, la vision de la situation demeure partielle.
Vous avez raison, monsieur le président, il y a dans cette loi un certain nombre de dispositions, par exemple sur les traitements numériques dans la santé, qui poursuivent un but d'intérêt général. Or le contrôle du Conseil constitutionnel isole, article par article, les dispositions d'un projet de loi. Certes, ce n'est que l'article 1 er - mais l'étude d'impact lui consacre bien plus qu'un 34 ème de sa dimension... Et nous considérons que cette disposition opère un véritable glissement sur la notion de loi dans une République.
Vous avez rappelé que le Sénat, à juste titre, a insisté pour que les lois de bioéthique soient rediscutées de façon régulière. C'est en effet tout à l'honneur de notre pays. Les états généraux de bioéthique n'ont pas d'égal ailleurs dans le monde. Il faut les maintenir. Or l'étude d'impact cite les avis du CCNE de 2017 et 2018, mais pas l'avis l'intermédiaire, qui fait l'état des lieux de la consultation. Or, dans cet avis intermédiaire, le CCNE dit qu'à l'issue d'une consultation très large et approfondie, beaucoup de participants étaient opposés à l'article 1 er . Autrement dit, grâce à ces instruments de participation des citoyens français, on a suscité des réactions que le Parlement, brutalement, s'apprête à fermer, simplement parce qu'une majorité s'est dégagée autour d'un programme électoral. Nous demandons à ce que ce type de loi, qui engage l'ensemble de la société, soit adopté au Parlement par une majorité des deux tiers. Et nous demandons une totale liberté de vote au sein des groupes parlementaires.
Mme Marie Mercier . - Soyez rassurés, nous sommes très libres !
Mme Pascale Morinière . - Quelques chiffres : le taux de fécondité est de 1,88 enfant par femme, alors que les femmes souhaitent 2,39 enfants. La différence signifie, en quelque sorte, qu'une femme sur deux souhaiterait un enfant de plus. À l'heure où l'on réforme les retraites, que c'est douloureux pour nous tous, je m'étonne que l'on ne prenne pas cette partie de la démographie en compte. Elle a pourtant un impact. L'âge moyen des femmes qui accouchent est de 30,5 ans ; l'âge moyen du premier enfant, de 28,5 ans. Ces chiffres continuent d'augmenter, alors que la fécondité est maximale entre 20 et 30 ans. L'intérêt général, ou le bien commun, ne serait-il pas que les jeunes femmes aient des enfants plus tôt, et qu'elles aient le nombre d'enfants qu'elles souhaitent ? Je ne dis pas qu'il faut faire plus d'enfants, je dis qu'il faut donner la possibilité aux couples d'avoir le nombre d'enfants qu'ils souhaitent. Les lois doivent faire en sorte de réaliser ces désirs d'enfant. Cela nous donnerait une démographie suffisamment vigoureuse pour assurer notre avenir.
M. Bertrand Lionel-Marie . - Georges Bernanos, dans La France contre les robots, écrit - ce qui me paraît pertinent quand on parle de l'autoconservation ovocytaire -, que « la société moderne est désormais un ensemble de problèmes techniques à résoudre ». On ne voit le problème que sous le petit angle technique, au lieu de le voir dans sa globalité.
Moins littéraire, mais tout aussi intéressant : une journaliste du magazine Elle a dit que « l'idée qu'il faut changer le corps des femmes pour le plier à la norme sociale du travail, plutôt que de changer la norme, est un choix politique contestable ».
Et l'avis du CCNE n° 126, du 15 juin 2017, s'opposait à l'autoconservation ovocytaire. Je le cite : « Différer un projet de grossesse à un âge tardif - connaissant les risques de ces grossesses tardives - peut difficilement être considéré comme participant à l'émancipation des femmes face aux limites biologiques. Outre le mésusage et les pressions socioprofessionnelles auxquels cette technique peut exposer, le bénéfice escompté au regard des moyens médicaux et économiques qui devraient être déployés apparaît très faible.». On ne comprend d'ailleurs pas très bien pourquoi le CCNE a changé d'avis entre 2017 et 2018. Le CCNE souligne aussi le risque des grossesses tardives. Et l'augmentation de la fréquence des grossesses tardives, à plus de 40 ans, ou ultra-tardives, à plus de 45 ans, dites à haut risque, pourrait être une conséquence dommageable de l'autoconservation ovocytaire. Bref, c'est une mauvaise réponse à une vraie question. La seule bonne réponse serait une réponse politique et sociale - ce qui relève de votre commission des affaires sociales !
Le magazine Bloomberg titrait récemment : « Freeze your eggs, free your career », c'est-à-dire : « Congelez vos ovocytes, libérez votre carrière ». On va vers une logique marchande - car ces techniques auront évidemment un coût. Je ne suis pas sûr que ce soit un progrès. Je suis même sûr du contraire !
Mme Anaïs Doisneau . - Madame la sénatrice, vous semblez, de façon très surprenante, balayer d'un revers de main un témoignage qui tente pourtant d'être aussi authentique que possible. Vous le qualifiez de relatif et subjectif. Comment pouvez-vous dire qu'il y a peut-être d'autres raisons psychiatriques ? J'essayais de montrer que les statistiques ne sont peut-être pas très judicieuses pour révéler le mal-être qu'induit la privation d'un père. D'un point de vue purement statistique, cela ne se verrait pas : c'est beaucoup trop intime. Nous montons une association, « La Voix des sans père », et nous constatons que tous ceux que nous rencontrons partagent des traits communs, conséquence de ce manque de père. Il ne s'agit pas d'un mal-être général, mais très précis.
M. Alain Milon , président . - Je connais bien Mme Mercier, qui est médecin et a rédigé des rapports sur la maltraitance de l'enfant. Je ne pense pas qu'elle ait voulu dire cela. Au contraire, elle a voulu dire que chacun avait le droit de penser comme il voulait, à partir du moment où cette pensée était justifiée ou justifiable.
Merci à tous. Vous avez parlé de choses illégales et légales, en faisant notamment référence à des juristes italiens. Il y a des personnages politiques qui sont légalement arrivés au pouvoir. D'autres, comme De Gaulle, illégalement !
Ce point de l'ordre du jour a fait l'objet d'une captation vidéo qui est disponible en ligne sur le site du Sénat .
Audition de M. Guillaume Drago,
président,
et Me Geoffroy de Vries, délégué
général,
de l'Institut Famille et République
M. Michel Amiel , président . - Nous poursuivons nos travaux sur le projet de loi relatif à la bioéthique en recevant MM. Guillaume Drago et Geoffroy de Vries de l'Institut Famille et République à la demande de notre collègue rapporteur Mme Muriel Jourda. Ils sont accompagnés de M. Jean Dupont-Cariot.
Me Geoffroy de Vries, délégué général de l'Institut Famille et République . - Merci de nous avoir conviés à la présente audition. L'Institut Famille et République est un cercle d'une centaine de juristes de toutes professions, qui travaille sur des questions relatives au droit de la famille et aux droits de l'homme. Nous intervenons ce jour sur la question de la liberté de conscience.
Notre République est fondée sur des principes constitutionnels dont font partie les libertés fondamentales. L'une de nos libertés les plus fondamentales est la liberté de pensée, ou d'opinion, qui veut que nous soyons tous libres de penser ce que nous voulons de telle ou telle question.
Cette liberté de pensée en crée d'autres : la liberté d'expression de sa pensée, la liberté de manifestation ainsi que la liberté de conscience. La liberté de conscience est la liberté de ne pas être contraint d'agir contre ses convictions intimes et fondamentales.
La liberté de conscience est en quelque sorte la petite soeur de la liberté de pensée. Elle est plus jeune, elle est moins connue, elle est sans doute aussi plus timide. Elle peine, souvent, à s'exprimer. Mais elle a toute sa part dans la grande famille des libertés fondamentales. Elle est reconnue par le Conseil constitutionnel. Elle est prévue par la Déclaration des droits de l'homme et du citoyen et par le préambule de la Constitution du 27 octobre 1946. Elle consiste à pouvoir objecter sa conscience personnelle pour refuser de procéder à un acte la heurtant profondément, sans être passible de sanctions.
Liberté et objection de conscience sont en réalité deux mots servant à signifier le même concept, les deux faces d'une même pièce. Cette liberté de conscience est davantage que la liberté d'opinion, car elle comporte une indéniable approche personnelle d'ordre plus fondamental et intime que la simple opinion. Elle implique un engagement de la personne tout entière, une intime conviction, une adhésion profonde, en son for intérieur, qui peut être de nature philosophique ou religieuse - mais non politique.
Ainsi, en tant que citoyen - y compris notaire, médecin ou officier d'état civil -, on peut, en son for intérieur, persister à être opposé à la déréglementation de la procréation médicalement assistée (PMA) ou à la recherche sur l'embryon, facilitées par le projet de loi, sans être concrètement confronté à un acte de ce type. Nous sommes là dans le cadre de la liberté de pensée ou d'opinion. Mais on peut aussi, in concreto, en tant que notaire ou médecin, être concerné directement par une demande de PMA pour un couple de femmes ou une femme seule sans pouvoir y renoncer légalement ; et donc être confronté à un conflit intérieur.
La liberté personnelle serait abolie si la personne concernée était contrainte d'agir contre sa conscience.
En réalité, il s'agit de concilier deux droits fondamentaux : en matière de PMA, par exemple, le droit à la PMA pour toutes les femmes, pour tous les couples - si le Parlement en décide ainsi - et le droit pour les notaires ou les médecins de pouvoir objecter leur liberté de conscience.
En tout état de cause, la PMA aura bien lieu. La reconnaissance d'une clause de conscience n'aura pas pour effet ou objet d'empêcher sa réalisation. Elle aura simplement pour effet de préserver la liberté du notaire ou du médecin en organisant son déport au profit d'un autre.
En réalité, la clause de conscience est d'inspiration républicaine. Elle ne s'oppose pas à ce que la loi permet, mais elle organise le déport de personnes qui ne peuvent adhérer à l'acte concerné. Elle permet, en somme, l'application de la loi, mais par un autre. C'est le contraire de la rébellion civique ou de la désobéissance civile. La clause de conscience est une garantie du caractère général de la loi.
Le projet de loi relatif à la bioéthique qui vous est soumis apporte des réponses très particulières à des situations très particulières et à des demandes minoritaires telles que l'extension de la PMA aux couples de femmes pour des raisons qui ne sont pas thérapeutiques, et ce en dérogeant aux principes de la filiation et de la médecine ainsi qu'au caractère général et à l'objectif de la loi.
La clause de conscience permet en réalité de rétablir le caractère général de la loi. Petite soeur de la liberté de pensée, la liberté de conscience demande à être reconnue et protégée. La liberté de conscience est à la loi de bioéthique ce que la soupape est à la marmite : un gage de fonctionnement et de sécurité.
M. Guillaume Drago, président de l'Institut Famille et République . - Je rappelle que la liberté de conscience est liée à la liberté d'opinion et qu'elle fait référence à plusieurs normes constitutionnelles qu'il faut avoir à l'esprit : le premier alinéa de l'article 1 er de la Constitution ; l'article 10 de la Déclaration des droits de l'homme et du citoyen de 1789 - « Nul ne doit être inquiété pour ses opinions, même religieuses, pourvu que leur manifestation ne trouble pas l'ordre public établi par la loi » - ; l'alinéa 5 du préambule de la Constitution de 1946 - « Nul ne peut être lésé, dans son travail ou son emploi, en raison de ses origines, de ses opinions ou de ses croyances » - et l'article 34 de la Constitution, dont l'un des alinéas signifie que la loi fixe les règles concernant les garanties fondamentales accordées aux citoyens pour l'exercice des libertés publiques. Il revient donc au législateur de garantir l'exercice effectif de ces libertés.
À ces règles constitutionnelles s'ajoute une jurisprudence constitutionnelle depuis la décision du Conseil constitutionnel du 23 novembre 1977 relative à la liberté de conscience des professeurs, qui a été confirmée ensuite à plusieurs reprises : en 1985 tout d'abord, puis à l'occasion de la loi du 4 juillet 2001 relative à l'interruption volontaire de grossesse et à la contraception.
Cette jurisprudence signifie que la liberté de conscience ne relève pas seulement du for intérieur, mais inclut aussi le droit d'extérioriser ses convictions et d'y conformer son attitude. Il ne s'agit donc pas seulement d'une liberté personnelle.
De ce point de vue, la jurisprudence de la Cour européenne des droits de l'homme (CEDH) souligne dans son arrêt du 26 mai 2011 que « les États sont tenus d'organiser leurs services de santé de manière à garantir que l'exercice effectif de la liberté de conscience des professionnels de la santé dans un contexte professionnel n'empêche pas les patients d'accéder à des services auxquels ils ont légalement droit ».
Il existe donc un équilibre fondamental entre l'exercice d'une profession, l'accès à un service public, mais aussi la nécessité pour le législateur d'organiser un exercice effectif de la liberté de conscience des professionnels. C'est la raison pour laquelle, dans le projet de loi relatif à la bioéthique, il nous paraît important d'instituer pour cette législation très largement dérogatoire à la loi commune une clause de conscience et un droit de retrait pour les professionnels de santé en cas d'assistance médicale à la procréation (AMP), en cas éventuel de gestation pour autrui (GPA) et en matière de recherche sur l'embryon, en renforçant ce qui est déjà inscrit dans le code de la santé publique.
Nous soutenons également la nécessité de reconnaître une clause de conscience pour les notaires, ou au moins une clause de déport concernant les actes authentifiant les déclarations de volonté des couples avant la naissance.
Nous soutiendrons également, comme nous l'avions fait en 2013 devant le Conseil constitutionnel à l'occasion d'un contentieux de question prioritaire de constitutionnalité (QPC), la même possibilité de déport pour les officiers et agents d'état civil en renvoyant au représentant de l'État dans le département, préfet ou sous-préfet, la désignation d'un autre officier d'état civil pour répondre aux demandes formulées d'enregistrement des actes.
La jurisprudence européenne, comme les résolutions de l'assemblée parlementaire du Conseil de l'Europe, insiste sur le fait que l'État doit lui-même agir expressément, explicitement, pour assurer l'équilibre entre ces prétentions en organisant dans le système de santé ou pour les autres professionnels la possibilité d'exercer leur droit à l'objection de conscience, tout en organisant l'accès aux services publics ou aux prestations prévu par la législation.
Il faut bien comprendre le sens de la demande de reconnaissance de ces différentes clauses de conscience.
Il s'agit d'équilibrer les droits des usagers de différents services publics avec les droits légitimes des professionnels concernés en présence d'une législation intrinsèquement dérogatoire au droit commun. Il s'agit seulement de permettre une forme de déport fondée sur une liberté constitutionnelle - la liberté de conscience -, qui n'est en aucun cas une forme de désobéissance civile.
Le professionnel qui invoque sa liberté de conscience ne s'oppose pas à la loi de la majorité, ne conteste pas la généralité de la loi, mais souligne son désaccord par un mécanisme de retrait, de déport, d'objection, qui est le contraire de la rébellion ou de la désobéissance civique. Ce n'est que si cette clause de conscience n'était pas reconnue que la tentation de la désobéissance à la loi pourrait voir le jour.
Me Jean Dupont-Cariot, membre de l'Institut Famille et République. - J'interviens en tant que notaire, en mon nom et au nom des quelques notaires qui m'ont fait part de leur malaise par rapport à ce sujet.
Je vous remercie de bien vouloir entendre les interrogations des notaires face au souhait du législateur de nous confier une nouvelle mission. Si cette mission constitue une marque de confiance à l'égard de notre profession, il est nécessaire d'en mesurer les conséquences.
En effet, le nouvel article 342-10 du code civil prévu ouvre le recours à la PMA aux couples de femmes et aux femmes non mariées. Ainsi, la PMA n'a plus seulement pour objet de pallier une infertilité, mais également de répondre à un projet parental. Pour ce faire, le législateur a envisagé de demander aux notaires de recevoir une déclaration anticipée qui sera faite par des couples de femmes ou des femmes non mariées désirant recourir à une AMP.
Quelles en seront les conséquences pour les notaires ?
Le notaire devra recevoir aux termes d'un acte authentique la volonté de deux femmes de recourir à l'AMP. Or la mission d'authentification d'un acte consiste non seulement à conférer l'authenticité à cet acte, mais également à en assurer l'efficacité.
Comment le notaire peut-il participer à la reconnaissance d'une filiation totalement dérogatoire au droit commun de la filiation ? La Cour de cassation a d'ailleurs rappelé en octobre 2019 que le droit de la filiation est impératif et d'ordre public. Le point important est bien cette reconnaissance conjointe prévue à l'article précédemment cité, qui établit la filiation de l'enfant non encore conçu.
Toutefois, le législateur prévoit dans le même article que le consentement est privé d'effet en cas de décès, d'introduction d'une demande de divorce, de cessation de communauté de vie, mais aussi en cas de révocation du consentement avant la réalisation de l'insémination. Ainsi, la caducité d'un acte authentique peut résulter de la seule volonté de l'une des parties à l'acte.
Par ailleurs, comment le notaire peut-il s'assurer que le consentement de l'un des déclarants n'est pas vicié - élément essentiel d'un acte authentique ? Sachant que l'acte n'indique pas laquelle des femmes accouchera, nous pouvons craindre que ne se produise au moment de l'insémination un désaccord entre elles et que l'une d'elles considère que son consentement a été vicié.
Sur ce point, deux objections s'imposent à nous en tant que notaires. Le notaire ne peut pas participer à l'authentification d'une filiation fictive au point de vue biologique et généalogique, dont l'objet n'est encore qu'à l'état de projet, à savoir celui d'un enfant non encore conçu.
De plus, le notaire ne peut pas authentifier une situation qui n'existe pas dans la réalité et qui n'est que fiction - aujourd'hui encore, un enfant ne peut naître que de la réunion d'un gamète masculin et d'un gamète féminin. A priori, deux femmes ne sauraient donc être mères du même enfant. En outre, n'y a-t-il pas risque que le notaire participe à une marchandisation du corps en établissant ces actes, puisque nous savons déjà qu'il existe une pénurie de gamètes en France ?
Par ailleurs, l'acte authentique fait foi à l'égard des tiers des faits que le notaire y a constatés. Face à l'enfant qui sera né de cette PMA, quelle sera la responsabilité du notaire qui aura reçu un acte authentique ayant pour conséquence de priver cet enfant de la possibilité de faire établir sa filiation paternelle ? Le notaire peut se voir reprocher d'avoir reçu un acte portant préjudice à un tiers. Et nous sommes tout à fait dans ce cas, le tiers étant l'enfant non encore conçu.
En effet, le législateur souhaite interdire toute action aux fins d'établissement ou de contestation de la filiation et prive donc l'enfant, qui serait ainsi reconnu par l'acte authentique, d'un droit fondamental dont jouissent les enfants nés dans d'autres conditions, ce qui crée pour lui une vraie injustice.
Vous comprendrez que cette mission que veut confier le législateur aux notaires crée pour un certain nombre d'entre nous un grand malaise dans l'exercice de leur profession.
Dans ces conditions, et compte tenu du fait que cette loi dérogatoire ne peut être appliquée sans scrupules, voire objections, je vous demande de reconnaître une liberté de conscience pour les notaires qui ne souhaiteraient pas authentifier ce type d'acte. Il pourrait ainsi être prévu, pour permettre l'application de cette loi si elle venait à être adoptée, d'organiser un déport du notaire en saisissant la Chambre des notaires et en demandant la nomination d'un notaire qui accepterait de recevoir cet acte.
C'est cette demande que je formule pour les confrères qui partagent cette interrogation sur la portée de cet acte dans notre exercice en tant qu'officiers publics et ministériels, c'est-à-dire l'exercice d'une liberté de conscience.
Mme Muriel Jourda , rapporteur . - Vous avez indiqué que cette liberté de conscience pourrait s'opposer à la mise en oeuvre d'un droit dérogatoire au droit commun. Dans la mesure où l'extension de l'AMP serait intégrée dans le code civil, en quoi s'agit-il d'un droit dérogatoire au droit commun ?
Sachant que la clause de conscience existe déjà, me semble-t-il, pour les professionnels de santé, est-il nécessaire de prévoir une législation spécifique pour qu'elle puisse être opposée ? Pour les notaires, je crois que la question se pose dans les mêmes termes.
M. Guillaume Drago . - Cette législation inscrite dans le code civil conduit à instaurer différents types de filiation et différents systèmes devant coexister. Le droit de la filiation, fondé sur les présomptions qui sont celles qui sont actuellement prévues dans le code civil, prévoira donc des systèmes différenciés.
Il est d'ailleurs tout à fait symptomatique que le projet initial, qui prévoyait un titre VII bis et un système dérogatoire - il souligne ainsi le caractère dérogatoire des dispositions prévues - ait été finalement intégré dans le droit commun via la coexistence de différents systèmes filiatifs. On ne peut donc pas considérer que cette législation est une législation qui proposerait un modèle unique en matière filiative.
À partir du moment où cette distinction est inscrite dans la loi, il conviendrait que les professionnels, qui auront à l'utiliser, aient la possibilité d'invoquer une clause de conscience.
Me Geoffroy de Vries . - Il n'existe pas de clause de conscience pour les notaires, comme il y en a pour le propriétaire foncier qui ne veut pas que la chasse soit pratiquée sur ses terres, les avocats, les journalistes ou les médecins dans le cas d'une IVG. La liberté de conscience est toutefois un principe fondamental reconnu par les lois de la République, la Déclaration des droits de l'homme et du citoyen de 1789 et la Convention européenne des droits de l'homme. Cependant, en pratique, pour pouvoir l'exercer, si l'on ne veut pas être contraint d'accomplir un acte qui heurte profondément sa conscience intérieure, il faut qu'une clause de conscience soit prévue dans la loi. C'est l'objet de notre discussion.
Mme Corinne Imbert , rapporteure . - Vous avez évoqué la clause de conscience dans la recherche pour l'embryon. Pourriez-vous nous préciser votre pensée ?
M. Guillaume Drago . - Cette clause de conscience en matière de recherche sur l'embryon a été introduite, à l'initiative du Sénat, dans le code de la santé publique par la loi du 7 juillet 2011 relative à la bioéthique, dont l'article 53 dispose « qu'aucun chercheur, aucun ingénieur, technicien ou auxiliaire de recherche quel qu'il soit, aucun médecin ou auxiliaire médical n'est tenu de participer à quelque titre que ce soit aux recherches sur des embryons humains ou sur des cellules souches embryonnaires ». Ce dispositif est donc très large, car il vise l'ensemble des professionnels de santé. Nous pourrions nous en inspirer pour définir une clause de conscience générale. Notre proposition n'est donc pas incongrue.
Le code de déontologie médicale comporte aussi une disposition prévoyant que les médecins peuvent refuser des soins pour des raisons professionnelles ou personnelles, hors cas d'urgence. C'est une clause de bon sens qui permet à un médecin de ne pas pratiquer un acte, hors cas d'urgence, s'il ne relève pas de sa spécialité. On ne peut pas demander à un généraliste de s'improviser rhumatologue ! Il ne s'agit donc pas vraiment d'une clause de conscience. La clause de conscience a été inscrite spécifiquement dans la loi de 1975 sur l'interruption volontaire de grossesse. Il ne faut donc pas confondre la clause de conscience et le déport pour des raisons professionnelles : leurs philosophies sont différentes. Le déport du code de déontologie ne serait pas adapté pour les dispositions de la loi de bioéthique que nous évoquons. Pour cela, il faut que la loi comporte une clause de conscience spécifique.
M. Olivier Henno , rapporteur . - Cette demande de clause de conscience correspond-elle au souhait d'évolution éthique de votre profession ou bien vise-t-elle spécifiquement la PMA pour les couples homosexuels et pour les femmes seules ?
Me Jean Dupont-Cariot . - Certains peuvent être, à titre personnel, favorables à la loi, d'autres non, mais, en tant que notaires, notre réflexion est de nature juridique : comment reconnaître une filiation, par un acte authentique, alors que l'enfant n'est pas encore né, qu'il ne s'agit encore que d'un projet ? Il faut aussi évoquer l'inégalité entre un enfant légitime, qui peut effectuer une action en recherche de paternité, et l'enfant conçu dans le cadre d'une PMA qui ne pourra le faire. On peut donc comprendre que certains notaires considèrent qu'ils ne peuvent conférer l'authenticité à un tel acte. Dans ce cas, ils pourront se déporter et un autre notaire le fera.
M. Guillaume Drago . - Notre proposition serait d'introduire une clause de conscience dans l'ordonnance du 28 juin 1945 relative à la discipline des notaires et de certains officiers ministériels, qui permettrait de ne pas bloquer la démarche : le notaire qui ne souhaiterait pas instrumenter demanderait au président de la chambre départementale ou interdépartementale de l'Ordre des notaires de désigner un autre notaire volontaire pour établir l'acte. Le notaire ne pourrait faire l'objet d'aucune sanction ou mesure discriminatoire dans le cas où son refus d'instrumenter serait fondé sur l'invocation de sa clause de conscience.
Me Geoffroy de Vries . - Pour les médecins, on pourrait ajouter après l'article L. 2141-12 du code de la santé publique un nouvel article, inspiré de l'article 53 de la loi de bioéthique de 2011.
M. Alain Milon , président . - Les médecins, en vertu du serment d'Hippocrate, ont pour mission première de donner la vie. L'IVG avait un autre but, et c'est pourquoi on a prévu une clause de conscience pour les médecins qui ne souhaitent pas la pratiquer. De la même façon, en 2011 - j'étais rapporteur de la loi au Sénat -, nous avions accordé la clause de conscience aux médecins et aux chercheurs ne souhaitant pas participer à des recherches sur les embryons ou les cellules souches embryonnaires parce que la recherche sur l'embryon entraîne sa destruction. Certains estimaient que cela pouvait être contraire au serment d'Hippocrate ; d'où, la clause de conscience.
Mais les notaires sont des officiers publics. Ils possèdent une délégation d'autorité de l'État pour appliquer la loi. Je comprends mal, dès lors, comment justifier une demande de dérogation pour ne pas appliquer la loi...
Me Geoffroy de Vries . - Vous dites que la première mission des médecins est de donner la vie. En l'occurrence, la PMA pour les femmes seules ou les couples de femmes n'a pas de visée thérapeutique, et si elle a pour effet de donner la vie, ce ne sera pas de manière naturelle, mais de manière quelque peu construite, pour ne pas dire déconstruite... On peut se demander comment évoluera l'enfant lorsqu'il grandira... Quant aux notaires, même s'ils sont dépositaires d'une mission d'autorité publique, il n'en demeure pas moins qu'ils pourraient être amenés à procéder à des actes qui heurtent profondément leur conscience. Il n'existe nul équivalent, nul cas où l'on soit contraint de faire quelque chose par la loi, si on ne l'a pas accepté dès le départ. Être notaire ne signifie pas accepter tout ce que le législateur peur lui demander de faire par la suite.
Me Jean Dupont-Cariot . - Le notaire, en tant qu'officier public, doit exécuter la loi. Le refus d'instrumenter est donc très encadré. Il vise des cas bien spécifiques, comme des actes concernant des fonds dont la provenance est douteuse, etc. C'est justement pour cette raison que nous demandons une clause de conscience. Certains notaires éprouvent un réel malaise au vu de la loi. Il ne s'agit pas de ne pas l'appliquer, car, grâce au déport, un autre notaire sera nommé qui recevra les actes. Les notaires réclament simplement une petite liberté de conscience dans l'exercice de leur profession.
M. Guillaume Drago . - Le Dr Galichon, qui devait faire partie de notre délégation, mais qui a été réquisitionné à cause des grèves, considère que la médecine est un art à visée thérapeutique et non une prestation de service pour répondre à des visées sociétales ou à des désirs personnels. La clause de conscience correspond à la demande d'exercer la médecine conformément à sa vocation professionnelle ou sociale.
M. Michel Amiel . - Je suis médecin. J'ai bien suivi votre raisonnement sur la liberté de pensée, la liberté de conscience et l'objection de conscience. Sans opposer les juristes aux médecins, je suis un peu surpris que vous mettiez sur le même plan l'objection de conscience pour les médecins et les officiers d'état civil. Je précise que je ne suis pas favorable à ce qu'on appelle couramment la PMA pour toutes, mais pour d'autres raisons que celles que vous invoquez. Je ne crois pas, non plus, qu'il faille interpréter la clause de conscience des médecins prévue dans le code de déontologie, comme vous le faites. Le code de déontologie dit simplement que l'on a le droit de ne pas effectuer un acte médical contre son gré, sans détailler. Je ne vois pas un médecin être assez fou pour effectuer un acte qu'il ne saurait pas réaliser !
J'en reviens à votre proposition : si je vous comprends bien, un officier d'état civil pourrait refuser l'inscription de l'enfant après la naissance ?
M. Michel Amiel . - Je veux aussi, comme Mme Jourda, vous demander en quoi cette loi est dérogatoire au droit commun de la filiation ?
Me Geoffroy de Vries . - Si je vous comprends bien, la liberté de conscience n'aurait donc pas la même importance selon qu'il s'agit d'un médecin, d'un officier d'état civil ou d'un maire ?
M. Michel Amiel . - Ce n'est pas une question d'importance. En tant que maire, je vois mal comment je pourrais refuser d'inscrire à l'état civil un enfant pour des raisons éthiques, même si je les partage, bien que pour d'autres motifs !
Me Geoffroy de Vries . - En tant que maire, vous serez donc conduit à reconnaître la filiation d'un enfant qui aurait deux mères et pas de père...
M. Michel Amiel . - Mais l'enfant existe !
Me Geoffroy de Vries. - Ce dont il est question, c'est de retranscrire les actes de filiation, et non de dire que l'enfant existe. Nous affirmons que cela peut heurter certains officiers d'état civil, qui pourraient revendiquer la possibilité de faire jouer une clause de conscience.
M. Michel Amiel . - Dès lors, que préconisez-vous ? Faudra-t-il envoyer les demandeurs chez le maire de la ville voisine ?
Me Geoffroy de Vries. - Comme un médecin, un notaire qui ferait jouer la clause de conscience serait remplacé par un collègue volontaire ou désigné par le président de la chambre professionnelle ; de même, le code général des collectivités territoriales prévoit que le préfet peut substituer un officier d'état civil à un autre, qui serait défaillant.
Nous considérons que la filiation issue d'une PMA organisée pour un couple de femmes est dérogatoire au droit commun, tout d'abord parce que le principe général est la filiation biologique : l'enfant est issu d'un homme et d'une femme ; dans son rapport, le Conseil d'État qualifie d'ailleurs la double filiation maternelle de « fiction juridique ». En outre, ce processus apparaît comme dérogatoire au principe même de la médecine, qui est, comme vous l'avez dit, monsieur le Président, de donner la vie. Nous estimons donc que cette loi est dérogatoire au droit commun de la filiation.
Me Jean Dupont-Cariot. - Je souhaite faire quelques rappels pour terminer. Tout d'abord, l'acte authentique est appelé, dans ce texte, à établir la filiation d'un enfant qui n'est pas encore conçu. Ensuite, j'insiste sur le fait que cet acte peut être dénoncé par une seule des deux personnes concernées. Enfin, je m'interroge sur la responsabilité vis-à-vis de l'enfant, qui, par cet acte authentique, se voit refuser par avance toute demande de recherche en paternité.
Ces trois éléments constituent pour moi autant de difficultés d'ordre juridique.
M. Michel Amiel . - Le troisième point pose en effet problème.
Ce point de l'ordre du jour a fait l'objet d'une captation vidéo qui est disponible en ligne sur le site du Sénat .
Mme Huguette Mauss,
présidente,
et de M. Jean-Pierre Bourély, secrétaire
général,
du Conseil national pour l'accès aux origines
personnelles (CNAOP)
Mme Catherine Deroche , présidente . - Je vous prie d'excuser le président Milon, qui nous rejoindra en cours de réunion.
Nous poursuivons nos travaux sur le projet de loi relatif à la bioéthique et nous entendons aujourd'hui Mme Huguette Mauss, présidente, et M. Jean-Pierre Bourély, secrétaire général du Conseil national pour l'accès aux origines personnelles (CNAOP).
Cette audition fait l'objet d'une captation vidéo retransmise en direct sur le site Internet du Sénat et consultable à la demande.
Le Conseil national pour l'accès aux origines personnelles a été créé par la loi du 22 janvier 2002. Il concerne les personnes pupilles de l'État ou adoptées qui ne connaissent pas l'identité de leurs parents de naissance, car ceux-ci ont demandé la préservation du secret de leur identité lors de l'accouchement, ou lorsqu'ils ont confié l'enfant à un service départemental de l'aide sociale à l'enfance (ASE) ou à un organisme autorisé pour l'adoption.
Certains de nos interlocuteurs se sont interrogés sur l'opportunité d'attribuer au CNAOP les missions confiées à la commission ad hoc créée par l'article 3 du projet de loi. Plus largement, vous pourrez nous faire part de l'expérience du CNAOP en matière d'accompagnement à l'accès aux origines personnelles.
Je vous laisse la parole pour un propos liminaire et, ensuite, vous pourrez échanger avec les quatre rapporteurs de cette commission, puis avec l'ensemble de nos collègues qui le souhaiteront.
Mme Huguette Mauss, présidente du Conseil national pour l'accès aux origines personnelles . - Le Conseil national pour l'accès aux origines personnelles existe depuis la loi de 2002. Son activité est importante et, au début, elle a même été très importante. La création du CNAOP a suscité un appel d'air. De nouveau, nous avons actuellement une augmentation du nombre des demandes de personnes qui souhaitent accéder à leurs origines personnelles. Nous dénombrions 500 demandes annuelles d'accès aux origines personnelles formulées par des personnes qui sont nés dans le secret, contre 700 aujourd'hui.
La loi de 2002 avait pour objet de faciliter l'accès à ses origines, à son histoire, avec le souci de préserver la santé de la mère et de l'enfant lors de la grossesse et de l'accouchement et d'éviter surtout des abandons sauvages. Elle a essayé d'instaurer un équilibre entre les intérêts de la mère et ceux de l'enfant par le biais de ce dispositif juridique propre à la France. Cette loi s'inscrit aussi dans la Convention internationale des droits de l'enfant. À plusieurs reprises, la jurisprudence a confirmé le bien-fondé de l'organisation et des prises de position relatives à l'accès aux origines pour les enfants nés dans le secret. À ce jour, aucune jurisprudence n'a remis en cause la loi précitée.
Pour accomplir ses missions, le CNAOP dispose à la fois d'une équipe au niveau national - un secrétariat général composé de sept personnes et dirigé par M. Bourély - et d'un réseau de correspondants départementaux. Dans chaque département, deux personnes au moins sont désignées par le président du conseil départemental pour être nos correspondants. Le CNAOP réalise donc un travail de maillage et d'écoute sur l'ensemble du territoire et à un double niveau. Tout d'abord, il oeuvre au moment de l'accouchement pour faire connaître aux femmes qui souhaitent accoucher dans le secret leurs droits, les aides auxquelles elles peuvent prétendre et la suite du processus. J'insiste sur cet aspect souvent méconnu dans l'accès aux origines. Les femmes qui accouchent et demandent le secret sont souvent dans une situation de détresse et ne maîtrisent pas du tout les dispositifs mis en place. Elles ignorent comment les choses s'organiseront lorsque l'enfant atteindra sa majorité, ou l'âge de discernement, et qu'il souhaitera accéder à ses origines.
Environ 100 000 à 150 000 personnes sont nées dans le secret depuis les années 1900 : l'expression alors consacrée « né sous X » est devenue « dans le secret de l'identité de la mère ». Seulement 5 à 10 % des personnes nées dans le secret ont demandé à accéder à leurs origines personnelles.
- Présidence de Mme Élisabeth Doineau, vice-présidente -
Mme Muriel Jourda , rapporteur . - Comment se passe l'accès aux origines ? Quelles recherches peuvent être faites ? Comment la personne a-t-elle connaissance de votre existence ? Comment la procédure se déroule-t-elle ensuite pour l'accès aux origines ?
Mme Huguette Mauss . - Le dispositif est très opérationnel. Les personnes qui souhaitent accéder à leurs origines nous contactent généralement par l'intermédiaire de notre site Internet. Là, elles peuvent télécharger des documents, y compris la demande.
D'autres personnes qui pensent être nées sous X, mais qui disposent de quelques informations sur l'identité de la mère, s'adressent souvent au service de l'aide sociale à l'enfance du département où ils sont nés. Il arrive en effet que certaines personnes disposent d'indications sur l'identité de la mère biologique.
Lorsqu'il y a saisine par un conseil départemental, nous sommes directement informés qu'une personne est à la recherche de ses origines. Un questionnaire plus complet lui est alors envoyé, puis les indications sont vérifiées. Nous commençons nos investigations auprès du conseil départemental, auprès de la maternité où est né l'enfant, mais aussi auprès des registres de l'état civil et d'un certain nombre de partenaires, notamment la justice. À ce stade-là, nous avons des échanges fournis avec les conseils départementaux, notamment nos correspondants dans les départements pour nourrir suffisamment le dossier.
Cette période est un temps important pour aider la personne à formaliser sa demande et l'accompagner, car il existe une appréhension à connaître ses origines, qui peut aussi prendre la forme de fantasmes à l'égard de la mère biologique ayant abandonné son enfant à la naissance.
Mme Muriel Jourda , rapporteur . - Concrètement, vos correspondants départementaux rencontrent-ils les personnes en attente de ces informations ? Arrive-t-il que certaines personnes renoncent en cours de route à connaître leurs origines ? Dans quelle proportion ?
Mme Huguette Mauss . - Nous sommes en mesure d'intervenir pour les personnes qui sont nées depuis 2002. Mais souvent, les personnes que nous rencontrons sont nées avant 2002 : des personnes âgées de 50 ans découvrent qu'elles ont été adoptées au moment, par exemple, du décès de leurs parents.
La recherche des origines se décompose en deux temps. La chargée de mission du CNAOP à Paris prend le temps d'entendre, d'expliquer aussi les procédures, les démarches qui seront réalisées avec les différents partenaires pour arriver à identifier et localiser la mère de naissance. Certaines demandes sont suspendues : des personnes éclairées par les correspondants du CNAOP ou les chargés de mission sur l'entourage, l'environnement, le contexte de la prise de relation avec la mère de naissance redoutent de se retrouver face à la mère inconnue. Il y a une attente et une crainte. Certaines personnes préfèrent avoir n'accès qu'à quelques renseignements, à des données non identifiantes telles que le contexte de la naissance ou les quelques caractéristiques de la mère biologique. Dans ce cas, nous suspendons la procédure, nous procédons à une clôture provisoire du dossier, et nous indiquons au demandeur que nous pourrons rouvrir le dossier, à sa demande, quand il le voudra.
Peu de dossiers sont suspendus en cours de route. Sur les 700 demandes que nous recevons chaque année, pour un tiers d'entre elles, nous n'arrivons pas à localiser et à identifier la mère de naissance. Parfois, nous la contactons la personne qui a accouché dans le secret, mais cette mère biologique refuse que le secret soit levé. Elle nous dit qu'un engagement avait été pris à son égard lors de l'accouchement et qu'elle ne se sent pas prête à assumer un contact avec l'enfant abandonné. Doit-on demander à la mère que nous avons identifiée si elle accepte ou non de lever le secret après son décès ? C'est une procédure à plusieurs détentes. Ce sont des mouvements difficiles à gérer, notamment avec l'enfant né dans le secret qui voit aussi les portes se fermer. Dans certains cas, nous suspendons donc le dossier, mais en accord avec l'enfant qui s'est adressé au CNAOP.
Mme Corinne Imbert , rapporteure . - Que pensez-vous du projet de loi tel qu'il est rédigé actuellement dans lequel il est fait mention de la création d'une commission pour la mise en oeuvre de l'accès aux origines des personnes nées d'un don de gamètes ? Avez-vous imaginé que la mission qui serait confiée à cette commission ad hoc puisse être remplie par le CNAOP ? Dans ce cas, avez-vous estimé les moyens supplémentaires à envisager pour mener à bien cette mission ? Pensez-vous que le traitement différencié des personnes issues d'un don par rapport aux personnes pupilles de l'État ou des personnes adoptées est justifié ? Pourriez-vous préciser ce qu'est la notion de données non identifiantes ?
Mme Huguette Mauss . - Sur la rédaction du projet de loi Bioéthique, le Conseil national pour l'accès aux origines personnelles n'est concerné que par l'article 9, qui porte sur l'information en cas de maladie génétique de la parentèle. Lorsqu'une maladie génétique est découverte dans la famille de la mère biologique, comment informer l'enfant né dans le secret qu'il est susceptible de développer la maladie ? Inversement, lorsqu'un enfant né dans le secret est porteur d'une maladie génétique, comment informer la mère biologique, notamment toute la parentèle, qu'elle est susceptible de développer cette maladie génétique ? Cette question a été soumise avant même la rédaction de la loi. Nous avons pris position et souhaitons que la transmission d'informations soit bien organisée entre l'enfant né dans le secret et la parentèle dans un souci de santé publique. L'objectif n'est pas contradictoire avec le maintien du secret. Nous estimons que cette connaissance réciproque doit se faire par le biais d'un généticien. À ce titre, le CNAOP peut jouer un rôle d'intermédiaire entre les généticiens sans pour autant lever le secret.
S'agissant de la deuxième question relative à la commission ad hoc pour les enfants nés de dons de gamètes, le vocable utilisé - accès aux origines personnelles - est très général. Or il cache des réalités très variées.
Pour les enfants nés de dons de gamètes, les conditions de la naissance sont très différentes de celles des enfants nés dans le secret, dans un contexte de naissances non voulues, de situations de détresse des femmes avec la volonté de tourner la page. Dans le cas de dons de gamètes, la démarche est beaucoup plus positive, voire altruiste de la part des donneurs, et il s'agit de la volonté d'un couple de former une famille en ayant recours à des dispositifs médicaux. Les problématiques sont différentes et l'accompagnement n'est pas non plus le même. Dans le cas des enfants nés par dons de gamètes, il y a moins de secrets. Les adoptions d'enfants nés dans le secret, même encore maintenant, sont dissimulées.
J'estime que le CNAOP ne peut répondre à la demande d'organiser l'accès aux origines pour les enfants nés par dons de gamètes. Les Centres d'étude et de conservation des oeufs et du sperme humains (CECOS) ont organisé ces collectes de gamètes. Il y a un acte médical, un accompagnement médical et une coordination de partenaires médicaux. Nous, nous sommes sur des considérations sociales. Je ne doute pas néanmoins que notre expérience et notre savoir-faire puissent aider à la construction des procédures sur le plan administratif. Nous accompagnons d'un point de vue psycho-administratif, pourrais-je dire, la personne qui souhaite accéder à ses origines, ainsi qu'une mère qui est toujours dans la défensive.
Actuellement, les réseaux sociaux et les tests ADN mettent sur la place publique des recherches et des possibilités de retrouver des origines par ce biais. C'est un risque parce des officines sont mercantiles. Nous sommes donc extrêmement attentifs quant aux démarches qui peuvent perturber les recherches des origines.
Les données non identifiantes sont toutes les données qui sont recueillies au moment de la naissance, mais qui ne touchent pas à l'identité, c'est-à-dire au nom, au prénom, à la date de naissance, à l'adresse de la personne. Cela concerne toutes les autres données : les conditions de l'accouchement, les informations données par la mère au moment de la naissance. Dans le cadre de la loi de 2002, un décret a permis en annexe d'établir un document dans lequel la correspondante du CNAOP au niveau du conseil départemental qui rencontre la femme au moment de l'accouchement consigne un certain nombre d'informations : la couleur des yeux, le type, tous les renseignements que la mère veut bien donner au moment de la naissance.
Il y a une démarche d'empathie de la part de la personne qui rencontre la femme qui accouche dans le secret pour obtenir le maximum d'informations dans l'intérêt de l'enfant, en lui précisant que le secret ne sera levé qu'avec son accord. Le principe de base du CNAOP, c'est la rencontre de deux volontés : celle de l'enfant, de retrouver sa mère biologique, et celle de la mère biologique de rencontrer son enfant. La convergence des volontés doit exister.
M. Bernard Jomier , rapporteur . - J'aurais souhaité que vous nous parliez des personnes ayant eu accès à leurs origines et des bénéfices qu'elles en retirent sur le plan psychologique, sur le plan de la santé ?
Vous nous avez fait part de votre volonté de concilier la volonté de l'adulte et celle de l'enfant. L'intérêt de l'enfant n'est-il pas une notion qui prime de plus en plus et devrait emporter la décision ?
Mme Huguette Mauss . - Une enquête de satisfaction a été réalisée en 2014 et 80 % des enfants sont satisfaits d'avoir eu accès à leurs origines. Néanmoins, il existe des nuances. Certains ont vécu des déceptions, ont vu leurs fantasmes et leurs espoirs brisés. Certains enfants adultes peuvent devenir agressifs avec les mères biologiques au motif qu'elles les ont abandonnés. Certaines mères biologiques sont harcelées par des enfants qui leur adressent beaucoup de reproches. Dans la grande majorité des cas, les personnes sont satisfaites de la rencontre. Cependant, il n'y a pas toujours de suite. Chaque histoire est particulière, mais nous essayons de mettre des chiffres sur ce que nous faisons et de regarder les grandes tendances. La moitié des mères que nous sollicitons refusent de lever le secret. Même vingt ou trente ans après, elles sont toujours dans ces mécanismes de défense. À long terme, les enfants estiment que la levée du secret les a aidés à se construire, à prendre de la distance et à ne plus vivre dans le fantasme.
L'intérêt supérieur de l'enfant est la question qui revient très régulièrement dans nos débats. Notre porte d'entrée, c'est l'enfant. Nous ne faisons jamais de démarche à la demande des mères biologiques. Nous faisons tout dans l'intérêt de l'enfant. C'est le droit de l'enfant à connaître ses origines. On ne peut cependant pas aller au-delà des volontés des personnes. Nous mettons à la disposition des enfants toutes les informations dont nous disposons et nous faisons en sorte que la mère accepte de lever son identité pour aller jusqu'au bout de la démarche. Pour nous aussi, il est difficile de ne pas aller jusqu'au bout de la démarche, mais on respecte les personnes, et nous essayons de ne jamais clôturer définitivement un dossier.
M. Olivier Henno , rapporteur . - Merci pour votre extrême humanité. Avant de poser ma question, permettez-moi de raconter l'expérience d'un proche. À l'âge de trente ans, après avoir été adopté, il a décidé de connaître ses origines et a interrogé le CNAOP. Sa mère qui avait souhaité un accouchement sous X a refusé de révéler son identité. L'enfant adulte a toutefois réussi à obtenir l'information de la part du responsable du département chargé de cette question. Il va mieux, mais a vécu une terrible dépression, car il n'a pu résoudre la question essentielle de savoir pourquoi il n'avait pas été désiré. La responsable du département a fini - c'est inimaginable ! - par révéler l'identité de la mère. Cet exemple permet de prendre conscience de l'idéalisation de la situation : la vie est parfois beaucoup plus complexe que ce que l'on imagine. Cette personne n'a pas réussi à répondre à la question essentielle : pourquoi n'a-t-elle pas été aimée ? C'est une question complètement différente de celle qui se pose dans le cadre d'un don.
Il me semble donc important que ce secret soit préservé, parfois même dans l'intérêt de l'enfant. On apprend que cela se passe bien dans 80 % des cas, mais ce chiffre est déclaratif : qui sait ce que cette information produit en réalité dans la psychologie personnelle des intéressés ?
Selon vous, la société ne pousse-t-elle pas les mères à révéler leur identité ? Pourra-t-elle tenir bon et défendre ce droit au secret qui me semble essentiel ?
Mme Huguette Mauss. - Nous sommes tous les jours confrontés au problème de la préservation du secret voulu par la mère : dans la moitié des dossiers, la mère refuse de lever le secret, et c'est toujours une interrogation pour nous. C'est pour cela que j'évoquais la procédure qui permet, tant que l'état de santé de la personne le permet, de prévoir une révélation de son identité après son décès. Or certaines personnes refusent de lever le secret, même après leur mort. C'est la situation la plus difficile pour nous, en particulier dans le cadre de maladies génétiques. Le contact physique est peut-être trop violent, pour l'un comme pour l'autre, mais, en cas de maladie génétique, c'est difficile à accepter, dès lors que l'on sait qu'il existe un risque pour la parentèle.
Parfois, en revanche, le secret préserve les intérêts de la mère biologique comme ceux de l'enfant. Nous adoptons une démarche d'accompagnement, et je suis admirative de l'équipe du CNAOP et de nos correspondants départementaux pour le temps et l'attention qu'ils consacrent à l'accompagnement des demandeurs dans leurs démarches afin de leur éviter de trop fantasmer sur l'autre partie et les conséquences qui en découleront.
M. Dominique de Legge . - S'agissant de l'accès aux origines, aujourd'hui, le droit, c'est de garder l'anonymat, mais le régime offre la possibilité, sous réserve du consentement de la mère, de le lever. La même question se pose en ce qui concerne la procréation médicalement assistée (PMA). Peut-on fonctionner avec deux régimes différents, l'un relevant des enfants nés sous X, dans lequel le droit au secret est défendu, et l'autre, qui crée un droit d'accès aux origines, concernant notamment les enfants nés par PMA ?
Mme Huguette Mauss. - Il n'y a pas que deux régimes : pensons, par exemple, aux enfants nés à l'étranger. L'accès aux origines est un droit inscrit dans les textes fondateurs du CNAOP et qui repose sur le respect des droits des personnes. Or, depuis 2002, la loi pose le principe selon lequel les femmes ont le droit d'accoucher dans le secret et de ne révéler leur identité qu'après consultation.
Dans le cadre de la loi Bioéthique, le dispositif pour identifier le don de gamètes est prévu d'emblée. Cela permettra de révéler les données identifiantes en plus des non identifiantes. Tant que la loi n'est pas votée, en effet, il n'existe pas d'obligation, pour le donneur de gamètes, d'accepter de donner son nom. Des informations non identifiantes seront révélées, mais pas les autres.
Je ne veux pas dresser de parallélisme entre les enfants nés dans le secret et ceux qui sont nés par don de gamètes. En effet, tout le dispositif est important, pas seulement le vocable et j'insiste, une fois encore, sur l'accompagnement psychologique des enfants nés dans le secret et de la mère. Le problème ne se pose pas dans les mêmes termes pour des donneurs de gamètes et les enfants nés par don.
C'est une position personnelle, mais les conditions de la naissance et de l'accès aux informations ne sont pas dans le même registre. Aujourd'hui, les réseaux sociaux et les tests ADN donnent des informations à tout-va, mais je mets en garde contre ces procédés qui peuvent être désastreux, aussi bien pour les enfants nés dans le secret que pour ceux nés par don de gamètes.
M. Yannick Vaugrenard . - Les enfants nés de dons de gamètes, comme les donneurs, peuvent également avoir besoin de soutien psychologique, mais les Cecos ne semblent pas armés pour cela. Selon vous, le CNAOP pourrait-il participer à cet accueil psychologique, à condition de disposer de moyens plus importants ?
En ce qui concerne les maladies génétiques, j'ai compris qu'il n'existait pas d'obligation d'information de la part de la femme qui a accouché sous X. Je me permets de dresser encore un parallèle avec le don de gamètes, mais il me semble que cela devrait au contraire être obligatoire, quel que soit le cas de figure. À défaut, nous sommes presque dans une situation de non-assistance à personne en danger. Ne pourrait-on pas obliger à partager cette information tout en conservant l'anonymat ?
Mme Huguette Mauss. - S'agissant de la capacité des Cecos à mener l'accompagnement psychologique, nous sommes, quant à nous, structurés autour des conseils départementaux, et j'ai déjà indiqué que le CNAOP était prêt à partager son expertise d'accompagnement dans le cadre de la commission ad hoc. Les collaborations peuvent être très importantes, mais, à mon sens, l'intensité de l'accompagnement psychologique sera moindre dans le cas des dons de gamètes que dans celui des enfants nés dans le secret. Nous sommes, en tout état de cause, prêts à partager notre expérience ; nous dispensons d'ailleurs chaque année des formations dans les départements pour animer notre réseau et partager nos pratiques.
Sur les maladies génétiques, j'entends votre question, mais l'article 9 a été rédigé de façon similaire à celui qui concerne le don de gamètes. Le législateur devait choisir entre « doit » et « peut », et il a choisi le second terme.
Lors de la naissance, nous insistons auprès de la mère pour que celle-ci laisse des informations dans le pli fermé sur son identité, dans un souci de santé publique.
Mme Élisabeth Doineau , présidente. - Merci madame la présidente, pour la clarté de vos propos ainsi que pour cette relation que vous avez su tisser avec les départements, notamment en matière de formation. Vous nous fournissez des éléments utiles, en particulier dans le cadre des conseils de famille.
Lors de nos autres auditions, nous avons beaucoup entendu que vous pourriez prendre la responsabilité de l'accompagnement, mais vos propos nous éclairent.
Ce point de l'ordre du jour a fait l'objet d'une captation vidéo qui est disponible en ligne sur le site du Sénat .
M. Jean-Marie Le Méné,
président de la
Fondation Jérôme Lejeune
M. Alain Milon , président . - Nous poursuivons nos travaux sur le projet de loi relatif à la bioéthique avec l'audition de M. Jean-Marie Le Méné, président de la Fondation Jérôme Lejeune. Avant de donner la parole à notre invité et alors que nos auditions touchent à leur fin, je rappelle que les auditions plénières ne sont pas nos seuls travaux et que les auditions des rapporteurs, qui sont nombreuses, sont ouvertes à l'ensemble des membres de la commission spéciale. Elles sont le lieu d'échanges aussi interactifs qu'approfondis, ce qui est moins le cas en formation plénière où le nombre des intervenants contraint parfois les débats. Les auditions des rapporteurs ne sont donc pas des sous-auditions.
Les demandes d'auditions sont très nombreuses. Je m'en réjouis, car elles témoignent de l'intérêt porté à nos travaux ; mais elles ne peuvent être toutes satisfaites. On peut le regretter, mais on peut noter également que les travaux du Sénat ne partent pas d'une table rase : ils bénéficient de l'ensemble des travaux antérieurs, notamment de ceux des États généraux de la bioéthique, qui ont permis une très large expression. Nos choix sont humains, ils sont donc imparfaits. En revanche, je récuse les procès d'intention sur les orientations supposées qui gouverneraient nos choix et dont j'observe qu'ils émanent de structures ayant des points de vue très différents. Le pluralisme, l'écoute et le respect sont la marque de nos travaux et j'entends, en ma qualité de président de cette commission spéciale, y veiller.
Ce n'est donc pas une brimade que de proposer une audition par un rapporteur. C'est néanmoins bien volontiers que nous accueillons, à sa demande, notre invité avec une captation vidéo.
M. Jean-Marie Le Méné, président de la Fondation Jérôme Lejeune . - Je vous suis très reconnaissant d'avoir accepté de m'auditionner. Je concentrerai mon propos sur deux sujets : le diagnostic préimplantatoire des aneuploïdies (DPI-A) et la recherche sur l'embryon.
J'ai quelques scrupules à commencer par un sujet qui n'est pas dans le texte transmis par l'Assemblée nationale, mais qui a été porté par un amendement centriste, rejeté par le Gouvernement et non adopté par l'Assemblée nationale, qui pourrait resurgir au Sénat. Cet amendement visait à étendre le diagnostic préimplantatoire (DPI) aux aneuploïdies et donc, notamment à la trisomie 21. Il s'agit, selon ses promoteurs, d'améliorer le taux d'implantation de l'embryon et de diminuer le nombre de fausses couches, dans le cadre de la procréation médicalement assistée (PMA).
Le DPI est légal pour les parents qui recourent à la PMA parce qu'ils sont porteurs d'une maladie génétique d'une particulière gravité. Mais la question d'étendre ce DPI à d'autres maladies d'origine génétique, mais non héréditaire - notamment en examinant les chromosomes de l'embryon -, fait débat depuis plusieurs années. Il s'agit du DPI-A, communément appelé « DPI de la trisomie 21 », mais qui recouvre aussi d'autres types de trisomie.
À l'occasion de cette troisième révision de la loi Bioéthique, la revendication d'étendre le DPI à la trisomie 21 a donc resurgi au motif d'améliorer les performances de la PMA.
Le premier argument utilisé par Mme Agnès Buzyn, ministre de la santé, pour rejeter l'amendement, c'est l'argument de l'eugénisme : elle a considéré que le DPI était eugéniste par nature. Je cite ses propos : « autoriser cette pratique conduirait manifestement à une dérive eugénique », « je sais depuis le début de l'élaboration de ce projet de loi que ce serait la question éthique la plus fondamentale et la plus complexe », « une telle décision est lourde à l'échelon collectif, je suis par conséquent très mal à l'aise. »
Son deuxième argument est scientifique, car l'extension du DPI repose sur des arguments scientifiques controversés. En effet, la diminution du nombre de fausses couches après un DPI-A n'est pas établie et la littérature scientifique en ce domaine est divisée ; il n'y a aucun consensus scientifique dans ce domaine, y compris dans les articles récents. Le Pr Bonnefont l'a confirmé lors de son audition par la mission d'information de l'Assemblée nationale sur la révision de la loi relative à la bioéthique: « l'augmentation des chances de grossesse après un test d'aneuploïdie n'a jamais été formellement démontrée. » Bien au contraire, il est courant et connu que certaines de ces anomalies se régularisent d'elles-mêmes dans les premiers jours du développement de l'embryon, avant l'implantation. Plusieurs travaux, y compris récents, sont parus sur cette autocorrection de l'embryon. Le Pr Bonnefont a aussi montré que ces techniques de DPI étaient délicates et qu'elles exposaient à des risques de faux positifs ou de faux négatifs : des aneuploïdies peuvent être limitées au placenta et laisser l'embryon sain. Enfin, le DPI-A peut abîmer, voire détruire, l'embryon. Il y a donc des incertitudes suffisantes pour ne pas élargir cette technique à d'autres pathologies, ainsi que l'a confirmé Mme Buzyn.
Le troisième argument de la ministre, auquel je suis assez sensible, est la question du financement et du coût de cette extension, car une généralisation serait inévitable. L'amendement ne proposait l'extension du DPI qu'aux couples qui ont recours à une PMA, mais qui étaient éligibles au DPI pour une maladie héréditaire, mais pas pour tous les couples ayant recours à la PMA. On va donc avoir deux types de couples qui ont recours à la PMA : ceux qui ont droit au DPI parce qu'ils ont une maladie héréditaire familiale - la myopathie par exemple - et les autres couples, qui sont beaucoup plus nombreux, et qui ont recours à la PMA pour des questions d'infertilité, mais qui ne sont pas éligibles au DPI. Je ne vois pas très bien quel argument on va pouvoir invoquer pour dire que l'extension du DPI ne sera ouverte qu'aux uns et pas aux autres ! Or ces deux groupes sont dans un statut d'égalité totale au regard de la trisomie 21, qui est certes une maladie génétique, mais pas une maladie héréditaire. Mme Buzyn s'est donc interrogée sur ce point : quelle garantie avons-nous, si nous passons ce cap, de ne pas aller au-delà ? Car les chiffres donnent le vertige : on passerait d'une moyenne annuelle de 250 couples concernés par un DPI à 150 000 ! Une PMA avec DPI coûte près de 20 000 euros, cela ferait donc une dépense de l'ordre de 3 milliards d'euros.
Cet amendement ouvrirait la boîte de Pandore : c'est toujours par la trisomie 21 que l'on commence, mais ensuite, dans la mesure où nous avons accès à l'ensemble du génome, il n'y a aucune raison de ne pas en profiter pour vendre d'autres diagnostics. Les laboratoires y trouveront leur compte ! Le Pr Bonnefont a également souligné « l'enjeu financier tout à fait intéressant pour les laboratoires, en particulier les établissements privés, qui vont développer ce type de tests ». « Faisons attention à ne pas nous laisser intoxiquer par des professionnels qui auraient des arrière-pensées plus financières que médicales », nous dit le Pr Bonnefont.
Permettez-moi, en tant que président de la Fondation Jérôme Lejeune, qui a créé la plus grande consultation médicale d'Europe spécialisée sur la trisomie 21 et d'autres pathologies avec retard mental et qui a relancé une recherche à visée thérapeutique, d'ajouter un élément important, qui est le fruit de notre expérience : on ne peut pas soutenir le projet d'une société inclusive et dégrader en même temps l'image de la trisomie 21 en la rendant responsable des déboires de la PMA. Car il s'agit bien de cela : éliminer des embryons qui sont susceptibles de dégrader les performances statistiques de la PMA. Or les dépistages conduisent d'ores et déjà, dans 96 % des cas, à éliminer cette population trisomique : en pratique, il ne naît plus d'enfants trisomiques 21, hormis bien sûr le souhait des parents. Nous sommes donc déjà dans une société qui élimine certains types d'anomalies génétiques ou certains types de disgrâces physiques détectées par des machines et des algorithmes : disons les choses comme elles sont ! Je le dis souvent : c'est la première fois dans l'histoire de la médecine que la médecine a rendu mortelle une pathologie qui ne l'est pas ; la trisomie 21 est une affection qui est certes grave, mais pas mortelle - nos patients les plus âgés ont entre 70 et 75 ans et leur espérance de vie continue de progresser. Comment peut-on espérer changer notre regard sur les personnes handicapées si l'eugénisme se renforce à chaque évolution technique, qui n'est pas forcément un progrès ?
Il existe un risque que cet amendement soit redéposé au Sénat parce que les principales institutions interrogées ou qui se sont prononcées lors du débat à l'Assemblée nationale sont favorables à cette extension du DPI : le Conseil d'État, le Comité consultatif national d'éthique (CCNE), l'Office parlementaire d'évaluation des choix scientifiques et technologiques (OPESCT), la mission d'information parlementaire, l'Agence de la biomédecine (ABM). Mme Buzyn a tenu ferme sur ce point : c'est une sage décision.
Nous recevons 10 000 patients chaque année, de la pédiatrie à la gériatrie. Le plus difficile n'est pas d'accueillir les patients et leurs familles. Comment progresser sur la thérapeutique quand on entend des propos tels que ceux qui ont été tenus à l'Assemblée nationale en séance publique ? Le député Philippe Berta a osé parler des enfants trisomiques en les comparant à des « légumes » ! Le député Philippe Vigier a eu ces mots terribles : « il faut traquer, oui je dis : traquer, les embryons porteurs d'anomalies chromosomiques. » Dans quel pays, à quelle époque vivons-nous ? Quel message les pouvoirs publics envoient-ils aux personnes que nous accueillons dans nos consultations ? Ces propos relèvent d'un racisme chromosomique. Ils sont méprisants, donc méprisables.
J'ai eu la chance, ou l'honneur, de suivre l'élaboration des quatre lois de bioéthique, mais de cinq révisions du cadre de la recherche sur l'embryon, car, étonnamment, il y a plus de révisions du cadre de la recherche sur l'embryon que de lois de bioéthique : en 1994, la loi interdisait la recherche sur l'embryon ; une dérogation temporaire a été ouverte en 2004 ; cette dérogation a été pérennisée en 2011 ; la recherche a été autorisée sous conditions en 2013 ; une dérogation à la dérogation, pour faciliter la recherche qui améliore la PMA, a été prévue en 2016 ; et finalement, toutes les conditions vont être supprimées en 2020. Deux de ces modifications du cadre juridique de la recherche sur l'embryon n'ont pas fait l'objet de lois de bioéthique, alors qu'elles auraient dû le faire.
Le sens de cette évolution témoigne d'un malentendu. Les parlementaires, les médias, les experts se félicitent régulièrement des garde-fous posés par les lois de bioéthique ; mais cette saga législative française d'un quart de siècle démontre exactement le contraire : on est passé de l'interdiction de la recherche sur l'embryon à l'interdiction de s'y opposer, du respect de l'embryon, qui était premier il y a 25 ans, au principe de son non-respect aujourd'hui. Les lois de bioéthique n'ont eu de cesse de déréguler la recherche sur l'embryon ; elles n'ont pas protégé la dignité de l'embryon, elles ont protégé l'intérêt des chercheurs.
La technoscience a fait croire à la médecine, qui a fait semblant de le croire, que si les embryons surnuméraires n'étaient pas utilisés, ils ne devaient pas être inutilisables, leurs cellules souches étant promises à un grand avenir thérapeutique. L'ABM, créée en 2004 à cet effet, a alors autorisé des recherches sur l'embryon. Mais rien ne s'est passé comme prévu : contrairement aux attentes triomphalistes, les espoirs placés dans les vertus curatives de l'embryon ont été déçus. C'est pourquoi, faute d'apporter la gloire, on demande à l'embryon humain de rapporter de l'argent. Objet de toutes les promesses, l'embryon est devenu l'objet de toutes les richesses. Hier, l'embryon devait nous guérir de tout et, aujourd'hui, il doit nous servir à tout.
C'est pourquoi il est aujourd'hui demandé au législateur de faire disparaître les dernières protections formelles de l'embryon qui sont présentées comme la cause des échecs de la recherche ; la recherche serait tellement brimée que les chercheurs n'arriveraient pas à trouver ; certains chercheurs expliquent d'ailleurs leurs insuccès par l'insécurité juridique due à la Fondation Jérôme Lejeune quand celle-ci recourt au juge pour faire respecter les lois. Moins la recherche sur l'embryon apporte la preuve de sa pertinence, et plus il faudrait l'affranchir de toute contrainte pour l'inscrire dans une finalité industrielle et commerciale. Certains scientifiques n'hésitent pas à dire qu'il ne faut pas « rater le virage industriel » !
Or, en quoi le fait de remplacer une autorisation de recherche par une simple déclaration du chercheur, de ne plus poursuivre de finalité médicale, de ne pas privilégier les cellules souches pluripotentes induites (induced pluripotent stem cells - iPS) comme des alternatives aux cellules embryonnaires et l'embryon animal comme alternative à l'embryon humain, de ne plus produire le consentement des parents, de ne plus rendre compte de la traçabilité des embryons, de repousser la limite de la conservation de l'embryon de sept à quatorze jours, serait-il favorable aux découvertes scientifiques ? Faudrait-il vraiment alléger toutes ces formalités absurdes établies par des législateurs absurdes ? En revanche, on voit bien que l'allégement législatif facilitera la production massive de cellules souches embryonnaires par des start-up ou des sociétés à but lucratif, dans une perspective utilitariste, dont d'ailleurs on ne comprend pas comment elle s'accommode du principe de non-patrimonialité du corps humain.
La recherche sur l'embryon n'est pas une question de techniques ou de procédures, c'est une question de principe. On attend du législateur, et certainement encore plus du Sénat, une réponse de sagesse. Le législateur va-t-il suivre la jurisprudence administrative qui a tendance à ne pas annuler des autorisations pourtant illégales délivrées par l'ABM à des scientifiques qui ne se cachent pas d'anticiper des transgressions nouvelles de la loi ?
- Présidence de Mme Catherine Deroche, vice-présidente -
M. Jean-Marie Le Méné . - L'article 17 du projet de loi supprime l'interdiction de créer des embryons transgéniques, ce qui permettra la création en laboratoire d'embryons génétiquement modifiés. Le ciseau génétique CRISPR-Cas9 - clustered regularly interspaced short palindromic repeats - associated protein 9 - courtes répétitions palindromiques groupées et régulièrement espacées - ainsi que la technique de transfert nucléaire dite de la « fécondation in vitro (FIV) à trois parents » pourront être expérimentés sur des embryons humains, par transgenèse ; cela se réalise d'ailleurs déjà sans attendre la modification de la loi. Le législateur va-t-il réguler ou simplement régulariser cette transgression déjà anticipée par les chercheurs ? Je me suis entretenu avec certains chercheurs concernés qui ne m'ont pas caché qu'ils étaient effectivement dans une transgression de la loi en vigueur. Ces techniques de transgenèse, et en particulier celle du CRISPR-Cas9, sont au coeur du débat sur l'embryon.
Bien entendu, aujourd'hui, il n'est pas envisagé de réimplanter l'embryon ainsi modifié parce qu'il existe encore un garde-fou ; mais il va sauter bien évidemment ! La vocation du non-transfert à des fins de gestation est un prétexte rassurant pour obtenir une transgression nouvelle. Il est bien connu que, pour obtenir une transgression nouvelle, il faut mettre un barrage à la transgression immédiatement suivante, comme cela a été le cas pour la PMA. La question se pose puisqu'il est question de demander une réinterprétation de l'article 13 de la Convention d'Oviedo, seul dispositif international qui protège l'embryon humain, afin d'autoriser l'embryon génétiquement modifié. Dans ces conditions, on a beau jeu de montrer la Chine du doigt alors que l'on prend la même voie ! Jamais la recherche sur l'embryon humain n'a été autant dérégulée.
On s'est ému à juste titre du forçage des gènes du moustique pour faire des moustiques qui ne se reproduisent pas. Mais ces gènes qui ne devaient pas se transmettre se sont transmis, et on a maintenant une population de moustiques qui présente trois types de génomes modifiés différents, apparus sans que l'on comprenne pourquoi et dont l'impact sur la santé publique est absolument inconnu. Qu'en sera-t-il si l'on accepte de modifier génétiquement l'embryon humain ? Il n'est pas envisagé de réimplanter l'embryon ainsi modifié, mais il existe néanmoins un régime de recherche, voté en 2016 à la sauvette, qui le prévoit : c'est le régime de recherche biomédicale sur l'embryon pour améliorer les techniques d'assistance médicale à la procréation (AMP).
Il n'est pas nécessaire d'être scientifique pour soutenir que l'embryon humain n'est pas un médicament, ni un réactif de paillasse, ni un substitut aux animaux de laboratoire, ni un être de non-droit voué à des oeuvres clandestines. S'il y a un domaine où l'activité humaine doit être responsable et solidaire, c'est bien à l'égard de la recherche, qui porte sur l'humain. « Traquer » tous les embryons humains trisomiques, comme l'a malheureusement dit un parlementaire, ou détruire des centaines d'embryons humains dans des projets de recherche ne paraît ni responsable ni solidaire.
Dans des autorisations de recherches récemment délivrées par l'ABM et publiées au Journal officiel, j'ai lu qu'un programme de recherche avait l'autorisation de détruire 70 embryons par an sur cinq ans, soit 350 embryons humains détruits pour une recherche et qu'un autre avait l'autorisation de détruire 150 embryons humains par an sur cinq ans, soit 750 embryons : c'est totalement déraisonnable !
« L'éthique va de pair avec la qualité scientifique. » Ce n'est pas de moi, c'est l'Institut national de la santé et de la recherche médicale (Inserm) qui l'écrit au sujet de l'expérimentation animale. Toute une littérature a été développée autour du respect des animaux de laboratoire qui impose de réduire le nombre des animaux, de les remplacer partout où c'est possible, d'obtenir une autorisation du ministère de la recherche pour tout projet de recherche sur l'animal ainsi qu'évaluation favorable du comité d'éthique - il y a 526 comités d'éthique sur l'expérimentation animale en France. Tout cela est extrêmement encadré. Mais s'agissant de l'embryon humain : on ne réduit pas le nombre des embryons utilisés - on les augmente dans des proportions délirantes -, on ne remplace pas le modèle de l'embryon humain par un modèle animal - on considère au contraire que la recherche sur l'embryon humain est une alternative à la recherche sur l'animal : les choses sont inversées ! -, on ne demandera plus d'autorisation - une simple déclaration suffira -, etc. Toute activité humaine doit être responsable et solidaire, c'est aujourd'hui le maître mot dans tous les domaines de l'activité humaine, il n'y a pas de raison que dans le domaine de la recherche sur l'embryon, ces qualificatifs soient oubliés.
Mme Corinne Imbert , rapporteure . - S'agissant du DPI-A, vos propos tournent essentiellement autour de la trisomie 21 : je trouve cela un peu réducteur, car il n'y a pas que la trisomie 21 et même, ce n'est souvent pas la trisomie 21 qui est le problème. Vous acceptez que l'on puisse faire un DPI pour rechercher une anomalie génétique, mais pourquoi ne pas rechercher une anomalie chromosomique, au-delà de la seule trisomie 21 ?
Sachez que nous sommes un certain nombre de parlementaires à prôner l'inclusion des personnes handicapées et ce serait faire un mauvais procès d'intention à cette commission spéciale, et au Sénat tout entier, que de prétendre le contraire.
M. Jean-Marie Le Méné . - Un sous-amendement du groupe centriste, présenté lors de l'examen à l'Assemblée nationale, permettait de rechercher toutes les anomalies chromosomiques à l'exclusion de la trisomie 21. Mme Buzyn a fait une réponse de médecin : on ne peut pas demander à un médecin de ne pas voir ce qu'il voit et l'obliger à mentir à son patient. Le sous-amendement n'a pas été adopté.
La trisomie 21 est une maladie génétique, chromosomique, mais pas héréditaire. Je ne suis pas un ardent défenseur du DPI, mais il se conçoit pour les maladies héréditaires, par exemple pour les familles qui ont eu plusieurs enfants atteints d'une myopathie. Mais ces personnes n'ont pas de risque particulier d'avoir une trisomie 21 : il ne serait pas juste de leur permettre de diagnostiquer une trisomie 21 et de l'interdire aux couples qui ont recours à la PMA sans DPI - ils sont 150 000 par an. Et si l'on recherche les accidents de la nature qui ne sont pas prévisibles héréditairement, on va tomber aussi sur le sexe. C'était l'argument principal que j'ai développé dans le livre que j'ai écrit sur le transhumanisme, en parlant des premières victimes du transhumanisme.
Il existe un marché mondial de la trisomie 21, de l'ordre de dix milliards de dollars et qui cible une clientèle féminine captive, à qui l'on fait peur d'avoir un enfant atteint de trisomie 21, une maladie qu'on ne devrait plus avoir. Il existe un ordre médical et scientifique établi pour lequel il n'y a plus de trisomie 21. Soyons clairs : il ne s'agit pas de philanthropie...
M. Guillaume Chevrollier . - Vous avez évoqué des intérêts particuliers. Pensez-vous que des intérêts financiers sont à l'oeuvre derrière ce projet de loi ? Si oui, pouvez-vous nous en dire plus ?
M. Jean-Marie Le Méné . - On est en train de passer à l'industrialisation et au recours au monde du privé. Des autorisations ont été délivrées par l'ABM pour fabriquer des cellules souches embryonnaires, mais pour le moment ces cellules souches embryonnaires ne sont absolument pas utilisées dans une perspective thérapeutique. D'abord, parce qu'il est extrêmement difficile de maîtriser leur développement. Ensuite, parce que ces cellules souches embryonnaires, greffées sur un organisme adulte à réparer ou à soigner, peuvent faire l'objet d'un rejet immunitaire. C'est pourquoi, depuis vingt ans, pratiquement rien ne se fait sur le plan thérapeutique. En revanche, ces cellules souches embryonnaires sont utilisées pour faire de la modélisation de pathologies et du screening moléculaire. Les laboratoires utilisent ce type de support alors qu'ils pourraient parfaitement utiliser d'autres types de cellules souches - des cellules souches animales ou des iPS - qui ne posent aucun problème éthique et qui présentent toutes les particularités requises pour se prêter à du ciblage moléculaire.
Depuis vingt ans, on a capitalisé sur l'embryon en se disant qu'après la thérapie génique, on allait basculer sur la thérapie cellulaire. Mais malgré tout l'argent, les subventions, les laboratoires, les matériels, les investissements, etc., la thérapie cellulaire ne porte pas ses fruits. Mais, comme il faut rentabiliser tout cela, on passe à l'ère industrielle : ces sociétés sont des sociétés lucratives, financées par le Téléthon. Je ne sais pas s'il y a des conflits d'intérêts : je n'ai pas investigué et je n'ai aucune raison de porter des soupçons sur qui que ce soit. Mais on a changé de logique : lorsque le Sénat et l'Assemblée nationale ont autorisé pour la première fois la recherche sur l'embryon, c'était dans le cadre d'un intérêt thérapeutique majeur. Mais tout s'est écroulé : l'intérêt thérapeutique majeur n'existe plus et même l'intérêt médical va être balayé. Seule reste la production de cellules embryonnaires que l'on va vendre. Or il s'agit d'éléments du corps humain auxquels s'applique le principe de non-patrimonialité inscrit au code civil et au code de la santé publique.
Mme Catherine Deroche , présidente . - Je vous remercie.
Ce point de l'ordre du jour a fait l'objet d'une captation vidéo qui est disponible en ligne sur le site du Sénat .
Mme Emmanuelle
Cortot-Boucher,
directrice générale de l'Agence de la
biomédecine
M. Alain Milon , président . - Nous poursuivons nos travaux sur le projet de loi relatif à la bioéthique avec l'audition de Mme Emmanuelle Cortot-Boucher, directrice générale de l'Agence de la biomédecine.
Cette audition fait l'objet d'une captation vidéo en vue de sa retransmission sur le site du Sénat.
Pour éclairer le débat sur la révision de la loi de bioéthique, plusieurs documents de référence sont disponibles : le rapport issu des États généraux de la bioéthique conduits sous l'égide du Comité consultatif national d'éthique (CCNE), le bilan de l'application de la loi de bioéthique par l'Office parlementaire des choix scientifiques et technologiques (Opecst) ou encore l'étude rédigée par le Conseil d'État. Le rapport sur l'application de la loi de bioéthique, publié en janvier 2018, a constitué le premier de ces documents publiés sur la réforme en cours. Il est très clair et très pédagogique sur les sujets parfois techniques dont l'Agence a la responsabilité. C'est pourquoi j'ai souhaité que notre commission vous entende aussi rapidement que votre nomination, effective depuis le 30 octobre dernier, le permettait.
Mme Emmanuelle Cortot-Boucher, directrice générale de l'Agence de la biomédecine . - Je vous remercie de m'avoir conviée pour évoquer le projet de loi relatif à la bioéthique. J'exerce mes fonctions depuis un mois et demi.
L'Agence de la biomédecine a été créée par la loi du 6 août 2004, dans la continuité de l'Établissement français des greffes. Ses activités se déploient dans quatre grands domaines qui ont en commun de requérir l'utilisation à des fins médicales ou scientifiques d'éléments ou de produits issus du corps humain : l'Agence de la biomédecine est ainsi compétente en matière de prélèvement et de greffe d'organes et de tissus, de prélèvement et de greffe de cellules souches hématopoïétiques - c'est-à-dire de moelle osseuse -, de procréation, d'embryologie et de génétique humaine. Ces domaines sont tous très largement régis par des dispositions issues des lois de bioéthique, et il est donc tout à fait naturel que l'Agence soit amenée à contribuer au débat en partageant l'expertise médicale, scientifique, juridique et éthique dont elle peut se prévaloir. Elle y contribue en restant fidèle au positionnement institutionnel qui est le sien : l'Agence est un établissement public administratif placé sous la tutelle du ministère de la santé ; ce statut exclut qu'elle puisse prendre position dans les débats de société, qui relèvent exclusivement de la représentation nationale ; en tant qu'organe administratif chargé d'une fonction d'expertise, l'Agence n'a pas vocation à y participer, et il est normal que, sur un certain nombre de sujets, elle choisisse délibérément de conserver une certaine neutralité.
L'Agence de la biomédecine a pris une part active au travail de préparation du projet de loi qui vous est soumis. Elle a répondu aux sollicitations des parlementaires.
Elle a produit trois documents, qui ont alimenté les travaux de réflexion : un état des lieux de l'encadrement juridique international, un rapport d'information au Parlement et au Gouvernement sur l'état des connaissances et des sciences - actualisé en décembre 2017 - et un bilan de l'application des précédentes lois de bioéthique. L'Agence de la biomédecine a par ailleurs suivi de très près les débats à l'Assemblée nationale en première lecture.
Le projet de loi prévoit l'ouverture de l'assistance médicale à la procréation (AMP) aux couples de femmes et aux femmes non mariées. Ce choix relève typiquement d'un débat de société dans lequel l'Agence de la biomédecine n'a pas d'observation particulière à faire valoir. Si l'évolution envisagée a lieu, l'Agence sera en mesure de soutenir sa mise en place dans les centres d'AMP et les centres de conservation des oeufs et du sperme en adaptant ses règles de bonnes pratiques. C'est le sens de l'ajout auquel les députés ont procédé au 4°bis de l'article L. 2141-1 du code de la santé publique.
Le projet de loi envisage d'instituer un droit d'accès à l'origine pour l'ensemble des enfants issus d'une AMP avec recours à un tiers donneur. Le système proposé prévoit, d'une part, la création d'une commission ad hoc placée auprès du ministre chargé de la santé qui aura pour mission de recevoir les demandes des enfants issus du don, ainsi que le cas échéant, celles des donneurs qui souhaiteraient savoir combien d'enfants ont été conçus à partir de leurs gamètes ; d'autre part, la mise en place d'un registre tenu par l'Agence de la biomédecine qui permettra, sur saisine de la commission ad hoc, d'avoir accès aux données nécessaires pour répondre à ces deux types de demandes. Pour rendre possible l'exercice de ce droit d'accès aux origines pour tous les enfants conçus avec recours à un tiers donneur, le projet de loi modifie le régime juridique du don de gamètes en prévoyant qu'il ne pourra plus être effectué que si le donneur accepte que son identité soit révélée aux enfants issus du don, s'ils en font la demande à leur majorité.
Le projet de loi prévoit donc un phasage en trois temps pour l'entrée en vigueur de ce nouveau régime : pendant une première période d'un an à compter de la promulgation de la loi, l'AMP sera élargie aux couples de femmes et aux femmes non mariées, mais continuera à se faire à partir de gamètes qui auront été recueillis sans que le donneur ait accepté le principe d'un droit d'accès à l'origine pour les enfants issus du don ; au cours d'une deuxième période, les dons ne pourront plus être effectués que si le donneur consent à ce que les enfants issus de ce don puissent, sur leur demande, avoir accès à son identité ; enfin, au cours de la troisième période, l'AMP ne pourra plus être réalisée qu'à partir de gamètes recueillis auprès de donneurs ayant consenti à la levée de l'anonymat avec la tenue d'un registre relatif aux donneurs de gamètes. C'est une nouvelle mission que le projet de loi envisage de confier à l'Agence de la biomédecine : l'outil élaboré devra être opérationnel à l'expiration d'un délai d'un an à compter de la promulgation de la loi et devra répondre à des exigences très fortes en termes de fiabilité, de résistance à l'obsolescence et de sécurité. Nos équipes ont d'ores et déjà commencé à réfléchir aux modalités pratiques de fonctionnement de ce registre en capitalisant notamment sur l'expérience acquise avec l'utilisation des autres traitements de données tenus par l'Agence de la biomédecine notamment dans le domaine de la greffe d'organes et de cellules.
L'Agence de la biomédecine se prépare aussi, dès à présent, à assurer la promotion du don de gamètes dans le contexte où il y aurait lieu de reconstituer un stock de gamètes recueillis sous l'empire du nouveau régime juridique. Si les dispositions envisagées sont adoptées, il s'agira sans aucun doute d'un changement de paradigme pour l'Agence, justifiant de repenser les méthodes de communication et les publics visés.
Dans le domaine du prélèvement et de la greffe d'organes, le projet de loi apporte des modifications importantes pour développer le don croisé, qui reste de faible importance en France. Il prévoit de faire disparaître la règle actuelle, qui limite à deux le nombre de paires donneur-receveur susceptibles d'être mises en relation, et de renvoyer au décret le soin de fixer un nombre plus élevé. Les chaînes ainsi constituées pourront en outre faire intervenir des donneurs décédés et les équipes de prélèvement et de greffe ne seront plus soumises à l'obligation de réaliser l'ensemble des opérations de manière simultanée, mais dans un délai de 24 heures. Par ailleurs, le projet de loi introduit des dispositions visant à faciliter le fonctionnement des comités d'experts chargés d'autoriser le prélèvement. Ces mesures sont cohérentes avec l'objectif du troisième plan Greffe adopté en 2017.
Dans le domaine du prélèvement et de la greffe de cellules souches hématopoïétiques, le projet de loi vise à tenir compte du développement de la greffe semi-compatible qui intervient dans un contexte familial et donne de bons résultats. L'Agence de la biomédecine doit suivre les donneurs de cellules, y compris lorsqu'ils sont apparentés. Tel est l'objet des dispositions introduites par le projet de loi à l'article L. 1418-1 du code de la santé publique. Le projet de loi ouvre aussi la possibilité pour un mineur de procéder à un don de moelle osseuse au profit de son père ou de sa mère. Compte tenu des risques inhérents à un tel don, un encadrement fort est prévu avec l'intervention d'un administrateur ad hoc qui représentera les intérêts propres du mineur, la saisine du président du tribunal de grande instance aux fins de recueillir le consentement de l'intéressé et l'obligation de vérifier avant de recourir à cette faculté, qu'elle est la seule option thérapeutique envisageable pour le père ou la mère de l'enfant.
Pour la recherche sur les cellules souches embryonnaires humaines (CSEh), le projet de loi prévoit de passer d'un régime d'autorisation à un régime de déclaration. Cette évolution, qui avait été suggérée par l'Agence de la biomédecine dans le rapport relatif à l'application de la précédente loi de bioéthique, tire les conséquences du fait que le travail sur ces cellules n'implique pas nécessairement la destruction d'un embryon et ne présente donc pas les mêmes risques sur le plan éthique que la recherche sur l'embryon lui-même. Le projet de loi propose toutefois une garantie : l'Agence de la biomédecine dispose d'un pouvoir d'opposition dont elle ne peut faire usage que sur le fondement d'un avis, rendu public, de son conseil d'orientation. Ces mesures visent à soutenir la recherche française sur les CSEh dont les très bons résultats doivent être consolidés : les équipes installées en France ont démontré leur capacité à mener des travaux de haut niveau et à publier dans des revues scientifiques à fort impact ; elles ont aussi fait la preuve de leur capacité à conduire des essais cliniques à partir de ces travaux. Les bénéfices concrets, pour les patients, des recherches qui sont conduites sur les CSEh paraissent désormais à portée de main.
En ce qui concerne la recherche sur l'embryon lui-même, le projet de loi maintient le principe d'une autorisation soumise à des conditions précises, notamment au respect de plusieurs principes éthiques. Il apporte en outre une précision nouvelle portant sur la durée de développement de l'embryon, fixée à quatorze jours, conformément au consensus international en la matière. Enfin, il clarifie la portée de l'interdiction, déjà présente dans la loi, de constituer un embryon chimérique : conformément à l'interprétation donnée par le Conseil d'État, le projet de loi précise que cette interdiction ne fait pas obstacle à l'introduction de CSEh dans un embryon animal, même si, d'un strict point de vue scientifique, une telle opération aboutit bien à la création d'une chimère.
Dans le domaine de la génétique, le projet de loi s'efforce de définir un cadre juridique conciliant deux exigences : tenir compte de l'évolution des techniques et protéger les personnes contre les risques que les tests génétiques peuvent leur faire courir si leurs résultats sont mal interprétés. En effet, les résultats des tests génétiques ont un caractère éminemment personnel, mais aussi une forte valeur prédictive - parfois à tort - et ils peuvent orienter les comportements individuels. Le projet de loi choisit de maintenir le principe de leur interdiction sauf à des fins scientifiques, médicales ou judiciaires et d'imposer le recours à un professionnel pour en interpréter les résultats. Pour rendre néanmoins tangibles les bénéfices issus des progrès de la génétique, le projet de loi précise les modalités d'information du patient et de sa parentèle en cas de découverte incidente faite à l'occasion d'un examen de génétique somatique. Il autorise aussi la réalisation d'examens génétiques sur une personne décédée ou une personne qui n'est pas en capacité de donner son consentement lorsqu'elle ne s'y est pas formellement opposée auparavant et organise les modalités d'information de la parentèle.
Dans ce projet de loi, l'Agence retrouve de nombreuses préoccupations exprimées par les professionnels avec lesquels elle est en contact régulier. L'Agence sera prête à mettre son expertise pour la mise en oeuvre des dispositions qui seront votées dans le cadre de la future loi de bioéthique.
M. Olivier Henno , rapporteur . - Hier, nous avons entendu l'Agence nationale de sécurité du médicament et des produits de santé (ANSM) sur la question du microbiote fécal et des innovations thérapeutiques qui lui sont liées. Notre éthique à la française est fondée sur le don anonyme et gratuit de sang, mais que penseriez-vous d'une éventuelle compensation, voire rémunération, du don ?
Que pensez-vous de l'idée d'étendre les compétences de l'Agence au champ de l'intelligence artificielle (IA) appliquée à la médecine ?
Mme Emmanuelle Cortot-Boucher . -La question de la rémunération du don peut se poser pour les organes, les cellules et les gamètes. Le principe de la gratuité du don est posé dans le code civil, et c'est sur ce principe qu'est bâti tout notre système de don, mais aussi la confiance du public dans ce système. Ce principe garantit que le donneur est libre et volontaire. Nous sommes tous très attachés à ce principe. Il est aussi garant de l'efficacité de nos collectes, car, dans les pays où le don est rémunéré, on a pu observer que certains donneurs ne donnaient plus. L'Agence défend donc ce principe de gratuité avec fermeté et signale toutes les initiatives qui s'en écarteraient.
Ce principe de gratuité va de pair avec le principe de neutralité financière du don pour le donneur, auquel nous sommes également très attachés : celui qui donne ne doit supporter aucun coût à raison de son don. L'Agence a élaboré un guide sur le sujet de la compensation financière, diffusé auprès de l'ensemble des centres hospitaliers.
L'IA est très prometteuse et ouvre des potentialités dans tous les domaines de la médecine - imagerie médicale, diagnostic, etc. Mais un tel champ de compétences irait bien au-delà de nos compétences traditionnelles définies par les activités de soin qui sont réalisées à partir d'éléments ou de produits issus du corps humain. L'Agence n'a pas d'expertise particulière à faire valoir sur un domaine aussi large que celui de l'IA appliquée à la médecine, et elle n'est pas armée en termes de moyens pour répondre à l'ensemble des questions qui seraient posées. Par exemple, l'Agence travaille avec l'École nationale supérieure et l'École d'économie de Paris pour modéliser les chaînes croisées de dons.
M. Bernard Jomier , rapporteur . - Je tiens à vous remercier ainsi que vos équipes pour leur disponibilité. Ce projet de loi ouvre le don à de nouvelles catégories de population : les mineurs pour le don de cellules souches hématopoïétiques et les majeurs protégés pour le don d'organe. Faut-il poursuivre sur d'autres domaines comme le don du sang ? Les donneurs ne me semblent pas suffisamment valorisés dans notre société : qu'en pensez-vous ?
La loi ne peut pas tout prévoir et l'Agence est parfois saisie de demandes de dérogations, ce qui aboutit souvent à faire évoluer la loi. Comment traitez-vous ces demandes ? Sont-elles transmises pour information au Parlement ?
Mme Emmanuelle Cortot-Boucher . - Le projet de loi ouvre le don aux mineurs et aux majeurs protégés. La question d'une extension au don de sang relèverait de l'Établissement français du sang ou du ministère de la santé et je ne m'aventurerais donc pas sur ce sujet.
La valorisation des donneurs est essentielle : il est fondamental que les donneurs aient des marques de reconnaissance et que l'on apprécie à sa juste valeur la générosité de leur geste. Nos actions de communication sont axées sur cette thématique ; nous avons développé le ruban vert comme signe de reconnaissance de tous ceux qui soutiennent le don d'organe ; nous participons à l'ouverture de lieux de mémoire consacrés aux donneurs dans les hôpitaux. Mais notre préoccupation est aussi de garantir que le donneur ne sera pas mû par une autre perspective que celle de sa pure générosité, car c'est la garantie de sa liberté, sans aucune pression, pas même sociale.
S'agissant des demandes de dérogation, nous appliquons la loi, dans le cadre de la jurisprudence du Conseil d'État. Cette jurisprudence nous impose parfois de tenir compte de circonstances particulières. Par exemple, en matière d'exportation de gamètes, alors même que la loi prévoit que leur exportation est interdite, une décision du Conseil d'État nous a enjoints d'autoriser cette exportation dans un cas très particulier, sous peine de porter une atteinte excessive au droit au respect de la vie familiale et privée de la personne concernée, garantie par la convention européenne des droits de l'homme et des libertés fondamentales.
L'Agence applique donc la loi, mais tient aussi compte, ponctuellement, de circonstances particulières qui peuvent aboutir, dans certains cas, à écarter la loi. Cela se fait bien entendu dans le cadre de la jurisprudence du Conseil d'État et avec beaucoup de précautions.
Mme Corinne Imbert , rapporteure . - Les règles de bonnes pratiques de l'Agence sont élaborées collégialement. Mais elles posent problème en matière de procréation médicalement assistée (PMA). Si un couple a déjà fait une tentative et souhaite procéder à une nouvelle tentative de stimulation chez la femme, les embryons doivent être obligatoirement détruits. Pourquoi détruire ces embryons sains ? Comment expliquez-vous cette règle ?
De nombreux professionnels sont favorables au diagnostic préimplantatoire aux aneuploïdies (DPI-A) qui ne figure pas dans le texte qui nous est soumis. Quelle est la position de l'Agence sur cette question ? Dans quel cadre pourrait-il être autorisé ? Devrait-il être réservé aux femmes qui ont fait plusieurs fausses couches ou aux femmes plus âgées qui produisent néanmoins beaucoup d'embryons ?
L'ouverture du dépistage prénatal permettrait la recherche d'un gène particulier responsable d'une maladie pour laquelle nous avons une thérapie. Qu'en pensez-vous ?
Mme Emmanuelle Cortot-Boucher . - Les règles de bonnes pratiques interdisent le cumul embryonnaire : on doit en effet utiliser les embryons déjà conçus avant de réaliser une nouvelle stimulation ovarienne afin d'éviter les embryons surnuméraires. Une dérogation, très encadrée, existe dans le cas où l'embryon déjà conçu aurait un défaut. Ce sont les dispositions législatives qui interdisent le cumul embryonnaire. Les règles de bonnes pratiques, validées par un arrêté, ne peuvent pas modifier la législation ; elles ne sont que réglementaires.
Le projet de loi ne propose pas d'extension du champ du DPI : aujourd'hui, celui-ci ne peut être effectué que lorsque le couple a de fortes chances de donner naissance à un enfant porteur d'une maladie incurable, ce qui suppose que cette maladie ait déjà été identifiée dans la famille. Cette question de l'extension du DPI pour détecter un nombre anormal de chromosomes avec un DPI-A ou des maladies génétiques autres que celles connues dans la famille a été discutée à l'Assemblée nationale en première lecture. Il s'agit d'une question de société sur laquelle l'Agence ne peut qu'apporter des éléments d'expertise médicale et scientifique. D'abord, nous ne disposons pas encore d'éléments très solides prouvant le lien entre le diagnostic préimplantatoire des aneuploïdies et l'élimination du risque de fausse couche : un programme hospitalier de recherche clinique va être lancé sur le sujet ; mais en l'état actuel des connaissances scientifiques, il n'est pas possible d'affirmer que l'extension du DPI aux aneuploïdies est un moyen d'augmenter l'efficacité des techniques d'AMP. Des mécanismes de correction naturelle peuvent jouer dans certains cas : des études américaines montrent qu'il existe de faux positifs qui se corrigent spontanément. Avec un DPI-A, nous risquons donc d'éliminer des embryons qui auraient été sains et d'aboutir à une perte de chance de procréer pour certaines femmes qui auraient peu d'embryons. Nous avons besoin d'éléments scientifiques plus étayés.
La situation d'un couple qui fait un DPI avant le transfert d'un embryon n'est pas la même que pour un couple qui fait un dépistage prénatal, car, dans ce deuxième cas, la grossesse a déjà commencé.
Enfin, il faut avoir en tête que l'extension du DPI aux aneuploïdies et à des maladies génétiques peut conduire à faire de l'AMP une voie plus sûre pour concevoir un enfant que la procréation naturelle. Dans la mesure où le projet de loi envisage de faire disparaître le critère de l'infertilité comme condition du recours à l'AMP, le risque existe, et doit être pris en compte, de voir des couples s'engager dans une démarche d'AMP au motif qu'elle garantirait de meilleures chances d'avoir un enfant sain.
Sur ces sujets, l'Agence de la biomédecine n'a pas de position particulière à faire valoir. Ce sont des questions de choix. Mais il faut prendre en compte ces risques divers et les peser - c'est le rôle du Parlement - pour prendre une décision et faire un choix collectif éclairé.
Mme Muriel Jourda , rapporteur . - Madame, vous avez cité la gratuité et le volontariat comme étant des principes forts de la bioéthique en France. Qu'en est-il de l'anonymat ?
Vous avez évoqué la possibilité que nous nous trouvions avec un stock assez bas de gamètes. Pourrions-nous envisager d'importer des gamètes de l'étranger ?
Enfin, l'âge limite pour pouvoir recourir à un AMP doit-il être fixé par la loi ou faire l'objet de rédactions de bonnes pratiques ?
Mme Emmanuelle Cortot-Boucher . - L'anonymat du donneur est un principe fort du système de dons de produits et d'éléments du corps humain en France. Votre question est liée à la création du droit d'accès aux origines pour les enfants issus du don.
Le principe d'anonymat demeure très important et l'Agence y est très attachée. Ce principe est respecté dans le cadre de la nouvelle loi, puisque les gamètes qui seront donnés aux couples ou aux femmes seules qui les attendent seront donnés par un donneur qui est anonyme pour le receveur, c'est-à-dire pour le couple ou la femme.
Le projet de loi crée en revanche un droit d'accès à l'origine pour l'enfant. Mais l'enfant n'est pas le receveur. L'enfant est un tiers. Cette levée de l'identité ne porte donc pas atteinte au principe de l'anonymat du don. L'anonymat du don a pour conséquence qu'un couple ne pourra pas choisir son donneur ni connaître son identité.
S'agissant des stocks de gamètes, le projet de loi conduit à instituer un nouveau régime de dons, impliquant l'obligation pour le donneur de consentir, si l'enfant issu du don le demande, à la révélation de son identité et à la transmission de données non identifiantes. Ce changement de régime juridique nous obligera à reconstituer un stock de gamètes.
Nous ne sommes pas les premiers à passer par ce type de changement. D'autres pays ont connu un changement de cette nature. Il a été observé qu'en ce cas le stock de gamètes prend un certain temps à se reconstituer, mais se reconstitue à un niveau qui n'est pas inférieur à celui qui était observé avant l'introduction d'un droit d'accès à l'origine. Ainsi, de nouvelles catégories de donneurs, plus jeunes, se sont mobilisées au Royaume-Uni.
En définitive, le niveau de dons n'a pas diminué, ou a retrouvé à tout le moins au bout de plusieurs années un niveau comparable à celui qui avait été observé antérieurement.
L'importation de gamètes est autorisée en France, mais dans des conditions extrêmement strictes. Elle est en effet soumise à l'autorisation de l'Agence de la biomédecine et doit être effectuée dans des conditions expressément prévues par le code de la santé publique, notamment pour la poursuite d'un projet parental précis. Le Gouvernement et le projet de loi n'envisagent pas de modifier ce cadre. L'importation de gamètes demeure donc encadrée et soumise à des conditions relativement limitées.
Pour ce qui concerne l'âge limite des donneurs, la loi ne prévoit pas, en l'état, de limites particulières. La question de l'âge doit être vue en liaison avec les professionnels, la limite devant être précisée soit dans le cadre des règles de bonnes pratiques soit dans un texte réglementaire. Un travail devra sans doute être conduit avec les professionnels sur ce sujet.
Mme Chantal Deseyne . - Ma question porte sur le transport des greffons, en particulier des greffons rénaux. Les deux facteurs les plus importants pour la réussite d'une greffe sont la compatibilité entre le donneur et le receveur et la rapidité d'exécution de la transportation. Or l'Agence de la biomédecine ne coordonne ni n'assure le transport des greffons, qui reste à la charge des établissements attributaires.
Comment pouvons-nous améliorer le transport des greffons en vue d'améliorer l'efficience médicale, et même économique, une personne greffée « coûtant » moins cher que quelqu'un qui doit être en dialyse ?
Mme Maryvonne Blondin . - Avez-vous donné un avis sur le liquide de la société Hemarina, HEMO2life, qui permet le transport de greffons dans de meilleures qualités d'oxygénation ?
Par ailleurs, je voudrais vous interroger sur un autre produit issu du corps humain : les bactériophages, qui servent à lutter contre l'antibiorésistance et qui sont utilisés en cas d'urgence vitale pour lutter contre les infections bactériennes telles que le staphylocoque doré.
Mme Emmanuelle Cortot-Boucher. - Le transport des greffons est organisé sous le contrôle des équipes de prélèvement par des transporteurs, notamment privés. L'Agence de la biomédecine n'a pas de compétences dans la supervision de ces transports et n'est pas armée matériellement pour s'en doter. Cela suppose en effet d'être au plus près du terrain afin d'organiser des conditions matérielles de transport très efficaces et d'être en contact avec les partenaires situés à proximité des centres de prélèvement afin d'organiser l'acheminement des greffons jusqu'à leur point d'arrivée.
Pour des considérations d'efficacité, il est important que cette activité soit organisée par des personnes situées au plus près des équipes de soin et de prélèvement.
L'Agence de la biomédecine s'assure que les greffons sont bien arrivés à destination, particulièrement en ce moment dans le contexte de la grève, en faisant remonter toute difficulté. Mais quant à assurer l'organisation pratique de ces transports, c'est une mission extrêmement précise, qui implique une connaissance des acteurs locaux et supposerait des moyens humains et matériels dont l'Agence ne dispose pas actuellement.
Le système actuel impliquant une organisation pratique par les centres hospitaliers et les équipes de prélèvement en liaison avec des acteurs et des entreprises locaux donne satisfaction. Nous n'avons pas connaissance de pertes de chances pour les malades qui seraient liées à des difficultés d'acheminement. L'Agence assure pour sa part un rôle de supervision distanciée, en ce sens qu'elle fait remonter toute difficulté particulière et prend les mesures qui s'imposent si un problème d'acheminement est constaté.
Concernant la molécule découverte récemment en Bretagne à partir de travaux sur le ver marin, cette molécule a fait l'objet d'une demande d'autorisation déposée à l'ANSM. Il ne s'agit pas en effet d'un produit du corps humain. Cet élément doit donc être autorisé par l'ANSM en tant que dispositif ou produit médical. L'Agence a donné un avis favorable à l'ANSM sur ce produit.
S'agissant des bactériophages, cette question n'intéresse pas directement l'Agence de la biomédecine, car il ne s'agit pas d'un élément issu du corps humain. Comme la molécule extraite du ver marin, ce produit doit faire l'objet d'une autorisation, le cas échéant, par l'ANSM. Nous ne sommes donc pas directement concernés par ce sujet.
Mme Élisabeth Doineau . - Vous avez dit que vous aviez grandement participé à l'élaboration du projet de loi, notamment par le bilan que vous avez pu faire de l'ancienne loi de bioéthique. Entre la loi initiale et celle qui est sortie de l'Assemblée nationale, certains éléments vous apparaissent-ils comme essentiels et devant être défendus au Sénat ?
Mme Emmanuelle Cortot-Boucher. - Nous retrouvons dans le projet de loi les principaux éléments avancés par les professionnels qui interviennent dans les champs de compétences qui sont les nôtres. L'Agence de la biomédecine y retrouve des éléments de réponse aux principales questions qui avaient été soulevées.
Nous n'avons donc pas de demande particulière à faire valoir devant le Sénat.
M. Alain Milon , président . - Merci beaucoup.
Ce point de l'ordre du jour a fait l'objet d'une captation vidéo qui est disponible en ligne sur le site internet du Sénat .
Mmes Nicole Belloubet, garde des sceaux, ministre de la justice,
et
Frédérique Vidal, ministre de l'enseignement supérieur,
de la recherche et de l'innovation, et M. Adrien Taquet,
secrétaire d'État auprès de la ministre des
solidarités et de la santé
M. Alain Milon , président . - Nous poursuivons nos travaux sur le projet de loi relatif à la bioéthique avec l'audition des membres du Gouvernement. Intervenant à la fin de nos travaux, il n'y a plus lieu de présenter les dispositions du texte, mais plutôt d'échanger avec les membres de la commission spéciale.
L'audition fait l'objet d'une captation en vidéo en vue de sa retransmission sur le site Internet du Sénat, où elle sera ensuite disponible à la demande.
La ministre des solidarités et de la santé est représentée par M. Adrien Taquet, secrétaire d'État. Mme Nicole Belloubet, qui doit quitter notre réunion de manière anticipée, interviendra en premier. Je demande donc à nos rapporteurs et à nos collègues ayant des questions sur les sujets intéressant la chancellerie, c'est-à-dire, pour l'essentiel, sur l'article 4 relatif à l'établissement de la filiation des enfants nés d'assistance médicale à la procréation (AMP) avec tiers donneur réalisée par un couple de femmes, de les poser prioritairement.
Mme Nicole Belloubet, garde des sceaux, ministre de la justice . - Je dois effectivement intervenir à 17 heures 30 dans l'hémicycle pour les conclusions de la commission mixte paritaire sur la proposition de loi visant à agir contre les violences au sein de la famille.
Le projet de loi instituant l'AMP avec un tiers donneur pour les couples de femmes, il convenait d'en tirer les conséquences sur la filiation, ce à quoi procède l'article 4, qui a suscité de nombreuses interrogations légitimes. Plusieurs hypothèses ont été soumises par le Gouvernement au Conseil d'État, différentes options étant envisageables : l'extension du dispositif de droit commun, la création d'un régime ad hoc applicable à tous les couples réalisant une AMP avec un tiers donneur et la création d'un dispositif réservé aux couples de femmes. Les débats ont été intenses entre les tenants des différentes solutions, chacune présentant des avantages et des inconvénients, relevés par les avis et les rapports préparatoires au projet de loi qui ont opté pour l'une ou l'autre option. Les pays étrangers qui ont ouvert l'AMP aux couples de femmes ont également retenu des solutions diverses, signe de la complexité du sujet.
Plusieurs principes et objectifs se sont imposés à la décision du Gouvernement. Les quatre principes retenus sont simples : offrir les mêmes droits aux enfants concernés - le Gouvernement a affirmé le choix de l'égalité -, apporter une sécurité juridique aux deux mères et à leurs enfants - évolution forte et juste, la filiation n'est pas établie sur la vraisemblance biologique, mais sur un engagement commun -, créer une procédure simple, sans démarche supplémentaire pour les couples de femmes, éviter, enfin, de modifier le droit applicable aux couples hétérosexuels en matière de filiation. Dès lors, le Gouvernement a opté pour la création d'un régime spécifique aux couples de femmes faisant appel à un tiers donneur dans le cadre d'une AMP.
Le débat sur l'article 4 a été intense à l'Assemblée nationale, en commission comme en séance publique, et a permis d'enrichir le texte. L'article comporte trois éléments. D'abord, les deux femmes devront consentir devant notaire (comme les couples hétérosexuels) à réaliser une AMP avec un tiers donneur et s'engager à cette occasion à devenir les mères de l'enfant. Ensuite, cette reconnaissance conjointe devra être produite, avec le certificat d'accouchement, à l'officier d'état civil, afin de permettre l'établissement de la filiation. Enfin, l'acte de naissance devra faire mention de la reconnaissance conjointe.
Le dispositif a évolué à l'Assemblée nationale, sans que soient remis en cause ses objectifs. La place du nouveau régime dans le code civil a ainsi été modifiée : initialement, le projet de loi créait un nouveau titre VII bis au sein du titre I er du code civil ; finalement un nouveau chapitre V sur le régime ad hoc relatif à l'AMP avec tiers donneur a été intégré au titre VII. Ce choix permet de mieux rendre compte du socle commun aux différents modes de filiation, sans bouleverser le droit de la filiation puisque les quatre premiers chapitres du titre premier concernant la filiation établie sur la vraisemblance biologique ne sont pas modifiés. En outre, les députés ont précisé la notion de reconnaissance conjointe. Initialement, il s'agissait d'une déclaration anticipée de volonté (DAV), mais le dispositif a fait l'objet de critiques quant à son caractère stigmatisant pour les couples de femmes. À la différence de la reconnaissance figurant à l'article 316 du code civil, qui intervient pendant la grossesse, voire après la naissance de l'enfant, la reconnaissance conjointe est réalisée avant la conception et ne concerne que les couples de femmes. Il ne peut y avoir de confusion possible entre les deux régimes.
Le Gouvernement a privilégié la voie de l'équité avec une solution juridique sûre et porteuse d'égalité. L'article 4 tire les conséquences indispensables à l'ouverture de l'AMP aux couples de femmes, afin de sécuriser la filiation, notamment à l'égard de la femme qui n'accouche pas. Le dispositif fait ainsi obstacle à l'éventuelle reconnaissance de l'enfant par un tiers. Il est simple et sécurisant pour les mères, comme pour les enfants.
Mme Muriel Jourda , rapporteur . - M. Touraine, rapporteur du texte à l'Assemblée nationale, a estimé que le projet de loi conduisait à « légitimer une procréation sans sexe pour tous ». Est-ce votre philosophie ?
L'article 4 du projet de loi prive les enfants de filiation paternelle. Un sondage réalisé en 2018 indique que 93 % des personnes interrogées considèrent que le père joue un rôle essentiel pour l'enfant. Que leur répondez-vous ? Ce même article ne fait plus référence à la femme qui accouche pour qualifier la mère. Cette rédaction n'est-elle pas porteuse d'un risque juridique ?
Le texte a retenu le terme de « femme non mariée » pour qualifier une femme seule, suscitant des critiques. Existe-t-il une raison juridique à ce choix ?
Enfin, ne pensez-vous pas qu'il existe un risque de confusion à avoir choisi le terme de reconnaissance pour deux procédures distinctes ?
Mme Nicole Belloubet, garde des sceaux . - La promotion de la procréation sans sexe ne ressort pas de ma philosophie. L'ouverture de l'AMP avec un tiers donneur aux couples de femmes reconnait un choix de vie et l'existence d'une pluralité de modèles familiaux. Dans ce contexte, nous sécurisons la filiation pour l'enfant et pour ses mères.
Je répondrai aux 93 % de sondés que vous mentionnez que nous prenons en considération la pluralité des familles. De nombreuses études menées sur les enfants sans père montrent qu'ils peuvent avoir des références masculines hors du cercle de la famille nucléaire et que leur situation familiale n'a aucune incidence sur leur développement psychologique.
S'agissant du risque de confusion entre les deux types de reconnaissance, je ne crois pas qu'il existe. La procédure de l'article 316 du code civil et la reconnaissance conjointe créée par le texte n'interviennent pas dans la même temporalité.
Nous avons effectivement eu, à l'Assemblée nationale, un débat fourni sur la référence à la femme qui accouche. Dans le cadre d'une AMP réalisée par un couple de femmes, l'une d'elles doit évidemment accoucher. Pour établir la filiation de l'enfant, l'officier d'état civil devra d'ailleurs disposer du certificat d'accouchement, ainsi que de la reconnaissance conjointe pour sécuriser la filiation avec l'autre mère. La rédaction de l'article pourra évoluer, dès lors que les droits des deux mères restent identiques.
Enfin, s'agissant de la notion de femme non mariée, il s'agit d'une demande du Conseil d'État, afin de respecter le cadre juridique de l'établissement de la filiation.
Mme Muriel Jourda , rapporteur . - Ne pouvait-on pas évoquer une femme célibataire ou une femme seule ?
Mme Nicole Belloubet, garde des sceaux . - Permettre à une femme mariée de pratiquer seule une AMP avec un tiers donneur aurait des conséquences juridiques complexes.
M. Adrien Taquet, secrétaire d'État auprès de la ministre des solidarités et de la santé . - L'intérêt supérieur de l'enfant est au coeur des préoccupations du Gouvernement. Les études menées depuis quarante ans sur les familles monoparentales ou formées par un couple de femmes montrent qu'il n'existe aucun effet délétère sur les enfants ni aucune conséquence sur la construction de leur sexualité. Ce n'est pas la structure, mais la dynamique familiale qui importe. La conférence de consensus sur les besoins fondamentaux de l'enfant, lancée par Laurence Rossignol lorsqu'elle était ministre des familles, de l'enfance et des droits des femmes, a établi que la sécurité constituait un méta-besoin de l'enfant. L'altérité peut se construire hors de la famille. L'adoption par une femme seule est autorisée depuis 1996 et depuis 2013 pour les couples de même sexe : des millions de femmes ont élevé des enfants seules, de manière subie ou choisie. Être parent est une histoire de désir et d'amour.
M. Bernard Jomier , rapporteur . - L'article 21 bis a été ajouté au texte par l'Assemblée nationale. Il prévoit la prise en charge des enfants présentant une variation du développement génital par les centres de référence des maladies rares du développement génital. Il a possiblement des répercussions sur l'état civil. Quelle sera l'articulation entre ce dispositif et l'inscription des enfants à l'état civil ?
Mme Nicole Belloubet, garde des sceaux . - Nous avons abordé le sujet de la prise en charge des enfants présentant une variation du développement génital sous différents angles. Initialement, nous avions songé à décaler de trois mois l'établissement de leur état civil, mais les associations nous ont indiqué que le délai resterait insuffisant. Leur prise en charge relève d'équipes pluridisciplinaires spécialisées. Les centres de référence constituent un appui important pour les familles, une concertation entre professionnels y propose des pistes thérapeutiques.
M. Adrien Taquet, secrétaire d'État . - Les parlementaires ont souhaité que les familles soient ainsi mieux accompagnées.
M. Alain Milon , président . - Il s'agit, en l'espèce, des seuls députés.
M. Bernard Jomier , rapporteur . - Nous avons bien compris l'objet de l'article, mais nous nous interrogeons sur ses conséquences en matière d'état civil.
Mme Nicole Belloubet, garde des sceaux . - Les rectifications simples d'état civil sont déjà possibles. Nous travaillons également à un possible report de la mention du sexe.
M. Michel Amiel . - Si l'égalité devant le fait d'avoir des enfants, principe que je ne soutiens pas, préside aux articles 1 er et 4 du projet de loi, pourquoi ne pas avoir étendu le texte, par cohérence avec le droit à l'enfant, à la gestation pour autrui (GPA) ?
M. Jacques Bigot . - Pour les couples hétérosexuels comme pour les couples de femmes, un consentement à l'AMP est prévu devant notaire. L'article 3 du projet de loi, pour sa part, affirme le droit, pour l'enfant né d'une AMP, d'accéder, à sa majorité, aux renseignements disponibles sur le donneur. Lorsque l'enfant sera issu d'un couple de femmes, ce sera simple, puisque le régime de filiation prévoit une reconnaissance conjointe figurant à l'état civil. En revanche, aucune mention équivalente n'existe sur l'acte d'état civil d'un enfant né d'une AMP au sein d'un couple hétérosexuel. Les parents peuvent donc garder confidentielle l'histoire de sa procréation. Par souci d'égalité entre les enfants, la procédure ne devrait-elle pas être identique ?
Mme Maryvonne Blondin . - La question de M. Jomier faisait référence à la circulaire d'octobre 2011 sur le report de la mention du sexe à l'état civil. Vous avez également évoqué la possibilité de modifier l'acte de naissance, mais le sexe de naissance demeure inscrit et cela peut s'avérer discriminant. Parfois, mais difficilement, une annulation est possible. Par ailleurs, les rectifications apposées à l'acte de naissance sont payantes, sauf erreur médicale. Je vous rappelle que la Cour européenne des droits de l'homme (CEDH) a été saisie, en octobre 2018, d'une action en responsabilité contre la France pour violation de ses obligations contre la torture envers les personnes intersexuées. Nos travaux sont attendus par toutes les organisations internationales. Clarifions le code civil.
M. Philippe Bas . - Madame Belloubet, vous rappelez deux principes : il ne faudrait modifier le droit que si c'est indispensable ou utile, et il faut faire progresser l'égalité. Mais est-ce que vous ne vous écartez pas de ces deux principes dans l'établissement de la filiation de l'enfant par rapport à sa mère biologique ? Vous créez deux régimes différents selon que la mère vit avec un homme ou avec une femme, avec des conséquences juridiques en chaîne en cas de contentieux sur la maternité...
Cette rupture d'égalité n'est pas nécessaire pour reconnaître une deuxième filiation maternelle pour un couple de femmes. On pourrait définir un régime de reconnaissance de filiation par rapport à la compagne de la mère.
Vous modifiez une règle fondamentale, à savoir l'accès à l'AMP pour les couples hétérosexuels pour lesquels il existait auparavant une condition d'infertilité. Pourquoi changer ce régime alors que le seul objectif politique est d'ouvrir l'AMP aux couples dont la fertilité n'est pas causée par une stérilité médicalement constatée et aux femmes seules ?
M. Olivier Henno , rapporteur . - Dans sa rédaction initiale, l'article 12 du projet de loi exclurait la possibilité de recourir à l'imagerie fonctionnelle pour l'expertise judiciaire. L'utilisation de l'imagerie est encadrée depuis 2011 et, semble-t-il, utilisée à bon escient par les juges. Quels risques avérés justifient cette interdiction ?
M. Guillaume Chevrollier . - Cette réforme aura des conséquences anthropologiques importantes. Comment pouvez-vous affirmer qu'elle n'aura pas de conséquences psychologiques sur les enfants privés de père ? Au nom du principe d'égalité, vous ouvrez la PMA aux couples de femmes et aux femmes seules. Comment, au nom de ce principe, êtes-vous sûre que les couples d'hommes ne revendiqueront pas la GPA ?
Mme Nicole Belloubet, garde des sceaux . - La première question rejoint la dernière... Monsieur Amiel, nous ne sommes pas allés jusqu'au bout avec la GPA, car le Conseil d'État l'affirme clairement : il n'y a pas de droit à l'enfant, l'enfant est inclus dans un projet familial et parental. Le principe d'égalité ne peut jouer de manière absolue, mais pour les personnes qui sont dans la même situation par rapport à la procréation. On peut ne pas appliquer à l'un des types de couples - couples d'hommes, de femmes, ou hétérosexuels - ce qu'on applique aux autres. Le Conseil d'État est très clair.
Ni le droit à l'enfant ni le principe d'égalité ne jouent pour la GPA. Dans le code civil, nous avons clairement inscrit l'interdiction de la GPA. On ne peut déduire de la PMA qu'il y aurait juridiquement une évolution vers la GPA.
Monsieur Bigot, vous évoquez une possible différenciation entre les enfants des couples de femmes, ayant accès à leurs origines, et ceux de couples hétérosexuels.
Dans le cas de couples hétérosexuels, nous avons fait le choix de laisser la famille déterminer le moment opportun pour dire à l'enfant quelles sont ses origines, sachant qu'il pourra faire une demande ensuite pour accéder à ses origines.
Madame Blondin, je vous entends évoquer les problèmes d'état civil et la jurisprudence de la CEDH. Il y a eu de nombreux débats à l'Assemblée nationale, et nous serons attentifs à ceux du Sénat. Il faut être vigilant et faciliter l'évolution des actes d'état civil. Je vois chaque semaine de nombreuses modifications d'état civil.
Monsieur Bas, il faut considérer l'égalité en matière de procréation par rapport aux couples. Un couple hétérosexuel est différent d'un couple homosexuel en ce qui concerne la procréation. Nous ne créons donc pas une rupture d'égalité. Selon vous, il n'est pas utile de reconnaître une double maternité. Nous avions cru utile, au contraire, de reconnaître au même moment la même qualité maternelle aux deux femmes du couple. Mais je ne suis pas sûre d'avoir totalement compris votre question.
Sur les incidences psychologiques pour les enfants, M. Taquet l'a rappelé, les différentes études nous laissent à penser qu'il n'y a pas d'incidence psychologique, les enfants pouvant trouver des figures masculines en dehors des parents.
Mme Frédérique Vidal, ministre de l'enseignement supérieur, de la recherche et de l'innovation . - On ne doit pas imposer aux couples hétérosexuels demandant une AMP avec tiers donneur de prouver une condition d'infertilité qui n'existe pas pour les couples homosexuels. Le parcours d'AMP est difficile. Par ailleurs, on demandait à ces couples une déclaration d'infertilité, et non des preuves scientifiques qu'il y avait un problème physiologique. Il nous a semblé plus simple d'enlever toute référence à l'infertilité.
Le Comité consultatif national d'éthique (CCNE) et l'Office parlementaire d'évaluation des choix scientifiques et technologiques (Opecst) ont recommandé de ne pas utiliser l'imagerie cérébrale fonctionnelle à des fins judiciaires. Tout est souvent une question d'interprétation. Ce n'est pas parce que l'on observe quelque chose sur l'imagerie que l'on peut catégoriser des personnes comme étant à risque criminel. Autant il est important de s'en servir pour déterminer des pathologies, autant on ne peut s'en servir par anticipation de faits qui ne se sont pas encore produits...
J'évoquerai le titre IV. Nous avons une première conviction, partagée par les parlementaires et la société : ce que la science sait rendre possible n'est pas nécessairement aligné sur ce que notre société souhaite. Résoudre cette tension entre ce que sait faire la recherche, avec les aspirations de notre société, c'est le coeur même de ces lois de bioéthique, régulièrement révisées. Le Sénat a joué un rôle considérable depuis 1994 dans la construction de ce droit. La communauté des chercheurs sait ce qu'elle doit au Sénat, notamment concernant l'autorisation de la recherche sur les cellules souches embryonnaires.
Nous ne devons pas sacrifier nos valeurs fondamentales à une quête éperdue et permanente du savoir, mais il ne faut pas non plus sacrifier l'espoir de développer la connaissance, de comprendre des thérapies innovantes, de guérir des maladies aujourd'hui incurables, sur des préjugés qui ne correspondent plus à l'état des connaissances.
Ce projet de loi réforme le cadre juridique de la recherche, avec de nouvelles facilités, mais aussi de nouvelles règles, compte tenu des avancées scientifiques sur la recherche de cellules souches embryonnaires ou de cellules souches induites.
Ce projet de loi autorise la recherche pour l'édition du génome de l'embryon sur des embryons ne faisant plus l'objet d'un projet parental. Un décret rappelle qu'aucun embryon n'est créé à des fins de recherche. Ce sont seulement des embryons conçus dans le cadre d'un projet parental qui sont, après arrêt de ce projet et demande aux parents, soit détruits, soit utilisés à des fins de recherche. La recherche sur l'édition du génome apporte des connaissances pour comprendre le rôle des différents gènes dans les mécanismes de différenciation cellulaire qui sont à l'oeuvre au cours du développement, mais aussi dans certains processus physiologiques - le vieillissement - ou pathologiques - le cancer...
Le projet de loi instaure une limite de 14 jours pour l'observation des embryons in vitro, afin qu'ils n'atteignent pas le stade de l'organogenèse. Cette limite n'existait pas auparavant, car on ne savait pas observer des embryons plus de quelques jours. Désormais, on sait le faire. Nous procédons d'une même logique : nous autorisons, mais lorsque les connaissances scientifiques l'exigent, nous mettons des limites.
Les interdits fondateurs de notre droit et les textes internationaux le confirment tous : on ne peut pas créer d'embryon à des fins de recherche ; on ne peut pas modifier le patrimoine génétique d'un embryon destiné à être implanté ; et on ne peut pas introduire de cellules animales dans un embryon humain. Ces trois principes ont été réaffirmés par le Gouvernement à l'Assemblée nationale.
Ce texte protège le statut particulier de l'embryon et par là même interdit le clonage et la modification de patrimoine génétique d'un embryon destiné à être réimplanté. Il ne nous prive pas des innovations thérapeutiques qui peuvent être mises au point, pour mieux comprendre les mécanismes de développement de différenciation cellulaire et de développement normal ou pathologique.
La question des chimères a suscité des interrogations. Dans le cadre de la loi de bioéthique actuelle, l'introduction d'une matière animale dans un embryon humain est interdite, mais rien n'est dit sur la réciproque. On pouvait introduire des cellules humaines dans un embryon animal. Cette possibilité nous permet de produire des modèles afin de comprendre les pathologies humaines et préparer des traitements. C'est pour cela que nous réaffirmons la première interdiction ; par contre, mettre des cellules humaines dans un embryon animal sera possible, mais soumis à un contrôle.
Les cellules souches embryonnaires sont capables de se transformer en n'importe quelles cellules. Elles sont à l'origine d'espoirs de thérapies cellulaires et de médecine régénérative pour des maladies comme Parkinson, le diabète ou l'insuffisance cardiaque.
Actuellement, les cellules souches sont soumises au même régime que les embryons. Or cela ne nous semble plus relever du même questionnement éthique. Auparavant, pour produire des cellules souches, il fallait détruire un embryon.
Désormais, on utilise des cellules souches dérivées d'embryons utilisés il y a plusieurs années. On n'a plus besoin d'avoir accès à un embryon. Nous différencions donc la recherche sur les embryons de celle sur les cellules souches embryonnaires, à moins que ces dernières ne découlent d'un embryon ; auquel cas, elles sont soumises au même régime que la recherche sur les embryons. Nous allégeons la recherche sur les cellules souches embryonnaires, en les soumettant à une simple déclaration au lieu d'un processus d'autorisation.
Les cellules souches pluripotentes induites sont des cellules adultes que le scientifique fait revenir à un état proche des cellules souches embryonnaires. À partir d'elles, on peut produire certaines cellules, mais pas toutes. Une question éthique se pose lorsqu'elles sont induites en gamètes, pouvant porter du matériel génétique et repartir dans un cycle de reproduction. La recherche sur ces cellules souches pluripotentes induites est, dans ce cas, soumise à un régime d'autorisation. Il n'est pas sûr que l'on pourra un jour remplacer les cellules souches embryonnaires par des cellules souches pluripotentes induites. Il est donc important de garder les deux types de recherche.
C'est en ouvrant de nouvelles voies, en traçant de nouvelles limites, en réaffirmant des lignes rouges, que ce texte dessine les contours d'une recherche libre et responsable. La recherche et la connaissance rendent notre avenir possible, mais le législateur doit définir le chemin à emprunter, l'horizon souhaité et la couleur de notre avenir.
M. Adrien Taquet, secrétaire d'État. - Un propos général n'ayant plus vraiment de sens à ce stade du débat, je reviendrai sur quelques sujets évoqués.
Monsieur Amiel, la loi de bioéthique n'est pas une loi d'égalité. Ce n'est pas cela qui nous guide. Nous voulons passer au prisme éthique les évolutions des techniques médicales. Il n'y a jamais eu, et il n'y aura pas effectivement de droit à l'enfant. Ce n'était pas le cas pour les couples hétérosexuels par le passé, cela ne sera pas le cas pour les couples homosexuels ou pour les femmes seules à l'avenir. Sinon l'enfant serait le simple produit d'un caprice ; ce serait dénigrer le projet parental. Or l'AMP est un parcours long, difficile, qui nécessite un accompagnement, que les députés ont renforcé.
Les familles homoparentales existent ; il serait hypocrite de ne pas le voir. Souvent, leur projet parental est bon, les enfants sont ardemment désirés. Si la famille est un point de repère, c'est d'abord une histoire et un parcours.
Connaître ses origines, c'est avoir une réponse légitime à la question : « d'où viens-je ? » Il faut rompre avec la dissimulation, avec la logique du secret qui abîme plus qu'elle ne protège.
Vous avez rencontré des enfants issus de l'AMP. Ces enfants ne sont pas à la recherche d'un père, mais d'un récit, d'une histoire importante pour se construire. La famille est le lieu où l'on doit se dire les choses. Pour une AMP avec don dans une famille hétérosexuelle, c'est aux parents de choisir de dévoiler les origines quand ils le souhaitent, dans l'intérêt de l'enfant. Nous voulons aussi permettre à chaque enfant d'accéder, à sa majorité, à ces informations. Un donneur n'est pas un parent. Ce n'est pas sa vocation, mais il est une pièce de l'identité de l'enfant.
Nous sortons le don du secret, mais pas de l'anonymat, pour le reconnaître dans son côté profondément humain, altruiste et solidaire. Nous réaffirmons également la force des institutions, qui ont vocation à encadrer et protéger chacun avec un seul choix, celui de la responsabilité individuelle et collective. Cet esprit de responsabilité nous guide dans nos débats et dans les choix que nous vous proposerons de faire dans d'autres domaines, tout aussi complexes et variés que l'auto-conservation de gamètes pour les femmes comme pour les hommes, l'amélioration de la qualité de la sécurité des pratiques pour les dons d'organes, le développement de la médecine génomique.
Je le redis, avec tous les ministres qui seront sur le banc du Gouvernement, l'intérêt supérieur de l'enfant a guidé nos réflexions, notamment dans le cas de l'AMP post mortem. Même si le principe d'égalité peut être mis en avant, nous avons décidé de faire primer l'intérêt supérieur de l'enfant. Que signifie naître dans le deuil ? Autant de sujets qui susciteront de riches débats.
Mme Corinne Imbert , rapporteure . - Madame Vidal, selon vous, le Gouvernement souhaite que ce projet de loi ne nous prive pas des évolutions scientifiques. Vous maintenez l'autorisation, par l'Agence nationale de sécurité du médicament et des produits de santé (ANSM), des recherches menées sur l'embryon dans le cadre de l'AMP, alors que la Cour des comptes critique une procédure lourde et complexe, qui décourage souvent les chercheurs. D'autant que ces recherches ne constituent pas le coeur de l'expertise de l'ANSM, qui doit s'en remettre, en pratique, à l'avis de l'Agence de la biomédecine. Souvent, l'avis de l'ANSM est conforme à celui de l'Agence de la biomédecine. Pourquoi ne pas confier tout simplement cette mission à cette dernière, qui dispose déjà de l'expertise nécessaire sur l'assistance médicale à la procréation et sur la recherche sur l'embryon, quitte à lui faire appliquer les dispositions spécifiques aux recherches cliniques ? Ne pourriez-vous pas justement renforcer les moyens de l'Agence de la biomédecine pour examiner ces projets de recherche clinique ? Vous souhaitiez supprimer sa mission en matière de nanotechnologies au motif qu'elle ne disposait pas d'expertise dans ce domaine, mais n'est-ce pas de votre responsabilité de lui donner les moyens humains et matériels de cette expertise ? Si je voulais être un peu taquine, je dirais que, si nous devions supprimer aux agences toutes les missions que l'État ne finance pas, il ne restera peut-être plus grand- chose...
Le droit était muet sur la possibilité d'utiliser des cellules humaines dans un embryon animal. Vous donnez cette possibilité dans le projet de loi, et affirmez que c'est soumis à un contrôle, mais de quel contrôle parlez-vous ? Vous supprimez l'interdiction générale de création d'embryons chimériques et transgéniques, et vous ne maintenez qu'une interdiction d'insérer dans un embryon humain des cellules animales. Vous autorisez donc l'adjonction de cellules souches humaines à des embryons animaux en vue de leur transfert chez la femelle : ne pensez-vous pas qu'il y a là un vrai risque de franchissement de la barrière des espèces ?
Mme Frédérique Vidal, ministre . - Actuellement, il n'y a aucune interdiction générale de créer des embryons chimériques. Seule une partie du projet de loi, concernant l'embryon humain, mentionne l'interdiction d'insérer des cellules animales dans des embryons humains.
Nous introduisons à un autre endroit du texte la possibilité, après une demande de déclaration d'usage des cellules souches embryonnaires, de les insérer dans des embryons animaux. On va donc vers un contrôle plus efficace que le flou préexistant. Honnêtement, c'est déjà très largement pratiqué en laboratoire, sans que l'on n'ait jamais observé de franchissement de barrière d'espèces. Il s'agit de regarder comment se développent des pathologies humaines dans le contexte d'un embryon animal.
On entre dans le cadre de la recherche interventionnelle sur la personne humaine lorsqu'on touche à des embryons qui ont vocation à être réimplantés. Cela ne relève pas de l'Agence de la biomédecine. Les questionnements de l'ANSM ne sont pas les mêmes que ceux de l'Agence de la biomédecine. Il ne s'agit pas de modifier génétiquement l'embryon qui sera réimplanté - c'est interdit, je le rappelle -, mais, par exemple, de l'enrober, au moment de la fécondation in vitro ou sur un embryon obtenu après fécondation in vitro, d'un certain nombre de molécules qui faciliteront la réimplantation de l'embryon chez la mère.
Je suis consciente des questions qui se posent sur les délais. L'Agence de la biomédecine s'est engagée à les tenir. Elle n'aura finalement à traiter que des questions sur lesquelles elle s'estime totalement légitime. Sur le sujet que vous évoquez, elle est obligée d'aller chercher de l'expertise ailleurs ; cela crée une forme d'embouteillage.
M. Olivier Henno , rapporteur . - Madame la ministre, vous avez évoqué la tension entre ce que sait faire la recherche et les limites que nous voulons lui fixer.
L'article 17 supprime l'interdiction générale de création des embryons chimériques. Même s'il s'agit de recherches sur des embryons non destinés à des fins de gestation, se pose la question de savoir jusqu'où l'on peut aller dans le franchissement de la barrière des espèces. Comment l'Agence de la biomédecine appréciera-t-elle si ces chimères présentent une proportion acceptable entre animal et humain ? Ne va-t-on pas plus loin que d'autres pays ?
Vous supprimez également l'interdiction de création d'embryons transgéniques. S'agit-il d'expérimenter sur des embryons surnuméraires la technique d'édition génomique CRISPR-Cas9 ? Est-ce dans le but de déterminer si nous pourrions, à terme, modifier le génome d'embryons destinés à être transférés à des fins de gestation. N'ouvririons-nous pas la porte à une remise en cause, dans le futur, de l'interdiction de modifier les caractéristiques transmissibles à la descendance ? J'ai le sentiment que l'article 17 implique un bouleversement de l'éthique « à la française ».
Mme Frédérique Vidal, ministre . - Je tiens à vous rassurer sur ces deux points.
L'article 17 supprime en effet l'interdiction générale de création des embryons chimériques, mais la modification d'un embryon humain par adjonction de cellules provenant d'autres espèces demeure interdite.
L'adjonction de cellules d'origine humaine dans des embryons d'origine animale se pratique dans tous les laboratoires du monde, y compris français, et ce n'était absolument pas interdit par la loi de bioéthique.
Le présent projet de loi précise qu'il n'est pas possible de modifier un embryon humain, et qu'il faut une autorisation pour modifier un embryon animal par adjonction de cellules embryonnaires humaines, comme c'est d'ailleurs le cas actuellement. Il s'agit de simples précisions, dans la mesure où il n'est jamais question, dans la loi de bioéthique, d'embryons animaux, mais seulement d'embryons humains. Nous avons levé un flou juridique sur lequel notre attention a été attirée.
On entend souvent parler de la possibilité de faire des greffes à partir d'organes animaux. Nous en sommes très loin, et encore plus éloignés du franchissement de la barrière d'espèce.
Pour ce qui concerne l'édition du génome, nous souhaitons en effet permettre l'utilisation de la technique d'édition génomique CRISPR-Cas9. Je rappelle que la convention d'Oviedo prévoit, et le présent projet de loi le réaffirme, qu'il est formellement interdit de réimplanter un embryon génétiquement modifié. Nous avons eu ce débat à l'Assemblée nationale. Des députés demandaient pourquoi nous nous priverions de la possibilité d'ôter un gène défectueux dominant et de laisser s'exprimer la copie de ce gène non défectueux, et donc, potentiellement, de guérir une maladie, dans la mesure où l'on sait le faire. Notre réponse a été négative, car nous ne souhaitons pas réimplanter d'embryons modifiés génétiquement via l'édition du génome.
Nous souhaitons, en revanche, autoriser l'édition du génome dans des embryons en vue d'étudier l'impact de ces modifications du génome. Pour ce faire, les embryons sont observés en culture pendant 14 jours, mais pas au-delà. Cette question a été longuement débattue, d'aucuns souhaitant prolonger ce délai jusqu'à 21 jours, d'autres préférant le raccourcir. Nous avons souhaité poser cette limite de 14 jours, car elle correspond à la limite de l'organogenèse : à partir du 15 e jour, en effet, on peut faire la différence dans un embryon entre les cellules qui seront à l'origine du système nerveux et les autres ; auparavant, ce n'est pas possible.
Mme Corinne Imbert , rapporteure . - Jusqu'à quel stade de développement peut-on étudier des embryons d'origine animale auxquels on a ajouté des cellules humaines ?
Mme Frédérique Vidal, ministre . - C'est extrêmement variable, et cela dépend du type de cellules qui sont ajoutées dans l'embryon. Parfois, une observation de quelques jours suffit. Encore une fois, c'est une pratique très courante - je pense aux recherches portant sur les caractéristiques des cellules immunitaires chez les souris. Il n'y aurait donc pas de raisons de faire un bond en arrière.
M. Bernard Jomier , rapporteur . - Comment avez-vous estimé la durée de conservation des embryons, que vous avez fixée à cinq ans ? S'agit-il de préserver un stock nécessaire afin de ne pas induire de rupture dans les activités de recherche sur l'embryon ?
Mme Frédérique Vidal, ministre . - Il ne me semble pas que cette durée de conservation ait été modifiée. Elle était déjà de 5 ans auparavant. Nous ne sommes pas en pénurie d'embryons confiés par des parents à la recherche. Par ailleurs, il n'y a presque plus de stocks d'embryons à une seule cellule.
M. Michel Amiel . - Vous disiez, monsieur le secrétaire d'État, que les lois de bioéthique servaient à accompagner sur le plan éthique les progrès de la science médicale, notamment biologiques, technologiques, thérapeutiques. Or la PMA n'est pas liée à une telle idée de progrès, même s'il y a des avancées en matière de recherche sur l'implantation de l'embryon. N'aurait-il pas été préférable de dissocier le débat sur la bioéthique - le sujet, évoqué par Mme Vidal, des cellules souches embryonnaires, par exemple - de celui sur la PMA.
Nous le verrons lors du débat en séance publique et dans le commentaire médiatique qui l'accompagnera, la discussion relative à la PMA risque en effet de confisquer l'espace consacré à la bioéthique proprement dite. Or je ne suis pas seul à penser que ce sujet relève non pas de la bioéthique, mais de l'éthique ou de la morale sociétale.
M. Bernard Bonne . - Monsieur le secrétaire d'État, on a parlé du droit à l'enfant et du droit de l'enfant tout à l'heure. On a dit que les enfants devaient avoir les mêmes droits, quelle que soit leur origine. Or un enfant né par PMA n'a pas les mêmes droits qu'un autre, né enfant dans un couple hétérosexuel, dans la mesure où il n'a pas de père, ou ne pourra le connaître qu'à l'âge de 18 ans. Dispose-t-on d'études montrant que les enfants nés par PMA connaissent le même développement que les autres ? Les seules études connues en la matière proviennent de pays étrangers.
On compare trop souvent les enfants qui n'ont pas eu de père à ceux dont les parents ont divorcé, ou qui vivent dans une famille monoparentale ou qui ont été adoptés. Sur quels éléments vous êtes-vous fondés pour dire que les enfants nés de PMA n'ont pas davantage de problèmes que les autres ? À égard, les résultats des études faites par les pédopsychiatres seront intéressants.
M. Jacques Bigot . - Madame la ministre, dans un avis récent, le CCNE s'est dit favorable à deux possibilités de dépistage en population générale : le dépistage préconceptionnel et les mutations actionnables. Le jury citoyen qu'il avait consulté y était également favorable. Or ce n'est pas proposé dans le présent projet de loi, alors même que c'était envisagé dans le rapport de M. Jean-Louis Touraine. Pour quel motif le Gouvernement ne l'a-t-il pas souhaité, alors même que ces dépistages existent dans d'autres pays ?
Mme Maryvonne Blondin . - Quel est votre avis sur les recherches en cours au sein de la station biologique de Roscoff sur la phagothérapie, un domaine de recherches que l'on pourrait intégrer dans l'article du projet de loi relatif au microbiote fécal. On a recours à la phagothérapie, qui a obtenu de l'ANSM une autorisation temporaire d'utilisation (ATU) lorsque le patient se trouve dans une impasse thérapeutique, et risque l'amputation ou la mort.
Nous pourrions encadrer cette pratique, peu coûteuse et naturelle, en prévoyant des procédures de culture des bactériophages. Nous pourrions également favoriser la recherche sur les anomalies du développement génital.
M. Yves Daudigny . - Les tenants de certains courants de pensée, auxquels je ne souscris pas, considèrent que l'embryon est un être humain à part entière. Comment répondre aux défenseurs de cette idée ?
Pour ce qui concerne la recherche sur l'embryon et les cellules souches, vous avez évoqué, madame la ministre, les espoirs qu'inspire la médecine régénérative. On sait aujourd'hui fabriquer de nouvelles cellules, mais pas l'architecture générale de l'organe. Ce domaine de la recherche doit-il avoir une vocation médicale affirmée, sachant qu'un chercheur ne sait pas, lorsqu'il commence sa recherche, ce qu'il va trouver ?
Mme Michelle Meunier . - Je suis d'accord avec vos propos, monsieur le secrétaire d'État, sur la recherche de l'histoire, plutôt que de l'identité et sur le nécessaire accompagnement. En la matière, le Conseil national d'accès aux origines personnelles (Cnaop) joue un rôle important, dont l'évolution est envisagée dans le cadre d'un regroupement de gouvernances. Aura-t-il toujours les moyens d'assurer ses missions ?
M. Guillaume Chevrollier . - Vous l'avez dit, madame la ministre, ce que la science sait rendre possible n'est pas forcément souhaitable. Je suis d'accord avec cette position. On a l'impression aujourd'hui d'un progressisme permanent : il faut réviser en permanence les textes sur la bioéthique. Vous proposez même dans le présent texte une révision tous les cinq ans, contre sept ans auparavant.
Il est prévu d'allonger la durée de culture des embryons de 7 à 14 jours, le quatorzième jour étant celui où s'opère la différenciation des tissus. Si l'on accepte cette recherche in vitro jusqu'à 14 jours et si, demain, on réussit à maintenir en vie l'embryon in vitro au-delà, à quel titre refuserait-on de faire des recherches au-delà de ce délai sur cet être humain en devenir, ce qui est, selon moi, la définition de l'embryon ? Cet allongement confortera l'instrumentalisation de l'embryon humain. Décidera-t-on, dans cinq ans, d'aller au-delà des 14 jours ?...
M. Olivier Henno , rapporteur . - L'article 11 prévoit que le patient est informé de l'utilisation des traitements algorithmiques au moment des résultats. Pourquoi ne pas l'en informer en amont ?
M. Alain Milon , président . - Je rappelle à Michel Amiel que la première loi de bioéthique a été provoquée par le débat sur la PMA.
M. Adrien Taquet, secrétaire d'État. - Une loi de bioéthique s'appréhende sous le prisme de nos principes éthiques, de l'évolution des différentes techniques et des transformations de la société. À cet égard, on peut considérer que la question de la PMA doit faire partie intégrante de la bioéthique. Vous craignez, monsieur Amiel, qu'elle ne phagocyte bon nombre d'autres sujets. Lors du débat à l'Assemblée nationale, elle a en effet pris une place importante, mais, progressivement, les parlementaires et la presse se sont intéressés aussi fortement aux autres points du texte.
Monsieur Bonne, depuis une cinquantaine d'années, environ 700 études ont été menées, surtout aux États-Unis et au Royaume-Uni, sur des enfants nés par PMA, mais aussi sur des enfants élevés par des couples homoparentaux ou des femmes seules. Certaines sont mentionnées dans l'étude d'impact. Aucune ne démontre quoi que ce soit d'atypique dans leur développement. À l'inverse, aucune étude, à notre connaissance, ne tend à démontrer le contraire.
Comme le disait Françoise Dolto - un propos repris par Boris Cyrulnik -, c'est probablement autant l'enfant qui choisit et qui construit les parents que l'inverse.
Madame Meunier , vous avez vu juste en faisant le lien entre nos différentes réformes. Dans le cadre de la stratégie nationale de prévention et de protection de l'enfance, que je mène et dont j'ai dévoilé les principales mesures en octobre dernier, est prévue une réflexion sur l'évolution de la gouvernance de la protection de l'enfance dans notre pays, celle-ci n'étant pas aussi efficace qu'elle devrait l'être. J'ai missionné l'Inspection générale des affaires sociales (IGAS) pour appréhender les implications techniques et juridiques de ce projet de rapprochement des différents organismes qui travaillent sur ces sujets.
Le Cnaop conservera les moyens d'exercer ses missions, et la nouvelle instance de gouvernance de la protection de l'enfance sera dotée de moyens supplémentaires. Chargé de l'accès aux origines des personnes nées sous le secret, pourra-t-il s'occuper de l'accès aux origines des personnes nées de dons ? Ses responsables ne sont pas favorables à une telle extension de leurs compétences. Nous avons fait le choix de confier à une commission ad hoc, adossée à l'Agence de la biomédecine et placée sous la responsabilité du ministère, la question du recueil des données concernant les enfants nés par tiers donneur, lesquels pourront solliciter l'accès à ces données à leur majorité.
Mme Frédérique Vidal, ministre . - S'agissant du dépistage préconceptionnel, celui-ci est d'ores et déjà autorisé en France dans le cadre d'une prise en charge médicale spécialisée et d'un conseil génétique. L'élargir à toute personne ou à tout couple fragiliserait nos principes et nos valeurs. La question se pose, notamment, de la définition de la liste de pathologies. Faut-il la définir par rapport à la gravité d'une maladie, alors même que les thérapies évoluent ? Par ailleurs, certaines pathologies ne sont pas causées par un seul gène défectueux. Partant, la revendication du droit à un enfant sain impliquerait de demander un séquençage complet du génome. Il faut également veiller à ne pas stigmatiser des couples qui souhaiteraient ne pas savoir, ou encore les personnes atteintes de ces maladies.
Pour toutes ces questions, qui heurtent nos valeurs éthiques, nous ne disposons pas de réponses précises, rassurantes et rationnelles. Voilà pourquoi nous n'avons pas souhaité étendre le dépistage préconceptionnel à des cas non prévus actuellement.
Pour ce qui concerne la révision des lois de bioéthique, tout d'abord, il est toujours possible de les modifier sans pour autant opérer une révision globale. Le Gouvernement souhaitait initialement une révision tous les 7 ans, mais l'Assemblée nationale a préféré retenir un délai de 5 ans, et nous l'avons suivie.
Madame Blondin, la phagothérapie n'a pas sa place dans une loi de bioéthique, les phages, les virus et les bactériophages n'étant pas des produits du corps humain. Je donnerai un exemple extrême : de même qu'il n'est pas besoin de prendre une loi de bioéthique pour procéder à une amputation, on peut recourir à la phagothérapie à la seule condition que ces actes soient contrôlés ; les autorisations sont d'ailleurs données au coup par coup et non de façon générale. Ces protocoles seront inclus dans le programme prioritaire de recherche sur la résistance aux antibiotiques.
Les recherches sur les anomalies du développement génital et sur les maladies rares ne relèvent pas davantage de la loi de bioéthique.
Monsieur Daudigny, le statut particulier de l'embryon est toujours reconnu dans ce projet de loi, comme il l'est dans les conventions signées par la France, dont la plus importante est la convention d'Oviedo. Pour être autorisée, la recherche sur un embryon doit respecter quatre critères : la pertinence scientifique ; l'inscription dans une finalité médicale ; l'utilisation exclusive de matériel humain, qu'il s'agisse d'embryons ou de cellules souches embryonnaires ; le respect de règles d'éthique, lesquelles sont de niveau international.
Le respect de ces critères conditionne également la publication des études afférentes à ces recherches dans les revues internationales, puisqu'il faut produire le numéro d'agrément pour pouvoir publier.
Pour ce qui concerne la durée d'observation, il est d'ores et déjà possible d'observer des embryons au-delà de 14 jours, mais nous proposons d'inscrire dans la loi une durée maximale. Ce faisant, nous nous référons non pas à ce que la science sait faire, mais à ce qui nous paraît souhaitable en termes de délai.
M. Adrien Taquet, secrétaire d'État . - Monsieur Henno, pour ce qui concerne l'utilisation des traitements algorithmiques, la temporalité de l'information délivrée au patient par le professionnel de santé dépend des catégories d'actes visés et des dispositions qui leur sont applicables. Dans le cas de l'implantation d'un dispositif médical, par exemple un pancréas artificiel, l'information préalable est absolument nécessaire. En revanche, si un dispositif d'intelligence artificielle a été utilisé dans le cadre d'un diagnostic, l'information sera donnée postérieurement.
Mme Frédérique Vidal, ministre . - En effet, il est très difficile de savoir à l'avance si l'on aura besoin d'utiliser un dispositif d'intelligence artificielle pour poser un diagnostic.
M. Alain Milon , président . - Je vais outrepasser mon devoir de réserve de président.
Lors de l'examen de la dernière loi de bioéthique, le Gouvernement et l'Assemblée nationale s'étaient prononcés contre l'obligation de révision tous les 5 ans. C'est le Sénat qui l'a imposée. On constate que les précédentes lois de bioéthique ont été révisées tous les 7 ans, car il fallait ensuite prendre les décrets d'application.
La dernière loi de bioéthique, adoptée voilà 7 ans, est finalement révisée au bout de 9 ans. Revenir à un délai de 5 ans n'est donc pas une mauvaise idée puisque cela permet de tenir compte de connaissances ou de pratiques qui avancent plus vite que prévu.
Par ailleurs, on peut toujours réviser une loi de bioéthique pour des domaines précis. Ainsi, 2 ans après la loi de bioéthique de 2011, le Sénat avait présenté une proposition de loi relative à la recherche sur l'embryon, adoptée ici et à l'Assemblée nationale.
On reproche à la loi de bioéthique de courir un peu après les scientifiques. Peu de pays dans le monde ont une telle législation, et nombreux sont ceux qui souhaiteraient en avoir une. J'ai rencontré des représentants du Sénat jordanien, qui nous disent vouloir suivre notre exemple.
Comme l'a expliqué M. le secrétaire d'État, la loi de bioéthique sert à constater les avancées scientifiques et à les borner pour éviter les initiatives de chercheurs « fous ». En outre, à chaque fois que la science avance, nous avons le devoir de regarder si ces progrès sont utiles et d'éviter ceux qui sont dangereux. Ces lois de bioéthique sont donc plus que nécessaires, tout en étant révisables.
Je vais être provocateur. Dans ma vie professionnelle, je n'ai jamais rencontré de personnes qui, exerçant leur droit à l'enfant, n'aient pas respecté le droit de l'enfant. Si l'on veut supprimer le droit à l'enfant, il faut revenir sur l'adoption, l'IVG et la pilule !
Mme Muriel Jourda , rapporteur . - Monsieur le secrétaire d'État, la commission ad hoc pour l'accès aux origines serait chargée de récolter auprès de l'Agence de la biomédecine des données identifiantes et non identifiantes pour les transmettre, ce qui est d'ordre purement administratif. Elle sera aussi chargée d'accompagner les enfants issus du don et les donneurs : comment se fera cet accompagnement ?
Que faire en cas de pénurie de gamètes liée, en cas d'extension de la PMA, à l'accroissement de la demande et, en cas de levée de l'anonymat, à la baisse du stock de gamètes, les deux phénomènes produisant un effet ciseau ? Peut-on songer à apporter des gamètes et comment s'assurer alors de l'accès aux origines ?
Mme Élisabeth Doineau . - La PMA post mortem n'a pas été évoquée à l'Assemblée nationale. En cas de décès du conjoint, la dynamique du projet de PMA du couple est rompue. Au vu de ces situations difficiles, il faudrait donner un cadre à la PMA post mortem. Une loi de bioéthique doit aussi servir à cela.
M. Adrien Taquet, secrétaire d'État . - S'agissant de la commission ad hoc chargée de l'accès aux origines, un décret en Conseil d'État précisera quels professionnels pourront être sollicités pour accompagner les enfants dans cette démarche. Intuitivement, on pense à des psychologues, des pédopsychiatres ou des assistants sociaux.
Mme Muriel Jourda , rapporteur . - Vous dites qu'il y aurait une appréciation de l'opportunité d'accéder à ses origines ?
M. Adrien Taquet, secrétaire d'État . - Non, vous avez raison, c'est un droit.
Sauf erreur, nous sommes le seul pays à proposer de façon concomitante l'ouverture de la PMA à toutes les femmes et la possibilité d'accéder à ses origines. Nous n'avons donc pas d'éléments de comparaison avec d'autres pays. Or nous sommes déjà en flux tendus sur les spermatozoïdes, et en pénurie d'ovocytes. Nous prenons donc très au sérieux le risque de pénurie de gamètes. Pour autant, nous n'allons pas en importer. Nous voulons renforcer les campagnes de communication pour recruter de nouveaux donneurs. L'Agence de la biomédecine s'y prépare. Ces campagnes sont assez confidentielles, mais incitent à faire preuve de solidarité et à donner. La nature des donneurs évoluera peut-être, avec la possibilité d'accéder aux origines après dix-huit ans. On l'a constaté dans les pays qui ont levé l'accès aux origines, après une baisse dans un premier temps, le nombre de donneurs y remonte, puis se stabilise, mais les motivations ont légèrement évolué.
Mme Frédérique Vidal, ministre . - Le moment où seront pris les décrets garantit qu'il n'y aura pas de destruction de gamètes. Quand on voit le très faible pourcentage de donneurs de gamètes, et le faible pourcentage de personnes sensibilisées à la possibilité pour elles de faire un don de gamètes, on comprend qu'il y a beaucoup à faire.
La PMA post mortem a suscité beaucoup de discussions, car elle soulève des questions abyssales. Nous ne l'avons finalement pas autorisée. D'abord, il y a la question du temps du deuil. Aussitôt après le décès, on se dit que c'est le rêve le plus cher que de faire naître un enfant. Mais, après un ou deux ans, le temps passant, la vie reprenant ses droits, est-on toujours dans la même envie ? Nous pourrions autoriser la PMA post mortem après une période de deuil. Mais comment estimer la durée d'un deuil ?
Puis, le projet parental était porté par le couple. Il ne reste que la femme. Peut-elle continuer à porter seule un projet parental conçu à deux ? Il faudrait aussi demander à l'autre parent s'il serait d'accord, au cas où il décède, pour qu'un enfant naisse après sa mort... Et il y a les pressions potentielles des familles, au moment du deuil, notamment de la famille du défunt. Comment, enfin, gérer les successions ?
En réalité, le nombre de demandes formulées auprès de la justice est extrêmement faible. Ce n'est donc pas la peine d'ouvrir la possibilité de généraliser la PMA post mortem, surtout au regard de la lourdeur du processus, pas seulement techniquement. On prononce rapidement les trois lettres P-M-A, mais c'est un projet sur le temps long !
M. Alain Milon , président . - Nous en rediscuterons en séance.
Ce point de l'ordre du jour a fait l'objet d'une captation vidéo qui est disponible en ligne sur le site du Sénat .
MM. Alexandre Urwicz, président,
et Fabien Joly, porte-parole
de l'Association des familles homoparentales (ADFH), Mme Marie-Claude
Picardat
et M. Dominique Boren, porte-parole de l'Association des
parents
et futurs parents gays et lesbiens (APGL), Mmes Catherine
Michaud, présidente de l'association GayLib, Laurène Chesnel,
déléguée Familles de l'Inter-LGBT et Véronique
Cerasoli,
administratrice et porte-parole de l'association SOS homophobie
M. Alain Milon , président . - Nous clôturons aujourd'hui nos auditions sur le projet de loi relatif à la bioéthique avec cette table ronde d'associations. Cette audition fait l'objet d'une captation en vidéo en vue de sa retransmission sur le site Internet du Sénat où elle sera ensuite disponible à la demande.
Nous accueillons MM. Alexandre Urwicz, président, et Fabien Joly, porte-parole de l'Association des familles homoparentales (ADFH), Mme Marie-Claude Picardat et M. Dominique Boren, porte-parole de l'Association des parents et futurs parents gays et lesbiens (APGL), Mme Catherine Michaud, présidente de l'association GayLib, Mme Laurène Chesnel, déléguée Familles de l'Inter-LGBT et Mme Véronique Cerasoli, administratrice et porte-parole de l'association SOS homophobie.
M. Alexandre Urwicz, président de l'Association des familles homoparentales (ADFH) . - Merci de recevoir l'ADFH. Nous vous avons adressé une note de 41 pages, et nous nous concentrerons ce matin sur quelques points importants.
Nous sommes extrêmement favorables à l'ouverture de la procréation médicalement assistée (PMA) à toutes les femmes, notamment faisant partie d'un couple de même sexe ou célibataires.
Nous sommes favorables à la disposition adoptée par l'Assemblée nationale établissant la reconnaissance conjointe anticipée (RCA), qui permet de respecter à la fois la solidarité des femmes en couple lorsqu'elles s'engagent dans une PMA, et de garantir que la filiation sera ancrée à la naissance, établie à la fois par l'accouchement de la mère, déclaré à l'état civil, et la cosignature de la conjointe lors de la RCA. Il est important de maintenir cette mesure.
Nous souhaiterions qu'elle soit étendue à tous les enfants nés par PMA avec don de gamètes. En l'état actuel du droit, il y a une rupture d'égalité entre les enfants conçus par don de gamètes avec des parents de même sexe et des enfants issus de parents hétérosexuels, au sens du dispositif du titre VII du code civil, se fondant sur un rapport pseudo-charnel des parents. Aucune information ne permet alors à l'enfant de savoir que son père n'est pas son géniteur.
En 1950, une circulaire demandait aux officiers d'état civil de ne pas envoyer la mention du jugement des enfants adoptés ; on faisait croire à l'enfant que ses parents étaient ses géniteurs. En 1966, l'État a refusé de continuer à perpétrer ce mensonge : les enfants avaient grandi et demandaient de la transparence sur leurs parents biologiques. On a alors demandé alors aux officiers d'état civil d'envoyer l'acte intégral avec toutes les mentions.
Aujourd'hui, cette situation est transposable aux enfants conçus par don de gamètes. Il est de l'intérêt supérieur de l'enfant de connaître son histoire personnelle et cet intérêt prévaut sur celui des parents de disposer d'un acte établissant une filiation pseudo-charnelle. Étendez les dispositions de l'article 4 à tous les enfants issus de dons, que ce soient des enfants de parents homosexuels, hétérosexuels, mariés ou non. C'est le sens de toutes les conventions internationales et de la Cour européenne des droits de l'homme (CEDH).
M. Fabien Joly, porte-parole de l'Association des familles homoparentales (ADFH) . - Le projet de loi adopté à l'Assemblée nationale, au-delà de limiter la RCA aux seuls couples de femmes, envisage mal deux autres sujets essentiels pour l'intérêt supérieur des enfants.
Les enfants nés avant l'entrée en vigueur de
la loi ont comme seule possibilité pour leur filiation la voie de
l'adoption. Mais même si le Gouvernement envisage de supprimer la
condition maritale préalable, certains enfants ne pourront être
adoptés si les parents sont séparés. La mère
ayant accouché pourra refuser que la mère sociale adopte.
Celle-ci n'aura aucun moyen juridique de faire établir une filiation
même si elle a participé au projet parental et à
l'éducation de l'enfant. C'est dramatique tant pour la mère que
pour l'enfant. Nous voulons que la filiation soit établie par la
possession d'état, jusqu'à dix ans après la
séparation avec la mère juridique, devant le notaire ou le
tribunal de grande instance
- futur tribunal judiciaire - pour
maintenir le lien avec l'enfant. C'est un manque du projet de loi.
Il est également impensable que les enfants nés par PMA avant la loi ne puissent bénéficier, avec l'accord du donneur, d'un accès à leurs origines, soit via des données non identifiantes, soit via l'identité du donneur. Nous souhaitons que cela ne repose pas sur le volontariat des donneurs, mais que la commission d'accès aux origines sollicite les donneurs pour savoir s'ils autorisent que des données identifiantes soient communiquées aux enfants le demandant. C'est une position équilibrée, entre le respect de l'anonymat et le droit des enfants à connaître leur histoire personnelle.
Mme Catherine Michaud, présidente de l'association GayLib . - GayLib est un mouvement LGBT associé aux radicaux, une association à caractère politique. Mon propos le sera également. Avec l'APGL, le Planning familial, SOS homophobie et Inter-LGBT, nous avons créé cet été le Collectif PMA.
Le projet de loi Bioéthique est attendu depuis très - trop ! - longtemps. Nous l'attendions de la précédente majorité, c'était une promesse non tenue du candidat Hollande. Cela aurait pu nous éviter d'avoir à nous marier et à adopter nos propres enfants pour être reconnues pleinement comme parents.
Nous n'allons pas refaire le match six ans plus tard, et force est de constater que ce texte est une avancée, même s'il n'est pas parfait.
Le moment venu, dans l'hémicycle, nous aurons besoin de tous les progressistes, quel que soit leur bord politique. Le Sénat est majoritairement à droite. Ne commettez pas les erreurs du passé sur le pacte civil de solidarité (Pacs), le mariage et l'adoption, souvent plus par posture que par conviction, peut-être même parfois par peur de ce que pourrait penser votre électorat en circonscription. Or, sur la PMA comme sur le mariage, sondage après sondage, les Français sont prêts depuis bien longtemps, bien avant le législateur.
Nos familles, nos conjoints et nos enfants sont une réalité intégrée à la société et à travers toute la France. Le schéma familial est divers, avec, à la fois, des familles traditionnelles, des familles recomposées, monoparentales, mais aussi homoparentales. Nous nous réjouissons de voir la PMA pour les femmes seules et en couple enfin à l'agenda parlementaire. C'est une liberté nouvelle, ouverte à toutes, de pouvoir faire un enfant, de disposer de son corps pour concevoir un enfant et de faire famille, quel que soit son régime matrimonial ou son orientation sexuelle. Cela restera dans l'histoire comme une grande avancée pour les femmes comme le droit de vote ou le droit à l'avortement.
Parmi les points positifs, outre l'aspect historique attendu de ce texte, il y a l'ouverture de la PMA aux femmes célibataires, marque importante de la confiance faite aux femmes dans un projet parental mûrement réfléchi. Cependant, nous n'acceptons pas les dispositions de filiation spécifique pour les couples de femmes. Nous ne pouvons pas comprendre que les lois de la République mettent en place un statut à part pour une catégorie de la population et ne protègent pas de la même manière tous ces enfants. Nous souhaitons que le droit commun soit élargi soit par reconnaissance, soit par présomption.
Par ailleurs, ce texte écarte les personnes transsexuelles de l'accès à la PMA, il refuse la PMA post mortem, ce qui est incohérent : une femme ne pourrait pas avoir un projet parental avec les gamètes de son défunt conjoint, mais aurait accès à la PMA avec un tiers donneur en tant que femme célibataire. Il manque aussi la promesse de campagne de reconnaître les enfants nés de GPA légalement à l'étranger.
Nos enfants doivent être protégés par la République comme tous les autres enfants. Ce projet de loi est une opportunité de mettre en exergue la valeur famille sous le triptyque de la République - Liberté, Égalité, Fraternité. Il y a la liberté de construire une famille, l'égalité enfin réelle, et la fraternité sans laquelle les deux autres ne seraient rien. J'ajouterais aussi le quatrième pilier de la République, la laïcité, chère aux radicaux. Les représentants des cultes ont été auditionnés, mais, depuis plus d'un siècle, le religieux ne doit pas influencer le législateur.
Ce texte va dans le sens de l'histoire. Il élargit quelque chose qui est déjà partiellement autorisé, légal, ouvert aux couples hétérosexuels. Tous les couples ne souhaitent pas forcément se marier, toutes les femmes ne souhaitent pas forcément avoir des enfants, donc il en est de même pour la PMA. Nous parlons ici de la liberté de pouvoir faire quelque chose par choix et non pas par contrainte de l'interdiction.
Le rôle du politique n'est pas de juger la société, mais de savoir l'observer pour mieux l'accompagner.
Mme Véronique Cerasoli, administratrice et porte-parole de l'association SOS homophobie . - Merci de nous avoir invités à cette table ronde. SOS homophobie lutte depuis vingt-cinq ans contre les discriminations envers les personnes LGBT, avec trois axes : le soutien aux victimes, la sensibilisation aux discriminations, notamment en milieu scolaire, et la défense de l'égalité des droits des personnes LGBT. Mon éclairage portera principalement sur le terrain juridique de construction de la loi et des techniques médicales.
Nous sommes un peu embarrassés de voir se focaliser l'attention des parlementaires, des médias et du public sur les quelques articles qui nous concernent, plutôt que sur la trentaine d'autres articles autrement plus engageants sur l'avenir et la bioéthique. Dont acte.
Je centrerai mon propos sur l'article 1 er , qui élargit l'accès à de nouveaux publics et l'article 4, qui en tire les conséquences en termes de filiation.
À l'article 1 er , nous sommes satisfaits de voir disparaître une discrimination fondée sur l'orientation sexuelle et le statut marital des femmes. Nous l'attendions depuis vingt-cinq ans. Il ne s'agit ni de développer une nouvelle technique, ni de créer de nouvelles situations, mais bien de mettre fin à une inégalité de traitement entre les personnes dans notre pays. Il s'agit aussi de protéger les enfants et les familles ayant eu recours à l'assistance médicale à la procréation (AMP). Cette inégalité a été créée ex nihilo par le législateur en 1994, qui a fait le choix - que d'autres pays n'ont pas fait - d'inclure la PMA dans une loi globale de bioéthique et d'en réserver l'accès aux seuls couples hétérosexuels. Cela exclut de facto les femmes seules et en couple avec une femme qui, jusque-là, pouvaient être accompagnées par leurs médecins et gynécologues dans un cadre médical sécurisé.
Cependant, une partie de nos concitoyens sont à nouveau discriminés par les choix actuels : les hommes transsexuels qui seraient en capacité de porter un enfant sont exclus de l'article 1 er . Or, nous considérons que les droits reproductifs appartiennent à la personne. Le sexe établi à l'état civil ne doit pas être une source d'empêchement à l'AMP. La loi qui, depuis 2016, permet un changement d'état civil sans stérilisation forcée obligerait à présent les hommes transsexuels à choisir entre leur changement d'état civil et la possibilité de porter un enfant. Au regard des débats à l'Assemblée nationale, nous avons malheureusement peu d'illusions sur le succès de voir notre demande d'inclusivité de toute personne en capacité de porter un enfant inclus à l'article 1 er .
Depuis 1994, des milliers de femmes ont été empêchées par la loi de bénéficier de l'accompagnement de leur médecin parce que célibataires ou lesbiennes. Par conséquent, elles ont adopté des stratégies de contournement, à savoir des PMA artisanales et des PMA à l'étranger, avec son lot d'insécurités et violences sanitaires - aléas de provenance et de stérilisation du sperme, traitements lourds et inutiles imposés par les cliniques étrangères - et les inégalités sociales et économiques liées au coût de l'AMP - déplacements, nuitées sur place, absences au travail...
Enfin, il y a une précarité juridique de la famille, une fois l'enfant né. Les premières victimes sont les femmes et leurs enfants, les compagnes, le reste de la famille. Jusqu'à l'établissement de la double filiation, plusieurs mois après la naissance de l'enfant dans le meilleur des cas, celui-ci n'a aucune existence légale.
Le Rapport annuel sur l'homophobie en France, publié par notre association, établit une corrélation entre les discriminations dans la loi et les comportements discriminatoires tolérés dans la société, jusqu'aux violences physiques et psychologiques qui touchent les personnes LGBT. En 2018, les violences contre les lesbiennes signalées à SOS homophobie ont augmenté de plus de 40 % en un an ; 365 actes nous ont été signalés, soit un par jour !
- Présidence de Mme Élisabeth Doineau, vice-présidente -
Mme Véronique Cerasoli . - Il est donc important de faire évoluer la loi afin que personne ne soit discriminé. Je tiens également à alerter sur la situation des personnes transsexuelles et intersexes dans notre pays.
S'agissant de l'article 4 du projet de loi, nous ne nous satisfaisons pas de l'option proposée par le Gouvernement et retenue par les députés qui consiste en l'établissement d'une filiation spécifique pour les couples de femmes, la reconnaissance conjointe anticipée : nous sommes en faveur de l'extension du droit commun.
La loi a pour ambition de mettre fin à une discrimination concernant les femmes lesbiennes, mais, dans le même temps, elle crée une nouvelle discrimination : la loi prend d'une main ce qu'elle donne de l'autre. Cette loi doit assurer la protection de tous les enfants et toutes les familles sans discrimination, sans hiérarchisation, sans création de documents ad hoc pour certains enfants en raison de leur mode de conception ou de l'orientation sexuelle de leurs parents. Ce choix d'une solution dérogatoire est d'autant moins compréhensible que tout existe déjà dans le droit : depuis 1994, il existe un système de filiation pour les couples hétérosexuels qui ont recours à une PMA avec donneur ; la double filiation maternelle existe ; la filiation d'un enfant avec un parent non géniteur existe.
Avec la reconnaissance conjointe anticipée, l'établissement de la filiation sera différent pour la femme qui accouche selon qu'elle est en couple avec un homme ou une femme, différent aussi pour le second parent non géniteur selon qu'il est un homme ou une femme, et l'acte de naissance de l'enfant sera différent selon que ses parents ont eu recours, à une PMA avec donneur ou pas et selon l'orientation sexuelle de ses parents. À même conception, même filiation : la femme qui accouche doit être mère par son accouchement, le second parent doit l'être par son consentement au don et présomption s'il est marié et par consentement au don et reconnaissance s'il n'est pas marié, qu'il soit homme ou femme. Sinon, cela revient à inscrire dans la loi une discrimination basée sur l'orientation sexuelle des personnes !
Pourtant l'extension du droit commun serait simple et n'enlèverait rien à personne. Alors pourquoi ce blocage ? En raison de l'hétéronormativité de notre droit, qui a été écrit par et pour les hommes.
Depuis des siècles, notre droit de la famille a été fondé sur le mariage, entre une femme et un homme jusqu'en 2013, et sur la volonté de contrôler le corps des femmes et la procréation. Le mariage pour tous n'est pas un mariage pour toutes, car le mariage entre deux femmes n'emporte pas les mêmes effets en termes de filiation qu'un mariage entre un homme et une femme. L'histoire, l'anthropologie, les sciences sociales montrent que les systèmes de parenté et donc de filiation sont soumis à des normes culturelles changeantes et aux évolutions politiques, économiques, sociales, religieuses et même biotechniques.
Reconnaissance ? Non, car il y a rupture d'égalité en droit des effets du mariage. Conjointe ? Non, car il y a rupture d'égalité entre les femmes qui accouchent. Anticipée ? Non, car il y a rupture d'égalité d'établissement de filiation entre des enfants selon que leur mère est en couple avec une femme ou pas.
Il n'existe pas de modèle universel ni unique de la famille, mais il n'y a qu'un seul type d'égalité et il n'y a pas d'égalité partielle : l'égalité est ou n'est pas.
Mme Laurène Chesnel, déléguée Familles de l'Inter-LGBT . - Je suis maman de deux petites filles conçues par PMA à l'hôpital universitaire de Bruxelles.
L'Inter-LGBT regroupe 70 associations de tous les domaines de la vie LGBT et plus largement des droits humains. Nous sommes organisateurs, chaque année, de la marche des fiertés de Paris et du Printemps des associations. Je regrette que l'association Les enfants d'Arc en ciel, qui accompagne chaque année des centaines de couples de femmes en PMA, n'ait pas été autorisée à venir à cette audition, cela aurait pourtant été très enrichissant.
Le projet de loi comporte de nombreux points positifs. Tout d'abord, l'ouverture de l'AMP aux couples de femmes, qui met fin à une situation cynique. Cette situation était financièrement inégale - une insémination coûte 1 000 euros et une FIV jusqu'à 8 000 euros par tentative. Elle engendre du stress, des problèmes de santé - le lien entre médecins, français et étranger, se fait difficilement -, mais aussi des difficultés dans le milieu professionnel en raison d'absences difficiles à justifier. Je me félicite de la clause de non-discrimination introduite par l'Assemblée nationale.
Les travaux de Susan Golombok sur le bien-être des enfants nés dans des couples de femmes ou chez des femmes célibataires montrent que nos enfants vont tout aussi bien que les autres.
L'ouverture de la PMA aux femmes célibataires est aussi une avancée. Celles-ci sont dans des situations très différentes ; leur profil socio-économique est aussi très différent ; elles sont très entourées et soutenues par leur famille et elles peuvent rencontrer un père par la suite. En effet, la fenêtre pour fonder une famille est étroite : le premier contrat à durée indéterminée est décroché vers 29 ans et, dès 35-38 ans, la fertilité diminue.
La question du remboursement est très importante. C'est conforme au principe de solidarité de notre système de santé et, s'il n'avait pas été prévu, cela aurait constitué une nouvelle discrimination. Le double don et l'accès aux origines constituent aussi des avancées. Mais le projet de loi souffre aussi de plusieurs manques.
Tout d'abord, la situation des veuves nous semble cruelle : elles ne pourront faire de PMA qu'avec un tiers donneur et non avec les gamètes de leur conjoint décédé. Les personnes transsexuelles et intersexes n'ont pas accès à l'autoconservation des gamètes même si elles suivent un traitement stérilisant, alors que la loi le leur permet en principe.
Nous attendons aussi des avancées sur les pratiques médicales, comme la méthode ROPA (réception d'ovocytes de la partenaire). À l'Assemblée nationale, il y a eu une confusion avec la notion de don. Il ne s'agit pas d'un don, puisque la génitrice deviendra bien mère. Dans un contexte de pénurie d'ovocytes, il est ridicule de faire attendre une personne sur une liste d'attente de plus de trois ans, alors qu'il y a des gamètes disponibles dans le couple.
Le dépistage des aneuploïdies est aujourd'hui proposé à la femme au 3 e mois de grossesse depuis 2009, mais il faudrait le proposer dès le stade de l'embryon en cas de FIV, car 40 % des FIV se soldent par un échec - embryons non viables ou interruption médicale de grossesse (IMG). Les femmes qui ne souhaiteraient pas faire ce dépistage ne le feraient pas. Cela a été présenté comme une grande nouveauté, mais cela ne l'est pas. Ce sera plus confortable pour les femmes.
Sur les donneurs et donneuses rares en raison de leurs caractéristiques ethniques, le registre national devrait être consultable par les médecins opérant des AMP afin de faire venir des gamètes d'un autre centre. Mais il faut aussi offrir la possibilité aux couples qui le souhaitent de refuser tout appariement basé sur leurs caractéristiques physiques.
Nous sommes en faveur d'une filiation de droit commun, identique à celles des couples hétérosexuels non mariés, avec la reconnaissance et la possibilité de déclarer judiciairement la parentalité. Il suffirait d'apporter à l'officier d'état civil le consentement devant notaire.
Les enfants des couples séparés avant l'adoption de l'enfant du conjoint sont actuellement dans une situation dramatique, sans aucune possibilité d'établissement de leur filiation. Notre piste préférée serait l'ouverture de la possession d'État. La voie belge serait également envisageable, avec l'adoption de l'enfant de l'ex-conjoint, ex-partenaire ou ex-concubin ; si la femme qui a l'autorité parentale refuse, le juge tranchera.
Nous n'avons pas de position particulière sur la gestation pour autrui (GPA) à l'étranger, sauf s'agissant de la régularisation de l'état civil des enfants. La Cour de cassation a modifié hier sa jurisprudence et permis la transcription des deux parents : peut-être faudrait-il l'entériner dans la loi ?
Nous avons aussi quelques autres préoccupations : le traitement algorithmique des données des personnes sans le consentement des personnes prévu à l'article 11 ; le statut de l'embryon à l'article 14 ; la nouvelle clause de conscience sur l'IMG à l'article 21 au lieu de renvoyer à la clause de conscience existante sur l'IVG ; la question des mineurs intersexes qui a connu une avancée notable à l'Assemblée nationale, mais il faudrait que la France se conforme à ses propres engagements et interdise les opérations non consenties par les enfants concernés.
En conclusion, je voudrais partager avec vous le témoignage d'une jeune femme, née en 1995 et membre de l'association Les enfants d'Arc en ciel : « Ai-je souffert de la situation de mes parents dans ma vie ? Non, j'ai une certitude que tout le monde n'a pas la chance d'avoir : je sais que mes parents m'ont ardemment désirée et que j'existe parce qu'elles se sont battues pour ça, cela a donné un premier sens à ma vie. Je n'ai pas souffert de l'homosexualité de mes parents, j'ai souffert de l'homophobie de notre société. La seconde question qu'on me pose : ai-je manqué d'un père ? La réponse est encore non. J'ai eu l'amour de deux femmes, j'ai eu des hommes dans mon entourage - mon parrain, mon grand-père, mes oncles, les pères de mes amis. Mais qu'est-ce qu'un père apporte de si spécifique qu'une mère ne peut apporter ? L'autorité ? Le goût pour le sport ? L'idée selon laquelle le père est indispensable sous-entend que l'homme est fondamentalement différent de la femme, au point qu'il apporterait par sa seule masculinité quelque chose de distinct. Mon expérience démontre que cette idée est fausse. Je vous demande, mesdames et messieurs les sénateurs de voter cet article du projet de loi : vivre dans un État qui ne reconnaît pas ma famille est une souffrance et voir de jeunes homosexuelles de mon âge douter de leurs capacités à être de bonnes mères à cause de l'homophobie ambiante est insupportable. Nos familles existent déjà par milliers. »
Mme Élisabeth Doineau , présidente . - Nous avons demandé à l'association Les enfants d'Arc en ciel, qui est membre de votre fédération, de se rapprocher de vous afin que vous puissiez porter leur voix. On ne peut donc pas dire que cette association n'a pas été autorisée à s'exprimer.
M. Dominique Boren, porte-parole de l'Association des parents et futurs parents gays et lesbiens (APGL) . - Je vous remercie de nous accueillir et de nous auditionner. C'est un plaisir de travailler avec l'association Les enfants d'Arc en ciel. C'est une voix importante à entendre, indépendamment de leur fédération.
Qui est l'APGL ?
L'APGL, créée en 1986, la plus ancienne et la plus large des associations homoparentales françaises, est à l'origine du terme « homoparentalité ». Son action est reconnue dans les domaines de l'homoparentalité et de la coparentalité.
Nous intervenons dans le champ associatif et social pour la reconnaissance et l'accompagnement des familles et des futures familles, mais également dans le champ du politique en défendant trois principes : toute personne LGBT a le droit de fonder une famille en France ; tous les parents doivent avoir les mêmes droits en termes de filiation, quels que soient leur statut - parents légaux ou sociaux - et leur contribution au projet parental ; les enfants des familles homoparentales doivent avoir le même lien sécurisé de filiation avec chacun de ses parents.
L'APGL a mené des combats contre les discriminations. Depuis la loi Taubira, elle intervient également dans le champ politique de manière plus institutionnalisée. Sa force repose sur ses adhérents, dont elle transmet les demandes aux pouvoirs publics. Grâce à sa mixité et à sa diversité, elle fait remonter des sujets qui concernent tous les types de familles.
Elle a donc toute légitimité pour demander une PMA encore plus égalitaire qu'elle ne l'est à l'issue de la première lecture du présent texte.
Nous organisons des débats ; nous avons aussi soutenu le film Mon enfant ma bataille, qui explique les parcours des familles ayant un projet de PMA. Nous sommes présents dans différentes instances de la République, notamment au sein de l'Union nationale des associations familiales (UNAF), du Haut Conseil de la famille, de l'enfance et de l'âge (HCFEA), entre autres, ce qui nous permet d'avoir une vue d'ensemble de ces questions. D'ailleurs, s'agissant de l'exclusion du droit commun de la filiation pour les seuls couples de femmes qui accéderaient à la PMA, nos interlocuteurs au sein de l'UNAF et du HCFEA ont été aussi surpris que nous par le vote des députés.
Depuis toujours, l'APGL défend le principe d'une inclusion des questions de famille et de parentalité dans les textes visant à accorder une égalité de droits aux personnes LGBT.
Malheureusement, nous avons dû faire face à une tendance de fond : les droits conférés aux personnes LGBT demeurent spécifiques, accordés « par la porte arrière » pour accéder au « salon » de l'égalité. Le Pacs nous avait ainsi été présenté comme une mesure d'égalité. Or, en 1999, les personnes hétérosexuelles avaient la possibilité de se marier, pas les homosexuelles. Par ailleurs, le Pacs ne couvrait pas le champ de la filiation. Les jurisprudences et les mentalités ont fait évoluer ce système, mais, au départ, celui-ci n'était pas satisfaisant en termes d'égalité des droits. Je crains que le présent projet de loi n'aboutisse au même résultat. Nous souhaitons cependant qu'il prévoie la plus large égalité possible.
Mme Marie-Claude Picardat. - En effet, nous souhaitons que la présente loi reflète l'esprit de notre République, c'est - à - dire qu'elle nous donne la liberté, l'égalité et qu'elle repose sur des principes de fraternité. Malheureusement, nous ne sommes pas tout à fait engagés dans cette direction. Or, quand la loi n'est pas tout à fait égalitaire, mais crée des sous-catégories de citoyens, elle suscite des souffrances. Nous l'avions expérimenté avec le Pacs, qui n'était pas un dispositif suffisant puisque les homosexuels étaient privés du mariage et de la filiation. Quant au mariage pour tous, s'il est égalitaire du point de vue de la conjugalité, il ne l'est pas sur le plan de la filiation.
Nous avions averti le législateur, à l'Assemblée nationale comme au Sénat, qu'une loi insuffisante pour établir la filiation dans le cadre du mariage entraînerait des souffrances. Ainsi, les premières familles à s'être présentées devant les tribunaux n'ont pas été traitées de manière égalitaire.
Nous sommes certes satisfaits que la PMA et le principe de son ouverture à toutes les femmes - seules ou en couple -, aient été adoptés. Nous apprécions aussi que le sujet de la filiation ait été pris en compte, y compris pour les homosexuels en dehors du mariage, ce qui a mis fin à une rupture d'égalité. Nous nous réjouissons que la sécurité sociale prenne en charge la PMA pour toutes les femmes, même si nous avons des questions à poser sur les modalités de cette prise en charge ; peut-être le législateur pourra-t-il nous éclairer et nous rassurer à cet égard ?
Nous approuvons, enfin, la possibilité qui est donnée d'accéder à des éléments de connaissance relatifs aux donneurs. Pour ma part, je récuse le terme « origines », parce que les origines d'un enfant ne se trouvent pas dans un matériel génétique, mais bien plutôt dans son histoire, qui lui est racontée par ses parents. Même si sa vie découle de ce capital génétique, celui-ci ne résume ni son histoire ni ses origines.
Nous savons que certains enfants demandent l'accès à ces éléments de connaissance, et il serait dommage de leur barrer la route. Le simple fait de donner cette possibilité suffit souvent à apaiser leur curiosité, et ils n'ont pas forcément besoin d'aller au-delà, comme nous l'apprennent certains retours d'expérience venus de pays étrangers.
Nous sommes dépités et étonnés, en revanche, que le législateur ait fait en sorte que nos revendications aient des conséquences négatives pour les couples hétérosexuels ayant recours à la PMA : ils « sortent du cadre » du présent texte, alors qu'ils n'avaient rien demandé et qu'ils étaient à l'abri de la loi française depuis des décennies. Nous avons en effet perçu comme une menace le fait d'inscrire le mode de conception sur l'état civil des enfants. Nous avons toujours été opposés à ce principe, qui n'a rien d'égalitaire selon nous. Par ailleurs, depuis sa mise en place, le dispositif de PMA pour les personnes hétérosexuelles a toujours bien fonctionné et n'a jamais donné lieu à contestations, notamment en termes de filiation. Pourquoi donc sortir ces personnes du droit commun ?
Notre revendication est républicaine, elle va dans le sens de l'égalité. Nous voulons pouvoir bénéficier, dans la plus large mesure, et si possible dans sa totalité, du droit commun de la filiation.
De manière inattendue, est imposé avec ce texte un mode d'établissement de la filiation qui serait réservé aux femmes homosexuelles et viendrait accoler les deux mères dans un même mouvement d'établissement de la filiation, comme si chacune ne pouvait pas l'établir indépendamment de l'autre. Au nom d'une prétendue avancée des droits, ce système prive la mère qui va accoucher de la possibilité de faire établir le lien de filiation avec son enfant par l'accouchement, comme peut le faire toute autre femme. Nous y sommes absolument opposés !
L'engagement dans la PMA n'est pas du tout équivalent à l'établissement de la filiation. Si la PMA engage les deux femmes de manière équivalente, leurs droits ne sont pas exactement les mêmes puisqu'elles n'établiront pas la filiation de façon identique. Nous avons des modèles à l'étranger, mais aussi en France : les femmes hétérosexuelles qui recourent à la PMA peuvent établir leur filiation par l'accouchement ; et pour les pères hétérosexuels qui n'ont pas de lien biologique avec l'enfant, l'établissement de la filiation est garanti par leur acceptation du principe de la PMA. Nous souhaitons bénéficier des mêmes droits.
Lorsque l'on crée des « petits » droits spécifiques, on est toujours obligé d'y revenir, mais entretemps sont survenus des souffrances et des dégâts dans les familles, sur le dos de nos enfants. C'est une façon pour le législateur d'introduire dans la loi des discriminations. En effet, en quoi la sexualité de la mère doit-elle induire une privation de droits qui sont ouverts aux autres femmes ?
En créant autant de droits et de régimes qu'il y a de situations spécifiques, on met les personnes dans des tiroirs et des catégories, et tout le monde s'y perd.
Le plus simple est d'ouvrir à tous les portes de notre grande maison républicaine, sans discrimination. Les personnes homosexuelles sont des citoyens à part entière et participent à l'effort national en travaillant, en faisant des enfants, en cotisant à la sécurité sociale, mais elles ne bénéficient pas pour le moment d'une entière égalité de droits. La deuxième mère, par exemple, doit pouvoir établir la filiation de manière simple, par un processus de reconnaissance reposant sur le fait qu'elle s'est engagée dans un processus de PMA, ce qui lui ouvre des droits et des devoirs. Nous ne voulons pas d'un droit qui ouvre d'autres formes d'exclusion.
Mme Muriel Jourda , rapporteur . -Vous avez tous évoqué la notion d'égalité, et son pendant qu'est la discrimination. Des ministres, ainsi que plusieurs juridictions, ont précisé que les femmes seules et les femmes homosexuelles n'étant pas dans la même situation que les couples hétérosexuels face à la procréation, le fait de les traiter différemment ne constituait pas une discrimination. Qu'en pensez-vous ?
Vous souhaitez bénéficier du droit commun en matière de PMA, c'est-à-dire celui dont relèvent les couples hétérosexuels. Dans ces couples, la femme devient la mère du fait de l'accouchement, et l'homme devient père par le biais de la présomption de paternité. Comment transcrire cette situation pour un couple homosexuel ?
Mme Laurène Chesnel . - La jurisprudence que vous évoquez est en train de changer. On a longtemps considéré que les couples hétérosexuels et les couples homosexuels étaient dans des situations différentes, ce qui justifiait de ne pas accorder le mariage à ces derniers. Cette conception a évolué. Il se passe la même chose pour les familles homoparentales : dans le cas du don, un parent a un lien biologique avec l'enfant, quand l'autre n'en a pas. Le sexe de ces parents constitue-t-il une différence de situation par rapport à des parents hétérosexuels ayant également recours au don ? Nous considérons que non, et le droit évolue dans ce sens : cette différence de situation n'est pas suffisante pour justifier des droits différents.
Mme Véronique Cerasoli . - Nous considérons quant à nous qu'il y a une similarité de situation face à la conception. Dans un couple ayant recours à la PMA avec donneur, la mère établit sa filiation par l'accouchement ; le second parent, qui n'est pas génétiquement lié à cet enfant, doit bénéficier, qu'il soit homme ou femme, d'une égalité de droits en termes d'établissement de la filiation.
Par ailleurs, dans le cadre d'une PMA, le mari de la femme qui accouche est présumé père. Deux femmes mariées doivent bénéficier d'une égalité de droits, la présomption de parentalité découlant du mariage avec la femme qui accouche. Pour les couples non mariés, elle doit découler d'une reconnaissance de filiation.
M. Alexandre Urwicz . - J'ai compris, madame le rapporteur, que vous faisiez référence à l'avis du Conseil d'État, dans lequel il est clairement indiqué qu'il n'est pas nécessaire d'ouvrir la PMA à toutes les femmes, et que cela ne constitue ni une atteinte à la liberté ni une discrimination.
L'égalité des droits, c'est un choix politique, un choix républicain. Si le mariage pour tous existe, c'est bien parce que le législateur a pris l'initiative de s'emparer du sujet et a fait voter la loi. Nous n'avons pas obtenu ce droit grâce à une décision de la Cour de cassation ou de la Cour européenne des droits de l'homme !
Selon nous, l'extension du droit commun est non pas une possibilité mais une imposture. Le titre VII du projet de loi, qui est basé sur le rapport charnel, ne fonctionne pas pour les couples lesbiens. On ne peut donc appliquer ces dispositions aux enfants de ces femmes.
Je vous demande, mesdames, messieurs les sénateurs, d'envisager les choses non sous le prisme de l'intérêt des parents, mais sous celui de l'intérêt supérieur de l'enfant, car c'est ce qui permettra aux enfants nés du don de bénéficier d'une égalité de droits, que leurs parents soient homosexuels ou hétérosexuels, mariés ou non, et de créer une modalité d'établissement de la filiation valable pour tous. On ne stigmatise pas les enfants adoptés ; pourquoi le ferait-on pour ceux nés du don ?
Nous disons clairement que nous voulons non pas de l'extension du droit commun, mais de l'extension de la reconnaissance conjointe anticipée pour tous les enfants. C'est cela, l'égalité !
Mme Marie-Claude Picardat . - Nous vivons un temps politique qui permet l'ouverture de droits à des personnes dont on reconnaît la spécificité.
De la même manière que l'on reconnaît aux hommes et aux femmes, qui sont pourtant différents, les mêmes droits, il faut agir ainsi pour les femmes homosexuelles ayant recours à la PMA. On ne doit pas valoir des spécificités là où elles n'ont pas lieu d'être ! La grossesse, l'accouchement et le processus médical sont les mêmes, que la femme soit homosexuelle ou hétérosexuelle. Pourquoi créer une divergence dans l'établissement du droit en raison de l'orientation sexuelle de la mère ? Il s'agit exactement d'une discrimination : on souligne une différence là où elle n'a pas lieu d'être, et l'on en fait découler des droits ou une absence de droits. Introduire une différence de traitement juridique au nom d'une différence d'orientation sexuelle est extrêmement problématique, surtout en termes d'établissement de la filiation par l'accouchement .
Pour la seconde mère, deux solutions sont possibles pour l'établissement de la filiation : celui-ci découle soit de l'engagement lié au mariage, soit d'une procédure de reconnaissance en mairie, mais sans inscription du mode de procréation dans l'acte d'état civil.
M. Bernard Jomier , rapporteur. - L'ouverture du droit à la PMA va se conjuguer avec le droit à l'accès aux origines, ce qui devrait entraîner une modification de la typologie des donneurs. Quelles sont vos propositions en la matière, et pour éviter une pénurie de gamètes ?
Madame Chesnel, vous avez émis des réserves sur le statut de l'embryon. Quelles sont-elles ?
Mme Marie-Claude Picardat . - L'État doit assumer ses responsabilités afin d'éviter une pénurie de gamètes.
S'agissant des dons de gamètes, de spermatozoïdes, d'embryons ou d'ovocytes, la situation est tendue car l'État ne fait pas de publicité, au sens large, sur ces questions. Il faut éduquer la population à la solidarité à l'égard des personnes qui ont besoin de ces dons, via des campagnes d'information.
Je pense que de nombreuses femmes homosexuelles se rendront encore à l'étranger, car les temps d'attente sont encore très longs. Mais celles qui se présenteront dans les centres d'études et de conservation des oeufs et du sperme (Cecos) seront-elles traitées à égalité avec les autres femmes, notamment en termes de délais ?
Les profils des donneurs changeront sans doute, mais cela ne justifie pas la destruction de gamètes qui a été un temps envisagée. Il faut respecter l'engagement pris envers les donneurs actuels et se donner le temps d'écouler ces gamètes, quitte à passer par une période transitoire où le don anonyme pourrait encore être proposé, le temps que de nouveaux donneurs prêts à partager plus d'informations puissent se manifester.
Mme Véronique Cerasoli. - On compte aujourd'hui environ 350 donneurs sur 67 millions de Français ! L'Institut national de la santé et de la recherche médicale (Inserm) avait montré que quelques dizaines de donneurs supplémentaires suffiraient à répondre à la demande anticipée.
Mme Catherine Michaud. - Il faut complètement abandonner l'idée de la destruction du stock de gamètes existant et aller chercher de nouveaux donneurs. Cela passe par des campagnes de communication à destination du grand public, similaires à ce qui se fait pour le don du sang. Rares sont les Français lambda qui ont été exposés aux campagnes existantes. Il faudrait rappeler qu'un homme peut donner ses gamètes dans une démarche altruiste.
M. Alexandre Urwicz. - Concernant l'impact du changement de régime d'anonymat, rappelons qu'au Royaume-Uni le nombre de donneurs a doublé dans les dix années suivant le changement similaire intervenu en 2005 ; en Suède, on a observé une baisse l'année suivant la réforme, mais le niveau antérieur a été retrouvé dès l'année suivante, et le nombre de donneurs n'a depuis lors cessé de progresser. Il n'y a donc pas d'inquiétude à avoir. Une phase transitoire peut être prévue, mais elle pourrait n'être que d'un an.
Mme Élisabeth Doineau , présidente . - La crainte relative au stock de gamètes doit en effet être mesurée. D'après le professeur Jean-Marc Ayoubi, il est important d'entraîner au don toutes les familles qui rencontrent des difficultés dans l'obtention de gamètes. Cette sensibilisation permet de recruter de nouveaux donneurs.
M. Alexandre Urwicz. - Vous avez raison. Dans le modèle actuel de don d'ovocytes, le délai moyen d'attente peut atteindre cinq ans, mais venir avec une autre femme prête à donner ses ovocytes à un centre de fertilité - mais non pas directement, puisque le principe reste l'anonymat - permet de réduire de moitié ce délai. On conclut ainsi presque un deal avec les personnes qui aident le centre de la sorte. Un tel système pourrait être envisagé pour le don de gamètes en cas de pénurie.
Quant au stock actuel, nous souhaiterions qu'on puisse contacter les donneurs pour leur demander s'ils consentent à entrer dans le nouveau régime ; cela pose plutôt des difficultés administratives qu'idéologiques, et permettrait d'éviter la destruction de tout le stock.
Mme Laurène Chesnel. - La pénurie concerne plus aujourd'hui les ovocytes que les gamètes. Ceux qui donnent aujourd'hui sont souvent des personnes elles-mêmes engagées dans un processus de PMA. Dès lors, on peut penser que l'ouverture de ce procédé aux femmes célibataires et lesbiennes conduira à une augmentation du don d'ovocytes. Aujourd'hui, le don de gamètes est effectué par 250 à 350 hommes par an, et 500 femmes donnent leurs ovocytes. Le stock de gamètes est extrêmement important : on compte 90 000 paillettes, soit treize ans de réponse à la demande actuelle. La demande doublerait avec l'ouverture de la PMA aux femmes célibataires et lesbiennes ; on aurait donc six ans et demi de stock dans ces circonstances.
Alors, pourquoi les futurs parents doivent-ils attendre ? Dans certains cas, on invoque la nécessaire maturation du projet parental, ce qui est peu compréhensible au vu des nombreuses années que les couples, notamment hétérosexuels, passent avant de s'engager dans cette procédure. En outre, les centres n'ont pas toujours les moyens de recontacter les donneurs. Surtout, chaque centre est autonome : il n'y a pas de répartition nationale du stock. Les problèmes organisationnels sont donc importants ; nous attendons beaucoup du rapport parlementaire qui doit être remis sur ce sujet.
Nous estimons, nous aussi, qu'il serait utile d'interroger les anciens donneurs afin d'éviter la destruction du stock. Certains centres envisagent déjà de le faire pour leur demander s'ils accepteraient que leurs gamètes soient utilisés par des couples homosexuels, approche extrêmement problématique du point de vue des discriminations !
Quant au statut de l'embryon, nous sommes gênés par le fait que l'article 14 du projet de loi ne porte que sur les embryons « conçus dans le respect des principes fondamentaux énoncés aux articles 16 à 16-8 du code civil ». Cela nous gêne, car ces articles ont pour objet les personnes nées vivantes et viables. Le raccourci employé à l'article 14 tend donc à donner une personnalité à l'embryon, ce qui est problématique, notamment par rapport au droit à l'IVG. Nous avons été alertés sur ce point par le Planning familial.
M. Olivier Henno , rapporteur . - Ma question porte sur l'accès aux origines pour les enfants qui naissent au terme d'un processus d'assistance médicale à la procréation. Cet accès est par nature différent de celui qui est offert aux enfants adoptés, dans la mesure où ceux qui sont issus d'une AMP sont désirés et aimés. Pensez-vous que le Conseil national pour l'accès aux origines personnelles (Cnaop) devrait être chargé de cet accès aux origines, ou bien faudrait-il confier cette responsabilité à une autre structure ?
Mme Laurène Chesnel. - Une autre commission est prévue dans le présent projet de loi. Il serait intéressant de lui donner des pouvoirs de recherche similaires à ceux du Cnaop pour les enfants déjà nés et de lui confier cette mission pour les enfants à naître. On pourrait tout confier au Cnaop, mais ce conseil y est réticent, car il ne dispose pas actuellement des moyens financiers nécessaires. Il faudra en tout cas s'inspirer de l'expérience du Cnaop, qui montre bien que l'on peut recontacter discrètement les anciens donneurs.
M. Alexandre Urwicz. - Le dispositif d'accès aux origines est très intéressant, mais une question se pose : comment l'enfant peut-il savoir s'il est issu d'un don ? Qui l'en informe ? Qui lui garantit cet accès à ses origines, à son histoire personnelle ? La réponse est donnée dans ce projet de loi uniquement aux enfants nés de couples lesbiens, et non à ceux de couples hétérosexuels, pour lesquels on en reste au modèle pseudo-charnel d'établissement de la filiation. Voudrez-vous, en tant que législateur, donner une même garantie d'accès aux origines à tous les enfants ? Sinon, discriminations et ruptures d'égalité perdureront.
Mme Véronique Cerasoli. - Nous ne comprenons pas le système de reconnaissance conjointe anticipée appliqué aux couples de femmes. Les enfants nés à la suite d'une AMP dans ces couples sont justement les seuls à ne pas avoir besoin d'une mention sur un document d'état civil pour savoir qu'ils ont été conçus grâce à un don. Nous y voyons un exemple de la primauté de l'hétéronormativité dans notre droit. Répondre à une demande d'accès aux origines par un système de filiation uniquement pour les familles fondées par deux femmes nous paraît une discrimination incompréhensible. Il n'y a pas de lien entre l'établissement de la filiation et l'accès aux origines.
Mme Marie-Claude Picardat. - Les enfants et les familles ont sans doute intérêt à pouvoir être accompagnés, même si je ne sais s'il vaut mieux confier cette tâche au Cnaop ou à un autre organisme. Cela dit, ce que l'on appelle la quête des origines n'est pas du tout une demande systématique des enfants. Ils peuvent savoir comment ils ont été conçus sans chercher à obtenir des renseignements sur les donneurs. L'AMP correspond à des situations familiales infiniment moins traumatisantes que l'adoption consécutive à un abandon, sauf parfois dans les couples hétérosexuels souffrant d'infertilité. L'enfant est informé de sa conception et a rarement des questions quant au donneur. Il est en revanche important que, s'il en a, elles puissent trouver des réponses, qui peuvent en fonction des individus aller jusqu'à des rencontres.
Mme Élisabeth Doineau , présidente . - Le Cnaop nous a décrit les situations qu'il rencontre dans l'accompagnement des enfants nés sous le secret. Certains ont besoin, à tout le moins, d'un accompagnement psychologique, surtout en cas de rencontre.
Mme Marie-Claude Picardat. - Les situations d'adoption et de don sont extrêmement différentes. Rencontrer un parent qui vous a abandonné est beaucoup plus traumatisant ; je le constate en tant que psychiatre. Il ne faut aucune stigmatisation, aucun forçage : on n'a pas à imposer sur l'état civil d'un enfant quelque chose qui n'y a pas sa place. L'essentiel est un accompagnement bienveillant de l'État, dans l'intérêt des familles et des enfants.
Mme Michelle Meunier . - Vous avez évoqué la mère qui accouche. Inscrire à l'état civil de l'enfant quelle mère l'a porté répondrait-il correctement à un souci de transparence, dans l'intérêt de l'enfant, ou bien cela entraînerait-il une stigmatisation ?
Mme Marie-Claude Picardat. - Envisagez-vous cette mention pour l'ensemble de la population, ou uniquement pour les femmes homosexuelles ? Nous voulons le droit commun : nous ne voulons pas porter seules un point de droit qui serait uniquement lié à l'orientation sexuelle. Si l'on en fait une mention obligatoire pour toutes les femmes qui accouchent, ce qui a été le cas un temps, c'est une chose, mais si cela ne concerne que les femmes homosexuelles, c'est une discrimination.
Mme Laurène Chesnel. - Concernant la différence qui pourrait exister, dans les couples de femmes, entre celle qui accouche et l'autre, il n'y a pas de mystère dans les couples : l'enfant sait qui est la mère qui l'a porté. Par ailleurs, l'accouchement ouvre des droits sociaux, notamment pour la retraite. En droit international privé, le statut de l'enfant est lié à la mère qui a accouché. Que se passerait-il s'il n'y avait plus de distinction entre les deux ? Dans les couples que nous avons rencontrés, la mère qui n'a pas accouché ne se sent pas moins mère, la différence ne les gêne pas.
Mme Véronique Cerasoli. - La simultanéité de l'établissement de la filiation entre les deux mères importe à nos yeux, ainsi que l'égalité en droits entre les deux parents, mais cela n'implique pas que l'accouchement soit effacé. Tout comme dans les familles hétérosexuelles, l'égalité des droits parentaux n'implique pas d'offrir au parent qui n'a pas accouché les droits sociaux qui découlent de l'accouchement. Changer sur ce point le droit de la filiation ne nous paraît donc pas pertinent.
M. Alexandre Urwicz. - Toutes les mentions relatives à des reconnaissances sont systématiquement portées en marge de l'acte intégral de naissance. Concernant la reconnaissance conjointe anticipée, nous proposons de modifier le texte pour préciser que l'établissement de la filiation de la mère ayant accouché est subordonné au cumul de deux modalités : la transmission du certificat d'accouchement et la signature de la reconnaissance conjointe anticipée. Ainsi, tout en préservant l'égalité de droits des deux parents, on ne renie à aucun moment la femme qui a accouché. Mme la garde des sceaux a même proposé que la mère ayant accouché soit nommée en premier dans l'acte de naissance ; nous n'y sommes pas opposés.
Mme Laurène Chesnel. - Lier la filiation à l'accouchement a représenté un progrès : auparavant, les femmes non mariées devaient reconnaître leur enfant. Cette réforme évite que des enfants ne se retrouvent par accident sans état civil : tous ont au moins une filiation établie à la naissance.
Par ailleurs, on a trop longtemps considéré que les lesbiennes n'avaient pas vraiment de sexualité ; ce stéréotype est assez fort. Ne pas reconnaître que nous portons nos enfants comme les autres femmes peut être interprété comme une atteinte à notre féminité.
Mme Marie-Claude Picardat. - Si une femme homosexuelle a recours à la PMA alors qu'elle est célibataire, il n'y aura pas de mention particulière ; seule celle qui est en couple avec une autre femme y sera sujette. On crée une situation incompréhensible de discrimination !
M. Yves Daudigny . - Je voudrais revenir sur le sujet de la PMA post mortem. En 2016, une veuve aurait été autorisée par la justice à importer d'Espagne le sperme de son mari décédé afin de concevoir un enfant. Cela fait-il jurisprudence ?
Mme Laurène Chesnel. - Je n'ai pas connaissance de cette décision de justice, mais elle ne m'étonnerait pas. Toutes les institutions consultées au sujet de la PMA post mortem y sont plutôt favorables. Il faut prendre garde aux évolutions prévues par le projet de loi sur le transfert éventuel des gamètes. Pourquoi obliger les gens à aller faire une démarche à l'étranger plutôt qu'en France ?
Mme Muriel Jourda , rapporteur . - Je précise que la femme qui a obtenu cette décision de justice était elle-même espagnole : il ne s'agit pas d'une situation où quelqu'un était forcé d'aller voir à l'étranger.
Mme Marie-Claude Picardat. - Ce n'est pas notre sujet, mais nous travaillons avec des associations qui s'y intéressent et y sont favorables.
Mme Élisabeth Doineau , présidente . - Souhaiteriez-vous apporter des précisions sur des sujets qui n'ont pas été abordés ?
Mme Marie-Claude Picardat. - Ce débat sur la filiation ne devrait pas être une occasion manquée pour l'ensemble des situations familiales. Le projet de loi valide deux points importants : la nécessité d'ouvrir la filiation hors mariage pour les couples de femmes et de sécuriser les liens familiaux dès la naissance de l'enfant, même si nous ne sommes pas d'accord avec les modalités proposées. On sortirait du moins de l'héritage de la loi ouvrant le mariage aux couples de personnes de même sexe, qui ne reconnaissait la famille homoparentale que dans le cadre du mariage et de l'adoption intrafamiliale, ce qui engendrait de nombreux écueils pour les familles séparées et les personnes qui ne veulent pas se marier. Il faut penser le droit de manière homogène en dépit de situations familiales différentes.
C'est encore une raison qui explique l'importance de rentrer dans le droit commun : cela résoudrait bien des situations qui causent aujourd'hui des souffrances infinies aux parents comme aux enfants. Il faudrait offrir des outils supplémentaires, dans le cadre de la reconnaissance conjointe anticipée, en direction des couples qui ont déjà eu recours à la PMA, notamment à l'étranger : établissement de la filiation par possession d'état ou modifications des règles régissant l'adoption intrafamiliale. Les promesses faites en 2013 quant au droit de la filiation n'ont pas été tenues, mais le débat politique est aujourd'hui beaucoup plus apaisé. C'est donc le moment d'élargir la réflexion sur la filiation et de faire entrer dans le droit commun l'établissement de la filiation hors mariage.
M. Fabien Joly. - Toutes nos associations sont d'accord pour concevoir un système protecteur pour l'enfant comme pour les femmes et les hommes qui en sont les parents, même si nous divergeons sur les modalités.
Notre association est favorable à la reconnaissance conjointe anticipée, mais aussi à son extension aux couples hétérosexuels et aux femmes célibataires. Nous voulons sécuriser les droits de ces femmes : si la RCA leur était étendue, elles seraient prémunies contre toute reconnaissance indue par un homme qui n'est pas lié génétiquement à l'enfant. Une telle reconnaissance paternelle serait impossible, car la RCA aurait induit l'établissement d'une seule filiation. Si cette femme refait sa vie avec quelqu'un, il serait toujours possible pour le nouveau conjoint d'adopter l'enfant.
La RCA ne concerne pas que les couples, elle concerne aussi les femmes célibataires, dont le projet parental, pour solitaire qu'il soit, n'en est pas moins réfléchi et mûri. Ces femmes doivent être sûres que la filiation exclusive envers elle sera garantie.
La ministre de la justice avait affirmé que les PMA faites à l'étranger après la loi pourraient bénéficier de la RCA. La Direction des affaires civiles et du Sceau a précisé qu'il suffirait que ces femmes signent un consentement devant notaire avant l'insémination. L'établissement de la filiation sera alors établi de la même manière que pour les femmes ayant accompli tout le processus en France.
Mme Véronique Cerasoli . - Je souligne le lien, documenté, entre discriminations dans le droit, les procédures ou la jurisprudence, et discriminations réelles et subies. Notre rapport annuel met bien ce lien en évidence. D'où l'importance d'un traitement égal : ne créons pas de différences là où elles n'ont pas lieu d'être.
Mme Marie-Claude Picardat . - Une association de mères seules s'oppose à l'inscription de la RCA à l'état civil. Inutile de parler à leur place.
Mme Laurène Chensel . - Le traitement des données de santé est aussi un sujet. Le respect du consentement doit être garanti, comme le prévoient la loi Kouchner et la loi Informatique et libertés. L'orientation sexuelle ou la séropositivité, révélées, peuvent avoir des conséquences dramatiques. La Haute Autorité de santé (HAS) doit être alertée dès qu'il y a un problème de transparence.
M. Alexandre Urwicz . - L'aspect médical est en effet fondamental. Voulez-vous créer une rupture d'égalité dans les chances de survie des enfants conçus par don ? Ce sera le cas, à l'heure de la médecine prédictive, si certains ont l'information sur leur origine et d'autres, non. Une maladie non détectable en 2019 peut le devenir en 2021... Et un droit n'est pas une obligation : nul n'est contraint d'aller ouvrir cette porte, mais il faut savoir qu'elle existe. Mettons tous les enfants à égalité !
Mme Élisabeth Doineau , présidente . - Merci. N'hésitez pas à nous transmettre par écrit toutes informations complémentaires utiles.
Ce point de l'ordre du jour a fait l'objet d'une captation vidéo qui est disponible en ligne sur le site du Sénat .
II. EXAMEN DU RAPPORT
Réunie les 7 et 8 janvier 2020 sous la présidence de M. Alain Milon, président, la commission spéciale procède à l'examen du rapport de M. Olivier Henno, Mme Corinne Imbert, M. Bernard Jomier et Mme Muriel Jourda, sur le projet de loi n° 63 (2019-2020) relatif à la bioéthique.
Mardi 7 janvier 2020
- Présidence de M. Alain Milon, président -
M. Alain Milon , président . - Mes chers collègues, je souhaite aux uns et aux autres une excellente année, pleine de succès.
Mme Muriel Jourda , rapporteur . - Je suis rapporteur du texte sur les quatre premiers articles. L'article 1 er étend l'assistance médicale à la procréation (AMP) aux femmes seules et aux couples de femmes. L'article 2 ouvre la possibilité d'une autoconservation de gamètes pour les femmes comme pour les hommes. L'article 3 donne droit aux personnes nées d'AMP avec tiers donneur d'accéder sans condition aux informations non identifiantes relatives à ce donneur et à son identité. L'article 4 crée un mode de filiation par déclaration anticipée de volonté permettant aux couples de femmes de devenir légalement parents de l'enfant dès la naissance de ce dernier.
Permettez-moi au préalable de formuler quelques observations d'ordre général.
D'abord, on peut se demander si ces articles ont leur place dans une loi de bioéthique ; les révisions périodiques de ces lois doivent en principe intervenir dès qu'une avancée scientifique nécessite de réexaminer ce que la science peut légalement faire. Or l'AMP est une technique ancienne.
Toutefois, il figure tout de même dans le texte au moins un principe de bioéthique : la non-marchandisation des produits du corps humain. En outre, les questions posées par les dispositions en discussion sont exactement celles qui se posent dans le cadre d'une loi de bioéthique : est-ce que cela doit être autorisé parce que c'est possible ?
La réponse à cette question est loin d'être évidente, car il faut légiférer en ayant à l'esprit l'intérêt de la société. Le droit de la filiation est, à cet égard, structurant pour la société, et il peut s'opposer aux désirs individuels. On le sait bien, la juxtaposition de désirs individuels ne constitue pas en soi une société ; au contraire. Par ailleurs, certains intérêts individuels peuvent être concurrents entre eux : l'intérêt des femmes qui veulent un enfant et l'intérêt des enfants peuvent s'opposer. On peut essayer de concilier ces intérêts contradictoires, mais ce n'est pas toujours possible et, dans certains cas, il faut faire prévaloir l'intérêt de l'un par rapport à celui de l'autre.
Ensuite, on ne peut pas, en une telle matière, s'abriter derrière le droit ni derrière l'opinion publique.
On a beaucoup entendu parler, au cours des dernières semaines, d'égalité et de discrimination. Ce sont des notions juridiques, qui ont été largement traitées par les juridictions pour ce qui concerne l'AMP et l'accès des femmes à cette technique. Or plusieurs décisions du Conseil constitutionnel et du Conseil d'État ont indiqué qu'aucun principe d'égalité n'imposait l'extension de l'AMP aux femmes seules et aux couples de femmes. La discrimination, l'inégalité, consiste à traiter différemment des situations identiques ; or il est évident que les femmes seules et les couples de femmes ne sont pas dans la même situation que les couples hétérosexuels. On ne peut donc pas s'abriter derrière le droit pour légiférer en matière d'AMP.
On ne peut non plus s'abriter derrière l'opinion. Il y a eu beaucoup de sondages à ce sujet, en particulier de l'Institut français d'opinion publique (IFOP). Ainsi, deux sondages de cet automne ont montré que les Français étaient à 65 % favorables à l'extension de l'AMP aux couples de femmes et à 61 % favorables au fait de favoriser la possibilité pour les couples d'hommes d'avoir un enfant, mais un troisième sondage indique que 83 % des Français sont favorables au droit des enfants d'avoir un père et une mère. Les Français sont donc favorables à la réforme, mais défavorables à ses conséquences...
Un autre argument n'a, selon moi, aucun impact : il consiste à dire que, ces situations existant déjà, pourquoi ne pas les autoriser ? Sans doute, ces situations - des femmes seules ou des couples de femmes ayant des enfants - existent, mais tout existe. La question n'est pas de savoir si cela existe, mais si nous devons l'institutionnaliser. Le fait que les choses se fassent à l'étranger n'est pas plus pertinent, car, de la même manière, tout se fait à l'étranger. Du reste, la GPA existe dans de nombreux pays, mais cela n'empêche pas les résidents de ces pays de faire du tourisme procréatif dans des pays moins-disants en matière d'éthique ; en effet, on trouvera toujours des pays où les choses sont moins chères, plus faciles, moins exigeantes. Nous devons déterminer ce que le droit français doit permettre.
J'en viens à la présentation des articles.
L'article 1 er étend, je le disais, l'AMP aux couples de femmes et aux femmes seules. Je le rappelle, l'AMP telle qu'elle existe actuellement consiste en un ensemble de droits permettant de remédier à l'infertilité biologique ou d'éviter la transmission d'une maladie grave aux enfants d'un couple. Cet article vise à étendre ce dispositif à toutes les femmes seules et à tous les couples de femmes, qu'il y ait ou non infertilité.
Autant le dire tout de suite, je nourris les plus grandes réserves à cette extension de l'AMP, en raison, en premier lieu, du rôle que l'on entend faire jouer à la médecine. L'AMP est actuellement proposée aux personnes infertiles qui devraient pouvoir avoir des enfants - elles doivent, par exemple, être en âge de procréer -, mais qui n'y parviennent pas, y compris si la médecine est incapable d'expliquer pourquoi. Ainsi, peu importe que l'on puisse ou non diagnostiquer les causes de l'infertilité, seul compte le résultat : on n'arrive pas à avoir des enfants. Par conséquent, même si la médecine ne peut soigner l'infertilité, elle peut y remédier. Il n'y a donc en la matière aucune discrimination, puisque les couples de femmes et les femmes seules ne sont pas en situation d'avoir des enfants. L'espèce humaine est une espèce à reproduction sexuée ; une femme seule ou deux femmes ne sont donc pas en mesure de procréer.
Avec une telle extension, on sort du rôle curatif de la médecine pour répondre à un désir individuel. La médecine devient donc la médecine des désirs et non la médecine du malade. Cela me semble très dangereux, car si l'on satisfait ce désir, il n'y a aucune raison de s'arrêter là, et on se dirige alors vers l'eugénisme et même le transhumanisme. Il peut s'agir, aujourd'hui, d'avoir un enfant, et, demain, d'avoir un enfant ayant telles ou telles caractéristiques... L'octroi d'un tel rôle à la médecine me semble dangereux pour l'avenir de l'homme.
En second lieu, on ne parle que du désir des femmes d'avoir des enfants, mais on évoque peu l'intérêt des enfants ; or cela doit être étudié et pris en compte. Le désir d'une femme d'avoir un enfant est bien sûr le même, quelles que soient sa situation de couple et son orientation sexuelle, et ce désir est tout à fait respectable. De même, la capacité d'amour, d'attention, de bienveillance et d'éducation ne dépend évidemment pas de la situation de couple d'une femme. Néanmoins la question est : si tout cela est nécessaire, est-ce suffisant ? En effet, la conséquence d'une telle évolution serait la privation de père, au quotidien et à long terme, car il n'y aurait plus de filiation paternelle possible ; on aura une ou deux mères et rien de plus. Or pense-t-on qu'un père sert à quelque chose ? Pour le coup, l'opinion est assez claire sur ce point : l'IFOP a posé cette question en juin 2018 : « un père est-il essentiel ? » - et la réponse a été « oui » à 93 %. Nous avons donc une indication claire, même si, je le répète, on ne peut pas s'appuyer sur un sondage pour déterminer si l'on doit ou non approuver cette extension.
J'irai plus loin en ce qui concerne la situation des femmes seules, car notre attention a été appelée à de nombreuses reprises sur ce point par les médecins, les associations ou encore le Comité consultatif national d'éthique pour les sciences de la vie et de la santé (CCNE).
Les enfants élevés par une femme seule, en effet, ne sont pas confrontés à l'altérité sexuée des parents. Les mères isolées ne bénéficient pas non plus de l'aide matérielle d'un père pour élever leurs enfants et les familles monoparentales sont souvent considérées comme un public à risques, vulnérable, par les politiques publiques. Le Président de la République a découvert, lors du Grand débat, que les familles monoparentales étaient dans une situation plus précaire que les autres, et pas seulement financièrement. Il est, en effet, plus difficile d'élever un enfant seul qu'à deux. Cela a été rappelé par de nombreuses personnes que nous avons reçues. Nous avons aussi auditionné, évidemment, une association de femmes seules qui ne se considéraient pas comme un public vulnérable. Elles nous ont expliqué qu'elles étaient seules parce que c'était leur choix et que leur situation n'était pas la même que celle des mères abandonnées. Toutefois, si elles revendiquent leur choix, elles nous ont expliqué aussi qu'il s'agissait d'un plan B, d'un choix contraint : faute de trouver le bon conjoint pour se marier et fonder une famille, elles ont décidé, l'horloge biologique tournant, d'avoir un enfant. Certes, elles ont préparé leur situation, mais toutes les familles monoparentales sont confrontées aux mêmes problématiques. Une étude de l'Unicef réalisée en 2014 sur le malaise des adolescents montrait l'importance, à cet égard, de l'absence de père ; Catherine Dolto avait déploré que l'on sous-estime le rôle éducatif du père. C'est pourquoi j'émets de vives réserves sur l'extension de l'AMP aux femmes seules.
J'ai aussi des réserves pour l'étendre aux couples de femmes, toujours dans l'intérêt de l'enfant. Les pédopsychiatres expliquent qu'être désiré, aimé, éduqué est important pour les enfants, mais l'amour ne suffit pas pour permettre à un enfant de se construire. Un enfant, en effet, se construit à travers l'altérité des sexes. Il se construit aussi sur le plan filiatif. Pour cela, il a besoin d'une filiation assimilable, cohérente et vraisemblable. Or, la procréation hétérosexuelle est la seule vraisemblable. Il faut pouvoir rendre ses origines crédibles. Elles ne le sont pas lorsque l'on indique que l'enfant est issu de deux personnes de même sexe. La construction de la filiation est plus difficile pour les enfants issus de l'AMP, comme on le constate déjà pour les enfants issus de couples hétérosexuels ayant eu recours à une AMP avec tiers donneur. Dans le cas des couples de femmes, on ajoute une difficulté, car la filiation n'est ni naturellement possible ni crédible. On risque donc de créer une inégalité entre les enfants qui pourront retracer leur filiation et les autres.
D'autres pédopsychiatres ont une autre vision : ils estiment que le rôle du père peut être joué par une femme et Mme Buzyn a ainsi dit qu'une mère peut être un père. Chacun jugera... Il me semble que les hommes et les femmes ne sont pas totalement interchangeables. La fonction paternelle est particulière. Deux femmes peuvent très bien élever un enfant, mais elles ne peuvent pas permettre sa construction psychique comme un couple hétérosexuel. Nous manquons malheureusement d'études sérieuses sur ce point. L'étude d'impact du Gouvernement n'est pas satisfaisante, car elle s'appuie sur des études souvent militantes et qui manquent de bases scientifiques, comme l'Académie de médecine l'a expliqué. On ne peut donc pas se fonder sur ces études pour se forger une conviction. En ce qui me concerne, j'ai été convaincue de l'importance pour un enfant de se construire sur le plan de la filiation par le témoignage, qui nous a été présenté par des associations homoparentales, d'une jeune femme d'une vingtaine d'années qui avait été élevée par deux femmes seules. Cette jeune femme expliquait que son éducation s'était bien passée, et qu'elle ne voyait pas l'usage d'un père ni sa spécificité... Cela m'a convaincue de l'inverse ! Si l'on en vient à nier l'autre partie de l'humanité parce que l'on n'en voit pas l'usage, alors je suis sceptique... Il ne me semble pas que le Parlement doive reconnaître une telle démarche. D'ailleurs, qu'il s'agisse des textes sur l'exercice conjoint de l'autorité parentale, la résidence alternée, le congé parental ou le congé de paternité, le législateur a toujours pris en considération l'intérêt de l'enfant et a estimé qu'il était dans son intérêt de faire une place plus importante au père. De nombreuses études montrent aussi que la socialisation de l'enfant est meilleure en cas d'intervention précoce du père dans sa vie, comme l'a relevé la délégation aux droits des femmes du Sénat dans son avis sur le partage plus équilibré du congé parental. Disons-le, l'absence de père est une mauvaise affaire pour l'enfant et sa présence peut améliorer sa vie. C'est toujours dans ce sens que le législateur a travaillé ces dernières années. Comme l'expliquait Jean Leonetti, nous devons arbitrer entre l'éthique de l'autonomie, le désir des femmes, et l'éthique de la vulnérabilité, l'intérêt de l'enfant à naître. Nous devons arbitrer en faveur de cette dernière.
Un autre élément à prendre en compte pour apprécier s'il est judicieux d'étendre l'AMP aux femmes seules est le droit de la filiation. Le droit de la filiation est un droit d'ordre public, qui a pour fonction de structurer la société et qui est donc supérieur aux intérêts des individus. Il sert de fondement à la prohibition de l'inceste, au droit de l'héritage, à la définition des droits et devoirs des parents et des enfants. Le titre VII du code civil est consacré à la filiation, le titre VIII à la filiation adoptive. Le titre VII est relatif à la filiation basée sur la procréation charnelle entre un père et une mère, c'est la filiation de la vraisemblance. La mère est la femme dont le nom figure sur l'acte de naissance. Si la femme est mariée, le mari est présumé être le père. Il y a une présomption de paternité. Si la mère n'est pas mariée, le père est celui qui reconnaît l'enfant. En cas de contestation de paternité, les tribunaux peuvent être saisis et des tests génétiques peuvent être réalisés. Le titre VIII définit la filiation adoptive, élective. On distingue la filiation simple et la filiation plénière. Actuellement, le recours à l'AMP repose sur le titre VII, car elle n'est ouverte qu'aux couples hétérosexuels. Le texte prévoit que les couples de femmes devront procéder à une reconnaissance anticipée de l'enfant à naître devant notaire, au moment même où ils annonceront leur intention de recourir à l'AMP. Toutefois, en droit de la filiation, la reconnaissance de maternité, ce n'est pas cela ! C'est la reconnaissance que l'on a participé à la procréation. En l'espèce, il est évident que la seconde femme n'a pu participer à la procréation ! Surtout, la volonté individuelle pure deviendrait un mode d'établissement de la filiation. Or, dans le droit commun, celle-ci ne suffit pas ; elle est toujours contrebalancée par la vraisemblance, la vérité ou une décision de justice, le droit de la filiation étant d'ordre public. En effet, si la volonté individuelle peut être très forte, elle peut aussi être volatile. On risque donc de fragiliser tout l'édifice de la filiation. D'où mes réserves sur l'élargissement de l'AMP aux femmes seules et aux couples de femmes.
J'en viens maintenant à la levée de l'anonymat des donneurs de gamètes. Aujourd'hui, le principe est celui de l'anonymat du don. Le donneur ne connaît pas les enfants nés grâce à son don et les enfants n'ont pas moyen de connaître leur père génétique. Des voix s'élèvent parmi les enfants issus d'un don pour lever cet anonymat, à l'image d'une association très active sur ce sujet. Sont-elles majoritaires ? Je ne le pense pas. L'association que j'évoque compte 169 membres, alors que l'on compte plus de 70 000 enfants issus de dons en France.
La levée de l'anonymat entraînerait aussi une autre difficulté. Il est très différent de donner ses gamètes en se disant que l'on n'aura à se soucier de rien ensuite ou en sachant que l'on pourra être contacté par la suite par l'enfant. Dans les pays qui ont levé l'anonymat, le nombre de donneurs a chuté, comme en Grande-Bretagne. Je crains aussi une marchandisation des gamètes, en contradiction avec le principe de non-marchandisation des produits du corps humain. Il est vrai qu'en Grande-Bretagne les dons ont fini par remonter, mais ce pays indemnise les donneurs, tout comme l'Espagne, tandis que la Belgique achète des gamètes à l'étranger pour faire face à la demande. C'est pourquoi je proposerai une autre formule : il s'agit de permettre, lorsqu'un enfant en fait la demande, de contacter le donneur pour savoir s'il est d'accord pour lever son anonymat. C'est un dispositif qui avait été préconisé par le Conseil d'État, qui est respectueux à la fois de la demande de l'enfant et du droit à la vie privée du donneur. Cela pourrait également s'appliquer aux dons passés.
Je terminerai par la question de l'autoconservation des gamètes.
L'autoconservation est possible en cas de problèmes de fertilité ou lorsque l'on fait un don de gamètes : à partir de trois recueils de sperme pour un homme, tandis que la femme peut garder la moitié des ovocytes au-delà de dix ovocytes donnés - jusqu'à cinq ovocytes donnés, tous les ovocytes sont destinés au don ; de six à dix ovocytes obtenus, au moins cinq ovocytes sont destinés au don ; au-delà de dix ovocytes obtenus, au moins la moitié des ovocytes est dirigée vers le don. Ce système constitue, en fait, une forme de contrepartie, et donc de rémunération, du don, et peut même s'apparenter, selon l'Académie de médecine, à un chantage à l'égard des femmes.
Faut-il autoriser la conservation de gamètes à des fins personnelles, comme le texte le prévoit ? En réalité, le problème se pose davantage pour les femmes que pour les hommes, car la période pendant laquelle elles peuvent avoir des enfants est plus restreinte. La question se pose. Plutôt que de concevoir des enfants lorsqu'elles ont l'âge de le faire, elles pourront ainsi choisir de décaler l'âge auquel elles ont un enfant, au prix d'une intervention chirurgicale, car la conservation des ovocytes réclame un mois de traitement, puis une intervention sous anesthésie générale, ce qui n'est pas anodin. Il est donc à craindre que le choix de conserver les ovocytes ne soit un choix dicté sous pression, pour pouvoir mener une carrière professionnelle. Pour ma part, je préférerais que l'on prenne mieux en compte la grossesse des femmes.
Aujourd'hui, c'est un lieu commun, les femmes qui ont des enfants sont davantage pénalisées qu'elles ne sont récompensées ; pourtant, leurs enfants sont aussi ceux des hommes et elles rendent un service à la société qui devrait être reconnu ; mais cet état d'esprit, hélas !, n'est pas celui qui prévaut chez les employeurs. Je préférerais donc que l'on développe une véritable politique familiale et que l'on construise des crèches plutôt que de parler de congeler des ovocytes.
Cela n'empêche pas la conservation des ovocytes - pour moi, ce n'était pas une priorité - dans la mesure où elle ne doit pas être exclusivement liée à la volonté que les femmes fassent carrière. Elle est surtout liée à la difficulté de trouver quelqu'un pour faire un enfant : les hommes ont du mal à s'engager et à faire des enfants, alors qu'ils ont une fertilité plus longue. L'allongement de la durée de vie induit en erreur sur l'allongement de la fertilité. Ne nous interdisons pas de congeler des ovocytes, mais sans leurrer les femmes : ce n'est pas parce qu'elles congèleront des ovocytes qu'elles auront des enfants.
M. Bernard Jomier , rapporteur . - Personne ne conteste que la suite du projet de loi s'inscrive bien dans le champ de la bioéthique.
J'ai apprécié la manière dont nous avons travaillé entre rapporteurs, avec le président de la commission, et les échanges très utiles que nous avons eus avec les personnes entendues. Les interventions de mes collègues sénateurs ont également bien enrichi notre réflexion.
Nous sommes tous animés d'une même volonté, et je vous partagerai quelques principes qui m'ont guidé et qui, je l'espère, infuseront ce texte.
Les nouvelles techniques médicales, qui sont une source de progrès et de mieux-être, apportent des avancées pour la santé et des moyens de prévention. On doit les aborder ainsi et identifier leurs apports positifs, avant d'examiner les risques et les dérives potentiels. Nous analyserons les risques de dérives, sous le prisme d'une bifurcation : soit il n'y a pas de risques, et la technique doit être mise en oeuvre au plus vite ; soit il y a un risque de dérive, et nous devons regarder si des garde-fous efficaces existent. Voilà l'enjeu.
Lorsqu'on regarde en arrière les précédents débats sur les lois de bioéthique, on voit que les craintes de dérives n'étaient pas justifiées - peut-être parce que le législateur a mis en place un dispositif efficace, selon lequel soit nous n'avons pas autorisé la technique, soit nous l'avons autorisée avec un cadre de gestion des risques et des évaluations.
In fine, nous analysons les termes du débat à partir de valeurs qu'il faut énoncer clairement. Comme dans l'ordre judiciaire, certains principes l'emportent sur d'autres. L'autonomie, la bienfaisance, la justice sont des valeurs cardinales. D'autres valeurs peuvent être invoquées : la vulnérabilité, l'égalité - elle est une demande croissante -, la dignité, la solidarité, le respect. Cela aboutit à des dilemmes éthiques qui se résolvent non pas par l'absolutisme d'une valeur sur les autres, mais par la recherche d'une modalité de résolution qui prend en compte ces différences. Je n'oppose pas l'autonomie et la vulnérabilité, et M. Leonetti ne le fait pas non plus dans son opuscule sur le sujet. Ces dernières années, l'éthique de l'autonomie s'est développée, notamment à propos du grand âge, âge auquel on est vulnérable. Nous ne devons pas rejeter les demandes d'autonomie sur des dons de parties du corps humain de la part de personnes non reconnues pour leur autonomie jusqu'à présent, comme les majeurs protégés ou les mineurs, même si ce sont des publics vulnérables.
Ce texte, dans la version issue des travaux de l'Assemblée nationale, est parfois plus marqué par la crainte et l'analyse des risques que des progrès attendus pour la société. Nous pouvons sans naïveté ni risque de dérives apporter du mieux à la population.
Je m'attacherai aux articles dont je suis rapporteur.
Le titre II du projet de loi comporte quelques dispositions intéressantes sur le don d'organes et de cellules. L'article 5 lève de façon bienvenue des contraintes pesant sur le développement du don croisé d'organes qui n'a donné lieu depuis son autorisation par la loi de 2011 qu'à douze greffes rénales en France - le rein étant le principal organe greffé. Le don croisé permet de répondre aux cas d'incompatibilité biologique entre un receveur et un proche prêt à lui faire don d'un organe, principalement d'un rein, rendant impossible la greffe. Je vous proposerai de rétablir dans la loi le nombre maximal de paires de donneurs et de receveurs impliqués dans une chaîne de dons en fixant ce nombre à six au lieu de quatre dans le projet de loi initial, alors que l'Assemblée nationale avait renvoyé cette question à un décret.
Cela permettra de ménager plus de souplesse tout en restant compatible avec les exigences de cette procédure lourde et complexe. Ces dispositions ne suffiront cependant pas à développer le don du vivant, qui ne représente qu'environ 500 à 600 greffes par an. Les listes de demandeurs de greffe d'organe s'allongent, avec, comme conséquence, 500 à 550 décès par an, faute de donneurs. Afin de créer un cadre plus incitatif, je vous propose un amendement qui pose les bases d'un « statut de donneur d'organes », dont le CCNE avait notamment préconisé la mise en place.
L'article 6 vise à prendre en compte le développement croissant des greffes de cellules souches hématopoïétiques à partir de donneurs haploidentiques. Il ouvre, en dernier ressort, la possibilité de recourir à un greffon en provenance d'un enfant mineur ou d'un majeur protégé au bénéfice de l'un de ses parents, tout en encadrant la procédure afin de renforcer la protection du donneur pressenti : un administrateur ad hoc serait nommé en dehors du cercle familial pour représenter l'enfant en lieu et place de ses parents ou pour donner son avis sur le prélèvement lorsque le majeur protégé fait l'objet d'une tutelle ou d'une habilitation familiale avec représentation à la personne et n'a pas la faculté de consentir lui-même. Les greffes nécessitent une similarité de poids entre le donneur et le receveur ; un enfant de dix ans ne peut pas donner à un parent. Les cas dont a été saisie l'Agence de la biomédecine sont des mineurs de 17 ans. Pour plus de simplicité, je vous proposer d'abaisser l'âge du consentement du mineur à 16 ans afin qu'il puisse exprimer lui-même son consentement devant le juge, sans recourir au truchement - peu utile à mes yeux à cet âge-là - d'une tierce personne.
L'article 7 vise à reconnaître une place plus importante au principe d'autonomie des majeurs protégés en faisant entrer dans le droit commun du don d'organes, de tissus et de cellules les majeurs qui font l'objet des mesures de protection juridique les plus légères, soit l'assistance ou la représentation aux biens. Il tend également à étendre le régime du consentement présumé en matière de prélèvement d'organe post mortem à tous les majeurs protégés. Sur ce dernier point, le Gouvernement me semble aller trop loin. Je vous propose d'exclure les personnes faisant l'objet d'une mesure de protection avec représentation à la personne, car elles sont hors d'état d'agir elles--mêmes du fait d'une altération de leurs facultés mentales ou corporelles et ne seront pas en capacité de comprendre le mécanisme du don présumé et encore moins de s'inscrire sur le registre national des refus ou d'exprimer leur refus à leur entourage.
Je vous proposerai également d'ouvrir le don du sang aux majeurs faisant l'objet d'une mesure de protection juridique avec représentation aux biens et assistance, en alignant le don du sang sur le régime des dons d'organes, de tissus et de cellules par donneur vivant proposé dans le cadre du projet de loi et aux mineurs de 17 ans, reprenant ainsi une disposition de la proposition de loi déposée par le député Damien Abad et adoptée à l'unanimité le 11 octobre 2018 à l'Assemblée nationale. Il est important de permettre aux majeurs protégés et aux mineurs de participer à la solidarité nationale par un geste citoyen.
Il paraît illogique de promouvoir le don du sang lors de la Journée Défense et citoyenneté à laquelle participent les jeunes de 16 à 18 ans, sans leur permettre ensuite de le mettre en pratique. Mon amendement s'appuie sur le cadre fixé par la directive européenne du 22 mars 2004, qui fixe les critères d'admissibilité pour les donneurs de sang : l'âge du don ne peut pas être inférieur à 17 ans et un consentement écrit de l'un des parents ou du tuteur légal est nécessaire dans ce cas.
La suppression, par l'article 20, de la proposition systématique d'un délai de réflexion en cas d'interruption médicale de grossesse (IMG) pour motif foetal et la clarification des dispositions applicables en matière d'IMG chez la femme mineure sont bienvenues. Elles permettent de garantir aux femmes enceintes les mêmes droits, en termes de consentement et d'autonomie, qu'en cas d'interruption volontaire de grossesse (IVG). De même, l'introduction dans la loi d'un encadrement des pratiques de réduction embryonnaire ou foetale en cas de grossesse multiple participe d'une sécurisation et d'une plus grande transparence de ces interventions réalisées pour prévenir des complications pour la mère, les embryons ou les foetus.
En revanche, rien ne justifie, comme cela est prévu par l'article 21, d'introduire une clause de conscience spécifique des professionnels de santé en matière d'IMG, dès lors qu'une clause de conscience générale, permettant de ne pas accomplir un acte contraire à ses convictions, bénéficie déjà aux professionnels de santé. Ceux qui souhaitent l'exercer n'ont pas attendu ce projet de loi pour le faire.
Pour mémoire, l'existence dans la loi d'une clause de conscience spécifique à l'IVG découle des débats parlementaires qui avaient entouré l'adoption de la loi du 17 janvier 1975 relative à l'interruption volontaire de grossesse : le Gouvernement avait alors consenti à cette disposition dans un souci d'apaisement.
En conséquence, je vous propose de supprimer la mention dans le projet de loi d'une clause de conscience spécifique des professionnels de santé en matière d'IMG, qui n'existe pas aujourd'hui, car elle est déjà couverte par la clause de conscience générale.
Sur les enfants présentant une variation du développement génital, l'article additionnel 21 bis a été introduit en séance publique à l'Assemblée nationale par l'adoption, avec l'avis favorable du Gouvernement, d'un amendement du groupe La République en Marche. S'il ne présente pas de lien très évident avec le texte d'origine, il met en lumière un sujet important : les enfants présentant des variations du développement génital, sur lesquels des interventions chirurgicales ou hormonales lourdes sont parfois opérées à un âge très précoce. Le dispositif proposé se contente de mettre dans le dur de la loi l'orientation de ces enfants vers les centres de référence de maladies rares du développement génital afin qu'ils puissent bénéficier de la meilleure prise en charge possible, c'est-à-dire d'une prise en charge auprès de professionnels spécialisés et expérimentés, qui proposent collégialement un diagnostic et des solutions thérapeutiques, en veillant à la bonne information et à l'accompagnement psycho-social de l'enfant et de sa famille. Il suit ainsi une recommandation émise par le CCNE dans son avis 132 publié en novembre dernier. Je proposerai deux amendements, l'un permettant d'intégrer le centre de référence des maladies endocriniennes de la croissance et du développement qui traite de cas d'hyperplasie congénitale des surrénales dans le dispositif ; et l'autre visant à soumettre le diagnostic et la prise en charge des variations du développement génital à un référentiel de bonnes pratiques unique, arrêté par la Haute Autorité de santé (HAS) en concertation avec les parties prenantes, dont les associations de patients. C'est également une recommandation formulée dans ledit avis du CCNE.
Je vous proposerai enfin plusieurs amendements précisant la rédaction de l'article 22, relatif à l'autoconservation pour motifs pathologiques de gamètes et de tissus germinaux, notamment pour protéger les personnes mineures souvent concernées par cette démarche.
M. Olivier Henno , rapporteur . - Les travaux de notre commission spéciale ont été d'une très grande densité, et j'ai été heureux de rencontrer l'excellence française dont les médias parlent trop peu : des grands médecins, des personnes engagées, éclairées, cherchant le progrès et son humanisation.
L'éthique à la française concerne les médecins, mais aussi les philosophes, les juristes, tous les citoyens, nous obligeant à l'humilité : ce n'est pas facile dans notre société de défiance, alors qu'il faut créer de la confiance. L'éthique à la française, ce n'est pas obéir aux seules aspirations individuelles comme dans les pays anglo-saxons ni être obsédé par un projet collectif comme en Asie, mais rechercher, à partir d'un socle de valeurs collectives, ce qui produira la norme.
Ce n'est pas non plus la peur de l'avenir, qu'il faut aborder de manière éclairée, en améliorant la vie sans la bouleverser. L'éthique à la française, c'est la gratuité du don, l'attention aux plus fragiles, la bienveillance. Ces principes ont guidé nos travaux, qui nous ont fait grandir sans faire disparaître notre part de doute.
La génétique - partie du projet de loi dont je suis rapporteur - connaît aujourd'hui une véritable révolution. Elle est source d'espoir pour le diagnostic et la prise en charge thérapeutique, notamment dans les domaines de la médecine prédictive et de la médecine de précision, en permettant, par exemple, la mise au point de thérapies géniques ciblées dans le traitement de maladies rares ou de cancers.
Le développement des techniques de séquençage haut débit et la démocratisation, sur le plan économique, de l'accès aux tests génétiques, font néanmoins craindre une mauvaise appréhension par la société de données génétiques de plus en plus massives, dont la signification diagnostique et les conséquences médicales restent encore en grande partie indéterminées.
Dans le prolongement d'une proposition de loi adoptée par le Sénat en juin 2018, l'article 8 autorise la réalisation, dans l'intérêt de la parentèle, d'un examen des caractéristiques génétiques sur une personne décédée, sauf opposition de cette personne manifestée de son vivant, lorsqu'est suspectée une anomalie génétique pouvant être responsable d'une affection grave justifiant des mesures de prévention ou de soins qui pourraient bénéficier aux membres de la famille.
Ces examens génétiques post mortem seront effectués à partir d'échantillons déjà conservés ou prélevés dans le cadre d'une autopsie. Les pratiques de conservation des échantillons biologiques prélevés par les laboratoires de biologie médicale pouvant varier d'un laboratoire à un autre, il pourrait être utile d'harmoniser ces pratiques par la publication de règles de bonnes pratiques en matière de conservation et de traçabilité de ces échantillons.
L'article 9 prévoit de renforcer la possibilité de transmettre une information génétique dans les situations de rupture du lien de filiation biologique, c'est-à-dire entre tiers donneurs et personnes nées d'un don, et entre parents de naissance et personnes nées dans le secret, dans un souci d'égalité d'accès de ces personnes, qui de fait n'ont pas connaissance des antécédents médicaux de leurs apparentés biologiques, aux mesures de prévention ou de soins, tout en préservant rigoureusement l'anonymat des personnes concernées.
Comme l'Assemblée nationale l'a fait pour les tiers donneurs et les personnes nées d'un don, je vous proposerai de rendre automatique l'alerte, par l'intermédiaire du médecin prescripteur de l'examen et du Conseil national d'accès aux origines personnelles (Cnaop), de la personne née dans le secret ou du parent de naissance sur l'existence d'une information génétique potentiellement majeure, voire vitale.
En aucun cas, ce mécanisme d'alerte ne donnera lieu à la révélation de l'identité de la personne initialement concernée par l'examen génétique ni à la révélation de l'anomalie génétique en cause ou des risques associés, dans le respect du droit de toute personne d'être tenue dans l'ignorance d'éventuelles prédispositions génétiques.
L'article 10 maintient le principe selon lequel un examen des caractéristiques génétiques ne peut être envisagé qu'en cas d'antécédent familial connu ou de symptôme d'une maladie d'origine potentiellement génétique. Rien n'est en revanche prévu pour prémunir les personnes ayant recours à des tests génétiques en accès libre, notamment sur Internet, contre les risques qu'emporte la délivrance d'une information génétique en dehors de toute consultation médicale, l'interdiction de ces tests restant aujourd'hui purement virtuelle.
En cohérence avec la position du CCNE sur ce sujet, je vous proposerai donc, à titre expérimental, d'ouvrir l'accès en population générale aux examens génétiques, notamment dans le cadre d'un dépistage préconceptionnel pour un couple s'engageant dans un projet parental, en l'absence d'antécédent familial connu ou de symptôme, pour la recherche de mutations génétiques dont la signification diagnostique est connue. Le Gouvernement pourra ainsi limiter les anomalies recherchées à une liste de mutations génétiques établie en concertation avec l'Agence de la biomédecine et la HAS.
En outre, face à l'ineffectivité de l'interdiction du recours aux tests génétiques en accès libre, il me semble indispensable d'autoriser, pour mieux l'encadrer, l'accès des examens génétiques à visée généalogique sous réserve du respect de conditions de nature à préserver les droits des personnes dans le traitement de données aussi sensibles. Conformes à un référentiel de qualité établi par l'agence de la biomédecine, ces tests ne pourront avoir pour objet de délivrer une information génétique d'ordre médical, ni ne pourront servir de fondement à des actions visant à faire valoir des droits patrimoniaux ou extrapatrimoniaux, notamment dans le cadre d'une démarche d'établissement d'un lien de filiation.
Sur l'intelligence artificielle et les neurosciences, le projet de loi crée, à l'article 11, un cadre juridique pour l'utilisation d'un traitement algorithmique de données massives lors de la réalisation d'un acte médical. Le développement de l'intelligence artificielle (IA) est très important en médecine. L'IA peut avoir des effets très positifs pour le patient, en améliorant l'efficacité des diagnostics ou en aidant à la prise de décision thérapeutique. Mais il ne s'agit pas d'une technique infaillible, elle peut présenter des biais. Les professionnels de santé doivent demeurer seuls décisionnaires en matière de soins apportés aux patients : c'est toujours l'humain qui doit décider en dernier ressort. Je présenterai donc plusieurs amendements pour renforcer les garanties applicables à l'utilisation de ces technologies, sans toutefois freiner son développement. Je vous proposerai ainsi que le patient soit informé en amont de l'utilisation d'un traitement algorithmique et qu'aucune décision médicale ne puisse exclusivement se fonder sur un traitement algorithmique. Ces principes ont fait consensus lors des auditions.
S'agissant des neurosciences, l'article 12 du projet de loi propose notamment de modifier l'article 16-14 du code civil, qui régit le recours aux techniques d'imagerie cérébrale, créé par la loi du 7 juillet 2011 relative à la bioéthique. Ces techniques ne sont aujourd'hui autorisées qu'à des fins médicales, de recherche scientifique ou dans le cadre d'expertises judiciaires, avec le consentement exprès de la personne concernée. Restons-en au droit en vigueur sur ce point qui me paraît satisfaisant, ce qu'avait également conclu le Conseil d'État dans son étude en 2018.
Enfin, l'article 13 du projet de loi confère au ministre de la santé le pouvoir d'interdire, en cas danger grave ou de suspicion de danger grave, les actes, procédés, techniques ou équipements qui ont pour objet de modifier l'activité cérébrale. Je vous propose d'exclure les équipements qui sont des dispositifs médicaux, car ils relèvent déjà des pouvoirs de police de l'Agence nationale de sécurité du médicament et des produits de santé (ANSM), qui peut interdire leur mise en service ou leur utilisation dans les mêmes hypothèses que celles de l'article 13.
Mme Corinne Imbert , rapporteure . - Je tiens à remercier mes collègues rapporteurs pour la qualité de nos échanges. Le travail que nous avons mené a été passionnant et nous conduit à nous interroger sur les limites acceptables du progrès scientifique. En tant que représentante du Sénat au CCNE, j'ai participé à la rédaction de son avis 129. Je me suis particulièrement intéressée à la question de l'équilibre qui doit être trouvé entre recherche et risques de dérives.
Le titre IV du projet de loi comporte une série de dispositions destinées à soutenir une recherche éthique et responsable au service de la santé humaine, notamment sur l'embryon et les cellules souches embryonnaires ou pluripotentes induites.
Le régime de déclaration préalable pour les recherches sur les cellules souches embryonnaires humaines, mis en place à l'article 14, permet d'acter la différence de nature de ces recherches avec celles sur l'embryon, qui continueront de faire l'objet d'un régime d'autorisation. Les cellules souches embryonnaires humaines n'ont en effet pas la capacité de former spontanément un embryon : les recherches portant sur ces cellules ne soulèvent donc pas les mêmes enjeux éthiques qu'une intervention sur l'embryon.
Afin de sécuriser sur le plan juridique les recherches menées sur les embryons surnuméraires, il me semble indispensable de préciser leurs prérequis, dont certains sont inadaptés au contexte scientifique actuel et donnent aujourd'hui lieu à des incertitudes ou des différences d'interprétation exploitées dans le cadre de contentieux quasi systématiques.
Il pourrait ainsi être utile de tenir compte du fait que les recherches sur l'embryon peuvent non seulement s'inscrire dans une finalité médicale déterminée, mais également poursuivre un objectif d'amélioration de la connaissance de la biologie humaine. C'est en particulier le cas dans le cadre de travaux de recherche fondamentale qui ne peuvent par définition anticiper avec précision les résultats de la recherche et les applications médicales qui pourraient en être tirées.
De même, le critère de l'absence de méthodologie alternative me paraît devoir être apprécié au regard de la pertinence scientifique des modèles alternatifs à la recherche sur l'embryon par rapport aux objectifs de la recherche considérée. Même si les cellules souches embryonnaires et les cellules souches pluripotentes induites permettent aujourd'hui d'envisager la constitution de modèles embryonnaires à usage scientifique susceptibles de mimer certaines étapes du développement embryonnaire, ces modèles ne permettent pas d'égaler les propriétés de l'embryon humain et donc de faire l'économie d'une recherche sur ce dernier.
Par ailleurs, avec le souci de permettre des avancées dans la compréhension du développement embryonnaire dans le respect des principes éthiques, je vous proposerai d'autoriser, à titre dérogatoire, le développement in vitro d'embryons jusqu'au 21 e jour suivant leur constitution dans le cadre de protocoles de recherche spécifiquement dédiés à l'étude des mécanismes de développement embryonnaire au stade de la gastrulation. La limite de culture in vitro de l'embryon à quatorze jours est en effet aujourd'hui réinterrogée par plusieurs pays qui envisagent de la repousser pour permettre des recherches indispensables à une meilleure connaissance du processus de différenciation des cellules souches embryonnaires.
Certaines recherches conduites sur les cellules souches embryonnaires ou les cellules souches pluripotentes induites requièrent une vigilance particulière. Il s'agit notamment de la possibilité de différencier ces cellules en gamètes, de les insérer dans des tissus extraembryonnaires afin de constituer des modèles mimant l'embryon ou de les insérer dans des embryons provenant d'autres espèces.
Ce dernier type de recherche n'est pas sans soulever d'importantes questions éthiques quant aux limites à poser au franchissement de la barrière des espèces. Par conséquent, il me semble indispensable de supprimer la possibilité, à l'article 14, de créer des embryons chimériques résultant de l'insertion de cellules souches embryonnaires humaines dans un embryon animal. En outre, il convient, à l'article 15, de poser deux verrous à la création d'embryons chimériques par l'adjonction à un embryon animal de cellules pluripotentes induites d'origine humaine : ces embryons ne pourront donner lieu à parturition, si bien qu'en cas de transfert chez la femelle, la gestation devra être obligatoirement interrompue dans un délai approuvé par l'Agence de la biomédecine, apprécié au regard des délais gestationnels propres aux espèces animales considérées ; la contribution des cellules d'origine humaine au développement de l'embryon chimérique ne saurait dépasser un seuil approuvé par l'Agence de la biomédecine.
Pour mémoire, le CCNE lui-même, dans son avis sur la révision de la loi de bioéthique, estimait qu'un encadrement des embryons chimériques résultant de l'insertion de cellules pluripotentes induites (iPS) humaines dans un embryon animal était nécessaire, « en particulier si les embryons chimériques sont transférés chez des femelles et donnent naissance à des animaux chimères avec le risque, chez le gros animal, que les cellules humaines se développent et induisent certaines caractéristiques humaines », notamment morphologiques ou neurologiques.
Concernant le diagnostic prénatal, l'article 19 vise à actualiser sa définition et à préciser la démarche de la femme enceinte et des couples en cas notamment de découvertes incidentes. Je vous proposerai d'étendre une définition encore restrictive au regard de la pratique réelle et des traitements et soins rendus possibles par le développement de la médecine foetale.
Concernant le diagnostic préimplantatoire (DPI), je vous propose de revenir sur la décision de l'Assemblée nationale, à l'article 19 bis A, d'abroger le DPI couplé à la recherche de compatibilité HLA (human leukocyte antigen) : cette technique, introduite par la loi du 6 août 2004 relative à la bioéthique et pérennisée en 2011, permet la naissance d'un enfant non seulement sain de la maladie de son aîné, mais également compatible en vue d'une greffe. Même si elle n'est plus pratiquée depuis 2014, cette technique, strictement encadrée, présente toujours un intérêt thérapeutique dans des situations certes très rares, mais humainement délicates.
Je vous présenterai également un amendement visant à autoriser, à titre expérimental et dans des indications ciblées, le recours au DPI pour la recherche d'aneuploïdies (DPI-A) c'est-à-dire d'anomalies chromosomiques. À l'heure actuelle, cette analyse in vitro des embryons avant leur implantation ne peut être réalisée que pour rechercher une pathologie génétique ciblée dont est porteur l'un des parents. Or, les sociétés savantes en médecine de la reproduction comme en cytogénétique mettent en avant l'intérêt d'y avoir recours dans des cas d'échecs répétés de fécondation in vitro (FIV) ou de fausses-couches à répétition. Cela vise à améliorer la prise en charge des femmes lors de ces parcours éprouvants d'AMP.
Enfin, en matière de gouvernance, je vous propose de confirmer le maintien souhaité par l'Assemblée nationale de la mission de l'Agence de la biomédecine dans le domaine des nanobiotechnologies et de maintenir également l'élaboration par l'agence d'un référentiel permettant d'évaluer la qualité des tests génétiques en accès libre, dans un contexte de recours croissant des Français aux tests ADN sur Internet.
M. Alain Milon , président . - Je remercie nos rapporteurs pour leur excellent travail.
M. Jacques Bigot . - Je remercie également nos quatre rapporteurs, notamment pour l'organisation des auditions qui ont permis d'éclairer notre commission. L'éthique ne relève pas de la médecine, mais du législateur, qui n'est pas toujours totalement averti. Comment faire évoluer les lois de bioéthique au regard des progrès, sans cesse plus importants, de la médecine génétique ? Se pose aussi, en filigrane, la question du maintien du niveau de la recherche génétique en France.
Comme l'a bien dit notre rapporteur Bernard Jomier, les craintes suscitées par les lois de bioéthique successives ne se sont pas réalisées et les protections posées ont été bonnes. Sur l'AMP et l'évolution de la parentalité, mesurons d'où nous venons, ainsi que ce qui se passe dans d'autres pays. Le rôle fondamental du père est aujourd'hui une évidence, mais tel n'a toujours pas été le cas, notamment dans nos sociétés patriarcales : le rôle du père a beaucoup changé depuis 1965. Voyons aussi les sociétés africaines au sein desquelles le grand-père joue un rôle majeur. N'ayons donc pas trop de certitudes. Un enfant privé de présence paternelle est-il en danger ? Je ne le pense pas. Souvenons-nous de ce qui se passait avant la réforme de la filiation de 1972 et celle du divorce de 1975. Prenons acte de ces évolutions.
Les familles monoparentales rencontrent des problèmes de ressources, de précarité et d'absence de choix de vie. Mais une femme seule peut très bien décider d'élever un enfant toute seule.
Le droit de la filiation est un droit d'ordre public qui a considérablement évolué. Jusqu'en 1972, la filiation légitime était fondée sur le mariage, avec la présomption irréfragable que le père était le mari de la femme, la filiation naturelle ne faisait pas entrer l'enfant dans la famille et la filiation adoptive pouvait concerner des enfants en très bas âge. Les évolutions de la science permettent aujourd'hui de satisfaire le désir d'enfant de couples qui ne peuvent pas avoir d'enfant. La question du désir d'enfant ne pose pas de problème. La vraie question est la suivante : quel est l'intérêt supérieur de l'enfant ? Il n'est pas possible de le définir avec certitude.
Devons-nous freiner ou accepter les évolutions permises par la science et attendues par une partie de la société ? Mon groupe considère que nous devons à notre société d'accompagner ces évolutions, en acceptant les freins nécessaires, mais sans en ajouter d'inutiles.
La société française accepte aujourd'hui le mariage pour tous et admet que ces couples aient des enfants. La situation juridique de ces enfants au sein de ces familles homoparentales va progressivement se stabiliser et nous pourrons évaluer leur situation dans quelques années. Elle ne devrait pas être plus compliquée que celle des enfants des couples séparés aujourd'hui. Vous mettez des freins conservateurs à l'évolution de notre société qui va pourtant plus vite que nous ne le pensons.
Dans son avis sur le présent projet de loi, le Conseil d'État a rappelé les trois principes qui s'appliquent en matière d'éthique : les principes de dignité, de liberté et de solidarité. L'extension de l'AMP risque-t-elle de mettre des enfants dans une situation indigne ? Je ne le pense pas. Peut-on préjuger de la liberté de l'enfant à naître d'avoir un père et une mère, ou deux pères, ou deux mères ? Nous n'en savons rien. Enfin, je considère, au nom de la solidarité, que ce qui se pratique ailleurs doit aussi pouvoir se pratiquer en France : mieux vaut autoriser nos concitoyens, afin de leur faire bénéficier de protections conformes aux valeurs de la République.
Je suis inquiet des propositions de suppression d'articles de notre rapporteur Muriel Jourda. Je suis moins inquiet s'agissant des propositions de nos trois autres rapporteurs et nous aurons un débat.
M. Guillaume Chevrollier . - Je tiens à féliciter nos rapporteurs et à remercier notre président qui a permis que toutes les sensibilités s'expriment, même si la loi ne pourra satisfaire tout le monde. Ce texte, qui traite de la condition humaine, de la filiation, du corps humain et de la bioéthique, nous pose des questions intimes, complexes et fondamentales.
J'ai déposé des amendements, car de trop nombreuses questions restent en suspens. En l'espèce, je suis favorable à l'application du principe de précaution, car nous devons dénoncer les menaces qui pèsent sur les êtres humains et pèseront sur les générations futures. La médecine est de plus en plus puissante et nous prenons de moins en moins de précautions pour limiter les tentations des chercheurs. Ce que la science veut ou peut n'est pas forcément souhaitable pour l'homme. Nous devons encadrer et orienter le progrès.
Sur l'AMP ouverte à toutes les femmes, nous nous apprêtons à modifier en profondeur la filiation et à supprimer le père du modèle légal filiatif, sans écouter les enfants ni reconnaître leurs droits. Or l'enfant a envie d'un père et d'une mère, et pas d'un père anonyme ou décédé. En outre, l'extension de l'AMP ne conduira-t-elle pas nécessairement à la gestation pour autrui (GPA), ne serait-ce qu'en vertu du principe d'égalité ?
Sur le plan scientifique, la manipulation des gènes est dangereuse ; nous n'avons que peu de visibilité sur le travail sur l'embryon ; nous n'avons eu aucun débat sur la FIV à trois parents ni sur le ciseau génétique Crispr-Cas9 ; la ministre s'est heureusement opposée à l'extension du DPI-A, qui aurait conduit à l'eugénisme absolu et au mythe de l'enfant sain. Dépensons de l'argent pour supprimer les maladies et non pas les malades !
Nous avançons les yeux fermés et légiférons sur des sujets que nous maîtrisons insuffisamment. Évitons de nous engager dans un projet aventureux. Sur ce sujet, je me considère comme un lanceur d'alerte.
M. Roger Karoutchi . - C'est avec la sérénité du profane que j'aborde ce texte. Sur ses aspects scientifiques, je m'en remettrai à l'avis des rapporteurs.
De mon point de vue, il n'appartient pas au législateur de se prononcer sur les sujets éthiques : ceux-ci ne relèvent pas du champ politique, mais du domaine personnel, moral ou religieux. Qui suis-je pour dire aux 67 millions de Français comment ils doivent vivre en famille ? Je viens d'un milieu extrêmement conservateur, très religieux et pratiquant, mais lorsque je vote la loi, je ne considère pas mon milieu d'origine, mais la société française. Je peux voter une loi que je n'accepterais pas pour moi-même.
La présomption du père me semble peu assurée, depuis Georges Feydeau et Eugène Labiche ! Je ne vois pas à quel titre je refuserais, par principe, l'extension de l'AMP, même si je suis favorable à l'édiction de garde-fous pour éviter les dérives et les détournements. Le droit a souvent été un cadre juridique imposé par la classe dirigeante, mais il évolue. Les principes humains doivent parfois primer les principes du droit. Je ne suivrai donc pas la position de notre rapporteur sur l'AMP.
M. Thani Mohamed Soilihi . - Nous devons faire preuve d'humilité sur ces sujets sensibles. Personne ne détient de vérité absolue.
Il n'est pas question de légaliser tout ce que la technique permet. Tout ce qui est possible techniquement n'est pas éthiquement souhaitable. Nos travaux nous ont parfois confortés dans nos positions et nous ont parfois fait changer d'avis sur certains points. Merci à nos rapporteurs pour leur excellent travail. L'extension de la PMA aux femmes seules et aux couples de femmes emporte des conséquences sur la filiation et sur les gamètes. Notre groupe, dans sa majorité, est favorable au texte issu des travaux de l'Assemblée nationale. Certains collègues ne partagent pas notre avis sur l'article 1 er - Michel Amiel le dira mieux que moi -, mais la majorité des membres du groupe votera le texte enrichi par le Sénat dans l'esprit de la bioéthique à la française.
Mme Marie-Pierre de la Gontrie . - Je partage la réflexion de Roger Karoutchi sur l'éthique, mais pas ses conclusions sur le rôle du législateur. Chacun connaît ici la phrase de Lacordaire : « Entre le fort et le faible, entre le riche et le pauvre, entre le maître et le serviteur, c'est la liberté qui opprime et la loi qui affranchit. » C'est notre responsabilité, dans ce domaine complexe, de dire ce qui est autorisé ou non.
Cette commission a travaillé dans des conditions de grande courtoisie. J'appartiens à une formation politique favorable à l'extension de la PMA, mais j'ai voulu examiner à fond cette question sans a priori ni certitudes.
C'est un choix politique au sens noble du terme : le droit n'impose ni le statu quo ni l'évolution, le Conseil d'État l'a dit. C'est la reconnaissance d'un pluralisme familial qui, comme Jacques Bigot l'a rappelé au sujet de la place du père, s'est imposé. Depuis 1966, une personne seule peut adopter ; la loi sur le mariage pour tous en 2013 a donné une reconnaissance légale aux couples homosexuels.
Muriel Jourda nous dit que ce n'est pas parce que quelque chose existe qu'il faut l'autoriser. Certes, mais entre 2 000 et 4 000 femmes ont recours à la PMA à l'étranger. La grossesse est suivie en France, l'enfant naît en France, la filiation est en France, mais la PMA est pratiquée à l'étranger, c'est tout de même particulier... Ce projet de loi veut reconnaître et normaliser l'existence de ces familles.
Il n'y a pas de droit à l'enfant. Le Conseil d'État l'a dit, l'enfant est un sujet. Il y a un droit à l'accès à une technique, mais rien ne dit que celle-ci fonctionnera. Il n'y a pas de droit à l'enfant, mais le désir d'enfanter existe. En parlant avec des gynécologues, j'ai pu me rendre compte à quel point la souffrance des femmes qui ne parviennent pas à enfanter était grande, ce n'est pas évident pour quelqu'un qui, comme moi, a eu la chance d'en avoir banalement - si j'ose dire.
Est-il légitime que la médecine intervienne hors du domaine de la pathologie ? Elle le fait déjà ! Comme Roger Karoutchi, je ne suis pas spécialiste. Une médecine de confort est apparue, comme la médecine esthétique - je suis une femme, je peux donc en parler... (Sourires.) Quel est l'intérêt de l'enfant ? En normalisant la situation de ces enfants issus de PMA, nous les sortons d'une certaine forme de clandestinité.
Certains disent : les couples de femmes n'ont qu'à adopter. D'abord, ce n'est pas la même chose de porter un enfant et d'adopter. Et puis, nous le savons tous, en France, il y a peu d'enfants adoptables.
Beaucoup d'organismes plus compétents que moi ont validé l'extension de la PMA : le conseil consultatif national d'éthique, la Commission nationale consultative des droits de l'homme (CNCDH) ; de même, beaucoup de médecins. De nombreux pays d'Europe y ont recours.
Je me demande, en écoutant les arguments qui y sont opposés, si accepter la PMA pour les couples de femmes et les femmes seules, ce ne serait pas faire le deuil du mythe de la famille parfaite, composée d'un papa, d'une maman, des enfants, du chien... Or, Roger Karoutchi l'a rappelé, cela fait longtemps que la famille ne ressemble plus à cela. Et quelle garantie avons-nous que ce modèle traditionnel rende les enfants heureux ?
Les études n'ont pas assez de recul, mais elles vont toutes dans le même sens : ce qui compte, c'est l'environnement familial et le désir d'enfant. Sur ce point, le professeur Nisand a eu un mot frappant : la seule différence avec les autres enfants, c'est qu'ils sont tous très désirés - or ce n'est pas le cas de tous les autres, malheureusement. On parle de l'absence du père ? Mais, aujourd'hui, un quart des familles sont monoparentales. Au moins, dans ce cas-ci, ne s'agit-il pas d'une monoparentalité subie.
Nous sommes favorables à la prise en charge par la sécurité sociale - c'est une question d'égalité - et favorables à l'accès aux origines, car c'est dans l'intérêt de l'enfant. La réforme de la filiation est le corollaire de ces changements et nous la soutenons donc. Enfin, l'autoconservation des gamètes n'est pas une question de confort. On a beaucoup lu sur le fait que des grands groupes américains, des Gafa, s'y intéressaient de près... Bien encadrée, l'autoconservation des gamètes permettra opportunément aux femmes d'avoir accès à la maternité dans des conditions plus souples qu'aujourd'hui.
M. Michel Amiel . - J'apprécie la modération des propos des uns et des autres dans ce débat complexe. Tous les arguments présentés pour ou contre l'extension de la PMA tiennent la route. Thani Mohamed-Soilihi l'a dit, la majorité de notre groupe est en faveur de cet article 1 er , mais ce n'est pas mon cas, comme d'autres collègues.
Ce qui me gêne le plus dans ce texte, c'est qu'il ne dissocie pas ce qui relève de l'éthique sociétale de ce qui relève de la bioéthique. Au moment du vote, je devrai choisir entre le soutien aux grands progrès qu'il propose dans ce domaine, et ma position en matière d'éthique sociétale.
J'ai abordé la question sans a priori, même si, étant médecin, j'y avais déjà un peu réfléchi. Finalement, le Gouvernement a voulu poser cela en termes politiques. Dans ces termes, il est très libéral d'ouvrir la PMA à toutes les femmes. Le manque de gamètes sera probablement aggravé par la levée de l'anonymat des donneurs...
M. Alain Milon , président . - Provisoirement.
M. Michel Amiel . - Je ne suis pas de votre avis. Ce qui deviendra possible ici continuera malgré tout à être fait ailleurs pour des raisons de faisabilité.
Deux éminents personnages m'ont conforté dans ma position. Jacques Testart est, avec René Frydman, l'un des pères du premier bébé-éprouvette. C'est un homme de gauche... vigoureuse. Dans un excellent article publié dans Charlie Hebdo, il dit que la PMA pour toutes conduit à une marchandisation du corps humain. La vie a profondément séparé ces deux scientifiques, mais je les ai rencontrés tous les deux et ils m'ont dit la même chose.
Sylviane Agacinski a publié un opuscule très bien documenté. Ce qui m'a gêné, c'est qu'on ait pu l'empêcher de s'exprimer à l'université, lieu du débat par excellence. Tout s'est passé comme si le débat était tranché et qu'il ne fallait pas s'opposer au sens de l'Histoire. C'est sans doute le sens de l'évolution actuelle, mais doit-on toujours s'y conformer ?
Dernier point, la reconnaissance d'un droit-créance. La société doit-elle avoir cette créance vis-à-vis d'un couple de femmes, l'accès à la procréation ? Pour moi, non. Mme de la Gontrie nous dit qu'être mère et porter un enfant sont des choses différentes. Il n'y aura donc pas égalité dans un couple de femmes entre la mère qui porte et la mère d'intention. Un film consacré au problème des mères porteuses, Diane a les épaules, l'a bien montré : porter son enfant, ce n'est pas la même chose qu'être mère par procuration.
M. Philippe Bas . - Il est difficile d'intervenir après ces rapports et ces prises de paroles très complets. Quand on touche à la liberté de chacun, renvoyer à la responsabilité individuelle est un bon réflexe : à chacun de juger de ce qui est bien et de ce qui est mal. Mais il y a une limite à ce principe : la loi reste une source d'inspiration pour les comportements, et elle apporte une protection aux plus faibles. Michel Amiel a raison : il est ennuyeux de mélanger bioéthique et éthique sociétale dans un même texte ; ce qui l'est encore plus, c'est de devoir traiter un texte qui, tel qu'issu des travaux de l'Assemblée nationale, rétroagit sur des points qui ont été réglés récemment ou qui n'ont pas besoin de législation pour l'être.
Si nous devons intervenir en matière d'AMP, nous ne devrions le faire qu'en tant que de besoin, sans remettre en cause la filiation maternelle liée à l'accouchement. La protection de l'enfant exige en effet que les contestations de la filiation et leurs causes soient les plus limitées possible. Or quoi de plus sûr que l'accouchement ? À mes yeux, c'est donc un recul de penser que la maternité d'intention serait supérieure à l'accouchement.
Je souscris aux propos de Roger Karoutchi : si l'accès à l'AMP est ouvert, encore faut-il qu'il le soit avec discernement, pour assurer les meilleures conditions, dans l'intérêt de l'enfant à naître.
Inutile de trop nous attarder : aucun de nous ne changera d'avis. La question posée au législateur est : faut-il organiser la naissance d'enfants sans père ? Je réponds que non. Plutôt qu'un bienfait, je vois dans l'absence totale de référence paternelle et même de lignée paternelle un manque pour l'enfant. Je crois que c'est très imprudent. Puisque la question de la liberté de chacun est posée, je pense que cela ne devrait pas en relever. J'admets qu'on puisse trouver comme moi que c'est un manque et non un bienfait, et accepter que cela relève de la liberté de chacun, comme le fait Roger Karoutchi. Mais ce n'est pas mon cas : je ne crois pas qu'il faille imposer aux médecins de procéder à l'AMP pour toutes les femmes, à l'assurance maladie de la financer aux frais de tous les Français si nous sommes convaincus que ce n'est pas une bonne chose.
Je suivrai notre rapporteur sur sa proposition de supprimer l'article 1 er . Si elle n'était pas suivie, je serais très vigilant sur l'encadrement à construire autour du texte inabouti issu de l'Assemblée nationale.
M. Yves Daudigny . - Le refus de la PMA pour toutes et de la congélation des ovocytes repose sur l'idée que leur interdiction serait une assurance contre la marchandisation, l'eugénisme et même la disparition de l'espèce humaine. Ces craintes sous-entendent que l'interdiction serait préférable à la régulation et que l'on ne saurait disposer de son propre corps. Elles reposent sur l'idée que la technologie n'est pas neutre et pourrait se retourner contre les humains. Je crois au contraire que si elles sont correctement régulées, elles renforceront la famille basée sur l'amour. Au nom de quoi s'opposer au fait que des femmes homosexuelles fondent une famille alors qu'elles ne font de tort à personne. Le faire au nom de la nature et du bien de l'enfant est fragile : cela supposerait une éthique de la nature qui identifie le naturel au moral - or c'est loin d'être évident, en particulier dans la famille. La technologie ne fragilise pas cette dernière. Nous pouvons l'utiliser pour fonder la parentalité sur une base plus solide que la seule procréation : procréer est un acte biologique, être parent est un acte social, affectif, institutionnel. La technologie peut-elle rendre les familles plus accueillantes ? Je pense que oui.
M. Daniel Chasseing . - Félicitons les rapporteurs. Muriel Jourda a bien expliqué les enjeux de l'article 1 er , à la lumière de l'avis de l'Académie de médecine et des psychiatres. J'ai pris connaissance de données rassurantes, même si certains considèrent qu'elles sont issues d'études pas toujours convaincantes. L'évolution des enfants élevés par des couples homosexuels ou par des femmes seules montre qu'ils n'ont pas plus de problèmes de comportement que les autres.
L'Académie de médecine approuve, dans son rapport, l'évolution qui figure dans le texte, et l'accès des personnes issues d'un don de gamètes à leurs origines. Elle ne s'oppose pas formellement à l'autoconservation des ovocytes, qui est une forme de confort et, de ce fait, ne doit peut-être pas être prise en charge par la sécurité sociale.
Les femmes seules peuvent déjà adopter. L'ouverture de la PMA pour elles est l'une des principales réformes qu'apporte ce texte. En général, la naissance d'un enfant résulte d'un projet partagé entre deux personnes. Une femme seule, cela n'est pas la même chose qu'une famille monoparentale, qui devient telle par accident. Il est vrai que les femmes seules qui s'orientent vers une PMA ont mûrement réfléchi leur projet, et qu'elles sont entourées de leur famille, et par des amis.
Je ne voterai pas les amendements de suppression de l'article 1 er , mais je suis d'accord pour limiter la prise en charge de la PMA aux situations résultant de problèmes médicaux et pour introduire le principe d'une évaluation psychologique préalable à la PMA, comme le propose l'amendement de M. Karoutchi. Je pense aussi que le don de gamètes doit faire l'objet de l'accord du conjoint. La conservation des gamètes dans des centres privés n'aboutira pas à ce qu'on pousse au don - d'ailleurs, il n'y aura pas assez de centres.
Mon amendement conditionne l'accès à l'identité du donneur à l'accord de celui-ci, tout en maintenant l'accès des enfants, à leur majorité, aux données non identifiantes.
M. Alain Milon , président . - La PMA perturbe moins notre société que ne la perturberont, dans l'avenir, les recherches sur l'embryon, les cellules-souches embryonnaires, la génétique et l'intelligence artificielle...
Mme Muriel Jourda , rapporteur . - On parle plus volontiers des mots de moins de trois syllabes ! Toutes les positions exprimées sont étayées et respectables. Nous sommes tous quelque peu conservateurs, monsieur Bigot. Aucun de nous ne chante : « du passé faisons table rase ! »
EXAMEN DES ARTICLES
Mme Muriel Jourda , rapporteur . - Les amendements identiques COM-167 , COM-20 , COM-89 rectifié bis, COM-100 et COM-106 suppriment cet article, qui étend l'AMP aux couples de femmes et aux femmes seules.
Mme Catherine Deroche . - C'est un article important, sur un sujet difficile, sur lequel les opinions, toutes différentes, doivent être également respectées. Si nous le votons, je ne vois pas pourquoi nous refuserions la GPA : en quoi le désir d'enfant d'une famille de femmes devrait-il primer celui d'une famille d'hommes ? Étant hostile à la GPA, je voterai ces amendements de suppression. Le don de sperme, qui est un don de patrimoine génétique, ne doit pas être banalisé.
Mme Laurence Rossignol . - Mon groupe ne votera pas ces amendements. Je comprends la première partie du propos du rapporteur, et ses inquiétudes. Mais faire intervenir l'intérêt supérieur de l'enfant pour répondre à la question éthique qui nous est posée, c'est se fourvoyer. D'ailleurs, « the best interest » prévu par la convention sur les droits de l'enfant doit plutôt être traduit par « le meilleur intérêt » : l'intérêt de l'enfant ne prime pas tout autre intérêt. Et je regrette qu'on n'en entende parler que pour ce type de débats, et moins lorsqu'il était question des violences corporelles, par exemple. L'article 1 er ne donnerait pas à tous les couples l'égalité devant le désir d'enfants. Il faut partir de la technique, celle de l'AMP, qui est légale et déjà ouverte aux couples hétérosexuels. Doit-elle leur être réservée ? Non. La GPA, elle, est illégale pour tous, car, en commercialisant le corps des femmes, elle s'apparente à du trafic d'êtres humains.
M. Joël Bigot . - La GPA pose un problème différent : le don de sperme n'est pas comparable à l'engagement que prend une femme de porter un enfant qui n'est pas le sien, et qu'elle n'élèvera pas ensuite. En France, nous n'y sommes pas prêts, notamment du point de vue de l'indemnisation. La double maternité qu'affirme la loi n'est pas négligeable. Ces situations existent déjà, avec des enfants conçus en Belgique, en Espagne ou ailleurs. En l'absence de reconnaissance d'une double filiation, quel sera le lien juridique avec la deuxième mère en cas de séparation ? La PMA étant une réalité, il faut l'accepter - et elle ne nous entraînera pas à la GPA, en tous cas dans l'immédiat.
Mme Catherine Di Folco . - La PMA n'est autorisée, actuellement, que pour les couples hétérosexuels infertiles.
M. Alain Milon , président . - Ou en difficulté.
Mme Catherine Di Folco . - Les propos de M. Bigot, qui parle de couples d'hommes et évoque la GPA, me semblent justifier la crainte, exprimée par Mme Deroche, que nous n'allions vers l'autorisation de la GPA.
Les amendements COM-167, COM-20, COM-89 rectifié bis, COM-100 et COM-106 ne sont pas adoptés.
Mme Muriel Jourda , rapporteur . - Mon amendement COM-170 est un amendement de repli. Pour moi, il n'est pas souhaitable d'ouvrir l'AMP à la monoparentalité, qu'elle soit choisie ou non. Je retire donc aux femmes seules l'accès à l'AMP. Il ne convient pas, non plus, d'ouvrir l'AMP aux couples hétérosexuels fertiles, qui n'en ont pas besoin. Je conserve donc, pour eux, le critère médical. Concernant l'âge, on s'en tenait pour l'instant aux recommandations de bonne pratique de l'Agence de la biomédecine. Le texte propose un décret en Conseil d'État, qui fixerait l'âge limite, pour le remboursement, à 43 ans. Je préfère un renvoi aux recommandations de bonne pratique, car les médecins disent qu'on peut très bien être fertile à 45 ans : ce couperet serait abrupt. Je maintiens l'interdiction du recours à un double don de gamètes, qui peut conduire à la GPA. Il faut aussi conserver le caractère médical de l'équipe pluridisciplinaire du centre, en y incluant un pédopsychiatre, qui serait plus utile qu'un infirmier ayant une compétence en psychiatrie. Il importe en effet que les parents sachent comment se comporter lors de la révélation à l'enfant de ses origines. La vérification de la motivation des demandeurs, en revanche, me paraît superflue ; je la remplace par le fait de s'assurer de la volonté des deux membres du couple à poursuivre leur projet parental par la voie de l'AMP après information sur l'adoption.
Je supprime aussi une précision apportée par l'Assemblée nationale, qui avait heurté les médecins, sur le fait que l'évaluation médicale ne peut conduire à débouter les demandeurs au regard notamment de leur orientation sexuelle, puisque cette infraction, la discrimination, existe déjà d'une manière générale, et qu'il était infamant à leur égard de les en soupçonner, alors qu'ils ont été unanimement favorables à l'extension de l'AMP aux couples de femmes. Je supprime aussi la précision sur le fait que le couple est incité à anticiper les conditions permettant d'informer l'enfant, avant sa majorité, de ce qu'il est issu d'un don, laquelle est ambiguë dans sa formulation et redondante dans son objectif avec l'information prévue par ailleurs sur l'accès à l'identité du tiers donneur. Enfin, je propose que les conditions actuelles de prise en charge par la sécurité sociale, avec exonération du ticket modérateur pour les seules AMP fondées sur un motif médical ou pathologique, soient maintenues.
Mme Catherine Deroche . - Ce qui est compliqué, c'est que je suis d'accord avec certains points, et moins avec d'autres.
M. Alain Milon , président. - Oui, c'est le problème. Je suis d'accord pour limiter l'exonération de ticket modérateur à l'acte de FIV. La grossesse doit être ensuite prise en charge comme n'importe quelle autre.
Pour le reste, je suis gêné par l'obligation de présence d'un pédopsychiatre. Il faut des pédopsychiatres ; malheureusement, aujourd'hui, en France, des régions entières en sont privées. Dans certaines facultés, à Rennes par exemple, il n'existe tout simplement pas de chaire de pédopsychiatrie. En attendant de former des pédopsychiatres - il faut quatorze ans pour cela -, une telle précision me semble donc inutile, pour ne pas dire dangereuse.
D'une manière générale, je trouve cet amendement très intéressant, mais je ne saurais le voter en l'état, parce qu'il ferme l'accès à l'AMP aux femmes seules.
M. Daniel Chasseing . - Je suis favorable à la disposition visée par le dernier paragraphe de l'objet, c'est-à-dire au maintien des conditions actuelles de prise en charge par la sécurité sociale, avec exonération du ticket modérateur, seules les AMP fondées sur un motif médical ou pathologique donnant lieu à prise en charge par l'assurance maladie.
Par ailleurs, je considère que les femmes seules doivent avoir le droit d'accéder à la PMA. Le projet d'une femme seule est en général très mûrement réfléchi ; en principe, cela se passe bien.
Mme Muriel Jourda , rapporteur . - S'agissant des pédopsychiatres, j'ai bien conscience que l'obligation créerait une difficulté. C'est pourquoi nous indiquons que l'équipe pluridisciplinaire peut être composée d'un psychologue de l'enfance en lieu et place d'un pédopsychiatre. Les premiers sont peut-être plus nombreux que les seconds...
Je présenterai de nombreux amendements de repli ; peut-être certains d'entre eux trouveront-ils grâce à vos yeux, mes chers collègues.
M. Philippe Bas . - Si j'ai bien compris, les amendements suivants se distinguent principalement de celui-ci en ce qu'ils admettent l'ouverture de l'assistance médicale à la procréation aux femmes seules ; mais ils maintiennent par ailleurs tout le reste du dispositif - il s'agit d'une position de repli de notre rapporteur.
Au fond, la question de principe que nous avons à trancher est la suivante : ouvrons-nous le dispositif aux femmes seules, ou pas ?
Il me paraît important de noter, en tout cas, qu'aucun des membres de la commission ne conteste qu'il est préférable de maintenir le régime actuel d'accès à l'assistance médicale à la procréation pour les couples formés d'un homme et d'une femme. Deux conditions, à l'exclusion de toute autre, régissent cet accès : l'infertilité du couple et le risque de transmission à l'enfant d'une pathologie grave. Autrement dit, les couples visés sont ceux qui rencontrent un problème médical particulier.
Sont proposées, par ailleurs, un certain nombre de modifications qui tendent à améliorer le texte de l'Assemblée nationale.
À ce stade, la principale question en débat entre nous est donc de savoir si l'on ouvre le dispositif aux couples formés de deux femmes ou aux femmes seules.
Mme Laurence Rossignol . - Cet amendement est riche : on y trouve à la fois l'exclusion des femmes seules et la non-prise en charge par la sécurité sociale de l'AMP pour toutes les situations autres que celles où le couple est infertile ou porteur d'une maladie grave.
Je peux comprendre qu'un certain nombre de nos collègues s'interrogent sur l'ouverture de la PMA aux couples de femmes et aux femmes seules, voire que, cessant de s'interroger, ils votent contre, comme quatorze d'entre nous l'ont fait tout à l'heure.
Pour autant, une fois votée ladite ouverture, je ne pense pas qu'il soit juste, en guise de repli, d'exclure le remboursement. Priver les couples de femmes et les femmes seules de remboursement ressemble à s'y méprendre à une mesure punitive, avec pour effet de les placer de nouveau dans la situation de devoir consentir un investissement financier spécifique.
Par ailleurs, en tant que législateur, lorsque je vote le budget de la sécurité sociale, il n'est pas rare que je vote pour des dépenses publiques dont je me demande, à titre personnel, si elles sont bien justifiées. Si je vote ces dépenses, c'est simplement parce qu'il faut admettre - telle est ma conception de la vie en collectivité - que nous ne vivons pas tous de la même façon. Ces différences ne doivent justifier aucune inégalité sociale ; or, en l'espèce, le non-remboursement introduirait une inégalité.
M. Alain Milon , président. - L'acte médical, la fécondation in vitro, existe en effet ; mais ce n'est pas, me semble-t-il, un acte de médecine. C'est un acte médical pur, pas un acte nécessaire pour dispenser un soin.
Je suis d'accord, par ailleurs, pour que la grossesse soit prise en charge intégralement. La grossesse, sans être une maladie, est et doit être prise en charge par la sécurité sociale.
M. Daniel Chasseing . - Le terme « punition » n'est pas acceptable.
M. Philippe Bas . - Si les actes médicaux accompagnant la grossesse sont pris en charge par l'assurance maladie, c'est tout de même parce que, bien qu'elle ne soit pas une maladie, des pathologies peuvent entraver son développement. À ce titre, un suivi médical est nécessaire. C'est la seule raison qui motive la prise en charge.
M. Alain Milon , président. - Pas seulement. En médecine et en droit de la sécurité sociale, on distingue bien les grossesses et les grossesses pathologiques, les secondes seules étant des maladies.
M. Philippe Bas . - Mais c'est la prévention des pathologies qui justifie l'obligation d'examens médicaux pris en charge par l'assurance maladie ; je suis formel. Concernant la solidarité en matière de financement de l'accès à l'assistance médicale à la procréation pour des raisons non médicales, si nos collègues souhaitent qu'elle s'exprime, très bien, mais ne l'imputons pas à l'assurance maladie ; créons bien plutôt un fonds de solidarité pour l'accès à l'AMP !
Mme Muriel Jourda , rapporteur. - Il ne s'agit évidemment pas d'une mesure punitive à l'égard des couples homosexuels. C'est tout simplement la conséquence directe du texte fondateur de la caisse primaire d'assurance maladie, qui prend en charge la maladie. La solidarité nationale prend en charge la maladie, et pas le type d'acte dont nous sommes en train de discuter.
M. Roger Karoutchi . - Dans cet amendement, dix-sept machins s'entremêlent. Mme Jourda, qui est une fine mouche, y reprend certains éléments des amendements que nous venons de rejeter : tout cela n'est pas clair. Autant je soutiendrai les amendements suivants de Mme Jourda, autant, ici, on rattrape par la bande ce que l'on n'a pas obtenu du premier coup.
Mme Catherine Di Folco . - Pas du tout !
Mme Muriel Jourda , rapporteur. - L'intérêt de cet amendement était de faire tomber tous les autres en votant sur tous les aspects du problème en même temps. C'est tout ! J'ai compris qu'il ne faut pas faire des mots de plus de trois syllabes ni des amendements de plus de trois lignes.
L'amendement COM-170 n'est pas adopté.
M. Alain Milon , président. - Nous passons aux amendements de « repli sur le repli ».
Mme Muriel Jourda , rapporteur. - L'amendement COM-171 est un amendement de « recroquevillement » : il s'agit d'autoriser l'accès à l'AMP aux femmes seules, aux couples de femmes et aux couples hétérosexuels infertiles ou qui présentent un risque de transmission d'une maladie d'une particulière gravité. Autrement dit, nous excluons de l'AMP les couples hétérosexuels qui n'ont pas de problème de fertilité.
L'amendement COM-171 est adopté et les amendements COM-60 , COM-21 rectifié, COM-22 rectifié, COM-47 , COM-1 , COM-62 , COM-136 , COM-2 , COM-19 rectifié et COM-23 rectifié deviennent sans objet.
Mme Marie-Pierre de la Gontrie . - J'eusse apprécié, monsieur le président, que vous nous précisiez que l'adoption de l'amendement de Mme la rapporteur ferait tomber les suivants, ce qui nous prive d'une discussion sur des sujets importants - vous me direz que je n'ai qu'à être meilleure en droit parlementaire.
M. Alain Milon , président . - Ces sujets pourront être évoqués en séance.
Mme Muriel Jourda , rapporteur. - L'amendement COM-3 vise à autoriser, pour les couples de femmes, la réception par l'une des deux membres du couple des ovocytes de sa partenaire. J'y suis opposée : cette disposition me semble créer une certaine confusion dans la filiation de l'enfant.
Mme Laurence Cohen . - Je ne vois pas pourquoi nous n'autoriserions pas cette pratique. Où est le blocage, madame la rapporteur ? Où est, en la matière, la discrimination ou l'inégalité ?
M. Alain Milon , président. - J'entends bien la question de Mme Cohen, et je comprends bien l'observation de Mme Jourda. Il faudrait peut-être préciser, pour les situations où un couple de femmes souhaite recourir à la PMA, que la femme qui accouchera ne peut recevoir les ovocytes de sa conjointe que si elle n'a pas elle-même d'ovocytes.
Mme Catherine Deroche . - L'objectif est de permettre un réel partage de la maternité : l'une porte l'enfant, l'autre donne l'ovocyte.
Mme Laurence Cohen . - Exactement !
Mme Catherine Deroche . - Chacune, ainsi, à sa part du bébé. J'y suis tout à fait défavorable.
Les amendements COM-3 et COM-88 ne sont pas adoptés.
Mme Muriel Jourda , rapporteur. - L'amendement COM-4 vise à inscrire dans la loi qu'aucune stimulation hormonale n'est proposée lorsqu'elle n'est pas nécessaire. J'y suis défavorable. Une telle décision relève non pas de la loi, mais d'un choix de technique médicale, et doit, à ce titre, rester entre les mains des médecins.
Mme Laurence Cohen . - J'entends l'argument ; nous le retirons.
L'amendement COM-4 est retiré.
Mme Muriel Jourda , rapporteur. - L'amendement COM-172 est un amendement de coordination.
M. Jacques Bigot . - Cet amendement pose malgré tout un problème. La situation à laquelle je pense est la suivante : celle d'un couple hétérosexuel engagé dans un projet de procréation par assistance médicale et dont l'homme décède sans que l'embryon ait été implanté. La femme s'entend dire qu'elle ne peut se faire implanter l'embryon, mais qu'elle peut le donner, ce qui pose, humainement, des problèmes terribles.
Cet amendement vise à ce qu'il soit demandé au couple, par anticipation, de décider.
Mme Muriel Jourda , rapporteur. - La situation que vous évoquez fera l'objet d'un traitement dans la suite de la discussion.
Le couple peut consentir de son vivant à ce que, si l'un de ses membres décède, l'embryon conservé soit accueilli par un autre couple ou par une autre femme. Dans le texte de l'Assemblée nationale, c'est le membre survivant qui donne son consentement. Or c'est le couple quand il est encore constitué qui doit donner son accord. Il s'agit vraiment d'un problème rédactionnel.
Quant au problème que vous évoquez, mon cher collègue, nous en débattrons plus tard dans le fil de la discussion.
M. Jacques Bigot . - Mais les amendements dont je parlais sont tombés !
L'amendement COM-172 est adopté.
Mme Muriel Jourda , rapporteur. - L'amendement COM-173 est également un amendement de coordination. Nous avons tout à l'heure adopté un amendement visant à maintenir, pour les couples hétérosexuels, les critères d'infertilité et de transmission d'une maladie grave. Il s'agit simplement d'en tirer les conséquences.
L'amendement COM-173 est adopté, ainsi que l'amendement de précision COM-174 et l'amendement rédactionnel COM-175 .
Mme Muriel Jourda , rapporteur. - Les activités de don de gamètes et d'accueil d'embryons sont réservées à un certain nombre de centres publics et privés à but non lucratif, ce qui exclut les centres d'AMP privés à but lucratif, lesquels sont autorisés à pratiquer les autres activités d'AMP.
Cette disposition est issue de la première loi de bioéthique, datant de 1994, et liée au principe de non-patrimonialité du corps humain, consacré par l'article 16-1 du code civil.
L'amendement COM-63 vise à ouvrir la conservation des embryons à des centres qui présentent par ailleurs toutes les conditions de sécurité sanitaire requises, ce qui pourrait contribuer à développer cette activité, pour le moment assez réduite.
L'Inspection générale des affaires sociales (IGAS), dans un rapport de 2011 - la situation n'a pas changé depuis lors -, avait jugé préférable de s'en tenir à l'existant. L'appariement doit rester sous la responsabilité d'un organisme sans lien financier avec le couple de receveurs. Le respect de la non-patrimonialité du corps humain exige de séparer les activités de don et d'accueil d'embryons des autres activités d'AMP, c'est-à-dire de la technique elle-même.
Reprenant le raisonnement de l'IGAS - il faut séparer le don de l'appariement, qui doit demeurer sous la responsabilité d'un organisme sans lien financier avec le couple de receveurs -, je suis défavorable à l'ouverture de la procédure d'accueil d'embryons aux centres privés à but lucratif.
Mme Marie-Pierre de la Gontrie . - Le manque d'établissements capables de conserver les embryons allonge considérablement les délais, évinçant certaines femmes pour des raisons d'âge et en incitant d'autres à partir à l'étranger, ce qui est problématique. Sous réserve de normes de sécurité claires et de règles d'agrément identiques pour tous, il paraît souhaitable d'élargir la liste des centres d'accueil.
M. Jacques Bigot . - En outre, il y a des difficultés spécifiques en outre-mer.
Mme Laurence Cohen . - Selon tous les professeurs de médecine que nous avons auditionnés, le fait qu'un établissement ait ou non un but lucratif n'a pas d'incidence. Cet amendement me paraît donc bienvenu.
M. Daniel Chasseing . - Au demeurant, le secteur public ne pourra pas absorber de nouvelles activités.
L'amendement COM-63 est adopté.
TABLEAU DES SORTS
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Article 1
er
|
|||
|
Mme Muriel JOURDA, rapporteur |
167 |
Suppression d'article |
Rejeté |
|
M. CHEVROLLIER |
20 |
Suppression d'article |
Rejeté |
|
M. RETAILLEAU |
89 rect. bis |
Suppression d'article |
Rejeté |
|
M. AMIEL |
100 |
Suppression d'article |
Rejeté |
|
M. REICHARDT |
106 |
Suppression d'article |
Rejeté |
|
Mme Muriel JOURDA, rapporteur |
170 |
Ouverture de l'accès à l'assistance médicale à la procréation aux couples de femmes |
Rejeté |
|
Mme Muriel JOURDA, rapporteur |
171 |
Ouverture de l'assistance médicale à la procréation aux couples de femmes et aux femmes non mariées, avec maintien d'un critère médical pour les autres demandes |
Adopté |
|
Mme de la GONTRIE |
60 |
Accès à l'AMP de toute personne en capacité de mener une grossesse |
Satisfait
|
|
M. CHEVROLLIER |
21 rect. |
Maintien du critère médical dans l'accès à l'AMP |
Satisfait
|
|
M. CHEVROLLIER |
22 rect. |
Suppression de l'ouverture de l'AMP aux femmes seules |
Satisfait
|
|
Mme LOPEZ |
47 |
Évaluation médicale et psychologique des demandeurs d'AMP |
Satisfait
|
|
Mme COHEN |
1 |
Non discrimination dans l'accès à l'AMP sur la base du changement de sexe à l'état civil |
Satisfait
|
|
M. Jacques BIGOT |
62 |
Autorisation de l'AMP post mortem |
Satisfait
|
|
Mme SCHILLINGER |
136 |
Autorisation de l'AMP post mortem |
Satisfait
|
|
Mme COHEN |
2 |
Autorisation de l'AMP post mortem |
Satisfait
|
|
Mme DOINEAU |
19 rect. |
Autorisation de l'AMP post mortem |
Satisfait
|
|
M. CHEVROLLIER |
23 rect. |
Limite d'âge fixée à 43 ans pour bénéficier d'une AMP |
Satisfait
|
|
Mme COHEN |
3 |
Autorisation du don d'ovocyte au sein d'un couple de femmes |
Rejeté |
|
Mme MEUNIER |
88 |
Autorisation du don d'ovocyte au sein d'un couple de femmes |
Rejeté |
|
Mme COHEN |
4 |
Recours à l'AMP sans stimulation hormonale |
Retiré |
|
Mme Muriel JOURDA, rapporteur |
172 |
Coordination |
Adopté |
|
Mme Muriel JOURDA, rapporteur |
173 |
Coordination |
Adopté |
|
Mme Muriel JOURDA, rapporteur |
174 |
Précision d'un renvoi |
Adopté |
|
Mme Muriel JOURDA, rapporteur |
175 |
Rédactionnel |
Adopté |
|
M. Jacques BIGOT |
63 |
Ouverture à tous les établissements de santé de la procédure d'accueil d'embryons |
|
Mme Muriel Jourda , rapporteur . - L'amendement de coordination COM-182 vise à tirer les conséquences du maintien du critère pathologique pour les couples hétérosexuels et à prévoir un suivi des couples receveurs et des enfants issus du don.
L'amendement COM-182 est adopté.
Mme Muriel Jourda , rapporteur . - L'équipe médicale pluridisciplinaire intervenant dans la procédure d'assistance médicale à la procréation « peut faire appel, si nécessaire, à d'autres spécialistes, notamment un médecin spécialisé en psychiatrie ou un psychologue ». Je propose d'en maintenir le caractère médical en retirant la référence à un « infirmier ayant une compétence en psychiatrie ».
Mme Marie-Pierre de la Gontrie . - Peut-on envisager un entretien psychiatrique pour les couples hétérosexuels qui envisagent de faire des enfants par voie naturelle ?
Mme Muriel Jourda , rapporteur . - En général, ils ne nous préviennent pas. Les entretiens visent, d'une part, à s'assurer que le couple est bien prêt à traverser ensemble l'épreuve difficile que constitue une procédure d'AMP et, d'autre part, à préparer les futurs parents à la manière dont ils devront évoquer sa conception avec l'enfant.
Mme Élisabeth Doineau . - J'imagine difficilement un couple hétérosexuel n'ayant pas de problème d'infertilité s'engager dans une procédure d'AMP, qui est un parcours du combattant. Quel est l'intérêt de préciser dans la loi qu'ils en sont exclus ?
L'amendement COM-176 est adopté.
Mme Muriel Jourda , rapporteur . - Selon le projet de loi et le droit existant, il appartient aux équipes médicales des centres d'AMP de « vérifier la motivation » des demandeurs. Comment un médecin pourrait-il juger de la légitimité d'un projet parental ?
L'amendement COM-177 est adopté.
Mme Muriel Jourda , rapporteur . - L'amendement COM-132 rectifié vise à réintroduire l'évaluation psychologique et sociale des demandeurs d'AMP, supprimée par l'Assemblée nationale. Pour ne pas trop alourdir la procédure, je propose plutôt une évaluation « psychologique et, en tant que de besoin, sociale ».
M. Roger Karoutchi . - D'accord.
L'amendement COM-132 rectifié est adopté.
Mme Muriel Jourda , rapporteur . - L'amendement COM-179 vise à supprimer la mention relative à l'absence de discrimination pouvant résulter de l'évaluation médicale. Il n'y a aucune raison de suspecter les médecins de vouloir déroger aux règles de non-discrimination.
L'amendement COM-179 est adopté.
Mme Muriel Jourda , rapporteur . - L'amendement COM-35 rectifié tend à prévoir la communication au donneur de gamètes du dossier remis au couple ou à la femme non mariée. Il ne semble pas nécessaire de préciser un tel principe, certes intéressant sur le fond, dans la loi. Retrait ou avis défavorable.
L'amendement COM-35 rectifié n'est pas adopté.
Mme Muriel Jourda , rapporteur . - La mention selon laquelle les membres du couple sont incités à anticiper l'information d'un enfant issu d'un don n'a aucune valeur normative ; l'amendement COM-178 vise donc à la supprimer.
Mme Marie-Pierre de la Gontrie . - Ce n'est pas cohérent avec votre position sur la présence du pédopsychiatre.
Mme Muriel Jourda , rapporteur . - Je ne conteste pas le principe sur le fond. Mais je ne vois pas l'intérêt d'introduire une proposition incitative dans la loi.
L'amendement COM-178 est adopté ; l'amendement COM-101 rectifié devient sans objet.
L'amendement rédactionnel COM-102 est adopté.
L'amendement de précision rédactionnelle COM-180 est adopté.
Mme Muriel Jourda , rapporteur . - L'amendement COM-181 vise à exclure de la prise en charge par l'assurance maladie les demandes d'AMP non fondées sur un critère médical.
L'amendement COM-181 est adopté ; l'amendement COM-25 rectifié devient sans objet.
Mme Muriel Jourda , rapporteur . - L'amendement COM-163 vise à supprimer une demande de rapport.
L'amendement COM-163 est adopté.
L'article 1 er est adopté dans la rédaction issue des travaux de la commission.
Article additionnel après l'article 1 er
Mme Muriel Jourda , rapporteur . - L'amendement COM-24 vise à prévoir une clause de conscience des médecins et professionnels de santé à l'égard de l'AMP. Or le code de la santé publique prévoit déjà une clause de conscience générale. La clause de conscience spécifique qui a été introduite pour l'interruption volontaire de grossesse tient au fait qu'il s'agit d'ôter la vie. Retrait ou avis défavorable.
Mme Laurence Rossignol . - Merci d'éviter les expressions comme « ôter la vie » à propos de l'IVG.
L'amendement COM-24 est retiré.
Mme Muriel Jourda , rapporteur . - L'amendement COM-157 tend à supprimer cet article, qui demande un rapport au Parlement.
L'amendement COM-157 est adopté ; en conséquence, l'article 1 er bis est supprimé.
Mme Muriel Jourda , rapporteur . - Les amendements identiques COM-26 et COM-90 rectifié bis tendent à supprimer la possibilité d'une autoconservation des gamètes pour les femmes qui souhaiteraient différer leur projet de maternité. Si l'autoconservation est, certes, loin d'être une solution miracle, les motifs pouvant conduire à y recourir sont multiples. Aujourd'hui, l'autoconservation n'est possible que dans le cadre d'une démarche de don, ce qui crée une forme de contrepartie, voire de chantage. En supprimant l'article 2, nous entérinerions cet état de fait. Retrait ou avis défavorable.
M. Guillaume Chevrollier . - L'autoconservation ovocytaire doit être réservée aux cas pathologiques. Je maintiens mon amendement.
Les amendements identiques COM-26 et COM 90 rectifié bis ne sont pas adoptés.
Mme Muriel Jourda , rapporteur . - L'amendement COM-109 concerne l'information du conjoint du donneur de gamètes. Avis favorable.
Mme Véronique Guillotin . - Quelle information doit être apportée au conjoint ?
Mme Muriel Jourda , rapporteur . - Il doit bénéficier de la même information que le donneur, dans la mesure où son consentement est sollicité pour le don de gamètes.
Mme Laurence Rossignol . - L'accord du conjoint n'est pas exigé.
Mme Muriel Jourda , rapporteur . - En l'état actuel du droit, si. Mais le texte adopté par l'Assemblée nationale supprime cette exigence. Nous souhaitons la rétablir.
L'amendement COM-109 est adopté.
Mme Muriel Jourda , rapporteur . - L'amendement COM-183 vise à rétablir le consentement du conjoint au don de gamètes. C'est d'autant plus utile que le texte ouvre aux enfants issus d'un don la possibilité d'accéder à l'identité du donneur.
L'amendement COM-183 est adopté ; les amendements COM-27 rectifié, COM-108 et COM-39 rectifié deviennent sans objet.
Mme Muriel Jourda , rapporteur . - L'Assemblée nationale a prévu une étude de suivi des donneurs de gamètes. Le code de la santé publique confie déjà à l'Agence de la biomédecine la mission de mettre en oeuvre un suivi de l'état de santé des donneurs. Un échantillon de personnes volontaires pourrait ne pas être totalement représentatif.
L'amendement COM-159 est adopté.
Mme Muriel Jourda , rapporteur . - Le Gouvernement a autorisé l'autoconservation des ovocytes « sans l'encourager » et a encadré les conditions d'âge, en réservant cette possibilité aux femmes âgées de trente-deux ans à trente-sept ans. Ces bornes d'âges sont jugées un peu restrictives par les professionnels de santé. L'amendement COM-162 rectifié vise à assouplir l'accès à cette technique. Des recommandations de bonnes pratiques, prises après avis de l'Agence de la biomédecine en associant les sociétés savantes concernées, pourraient encadrer le dispositif.
Mme Marie-Pierre de la Gontrie . - Notre amendement tendant à modifier les bornes d'âges avait le même objet que l'amendement COM-162 rectifié. Pourtant, seul le nôtre a été déclaré irrecevable en application de l'article 40 de la Constitution...
Mme Muriel Jourda , rapporteur . - Je propose que les conditions d'âge soient fixées par décret en Conseil d'État. Je prévois donc tout de même des bornes.
Mme Marie-Pierre de la Gontrie . - Les choix qui figurent dans l'étude d'impact ne sont pas satisfaisants : la tranche d'âge retenue est très restreinte.
Mme Muriel Jourda , rapporteur . - Entre trente-deux ans et trente-sept ans, c'est le moment où les femmes commencent à s'inquiéter de ne pas avoir d'enfants.
Mme Marie-Pierre de la Gontrie . - Et où la fertilité décroît !
Mme Muriel Jourda , rapporteur . - En effet. Le dispositif est un peu ambigu : on autorise « sans encourager » l'autoconservation, mais pas au moment le plus propice.
M. Yves Daudigny . - Il y a peut-être des arrière-pensées relatives aux femmes de vingt-cinq ans ayant un plan de carrière.
L'amendement COM-162 rectifié est adopté.
Mme Muriel Jourda , rapporteur . - L'amendement COM-44 vise à supprimer la condition d'âge pour les femmes majeures souffrant de certaines pathologies définies par décret. Le code de la santé publique ouvre déjà une possibilité de conserver ses gamètes pour des personnes « dont la prise en charge médicale est susceptible d'altérer la fertilité ou dont la fertilité risque d'être prématurément altérée ». Aucune condition d'âge n'est fixée. Dans près de la moitié des cas, les gamètes ou tissus germinaux sont prélevés sur des personnes mineures. Cela concerne les cancers, mais aussi certaines pathologies comme l'endométriose ou les kystes de l'ovaire. Retrait.
L'amendement COM-44 est retiré.
Mme Muriel Jourda , rapporteur . - Les auteurs de l'amendement COM-133 rectifié proposent que l'équipe clinicobiologique pluridisciplinaire informe les personnes souhaitant effectuer une autoconservation sur l'infertilité et les risques liés aux grossesses tardives. Le projet de loi prévoit déjà une information préalable sur « les conditions, les risques et les limites de la démarche et de ses suites ». Avis de sagesse.
L'amendement COM-133 rectifié est adopté.
Mme Muriel Jourda , rapporteur . - L'amendement COM-87 rectifié bis vise à permettre à l'ensemble des établissements de santé autorisés, publics ou privés, de procéder à la conservation de gamètes. Les quarante-trois centres d'AMP privés réalisent en effet 51 % des tentatives d'AMP et constituent parfois la seule offre médicale disponible. Le secteur privé lucratif assure déjà l'activité d'autoconservation pour des motifs pathologiques. Avis favorable.
L'amendement COM-87 rectifié bis est adopté ; les amendements COM-42 rectifié, COM-45 , COM-65 et COM-134 rectifié deviennent sans objet.
L'amendement COM-66 est retiré.
Mme Muriel Jourda , rapporteur . - L'amendement COM-140 vise à faire en sorte que les frais liés à l'autoconservation de gamètes ne puissent pas être pris en charge ou compensés par l'employeur ou par toute personne physique de droit public ou privé avec laquelle la personne concernée serait dans une situation de dépendance économique.
Mme Marie-Pierre de la Gontrie . - Et si la personne est en situation de dépendance économique par rapport à son conjoint ?
L'amendement COM-140 est adopté.
Mme Muriel Jourda , rapporteur . - L'amendement COM-135 rectifié concerne le consentement au devenir des gamètes conservés en cas de décès lors de la consultation annuelle. Avis favorable.
L'amendement COM-135 rectifié est adopté.
Mme Muriel Jourda , rapporteur . - Le texte pose le principe d'une confirmation par écrit du premier consentement donné par une personne sur le devenir de ses gamètes après un délai de réflexion de trois mois. L'amendement COM-161 tend à alléger la procédure en prévoyant que le silence de la personne à l'issue du délai vaut confirmation du consentement.
L'amendement COM-161 est adopté.
Mme Muriel Jourda , rapporteur . - Selon le texte adopté par l'Assemblée nationale, si la personne dont les gamètes sont conservés est « perdue de vue » pendant dix ans ou décède, il est mis fin à la conservation en l'absence de consentement à ce que ces gamètes fassent l'objet d'un don ou de recherches. L'amendement COM-168 vise à préciser que le consentement au recueil, au prélèvement et à la conservation doit être établi ab initio.
L'amendement COM-168 est adopté.
Mme Muriel Jourda , rapporteur . - L'amendement COM-160 tend à interdire l'importation ou l'exportation de gamètes à titre onéreux.
L'amendement COM-160 est adopté.
Mme Muriel Jourda , rapporteur . - L'amendement COM-164 prévoit un dispositif transitoire pour que les gamètes conservés selon l'ancien système bénéficient des règles de consultation qui s'appliquent au nouveau.
L'amendement COM-164 est adopté.
Mme Muriel Jourda , rapporteur . - L'amendement COM-67 tend à autoriser à titre dérogatoire des établissements privés à pratiquer l'activité de don de gamètes. J'y suis plutôt défavorable pour les établissements privés à but lucratif. Mais vous leur avez permis tout à l'heure d'accueillir des embryons. C'est donc un avis de sagesse.
Mme Marie-Pierre de la Gontrie . - Cet amendement, qui é été déposé par notre collègue Catherine Conconne, est avant tout motivé par la situation en outre-mer.
L'amendement COM-67 est adopté.
L'article 2 est adopté dans la rédaction issue des travaux de la commission.
Mme Muriel Jourda , rapporteur . - L'amendement COM-158 vise à supprimer cet article, qui est dépourvu de portée normative.
L'amendement COM-158 est adopté ; en conséquence, l'article 2 bis est supprimé.
Article additionnel avant l'article 3
Mme Muriel Jourda , rapporteur . - L'amendement COM-28 vise à inscrire dans le code civil le principe de primauté de l'intérêt supérieur de l'enfant. Or ce principe a déjà une valeur conventionnelle et constitutionnelle, donc supérieure à la loi, du fait de la Convention relative aux droits de l'enfant et à la jurisprudence du Conseil constitutionnel. Intégrer l'intérêt supérieur de l'enfant dans le code civil n'aurait donc qu'une portée symbolique. Par ailleurs, les notions de « considération primordiale » et de « primauté » ne sont pas équivalentes : la « considération primordiale » désigne une considération de la plus grande importance quand « primauté » signifie qu'il s'agit d'une considération de premier rang. L'intérêt supérieur de l'enfant doit être mis en équilibre avec les autres intérêts en présence. Retrait ou avis défavorable.
L'amendement COM-28 n'est pas adopté.
Mme Muriel Jourda , rapporteur . - L'amendement COM-103 rectifié vise à conditionner la levée de l'anonymat du donneur de gamètes sur demande de l'enfant issu de ce don à l'accord du donneur. Je sollicite le retrait de cet amendement au profit du mien, qui ne tend pas à rédiger intégralement l'article. Sur le fond, nous avons la même approche.
M. Daniel Chasseing . - L'enfant aura systématiquement accès à sa majorité aux données non identifiantes du donneur, mais la communication des données identifiantes du donneur ne pourra s'effectuer qu'avec l'accord de ce dernier. C'est le sens de mon amendement.
L'amendement COM-103 rectifié est retiré.
Mme Muriel Jourda , rapporteur . - L'amendement COM-264 vise à mieux équilibrer la levée de l'anonymat entre les intérêts de l'enfant et ceux du donneur.
L'amendement COM-264 est adopté.
L'amendement de coordination COM-232 est adopté.
Mme Muriel Jourda , rapporteur . - L'amendement COM-234 a pour objet de permettre au donneur de gamètes ou à la personne issue du don de gamètes d'actualiser ses données médicales.
L'amendement COM-234 est adopté.
L'amendement de précision COM-235 est adopté.
Mme Muriel Jourda , rapporteur . - Le texte adopté par l'Assemblée nationale prévoit que l'état général du donneur figure parmi les données non identifiantes. Cela peut créer une confusion avec les données médicales, qui sont communicables à un médecin dès la naissance de l'enfant. L'amendement COM-236 vise à supprimer la référence aux « données relatives à l'état général ». Je propose également de préciser que la rédaction des motivations du don s'effectuera en concertation avec le médecin.
L'amendement COM-236 est adopté ; les amendements COM-38 rectifié et COM-69 deviennent sans objet.
Mme Muriel Jourda , rapporteur . - L'adoption de l'amendement COM-68 aurait pour effet d'assimiler la situation du donneur de gamètes à celle de la femme qui accouche sous X. Un donneur de gamètes ne doit pas être confondu avec un parent. Il ne paraît pas judicieux de laisser un dossier très personnel à un enfant issu d'un don. Avis défavorable.
M. Jacques Bigot . - Le texte prévoit que le donneur peut indiquer les « motivations » de son don. Dans un tel document, il peut raconter beaucoup de choses...
Mme Muriel Jourda , rapporteur . - La motivation sera rédigée avec le médecin, dont on peut espérer qu'il modèrera l'enthousiasme littéraire du donneur.
L'amendement COM-68 n'est pas adopté.
Mme Muriel Jourda , rapporteur . - L'amendement COM-237 reprend une recommandation de la CNIL. Il précise la durée maximale de conservation des données relatives aux donneurs, à leurs dons et aux personnes issues de leurs dons qui sont conservées par l'Agence de la biomédecine. Cette durée ne pourra être supérieure à 120 ans.
M. Roger Karoutchi . - Pourquoi 120 ans ?
Mme Muriel Jourda , rapporteur . - Il faut prévoir un délai suffisamment long pour permettre aux enfants recherchant ces informations de se manifester. On peut conserver les gamètes très longtemps et il faut aussi tenir compte de l'allongement de la durée de la vie.
L'amendement COM-237 est adopté.
Mme Muriel Jourda , rapporteur . - L'amendement COM-239 vise à confier au Conseil national pour l'accès aux origines personnelles (CNAOP) les missions relatives à l'accès aux origines des personnes conçues par assistance médicale à la procréation avec tiers donneur que le Gouvernement souhaite confier à une commission ad hoc distincte, dans le cadre de la levée de l'anonymat des donneurs. Le CNAOP existe depuis dix-sept ans et a acquis une expérience forte en matière d'accès aux origines et d'accompagnement des personnes adoptées et des pupilles de l'État. Le Gouvernement a fait le choix de créer une nouvelle commission, semble-t-il devant la réticence du CNAOP lui-même qui, par la voix de sa présidente, a mis en avant le fait que le Conseil ne travaillait pas avec des médecins, mais avec le réseau des services départementaux de l'aide sociale à l'enfance, et que les publics concernés étaient très différents.
Toutefois, il semble judicieux de mutualiser les moyens et de mettre en valeur l'expérience acquise par le CNAOP. Une formation distincte adaptée aux nouvelles missions serait constituée en son sein et les moyens supplémentaires, prévus initialement pour la commission ad hoc, pourraient lui être affectés pour l'aider à développer ses nouvelles compétences.
M. Jacques Bigot . - La femme qui a accouché sous X souhaite rester anonyme et son identité n'est pas toujours connue. Les personnes qui choisissent de donner leurs gamètes le font dans des centres spécialisés, en déclinant leur identité. Les logiques sont très différentes. Je comprends donc l'argumentation de la présidente du CNAOP. Il faut distinguer les missions. L'accès aux origines pour des enfants nés sous X ne relève pas de la même démarche que pour des enfants nés d'un don de gamètes.
Mme Laurence Rossignol . - Mais cela ne justifie pas l'existence de deux organismes distincts. Un même organisme peut assurer les deux missions avec deux formations distinctes.
M. Alain Milon , président . - Absolument !
Mme Élisabeth Doineau . - Je voterai cet amendement. Lors de l'audition de la présidente du CNAOP, on a senti une réticence à assumer cette nouvelle charge, car les problématiques sont différentes. Le CNAOP est chargé d'instruire les demandes d'enfants nés sous X, non désirés, alors que les enfants nés grâce à des dons ont été désirés. Toutefois, la présidente a aussi indiqué qu'elle pourrait faire bénéficier la nouvelle commission de l'expertise du CNAOP. Il serait donc judicieux que les demandes d'accès aux origines relèvent du même organisme, quitte à créer deux formations distinctes en son sein.
L'amendement COM-239 est adopté.
Mme Muriel Jourda , rapporteur . - L'amendement COM-238 vise à supprimer toute possibilité pour le donneur d'obtenir des informations sur les enfants nés grâce à ses dons : leur nombre, leur sexe, etc. Cette possibilité de s'informer du « résultat » de ses dons est de de nature à créer un lien ambigu entre le donneur et les personnes issues de ses dons et à transformer le caractère purement altruiste du don.
L'amendement COM-238 est adopté.
Mme Muriel Jourda , rapporteur . - Les amendements identiques COM-265 et COM-252 visent à donner la possibilité aux personnes issues d'une AMP avec donneur sous l'empire de la législation actuelle de saisir l'organisme chargé de l'accès aux données non identifiantes et à l'identité des donneurs afin qu'il contacte le donneur et l'interroge sur sa volonté, ou non, de communiquer ses informations personnelles. Cet amendement offrirait ainsi une réponse aux personnes qui sont actuellement en quête de leurs origines et auxquelles le projet de loi n'apporte pas de solution. Si l'on donne cette possibilité aux enfants issus d'un don qui naitront après le vote de la loi, pourquoi la refuser systématiquement aux enfants issus du don dans le régime actuel ? Il s'agit de ménager un juste équilibre entre la vie privée des anciens donneurs et la possibilité pour les enfants issus de leurs dons de connaître leurs origines.
Mme Laurence Rossignol . - J'avais déposé un amendement similaire, mais il a été déclaré irrecevable en application de l'article 40 de la Constitution.
M. Alain Milon , président . - Votre amendement avait un champ plus large et un caractère systématique pour l'ensemble des donneurs. Il avait donc une incidence financière. Les amendements présentés ici sont plus restreints, car ils ne visent que les enfants qui le demandent.
Les amendements COM-265 et COM-252 sont adoptés.
L'amendement de coordination COM-233 est adopté.
Mme Muriel Jourda , rapporteur . - L'amendement COM-36 rectifié tend à préciser que le donneur peut avoir accès à ses données non identifiantes et identifiantes. Cet amendement est satisfait par l'article 15 du Règlement général sur la protection des données (RGPD). Retrait ?
L'amendement COM-36 rectifié est retiré.
Mme Muriel Jourda , rapporteur . - Avec le texte, on passe d'un régime d'anonymat du don à un régime où il est possible de lever l'anonymat. Trois phases sont prévues. D'abord, une phase d'environ un an, pendant laquelle seront créées la base de données auprès de l'Agence de biomédecine et la commission ad hoc. Ensuite, une deuxième phase, dont la durée serait déterminée par décret, pendant laquelle de nouveaux donneurs autorisant l'accès à leurs données personnelles seront recrutés tandis que les anciens donneurs pourront se manifester auprès des Centres d'étude de conservation des oeufs et du sperme humains (Cecos) pour accepter de se soumettre au nouveau régime ; pendant la constitution de ces stocks de gamètes et d'embryons « nouveau régime », les AMP continueront à être réalisées avec les stocks collectés sous le régime de l'anonymat. Enfin, une troisième phase, pendant laquelle ne seront plus utilisés que les gamètes et embryons de donneurs ayant accepté de donner accès à leur identité et leurs données non identifiantes, les stocks constitués sous l'ancien régime de l'anonymat étant alors détruits.
L'amendement COM-5 vise à fixer, dans la loi, la durée de la deuxième phase à cinq ans. L'objectif est d'éviter la destruction de gamètes et d'embryons, en laissant aux anciens donneurs le plus de temps possible pour transmettre leur accord pour basculer dans le nouveau régime. J'émets un avis défavorable, car cette solution semble trop rigide. Nul ne sait, en effet, combien de temps il faudra pour constituer le nouveau stock de gamètes ; cela dépendra grandement du nombre de donneurs qui accepteront de donner accès à leurs données personnelles. Il faut laisser de la souplesse au Gouvernement pour choisir la date de bascule appropriée en fonction du niveau du stock de gamètes et d'embryons constitué sous l'empire de la nouvelle loi.
L'amendement COM-74 vise, au contraire, à supprimer la deuxième phase. Avis défavorable.
L'amendement COM-5 n'est pas adopté, non plus que l'amendement COM-74.
L'amendement rédactionnel COM-240 est adopté.
Mme Muriel Jourda , rapporteur . - L'amendement COM-245 s'inspire d'une recommandation de la Fédération française des Cecos. Il s'agit de prévoir, pendant la période transitoire, que les donneurs qui ont donné leurs gamètes et leurs embryons avant l'ouverture de l'AMP aux couples de femmes et aux femmes non mariées devront donner leur consentement exprès pour qu'ils soient utilisés en faveur de ces nouvelles bénéficiaires.
M. Alain Milon , président . - Je ne suis pas très favorable à cet amendement.
Mme Muriel Jourda , rapporteur . - Les donneurs n'ont pas donné leur accord pour une utilisation de leurs gamètes par des femmes seules ou par des couples de femmes, car ce n'était pas autorisé dans l'ancien cadre.
M. Alain Milon , président . - Ils ont donné leurs gamètes... Cela suffit. Quand je donne mon sang, je ne sais pas à qui je le donne !
M. Philippe Bas . - Il ne s'agit pas de dire que les donneurs ont refusé de donner leurs gamètes à des femmes seules ou à des couples de femmes. Simplement, ils n'ont pas exprimé leur consentement. Il convient de leur laisser la liberté de dire s'ils sont d'accord ou non.
Mme Marie-Pierre de la Gontrie . - Peu importe la rhétorique, cet amendement est restrictif ! J'y suis aussi défavorable.
M. Thani Mohamed Soilihi . - L'idéal serait de trouver une solution pour que ceux qui ont fait un don dans l'ancien régime et qui s'opposeraient à l'usage de leur don dans les nouvelles conditions puissent exprimer leur désaccord. La loi crée un nouveau régime. Si les donneurs y sont opposés, il faut qu'ils puissent se manifester, mais la loi n'a pas à préjuger de leur opinion.
Mme Laurence Rossignol . - Il appartiendrait donc aux anciens donneurs de faire une démarche volontaire pour exprimer leur désaccord.
M. Thani Mohamed Soilihi . - Oui.
Mme Laurence Rossignol . - Cela semble difficile...
M. Bernard Jomier . - L'amendement, tel qu'il est rédigé, requiert un accord exprès des donneurs pour que leurs gamètes soient utilisés au bénéfice des femmes seules ou des couples de femmes. Cela exclut, de fait, les donneurs de l'ancien système qui n'auront pas donné leur consentement exprès. Rien ne garantit qu'ils seront contactés. Lorsque l'on fait un don, son bénéfice est fixé par les règles légales en vigueur. Il n'appartient pas au donneur de choisir le bénéficiaire. Si l'on introduit des critères de sélection, on introduit une hiérarchie ou une discrimination entre les bénéficiaires potentiels. C'est une entorse au principe d'égalité. L'amendement est donc, au minimum, mal rédigé.
L'amendement COM-245 n'est pas adopté.
Mme Muriel Jourda , rapporteur . - L'amendement COM-75 était lié à un amendement déclaré irrecevable en application de l'article 40 de la Constitution, qui confiait comme mission à la commission ad hoc de rechercher tous les anciens donneurs pour solliciter leur consentement à se soumettre au nouveau régime. L'amendement vise à fixer la date à laquelle il sera mis fin à la conservation des gamètes et embryons dont les donneurs n'ont pas donné leur consentement à la communication de leurs données personnelles. L'idée était de laisser plus de temps à la commission pour accomplir cette nouvelle mission. Mais, en l'absence de possibilité de confier cette mission à la commission, cet objectif n'a plus de sens. Retrait sinon avis défavorable.
L'amendement COM-75 n'est pas adopté.
Mme Muriel Jourda , rapporteur . - L'article L. 1244-2 du code de la santé publique prévoit que si les donneurs font partie d'un couple, l'autre membre du couple doit également consentir au don de gamètes. Par cohérence, il semble donc nécessaire de solliciter l'accord de ces personnes pour la communication des données non identifiantes aux personnes issues du don. Tel est l'objet de l'amendement COM-242 rectifié. L'accord de l'autre membre du couple serait également sollicité en cas de demande d'accès à l'identité du donneur. En cas de séparation du couple, le consentement de l'ancien conjoint, pacsé ou concubin ne serait plus nécessaire.
Mme Maryvonne Blondin . - Il s'agit des couples mariés ?
Mme Muriel Jourda , rapporteur . - Pas nécessairement. Dans le code de la santé publique, l'extension du mot « couple » est très large et désigne deux personnes liées, quel que soit le statut : mariage, PACS, concubinage, etc.
L'amendement COM-242 rectifié est adopté.
Mme Muriel Jourda , rapporteur . - L'amendement COM-40 rectifié bis vise à ce que les données non identifiantes puissent être communiquées en cas de désaccord au sein d'un couple de donneurs en cas de don d'embryon. Cet amendement ne semble pas compatible avec l'alinéa 8 de l'article qui prévoit que « lorsque le tiers donneur est un couple, son consentement s'entend du consentement exprès de chacun de ses membres. » Par ailleurs, cet amendement reviendrait à imposer à l'un des donneurs la transmission de ses données non identifiantes. Avis défavorable.
L'amendement COM-40 rectifié bis n'est pas adopté.
L'amendement de coordination COM-243 est adopté.
Mme Muriel Jourda , rapporteur . - L'amendement COM-241 supprime une demande de rapport.
L'amendement COM-241 est adopté.
L'article 3 est adopté dans la rédaction issue des travaux de la commission.
Mme Muriel Jourda , rapporteur . - Les amendements identiques COM-253 , COM-29 , COM-91 rectifié bis et COM-107 visent à supprimer la filiation d'un enfant né d'une AMP demandée par un couple de femmes. Dans la mesure où vous n'avez pas voté la suppression de l'extension de l'AMP, l'amendement COM-253 n'a plus de sens et je le retire.
L'amendement COM-253 est retiré, ainsi que les amendements COM-29 et COM-91 rectifié bis. L'amendement COM-107 n'est pas adopté.
Mme Muriel Jourda , rapporteur . - Le sujet que nous abordons avec l'amendement COM-254 rectifié est vaste. Il s'agit de l'établissement de la filiation pour les couples de femmes.
La version de l'Assemblée nationale établit la filiation, pour les couples de femmes, par la double reconnaissance qu'elles font de l'enfant devant le notaire lorsqu'elles consentent à l'assistance médicale à la procréation.
Le fondement de la filiation, en droit français, est, d'une part, le titre VII du code civil, sur la filiation relative à la procréation charnelle, et, d'autre part, le titre VIII, sur la filiation relative à l'adoption, ou filiation élective, ce qu'on appelle aujourd'hui le « parent d'intention », c'est-à-dire le choix d'être parent.
Le droit de la filiation est un droit d'ordre public ; il est structurant pour la société et, comme tel, n'est pas à disposition des membres de cette société, car on ne saurait soumettre un droit d'ordre public à la seule volonté. La volonté a évidemment un rôle à jouer dans la filiation ; néanmoins, par définition, nous ne pouvons pas décider des liens de filiation qui s'établissent entre nous, nos parents, nos enfants, notre famille : c'est l'État qui en décide, instaurant des critères et des garde-fous.
En l'occurrence, une femme décide de devenir mère - le critère est sa volonté. La volonté est par essence volatile ; elle a davantage sa place dans le cadre de relations contractuelles que là où il s'agit de créer des liens soumis à un droit institutionnel, structurant et d'ordre public tel que le droit de la filiation. De ce point de vue, l'introduction du critère de la volonté pure dans le droit de la filiation me semble destructrice ; ce critère finira par se substituer aux règles de la procréation charnelle, qui impliquent qu'on puisse vérifier la réalité d'un lien de filiation.
Ce mode d'établissement de la filiation ne me paraît donc pas avoir sa place au sein d'un droit d'ordre public. De surcroît, la reconnaissance, en droit de la filiation, est une façon d'indiquer que l'on a participé à la procréation de l'enfant, ce qui n'est pas le cas pour la femme qui n'a pas porté l'enfant.
Dans la mesure où notre rôle n'est pas de conduire une réforme du droit de la filiation, ce qui serait un travail titanesque, mais d'étendre la procédure d'AMP à un nouveau public, nous n'avons ni à créer un nouveau droit de la filiation ni à détruire ce qui existe. Il nous faut trouver une façon d'établir les liens de filiation propres aux couples de femmes qui respecte à la fois l'édifice institutionnel du droit de la filiation et les droits nouveaux qui s'y sont greffés.
Aujourd'hui, la filiation des couples hétérosexuels qui ont recours à l'AMP est établie sur le fondement du titre VII relatif à la procréation charnelle, qu'on peut aisément leur appliquer : il n'y a ni contentieux ni difficulté. Il n'y a pas de raison, au motif que nous adjoignons un nouveau public au dispositif de l'AMP, de détruire l'existant. Comme le dit le proverbe américain, « si ce n'est pas cassé, ne le répare pas ».
Et je ne vois pas non plus l'intérêt de créer, pour les femmes, un nouveau système s'éloignant totalement du droit de la filiation actuel.
Je propose donc, au sein du droit de la filiation existant, de procéder aux ajustements nécessaires à l'établissement d'une filiation crédible, cohérente et efficace pour les couples de femmes qui auront accès à l'AMP, mais sans aller plus loin.
Tel est l'objet de mon amendement. Il s'agit d'abord de constater - ce principe fonctionne, et n'a pas de raison de ne pas être utilisé - que la femme qui accouche est la mère de l'enfant, celle dont le nom est indiqué sur l'acte de naissance de l'enfant. Ce principe de procréation charnelle fonctionne efficacement ; nous n'avons aucune raison d'y renoncer.
Il s'agit ensuite d'établir la filiation de l'autre femme. Nous ne pouvons pas procéder comme pour la procréation charnelle, pour des raisons qui me semblent évidentes. La filiation de l'autre mère, qui décide de devenir mère en même temps que la femme qui accouche, est une filiation élective, une maternité d'intention : elle choisit de devenir mère. Or, la filiation élective, c'est l'adoption : cela existe déjà dans le droit français.
La difficulté est que la filiation, dans ce système, n'est pas très sécurisée : celle qui doit adopter doit avoir l'accord de la femme qui a accouché ; sa filiation est dépendante de la volonté de l'autre. C'est ce à quoi on peut trouver à redire.
Je vous propose donc, d'une part, de constater que la femme qui accouche est la mère de l'enfant, et, d'autre part, que, lorsque les deux femmes consentent devant notaire à l'AMP, un autre acte soit établi, valant consentement à l'adoption. Ce consentement à l'adoption de la femme qui n'accouche pas sera donc acquis en même temps que le consentement à l'AMP est donné.
Je vous propose aussi d'introduire dans la loi un mécanisme qui existe déjà pour les couples hétérosexuels non mariés : si la femme qui doit adopter ne le fait pas, l'adoption peut être prononcée à la requête de la femme qui accouche. La femme qui adopte est donc certaine du consentement de l'autre mère, et la femme qui accouche peut faire prononcer l'adoption même si l'autre vient à manquer à ses obligations.
Pour les couples mariés, il existe une présomption de paternité : automatiquement, le père devient le père. Mais, s'agissant des couples non mariés, le père doit reconnaître l'enfant ; et, si l'homme ne le fait pas, la femme peut saisir le tribunal pour faire reconnaître judiciairement la paternité. Le même système s'appliquerait au cas que nous avons à traiter. Il me semble qu'ainsi la filiation est sécurisée, et que le droit est conforme à la réalité : une femme procrée et l'autre choisit de devenir mère, via la filiation élective. Et les deux femmes, avec le dispositif que je propose, seraient assurées que l'adoption peut être prononcée y compris si l'une d'entre elles manque à ses obligations.
Cette proposition nécessite un aménagement du droit de l'adoption. Notre collègue Corinne Imbert a rendu au mois d'octobre, avec la députée Monique Limon, un rapport qui préconise d'ouvrir l'adoption à tous les couples. Je propose quant à moi de mettre en cohérence le public de l'AMP avec le public de l'adoption ainsi élargi. Je propose également de simplifier la procédure lorsque l'enfant est issu d'une AMP avec donneur : on se passerait complètement de la condition d'accueil au foyer de l'adoptant, puisqu'il s'agit, en l'espèce, d'adopter non pas l'enfant du conjoint, mais l'enfant qu'on a décidé d'avoir à deux sans pouvoir le faire suivant les modes de procréation charnelle. Je propose enfin que le tribunal de grande instance rende son jugement sous le délai d'un mois - la procédure serait ainsi plus rapide encore que celle qui s'applique à l'homme qui n'aurait pas reconnu son enfant malgré son engagement.
L'adoption créerait un lien de filiation exactement identique à celui du titre VII ; ce lien serait créé du jour de la requête en adoption. Concrètement, la filiation pourrait être établie, rétroactivement, au jour de la naissance, une fois la décision prononcée.
Voilà ma proposition ; je sais qu'elle est sensiblement différente de celle de l'Assemblée nationale. J'ai essayé de travailler sans idéologie. Vous avez compris que je ne suis pas favorable à cette extension ; mais, une fois qu'il en a été décidé, il faut instaurer un lien de filiation pour la seconde mère sécurisé, mais crédible. Ce lien doit correspondre à une réalité ; il ne saurait par conséquent prendre la forme d'une reconnaissance, laquelle reviendrait à dire à l'enfant qu'il a été fait par deux femmes, ce qui est rigoureusement impossible. Le mensonge institutionnel n'est jamais bon.
M. Daniel Chasseing . - Je n'ai pas très bien compris l'argument.
Deux femmes vont devant notaire pour signer une reconnaissance de filiation conjointe anticipée, mettant sur un pied d'égalité celle qui accouchera et celle qui s'engagera à remplir le rôle parental, ainsi que cela existe pour les couples hétérosexuels ; il faudra ensuite, lors de la déclaration en mairie, produire le certificat d'accouchement en même temps que la déclaration anticipée. Y a-t-il vraiment besoin d'ajouter l'adoption dans ce dispositif ?
Mme Muriel Jourda , rapporteur . - Je n'ajoute pas l'adoption ; je substitue à la procédure de reconnaissance une autre procédure qui me paraît beaucoup plus respectueuse tant du droit de la filiation que de la réalité de cette double maternité.
M. Jacques Bigot . - Sans partager du tout la position de Muriel Jourda, je mesure la difficulté qu'il y a, lorsqu'on est contre la PMA pour les femmes, à construire pour elles un droit de la filiation.
L'article 310-1 du code civil dispose que la filiation est légalement établie, par l'effet de la loi, par la reconnaissance volontaire ou la possession d'état constatée par un acte de notoriété. S'agissant de la reconnaissance, les cas sont nombreux de compagnons d'une mère dont l'enfant est issu d'une première relation et qui reconnaissent l'enfant comme étant le leur. Tout le monde l'accepte, à telle enseigne, d'ailleurs, que, si cet homme s'est comporté comme un véritable père, cette reconnaissance ne peut plus être contestée au bout de quelques années- une possession d'état lui est alors reconnue.
Tout a d'abord été construit à partir de la présomption de paternité, avant que soit introduite une autre notion, celle de reconnaissance préalable par le père. Ce dernier peut déclarer qu'il connaît une femme qui va accoucher et qu'il sera le père de son enfant le jour où elle accouchera. D'office, sa filiation sera alors établie.
C'est sur cette base que le législateur a construit la notion de reconnaissance conjointe liée à un acte notarié. L'idée que l'enfant aura deux mères peut certes bouleverser. Mais à partir du moment où l'on accepte d'étendre la PMA aux couples de femmes, la logique veut que l'on en reste à cette reconnaissance conjointe faite devant notaire, officier ministériel, et transmise à l'officier d'état civil. La simplicité de cette démonstration, au regard de la complexité de l'amendement, me paraît militer pour le rejet de ce dernier.
Mme Muriel Jourda , rapporteur. - Je n'en disconviens pas : il existe des reconnaissances mensongères ; mais elles sont du moins vraisemblables, parce qu'elles ne sont possibles que dans le cadre législatif du titre VII du code civil, sur la procréation charnelle, qui ne peut être étendu aux couples de femmes.
Je souhaite simplement que l'on respecte les fondements de la filiation : la reconnaissance, dans le code civil, c'est l'aveu d'une participation à la conception. Utiliser une notion propre à la procréation charnelle pour l'appliquer à une filiation élective, c'est pratiquer la plus totale confusion des genres. Une telle formule a peut-être l'avantage de la simplicité, mais cette simplicité est créatrice de confusion. La reconnaissance n'a jamais été au fondement d'une filiation d'intention pure : c'est - je l'ai dit - l'aveu d'une participation à la procréation.
Je ne vois pas comment on peut mêler à ce point deux types de filiation sans créer une confusion totale, y compris dans l'esprit de l'enfant, qui comprendra qu'il a été procréé par deux femmes, ce qui n'est évidemment pas possible.
Mme Laurence Rossignol . - Il faut reconnaître à Muriel Jourda le mérite d'essayer d'élaborer un système cohérent.
Cette affaire ne relève pas tant d'un problème juridique que d'un problème de choix politique. Ce que demandent les couples lesbiens, c'est que la deuxième mère ne soit pas renvoyée à l'adoption ; or notre rapporteur propose précisément de maintenir la deuxième mère dans un statut d'adoptante.
C'est d'ailleurs ce qu'a prévu la loi Taubira - j'en suis presque amusée : l'épouse peut adopter l'enfant de sa conjointe. Au moment du vote de la loi Taubira, souvenez-vous, mes chers collègues, des réactions d'hostilité à cette disposition ! Je constate que les choses, au fil du temps, évoluent.
L'amendement du rapporteur est inutile et incohérent au regard de ce que nous avons voté à l'article premier. De surcroît, il n'est pas durable, car nous avons besoin d'une grande réforme de l'adoption.
M. Philippe Bas . - Du point de vue des droits et des devoirs, la mère adoptive n'est pas moins mère que la mère qui a accouché. De ce point de vue, la proposition de notre rapporteur est d'effet équivalent à la disposition adoptée par l'Assemblée nationale, mais elle s'en distingue cependant.
Le texte de l'Assemblée nationale distingue en effet la mère qui accouche selon qu'elle appartient à un couple hétérosexuel ou homosexuel - dans ce dernier cas, elle n'est que l'une des deux femmes ayant fait acte de reconnaissance. Cette disposition est fragile et crée une discrimination entre couples selon qu'ils sont hétérosexuels ou homosexuels. La commodité de ce dispositif est acquise au prix de graves distorsions de notre droit de la filiation. Pourquoi renoncer au principe de filiation robuste selon lequel la femme qui accouche est mère ?
Concernant l'autre parent, nous souhaitons lui accorder les mêmes droits et devoirs, en s'inspirant de ce qui existe pour les pères. Nous disposons de trois moyens pour ce faire.
La présomption de maternité pourrait s'appliquer aux couples de femmes mariées, mais elle présente l'inconvénient de ne pas dire la vérité à l'enfant ; la vérité doit en effet être dite à l'enfant le plus tôt possible.
La reconnaissance pourrait également être utilisée comme le propose l'Assemblée nationale. Mais, en droit, la reconnaissance est l'acte par lequel l'auteur de la vie de l'enfant assume sa responsabilité à l'égard de cet enfant ; en utilisant la notion de reconnaissance avec un autre sens, nous risquons de fragiliser cette notion juridique et ce truchement ne dit pas non plus la vérité à l'enfant.
La seule démarche de vérité à l'égard de l'enfant qui est à notre disposition est donc celle de l'adoption, avec les assouplissements proposés par notre rapporteur. Ce faisant, nous ne sommes nullement en retrait par rapport à l'objectif de l'Assemblée nationale en termes d'égalité des droits et des devoirs des deux femmes à l'égard de l'enfant - ce qui est particulièrement important en cas de divorce.
M. Daniel Chasseing . - Les couples de femmes qui vivaient dans la clandestinité peuvent maintenant adopter. La reconnaissance conjointe anticipée de filiation les met maintenant sur un pied d'égalité. Alors, pourquoi aller vers l'adoption ?
L'amendement COM-254 rectifié n'est pas adopté.
Mme Muriel Jourda , rapporteur . - L'amendement COM-6 vise à établir une double filiation maternelle ou paternelle par la reconnaissance volontaire. J'y suis opposée, car cela est en contradiction avec le système français de la filiation. En outre, l'amendement contredit le titre dans lequel il s'insère. Enfin, un enfant qui aurait une double filiation paternelle hors adoption ne pourrait être conçu que par la voie d'une gestation pour autrui, ce qui est interdit par la loi française.
L'amendement COM-6 n'est pas adopté.
Mme Muriel Jourda , rapporteur . - L'amendement COM-7 vise à établir une double filiation maternelle ou paternelle par la possession d'état. Or cette notion est liée à la filiation charnelle et n'est donc pas adaptée. J'y suis défavorable.
L'amendement COM-7 n'est pas adopté.
Mme Muriel Jourda , rapporteur . - L'amendement COM-8 vise à établir la filiation par présomption pour l'épouse de la femme qui accouche. J'y suis défavorable.
L'amendement COM-8 n'est pas adopté.
Mme Muriel Jourda , rapporteur . - L'amendement COM-71 impose de mentionner dans l'acte de naissance de l'enfant le consentement à l'AMP avec donneur. Le projet du Gouvernement prévoit que l'officier d'état civil mentionne la reconnaissance conjointe dans l'acte de naissance de l'enfant, car c'est l'acte qui établit la filiation de l'enfant. En revanche, le consentement à l'AMP n'établit pas la filiation de l'enfant qui en est issu, c'est la reconnaissance. Il n'y a donc aucune raison de faire figurer sur l'acte de naissance le mode de conception de l'enfant. J'y suis défavorable.
M. Jacques Bigot . - Cet amendement rejoint l'amendement suivant, dont notre président est l'auteur. Dans les couples hétérosexuels ayant eu recours à l'AMP, la filiation peut sembler normale et les parents ne sont pas obligés de dévoiler à leur enfant le mode de sa conception. En revanche, dans un couple de femmes, cela n'est pas possible. Cela crée une forme d'inégalité. Nous proposons donc une reconnaissance conjointe pour ces deux cas de figure. À côté de la filiation naturelle et de la filiation adoptive, il y a le projet médical qui permet néanmoins d'avoir un enfant.
L'amendement COM-71 n'est pas adopté.
Mme Muriel Jourda , rapporteur . - L'amendement COM-251 de notre président Alain Milon vise à créer un régime spécifique d'établissement de la filiation pour tous les couples et les femmes seules ayant recours à l'AMP. Je suis défavorable à l'établissement de la filiation sur la base de la seule volonté. En outre, rien ne vient justifier juridiquement un changement de régime pour les couples de sexe différent qui ont recours à une AMP avec donneur : pourquoi changer ce qui fonctionne bien et sans contentieux ? Une telle mention viendrait souligner le caractère infertile du couple, ce qui constitue une atteinte injustifiée à la vie privée. Rien ne vient non plus justifier que l'accouchement ne suffise plus pour établir la filiation de la mère : quelle régression pour les femmes ! L'accouchement doit suffire à établir la filiation. J'y suis donc défavorable.
L'amendement COM-251 n'est pas adopté.
Mme Muriel Jourda , rapporteur . - L'amendement COM-72 vise à établir la filiation de la mère d'intention par la reconnaissance volontaire pour un enfant conçu par AMP avant l'entrée en vigueur de la loi, en France ou à l'étranger. Il s'agirait donc d'une filiation rétroactive. En premier lieu, je ne vois pas très bien comment les deux femmes pourraient prouver qu'elles ont eu recours à une AMP en France alors que cela n'était pas autorisé. En second lieu, pour les enfants conçus par AMP à l'étranger, je suis défavorable à la reconnaissance volontaire. En outre, il me semble délicat d'établir une filiation rétroactive : cela pourrait aboutir à imposer, parfois contre l'avis de la mère seule parent de l'enfant, l'établissement d'une autre filiation qui n'était pas possible ni envisageable à l'époque. Avis défavorable.
Mme Marie-Pierre de la Gontrie . - Le problème est plus important que cela. Pendant longtemps, les mères n'avaient pas de solution et des enfants sont aujourd'hui sans statut. Préoccupons-nous des enfants qui existent !
Mme Muriel Jourda , rapporteur . - Les enfants ont une mère qui a accouché et, depuis 2014, la Cour de cassation autorise l'adoption de l'enfant par la conjointe.
Mme Marie-Pierre de la Gontrie . - Pour cela il faut être marié. C'est un sujet sur lequel nous devrons revenir.
M. Thani Mohamed Soilihi . - Je comprends l'intention des auteurs de l'amendement, mais je suis gêné par la construction juridique, notamment rétroactive, proposée.
L'amendement COM-72 n'est pas adopté.
L'article 4 est adopté sans modification.
Article additionnel après l'article 4
Mme Muriel Jourda , rapporteur . - L'amendement COM-99 rectifié ter interdit la transcription totale de l'acte de naissance étranger d'un enfant français conçu par GPA. La GPA est certes interdite en droit français, mais la transcription des actes d'état civil permet de contourner la loi française. Je suis favorable à l'adoption de cet amendement pour un motif de fond, mais aussi pour amener le Gouvernement à redire devant le Sénat que ce projet de loi n'est pas un acte préparatoire à la légalisation de la GPA. Nous pourrions ainsi, tous, donner nos positions respectives sur cette importante question.
Mme Laurence Rossignol . - Si cet amendement n'est pas adopté par notre commission, il pourra être redéposé en vue de la séance publique.
Mme Muriel Jourda , rapporteur . - Bien évidemment, mais s'il est adopté par notre commission, le Gouvernement sera obligé de réagir.
M. Roger Karoutchi . - S'agissant des amendements, la réforme de 2008 n'est pas allée jusqu'au bout : nous pourrions même éviter d'avoir à nous réunir en commission comme ce soir !
Je voterai cet amendement, car, adopté par la commission, il aura plus de force. Bien que plutôt favorable à l'extension de l'AMP, je suis très défavorable à la GPA. Chacun se renvoie la balle pour ne pas décider, mais la loi c'est la loi et nous devons dire clairement que nous sommes hostiles à la GPA, en commission comme en séance publique. Le Gouvernement devra alors prendre une position claire au regard des décisions de justice.
M. Thani Mohamed Soilihi . - Même si l'amendement était adopté par notre commission, le Gouvernement pourrait attendre la deuxième lecture à l'Assemblée nationale pour revoir sa copie.
Mme Laurence Rossignol . - Cela serait délicat, car des sénateurs déposeront vraisemblablement des amendements de suppression de cette disposition.
Mme Marie-Pierre de la Gontrie . - Je suis personnellement hostile à la GPA, mais favorable à la transcription des actes de naissance des enfants nés par GPA à l'étranger, dans l'intérêt des enfants - et j'assume cette contradiction. Or cet amendement ne traite absolument pas de l'intérêt de l'enfant, qui est pourtant premier. Cet amendement est un cheval de Troie : il vise à priver de droits ces enfants, sans rien apporter d'autre, car la GPA est déjà interdite dans notre droit.
M. Olivier Henno . - Ma position est assez voisine. Je suis contre la GPA, mais un couple de mes amis a effectué une GPA au Canada. Le législateur doit donner un statut à cet enfant, comme n'importe quel autre enfant français.
Mme Laurence Rossignol . - La filiation paternelle des enfants nés de GPA ne pose pas de difficulté puisqu'ils ont un père biologique : ce ne sont donc pas des enfants fantômes de l'état civil. La question de la sécurisation de leur statut me semble agitée de façon exagérément anxiogène. Rappelons toutefois que la jurisprudence Mennesson reste relativement restrictive et l'on ne peut pas en déduire que tous les couples d'hommes qui réalisent une GPA à l'étranger pourront obtenir la transcription de l'état civil de leur enfant.
Je suis favorable au contenu de l'amendement, mais je ne le voterai pas, car je ne partage pas les convictions de ses auteurs, notamment au sujet, essentiel, du droit à l'avortement.
M. Daniel Chasseing . - Je suis également contre la GPA. Ce texte prévoit-il la reconnaissance des enfants de mères porteuses nés à l'étranger ?
Mme Marie-Pierre de la Gontrie . - Cet amendement est un cavalier.
Mme Catherine Di Folco . - Je fais partie des cosignataires de cet amendement, sans pour autant être contre l'avortement. Je m'insurge contre les propos catégoriques de Mme Rossignol qui ne semble pas connaître toutes mes convictions.
M. Yves Daudigny . - La GPA n'est pas le sujet, elle est interdite en France. Mais le 18 décembre dernier, la Cour de cassation a validé le principe de la transcription de l'état civil d'un enfant né par GPA à l'étranger dans un couple d'hommes, reconnaissant le père biologique et le père d'intention. C'est une décision inédite. Je me trouve dans une position proche de celle de Mme de la Gontrie et je ne voterai pas cet amendement.
M. Alain Milon , président . - Je voterai cet amendement afin que nous puissions avoir un débat sur la GPA : j'y suis extrêmement favorable, car je suis contre la marchandisation du corps humain. Avec Michèle André, j'avais déposé une proposition de loi sur la GPA éthique. Nous pourrons ainsi évoquer la GPA telle que je la conçois.
Mme Marie-Pierre de la Gontrie . - Dialectique subtile !
M. Bernard Jomier . - Le Gouvernement a exclu la GPA du champ du présent projet de loi : cet amendement n'est-il pas irrecevable en application de l'article 45 de la Constitution ?
M. Alain Milon , président . - Il présente un lien avec les questions de filiation.
L'amendement COM-99 rectifié ter est adopté et devient article additionnel.
Article additionnel avant l'article 5
M. Bernard Jomier , rapporteur . - La loi de bioéthique de 2004 a érigé à l'article L. 1231-1 A du code de la santé publique le prélèvement et la greffe d'organes au rang de « priorité nationale ».
L'amendement COM-141 complète cette disposition en affirmant le principe d'un statut de donneur d'organes. Cette proposition avait été envisagée par le CCNE, dans son avis 129, en vue de développer le don d'organes en France, en particulier le don du vivant.
Il s'agit, d'une part, d'ouvrir droit à une forme de reconnaissance symbolique et, d'autre part, de reconnaître explicitement le principe de neutralité financière du don pour le donneur d'organes.
Ce principe est encadré, à l'heure actuelle, par plusieurs dispositions de nature législative ou réglementaire, comme celles qui prévoient la prise en charge intégrale des frais afférents au prélèvement ou à la collecte par l'établissement de santé chargé d'effectuer le prélèvement ou la collecte (article L. 1211-4 du code de la sécurité sociale), l'exonération du forfait journalier hospitalier ou du ticket modérateur, mais aussi la prise en charge de frais d'examens, de transport, d'hébergement ou encore la compensation de la perte de revenu (articles R. 1211-2 et suivants du code de la santé publique).
Pour autant, ces dispositions sont trop peu connues et les démarches demeurent trop souvent complexes pour les donneurs, comme le soulignent des associations ainsi que le CCNE. L'Agence de la biomédecine a ainsi publié un « Guide de prise en charge financière des donneurs vivants d'éléments du corps humain » visant à permettre « une amélioration des pratiques de prise en charge financière des donneurs vivants ». Ce guide érige le principe de « neutralité financière du don » comme une obligation. Il est donc proposé de l'affirmer au plan législatif pour lui donner toute la visibilité nécessaire et en faire une priorité dans la politique de promotion du don.
L'amendement COM-141 est adopté et devient article additionnel.
M. Bernard Jomier , rapporteur . - L'amendement COM-142 réintroduit au niveau de la loi le nombre maximal de paires de donneurs-receveurs pouvant être impliquées dans un don croisé d'organes, en portant ce nombre à six au lieu de quatre dans le projet de loi initial.
En effet, l'équilibre du texte adopté par l'Assemblée nationale n'est pas satisfaisant : tout en renvoyant au décret en Conseil d'État la fixation du nombre maximal de paires, les députés ont tenu à prévoir une information du Parlement en cas de modification.
Il apparaît préférable de fixer ce nombre dans la loi, tout en ménageant une souplesse par rapport au texte initial.
D'après l'étude d'impact et au vu des expériences internationales et des auditions auxquelles nous avons procédé, la « taille souhaitable » des chaînes, permettant leur réalisation dans de bonnes conditions d'un point de vue logistique et permettant de limiter les ruptures de chaîne, se situerait entre 4 et 6 paires, avec un nombre moyen de 4,6 paires rapporté dans l'expérience américaine qui est la plus importante.
D'après l'Agence de la biomédecine, ce nombre resterait compatible avec le délai de réalisation des opérations de prélèvement, fixé à 24 heures par le projet de loi.
L'amendement COM-142 est adopté.
L'amendement COM-184 de coordination est adopté.
L'article 5 est adopté dans la rédaction issue des travaux de la commission.
Articles additionnels après l'article 5
M. Bernard Jomier , rapporteur . - L'amendement COM-9 prévoit de sanctionner le fait de mettre en relation les donneurs et les receveurs d'organes.
La loi encadre très strictement le don du vivant en France, qui ne peut intervenir que d'une personne de l'entourage familial ou affectif du receveur. Le donneur est informé par un comité d'experts et doit exprimer son consentement au don devant un magistrat, afin d'éviter que sa décision résulte de pressions.
Passer outre cette procédure est déjà punie par le code pénal (article 511-3). L'article 511-2 du code pénal punit en outre de sept ans d'emprisonnement et 100 000 euros d'amende « le fait d'apporter son entremise pour favoriser l'obtention d'un organe contre le paiement de celui-ci, ou de céder à titre onéreux un tel organe du corps d'autrui ».
Il me semble donc que l'arsenal de sanctions est déjà complet.
En outre, l'Agence de la biomédecine a dans ses missions celles de faire un état des lieux d'éventuels trafics d'organes et des mesures de lutte contre ces trafics. Mon avis est donc défavorable.
L'amendement COM-9 n'est pas adopté.
M. Bernard Jomier , rapporteur . - La loi de bioéthique de 2011 a prévu que les professionnels de santé peuvent porter sur la carte vitale, avec le consentement de la personne, la mention : « A été informé de la législation relative au don d'organes ». Cela s'inscrit dans les informations nécessaires aux interventions urgentes. Il en est de même d'ailleurs pour le dossier médical partagé qui comporte une disposition similaire.
Mais en effet, cela ne fait pas état de la position de la personne.
Depuis la loi Caillavet, et comme l'a réaffirmé la loi de modernisation de notre système de santé de janvier 2016, toute personne est présumée consentir au prélèvement d'organes.
C'est principalement l'inscription sur le registre national des refus, tenu par l'Agence de la biomédecine, qui permet aux équipes médicales de savoir que la personne entend s'opposer à ce prélèvement.
Je ne pense pas qu'il soit indispensable de porter cette précision sur la carte vitale. Ce pourrait être source de confusion d'avoir, le cas échéant, des mentions contradictoires entre les différents supports puisqu'une personne peut inscrire ou retirer son nom à tout moment du registre des refus. Je donne donc un avis défavorable à cet amendement.
L'amendement COM-10 n'est pas adopté.
M. Bernard Jomier , rapporteur . - Les amendements COM-46 et COM-48 visent à créer un « registre national de patients transplantés à l'étranger » qui serait géré par l'Agence de la biomédecine afin de mieux contrôler le « tourisme de la transplantation ».
L'agence réalise déjà tous les deux ans une enquête auprès des établissements de santé sur ce sujet et doit rendre compte dans son rapport annuel d'un « état des lieux d'éventuels trafics d'organes ou de gamètes et des mesures de lutte contre ces trafics » (article L. 1418-1-1 du code de la santé publique).
Par ailleurs, la France vient de ratifier en novembre 2019 la convention de St Jacques-de-Compostelle contre le trafic d'organes humains qui permet de renforcer les moyens de lutte contre les prélèvements illicites d'organes et la traçabilité en ce domaine.
Le cadre juridique me semble donc déjà suffisant, d'autant que ce type de trafic n'existe pas en France. C'est donc un avis défavorable.
Les amendements COM-46 et COM-48 ne sont pas adoptés.
M. Bernard Jomier , rapporteur . - L'amendement COM-49 prévoit le renforcement des sanctions applicables en cas de prélèvement illicite d'organe. L'article 511-3 du code pénal punit de sept ans d'emprisonnement et de 100 000 euros d'amende le fait de prélever un organe sur une personne vivante majeure, y compris dans une finalité thérapeutique, sans que le consentement de celle-ci ait été recueilli dans les conditions prévues par la loi.
Ces conditions ne seraient pas pertinentes dans le cas d'une personne décédée visé par l'amendement.
Par ailleurs, ces sanctions sont déjà lourdes et le fait de passer le montant de l'amende de 100 à 150 000 euros ne me paraît pas susceptible de rendre la disposition plus efficace ou dissuasive. J'émets donc un avis défavorable.
L'amendement COM-49 n'est pas adopté.
M. Bernard Jomier , rapporteur . - L'amendement COM-50 prévoit de créer une enquête annuelle de l'Agence de la biomédecine sur les patients ayant eu recours à une transplantation à l'étranger. Cette enquête est déjà réalisée tous les deux ans par l'Agence de la biomédecine dans le cadre du suivi du trafic d'organes qui lui est imposé par l'article L. 1418-1-1 du code de la santé publique issus de la précédente loi de bioéthique.
Il ne me semble donc pas utile de le préciser à un autre endroit du texte.
L'amendement COM-50 n'est pas adopté.
M. Bernard Jomier , rapporteur . - L'amendement COM-51 vise à établir le suivi des patients réalisant une transplantation d'organes à l'étranger. Il s'inscrit dans le même esprit que les autres puisque son objectif est d'améliorer le contrôle des transplantations d'organes réalisées à l'étranger.
Or, l'Agence de la biomédecine assume déjà cette responsabilité, notre arsenal pénal permet de réprimer les dérives qui seraient constatées et la convention de St Jacques de Compostelle renforce la lutte au niveau international contre le trafic d'organes.
L'amendement COM-51 n'est pas adopté.
Article 5 bis
L'article 5 bis est adopté.
Article 6
Les amendements de coordination COM-139 et COM-247 sont adoptés.
M. Bernard Jomier , rapporteur . - L'amendement COM-248 vise à abaisser l'âge du consentement afin qu'un mineur de seize ans puisse lui-même consentir au prélèvement de cellule souches hématopoïétiques (CSH) au bénéfice de l'un de ses parents.
Cette proposition est née d'un double constat :
- comme l'a souligné la présidente de la Fédération nationale des administrateurs ad hoc (FENAAH), à partir du moment où les deux parents sont en accord pour ce don, ainsi que l'enfant donateur lui-même, il n'y a pas réellement de conflit d'intérêts et la désignation d'un administrateur ad hoc ne se justifie nullement ; un enfant aura naturellement envie de « sauver » un parent en danger de mort et ce n'est pas la nomination d'un mandataire ad hoc qui pourrait écarter la pression qui pèse de toute manière sur l'enfant. Un accompagnement psychologique adapté serait plus propice à résoudre les difficultés que la désignation d'un administrateur ad hoc peu formé à ce genre de situation ;
- à ce jour, selon les informations obtenues de l'Agence de la biomédecine, seules deux demandes de dérogation relatives à des dons de cellule souches hématopoïétiques d'un mineur vers l'un de ses parents ont été formulées et il s'agissait dans les deux cas de mineurs de plus de dix-sept ans.
Cet âge s'explique par la nécessaire adéquation qui doit exister entre le poids du donneur pressenti et celui du patient. En effet, la quantité de cellules souches hématopoïétiques prélevée est fonction du poids du donneur tandis que la quantité de cellules souches nécessaires au malade pour assurer une bonne prise de greffe est calculée en fonction du poids de celui-ci. Je rappelle que le prélèvement de cellules souches hématopoïétiques au bénéfice d'un parent ne concernera pas les jeunes enfants.
Compte tenu de ce constat, il me paraît souhaitable d'abaisser l'âge du consentement, afin que dès seize ans, un adolescent, qui a alors la « faculté de consentir », puisse lui-même assumer la décision et l'exprimer directement.
Par cohérence, cet abaissement de l'âge de consentement jouerait également dans les autres hypothèses de dons intrafamiliaux de CSH.
Pour garder une certaine souplesse et permettre de faire
face aux situations
- a priori rares - où les donneurs
pressentis seraient des mineurs de moins de seize ans, je n'ai pas
souhaité interdire les prélèvements en deçà
de seize ans ; l'intervention d'un administrateur ad hoc est maintenue
dans ce cas.
M. Alain Milon , président . - Il s'agit d'un amendement important.
L'amendement COM-248 est adopté.
L'article 6 est adopté dans la rédaction issue des travaux de la commission.
M. Bernard Jomier , rapporteur . - L'amendement COM-249 vise à n'appliquer le droit commun du prélèvement post mortem qu'aux majeurs faisant l'objet d'une protection juridique avec représentation aux biens ou assistance.
En effet, il est peu probable qu'un majeur dont, par définition, les facultés mentales ou corporelles sont altérées et l'empêchent de pourvoir seul à ses intérêts, ait la capacité d'autonomie, voire de discernement, pour être informé du système du consentement présumé, en comprendre les enjeux et s'inscrire sur le registre national des refus - un dispositif par ailleurs peu connu de la population en général malgré les campagnes d'information de l'Agence de biomédecine - ou exprimer un refus à son entourage.
D'autre part, ce choix de s'inscrire ou non de son vivant sur le registre national des refus et de laisser ou non prélever ses organes après sa mort, est un choix éminemment personnel. Il n'est pas pris dans l'intérêt de la personne, mais dans un but purement altruiste. Il ne semble pas, de ce fait, relever de la mission d'un représentant légal.
Dans ces conditions, je considère qu'il n'y a pas lieu de laisser pratiquer des prélèvements d'organes après la mort des majeurs faisant l'objet d'une protection juridique avec représentation à la personne, ce d'autant plus que le contrôle a minima prévu dans le droit actuel - le consentement écrit du tuteur - ne peut être maintenu compte tenu de la cessation de sa mission au décès du majeur protégé en application de l'article 418 du code civil.
L'amendement COM-249 est adopté.
L'amendement COM-266 de coordination est adopté.
L'article 7 est adopté dans la rédaction issue des travaux de la commission.
Article additionnel après l'article 7
M. Bernard Jomier , rapporteur . - Cet amendement vise à ouvrir le don du sang aux majeurs faisant l'objet d'une mesure de protection juridique avec représentation aux biens et assistance, alignant ainsi le don du sang sur le régime des dons d'organes, de tissus et de cellules par donneur vivant proposé dans le cadre du projet de loi ainsi qu'aux mineurs de 17 ans, reprenant ainsi une disposition de la proposition de loi visant à la consolidation du modèle français du don du sang déposée par le député M. Damien Abad et votée à l'unanimité le 11 octobre 2018 à l'Assemblée nationale.
Il s'agit de permettre aux majeurs protégés et aux mineurs de participer ainsi à la solidarité nationale.
S'agissant des mineurs, le don du sang est promu comme geste citoyen lors de la Journée défense et citoyenneté à laquelle participent les jeunes de 16 à 18 ans.
L'âge du don ne peut toutefois pas être inférieur à 17 ans en raison d'une directive européenne du 22 mars 2004 qui fixe les critères d'admissibilité pour les donneurs de sang ; cette directive exige également un consentement écrit de l'un des parents ou du tuteur légal en cas de don de 17 à 18 ans.
Ces dispositions présentent un lien avec le texte en discussion dans la mesure où celui-ci comprend déjà des mesures relatives au consentement des mineurs et majeurs protégés à donner des éléments et produits du corps humain.
Mme Catherine Di Folco . - Il me semble que l'amendement COM-249 implique qu'un majeur protégé décédé ne peut pas être donneur d'organes.
M. Bernard Jomier , rapporteur . - Cette hypothèse ne s'applique que s'il s'agit d'un majeur bénéficiant d'une mesure de protection avec représentation à la personne.
Mme Catherine Di Folco . - En revanche, l'amendement COM-250 prévoit qu'un majeur protégé puisse donner son sang.
M. Bernard Jomier , rapporteur . - Oui, sauf s'il s'agit, comme précédemment, d'un majeur bénéficiant d'une mesure de protection avec représentation à la personne. J'estime que ces personnes ne sont pas en capacité de fournir un consentement libre et éclairé et que cette décision ne peut donc pas être transférée au tuteur légal. L'équilibre consacré par cet amendement permet de répondre à une revendication forte de la part des associations de personnes en situation de handicap qui consiste à améliorer l'exercice de la citoyenneté. De nombreuses mesures ont été prises en ce sens ces dernières années, comme l'amélioration de l'exercice du droit de vote. À cet égard, ouvrir à une partie des majeurs protégés le don du sang et le don d'organes post mortem participe de cette logique.
L'amendement COM-250 est adopté et devient article additionnel.
Article 8
L'amendement COM-223 de correction rédactionnelle est adopté.
M. Olivier Henno , rapporteur . - Les pratiques de conservation des échantillons biologiques prélevés par les laboratoires de biologie médicale peuvent varier d'un laboratoire à un autre. Or le bon état de conservation et la traçabilité de ces échantillons seront déterminants pour donner un caractère pleinement opérationnel à la possibilité de réaliser des examens génétiques post-mortem. L'amendement COM-185 propose donc d'harmoniser ces pratiques par la publication de règles communes en fonction des différentes situations.
L'amendement COM-185 est adopté.
L'article 8 est adopté dans la rédaction issue des travaux de la commission.
M. Olivier Henno , rapporteur . - L'amendement COM-186 porte sur la communication des coordonnées du médecin prescripteur aux apparentés informés par un proche de l'existence d'une information génétique susceptible de les concerner.
L'amendement COM-186 est adopté.
L'amendement rédactionnel COM-187 est adopté.
M. Olivier Henno , rapporteur . - L'amendement COM-188 de clarification concerne l'hypothèse où une personne a fait l'objet d'un examen des caractéristiques génétiques mais est décédée avant l'annonce du résultat ou avant d'avoir pu informer ses apparentés. Il tend à préciser que le médecin ne peut procéder à l'information de la parentèle de l'existence d'une information médicale d'ordre génétique pouvant les concerner que dans les cas où la personne s'était auparavant opposée à être informée du résultat ou s'était opposée à ce que les membres de sa famille potentiellement concernés bénéficient de cette information.
L'amendement COM-188 est adopté.
M. Olivier Henno , rapporteur . - L'amendement COM-227 vise à préciser que le responsable du centre d'AMP transmettra à la personne qui a choisi d'aller en consultation de génétique médicale les coordonnées du médecin prescripteur afin de permettre à son médecin de connaître l'anomalie qu'il convient de rechercher.
L'amendement COM-227 est adopté.
M. Olivier Henno , rapporteur . - L'amendement COM-189 vise à rendre obligatoire, en cas de détection d'une anomalie génétique chez une personne née sous le secret ou le parent de naissance d'une personne née sous le secret, la saisine par le médecin prescripteur du Cnaop afin que celui-ci procède à l'information de la personne entretenant un lien biologique avec la personne chez qui l'anomalie a été découverte. Il convient de rappeler que la nature de l'anomalie et les risques associés ne sont jamais communiqués par le Cnaop et que la personne contactée par le Cnaop conserve le droit « de ne pas savoir », en refusant de se rendre à une consultation de génétique, en application des règles de droit commun en matière d'examen des caractéristiques génétiques.
L'amendement COM-189 est adopté.
M. Olivier Henno , rapporteur . - Le Cnaop a rappelé qu'il lui est nécessaire de pouvoir utiliser le numéro d'inscription des personnes au répertoire national d'identification des personnes physiques et consulter ce répertoire, afin de pouvoir accomplir ses missions en matière d'information des personnes nées dans le secret ou des parents de naissance d'enfants nés sous le secret sur l'existence d'une information médicale d'ordre génétique pouvant les concerner. L'amendement COM-224 prévoit que cet accès ne pourra être effectué que dans des conditions fixées par décret en Conseil d'État, pris après avis de la CNIL.
L'amendement COM-224 est adopté.
L'article 9 est adopté dans la rédaction issue des travaux de la commission.
M. Olivier Henno , rapporteur . - L'amendement COM-83 tend à la suppression de l'interdiction de la publicité en faveur des tests génétiques. La publicité en faveur de tests génétiques commerciaux disponibles en accès libre relève de pratiques commerciales trompeuses qui font déjà l'objet de sanctions pénales en application de l'article L. 121-4 du code de la consommation. Il n'est donc en effet pas indispensable de préciser cette interdiction dans le code de la santé publique.
L'amendement COM-83 est adopté et l'amendement COM-228 devient sans objet.
L'article 10 est adopté dans la rédaction issue des travaux de la commission.
Article additionnel après l'article 10
M. Olivier Henno , rapporteur . - L'amendement COM-11 tend à l'interdiction des tests génétiques commerciaux en accès libre. La réalisation de tests génétiques commerciaux en dehors du cadre légal, de même que la publicité en faveur de tels tests font déjà l'objet de dispositifs de sanctions pénales inscrits dans le code pénal et le code de la consommation. S'il est vrai que l'interdiction des tests génétiques en accès libre ne s'est traduit jusqu'ici par aucune poursuite ni condamnation, se contenter de répéter cette interdiction dans le code de la santé publique ne changera rien au recours croissant des Français à ces tests. Il convient donc d'y apporter une réponse pragmatique afin de réellement protéger nos concitoyens des dangers que peut représenter la réalisation de tests ADN en dehors d'une consultation de génétique médicale. C'est pourquoi je vous propose, par mon amendement COM-190, deux mesures pour répondre à l'intérêt grandissant des Français pour les examens génétiques. Il s'agit tout d'abord de tenir compte de l'enthousiasme des Français pour les tests génétiques à visée généalogique en les soumettant à une procédure de labellisation et en interdisant toute communication d'informations à caractère médical dans le cadre de ces tests. Il vise par ailleurs à autoriser à titre expérimental les examens de génétique en population générale ou à l'occasion d'un dépistage préconceptionnel dans le strict cadre d'une consultation de génétique médicale, conformément aux recommandations du CCNE.
J'émets un avis défavorable à l'adoption de cet amendement.
L'amendement COM-11 n'est pas adopté.
M. Olivier Henno , rapporteur . - L'amendement COM-190 que je vous soumets tend à encadrer l'utilisation d'examens génétiques à visée généalogique. Face à l'ineffectivité de l'interdiction absolue des tests génétiques commerciaux, cet amendement fait le choix d'une solution pragmatique, afin de tenir compte de l'enthousiasme des Français pour ces tests, tout en les prémunissant contre des risques majeurs tels que la cession de données génétiques personnelles à des sociétés étrangères en dehors de tout contrôle et la délivrance d'informations génétiques d'ordre médical sans conseil délivré par des professionnels qualifiés. Il est ainsi proposé de n'autoriser que le recours aux tests génétiques exclusivement à visée généalogique, sous réserve que ces tests remplissent notamment plusieurs conditions. Tout d'abord leur conformité à un référentiel de qualité élaboré par l'agence de la biomédecine devra être attestée selon une procédure d'évaluation définie par voie réglementaire. On peut par exemple imaginer la délivrance d'une attestation de conformité par le comité français d'accréditation ou une procédure d'autoévaluation. Ces tests ne pourront par ailleurs avoir pour objet de délivrer une information génétique d'ordre médical. Ils ne pourront, en conséquence, faire l'objet d'une prise en charge par la solidarité nationale. Enfin, les informations tirées de ces tests ne pourront servir de fondement à des actions visant à faire valoir des droits patrimoniaux ou extra-patrimoniaux, notamment dans le cadre d'une démarche d'établissement d'un lien de filiation. Cet amendement permet de satisfaire l'amendement COM-53 de nos collègues Vincent Éblé et Jacques Bigot, qui poursuit le même objectif. Pour rappel, la France et la Pologne sont les seuls pays en Europe à interdire les tests génétiques à visée généalogique.
L'amendement COM-190 est adopté et l'amendement COM-53 est retiré.
TABLEAU DES SORTS
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Article 1
er
|
|||
|
Mme Muriel JOURDA, rapporteur |
182 |
Coordination avec le maintien du critère pathologique dans l'accès à l'AMP |
Adopté |
|
Mme Muriel JOURDA, rapporteur |
176 |
Composition de l'équipe médicale pluridisciplinaire |
Adopté |
|
Mme Muriel JOURDA, rapporteur |
177 |
Information des demandeurs sur les possibilités en matière d'adoption |
Adopté |
|
M. KAROUTCHI |
132 rect. |
Évaluation psychologique et sociale des demandeurs d'AMP |
Adopté avec modification |
|
Mme Muriel JOURDA, rapporteur |
179 |
Suppression de la mention sur l'absence de discrimination pouvant résulter de l'évaluation médicale |
Adopté |
|
M. CHASSEING |
35 rect. |
Transmission du dossier guide au donneur de gamète |
Rejeté |
|
Mme Muriel JOURDA, rapporteur |
178 |
Suppression de la mention selon laquelle les membres du couple sont incités à anticiper l'information d'un enfant issu d'un don |
Adopté |
|
M. MOHAMED SOILIHI |
101 rect. |
Cohérence rédactionnelle |
Satisfait
|
|
M. MOHAMED SOILIHI |
102 |
Rédactionnel |
Adopté |
|
Mme Muriel JOURDA, rapporteur |
180 |
Rédactionnel |
Adopté |
|
Mme Muriel JOURDA, rapporteur |
181 |
Prise en charge des seules AMP réalisées sur critère médical |
Adopté |
|
M. CHEVROLLIER |
25 rect. |
Conditions de prise en charge de l'AMP |
Satisfait
|
|
Mme Muriel JOURDA, rapporteur |
163 |
Suppression de la demande de rapport d'évaluation spécifique à cet article |
Adopté |
|
Article additionnel après l'article 1 er |
|||
|
M. CHEVROLLIER |
24 |
Clause de conscience des médecins et professionnels de santé à l'égard de l'AMP |
Retiré |
|
Article 1
er
bis
(nouveau)
|
|||
|
Mme Muriel JOURDA, rapporteur |
157 |
Suppression d'article |
Adopté |
|
Article 2
|
|||
|
M. CHEVROLLIER |
26 |
Suppression d'article |
Rejeté |
|
M. RETAILLEAU |
90 rect. bis |
Suppression d'article |
Rejeté |
|
M. REICHARDT |
109 |
Information de l'autre membre du couple sur les dispositions relatives au don |
Adopté |
|
Mme Muriel JOURDA, rapporteur |
183 |
Maintien du consentement du conjoint au don de gamètes |
Adopté |
|
M. CHEVROLLIER |
27 rect. |
Maintien du consentement du conjoint au don de gamètes |
Satisfait
|
|
M. REICHARDT |
108 |
Maintien du consentement du conjoint au don de gamètes |
Satisfait
|
|
M. CHASSEING |
39 rect. |
Maintien du consentement du conjoint au don de gamètes |
Satisfait
|
|
Mme Muriel JOURDA, rapporteur |
159 |
Suppression de l'étude du suivi des donneurs |
Adopté |
|
Mme Muriel JOURDA, rapporteur |
162 rect. |
Renvoi à une recommandation de bonnes pratiques des conditions d'âge pour accéder à l'autoconservation |
Adopté |
|
Mme GUILLOTIN |
44 |
Suppression de la condition d'âge pour des femmes souffrant de certaines pathologies |
Retiré |
|
M. KAROUTCHI |
133 rect. |
Information sur l'infertilité et les risques liés aux grossesses tardives |
Adopté |
|
M. MOHAMED SOILIHI |
87 rect. bis |
Ouverture de la possibilité d'autoconservation dans les établissements de santé privés lucratifs |
Adopté |
|
M. CHASSEING |
42 rect. |
Ouverture de la possibilité d'autoconservation dans les établissements de santé privés lucratifs |
Satisfait
|
|
Mme GUILLOTIN |
45 |
Ouverture de la possibilité d'autoconservation dans les établissements de santé privés lucratifs |
Satisfait
|
|
Mme de la GONTRIE |
65 |
Ouverture de la possibilité d'autoconservation dans les établissements de santé privés lucratifs |
Satisfait
|
|
M. KAROUTCHI |
134 rect. |
Ouverture de la possibilité d'autoconservation dans les établissements de santé privés lucratifs |
Satisfait
|
|
Mme CONCONNE |
66 |
Autorisation à titre dérogatoire des établissements privés à pratiquer l'autoconservation |
Satisfait
|
|
Mme Muriel JOURDA, rapporteur |
140 |
Non prise en charge par l'employeur des frais liés à l'autoconservation |
Adopté |
|
M. KAROUTCHI |
135 rect. |
Consentement au devenir des gamètes conservés en cas de décès lors de la consultation annuelle |
Adopté |
|
Mme Muriel JOURDA, rapporteur |
161 |
Allègement de la procédure de confirmation du consentement |
Adopté |
|
Mme Muriel JOURDA, rapporteur |
168 |
Recueil du consentement sur le devenir des gamètes en cas de décès |
Adopté |
|
Mme Muriel JOURDA, rapporteur |
160 |
Encadrement des conditions d'importation et d'exportation de gamètes |
Adopté |
|
Mme Muriel JOURDA, rapporteur |
164 |
Dispositions transitoires |
Adopté |
|
Mme CONCONNE |
67 rect. |
Autorisation à titre dérogatoire des établissements privés à pratiquer l'activité de don de gamètes |
Adopté |
|
Article
2
bis
(nouveau)
|
|||
|
Mme Muriel JOURDA, rapporteur |
158 |
Suppression d'article |
Adopté |
|
Article additionnel avant l'article 3 |
|||
|
M. CHEVROLLIER |
28 |
Inscription de la primauté de l'intérêt supérieur de l'enfant dans le code civil |
Rejeté |
|
Article 3
|
|||
|
M. CHASSEING |
103 rect. |
Sollicitation de l'accord du donneur avant de communiquer son identité |
Retiré |
|
Mme Muriel JOURDA, rapporteur |
264 |
Sollicitation de l'accord du donneur avant de communiquer son identité |
Adopté |
|
Mme Muriel JOURDA, rapporteur |
232 |
Amendement de coordination |
Adopté |
|
Mme Muriel JOURDA, rapporteur |
234 |
Actualisation des données médicales par le donneur et la personne issue du don |
Adopté |
|
Mme Muriel JOURDA, rapporteur |
235 |
Amendement de précision |
Adopté |
|
Mme Muriel JOURDA, rapporteur |
236 |
Suppression de l'état général des données non identifiantes et rédaction des motivations en concertation avec le médecin |
Adopté |
|
M. CHASSEING |
38 rect. |
Inclusion des antécédents médicaux dans les données non identifiantes |
Satisfait
|
|
Mme de la GONTRIE |
69 |
Inclusion des antécédents médicaux dans les données non identifiantes |
Satisfait
|
|
Mme de la GONTRIE |
68 |
Possibilité pour le donneur de laisser tout élément ou information à destination de la personne issue de son don |
Rejeté |
|
Mme Muriel JOURDA, rapporteur |
237 |
Fixation de la durée maximale de conservation des données relatives aux donneurs, à leurs dons et aux personnes issues de leurs dons auprès de l'Agence de biomédecine |
Adopté |
|
Mme Muriel JOURDA, rapporteur |
239 |
Transfert des missions confiées à la commission ad hoc au CNAOP |
Adopté |
|
Mme Muriel JOURDA, rapporteur |
238 |
Suppression de la possibilité pour le donneur de demander des informations sur les enfants nés grâce à ses dons |
Adopté |
|
Mme Muriel JOURDA, rapporteur |
265 |
Sollicitation des anciens donneurs pour demander leur consentement à la communication de leurs données non identifiantes et de leur identité en cas de demande |
Adopté |
|
M. MILON |
252 |
Sollicitation des anciens donneurs pour demander leur consentement à la communication de leurs données non identifiantes et de leur identité en cas de demande |
Adopté |
|
Mme Muriel JOURDA, rapporteur |
233 |
Amendement de coordination |
Adopté |
|
M. CHASSEING |
36 rect. |
Communication au donneur de ses données non identifiantes et identifiantes |
Retiré |
|
Mme COHEN |
5 |
Durée de cinq ans de la période transitoire |
Rejeté |
|
M. Jacques BIGOT |
74 |
Durée d'un an maximum de la période transitoire |
Rejeté |
|
Mme Muriel JOURDA, rapporteur |
240 |
Amendement rédactionnel |
Adopté |
|
Mme Muriel JOURDA, rapporteur |
245 |
Accord des anciens donneurs à l'utilisation de leurs gamètes et embryons pour réaliser des AMP en faveur de deux femmes ou d'une femme non mariée |
Rejeté |
|
M. Jacques BIGOT |
75 |
Fixation de la date à compter de laquelle il est mis fin à la conservation des gamètes et embryons issus de dons réalisés avant l'entrée en vigueur de la loi |
Rejeté |
|
Mme Muriel JOURDA, rapporteur |
242 rect. |
Accord de l'autre membre du couple en cas de consentement du donneur à la communication de ses données non identifiantes ou de son identité |
Adopté |
|
M. CHASSEING |
40 rect. bis |
Transmission des données non identifiantes en cas de désaccord au sein d'un couple de donneurs |
Rejeté |
|
Mme Muriel JOURDA, rapporteur |
243 |
Amendement de coordination |
Adopté |
|
Mme Muriel JOURDA, rapporteur |
241 |
Suppression de la demande de rapport au Gouvernement |
Adopté |
|
Article 4
|
|||
|
Mme Muriel JOURDA, rapporteur |
253 |
Suppression de l'article |
Retiré |
|
M. CHEVROLLIER |
29 |
Suppression de l'article |
Retiré |
|
M. RETAILLEAU |
91 rect. bis |
Suppression de l'article |
Retiré |
|
M. REICHARDT |
107 |
Suppression de l'article |
Rejeté |
|
Mme Muriel JOURDA, rapporteur |
254 rect. bis |
Établissement de la filiation par l'adoption pour la mère d'intention qui recourt à une AMP |
Rejeté |
|
Mme COHEN |
6 |
Établissement d'une double filiation maternelle ou paternelle par la reconnaissance volontaire |
Rejeté |
|
Mme COHEN |
7 |
Établissement d'une double filiation maternelle ou paternelle par la possession d'état |
Rejeté |
|
Mme COHEN |
8 |
Établissement de la filiation par présomption pour l'épouse de la femme qui accouche |
Rejeté |
|
Mme ROSSIGNOL |
71 |
Mention du consentement à l'AMP avec donneur dans l'acte de naissance de l'enfant |
Rejeté |
|
M. MILON |
251 |
Régime spécifique d'établissement de la filiation pour tous les couples et les femmes seules ayant recours à l'AMP |
Rejeté |
|
Mme de la GONTRIE |
72 |
Établissement de la filiation de la mère d'intention par la reconnaissance volontaire pour un enfant conçu par AMP avant l'entrée en vigueur de la présente loi |
Rejeté |
|
Article additionnel après l'article 4 (nouveau) |
|||
|
M. RETAILLEAU |
99 rect. ter |
Interdiction de la transcription totale de l'acte de naissance étranger d'un enfant français conçu par GPA dans certaines hypothèses |
Adopté |
|
Article additionnel avant l'article 5 |
|||
|
M. JOMIER, rapporteur |
141 |
Statut de donneur d'organes |
Adopté |
|
Article 5
|
|||
|
M. JOMIER, rapporteur |
142 |
Fixation à 6 du nombre maximal de paires impliquées dans un don croisé |
Adopté |
|
M. JOMIER, rapporteur |
184 |
Coordination |
Adopté |
|
Article additionnel après l'article 5 |
|||
|
Mme COHEN |
9 |
Sanction contre le fait de mettre en relation des donneurs et receveurs d'organes |
Rejeté |
|
Mme COHEN |
10 |
Mention sur la carte vitale de la volonté en matière de don d'organes |
Rejeté |
|
Mme LOPEZ |
46 |
Création d'un registre national de patients transplantés à l'étranger |
Rejeté |
|
Mme PUISSAT |
48 |
Création d'un registre national de patients transplantés à l'étranger |
Rejeté |
|
Mme PUISSAT |
49 |
Renforcement des sanctions applicables en cas de prélèvement illicite d'organe |
Rejeté |
|
Mme PUISSAT |
50 |
Enquête annuelle de l'Agence de la biomédecine sur les patients ayant eu recours à une transplantation à l'étranger |
Rejeté |
|
Mme PUISSAT |
51 |
Suivi des patients réalisant une transplantation d'organes à l'étranger |
Rejeté |
|
Article 6
|
|||
|
M. JOMIER, rapporteur |
139 |
Amendement de coordination |
Adopté |
|
M. JOMIER, rapporteur |
247 |
Amendement de coordination |
Adopté |
|
M. JOMIER, rapporteur |
248 |
Abaissement de l'âge de consentement pour le don de cellule souche hématopoïétique à 16 ans |
Adopté |
|
Article 7
|
|||
|
M. JOMIER, rapporteur |
249 |
Exclusion des majeurs faisant l'objet d'une protection juridique avec représentation à la personne du prélèvement post mortem |
Adopté |
|
M. JOMIER, rapporteur |
266 |
Amendement de coordination |
Adopté |
|
Article additionnel après l'article 7 (nouveau) |
|||
|
M. JOMIER, rapporteur |
250 |
Levée partielle de l'interdiction du don du sang applicable aux majeurs protégés et abaissement de l'âge de don du sang pour mineurs |
Adopté |
|
Article 8
|
|||
|
M. HENNO, rapporteur |
223 |
Corrections et harmonisations rédactionnelles |
Adopté |
|
M. HENNO, rapporteur |
185 |
Renvoi à un arrêté du ministre chargé de la santé pour la fixation de règles de bonne pratique en matière de conservation et de traçabilité d'échantillons biologiques humains |
Adopté |
|
Article 9
|
|||
|
M. HENNO, rapporteur |
186 |
Communication des coordonnées du médecin prescripteur aux apparentés informés par un proche de l'existence d'une information génétique susceptible de les concerner |
Adopté |
|
M. HENNO, rapporteur |
187 |
Amendement rédactionnel |
Adopté |
|
M. HENNO, rapporteur |
188 |
Amendement de clarification |
Adopté |
|
M. HENNO, rapporteur |
227 |
Transmission des coordonnées du médecin prescripteur par le responsable du centre d'AMP au médecin consulté par un tiers donneur ou une personne née d'un don |
Adopté |
|
M. HENNO, rapporteur |
189 |
Modalités de transmission d'une information génétique entre personnes concernées par une rupture du lien de filiation biologique dans le cadre d'une naissance dans le secret |
Adopté |
|
M. HENNO, rapporteur |
224 |
Accès du Cnaop au répertoire national d'identification des personnes physiques |
Adopté |
|
Article 10
|
|||
|
M. AMIEL |
83 |
Suppression de l'interdiction de la publicité en faveur des tests génétiques |
Adopté |
|
M. HENNO, rapporteur |
228 |
Amendement de clarification rédactionnelle |
Satisfait
|
|
Article additionnel après l'article 10 |
|||
|
Mme COHEN |
11 |
Interdiction des tests génétiques commerciaux en accès libre |
Rejeté |
|
M. HENNO, rapporteur |
190 |
Encadrement de l'utilisation d'examens génétiques à visée généalogique |
Adopté |
|
M. ÉBLÉ |
53 |
Encadrement des examens génétiques à visée généalogique |
Retiré |
Mercredi 8 janvier 2020
Articles additionnels après l'article 10
M. Olivier Henno , rapporteur . - L'amendement COM-191 ouvre l'accès à titre expérimental aux examens génétiques en population générale et dans le cadre du dépistage préconceptionnel. Il suit une recommandation du Comité consultatif national d'éthique (CCNE) formulée dans son avis 129 en autorisant, à titre expérimental, la prescription en population générale et dans le cadre du dépistage préconceptionnel d'examens des caractéristiques génétiques en l'absence d'antécédent familial ou de contexte clinique justifiant la recherche d'une anomalie génétique prédéterminée, sous réserve que les conditions suivantes soient remplies : le consentement de la personne doit être obtenu dans les mêmes conditions que celles prévues par l'article 16-10 du code civil, après que la personne a reçu les informations listées par le même article, à l'exception de l'indication de l'examen ; l'examen réalisé dans le cadre de cette expérimentation ne peut faire l'objet d'une prise en charge par la solidarité nationale. Il devra être pris en charge directement par la personne et, le cas échéant, par son organisme complémentaire d'assurance maladie.
Cet amendement satisfait les amendements COM-56 et COM-57 de nos collègues socialistes, qui poursuivent les mêmes objectifs.
L'amendement COM-191 est adopté et devient article additionnel. Les amendements COM-56 et COM-57 deviennent sans objet.
M. Olivier Henno , rapporteur . - L'amendement COM-256 supprime les termes de « données massives », prévoit l'information du patient par le professionnel de santé sur le recours à un traitement algorithmique et que le professionnel de santé explique au patient sous une forme intelligible la manière dont le traitement serait mis en oeuvre à son égard.
L'amendement COM-256 est adopté.
M. Olivier Henno , rapporteur . - L'amendement COM-257 fait assurer la supervision du traitement algorithmique par le professionnel de santé.
L'amendement COM-257 est adopté.
M. Olivier Henno , rapporteur . - L'amendement COM-258 consacre le principe selon lequel « aucune décision médicale ne peut être prise sur le seul fondement d'un traitement algorithmique. »
L'amendement COM-258 est adopté.
M. Olivier Henno , rapporteur . - L'amendement COM-12 , en discussion commune avec l'amendement COM-259 , propose que des référentiels de bonnes pratiques permettent d'établir la traçabilité d'un traitement algorithmique, en concertation avec les associations des usagers du système de santé. Associer les usagers est une idée très intéressante. Je vous propose plutôt que le décret que je prévois à mon amendement COM-260 sur les conditions d'application de l'article soit pris après concertation de l'Union nationale des associations agréées d'usagers du système de santé. Cette union, créée sur le fondement de l'article L. 114-6 du code de la santé publique, regroupe 72 associations agréées. L'amendement COM-12 pourrait ainsi être retiré et devenir un sous-amendement à l'amendement COM-260.
M. Alain Milon , président. - Les auteurs de l'amendement étant absents, l'avis est donc défavorable. L'amendement pourra être déposé pour la séance publique.
L'amendement COM-12 n'est pas adopté.
L'amendement COM-259 est adopté.
M. Olivier Henno , rapporteur . - L'amendement COM-260 renvoie au pouvoir réglementaire les conditions de mise en oeuvre du nouvel article L. 4001-3 du code de la santé publique, après avis de la Commission nationale de l'informatique et des libertés (CNIL).
Mme Maryvonne Blondin . - Il y a donc bien un avis de la CNIL ?
M. Alain Milon , président . - Tout à fait.
L'amendement COM-260 est adopté.
L'article 11 est adopté dans la rédaction issue des travaux de la commission.
M. Olivier Henno , rapporteur . - L'amendement COM-261 maintient le droit en vigueur sur le recours aux techniques d'imagerie cérébrale. L'article 12 exclut l'utilisation de l'imagerie cérébrale fonctionnelle pour les expertises judiciaires, afin de répondre aux critiques de l'Office parlementaire d'évaluation des choix scientifiques et technologiques (Opecst) et du CCNE.
L'amendement COM-261 est adopté.
L'article 12 est adopté dans la rédaction issue des travaux de la commission.
M. Olivier Henno , rapporteur . - L'amendement COM-262 exclut du champ de l'article 13 du projet de loi les équipements qui sont des dispositifs médicaux.
L'amendement COM-262 est adopté.
L'article 13 est adopté dans la rédaction issue des travaux de la commission.
Mme Corinne Imbert , rapporteure . - L'amendement COM-92 rectifié bis supprimerait l'article 14 du projet de loi ; cela empêcherait des adaptations du cadre juridique des recherches sur l'embryon et les cellules souches embryonnaires humaines rendues nécessaires par l'évolution des connaissances et des techniques, ne serait-ce que pour instituer un délai limite de culture in vitro des embryons surnuméraires, délai qui n'existe pas aujourd'hui. Avis défavorable.
L'amendement COM-92 rectifié bis n'est pas adopté.
Mme Corinne Imbert , rapporteure . - L'amendement COM-115 rectifié réécrit l'article 14 pour le cantonner à une disposition tendant à suspendre les recherches sur l'embryon et les cellules souches embryonnaires humaines dans l'attente d'une évaluation de ces recherches par l'Opecst. Une telle suspension accentuerait le retard qu'accuse déjà notre pays dans ce domaine. En outre, l'Agence de la biomédecine est déjà chargée par la loi d'évaluer, dans son rapport annuel d'activité, l'état d'avancement des recherches sur l'embryon et les cellules souches embryonnaires, en incluant un comparatif avec les recherches concernant les cellules souches adultes, les cellules pluripotentes induites et les cellules issues du sang de cordon, du cordon ombilical et du placenta, ainsi qu'un comparatif avec la recherche internationale. Avis défavorable.
L'amendement COM-115 rectifié n'est pas adopté.
Mme Corinne Imbert , rapporteure . - L'amendement COM-34 rectifié a trait à l'intégrité des embryons humains sujets de recherches menées dans le cadre de l'assistance médicale à la procréation (AMP). Les recherches menées dans le cadre de l'AMP et réalisées sur des gamètes ou un embryon conçu in vitro visent principalement à améliorer le taux de fécondation des ovocytes par les spermatozoïdes, à améliorer la maturation de l'embryon in vitro avant transfert ou à faciliter son implantation dans l'utérus. Certaines recherches consistent ainsi à élaborer un milieu de préparation et de culture de l'embryon favorisant sa maturation et sa capacité d'implantation à la surface utérine, et peuvent donc avoir un « effet » sur l'embryon et être regardées comme une intervention du point de vue de l'embryon. Avis défavorable.
L'amendement COM-34 rectifié n'est pas adopté.
Mme Corinne Imbert , rapporteure . - Les recherches menées sur l'embryon dans le cadre de l'AMP sont nécessairement considérées comme des recherches interventionnelles pour deux raisons : elles impliquent un transfert dans l'utérus de la femme et comportent donc à tout le moins une intervention sur cette dernière. Par ailleurs, plusieurs recherches autorisées dans ce cadre par l'Agence nationale de sécurité du médicament et des produits de santé (ANSM) ont pu impliquer des interventions sur l'embryon par l'expérimentation de milieux de culture favorisant une meilleure maturation ou une meilleure implantation, mais également des interventions sur la femme au travers de stimulations ovariennes ou hormonales ; les recherches non interventionnelles ne font pas l'objet d'une autorisation expresse de l'ANSM, elles ne sont soumises qu'à un avis conforme d'un comité de protection des personnes (CPP) ou, pour certaines recherches impliquant l'utilisation de données de santé, à un avis de la CNIL.
L'assimilation des recherches menées sur l'embryon dans le cadre de l'AMP à des recherches non interventionnelles, proposée par l'amendement COM-112 rectifié, aurait alors pour conséquence d'être moins protectrice de l'embryon concerné. L'article 14 du projet de loi précise déjà qu'aucune intervention de nature à modifier le génome des gamètes ou de l'embryon n'est possible dans le cadre de ce type de recherche. Avis défavorable.
L'amendement COM-112 rectifié n'est pas adopté.
Mme Corinne Imbert , rapporteure . - L'amendement COM-113 rectifié met en place une mission d'information sur les recherches menées dans le cadre de l'AMP, dans quel cadre ? S'agit-il d'une mission parlementaire ou d'une mission gouvernementale ? S'il est envisagé une mission d'information parlementaire sur les recherches menées dans le cadre de l'AMP, la création d'une telle mission est de l'initiative propre d'une assemblée parlementaire et non du domaine de la loi. Du reste, les résultats des essais cliniques autorisés par l'ANSM et conduits dans le cadre d'une procédure d'AMP sont ou seront disponibles, en fonction de l'état d'avancement de la recherche, sur le site clinicaltrials.gov. Je vous invite à le consulter, même s'il est rédigé en anglais.
L'amendement COM-113 rectifié est retiré.
Mme Corinne Imbert , rapporteure . - L'amendement COM-192 apporte des précisions de terminologie sur les recherches menées sur l'embryon dans le cadre de l'AMP. Il convient de prévenir toute confusion entre les essais cliniques réalisés sur des personnes relevant du cadre des recherches impliquant la personne humaine et les recherches menées sur un gamète ou un embryon.
L'amendement COM-192 est adopté ; l'amendement COM-114 rectifié devient sans objet.
Mme Corinne Imbert , rapporteure . - L'amendement COM-226 précise l'interdiction de création d'embryons à des fins de recherche.
M. Alain Milon , président . - C'est un sujet important.
L'amendement COM-226 est adopté.
Mme Corinne Imbert , rapporteure . - L'amendement COM-193 élargit le prérequis de finalité médicale applicable aux recherches sur l'embryon. Toute recherche sur l'embryon participe potentiellement de l'ambition de réaliser des progrès médicaux sans qu'il puisse être démontré avec précision et ab initio l'intérêt d'une recherche fondamentale en termes thérapeutiques. Afin de sécuriser, sur le plan juridique, les décisions d'autorisation des protocoles de recherche sur l'embryon, il convient d'ajouter au prérequis de la finalité médicale l'objectif d'amélioration de la connaissance de la biologie humaine, plus pertinent en matière de recherche fondamentale.
L'amendement COM-193 est adopté.
Mme Corinne Imbert , rapporteure . - L'amendement COM-194 précise le prérequis d'absence de méthodologie alternative pour la mise en oeuvre de recherches sur l'embryon. Afin de sécuriser, sur le plan juridique, les décisions d'autorisation des protocoles de recherche sur l'embryon, précisons qu'une méthode alternative au recours aux embryons n'est recevable que s'il est démontré qu'elle présente une pertinence scientifique comparable avec l'embryon humain.
L'amendement COM-194 est adopté ; l'amendement COM-76 devient sans objet.
Mme Corinne Imbert , rapporteure . - L'amendement COM-58 est relatif aux principes éthiques applicables à la recherche sur l'embryon et les cellules souches embryonnaires humaines. Les embryons surnuméraires et les cellules souches embryonnaires humaines sur lesquels des recherches peuvent être pratiquées doivent avoir été produits dans un cadre respectueux de la dignité du corps humain, défini par les articles 16 à 16-8 du code civil, qui implique notamment le respect du consentement des personnes à partir desquelles l'embryon a été produit ou encore l'interdiction des pratiques eugéniques ou de la commercialisation d'éléments dérivés du corps humain. Conservons la référence aux principes éthiques inscrits dans le code civil. Avis défavorable.
L'amendement COM-58 n'est pas adopté.
Mme Corinne Imbert , rapporteure . - L'amendement COM-116 rectifié supprime un alinéa interdisant le transfert à des fins de gestation d'embryons faisant l'objet d'une recherche et limitant la durée de développement in vitro d'embryons surnuméraires. Il supprimerait des garanties auxquelles l'auteur semble pourtant attaché. Dans ces conditions, des embryons surnuméraires ayant fait l'objet d'une recherche pourraient être implantés ou pourraient être cultivés in vitro bien au-delà de 7 jours, puisque l'évolution des techniques permet désormais d'aller au-delà de 14 jours. Avis défavorable.
L'amendement COM-116 rectifié n'est pas adopté.
L'amendement de précision rédactionnelle COM-225 est adopté.
Mme Corinne Imbert , rapporteure . - Je souscris à l'esprit de l'amendement COM-77 , qui étend le délai limite de culture d'embryons in vitro à des fins de recherche, mais je propose à ses auteurs de se rallier à mon amendement COM-195 , qui, dans sa rédaction, envisage l'extension à 21 jours du délai limite de culture d'embryons in vitro à des fins de recherche dans un cadre dérogatoire. Ce délai serait étendu uniquement pour des protocoles de recherche spécifiquement dédiés à l'étude des mécanismes de développement embryonnaire au stade de la gastrulation, entre les 15 e et 21 e jours de développement de l'embryon. Les recherches qui peuvent être menées sur des embryons jusqu'au 14 e jour de développement ne pourront ainsi être conduites sur des embryons au-delà du 14 e jour. Retrait au profit de l'amendement COM-195, ou à défaut, avis défavorable.
M. Bernard Jomier . - L'amendement de Mme Imbert ne limite l'objet des recherches qu'aux études sur la gastrulation. Actuellement, les recherches sont arrêtées, par consensus, au 14 e jour, mais on manque d'informations sur la formation des organes entre le 14 e et le 21 e jour. Pourquoi se limiter aux recherches sur la gastrulation et non ouvrir à d'autres recherches collatérales ? Lors de cette phase de développement embryonnaire, on peut détecter d'autres problèmes...
Mme Corinne Imbert , rapporteure . - J'ai une position de prudence. Actuellement, le délai de 14 jours fait consensus parmi les scientifiques, mais n'est pas reconnu par la loi. Les techniques permettent désormais de faire vivre un embryon plus de 7 jours. Je propose donc une dérogation pour les protocoles de recherche entre le 14 e et le 21 e jour.
M. Bernard Jomier . - Nous en reparlerons...
M. Jacques Bigot . - Nous préférons le retirer, quitte à y revenir en séance publique.
M. Yannick Vaugrenard . - Que signifie la gastrulation ?
M. Bernard Jomier . - Il s'agit d'un des stades du développement embryonnaire.
Mme Catherine Deroche . - Pourquoi la demande, formulée par les chercheurs, d'un prolongement de la limite de développement au-delà de laquelle il n'est plus possible de faire des recherches sur l'embryon n'a-t-elle pas été prise en compte par l'Assemblée nationale ? Comment le débat s'y est-il déroulé ?
M. Michel Amiel . - Ce sujet extrêmement sensible, celui du statut de l'embryon, exige en effet la plus grande prudence, ce qui explique une certaine frilosité. Il n'existe pas aujourd'hui de statut de l'embryon, et c'est très bien comme cela ; un tel statut remettrait en question, en effet, les lois sur l'IVG.
Au-delà des mots techniques, morula, gastrula, des choses décisives se jouent aussi entre le 14 e et le 21 e jour ; on parle d'embryogenèse et d'organogenèse. Il ne me paraît donc pas scabreux d'étendre la limite à vingt-et-un jours. Cette extension d'une semaine ne me choque pas, et je n'ai pas du tout le sentiment que, en prenant une telle décision, nous jouerions aux apprentis sorciers, dans la mesure où, sur cette semaine décisive en embryologie, les inconnues sont très nombreuses.
Mme Corinne Imbert , rapporteure . - Je réponds à Catherine Deroche : le Gouvernement s'est fondé sur le consensus international et sur l'avis du Conseil d'État ; mais cette phase du développement embryonnaire, du 15 e au 21 e jour, est bel et bien décisive ; l'ouverture de possibilités de recherche, par voie dérogatoire, me semble envisageable.
L'amendement COM-77 est retiré et l'amendement COM-195 est adopté.
Mme Corinne Imbert , rapporteure . - L'amendement COM-33 rectifié tend à interdire l'expérimentation de l'utérus artificiel. La rédaction proposée vise l'alinéa 16 ; or cet alinéa prévoit déjà explicitement que les embryons surnuméraires ayant fait l'objet d'une recherche ne peuvent pas être transférés à des fins de gestation - ils ne pourront donc pas être transférés dans un utérus artificiel.
Je ne nie pas que de telles expérimentations soulèvent des enjeux éthiques, mais la modification proposée n'est pas pertinente. Avis défavorable.
M. Guillaume Chevrollier . - Je le retire.
L'amendement COM-33 rectifié est retiré.
Mme Corinne Imbert , rapporteure . - L'amendement COM-196 supprime la précision relative à la possibilité pour les recherches sur l'embryon de porter sur les causes de l'infertilité, introduite par l'Assemblée nationale.
L'amendement COM-196 est adopté ; l'amendement COM-129 rectifié devient sans objet.
Mme Corinne Imbert , rapporteure . - S'agissant de l'amendement COM-104 rectifié, l'ordre des primates inclut les singes et les hominidés, dont l'être humain. Pour ce qui concerne la partie portant sur les embryons humains, l'amendement est satisfait par l'alinéa 16 de l'article 14, qui interdit le transfert à des fins de gestation des embryons humains surnuméraires sur lesquels une recherche a été réalisée, et limite à quatorze jours leur développement in vitro.
Quant aux embryons de primates non humains, ils ne font pas partie du champ du projet de loi initial, qui ne comporte aucune disposition relative aux droits des animaux faisant l'objet de recherches scientifiques. Avis défavorable.
L'amendement COM-104 rectifié n'est pas adopté.
Mme Corinne Imbert , rapporteure . - L'amendement COM-32 rectifié restreint l'importation de cellules souches embryonnaires humaines aux seules lignées provenant de pays signataires de la convention d'Oviedo. Or le code de la santé publique prévoit déjà que l'importation de cellules souches embryonnaires humaines ne peut être autorisée par l'Agence de la biomédecine que si ces cellules souches ont été obtenues dans le respect des principes éthiques fondamentaux prévus par notre législation, principes qui découlent précisément de la convention d'Oviedo. Le fait que des lignées soient importées de pays n'ayant pas signé ou ratifié la convention d'Oviedo n'implique pas nécessairement que ces lignées aient été produites en méconnaissance des principes éthiques posés par cette convention. Avis défavorable.
M. Guillaume Chevrollier . - Est-on certain que le projet de loi respecte la signature de la France dans le cadre de cette convention d'Oviedo ?
Mme Corinne Imbert , rapporteure . - Oui, heureusement !
L'amendement COM-32 rectifié n'est pas adopté.
Mme Corinne Imbert , rapporteure . - L'amendement COM-119 rectifié limite les recherches sur les cellules souches embryonnaires humaines aux seules lignées déjà existantes. Son adoption aurait pour effet de limiter considérablement les potentialités de telles recherches, en excluant la possibilité de mener des recherches sur des cellules souches embryonnaires dérivées d'embryons présentant des caractéristiques jusqu'alors inconnues, par exemple une anomalie génétique nouvellement identifiée. Avis défavorable.
L'amendement COM-119 rectifié n'est pas adopté.
Mme Corinne Imbert , rapporteure . - L'amendement COM-120 rectifié concerne la publicité des lignées de cellules souches embryonnaires humaines dérivées ou importées en France. Toutes les lignées de cellules souches embryonnaires humaines disponibles en Europe, et donc en France, sont répertoriées dans le registre européen hPSCReg (Human Pluripotent Stem Cell Registry), accessible à tous sur Internet. Il n'y a donc pas lieu d'acter par décret l'existence de ces lignées. Avis défavorable.
L'amendement COM-120 rectifié n'est pas adopté.
Mme Corinne Imbert , rapporteure . - L'amendement COM-197 élargit le prérequis de finalité médicale applicable aux recherches sur les cellules souches embryonnaires humaines - je propose d'ajouter que le directeur de l'Agence de la biomédecine s'oppose à la réalisation du protocole de recherche si celui-ci ne vise pas à améliorer la connaissance de la biologie humaine. Il s'agit de mieux sécuriser le dispositif sur le plan juridique.
L'amendement COM-197 est adopté ; l'amendement COM-54 devient sans objet.
Mme Corinne Imbert , rapporteure . - L'amendement COM-121 rectifié maintient le prérequis d'absence de méthodologie alternative pour la réalisation de recherches sur des cellules souches embryonnaires humaines.
Ce prérequis n'est pertinent que pour les recherches sur l'embryon ; il ne l'est pas pour les recherches sur les cellules souches embryonnaires humaines, celles-ci ne présentant plus les mêmes propriétés qu'un embryon - elles n'ont pas la capacité de former spontanément un nouvel embryon. Avis défavorable.
L'amendement COM-121 rectifié n'est pas adopté.
Mme Corinne Imbert , rapporteure . - L'amendement COM-122 rectifié supprime la référence aux recherches éthiquement sensibles pratiquées à partir de cellules souches embryonnaires humaines.
La suppression de l'alinéa relatif à ces recherches aurait en réalité pour effet de les faciliter, puisqu'elles ne seraient plus soumises à un avis préalable du conseil d'orientation de l'Agence de la biomédecine, cet avis précédant toute décision d'opposition du directeur général de l'agence. L'article 14 du projet de loi prévoit en effet une procédure spécifique pour les recherches ayant pour objet la différenciation de cellules souches embryonnaires humaines en gamètes ou la constitution de modèles embryonnaires à usage scientifique, avec, compte tenu de leur sensibilité sur le plan éthique, une vigilance particulière du conseil d'orientation de l'agence. Avis défavorable.
L'amendement COM-122 rectifié est retiré.
Mme Corinne Imbert , rapporteure . - L'amendement COM-201 supprime la possibilité de créer des embryons chimériques par l'insertion de cellules souches embryonnaires humaines dans un embryon animal.
Mme Laurence Cohen . - Très bien !
L'amendement COM-201 est adopté ; l'amendement COM-130 rectifié devient sans objet.
Mme Corinne Imbert , rapporteure . - L'amendement COM-93 rectifié vise à interdire la création d'embryons chimériques par adjonction à un gamète animal de cellules ou de matériel génétique d'origine humaine.
Mon amendement COM-201, que vous venez d'adopter, prévoit déjà de ne pas aller au-delà de ce qui est possible aujourd'hui en matière d'embryons chimériques, en interdisant l'insertion de cellules souches embryonnaires humaines dans un embryon animal. Je proposerai en outre d'encadrer, à l'article 15, l'insertion de cellules souches pluripotentes induites humaines dans un embryon animal. Demande de retrait ou, à défaut, avis défavorable.
L'amendement COM-93 rectifié n'est pas adopté.
L'amendement de coordination COM-267 est adopté.
L'article 14 est adopté dans la rédaction issue des travaux de la commission.
Mme Corinne Imbert , rapporteure . - L'amendement COM-125 rectifié vise à supprimer les dispositions encadrant les recherches sur les cellules souches pluripotentes induites (iPS), ce qui reviendrait à laisser la voie libre à des recherches présentant des risques éthiques sérieux, puisque ces cellules ne font aujourd'hui l'objet d'aucun cadre juridique. Avis défavorable.
L'amendement COM-125 rectifié n'est pas adopté.
Mme Corinne Imbert , rapporteure . - L'amendement COM-123 rectifié a pour objet de préciser que les cellules iPS sont utilisées pour la recherche pharmacologique. Une telle précision n'a rien d'exclusif et n'aura pas pour conséquence d'empêcher quelque protocole de recherche que ce soit portant, à des fins de recherche pharmacologique, sur des cellules souches embryonnaires humaines.
Par ailleurs, les cellules souches embryonnaires et les cellules souches pluripotentes induites ne sont pas strictement équivalentes : les secondes sont obtenues par reprogrammation, cette procédure étant susceptible d'entraîner des altérations génétiques ou épigénétiques. Avis défavorable.
L'amendement COM-123 rectifié n'est pas adopté.
Mme Corinne Imbert , rapporteure . - L'amendement COM-124 rectifié supprime l'encadrement de certaines recherches éthiquement sensibles sur les cellules souches pluripotentes induites.
La suppression de l'alinéa visé aurait pour conséquence de ne pas soumettre à déclaration préalable certaines recherches sensibles sur le plan éthique impliquant des cellules souches pluripotentes induites, comme la différenciation en gamètes ou la constitution de modèles mimant l'embryon.
En adoptant cet amendement, nous autoriserions d'emblée ces recherches au lieu de les empêcher, puisque les cellules iPS ne font aujourd'hui l'objet d'aucun encadrement ; le directeur général de l'Agence de la biomédecine se verrait ainsi privé de la possibilité de s'y opposer après avis du conseil d'orientation de l'Agence.
J'en demande le retrait au profit de l'amendement COM-199 - je propose en effet de définir un cadre plus exigeant pour les recherches sensibles sur le plan éthique.
L'amendement COM-124 rectifié est retiré ; l'amendement COM-199 est adopté ; les amendements COM-94 rectifié, COM-131 rectifié, COM-96 rectifié et COM-95 rectifié deviennent sans objet.
Mme Corinne Imbert , rapporteure . - L'amendement COM-59 supprime la référence aux principes éthiques applicables aux recherches sur les cellules souches pluripotentes induites. Pour les mêmes raisons qui ont présidé tout à l'heure, à l'article 14, au rejet de l'amendement COM-158, l'objectif poursuivi étant le même, j'émets un avis défavorable sur cet amendement.
L'amendement COM-59 n'est pas adopté.
L'article 15 est adopté dans la rédaction issue des travaux de la commission.
Mme Corinne Imbert , rapporteure . - L'amendement COM-31 rectifié vise à inscrire dans les protocoles de recherche sur l'embryon le consentement écrit du couple cédant ses embryons à la recherche.
Ne pourront par définition être utilisés pour la recherche que les embryons cédés par les couples ayant consenti par écrit à cette utilisation. L'Agence de la biomédecine ne peut évidemment pas allouer à des protocoles de recherche des embryons qui n'auraient pas été délibérément cédés à la recherche par les couples. Avis défavorable.
L'amendement COM-31 rectifié est retiré.
Mme Corinne Imbert , rapporteure . - L'amendement COM-202 allège la procédure de confirmation du consentement s'agissant du devenir des embryons ne faisant plus l'objet d'un projet parental, en coordination avec ce que nous avons voté hier à l'article 2.
L'amendement COM-202 est adopté.
Mme Corinne Imbert , rapporteure . - L'amendement COM-30 rectifié a pour objet l'inscription dans les protocoles de recherche sur l'embryon du consentement écrit du couple cédant ses embryons à la recherche. Même avis, défavorable, que sur l'amendement COM-31 rectifié, pour les mêmes raisons.
L'amendement COM-30 rectifié n'est pas adopté.
Mme Corinne Imbert , rapporteure . - L'amendement COM-84 rectifié traite des possibilités de devenir des embryons conservés examinées lors de la consultation annuelle pour la détermination de directives anticipées en cas de décès de l'un des membres du couple.
Y serait introduite la possibilité de mettre fin à la conservation des embryons. C'est une précision pertinente ; avis favorable.
L'amendement COM-84 rectifié est adopté.
Mme Corinne Imbert , rapporteure . - L'amendement COM-203 allonge à dix ans le délai de conservation des embryons cédés à la recherche non inclus dans un protocole de recherche.
Une durée de conservation de cinq ans ne permet pas de tenir compte des contraintes qui pèsent aujourd'hui sur le montage et la mise en oeuvre d'un protocole de recherche, ces opérations pouvant nécessiter bien plus de cinq années, en particulier lorsqu'une autorisation de recherche fait l'objet d'une contestation en justice.
L'amendement COM-203 est adopté.
L'article 16 est adopté dans la rédaction issue des travaux de la commission.
M. Olivier Henno , rapporteur . - L'article 17 est étroitement lié à l'article 15.
L'amendement COM-97 rectifié maintient l'interdiction de créer des embryons chimériques. À l'heure actuelle, l'interdiction, parce qu'elle prend place dans un chapitre consacré aux recherches sur l'embryon humain et les cellules souches embryonnaires humaines, ne s'applique qu'à la modification d'un embryon humain par adjonction de cellules provenant d'autres espèces et à la modification d'un embryon animal par adjonction de cellules souches embryonnaires humaines.
En revanche, est possible la création d'embryons chimériques résultant de l'adjonction de cellules souches pluripotentes induites humaines à un embryon animal. En effet, les cellules iPS ne font pour le moment l'objet d'aucun encadrement dans le code de la santé publique.
En coordination avec un amendement de Mme Imbert à l'article 15, il est donc proposé de maintenir l'interdiction dans son champ actuel et d'encadrer strictement les embryons chimériques recourant aux cellules iPS, afin de prévenir les risques de franchissement de la barrière des espèces.
Je propose donc aux auteurs de cet amendement de le retirer pour se rallier à l'amendement COM-200 , qui clarifie l'interdiction dont je viens de parler.
L'amendement COM-97 rectifié n'est pas adopté ; l'amendement COM-200 est adopté.
M. Olivier Henno , rapporteur . - L'amendement COM-128 rectifié est satisfait.
En effet, trois dispositions prévoient déjà l'interdiction de modifier le patrimoine génétique des enfants à naître : l'article 16-4 du code civil prévoit qu'aucune transformation ne peut être apportée aux caractères génétiques dans le but de modifier la descendance de la personne ; dans sa version issue de l'Assemblée nationale, l'article L. 2141-3-1 inséré dans le code de la santé publique par l'article 14 du projet de loi prévoit qu'aucune intervention ayant pour objet de modifier le génome des gamètes ou de l'embryon ne peut être entreprise dans une recherche menée dans le cadre de l'assistance médicale à la procréation ; l'article L. 2151-5 du code de la santé publique interdit le transfert à des fins de gestation des embryons surnuméraires sur lesquels une recherche aurait été réalisée - il interdit donc le transfert de tout embryon sur lequel une technique d'édition génomique telle que le CRISPR-Cas9 aurait été expérimentée.
L'amendement COM-128 rectifié est retiré.
M. Olivier Henno , rapporteur . - L'amendement COM-204 vise à corriger une erreur matérielle.
L'amendement COM-204 est adopté.
L'article 17 est adopté dans la rédaction issue des travaux de la commission.
M. Olivier Henno , rapporteur . - L'amendement COM-206 vise à préserver les droits de la personne en matière d'effacement de ses données ou de retrait de son consentement à l'utilisation de ses données dans le cadre d'une recherche.
L'amendement COM-206 est adopté.
M. Olivier Henno , rapporteur . - L'amendement COM-55 rectifié a pour objet l'information de la parentèle en cas de découverte d'anomalies génétiques à l'occasion d'un examen génétique réalisé dans le cadre d'une recherche scientifique.
Ainsi une découverte incidente peut-elle bénéficier aux membres de la famille d'une personne et non uniquement à la personne concernée. Avis favorable.
L'amendement COM-55 rectifié est adopté, ainsi que les amendements rédactionnels COM-205 et COM-207 .
L'article 18 est adopté dans la rédaction issue des travaux de la commission.
Mme Corinne Imbert , rapporteure . - Si l'article 19 actualise la définition de la médecine foetale, celle-ci demeure encore restrictive en ne faisant référence qu'au traitement des affections de particulière gravité alors que les centres pluridisciplinaires de diagnostic prénatal assurent également la prise en charge in utero de pathologies ou malformations présentant des degrés divers de gravité, sans que l'IMG soit la seule issue. Mon amendement COM-165 élargit donc le champ de la définition proposée, suivant une préconisation du CCNE et des professionnels concernés, en visant plus généralement la prise en charge in utero d'affections susceptibles d'avoir un impact sur le devenir du foetus ou de l'enfant à naître.
L'amendement COM-165 est adopté ; l'amendement COM-52 devient sans objet.
Les amendements rédactionnels COM-143 et COM-169 sont adoptés.
Mme Corinne Imbert , rapporteure . - L'amendement COM-98 rectifié bis conduit à réintroduire un délai de réflexion avant une éventuelle IMG. Ce délai est supprimé par l'article 20, qui rappelle également le cadre accompagnant la femme enceinte dans ces situations douloureuses, avec l'intervention d'une équipe pluridisciplinaire pouvant notamment comporter un psychologue. Je fais confiance aux équipes des centres de diagnostic prénatal (DPN) pour apporter une information complète afin de permettre un choix éclairé : telle est précisément leur mission, et le code de la santé publique prévoit déjà que le médecin prescripteur doit apporter à la femme enceinte « toute l'information nécessaire à la compréhension » des résultats. Les alinéas 17 et 18 de l'article 19 renvoient à des recommandations de bonnes pratiques les modalités de communication des résultats ou encore celles de fonctionnement des centres de DPN. En l'état, l'amendement pourrait être plus contraignant au détriment de la femme enceinte en imposant une décision au terme d'un délai d'une semaine - le délai de réflexion avant IMG était d'au moins sept jours - et sans prise en compte des cas d'urgence mettant en péril la santé de la femme. Son imputation dans le texte ne viserait en outre que les conséquences des résultats des caractéristiques génétiques, qui ne constituent pas l'ensemble des situations de diagnostic prénatal.
M. Alain Milon , président . - En outre, le délai de réflexion peut être demandé par la personne concernée.
Mme Marie-Pierre de la Gontrie . - Cela fera polémique en séance. Il est pénible de revenir à ce sujet.
M. Alain Milon , président . - Chacun a le droit d'exprimer ses idées...
Mme Muriel Jourda . - Et celle-ci n'est pas plus pénible qu'une autre !
Mme Marie-Pierre de la Gontrie . - À chaque étape, on revient sur l'IVG, ce qui donne le sentiment que ce n'est jamais acquis.
M. Roger Karoutchi . - Je ne suis pas d'accord. Dire qu'une semaine de réflexion, c'est remettre en cause l'IVG, c'est exagéré. Et l'on doit pouvoir s'exprimer dans une enceinte parlementaire - sinon, à quoi bon un Parlement ?
Mme Laurence Rossignol . - Hier soir, à propos de l'amendement sur les enfants nés par GPA à l'étranger, dont je partageais le contenu, tout en étant gênée par ses signataires, j'ai évoqué de fortes divergences avec ces derniers sur l'IVG. L'une de nos collègues l'a mal pris et m'a accusée de lui faire un procès d'intention.
Mme Catherine Di Folco . - Je suis là...
Mme Laurence Rossignol . - J'observe que les mêmes signataires témoignent par cet amendement qu'ils sont en effet des adversaires de l'IVG.
Mme Catherine Di Folco . - Pas du tout !
Mme Marie-Pierre de la Gontrie . - Vous savez bien que la question du délai renvoie à celle de l'accès !
M. Alain Milon , président . - L'IVG est un sujet spécifique, qui n'a rien à voir avec celui de cet amendement. Je rejoins M. Karoutchi : si les parlementaires ne peuvent formuler leur idéal, ou leur avis, inutile d'avoir un Parlement ! Ensuite, la majorité s'exprime. En l'espèce, celle-ci est transpartisane.
L'amendement COM-98 rectifié bis n'est pas adopté.
L'article 19 est adopté dans la rédaction issue des travaux de la commission.
Mme Corinne Imbert , rapporteure . - L'Assemblée nationale a supprimé la technique dite du double diagnostic préimplantatoire (DPI-HLA), instaurée à titre expérimental par la loi de 2004 avant d'être pérennisée par la loi de 2011, en sollicitant en parallèle un rapport sur les progrès réalisés dans la collecte et le stockage d'unités de sang placentaire. La technique du DPI-HLA vise à ce que l'enfant à naître, en plus d'être indemne de l'anomalie génétique grave affectant un frère ou une soeur, présente des caractéristiques d'HLA - antigènes des leucocytes humains - compatibles avec l'aîné malade pour que les cellules souches du sang de cordon ombilical soient susceptibles de lui être greffées. Certes, cette technique, lourde et complexe, avec une faible probabilité de succès, n'est plus pratiquée depuis 2014 par l'hôpital Antoine Béclère, qui était le seul à l'avoir mise en oeuvre. Entre 2006 et 2014, 38 demandes d'autorisation ont été accordées par l'Agence de la biomédecine. La démarche a été entreprise par 25 couples. Au final, les naissances obtenues dans ce cadre ont permis d'envisager la greffe de trois enfants.
Les questionnements éthiques entourant cette pratique sont bien évidemment sensibles. Pour autant, les raisons ayant conduit le Parlement en 2004 comme en 2011 à autoriser cette procédure exceptionnelle, selon des modalités strictement encadrées, appellent à mettre en doute les motifs invoqués pour supprimer aujourd'hui la possibilité de sa mise en oeuvre. Comme l'ont confirmé des experts, le recours à une greffe intrafamiliale demeure une option thérapeutique pertinente pour certaines maladies rares et même la plus efficace. L'évolution des thérapeutiques disponibles depuis 2011 n'a donc pas rendu caduc, en solution de dernier recours, comme le prévoit expressément la loi, le DPI-HLA, au point de justifier son abrogation. Mon amendement COM-145 propose donc de rétablir cette disposition instaurée par le législateur en 2004 et dont l'application a été confirmée en 2011.
L'amendement COM-145 est adopté ; les amendements COM-79 et COM-138 deviennent sans objet.
L'article 19 bis A est supprimé.
Mme Corinne Imbert , rapporteure . - Mon amendement COM-146 supprime cet article, introduit par l'Assemblée nationale, qui sollicite la réalisation, par l'Agence de la biomédecine, d'un état des lieux du diagnostic préimplantatoire et du diagnostic prénatal, avant le réexamen de la loi prévu dans un délai de cinq ans par l'article 32. Cette demande paraît inutile dès lors que l'Agence de la biomédecine réalise, dans son rapport médical et scientifique, des rapports d'activité ciblés sur le diagnostic préimplantatoire d'une part et le diagnostic prénatal d'autre part, qui comportent de nombreuses données.
L'amendement COM-146 est adopté ; l'article 19 bis est supprimé.
Articles additionnels après l'article 19 bis (nouveau)
Mme Corinne Imbert , rapporteure . - Mon amendement COM-166 autorise, à titre expérimental et sous conditions, le diagnostic préimplantatoire avec recherche d'aneuploïdies (DPI-A) c'est-à-dire d'anomalies chromosomiques. À l'heure actuelle, le DPI, c'est-à-dire l'analyse in vitro des embryons avant implantation, ne peut être réalisé que lorsqu'une maladie génétique d'une particulière gravité reconnue comme incurable a été diagnostiquée chez l'un des parents ou l'un de ses ascendants immédiats. Il est alors exclusivement ciblé sur l'anomalie responsable de cette pathologie. Les professionnels et sociétés savantes spécialisés en AMP, ainsi qu'en cytogénétique, préconisent le recours possible au DPI dans d'autres indications médicales très ciblées, dans le but d'améliorer la prise en charge de femmes ayant des antécédents d'échec d'implantation embryonnaire ou fausses-couches à répétition ou dont l'âge les prédispose à des anomalies responsables d'échec d'implantation ou d'échec de développement embryonnaire. Dans ces indications, le recours au DPI-A ne permettrait pas d'augmenter les taux de naissance en AMP, mais d'y arriver plus rapidement en implantant en premier l'embryon le plus susceptible de se développer, afin d'éviter plusieurs échecs douloureux. L'ouverture à titre expérimental de ce dispositif tel que préconisé par cet amendement ne serait pas incompatible avec la poursuite de la recherche clinique annoncée par la ministre de la santé, visant à mieux cibler les conditions et les indications médicales pertinentes pour le recours au DPI-A. L'amendement ouvre la voie à des avancées sur ce sujet, de nature à contribuer à améliorer l'efficience de la démarche d'AMP.
M. Bernard Jomier . - Pour que l'amendement soit recevable, la rapporteure a précisé qu'il n'y aurait pas de prise en charge. On n'imagine pas, pourtant, que cela soit le cas. Notre amendement, similaire, prévoyait un régime expérimental, et renvoyait à un décret en Conseil d'État la définition des modalités, ce qui laissait la porte ouverte à une prise en charge.
Mme Corinne Imbert , rapporteure . - En effet, mais mon amendement a pour avantage d'inscrire le DPI-A dans la loi...
M. Bernard Jomier . - Le nôtre a bien passé l'article 40 ! Mais nous nous rallions au vôtre.
M. Alain Milon , président . - C'est qu'il prévoyait un dispositif expérimental. Nous en reparlerons en séance.
L'amendement COM-166 est adopté, et devient article additionnel. L'amendement COM-80 rectifié est retiré.
Mme Corinne Imbert , rapporteure . - Certaines maladies génétiques d'une particulière gravité font aujourd'hui l'objet de thérapies géniques prometteuses qui représentent un véritable gain de chances pour les personnes concernées lorsque ces thérapies sont administrées à un stade précoce chez le jeune enfant, idéalement avant l'apparition des premiers symptômes. C'est le cas notamment pour certaines formes de l'amyotrophie spinale infantile. Le dépistage de ces maladies génétiques ne peut néanmoins être réalisé par le biais des tests enzymatiques biochimiques réalisés en première intention dans le cadre du dépistage néonatal à partir d'une goutte de sang prélevé sur le nouveau-né - c'est le test de Guthrie. Le dépistage de ces maladies nécessite en effet un examen ciblé de génétique moléculaire qui permet d'identifier la présence d'une mutation génétique bien précise. Ce test, peu coûteux, permettrait de faire bénéficier les nouveau-nés concernés par cette mutation de traitements qui amélioreraient significativement leurs espérance et qualité de vie. Dans la mesure où les examens génétiques ne peuvent être pratiqués qu'en cas de symptôme déjà présent de la maladie ou d'antécédent familial, il est nécessaire de déroger aux articles 16-10 du code civil et L. 1131-1 du code de la santé publique pour les pratiquer en première intention. C'est l'objet de mon amendement COM-198 .
L'amendement COM-198 est adopté, et devient article additionnel.
M. Bernard Jomier , rapporteur . - Mon amendement COM-208 prévoit la possibilité pour la femme de désigner une sage-femme comme professionnel de santé participant à l'examen pluridisciplinaire des demandes d'IMG.
L'amendement COM-208 est adopté.
M. Bernard Jomier , rapporteur . - L'amendement COM-85 caractérise le danger présenté par une grossesse multiple pour les embryons ou les foetus. Une grossesse multiple peut mettre en péril la santé des embryons ou des foetus dès lors que le risque de grande prématurité est accru et peut avoir des conséquences à long terme sur l'état de santé de l'embryon ou du foetus, avec un risque de mortalité périnatale ou de séquelles neuro-développementales. Toutefois, ces embryons et foetus étant initialement sains, il peut être utile de préciser que c'est leur devenir qui est mis en péril. Avis favorable.
L'amendement COM-85 est adopté. L'amendement rédactionnel COM-209 est adopté.
M. Bernard Jomier , rapporteur . - Mon amendement COM-210 prévoit la possibilité pour la femme de désigner un médecin ou une sage-femme pour participer à l'examen pluridisciplinaire d'une demande de réduction embryonnaire ou foetale.
L'amendement COM-210 est adopté.
L'article 20 est adopté dans la rédaction issue des travaux de la commission.
M. Bernard Jomier , rapporteur . - L'amendement COM-13 supprime la clause spécifique de conscience des professionnels de santé en matière d'IMG. Je propose aux auteurs de l'amendement de se rallier à l'amendement COM-211, qui supprime lui aussi la clause de conscience spécifique des professionnels de santé en matière d'IMG, tout en maintenant au niveau législatif l'obligation pour le médecin qui refuse de pratiquer l'IMG de référer la patiente à un praticien susceptible de réaliser l'intervention. Dans l'intérêt de la femme et de son accès aux soins, il est utile d'inscrire cette obligation dans la loi.
M. Bernard Bonne . - Je croyais qu'elle y figurait déjà.
M. Alain Milon , président . - Pour l'IVG.
Mme Marie-Pierre de la Gontrie . - Là, il s'agit d'IMG.
Mme Laurence Cohen . - En quoi votre amendement est-il plus complet ?
M. Bernard Jomier , rapporteur . - Contrairement au vôtre, mon amendement ne supprime pas l'obligation, pour un médecin qui refuse de pratiquer une IMG, d'indiquer à la patiente un confrère acceptant de le faire.
Mme Laurence Rossignol . - Cette obligation n'est pas dans la clause de conscience générale ?
M. Bernard Jomier , rapporteur . - Non, elle ne figure que dans la clause de conscience spécifique pour l'IVG.
L'amendement COM-13 n'est pas adopté. L'amendement COM-211 est adopté.
L'article 21 est adopté dans la rédaction issue des travaux de la commission.
M. Bernard Jomier , rapporteur . - L'amendement COM-246 intègre dans le dispositif tous les centres de référence des maladies rares compétents pour la prise en charge des enfants présentant une variation du développement génital, en particulier le Centre de référence des maladies rares endocriniennes de la croissance et du développement (CERMERCD), qui s'occupe des cas d'hyperplasie congénitale des surrénales.
Il étend à dix-huit mois le délai de remise du rapport au Parlement afin de permettre aux centres de référence d'avoir un regard sur une année complète d'activité. Il supprime également une précision inutile relative à la possibilité que le rapport fasse l'objet d'un débat parlementaire.
L'amendement COM-246 est adopté.
M. Bernard Jomier , rapporteur . - L'amendement COM-263 inscrit dans la loi une recommandation du CCNE dans son avis 132 « Questions éthiques soulevées par la situation des personnes ayant des variations du développement sexuel », à savoir l'élaboration de bonnes pratiques pour le diagnostic et la prise en charge des variations génitales.
L'amendement COM-263 est adopté.
L'article 21 bis est adopté dans la rédaction issue des travaux de la commission.
Article additionnel après l'article 21 bis (nouveau)
M. Bernard Jomier , rapporteur . - L'amendement COM-14 rectifié inscrit dans la loi que les actes de conformation sexuée sont dépourvus de nécessité médicale et en conséquence, qu'ils sont interdits sauf urgence vitale ou consentement personnel du patient, même mineur. Il se fonde sur des recommandations internationales qui tendent à exclure les interventions chirurgicales ou hormonales opérées sur des patients avant leur âge de discernement.
Il n'y a pas de consensus en France sur la question, ainsi que l'a relevé le CCNE dans son avis 132 de novembre dernier. La plupart des associations de personnes nées avec une variation sexuelle nient le bénéfice, pour la construction de l'identité sexuelle de l'enfant, d'une intervention chirurgicale ou d'un traitement hormonal, et dénoncent leurs séquelles physiques et psychiques à long terme. Ils évoquent des mutilations opérées sur des organes sains pour des raisons esthétiques ou purement sociales, et non pas médicales.
Mais la position des patients atteints d'hyperplasie congénitale des surrénales semble différente (Mme Maryvonne Blondin le confirme.) : les représentantes de l'association Surrénales que j'ai entendues se sont déclarées pour une intervention précoce. Mais dans leur cas, il n'existe pas de doute sur le sexe de l'enfant et le traitement hormonal est la plupart du temps vital, en raison des pertes de sel et des risques de déshydratation que cela fait courir à l'enfant.
La plupart des chirurgiens et endocrinologues français, quant à eux, justifient les interventions par leurs fins réparatrices et fonctionnelles et font valoir, pour certains, que, pour des raisons psychologiques, le corps doit, dans la mesure du possible, correspondre à une identité sexuelle. Une intervention précoce permet selon eux une minimisation des conséquences psychologiques pour l'enfant et son entourage.
En tout état de cause, le cadre législatif actuel - l'article 16-3 du code civil - interdit déjà les opérations chirurgicales et les traitements irréversibles pratiqués de manière précoce sur un enfant quand il n'y a pas de nécessité médicale, voire en cas d'opération mutilante, lorsqu'il n'y a pas de motif médical très sérieux, selon les termes de l'article R. 4127-41 du code de la santé publique.
L'amendement dont nous discutons viendrait réduire les opérations précoces aux seuls cas d'urgence vitale, excluant ainsi les autres cas de nécessité médicale, dont les opérations visant à éviter des pertes de chance fonctionnelle, sans laisser aucune marge d'appréciation aux médecins eux-mêmes.
Je partage l'intention des auteurs de l'amendement et je partage leurs objectifs, mais cet amendement n'apporte pas la bonne solution.
Mon amendement précédent COM-263 permet de mieux traiter cette question, en renvoyant le diagnostic et la prise en charge d'une variation génitale à un référentiel de bonnes pratiques discuté en concertation avec les associations de patients, qui pourrait comprendre un cadre commun des interventions précoces. Avis défavorable.
Mme Maryvonne Blondin . - C'est effectivement un sujet important, dont je parlerai en séance publique. Le référentiel évoqué par le rapporteur devra bien être mis en place avec tous les acteurs ; or il y a des divergences entre les différentes associations de patients. La Cour européenne des droits de l'homme (CEDH) et le défenseur des droits soutiennent la position qui consiste à n'agir dans ce domaine qu'en cas d'urgence vitale.
Mme Laurence Cohen . - Le sujet mérite qu'on s'y attarde. Je maintiendrai donc mon amendement afin de pouvoir l'aborder en séance publique.
L'amendement COM-14 rectifié n'est pas adopté.
M. Bernard Jomier , rapporteur . - L'amendement COM-148 prévoit une information ad hoc des personnes engagées dans un traitement sur la conversation des tissus germinaux.
L'amendement COM-148 est adopté.
M. Bernard Jomier , rapporteur . - L'amendement COM-150 améliore l'expression du consentement d'une personne mineure sur la préservation des tissus germinaux.
L'amendement COM-150 est adopté.
M. Bernard Jomier , rapporteur . - L'amendement COM-147 prévoit le suivi des personnes ayant recours à l'autoconservation de leurs gamètes ou tissus germinaux pour des raisons pathologiques.
En effet, les seules données existantes, retracées par l'Agence de la biomédecine dans son rapport médical et scientifique, sont relatives aux personnes ayant ensuite recours à une procédure d'AMP.
Un suivi plus large des patients après guérison permettrait d'évaluer le réel impact des traitements reçus ou de la maladie sur le fonctionnement des gonades et de la fertilité pour mieux cibler les indications de cette conservation.
L'amendement COM-147 est adopté, ainsi que l'amendement rédactionnel COM-151 .
M. Bernard Jomier , rapporteur . - Rien ne s'oppose dans cet article à la préservation de la fertilité des personnes ayant changé de sexe, mais les conditions d'accès à l'AMP peuvent être jusqu'à présent un facteur bloquant dans la suite de la démarche.
La précision proposée à l'amendement COM-17 ne me semble pas indispensable ; mais je ne vois pas non plus de raison de m'opposer à l'inscription dans la loi d'une possibilité qui n'a en effet pas lieu d'être écartée. Sagesse.
M. Alain Milon , président . - Ce n'est pas évident. Je m'exprimerai sur ce sujet en séance. Nous avons connu le cas, en Espagne, d'une femme voulant devenir un homme, mais qui, auparavant, avait mené une grossesse, alors qu'elle était déjà en traitement hormonal.
L'amendement COM-17 est adopté.
M. Bernard Jomier , rapporteur . - L'amendement COM-152 précise l'encadrement de la fin de conservation des gamètes ou tissus d'une personne mineure.
L'amendement COM-152 est adopté, ainsi que l'amendement COM-155 .
M. Bernard Jomier , rapporteur . - L'amendement COM-154 modifie les conditions selon lesquelles il est mis fin à la conservation des gamètes, de manière à préserver la possibilité de leur utilisation à un âge où la grossesse est possible - nous avons fixé cet âge à vingt ans après la majorité.
L'amendement COM-154 est adopté.
L'article 22 est adopté dans la rédaction issue des travaux de la commission.
Mme Corinne Imbert , rapporteure . - L'amendement COM-15 supprime l'article élargissant les compétences des conseillers en génétique. Ces derniers exercent toujours sous la responsabilité d'un médecin qualifié et sollicitent régulièrement son avis dans le cadre des prises en charge qu'ils assurent. Rien ne s'oppose donc à ce que les conseillers en génétique puissent non seulement prescrire des examens de génétique, mais également en communiquer les résultats, pour autant que cette communication soit réalisée avec l'accord du médecin généticien. Avis défavorable.
L'amendement COM-15 n'est pas adopté.
Mme Corinne Imbert , rapporteure . - Le texte adopté par l'Assemblée nationale cantonne les conseillers en génétique à l'annonce de résultats ne révélant pas d'anomalie génétique : cette situation aurait pour effet de créer une asymétrie entre les patients convoqués pour une consultation avec un conseiller en génétique, qui anticiperont un diagnostic rassurant en l'absence d'anomalie génétique, et ceux convoqués pour une consultation avec un médecin généticien, qui s'attendront d'emblée à un diagnostic problématique.
L'amendement COM-212 ouvre la possibilité aux conseillers en génétique d'annoncer le résultat de tout examen de génétique, sous réserve que cette communication soit réalisée avec l'accord et sous la supervision du médecin généticien.
L'amendement COM-212 est adopté.
L'article 23 est adopté dans la rédaction issue des travaux de la commission.
Article 24
L'amendement rédactionnel COM-213 est adopté.
L'article 24 est adopté dans la rédaction issue des travaux de la commission.
TABLEAU DES SORTS
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Article additionnel après l'article 10 |
|||
|
M. HENNO, rapporteur |
191 |
Ouverture de l'accès à titre expérimental aux examens génétiques en population générale et dans le cadre du dépistage préconceptionnel |
Adopté |
|
M. Jacques BIGOT |
56 |
Accès aux examens génétiques en population générale |
Satisfait
|
|
M. Jacques BIGOT |
57 |
Possibilité de recourir aux examens génétiques dans le cadre d'un dépistage préconceptionnel |
Satisfait
|
|
Article 11
|
|||
|
M. HENNO, rapporteur |
256 |
Information préalable du patient du recours à un traitement algorithmique |
Adopté |
|
M. HENNO, rapporteur |
257 |
Supervision du traitement algorithmique par le professionnel de santé |
Adopté |
|
M. HENNO, rapporteur |
258 |
Consécration du principe selon lequel aucune décision ne peut être prise sur le seul fondement d'un traitement algorithmique |
Adopté |
|
Mme COHEN |
12 |
Définition de référentiels de bonnes pratiques et concertation avec les associations des usagers du système de santé |
Rejeté |
|
M. HENNO, rapporteur |
259 |
Obligation de transparence du fonctionnement d'un traitement algorithmique |
Adopté |
|
M. HENNO, rapporteur |
260 |
Renvoi au pouvoir réglementaire des conditions de mise en oeuvre du nouvel article L. 4001-3 du code de la santé publique |
Adopté |
|
Article 12
|
|||
|
M. HENNO, rapporteur |
261 |
Maintien du droit en vigueur sur le recours aux techniques d'imagerie cérébrale |
Adopté |
|
Article 13
|
|||
|
M. HENNO, rapporteur |
262 |
Exclusion des dispositifs médicaux du champ d'application de l'article 13 |
Adopté |
|
Article 14
|
|||
|
M. RETAILLEAU |
92 rect. bis |
Suppression de l'article |
Rejeté |
|
M. CHEVROLLIER |
115 rect. |
Réécriture globale de l'article 14 pour suspendre les recherches sur l'embryon et les cellules souches embryonnaires humaines dans l'attente d'une évaluation de ces recherches par l'Opecst |
Rejeté |
|
M. CHEVROLLIER |
34 rect. |
Intégrité des embryons humains sujets de recherches menées dans le cadre de l'assistance médicale à la procréation |
Rejeté |
|
M. CHEVROLLIER |
112 rect. |
Assimilation des recherches menées dans le cadre d'une assistance médicale à la procréation à des recherches non interventionnelles |
Rejeté |
|
M. CHEVROLLIER |
113 rect. |
Mise en place d'une mission d'information sur les recherches menées dans le cadre de l'assistance médicale à la procréation |
Retiré |
|
Mme IMBERT, rapporteure |
192 |
Précision de terminologie concernant les recherches menées sur l'embryon dans le cadre de l'assistance médicale à la procréation |
Adopté |
|
M. CHEVROLLIER |
114 rect. |
Publicité des autorisations de recherches menées dans le cadre d'une assistance médicale à la procréation |
Satisfait
|
|
Mme IMBERT, rapporteure |
226 |
Précision de l'interdiction de création d'embryons à des fins de recherche |
Adopté |
|
Mme IMBERT, rapporteure |
193 |
Précision du prérequis de finalité médicale applicable aux recherches sur l'embryon |
Adopté |
|
Mme IMBERT, rapporteure |
194 |
Précision du prérequis d'absence de méthodologie alternative pour la mise en oeuvre de recherches sur l'embryon |
Adopté |
|
M. DAUDIGNY |
76 |
Clarification du prérequis relatif à la méthodologie alternative en matière de recherche sur l'embryon |
Satisfait
|
|
M. DAUDIGNY |
58 |
Principes éthiques applicables à la recherche sur l'embryon et les cellules souches embryonnaires humaines |
Rejeté |
|
M. CHEVROLLIER |
116 rect. |
Suppression d'un alinéa interdisant le transfert à des fins de gestation d'embryons faisant l'objet d'une recherche et limitant la durée de développement in vitro d'embryons surnuméraires |
Rejeté |
|
Mme IMBERT, rapporteure |
225 |
Amendement de précision rédactionnelle |
Adopté |
|
M. JOMIER |
77 |
Extension du délai limite de culture d'embryons in vitro à des fins de recherche |
Retiré |
|
Mme IMBERT, rapporteure |
195 |
Extension à 21 jours, à titre dérogatoire, de la durée limite de développement in vitro d'embryons dans le cadre de protocoles de recherche dédiés à l'étude des mécanismes du développement embryonnaire au stade de la gastrulation |
Adopté |
|
M. CHEVROLLIER |
33 rect. |
Interdiction de l'expérimentation de l'utérus artificiel |
Retiré |
|
Mme IMBERT, rapporteure |
196 |
Suppression de la précision relative à la possibilité pour les recherches sur l'embryon de porter sur les causes de l'infertilité |
Adopté |
|
M. CHEVROLLIER |
129 rect. |
Interdiction de recherches sur l'embryon portant sur les causes de l'infertilité |
Satisfait
|
|
Mme LASSARADE |
104 rect. |
Interdiction de transfert à des fins de gestation des embryons de primates ayant fait l'objet d'une recherche |
Rejeté |
|
M. CHEVROLLIER |
32 rect. |
Encadrement de l'importation de cellules souches embryonnaires humaines |
Rejeté |
|
M. CHEVROLLIER |
119 rect. |
Limitation des recherches sur les cellules souches embryonnaires humaines aux seules lignées déjà existantes |
Rejeté |
|
M. CHEVROLLIER |
120 rect. |
Publicité des lignées de cellules souches embryonnaires humaines dérivées ou importées en France |
Rejeté |
|
Mme IMBERT, rapporteure |
197 |
Précision du prérequis de finalité médicale applicable aux recherches sur les cellules souches embryonnaires humaines |
Adopté |
|
M. DAUDIGNY |
54 |
Clarification des prérequis pour la réalisation de recherches sur les cellules souches embryonnaires humaines |
Satisfait
|
|
M. CHEVROLLIER |
121 rect. |
Maintien du prérequis de l'absence de méthodologie alternative pour la réalisation de recherches sur des cellules souches embryonnaires humaines |
Rejeté |
|
M. CHEVROLLIER |
122 rect. |
Suppression de la référence aux recherches éthiquement sensibles pratiquées à partir de cellules souches embryonnaires humaines |
Retiré |
|
Mme IMBERT, rapporteure |
201 |
Suppression de la possibilité de créer des embryons chimériques par l'insertion de cellules souches embryonnaires humaines dans un embryon animal |
Adopté |
|
M. CHEVROLLIER |
130 rect. |
Interdiction de la création d'embryons chimériques résultant de l'insertion de cellules souches embryonnaires humaines dans un embryon animal |
Satisfait
|
|
M. RETAILLEAU |
93 rect. |
Interdiction de création d'embryons chimériques par adjonction à un gamète animal de cellules ou de matériel génétique d'origine humaine |
Rejeté |
|
Mme IMBERT, rapporteure |
267 |
Diverses coordinations |
Adopté |
|
Article 15
|
|||
|
M. CHEVROLLIER |
125 rect. |
Suppression des dispositions encadrant les recherches sur les cellules souches pluripotentes induites |
Rejeté |
|
M. CHEVROLLIER |
123 rect. |
Utilisation des cellules pluripotentes induites à des fins de recherche pharmacologique |
Rejeté |
|
M. CHEVROLLIER |
124 rect. |
Suppression de l'encadrement de certaines recherches éthiquement sensibles sur les cellules souches pluripotentes induites |
Retiré |
|
Mme IMBERT, rapporteure |
199 |
Encadrement de l'insertion de cellules souches pluripotentes induites humaines dans un embryon animal |
Adopté |
|
M. RETAILLEAU |
94 rect. |
Soumission à déclaration préalable auprès de l'agence de la biomédecine de l'ensemble des protocoles de recherche sur les cellules souches pluripotentes induites |
Satisfait
|
|
M. CHEVROLLIER |
131 rect. |
Suppression de la possibilité d'insérer des cellules souches pluripotentes induites humaines dans un embryon animal |
Satisfait
|
|
M. RETAILLEAU |
96 rect. |
Institution de prérequis préalables à la mise en oeuvre de protocoles de recherche impliquant des cellules souches pluripotentes induites |
Satisfait
|
|
M. RETAILLEAU |
95 rect. |
Suppression de la possibilité d'insérer des cellules souches pluripotentes induites humaines dans un embryon animal |
Satisfait
|
|
M. DAUDIGNY |
59 |
Principes éthiques applicables aux recherches sur les cellules souches pluripotentes induites |
Rejeté |
|
Article 16
|
|||
|
M. CHEVROLLIER |
31 rect. |
Inscription dans les protocoles de recherche sur l'embryon du consentement écrit du couple cédant leurs embryons à la recherche |
Retiré |
|
Mme IMBERT, rapporteure |
202 |
Allègement de la procédure de confirmation du consentement sur le devenir des embryons ne faisant plus l'objet d'un projet parental |
Adopté |
|
M. CHEVROLLIER |
30 rect. |
Inscription dans les protocoles de recherche sur l'embryon du consentement écrit du couple cédant leurs embryons à la recherche |
Rejeté |
|
M. AMIEL |
84 rect. |
Détermination du devenir des embryons conservés en cas de décès de l'un des membres du couple |
Adopté |
|
Mme IMBERT, rapporteure |
203 |
Allongement à dix ans du délai de conservation des embryons cédés à la recherche non inclus dans un protocole de recherche |
Adopté |
|
Article 17
|
|||
|
M. RETAILLEAU |
97 rect. |
Maintien de l'interdiction de création d'embryons chimériques |
Rejeté |
|
M. HENNO, rapporteur |
200 |
Clarification de l'interdiction de création d'embryons chimériques |
Adopté |
|
M. CHEVROLLIER |
128 rect. |
Interdiction de la création d'embryons génétiquement modifiés |
Retiré |
|
M. HENNO, rapporteur |
204 |
Correction d'une erreur matérielle |
Adopté |
|
Article 18
|
|||
|
M. HENNO, rapporteur |
206 |
Préservation des droits de la personne en matière d'effacement de ses données ou de retrait de son consentement à l'utilisation de ses données dans le cadre d'une recherche |
Adopté |
|
M. Jacques BIGOT |
55 rect. |
Information de la parentèle en cas de découverte d'anomalies génétiques à l'occasion d'un examen génétique réalisé dans le cadre d'une recherche scientifique |
Adopté |
|
M. HENNO, rapporteur |
205 |
Harmonisation rédactionnelle |
Adopté |
|
M. HENNO, rapporteur |
207 |
Amendement de clarification rédactionnelle |
Adopté |
|
Article 19
|
|||
|
Mme IMBERT, rapporteure |
165 |
Définition de la médecine foetale |
Adopté |
|
M. Jacques BIGOT |
52 |
Suppression de la référence à une affection de particulière gravité |
Satisfait
|
|
Mme IMBERT, rapporteure |
143 |
Rédactionnel |
Adopté |
|
Mme IMBERT, rapporteure |
169 |
Clarification rédactionnelle |
Adopté |
|
M. RETAILLEAU |
98 rect. bis |
Délai de réflexion après l'annonce d'un résultat |
Rejeté |
|
Article
19
bis
A (nouveau)
|
|||
|
Mme IMBERT, rapporteure |
145 |
Suppression d'article |
Adopté |
|
M. JOMIER |
79 |
Suppression de l'abrogation du double diagnostic préimplantatoire |
Satisfait
|
|
Mme SCHILLINGER |
138 |
Suppression de l'abrogation du double diagnostic préimplantatoire |
Satisfait
|
|
Article
19
bis
(nouveau)
|
|||
|
Mme IMBERT, rapporteure |
146 |
Suppression d'article |
Adopté |
|
Article additionnel après l'article 19 bis (nouveau) |
|||
|
Mme IMBERT, rapporteure |
166 |
Autorisation sous condition du diagnostic préimplantatoire |
Adopté |
|
M. JOMIER |
80 rect. |
Expérimentation du diagnostic préimplantatoire avec recherche d'aneuploïdies |
Retiré |
|
Mme IMBERT, rapporteure |
198 |
Possibilité de recherche en première intention chez le nouveau-né d'anomalies génétiques graves susceptibles de mesures de prévention ou de soins |
Adopté |
|
Article 20
|
|||
|
M. JOMIER, rapporteur |
208 |
Possibilité pour la femme de désigner une sage-femme comme professionnel de santé participant à l'examen pluridisciplinaire des demandes d'interruption médicale de grossesse |
Adopté |
|
Mme SCHILLINGER |
85 |
Caractérisation du danger présenté par une grossesse multiple pour les embryons ou les foetus |
Adopté |
|
M. JOMIER, rapporteur |
209 |
Harmonisation rédactionnelle |
Adopté |
|
M. JOMIER, rapporteur |
210 |
Possibilité pour la femme de désigner un médecin ou une sage-femme pour participer à l'examen pluridisciplinaire d'une demande de réduction embryonnaire ou foetale |
Adopté |
|
Article 21
|
|||
|
Mme COHEN |
13 |
Suppression de la clause spécifique de conscience des professionnels de santé en matière d'interruption médicale de grossesse |
Rejeté |
|
M. JOMIER, rapporteur |
211 |
Suppression de la clause de conscience spécifique des professionnels de santé en matière d'interruption médicale de grossesse |
Adopté |
|
Article
21
bis
(nouveau)
|
|||
|
M. JOMIER, rapporteur |
246 |
Intégration au dispositif de tous les centres de référence des maladies rares compétents et extension du délai de remise du rapport au Parlement |
Adopté |
|
M. JOMIER, rapporteur |
263 |
Élaboration de recommandations de bonnes pratiques pour le diagnostic et la prise en charge des variations génitales par la HAS |
Adopté |
|
Article additionnel après l'article 21 bis (nouveau) |
|||
|
Mme COHEN |
14 rect. |
Interdiction des actes médicaux de conformation sexuée, sauf cas d'urgence vitale ou consentement personnel du patient même mineur |
Rejeté |
|
Article 22
|
|||
|
M. JOMIER, rapporteur |
148 |
Information des personnes à leur majorité |
Adopté |
|
M. JOMIER, rapporteur |
150 |
Expression du consentement d'une personne mineure |
Adopté |
|
M. JOMIER, rapporteur |
147 |
Étude de suivi des personnes concernées |
Adopté |
|
M. JOMIER, rapporteur |
151 |
Rédactionnel |
Adopté |
|
Mme COHEN |
17 |
Préservation de la fertilité pour les personnes ayant changé de sexe à l'état civil |
Adopté |
|
M. JOMIER, rapporteur |
152 |
Précision encadrant la fin de conservation des gamètes ou tissus d'une personne mineure |
Adopté |
|
M. JOMIER, rapporteur |
155 |
Allègement de la procédure de confirmation du consentement |
Adopté |
|
M. JOMIER, rapporteur |
154 |
Modifications des conditions selon lesquelles il est mis fin à la conservation des gamètes |
Adopté |
|
Article 23
|
|||
|
Mme COHEN |
15 |
Suppression de l'article élargissant les compétences des conseillers en génétique |
Rejeté |
|
Mme IMBERT, rapporteure |
212 |
Possibilité pour les conseillers en génétique de communiquer les résultats d'un examen génétique avec l'autorisation et sous la supervision du médecin généticien |
Adopté |
|
Article 24
|
|||
|
Mme IMBERT, rapporteure |
213 |
Amendement rédactionnel |
Adopté |
- Présidence de Mme Catherine Deroche, vice-présidente -
Mme Catherine Deroche , présidente . - Nous reprenons l'examen du rapport et du texte de la commission spéciale.
Mme Corinne Imbert , rapporteure . - L'amendement COM-214 vise à clarifier la frontière entre génétique somatique et génétique constitutionnelle en précisant que les examens de génétique somatique consistent à rechercher en première intention et à analyser des caractéristiques génétiques ni héritées ni transmissibles. En cas de découverte incidente de caractéristiques génétiques constitutionnelles, la personne sera toujours invitée à se rendre à une consultation de génétique constitutionnelle.
Mme Véronique Guillotin . - Pourriez-vous nous en dire plus ?
Mme Corinne Imbert , rapporteure . - Les tests génétiques tumoraux, notamment dans le cadre de cancers des ovaires, visent à établir la signature génétique d'une tumeur, qui est en théorie somatique, donc non transmissible, mais qui peut également se révéler constitutionnelle, donc transmissible.
L'amendement COM-214 est adopté.
Mme Corinne Imbert , rapporteure . - L'amendement COM-215 tend à ouvrir la possibilité de rechercher des altérations génétiques somatiques au niveau des cellules germinales, dont la présence ne peut pas être exclue. Certaines tumeurs germinales peuvent résulter d'altérations de ces cellules à partir desquelles un examen des caractéristiques génétiques somatiques pourra à titre exceptionnel être envisagé.
L'amendement COM-215 est adopté.
L'amendement rédactionnel COM-216 est adopté.
L'article 25 est adopté dans la rédaction issue des travaux de la commission.
M. Olivier Henno , rapporteur . - L'amendement COM-217 vise à appliquer les règles éthiques françaises, notamment l'anonymat du don, à l'exception du don intrafamilial, pour le microbiote fécal.
L'amendement COM-217 est adopté.
L'article 26 est adopté dans la rédaction issue des travaux de la commission.
Article 27
L'article 27 est adopté sans modification.
Article 28
L'article 28 est adopté sans modification.
Mme Muriel Jourda , rapporteur . - Mon amendement COM-244 et l'amendement COM-18 , qui sont identiques, visent à supprimer l'article 29 A. Compte tenu de l'existence de l'Office parlementaire d'évaluation des choix scientifiques et technologiques (Opesct), la création d'une délégation parlementaire à la bioéthique dans chaque assemblée ne semble pas justifiée. Comme le soulignaient Roger Karoutchi et Alain Richard, mieux vaut éviter la dispersion des sénateurs et la « polysynodie » des structures.
Les amendements COM-244 et COM-18 sont adoptés ; en conséquence, l'article 29 A est supprimé
Mme Catherine Deroche , présidente . - L'amendement COM-105 rectifié est irrecevable en application de l'article 45 de la Constitution : aucune disposition du projet de loi initial ne concerne les droits des animaux ni les conditions dans lesquelles des expérimentations peuvent être menées sur des animaux.
À cette occasion, j'indique à la commission que le projet de « périmètre » défini en application du vade mecum adopté par la conférence des présidents le 20 mars 2019 est disponible sur Demeter. Si vous n'avez pas d'observations, il sera diffusé à l'ensemble de nos collègues dès la fin de la réunion.
Mme Corinne Imbert , rapporteure . - L'amendement COM-229 tend, par coordination, à supprimer les références aux délégations parlementaires à la bioéthique.
L'amendement COM-229 est adopté.
Mme Corinne Imbert , rapporteure . - L'amendement COM-16 tend à porter de quarante à quarante-six le nombre de membres du Comité consultatif national d'éthique (CCNE) pour y inclure des représentants d'associations, notamment des associations d'usagers du système de santé, d'associations familiales et d'associations de personnes handicapées. Même si elle n'est pas expressément prévue, la présence au sein des membres du CCNE de représentants du monde associatif est possible, notamment au sein du collège des personnes qualifiées. Il y a aujourd'hui la présidente d'ATD Quart Monde France et d'autres personnalités impliquées dans le milieu associatif. Retrait ou avis défavorable.
Mme Laurence Cohen . - Lors des auditions, nous avons ressenti une forte volonté d'implication. Même si une telle possibilité existe déjà, le fait de l'inscrire dans la loi la conforte.
L'amendement COM-16 n'est pas adopté.
Mme Corinne Imbert , rapporteure . - L'amendement COM-61 et mon amendement COM-218 visent à supprimer les conditions d'équilibre politique dans la désignation des parlementaires membres du CCNE.
Les amendements identiques COM-61 et COM-218 sont adoptés.
L'article 29 est adopté dans la rédaction issue des travaux de la commission.
Mme Corinne Imbert , rapporteure . - L'amendement COM-78 et mon amendement COM-219 ont pour objet le maintien de l'élaboration par l'Agence de la biomédecine d'un référentiel d'évaluation de la qualité des tests génétiques en accès libre.
Les amendements identiques COM-78 et COM-219 sont adoptés.
L'amendement de coordination COM-220 est adopté.
Mme Corinne Imbert , rapporteure . - L'amendement COM-221 tend à l'inclusion d'une analyse des décisions d'opposition de l'Agence de la biomédecine à certains protocoles de recherche sur les cellules souches embryonnaires et pluripotentes induites.
L'amendement COM-221 est adopté.
Mme Corinne Imbert , rapporteure . - L'amendement COM-222 vise à rétablir le principe de parité entre les représentants institutionnels et les personnalités qualifiées au sein du conseil d'administration de l'Agence de la biomédecine.
L'amendement COM-222 est adopté.
Mme Corinne Imbert , rapporteure . - Les auteurs de l'amendement COM-81 proposent l'introduction dans le rapport annuel de l'activité de l'Agence de la biomédecine d'un volet consacré aux évolutions législatives et réglementaires qui pourraient être justifiées par l'évolution des connaissances et des techniques, mais aussi par des situations qui ne seraient pas couvertes par le droit en vigueur et nécessiteraient des autorisations de dérogation. Cette évolution est bienvenue. Elle permettrait une réactualisation plus réactive de notre législation dans des domaines où il est parfois délicat d'attendre tous les cinq ans. Avis favorable.
L'amendement COM-81 est adopté.
L'article 30 est adopté dans la rédaction issue des travaux de la commission.
M. Olivier Henno , rapporteur . - L'amendement COM-231 tend à supprimer l'habilitation à légiférer par ordonnance pour l'investigation clinique en matière de dispositif médical.
L'amendement COM-231 est adopté.
L'article 31 est adopté dans la rédaction issue des travaux de la commission.
Article 32
L'amendement rédactionnel COM-255 est adopté.
Mme Muriel Jourda , rapporteur . - Les auteurs de l'amendement COM-73 souhaitent exclure le titre I er du champ de la clause de révision de la loi de bioéthique. Outre que le titre I er ne concerne pas seulement l'assistance médicale à la procréation - il concerne également l'autoconservation des gamètes et l'accès aux origines -, je ne vois pas pourquoi l'on exclurait certaines dispositions de cette clause. Avis défavorable.
M. Jacques Bigot . - Les règles de bioéthique évoluent avec les connaissances médicales. Aucune évolution des connaissances médicales susceptible de justifier une modification législative dans cinq ans n'est attendue sur la procréation médicalement assistée (PMA). Rien ne justifie donc d'annoncer par avance qu'il faudra revoir la loi en la matière.
Mme Marie-Pierre de la Gontrie . - L'affirmation du caractère « révisable » du projet de loi peut laisser entendre que l'autorisation de la PMA serait réversible. S'il est toujours possible de réviser une loi quand c'est nécessaire, prévoir par principe de revenir sur un droit nouveau tous les cinq ans me pose problème.
Mme Laurence Rossignol . - Nous avons été plusieurs, dont Muriel Jourda, à nous étonner de la présence de l'extension de la PMA dans un projet de loi relatif à la bioéthique. Une telle réforme aurait été plus à sa place dans un texte portant sur le droit civil. Nul ne songerait à conclure une loi sur la filiation en prévoyant de la réviser cinq ans plus tard. Soyons cohérents : si nous considérons que la PMA n'a rien à faire dans un texte sur la bioéthique, ne la soumettons pas aux révisions de la loi de bioéthique.
M. Philippe Bas . - Y a-t-il des matières qui justifient un réexamen régulier de la loi, et d'autres non ? C'est un débat intéressant. Le gouvernement de 2004 avait choisi d'inscrire dans le premier texte de révision des lois de bioéthique que celles-ci ne seraient plus révisées à échéance régulière, les principes de bioéthique n'étant pas contingents à l'état de la science. Le législateur avait rétabli une clause de révision. Cela ne me semblait pas justifié. Le Parlement peut toujours se saisir de dispositions législatives qu'il souhaite faire évoluer. Je ne vois pas en quoi il serait plus nécessaire de réviser tous les cinq ans le volet de recherche biomédicale que celui relatif aux questions de société. La distinction proposée dans l'amendement ne se justifie pas. Et prévoir un réexamen de la loi dans cinq ans ne préjuge en rien des décisions que le législateur prendra à ce moment-là.
M. Bernard Jomier . - Présent dans chaque loi de bioéthique, le principe de révision ne vaut pas nécessairement révision de tous les items. C'est le Gouvernement qui décide de la liste des sujets figurant dans un projet de loi. Toutes les dispositions dont nous débattons aujourd'hui ne seront pas automatiquement révisées dans cinq ans. Si le Gouvernement veut rouvrir tel ou tel débat, il a de multiples outils pour le faire. Cet amendement a toutefois le mérite de confirmer que les premiers articles du projet de loi étaient bien des articles parasites.
Mme Muriel Jourda , rapporteur . - La question de savoir pourquoi de telles dispositions figuraient dans un projet de loi relatif à la bioéthique a effectivement pu se poser. Mais je considère qu'elles y ont leur place. Elles renvoient à un débat essentiel en matière de bioéthique : ce n'est pas parce que des choses sont possibles qu'il faut forcément les faire. En outre, la clause de révision qui existait dans le texte de 2011 n'a pas abouti, tant s'en faut, à une restriction des droits.
M. Jacques Bigot . - Je réfléchirai avec mes collègues au dépôt d'un amendement de suppression du premier alinéa de l'article 32, dont la rédaction - la présente loi « fait l'objet d'un nouvel examen d'ensemble » - est problématique.
Mme Laurence Cohen . - Nous souscrivons à l'analyse de Jacques Bigot, et nous voterons cet amendement.
Mme Catherine Deroche , présidente . - En 2011, le texte initial ne prévoyait pas de clause de révision. C'est le Sénat qui en avait introduit une, contre l'avis du gouvernement de l'époque.
L'amendement COM-73 n'est pas adopté.
L'article 32 est adopté dans la rédaction issue des travaux de la commission.
Mme Corinne Imbert , rapporteure . - L'amendement COM-156 tend à supprimer cet article, qui prévoit la remise d'un rapport.
L'amendement COM-156 est adopté ; en conséquence, l'article 33 est supprimé.
M. Olivier Henno , rapporteur . - L'amendement COM-230 vise à supprimer cet article, pour les mêmes raisons.
L'amendement COM-230 est adopté ; en conséquence, l'article 34 est supprimé.
Le projet de loi est adopté dans la rédaction issue des travaux de la commission.
TABLEAU DES SORTS
|
Auteur |
N° |
Objet |
Sort de l'amendement |
|
Article 25
|
|||
|
Mme IMBERT, rapporteure |
214 |
Clarification de la frontière entre génétique somatique et génétique constitutionnelle |
Adopté |
|
Mme IMBERT, rapporteure |
215 |
Possibilité de rechercher des altérations génétiques somatiques au niveau de cellules germinales |
Adopté |
|
Mme IMBERT, rapporteure |
216 |
Harmonisation rédactionnelle |
Adopté |
|
Article 26
|
|||
|
M. HENNO, rapporteur |
217 |
Règles d'anonymat du don applicables en matière de transplantation de microbiote fécal |
Adopté |
|
Article 29 A (nouveau)
|
|||
|
Mme Muriel JOURDA, rapporteur |
244 |
Suppression d'article |
Adopté |
|
Mme COHEN |
18 |
Suppression d'article |
Adopté |
|
Article 29
|
|||
|
Mme LASSARADE |
105 rect. |
Prise en compte par le CCNE des enjeux s'attachant aux expérimentations conduites sur des animaux |
Irrecevable au titre de l'article 45 |
|
Mme IMBERT, rapporteure |
229 |
Suppression des références aux délégations parlementaires à la bioéthique |
Adopté |
|
Mme COHEN |
16 |
Augmentation du nombre de membres du CCNE pour inclure des représentants d'associations |
Rejeté |
|
M. Jacques BIGOT |
61 |
Suppression des conditions d'équilibre politique dans la désignation des parlementaires membres du CCNE |
Adopté |
|
Mme IMBERT, rapporteure |
218 |
Suppression des conditions d'équilibre politique dans la désignation des parlementaires membres du CCNE |
Adopté |
|
Article 30
|
|||
|
M. JOMIER |
78 |
Maintien de l'élaboration par l'agence de la biomédecine d'un référentiel d'évaluation de la qualité des tests génétiques en accès libre |
Adopté |
|
Mme IMBERT, rapporteure |
219 |
Maintien de l'élaboration par l'agence de la biomédecine d'un référentiel d'évaluation de la qualité des tests génétiques en accès libre |
Adopté |
|
Mme IMBERT, rapporteure |
220 |
Amendement de coordination et de correction d'une erreur matérielle |
Adopté |
|
Mme IMBERT, rapporteure |
221 |
Inclusion d'une analyse des décisions d'opposition de l'agence de la biomédecine à certains protocoles de recherche sur les cellules souches embryonnaires et pluripotentes induites |
Adopté |
|
Mme IMBERT, rapporteure |
222 |
Rétablissement du principe de parité au sein du conseil d'administration de l'agence de la biomédecine entre les représentants institutionnels et les personnalités qualifiées |
Adopté |
|
M. JOMIER |
81 |
Transmission annuelle au Parlement par l'agence de la biomédecine de recommandations d'évolutions législatives dans le domaine de la bioéthique |
Adopté |
|
Article 31
|
|||
|
M. HENNO, rapporteur |
231 |
Suppression de l'habilitation à légiférer par ordonnance sur l'investigation clinique en matière de dispositif médical |
Adopté |
|
Article 32
|
|||
|
Mme Muriel JOURDA, rapporteur |
255 |
Amendement rédactionnel |
Adopté |
|
M. Jacques BIGOT |
73 |
Exclusion du titre I er du champ de la clause de révision de la loi de bioéthique |
Rejeté |
|
Article 33 (nouveau)
|
|||
|
Mme IMBERT, rapporteure |
156 |
Suppression d'article |
Adopté |
|
Article 34 (nouveau)
|
|||
|
M. HENNO, rapporteur |
230 |
Suppression d'article |
Adopté |
LISTE DES PERSONNES
ENTENDUES
PAR LES RAPPORTEURS ET CONTRIBUTIONS ÉCRITES
Auditions de M. Olivier Henno
David Gruson , membre du comité de direction de la chaire santé de Sciences Po, professeur associé à la faculté de médecine Paris-Descartes, fondateur de l'initiative « Ethik IA »
Anaïs Person , initiative Ethik IA, chef de projet déléguée à la protection des données (société Seraphin.legal)
Claude Kirchner , président de la commission de réflexion sur l'éthique de la recherche en sciences et technologies du numérique d'Allistene (Cerna)
Jean Charlet , chargé de mission recherche à l'Assistance publique - Hôpitaux de Paris (AP-HP)
Sonia Desmoulin-Canselier , docteur en droit privé et chargée de recherche au Centre national de la recherche scientifique (CNRS)
Bénédicte Bévière-Boyer , maître de conférences en droit privé et sciences criminelles à l'université Paris 8, directrice adjointe du Centre de recherche en droit privé et droit de la santé
Association française des hébergeurs agréés de données de santé à caractère personnel (AFHADS)
Bruno Virieux , directeur « Data science »
Direction de la recherche, des études, de l'évaluation et des statistiques (Drees) du ministère des solidarités et de la santé
Jean-Marc Aubert , directeur
Institut national des données de santé (prochainement dénommé Plateforme des données de santé)
Stéphanie Combes , cheffe de projet « Health Data Hub » à la direction de la recherche, des études, de l'évaluation et des statistiques (Drees)
Marc Cuggia , professeur d'informatique médicale et praticien hospitalier au centre hospitalier universitaire (CHU) de Rennes
Agence nationale de sécurité du médicament et des produits de santé (ANSM)
Christelle Ratignier-Carbonneil , directrice générale adjointe
Carole Le-Saulnier , directrice des affaires juridiques et réglementaires
Intitut national de la santé et de la recherche médicale (Inserm) - Alliance pour les sciences de la vie et de la santé (Aviesan)
Étienne Hirsch , directeur de l'institut thématique « Neurosciences, neurologie et psychiatrie » de l'Inserm, directeur de l'institut thématique multi-organismes « Neurosciences, sciences cognitives, neurologie et psychiatrie » d'Aviesan
Bernard Poulain , directeur adjoint scientifique « Neurosciences, cognition » de l'institut national des sciences biologiques, directeur de l'institut thématique multi-organismes « Neurosciences, sciences cognitives, neurologie et psychiatrie » d'Aviesan
Anne-Sophie Etzol , chargée des relations institutionnelles, département « Information scientifique et communication » de l'Inserm
Stanislas Dehaene , professeur au Collège de France, chaire de psychologie cognitive expérimentale
Haute Autorité de santé (HAS)
Isabelle Adenot , membre du collège de la HAS, présidente de la commission nationale d'évaluation des dispositifs médicaux et des technologies de santé (CNEDiMTS)
Denis-Jean David , adjoint au chef du service d'évaluation des actes professionnels, direction de l'évaluation médicale, économique et de santé publique
Contributions écrites
Olivier Oullier
, président de la
société de neuroinformatique EMOTIV, professeur à
l'université d'Aix-Marseille, membre du comité scientifique de
l'Office parlementaire d'évaluation des choix scientifiques et
technologiques (Opecst)
Laura Pignatel
, laboratoire de
droit privé et de sciences criminelles, faculté de droit et de
science politique, université d'Aix-Marseille
Luc Buée , président de la Société des neurosciences, directeur de recherches au Centre national de la recherche scientifique
Auditions de Mme Corinne Imbert
Agence de la biomédecine
Philippe Jonveaux, directeur de la procréation, de l'embryologie et de la génétique humaines
Pascale Levy, chef du pôle diagnostic à la direction de la procréation, de l'embryologie et de la génétique humaines
Samuel Arrabal, chef du pôle recherche
Jean-Paul Bonnefont , professeur de biologie moléculaire à l'université Paris-Descartes, responsable du laboratoire hospitalier de génétique moléculaire de l'hôpital Necker - Enfants malades
Pierre Savatier , chef d'équipe du Stem-cell and Brain Research Institute (Lyon), spécialiste des embryons chimériques
Laurence Brunet , chercheure à l'Institut des sciences juridiques et philosophiques de la Sorbonne (Paris 1 Panthéon-Sorbonne)
Pierre Jouannet , membre de l'Académie nationale de médecine et du comité d'éthique de l'Inserm, spécialiste de la biologie de la reproduction
Mickaël Grynberg, chef du service de médecine de la reproduction et préservation de la fertilité de l'hôpital Antoine-Béclère
Julie Steffann , service de génétique moléculaire à l'hôpital Necker-Enfants malades
Marie-Pierre Bichet , présidente de l'Association française de la maladie de Fanconi (AFMF) et vice-présente de l'Alliance Maladies rares
Christian Cottet , directeur général, et François Lamy , vice-président chargé de la recherche, de l'AFM-Téléthon
Hélène Bérrué-Gaillard, référente recherche au sein de l'Alliance Maladies rares
Auditions de M. Bernard Jomier
Fédération des associations pour le don d'organes et de tissus humains (France ADOT)
Marie-Claire Paulet, présidente nationale
Association Renaloo
Magali Leo, responsable du pôle Plaidoyer
Lionel Rostaing , néphrologue au CHU de Grenoble, membre du comité médical et scientifique
Agence de la biomédecine
Olivier Bastien,
directeur du
prélèvement et des greffes d'organes et de
tissus
Evelyne Marry
, directrice du
prélèvement et des greffes de cellules souches
hématopoïétiques
Société française de médecine des prélèvements d'organes et de tissus (SFMPOT)
Julien Rogier , président
Jean-Marie Jouannic, chef du service de médecine foetale de l'hôpital Armand Trousseau
Stéphanie Staraci, psychologue à l'hôpital de la Pitié-Salpêtrière (unité fonctionnelle « Consultation génétique cardiologie - maladies rares »)
Michèle Goussot-Souchet, sage-femme à l'hôpital Cochin Port-Royal
Adrien Gantois, président du Collège national des sages-femmes de France
Association Agapa
Sabine Jourdan, bénévole accompagnante, responsable des groupes de parole
Anne-Claude Duvert, bénévole accompagnante, animatrice de groupes de parole et de cafés-rencontres
Association Petite Émilie
Clarisse Beauvois , présidente de l'association
Laurence Pavie, vice-présidente
Centre de référence des maladies rares du développement génital (DEV GEN)
Pierre Mouriquand , chef du service de chirurgie uro-viscérale de l'enfant du CHU de Lyon
Rémi Besson , chirurgien pédiatre urologue au CHU de Lille
Nicolas Kalfa , chirurgien au CHU de Montpellier
Claire Bouvattier , pédiatre gynécologue à l'hôpital Bicêtre
Laurence Brunet , juriste et éthicienne au centre de référence des maladies rares du développement génital à l'hôpital Bicêtre
Lise Dieu-Phuong Duranteau , endocrinologue-gynécologue, responsable de l'Unité de gynécologie adolescente et jeune adulte au sein des hôpitaux universitaires Paris Saclay
Marc Fellous, médecin généticien
Association « Maison intersexualité et hermaphrodisme Europe » (Amihe)
Sylvaine Telesfort , présidente
Groupe de soutien au syndrome de l'insensibilité aux androgènes (GSSIA)
Evelyne Danne , présidente fondatrice
Association Surrénales
Nathalie Colin , présidente
Claudine Colin , ex-présidente
Collectif Intersexes et AlliéEs (CIA)
Mathilde Abel , représentante
Benjamin Moron-Puech , enseignant-chercheur en droit à l'université Panthéon-Assas, Association GISS | Alter Corpus
Contributions écrites
Fédération nationale des administrateurs ad hoc (Fenaah)
Josiane Bigot , présidente
Juliane Léger , coordinateur du Centre de référénce des maladies endocriniennes de la croissance et du développement
Auditions de Mme Muriel Jourda
Association Procréation Médicalement Anonyme (PMAnonyme)
Clément Sillau-Roussial , vice-président
Sylvie Jouanny , porte-parole des parents
Association Origines
Arthur Kermalvezen , président et fondateur
Audrey Kermalvezen , cofondatrice
Collectif pour le droit aux origines
Pierre Verdier, avocat, président de la Coordination des actions pour le droit à la connaissance des origines (Cadco)
Association Dons de gamètes solidaires
Frédéric Letellier-Cohen, vice-président
Irène Théry, sociologue du droit, de la famille et de la vie privée, directrice d'études à L'École des hautes études en sciences sociales (EHESS)
Frédéric Worms , professeur de philosophie à l'Ecole normale supérieure, membre du Comité consultatif national d'éthique (CCNE)
Collectif Centres privés AMP
Anne Grelat , gynécologue spécialisée en AMP
Bertrand Keppi, biologiste médical spécialisé en AMP
Association des enfants du don (ADEDD)
Christophe Masle , président de France AMP (fédération des ADEDD)
Adèle Bourdelet , présidente de l'ADEDD Ile-de-France et Normandie
Collectif des maires pour l'enfance
Franck Meyer , président
Ministère de la justice - Direction des affaires civiles et du Sceau (DACS)
Jean-François de Montgolfier , directeur
Marie-Charlotte Dalle , sous-directrice du droit civil
Mélanie Bessaud , chef du bureau du droit des personnes et de la famille
Sabine Carré , rédactrice au bureau du droit des personnes et de la famille
Pierre Lévy-Soussan, pédopsychiatre, psychanalyste
Christian Flavigny, pédopsychiatre
Lisa Carayon, maîtresse de conférences, Université Paris 13 - Paris Nord
Aline Cheynet de Beaupré, professeur de droit privé, Université d'Orléans
Victor Deschamps, maître de conférences en droit privé, Université Paris 2 Panthéon-Assas
Anne-Marie Leroyer, professeur, droit privé et sciences criminelles, Université Paris 1 Panthéon-Sorbonne
Astrid Marais, professeure, droit privé et sciences criminelles, Université Paris 8
Aude Mirkovic, maître de conférences en droit privé, habilitée à diriger des recherches (HDR)
Conseil supérieur du notariat (CSN)
Jean-François Humbert , directeur
François Devos, directeur des affaires juridiques
Christine Mandelli , administrateur, chargée des relations avec les institutions
Conseil national des barreaux
Régine Barthélémy, avocate
Anne-Charlotte Varin, directrice des affaires publique s
Barreau de Paris
Emilie Chandler, avocate, membre du Conseil de l'ordre
Julien Aubignat, responsable des affaires publiques
Catherine Jousselme , professeur de psychiatrie de l'enfant et de l'adolescent (Université Paris-Sud), chef de service et chef du pôle universitaire du centre hospitalier Fondation Vallée Gentilly
Cour de cassation
François Molins, procureur général
Rachel Le Cotty, conseiller référendaire à la première chambre civile
Domitille Duval-Arnould, conseiller à la première chambre civile
Contributions écrites
Jacques Toubon , Défenseur des droits
Association des maires de France et des présidents d'intercommunalités (AMF)
Claire Neirinck , professeur émérite de droit privé et de sciences criminelles de l'Université de Toulouse 1 Capitole
Auditions communes
de M. Olivier Henno et Mme
Corinne Imbert
Association française des conseillers en génétique (AFCG)
Marie-Antoinette Voelckel , présidente
Emmanuelle Haquet , vice-présidente
Emilie Consolino , responsable de la communication
Antoine De Pauw , webmaster et membre du bureau du Groupe génétique et cancer (GGC)
Stéphane Bézieau , président de la Fédération française de génétique humaine
Massimiliano Rossi , président de l'Association francophone de génétique clinique
Martine Doco-Fenzy , présidente de l'Association des cytogénéticiens de langue française
Dominique Stoppa-Lyonnet , généticien clinique, chef de service du Pôle de médecine diagnostique et théranostique à l'Institut Curie
Pascal Pujol , président de la Société française de médecine prédictive et personnalisée
Auditions communes
de M. Olivier Henno et Mme
Muriel Jourda
Geneanet
Jacques Le Marois, président-directeur général
Christophe Becker , directeur
Fédération française de généalogie
Valérie Arnold-Gautier , présidente
Alain Rossi , vice-président
Thierry Chestier , secrétaire général
Filae
Toussaint Roze , président fondateur
Emmanuel Condamine , directeur général
DNA Pass
Nathalie Jovanovic-Floricourt , présidente
MyHeritage
Gilad Japhet , fondateur, chief executive officer
Florence Maisel , conseil en affaires publiques de MyHeritage en France ( managing partner , Interel)
Commission nationale de l'informatique et des libertés (CNIL)
Hélène Guimiot-Breaud , cheffe du service santé
Erik Boucher de Crèvecoeur , ingénieur expert à la Direction des technologies et de l'innovation
Tiphaine Havel , conseillère pour les questions institutionnelles et parlementaires
Auditions communes
de M. Bernard Jomier et Mme
Muriel Jourda
Société de médecine de la reproduction (SMR)
Nathalie Massin, présidente
Groupe de recherche et d'étude sur la cryoconservation de l'ovaire et du testicule (Grecot)
Catherine Poirot, présidente
Société française de gynécologie (SFG)
Joëlle Bellaisch-Allart, présidente
Groupe d'Etude de la Fécondation in vitro en France
Géraldine Porcu Buisson , présidente
Groupe Etude pour le don d'ovocyte (GEDO)
Hélène Letur , présidente
Syndicat national des gynécologues et obstétriciens de France (Syngof)
Arnaud Grisey, vice-président du Conseil national professionnel de gynécologie-obstétrique et gynécologie médicale
Fédération nationale des collèges de gynécologie médicale (FNCGM)
Pia de Reilhac , présidente
Marie de Crécy , membre du conseil d'administration de la FNCGM et du collège de gynécologie de Paris Île-de-France
Conseil national de l'ordre de médecins
Jean-Marcel Mourgues, vice-président de l'Ordre
Anne-Marie Trarieux, présidente de la section éthique et déontologie
Réseau fertilité France
Larissa Meyer, présidente
Association MAIA
Laëtitia Poisson Deleglise, présidente
Deborah Schouhmann Antonio , thérapeute conseil
Collectif BAMP
Virginie Rio, présidente
Stéphanie Krystlik, membre du conseil d'administration
Association Mam'ensolo
Anne-Sophie Duperray , cofondatrice
N.
Planning familial
Véronique Séhier , coprésidente du Planning familial
Elisabeth Devauchelle, vice-présidente de l'association départementale du Planning familial du Puy-de-Dôme, membre du groupe de travail national sur la loi bioéthique
Union nationale des associations familiales (UNAF)
Marie-Andrée Blanc, présidente
Guillemette Leneveu, directrice générale
Claire Ménard, chargée des relations parlementaires
Auditions communes
de M. Olivier Henno, Mme
Corinne Imbert,
M. Bernard Jomier et Mme Muriel Jourda
Direction Générale de la Santé (DGS)
Jérôme Salomon, directeur général
Maurice-Pierre Planel, directeur général adjoint
Hélène Monasse, sous-directrice de politique des produits de santé et de la qualité des pratiques et des soins
Muriel Cohen, adjointe à la cheffe du bureau bioéthique, éléments et produits du corps humain
Alix Lemarie, chargée de mission au sein du bureau bioéthique, éléments et produits du corps humain
Sarah Rueda, chargée de mission à la division droits des usagers, affaires juridiques et éthiques
* 1 Rapport de Jean-François Eliaou, député et Annie Delmont-Koropoulis, sénatrice, 25 octobre 2018.
* 2 Texte n° 114 adopté par le Sénat le 6 juin 2018 sur la proposition de loi relative à l'autorisation des examens des caractéristiques génétiques sur les personnes décédées.
* 3 Article L. 2131-1 du code de la santé publique.
* 4 Selon l'expression reprise dans le rapport final des États généraux de la bioéthique, juillet 2009.
* 5 Loi n° 94-654 du 29 juillet 1994 relative au don et à l'utilisation des éléments et produits du corps humain, à l'assistance médicale à la procréation et au diagnostic prénatal.
* 6 Si la première FIV a été réalisée au Royaume-Uni en 1978, la première FIV par ICSI a été effectuée en Belgique en 1992.
* 7 Dans son avis n° 129 ainsi que dans l'avis n° 113 sur « La demande d'assistance médicale à la procréation après le décès de l'homme faisant partie du couple » du 10 février 2011.
* 8 Dans ces décisions n os 420468 et 420469, le Conseil d'Etat a considéré, « alors même que le vieillissement n'entraîne pas systématiquement chez l'homme un arrêt du fonctionnement gonadique, [que] l'Agence de la biomédecine a pu légalement fixer, compte tenu du large consensus existant dans la communauté scientifique et médicale, à 59 ans révolus, en principe, l'âge de procréer au sens et pour l'application de l'article L. 2141-2 du code de la santé publique. » Les demandeurs étaient âgés de 61 et 63 ans à la date de prélèvement de gamètes.
Comme l'a indiqué l'Agence de la biomédecine, une requête a par ailleurs été déposée devant le tribunal administratif de Montreuil concernant un refus d'exportation de gamètes en raison de l'âge trop élevé de la femme.
* 9 Cf. ci-dessus.
* 10 Conseil d'Etat, 28 septembre 2018 (refus du centre d'AMP du CHU de Toulouse d'autoriser une AMP à un couple homosexuel).
* 11 Conseil d'Etat (référé), 31 mai 2016 (refus de l'AP-HP de permettre l'exportation des gamètes du mari décédé de la requérante vers un établissement de santé espagnol) ou Conseil d'Etat (référé), 4 décembre 2018 (refus du Cecos de l'hôpital Cochin de Paris de permettre l'exportation des gamètes du fis décédé de la requérante vers un établissement de santé situé en Israël).
* 12 Loi n° 2019-222 du 23 mars 2019 de programmation 2018-2022 et de réforme pour la justice.
* 13 Cet article L. 160-14 du code de la sécurité sociale liste les cas dans lesquels la participation de l'assuré peut être limitée ou supprimée dans des conditions fixées par décret en Conseil d'État, après avis de l'Union nationale des caisses d'assurance maladie (Uncam) et de l'Union nationale des organismes d'assurance maladie complémentaire (Unocam).
* 14 Sur la base d'une décision de l'Uncam du 11 mars 2005.
* 15 La Cour des comptes a évalué en effet à environ 295 millions d'euros le coût global de l'AMP pour l'assurance maladie, incluant les remboursements de tentatives réalisées à l'étranger lorsque le couple remplit les conditions pour en bénéficier en France (à hauteur de 1,9 million d'euros). Elle notait toutefois qu'aucune autorité administrative ne dispose de vision consolidée de ces dépenses (Rapport sur l'application des lois de financement de la sécurité sociale, septembre 2019).
* 16 Le rapporteur a toutefois indiqué en séance publique y donner, à titre personnel, un avis de sagesse.
* 17 Le rapporteur a toutefois indiqué en séance publique y donner, à titre personnel, un avis de sagesse.
* 18 Décision n° 2013-669 DC du 17 mai 2013 sur la loi ouvrant le mariage aux couples de personnes de même sexe.
* 19 Tribune publiée dans Le Figaro le 2 décembre 2019.
* 20 Aux termes de l'article L. 111-2-1 du code de la sécurité sociale.
* 21 Loi n° 2002-305 du 4 mars 2002 relative à l'autorité parentale.
* 22 Loi n° 2014-873 du 4 août 2014 pour l'égalité réelle entre les femmes et les hommes.
* 23 Avis n° 794 (2012-2013) de Mme Michelle Meunier, fait au nom de la commission des affaires sociales, déposé le 23 juillet 2013.
* 24 Avis n° 126 du CCNE du 15 juin 2017 sur les demandes sociétales de recours à l'assistance médicale à la procréation (AMP).
* 25 La loi « bioéthique » de 2011 a supprimé le régime des agréments dans les domaines de l'AMP, renvoyant l'appréciation de la formation et de l'expérience des professionnels de santé du secteur aux ARS à l'occasion de l'instruction des demandes d'autorisation.
* 2627 « Chapitre IX - L'assistance médicale à la procréation : une efficience à renforcer », rapport sur l'application des lois de financement de la sécurité sociale, Cour des comptes, septembre 2019.
* 28 Cf. article 22 du projet de loi et article L. 2141-11 du code de la santé publique. Pour les hommes, l'autoconservation de sperme est aussi possible avant une vasectomie, c'est-à-dire une stérilisation masculine irréversible.
* 29 Selon les conditions prévues par l'arrêté du 30 juin 2017 relatif aux règles de bonnes pratiques cliniques et biologiques d'assistance médicale à la procréation. La conservation de sperme est possible lorsque l'on craint un échec de recueil le jour de la FIV, en vue de s'assurer que celle-ci sera réalisable le jour de la ponction ovocytaire.
* 30 Art. L. 1244-2 du code de la santé publique.
* 31 Décret n° 2015-1281 du 13 octobre 2015 relatif au don de gamètes.
* 32 La conservation des ovocytes, Bull. Académie nationale de médecine, 2017, séance du 13 juin 2017.
* 33 Les chances d'obtenir une naissance en congelant 10 ovocytes sont de 50 % pour les femmes de 37 ans, et de 30 % pour celles de 40 ans.
* 34 Selon les dispositions régissant le service hospitalier fixées par l'article L. 6112-2 du code de la santé publique.
* 35 Cf. le II de l'article L. 6154-2 du code de la santé publique.
* 36 D'après le rapport précité de l'Académie de médecine, toutes les techniques d'assistance médicale à la procréation voient leur taux de succès diminuer dès que la femme atteint 35 ans : ils passent de 30,1 % à 34 ans, 23,6 % à 38 ans, 16,5 % à 43 ans. En revanche, quand les ovocytes proviennent de tiers donneuses réglementairement âgées de moins de 37 ans, le taux de succès de l'AMP reste encore de 46 % au-delà de 40 ans.
* 37 L'arrêté du 30 juin 2017 prévoit une sélection clinique et biologique des donneurs de gamètes qui comprend un entretien individuel et des examens biologiques (tests de sécurité sanitaire : sérologies CMV et HTLV 1 et 2 etc. et un bilan génétique afin de permettre d'identifier les facteurs de risque de transmission à l'enfant d'une anomalie génétique).
* 38 D'après les indications de l'Agence de la biomédecine, congeler entre 8 et 10 ovocytes pour une femme de moins de 35 ans donnerait 40 à 60 % de chances de naissance vivante par ovocyte décongelé, tandis qu'après 35 ans, ces taux diminuent à 30-40 %. Un modèle mathématique a été développé pour prédire la probabilité de naissance vivante, basée sur le nombre d'ovocytes congelés et l'âge de la femme : dans ce modèle, à 34, 37 et 42 ans (pour des femmes avec une réserve ovarienne normale), il faut congeler respectivement 10, 20 et 61 ovocytes pour avoir 75 % de chances d'obtenir au moins une naissance vivante.
* 39 Plusieurs autres amendements déposés par des sénateurs de différents groupes avaient un objet similaire : les amendements COM-45, COM-42, COM-65 et COM-134.
* 40 Similaire aux amendements COM-27, COM-108 et COM-39.
* 41 Centre d'études et de conservation des oeufs et du sperme humain.
* 42 Ce principe général est décliné pour le don d'embryons à l'article L. 2141-6 du code de la santé publique.
* 43 Décision n° 94-343/344 DC du 27 juillet 1994 relative à la loi relative au respect du corps humain et loi relative au don et à l'utilisation des éléments et produits du corps humain, à l'assistance médicale à la procréation et au diagnostic prénatal.
* 44 Conseil d'Etat, 28 décembre 2017, n°396571, 10 ème - 9 ème chambres réunies.
* 45 Art. R. 1211-25 du code de la santé publique et arrêté du 30 juin 2017 modifiant l'arrêté du 11 avril 2008 modifié relatif aux règles de bonnes pratiques cliniques et biologiques d'assistance médicale à la procréation.
* 46 Conseil d'État, 12 novembre 2015, n° 372121.
* 47 Rapport n° 236 (1993-1994) de M. Jean Chérioux, fait au nom de la commission des affaires sociales, déposé le 12 janvier 1994.
* 48 Selon les termes de Mme Audrey Kermalvezen, co-fondatrice de l'association Origines.
* 49 La commission a adopté un article 10 bis qui vise à les autoriser.
* 50 Voir la brochure « Mon histoire à moi » :
https://www.cecos.org/wp-content/uploads/2019/08/mon_histoire_a_moi.pdf
* 51 Voir l'audition de Marie-Claude Picardat, ancienne co-présidente de l'Association des parents et futurs parents gays et lesbiens (APGL) le 19 décembre 2019 : http://www.senat.fr/compte-rendu-commissions/20191216/cs_bioethique.html#toc4.
* 52 Pr Jean-François Mattei, vice-président de l'Académie nationale de médecine : « Si l'enfant devenu jeune adulte retrouve l'identité de son donneur-géniteur, personne ne sait comment ce lien biologique masculin direct pourrait être investi ».
* 53 Arrêt Odièvre c. France du 13 février 2003, Req.° 42326/98.
* 54 Deux contentieux sont en cours contre la France devant la CEDH.
* 55 La notion de tiers donneur, définie dans le cadre du projet de loi, à l'article L. 2143-1 comprend les couples, en cas de dons d'embryons.
* 56 Voir les sites de la société danoise Cryos International ou de la société espagnole IVI.
* 57 Ou à 16 ans en cas d'émancipation en application de l'article 413-6 du code civil.
* 58 Et non son adresse, puisqu'il n'y a pas de « droit à la rencontre ».
* 59 Délibération n°2019-097 du 11 juillet 2019.
* 60 Par exemple, le registre des donneurs volontaires de moelle osseuse, la liste nationale d'attente de greffe ou le registre national des refus.
* 61 Créé par la loi n° 2002-93 du 22 janvier 2002 relative à l'accès aux origines des personnes adoptées et pupilles de l'Etat.
* 62 Entre la promulgation de la loi et le premier jour du treizième mois suivant la promulgation de la loi.
* 63 Selon une étude effectuée par l'équipe de recherche du Professeur Louis Bujan du CECOS de Toulouse, environ 50 % des candidats actuels au don de gamètes accepteraient de donner sous le régime envisagé par le présent projet de loi. Cela signifie que la moitié des donneurs serait à renouveler.
* 64 « Gare à la pénurie de gamètes » Tribune de Mme Marie-Xavière Catto, Maître de conférences à l'Université Paris 1, Le Monde du 5 août 2019.
* 65 Réponses au questionnaire du Planning familial.
* 66 Tribune de M. Israël Nisand et Mmes Maud Nisand et Brigitte Letombe publiée dans le Figaro le 2 décembre 2019.
* 67 Réponses au questionnaire de la Fédération française des CECOS.
* 68 Audition du 20 novembre 2019
http://www.senat.fr/compte-rendu-commissions/20191118/cs_bioethique.html#toc2
* 69 Audition du 18 décembre 2019 :
http://www.senat.fr/compte-rendu-commissions/20191216/cs_bioethique.html#toc2
* 70 Amendement n° 380 de M. Jean François Mbaye.
* 71 Amendement n° 2296 de la rapporteure, Mme Coralie Dubost.
* 72 Amendement n° 1986 de Mme Marie Tamarelle-Verhaeghe.
* 73 Amendement n° 2330 des députés du groupe La République en marche.
* 74 Amendements n° 1585 de M. Jean-Louis Touraine et 2007 de M. Bruno Fuchs.
* 75 Amendement n° 2541 de la rapporteure, Mme Coralie Dubost.
* 76 Réponse au CCNE au questionnaire : « Une difficulté réside dans la temporalité entre le consentement du donneur et le moment où les enfants feront leur demande : le donneur accepte que 18 ans après son don, son identité soit communiquée aux enfants issus de son don, si ces derniers le souhaitent. La projection dans un temps aussi long peut être problématique. Le donneur peut avoir lui-même évolué sur la question, avoir éventuellement changé d'avis ou posé des difficultés dans sa vie présente .
* 77 « Révision de la loi de bioéthique : quelles options pour demain ? », Conseil d'État, section du rapport et des études, étude adoptée en assemblée générale le 28 juin 2018.
* 78 Table ronde du 28 novembre 2019 avec Mme Marie Mesnil, MM. Hugues Fulchiron et Jean-René Binet : http://www.senat.fr/compte-rendu-commissions/20191125/cs_bioethique.html#toc4.
* 79 Ni le CNAOP, ni la commission ad hoc envisagée n'ont de personnalité juridique distincte de l'État, ce qui permet un transfert des missions dans le respect de l'article 40 de la Constitution.
* 80 Ou concubin ou pacsé.
* 81 Voir l'étude du Conseil d'Etat du 28 juin 2018 « Révision de la loi de bioéthique : quelles options pour demain ? » et les réponses au questionnaire de Mme Marie Mesnil, maîtresse de conférences en droit privé à l'Université de Rennes 1.
* 82 Mme Catherine Jousselme, professeur de psychiatrie de l'enfant et de l'adolescent, a estimé « important de prévoir un accompagnant des donneurs dans la rédaction ».
* 83 Vocabulaire juridique , Gérard Cornu, Association Henri Capitant, quadrige, douzième édition mise à jour en 2018.
* 84 Loi n° 94-653 du 29 juillet 1994 relative au respect du corps humain.
* 85 Pater is est quem nuptiæ demonstrant, le père est celui que le mariage désigne.
* 86 La mère peut aussi reconnaître l'enfant avant ou après la naissance. Si, lors de l'accouchement, la mère a demandé que le secret de son admission et son identité soit préservés, elle peut, dans les deux mois suivant la naissance de l'enfant, reconnaître l'enfant et demander qu'il lui soit remis (articles 316 et 351 du code civil).
* 87 L'article 311-1 du code civil dispose que « la possession d'état s'établit par une réunion suffisante de faits qui révèlent le lien de filiation et de parenté entre une personne et la famille à laquelle elle est dite appartenir (...) » et l'article 311-2 du même code qu'elle « doit être continue, paisible, publique et non équivoque ».
* 88 Deux types d'actions judiciaires sont possibles : les actions en établissement de la filiation (recherche en paternité ou maternité) et les actions en contestation de la filiation.
* 89 Défendre les principes, veiller à l'intérêt des enfants - Quelle réponse apporter au contournement du droit français par le recours à l'AMP et à la GPA à l'étranger ? Rapport d'information n° 409 (2015-2016) de M. Yves Détraigne et Mme Catherine Tasca fait au nom de la commission des lois, déposé le 17 février 2016, p. 21. Ce rapport est consultable à l'adresse suivante : https://www.senat.fr/notice-rapport/2015/r15-409-notice.html
* 90 La Cour de cassation a d'ailleurs rappelé que ces textes ne régissent que les procréations médicalement assistées avec donneur, dans un arrêt n° 229 du 16 mars 2016 de sa première chambre civile (15-13.247).
* 91 Cet acte donne lieu à la perception d'un émolument fixe de 76,92 euros pour le notaire (article A. 444-84 du code de commerce). Il est en revanche exonéré de frais d'enregistrement (article 847 bis du code général des impôts).
* 92 Il faut noter que la possession d'état ne pourrait pas être utilisée dans le cadre d'une AMP avec tiers donneur, car le couple est contraint de reconnaître l'enfant.
* 93 « L'article 311-20 (...) n'a pas pour objet d'établir la filiation ; il fait du consentement à l'AMP la condition sine qua non pour qu'ensuite celle-ci puisse avoir lieu. C'est donc un article sur le consentement, pas sur le mode d'établissement de la filiation. Certes, il vient ensuite sécuriser la filiation, mais il ne l'établit pas. Ce qui établit la filiation, ce sont évidemment les règles normales du titre VII, telles qu'elles sont déjà prévues », Nicole Belloubet, compte rendu des débats à l'Assemblée nationale, deuxième séance publique du mercredi 2 octobre 2019.
* 94 Étude d'impact, projet de loi relatif à la bioéthique, 23 juillet 2019, p. 181.
Ce document est consultable à l'adresse suivante :
http://www.assemblee-nationale.fr/dyn/15/dossiers/bioethique_2?etape=15-AN1-DEPOT
* 95 « (...) aucune disposition ni aucun principe à valeur constitutionnelle ne prohibe les interdictions prescrites par le législateur d'établir un lien de filiation entre l'enfant issu de la procréation et l'auteur du don et d'exercer une action en responsabilité à l'encontre de celui-ci », Conseil constitutionnel, décision n° 93-343/344 DC du 27 juillet 1994 sur la loi relative au respect du corps humain et la loi relative au don et à l'utilisation des éléments et produits du corps humain, à l'assistance médicale à la procréation et au diagnostic prénatal, considérant n° 17.
* 96 Voir commentaire de l'article 3 du projet de loi.
* 97 Conseil constitutionnel, décision n° 2013-669 DC du 17 mai 2013 sur la loi ouvrant le mariage aux couples de personnes de même sexe, considérant 56.
* 98 Cour de cassation, avis n° 15003 du 7 mars 2018, première chambre civile (demande d'avis n° F 17-70.039).
* 99 Ibid. rapport précité , p. 24.
* 100 La fraude à la loi se caractérise par un acte régulier en soi, accompli dans l'intention d'obtenir ce que la loi française prohibe, par des moyens détournés et formellement légaux, en France ou à l'étranger.
* 101 Cour de cassation, assemblée plénière, avis n° 15011 du 22 septembre 2014 (demande n° 1470006).
* 102 Ibid. supra, p. 24 .
* 103 Bilan de la dépêche de la Direction des affaires civiles et du sceau du 14 février 2019 sollicitant auprès des Procureurs généraux des éléments chiffrés relatifs aux procédures d'adoption depuis le mois de septembre 2014, Ministère de la justice, étude d'impact du projet de loi, p. 182.
* 104 La transcription n'est pas obligatoire. Un acte d'état civil étranger produit des effets relatifs en France. Dans la vie quotidienne, les effets sont plein et entiers (décisions à prendre pour l'enfant, représentation légale de l'enfant ne posent en général pas de difficulté). En revanche, certains droits découlant de l'établissement et de la reconnaissance de la filiation sont fermés (la succession par exemple).
* 105 « Tout acte de l'état civil des Français et des étrangers fait en pays étranger et rédigé dans les formes usitées dans ce pays fait foi, sauf si d'autres actes ou pièces détenus, des données extérieures ou des éléments tirés de l'acte lui-même établissent, le cas échéant après toutes vérifications utiles, que cet acte est irrégulier, falsifié ou que les faits qui y sont déclarés ne correspondent pas à la réalité. »
* 106 Cour de cassation, première chambre civile, arrêt n° 824 du 5 juillet 2017 n° (15-28.597).
* 107 Cour de cassation, première chambre civile, 18 novembre 2019, n°s 18-50.007 et 18-14.751, considérant 8 : « en présence d'une action aux fins de transcription de l'acte de naissance étranger d'un enfant, ni la circonstance que l'enfant soit né d'une assistance médicale à la procréation ni celle que cet acte désigne la mère ayant accouché et une autre femme en qualité de mère ou de parent ne constituent un obstacle à sa transcription sur les registres français de l'état civil, lorsque l'acte est probant au sens de l'article 47 du code civil ».
* 108 Voir commentaire de l'article 4 bis .
* 109 Révision de la loi de bioéthique, quelles options pour demain ? Conseil d'État, étude à la demande du Premier ministre, adoptée en assemblée générale le 28 juin 2018, p. 61. Cette étude est consultable à l'adresse suivante :
* 110 Voir par exemple les réponses de Nicole Belloubet lors de la première séance publique du 3 octobre 2019 à l'Assemblée nationale.
* 111 Comme l'ont indiqué les services de la Chancellerie lors de leur audition par le rapporteur.
* 112 Voir commentaire de l'article 1 er sur les raisons du choix des termes de « femme non mariée ».
* 113 Voir commentaire de l'article 3.
* 114 Filiation, origines, parentalité, Le droit face aux nouvelles valeurs de responsabilité générationnelle. Rapport du groupe de travail Filiation, origines, parentalité, remis en 2014 aux ministres des affaires sociales et de la santé et au ministre délégué chargé de la famille, Irène Théry, présidente et Anne-Marie Leroyer, rapporteur.
* 115 Article 55 du code civil.
* 116 Voir le compte rendu des débats à l'Assemblée nationale, troisième séance publique du jeudi 3 octobre 2019.
* 117 « Tous les enfants dont la filiation est légalement établie ont les mêmes droits et les mêmes devoirs dans leurs rapports avec leur père et mère. Ils entrent dans la famille de chacun d'eux. »
* 118 « L'adopté a, dans la famille de l'adoptant, les mêmes droits et les mêmes obligations qu'un enfant dont la filiation est établie en application du titre VII du présent livre. »
* 119 La loi de pays n° 2012-2 du 20 janvier 2012 relative au transfert à la Nouvelle-Calédonie des compétences de l'État en matière de droit civil, de règles concernant l'état civil et de droit commerciale a transféré le droit civil et les règles concernant l'état civil à la Nouvelle-Calédonie.
* 120 Objet de l'amendement n° 2266, dont le texte est consultable à l'adresse suivante :
http://www.assemblee-nationale.fr/dyn/15/amendements/2187/CSBIOETH/2266
* 121 Conseil constitutionnel, décision n° 2018-768 QPC du 21 mars 2019.
* 122 Comme l'ont montré les auditions des docteurs Flavigny et Lévy-Soussan.
* 123 Comme l'a montré l'audition du docteur Jousselme.
* 124 Comme le montrent les travaux de l'Académie de médecine et l'audition du docteur Lévy-Soussan.
* 125 Sexe, mensonge et quiproquo. À propos de la filiation d'un enfant procréé par un couple de même sexe , Astrid Marais, La Semaine Juridique Edition Générale n°48, 25 novembre 2019.
* 126 Étude précitée, p. 61.
* 127 En pratique, si un accouchement dans le secret intervenait au sein d'un couple de femmes, la mère n'ayant pas accouché pourrait remettre la reconnaissance conjointe au procureur de la République pour qu'il la communique à l'officier de l'état civil et que celui-ci la porte en marge de l'acte de naissance de l'enfant. Une fois l'enfant identifié par la seconde femme, la filiation pourrait alors être établie.
* 128 Voir commentaire de l'article 1 er .
* 129 Comme le propose le récent rapport sur l'adoption remis par nos collègues respectivement sénatrice et députée, Corinne Imbert et Monique Limon, au Gouvernement (octobre 2019).
* 130 « Toute convention portant sur la procréation ou la gestation pour le compte d'autrui est nulle ». Cet interdit est, aux termes de l'article 16-9 du code civil, d'ordre public.
* 131 Cour de cassation, première chambre civile, arrêts du 13 septembre 2013, pourvois n° 12-18315 et 12-30138 : « la naissance est l'aboutissement, en fraude à la loi française, d'un processus d'ensemble comportant une convention de gestation pour le compte d'autrui, convention qui, fût-elle licite à l'étranger, est nulle d'une nullité d'ordre public ».
* 132 Cour de cassation, première chambre civile, arrêts du 5 juillet 2017, pourvois n° 15-28.597, 16-16.901, 16-50.025 et 16-16.455.
* 133 Cour européenne des droits de l'homme, grande chambre, avis consultatif relatif à la reconnaissance en droit interne d'un lien de filiation entre un enfant né d'une gestation pour autrui pratiquée à l'étranger et la mère d'intention, 10 avril 2019, demandé par la Cour de cassation française (demande n° P16-2018-001).
* 134 Cour européenne des droits de l'homme, cinquième section, 19 novembre 2019, requêtes n os 1462/18 et 17348/18, C. contre la France et E. contre la France.
* 135 L'adoption de l'enfant du conjoint n'est possible de manière certaine en France lorsque l'enfant est issu d'une GPA que depuis les arrêts de la Cour de cassation précités du 5 juillet 2017.
* 136 Ibid. considérant 43.
* 137 Cour de cassation, première chambre civile, arrêts 18 décembre 2019, pourvois n° 18-12.327 et 18-11.815.
* 138 « Tout acte de l'état civil des Français et des étrangers fait en pays étranger et rédigé dans les formes usitées dans ce pays fait foi, sauf si d'autres actes ou pièces détenus, des données extérieures ou des éléments tirés de l'acte lui-même établissent, le cas échéant après toutes vérifications utiles, que cet acte est irrégulier, falsifié ou que les faits qui y sont déclarés ne correspondent pas à la réalité. »
* 139 Cour de cassation, assemblée plénière, 4 octobre 2019, pourvoi n° 10-19.053.
* 140 En termes de reconnaissance symbolique, on peut relever, parmi les dispositions existant déjà, l'article L. 1233-3 du code de la santé publique, issu de la loi de bioéthique de 2004, prévoyant la création d'un « lieu de mémoire destiné à l'expression de la reconnaissance aux donneurs d'éléments de leur corps en vue de greffe » dans les établissements de santé effectuant les prélèvements, ou encore la « Journée nationale de réflexion sur le don d'organe et la greffe et de reconnaissance envers les donneurs » instituée par l'article 10 de la loi de bioéthique de 2011.
* 141 Notamment des dispositions prévoyant la prise en charge intégrale des frais afférents au prélèvement ou à la collecte par l'établissement de santé chargé d'effectuer le prélèvement ou la collecte (article L. 1211-4 du code de la sécurité sociale), l'exonération du forfait journalier hospitalier ou du ticket modérateur, la prise en charge de frais d'examens, de transport, d'hébergement ou la compensation de la perte de revenu (articles R. 1211-2 et suivants du code de la santé publique).
* 142 Ces donneurs doivent être majeurs. L'article L. 1231-2 du code de la santé publique dispose en effet qu'« aucun prélèvement d'organes, en vue d'un don, ne peut avoir lieu sur une personne vivante mineure ou sur une personne vivante majeure faisant l'objet d'une mesure de protection légale ».
* 143 D'après l'Agence de la biomédecine, rapport sur l'application de la loi de bioéthique, janvier 2018.
* 144 A l'instar de l'Espagne, du Luxembourg, de l'Irlande, de l'Italie, des Pays-Bas ou du Royaume-Uni (source : OPECST, L'évaluation de l'application de la loi n° 2011-814 du 7 juillet 2011 relative à la bioéthique, octobre 2018).
* 145 Source : Renaloo. Contribution aux Etats Généraux de la bioéthique 2018.
* 146 Rapport de janvier 2018 sur l'application de la loi de bioéthique.
* 147 L'article L. 161-31 du code de la sécurité sociale, s'agissant de la carte Vitale, prévoit que « Les professionnels de santé peuvent porter sur le volet, avec le consentement exprès du titulaire de la carte, les informations nécessaires aux interventions urgentes ainsi que la mention : " A été informé de la législation relative au don d'organes". »
* 148 A l'article L. 1111-14 du code de la santé publique. Cette disposition a été modifiée par la loi « santé » de janvier 2016 qui a prévu, à l'article L. 1111-15 du même code que le dossier médical partagé comporte un volet relatif au don d'organes ou de tissus.
* 149 Art. L. 312-17-2 du code de l'éducation.
* 150 Sang qui circule dans tout le corps.
* 151 Le système HLA ( human leukocyte antigen ) correspond à la carte d'identité génétique de chaque individu et permet de vérifier les éventuelles incompatibilités et risques de rejet de greffe.
* 152 Les CSH peuvent également parfois venir de sang placentaire, appelé aussi sang de cordon.
* 153 https://www.dondemoelleosseuse.fr.
* 154 Art. L. 1231-2 du code de la santé publique.
* 155 La loi n°76-1181 du 22 décembre 1976 relative aux prélèvements d'organes dite « Caillavet » était moins restrictive car elle prévoyait le prélèvement de tout organe transplantable au bénéfice d'un frère ou d'une soeur.
* 156 Loi n° 94-654 du 29 juillet 1994 relative au don et à l'utilisation des éléments et produits du corps humain, à l'assistance médicale à la procréation et au diagnostic prénatal.
* 157 Ancien article L.671-5 du code de la santé publique.
* 158 Loi n° 2004-800 du 6 août 2004 relative à la bioéthique.
* 159 Loi n° 2011-814 du 7 juillet 2011 relative à la bioéthique.
* 160 Anciennement, président du tribunal de grande instance.
* 161 Il s'agit très majoritairement de majeurs en tutelle, mais également des majeurs qui font l'objet d'habilitation familiale ou de mandat de protection future.
* 162 Loi n° 2007-308 du 5 mars 2007 portant réforme de la protection juridique des majeurs.
* 163 Voir article 30.
* 164 Voir 8° de l'article L. 1418-1 du code de la santé publique.
* 165 Amendements n° 2148, 2149 et 2150 de M. Hervé Saulignac.
* 166 Voir l'article 388-2 du code civil.
* 167 Par référence au dispositif prévu pour le majeur qui fait l'objet d'une mesure de protection juridique avec représentation à la personne.
* 168 En application de l'article L. 1231-3 du code de la santé publique.
* 169 Article 425 du code civil.
* 170 Cette assistance peut prendre la forme d'une apposition de signature à côté de celle de la personne protégée.
* 171 Hormis pour les décisions strictement personnelles de l'article 459 du code civil :
* 172 380 000 personnes sous tutelle et environ 15 000 personnes au titre des habilitations familiales comprenant des mesures de représentation à la personne, selon la direction générale de la santé.
* 173 La même interdiction existe en matière de don du sang ou de ses composants à l'article L. 1221-5 du code de la santé publique. Voir commentaire de l'article 7 bis .
* 174 Depuis la loi n° 2007-308 du 5 mars 2007 portant réforme de la protection juridique des majeurs, l'expression « protection juridique » est préférée à l'expression « protection légale ».
* 175 Pendant longtemps, ces greffes en domino ont concerné les malades atteints de la mucoviscidose sur lesquels, pour des raisons techniques, les chirurgiens préféraient pratiquer une greffe cardio-pulmonaire même lorsque l'organe cardiaque ne présentait aucune défaillance. L'ancien coeur du receveur pouvait alors être proposé à un patient en attente de greffe cardiaque. Aujourd'hui, ce type de greffe continue à être pratiqué, par exemple, en cas de neuropathie amyloïde : le foie du patient atteint de cette maladie neurologique doit être retiré, mais reste fonctionnel, et peut être transplanté chez un autre patient qui présente un besoin urgent de greffe hépatique : malgré le risque de développer une neuropathie amyloïde au bout de plusieurs années, le bilan des risques et des avantages d'une telle greffe reste positif.
* 176 Art. L. 1232-1 du code de la santé publique.
* 177 Loi n° 2007-308 susmentionnée.
* 178 Voir le commentaire de l'article 6.
* 179 Loi n° 2019-222 du 23 mars 2019 de programmation 2018-2022 et de réforme pour la justice.
* 180 Que ce soit dans le cadre d'une sauvegarde de justice, d'une curatelle, d'une tutelle aux biens, d'un mandat de protection future ou d'une habilitation familiale.
* 181 Alinéa 3 de l'article L. 1232-1 du code de la santé publique.
* 182 En Ile-de-France, dans la semaine du 14 au 21 novembre 2015, les dons ont progressé de 280 % par rapport à l'année précédente.
* 183 Texte n° 40 (2018-2019) transmis au Sénat le 12 octobre 2018.
* 184 Directive 2004/33/CE de la commission du 22 mars 2004 portant application de la directive 2002/98/CE du Parlement européen et du Conseil concernant certaines exigences techniques relatives au sang et aux composants sanguins.
* 185 Titre III « Examen des caractéristiques génétiques, identification par empreintes génétiques et profession de conseiller en génétique » du livre I er de la première partie du code de la santé publique.
* 186 Désignée par le patient dans le cas où celui-ci serait hors d'état d'exprimer sa volonté en application l'article L. 1111-6 du code de la santé publique.
* 187 CCNE, Réflexion éthique sur l'évolution des tests génétiques liée au séquençage de l'ADN humain à très haut débit , avis n° 124, 21 janvier 2016.
* 188 Relatif à l'examen des caractéristiques génétiques de personnes dont il est impossible de recueillir le consentement.
* 189 Cet article impose un consentement exprès préalable à toute identification par des empreintes génétiques entreprise à des fins médicales ou de recherche, ou dans le cadre d'une procédure judiciaire.
* 190 Selon l'étude d'impact annexée au projet de loi, il s'agira des membres de la famille mentionnés au dossier de soins, dont le dossier médical prévu à l'article L. 1111-4 du code de la santé publique ou au dossier de soins infirmiers prévu à l'article R. 4311-4 du même code. Le membre de la famille doit donc avoir un lien génétique avec le sujet de l'examen ; dans le cas contraire, le dispositif prendra fin.
* 191 Agence de la biomédecine, Rapport sur l'application de la loi de bioéthique , janvier 2018, p. 46.
* 192 Avis n° 124.
* 193 Arrêté du 26 novembre 1999 relatif à la bonne exécution des analyses de biologie médicale.
* 194 Loi n° 2004-800 du 6 août 2004 relative à la bioéthique. Figurant initialement à l'article L. 1131-1 du code de la santé publique, le dispositif d'information de la parentèle a été « déplacé » à l'article L. 1131-1-2 par la loi n° 2011-814 du 7 juillet 2011 relative à la bioéthique.
* 195 Dont la ratification a été autorisée par l'article 1 er de la loi relative à la bioéthique de 2011.
* 196 Qui désigne tant la personne dont les gamètes ont été recueillis ou prélevés que le couple ou la femme ayant consenti à ce qu'un ou plusieurs de ses embryons soient accueillis par un autre couple ou une autre femme.
* 197 Mentionné au 2° de l'article L. 147-2 du code de l'action sociale et des familles.
* 198 Mentionnés au 1° de l'article L. 147-2 du code de l'action sociale et des familles.
* 199 Lorsque la personne ayant fait l'objet de l'examen génétique délègue au médecin prescripteur le soin d'informer la parentèle.
* 200 Lorsque la personne ayant fait l'objet de l'examen génétique est un majeur protégé ou une personne hors d'état d'exprimer sa volonté.
* 201 Lorsque la personne ayant fait l'objet de l'examen génétique est décédée avant l'annonce du résultat ou avant d'avoir pu informer les membres de sa famille.
* 202 « Whole Exome Sequencing - WES » ou « Whole Genome Sequencing - WGS ».
* 203 Ensemble des gènes du génome qui code pour la synthèse des protéines, qui peut ainsi être regardé comme l'« ADN codant ».
* 204 Elle correspond à la portion d'individus porteurs du variant génétique (génotype) qui manifestent la maladie (phénotype ou morphotype de la maladie).
* 205 Conseil d'État, Révision de la loi de bioéthique : quelles options pour demain ? , étude à la demande du Premier ministre adoptée en assemblée générale le 28 juin 2018.
* 206 Mention dans le consentement écrit de l'indication ou de l'objectif de l'examen et caractère révocable, en tout ou partie, du consentement.
* 207 Article 226-28-1 du code pénal.
* 208 En France, les tests conduisant à l'établissement ou à la contestation d'un lien de filiation ne peuvent être réalisés qu'en exécution d'une mesure d'instruction ordonnée par le juge, avec le consentement du parent présumé, en application de l'article 16-11 du code civil.
* 209 9° de l'article L. 1418-1 du code de la santé publique.
* 210 Plusieurs articles de presse soulèvent la question de la cession de données génétiques de clients par des sociétés de tests ADN à des institutions judiciaires comme le Federal Bureau of Investigation (FBI) aux États-Unis pour la recherche de suspects.
* 211 Comité consultatif national d'éthique pour les sciences de la vie et de la santé, Contribution du comité consultatif national d'éthique à la révision de la loi de bioéthique 2018-2019 , avis 129, 18 septembre 2018.
* 212 Cette définition a été introduite par le scientifique Marvin Minsky en 1956.
* 213 Comment permettre à l'homme de garder la main ? Les enjeux éthiques des algorithmes et de l'intelligence artificielle, Synthèse du débat public animé par la Commission nationale de l'informatique et des libertés dans le cadre de la mission de réflexion éthique confiée par la loi pour une République numérique, décembre 2017. Ce document est consultable à l'adresse suivante : https://www.cnil.fr/sites/default/files/atoms/files/cnil_rapport_garder_la_main_web.pdf
* 214 Ibid . supra .
* 215 Numérique & Santé, quels enjeux éthiques pour quelles régulations ? Rapport du groupe de travail dirigé notamment par David Gruson, membre du comité de direction de la chaire santé de Sciences Po, professeur associé à la faculté de médecine Paris-Descartes et par Claude Kirchner, président de la commission de réflexion sur l'éthique de la recherche en sciences et technologies du numérique d'Allistene (Cerna), tous deux entendus lors des auditions. Ce rapport est consultable à l'adresse suivante : https://www.ccne-ethique.fr/sites/default/files/publications/rapport_numerique_et_sante_19112018.pdf
* 216 Article 9 du règlement (UE) 2016/679 du 27 avril 2016 relatif à la protection des personnes physiques à l'égard du traitement des données à caractère personnel et à la libre circulation de ces données (RGPD) et 6 de la loi n° 78-17 du 6 janvier 1978 relative à l'informatique, aux fichiers et aux libertés.
* 217 La personne doit être informée de la possibilité de réutiliser ultérieurement ses données pour une autre finalité au moment de leur collecte (article 13 du RGPD) ou au moment de leur réutilisation (article 14 du RGPD), sauf, dans ce dernier cas, si cette information se révèle impossible ou exigerait des efforts disproportionnés en particulier pour le traitement à des fins de recherche scientifique.
* 218 « Le fait, par toute personne détentrice de données à caractère personnel à l'occasion de leur enregistrement, de leur classement, de leur transmission ou de toute autre forme de traitement, de détourner ces informations de leur finalité telle que définie par la disposition législative, l'acte réglementaire ou la décision de la Commission nationale de l'informatique et des libertés autorisant le traitement automatisé, ou par les déclarations préalables à la mise en oeuvre de ce traitement, est puni de cinq ans d'emprisonnement et de 300 000 euros d'amende. »
* 219 Révision de la loi de bioéthique, quelles options pour demain ? Conseil d'État, étude à la demande du Premier ministre, adoptée en assemblée générale le 28 juin 2018. Ce document est consultable à l'adresse suivante :
https://www.conseil-etat.fr/ressources/etudes-publications/rapports-etudes/etudes/revision-de-la-loi-de-bioethique-quelles-options-pour-demain
* 220 Article 1242 du code civil : « On est responsable non seulement du dommage que l'on cause par son propre fait, mais encore de celui qui est causé par le fait des personnes dont on doit répondre, ou des choses que l'on a sous sa garde. »
* 221 Articles 1245 et suivants du code civil.
* 222 Cour de justice de l'Union européenne, 7 décembre 2017, Syndicat national de l'industrie des technologies médicales, n° C-329/16 : « un logiciel dont l'une des fonctionnalités permet l'exploitation de données propres à un patient, aux fins, notamment, de détecter les contre-indications, les interactions médicamenteuses et les posologies excessives, constitue, pour ce qui est de cette fonctionnalité, un dispositif médical (...) et ce même si un tel logiciel n'agit pas directement dans ou sur le corps humain ».
* 223 Les termes choisis sont volontairement larges, mais les principales hypothèses visées sont celles du médecin et de son patient. Le cas échéant, ce périmètre permet d'inclure les autres professionnels de santé qui auraient recours à un traitement algorithmique à des fins médicales.
* 224 Article 47 de la loi n° 78-17 du 6 janvier 1978 relative à l'informatique, aux fichiers et aux libertés.
* 225 Article 4-3 de la loi n° 2016-1547 du 18 novembre 2016 de modernisation de la justice du XXI e siècle.
* 226 Commission nationale de l'informatique et des libertés (CNIL), délibération n° 2019-097 du 11 juillet 2019 portant avis sur un projet de loi relatif à la bioéthique. Cet avis est consultable à l'adresse suivante : https://www.legifrance.gouv.fr/affichCnil.do?id=CNILTEXT000038848207
* 227 Dans son avis n° 129 du 18 septembre 2018.
* 228 Rapport n° 2243 pré-cité, p. 233.
* 229 Révision de la loi de bioéthique, quelles options pour demain ? Conseil d'État, étude à la demande du Premier ministre, adoptée en assemblée générale le 28 juin 2018, p. 207. Cette étude est consultable à l'adresse suivante :
* 230 L'article R. 4127-5 du code de la santé publique dispose que « Le médecin ne peut aliéner son indépendance professionnelle sous quelque forme que ce soit. »
* 231 Les articles L. 4161-1 et suivants du code de la santé publique répriment l'exercice illégal de la médecine.
* 232 Étude du Conseil d'État publiée en 2018 précitée, p. 207.
* 233 Avis n° 129, contribution du comité consultatif national d'éthique à la révision de la loi de bioéthique, rendu public le 25 septembre 2018. Cet avis est consultable à l'adresse suivante :
* 234 Le scanner cérébral ou tomodensitométrie (TDM) cérébrale est un examen permettant de reconstituer des images du cerveau sous forme de fines coupes axiales en deux ou trois dimensions.
* 235 L'imagerie par résonance magnétique (IRM) est un examen basé sur l'utilisation de champs électromagnétiques. Il donne des images du corps en deux ou trois dimensions.
* 236 La tomographie par émission de positons (TEP) est une méthode d'imagerie médicale qui permet de mesurer en trois dimensions l'activité métabolique d'un organe grâce aux émissions produites par les positons issus de la désintégration d'un produit radioactif injecté au préalable.
* 237 L'électroencéphalographie permet d'enregistrer l'activité électrique spontanée des neurones du cerveau. Le tracé ainsi obtenu est appelé électroencéphalogramme.
* 238 Ibid . supra .
* 239 Avis n° 381 (2010-2011) de M. François-Noël Buffet, fait au nom de la commission des lois, déposé le 29 mars 2011, sur le projet de loi relatif à la bioéthique. Ce rapport est accessible à l'adresse suivante : https://www.senat.fr/rap/a10-381/a10-381.html
* 240 Cet article dispose notamment que : « N'est pas pénalement responsable la personne qui était atteinte, au moment des faits, d'un trouble psychique ou neuropsychique ayant aboli son discernement ou le contrôle de ses actes (...) ».
* 241 Articles 143 et suivants du code de procédure civile.
* 242 Articles 156 et suivants du code de procédure pénale.
* 243 Imagerie à résonance magnétique fonctionnelle.
* 244 Rapport n° 80 (2018-2019) de M. Jean-François Eliaou, député et Mme Annie Delmont-Koropoulis, sénatrice, fait au nom de l'Office parlementaire d'évaluation des choix scientifiques et technologiques, déposé le 25 octobre 2018, sur l'évaluation de l'application de la loi n° 2011-814 du 7 juillet 2011 relative à la bioéthique. Ce rapport est consultable à l'adresse suivante : http://www.senat.fr/rap/r18-080/r18-080.html#_Toc530050027
* 245 Révision de la loi de bioéthique, quelles options pour demain ? Conseil d'État, étude à la demande du Premier ministre, adoptée en assemblée générale le 28 juin 2018, p. 188. Cette étude est consultable à l'adresse suivante :
* 246 Décisions n° 2004-492 DC du 2 mars 2004 sur la loi portant adaptation de la justice aux évolutions de la criminalité, cons. 110.
* 247 Les cahiers du Conseil constitutionnel, cahier n° 16, commentaire de la décision n° 2004-492 DC du 2 mars 2004 sur la loi portant adaptation de la justice aux évolutions de la criminalité.
* 248 Usages et interprétations judiciaires des images cérébrales, Sonia Desmoulin-Canselier, Revue de science criminelle et de droit comparé, Dalloz, 2018, p. 343.
* 249 Étude du Conseil d'État précitée, p. 189.
* 250 Pour un exemple s'agissant d'une expertise psychocriminologique, voir Cour de cassation, chambre criminelle, 29 janvier 2003, n° 02-86.774.
* 251 Voir commentaire de l'article 13.
* 252 Stimulation externe au moyen de courants électriques, magnétiques ou électromagnétiques.
* 253 Consiste à implanter une électrode de stimulation dans le cerveau du patient pour en activer certaines zones.
* 254 Titre II du livre I er de la cinquième partie du code de la santé publique.
* 255 Titre I er du livre II de la cinquième partie du code de la santé publique.
* 256 Recours aux techniques biomédicales en vue de « neuro-amélioration » chez la personne non-malade : enjeux éthiques Comité consultatif nationale d'éthique pour les sciences de la vie et de la santé, avis n° 122, 12 février 2012. Cet avis est consultable à l'adresse suivante :
https://www.ccne-ethique.fr/sites/default/files/publications/ccne.avis_ndeg122.pdf
* 257 Avis n° 129 précité, p. 94.
* 258 « Toute expérimentation sur l'embryon est interdite » (article L. 152-8 du code de la santé publique, abrogé pour renumérotation par l'ordonnance n° 200-548 du 15 juin 2000).
* 259 Loi n° 94-654 du 29 juillet 1994 relative au don et à l'utilisation des éléments et produits du corps humain, à l'assistance médicale à la procréation et au diagnostic prénatal.
* 260 Loi n° 2004-800 du 6 août 2004 relative à la bioéthique.
* 261 Loi n° 2011-814 du 7 juillet 2011 relative à la bioéthique.
* 262 Pertinence scientifique ; permettre des progrès médicaux majeurs ; impossibilité de parvenir au résultat escompté par d'autres moyens ; respect des principes éthiques relatifs à ce type de recherche.
* 263 Loi n° 2013-715 du 6 août 2013 tendant à modifier la loi n° 2011-814 du 7 juillet 2011 relative à la bioéthique en autorisant sous certaines conditions la recherche sur l'embryon et les cellules souches embryonnaires.
* 264 La proposition de loi avait été déposée par notre ancien collègue Jacques Mézard et des membres du groupe RDSE.
* 265 Loi n° 2016-41 du 26 janvier 2016 de modernisation de notre système de santé.
* 266 http://hpscreg.eu/ .
* 267 Premier alinéa de l'article L. 2151-2 du code de la santé publique.
* 268 Second alinéa de l'article L. 2151-2 du code de la santé publique dans sa rédaction prévue par l'article 17 du projet de loi.
* 269 Art. L. 2151-3 du code de la santé publique.
* 270 Art. L. 2151-4 du code de la santé publique.
* 271 Art. 16 du code civil.
* 272 Art. 16 du code civil.
* 273 Articles 16-1 et 16-5 du code civil.
* 274 Article 16-3 du code civil.
* 275 Article 16-4 du code civil.
* 276 Article 16-4 du code civil.
* 277 Article 16-4 du code civil.
* 278 Article 16-6 du code civil.
* 279 Article 16-8 du code civil.
* 280 Décision n° 2013-674 du 1 er août 2013 relative à la loi tendant à modifier la loi n° 2011-814 du 7 juillet 2011 relative à la bioéthique en autorisant sous certaines conditions la recherche sur l'embryon et les cellules souches embryonnaires.
* 281 Dernier alinéa de l'article L. 2131-4 du code de la santé publique.
* 282 Avant-dernier alinéa de l'article L. 2141-3 du code de la santé publique.
* 283 Donnant lieu aux trois feuillets : neurectoderme, endoderme et mésoderme.
* 284 Actuel article L. 2151-6 du même code.
* 285 En application de l'article L. 2142-1 du code de la santé publique.
* 286 En application du 2° du II de l'article L. 2141-4 du code de la santé publique.
* 287 Pour ces recherches, une décision d'opposition du directeur général de l'agence devra être précédée d'un avis public de son conseil d'orientation.
* 288 « Clustered Regular Interspaced Short Palindromic Repeats » (« courtes répétitions palindromiques regroupées et régulièrement espacées »), correspondant aux « ciseaux génétiques ». Cas9 : « CRISPR associated protein 9 ».
* 289 Article 18 de la convention d'Oviedo, 2 nd alinéa : « La constitution d'embryons humains aux fins de recherche est interdite. »
* 290 Le plus souvent les fibroblastes de la peau et les cellules du sang.
* 291 66 % des embryons ne faisant plus l'objet d'un projet parental ; 9 % du total des embryons en cours de conservation.
* 292 34 % des embryons ne faisant plus l'objet d'un projet parental ; 5 % du total des embryons en cours de conservation.
* 293 Les centres sont ainsi souvent amenés à renvoyer une lettre recommandée avec accusé de réception lorsque la confirmation n'a pas été donnée.
* 294 Devenant l'article L. 2151-9 du même code, par l'article 14 du projet de loi.
* 295 Conseil d'État, Révision de la loi de bioéthique : quelles options pour demain ? , étude à la demande du Premier ministre adoptée en assemblée générale le 28 juin 2018.
* 296 Compte tenu des restrictions applicables en la matière aux États-Unis.
* 297 Posé par l'article 16-10 du code civil.
* 298 Des échantillons biologiques ont en effet pu être prélevés à l'issue de soins médicaux ou dans le cadre de recherches cliniques.
* 299 Conseil d'État, Révision de la loi de bioéthique : quelles options pour demain ? , étude à la demande du Premier ministre adoptée en assemblée générale le 28 juin 2018.
* 300 L'article 9 du projet de loi prévoit que l'article L. 1131-1-1 du code de la santé publique est désormais consacré aux modalités de transmission d'une information d'ordre médical entre des personnes dont le lien de filiation biologique a été rompu (tiers donneur et personne née d'un don).
* 301 La CNIL considère ainsi que le droit en vigueur semble écarter du dispositif dérogatoire au consentement les examens génétiques présentant un risque de ré-identification élevé, comme les séquençages complets du génome.
* 302 Loi n° 78-17 du 6 janvier 1978 relative à l'informatique, aux fichiers et aux libertés.
* 303 Délibération n° 2019-097 du 11 juillet 2019 portant avis sur un projet de loi relatif à la bioéthique.
* 304 Art. L. 1111-6 du code de la santé publique : « Elle rend compte de la volonté de la personne. »
* 305 « [...] La personne protégée prend seule les décisions relatives à sa personne dans la mesure où son état le permet.
Lorsque l'état de la personne protégée ne lui permet pas de prendre seule une décision personnelle éclairée, le juge ou le conseil de famille s'il a été constitué peut prévoir qu'elle bénéficiera, pour l'ensemble des actes relatifs à sa personne ou ceux d'entre eux qu'il énumère, de l'assistance de la personne chargée de sa protection. [...] »
* 306 Comme le souligne l'étude d'impact, les risques liés aux gestes de chirurgie foetale ont notamment diminué grâce à la foetoscopie (introduction d'une fibre optique et d'instruments dans l'utérus à travers la paroi abdominale maternelle par une ou deux mini-incisions) et au guidage par échographie.
* 307 Décret n° 2014-32 du 14 janvier 2014 relatif aux diagnostics anténataux.
* 308 Rapport médical et scientifique.
* 309 Dans les deux tiers des cas, l'examen est prescrit dans le cadre d'un antécédent familial, dans les autres cas c'est un signe d'appel échographique qui justifie l'examen.
* 310 Cette utilisation s'inscrit essentiellement, à ce jour, dans le cadre d'un signe d'appel échographique. Contrairement au caryotype, cet examen n'est pas à la nomenclature des actes de biologie médicale. Il peut être réalisé par des laboratoires autorisés pour le diagnostic génétique moléculaire prénatal ou le diagnostic cytogénétique prénatal.
* 311 A l'heure actuelle, ces bonnes pratiques sont fixées par l'arrêté du 1 er juin 2015 déterminant les recommandations de bonnes pratiques relatives aux modalités d'accès, de prise en charge des femmes enceintes et des couples, d'organisation et de fonctionnement des centres pluridisciplinaires de diagnostic prénatal en matière de diagnostic prénatal et de diagnostic préimplantatoire.
* 312 Un amendement du rapporteur Jean-François Eliaou, visant à supprimer ce même dispositif, avait été rejeté lors de l'examen du texte par la commission spéciale, faisant alors l'objet d'un avis défavorable du Gouvernement.
* 313 Ce dispositif avait été introduit dans le projet de loi à l'initiative du rapporteur du texte à l'Assemblée nationale.
* 314 Human leukocyte antigens (HLA) : antigènes des leucocytes humains.
* 315 Selon les indications de l'Agence de la biomédecine.
* 316 Source : Agence de la biomédecine, rapport sur l'application de la loi de bioéthique, janvier 2018.
* 317 L'évaluation de l'application de la loi n° 2011-814 du 7 juillet 2011 relative à la bioéthique, Office parlementaire d'évaluation des choix scientifiques et technologiques, 25 octobre 2018.
* 318 L'article R. 160-2 du code de la sécurité sociale prévoit que les caisses d'assurance maladie ne peuvent procéder au remboursement des frais de soins dispensés à l'étranger que sur autorisation préalable et lorsque la prise en charge des soins envisagés est prévue par la réglementation française.
* 319 D'après les indications de la direction générale de la santé, le typage HLA est autorisé dans onze pays en Europe : Belgique, Danemark, Espagne, France, Grèce, Norvège, Portugal, République tchèque, République du Bélarus, Royaume-Uni, Suède.
* 320 Art. L. 1418-1 du code de la santé publique.
* 321 Loi n° 94-654 du 29 juillet 1994 relative au don et à l'utilisation des éléments et produits du corps humain, à l'assistance médicale à la procréation et au diagnostic prénatal.
* 322 Il s'agit des centres de Strasbourg, Montpellier, Nantes et Paris ; le cinquième, situé à Grenoble, a été autorisé en octobre 2017. Les praticiens de ces centres sont agréés individuellement par l'Agence.
* 323 Une contribution commune a été relayée en ce sens par la Société de médecine de la reproduction, la Société française de gynécologie, le Collège national des gynécologues et obstétriciens français, le groupe d'études de la FIV en France et le groupe d'étude pour le don d'ovocyte.
* 324 Compte rendu de la table ronde du 27 novembre 2019.
* 325 Un embryon peut en effet comporter à la fois des cellules présentant des anomalies chromosomiques et d'autres non, d'où une situation dite de « mosaïque ».
* 326 Les spécialistes entendus ont indiqué en effet que le risque d'anomalies chromosomiques était corrélé à l'âge de la femme, avec un risque plus élevé à partir de 35 ans.
* 327 Amyotrophie spinale proximale liée au gène SMN1.
* 328 « Le défi de la prise en charge des VDS la relation enfant- parents » par le Pr Rémi Besson du centre hospitalo-universitaire de Lille.
* 329 « Révision de la loi de bioéthique : quelles options pour demain ? », Conseil d'État, section du rapport et des études, 28 juin 2018.
* 330 Avis du Défenseur des droits n° 19-13 du 20 décembre 2019.
* 331 Rapport d'information de Mmes Maryvonne Blondin et Corinne Bouchoux, fait au nom de la délégation aux droits des femmes n° 441 (2016-2017) - 23 février 2017.
* 332 Sentiment d'inadéquation de la personne entre le sexe social de sa naissance et son identité de genre, soit le sexe auquel elle se sent appartenir.
* 333 « Le droit des enfants à l'intégrité physique », Résolution 1952 (2013).
* 334 « Droits de l'homme et personnes intersexes », document thématique, juin 2015.
* 335 Amendement n° 2334 de M. Raphaël Gérard et les autres membres du groupe La République en Marche, sous-amendé par les amendements n° 2624 de M. Jean-Louis Touraine et Mme Laurence Vanceunebrock-Mialon et n° 2636 de Mme Maud Petit.
* 336 Cette question, de même que la fin de vie, était dans le périmètre de la demande d'étude du Premier ministre au Conseil d'Etat, formulée par sa lettre du 6 décembre 2017.
* 337 « Les actes de prévention, d'investigation ou de traitements et de soins ne doivent pas, en l'état des connaissances médicales, lui faire courir de risques disproportionnés par rapport au bénéfice escompté ».
* 338 Précision apportée par les amendements n° 2624 de M. Jean-Louis Touraine et Mme Laurence Vanceunebrock-Mialon et n° 2636 de Mme Maud Petit.
* 339 Note d'information interministérielle n° DGOS/DIR/DGRI/2018/218 du 19 septembre 2018 relative aux filières de santé, aux centres de référence et aux plateformes d'expertise et outre-mer dédiés aux maladies rares.
* 340 Arrêté du 9 mai 2017 de labellisation de centres de références pour une maladie rare ou un groupe de maladies rares.
* 341 Chiffre communiqué par l'association Surrénales.
* 342 Avis n° 132 du 19 septembre 2019 « Questions éthiques soulevées par la situation des personnes ayant des variations du développement sexuel ».
* 343 Avis n° 132 précité.
* 344 Article 3 de l'ordonnance n° 2008-480 du 22 mai 2008 transposant en matière de don de gamètes et d'assistance médicale à la procréation la directive 2004/23/ CE du Parlement européen et du Conseil du 31 mars 2004.
* 345 Source : rapport médical et scientifique.
* 346 En particulier des tissus ovariens, par le prélèvement du fragment d'ovaire contenant la réserve de follicules, ou des tissus testiculaires chez des patients prépubères.
* 347 Groupe d'étude sur la cryoconservation de l'ovaire et du testicule.
* 348 Certaines greffes ont également permis la naissance d'enfants sans qu'il soit nécessaire de recourir à une AMP. Ces données ne font toutefois pas l'objet d'un recueil par l'Agence de la biomédecine.
* 349 La rédaction de ces articles est issue du décret n° 2016-273 du 4 mars 2016 relatif à l'assistance médicale à la procréation, transposant des directives européennes.
* 350 Seul l'arrêté du 17 juin 2017 relatif aux règles de bonnes pratiques cliniques et biologiques d'assistance médicale à la procréation a prévu en ce sens qu'au moment du consentement, « le patient et/ou son représentant légal s'engage à tenir l'équipe informée de tout changement sur sa situation personnelle et son adresse et à répondre aux courriers adressés annuellement par le laboratoire. »
* 351 Biologistes des laboratoires d'étude de la fécondation et de la conservation de l'oeuf.
* 352 Le rapporteur a indiqué toutefois en séance publique y donner à titre personnel un avis de sagesse.
* 353 Aux termes de l'article L. 458 du code civil, « sont réputés strictement personnels la déclaration de naissance d'un enfant, sa reconnaissance, les actes de l'autorité parentale relatifs à la personne d'un enfant, la déclaration du choix ou du changement du nom d'un enfant et le consentement donné à sa propre adoption ou à celle de son enfant. »
* 354 « Préservation de la fertilité et cancer », rapport conjoint de l'Institut national du Cancer (INCa) et de l'Agence de la biomédecine, 19 février 2013.
* 355 L'article R. 2141-17 du code de la santé publique prévoit à l'heure actuelle qu'il est mis fin à la conservation si la personne n'ayant pas répondu à la consultation « n'est plus en âge de procréer ». La limite d'âge pour l'accès à l'AMP ne serait toutefois plus pertinent dès lors que la finalité de la conservation est étendue à la restauration d'une fonction hormonale.
* 356 Loi n° 2004-806 du 9 août 2004 relative à la politique de santé publique.
* 357 Arrêté du 10 avril 2008 relatif à l'autorisation d'exercice de la profession de conseiller en génétique.
* 358 Article 2 du décret n° 2007-1429 du 3 octobre 2007 relatif à la profession de conseiller en génétique et modifiant le code de la santé publique.
* 359 Bien qu'il ait été retiré dans un premier temps par Mme Mauborgne, l'amendement a été ensuite repris par le député Philippe Vigier (UDI).
* 360 Par prélèvement de cellules chorioniques, constitutives du futur placenta.
* 361 Réécrit par l'article 10 du projet de loi.
* 362 Réaction du greffon contre l'hôte (« Graft versus host disease »).
* 363 Qui définit un médicament comme « toute substance ou composition présentée comme possédant des propriétés curatives ou préventives à l'égard des maladies humaines ou animales, ainsi que toute substance ou composition pouvant être utilisée chez l'homme ou chez l'animal ou pouvant leur être administrée, en vue d'établir un diagnostic médical ou de restaurer, corriger ou modifier leurs fonctions physiologiques en exerçant une action pharmacologique, immunologique ou métabolique. »
* 364 Art. L. 5121-1 du code de la santé publique.
* 365 Art. L. 5121-5 du code de la santé publique.
* 366 Art. L. 5121-1-1 du code de la santé publique.
* 367 Art. L. 5121-12 du code de la santé publique.
* 368 Loi n° 2011-302 du 22 mars 2011 portant diverses dispositions d'adaptation de la législation au droit de l'Union européenne en matière de santé, de travail et de communications électroniques.
* 369 Directive 2001/83/CE du Parlement européen et du Conseil du 6 novembre 2001 modifié.
* 370 Et sur la base des précisions apportées par le décret n° 2016-1536 du 15 novembre 2016 relatif aux médicaments de thérapie innovante, qui a précisé aux articles R. 4211-37 à R. 4211-42 les conditions d'autorisation des établissements (en termes d'aménagement des locaux, de qualification du personnel ou encore de conformité du matériel).
* 371 L'article R. 2141-1 du code de la santé publique précise que « Les procédés biologiques utilisés en assistance médicale à la procréation (...) s'entendent des méthodes de préparation et de conservation des gamètes et tissus germinaux, de fécondation in vitro et de conservation des embryons, que ce soit à des fins d'assistance médicale à la procréation ou de préservation de la fertilité. »
L'arrêté de 2012 a défini comme suit ces procédés biologiques régulièrement utilisés à la date de publication de la loi du 7 juillet 2011 : 1° Préparation de sperme en vue d'assistance médicale à la procréation ; 2° Fécondation in vitro sans micromanipulation ; 3° Fécondation in vitro avec micromanipulation ; 4° Congélation des gamètes ; 5° Congélation des tissus germinaux ; 6° Congélation des zygotes et des embryons ; 7° Maturation in vitro des ovocytes.
* 372 Loi n° 2017-220 du 23 février 2017 ratifiant l'ordonnance n° 2016-966 du 15 juillet 2016 portant simplification de procédures mises en oeuvre par l'Agence nationale de sécurité du médicament et des produits de santé et comportant diverses dispositions relatives aux produits de santé.
* 373 Rapport d'information fait au nom de la mission d'information sur la révision de la loi relative à la bioéthique, n° 1572, enregistré à la Présidence de l'Assemblée nationale le 15 janvier 2019.
* 374 Article 6 ter de l'ordonnance n° 58-1100 du 17 novembre 1958 relative au fonctionnement des assemblées parlementaires.
* 375 Voir les articles 21 de la loi n° 94-654 du 29 juillet 1994 relative au don et à l'utilisation des éléments et produits du corps humain, à l'assistance médicale à la procréation et au diagnostic prénatal et 47 de la loi n° 2011-814 du 7 juillet 2011 relative à la bioéthique.
* 376 Rapport n° 80 (2018-2019) de M. Jean-François Eliaou, député et Mme Annie Delmont-Koropoulis, sénatrice, fait au nom de l'Office parlementaire d'évaluation des choix scientifiques et technologiques, déposé le 25 octobre 2018
* 377 Loi n° 2016-87 du 2 février 2016 créant de nouveaux droits en faveur des malades et des personnes en fin de vie.
* 378 Rapport n° 644 (2018-2019) de Mme Annie Delmont-Koropoulis, sénatrice et M. Jean-François Eliaou, député, fait au nom de l'Office parlementaire d'évaluation des choix scientifiques et technologiques, déposé le 9 juillet 2019.
* 379 C'est dans cet esprit qu'il a récemment refusé la création d'une délégation parlementaire à la sécurité économique ; voir le rapport n° 254 (2018-2019) de MM. Michel Canevet, Jean-François Husson et Mme Élisabeth Lamure, fait au nom de la commission spéciale, déposé le 17 janvier 2019.
* 380 Compte rendu de la réunion du Bureau du Sénat du mercredi 11 mars 2015 : https://www.senat.fr/role/fiche/bur_cr_reunion110315.html
* 381 Art. R. 1412-3 du code de la santé publique.
* 382 S'agissant des organes consultés par le CCNE sur l'organisation des états généraux devant précéder tout projet de réforme en matière de bioéthique et sur la question de savoir s'il est opportun pour le Gouvernement de mobiliser la commission nationale du débat public.
* 383 Loi n° 2018-699 du 3 août 2018 visant à garantir la présence des parlementaires dans certains organismes extérieurs au Parlement et à simplifier les modalités de leur nomination.
* 384 Il en avait été de même avec la loi de bioéthique de juillet 2011, étendue adaptée à la Nouvelle-Calédonie, la Polynésie Française et Wallis-et-Futuna par l'ordonnance du 18 avril 2012.
* 385 Un dispositif médical de diagnostic in vitro est défini comme tout dispositif médical, utilisé seul ou en association, destiné par le fabricant à être utilisé in vitro dans l'examen d'échantillons provenant du corps humain, uniquement ou principalement dans le but de fournir des informations sur un état physiologique ou pathologique, des déficiences congénitales physiques ou mentales, la prédisposition à une affection ou à une maladie, permettant de prévoir la réponse ou les réactions à un traitement ou de définir ou contrôler des mesures thérapeutiques.
* 386 Directives 90/385 du 20 juin 1990 et 93/42/CEE du 14 juin 1993.
* 387 Directive 98/79CE du 27 octobre 1998.
* 388 Relèvent de la classe III d'après le règlement 2017/745 « les dispositifs non invasifs consistant en une substance ou un mélange de substances et destinés à une utilisation in vitro en contact direct avec des cellules, tissus ou organes humains prélevés dans le corps humain ou utilisés in vitro avec des embryons humains avant leur implantation ou leur administration dans le corps. »
* 389 La mise en cohérence de la législation nationale avec le règlement de 2007 a été principalement faite par la loi n° 2011-302 du 22 mars 2011 portant diverses dispositions d'adaptation de la législation au droit de l'Union européenne en matière de santé, de travail et de communications électroniques.
* 390 Proposition de loi n° 105 relative à l'évaluation éthique de la recherche impliquant la personne humaine, présentée par Catherine Deroche, enregistrée le 7 novembre 2019.
* 391 Loi n° 2012-300 du 5 mars 2012 relative aux recherches impliquant la personne humaine.
* 392 Cette habilitation votée contre l'avis du Sénat a donné lieu à l'ordonnance n° 2016-800 du 16 juin 2016 relative aux recherches impliquant la personne humaine.
* 393 Aucun principe de révision ne figurait dans la loi n° 94-653 du 29 juillet 1994 relative au respect du corps humain, qui fixât les grands principes tels l'inviolabilité ou la non patrimonialité du corps humain.
* 394 La rapporteure Laëtitia Romeiro Dias a indiqué toutefois y être favorable à titre personnel.
* 395 « La politique des greffes : une chaîne de la greffe fragile à mieux organiser », Cour des comptes, rapport sur l'application des lois de sécurité sociale, septembre 2019.